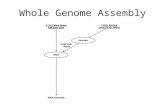
Whole Genome Assembly
32
Whole Genome Assembly
-
Upload
richard-maxwell -
Category
Documents
-
view
49 -
download
2
description
Whole Genome Assembly. WGA. 1. Screener 2. Overlapper 3. Unitigger, 4. Scaffolder, 5. Repeat Resolver. Overlapper. ...looks for end-to end overlaps of at least 40 bp with no more than 6% differences in match. What’s the significance?. ...a one in 10 17 event. - PowerPoint PPT Presentation
Transcript of Whole Genome Assembly

Whole Genome Assembly

WGA
1. Screener
2. Overlapper
3. Unitigger,
4. Scaffolder,
5. Repeat Resolver.

Overlapper
...looks for end-to end overlaps of at least 40 bp with no more than 6% differences in match.
What’s the significance? ...a one in 1017 event.
Sequencing Fidelity: 99.96%

However
...the Screener doesn’t include all of the “low frequency” level repeats,
...so, a majority of the Overlapper outputs are bogus.

Unitigger
...differentiates between a true overlap, and an overlap that includes more than one loci.

8X
...over-collapsed.
...in a world where real data matches expected data, each loci would have 8X coverage,
...if there were repeats, then contigs would be “over-represented”, on average 8 more per repeat.

What Now?
... uniquely assembled contigs (unitigs) are readily identifiable,
– all of the assembled sequences match over all of the known sequence,
- and -
...are consistent with an 8x coverage.

Unitigs
...contig cluster is consistent with expected size,
...no dissimilar sequences between any members.
...all other contigs are sent to the Discriminator.

Discriminator
...parses the “over-collapsed” contig by using sequence outside of the overlap region

Discriminator
...may yield unitigs.

Unitigger Output
...correctly assembled contigs covering 73.6% of the genome.

Repeat Resolver
...most of the remaining gaps were due to repeats.
1. Allow “low Discriminator Value” contigs to fill gaps,
2. Find BAC sequences that unambiguously match outside the nearest unitig,
– 1 in 107 chance of being wrong,
3. Ensure the mate end sequence of candidate BACs match.

If that Doesn’t Work
...find a mate-pair that spans the gap, and sequence it,
Chromosome Walking
...make sequencing primer from BES...

Scaffolder
...contigs the contigs,
– uses mate-pair information.

WGA Result
...91% sequence, 9% gaps,

Compartmentalized Shotgun Assembly
Mapping

Scaffolds

Sequence Tagged Sites STS
...PCR primers are designed for unique regions of the genome or chromosome,
...the chromosome is cut ,
...assay two PCR products, frequency of co-amplification indicates .

Sequence Tagged Sites STS

Compartmentalized Shotgun Assembly
...ideally 24,
...really 3845.

92.2 % Sequence
7.8 % Gaps
CSA
91 % Sequence
9 % Gaps
WPA

PFP
Chromosome 21
CSA
Green: Same Order,
Orientation Yellow: Same
Orientation
Red: Out of Order, Orientation
Blue: GapsViolations:Red : misorientedYellow: distance

Chromosome 8
PFP
CSA

PFP
CSA

Major Public Sequence Databases

• 281 Curated Data Bases,
• “... facilitating Biological Discovery”.

What Do We Know?(based on functional group analysis)
Science 291 (5507), 1304-1351

Functional Groups
1st GenBank NR protein database was partitioned into clusters using BLASTP,

Describing Aligned Sequences
2nd Statistical descriptions of the cluster are developed and tested,
• Hidden-Markov Markers: statistical descriptions of aligned sequences.

Functional Group Annotation
3rd Categorization was done by manual review of the family and subfamily names,
...by examining SwissProt and GenBank records,
...and by review of the literature as well as resources on the World Wide Web.
http://www.expasy.ch/cgi-bin/niceprot.pl?P29965

Outcomes?
• A relatively small number of structural and functional domains are used in a large number of different proteins,
• Pfam: 527 families,
• average length is 275 residues,
• 456 had “annotated functions”.
Nucleic Acids Research 26, 320-322

New Genes
4th Newly sequenced genes are virtually translated, and the
predicted proteins are assayed against raw and HMM databases,
...significance cut-off levels are determined for each functional
group family.