
WHO GENERAL GUIDANCE ON VARIATIONS TO … variations to multisource 5 pharmaceutical products 6 7...
24
Working document QAS/14.575 February 2014 Draft for comment 1 2 WHO GENERAL GUIDANCE ON 3 VARIATIONS TO MULTISOURCE 4 PHARMACEUTICAL PRODUCTS 5 6 (February 2014) 7 8 DRAFT FOR COMMENT 9 10 11 12 13 14 15 16 17 18 © World Health Organization 2014 19 All rights reserved. 20 This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft 21 may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in 22 any form or by any means outside these individuals and organizations (including the organizations' concerned staff and 23 member organizations) without the permission of the World Health Organization. The draft should not be displayed on any 24 website. 25 Please send any request for permission to: 26 Dr Sabine Kopp, Group Lead, Medicines Quality Assurance, Technologies, Standards and Norms, Department of Essential 27 Medicines and Health Products, World Health Organization, CH-1211 Geneva 27, Switzerland. Fax: (41-22) 791 4730; 28 email: [email protected]. 29 The designations employed and the presentation of the material in this draft do not imply the expression of any opinion 30 whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or 31 of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate 32 border lines for which there may not yet be full agreement. 33 The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or 34 recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors 35 and omissions excepted, the names of proprietary products are distinguished by initial capital letters. 36 All reasonable precautions have been taken by the World Health Organization to verify the information contained in this 37 draft. However, the printed material is being distributed without warranty of any kind, either expressed or implied. The 38 responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health 39 Organization be liable for damages arising from its use. 40 This draft does not necessarily represent the decisions or the stated policy of the World Health Organization. 41 42 Should you have any comments on the attached revision, please send these to Dr S. Kopp, Group Lead, Medicines Quality Assurance, Technologies, Standards and Norms ([email protected]) with a copy to Ms Marie Gaspard ([email protected]) by 7 April 2014. Our working documents will be sent out electronically only and will also be placed on the Medicines website for comment under “Current projects”. If you do not already receive our draft working documents please let us have your email address (to [email protected]) and we will add it to our electronic mailing list.
Transcript of WHO GENERAL GUIDANCE ON VARIATIONS TO … variations to multisource 5 pharmaceutical products 6 7...

Working document QAS/14.575
February 2014
Draft for comment
1
2
WHO GENERAL GUIDANCE ON 3
VARIATIONS TO MULTISOURCE 4
PHARMACEUTICAL PRODUCTS 5
6
(February 2014) 7
8
DRAFT FOR COMMENT 9
10
11
12
13
14
15
16
17
18
© World Health Organization 2014 19
All rights reserved. 20
This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft 21 may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in 22 any form or by any means outside these individuals and organizations (including the organizations' concerned staff and 23 member organizations) without the permission of the World Health Organization. The draft should not be displayed on any 24 website. 25
Please send any request for permission to: 26
Dr Sabine Kopp, Group Lead, Medicines Quality Assurance, Technologies, Standards and Norms, Department of Essential 27 Medicines and Health Products, World Health Organization, CH-1211 Geneva 27, Switzerland. Fax: (41-22) 791 4730; 28 email: [email protected]. 29
The designations employed and the presentation of the material in this draft do not imply the expression of any opinion 30 whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or 31 of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate 32 border lines for which there may not yet be full agreement. 33
The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or 34 recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors 35 and omissions excepted, the names of proprietary products are distinguished by initial capital letters. 36
All reasonable precautions have been taken by the World Health Organization to verify the information contained in this 37 draft. However, the printed material is being distributed without warranty of any kind, either expressed or implied. The 38 responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health 39 Organization be liable for damages arising from its use. 40
This draft does not necessarily represent the decisions or the stated policy of the World Health Organization. 41 42
Should you have any comments on the attached revision, please send these to Dr S. Kopp, Group
Lead, Medicines Quality Assurance, Technologies, Standards and Norms ([email protected]) with a
copy to Ms Marie Gaspard ([email protected]) by 7 April 2014.
Our working documents will be sent out electronically only and will also be placed on the
Medicines website for comment under “Current projects”. If you do not already receive our
draft working documents please let us have your email address (to [email protected]) and we
will add it to our electronic mailing list.
Working document QAS/14.575
page 2
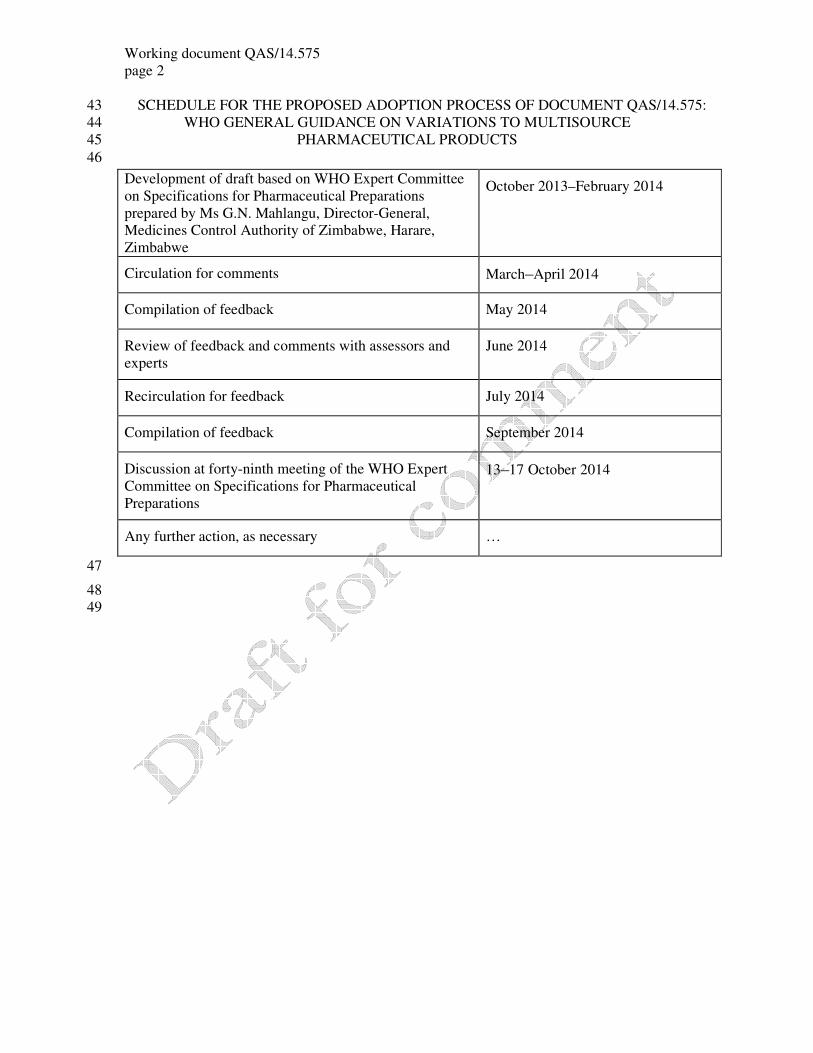
SCHEDULE FOR THE PROPOSED ADOPTION PROCESS OF DOCUMENT QAS/14.575: 43
WHO GENERAL GUIDANCE ON VARIATIONS TO MULTISOURCE 44
PHARMACEUTICAL PRODUCTS 45
46
47
48
49
Development of draft based on WHO Expert Committee
on Specifications for Pharmaceutical Preparations
prepared by Ms G.N. Mahlangu, Director-General,
Medicines Control Authority of Zimbabwe, Harare,
Zimbabwe
October 2013–February 2014
Circulation for comments March–April 2014
Compilation of feedback May 2014
Review of feedback and comments with assessors and
experts
June 2014
Recirculation for feedback July 2014
Compilation of feedback September 2014
Discussion at forty-ninth meeting of the WHO Expert
Committee on Specifications for Pharmaceutical
Preparations
13–17 October 2014
Any further action, as necessary …

Working document QAS/14.575
page 3
Contents 50
51
page 52
53
1. Introduction ……………………………………………………………………………... 54
1.1 Objectives ………………………………………………………………………. 55
1.2 Scope and application of the guideline …………………………………………. 56
2. Glossary …………………………………………………………………………………. 57
3. General considerations ………………………………………………………………….. 58
3.1 Reporting types …………………………………………………………………. 59
3.1.1 Notification ……………………………………………………………… 60
3.1.2 Minor variation ………………………………………………………….. 61
3.1.3 Major variation ………………………………………………………….. 62
3.2 New applications and extension applications …………………………………… 63
3.3 Labelling information …………………………………………………………… 64
3.4 Conditions to be fulfilled ……………………………………………………….. 65
3.5 Documentation required ………………………………………………………… 66
4. General stability considerations ………………………………………………………… 67
5. Comparative studies …………………………………………………………………….. 68
5.1 Comparative in vivo studies …………………………………………………….. 69
5.2 Comparative in vitro studies …………………………………………………….. 70
5.3 Variations (changes) to pharmaceutical aspects of registered products which … 71
may be made without prior approval 72
5.3.1 Annual notification ………………………………………………………. 73
5.3.2 Immediate notification – “Do and tell” ………………………………….. 74
5.4 Variations (changes) to pharmaceutical aspects of registered products which … 75
require prior approval before implementation 76
5.4.1 Minor variation …………………………………………………………... 77
5.4.2 Major variation …………………………………………………………… 78
79
80
81
82
83

Working document QAS/14.575
page 4
1. INTRODUCTION 84
85
This guidance document is intended to provide supportive information on how to present an 86
application to implement a change to a registered multisource medicinal product. A marketing 87
authorization holder or applicant is responsible for the safety, efficacy and quality of a 88
medicinal product that is placed on the market throughout its life-cycle. As such, changes are 89
required or necessary for an approved or registered product to account for technical and 90
scientific progress, to improve the quality of the medicinal product, to meet market 91
requirements such as scale-up or additional manufacturing sites, or updates to product 92
information (e.g. updates to information on adverse reactions). The common areas for change 93
are pharmaceutical aspects (quality control, source of raw materials, manufacturing, shelf-life 94
etc.) and product information. Such changes, regardless of the nature of the change, are 95
referred to as variations and may require the approval of national medicines regulatory 96
authority (NMRA) prior to implementation. 97
98
Technical requirements for the different types of variations are set out in these guidelines in 99
order to facilitate the submission of appropriate documentation by applicants and their 100
assessment by NMRAs. 101
102
1.1 Objectives 103
104
This guidance document is intended to assist applicants with the classification of changes 105
made to a registered or approved finished pharmaceutical product (FPP) and provide 106
guidance on the technical and other general data requirements to support changes to the 107
quality attributes of the active pharmaceutical ingredient (API) or FPP. Variation applications 108
are categorized into major variation, minor variation (prior approval) and minor variation 109
(notification). 110
111
1.2 Scope and application of the guideline 112
113
These guidelines can be used by both NMRAs and applicants with respect to changes to the 114
quality sections of product dossiers for an API or an FPP. This guidance should be read in 115
conjunction with the Guidelines on submission of documentation for a multisource finished product 116

Working document QAS/14.575
page 5
(1) and Guidance on variations (2) as well as other related WHO guidelines or applicable 117
national guidelines. 118
119
The NMRAs reserve the rights to request for additional information where necessary, in line 120
with national requirements. 121
122
This guidance document is applicable only to APIs and excipients manufactured by chemical 123
synthesis or semisynthetic processes and FPPs containing such APIs and excipients. APIs 124
produced by fermentation and APIs of biological, biotechnological or herbal origin are treated 125
as special cases. 126
127
It is recommended that the principles established in this guidance document be applied to 128
similar quality changes that occur during the development of the product and that the 129
recommended supporting data be included with the initial application for registration of a 130
medicinal product. 131
132
When a variation leads to a revision of the package insert, the patient information leaflet 133
(PIL) and labelling,1 the updated product information should be submitted as part of the 134
application. 135
136
For variations that require generation of stability data on the API or FPP, the stability studies 137
required, including commitment batches, should always be continued to cover the currently 138
accepted retest or shelf-life period. The NMRAs should be informed immediately if any 139
problems with the stability of APIs or FPPs occur during storage, e.g. if found to be outside 140
specifications or potentially outside specifications. 141
142
Applicants should be aware that some variations might require the submission of additional 143
consequential variations. Therefore for any given change the applicant should consider 144
whether one or more variations might be required to be submitted. 145
146
If changes to the dossier only concern editorial changes, such changes need not be submitted 147
as a separate variation but can be included as a notification together with a subsequent 148
1 Different regions/countries use different terminology for product information. In this document package insert,
PIL and label are used to refer to product information.

Working document QAS/14.575
page 6
variation concerning that part of the dossier. In such a case a declaration should be provided 149
that the contents of the associated sections of the dossier have not been changed by the 150
editorial changes beyond the substance of the variation submitted. 151
152
2. GLOSSARY 153
154
The definitions provided below apply to the terms used in this guidance. They may have 155
different meanings in other contexts and documents. 156
157
active pharmaceutical ingredient (API) 158
A substance used in the FPP, intended to furnish pharmacological activity or to otherwise 159
have direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease, or to 160
have direct effect in restoring, correcting or modifying physiological functions in human 161
beings. 162
163
active pharmaceutical ingredient (API) starting material 164
A raw material, intermediate, or an API that is used in the production of an API and that is 165
incorporated as a significant structural fragment into the structure of the API. An API starting 166
material can be an article of commerce, a material purchased from one or more suppliers 167
under contract or commercial agreement, or produced in-house. 168
169
applicant 170
For the purposes of this document, the term applicant refers to any person or entity that holds 171
the legal responsibility for the product on the market by submission of the required 172
documentation on a product that has been listed after evaluation as registered or approved. 173
174
biobatch 175
The batch used to establish bioequivalence or similarity to the comparator product as 176
determined in bioequivalence or biowaiver studies, respectively. 177
178
final intermediate 179
Product formed in the last reaction in the synthetic pathway that undergoes synthetic 180
transformation to the API or the crude API. Purification is not considered to be a synthetic 181
transformation. 182

Working document QAS/14.575
page 7
183
finished pharmaceutical product (FPP) 184
A finished dosage form of a pharmaceutical product, which has undergone all stages of 185
manufacture including packaging in its final container and labelling. 186
187
in-process control 188
Check performed during manufacture to monitor or to adjust the process in order to ensure 189
that the final product conforms to its specifications. 190
191
manufacturer 192
A company that carries out operations such as production, packaging, repackaging, labelling 193
and relabelling of pharmaceuticals. 194
195
multisource (generic) pharmaceutical product 196
Pharmaceutically equivalent products that may or may not be therapeutically equivalent. 197
198
officially recognized pharmacopoeia (or compendium) Those pharmacopoeias recognized by 199
the national regulatory agencies (e.g. national pharmacopoeia (if applicable), The 200
International Pharmacopoeia (Ph.Int.), the European Pharmacopoeia (PhEur.), the British 201
Pharmacopoeia (BP), the Japanese Pharmacopoeia (JP) and the United States Pharmacopeia 202
(USP)). 203
204
pilot-scale batch 205
A batch of an API or FPP manufactured by a procedure fully representative of and simulating 206
that to be applied to a full production-scale batch. For example, for solid oral dosage forms, a 207
pilot scale is generally, at a minimum, one-tenth that of a full production scale or 100 000 208
tablets or capsules, whichever is the larger, unless otherwise adequately justified. 209
210
production batch 211
A batch of an API or FPP manufactured at production scale by using production equipment in 212
a production facility as specified in the application. 213
214
register 215
A list of all the pharmaceutical products authorized for marketing in a particular country. The 216

Working document QAS/14.575
page 8
medicines regulatory authority of the country in question maintains the register. 217
218
registered medicines products 219
Pharmaceutical products that have a marketing authorization. 220
221
stringent regulatory authority (SRA) 222
A stringent regulatory authority is: 223
• the medicines regulatory authority in a country which is: (a) a member of the 224
International Conference on Harmonisation (ICH) (European Union (EU), Japan and the 225
United States of America); or (b) an ICH Observer, being the European Free Trade 226
Association (EFTA) as represented by Swiss Medic and Health Canada (as may be 227
updated from time to time); or (c) a regulatory authority associated with an ICH member 228
through a legally-binding, mutual recognition agreement including Australia, Iceland, 229
Liechtenstein and Norway (as may be updated from time to time); 230
• only in relation to good manufacturing practices (GMP) inspections: a medicines 231
regulatory authority that is a member of the Pharmaceutical Inspection Co-operation 232
Scheme (PIC/S) as specified at http://www.picscheme.org. 233
234
validation 235
The demonstration, with documentary evidence, that any procedure, process, equipment, 236
material, activity or system actually leads to the expected results. 237
238
variation 239
A change to any aspect of a pharmaceutical product, including but not limited to a change to 240
formulation, method and site of manufacture, specifications for the finished product and 241
ingredients, container and container labelling and product information. 242
243
3. GENERAL CONSIDERATIONS 244
245
3.1 Reporting types 246
247
The definitions outlined in the following reporting types are intended to provide guidance 248
with respect to the classification of quality-related changes. Specific examples of changes are 249
provided in these guidelines. However, it should be noted that a change not covered by these 250

Working document QAS/14.575
page 9
guidelines, should be considered as a major change by default. Whenever the applicant is 251
unclear about the classification of a particular change, the respective NMRA should be 252
contacted. It remains the responsibility of the applicant to submit relevant documentation to 253
justify that the change will not have a negative impact on the quality of the product. 254
255
Individual changes normally require the submission of separate variations. Grouping of 256
variations is acceptable only under the following circumstances: 257
258
• when variations are consequential to each other, e.g. introduction of a new impurity 259
specification that requires a new analytical procedure; 260
• when the same change affects multiple FPPs, e.g. addition of a new API 261
manufacturing site for multiple FPPs; 262
• when all the changes fall under annual notification. 263
264
Applicants are also advised to exercise caution whenever several changes to the same FPP are 265
envisaged. Although each of the individual changes may be classified as a particular reporting 266
type, classification within a higher-risk category may be warranted as a result of the 267
composite effect of these changes. In all such cases, applicants are advised to contact the 268
NMRA prior to submission of the variation application to obtain guidance on classifying such 269
changes. 270
271
3.1.1 Notifications 272
273
Notifications are changes that could have minimal or no adverse effects on the overall safety, 274
efficacy and quality of the FPP. Such notifications may not require prior acceptance, but must 275
be notified to the NMRA immediately after implementation, i.e. immediate notification (IN), 276
or within 12 months following implementation, i.e. annual notification (AN). 277
278
3.1.2 Minor variation 279
280
Minor variations are changes that may have minor effects on the overall safety, efficacy and 281
quality of the FPP. Applicants must satisfy themselves that they meet all of the prescribed 282
conditions for the change and submit all required documentation with the variation 283
application. 284

Working document QAS/14.575
page 10
285
Prior approval by the NMRA may be required, before the changes can be implemented. The 286
“timeline” and “implementation of the variation” are subject to the NMRA’s specific 287
proposals and should be made publicly available. 288
289
3.1.3 Major variation 290
291
Major variations are changes that could have major effects on the overall safety, efficacy and 292
quality of the FPP. In general, a change that is supported by extensive documentation and/or 293
requires extensive assessment of the supporting documentation would be considered as major 294
variation, e.g. a change supported by in vivo studies. Prior approval by the NMRA is required 295
before the changes can be implemented. The “timeline” and “implementation of the 296
variation” are subject to the NMRA’s specific proposals. 297
298
The NMRA reserves the right to recategorize the application type, where deemed appropriate. 299
Subject to country specific procedure, recategorization may require the applicant to resubmit 300
a new application or additional data according to the correct category. 301
302
3.2 New applications and extension applications 303
304
Certain changes are so fundamental that they alter the terms of the accepted dossier and 305
consequently cannot be considered as changes. In these cases a new dossier must be 306
submitted in line with applicable national requirements for applications for registration of 307
medicines. 308
309
Examples of such changes are listed below: 310
311
1. Change of the API to a different API. 312
2. Inclusion of an additional API in a multicomponent product. 313
3. Removal of one API from a multicomponent product. 314
4. Change in the dose and/or strength of one or more APIs. 315
5. Change from an immediate-release product to an extended or delayed-release dosage form or 316
vice versa. 317
6. Change from a liquid to a powder for reconstitution or vice versa. 318
7. Changes in the route of administration. 319

Working document QAS/14.575
page 11
320
3.3 Labelling information 321
322
For any change to labelling information (package insert, PIL, labels), the NMRA must be 323
notified and submission of the revised labelling information is expected as per country 324
specific proposal or requirements. 325
326
3.4 Conditions to be fulfilled 327
328
For each variation, attempts have been made to identify particular circumstances where lower 329
reporting requirements, e.g. notifications, are possible. A change that does not meet all of the 330
conditions stipulated under general and specific circumstances for the notifications are 331
automatically considered at the next higher level of change, i.e. require prior approval before 332
implementation. 333
334
3.5 Documentation required 335
336
All data recommended to support a change should be provided with the submission. 337
338
When recommended supporting data cannot be submitted, a detailed rationale should be 339
provided. Regardless of the documents specified, applicants should ensure that they have 340
provided all relevant information to support the variation. Additional documentation may be 341
required. For all changes it remains the responsibility of the applicant to provide all necessary 342
documents to demonstrate that the change does not have a negative impact on the safety, 343
efficacy or quality of the FPP. 344
345
Where applicable, the following should be included in the application for variations requiring 346
prior approval: 347
348
• a covering letter (including a list of changes describing each in sufficient detail to allow for a 349
quick assessment as to whether the appropriate reporting category has been used); 350
• section of the original dossier affected by the change(s); 351
• current and proposed condition(s); 352
• reason for the change(s); 353

Working document QAS/14.575
page 12
• where relevant, a side-by-side comparison of the previously approved and the proposed 354
information; 355
• replacement of the relevant sections of the dossier as per acceptable dossier format for the 356
respective NMRAs with the proposed changes clearly annotated; 357
• copies of package insert, PIL and labels, if relevant; 358
• registration status and date of the proposed change(s) in other countries/agencies that had 359
approved the variation(s), especially the country of origin and the reference agencies. 360
361
It should be noted that the NMRA reserves the right to request further information not 362
explicitly described in these guidelines. 363
364
Alternative approaches to the principles and practices described in this document may be 365
acceptable provided they are supported by adequate scientific justification. It is also important 366
to note that the NMRA may request information or material, or defines conditions not 367
specifically described in this guidance, in order to adequately assess the safety, efficacy and 368
quality of an FPP. 369
370
4. GENERAL STABILITY CONSIDERATIONS 371
372
The effect of the changes on an approved medicinal product on the stability of the medicinal 373
product should be evaluated. For general guidance on conducting stability studies, applicants 374
are referred to the WHO Guideline on Stability Studies [3]. For variation submissions, the 375
following points also should be considered: 376
• In most cases (except those involving scale up), stability data from pilot scale batches will be 377
acceptable to support the proposed change. 378
• Where stability data show a trend toward potency loss or degradant increase under accelerated 379
conditions, it is recommended that historical accelerated stability data from a representative 380
pre-change batch be submitted for comparison. It is also recommended that under these 381
circumstances, all available long-term data on test batches from ongoing studies be provided 382
in the supplement. Submission of historical accelerated and available long-term data would 383
facilitate review and approval of the supplement. 384
• A commitment should be included to conduct long-term stability studies through the 385
expiration-dating period, according to the approved protocol, on the first or first three 386
production batches and to report the results in the annual reports. 387
388

Working document QAS/14.575
page 13
5. COMPARATIVE STUDIES 389
390
5.1 Comparative in vivo studies 391
392
A number of changes outlined include recommendations for supporting comparative in vivo 393
studies (e.g. comparative bioavailability studies). 394
395
Applicants should consult the ICH Q5E guideline and applicable WHO guidance documents 396
when conducting comparative in vivo studies. 397
398
5.2 Comparative in vitro studies 399
400
A number of changes outlined include recommendations for supporting comparative in vitro 401
studies (e.g. comparative dissolution studies). Where an in vitro comparison is recommended 402
to support a variation, the comparison should be made to the product manufactured according 403
to the same formulation and manufacturing process used in the pivotal clinical and/or 404
comparative bioavailability studies approved for the original drug submission (e.g. including 405
batch formula, manufacturing process). This is referred to as the "approved product" in the 406
appendices. 407
408
Alternative approaches to this recommendation may be acceptable if scientifically justified. 409
For example, a comparison to an applicant's marketed product (rather than the product used in 410
the pivotal clinical and/or comparative bioavailability studies) could be justified if a 411
significant body of information has been established for the marketed drug product. For the 412
purposes of this document, a significant body of information for the marketed drug product is 413
likely to exist after a reasonable number of batches of the drug product will be marketed 414
during the specified period of time (e.g. a minimum of 10 batches). 415
416
Applicants should refer to the General Chapters available in the current Schedule B 417
pharmacopoeia for general dissolution and drug release specifications (e.g. United States 418
Pharmacopeia (USP) <711>, USP <724>, European Pharmacopoeia (Ph.Eur.) 2.9.3). 419
420
5.3 Variations (changes) to pharmaceutical aspects of registered products which may be 421
made without prior approval 422

Working document QAS/14.575
page 14
423
5.3.1 Annual notification 424
425
Changes that may be applied without prior approval but included in annual notifications (AN) 426
are listed below: 427
428
1. Changes in batch size of the API or intermediate involving up to 10-fold compared to the 429
currently accepted batch size on condition that (a) the product continues to meet specifications, (b) the 430
dissolution profile is not significantly altered, (c) a stability study has been commenced on at least one 431
full-scale production batch and (d) the change does not concern a sterile API. 432
433
2. Additional tests and limits for starting materials or finished products on condition that these 434
do not reflect a change in processing, e.g. from a fine to microfine particle size. 435
436
3. A change in the content of an excipient of up to ±5%. 437
438
4. Alteration of the quantitative composition of a tablet or capsule coating amounting to less than 439
2% of the total weight of the tablet or capsule. On condition that (a) the coating has no modified- 440
release properties, (b) there is no API in the coating, (c) any new colours are permitted by the 441
European Commission’s List of Permitted Food Colours (4), or the United States Food and Drug 442
Agency’s (US-FDA) Summary of Color Additives for Use in the United States in Foods, Drugs, 443
Cosmetics, and Medical Devices (5) and (d) the change is notified. 444
445
5. Changes to the volume of granulating fluid of up to ±15%, provided that (a) the product 446
continues to meet specifications and (b) the dissolution profile is not significantly altered. 447
448
6. Changes to the quantitative content of agents whose only function is to make the product 449
viscous. On condition that (a) it has been demonstrated that any solid material present is at least 450
equally well suspended and (b) a stability study has been commenced on at least two batches of the 451
altered product. 452
453
7. Change in weight of tablet coatings or capsule shells involving immediate-release oral FPPs. 454
On condition that (a) multipoint in vitro dissolution profiles of the proposed version of the product 455
(determined in the routine release medium on at least two batches of pilot- or production-scale) are 456
similar to the dissolution profiles of the biobatch, (b) coating is not a critical factor for the release 457
mechanism and (c) specifications for the FPP are updated only with respect to weight and dimensions, 458

Working document QAS/14.575
page 15
if applicable. 459
460
8. Alteration of methods of manufacture and manufacturing equipment on condition that (a) the 461
product is not a slow- or otherwise modified-release product, (b) a new stability study has been 462
commenced on at least two batches of the altered product, (c) no change in the specifications of the 463
intermediates or the FPP, (d) the dissolution profiles are similar to those of the biobatch and (e) the 464
manufacturing processes for the currently accepted and proposed products use the same principles 465
(e.g. a change from wet to dry granulation, from direct compression to wet or dry granulation, or vice 466
versa, would be considered a change in manufacturing principle), the same processing intermediates 467
and there are no changes to any manufacturing solvent used in the process. 468
469
9. Change in the batch size of the FPP involving downscaling. On condition that (a) the change 470
does not affect the reproducibility and/or consistency of the product, (b) the change pertains only to 471
immediate-release oral pharmaceutical forms and to non-sterile liquid forms and (c) changes to the 472
manufacturing method and/or to the in-process controls are only those necessitated by the change in 473
batch size, e.g. use of different-sized equipment. 474
475
10. Change to in-process tests or limits applied during the manufacture of the FPP or intermediate 476
involving (a) tightening of in-process limits, (b) deletion of a test and (c) addition of new tests and 477
limits. 478
479
11. Additional tests and limits for starting materials or finished products on condition that these 480
do not reflect a change in processing, e.g. from a fine to microfine particle size. 481
482
12. Change in specifications of an excipient to comply with an officially-recognized 483
pharmacopoeia provided that there is no change to the specifications other than those required to 484
comply with the pharmacopoeia (e.g. no change in particle size distribution). 485
486
13. Update to the specifications to comply with an officially-recognized pharmacopoeial 487
monograph as a result of an update to this monograph to which the FPP is controlled 488
489
14. Change in the analytical procedures for the FPP involving updating the analytical procedure 490
with an officially-recognized pharmacopoeial monograph as a result of an update to that monograph. 491
492
493
5.3.2 Immediate notification – “do and tell” 494
495

Working document QAS/14.575
page 16
496
497
Applicants must satisfy themselves that they meet all of the prescribed conditions for the 498
change and submit all required documentation with the notification application. Such changes 499
can be implemented immediately at the time of submission and they can be considered 500
accepted if an objection is not issued by the NMRA within a reasonable period subject to 501
country specific proposal after the date of acknowledgement of receipt of the application. 502
503
Examples of such changes are listed below: 504
505
1. Change to the marketing authorization holder (name, address and/or legal entity). On 506
condition that there is no change to the product, including sites of manufacture. 507
508
2. Change in the name and/or corporate address of the supplier of the FPP on condition 509
that the supplier of the product remains the same legal entity. 510
511
3. Deletion of a manufacturing site or manufacturer involving production or testing of 512
the API/FPP intermediate or API/FPP provided that at least one other site continues to 513
perform the same function(s) as the site(s) intended to be deleted and that the deletion of the 514
site is not a result of critical deficiencies in manufacturing. 515
516
4. Change in the manufacturing process of the API. On condition (a) that there is no 517
change in the physical state (e.g. crystalline, amorphous) of the API, (b) for low solubility 518
APIs, there is no change in the polymorphic form and whenever particle size is critical 519
(including low solubility APIs) there is no significant change in the particle size distribution 520
compared to that of the API lot used in the preparation of the biobatch, (c) where materials of 521
human or animal origin are used in the process, the manufacturer does not use any new 522
process for which assessment of viral safety data or transmissible spongiform encephalopathy 523
(TSE) risk assessment is required, (d) no change in qualitative and quantitative impurity 524
profile or in physicochemical properties of the API, (e) the change does not affect the 525
sterilization procedures of a sterile API, (f) the change involves only steps before the final 526
intermediate and (g) the change does not require revision of the starting material, intermediate 527
or API specifications. 528

Working document QAS/14.575
page 17
529
5. Change in batch size of the API or intermediate involving downscaling on condition 530
that (a) no changes to the manufacturing process other than those necessitated by changes in 531
scale (e.g. use of a different size of equipment) and (b) the change does not affect the 532
reproducibility of the process. 533
534
6. Changes to the test parameters, acceptance criteria or analytical procedures of the API 535
manufacturer that do not require a change to the FPP manufacturer’s API specifications. 536
537
7. Change to the test parameters or acceptance criteria of the API specifications of the 538
FPP manufacturer involving (a) replacement of a test parameter, (b) relaxation of an 539
acceptance criterion and (c) addition of test parameter. On condition that (a) for insoluble 540
APIs there is no change in the polymorphic form and whenever particle size is critical 541
(including low-solubility APIs), there is no change in particle size distribution acceptance 542
criteria, (b) no additional impurity found over the International Conference on Harmonisation 543
of Technical Requirements for Registration of Human Medicines (ICH) identification 544
threshold and (c) the change does not concern sterility testing. 545
546
8. Change to the analytical procedures used to control the API by the FPP manufacturer 547
involving change from a currently accepted in-house analytical procedure to an analytical 548
procedure in an officially-recognized pharmacopoeia or from the analytical procedure in one 549
officially-recognized pharmacopoeia to an analytical procedure in another officially-550
recognized pharmacopoeia. 551
552
9. Changes to the container-closure system in immediate contact with the product or 553
additional types of container-closure on condition that (a) the product is not a sterile product, 554
(b) the new system offers equal or better protection to the product, (c) stability data are 555
available on two batches of the product in the new container for at least three months under 556
accelerated conditions (as defined in relevant guidelines) or one year under non-accelerated 557
conditions, (d) a stability study has been commenced on at least two batches of the altered 558
product for the full duration of the shelf-life and (e) the change is notified. Changes may not 559
be made to labelling without prior approval. 560
561
10. Reduction in the retest period or shelf-life of the API provided the change is not 562

Working document QAS/14.575
page 18
necessitated by unexpected events arising during manufacture or because of stability 563
concerns. 564
565
11. Change in the composition of a solution dosage form. On condition that (a) the 566
affected excipient(s) does/do not function to affect the solubility and/or the absorption of the 567
API, (b) the affected excipient(s) does/do not function as a preservative or preservative 568
enhancer, (c) no change in the specifications of the affected excipient(s) or the FPP, (d) no 569
change in the physical characteristics of the FPP (e.g. viscosity, osmolality, pH), (e) the 570
change does not concern a sterile FPP and (f) the excipients are qualitatively the same. The 571
change in the amount (or concentration) of each excipient is within ±10% of the amount (or 572
concentration) of each excipient in the originally approved product. 573
574
12. Changes to flavours, perfumes or colours on condition that (a) any new colours are 575
permitted by the European Commission’s List of Permitted Food Colours (4), or the United States 576
Food and Drug Agency’s (US-FDA) Summary of Color Additives for Use in the United States in 577
Foods, Drugs, Cosmetics, and Medical Devices (5), (b) the change is notified, (c) stability data are 578
available on two batches of the altered product for at least three months under accelerated 579
conditions (as defined in relevant guidelines) or one year under non-accelerated conditions 580
and (d) a new stability study has been commenced on at least two batches of the altered 581
product for the full duration of the shelf-life. 582
583
13. A change or addition of imprints, embossing or other markings on solid dosage forms, 584
including replacement or addition of inks used for product markings and change in scoring 585
configuration. On condition that (a) these do not imply an unapproved indication or patient 586
population, (b) no unapproved colour (as defined above) is introduced, (c) any changes to 587
scoring are consistent with the dose schedules in the approved product information and (d) 588
the change is notified. 589
590
14. Change in dimensions without change in qualitative or quantitative composition and 591
mean mass of tablets, capsules, suppositories and pessaries. On condition that (a) 592
specifications for the FPP are updated only with respect to dimensions of the FPP and (b) 593
multipoint in vitro dissolution profiles of the current and proposed versions of the product 594
(determined in the routine release medium, on at least one batch of pilot- or production-scale) 595
are comparable. 596

Working document QAS/14.575
page 19
597
15. Addition or replacement of a manufacturing site for part or all of the manufacturing 598
process for an FPP involving (a) secondary packaging of all types of FPPs, (b) primary 599
packaging site of solid FPPs (e.g. tablets, capsules), semi-solid FPPs (e.g. ointments, creams) 600
and solution liquid FPPs and (c) primary packaging site of other liquid FPPs (suspensions, 601
emulsions). On condition that (a) satisfactory good manufacturing practices (GMP) inspection 602
in the last three years and (b) site appropriately authorized by an NMRA (to manufacture the 603
pharmaceutical form and the product concerned). 604
605
16. Change in the batch size of the FPP involving up to and including a factor of 10 606
compared to the biobatch. On condition that (a) the change does not affect the reproducibility 607
and/or consistency of the product, (b) the change pertains only to immediate-release oral 608
pharmaceutical forms and to non-sterile liquid forms, (c) changes to the manufacturing 609
method and/or to the in-process controls are only those necessitated by the change in batch 610
size, e.g. use of different-sized equipment, (d) a validation protocol is available or validation 611
of the manufacture of three production-scale batches has been successfully undertaken in 612
accordance with the current validation protocol and (f) the biobatch size was at least 100 000 613
units in the case of solid oral dosage forms. 614
615
17. Change to in-process tests or limits applied during the manufacture of the FPP or 616
intermediate involving revision or replacement of a test. 617
618
18. Change in the specifications of the FPP involving test parameters and acceptance 619
criteria: (a) relaxation of an acceptance criterion; or (b) replacement of a test parameter 620
provided that the change to the specifications does not affect the stability and the performance 621
of the product and the change does not concern sterility testing. 622
623
19. Analytical methodology for the finished product on condition that (a) validation shows 624
that the new method is equivalent to or better than the existing method and (b) major changes 625
(e.g. ultra violet assay to high-pressure liquid chromatography (HPLC)) are notified. 626
627
20. Change in the package size involving: (a) change in the number of units (e.g. tablets, 628
ampoules, etc.) in a package; and (b) change in the fill weight or fill volume of non-parenteral 629
multidose products. On condition that (a) the change is consistent with the posology and 630

Working document QAS/14.575
page 20
treatment duration accepted in the package insert and (b) no change in the primary packaging 631
material. 632
633
21. Changes to the container-closure system in immediate contact with the product or 634
additional types of container-closure. On condition that (a) the product is not a sterile product, 635
(b) the new system offers equal or better protection to the product, (c) stability data are 636
available on two batches of the product in the new container for at least three months under 637
accelerated conditions (as defined in relevant guidelines) or one year under non-accelerated 638
conditions, (d) a stability study has been commenced on at least two batches of the altered 639
product for the full duration of the shelf-life and (e) the change is notified. Changes may not 640
be made to labelling without prior approval. 641
642
22. Change in any part of the (primary) packaging material not in contact with the FPP 643
formulation (e.g. colour of flip-off caps, colour code rings on ampoules or change of needle 644
shield), provided the change does not concern a fundamental part of the packaging material, 645
which affects the delivery, use, safety or stability of the FPP. 646
647
23. Reduction in the shelf-life of the FPP (as packaged for sale) or in the in-use period of 648
the FPP (after first opening or after reconstitution or dilution). 649
650
New sites of manufacture require prior approval because the NMRA should see evidence of 651
compliance with GMP, e.g. a WHO-type certificate of pharmaceutical product (6). Changes 652
or additions to pack size also require prior approval because the new size must be consistent 653
with the approved uses of the product. 654
655
Changes may not be made to labelling without prior approval, except for changes to layout 656
without alteration of text or meaning. Pictures or diagrams may not be added without prior 657
approval because they may imply an unapproved indication. 658
659
Notifications of variations, and applications to vary, must be accompanied by this statement: 660
661
“No variations have been made other than (1) those notified herewith and (2) changes 662
which are permitted without notification or prior approval according to the guidelines of 663
the medicines regulatory authority of ...........” 664

Working document QAS/14.575
page 21
5.4 Variations (changes) to pharmaceutical aspects of registered products which require 665
prior approval before implementation 666
667
5.4.1 Minor variation 668
669
Below is a list of variations considered minor. In addition to the general documents stated in 670
section 3.5. Documentation required, specific documentation in support of each type of 671
variation should be provided. 672
673
1. Replacement or addition of a new manufacturing site or manufacturer of an API involving 674
API testing only provided the transfer of analytical methods has been successfully undertaken and 675
no change in the FPP manufacturer’s API specifications. 676
677
2. Change of drug product name. 678
679
3. Change of the specification of drug substance a) specification limits are tightened and b) 680
addition of new test parameter and limits. 681
682
4. Change in product labelling should be in accordance to country specific labelling requirement 683
and this includes: 684
685
686
a) change of the layout/artwork without altering meaning; 687
b) addition/deletion/replacement of pictures, diagrams, bar code, logos and/or texts that do not 688
imply an unapproved indication; 689
c) addition/strengthening of warnings, precautions, contraindications and/or adverse 690
events/effects to the approved product labelling; 691
d) tightening of product’s target population; 692
e) deletion of indication. 693
694
5. Change in batch size of the API or intermediate involving more than 10-fold increase 695
compared to the currently accepted batch size. 696
697
6. Change to the specifications or analytical procedures applied to materials used in the 698
manufacture of the API (e.g. raw materials, starting materials, reaction intermediates, solvents, 699
reagents, catalysts) involving addition or replacement of a specification parameter as a result of a 700

Working document QAS/14.575
page 22
safety or quality issue. 701
702
7. Change of the specification of drug substance a) specification limits are tightened and b) 703
addition of new test parameter and limits. 704
705
8. Change to the test parameters or acceptance criteria of the API specifications of the FPP 706
manufacturer a) addition of a test parameter, b) replacement of test parameter and c) relaxation of 707
an acceptance criterion. 708
709
9. Change to the analytical procedures used to control the API by the FPP manufacturer 710
involving modification or replacement of an analytical procedure. 711
712
10. Change in the immediate packaging (primary and functional secondary components) for the 713
storage and shipment of the API provided the change is not due to instability issues. 714
715
11. Change in the retest period or shelf-life of the API involving extension provided (a) no change 716
to the primary packaging in direct contact with the API or to the recommended condition of 717
storage and (b) stability data were generated in accordance with the currently accepted stability 718
protocol. 719
720
12. Any change in the labelled storage conditions of the API provided (a) the stability studies 721
must show compliance with specification and (b) no change in shelf-life/retest period. 722
723
13. Change or addition of imprints, embossing or other markings, including replacement or 724
addition of inks used for product markings and change in scoring configuration involving addition 725
of a score line. On condition that (a) the change does not affect the stability or performance 726
characteristics (e.g. release rate) of the FPP, (b) changes to the FPP specifications are those 727
necessitated only by the change to the appearance or to the scoring and (c) addition or deletion of 728
a score line from a generic product is consistent with a similar change in the comparator product 729
or was requested by NMRA. 730
731
14. Change in dimensions without change in qualitative or quantitative composition and mean 732
mass of gastroresistant, modified or prolonged-release FPPs and scored tablets. On condition that 733
specifications for the FPP are updated only with respect to dimensions of the FPP and multipoint 734
in vitro dissolution profiles of the current and proposed versions of the product (determined in the 735
routine release medium, on at least one batch of pilot- or production-scale) are comparable. 736
737

Working document QAS/14.575
page 23
15. Addition or replacement of a manufacturing site for part or all of the manufacturing process 738
for an FPP involving all other manufacturing operations except batch control and/or release 739
testing. On condition that (a) no change in the batch formula, description of manufacturing 740
process and process controls, equipment class and process controls, controls of critical steps and 741
intermediates or FPP specifications, (b) satisfactory inspection in the last three years, (c) site 742
appropriately authorized by an NMRA (to manufacture the pharmaceutical form and the product 743
concerned) and (d) validation protocol is available or validation of the manufacturing process at 744
the new site has been successfully carried out on at least three production-scale batches in 745
accordance with the current protocol. 746
747
16. Change in the manufacturing process of the FPP. 748
749
17. Change in the specifications or analytical procedures for an excipient involving change or 750
replacement of an analytical procedure. 751
752
18. Replacement or addition of a primary packaging type provided the change does not concern a 753
sterile FPP. 754
755
19. Change in qualitative and/or quantitative composition of the immediate packaging material for 756
semisolid and liquid FPPs. On condition that (a) the change does not concern a sterile FPP, (b) no 757
change in the packaging type and material (an example of an allowable change is blister to blister) 758
and (c) the relevant properties of the proposed packaging are at least equivalent to those of the 759
currently accepted material. 760
761
20. Extension in the shelf-life of the FPP (as packaged for sale) provided there is no change to the 762
primary packaging type in direct contact with the FPP and to the recommended conditions of 763
storage and stability data were generated in accordance with the currently accepted stability 764
protocol. 765
766
21. Extension in the in-use period of the FPP (after first opening or after reconstitution or 767
dilution). 768
769
22. Change in the labelled storage conditions of the FPP (as packaged for sale), the product 770
during the in-use period or the product after reconstitution or dilution. 771
772
773
5.4.2 Major variation 774

Working document QAS/14.575
page 24
775
776
Variations, which fail to fulfil the conditions for notifications or minor variations 777
automatically, become major variations. In addition, variations, which are not covered by 778
these guidelines, should be considered as a major change by default. 779
780
References 781
782
1. Guidelines on submission of documentation for a multisource (generic) finished 783
pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part. 784
In: WHO Expert Committee on Specifications for Pharmaceutical Preparations. Forty-sixth 785
report. Geneva, World Health Organization. Technical Report Series, No. 970, 2012, Annex 4. 786
787
2. Guidance on variations to a prequalified dossier. In: WHO Expert Committee on Specifications 788
for Pharmaceutical Preparations. Forty-first report. Geneva, World Health Organization. 789
Technical Report Series, No. 943, 2007, Annex 6. 790
791
3. Guidelines for stability testing of active pharmaceutical ingredients and finished pharmaceutical 792
products. In: WHO Expert Committee on Specifications for Pharmaceutical Preparations. 793
Forty-third report. Geneva, World Health Organization. Technical Report Series, No. 953, 794
2009, Annex 2. 795
796
4. European Commission. List of permitted food colours: EC Directive 94/36/EC. Official Journal 797
of the European Communities 1994; L237. 798
799
5. FDA. Summary of Color Additives for Use in the United States in Foods, Drugs, Cosmetics, 800
and Medical Devices. 2010. 801
802
6. Guidelines for implementation of the WHO Certification Scheme on the Quality of 803
Pharmaceutical Products Moving in International. In: WHO Expert Committee on Specifications 804
for Pharmaceutical Preparations. Thirty-fourth report. Geneva, World Health Organization. 805
Technical Report Series, No. 863, 1996, Annex 10. 806
807
808
*** 809


















