White reflex behind the pupil
64
W HITE REFLEX BEHIND THE PUPIL Contd… Guide: Dr.Sulatha Bhandary Presenter : Dr. Aditi Singh
-
Upload
aditi-singh -
Category
Health & Medicine
-
view
129 -
download
1
Transcript of White reflex behind the pupil

WHITE REFLEX BEHIND THE PUPIL
Contd…
Guide: Dr.Sulatha Bhandary
Presenter : Dr. Aditi Singh

PERSISTENT HYPERPLASTIC PRIMARY VITREOUS
(PHPV) OR PERSISTENCE OF THE FETAL
VASCULATURE (PFV)
PHPV is a congenital malformation
Caused by the arrest of normal regression of embryonic vitreous and hyaloid vasculature.
That normally starts at 9 weeks of gestation.

CLINICAL FEATURES:
unilateral, non hereditary.
full term infants
microphthalmia
most common sign is leukocoria,

Subclassified into three types:
Purely anterior presentation:
Unilateral leukocoria,
Retrolental mass into which ciliary processes are inserted.
Complications-cataract, glaucoma and a retrolenticularmembrane.
Treatment: vitreoretinal surgery
cataract surgery

Purely posterior presentation
Leukocoria, strabismus or nystagmus.
Dense white membrane or prominent retinal fold ,extends from optic disc to the ora serrata, can be associated with retinal detachment.
Treatment: not possible.
Combination of anterior and posterior presentations is the most commonly seen.
Complications:
early cataract and closed-angle glaucoma.
Long term: terminal glaucoma, intra-ocular hemorrhage, retinal
detachment, uveitis or phthisis bulbi.

DIFFERENTIATED FROM RETINOBLASTOMA
(A) Axial ultrasound image with color Doppler shows a vessel running through the vitreous of both globes, from the posterior surface of the lens capsule to the optic disc (arrowhead). Echogenic foci suggesting hemorrhage are also seen (arrows).
(B) On pulsed Doppler examination, the vessel shows arterial flowIndian J Ophthalmol. 2009 Jan-Feb; 57(1): 53–54.
PMCID: PMC2661510
Bilateral persistent hyperplastic primary vitreous

Axial CT image shows diffusely hyperdense attenuation of vitreous in both globes, suggesting hemorrhage.
Subtle linear structures are also seen (arrowheads), representing hyaloidartery in Cloquet's canal
Absence of calcification
Indian J Ophthalmol. 2009 Jan-Feb; 57(1):
53–54.
PMCID: PMC2661510
Bilateral persistent hyperplastic primary
vitreous

COATS’ DISEASE (PRIMARY RETINAL
TELANGIECTASIA)(FIRST DESCRIBED IN 1908 BY GEORGE COATS)
Shields et al. defined Coats’ disease as ‘Idiopathic retinal teleangiectasia associated with intraretinal exudation and frequent exudative retinal detachment without signs of appreciable retinal or vitreal traction’ .
diagnosed in the first decade of life
mild forms -can present well into adulthood
Inheritenace-
mostly sporadic
may be a genetic cause

> 75 % of patients are male
95% of the cases are unilateral.
no clear racial predilection.
The occurrence of retinal telangiectasis and exudation bilaterally, in female patients, or in adults should prompt a search for non-Coats causes of these findings.

PRESENTATION AND CLINICAL FINDINGS:
The most common presenting signs of Coats’ disease are –
decreased visual acuity, strabismus and leukocoria
.
rarely reveals anterior segment findings
severe cases may show-
corneal edema
anterior chamber shallowing
cells and flare in the anterior chamber
neovascular glaucoma.

posterior segment findings range from –
mild peripheral telangiectasia of the retinal vasculature to florid exudation with total retinal detachment.
more than half the telangiectasias are found anterior to the equator.
the telangiectatic vessels appear to have a predilection for the temporal and inferior quadrants.
telangiectasias may be accompanied by grape-like clusters of microaneurysms, fusiform dilation of retinal arterioles (“light bulb aneurysms”), sheathing of retinal vessels, or venous beading.

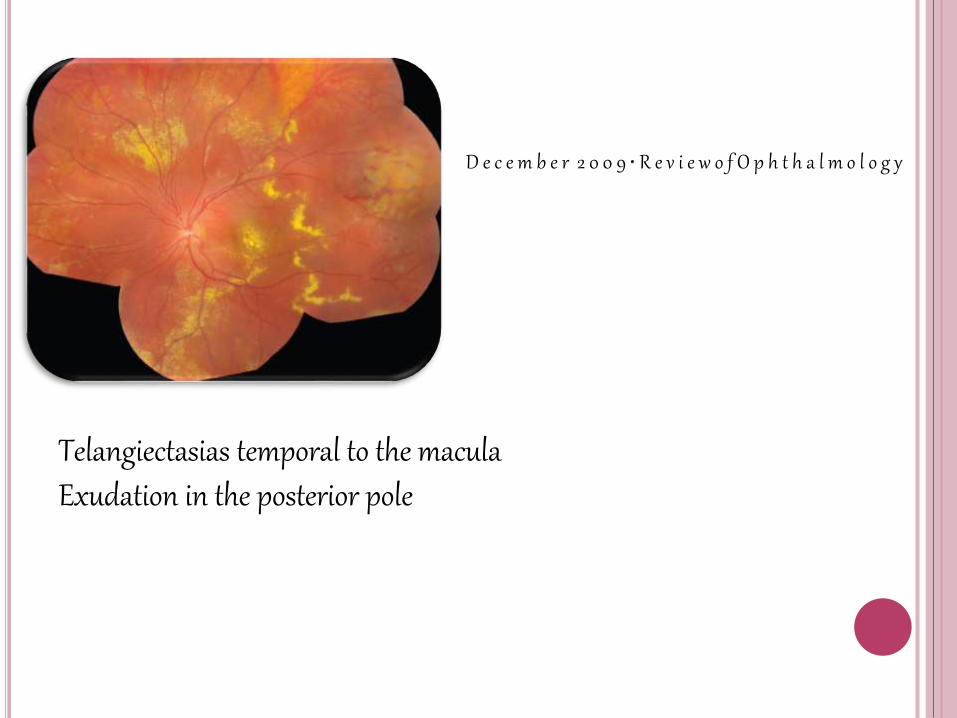
Despite the peripheral location of the telangiectasias, the intraretinal and subretinal exudates often migrate toward the macula, leading to severe visual loss
In cases of massive exudation, a dense nodule may form in the macula.
Formation of a fibrotic nodulein the central macula is associated with aparticularly poor visual prognosis.
D e c e m b e r 2 0 0 9 • R e v i e w o f O p h t h a l m o l o g y
D e c e m b e r 2 0 0 9 • R e v i e w o f O p h t h a l m o l o g y
Telangiectasias temporal to the macula Exudation in the posterior pole

Classification( Shields et al)in 2000
Stage -1. Retinal telangiectasia only
2 .Telangiectasia and exudation(2A, extrafoveal exudation; 2B, foveal exudation)
3a .Exudative subtotal retinal detachment
3b. Exudative total retinal detachment
4. Total retinal detachment and glaucoma
5.Advanced end-stage disease

ANCILLARY TESTING
Fluorescein angiography
Ultrasound
Computerized tomography (CT)
Magnetic resonance imaging (MRI)

FLUORESCEIN ANGIOGRAPHY
areas of telangiectasia(early hyperfluroscence)
may also show microaneurysms,
areas of capillary nonperfusion,
or retinal neovascularization
Areas of anomalous vasculature will leak in the late phases of the angiogram
D e c e m b e r 2 0 0 9 • R e v i e w o f O p h t h a l m o l o g y

ULTRASOUND
subretinal opacities due to cholesterolosis present from the exudates
retinal detachment
Optical coherence tomography
evaluate the extent of intraretinal and subretinal fluid
exudates or fibrosis in the macula

retinoblastoma coats
CT solid tumours
and calcifications
scan is clear of these
lesions
MRI T 1 -weighted image
will show a
hyperintense
mass,
T 2 -weighted image
shows a hypointense
mass
exudate in Coats’
disease is hyperintense
on both T 1 -
weighted
T 2 -weighted MRI
images.
MRI(use of gadolinium
)
enhances
the solid tumours
not seen
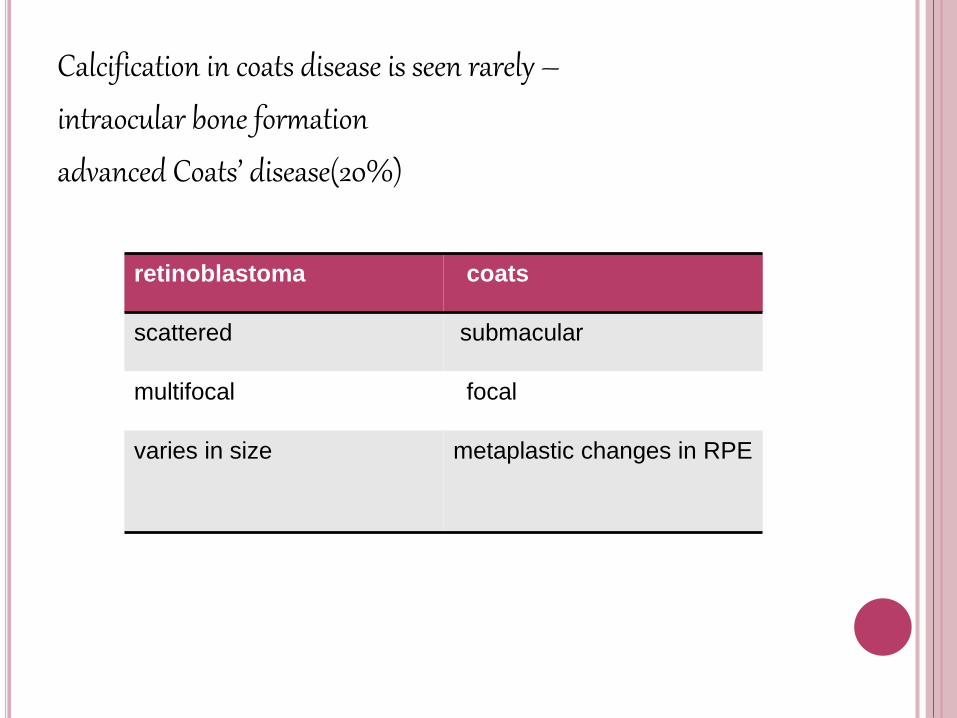
Calcification in coats disease is seen rarely –
intraocular bone formation
advanced Coats’ disease(20%)
retinoblastoma coats
scattered submacular
multifocal focal
varies in size metaplastic changes in RPE

TREATMENT(OPHTHALMOLOGICA 2012;227:175–182):
Stage Treatment
Mild disease (1, 2) without progression Observation – no treatment
Mild disease (1, 2) with progression Laser photocoagulation/cryotherapy
Advanced disease (3, 4) Vitreoretinal surgery
Advanced end-stage disease (5) Observation – no treatment
(with comfortable eye)
Advanced end-stage disease (5) Enucleation
(painful eye)
Adjuvant therapy Intravitreal triamcinolone
( long term saftey unknown ) anti -VEGF.
is unkno

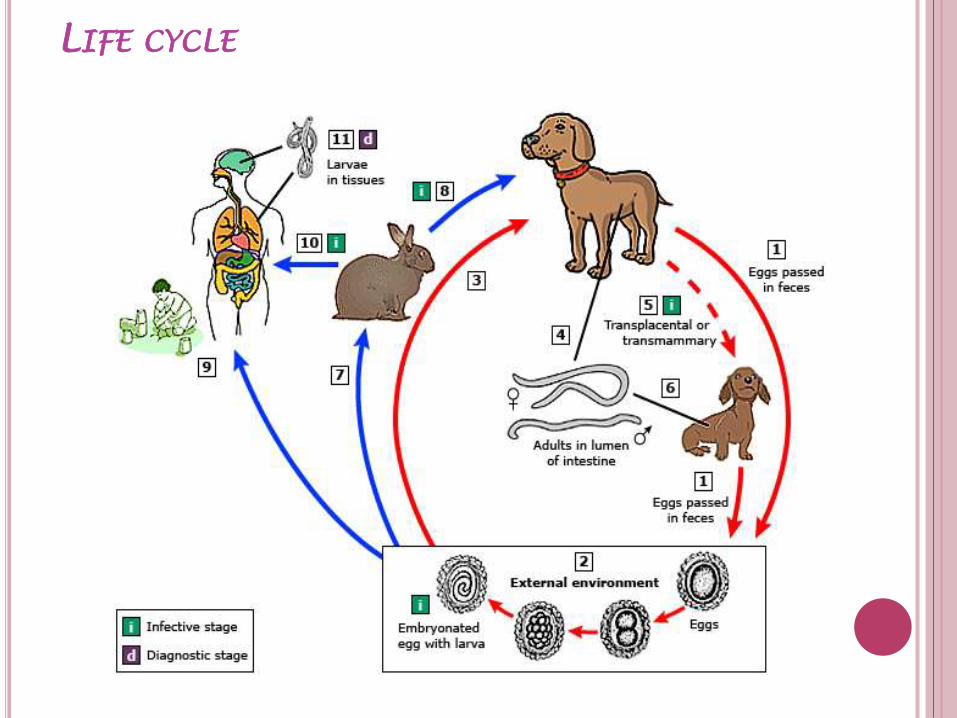
TOXOCARIASIS: VISCERAL AND OCULAR LARVA
MIGRANS
Epidemiology
occurs worldwide
tropical regions
among rural populations
Transmission
ingestion of embryonated eggs (contaminated food or soil )
disease of young children
LIFE CYCLE

CLINICAL MANIFESTATIONS
Ocular larva migrans (OLM)
sole manifestation(without VLM)
unilateral.
hosp no.2192672
I.Diffuse chronic endophthalmitis (age 2–9 years):
decreased vision with floaters
chronic anterior uveitis , vitritis
snowbanking, macular edema, exudative retinal detachment
complications include tractional retinal detachment, cycliticmembrane, cataract, hypotony
poor prognosis

II.Posterior pole granuloma (age 6–14 years):
decreased visual acuity
relatively clear media
yellow-white granuloma 1–2 DD at the macula/papillomacularbundle
with retinal traction and vitreous bands
Central and peripheric subretinal granulomas with
two fibrous traction bands proliferating to the
periphery, detected by an examination of the eye
fundus in a 16-year-old adolescent with ocular larva
migrans

III.Peripheral granuloma (age 6 years–adult):
usually asymptomatic
yellow-white granuloma anterior to the equator with vitreous bands.
traction may cause macular heterotopia or retinal detachment (tractional or rhegmatogenous).
Atypical presentations :
1.Inflammation and swelling of ONH
2.Motile subretinal nematode
3.Diffuse chorioretinitis
Hosp.no :2113778

Visceral larva migrans
young children
results in hepatitis and pneumonitis as the larvae migrate through the liver and lungs
can also affect CNS,muscles and the heart.

DIAGNOSIS OF OLM
usually occurs in isolation.
diagnosis is usually based on history , clinical appearance.
A strong clinical suspicion and positive history such as-
Close contact with cats and dogs,
Poor hygiene and habits as geophagia.
Systemic symptoms as abdominal pain may be occasionally present(though not always).

There are several immunological tests used to diagnose visceral larva migrans;
not as reliable for ocular larva migrans.
Specific titres for Toxocara are assessed with ELISA , which uses antigens secreted by the larva to diagnose infection.
The sensitivity of ELISA is 75 % and its specificity is more than 90%.
Patients with ocular larva migrans usually have low or even negative titres.

Goldmann-Witmer coefficient
(level of specific IgG in aqueous humour/level of specific IgG in serum)/(total IgG in aqueous humour/total IgG in serum) >3.0
is suggestive of intraocular antibody production,
Can help to establish the diagnosis .
OCT and B-scan ultrasonography can be performed .

IMAGING STUDIES
OCT:
OCT of a toxocara granuloma –
a reflective mass above the level of the RPE.
detect the presence of intraretinal, subretinal or sub- RPE fluid
In a case of diagnosed OLM a granulomatous mass was present superior-temporal to the right optic discOCT of this large granuloma revealed an elevated lesion with associated intraretinal cyst-like formations
In vivo diagnostic imaging of ocular toxocariasis( Clin Exp Optom 2009; 92: 2: 146–149)

B SCAN:
can be used to demonstrate -
vitreous traction
subclinical retinal detachment
hosp no.:2113778
access structures not visible ophthalmoscopically
rule out the presence of calcification
vitreous inflammation may present as low amplitude echoes

TREATMENT
albendazole (adults: 800 mg orally twice daily for two weeks; children: 400 mg orally twice daily for two weeks).
concomitant prednisolone(1.5 mg/kg for adults; 1.0 mg/kg for adults) tapered over a few months.
Mebendazole is an alternative to albendazole (100 to 200 mg orally twice daily for five days),
Diethylcarbamazine (DEC) (3 to 4 mg/kg/day for 21 days, starting at 25 mg/day for adults) has greater side effects than albendazole .
In complicated cases, surgical intervention may be warranted.

7 Yr/M Hosp .No.: 2113778
Presenting complaint-(LE) DOV and floaters.
Vision (LE)-6/18,N10
Anterior segment- (LE) sluggish pupillary reaction
cells in anterior vitreous
Posterior segment:

Toxoplasma IgM : negative
Mantoux: negative.
Chest xray: normal
Blood investigations : normal.
Started on course of albendazole,tapering dose of steroids and epitoin
Vision improved to 6/9 ,N6 after 4 months.
6/6 ,N6 after 12 months.

Differentiated from retinoblastoma:
In acute nematode endophthalmitis- severe inflammation, cellular reaction and vitreous tractional bands.
Vitreoretinal tractions and secondary cataracts are rare in
retinoblastoma.
Toxocara granuloma ,shows retinal contraction and distortion
around the mass, this is not seen in comparable size
retinoblastoma mass.

RETINOPATHY OF PREMATURITY
Retinopathy of prematurity (ROP),formerly known as retrolental
fibroplasia because of its end-stage appearance, is a developmental
vascular proliferative disorder that occurs in the retina of preterm
infants with incomplete retinal vascularization

Risk factors
The most important risk factor for developing ROP is prematurity.
Low birth weight
low gestational age
assisted ventilation for longer than one week
surfactant therapy, high blood transfusion volume
hyperglycemia, and insulin therapy

The five stages indicate the increasing severity of disease:
Stage 1 : (demarcation line)flat white line that demarcates the vascular and avascular retina.
Stage 2: (ridge )in region of demarcation line ,has height and width,extend above the plane of the retina.
Stage 3: (extraretinal fibrovascular proliferation)new blood vessels and fibrous tissue grow along the ridge and often extend into the vitreous .
Stage 4 :partial retinal detachment.
4A and 4B,:detachment excluding or including the macula, respectively
Stage 5 : total retinal detachment
.

Screening criteria
All infants with-
birth weight ≤1500 g or
a gestational age (GA) of less than 30 weeks,
as well as those with birth weight between 1500 g and 2000 g or
a GA of more than 30 weeks whose clinical course places them at increased risk for ROP (as determined by the attending clinician)
as per the 2006 Joint Statement of the American Academy of Pediatrics (AAP) Section on Ophthalmology, the American Academy of Ophthalmology (AAO), and the American Association for Pediatric Ophthalmology and Strabismus (AAPOS).

TREATMENT
Standard treatment consists of ablation of the peripheral avascular retina by laser photocoagulation or cryotherapy .
Photocoagulation
Laser photocoagulation, using the diode or argon laser, has
become standard treatment for ROP.
Cryotherapy
Cryotherapy was the only proven treatment for ROP until the early
1990s, but it has been replaced by laser photocoagulation as
standard therapy.

OPTIC DISC ABNORMALITIES
Optic disc coloboma
sharply defined, white, inferiorly decentered excavation of the optic disc
inferior neuroretinal rim is thin or absent.
defect may extend inferiorly
involve the adjacent retina and choroid
unilateral or bilateral
with equal frequency and may be sporadic or inherited

CLINICAL FEATURES:
Visual loss is variable and difficult to predict based upon disc appearance.
Colobomas of the iris and ciliary body often coexist.
Other coexisting ocular malformations –
orbital cyst ,
iris heterochromia , and
retinal venous malformations

ASSOCIATED SYSTEMIC MANIFESTATIONS:
renal coloboma (papillorenal) syndrome an autosomaldominant trait.
renal manifestations -vesicoureteral reflux, renal hypoplasia, renal failure, and chronic nephritis.
CHARGE syndrome , Walker-Warburg syndrome , focal dermal hypoplasia , Aicardi syndrome , Goldenhar syndrome , linear sebaceous nevus syndrome , and Noonan syndrome
Difference from retinoblastoma:
Ophthalmoscopy: clearly defined depressed lesion
Coexisting iris coloboma.

MYELINATED NERVE FIBRES:
Myelinated nerve fibers appear as striated white patches with feathery borders .
caused by differential myelination of individual axons
ophthalmoscopically evident in 0.3 to 0.6 percent of the population and are seen in 1 percent at postmortemexamination .

bilateral in 17 to 20 percent
they are continuous with the disc in 81percent.
Hosp no: 2034628

CLINICAL FEATURES:
Visual acuity - normal
enlarged blind spots and scotomas can occur
high myopia and amblyopia can occur
may progress after birth
rarely may be acquired
may be inherited in an autosomal dominant fashion (component of the Gorlin syndrome )
No particular ophthalmologic follow-up is necessary.
Differentiated from retinoblastoma by its typical clinical appearance.
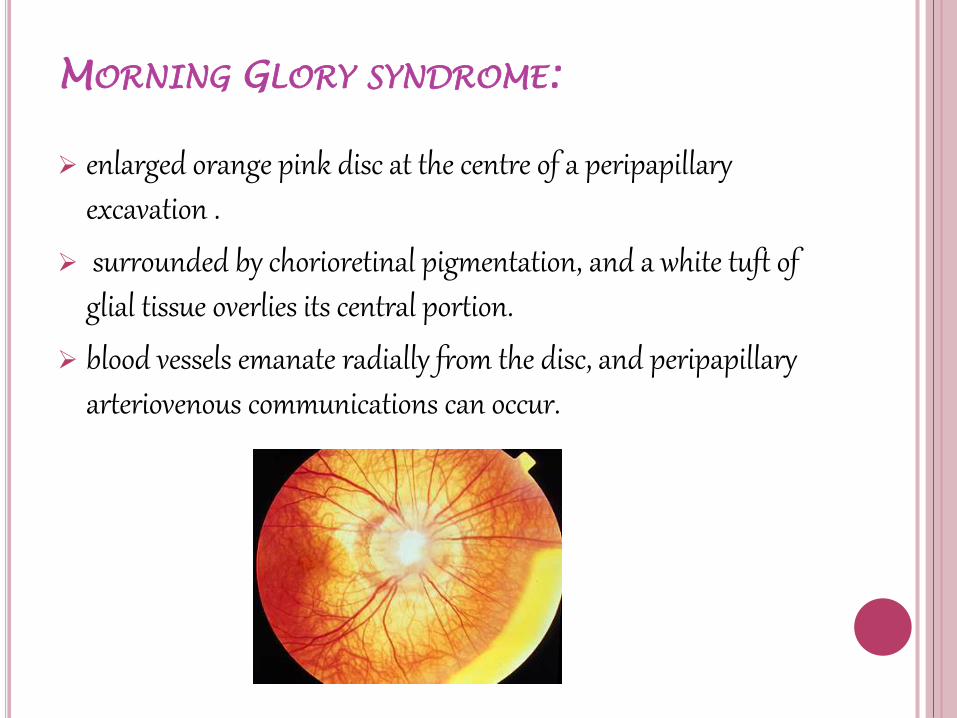
MORNING GLORY SYNDROME:
enlarged orange pink disc at the centre of a peripapillary excavation .
surrounded by chorioretinal pigmentation, and a white tuft of glial tissue overlies its central portion.
blood vessels emanate radially from the disc, and peripapillaryarteriovenous communications can occur.

Demogrphy:
usually unilateral
more common in females, rare in African-Americans .
Clinical featurs:
They can be associated with congenital cataracts
Visual acuity typically is 20/200 to finger counting
Acquired visual loss can occur from-
serous retinal detachment
retinal folds and
subretinal neovascularization.

VITREORETINAL DYSPLASIA
Caused by faulty differenciation of retina and vitreous.
In isolation or assosiated with systemic abnormalities like-
Norrie disease,incontinentia pigmenti ,Warburg syndrome.trisomy 13, trisomy 18
Signs:
Congenital blindness with roving eye movments in bilateral cases.
Leukocoria.
Microphthalmos,shallow anterior chamber and elongated ciliaryprocesses.

Norrie disease : X-linked disorder associated with microcephaly, congenital blindness, deafness, and progressive neuropsychiatric illness .
Incontinentia pigmenti : XL dominant. Lethal in utero for boys.
vesicobullous rash on trunk, malformed teeth ,hair ,nails and bones.
Warburg syndrome: AR. Presents with congenital muscular dystrophy.
Neonatal death is common.

Medulloepithelioma
non hereditary tumor
usually unilateral
derived from the immature embryonic medullary epithelium usually the ciliary body.
Clinical feature:
usually occur in early childhood
with leukocoria and poor vision and
typically a vascularized iris mass, this greyish or salmon-colored.

tumor may be polycystic
cystic fragments may be found liberated into the aqueous humor or vitreous body
growth is slow
60 to 90% -malignant, locally invasive and distant metastasis is rare
smaller tumors-local resection
recurrence-enucleation

Differences from retinoblastoma:
Not familial
Almost always unilateral
Often multicystic
Involves ciliary body region rather than posterior
fundus
Can appear as tumourous cyclitic membrane(unusual in
retinoblastoma).

Retinal Astrocytomas:
Definition:
Benign neuroglial tumor that arises from retinal astrocytes.
Key Feature:
Translucent to opaque white inner retinal tumor.
Associated feature:
Tuberous sclerosis in many affected individuals, especially those who have bilateral, multifocal retinal lesions.

Epidemiology and pathogenesis
arises early in life
frequently detected in childhood or adolescence.
affects both sexes equally.
risk factors for development of an astrocytoma of the retina include tuberous sclerosis and possibly neurofibromatosis .
Hosp. no:22238383
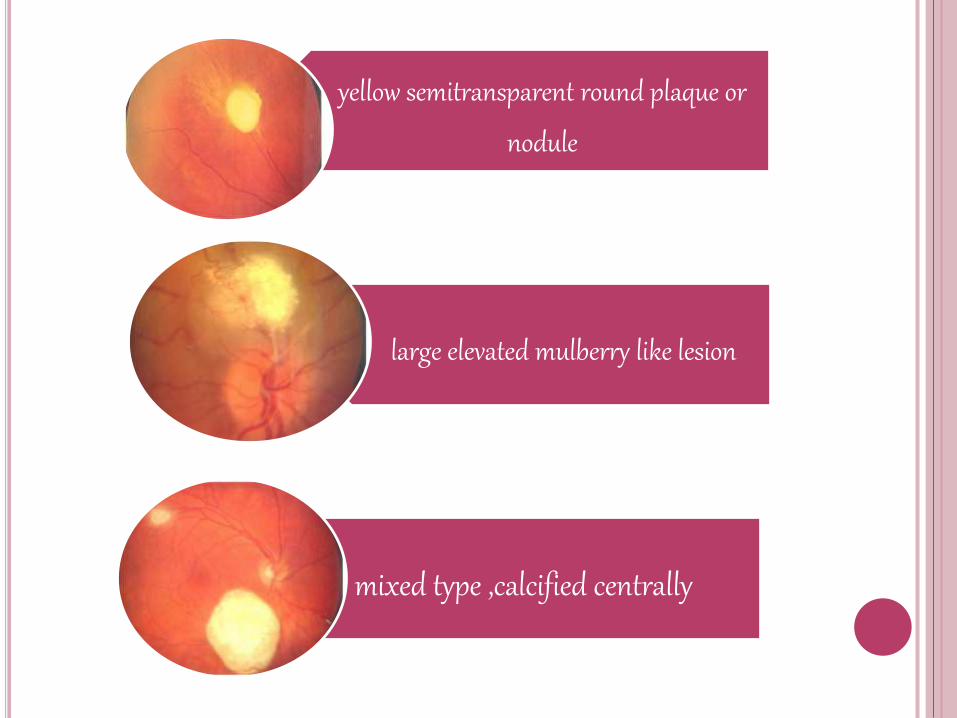
yellow semitransparent round plaque or
nodule
large elevated mulberry like lesion
mixed type ,calcified centrally
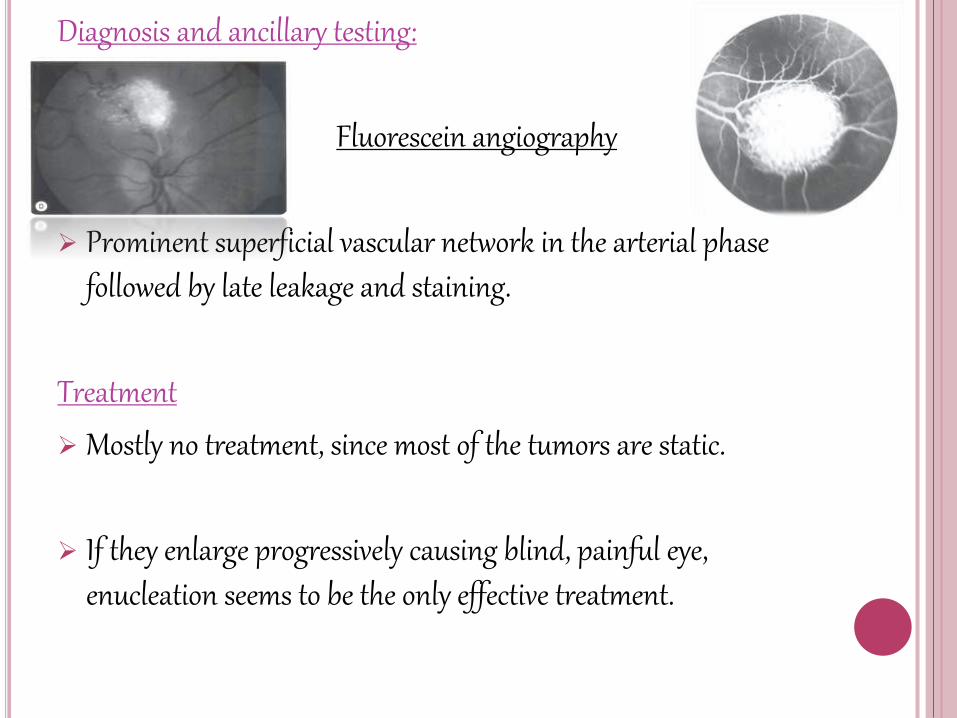
Diagnosis and ancillary testing:
Fluorescein angiography
Prominent superficial vascular network in the arterial phase followed by late leakage and staining.
Treatment
Mostly no treatment, since most of the tumors are static.
If they enlarge progressively causing blind, painful eye, enucleation seems to be the only effective treatment.

Differences from retinoblastoma:
Calcification in astrocytoma is-usually glistining yellow
retinoblastoma is – dull chalky white.
Clinical evidence of tuberous sclerosis and possibly neurofibromatosis support the diagnosis of astrocytichamartoma.

Vitreous hemorrhage
Vitreous hemorrhage causes leukocoria when there is extensive organization of the blood into a clot before degradation .
Conditions:
• Hemorrhagic disease of the newborn
• Advanced ROP
• Persistent fetal vasculature
• Trauma
• Leukemia or other blood dyscrasias
Differences from retinoblastoma:
• Ophthalmoscopy : intravitreal location without retinal
involvement
• B-scan: does not disclose a tumor pattern.

Congenital cataract:
two third of the cases are bilateral.
various causes –genetic mutations,chromosomalabnormalities,metabolic disorders ,intrautrine insult etc.
Management:
Laboratory evaluation
may not be necessary if the history and examination reveal a definitive etiology for the cataract (eg, a family history of heritable cataracts, associated ocular disease or trauma, or obvious syndrome/chromosomal defect).

Laboratory evaluation - Urine for reducing substances after ingestion of galactose-containing
milk (eg, human or cow's milk), and possibly urine amino acids and red cell galactokinase
Toxoplasmosis, rubella, CMV, herpes simplex, and varicella titers and syphilis serology
Calcium, phosphate, and blood sugar (to exclude metabolic disorders, such as diabetes, hypoparathyroidism)
Karyotype and/or other genetic testing
Surgical management:Depends upon the age of the child, laterality and density of the cataract.

BIBLIOGRAPHY1.Persistent hyperplastic primary vitreous Mira Silbert, Andrew S. Gurwood The EyeInstitute, 1200 W.
Godrey Avenue, Philadelphia, PA 19141, USA Accepted 1 September 2000. Available online 21 December Clinical Eye and Vision CareVolume 12, Issues 3–4, December 2000, Pages 100-104.
2.Shields JA, Shields CL, Honavar SG, Demirci H: Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001; 131: 561–571.
3.Jones JH, Kroll AJ, Lou PL, Ryan EA: Coats’ disease. Int Ophthalmol Clin 2001; 41: 189– 198.
4. Shields JA, Shields CL, Honavar SG, Demirci H: Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001; 131: 561–571.
5.Berger W, van de Pol D, Bächner D, Oerlemans F, Winkens H, Hameister H, Wieringa
B, Hendriks W, Ropers HH: An animal model for Norrie disease (ND): gene targeting
of the mouse ND gene. Hum Mol Genet 1996; 5: 51–59.
6.Black GC, Perveen R, Bonshek R, Cahill M,Clayton-Smith J, Lloyd IC, McLeod D: Coats’
disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet 1999; 8: 2031–2035.
7. Dickinson JL, Sale MM, Passmore A, FitzGerald LM, Wheatley CM, Burdon KP, Craig JE, TengtrisornS, Carden SM, Maclean H, Mackey DA: Mutations in the NDPgene: contribution to Norrie disease, familialexudative vitreoretinopathy and retinopathyof prematurity. Clin Experiment Ophthalmol2006; 34: 682–688.

8.Edward DP, Mafee MF, Garcia-Valenzuela E, Weiss RA: Coats’ disease and persistent hyperplasticprimary vitreous. Role of MR imaging and CT. Radiol Clin North Am 1998; 36: 1119–1131.
9. Potter PD, Shields CL, Shields JA, Flanders AE: The role of magnetic resonance imaging
in children with intraocular tumors and simulating lesions. Ophthalmology 1996; 103: 1774–1183.
10.Senft SH, Hidayat AA, Cavender JC: Atypical presentation of Coats disease. Retina 1994; 14: 36–38.
11.Lai WW, Edward DP, Weiss RA, Mafee MF, Tso MO: Magnetic resonance imaging findingsin a case of advanced Coats’ disease. Ophthalmic Surg Lasers 1996; 27: 234–238.
12.Singh A, Cunningham ET, Stewart J. Detection and treatment of ocular toxocariasis. Rev Ophthalmol 2007; 14: 55–58.
13.Despommier D. Toxocariasis: Clinical aspects, epidemiology, medical ecology, and molecular aspects. Clin Microbiol Rev 2003; 16: 265–272.
14.subretinal toxocara granuloma: case report. Arq Bras Oftalmol 2006; 69: 403–405.
15.Afshari M, Hart L, Afshari N, Mukai S. Ophthalmic ultrasonography in children. IntOphthalmol Clin 2001; 41: 153–164.

16.Jack J Kanski,Bard Bowling.Clinical Ophthalmology.A SYSTEMIC APPROACH. VIIth edition.
17.Harley’s Pediatric Ophthalmology. Vth edition
18.Retina .stephen Ryan .III rd edition
19. Retina, Vitreous, Macula by Jerry A. Shield, David R. Guyer Lawrence A. YannuzziStanley ,Chang W. Richard Green