
WGS for the 100,000 Genomes · Population and Medical Genomics Group Applying Genomics to Cancer,...
21
© 2014 Illumina, Inc. All rights reserved. Illumina, 24sure, BaseSpace, BeadArray, BlueFish, BlueFuse, BlueGnome, cBot, CSPro, CytoChip, DesignStudio, Epicentre, GAIIx, Genetic Energy, Genome Analyzer, GenomeStudio, GoldenGate, HiScan, HiSeq, HiSeq X, Infinium, iScan, iSelect, ForenSeq, MiSeq, MiSeqDx, MiSeqFGx, NeoPrep, Nextera, NextBio, NextSeq, Powered by Illumina, SeqMonitor, SureMDA, TruGenome, TruSeq, TruSight, Understand Your Genome, UYG, VeraCode, verifi, VeriSeq, the pumpkin orange color, and the streaming bases design are trademarks of Illumina, Inc. and/or its affiliate(s) in the U.S. and/or other countries. All other names, logos, and other trademarks are the property of their respective owners. WGS for the 100,000 Genomes Mark T. Ross Population and Medical Genomics Group Applying Genomics to Cancer, 21 Sept 2015
Transcript of WGS for the 100,000 Genomes · Population and Medical Genomics Group Applying Genomics to Cancer,...

© 2014 Illumina, Inc. All rights reserved.
Illumina, 24sure, BaseSpace, BeadArray, BlueFish, BlueFuse, BlueGnome, cBot, CSPro, CytoChip, DesignStudio, Epicentre, GAIIx, Genetic Energy, Genome Analyzer, GenomeStudio, GoldenGate, HiScan, HiSeq, HiSeq X, Infinium, iScan, iSelect, ForenSeq, MiSeq,
MiSeqDx, MiSeqFGx, NeoPrep, Nextera, NextBio, NextSeq, Powered by Illumina, SeqMonitor, SureMDA, TruGenome, TruSeq, TruSight, Understand Your Genome, UYG, VeraCode, verifi, VeriSeq, the pumpkin orange color, and the streaming bases design are
trademarks of Illumina, Inc. and/or its affiliate(s) in the U.S. and/or other countries. All other names, logos, and other trademarks are the property of their respective owners.
WGS for the 100,000
Genomes
Mark T. Ross
Population and Medical Genomics Group
Applying Genomics to Cancer, 21 Sept 2015

2
100,000 genomes
Cancer and rare genetic disease
ISO-accredited workflow (2016)
Data delivered electronically,
stored securely and analysed in
England Data Centre
Combine with extracted clinical
information for analysis,
interpretation, and aggregation
Genomics England Partnership
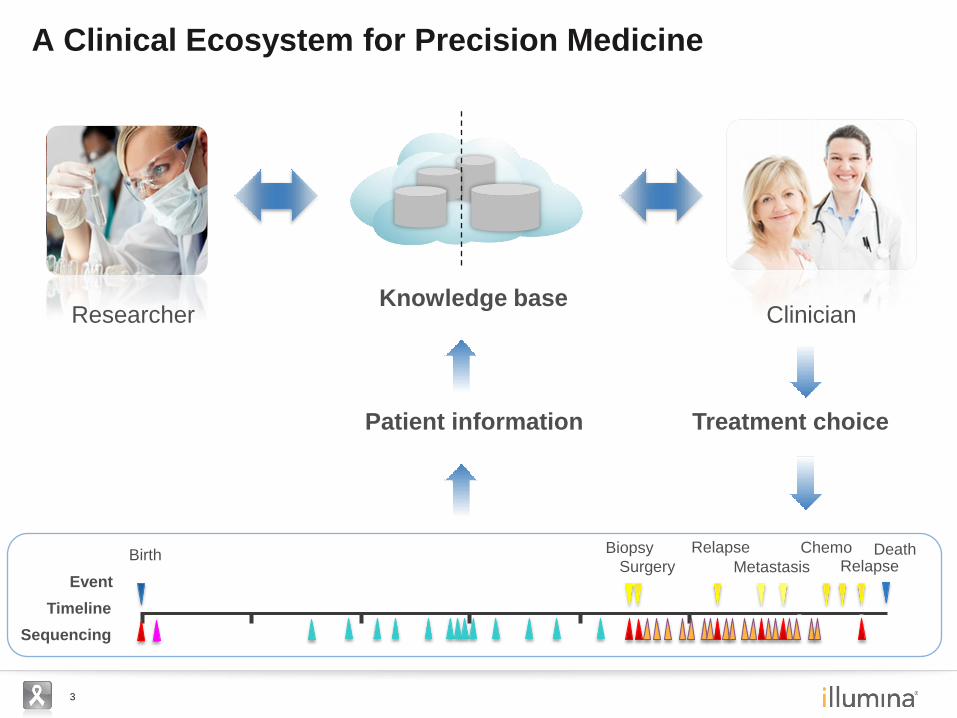
3
A Clinical Ecosystem for Precision Medicine
Patient information Treatment choice
Knowledge base Researcher Clinician
Timeline
Birth Biopsy
Surgery
Relapse
Event Metastasis
Chemo Relapse
Sequencing
Death

4
Fast WGS for genetic diagnosis of intensive care patient
Undiagnosed condition: – Male child presents at 5 months with developmental regression,
hypotonia, and seizures
One WGS test: DNA sample to answer in 4 days
Filter annotations rapidly using generic queries: Number of transcripts with functional variants: 13,367
‘With 2 variants’** and <5% allele frequency: 1,458
And predicted to be functional: 287
And in a gene linked to disease: 35
And predicted to be deleterious: 21
Evolutionarily conserved: 6
Apply control genomes filter: 1
Confirm Menkes diagnosis – A novel hemizygous variant in ATP7A, a gene with mutations
known to disrupt copper metabolism
Kingsmore et al. (Children’s Mercy Hospital)
** shorthand for homozygous., compound heterozygous, hemizygous positions

5
miR-26a-2 PTEN
PDGFRA
c-kit
PI3K AKT Growth
44-year old with glioblastoma recurrence
Not responding to treatments
Sequence genomes from three biopsies and normal genome
WGS of brain tumour defines new treatment options
Swanton et al. (CRUK London Research Institute)
DNA rearrangements amplify
growth regulator genes
PDGFRA gene
normal
amplified
New treatment indicated
(10 days from start)
PDGFRA
c-kit
Amplify
+ + +
+
Growth
C-kit
miR-26a-2
Cancer
+
Cancer
Imatinib
Sunitinib
Pazopanib

6
Trinucleotide contexts around somatic mutations reveal signatures of exposure
Mutation context spectra reveal catastrophic events - kataegis
Genome-wide mutation signatures in cancer
Alexandrov et al. 2013 Sanger
Smoking: Most are NCN>NAN
UV: Most are TCN>TTN
Smoking
UV light
Alexandrov et al. 2013
Sequence context
Nik-Zainal et al. 2012
Context and density
C>T, 14Mb on chr 6 of a BRCA1 mutated tumour

7
Why genomes?
Working with clinical samples
Maximise throughput, coverage and accuracy
Shrink the data footprint
Annotation, interpretation, reporting
Scale up: a fast, convenient, high throughput ISO workflow
Aggregate the information for maximum value
Evolving the technology for medicine
Sequence Analyse Annotate Interpret Answer Sample

8
Simple library prep: reproducible, automated, low cost, low hands-on, fast (4 hrs)
Minimum bias: PCR-free, recovers all genome (reference and non-reference)
Low input DNA: maximises access to clinical samples
Maximum coverage: (depth dependent)
– 30x genome (110 Gb): >96% of hg19 genome ~98% of e! exons
All variant types: all variant types can be detected; both novel and known
Questions/trade-offs:
– Cost, data volume/management, study size (n)
– Sensitivity, diagnostic yield, long term value
Why focus on whole genomes?
Large
SVs
Balanced
translocation
Distant
consanguinity
Uniparental
disomy
Coding
variants
Non-coding
variants
Panel Knowns Knowns No No Yes Knowns
Exome Partial No Partial Partial Yes No
Genome Yes Yes Yes Yes Yes Yes
The complete, accurate genetic make-up of an individual

9
Improving coverage and reducing bias
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1ug PCRv2
1ug PCR
500ng PCR Free
100ng PCR free Sample 1
100ng PCR free Sample 2
HiSeqX PCRFree (V2)
No
rmalised
Gen
om
e C
ov
era
ge
1 ug PCR v2 clusters
1 ug PCR
500 ng PCR-free (gel)
PCR-free (beads)
PCR-free (beads)
PCR-free on X (beads)
ARX exon 2, 77% GC / HiSeq X
Nano v1
Nano v2
PCR-free

10
Improving technology
Patterned flowcells for higher density (2x)
Faster imaging for greater speed (6x)
Improved SBS chemistry for accuracy &
readlength (99.8-99.9% cycle efficiency)
Increasing data rate & reducing cost
0
2
4
6
8
10
2010 2011 2012 2013 2014
WGSPrice($k/30x)
0
100
200
300
400
500
600
2010 2011 2012 2013 2014
DataRate(Gb/day)
2 um features 0.4 um features
(images to same scale)

11
Assessing accuracy
Variant calling accuracy based on Pt truth sets
– enable objective algorithm performance assessment & improvement
Sensitive to depth and quality of alignment
Very low false positive SNP or indel call rate
Pipeline SNV (0 bp) Indel (1-50 bp) Time
(hr)**
Sn (%) Sp (%) Sn (%) Sp (%)
HAS (Isaac) (H2’15) 96.9 99.8 92.3 98.3 5
GATK 3.2 98.1 99.9 88.6 98.9 38
**Using 40 CPU, Intel Xeon @ 2.80 GHz, 132 GB RAM**
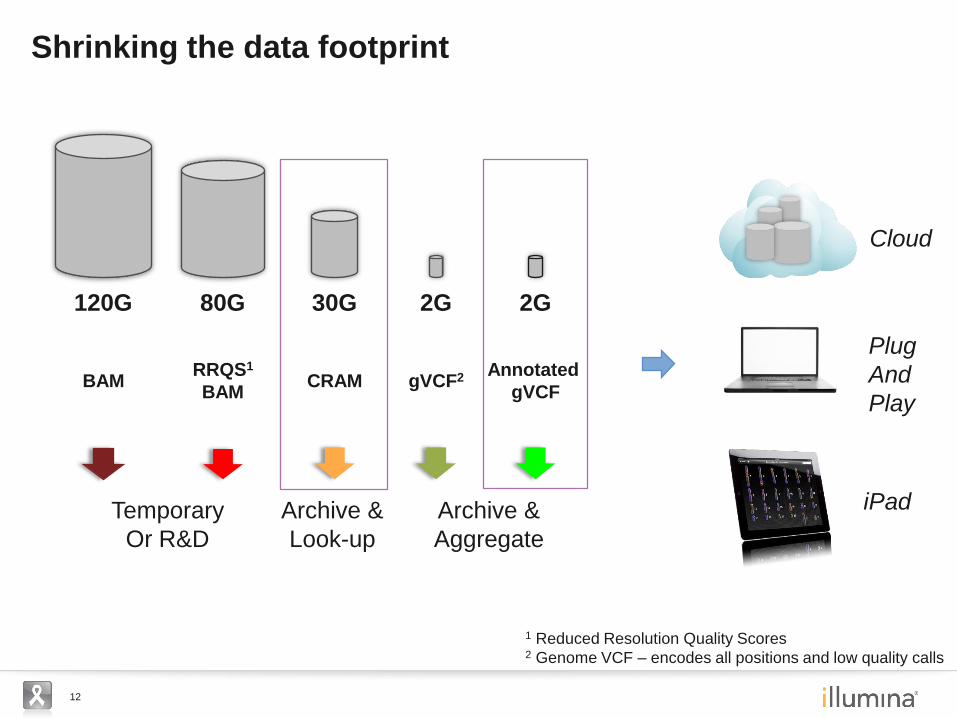
12
Shrinking the data footprint
Archive &
Aggregate
1 Reduced Resolution Quality Scores 2 Genome VCF – encodes all positions and low quality calls
Archive &
Look-up
Temporary
Or R&D
iPad
Plug
And
Play
Cloud
gVCF2
2G
Annotated
gVCF
2G
CRAM
30G
RRQS1
BAM
80G 120G
BAM

13
Adding comprehensive annotation
Capture publicly available/licensed information (Ve!P-based)
– Genes: Nomenclature, coordinates, transcripts (ensembl, NCBI)
– Functional effects: VEP consequence, regulatory regions (ensembl, UCSC, Encode)
– Population information: 1000-genomes, EVS
– Disease association: Clinvar, COSMIC, PharmGKB (+ manual review)
Accelerate and improve efficiency of annotation process
– Streamline reporting algorithms and data storage structures
– Speed: From 11 hr 48 min to 3 min per genome
– Footprint: From 2+8 Gb Cache+RAM to 0.23+0.3 Gb per genome
How do we create medically useful genomes?

14
Tumour and normal genomes
Extra depth of the tumour genome sequence
Sample purity, quality and quantity
Variability in fixation and extraction processes
Evolution of disease and heterogeneity
Somatic variant calling, annotation, interpretation, reporting
Evolving the technology for cancer medicine
Sequence Analyse Annotate Interpret Answer Sample
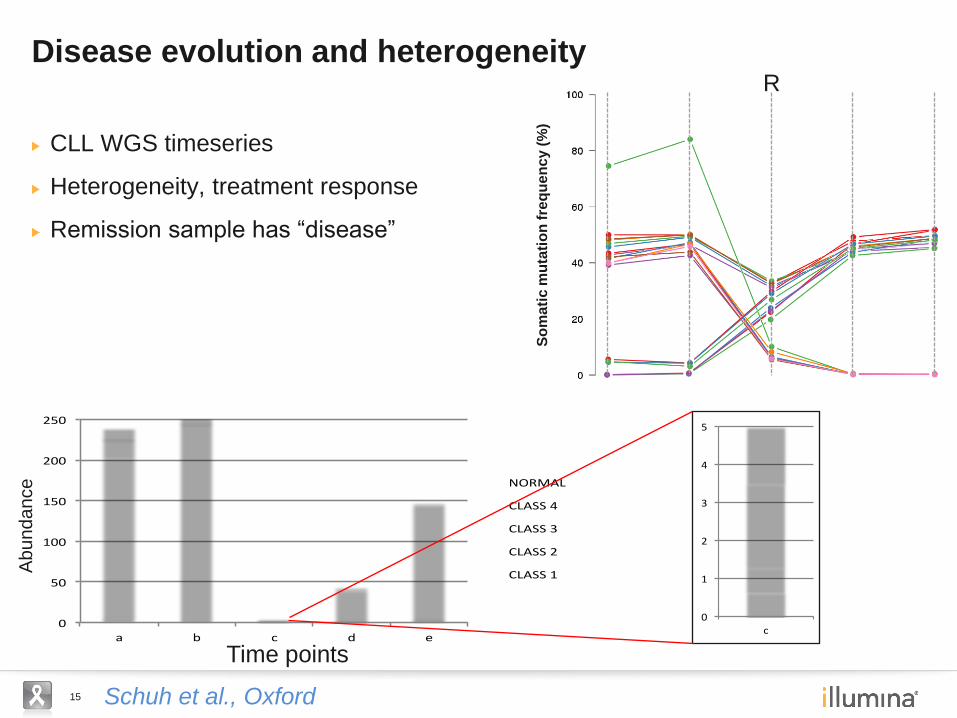
15 Schuh et al., Oxford
CLL WGS timeseries
Heterogeneity, treatment response
Remission sample has “disease”
Disease evolution and heterogeneity
So
ma
tic
mu
tati
on
fre
qu
en
cy (
%)
0
50
100
150
200
250
a b c d e
NORMAL
CLASS4
CLASS3
CLASS2
CLASS1
Time points
Abundance
0
1
2
3
4
5
c
NORMAL
CLASS4
CLASS3
CLASS2
CLASS1
R
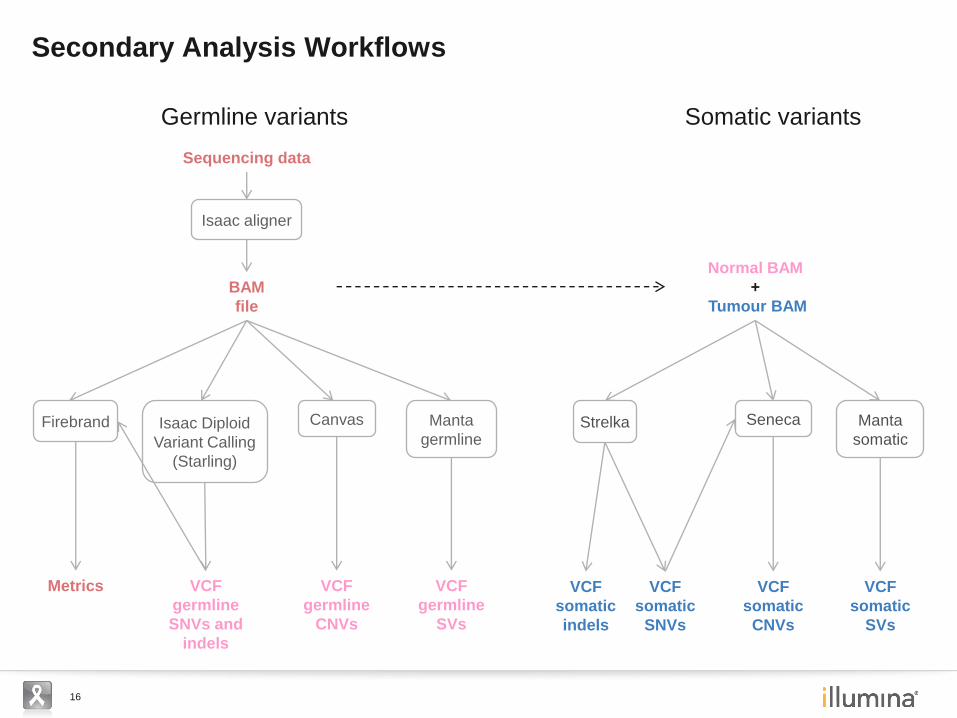
16
Secondary Analysis Workflows
Isaac aligner
BAM
file
Sequencing data
Isaac Diploid
Variant Calling
(Starling)
VCF
germline
SNVs and
indels
Canvas
VCF
germline
CNVs
Manta
germline
VCF
germline
SVs
Firebrand
Metrics
Normal BAM
+
Tumour BAM
Seneca
VCF
somatic
CNVs
Manta
somatic
VCF
somatic
SVs
Strelka
VCF
somatic
indels
VCF
somatic
SNVs
Germline variants Somatic variants
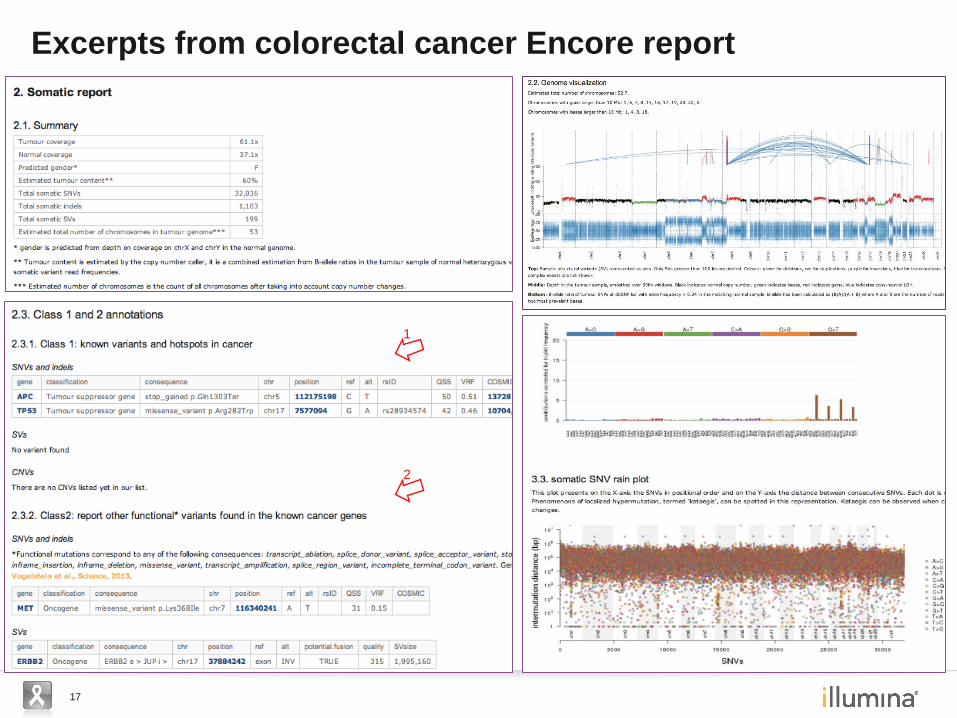
17
Excerpts from colorectal cancer Encore report
1
2

18
Workflow: DNA to annotated Genome
Sample Accession
Sample Quant Quality Control, FFPE
check Library Prep
Library Quality Control qPCR
X10 Sequence Run + QC
Genome build, tumour / normal subtractiongVCF
annotation HiSeq Analysis Software
Analysis Quality Control, identity
check, contamination screen
Network Delivery
Automation + 96 well format
Flowcell prep Genotype
pre
LIMs
Project configured
Track lab and analysis processes
Project management, Pipeline
automation
Library Amp
(if needed)/
Genotype post
Pre-PCR lab
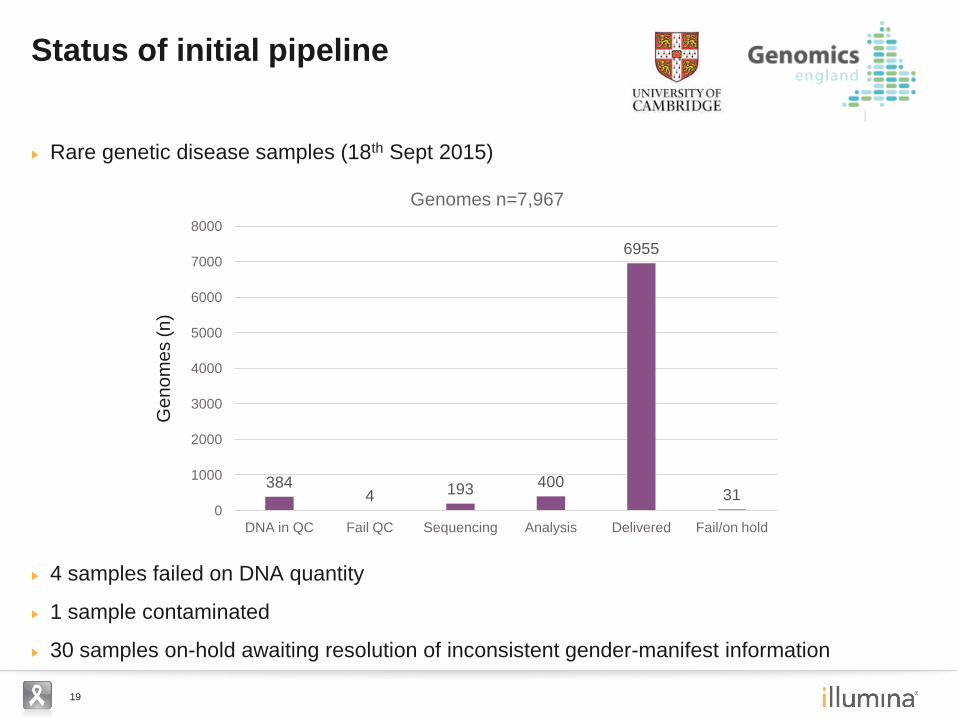
19
Rare genetic disease samples (18th Sept 2015)
4 samples failed on DNA quantity
1 sample contaminated
30 samples on-hold awaiting resolution of inconsistent gender-manifest information
Status of initial pipeline
Ge
no
me
s (
n)
384 4 193 400
6955
31 0
1000
2000
3000
4000
5000
6000
7000
8000
DNA in QC Fail QC Sequencing Analysis Delivered Fail/on hold
Genomes n=7,967

20
Evaluate utility of genome sequencing in healthcare (Genomics England)
A powerful lens for the patient and personalised medicine
Early applications in rare genetic disease and cancer (40% of population)
Population-level screening for systematic studies
Aggregate G, and G+P data to add value (standard, secure, accessible)
Integrate with targeted testing, e.g. pre-natal, MRD and other tests
Future Prospects
Researcher
Treatment choice
Clinician
Patient
Knowledge
Information
21
Acknowledgements
David Bentley
Sean Humphray
Elliott Margulies
Mike Eberle
Ryan Taft
Lisa Murray
Klaus Maisinger
Come Raczy
Semyon Kruglyak
Stewart MacArthur
Philip Tedder
John Peden
Roman Petrovski
Kevin Hall
Keira Cheetham
Jennifer Becq
Miao He
Russell Grocock
Peter Saffrey
Josh Bernd
Richard Shaw
Chris Saunders
Shankar Ajay
Pedro Cruz
Jason Betley
Jacqueline Weir
Zoya Kingsbury
Core Sequencing Group
David Quackenbush
Eddy Kim
Van Lee-Pham
Anna Powell
Francisco Garcia
Kirby Bloom
Tina Hambuch
Erica Ramos
& many others
Illumina The team at Genomics England
Stephen Kingsmore and the Children’s Mercy
Hospital, Kansas City team
Charles Swanton and team, CRUK London
Research Institute
Anna Schuh, Jenny Taylor and the WGS500
consortia, Oxford
Lisa Russell, Christine Harrison and team,
LRCG Newcastle
Mike Stratton and the Cancer Genome Project
team, Wellcome Trust Sanger Institute, Hinxton
Gil McVean and Zamin Iqbal, WTCHG, Oxford
Jan Veldink and team, UMC Utrecht
Willem Ouwehand, Lucy Raymond and the
UCAM Project team, Haematology,
Addenbrookes, Cambridge
Andrew Beggs and team, Cancer Sciences,
Birmingham
Collaborators


















