
Weakly Anti-inflammatory Limonoids from the Seeds of Xylocarpus rumphii
7
Weakly Anti-inflammatory Limonoids from the Seeds of Xylocarpus rumphii Chanin Sarigaputi, † Damrong Sommit, ‡ Thapong Teerawatananond, § and Khanitha Pudhom* ,§ † Program in Biotechnology and § Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand ‡ Department of Chemistry, Faculty of Science, Mahanakorn University, Bangkok 10530, Thailand * S Supporting Information ABSTRACT: Seven new limonoids, namely, xylorumphiins E-J(1-2 and 4-7) and 2-hydroxyxylorumphiin F (3), along with three known derivatives (8-10), were isolated from the seeds of Xylocarpus rumphii. 2-Hydroxyxylorumphiin F (3) and xylorumphiin I (6) displayed moderate inhibitory activity against nitric oxide production from lipopolysaccharide-activated macrophages with IC 50 values of 24.5 and 31.3 μM, respectively. R esearch on limonoids is of growing interest due to the range of biological activities, such as insect antifeedant, growth regulation, antibacterial, antifungal, antimalarial, anti- cancer, and antiviral activities. 1-3 The mangroves of the genus Xylocarpus are known to produce a large number of limonoids, particularly mexicanolides and phragmalins. 4-8 In our continu- ing search for new biologically active limonoids from the plants in this genus, we have reported the isolation of a number of limonoids from the seed kernels of all three species X. granatum, X. moluccensis, and X. rumphii, collected from several areas of the mangrove forests in Thailand. 9-13 In this study, we report the isolation and structural determination of six new mexicanolides (1-6) and a new phragmalin (7), along with three known limonoids, xyloccensins X, E, and K (8-10), from the seeds of X. rumphii collected from Kudee Island, Thailand. The structures of these compounds were established via spectroscopic data or by comparison with literature data. 9-14 Their anti-inflammatory activities were also evaluated via suppression of nitric oxide (NO) production in activated macrophages. ■ RESULTS AND DISCUSSION Xylorumphiin E (1), a white, amorphous powder, had the molecular formula C 35 H 48 O 11 , as established by 13 C NMR data and an HRESIMS m/z 667.2996 [M + Na] + ion (calcd 667.3089), corresponding to 12 indices of hydrogen deficiency. From the 1 H and 13 C NMR spectroscopic data (Tables 1 and 3), it was clear that six of the 12 indices of hydrogen deficiency came from two carbon-carbon double bonds (furan ring) and four ester carbonyl carbons. Therefore, it required six additional rings in the structure of 1. The NMR data of 1 and its 2D correlations (Figure 1) indicated the presence of four tertiary methyls [δ H 0.75 s, 1.04 s (×2), and 1.19 s; δ C 21.0, 22.1, 22.2, and 24.6], a typical β-substituted furan moiety [δ H 6.39, 7.39, and 7.54 s; δ C 110.0, 121.0, 141.5, and 142.9], a methoxycarbonyl (δ H 3.70 s; δ C 51.9 and 173.9), and two isobutyryl groups [δ H 1.04 br s (6H), 1.10 d (J = 6.8 Hz), 1.21 d(J = 6.8 Hz), 2.53 m, and 2.66 m; δ C 17.5, 18.3, 19.1, 20.0, 33.3, 33.9, 174.9, and 177.0]. The aforementioned data strongly suggested that 1 was a mexicanolide-type limonoid. The NMR data of 1 were similar to those of xylorumphiin A, 11 except for the presence of a methine (δ H 2.61 m; δ C 57.1) in place of an oxygenated carbon in xylorumphiin A. The methine function- ality was assigned to C-2 by the 1 H- 1 H COSY correlations from its proton at δ H 2.61 to H-3 and H-30 and by its HMBC correlations to C-1, C-3, and C-30 (Figure 1a). The quaternary carbon resonance at δ C 106.7 was assigned to C-1, a hemiacetal carbon, by its HMBC correlation with a proton resonance at δ H 3.57 (1-OH) that did not show correlation with any carbon in the HSQC spectrum. The HMBC correlations from H-3 [δ H 5.09 d (J = 9.2 Hz)] and H-30 (δ H 6.17 br s) to the carbonyl Received: April 29, 2014 Article pubs.acs.org/jnp © XXXX American Chemical Society and American Society of Pharmacognosy A dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX-XXX
Transcript of Weakly Anti-inflammatory Limonoids from the Seeds of Xylocarpus rumphii

Weakly Anti-inflammatory Limonoids from the Seeds of XylocarpusrumphiiChanin Sarigaputi,† Damrong Sommit,‡ Thapong Teerawatananond,§ and Khanitha Pudhom*,§
†Program in Biotechnology and §Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand‡Department of Chemistry, Faculty of Science, Mahanakorn University, Bangkok 10530, Thailand
*S Supporting Information
ABSTRACT: Seven new limonoids, namely, xylorumphiins E−J (1−2 and 4−7) and 2-hydroxyxylorumphiin F (3), along withthree known derivatives (8−10), were isolated from the seeds of Xylocarpus rumphii. 2-Hydroxyxylorumphiin F (3) andxylorumphiin I (6) displayed moderate inhibitory activity against nitric oxide production from lipopolysaccharide-activatedmacrophages with IC50 values of 24.5 and 31.3 μM, respectively.
Research on limonoids is of growing interest due to therange of biological activities, such as insect antifeedant,
growth regulation, antibacterial, antifungal, antimalarial, anti-cancer, and antiviral activities.1−3 The mangroves of the genusXylocarpus are known to produce a large number of limonoids,particularly mexicanolides and phragmalins.4−8 In our continu-ing search for new biologically active limonoids from the plantsin this genus, we have reported the isolation of a number oflimonoids from the seed kernels of all three species X.granatum, X. moluccensis, and X. rumphii, collected from severalareas of the mangrove forests in Thailand.9−13 In this study, wereport the isolation and structural determination of six newmexicanolides (1−6) and a new phragmalin (7), along withthree known limonoids, xyloccensins X, E, and K (8−10), fromthe seeds of X. rumphii collected from Kudee Island, Thailand.The structures of these compounds were established viaspectroscopic data or by comparison with literature data.9−14
Their anti-inflammatory activities were also evaluated viasuppression of nitric oxide (NO) production in activatedmacrophages.
■ RESULTS AND DISCUSSION
Xylorumphiin E (1), a white, amorphous powder, had themolecular formula C35H48O11, as established by 13C NMR dataand an HRESIMS m/z 667.2996 [M + Na]+ ion (calcd667.3089), corresponding to 12 indices of hydrogen deficiency.From the 1H and 13C NMR spectroscopic data (Tables 1 and
3), it was clear that six of the 12 indices of hydrogen deficiencycame from two carbon−carbon double bonds (furan ring) andfour ester carbonyl carbons. Therefore, it required six additionalrings in the structure of 1. The NMR data of 1 and its 2Dcorrelations (Figure 1) indicated the presence of four tertiarymethyls [δH 0.75 s, 1.04 s (×2), and 1.19 s; δC 21.0, 22.1, 22.2,and 24.6], a typical β-substituted furan moiety [δH 6.39, 7.39,and 7.54 s; δC 110.0, 121.0, 141.5, and 142.9], amethoxycarbonyl (δH 3.70 s; δC 51.9 and 173.9), and twoisobutyryl groups [δH 1.04 br s (6H), 1.10 d (J = 6.8 Hz), 1.21d (J = 6.8 Hz), 2.53 m, and 2.66 m; δC 17.5, 18.3, 19.1, 20.0,33.3, 33.9, 174.9, and 177.0]. The aforementioned data stronglysuggested that 1 was a mexicanolide-type limonoid. The NMRdata of 1 were similar to those of xylorumphiin A,11 except forthe presence of a methine (δH 2.61 m; δC 57.1) in place of anoxygenated carbon in xylorumphiin A. The methine function-ality was assigned to C-2 by the 1H−1H COSY correlationsfrom its proton at δH 2.61 to H-3 and H-30 and by its HMBCcorrelations to C-1, C-3, and C-30 (Figure 1a). The quaternarycarbon resonance at δC 106.7 was assigned to C-1, a hemiacetalcarbon, by its HMBC correlation with a proton resonance at δH3.57 (1-OH) that did not show correlation with any carbon inthe HSQC spectrum. The HMBC correlations from H-3 [δH5.09 d (J = 9.2 Hz)] and H-30 (δH 6.17 br s) to the carbonyl
Received: April 29, 2014
Article
pubs.acs.org/jnp
© XXXX American Chemical Society andAmerican Society of Pharmacognosy A dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXX

carbons (C-1′ and C-1″) of the isobutyryl groups confirmedtheir location at C-3 and C-30, respectively. The relativeconfiguration of 1 was established by NOE interactions (Figure1b). The NOESY spectrum showed close similarity to reportedNOE data of xylorumphiin A.11 The NOE cross-peaks observedfrom H-3 to H-2 and H3-29 indicated an α-orientation of theseprotons and hence a 3β-isobutyryl group, whereas the lack of aninteraction from H-3 to H-5 indicated a β-orientation of H-5.The α-orientation of the 30-isobutyryl group was deduced bythe NOE cross-peak between H-5 and H-30, without theinteractions between H-3/H-30 and/or H-2/H-30. On thebasis of the above results, the structure of 1 was elucidated asshown.Xylorumphiin F (2), a white, amorphous powder, gave a
molecular formula of C36H50O11 as determined by 13C NMRdata and an HRESIMS ion at m/z 657.3270 [M − H]− (calcd657.3269). The MS and NMR data suggested the presence ofan additional −CH2− unit in 2 compared to xyrolumphiin E(1).11 The NMR spectroscopic data of 2 (Tables 1 and 3) weresimilar to those of 1 except for the replacement of an isobutyrylgroup in 1 by a 2-methylbutyryl group at C-30. The existenceof the 2-methylbutyryl group was confirmed by 1H−1H COSYcorrelations between H3-5″/H-2″, H-2″/H2-3″, and H2-3″/H3-4″. The HMBC correlation from H-30 (δH 6.17 br s) to thecarbonyl carbon (δC 174.9) of the 2-methylbutyryl groupdefined its location at C-30. The absolute configuration at C-2″in the 2-methylbutyryl group could be determined according tothe specific rotation of the corresponding acid derived from thealkaline hydrolysis of 2 ([α]D −14.3 for (R)-2-methylbutyricacid and [α]D +19.2 for (S)-2-methylbutyric acid).15,16
Although a 1:1 mixture with isobutanoic acid was obtainedfrom the hydrolysis, isobutanoic acid is optically active.Therefore, the absolute configuration at C-2″ in the 2-methylbutyryl group was assigned as S from the [α]20D valueof +10 (c 0.05, MeOH) of this mixture. Both compounds 1 and2 shared the same relative configuration, as confirmed bysimilar NOE correlations.2-Hydroxyxylorumphiin F (3), a white, amorphous powder,
was assigned a molecular formula of C36H50O12 by13C NMR
data and an HRESIMS m/z 697.3169 [M + Na]+ ion (calcd697.3194). Its 1H and 13C NMR data (Tables 1 and 3) closelyresembled those of 2, except for the presence of an oxygenatedtertiary carbon (δC 82.1) instead of a methine group in 2. Thiscarbon was assigned to C-2 due to its HMBC correlation withH-3 (δH 4.89 s). A proton resonance at δH 3.21 that did notshow correlation with any carbon in the HSQC spectrum wasassigned to 2-OH by its HMBC correlations to C-1, C-2, C-3,and C-30 (Figure S1a). Similar NOE correlations suggestedthat compound 3 possessed the same relative configuration asthose of 1 and 2. The key NOE cross-peak observed in 3 from2-OH to H-3, along with the lack of correlation from 2-OH toH-30, confirmed the α-orientation of 2-OH (Figure S1b). Theabsolute configuration at C-2″ in the 2-methylbutyryl group
Table 1. 1H NMR Spectroscopic Data of 1−4a
1 2 3 4
position(mult., J in
Hz)(mult., J in
Hz)(mult., J in
Hz)(mult., J in
Hz)
2 2.61, m 2.59, m3 5.09, d (9.2) 5.11, d (9.6) 4.89, s 4.89, s5 2.59, m 2.61, m 2.64, m 2.64, m6 2.32, d (10.0) 2.33, d (9.6) 2.37, dd (9.6,
16.4)2.35, m
2.28, br s 2.25, d (16.4) 2.27, br s 2.24, m9 1.44, d (12.4) 1.42, m 1.51, m 1.48, dd (2.8,
12.8)11 1.92, d (14.0) 1.92, dd (3.6,
13.2)1.89, m 1.89, m
1.67, m 1.66, m 1.69, m 1.69, m12 1.83, d (16.4) 1.83, d (16.4) 1.83, m 1.85, m
1.33, m 1.31, m 1.35, m 1.33, m14 2.19, d (10.0) 2.19, d (9.6) 2.75, dd (9.2,
19.6)2.22, d (9.6)
15 3.25, d (20.0) 3.26, d (20.0) 2.22, d (9.2) 3.15, d (20.0)2.73, dd (10.0,20.0)
2.73, dd (9.6,20.0)
3.13, d (19.6) 2.74, dd (9.6,20.0)
17 5.26, s 5.25, s 5.18, s 5.18, s18 1.04, br s 1.05, s 1.03, s 1.05, s19 1.04, br s 1.04, s 1.12, s 1.12, s21 7.54, s 7.53, s 7.55, s 7.53, s22 6.39, s 6.39, s 6.40, s 6.39, s23 7.39, s 7.39, s 7.40, s 7.40, s28 0.75, s 0.75, s 0.73, s 0.73, s29 1.19, s 1.19, s 1.25, s 1.25, s30 6.17, br s 6.17, br s 6.23, s 6.24, d (5.2)7-OMe 3.70, s 3.70, s 3.21, s 3.69, s1-OH 3.57, s 4.21, s 4.25, s2-OH 3.21, s3-Acyl2′ 2.53, m 2.65, m 2.61, m 2.35, m3′ 1.10, d (6.8) 1.11, d (6.8) 1.09, d (6.8) 1.41, m
1.65, m4′ 1.21, d (6.8) 1.21 d (6.8) 1.22, d (6.8) 0.90, t (7.6)5′ 1.21, d (7.2)30-Acyl2″ 2.66, m 2.41, m 2.43, m 2.45, m3″ 1.04, br s 1.66, m 1.67, m 1.28, m
1.42, m 1.28, m 1.67, m4″ 1.04, br s 0.90, t (7.6) 0.91, t (7.6) 0.91, t (7.6)5″ 1.20, d (7.2) 1.08, d (6.4) 1.08, d (7.2)
aData were measured in CDCl3 at 400 MHz.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXB

was determined as S, the same as that in 2, from the [α]20Dvalue of +12 (c 0.05, MeOH) of the corresponding acid.Xylorumphiin G (4) was obtained as colorless crystals. The
molecular formula C37H52O12 was assigned by13C NMR and an
HRESIMS ion at m/z 687.3390 [M − H]− (calcd 687.3381).The NMR data (Tables 1 and 3) indicated that 4 was similar to3, except for the presence of a 2-methylbutyryl group in placeof the isobutyryl group in 3. The HMBC correlations from H-3
and H-30 to the respective carbonyl carbon of the 2-methylbutyryl groups placed these acyl groups at C-3 and C-30. A proton at δH 4.25, which did not display correlation witha carbon in the HSQC spectrum, was assigned to 1-OH; thesecond 2-methylbutyryl group was thus assumed to be attachedat C-3. In order to clarify the full structure and establish therelative configuration of 4, single-crystal X-ray diffractionanalysis was utilized. Thus, the 2-methylbutyryl groups at C-3and C-30 were α-oriented (Figure 2). The absoluteconfigurations at C-2′ and C-2″ in the respective 2-methylbutyryl groups were determined as S due to the [α]20Dvalue of +15 (c 0.03, MeOH) of the corresponding acidobtained from alkaline hydrolysis.Xylorumphiin H (5), a white, amorphous powder, had a
molecular formula of C33H44O12 as determined by 13C NMRdata and HRESIMS based on the molecular ion m/z 631.2781[M − H]− (calcd 631.2749). The NMR spectroscopic data of 5(Tables 2 and 3) were similar to those of xylorumphiin A,11
with the only difference being the presence of an acetyl ratherthan an isobutyryl group at C-3. This deduction was validatedby the HMBC correlation from H-3 to the acetyl carbonyl (δC171.3). The relative configuration of 5 was identical to that ofxylorumphiin A based on analysis of the NOESY data.Xylorumphiin I (6) was obtained as a white, amorphous
powder. It gave a molecular formula of C37H50O12 asestablished by 13C NMR and an HRESIMS ion at m/z685.3266 [M − H]− (calcd 685.3219). The NMR spectro-scopic data of 6 (Tables 2 and 3) were similar to those of 4,except for the presence of a Δ14,15 double bond (δH 6.01 s; δC118.3 and 158.6). The location of the Δ14,15 double bond wascorroborated by HMBC correlations from H-15 to C-8 and C-16 (Figure 3). The presence of two 2-methylbutyryl groups wasconfirmed by 1H−1H COSY correlations. Their positions at C-3 and C-30 were validated by the HMBC correlations from H-3and H-30 to the respective 2-methylbutyryl carbonyls, C-1′ andC-1″. The absolute configurations at C-2′ and C-2″ in therespective 2-methylbutyryl groups were assigned as S from the[α]20D value of +18 (c 0.05, MeOH) of the corresponding acid.The similar NOE correlations suggested that compound 6possessed the same relative configuration as those of 3−5.Xylorumphiin J (7), a white solid, had the molecular formula
C35H42O15 as established by 13C NMR and HRESIMS (m/z701.2471 [M − H]−, calcd 701.2440) data. Analysis of 1D and2D NMR spectroscopic data revealed that 7 was a phragmalinortho ester. The existence of an ortho acetate group wascharacterized by a methyl singlet at δH 1.73, showing an HMBCcorrelation with a quaternary carbon at δC 119.4. This wasfurther supported by the presence of three oxygenated tertiarycarbons at δC 85.4, 85.1 and 86.8, assigned as C-1, C-8, and C-9, respectively, by HMBC correlations of H-14/C-8, H-30/C-8,H3-19/C-1, H3-19/C-9, and H2-29/C-1 (Figure 4a). These datasuggested that 7 was a phragmalin 1,8,9-ortho acetate. TheNMR data of 7 (Tables 2 and 3) were similar to those ofxyloccensin E,13 except for the presence of an oxygenatedmethine (δH 5.06 br s; δC 64.2) instead of the C-15 methylenegroup in xyloccensin E. HMBC cross-peaks from a hydroxyproton at δH 3.12, which was not correlated with any carbon inthe HSQC spectrum, to C-14, C-15, and C-16 confirmed thelocation of this hydroxy group at C-15. Similar NOEcorrelations suggested that compound 7 possessed the samerelative configuration as xyloccensin E.13 The α-orientation ofH-15 was established by its NOE correlation with H-14 (Figure4b).
Table 2. 1H NMR Spectroscopic Data of 5−8a
5 6 7 8
position (mult., J in Hz)(mult., J in
Hz) (mult., J in Hz)(mult., J in
Hz)
23 4.85, s 4.83, s 5.11, s 4.80, s5 2.61, m 2.66, d
(8.8)2.99, d (10.0) 2.66, br d
(9.2)6 2.36, dd (9.2,
16.4)2.18, m 2.46, dd (10.0,
16.4)2.36, m
2.25, d (16.4) 2.35, m 2.24 m 2.16, d(15.2)
9 1.48, dd (2.4,12.8)
2.20, m 2.21, d(10.0)
11 1.90, dd (3.2,13.2)
2.35, m 2.06, m 2.34, m
1.69, m 1.81, m 1.67, m 1.80, m12 1.83, m 2.04, m 1.51, m 2.05, m
1.33, m 1.40, m 1.20, m 1.39, m14 2.21, d (9.2) 2.09, d (2.4)15 3.11, d (19.6) 6.01, s 5.06, br s 6.07, s
2.74, d (9.2,19.6)
17 5.21, s 4.92, s 5.58, s 4.91, s18 1.05, s 1.22, s 1.05, s 1.21, s19 1.12, s 1.14, s 1.16, s 1.14, s21 7.55, s 7.48, s 7.56, s 7.48, s22 6.39, s 6.41, s 6.48, s 6.41, s23 7.40, s 7.41, s 7.42, s 7.41, s28 0.72, s 0.78, s 0.89, s 0.79, s29 1.24, s 1.30, s 1.98, d (11.2) 1.31, s
1.68, d (11.2)30 6.19, s 5.61, s 6.32, s 5.58, s32 1.73, s7-OMe 3.70, s 3.69, s 3.68, s 3.68, s1-OH 4.25, s 4.52, s 4.66, s2-OH 3.27, s 3.90, s 4.13, s15-OH 3.12, br s3-Acyl2′ 2.07, 5 2.30, m 2.26, s 2.29, m3′ 1.66, m 1.65, m
1.40, m 1.40, m4′ 0.90, t (7.2) 0.90, t (7.2)5′ 1.15, d
(7.2)1.14, d (6.8)
30-Acyl2″ 2.65, m 2.30, m 1.94, s 2.54, m3″ 1.09, d (6.4) 1.66, m 1.12, d (7.2)
1.40, m4″ 1.11, d (6.4) 0.92, t (7.6) 1.15, d (6.8)5″ 1.10, d
(7.2)2-Acyl2‴ 2.16, s
aData were measured in CDCl3 at 400 MHz.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXC

Xyloccensin X (8) was obtained as colorless crystals, mp214−216 °C. Compound 8 was first reported in 2006 as amixture with xyloccensin Y.17 In the current study, the 1H and13C NMR data for compound 8 (Tables 2 and 3) were assigned
unambiguously.All isolated compounds were evaluated for their anti-
inflammatory activities by monitoring the inhibition of LPS-induced NO production in J774.A1 macrophages. Only 2-hydroxyxylorumphiin F (3) and xylorumphiin J (6) exhibitedmoderate anti-inflammatory activities, with IC50 values of 24.5and 31.3 μM, respectively, while the remaining compounds didnot show any significant effects at a dose of 50 μM.
■ EXPERIMENTAL SECTION
General Experimental Procedures. Melting points weredetermined on a Stuart Scientific melting point apparatus and areuncorrected. Optical rotations were measured on a PerkinElmer 341polarimeter at 20 °C. UV spectra were recorded on a Shimadzu UV-160 UV−visible spectrometer. IR spectra were measured on a Nicolet6700 FT-IR spectrophotometer. NMR spectra were acquired on aVarian Mercury-400 Plus NMR spectrometer with TMS as internalstandard. HRESIMS was carried out on a micrOTOF-Q II ESI massspectrometer. Single-crystal X-ray diffraction analysis was performedon an Oxford Gemini S Ultra diffractometer.
Plant Material. The fruits of X. rumphii were collected in February2011 from Kudee Island, Thailand, and authenticated by the RoyalForest Department, Bangkok, Thailand. A voucher specimen (BKF
Table 3. 13C NMR Spectroscopic Data of 1−8a
position 1 2 3 4 5 6 7 8
1 106.7 106.6 107.2 107.2 107.1 108.4 85.4 108.52 57.1 57.1 82.1 82.1 82.2 80.9 86.0 81.03 73.4 73.5 80.3 80.3 80.5 82.4 81.1 82.64 38.1 38.0 38.9 38.9 38.7 38.7 46.2 38.75 40.5 40.4 40.3 40.4 40.3 40.3 35.5 40.36 32.5 32.5 32.3 32.3 32.3 31.9 33.3 31.97 173.9 173.9 173.9 173.8 173.9 173.7 172.6 173.78 82.4 82.3 81.1 81.1 81.0 80.4 85.1 80.49 64.4 63.4 63.3 63.3 63.3 51.5 86.8 51.510 43.7 43.7 42.6 42.6 42.6 42.2 45.8 42.311 19.6 19.6 19.7 19.7 19.7 15.1 25.5 15.112 35.9 35.9 35.8 35.9 35.9 25.1 28.8 25.013 36.3 36.3 36.3 36.3 36.2 38.9 36.2 38.914 46.6 46.6 46.5 46.5 46.4 158.6 53.0 158.515 29.0 29.0 29.0 29.1 29.0 118.3 64.2 118.416 169.9 170.0 169.6 169.5 169.5 163.1 174.2 163.117 77.1 77.1 77.2 77.0 77.1 81.2 78.8 81.318 21.0 22.2 22.2 22.1 22.2 19.6 19.2 19.619 22.2 21.0 21.0 21.0 21.0 20.6 16.7 42.320 121.0 121.0 120.8 120.9 120.8 119.9 120.5 120.021 141.5 141.5 141.6 141.5 141.6 141.3 141.0 141.322 110.0 110.0 109.9 109.3 110.0 109.9 109.6 109.923 142.9 143.0 143.1 143.1 143.0 142.9 143.2 142.928 24.6 22.1 24.2 24.2 23.8 24.5 14.6 24.529 22.1 24.6 22.0 22.0 21.9 21.7 40.1 21.730 75.9 75.8 76.7 75.6 75.6 75.4 69.8 75.431 119.432 20.97-OMe 51.9 51.9 52.0 51.9 51.9 51.9 52.0 51.93-Acyl1′ 177.0 177.2 177.6 177.1 171.3 176.7 170.0 176.82′ 33.3 33.9 33.9 40.5 21.3 40.9 21.0 41.03′ 18.3 18.3 18.2 16.8 26.1 26.14′ 20.0 20.0 20.2 25.3 11.4 11.45′ 11.2 16.2 16.130-Acyl1″ 174.9 174.9 174.3 174.1 174.5 174.7 168.2 175.22″ 33.9 40.6 40.7 40.6 34.0 40.9 21.0 34.33″ 19.1 25.3 27.2 27.0 17.9 26.4 18.84″ 17.5 11.2 12.0 11.9 19.5 11.6 19.05″ 16.4 14.9 14.8 15.72-Acyl1‴ 170.42‴ 21.7
aData were measured in CDCl3 at 100 MHz.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXD

No. 1638) was deposited at the Forest Herbarium, Royal ForestDepartment, Bangkok, Thailand.
Extraction and Isolation. The air-dried, powdered seeds of X.rumphii (5 kg) were extracted three times with MeOH (10 L, each for3 days) at room temperature. The extract was concentrated underreduced pressure. The combined MeOH extract was partitionedbetween EtOAc and H2O to obtain the EtOAc crude extract (107 g).The EtOAc extract was fractionated over a column of silica gel with agradient of EtOAc−n-hexane (from 1:9 to 1:0) to give 14 fractions,A−N. Fraction G (7.84 g) was subjected to passage over a column ofSephadex LH20 (MeOH) to afford four fractions (G1−G4), andfraction G2 (376.31 mg) was rechromatographed over silica gel elutingwith an EtOAc−n-hexane gradient (from 3:7 to 1:1) to yield sevensubfractions (G2a−G2g). Fraction G2a (47.9 mg) was purified bysilica gel CC (EtOAc−n-hexane, from 2:3 to 1:1) to give 4 (4.3 mg).Fraction G2e (74.6 mg) was chromatographed over a silica gel columnwith MeOH−CH2Cl2 (2:98), and the major fraction (G2e.4) wasrechromatographed with EtOAc−n-hexane (from 3:7 to 1:1) to obtaincompounds 6 (7.7 mg) and 8 (19.3 mg). Fraction G2f (22.0 mg) waspurified by CC over silica gel with EtOAc−n-hexane (2:3) to givecompounds 1 (2.9 mg) and 3 (11.3 mg). Using the same procedure,fraction G2g (19.7 mg) yielded compound 2 (11.2 mg). Fraction J wasrecrystallized from MeOH to give xyloccensin E (9, 178.8 mg).Fraction K (3.44 g) was subjected to a column of Sephadex LH-20with MeOH to afford six fractions, K1−K6. Fraction K2 (115.6 mg)was further purified by silica gel CC eluted with MeOH−CH2Cl2(4:96) to yield compounds 5 (7.6 mg) and 7 (9.9 mg). Fraction K4was recrystallized from MeOH to afford xyloccensin K (10, 85.3 mg).
Xylorumphiin E (1): white, amorphous powder; [α]20D −13 (c 0.03,MeOH); UV (MeOH) λmax (log ε) 209 (3.24) nm; IR (neat) νmax3370 (br), 2974, 2933, 2878, 1715, 1453, 1377, 1295, 1264, 1195,
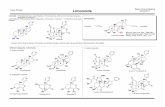
Figure 1. (a) Selected HMBC and COSY correlations of 1. (b) Diagnostic NOE correlations of 1.
Figure 2. ORTEP drawing of 4.
Figure 3. Selected HMBC and COSY correlations of 6.
Figure 4. (a) Selected HMBC and COSY correlations of 7. (b) Diagnostic NOE correlations of 7.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXE

1143, 1061 cm−1; 1H and 13C NMR spectroscopic data (see Tables 1and 3); HRESIMS m/z 667.2996 [M + Na]+ (calcd for C35H48O11Na,667.3089).Xylorumphiin F (2): white, amorphous powder; [α]20D +18 (c 0.1,
MeOH); UV (MeOH) λmax (log ε) 208 (3.50) nm; IR (neat) νmax3424 (br), 2970, 2928, 2863, 1726, 1461, 1383, 1302, 1189, 1140,1059 cm−1; 1H and 13C NMR spectroscopic data (see Tables 1 and 3);HRESIMS m/z 657.3270 [M − H]− (calcd for C36H49O11, 657.3269).2-Hydroxyxylorumphiin F (3): white, amorphous powder; [α]20D
−19 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 209 (3.61) nm; IR(neat) νmax 3404, 2970, 2934, 2876, 1720, 1454, 1379, 1392, 1227,1143, 1062 cm−1; 1H and 13C NMR spectroscopic data (see Tables 1and 3); HRESIMS m/z 697.3169 [M + Na]+ (calcd for C36H50O12Na,697.3194).Xylorumphiin G (4): colorless crystals (MeOH); mp 162−164 °C;
[α]20D −68 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (3.51) nm;IR (neat) νmax 3410 (br), 2967, 2924, 2883, 1720, 1457, 1377, 1292,1189, 1147, 1066 cm−1; 1H and 13C NMR spectroscopic data (seeTables 1 and 3); HRESIMS m/z 687.3390 [M − H]− (calcd forC37H51O12, 687.3381).Xylorumphiin H (5): white, amorphous powder; [α]20D +12 (c 0.1,
MeOH); UV (MeOH) λmax (log ε) 210 (3.64) nm; IR (neat) νmax3440 (br), 2967, 2931, 2883, 1717, 1461, 1370, 1260, 1224, 1137,1014 cm−1; 1H and 13C NMR spectroscopic data (see Tables 2 and 3);HRESIMS m/z 631.2781 [M − H]− (calcd for C33H43O12, 631.2749).Xylorumphiin I (6): white, amorphous powder; [α]20D +4 (c 0.1,
MeOH); UV (MeOH) λmax (log ε) 209 (4.16) nm; IR (neat) νmax3375 (br), 2973, 2934, 2876, 1720, 1461, 1377, 1289, 1260, 1143,1062 cm−1; 1H and 13C NMR spectroscopic data (see Tables 2 and 3);HRESIMS m/z 685.3266 [M − H]− (calcd for C37H49O12, 685.3319).Xylorumphiin J (7): white solid; decomposed at 238 °C; [α]20D −7
(c 0.1, MeOH); UV (MeOH) λmax (log ε) 210 (3.64) nm; IR (neat)νmax 3492, 2957, 2924, 2850, 1730, 1460, 1428, 1370, 1311, 1240,1088 cm−1; 1H and 13C NMR spectroscopic data (see Tables 2 and 3);HRESIMS m/z 701.2471 [M − H]− (calcd for C35H41O15, 701.2440).X-ray Crystallographic Analysis of 4. The single-crystal X-ray
diffraction data were collected at 296 K on a Bruker APEX-II CCDdiffractometer with Mo Kα radiation (λ = 0.710 73 Å). The crystalstructure was solved by direct methods using SHELXS-9718 andrefined with full-matrix least-squares on all F2 data using SHELXS-97to final R values.19 All non-hydrogen atoms were anisotropicallyrefined, except for the oxygen C-3A atom, which was refinedisotropically. The C-3A atom is disordered over two positions withsite occupancies of 0.52 and 0.48. All hydrogen atoms were added atcalculated positions and refined using a rigid model, with C−H = 0.93Å (aromatic), 0.97 Å (CH2), and 0.98 Å (CH3) and O−H = 0.82 Å.Crystallographic data for 4 have been deposited with the CambridgeCrystallographic Data Centre under the deposition number CCDC998571.Crystal data of 4: colorless crystal; C37H50O12, Mr = 688.80, prism,
a = 12.1699(10) Å, b = 12.3165(16) Å, c = 24.075(3) Å, space groupP212121, Z = 4, V = 3608.6(7) Å3, μ(Mo Kα) = 0.07 mm−1, andF(000) = 1200. Crystal dimensions: 0.38 × 0.22 × 0.20 mm3.Independent reflections: 3394 (Rint = 0.035).Absolute Configuration of C-2 of the 2-Methylbutyryl
Group of Xylorumphiins F, G, and I (2, 4, and 6) and 2-Hydroxyxylorumphiin F (3). Each compound (3 mg) was dissolvedin EtOH (0.5 mL) and treated with 10% aqueous KOH solution (1mL). After stirring overnight, the mixture was concentrated andwashed with EtOAc (×3), and the aqueous layer was acidified withHCl to pH 3.0 and extracted with CH2Cl2 (×3). The combinedorganic layer was subjected to Sephadex LH-20 CC (CH2Cl2−MeOH,1:1) to yield 2S-methylbutanoic acid, [α]20D +10, +12, +15, and +18,for compounds 2−4 and 6, respectively, lit. [α]25D +19.2.16
Anti-inflammatory Bioassay. The inhibitory effects of sampleson NO production were evaluated in LPS-activated murine macro-phage J774.A1 cells according to a reported method.20 The cell lineswere seeded in 24-well plates with 1 × 105 cells/well and allowed toadhere for 24 h at 37 °C in 5% CO2. The cells were pretreated withvarious concentrations of test compounds or vehicle (DMSO) for 2 h
and then activated with 1 μg/mL of LPS for 20 h. Indomethacin (IC50= 28.56 μM) was used as a positive control. The culture supernatant(50 μL) of each well was collected, and the concentration of NO wasfurther measured by using Griess reagent. The absorbance wasmeasured at 540 nm with a microplate reader. The nitrite level in thesamples was calculated from the standard curve created from knownconcentrations of sodium nitrite.
■ ASSOCIATED CONTENT*S Supporting Information1D and 2D NMR spectra of compounds 1−7 and CIF file forcompound 4. These materials are available free of charge via theInternet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*Tel: +66-2-2187641. E-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThe authors are grateful for the financial support from ThailandResearch Fund and Chulalongkorn University through theRoyal Golden Jubilee Ph.D. Program (PHD/0009/2553) andthe 90th Anniversary of Chulalongkorn University Fund(Ratchadaphiseksomphot Endownment Fund). This researchhas also been partially supported by the Ratchadaphiseksom-phot Endowment Fund of Chulalongkorn University(RES560530208-AS).
■ REFERENCES(1) Koul, O.; Sing, G.; Singh, R.; Daniewski, W. M.; Berlozecki, S. J.Biosci. (Bangalore, India) 2004, 29, 409−416.(2) Endo, T. M.; Shimada, T.; Moriguchi, T.; Hidaki, T.; Matsumoto,R.; Hasegawa, S.; Omura, M. Plant Biotechnol. 2002, 19, 397−403.(3) Nakagawa, H.; Duan, H.; Takaishi, Y. Chem. Pharm. Bull. 2001,49, 649−651.(4) Zhou, Y.; Cheng, F.; Wu, J.; Zou, K. J. Nat. Prod. 2006, 69, 1083−1085.(5) Cui, J.; Wu, J.; Deng, Z.; Proksch, P.; Lin, W. J. Nat. Prod. 2007,70, 772−778.(6) Yin, S.; Wang, X.-N.; Fan, C.-Q.; Lin, L.-P.; Ding, J.; Yue, J.-M. J.Nat. Prod. 2007, 70, 682−685.(7) Li, M.-N.; Yang, X.-B.; Pan, J.-U.; Feng, G.; Xiao, Q.; Sinkkonen,J.; Satyanandamurty, T.; Wu, J. J. Nat. Prod. 2009, 72, 2110−2114.(8) Li, M.-N.; Yang, S.-X.; Pan, J.-U.; Xiao, Q.; Satyanandamurty, T.;Wu, J. J. Nat. Prod. 2009, 72, 1657−1662.(9) Pudhom, K.; Sommit, D.; Nuclear, P.; Ngamrojanavanich, N.;Petsom, A. J. Nat. Prod. 2009, 72, 2188−2191.(10) Pudhom, K.; Sommit, D.; Nuclear, P.; Ngamrojanavanich, N.;Petsom, A. J. Nat. Prod. 2010, 73, 263−266.(11) Sarigaputi, C.; Nuanyai, T.; Teerawatananond, T.; Pengpreecha,S.; Muangsin, N.; Pudhom, K. J. Nat. Prod. 2010, 73, 1456−1459.(12) Ravangpai, W.; Sommit, D.; Teerawatananond, T.; Sinpranee,N.; Palaga, T.; Pengpreecha, S.; Muangsin, N.; Pudhom, K. Bioorg.Med. Lett. 2011, 21, 4485−4489.(13) Sarigaputi, C.; Teerawatananond, T.; Pengpreecha, S.;Muangsin, N.; Pudhom, K. Acta Crystallogr. 2010, E66, o1348−o1349.(14) Kokpol, U.; Chavasiri, W.; Tip-Pyang, S.; Veerachato, G.; Zhao,F.; Simpson, J.; Weavers, R. T. Phytochemistry 1996, 41, 903−905.(15) Meyers, A. I.; Knaus, G.; Kamata, K. J. Am. Chem. Soc. 1974, 96,268−270.(16) Brechbuhler, S.; Buchi, G.; Milne, G. J. Org. Chem. 1967, 32,2641−2642.(17) Roy, A. D.; Kumar, R.; Gupta, P.; Khaliq, T.; Narender, T.;Aggarwal, V.; Roy, R. Magn. Reson. Chem. 2006, 11, 1054−1057.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXF

(18) Sheldrick, G. M. SHELXS-97, Program for Crystal StructureSolution; University of Gottingen: Germany, 1997.(19) Sheldrick, G. M. SHELXS-97, Program for Crystal StructureRefinement; University of Gottingen: Germany, 1997.(20) Khan, S.; Shin, E. M.; Choi, R. J.; Jung, Y. H.; Kim, J.; Tosun, A.;Kim, Y. S. J. Cell. Biochem. 2011, 112, 2179−2188.
Journal of Natural Products Article
dx.doi.org/10.1021/np5003687 | J. Nat. Prod. XXXX, XXX, XXX−XXXG
















![Tracing the biosynthetic origin of limonoids and their ...bmcplantbiol.biomedcentral.com/track/pdf/10.1186/s12870-018-1447-6the neem tree [11, 12]. The pharmaceutical applications](https://static.fdocuments.us/doc/165x107/608e9a034b4e273b9347b18b/tracing-the-biosynthetic-origin-of-limonoids-and-their-the-neem-tree-11-12.jpg)
