
Università degli Studi di Trieste - Home | OpenstarTs
114
Università degli Studi di Trieste PhD program in MOLECULAR MEDICINE Scientific field: Neuroscience PhD Thesis Study of the regulation of BDNF protein synthesis and BDNF protein levels for clinical applications Emiliano Leone Coordinator: Chiar.mo Prof. Giannino Del Sal Supervisor: Chiar.mo Prof. Enrico Tongiorgi External supervisor: Chiar.mo Prof. Marco Canossa Academic year 2005-2007 (XX cycle)
Transcript of Università degli Studi di Trieste - Home | OpenstarTs

Università degli Studi di Trieste
PhD program in MOLECULAR MEDICINE
Scientific field: Neuroscience
PhD Thesis
Study of the regulation of BDNF protein synthesis and BDNF protein levels for clinical applications
Emiliano Leone
Coordinator: Chiar.mo Prof. Giannino Del Sal
Supervisor: Chiar.mo Prof. Enrico Tongiorgi
External supervisor: Chiar.mo Prof. Marco Canossa
Academic year 2005-2007 (XX cycle)

Names and full addresses of the committee Supervisor: Prof. Enrico Tongiorgi Dipartimento di Biologia - Università degli studi di Trieste Via Giorgieri 10 – 34100 Trieste External supervisor: Prof. Marco Canossa Dipartimento di Farmacologia - Università degli studi di Bologna Via Irnerio 48 - 40126 Bologna President of the committee: Prof. Francesco Tedesco Dipartimento di Fisiologia e Patologia - Università degli studi di Trieste Via Fleming, 22 - 34100 Trieste Committe members: Prof. Stefano Gustincich Settore di Neurobiologia – S.I.S.S.A. Trieste AREA Science Park, Ss 14, Km 163.5 - 34012 Basovizza (TS) Prof. Massimo Levrero Dipartimento di Medicina Interna - Università degli Studi di ROMA "La Sapienza" Regina Elena Cancer Institute, Via delle Messi d'Oro 156 - 00158 Roma

TTaabbllee ooff ccoonntteennttss
11 INTRODUCTIONINTRODUCTION ...................................................................................................................................................................................................................... 11
2.1 Neuropsychiatric disorders ....................................................................................... 1 2.2 The cognitive processes .............................................................................................. 4 2.3 The Neurotrophins .................................................................................................... 12 2.4 Brain-derived neuotrophic factor ............................................................................ 17
22 AIM OF THE THESISAIM OF THE THESIS .................................................................................................................................................................................................. 2277
33 MATHERIALS AND METHODSMATHERIALS AND METHODS .............................................................................................................................................................. 2288
44 RESULTSRESULTS ............................................................................................................................................................................................................................................ 4444
4.1 Preliminary studies ................................................................................................... 44 4.2 Cellular and molecular mechanisms of cognitive processes ................................. 48 4.3 proBDNF/BDNF ratio in the serum of schizophrenic patients ............................ 55 4.4 Development of an ELISA kit for the dosage of proBDNF/BDNF ratio .............. 63
55 DISCUSSIONDISCUSSION ................................................................................................................................................................................................................................ 7766
66 CONCLUSIONSCONCLUSIONS ........................................................................................................................................................................................................................ 8866
ACKNOWLEDGEMENTSACKNOWLEDGEMENTS ........................................................................................................................................................................................................ 8888
REFERENCESREFERENCES ............................................................................................................................................................................................................................................ 8899

ABSTRACT Brain Derived Neurotrophic Factor (BDNF), a member of the family of neurotrophins, is
a key molecule involved in growth, development and modulation of neuronal system.
Due to its critical role in a large number of neuronal processes including synaptic
plasticity, BDNF altered function and levels are associated even more strongly with
many different neuropsychiatric disorders such as schizophrenia and the associated
cognitive impairment.
In this PhD thesis, we demonstrated that BDNF, which mRNA is transported into
dendrites in response to neuronal activity, is capable to be translated directly in the
dendritic compartment after electrical stimulation. Considering that local protein
synthesis (LPS) in dendrites is commonly accepted as a mechanism strongly involved in
synaptic plasticity and cognitive processes, we designed a cellular assay based on the
detection of BDNF LPS, for the identification of developing compounds able to improve
cognitive performances in patients affected by neuropsychiatric disorders.
In addition, we studied BDNF protein levels in serum of schizophrenic subjects, finding
an alteration of the ratio between BDNF and its precursor form (proBDNF), known to
elicits biological effects opposite to mature BDNF on neuronal system. Based on this
evidence, we developed an ELISA kit for the measurement of proBDNF/BDNF ratio
that could represent a powerful tool for diagnosis of psychosis and for the follow-up of
the medical treatment.

List of papers
1. Leone E., Carlino D., De Vanna M., Tongiorgi E. - The ratio between BDNF
precursor and its mature form is decreased in serum of schizophrenic patients. –
Submitted to Journal of Neurochemistry.
2. Leone E., Vicario A., Baj G. – Business Plan “Neuroscrin” – selected as one of the
best ten projects of the university of Trieste “Start Cup 2006” competition.
3. Vicario A., Baj G., Leone E. and Tongiorgi E. (2006) – Evolutionary conserved
mechanisms of RNA trafficking in neurons and the regulation of spine morphology –
Caryologia Vol. 59 n. 4: 413-423.
4. Baj G., Leone E., Gan W.B., Chao M. V. and Tongiorgi E. - BDNF mRNA splice
variants represent a spatial code to regulate local plasticity of dendrites and spines in
developing and mature neurons – Submitted to PNAS.

List of abbreviations
MDD: major depressive disorder
AD: Alzheimer’s disease
ADHD: attention deficit hyperactivity disorder
ANOVA: Analysis of variance
BD: bipolar disorder
BDNF: brain-derived neurotrophic factor
BMP: bone morphogenetic protein
BORB: Birmingham Object Recognition Battery
CA1:Cornus ammoni 1
CA3: Cornus ammoni 3
CaMKIIα: Calcium Calmodulin Kinase alpha
cAMP: Adenosine 3'5' Cyclic Monophosphate
CDS: Coding Sequence
CNS: central nervous system
CPT: Continuous Performance Test
CREB: cAMP response element-binding
DEPC: diethil pyrocarbonate
DG: dentate gyrus
DIV: Days In Vitro
D-MEM: Dulbecco’s Modified Eagle Medium
dNTP: Deoxynucleotide Triphosphate
DSM-IV: Diagnostic and Statistical Manual of Mental Disorders
EDTA: ethylenediaminetetraacetic acid
EGF: epidermal growth factor
ELISA: Enzyme-Linked Immunosorbent Assay
ER: Endosplamic Reticulum
ERK: Extracellular Regulated Kinase
FGF: fibroblast growth factor
FRAP: fluorescence recovery after photobleaching
GFP: Green Fluorescence Protein

HRP: horse radish peroxidase
IEGs: Immediate-Early Genes
IGF: insulin-like growth factor
ISH: In situ hybridisation
LPS: Local Protein Synthesis
LTD: Long Term Depression
LTP: Long Term Potentiation
LTP1: early-LTP
LTP2: intermediate Late-LTP
LTP3: late-LTP
MCID: Micro Computer Imaging Device
MEM: Minimum Essential Medium
mGluR: metabotropic glutamate receptor
MRI: magnetic resonance imaging
mRNA: messenger Ribonucleic Acid
NGF: nerve growth factor
NMDA: N-methyi-D-Aspartate
NT: neurotrophin
NTR: neurotrophin receptor
p75NTR: p75 Neurotrophin Receptor PBS: Phosphate Buffered Saline
PCR: Polymerase Chain Reaction
PDGF: platelet-derived growth factor
PFA: Paraformaldheyde
PNS: Periferal Nervous System
ROD: Relative Optical Density
RT: Room Temperature
RT-PCR: Reverse Transcriptase-Polymerase Chain Reaction
SDS: Sodium Dodecyl Sulfate
SSC: Standard Sodium Citrate
TGF: transforming growth factor
TGN: trans-Golgi network

TMT: Trail Making Test
Trk: Tropomiosin receptor kinase
tRNA: transfer Ribonucleic Acid
UTR: Untranslated Region
VOSP: Visual Object Space Perception

PREFAX
Although human interest in the nature of the mind sure goes back to long time ago, the
systematic scientific search for the causes of mental disorders did not begin until the
latest part of the 19th century. Even if from different perspectives, pionieristic works of
renowned scientists like Emil Kraepelin, Sigmund Freud or Adolph Meyer began to shed
light on previously not understood aspects of the human brain, including mental
illnesses. During the following years, an increasing number of neurologists and
psychologists focused their attention on psychosis, providing a deeper knowledge of the
mechanisms involved in the disorders.
In parallel with the scientific findings, incidence and prevalence of these mental disturbs
have rapidly grown, probably due to the even more stressful 20th century lifestyle and
other environmental factors. Today, neuropsychiatric disorders represent the second
largest cause of morbidity and premature mortality worldwide. The World Health
Organization has estimated that, collectively, neuropsychiatric disorders comprise 13%
of all reported diseases, having a terrific impact on life quality of the patients affected.
These disorders include major depression, anxiety, schizophrenia, bipolar disorder,
obsessive-compulsive disorder, alcohol and substance abuse, and attention-deficit
hyperactivity disorder and account for 50% of the disability in less developed and
developing countries. Statistics can become even more alarming, considering that
approximately one in five of worldwide population will experience an episode of a
psychiatric illness such as schizophrenia, a mood disorder (depression and bipolar
disorder) or anxiety along his life. The prevalence of these disorders, and their personal
and social costs, has fuelled a half century of research aimed at elucidating the etiologies
and pathophysiological mechanisms of these devastating disorders with the ultimate goal
of designing pharmacotherapies that can correct the underlying neurochemical defects.
However, despite of the outstanding efforts employed, many drugs in use today for
treating neuropsychiatric disorders are at best refinements of compounds identified over
40 years ago and found to be effective by empirical observations. As the biochemical

mechanisms of action of the effective agents were elucidated, theories were postulated
concerning the neurochemical bases for the disorders. Thus, in the 1960s, the dopamine
hypothesis of schizophrenia and the monoamine theory of depression were introduced,
based in large part upon the abilities of antischizophrenic drugs to block dopamine
receptors and antidepressant drugs to increase synaptic levels of monoamine
neurotransmitters. These concepts have continued to address drug development efforts
until today. Recent results, particularly the identification of gene polymorphisms
influencing a multitude of biochemical pathways, have revealed a molecular complexity
of these disorders that was unappreciated until the past decade. It is increasingly clear
that neuropsychiatric disorders arise from interactions of multiple predisposing genes of
variable penetrance overlaid by diverse experiential and environmental influences.
Elucidating the role of these genes in the pathogenesis and in the pathophysiology of
psychosis could lead to an outstanding step forward in the diagnosis and the treatment of
these disorders, including some common core features as cognitive impairment.

Introduction
1. INTRODUCTION
1.1 Neuropsychiatric disorders
Neuropsychiatric disorders are a broad range of psychosis that include depression,
bipolar disorders, anxiety, obsessive-compulsive disorder, attention deficit hyperactivity
disorder, alcohol and drugs abuse, schizophrenia and other alterated mental conditions.
To introduce these disorders, sharing some common aspects but also many peculiar
features, it is important to underline their vast diffusion and to briefly summarize their
characteristics. Thus, considering the incidence, the prevalence, the social costs and the
amount of scientific information, it is possible to individuate four principal psychoses:
Depression:
Major depressive disorder (MDD) is a serious disorder that affects approximately 17%
of the population at some point in life, resulting in major social and economic
consequences (Hirschfeld 2002). According to the Diagnostic and Statistical Manual of
Mental Disorders (DSM-IV), MDD is a heterogeneous disorder that manifests with
symptoms at the psychological, behavioural and physiological levels. There is still very
little known about the neurobiological alterations that underlie the pathophysiology or
treatment of MDD. Several lines of evidence suggest that depression in most people is
caused by interactions between a genetic predisposition and some environmental factors
(Costello et al. 2002; Merikangas et al. 2002; Nestler et al. 2002; Nestler et al. 2002).
Bipolar disorder:
Bipolar disorder (BD), also known as manic-depressive illness, is a brain disorder that
causes unusual shifts in a person’s mood, energy, and ability to function. More than 2
million American adults, or about 1% of the population age 18 and older in any given
year, have BD (Spearing 2001). BD typically develops in late adolescence or early
adulthood. However, some people have their first symptoms during childhood, and some
develop them later in life. BD is often not recognized as an illness, and people may
1

Introduction
suffer for years before it is properly diagnosed and treated. Like diabetes or heart
disease, BD is a long-term illness that must be carefully managed throughout a person’s
lifetime.
Attention deficit hyperactivity disorder (ADHD):
Attention deficit hyperactivity disorder (ADHD) is a behavioural diagnosis based on the
presence of developmentally inappropriate levels of impulsivity, overactivity and
inattentiveness. The overall prevalence for ADHD among children is estimated at around
3–10% (Burd et al. 2003; Faraone et al. 2003; Ford et al. 2003), depending on the
measure used and the population sampled. Boys are five times more often affected than
girls (Ford et al. 2003). The disorder persists into adult life in around two-thirds of cases,
either as the full condition or persistence of some symptoms associated with
impairments in psychosocial functioning (Faraone et al. 2004; Asherson 2005).
Schizophrenia:
Schizophrenia is a chronic and debilitating mental illness characterized by loss of
contact with reality (psychosis). It affects 1% of the world's population, or around 50
million people, across all countries and socio-economic classes. The incidence of
schizophrenia is concentrated in the 15–24 age group for both sexes, whilst the incidence
rate for males in this age group is 0.06% compared to 0.04% for the female population.
After the age of 25, the incidence of schizophrenia drops rapidly for both sexes,
remaining relatively constant at 0.005% in those over the age of 45. The US has by far
the largest prevalent schizophrenic population, consisiting of nearly 3.5 million patients.
The aetiology of schizophrenia is complex, with both genetic and environmental
components and the molecular basis of the disorder is still poorly understood. Familial
studies have shown that the risk of developing schizophrenia is greater in parents,
children and siblings of patients with the disorder than in the general population with the
risk being highest in the monozygotic twin of an affected individual (Murphy et al.
1996). A genetic basis for schizophrenia, however, cannot be inferred from the increased
incidence in families as not all conditions that run in families are genetic. Recent studies
2

Introduction
using magnetic resonance imaging (MRI) techniques have revealed anatomical
abnormalities in some schizophrenic brains, a reduction in the volume of the thalamus,
amygdala, hippocampus and an increase in ventricle size (Andreasen et al. 1994; Nelson
et al. 1998). It is thought that these structural changes result from the combined actions
of genes with other environmental factors such as a viral infection or perinatal injury.
There is currently no simple laboratory test to make a diagnosis of schizophrenia which
is based on symptomatic evaluation of the patient using certain tests to help rule out
general medical conditions which could cause psychotic symptoms. However, the
diagnosis of schizophrenia can be extremely difficult, with many patients going
unrecognized for years. The symptoms necessary for definitive diagnosis may go
unnoticed, or do not show themselves fully until the illness is advanced. Many
physicians, well aware of the stigma that still surrounds schizophrenia, do not like to
make a definitive diagnosis until they are sure that it is the correct one, with many
adopting a 'watchful waiting' approach. In addition, up to 10% of the schizophrenic
population may never make themselves eligible for diagnosis and this group of
schizophrenics includes those patients whose symptoms never become sufficiently
serious to warrant a visit to the physician, pre-diagnosis suicides and the schizophrenic
homeless population.
Symptoms of schizophrenia include:
• positive symptoms, such as delusions (false beliefs based on incorrect
perception of reality), hallucinations (sensory perceptions that occur in the
absence of external stimuli), and disorganized speech or behaviour.
• negative (or deficit) symptoms such as affective flattening, alogia and
avolition.
Pharmacological treatment of schizophrenia could be divided in two main classes of
compounds called typical and atypical antipsychotics drugs. Typical antipsychotics,
although having effects on the positive symptoms of schizophrenia, also cause unwanted
side effects including extra pyramidal symptoms. This class of compounds has a high
3

Introduction
affinity for D2 receptors. Atypical antipsychotics, principally targeting a wider range of
receptors, are generally more effective against the negative symptoms of schizophrenia,
with less effects on extra pyramidal symptoms. However, many atypical antipsychotics
also cause unwanted side effects including elevations in prolactin and weight gain.
In addition, a core component of the disorder is cognitive deficit, a peculiarity shared
with almost all neuropsychiatric disorders. Schizophrenia patients have been
demonstrated to show impairments in cognitive performances (Keefe et al. 2005),
leading to a significant aggravation of life conditions and quality. Despite this and the
large number of studies addressing cognition, this aspect of the disorder remains the
most difficult to treat.
1.2 The cognitive processes
Defining cognitive processes is not so easy, not only because of the high complexity of
the phenomenon and its poor comprehension, but also because it depends on the
discipline that is trying to describe it. From a psychological point of view, the term
cognition can be referred to abstract concepts such as mind, reasoning, perception,
intelligence, learning, and many others that are related to numerous capabilities of the
human brain. Consequently, this description tends to apply to processes such as memory,
attention, perception, action, problem solving and mental imagery. However, cognitive
processes is the objects of numerous studies in various other fields as for instance
neurology, neuroscience, philosophy, systemics and in more recent years molecular and
cellular cognition, a new scientific branch that integrate molecular, cellular, and
behavioral mechanisms, trying to find common aspects and treatments for cognitive
disorders.
Even if an exhaustive cellular and molecular explanation of cognitive process is far to be
reached, it is possible to identify synaptic plasticity, the ability of the brain to change the
strength of synaptic connections in response to different stimuli, as a common
mechanism to several mental faculties.
4

Introduction
Based on very early experimental evidence that leaded Donald Hebb in 1949 (Hebb
1949) to postulate its famous theory (“cells that fire together, wire together”), a huge
number of studies tried to describe synaptic remodelling and its relathionship with
cognition. It is now generally accepted that synaptic plasticity is the mechanism that
underlies learning and memory formation. To study the molecular and cellular bases of
these processes, scientific research identified some cellular models of synaptic plasticity,
whose the best suited seems to be Long Term Potentiation or LTP.
Cellular mechanisms of cognitive processes: Long Term Potentiation
Firstly described as a synaptic potentiation of greater magnitude and persistence due to a
repeated delivery of conditioning stimuli in the DG of the hippocampus (Bliss et al.
1993), Long Term Potentiation (or its opposite equivalent Long Term Depression) has
rapidly arisen as a fundamental tool to investigate the common aspects of mental
processes.
Experimentally, LTP can be induced in a variety of ways; however, the most widespread
method is high-frequency (tetanic) and/or patterned electrical stimulation of afferent
fibers. Depending on the receptors implicated, it can be distinct in two different forms:
NMDA receptor-dependent (CA3–CA1 synapses in the hippocampus) (Palmer et al.
2004) and NMDA receptor-independent (DG–CA3 mossy fiber synapses) (Nicoll et al.
2005). Following the induction, from a temporal and functional point of view, it is
possible to divide LTP in three different phases (Abraham 1991), as schematically
represented in figure 1:
• LTP1 or Early-LTP: depending on post-translational modification of existing
proteins.
• LTP2 or intermediate Late-LTP: depending on new protein synthesis.
• LTP3 or Late-LTP: depending on new gene transcription and new protein
synthesis.
5

Introduction
Several studies have demonstrated that the switch from the early to the late phase of LTP
and its maintenance requires de novo protein synthesis. Indeed, blocking protein
synthesis in hippocampal CA1 region (Frey et al. 1988), in Mossy-fiber CA3 synapses
(Huang et al. 1994), 1994) or in C-fiber evoked field potentials in spinal dorsal horn (Hu
et al. 2003) results in a inhibition of late phase-LTP.
Figure 1. Early and Late-phase LTP. The early phase (LTP1) involves post-translational modification of existing proteins, the intermediate phase (LTP2) depends on new protein synthesis, while the late phase (LTP3) requires not only new protein synthesis but also specific gene transcription. Each phase is regulated by intracellular calcium (Raymond 2007).
6

Introduction
Once switched from the early to the late phase, LTP is also depending upon new gene
transcription for its maintenance. A miscellany of Immediate-Early Genes (IEGs)
encoding transcription factor and non-transcription factor proteins are induced following
LTP induction (Williams et al. 1995; Tsui et al. 1996; Lanahan et al. 1997; Bozon et al.
2003; Steward et al. 2007). Transcription factor IEGs such as zif268 (a.k.a. egr-1, ngfi-a,
krox24) and nurr1 regulate late response genes, although the targets of these genes have
yet to be identified. The non-transcription factor genes encode synaptic proteins such as
activity-regulated cytoskeleton-associated protein (Arc; a.k.a. activity-regulated gene,
Arg3.1) and Homer1a, as well as secreted proteins such as tissue plasminogen activator
and neuronal activity-regulated pentraxin. However, LTP maintenance needs not only
new gene transcription but also new protein synthesis.
Once established the importance of new protein synthesis in a cellular analogue of
learning and memory as LTP, it remains to be clarified how nervous system could assure
spatial and temporal specificity of protein synthesis only to activated synapses. At
present, three models are believed to be the most suited to explain the phenomenon
(Jiang et al. 2002). The first one proposes a specific targeting of new synthesized
proteins from the cell nucleus to activated synapses, the second one suggests a specific
marking of activated synapses in order to deliver new synthesized proteins to them (or a
specific capture ability), and finally the third one indicates local protein synthesis in
dendrites as the responsible for specificity (see figure 2).
Although the first two models might play a secondary role as well, a wide number of
scientific evidence demonstrates that local protein synthesis is the central mechanism
used by neurons to solve the problem.
7

Introduction
Figure 2. The three models proposed for synaptic activity-induced protein synthesis in postsynaptic neurons (Jiang et al. 2002).
Molecular mechanisms of cognitive processes: local protein synthesis in dendrites The discovery of polyribosomes in neuronal dendrites, and the preferential localization
of them at the bases of dendritic spines (Steward et al. 1982), firstly suggested the idea
of local protein synthesis. In the next years, an increasing number of studies revealed the
presence of almost every translation machinery component in the dendrites, including
tRNAs, initiation and elongation factors, endoplasmic reticulum, Golgi cisternae and
elements of the co-translational signal recognition mechanism (Tiedge et al. 1996;
Gardiol et al. 1999; Horton et al. 2003), as well as a restricted group of mRNAs
encoding for proteins considered crucial for synaptic plasticity (Steward 1997; Tongiorgi
et al. 1997; Kuhl et al. 1998). Based on this data, and on an early study by Feig and
Lipton (Feig et al. 1993) in which electrical stimulation was found to induce new protein
synthesis (labelled with [3H] leucines) in the dendrites but not in the cell body, several
works in the last ten years focused their attention on local protein synthesis using
different techniques. In one of these studies, production of genetically modified mice
with a mutation able to disrupt the dendritic targeting of CaMKIIα mRNA (an important
gene involved in plasticity) produced a reduction in LTP and an impairment of spatial
8

Introduction
memory, associative fear conditioning and object recognition memory (Miller et al.
2002). Another research group, using the 5’and 3’untranslated regions of the same gene
flanking the coding sequence of green fluorescent protein, created a reporter protein with
both dendritic localization and translational regulation (Aakalu et al. 2001). Stimulation
of hippocampal neurons able to induce LTP, determined an increase in new dendritic
protein translation revealed with FRAP (fluorescence recovery after photobleaching).
Moreover, the stimulation of protein synthesis was blocked by a well-known inhibitor of
protein synthesis (anisomycin) and not observed in untreated neurons. Similarly,
choosing a mechanical method to isolate dendrites from the cell body, Kang and
Schuman (Kang et al. 1996) demonstrated that LTP requires protein synthesis and still
persists after removal of the cell body, strongly suggesting that dendritic local protein
synthesis is capable to support LTP. Different stimulation protocols leads to the same
conclusion deducted in this experiment (Cracco et al. 2005; Vickers et al. 2005). Indeed,
mechanical isolated dendrites from the CA1 region of rat hippocampus treated with
anysomicin or rapamycin, and then stimulated with two tetanic trains in order to induce
LTP, showed a decline in potentiation with respect to untreated dendrites. Local protein
synthesis has been reported to be important also in Long Term Depression (LTD). In an
elegant work, bath application of an agonist of metabotropic glutamate receptor
(mGluR) was used to induce LTD in hippocampal slices (Huber et al. 2000), and the
depression was shown to be not perturbed in isolated dendrites where the cell body was
mechanically removed. Furthermore, administration of anisomycin or mRNA cap
analogues resulted in inhibition of the establishment of LTD, whilst inhibition of
transcription did not affect it.
Thus, it is becoming increasingly clear how local protein synthesis is a crucial
mechanism for cognitive processes. Although probably other mechanisms are involved
in induction, maintenance and modulation of LTP and synaptic plasticity, experimental
evidence strongly suggests that dendritic synthesis of new proteins is essential for
memory consolidation.
9

Introduction
Alterations of cognitive processes and its role in neuropsichiatric disorders
Even considering only the molecular and cellular mechanisms that control learning and
memory formation described above, it appears clear that cognitive processes is a high
complex phenomenon in which interactions between different neuronal districts and
brain areas need to be extremely fine regulated. Hence, the alteration of only one of the
numerous pathways implicated may lead to a dramatic effect on the overall capability of
cognitive performance.
Cognitive impairment has been extensively described both by psychiatry and
neurobiology. Psychiatry has tried to define it as a difficulty in learning, memory
formation and storage, reasoning, judgment, intuition, lack of awareness and insight.
Conversely, instead of giving a very general description, neurobiology has investigated
restricted aspects of cognitive deficit, looking for the fundamental mechanisms of the
impairment.
Numerous attitudinal and psychological tests are currently used to estimate the degree of
the dysfunction (table 1).
It is possible to find alterations of the cognitive pathways in almost all neuropsychiatric
disorder. From its earliest conceptualizations, cognitive dysfunction has been viewed as
a core feature of schizophrenia (Kraepelin, 1919). Over the past 15 years, a wealth of
evidence has provided support for this view, suggesting that neurocognitive deficit is a
hallmark of schizophrenia evident, by current estimates, in at least 70% of patients
(Palmer et al., 1997), both at disease onset (Saykin et al., 1994) and after many years of
treatment (Harvey et al., 1999). In these patients, basic cognitive functions such as
learning and memory are altered and often compromised.
Compared to schizophrenic patients, cognitive impairment is observed in a lesser degree
in bipolar patients. Nevertheless, the pattern of cognitive dysfunctions is similar in both
groups (Martinez-Aran et al. 2002; Daban et al. 2006) and seem to persist in the long-
term patients (Balanza-Martinez et al. 2005) in relationship with the history of psychotic
symptoms (Martinez-Aran et al. 2008).
10

Introduction
Screening
Mini-Mental State Examination
Attention and executive functions
Trail Making Test (TMT)
Language
Street Completion Test
BORB
VOSP
Memory
Digit span
Corsi span
Speech Test
Attention
Spinnler Matrix Attention Test
Continuous Performance Test (CPT)
Bourbon-Wiersma Test
Table 1. List of the most commonly used neuropsychological tests for the evaluation of cognitive performances. Tests are grouped by the cognitive function that they measure (screening, language, memory and attention).
Also depression (Mitchell et al. 1996; Potter et al. 2007) and Attention Deficit
Hyperactivity Disorder (Willcutt et al. 2005) are characterized by compromised mental
performances. In particular, spatial storage and spatial “central executive” components
appear most strongly compromised in ADHD, although verbal storage and verbal
“central executive” functioning are affected as well (Martinussen et al. 2005).
Various molecular pathways seem to be involved in cognitive impairment. However,
one of the most intriguingly class of molecules appears to be the neurotrophin family.
11

Introduction
1.3 The neurotrophins
Working on a mouse sarcoma tumour implanted close to the spinal cord of developing
chicken, Levi-Montalcini and Hamburger (1953) discovered that it secreted a soluble
factor inducing the hypertrophy and fiber outgrowth of sympathetic neurons.
Subsequently, in vitro studies revealed that this factor was a relatively low molecular
weight protein factor, which was later identified as nerve growth factor or NGF (Levi-
Montalcini et al. 1954; Cohen et al. 1957; Levi-Montalcini et al. 1963). This finding
opened a new field of research in neurobiology devoted to the identification and
elucidation of the cellular functions of the so called neurotrophic factors (in which is
possible to include also other molecules such as: transforming growth factor-_ (TGF-_),
insulin-like growth factor (IGF), epidermal growth factor (EGF), fibroblast growth
factor (FGF), interleukin-6, bone morphogenetic protein (BMP), platelet-derived growth
factor (PDGF)). Among this protein family, the neurotrophins (NTs) is of fundamental
importance, because of their widespread expression in nearly all neuronal populations in
the CNS and PNS and because of their well-known physiological role in neuronal
survival, process outgrowth and regulation of synaptic plasticity.
Two decades after the identification of NGF as the prototypic NT for PNS neurons,
another neurotrophic factor was isolated from pig brain, which was named brain-derived
neurotrophic factor (BDNF) and was found to be highly homologous in protein sequence
to NGF (Barde et al. 1982; Leibrock et al. 1989). Since then, four additional members of
the family of NTs have been identified (neurotrophin-3 (NT-3), neurotrophin-4/5 (NT-
4/5), neurotrophin-6 (NT-6) and neurotrophin-7 (NT-7)). NT-6 and NT-7 have been
found only in fish (Gotz et al. 1994; Lai et al. 1998).
All NTs are generated as pre-pro-neurotrophin precursors (approximately 240–260
amino acids long), which are further processed until they are eventually secreted as
mature homodimeric proteins into the extracellular space (length of monomer: 118–129
amino acids). The road to reach the extracellular space is characterized by some
common steps. The mRNA is addressed to ER, the synthesized pro-protein is then
packed in non-clathrin-coated transport vesicles and finally accumulates in the
12

Introduction
membrane stacks of the trans-Golgi network (TGN). Here, NTs can be sorted to the
constitutive pathway, where secretory vesicles are transported by default to cell
periphery, or to the regulated pathway, dependent upon calcium regulation (figure 3).
Figure 3. Schematic representation of BDNF traffic (Lessmann et al. 2003).
Once secreted, NTs can bind to two types of cell surface receptors, the Trk tyrosine
kinases and the p75 neurotrophin receptor (p75NTR). Each Trk receptor (Trk A, Trk B
and Trk C) is preferentially activated by one or more NTs and is responsible for
mediating most cellular responses. More specifically TrkA is activated by NGF, TrkB
by BDNF and NT-4/5, and TrkC by NT-3 (figure 4).
13

Introduction
Figure 4. NTs binding to their receptor.
Binding of the NT initiates Trk dimerization, transphosphorylation of tyrosine residues
in its cytoplasmic domain and kinase activation (see figure 5). The phosphotyrosine
residues function as binding sites for recruiting specific cytoplasmic signalling proteins.
These proteins may in turn be activated by phosphorylation. Contrarily to Trk receptors,
p75NTR can be activated by the binding with all the four neurotrophins with
approximately equal affinity, even if it is lower than Trk receptors. While Trk receptors
transmit positive signals, p75NTR transmits both positive and negative signals. The
signals generated by the two neurotrophin receptors can either augment or oppose each
other, regulating almost all aspects of neuronal development and function, including
precursor proliferation and commitment, cell survival, axon and dendrite growth,
membrane trafficking, synapse formation and function, as well as glial differentiation
and interactions with neurons. Moreover, recent studies have revealed a diversity of roles
for these factors outside the nervous system, most notably in cardiac development,
neovascularization and immune system function (Donovan et al. 2000; Kermani et al.
2005).
14

Introduction
Figure 5. Simplified diagram of NTs intracellular signalling pathways (Reichardt 2006).
For exhaustive reviews on neurotrophins, see for instance: (Kaplan et al. 2000; Roux et
al. 2002; Lessmann et al. 2003; Reichardt 2006).
Neurotrophins in learning and memory
As mentioned before, one of the most important cerebral functions regulated by
neurotrophins is cognitive processes, especially for what concern learning and memory.
The first reports indicating that neurotrophic factors modulated this process appeared
more than a decade ago when the work of two groups demonstrated that epidermal
growth factor and fibroblast growth factor enhanced LTP in CA1 (Terlau et al. 1989;
Terlau et al. 1990). Based on these studies, later experiments underlined also the
implication of NGF, NT-3 and BDNF.
15

Introduction
NGF did not induce LTP in CA1 (Tancredi et al. 1993), possibly because of the low
TrkA density in this area, but indirect evidence suggests that NGF may play a role in
expression of LTP in perforant path granule cell synapses where expression of the
neurotrophin and its receptor are relatively high (Mrzljak et al. 1993). Furthermore, the
involvement of NGF in synaptic plasticity was demonstrated using behavioural tasks.
Rats implanted with cannulae in the CA1 area of the dorsal hippocampus and trained in
one-trial step-down inhibitory avoidance showed that NGF administration blocks short-
term memory and improves long-term memory (Walz et al. 2005), while infusion of
NGF into the lateral ventricles of aged rats demonstrated its potential role in improving
spatial but not episodic memory (Niewiadomska et al. 2006).
NT-3 was shown to induce potentiation in hippocampus, a finding which was replicated
in isolated spinal cord (Arvanov et al. 2000). However, NT-3 was found to be not
necessary for LTP induction (Ma et al. 1999), even if more recently using conditional
knock-out mice, it was detected a selective impairment in LTP in the lateral, but not
medial perforant path-granule neuron synapses. Mice also exhibited deficits in spatial
memory tasks (Shimazu et al. 2006) .
However, the neurotrophin that seems to be more implicated in cognitive processes is
BDNF. Acute application of exogenous BDNF enhances synaptic transmission and
induces a form of potentiation that resembles LTP in CA1 cultures (Kang et al. 1995)
and CA1 slices (Figurov et al. 1996). The focus of attention on BDNF-induced
potentiation has intensified with the finding that, in addition to its stimulatory effect in
vitro, intrahippocampal infusion of BDNF into anesthetized rats leads to potentiation of
the synaptic response in dentate gyrus (Messaoudi et al. 1998) and is peculiar of the
transcription-dependent late phase of LTP (Messaoudi et al. 2002), driving the switch
from the early phase (Pang et al. 2004). Moreover, BDNF is necessary and sufficient for
the persistence of long term memory storage, as recently demonstrated (Bekinschtein et
al. 2008). The fundamental role in memory consolidation seems to be dependent upon
the transcription of the Immediate Early Gene Arc. Indeed, Ying and colleagues (Ying et
al. 2002) examined the expression of Arc mRNA following BDNF-LTP and it resulted
to be sharply upregulated. In situ hybridization revealed enhanced Arc expression across
16

Introduction
the granule cell layer and molecular layer of the dentate gyrus, indicating delivery of
transcripts to dendritic processes.
Several scientific evidences deriving from behavioural experiments support the data
emerging from cellular and molecular studies. Increases in BDNF mRNA and protein
have been recorded in hippocampus after training in a spatial learning task (Gooney et
al. 2002) and in dentate gyrus after training in a passive avoidance test (Ma et al. 1998),
paralleling the changes observed after induction of LTP. Consistently, a rapid and
selective induction of BDNF expression has been reported in amygdala during
hippocampus-dependent contextual learning (Hall et al. 2000). In addition, it has been
shown that behaviour in the water maze test is impaired in mutant mice carrying a
deletion of one copy of the BDNF gene (Linnarsson et al. 1997), in rats that had
received an intracerebroventricular infusion of anti-BDNF antibody (Mu et al. 1999) or
with hippocampal-specific deletion of BDNF (Heldt et al. 2007).
Considered together, these findings suggest that a deep comprehension of BDNF
regulation and functions might be of extreme importance in order to elucidate the basic
aspects of cognitive processes and consequently, the physiopathology of cognitive
impairment.
1.4 Brain-derived neurotrophic factor
Distribution and functions
Brain-derived Neurotrophic Factor (BDNF), firstly discovered in 1982 by Barde and
colleagues (Barde et al. 1982), is a member of the family of neurotrophins that is highly
conserved among several species (Hofer et al. 1990; Rosenthal et al. 1991), is the most
studied neurotrophin after Nerve Growth Factor (NGF) and is the most abundant in the
brain. BDNF is widely expressed in the central nervous system, especially in the
hippocampal formation, cerebral cortex, and amygdaloid complex (Ernfors et al. 1990;
Ernfors et al. 1990; Hofer et al. 1990; Phillips et al. 1990; Wetmore et al. 1990), and its
expression increases until reaching a maximal level after birth (Maisonpierre et al. 1990;
17

Introduction
Friedman et al. 1991; Friedman et al. 1991; Schecterson et al. 1992). Then, the
expression of BDNF seems not to decline with age (Lapchak et al. 1993; Narisawa-Saito
et al. 1996; Katoh-Semba et al. 1998). However, BDNF is not only widespread
distributed in the central nervous system, but its mRNA has been observed in numerous
other tissues as colon, intestine, leukocyte, ovary, spleen, thymus, testis, pancreas and
blood (Rosenfeld et al. 1995; Liu et al. 2005; Pruunsild et al. 2007). BDNF almost
ubiquitary distribution underlines the importance of these molecule in a large number of
different cellular processes, especially for what concerns neuronal functions. Indeed, this
neurotrophin is strongly involved in:
• cell proliferation (Zhang et al. 2003; de Groot et al. 2006; Hu et al. 2006)
• cell survival/cell death (Woo et al. 2005; Pyle et al. 2006; Lipsky et al. 2007)
• differentiation/phenotype maintenance (Schuhmann et al. 2005; Young et al.
2007)
• dendrites and axon arborisation (Lom et al. 2002; Kumar et al. 2005)
• spines maturation (Murphy et al. 1998; Amaral et al. 2007)
• synapse maturation (Hu et al. 2005; Shen et al. 2006)
• synaptic transmission (LTP & LTD) (Aicardi et al. 2004; Lu et al. 2007)
BDNF gene structure
All genes encoding neurotrophins have a basically similar structure but among all
discovered neurotrophins the genomic structure of the BDNF gene is unusually complex.
Despite the fact that the gene organization and regulation of human and rodent BDNF
gene expression have received close attention during the last decade, knowledge of the
structural organization of mouse, rat and human BDNF gene has been well characterized
only in the last years (Liu et al. 2005; Pruunsild et al. 2007). Each BDNF transcripts is
composed by non coding exons spliced upstream of a common 3’ exon that encompasses
the whole coding sequence. The detailed characterization of the rodents and human
BDNF gene has revealed the existence of four exon clusters, each of them regulated by
different promoters located upstream of these alternatively spliced exons. These
18

Introduction
promoters enable a precise regulation of BDNF expression in different cell types or in
response to different stimuli.
In detail human BDNF gene spans about 70 kb and encode for 11 exons (Figure 6).
MOUSE AND RAT BDNF GENE
HUMAN GENE
Figure 6. Comparison between rodent and human BDNF gene. Each roman number represents a different exon, grouped in clusters delimitated by the double vertical dash (//). Exons Vh and VIIIh are uniquely present in the human gene (Pruunsild et al. 2007).
Exons II, III, IV, V, Vh, VI, and VIIIh are untranslated exons and translation of the
transcripts containing these exons starts from the ATG positioned in exon IX. Exons I,
VII, and VIII contain in-frame ATG codons that could be used as alternative translation
start sites, leading to the production of protein variants differing for the length of the N-
terminus. Two exons, VIII and VIIIh, are rarely used and always in combination with
exon V as the 5′ exon. Exon IX, which is always translated, can undergo to alternative
splicing events, generating transcripts containing variants of exon IX. These variants are
named “a”, “b”, “c” and “d” or can result from an arrangement of these regions.
The biological meaning of this complexity has been extensively investigated in our lab.
Tongiorgi and colleagues demonstrated that BDNF mRNAs are targeted to the distal
dendrites (>100 micron from the soma) in response to titanic electrical activity or other
stimuli as BDNF itself (Tongiorgi et al. 1997; Righi et al. 2000). The dendritic targeting
19

Introduction
was also demonstrated in vivo in animal models of epilepsy (Tongiorgi et al. 2004).
Interstingly, using in situ hybridization, it was noticed that different transcripts seem to
have different spatial and temporal localization in the neuronal districts (Chiaruttini et
al. 2008), suggesting the existence of a spatiotemporal code that links different splice
variants to different localizations and biological functions. To support this hypothesis,
very recent submitted data revealed that some mRNAs are implicated in cell survival,
whilst other splice variants are involved in the regulation of dendritogenesis and
spinogenesis.
BDNF protein: the importance of the pro region
As the other members of the neurototrophin family, with whom it shares numerous
structural features, human BDNF protein is firstly translated in an immature form of
about 32 kDa (proBDNF) that undergoes N-glycosylation and glycosulfation on residues
located within the pro-domain of the precursor (Mowla et al. 2001). The pro-domain and
the mature region are composed by 112 and 119 amminoacids respectively.
Despite of the high complexity of BDNF gene (see previous chapter), analyses of the
predicted translation products of the transcripts revealed they can generate only four
different protein variants (see figure 7)(Liu et al. 2005). Most of the BDNF mRNAs are
translated in the previously described protein, named proBDNF1. The transcript
containing BDNF exon I also encodes a 50-UTR inframe methionine that may serve as
an alternative translational initiation site for a proBDNF2 that would contain eight
additional N-terminal amino acids (MFHQVRRV). The transcript called BDNF VIIb
would be also capable to produce proBDNF3 with an addition of seven amminoacids to
the N-terminus of proBDNF2. Furthermore, BDNF VII mRNA could undergo specific
splicing event that leads to the deletion of 48 amminoacids (154-201) within the coding
region, generating proBDNF4. Finally, in a recent work, it has been hypothesized that
exon VIII would produce a proBDNF variant with an addition of 73 amminoacids to the
N-terminus of proBDNF2 (Pruunsild et al. 2007). Possible different localization and
functions of proBDNF variants are still unknown.
20

Introduction
To reach the mature form of about 14 kDa, proBDNF variants need to be cleaved by
specific proteases at the N-terminal Arg-Gly-Leu-Thr57-2-Ser-Leu site and cleavage is
abolished when Arg54 is changed to Ala (R54A). ProBDNF is also able to generate a
truncated form of about 28 kDa that arises by a different processing mechanism than
mature BDNF and it’s not an obligate intermediate (Mowla et al. 2001). Proteolitic
cleavage can occur either intracellularly or extracellularly. Intracellular cleavage is
primarily leaded by furin, a protease owning to the family of convertases and it can takes
place within the trans-Golgi network and/or immature secretory vesicles (Seidah et al.
1996). Differently, the extracellular processing of proBDNF can be regulated selectively
by plasmin and matrix metalloproteinases (Lee et al. 2001). Wherever it is processed,
proBDNF distribution in the brain seems to almost overlap with mature BDNF
localization (Zhou et al. 2004).
proBDNF1
Figure 7. The four proBDNF variants. Boxes of the same colour represent the same amminoacidic sequence.
Intriguingly, data emerging from recent works suggests that proBDNF is not only a
precursor of the mature and active form of BDNF, but it also seems to elicit important
biological functions. This hypothesis firstly derives from the evidence that proNGF,
another neurotrophin precursor, is able to bind p75NTR with high affinity and to
promote neuronal apoptosis (Lee et al. 2001). Based on this study, Woo and colleagues
proBDNF2 (ex I)
NH2
NH2
proBDNF3 (ex VIIB) NH2
pro-BDNF4 (Δ48aa)
NH2
Pro region Mature region
21

Introduction
(Woo et al. 2005) demonstrated that also proBDNF is capable to induce apoptosis in
cultured sympathetic neurons through the activation of a complex formed by p75NTR
and sortilin. Both the receptors were found to be necessary for the induction of cell
death. Furthermore, proBDNF was also linked to synaptic plasticity and in particular it
was established that it is able to enhance LTD following binding to p75NTR.
Taken together, these findings reveal a previously unknown functional role of proBDNF
opposite to the role played by mature BDNF. Therefore, cell survival and synaptic
plasticity seems to depend on the right balancing of these two forms.
BDNF in neuropsychiatric disorders and cognitive deficit
After analyzing BDNF and proBDNF biological functions, it appears clear that this
molecule play a fundamental role in brain health and in cognitive processes. As already
discussed, the versatility of BDNF is emphasized by its contribution to a range of
adaptive neuronal responses: LTP, LTD, certain forms of short-term synaptic plasticity,
as well as homeostatic regulation of intrinsic neuronal excitability (Desai et al. 1999;
Asztely et al. 2000; Ikegaya et al. 2002; Santi et al. 2006). Thus, it seems reasonable
that even a single alteration of its highly complex regulation may lead to a pathological
condition.
Numerous experimental evidences indicate that an alteration in BDNF mRNA and
protein levels could lead to psychotic condition, both in post-mortem tissues and in
animal models of schizophrenia. In particular, mRNAs levels of BDNF and its receptor
TrkB have been found to be decreased in prefrontal cortex of subjects with
schizophrenia (Weickert et al. 2003; Weickert et al. 2005), whilst there is an increase of
the BDNF mRNA levels in cerebellar cortex (Paz et al. 2006). Similarly, BDNF protein
levels have been reported to be decreased in hippocampus and increased in cortical areas
of affected patients (Durany et al. 2001), as well as there is an alteration of BDNF
pathway in animal models of schizophrenia (Lipska et al. 2001; Ashe et al. 2002).
Interestingly, BDNF was also found to be linked with cognitive impairment typical of
many schizophrenic patients (Ho et al. 2006). Ho and colleagues demonstrated that in a
schizophrenic population, patients carrying the BDNF gene Val66Met polymorphism
22

Introduction
(that changes a valine into a methionine in position 66) performed poorly in verbal
memory and visuospatial abilities tests, suggesting that this polymorphism could be
implicated in the pathogenesis of cognitive impairment in schizophrenia.
Even if schizophrenia seems to be the neuropsychiatric disorder in which BDNF is
mostly involved in its pathogenesis, it is possible to find a significative role of this
neurotrophin also in bipolar disorders and ADHD. Genetic association studies
enlightened that Val66Val allele frequency is higher in patients with bipolar disorders
compared to a group of healthy volunteers (Lohoff et al. 2005). Moreover, the
alternative Val66Met polymorphism was also found to be protective against early onset
of the psychosis (Geller et al. 2004), confirming indirectly that Val66Val allele seems to
be a risk factor for developing bipolar disorders. For what concern ADHD, it is known
that BDNF heterozygous null mutants (Kernie et al. 2000) and BDNF conditional
knockout mice (Rios et al. 2001) exhibit increased locomotor hyperactivity, which
mimics the fundamental behavioural characteristics of ADHD. However, association
studies did not reveal any linkage between the disorder and BDNF polymorphisms (Lee
et al. 2007; Schimmelmann et al. 2007).
Conversely, the role of BDNF in major depression is still ambiguous. Analysis of post-
mortem hippocampus showed that BDNF expression is decreased in depressed suicide
patients (Dwivedi et al. 2003), as well as the kinase ERK (Extracellular Regulated
Kinase) that regulates a fundamental BDNF signalling pathway (Dwivedi et al. 2001).
Other evidence suggesting that BDNF is involved in the pathophysiology of depression
derives from the observation of the effects of antidepressant drugs. Indeed, it has been
extensively demonstrated that different classes of antidepressants significantly increased
the expression of BDNF in the major subfields of the hippocampus, including the
granule cell layer and the CA1 and CA3 pyramidal cell layers (Nibuya et al. 1995;
Nibuya et al. 1996). The upregulation of BDNF was found also in post-mortem tissues
of patients treated with antidepressant nearly to the time of death (Karege et al. 2005).
Nevertheless, behavioural studies are not so convincing. Although various studies found
data indicating some depressive-like behaviours in mice lacking BDNF or its receptor
23

Introduction
TrkB in the forebrain, the effects were small or sometimes not significative (Zorner et al.
2003; Monteggia et al. 2007).
The controversial data about the role of BDNF in depression was proposed to be linked
with the biological effects of its precursor. Considering that proBDNF enhances LTD
(Woo et al. 2005) and LTD is related with stress-induced depressive-like behaviours
(Holderbach et al. 2007), it has been suggested that the correct modulation of the ratio
between mature BDNF and its precursor may play an important part in the pathogenesis
of depression (Martinowich et al. 2007). This consideration has been proved to be true in
Alzheimer’s disease, a pathology that shares many common aspects with
neuropsychiatric disorders. Using post-mortem tissues of the parietal cortex of
Alzheimer patients, Michalsky and colleagues demonstrated by Western blot that there is
a selective reduction of proBDNF in affected subjects (Michalski et al. 2003).
Furthermore, in another study, the same group underlined that this reduction occurs in
the early stage of the disease and it is positive correlated with loss of cognitive functions
(Peng et al. 2005).
Reviewing data about BDNF and its relationship with neuropsychiatric disorders
emphasizes how little is still known. New scientific findings have shifted the attention
towards the importance of proBDNF biological functions in these disorders, forcing
scientists to challenge with the high complex modulation of this protein. Therefore, it is
very likely that BDNF may be implicated in many more aspects of psychosis not deeply
investigated yet, as for what concern cognitive impairment. Due to the numerous data
that indicates the involvement of the neurotrophin in various aspects of psychoses,
BDNF has always been considered a putative diagnostic and therapeutic marker of
mental health. Hence, examining BDNF levels in tissues from which it is easy to obtain
a sample as human serum, could represent a powerful tool for clinical applications.
24

Introduction
BDNF in the serum: a possible marker of neuropsychiatric disorders and cognitive
deficit?
BDNF was firstly identified within the human serum in 1995 by Rosenfeld (Rosenfeld et
al. 1995), even if in an earlier experiment Yamamoto isolated its cDNA from human
platelets (Yamamoto et al. 1990). These findings rapidly leaded to the development of
an ELISA-based test to measure BDNF levels in human blood (Pliego-Rivero et al.
1997). Successively, it was shown that blood BDNF is essentially stored in blood
platelets, from which it can be released into the plasma through activation or clotting
processes (Radka et al. 1996; Fujimura et al. 2002). Moreover, it was reported that
BDNF could cross the blood brain barrier (Pan et al. 1998), and that BDNF levels in
brain and serum underwent similar changes during maturation and aging processes in
rats (Karege et al. 2002), suggesting that serum BDNF levels may reflect the BDNF
levels in brain.. Alternative sources of blood BDNF were identified in endothelial cells
(Nakahashi et al. 2000) and lymphocytes (Noga et al. 2003) but their contribution is
believed to be marginal compared with the bulk release from platelets.
The concrete possibility that serum BDNF might proportionally reflect the amount of
BDNF in the CNS quickly attracted numerous investigations due to the easy
accessibilities of the samples and the fast and high reproducible measurement method
for estimate BDNF levels, based on ELISA tests. At the moment, a huge number of
studies have reported altered levels of this neurotrophin in serum of psychotic subjects.
Indeed, it has been reported that serum BDNF levels were significantly decreased in
antidepressant-free patients with Major Depression (Karege et al. 2002; Shimizu et al.
2003). Similarly, serum BDNF levels are reduced in bipolar disorders (Cunha et al.
2006; Monteleone et al. 2008), whilst nothing is still known about BDNF serum levels
in ADHD or in cognitive deficit. Regarding schizophrenia, data emerging from these
experiments are controversial. Surprisingly, some studies reported no variation of BDNF
levels in serum of patients compared to healthy human volunteers (Shimizu et al. 2003;
Jockers-Scherubl et al. 2004; Huang et al. 2006), while other authors found a
significative decrease (Toyooka et al. 2002; Pirildar et al. 2004; Tan et al. 2005; Grillo
et al. 2007). Furthermore, a very recent study detected an increase of the total amount of
25

Introduction
26
serum BDNF (Gama et al. 2007). Data obtained in these experiments are summarized in
Table 2:
Paper Healthy control (ng/ml)
Schizophrenic patients (ng/ml)
Variation
Toyooka et al. 2002 11,4±7,7 6,3±3,4 Decrease
Shimizu et al. 2003
28.5±9,1 27.9±12,3 No variation
Pirildar et al. 2004
26,8±9.3 14,19±8,12 Decrease
Jockers-Scherubl et al. 2004
13,2± 5,2 13,1±5,9 No variation
Tan et al. 2005
9,9±4,3 7,3±2,6 Decrease
Huang et al. 2006
14,17 ± 6,86 14,20 ± 6,92 No variation
Gama et al. 2007
0,19±0,08 (pg/µg protein)
1,21±0,98 (pg/µg protein)
Increase
Grillo et al. 2007
16,9 ± 2,6 10,1 ± 5,2 Decrease
Table 2. BDNF serum levels in healthy human volunteers and schizophrenic patients, as reported in recent publications. All values are intended as mean ± standard deviation.
The controversial results in the dosage of BDNF in human serum of schizophrenic
patients suggests that a simple measure of total BDNF could not be sufficient in some
circumstances to established a relationship between serum levels and the pathology,
considering the complexity of BDNF synthesis, functions and regulation. As discussed
in the previous chapter, measurement of proBDNF and the ratio between itself and the
mature form might be more informative rather than the simple estimate of the total
amount of BDNF. Addressing these questions could help us to reach a better
comprehension of the phenomenon, as well as to open new research perspectives in the
field.

Aim of the thesis
2. AIM OF THE STUDY
The role of BDNF in neuropsychiatric disorders and the related cognitive impairment
has been extensively discussed in the previous section. Existing data suggests that the
common cellular mechanism that could be altered is the regulation of BDNF protein
synthesis. Therefore, understanding the molecular basis of the cognitive processes as the
local protein synthesis in dendrites, trying to correlates them with BDNF protein levels
in neuropsychiatric disorders and cognitive impairment, may represent the first step for
the development of new powerful tools for compound screening, as well as for the
diagnosis and the follow-up of medical treatment of these disorders.
It is possible to summarize this PhD project in two aims, both correlated with industrial
and clinical applications:
Aim 1) to study the cellular mechanisms of BDNF translation in dendrites;
Industrial application: development of a cellular assay for the in vitro screening of
compounds to treat cognitive deficit.
Aim 2) to study the correlation between neuropsychiatric disorders, cognitive deficit and
levels of BDNF in the serum.
Clinical application: development of an ELISA kit for the dosage of BDNF in human
serum as a biomarker of human cognitive deficits.
27

Materials and methods
3. MATERIALS AND METHODS
To allow an easier consultation of “Materials and methods”, this section will be divided
in chapters based on the structure of the next section (“results”).
3.1 Preliminary experiments
Materials
A 30 base Arc oligoprobe (5’ ATA CAG TGT CTG GTA CAG GTC CCG CTT ACG
3’), used previously by Pinaud and colleagues (Pinaud et al. 2001), was synthesised by
Invitrogen (Paisley, UK). dATPα 35S was obtained from Amersham (Chalfont St.Giles,
UK). Cobalt chloride, 5x reaction buffer and terminal transferase enzyme were
purchased from Roche (Lewes, UK).
Animal treatment
Male Sprague-Dawley rats with an arrival weight range of 160-220g were supplied by
Charles River, UK. These animals were group-housed with 5 rats per cage. All animals
were treated according to the Home Office guidelines and their use in any scientific
procedure was fully documented. Animals were given a five-day acclimatisation period
before their use in any procedure was permitted.
The dose volume used in this study was 2ml/kg and the dose was subcutaneously
injected (s.c.). 50.7 mg xanomeline were dissolved in 3.31 ml of 0.9% saline to give a
concentration of 10mg/kg. Two dilutions of this top dose were made to give 1 and
3mg/kg respectively. 0.9% saline was used as the vehicle. Animals were weighed and
then administered with drug (1, 3 or 10mg/kg) or vehicle (n=6).
1 hour post dose, animals were sacrificed, their brains removed and frozen in isopentane
at -40oC.
28

Materials and methods
Preparation of sections
Coronal sections of 14µm were cut through the prefrontal cortex (bregma 1.78 - 1.54
mm) and the dorsal hippocampus (bregma -1.58 - -1.82 mm) using a thaw cryostat
mounted onto Polylysine slides (BDH, Poole, UK) and left to dry. Slides were stored at -
70oC.
Radioactive in situ hybridisation
Preparation of 4% Paraformaldehyde (PFA)
32 g of PFA were dissolved in 800 ml of DEPC treated phosphate buffered saline (PBS)
by heating to 55oC with continuous stirring and adding a pellet of sodium hydroxide.
The solution was cooled, filtered and the pH was adjusted to 7.4.
Preparation of hybridisation buffer
Hybridisation buffer was made up as follows:
- 10% dextran sulphate
- 1x Denhardt’s solution
- tRNA 0.1 mg/ml (from yeast)
- 0.1% SDS
- 0.1% Sarkosyl
- 50% deionised Formamide
- EDTA 4mM
- Tris-HCl pH 7.5 80mM
- polyA 0.1 mg/ml
- DNA 0.1 mg/ml (salmon sperm)
29

Materials and methods
3’end labelling of Arc oligo with terminal transferase
The 3’ end labelling was set up by adding the following components to a sterile 1.5 ml
Eppendorf tube:
- 2µl of 100 ng/µl Arc oligo
- 8µl of 15 mM cobalt chloride
- 8µl of 5x reaction buffer
- 10µl of sterile water
- 10µl of dATPα 35S
- 2µl of terminal transferase enzyme (40u)
The reaction mixture was vortexed briefly before incubation in a waterbath at 37oC for 1
hour. The reaction was stopped by adding 60µl of sterile water. The oligoprobe was
purified by applying the diluted reaction mixture to a Roche quickspin column and
centrifuging at 2000 rpm for 4 minutes.
Counting of radioactivity incorporated
To verify the radioactivity incorporated, 1µl of purified oligoprobe was spotted onto a
Whatman filter circle and placed in a scintillation vial with 5ml of scintillation fluid. The
vial was subsequently placed in a scintillation counter for measurement of radioactivity.
Counts should be in the range of 50 000 - 300 000 dpm/ul.
Pre-treatment of the slides
Slides were removed from the -70oC freezer and thawed at room temperature. For each
animal, four slides were used for ISH, two containing sections through the prefrontal
cortex and two through the dorsal hippocampus. Slides were fixed in ice cold 4% PFA
for 10 minutes and then washed twice for 5 minutes in DEPC treated PBS. Following
washing, slides were laid out in trays ready for hybridisation.
30

Materials and methods
Hybridisation
1 ul of the labelled purified oligoprobe was used per 100 ul of hybridisation buffer and
100 ul of buffer containing probe was used per slide. For each pair of slides, one was
hybridised with the antisense probe and the other hybridised with the sense probe. The
hybridisation buffer and probe mixture was vortexed thoroughly and left to stand for ten
minutes before application to slides. Slides were coverslipped and incubated in a humid
chamber overnight at 43oC.
Post hybridisation washes
Coverslips were removed gently by immersing slides in 2 x SCC (0.3M NaCl, 0.3M
sodium citrate) at room temperature. Slides were washed at 55oC in decreasing
concentrations of SSC buffer: 4x SSC 2 x 15 min, 2x SSC 2 x 15 min and 1x SSC 2 x 15
min. Slides were then dehydrated through increasing concentrations of ethanol (70, 90
and 100%). After washing, the slides were left to dry thoroughly for several hours. Once
dry, slides were placed in autoradiography cassettes with Kodak biomax MR film for
one week with a 14C autoradiographic scale (Amersham, Chalfont St.Giles, UK).
Quantification of Arc mRNA
Following development of films, images from each film were digitised and captured on a
computer screen. The software program MCID Elite (Micro Computer Imaging Device,
Imaging Research Inc., UK) was used to measure the expression of Arc mRNA in rat
prefrontal cortex and hippocampus. A standard curve was set up using the 14C
autoradiographic scales and Relative Optical Density (ROD) was converted to nCi/g
before measuring Arc expression. In the prefrontal cortex, the superficial and the deep
layers were measured by placing a counting box of defined area onto each digitised
image. For the deep layer, a 200x800µm rectangular box was used, while for the
superficial layer the box size was 400x400µm (Fig. 4).
In the hippocampus, measurements were taken from CA1 and CA3 regions, as well as
from the whole anatomical area. All hippocampal counting was made by free hand
drawing around the regions of interest. Measurements were taken from both sides of the
31

Materials and methods
brain and each slide contained 2 sections so for each animal, 4 counts were obtained in
total. 4 corresponding measurements were taken from each control slide and these non
specific values subtracted before means were calculated for each group.
Statistical analysis
All data was expressed as the mean expression value in nCi/g for each brain area after
vehicle or xanomeline (1, 3, 10 mg/kg) administration. The data was analysed with the
software STATISTICA 6 to assess the statistical significance. An ANOVA with Fishers
LSD test was performed for both the prefrontal and hippocampal data. P-values of 0.05
or less were considered to be statistically significant.
3.2 Cellular and molecular mechanisms of cognitive processes
Primary cultures of rat hippocampal neurons
Primary hippocampal neurons from postnatal Sprague–Dawley rats were made
according to the method of Malgaroli and Tsien (Malgaroli et al. 1992) with slight
modifications (Tongiorgi et al. 1997). Hippocampi were dissected from 1 to 2-d-old
animals (P1-P2). All the dissection was performed in 200 μm kynurenic acid (Sigma,
St.Louis, MO) and 25 μm 2-amino-5-phosphonovalerate (APV; Tocris Cookson, Bristol,
UK) on ice. The hippocampi were treated with trypsin (2mg/ml; Sigma) and DNase I
(0,3mg/ml; Sigma) for 10 min at 37°C to isolate the single cells. Cells were cultured for
8 d in 5% CO2-humidified incubator, in MEM and Glutamax I with 10% FBS, 7mg/ml
vitamin B12, 30 μg/ml insulin, and 100 μg/ml bovine transferrin and 10 μM cytosine
arabinoside (AraC) to halt the proliferation of non-neuronal cells. The cells were then
plated in 24-well chambers at 2 x 105 cells/well. The medium was changed every two
days from the second day in culture onwards.
Post-natal Sprague–Dawley rats were supplied by the Experimental Animal Center
(CSPA-University of Trieste). Procedures involving animals and their care were carried
out in accordance with national (Decreto Legge N116, Gazzetta Ufficiale, suppl 40, 18-
2-1992) and international laws and policies (European Community Council Directive
32

Materials and methods
86/609,Oja L 358, 1, December 12, 1987; National Institutes of Health Guide for the
Care and Use of Laboratory Animals, US National Research Council, 1996). All efforts
were made to minimize animal suffering and to reduce the number of animals.
BDNF-GFP chimeric constructs
The BDNF isoforms-GFP chimeras were previously cloned in our lab by Cristina
Chiarruttini, a former PhD student, as described below. RNA extraction from total brain
was performed following the method described by Chomczynski and Sacchi
(Chomczynski et al. 1987). Since exon I is slightly expressed in the hippocampus
(Timmusk et al. 1993), RNA for exon I has been extracted from the whole brain of a rat
treated for 3 hs with kainic acid (12 mg/Kg). After extraction 1 μg of total RNA was
subjected to reverse transcription (RT-PCR). The RNA was added to the RT-PCR mix
containing 5X first strand buffer, 250mM Tris-HCl pH 8.3, 375mM KCl, 15mM MgCl2,
10 mM DTT, 2 units of RNase inhibitor (Roche Diagnostics), 100ng Primer random
p(dN)6 (Roche Diagnostics), 200μM dNTPs mix (Promega), 200 units of SuperScriptII,
Life Technologies, Invitrogen. Then the mix has been incubated at 42°C for 50 min.2 μl
of the RT-PCR reaction have been added to the PCR mix: 10X Reaction Buffer, 500mM
KCl, 100mM Tris-HCl, pH 9, 1,5mM MgCl2, 200μM dNTPs mix, Taq DNA polymerase
0,04units/reaction (all from Promega), specific forward and reverse primers 500nM
(Celbio) to amplify the different BDNF isoforms. Specific forward primers for each of
the 5’ exons have been used with a common reverse primer which overlaps to the end of
the CDS
33

Materials and methods
The forward primers are the following:
Exon1 Forw 5’- AATTCTCGAGGGTCTTCCCGCCCTAGCCTGAC
Exon2c Forw 5’- AATTCTCGAGCGGAGCGTTTGGAGAGCCAGCC
Exon4 Forw 5’- AATTCTCGAGTGAAGGCGTGCGAGTATTACCTCC
Exon6 Forw 5’- AATTCTCGAGTCGCACGGTCCCCATTGCGCC
CDS Forw 5’- GCGCTCGAGATGACCATCCTTTTCCTTAC
The sequence underlined corresponds to the XhoI restriction site for the cloning in
pEGFP-N1 plasmid (Clontech). The common reverse primer is the following:
CDS Rev 5’- GTCACACGTGTCCCCTTTTAATGGTCAGT
The sequence underlined corresponds to the SacII restriction site for the cloning in
pEGFP-N1. All the PCR products were cloned in the XhoI and SacII cloning sites of the
pEGFP-N1 plasmid in frame with the GFP gene.
Neurons transfection
To transfect the different constructs we chose the method of the Lipofectamine 2000TM
(Life Technology, Invitrogen). For each well of cells to be transfected, 1 μg of DNA has
been diluted in 50 μl of MEM without serum and antibiotics. At the same time 2µl of the
LipofectamineTM solution (1mg/ml) have been incubated in 50 μl of MEM. After 5 min
the two solutions have been mixed, let for at least 20 min at RT and then added to the
cells. 24 hrs after transfection the Lipofectamine-DNA mix has been carefully removed
washing the cells twice with PBS.
Electrical stimulation
In all the experiments of electrical stimulation, neurons were depolarized at RT with
oxygenated K-medium (10 mM KCl, 1.8 mM CaCl2z2H2O, 0.8 mM MgSO4z7H2O,
34

Materials and methods
101 mM NaCl, 26 mM NaHCO3, 1 mM NaH2PO4z2H2O, 0.7% D-glucose, 15 mM
HEPES, pH 7.4).
Immunofluorescence
To analyze the expression of the GFP-chimeras in hippocampal neurons
immunofluorescence has been performed. Cells were fixed for 15 min with PFA
4%/PBS at RT, washed twice in PBS and permeabilized with Triton X-100 0,5% in PBS
(Sigma) for 15 minutes. After two washes in PBS, hippocampal neurons transfected with
the BDNF-GFP chimeras were incubated 1 hour at RT with anti-GFP antibody (BD
Living colour) diluted 1:100 in PBS, washed twice and incubated for 1h at RT with the
secondary antibody 1:100 (ALEXA 488 anti-mouse, Invitrogen). Subsequently, after
another set of washes, they were mounted on coverslips using the mowiol (Sigma)
mounting gel.
Dendrites transection and electrical stimulation
Once identified a healthy apical dendrite of a pyramidal neuron transfected with BDNF-
GFP contructs, complete transection of the dendrite was performed lowering down
carefully a micropipette until the dendrite was pinched. After allowing the micropipette
to rest for 1-2 minutes, dendrite was mechanically separated from the cell body with
slow and repeated movements of the micropipette. This procedure, due to its high
invasivity, frequently determined dendrite disruption. For this reason, only healthy
dendrites after transection were utilized for our experiments.
Severed dendrites were depolarized for 15 min at RT with oxygenated K-medium and
subsequently images were acquired each 30 seconds for one hour.
Image acquisition, manipulation and measurements
Neurons and dendrites images were acquired by a Nikon AXM1200 digital camera on a
Nikon E800 Microscope with interference contrast-equipped lens and with Fluorescence
lamp. For immunofluorescence experiments, all images were acquired with the same
exposure time and background noise was subtracted.
35

Materials and methods
Images manipulation was performed using NIH ImageJ software. In particular, dendites
pictures were stretched using Image J plug-in “Straighten” (Kocsis et al. 1991), allowing
to take measurements not depending by free-hand drawing but determined by a
rectangular box.
For the measurements of the maximal dendritic length reached by GFP chimeras in
localization experiments, the starting point was considered the intersection of the cell
body with the beginning of the dendrite. After calibration, a rectangular box was traced
from this point until the last fluorescent signal detectable along the dendrite, indicating
the distance in microns from the soma.
For the definition of the minimal time of KCl stimulation and the demonstration of
BDNF LPS, fluorescent intensity in intact or severed dendrites was measured as mean of
grey values inside a constant rectangular box along the dendrites until the last detectable
BDNF signal. For BDNF LPS demonstration, we calculated a normalized difference
score (Δfluo/fluo), that indicates the change in dendritic fluorescence as a function of
time.
Statistical analysis
Comparisons between two or more groups were performed with Student’s t-test or one-
way ANOVA using Sigma Stat 3.1 software. A p-value of 0,05 was considered to be
significative.
3.3 ProBDNF/BDNF ratio in the serum of schizophrenic patients
Subjects
40 schizophrenia patients (14 males and 26 females) and 40 healthy volunteers (20
males and 20 females) were enrolled for the study. All the patients included in the study
met DSM-IV (American Psychiatric Association, 1994) criteria for schizophrenia. The
diagnosis of schizophrenia was made independently by at least two experts. Average age
of schizophrenic subjects and control group were 47.2 ± 10.5 and 43.1 ± 8.4
36

Materials and methods
respectively. To assess whether the pharmacological treatment influenced BDNF levels,
we enrolled patients under atypical (32 subjects) and typical (8) antipsychotic drugs
treatment. Blood sampling was constantly performed in the early morning around 8 am.
Measurement of total serum BDNF
For serum samples preparation, blood (5ml) was collected in anticoagulant-free tubes
and maintained at room temperature for 1 hour, followed by 1 hour at 4 ºC to allow
complete release of BDNF from platelets. Finally, serum was isolated by centrifugation
at 2000 g for 10 min at 4 ºC. BDNF serum samples were stored within a period of 2
months at -20 ºC until the collection of the whole samples of the different groups and
then analyzed in a single experiment repeated three times. Total serum BDNF was
quantified using a specific ELISA kit following manufacturer protocol (BDNF Emax
immunoassay system, Promega Co, USA). To determine serum BDNF values, it was
used a microplate reader (Anthos Labtec Instrument, Chatel-St-Denis, CH) set at 450
nm.
Western blotting
Before testing, human albumin and IgG were depleted from serum samples using a
specific kit (Qproteome Albumin/IgG Depletion Kit, Qiagen, USA) to allow a better
detection of specific signal, considerably lower than albumin and IgG. Total proteins
amount in purified serum samples was determined using Bradford assay (Sigma-Aldrich,
St. Louis, MO, USA) Samples (30 µg) were then separated by electrophoresis on 12%
sodium dodecyl sulphate-polyacrylamide gels at 120 V for about 90 minutes and
transferred onto nitrocellulose membranes (Protran Nitrocellulose Transfer Membrane,
Whatman, DE) in transfer buffer [39 mM glycine, 48 mM Tris, 0.037% (v/v) SDS, 20%
(v/v) methanol] at 100 mA for 1 hour. After an incubation of 1 hour at room temperature
in blocking solution (4% (v/v) non-fat milk powder, 0,05% (v/v) tween-20 in phosphate-
buffered saline) to block non specific signal, membranes were incubated overnight at 4
ºC with an anti-BDNF antibody (N-20 anti-BDNF, Santa Cruz Biotechnology, Santa
Cruz, CA, USA; diluted 1:500), capable to detect both the mature and the precursor form
37

Materials and methods
of BDNF (Michalski et al. 2003; Peng et al. 2005). Following washing in blocking
solution, incubation with anti-rabbit secondary antibody (Polyclonal Goat Anti-Rabbit
Immunoglobulins/HRP, DakoCytomation, DK, diluted 1:10,000) for 1 hour at room
temperature and another set of washes, immunoreactive proteins were detected using an
electrochemiluminescence system (Amersham Biosciences).
Cognitive tests
Cognitive tests were performed to evaluate cognitive performances in schizophrenic
patients. A brief description of the tests is reported below:
Speech test (bisillabic words)
This test is widely used for measurement of working memory. Each trial was composed
by a set of bisillabic words (from 2 to 9) of increasing length. For each length, there
were presented 3 word sequences. The patient had to repeat all the elements of the
sequences in the right order. After a correct answer, the sequence length was increased
until a mistake. Score was calculated as the maximum sequence length correctly
repeated by the subject.
Trail making test
The test consisted of two parts, A and B. Part A consistsed of encircled numbers from 1
to 25 randomly spread across a sheet of paper. The object of the test was for the subject
to connect the numbers in order, beginning with 1 and ending with 25, in as little time as
possible. Part B required the subject to connect numbers and letters in an alternating
pattern (1-A-2-B-3-C, etc.) in as little time as possible. Scores were calculated by adding
the time spent by the subject to complete Part A with the time spent to complete Part B.
This test was used to evaluate attention deficit of the subjects.
Digit span test
Digit span is a common measure of short-term memory. The test required subjects to
repeat a string of number starting from one digit. After each correct answer, another
38

Materials and methods
single digit was added to the string until patients did not make mistakes. Scores were
calculated as the number of digit remembered correctly.
Matrix test
The test evaluates the screening abilities in a specific visual context. Each trial was
composed by three matrix of increasing difficulty with 13 rows of 10 digits (from 0 to 9)
each. In a maximal time of 60 seconds, the score was calculated as the total number of
correct rows filled by the patient (for a maximal score of 60).
Densitometry and statistical analysis
Densitometry of immunoreactive bands was determined by scanning films using an
Epson Scanner (Epson perfection V500 photo) and NIH Image J software.
For measurement of total serum BDNF and proBDNF/BDNF ratio, each set of data was
statistically analyzed using one-way ANOVA and pairwise multiple comparison
procedure (Holm-Sidak). To correlate cognitive performances with proBDNF/BDNF
ratio, we utilized Spearman correlation.
Each statistical analysis was performed using Sigma Stat 3.1 software. A P value of 0.05
was set as level of statistical significance.
3.4 Development of an ELISA kit for the dosage of proBDNF/BDNF ratio
Antibodies design and antigenic peptides synthesis
Identification of the antigenic sites of BDNF was performed using two different
softwares (JaMBW, Peptideselect design tool-Invitrogen) for the evaluation of antigenic
and hydrophobic indexes. To estimate the antigen exposure on the surface of the
molecule, we analyzed a cristallografic structure of BDNF (Robinson et al. 1995) with
Protein Explorer software. The synthesis of the immunogenic peptides, based on the
antigenic sequences previously identified, was externalized (EzBioLab, USA). In details,
we designed eight antibodies reactive against the following BDNF sequences:
39

Materials and methods
Mat 1-12: HSDPARRGELSV-C
Mat 91-102: TMDSKKRIGWRF-C
Pro total: DEDQKVRPNEENNKD-C
Pro1: TMVISYFGC
Pro2: C-FHQVRRV
Pro3: C-FHQVRRV
Pro4 inside del: MQSREEW-C
Pro 4 outside del: EKVPVSKGQLKQY-C
Cysteine was added for allow peptide cross-linking to a resin for the purification of the
specific IgGs.
Antibodies production
For this step, we adopted a double strategy based on the home made production and on
the externalization of the entire process (Pacific Immunology, USA).
The protocol for antibodies production of Pacific Immunology is property of the
company and it was not revealed.
For the home made production, antigenic peptides were conjugated with Keyhole
Limpet Hemocyanin (KLH), a carrier-protein that increases peptides antigenicity.
Briefly, 6 mg of lyophilized KLH were dissolved in170 µl of borate buffer 0,1 M pH 10
and mixed in agitation with 2 mg of peptide previously dissolved in 50 µl of borate
buffer. 80 µl of glutaraldheyde 0,3% in borate buffer were added drop by drop and
solution was left 1 hour at RT. Subsequently, there were added 50 µl of glycine and the
solution was incubated another hour at RT. Finally, PBS was added up to 2 ml and the
solution was dialyzed overnight at 4º.
After conjugation, 200 µg of immunogenic peptide were mixed in 1ml of physiological
solution with Freund’s adjuvant and injected intramuscular in New Zeeland White
Rabbits after local anesthetization with lidocaine. Collection of iperimmune serum was
obtained from posterior marginal vein of rabbit’s ear. We collected 20 ml of iperimmune
40

Materials and methods
serum for the first bleed and a maximum of 50 ml for the following bleeds. After the
tenth bleed, rabbits were exanguinated obtaining a final bleed of about 100 ml.
Antibodies purification
Purification of the specific IgGs was performed in column, immobilizing the peptide on
a suitable resin (Sulfolink Coupling Gel, Pierce, USA). Immunoglobulin purification
was made accordingly to the manufacturer protocol and checked on SDS 12%
polyacrylamide gel.
Cell cultures
Human epithelial kidney cell line 293T (HEK) were cultured in Minimum Essential
Medium with Earle’s salts and Glutamax I (MEM, Life Technologies, Invitrogen; (Eagle
1959)) with 10% foetal bovine serum (FBS) in 5% CO2-humidified incubator.
For transfected cells, the day before transfection, trypsinized and counted cells were
plated at 5 x 105 cells per well in 6-well plates, in order to have the cell’s monolayer at
90-95% of confluence on the day of transfection. After 24 hrs they were analyzed by
fluorescence and subsequently, after a quick wash in PBS, cells were lysate on ice in
lysis buffer (137mM NaCl, 20mM Tris HCl pH 8.0, 1% NP-40, 10% glycerol, 1mM
PMSF, 10µg/ml aprotinin, 1µg/ml leupeptin, 0.5mM sodium vanadate) and protein
concentration was quantified by Bradford method according to the manufacturer
protocol (Sigma, USA).
SK-N-BE derived from an human adrenal neuroblastoma with n-myc 100 x amplified
(high growth rate).The neuroblastoma cell lines were cultured in Dulbecco’s modified
Eagle’s medium (D-MEM), 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100
U/ml penicillin, and 100 g/ml streptomycin at 37°C with 5% CO2. In order to induce
permanent neuronal differentiation, 5 µM retinoic acid (RA) final concentration has
been added to cultures at the time of plating, and replaced every 48 hr. Transfection and
lysis were performed equally to HEK 293T protocol.
41

Materials and methods
Cell transfection
Cells were transfected with Lipofectamine 2000 TM method. For more details, see the
correspondent paragraph in chapter 3.2
Dot blot
Immunogenic peptides or SN-K-BE lysate were spotted at different concentrations (0,1;
0,5; 1 and 5 µg) on Protran Nitrocellulose Transfer Membrane (Whatman, Germany)
and left to dry 30 minutes at RT prior the immobilization with glutaraldheyde 0,3 % in
PBS for 1 hour. After three PBS washes, membranes were blocked for 1 hour at RT with
blocking solution (4% non-fat milk powder, 0,05% (v/v) tween-20 in PBS). Then,
membranes were incubated 1 hour at RT with the correspondent antisera diluted 1:100 in
blocking solution, washed three times and incubated for another hour at RT with
secondary antibody (Polyclonal Goat Anti-Rabbit Immunoglobulins/AP,
DakoCytomation, DK, diluted 1:1500). After another set of washes, membranes were
developed with NBT/BCIP solution (Sigma, USA).
Western blot
Western blot protocol was performed as described in chapter 3.3.
For transfected HEK 293T western blot experiments, we performed serial dilutions of
primary antibodies to establish the suitable concentration. In particular, Mat 1-12
optimal dilution was found to be 1:1500 dilution and pro total optimal dilution was
determined as 1:1000. The other immunoglobulins against the different proBDNF
variants were tested in a range of concentrations starting from 1:100 until 1:5000. Anti-
GFP antibody was diluted 1:1000 and Alomone anti-BDNF and anti-proBDNF were
used at a dilution of 1:1000 and 1:500 respectively.
For SN-K-BE experiments, custom and Alomone anti-BDNF antibodies were used at the
same concentrations utilized for HEK 293T experiments.
42

Materials and methods
43
ELISA
The day before the experiments, ELISA plates (Corning, USA) were coated with the
antigens overnight at 4º. The day after, plates were washed with PBS and blocked 1 hour
with polypep (Sigma, USA) 1% in PBS at RT. After blocking, plates were incubated 1
hour at RT with the different polyclonal antibodies against BDNF. For competition
studies, soluble antigens were addede to the plates contemporary to the primary
antibody. Plates were then washed five times with PBS and incubated with secondary
antibody conjugated with HRP (Euroclone, Italy) diluted 1:1000. After another set of
washes, plates were developed with TMB (Euroclone, Italy) for 30 minutes at RT.
Samples absorbance was read t 450 nm with a microplate reader (Anthos Labtec
Instrument, Chatel-St-Denis, CH).

Results
4. RESULTS
4.1 Preliminary experiments
The PhD project intended to evaluate possible biomarkers of cognitive processes, in
order to identify the most suitable for the development of an in vitro assay capable to
individuate cognition enhancing properties of new compounds. Considering that Arc is
an immediate early gene usually present at very low basal levels, is robustly induced
upon strong neuronal activity, and the newly synthesized mRNA is rapidly delivered
into dendrites following NMDA receptor activation or spatial memory tasks (Steward et
al. 1998; Guzowski et al. 1999), we firstly hypothesized that this gene could be
appropriate to our purposes. Therefore, we tried to link Arc with cognition through the
measurement of its mRNA expression in rats after the administration of xanomeline, a
selective agonist of the muscarinic acetylcholine receptor type 1 (M1 receptor). M1
receptor was chosen because its activation is strongly involved in cognitive processes
(Anagnostaras et al. 2002), especially for what concern the cognitive impairment
associated with neuropsychiatric disorders such as schizophrenia (Dean 2004).
Test of Arc oligoprobe for radioactive in situ hybridisation
Arc mRNA expression was tested through radioactive in situ hybridisation. To ensure
the quality and the specificity of the oligoprobe before using it for the in situ
hybridisation, a small amount was hybridised to rat brain sections from untreated control
animals.
A typical Arc expression pattern with minimal background was revealed in the forebrain
of control animals. Other brain sections, hybridised with a Arc sense probe, revealed a
barely undetectable signal (figure 8).
44

Results
Arc oligoprobe test in the prefrontal cortex
Antisense probe
Antisense probe
Sense probe
Sense probe
Arc oligoprobe test in the hippocampus
Figure 8. Arc oligoprobe test in rat prefrontal cortex and the hippocampus. Sense probes did not have any reactivity in control slides (right panel). Antisense probes detected the expected Arc expression pattern in the different brain area (left panel).
Arc expression following administration of the M1 receptor agonist xanomeline
To determine whether Arc mRNA expression increases after xanomeline injection, a
total of 10 animals for each dose of the compound (1, 3, 10 mg/kg) and for the control
(injected with vehicle) were sacrificed 1 hour post dose. Two different brain areas
related with cognitive functions, prefrontal cortex (pfctx) and hippocampus, were
investigated.
Prefrontal cortex (pfctx)
The expression of Arc mRNA was shown to be statistically significant increased in the
deep layer of the prefrontal cortex following acute administration of xanomeline (figure
9). The Arc response appeared dose-related in this brain region (significative only for 3
and 10 mg/kg of the agonist). Expression in the superficial layer was not significantly
increased.
45

Results
nCi/g
0
5
10
15
20
25
30
35 pfctx deep layer pfctx
* *
Vehicle as
Vehicle sense 10 mg/kg xan sense
10 mg/kg xan as
v 1 3 10 v 1 3 10 mg/kg
Figure 9. Arc expression in the prefrontal cortex (pfctx) after administration of xanomeline. In the left panel, it is shown Arc expression in control (left) and in animal treated (right) sections, both for the antisense (up) and the negative control sense (down) probes. In the right panel, the graph reports Arc expression densitometry in prefrontal cortex and in the deep layer of prefrontal cortex after administration of different doses of xanomeline. There is a significative increase (p<0,05) in the deep layer of the pfctx in a dose-dependent manner (for 3 and 10 mg/kg of xanomeline). For densitometry measurements, a standard curve was set up using the 14C autoradiographic scales and Relative Optical Density (ROD) was converted to nCi/g before measuring Arc expression. As=antisense, xan=xanomeline.
Hippocampus
The expression of Arc mRNA was measured in the hippocampal CA1 and CA3 fields as
well as the dentate gyrus in response to xanomeline administration (1, 3, 10 mg/kg).
Although an increase in Arc expression at 1 and 3 mg/kg was observed in the CA1 field,
these increases were not significant and no changes in Arc expression were observed
either in the CA3 field or the dentate gyrus (figure 10)
46

Results
47
nCi/g
05
1015
2025
3035
4045
50CA1 CA3 DG
Vehicle as 10 mg/kg xan as
Vehicle sense 10 mg/kg xan sense
v 1 3 10 v 1 3 10 v 1 3 10 mg/kg
Figure 10. Arc expression in the hippocampus after administration of xanomeline. In the left panel, it is show Arc expression in control (left) and in animal treated (right) slides, both for the antisense (up) and the negative control sense (down) probes. In the right panel, the graph reports Arc expression densitometry in hippocampus CA1, CA3 and DG areas after administration of different doses of xanomeline.There is no significative increase in any brain area investigated. For densitometry measurements, a standard curve was set up using the 14C autoradiographic scales and Relative Optical Density (ROD) was converted to nCi/g before measuring Arc expression. As=antisense, xan=xanomeline.
The controversial results obtained with Arc, convinced our group to shift towards other
possible biomarkers of cognitive processes. Based on the literature data described in the
introduction, as well as on the previous studies carried out in our laboratory, we decided
to investigate the role of BDNF in mental processes and its possible use as a biomarker
of cognitive deficit.
The study was divided in two parallel approaches, one focused on the investigation of
the cellular and molecular mechanisms of cognition driven by BDNF (AIM 1), and the
other based on the analysis of BDNF protein levels in the human serum and their
relationship with cognitive performances (AIM 2). Possibly, each of the two projects
should lead to industrial and clinical applications.
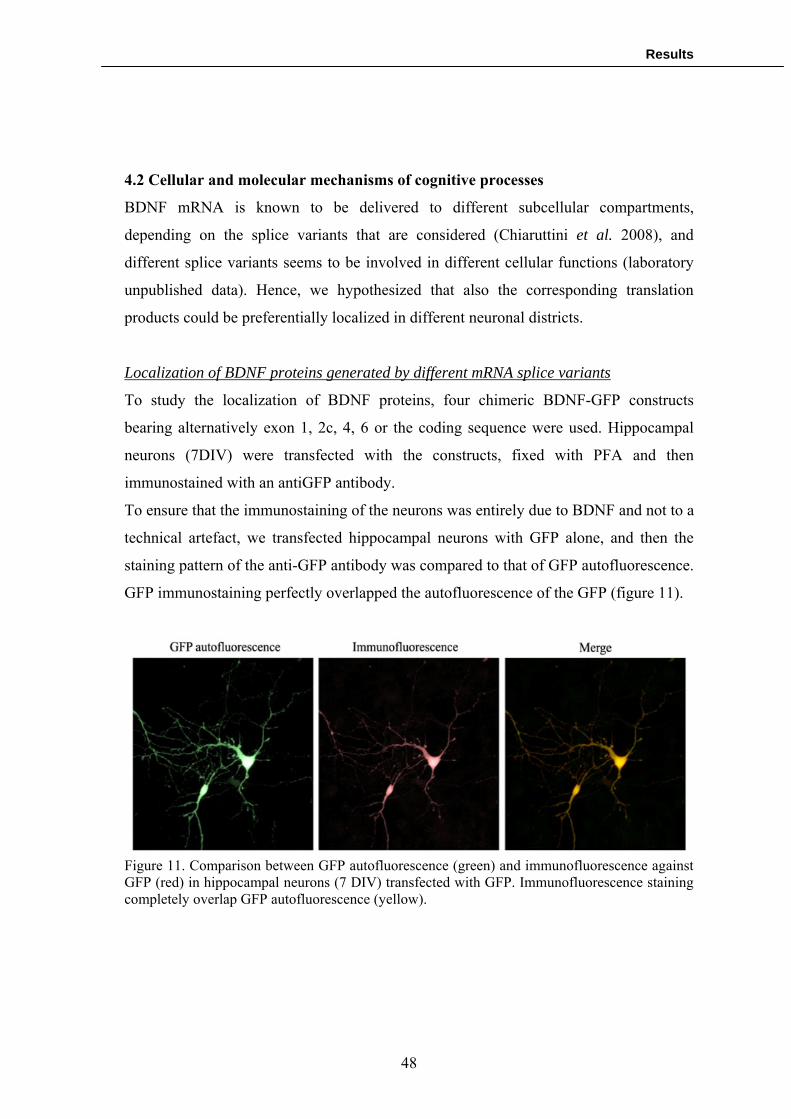
Results
4.2 Cellular and molecular mechanisms of cognitive processes
BDNF mRNA is known to be delivered to different subcellular compartments,
depending on the splice variants that are considered (Chiaruttini et al. 2008), and
different splice variants seems to be involved in different cellular functions (laboratory
unpublished data). Hence, we hypothesized that also the corresponding translation
products could be preferentially localized in different neuronal districts.
Localization of BDNF proteins generated by different mRNA splice variants
To study the localization of BDNF proteins, four chimeric BDNF-GFP constructs
bearing alternatively exon 1, 2c, 4, 6 or the coding sequence were used. Hippocampal
neurons (7DIV) were transfected with the constructs, fixed with PFA and then
immunostained with an antiGFP antibody.
To ensure that the immunostaining of the neurons was entirely due to BDNF and not to a
technical artefact, we transfected hippocampal neurons with GFP alone, and then the
staining pattern of the anti-GFP antibody was compared to that of GFP autofluorescence.
GFP immunostaining perfectly overlapped the autofluorescence of the GFP (figure 11).
Figure 11. Comparison between GFP autofluorescence (green) and immunofluorescence against GFP (red) in hippocampal neurons (7 DIV) transfected with GFP. Immunofluorescence staining completely overlap GFP autofluorescence (yellow).
48

Results
The immunofluorescence for the BDNF-GFP chimeric constructs using an anti-GFP
antibody, revealed that they are differently distributed within the soma or the dendrites
(figure 12).
Figure 12. Subcellular localization of BDNF proteins translated from 5 different BDNF mRNA splice variants. Hippocampal neurons (n=20 for each group) were transfected with BDNF-GFP plasmids containing either exon 1, 2C, 4, 6 or CDS (coding sequence) and immunostained with antiGFP antibody. In the upper panel, there is a comparison between the length of the dendritic staining of different BDNF proteins and mRNAs splice variants. The maximal distance from the soma reached by the dendritic staining was measured starting from the beginning of the dendrite (white arrows) until the last BDNF signal along the dendrite could be detected (red arrows for the protein, black arrows for the mRNA). In the lower panel, the probability plots (percentage of transfected neurons where BDNF signal reaches an established dendritic length) of dendritic staining shows a clear correlation between the localization of BDNF-GFP proteins and mRNAs. Exon 1 and 4 revealed to have a somatodendritic distribution, while exon 2c, 6 and CDS were preferentially localized in the distal dendrites.
Exon 1 and exon 4 BDNF-GFP chimaeras are predominantly restricted to the
somatodendritic subfield (50% of the protein within the first 15 µm or 45 µm
49

Results
respectively), while exon 2c, exon 6 and CDS BDNF-GFP constructs are delivered
throughout the proximal and the distal dendrites (50% of the protein within the first 70
µm for exon 2c and 6, 85 µm for CDS).
The protein localization is substantially comparable to the distribution of the
correspondent mRNA splice variants, except for exon 4 in which the mRNA seems to be
distributed closer to the soma than the corresponding chimeric protein (50% of the
mRNA is localized within the first 15 µm, whilst the protein is distributed around the
proximal 45 µm of the dendrite. However, both the mRNA and the protein originating
from exon4 variant could be considered to have somatodendritic localization.
Demonstration of BDNF Local Protein Synthesis in dendrites
Once established that BDNF splice variants are differentially distributed and the protein
products that they generate have a distribution similar to the cognate mRNA, we
investigated whether these mRNAs could be translated locally into dendrites following
electrical stimulation. To determine the minimum time required to induce a detectable
increase in protein synthesis, we stimulated with KCl hippocampal neurons transfected
with exon 6 BDNF-GFP plasmid and immunofluorescence was measured at different
times (3, 10, 15, 30, 45 and 60 minutes). Results demonstrated that the total fluorescent
intensity along the whole dendrite was maximal after 15 minutes of stimulation (figure
13) and the declined rapidly at subsequent times.
50

Results
Time of KCl stimulation (min.)
not stim. 10' KCl 15' KCl 30'KCl 45'KCl 60'KCl
Mea
n of
gre
y va
lues
(0-2
55)
0
10
20
30
40
50
*
Figure 13. Definition of the minimum time of KCl stimulation required to induce an increase in BDNF protein synthesis. Hippocampal neurons (n=20) were transfected with exon 6 BDNF-GFP, stimulated with KCl for different times and fixed prior the immunofluorescence detection of BDNF expression (determined as mean of gray values along the whole dendrite). It was noticed a significative increase of protein signal (p<0.05) after 15’ of KCl stimulation.
To demonstrate that BDNF mRNAs can be translated within the dendritic compartment,
first hippocampal neurons were transfected with either a somatodendritic or a distal
dendritic chimerical construct (exon 4 or exon 6 BDNF-GFP), and then the dendrites
were separated mechanically from the soma using a micropipette. For this set of
experiments, only healthy severed dendrites were considered. This preparation allowed
to examine unambiguously the local synthesis of proteins in dendrites, without
contribution of protein transport from the soma.
Stimulation of healthy transected dendrites with KCl for 15 minutes produced an
increase in protein synthesis reaching the peak 20 minutes after the stimulation (figure
14).
51

Results
Figure 14. Demonstration of exon 6 BDNF-GFP Local Protein Synthesis in cut dendrites. Upper panel shows a severed dendrite from a neuron transfected with exon 6 BDNF-GFP construct at different times after stimulation with KCl. The increase in protein synthesis is especially focused in “hot spots”. The graph in the lower panel compares the normalized fluorescence intensity along the whole dendrite in not stimulated and electrically stimulated dendrites at different time points. Fluorescence intensity is the mean of 7 severed dendrites.
The increment in BDNF-GFP fluorescence was detectable only in dendrites of neurons
transfected with exon 6 BDNF-GFP construct and not in dendrites from neurons
transfected with exon 4 BDNF-GFP (figure 15).
52

Results
Figure 15. Study of the local protein synthesis of exon 4 BDNF-GFP. Upper panel shows a severed dendrite from a neuron transfected with exon 4 BDNF-GFP construct at different times after stimulation with KCl. There is no increase in the fluorescent signal, indicating that this plice variant is not able to be synthesized locally. The graph in the lower panel compares the normalized fluorescence intensity along the whole dendrite in not stimulated and electrically stimulated dendrites at different time points. Fluorescence intensity is the mean of 7 severed dendrites. BDNF protein synthesis occurred mainly in spots located at different distances along
the dendrites. The spot intensity was found to be maximal 15 minutes after stimulus,
rapidly decaying within the next 15 minutes (figure 16). Treatment of the dendrites with
an inhibitor of protein synthesis (cyclohexymide) prior the stimulation completely
blocked the signal increase, indicating that it was due to effective protein synthesis
rather than protein redistribution.
53

Results
Taken together, these findings demonstrate that BDNF mRNA can be directly translated
into dendrites.
Figure 16. In the upper panel it is possible to appreciate the fluorescence intensity of a single “hot spot” of exon 6 BDNF-GFP synthesis in a severed dendrite at different times of KCl stimulation. The graph in the lower panel represents the mean of fluorescence intensity of 10 “hot spots”.
Future perspectives: LPS and industrial applications
The results showing that we are able to detect BDNF local protein synthesis, which
represents one of the major mechanisms involved in cognitive processes, suggested us
the possibility of utilize this assay for industrial applications. In particular, LPS might
result an important biomarker for cognition enhancing properties of developing
54

Results
compounds. Considering that drug screening process is always looking for innovative
biomarkers, we are developing a semi-automated method capable to detect LPS that is
the core of an industrial project in the drug screening field (figure 17). At the moment,
we are optimizing the creation of the physical support needed for the screening.
This industrial project is supported by a business plan that was awarded in the “StartCup
2007” competition as one of the best ten projects of the university of Trieste (NeuroScrin
Project).
Figure 17. Schematic representation of the semi-automated method for the detection of LPS in drug screening process. Hippocampal neurons transfected with a fluorescent reporter are plated into a square canal, forcing the neurite to grow in a upper layer (left panel). Afterwards, neurons are separated from the soma using an automated “dendrite cutter” and stimulated with developing compounds for the automated detection of the LPS signal.
4.3 ProBDNF/BDNF ratio in the serum of schizophrenic patients.
Simultaneously to the investigation of the cellular and molecular mechanisms of
cognitive processeses, the PhD project focused on BDNF protein levels in human serum
as another potential biomarker of cognition and mental health. To provide a proof of
concept of this idea, we decided to examine a group of patients suffering of
55

Results
schizophrenia, the neuropsychiatric disorder with the highest incidence of cognitive
impairment, studying the correlation between BDNF serum levels and their cognitive
performances.
Total BDNF levels in the serum of schizophrenic patients
To elucidate whether there is an alterations of total serum BDNF in schizophrenic
patients, we analyzed with a commercially available ELISA kit two groups of subjects
consisting in 40 healthy human volunteers and 40 age matched schizophrenic patients.
Blood samples collected, after coagulation at room temperature, were left 1 hour on ice
to induce a stress response capable to induce BDNF release in the serum from platelets.
Table 3 shows the demographic data and the BDNF serum levels of the two samples,
underlining that there is no statistically significative difference between the control
group and schizophrenic patients (p=0.558, One-way ANOVA).
Healthy human volunteers (n=40)
Schizophrenic patients (n=40)
Age 43.1 ± 8.4 47.2 ± 10.5
Gender (M/F) 20 M / 20 F 14 M / 26 F
Antipsychotic treatment
No 32 atypical 8 typical
BDNF serum values (ng/ml)
25.2 ± 7.74 M / 28.9 ± 4.2 F total mean: 26.5 ± 8.12
24.3 ± 5.8 M/ 27.3 ± 6.5 F total mean: 25.3 ± 6.54
Table 3. Demographic data and BDNF serum values of the sample. All values are intended as mean ± standard deviation.
Serum concentration of total BDNF in control group was 26.5 ± 8.12 ng/ml (mean ±
standard deviation), while in schizophrenic subjects corresponded to 25.3 ± 6.54 ng/ml.
The median of the two groups (28.8 and 26.06 ng/ml respectively) and values
distribution are reported in Figure 18. Moreover, as reported previously (Ziegenhorn et
56

Results
al. 2007), we did not detect significant gender difference between BDNF serum values
of male (25.2 ± 7.74 ng/ml) and female (28.9 ± 4.2 ng/ml) healthy donors (p=0,144,
One-way ANOVA).
Healthy volunteers Schizophrenic patients
BD
NF
seru
m le
vels
(ng/
ml)
0
10
20
30
40
50p=0.558
26.0628.8
Figure 18. Box plot of BDNF serum values (ng/ml) in healthy human volunteers (n=40) and schizophrenic patients (n=40). There is no statistically significative difference (p=0.558) between the two groups of subjects. The upper line of the box corresponds to the 75th percentile, the middle bold line is the median value and the lower line marks the 25th percentile. Whiskers (error bars) above and below the box indicate the 90th and 10th percentiles respectively.
Measurement of the ratio proBDNF/BDNF in the serum of schizophrenic subjects
Since we did not find any alteration of the total BDNF in the serum of schizophrenic
patients, we hypothesized that the correct balance between proBDNF and mature
BDNF, which elicit opposite biological effects on the nervous system, might be altered
in schizophrenia, possibly leading to the cognitive deficit related with this disorder.
Consequently, we firstly demonstrated through Western Blot technique using an
antibody able to recognize both the precursor and the mature form (N-20, Santa Cruz),
that proBDNF is present at detectable levels in human serum. To allow the detection of
57

Results
proBDNF, it was necessary to clear the samples from albumin and IgG, which constitute
the predominant portion of the protein population in human serum (see Matherials and
Methods, chapter 3.3). Thanks to this purification step, we were able to load a sufficient
amount of sample in order to recognize proBDNF band. Competition experiment, in
which the antibody was pre-incubated with the peptide used to produce the
immunoglobulin, confirmed the specificity of the signal (figure 19).
Figure 19. Specificity of N-20 antiBDNF antibody. The immunoglobulin was able to recognize recombinant BDNF (250 pg), recombinant BDNF (250 pg) and the two forms of BDNF in 30 µg of human serum proteins (left panel, from left to right). Detection of recombinant BDNF and proBDNF, as well as BDNF bands in the serum were abolished by incubation of the primary antibody with a 50-fold excess of specific blocking peptide (right panel)
Once established that proBDNF is measurable in human serum, we examined by
Western Blot the serum samples of both schizophrenic and healthy volunteers group
(figure 20). Band densitometry (with background noise subtraction) allowed us to
measure the ratio between proBDNF and mature BDNF, finding a significant reduction
of this ratio in psychotic subjects compared to the control group.
58

Results
healthy volunteers schizophrenic patients
proB
DN
F/B
DN
F ra
tio
0,00
0,05
0,10
0,15
0,20
0,25
0,04
0,19
*
Figure 20. Western blot of serum samples from schizophrenic subjects (upper panel, n=20) and healthy volunteers (lower panel, n=20). In each lane it was loaded the same amount of serum proteins (30 µg) depleted from albumin and IgG. The amount was estimated through Bradford method. Pro and mature BDNF are at the expected molecular weight of 32 and 14 kDa. The bar graph below reports proBDNF/BDNF ratio in control and schizophrenic subjects, evinced by the densitometry of the immunoreactive bands. The ratio proBDNF/BDNF in human serum is significantly decreased (p<0.05, One-way ANOVA) in schizophrenic patients (0,04) compared to healthy controls (0,19).
Correlation between proBDNF/BDNF ratio and cognitive performances
Considering that BDNF is strongly involved in cognitive processes, we utilized some of
the most commonly used neuropsychological tests to established whether there is a
correlation between the decrease of the ratio proBDNF/BDNF and the severity of
cognitive impairment in schizophrenic patients. All subjects with cognitive dysfunctions
reported in their clinical history, were examined with four different tests (TMT or Trial
59

Results
Making Test A and B, Digit Span, Speech Test of bisillabic words, Spinnler Matrix
Attention Test) for the evaluation of attention, short term memory, working memory and
screening abilities respectively (table 4).
.
Speech Test
(working memory)
TMT A, B
(attention)
Digit Span
(short-term
memory)
Matrix Test
60/60 (screening
abiity)
ProBDNF/BDNF ratio
5 n.s. (not solved) 7 38 0,00
4 30s 10 53 0,07 4 n.s. 2 6 0,10 4 34s 4 51 0,00 3 n.s. 2 25 0,00 4 31s 10 58 0,03 5 60s 10 43 0.00 4 n.s. 4 15 0.13 4 30s 4 49 0.03 5 43s 4 46 0,07 6 25s 10 43 0,21 4 20s 7 32 0,00 5 51s 9 60 0,10 4 n.s. 4 35 0,18
5 n.s. 9 50 0,02 5 25s 9 49 0,00 4 44s 4 60 0,00 5 35s 11 60 0,00 4 56s 9 47 0,00 4 2,35s 4 23 0,00
Table 4. Cognitive performances of schizophrenic patients and corresponding ratio of proBDNF/BDNF in the serum. The table reports the score for each test (Speech Test, TMT A and B, Digit Span, Matrix Test) used for estimate attention, short term memory and working memory abilities of the subjects.
60

Results
Subsequently, the cognitive performances and the ratio proBDNF/BDNF were correlated
with Spearman rank correlation. We analyzed separately each cognitive test, finding no
positive or negative correlation between test scores and proBDNF/BDNF ratio levels
(figure 21).
Speech Test3,5 4,0 4,5 5,0 5,5 6,0 6,5
proB
DN
F/B
DN
F ra
tio
0,00
0,05
0,10
0,15
0,20
0,25
TMT Test0 50 100 150 200 250 300 350
proB
DN
F/B
DN
F ra
tio
0,00
0,05
0,10
0,15
0,20
0,25
Digit Span Test0 2 4 6 8 10 12
proB
DN
F/B
DN
F ra
tio
0,00
0,05
0,10
0,15
0,20
0,25
Spinnler Matrix Attention Test 0 10 20 30 40 50 60 70
proB
DN
F/B
DN
F ra
tio
0,00
0,05
0,10
0,15
0,20
0,25
Figure 21. Correlation between cognitive tests and ratio proBDNF/BDNF in schizophrenic subjects (n=20). Spearman rank correlation revealed that there was no positive or negative correlation (p>0,05) with proBDNF/BDNF ratio for Speech Test (upper panel, left), TMT Test (upper panel, right), Digit Span Test (lower panel, left) and Spinnler Matrix Attention Test (lower panel, right).
Considering that our population was composed by patients treated with different
antipsychotics that could be divided in two main compound classes (typical and atypical
antipsychotics), we evaluated if the results were influenced by the pharmacological
treatment. Although proBDNF/BDNF ratio in patients treated with atypical
antipsychotics was double in respect of patients treated with typical antipsychotics, the
61

Results
limited number of subjects analyzed did not allowed us to reach a statistical significance
(figure 22).
typical antipsychotics atypical antipsychotics
proB
DN
F/B
DN
F ra
tio
0,0
0,1
0,2
0,3
0,4
0,5p>0,05
Figure 22. Comparison of the ratio between proBDNF and BDNF in schizophrenic patients treated with typical or atypical antipsychotics. There are no significative alterations (p>0,05) between the two groups.
Even though we did not find any correlation with cognitive impairment or
pharmacological treatment, experimental evidence of a reduced ratio proBDNF/BDNF
in the serum of schizophrenic subjects convinced us of the importance of this
measurement at least in schizophrenia.
However, the available ELISA kits allow only the detection of total BDNF, which we
showed to have no clinical relevance in schizophrenia. Therefore, we started to develop
a novel diagnostic kit based on an ELISA platform able to discriminate between the
precursor and the mature form of BDNF in a rapid and reproducible way.
62

Results
4.4 Development of an ELISA kit for the dosage of proBDNF/BDNF ratio
The first step for the development of the ELISA kit was the design of polyclonal
antibodies. In addition to the antibodies against the BDNF pro and mature form, we
planned the production of four antibodies designed to recognize specifically the four
different proBDNF protein variants (Pro1, Pro2, Pro3 and Pro 4), in order to investigate
whether single protein variants might be specifically involved in neuropsychiatric
disorders and cognitive deficit.
Antibodies design
To identify BDNF antigenic sites, we evaluated numerous parameters such as
hydrophobicity and antigenicity index using two different appropriate softwares
(JaMBW, Peptideselect design tool-Invitrogen). Once recognized the most promising
sites, we checked their effective exposure on the molecule surface through the analysis
of the three-dimensional crystallographic structure of mature BDNF (Robinson et al.
1995; Robinson et al. 1999). Unfortunately, the three dimensional structure of proBDNF
is not available yet.
We were able to identify three different high antigenic sites on the mature form, thus we
decided to utilize each of the three sequences for the antibodies production to ensure the
maximal chances of success. In summary, we designed eight peptides to create eight
antibodies able to discriminate between mature BDNF, proBDNF and the four proBDNF
variants (figure 23). The antibody for the detection of proBDNF4, which is carachterized
by the presence of a deletion within the mature region, was targeted against a peptide
containing 5 amminoacids before and five amminoacids after the deletion region,
recognizing a unique sequence present only in proBDNF4 variant.
63

Results
Figure 23. Schematic representation of the plan for the production of the antiBDNF antibodies
3 - Pro total
7 - Pro 4 inside del 8 - Pro 4 outside del
1 - Mat 1-12 2 - Mat 91-102
Antibodies against the mature form:
Antibody against the whole pro variants:
4 - Pro1 5 - Pro2 6 - Pro3
Antibodies against the single pro variants:
Antibody against proBDNF4:
pro-BDNF1
pro-B(ex1) DNF2
pro-BDNF3 (ex6B)
NH2
NH2
7NH2
2
pro-BDNF4
3 4
5
6
8 (Δ48aa)
NH2
NH2
1Mature BDNF
Pro region Mature region
64

Results
Antibodies production
Each polyclonal antibody was produced starting from the synthesis of a peptide
corresponding to the antigenic sequence identified as described in the previous
paragraph (EzBiolab, USA). To maximize our rate of success we followed a double-
chance strategy for the production of the antibodies based either on the home-made
production or on the externalization of the process. Combination of these two strategies
allowed us to choose the best antiserum for each of the antibodies planned.
Antibodies validation
After the final bleed, each antiserum was validated through Dot Blot, Western Blot and
ELISA techniques.
Dot blot:
We tested in Dot Blot the eight antisera made with both strategies (a total of 16
antibodies), in order to establish a battery of the eight best produced. Antisera and their
corresponding pre-immune antisera were tested against their antigenic peptides and on a
SN-K-BE (human neuroblastoma cell line) lysate.
We were able to select one antiserum for each of the antibodies designed capable to
recognize the corresponding peptide spotted at different concentrations (0,1 µg; 0,5 µg;
1 µg; 5 µg, see figure 24). Except for Pro total that seemed to have lower affinity for the
antigen than the others (the signal is detectable only for 1 and 5 µg spots), all antisera
had a strong reactivity (figure 24). Pre-immune antisera revealed to be almost no
reactive against the peptides (figure 24), whilst there was a mild response against lysates
from human neuroblastoma SN-K-BE, a cell line that express BDNF at high levels (data
not shown).
65
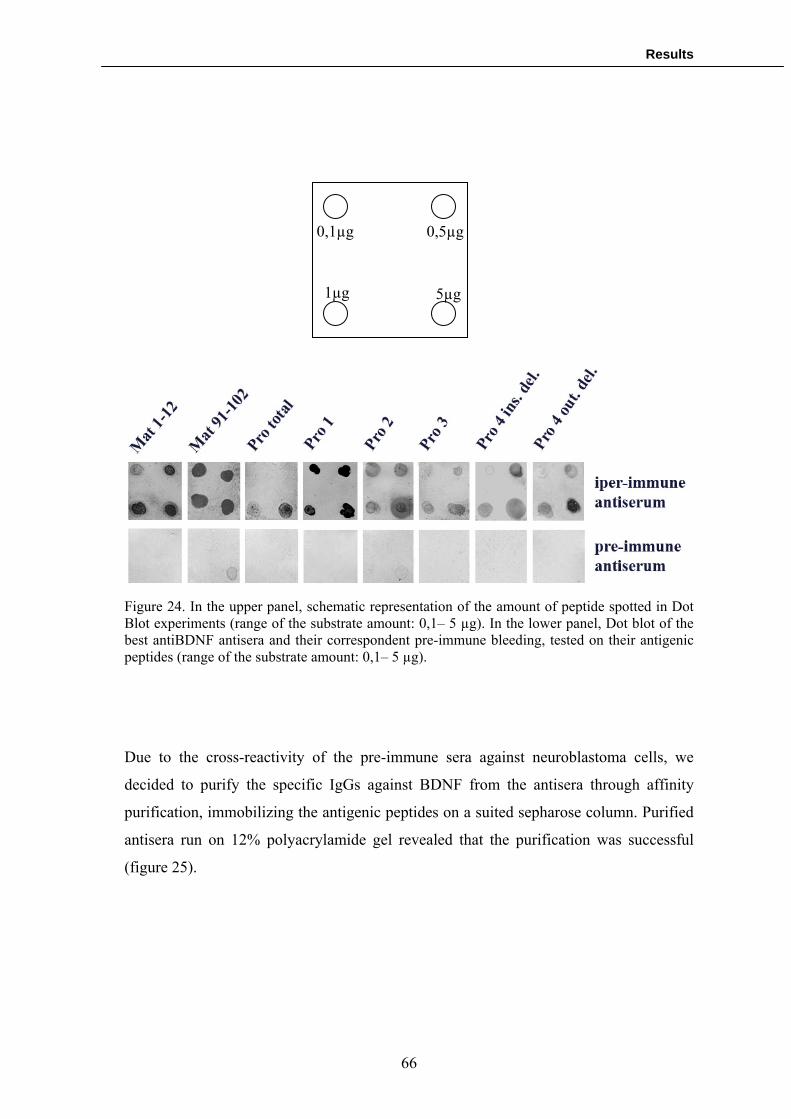
Results
0,1µg 0,5µg
1µg 5µg
Figure 24. In the upper panel, schematic representation of the amount of peptide spotted in Dot Blot experiments (range of the substrate amount: 0,1– 5 µg). In the lower panel, Dot blot of the best antiBDNF antisera and their correspondent pre-immune bleeding, tested on their antigenic peptides (range of the substrate amount: 0,1– 5 µg).
Due to the cross-reactivity of the pre-immune sera against neuroblastoma cells, we
decided to purify the specific IgGs against BDNF from the antisera through affinity
purification, immobilizing the antigenic peptides on a suited sepharose column. Purified
antisera run on 12% polyacrylamide gel revealed that the purification was successful
(figure 25).
66

Results
Figure 25. Purification of Mat 1-12 antiBDNF antibody. 12 % polyacrylamide gel stained with Coomassie Blue of the first two fractions eluited from the purification column. The heavy and the light chains of IgGs are at the expected molecular weight.
Western blot:
Purified polyclonal antibodies were tested for BDNF detection both in transfected and
non transfected cells. To express exogenous BDNF, we utilized HEK 293-T kidney cell
line because of their high transfection rate and the barely undetectable levels of BDNF
mRNAs in microarray platforms (pubmed GEO profiles; GDS1852 record). Absence of
BDNF protein was confirmed by Western blot using commercially available antiBDNF
antibodies (figure 26, lane 4).
To validate the antibodies against the mature form (Mat 1-12, Mat 91-102 and Pro 4
inside), the pro form (pro total) and the Pro1 variant of BDNF, the cell line was
transfected with a construct encoding for the human proBDNF protein in fusion with
GFP (CDS BDNF-GFP) with all the epitopes recognized by the different antibodies (see
Introduction, chapter 1.4).
Among the antibodies against the mature region, Mat 1-12 revealed to have strong
affinity in western blot for BDNF-GFP protein (figure 26).
67

Results
1 2 3 4 5 6 7 8
Figure 26. Western Blot validation of Mat 1-12 anti-BDNF antibody in lysate (30 µg) of HEK 293-T cells transfected with CDS BDNF-GFP plasmid. BDNF-GFP protein is expected at about 62 kDa (32 kDa for BDNF + 30 kDa for GFP). 1, 2, 3, 4 and 5 columns: controls. As expected, anti-GFP antibody did not detected BDNF in HEK 293 lysate and recognized GFP and BDNF-GFP in transfected HEK 293 cell lysate (columns 1, 2 and 3). Also Alomone anti-BDNF detected BDNF-GFP in transfected cell lysate (column 5), with no signal in non transfected HEK 293 (column 4). Mat 1-12 antibody recognized the same bands of Alomone anti-BDNF with higher affinity (columns 6 and 7), with no signal for the correspondent pre-immune antiserum (column 8).
In contrast, the antibody against the pro form of BDNF (pro total) demonstrated a
weaker affinity for BDNF-GFP protein (figure 27), whilst pro1 immunoglobulin failed
to detect the expected band (data not shown).
68

Results
1 2 3 4 5 6 7 8
Figure 27. Western Blot validation of pro total anti-BDNF antibody in lysate (30 µg) of HEK 293-T cells transfected with CDS BDNF-GFP plasmid. BDNF-GFP protein is expected at about 62 kDa (32 kDa for BDNF + 30 kDa for GFP). 1, 2, 3, 4 and 5 columns: controls. Anti-GFP antibody did not detected BDNF in HEK 293 lysate and recognized GFP and BDNF-GFP in transfected HEK 293 cell lysate (columns 1, 2 and 3). Alomone anti-proBDNF detected BDNF-GFP in transfected cell lysate (column 5), with no signal in non transfected HEK 293 (column 4). Pro total antibody recognized the same bands of Alomone anti-BDNF with similar affinity (columns 6 and 7), even though there was a not specific band at about 70 kDa. Correspondent pre-immune antiserum did not show reactivity against BDNF-GFP (column 8). The antibody pro1, raised against an antigen common to all the different proBDNF
variants pro2, pro3 and pro4 immunoglobulins, failed to recognize BDNF when tested
on HEK 293-T cells transfected with exon 1 (pro2) or exon VII (pro3 and pro4) BDNF-
GFP plasmids, (data not shown).
69

Results
To ensure that data obtained from the previous experiments were not due to technical
artefacts, we validated the antibodies in the SN-K-BE neuroblastoma cell line that was
already used for the dot blot experiments.
Mat 1-12 confirmed to have high affinity for BDNF, both in SN-K-BE lysate and in a
preparation of purified recombinant human BDNF. Conversely, the antibody against the
pro region (pro total) revealed an unexpected cross-reactivity with mature BDNF (figure
28).
1 2 3 4 5 6
Figure 28. Western Blot validation of Mat 1-12 and pro total anti-BDNF antibody in SN-K-BE cell lysate (30 µg) and on recombinant human BDNF protein. Anti-BDNF Alomone and mat 1-12 demonstrated high reactivity against recombinant BDNF and proBDNF (columns 1 and 3), while anti-BDNF Alomone showed strongest reactivity against mature BDNF of SN-K-BE cell lysate than Mat 1-12 (columns 2 and 4). Neither of the antibodies detected proBDNF in the lysate. Pro total revealed to have an unwanted cross-reactivity against recombinant mature BDNF and mature BDNF of SN-K-BE cell lysate (columns 5 and 6).
The antibodies against the four pro BDNF variants failed to detect BDNF both in the
neuroblastoma cell lysate and in the preparation of purified recombinant human BDNF.
.ELISA
70
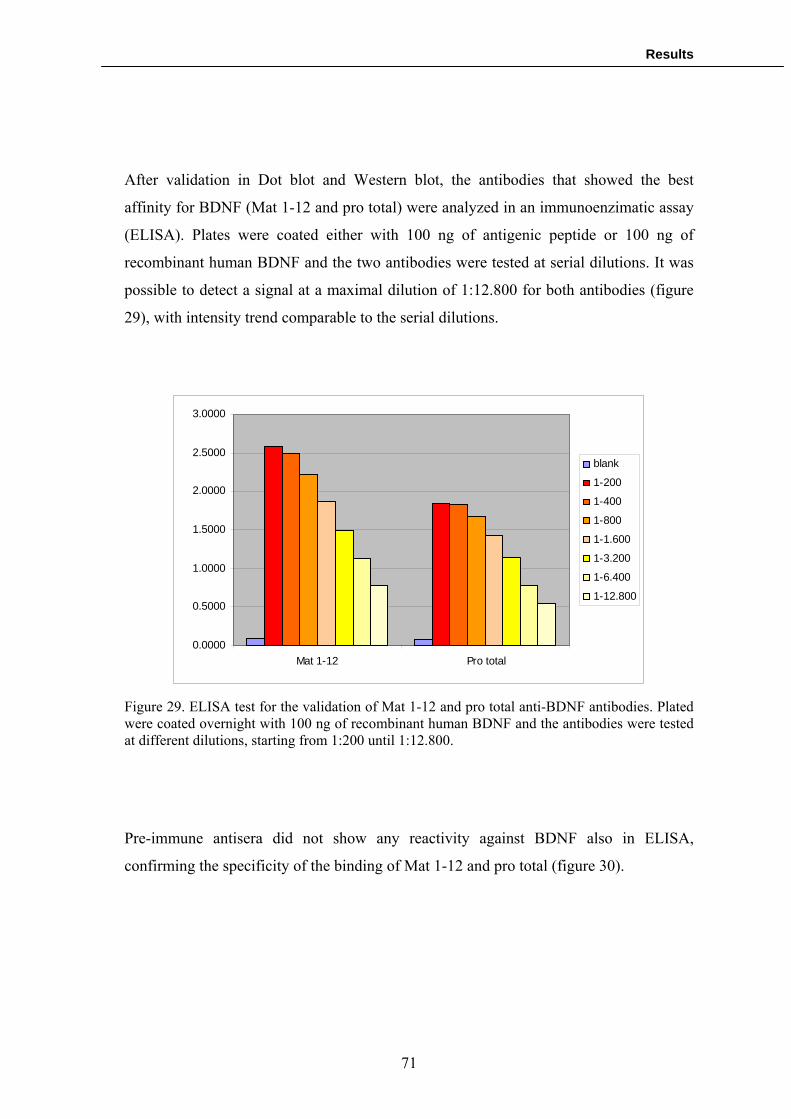
Results
After validation in Dot blot and Western blot, the antibodies that showed the best
affinity for BDNF (Mat 1-12 and pro total) were analyzed in an immunoenzimatic assay
(ELISA). Plates were coated either with 100 ng of antigenic peptide or 100 ng of
recombinant human BDNF and the two antibodies were tested at serial dilutions. It was
possible to detect a signal at a maximal dilution of 1:12.800 for both antibodies (figure
29), with intensity trend comparable to the serial dilutions.
0.0000
0.5000
1.0000
1.5000
2.0000
2.5000
3.0000
Mat 1-12 Pro total
blank
1-200
1-400
1-800
1-1.600
1-3.200
1-6.400
1-12.800
Figure 29. ELISA test for the validation of Mat 1-12 and pro total anti-BDNF antibodies. Plated were coated overnight with 100 ng of recombinant human BDNF and the antibodies were tested at different dilutions, starting from 1:200 until 1:12.800.
Pre-immune antisera did not show any reactivity against BDNF also in ELISA,
confirming the specificity of the binding of Mat 1-12 and pro total (figure 30).
71

Results
0.0000
0.5000
1.0000
1.5000
2.0000
2.5000
3.0000
Mat 1-12 pre-imm. Pro total pre-imm.
blank
1 -50
1-100
1-200
1-400
1-800
1-1.600
1-3.200
Figure 30 ELISA test of pre-immune antisera of Mat 1-12 and pro total. Plated were coated overnight with 100 ng of recombinant human BDNF and the antibodies were tested at different dilutions, starting from 1:50 until 1:3.200. Pre-immune antisera did not reveal any reactivity against BDNF.
These data underlined the good affinity of Mat 1-12 for BDNF and confirmed that pro
total seems to have an undesired cross-reactivity with recombinant mature BDNF.
Development of the ELISA kit for the dosage of the ratio proBDNF/BDNF
Antibodies validation disclosed that pro total has an unwanted cross-reactivity with
mature BDNF, forcing us to follow another strategy for the development of the ELISA
kit for the dosage of the ratio proBDNF/BDNF (see future perspectives).
At the same time, we performed some pilot studies based on Mat 1-12 to determine the
most suitable ELISA platform for the assay. Two possible platforms were analyzed.
Firstly, we investigate whether a competitive structure of the test, in which the amount
of BDNF in the samples is coated to the ELISA plates and compete with recombinant
BDNF for the binding with the anti-BDNF antibody, might be suitable for the
determination of BDNF quantity in the sample. The reason of this choice was related to
72

Results
the possibility to obtain an assay different from those (ELISA “sandwich”) that are
already present on the market for the detection of total BDNF.
Hence, we started to produce a BDNF calibration curve to quantify the ability of the
antibody to compete for BDNF. Plates were coated with 100 ng of recombinant BDNF
and Mat 1-12 antibody was incubated in the plates with different concentration of
soluble recombinant BDNF or soluble antigenic peptide. It was possible to obtain a
satisfactory competition curve, in which the affinity of the antibody for coated BDNF
decreases as the concentration of soluble competitor increases, only for the antigenic
peptide and not for BDNF (figure 31). Indeed, the competition curve decreased until
90% with soluble peptide (optimal result), whilst it reached only the 50% with soluble
BDNF. These results convinced us to choose for the other ELISA platform.
Mat 1-12 + BDNF Mat 1-12 + antigenic peptde
Figure 31. ELISA competition experiments for Mat 1-12 (coating: 100ng of BDNF). The two graphs reports the competition curves of Mat 1-12 against the antigenic peptide (right panel) and recombinant BDNF (left panel). X axis represents the ng of soluble BDNF, Y axis represents the normalized percentage of coated BDNF bound by Mat 1-12. 100 ng of antigenic peptide demonstrated to be able to saturate almost completely Mat 1-12 with a satisfactory competition curve. The competition curve for Mat 1-12 + BDNF revealed to be not satisfactory, because the trend reached a plateau for 10 ng of soluble BDNF and a 50% saturation of Mat 1-12.
0102030405060708090
100110
0.001 0.01 0.1 1 10 100ng BDNF
0102030405060708090
100110
0.001 0.01 0.1 1 10 100
% B/Bo% B/Bo
ng BDNF
73

Results
Future perspectives
Once established that an ELISA test based on a competitive structure might be not
suitable to our purposes, we designed a new platform based on ELISA sandwich. In this
test, we planned to use as “detection” antibody for the dosage of total BDNF Mat 1-12,
that demonstrated good affinity and specificity for this neurotrophin.
Considering that pro total antibody reactivity was not entirely specific, it was necessary
to produce a new “detection” antibody for the dosage of proBDNF. Therefore, in order
to obtain a more specific detection antibody, we decided to start the production of a
monoclonal antibody against the pro region.
In addition, we planned the production of another monoclonal antibody also for the
mature region. Thanks to this strategy, we will be able to utilize this monoclonal as the
“capture” antibody for the kit (figure 32), ensuring a better binding specificity than a
polyclonal immunoglobulin. Another advantage of the monoclonal antibodies is that
they can be reproduced in a highly standardized manner, which is an essential feature for
industrial projects.
Figure 32. Schematic representation of the ELISA sandwich for the dosage of proBDNF/BDNF ratio. “Capture” antibodies is the monoclonal anti-BDNF, while the two “detection” antibodies are respectively Mat 1-12 and monoclonal anti-proBDNF.
74

Results
75
Using Mat 1-12 and monoclonal anti proBDNF antibodies as “detection”, we will be
able to estimate proBDNF/BDNF ratio in tested samples. To measure the amount of
mature BDNF, it will be sufficient to subtract the amount of proBDNF detected with
monoclonal anti-proBDNF to the BDNF total amount revealed by Mat 1-12, because
Mat 1-12 recognizes both mature BDNF and proBDNF.
At the moment, we are validating the recently produced monoclonal anti-BDNF and
anti-proBDNF antibodies.

Discussion
5. DISCUSSION
5.1 Preliminary experiments
Preliminary experiments were designed in order to identify novel biomarkers of
cognitive processes that could constitute the scientific basement of clinical and industrial
assays suitable for the screening of cognition enhancing properties of developing
compounds. For this purpose, we focused our attention on Arc, an IEG involved in
synaptic plasticity and long term memory formation. To assess the potential use of Arc
as a marker of synaptic plasticity, this study utilised the M1 agonist xanomeline and the
technique of quantitative in situ hybridisation to analyse Arc expression and its
distribution in the brain of rats treated with the cognitive enhancer xanomeline, an
agonist of M1 receptor.
We decided to measure Arc mRNA expression and brain distribution following
administration of xanomeline for many reasons. It is well established that the muscarinic
cholinergic system is involved in cognitive processes and muscarinic M1 preferring
agonists have been targeted for the treatment of cognitive deficits in disorders such as
Alzheimer’s disease (AD) and have been demonstrated to improve cognition
performances in animal models as well as in the clinic. In particular, xanomeline has
been shown to have pro cognitive effects in the 8 arm radial maze, an established animal
model of spatial learning (Bymaster et al. 2002). Furthermore, recent data examining the
effects of pilocarpine, an M1/M3 agonist, on the expression of the IEG Arc in the rat
forebrain, suggests that a correlation exists between mAChR controlled signalling
cascades, the induction of Arc and synaptic plasticity (Bymaster et al. 2002). Thus,
administration of a compound capable to ameliorate cognitive performances, followed
by the evaluation of Arc expression in brain areas specifically involved in cognitive
processes (prefrontal cortex and hippocampus), allowed us to investigate Arc expression
as a potential cognition biomarker.
Results revealed a significant increase in Arc expression in the deep layers of the rat
prefrontal cortex, 1 hour following xanomeline administration, at 3 and 10 mg/kg.
76

Discussion
Significant changes in Arc expression were not detected in the superficial layers of the
prefrontal cortex, or in any of the different areas of dorsal hippocampus studied (CA1,
CA3 and DG). The increased Arc expression in the prefrontal cortex (deep layers)
correlates with results from a parallel study using immunohistochemistry to examine the
response of the IEG c-Fos to acute administration of xanomeline, where a robust
increase in c-Fos expression in cortical areas (prefrontal cortex, somatosensory cortex,
cingulate cortex) was observed (unpublished data).
Although Arc expression was unchanged in the hippocampus, it should be noted that
these animals were not habituated to handling and it is conceivable that hippocampal
effects may have been obscured by stress. Indeed, the expression of IEGs is known to be
affected by stress (Herrera et al. 1996) and in house studies have shown that handling
and mock dosing for three days prior to the actual day of dosing may decrease basal
levels of Arc (unpublished data).
However, considering the controversial data about Arc expression in prefrontal cortex
and hippocampus that did not ensure the use of Arc as a reliable biomarker of cognitive
processes, we chose to abandon the study of this gene and test another biomarker of
cognition.
5.2 Cellular and molecular mechanisms of cognitive processes
To individuate a new biomarker candidate of cognitive processes, we based our
considerations on scientific literature and experimental evidence previously
demonstrated in our laboratory. Consequently we reasoned that Brain-derived
Neurotrophic Factor, due to its important role in neuroplasticity, could represent an
interesting cognition biomarker.
Indeed, BDNF mRNA upregulation is consistently involved in long-term synaptic
plasticity, considering that its expression is enhanced by the LTP-inducing tetanic
stimulation in CA1 (Patterson et al. 1992) and dentate gyrus (Castren et al. 1993;
Dragunow et al. 1993). To support this linkage, numerous studies demonstrated that the
tetanic stimulation-dependent BDNF transcription requires CREB, a cAMP-dependent
77

Discussion
transcription factor which is strongly implicated in LTP and memory in different animal
species (Shieh et al. 1998; Tao et al. 1998).
Furthermore, BDFN expression revealed to be increased also in in vivo experiments with
animal models. Radial arm maze training in rats for spatial learning and memory
provokes a significant increase in the BDNF mRNA expression in the hippocampus
(Mizuno et al. 2000; Mizuno et al. 2003). A rapid increase in BDNF mRNA expression
has been noticed also in inhibitory avoidance training in dentate gyrus of the
hippocampus (Ma et al. 1998; Alonso et al. 2002), and during hippocampus-dependent
contextual learning (Hall et al. 2000). In addition, rats performance in Morris water
maze and their exposure to enriched environment rapidly and selectively induce BDNF
expression (Falkenberg et al. 1992).
Previous experiments in our laboratory convinced us to study BDNF regulation for
industrial and clinical applications. In fact, it was demonstrated in rats that
administration of pilocarpine, which stimulate muscarinic acetylcholine receptor
similarly to xanomeline, induces dendritic targeting of BDNF mRNA (Tongiorgi et al.
2004). The dendritic targeting of BDNF mRNA, which is peculiar of some splice
variants such as exon 2c and exon 6 (Chiaruttini et al. 2008), suggested us the idea of an
extremely fine regulation of BDNF protein synthesis, where the dendritic mRNAs might
be translated directly into dendrites to assure spatial and temporal specificity to synaptic
plasticity.
Before studying BDNF protein synthesis, a mechanism strongly involved in cognition
(see Introduction, chapter 1.2), we investigated whether BDNF proteins, translated
starting from the different mRNA splice variants, were localized in different neuronal
compartments. We hypothesized that, if this happens, it is likely that dendritic BDNF
proteins could be synthesized locally. Immunofluorescence experiments revealed that
there is a differential distribution of BDNF proteins, and this distribution matches with
the localization of BDNF mRNA slice variants (somatodendritic for exons 1 and 4, distal
dendritic for exons 2c, 6 and CDS). Interestingly, other data from our laboratory
(submitted article) showed that silencing of distal dendritic splice variants affects normal
78

Discussion
dendritic arborisation and neuronal shaping, suggesting their involvement in synaptic
plasticity.
Based on this evidence, we overexpressed alternatively somatodendritic exon 4 or distal
dendritic exon 6 to demonstrate BDNF local protein synthesis after electrical
stimulation. Dendrites were mechanically separated from the soma to avoid protein
transport, ensuring us the specificity of protein translation.
Detection of protein translation in severed dendrites after KCl stimulation, estimated by
fluorescence densitometry, represents the first unambiguous demonstration of BDNF
local protein synthesis in dendrites. The increment revealed to be rapid reaching a peek
at 15 minutes after stimulation, possibly indicating that LPS is important for a rapid
delivery of BDNF to activated synapses but does not contribute to the immediate
response (30 sec.- 3 min.) leading to BDNF secretion (Santi et al. 2006). Notably, this
mechanism is peculiar of exon 6 and not of exon 4, supporting the idea of an
involvement in synaptic plasticity of BDNF splice variants distributed along the distal
dendrites.
Moreover, it is important to underline that the synthesis of new BDNF protein is
particularly focused in “hot spots” that seem to be preferentially distributed along the
apical dendrite of pyramidal neurons, in proximity of the branching points. This
observation suggests the existence of translation sites along the dendrites that target new
BDNF proteins to nearby synapses. Accordingly, it has been demonstrated that the
dendritic distribution of Golgi-like apparatus (Golgi outposts), necessary for the post-
translational modifications of BDNF, is highly comparable with “hot spots” localization,
supporting our observations (Horton et al. 2003).
This set of experiments allowed us to better understand some fundamental aspects of
BDNF regulation and to identify this neurotrophin as a potential biomarker of cognitive
processes. In addition, the high fidelity of the method for LPS detection convinced us to
plan an industrial assay for drug screening process, capable to individuate new
compounds able to ameliorate cognitive functions in psychotics patients (see Results,
chapter 4.2). The semi-automated measurement of LPS after neuronal stimulation with
79

Discussion
developing drugs could represent a powerful tool for industrial drug screening process at
the pre-clinical level.
5.3 ProBDNF/BDNF ratio in the serum of schizophrenic patients
After the individuation of BDNF as a biomarker of synaptic plasticity and the
development of an industrial assay for drug screening, we investigated whether BDNF
could be also utilized in clinical assays to evaluate neuropsychiatric disorders and
associated cognitive deficit. Based on the scientific literature (see Introduction, chapter
1.4), we focused our attention of BDNF protein levels in human serum, an accessible
tissue sample.
For this purpose, we collected serum samples from a population of healthy volunteers
and from subjects affected by schizophrenia, the neuropsychiatric disorder with the
highest incidence of cognitive deficit (Kurtz 2005). We selected our groups as much
homogeneous as possible, avoiding possible BDNF variations due to age or sex reported
in some studies (Lommatzsch et al. 2005). In addition, blood sampling was performed
constantly in the early morning, to exclude influences due to hormonal or circadian
changes. To investigate whether there is an alteration of BDNF levels due to
pharmacological treatment, we collected serum from patients treated both with typical
and atypical antipsychotic drugs.
Considering the controversial results about total BDNF serum levels in schizophrenia
(see Introduction, chapter 1.4), we firstly determined if there was a significant alteration
in schizophrenia compared to BDNF serum levels in control group. For this and the
following experiments, samples were processed within six months from the collection,
eliminating the possibility of BDNF degradation due to long term storage or freeze-thaw
cycles (Trajkovska et al. 2007). Using a commercially available ELISA kit, we did not
find any variation of BDNF serum levels between schizophrenic and control groups.
Nevertheless, considering the biological role played by proBDNF in modulating
neuronal system, we decided to investigate whether there were alterations of the
balancing between mature BDNF and its pro form.
80

Discussion
Firstly, we demonstrated the existence of proBDNF in human serum using an antibody
(anti-BDNF N-20, Santa Cruz) commonly used to detect pro and mature BDNF in other
tissues or cell lysates (Marcinkiewicz et al. 1998; Michalski et al. 2003). Western blot
detection revealed a predominant band correspondent to mature BDNF and a much
lower signal for proBDNF. At best of our knowledge, this is the first demonstration of
the presence of proBDNF at detectable levels in human serum.
Subsequently, we selected 20 schizophrenic patients with diagnosed cognitive deficit
and we measured their ratio proBDNF/BDNF by densitometry of the respective bands. It
was decided to analyze the ratio instead of the single amounts of pro and mature BDNF
for two reasons. At first, scientific literature suggests that neuronal modulation is
influenced by the correct balancing of positive and negative signals determined by the
two forms (Lu 2003). Therefore, estimation of a single BDNF form might not reflect the
overall effects of BDNF modulation of neuronal circuits. In addition, the ratio
proBDNF/BDNF is independent by the quantity of serum proteins loaded in the gel.
From a technical point of view, even though we ensured to load the same amount of
serum proteins for each sample with Bradford method, this measure allowed us to feel
more confident about the data obtained.
Comparison of serum proBDNF/BDNF ratio between schizophrenic subjects and
healthy volunteers revealed a significant decrease in schizophrenic patients. These
differences are unlikely to result from the difference in binding affinities of antibody N-
20 for proBDNF and mature BDNF as N-20 recognizes 20 amino acids at the N-
terminus of mature BDNF that are also present in proBDNF.
Considering that mature BDNF is involved in neuronal survival and LTP, we would
have expected an increment of proBDNF/BDNF ratio in schizophrenic patients, linked
with apoptotic effects and LTD facilitation of proBDNF. However, some studies with
magnetic resonance imaging (MRI) enlightened a volume increase in certain brain areas
such as caudate, putamen and pallidum of the bilateral dorsal striatum of schizophrenic
patients (Goldman et al. 2008), suggesting that the increase could be mediated by
survival and proliferation effects due to endogenous signalling systems including those
mediated by mature BDNF. In addition, studies in experimental models of epilepsy have
81

Discussion
suggested that a large excess of mature BDNF could determine effects opposite to its
normal positive modulation (Binder et al. 2001; Simonato et al. 2006). Indeed, increase
in BDNF levels is considered to be strongly involved in hyperexcitable neuronal state
typical of epilepsy, and in particular in animal models of epilepsy, it was observed a
selective overexpression of mature BDNF while proBDNF remained at controls levels
(Langone, personal communication). Hyperexcitability due to the excess of mature
BDNF is determined by various different mechanisms, included enhanced glutamate-
mediated synaptic transmission and BDNF itself is known to potentiate glutamatergic
transmission (Binder et al. 2001). Excessive release of glutamate was demonstrated to be
associated with excitotoxicity, dendritotoxicity and spine loss (Swann et al. 2000;
Henshall 2007). Considering these findings, it seems reasonable to hypothesize that an
excess of positive modulation due to an overproduction of mature BDNF might be
involved in cellular responses leading to a psychotic condition, such as for schizophrenic
patients. Hence, treatments able to restore the normal balance of BDNF protein variants
should be of benefit for these patients.
From a molecular point of view, altered levels of proBDNF/BDNF ratio in psychotic
subjects could be due to differences in the processing of proBDNF. Enhanced proteolitic
cleavage of proBDNF, derived from enzymatic dysfunctions, may lead to a decrement of
the ratio in the serum of schizophrenic patients. At the moment, there are no studies
investigating possible alterations of proteolitic enzymes in schizophrenia. Nevertheless,
it was demonstrated that mice lacking tissue plasminogen activator or plasminogen,
members of the plasmin cascade leading to proteolitic processing of proBDNF, are
protected by stress-induced reduction of dendritic spines and poor cognitive
performances (Pawlak et al. 2005). These findings suggest that proBDNF is protective
against stress-induced effects leading to depression-like disorders, correlating with
decreased proBDNF/BDNF ratio in schizophrenic patients.
Psychotic subjects were also tested for their cognitive performances with four different
tests, to estimate cognitive functions such as attention, screening abilities, short term
memory and working memory. These tests were chosen among others for their
widespread employment in psychiatric field. A similar work was performed on parietal
82

Discussion
cortex of Alzheimer patients, demonstrating a correlation between proBDFN levels and
the severity of cognitive impairment (Peng et al. 2005).
We were not able to identify any correlation between proBDNF/BDNF ratio and
cognitive performances of schizophrenic subjects. However, it is possible that other test
addressing different cognitive functions could reveal a correlation with the
proBDNF/BDNF ratio.
Finally, we found no significant differences in serum pro BDNF/BDNF ratio between
patients treated with typical or atypical antipsychotic. This could be due to the small
number of samples examined (8 typical and 12 atypical), considering that it seems to be
noticed an increasing trend in subjects under typical antipsychotic treatment. In the next
future, we intended to increase our sample population, screening the donors also with
other neuropsychiatric tests, in order to understand if the ratio proBDNF/BDNF is
correlated with cognitive performances and if it is influenced by antipsychotic treatment.
5.4 Development of an ELISA kit for the dosage of proBDNF/BDNF ratio
Our observations of a decrease in serum proBDNF/BDNF ratio of schizophrenic patients
lead us to plan the development of a fast and reproducible Elisa assay for its
quantification. The ELISA kit is predicted to be an important clinical support for the
diagnosis of psychosis and possibly for the follow-up of antipsychotic treatment, as
discussed in the previous chapter. At the moment, available commercial ELISA kits are
capable to detect only the total amount of BDNF, without distinguish between pro and
mature form.
From the technical point of view, polyclonal antibody design was simple for what
concern the identification of antigenic sites within BDNF mature region (3 antigenic
sites found), whilst it was more problematic for proBDNF. We were able to identify a
unique antigenic site, with excellent parameters of antigenicity and hydrophobicity, for
the production of an antibody against the pro region. Similarly, proBDNF variants
forced us to design the immunoglobulins on very small N-terminal fractions of the
protein, avoiding any possibility of discrimination between different antigenic sites.
83

Discussion
Once designed the immunogenic peptides, we decided to adopt a double strategy for the
antibody generation characterized by the home made production or the externalization of
the whole process. The strategy revealed to be successful, allowing us to obtain the
entire set of antibodies validated in dot blot.
Unfortunately, Western blot validation showed that antibodies against total proBDNF
and the single proBDNF variants failed to recognize with specificity their targets in
different cellular lysates. Pro total, the antibody against the pro region, revealed to have
a weak immunoaffinity for proBDNF and an unexpected cross-reactivity with mature
BDNF, both in cell lysates and in a preparation of the purified recombinant protein.
Conversely, the antibodies against the proBDNF variants generally showed no reactivity.
This could be due to the nature of the N-terminal region that they recognize. Pro1 and
Pro2 are directed against sequences predicted to be part of a signal peptide by our
analysis with SignalIP 3.0 software (Emanuelsson et al. 2007). Usually, signal peptides
are cleaved immediately after protein translation, which would account for the lack of
detection of these two proBDNF variants by the antibodies. Conversely, Pro3 was
predicted to detect a sequence inside a signal anchor that inserts the nascent protein in
the ER membrane without any cleavage of the peptide. This observation is confusing,
because BDNF is a secreted protein sorted in vesicle and there are no evidences of its
presence in the ER membrane. Moreover, if the signal anchor is not cleaved, pro3
antibody should be able to detect pro3 protein variant, considering that it showed a
satisfactory immunoreactivity in Dot blot experiments. Finally, pro4 might have failed to
detect BDNF in Western blot because it is reported that the mRNA is expressed at very
low levels in neurons (Liu et al. 2005), leading to the translation of a probably
undetectable amount of protein.
Our best antibody, Mat 1-12 against the mature region, was satisfactory validated in Dot
blot, western blot and ELISA. Once established the necessity of an ELISA sandwich
structure of the kit, which is the common format adopted by the commercial kits for the
detection of total BDNF, we decided to utilize Mat 1-12 as the “detection” antibody for
the measurement of mature BDNF. The other “detection” antibody for the pro form of
BDNF is a monoclonal immunoglobulin that is currently under validation. Similarly, the
84

Discussion
85
“capture” antibody needed for the immobilization of total BDNF will be another
monoclonal antibody under validation. Monoclonal antibodies were produced to
guarantee higher specificity and higher affinity of the binding than polyclonal
immunoglobulins and a better reproducibility of the antibody production, which is
essential for an industrial project. Measurement of proBDNF/BDNF ratio will be
determined by a double detection of proBDNF and total BDNF in different wells.
Mature BDNF will be deducted by subtraction of proBDNF signal from total BDNF
amount.
Prior the kit release on the market, we scheduled other clinical experiments. In
particular, we are interested to understand if the ratio proBDNF/BDNF is altered in other
neuropsychiatric disorders and if it is possible to individuate a strictly defined diagnostic
range for clinical evaluation of psychotic subjects. Furthermore, we are interested to
investigate if the ratio is altered after pharmacological treatment in the different
neuropsychiatric disorders. If we were able to detect a variation, the kit might be
extremely useful to support the follow-up of patients’ medical treatment, evaluating their
response to antipsychotics. At the moment, we are collecting serum samples of patients
with depression, bipolar disorder and Attention Deficit Hyperactivity Disorder for these
studies.

Conclusions
6. CONCLUSIONS
The PhD project philosophy was based upon the study of the basic cellular and
molecular mechanisms of neuropsychiatric disorders and cognition, aimed to the
development of industrial and clinical applications. Our approach was characterized by
experiments performed both in vitro and in vivo, in order to achieve a better
comprehension of the phenomena and to design industrial and clinical assays with the
highest probability of success.
Briefly, the aims of the thesis were:
Aim 1) to study the cellular mechanisms of BDNF translation in dendrites;
Industrial application: development of a cellular assay for the in vitro screening of
compounds to treat cognitive deficit.
Aim 2) to study the correlation between neuropsychiatric disorders, cognitive deficit and
levels of BDNF in the serum.
Clinical application: development of an ELISA kit for the dosage of BDNF in human
serum as a biomarker of human cognitive deficits.
Aim 1 allowed us to consistently identify BDNF as a biomarker of synaptic plasticity
and cognitive processes. In particular, we gave the first demonstration of BDNF local
protein synthesis in severed dendrites after electrical stimulation. The development of a
cellular assay for the drug screening process, in which we utilize BDNF local protein
synthesis as a biomarker of cognition enhancing properties of novel compounds, is the
main core of a business plan for the start-up of a spin-off company. At the moment, we
are validating the physical support for local protein synthesis detection.
Aim 2, based on BDNF protein levels, was characterized by the demonstration of
proBDNF existence in human serum. Especially, we demonstrated that, in schizophrenic
subjects, there are no alterations in serum total BDNF compared to a control group,
whilst there is a significant reduction of the proBDNF/BDNF ratio.
86

Conclusions
87
Considering this observation, we planned to develop an ELISA kit capable to estimate
proBDNF/BDNF ratio for the diagnosis and for the follow-up of the medical treatment
of patients affected by neuropsychatric disorders. The assay is different from the
commercially available kits for BDNF detection, unable to discriminate between mature
and pro form. At the moment, we are validating an ELISA “sandwich” platform.

Acknowledgments
ACKNOWLEDGMENTS
This work was partially supported by GlaxoSmithKline (Harlow, UK) and Euroclone
SpA (Area Science Park, Trieste), and partially carried out in their laboratories. We
thank Psychiatric Clinic of Trieste and in particular Prof. Maurizio De Vanna, Dott.
Davide Carlino and Dott.ssa Raffaella Marin for providing serum samples of
schizophrenic subjects and evaluating their cognitive performances.
I warmly thank Prof. Tongiorgi, both from the professional and human point of view, for
giving me the opportunity to work on very exciting scientific themes and supporting my
aspiration of working on applicative projects. His scientific and human advices are
fundamental for this PhD and for my future career.
Finally, I wish to thank all the people of my lab for three years spent together…
88

References
REFERENCES
Aakalu G., Smith W. B., Nguyen N., Jiang C. and Schuman E. M. (2001) Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30(2), 489-502.
Abraham W. C. a. O., S. (1991). Kindling and Synaptic Plasticity. The Legacy of Graham Goddard, Birkhauser.
Aicardi G., Argilli E., Cappello S., Santi S., Riccio M., Thoenen H. and Canossa M. (2004) Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 101(44), 15788-92.
Alonso M., Vianna M. R., Izquierdo I. and Medina J. H. (2002) Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol 22(5-6), 663-74.
Amaral M. D. and Pozzo-Miller L. (2007) TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. J Neurosci 27(19), 5179-89.
Anagnostaras S. G., Schallert T. and Robinson T. E. (2002) Memory processes governing amphetamine-induced psychomotor sensitization. Neuropsychopharmacology 26(6), 703-15.
Andreasen N. C., Arndt S., Swayze V., 2nd, Cizadlo T., Flaum M., O'Leary D., Ehrhardt J. C. and Yuh W. T. (1994) Thalamic abnormalities in schizophrenia visualized through magnetic resonance image averaging. Science 266(5183), 294-8.
Arvanov V. L., Seebach B. S. and Mendell L. M. (2000) NT-3 evokes an LTP-like facilitation of AMPA/kainate receptor-mediated synaptic transmission in the neonatal rat spinal cord. J Neurophysiol 84(2), 752-8.
Ashe P. C., Chlan-Fourney J., Juorio A. V. and Li X. M. (2002) Brain-derived neurotrophic factor (BDNF) mRNA in rats with neonatal ibotenic acid lesions of the ventral hippocampus. Brain Res 956(1), 126-35.
Asherson P. (2005) Clinical assessment and treatment of attention deficit hyperactivity disorder in adults. Expert Rev Neurother 5(4), 525-39.
Asztely F., Kokaia M., Olofsdotter K., Ortegren U. and Lindvall O. (2000) Afferent-specific modulation of short-term synaptic plasticity by neurotrophins in dentate gyrus. Eur J Neurosci 12(2), 662-9.
Balanza-Martinez V., Tabares-Seisdedos R., Selva-Vera G., Martinez-Aran A., Torrent C., Salazar-Fraile J., Leal-Cercos C., Vieta E. and Gomez-Beneyto M. (2005) Persistent cognitive dysfunctions in bipolar I disorder and schizophrenic patients: a 3-year follow-up study. Psychother Psychosom 74(2), 113-9.
Barde Y. A., Edgar D. and Thoenen H. (1982) Purification of a new neurotrophic factor from mammalian brain. Embo J 1(5), 549-53.
Bekinschtein P., Cammarota M., Katche C., Slipczuk L., Rossato J. I., Goldin A., Izquierdo I. and Medina J. H. (2008) BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci U S A.
89

References
Binder D. K., Croll S. D., Gall C. M. and Scharfman H. E. (2001) BDNF and epilepsy:
too much of a good thing? Trends Neurosci 24(1), 47-53. Bliss T. V. and Collingridge G. L. (1993) A synaptic model of memory: long-term
potentiation in the hippocampus. Nature 361(6407), 31-9. Bozon B., Kelly A., Josselyn S. A., Silva A. J., Davis S. and Laroche S. (2003) MAPK,
CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci 358(1432), 805-14.
Burd L., Klug M. G., Coumbe M. J. and Kerbeshian J. (2003) Children and adolescents with attention deficit-hyperactivity disorder: 1. Prevalence and cost of care. J Child Neurol 18(8), 555-61.
Bymaster F. P., Felder C., Ahmed S. and McKinzie D. (2002) Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord 1(2), 163-81.
Castren E., Pitkanen M., Sirvio J., Parsadanian A., Lindholm D., Thoenen H. and Riekkinen P. J. (1993) The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. Neuroreport 4(7), 895-8.
Chiaruttini C., Sonego M., Baj G., Simonato M. and Tongiorgi E. (2008) BDNF mRNA splice variants display activity-dependent targeting to distinct hippocampal laminae. Mol Cell Neurosci 37(1), 11-9.
Chomczynski P. and Sacchi N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162(1), 156-9.
Cohen S. and Levi-Montalcini R. (1957) Purification and properties of a nerve growth-promoting factor isolated from mouse sarcoma 180. Cancer Res 17(1), 15-20.
Costello E. J., Pine D. S., Hammen C., March J. S., Plotsky P. M., Weissman M. M., Biederman J., Goldsmith H. H., Kaufman J., Lewinsohn P. M., Hellander M., Hoagwood K., Koretz D. S., Nelson C. A. and Leckman J. F. (2002) Development and natural history of mood disorders. Biol Psychiatry 52(6), 529-42.
Cracco J. B., Serrano P., Moskowitz S. I., Bergold P. J. and Sacktor T. C. (2005) Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus 15(5), 551-6.
Cunha A. B., Frey B. N., Andreazza A. C., Goi J. D., Rosa A. R., Goncalves C. A., Santin A. and Kapczinski F. (2006) Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neurosci Lett 398(3), 215-9.
Daban C., Martinez-Aran A., Torrent C., Tabares-Seisdedos R., Balanza-Martinez V., Salazar-Fraile J., Selva-Vera G. and Vieta E. (2006) Specificity of cognitive deficits in bipolar disorder versus schizophrenia. A systematic review. Psychother Psychosom 75(2), 72-84.
de Groot D. M., Coenen A. J., Verhofstad A., van Herp F. and Martens G. J. (2006) In vivo induction of glial cell proliferation and axonal outgrowth and myelination by brain-derived neurotrophic factor. Mol Endocrinol 20(11), 2987-98.
Dean B. (2004) M1 receptor agonism, a possible treatment for cognitive deficits in schizophrenia. Neuropsychopharmacology 29(8), 1583-4; author reply 1585-6.
90

References
Desai N. S., Rutherford L. C. and Turrigiano G. G. (1999) BDNF regulates the intrinsic
excitability of cortical neurons. Learn Mem 6(3), 284-91. Donovan M. J., Lin M. I., Wiegn P., Ringstedt T., Kraemer R., Hahn R., Wang S.,
Ibanez C. F., Rafii S. and Hempstead B. L. (2000) Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development 127(21), 4531-40.
Dragunow M., Beilharz E., Mason B., Lawlor P., Abraham W. and Gluckman P. (1993) Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett 160(2), 232-6.
Durany N., Michel T., Zochling R., Boissl K. W., Cruz-Sanchez F. F., Riederer P. and Thome J. (2001) Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res 52(1-2), 79-86.
Dwivedi Y., Rizavi H. S., Conley R. R., Roberts R. C., Tamminga C. A. and Pandey G. N. (2003) Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60(8), 804-15.
Dwivedi Y., Rizavi H. S., Roberts R. C., Conley R. C., Tamminga C. A. and Pandey G. N. (2001) Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J Neurochem 77(3), 916-28.
Eagle H. (1959) Amino acid metabolism in mammalian cell cultures. Science 130(3373), 432-7.
Emanuelsson O., Brunak S., von Heijne G. and Nielsen H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2(4), 953-71.
Ernfors P., Ibanez C. F., Ebendal T., Olson L. and Persson H. (1990) Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci U S A 87(14), 5454-8.
Ernfors P., Wetmore C., Olson L. and Persson H. (1990) Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron 5(4), 511-26.
Falkenberg T., Mohammed A. K., Henriksson B., Persson H., Winblad B. and Lindefors N. (1992) Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett 138(1), 153-6.
Faraone S. V., Sergeant J., Gillberg C. and Biederman J. (2003) The worldwide prevalence of ADHD: is it an American condition? World Psychiatry 2(2), 104-113.
Faraone S. V., Spencer T. J., Montano C. B. and Biederman J. (2004) Attention-deficit/hyperactivity disorder in adults: a survey of current practice in psychiatry and primary care. Arch Intern Med 164(11), 1221-6.
Feig S. and Lipton P. (1993) Pairing the cholinergic agonist carbachol with patterned Schaffer collateral stimulation initiates protein synthesis in hippocampal CA1 pyramidal cell dendrites via a muscarinic, NMDA-dependent mechanism. J Neurosci 13(3), 1010-21.
91

References
Figurov A., Pozzo-Miller L. D., Olafsson P., Wang T. and Lu B. (1996) Regulation of
synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381(6584), 706-9.
Ford T., Goodman R. and Meltzer H. (2003) The British Child and Adolescent Mental Health Survey 1999: the prevalence of DSM-IV disorders. J Am Acad Child Adolesc Psychiatry 42(10), 1203-11.
Frey U., Krug M., Reymann K. G. and Matthies H. (1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452(1-2), 57-65.
Friedman W. J., Ernfors P. and Persson H. (1991) Transient and persistent expression of NT-3/HDNF mRNA in the rat brain during postnatal development. J Neurosci 11(6), 1577-84.
Friedman W. J., Olson L. and Persson H. (1991) Cells that Express Brain-Derived Neurotrophic Factor mRNA in the Developing Postnatal Rat Brain. Eur J Neurosci 3(7), 688-697.
Fujimura H., Altar C. A., Chen R., Nakamura T., Nakahashi T., Kambayashi J., Sun B. and Tandon N. N. (2002) Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost 87(4), 728-34.
Gama C. S., Andreazza A. C., Kunz M., Berk M., Belmonte-de-Abreu P. S. and Kapczinski F. (2007) Serum levels of brain-derived neurotrophic factor in patients with schizophrenia and bipolar disorder. Neurosci Lett 420(1), 45-48.
Gardiol A., Racca C. and Triller A. (1999) Dendritic and postsynaptic protein synthetic machinery. J Neurosci 19(1), 168-79.
Geller B., Badner J. A., Tillman R., Christian S. L., Bolhofner K. and Cook E. H., Jr. (2004) Linkage disequilibrium of the brain-derived neurotrophic factor Val66Met polymorphism in children with a prepubertal and early adolescent bipolar disorder phenotype. Am J Psychiatry 161(9), 1698-700.
Goldman A. L., Pezawas L., Mattay V. S., Fischl B., Verchinski B. A., Zoltick B., Weinberger D. R. and Meyer-Lindenberg A. (2008) Heritability of brain morphology related to schizophrenia: a large-scale automated magnetic resonance imaging segmentation study. Biol Psychiatry 63(5), 475-83.
Gooney M., Shaw K., Kelly A., O'Mara S. M. and Lynch M. A. (2002) Long-term potentiation and spatial learning are associated with increased phosphorylation of TrkB and extracellular signal-regulated kinase (ERK) in the dentate gyrus: evidence for a role for brain-derived neurotrophic factor. Behav Neurosci 116(3), 455-63.
Gotz R., Koster R., Winkler C., Raulf F., Lottspeich F., Schartl M. and Thoenen H. (1994) Neurotrophin-6 is a new member of the nerve growth factor family. Nature 372(6503), 266-9.
Grillo R. W., Ottoni G. L., Leke R., Souza D. O., Portela L. V. and Lara D. R. (2007) Reduced serum BDNF levels in schizophrenic patients on clozapine or typical antipsychotics. J Psychiatr Res 41(1-2), 31-5.
Guzowski J. F., McNaughton B. L., Barnes C. A. and Worley P. F. (1999) Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci 2(12), 1120-4.
92

References
Hall J., Thomas K. L. and Everitt B. J. (2000) Rapid and selective induction of BDNF
expression in the hippocampus during contextual learning. Nat Neurosci 3(6), 533-5.
Hebb D. O. (1949). The organization of behaviour. New York, Wiley. Heldt S. A., Stanek L., Chhatwal J. P. and Ressler K. J. (2007) Hippocampus-specific
deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry 12(7), 656-70.
Henshall D. C. (2007) Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem Soc Trans 35(Pt 2), 421-3.
Herrera D. G. and Robertson H. A. (1996) Activation of c-fos in the brain. Prog Neurobiol 50(2-3), 83-107.
Hirschfeld R. M. (2002). Risk factors for major depression and bipolar disorder. Neuropsychopharmacology—The Fifth Generation of Progress. Philadelphia, Lippincott Williams and Wilkins: 1017-1025.
Ho B. C., Milev P., O'Leary D. S., Librant A., Andreasen N. C. and Wassink T. H. (2006) Cognitive and magnetic resonance imaging brain morphometric correlates of brain-derived neurotrophic factor Val66Met gene polymorphism in patients with schizophrenia and healthy volunteers. Arch Gen Psychiatry 63(7), 731-40.
Hofer M., Pagliusi S. R., Hohn A., Leibrock J. and Barde Y. A. (1990) Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. Embo J 9(8), 2459-64.
Holderbach R., Clark K., Moreau J. L., Bischofberger J. and Normann C. (2007) Enhanced long-term synaptic depression in an animal model of depression. Biol Psychiatry 62(1), 92-100.
Horton A. C. and Ehlers M. D. (2003) Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23(15), 6188-99.
Hu B., Nikolakopoulou A. M. and Cohen-Cory S. (2005) BDNF stabilizes synapses and maintains the structural complexity of optic axons in vivo. Development 132(19), 4285-98.
Hu N. W., Zhang H. M., Hu X. D., Li M. T., Zhang T., Zhou L. J. and Liu X. G. (2003) Protein synthesis inhibition blocks the late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. J Neurophysiol 89(5), 2354-9.
Hu Y., Sun C. Y., Wang H. F., Guo T., Wei W. N., Wang Y. D., He W. J., Wu T., Tan H. and Wu T. C. (2006) Brain-derived neurotrophic factor promotes growth and migration of multiple myeloma cells. Cancer Genet Cytogenet 169(1), 12-20.
Huang T. L. and Lee C. T. (2006) Associations between serum brain-derived neurotrophic factor levels and clinical phenotypes in schizophrenia patients. J Psychiatr Res 40(7), 664-8.
Huang Y. Y., Li X. C. and Kandel E. R. (1994) cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell 79(1), 69-79.
Huber K. M., Kayser M. S. and Bear M. F. (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288(5469), 1254-7.
93

References
Ikegaya Y., Ishizaka Y. and Matsuki N. (2002) BDNF attenuates hippocampal LTD via
activation of phospholipase C: implications for a vertical shift in the frequency-response curve of synaptic plasticity. Eur J Neurosci 16(1), 145-8.
Jiang C. and Schuman E. M. (2002) Regulation and function of local protein synthesis in neuronal dendrites. Trends Biochem Sci 27(10), 506-13.
Jockers-Scherubl M. C., Danker-Hopfe H., Mahlberg R., Selig F., Rentzsch J., Schurer F., Lang U. E. and Hellweg R. (2004) Brain-derived neurotrophic factor serum concentrations are increased in drug-naive schizophrenic patients with chronic cannabis abuse and multiple substance abuse. Neurosci Lett 371(1), 79-83.
Kang H. and Schuman E. M. (1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267(5204), 1658-62.
Kang H. and Schuman E. M. (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273(5280), 1402-6.
Kaplan D. R. and Miller F. D. (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10(3), 381-91.
Karege F., Perret G., Bondolfi G., Schwald M., Bertschy G. and Aubry J. M. (2002) Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109(2), 143-8.
Karege F., Schwald M. and Cisse M. (2002) Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci Lett 328(3), 261-4.
Karege F., Vaudan G., Schwald M., Perroud N. and La Harpe R. (2005) Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136(1-2), 29-37.
Katoh-Semba R., Semba R., Takeuchi I. K. and Kato K. (1998) Age-related changes in levels of brain-derived neurotrophic factor in selected brain regions of rats, normal mice and senescence-accelerated mice: a comparison to those of nerve growth factor and neurotrophin-3. Neurosci Res 31(3), 227-34.
Keefe R. S., Eesley C. E. and Poe M. P. (2005) Defining a cognitive function decrement in schizophrenia. Biol Psychiatry 57(6), 688-91.
Kermani P., Rafii D., Jin D. K., Whitlock P., Schaffer W., Chiang A., Vincent L., Friedrich M., Shido K., Hackett N. R., Crystal R. G., Rafii S. and Hempstead B. L. (2005) Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest 115(3), 653-63.
Kernie S. G., Liebl D. J. and Parada L. F. (2000) BDNF regulates eating behavior and locomotor activity in mice. Embo J 19(6), 1290-300.
Kocsis E., Trus B. L., Steer C. J., Bisher M. E. and Steven A. C. (1991) Image averaging of flexible fibrous macromolecules: the clathrin triskelion has an elastic proximal segment. J Struct Biol 107(1), 6-14.
Kuhl D. and Skehel P. (1998) Dendritic localization of mRNAs. Curr Opin Neurobiol 8(5), 600-6.
94

References
Kumar V., Zhang M. X., Swank M. W., Kunz J. and Wu G. Y. (2005) Regulation of
dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci 25(49), 11288-99.
Kurtz M. M. (2005) Neurocognitive impairment across the lifespan in schizophrenia: an update. Schizophr Res 74(1), 15-26.
Lai K. O., Fu W. Y., Ip F. C. and Ip N. Y. (1998) Cloning and expression of a novel neurotrophin, NT-7, from carp. Mol Cell Neurosci 11(1-2), 64-76.
Lanahan A., Lyford G., Stevenson G. S., Worley P. F. and Barnes C. A. (1997) Selective alteration of long-term potentiation-induced transcriptional response in hippocampus of aged, memory-impaired rats. J Neurosci 17(8), 2876-85.
Lapchak P. A., Araujo D. M., Beck K. D., Finch C. E., Johnson S. A. and Hefti F. (1993) BDNF and trkB mRNA expression in the hippocampal formation of aging rats. Neurobiol Aging 14(2), 121-6.
Lee J., Laurin N., Crosbie J., Ickowicz A., Pathare T., Malone M., Tannock R., Kennedy J. L., Schachar R. and Barr C. L. (2007) Association study of the brain-derived neurotropic factor (BDNF) gene in attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 144(8), 976-81.
Lee R., Kermani P., Teng K. K. and Hempstead B. L. (2001) Regulation of cell survival by secreted proneurotrophins. Science 294(5548), 1945-8.
Leibrock J., Lottspeich F., Hohn A., Hofer M., Hengerer B., Masiakowski P., Thoenen H. and Barde Y. A. (1989) Molecular cloning and expression of brain-derived neurotrophic factor. Nature 341(6238), 149-52.
Lessmann V., Gottmann K. and Malcangio M. (2003) Neurotrophin secretion: current facts and future prospects. Prog Neurobiol 69(5), 341-74.
Levi-Montalcini R. and Angeletti P. U. (1963) Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol 7, 653-9.
Levi-Montalcini R., Meyer H. and Hamburger V. (1954) In vitro experiments on the effects of mouse sarcomas 180 and 37 on the spinal and sympathetic ganglia of the chick embryo. Cancer Res 14(1), 49-57.
Linnarsson S., Bjorklund A. and Ernfors P. (1997) Learning deficit in BDNF mutant mice. Eur J Neurosci 9(12), 2581-7.
Lipska B. K., Khaing Z. Z., Weickert C. S. and Weinberger D. R. (2001) BDNF mRNA expression in rat hippocampus and prefrontal cortex: effects of neonatal ventral hippocampal damage and antipsychotic drugs. Eur J Neurosci 14(1), 135-44.
Lipsky R. H. and Marini A. M. (2007) Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci 1122, 130-43.
Liu Q. R., Walther D., Drgon T., Polesskaya O., Lesnick T. G., Strain K. J., de Andrade M., Bower J. H., Maraganore D. M. and Uhl G. R. (2005) Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson's Disease. Am J Med Genet B Neuropsychiatr Genet 134(1), 93-103.
Lohoff F. W., Sander T., Ferraro T. N., Dahl J. P., Gallinat J. and Berrettini W. H. (2005) Confirmation of association between the Val66Met polymorphism in the
95

References
brain-derived neurotrophic factor (BDNF) gene and bipolar I disorder. Am J Med Genet B Neuropsychiatr Genet 139(1), 51-3.
Lom B., Cogen J., Sanchez A. L., Vu T. and Cohen-Cory S. (2002) Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J Neurosci 22(17), 7639-49.
Lommatzsch M., Zingler D., Schuhbaeck K., Schloetcke K., Zingler C., Schuff-Werner P. and Virchow J. C. (2005) The impact of age, weight and gender on BDNF levels in human platelets and plasma. Neurobiol Aging 26(1), 115-23.
Lu B. (2003) Pro-region of neurotrophins: role in synaptic modulation. Neuron 39(5), 735-8.
Lu Y., Christian K. and Lu B. (2007) BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem.
Ma L., Reis G., Parada L. F. and Schuman E. M. (1999) Neuronal NT-3 is not required for synaptic transmission or long-term potentiation in area CA1 of the adult rat hippocampus. Learn Mem 6(3), 267-75.
Ma Y. L., Wang H. L., Wu H. C., Wei C. L. and Lee E. H. (1998) Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience 82(4), 957-67.
Maisonpierre P. C., Belluscio L., Friedman B., Alderson R. F., Wiegand S. J., Furth M. E., Lindsay R. M. and Yancopoulos G. D. (1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5(4), 501-9.
Malgaroli A. and Tsien R. W. (1992) Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature 357(6374), 134-9.
Marcinkiewicz M., Savaria D. and Marcinkiewicz J. (1998) The pro-protein convertase PC1 is induced in the transected sciatic nerve and is present in cultured Schwann cells: comparison with PC5, furin and PC7, implication in pro-BDNF processing. Brain Res Mol Brain Res 59(2), 229-46.
Martinez-Aran A., Penades R., Vieta E., Colom F., Reinares M., Benabarre A., Salamero M. and Gasto C. (2002) Executive function in patients with remitted bipolar disorder and schizophrenia and its relationship with functional outcome. Psychother Psychosom 71(1), 39-46.
Martinez-Aran A., Torrent C., Tabares-Seisdedos R., Salamero M., Daban C., Balanza-Martinez V., Sanchez-Moreno J., Manuel Goikolea J., Benabarre A., Colom F. and Vieta E. (2008) Neurocognitive Impairment in Bipolar Patients With and Without History of Psychosis. J Clin Psychiatry, e1-e7.
Martinowich K., Manji H. and Lu B. (2007) New insights into BDNF function in depression and anxiety. Nat Neurosci 10(9), 1089-93.
Martinussen R., Hayden J., Hogg-Johnson S. and Tannock R. (2005) A meta-analysis of working memory impairments in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 44(4), 377-84.
Merikangas K. R., Chakravarti A., Moldin S. O., Araj H., Blangero J. C., Burmeister M., Crabbe J., Jr., Depaulo J. R., Jr., Foulks E., Freimer N. B., Koretz D. S.,
96

References
Lichtenstein W., Mignot E., Reiss A. L., Risch N. J. and Takahashi J. S. (2002) Future of genetics of mood disorders research. Biol Psychiatry 52(6), 457-77.
Messaoudi E., Bardsen K., Srebro B. and Bramham C. R. (1998) Acute intrahippocampal infusion of BDNF induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J Neurophysiol 79(1), 496-9.
Messaoudi E., Ying S. W., Kanhema T., Croll S. D. and Bramham C. R. (2002) Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci 22(17), 7453-61.
Michalski B. and Fahnestock M. (2003) Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer's disease. Brain Res Mol Brain Res 111(1-2), 148-54.
Miller S., Yasuda M., Coats J. K., Jones Y., Martone M. E. and Mayford M. (2002) Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron 36(3), 507-19.
Mitchell A. J. and Dening T. R. (1996) Depression-related cognitive impairment: possibilities for its pharmacological treatment. J Affect Disord 36(3-4), 79-87.
Mizuno M., Yamada K., Olariu A., Nawa H. and Nabeshima T. (2000) Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J Neurosci 20(18), 7116-21.
Mizuno M., Yamada K., Takei N., Tran M. H., He J., Nakajima A., Nawa H. and Nabeshima T. (2003) Phosphatidylinositol 3-kinase: a molecule mediating BDNF-dependent spatial memory formation. Mol Psychiatry 8(2), 217-24.
Monteggia L. M., Luikart B., Barrot M., Theobold D., Malkovska I., Nef S., Parada L. F. and Nestler E. J. (2007) Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry 61(2), 187-97.
Monteleone P., Serritella C., Martiadis V. and Maj M. (2008) Decreased levels of serum brain-derived neurotrophic factor in both depressed and euthymic patients with unipolar depression and in euthymic patients with bipolar I and II disorders. Bipolar Disord 10(1), 95-100.
Mowla S. J., Farhadi H. F., Pareek S., Atwal J. K., Morris S. J., Seidah N. G. and Murphy R. A. (2001) Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem 276(16), 12660-6.
Mrzljak L. and Goldman-Rakic P. S. (1993) Low-affinity nerve growth factor receptor (p75NGFR)- and choline acetyltransferase (ChAT)-immunoreactive axons in the cerebral cortex and hippocampus of adult macaque monkeys and humans. Cereb Cortex 3(2), 133-47.
Mu J. S., Li W. P., Yao Z. B. and Zhou X. F. (1999) Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res 835(2), 259-65.
Murphy D. D., Cole N. B. and Segal M. (1998) Brain-derived neurotrophic factor mediates estradiol-induced dendritic spine formation in hippocampal neurons. Proc Natl Acad Sci U S A 95(19), 11412-7.
Murphy K. C., Cardno A. G. and McGuffin P. (1996) The molecular genetics of schizophrenia. J Mol Neurosci 7(2), 147-57.
97

References
Nakahashi T., Fujimura H., Altar C. A., Li J., Kambayashi J., Tandon N. N. and Sun B.
(2000) Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett 470(2), 113-7.
Narisawa-Saito M. and Nawa H. (1996) Differential regulation of hippocampal neurotrophins during aging in rats. J Neurochem 67(3), 1124-31.
Nelson M. D., Saykin A. J., Flashman L. A. and Riordan H. J. (1998) Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging: a meta-analytic study. Arch Gen Psychiatry 55(5), 433-40.
Nestler E. J., Barrot M., DiLeone R. J., Eisch A. J., Gold S. J. and Monteggia L. M. (2002) Neurobiology of depression. Neuron 34(1), 13-25.
Nestler E. J., Gould E., Manji H., Buncan M., Duman R. S., Greshenfeld H. K., Hen R., Koester S., Lederhendler I., Meaney M., Robbins T., Winsky L. and Zalcman S. (2002) Preclinical models: status of basic research in depression. Biol Psychiatry 52(6), 503-28.
Nibuya M., Morinobu S. and Duman R. S. (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15(11), 7539-47.
Nibuya M., Nestler E. J. and Duman R. S. (1996) Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16(7), 2365-72.
Nicoll R. A. and Schmitz D. (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6(11), 863-76.
Niewiadomska G., Baksalerska-Pazera M., Gasiorowska A. and Mietelska A. (2006) Nerve growth factor differentially affects spatial and recognition memory in aged rats. Neurochem Res 31(12), 1481-90.
Noga O., Englmann C., Hanf G., Grutzkau A., Seybold J. and Kunkel G. (2003) The production, storage and release of the neurotrophins nerve growth factor, brain-derived neurotrophic factor and neurotrophin-3 by human peripheral eosinophils in allergics and non-allergics. Clin Exp Allergy 33(5), 649-54.
Palmer M. J., Isaac J. T. and Collingridge G. L. (2004) Multiple, developmentally regulated expression mechanisms of long-term potentiation at CA1 synapses. J Neurosci 24(21), 4903-11.
Pan W., Banks W. A., Fasold M. B., Bluth J. and Kastin A. J. (1998) Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 37(12), 1553-61.
Pang P. T., Teng H. K., Zaitsev E., Woo N. T., Sakata K., Zhen S., Teng K. K., Yung W. H., Hempstead B. L. and Lu B. (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306(5695), 487-91.
Patterson S. L., Grover L. M., Schwartzkroin P. A. and Bothwell M. (1992) Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron 9(6), 1081-8.
Pawlak R., Rao B. S., Melchor J. P., Chattarji S., McEwen B. and Strickland S. (2005) Tissue plasminogen activator and plasminogen mediate stress-induced decline of
98

References
neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci U S A 102(50), 18201-6.
Paz R. D., Andreasen N. C., Daoud S. Z., Conley R., Roberts R., Bustillo J. and Perrone-Bizzozero N. I. (2006) Increased expression of activity-dependent genes in cerebellar glutamatergic neurons of patients with schizophrenia. Am J Psychiatry 163(10), 1829-31.
Peng S., Wuu J., Mufson E. J. and Fahnestock M. (2005) Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem 93(6), 1412-21.
Phillips H. S., Hains J. M., Laramee G. R., Rosenthal A. and Winslow J. W. (1990) Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science 250(4978), 290-4.
Pinaud R., Penner M. R., Robertson H. A. and Currie R. W. (2001) Upregulation of the immediate early gene arc in the brains of rats exposed to environmental enrichment: implications for molecular plasticity. Brain Res Mol Brain Res 91(1-2), 50-6.
Pirildar S., Gonul A. S., Taneli F. and Akdeniz F. (2004) Low serum levels of brain-derived neurotrophic factor in patients with schizophrenia do not elevate after antipsychotic treatment. Prog Neuropsychopharmacol Biol Psychiatry 28(4), 709-13.
Pliego-Rivero F. B., Bayatti N., Giannakoulopoulos X., Glover V., Bradford H. F., Stern G. and Sandler M. (1997) Brain-derived neurotrophic factor in human platelets. Biochem Pharmacol 54(1), 207-9.
Potter G. G. and Steffens D. C. (2007) Contribution of depression to cognitive impairment and dementia in older adults. Neurologist 13(3), 105-17.
Pruunsild P., Kazantseva A., Aid T., Palm K. and Timmusk T. (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90(3), 397-406.
Pyle A. D., Lock L. F. and Donovan P. J. (2006) Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol 24(3), 344-50.
Radka S. F., Holst P. A., Fritsche M. and Altar C. A. (1996) Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res 709(1), 122-301.
Raymond C. R. (2007) LTP forms 1, 2 and 3: different mechanisms for the "long" in long-term potentiation. Trends Neurosci 30(4), 167-75.
Reichardt L. F. (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361(1473), 1545-64.
Righi M., Tongiorgi E. and Cattaneo A. (2000) Brain-derived neurotrophic factor (BDNF) induces dendritic targeting of BDNF and tyrosine kinase B mRNAs in hippocampal neurons through a phosphatidylinositol-3 kinase-dependent pathway. J Neurosci 20(9), 3165-74.
Rios M., Fan G., Fekete C., Kelly J., Bates B., Kuehn R., Lechan R. M. and Jaenisch R. (2001) Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15(10), 1748-57.
99

References
Robinson R. C., Radziejewski C., Spraggon G., Greenwald J., Kostura M. R., Burtnick
L. D., Stuart D. I., Choe S. and Jones E. Y. (1999) The structures of the neurotrophin 4 homodimer and the brain-derived neurotrophic factor/neurotrophin 4 heterodimer reveal a common Trk-binding site. Protein Sci 8(12), 2589-97.
Robinson R. C., Radziejewski C., Stuart D. I. and Jones E. Y. (1995) Structure of the brain-derived neurotrophic factor/neurotrophin 3 heterodimer. Biochemistry 34(13), 4139-46.
Rosenfeld R. D., Zeni L., Haniu M., Talvenheimo J., Radka S. F., Bennett L., Miller J. A. and Welcher A. A. (1995) Purification and identification of brain-derived neurotrophic factor from human serum. Protein Expr Purif 6(4), 465-71.
Rosenthal A., Goeddel D. V., Nguyen T., Martin E., Burton L. E., Shih A., Laramee G. R., Wurm F., Mason A., Nikolics K. and et al. (1991) Primary structure and biological activity of human brain-derived neurotrophic factor. Endocrinology 129(3), 1289-94.
Roux P. P. and Barker P. A. (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67(3), 203-33.
Santi S., Cappello S., Riccio M., Bergami M., Aicardi G., Schenk U., Matteoli M. and Canossa M. (2006) Hippocampal neurons recycle BDNF for activity-dependent secretion and LTP maintenance. Embo J 25(18), 4372-80.
Schecterson L. C. and Bothwell M. (1992) Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron 9(3), 449-63.
Schimmelmann B. G., Friedel S., Dempfle A., Warnke A., Lesch K. P., Walitza S., Renner T. J., Romanos M., Herpertz-Dahlmann B., Linder M., Schafer H., Seitz C., Palmason H., Freitag C., Meyer J., Konrad K., Hinney A. and Hebebrand J. (2007) No evidence for preferential transmission of common valine allele of the Val66Met polymorphism of the brain-derived neurotrophic factor gene (BDNF) in ADHD. J Neural Transm 114(4), 523-6.
Schuhmann B., Dietrich A., Sel S., Hahn C., Klingenspor M., Lommatzsch M., Gudermann T., Braun A., Renz H. and Nockher W. A. (2005) A role for brain-derived neurotrophic factor in B cell development. J Neuroimmunol 163(1-2), 15-23.
Seidah N. G., Benjannet S., Pareek S., Chretien M. and Murphy R. A. (1996) Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett 379(3), 247-50.
Shen W., Wu B., Zhang Z., Dou Y., Rao Z. R., Chen Y. R. and Duan S. (2006) Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron 50(3), 401-14.
Shieh P. B., Hu S. C., Bobb K., Timmusk T. and Ghosh A. (1998) Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20(4), 727-40.
Shimazu K., Zhao M., Sakata K., Akbarian S., Bates B., Jaenisch R. and Lu B. (2006) NT-3 facilitates hippocampal plasticity and learning and memory by regulating neurogenesis. Learn Mem 13(3), 307-15.
100

References
Shimizu E., Hashimoto K., Okamura N., Koike K., Komatsu N., Kumakiri C., Nakazato
M., Watanabe H., Shinoda N., Okada S. and Iyo M. (2003) Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry 54(1), 70-5.
Shimizu E., Hashimoto K., Watanabe H., Komatsu N., Okamura N., Koike K., Shinoda N., Nakazato M., Kumakiri C., Okada S. and Iyo M. (2003) Serum brain-derived neurotrophic factor (BDNF) levels in schizophrenia are indistinguishable from controls. Neurosci Lett 351(2), 111-4.
Simonato M., Tongiorgi E. and Kokaia M. (2006) Angels and demons: neurotrophic factors and epilepsy. Trends Pharmacol Sci 27(12), 631-8.
Spearing M. (2001). Bipolar Disorder. Washinghton, DC, NIH. Steward O. (1997) mRNA localization in neurons: a multipurpose mechanism? Neuron
18(1), 9-12. Steward O., Huang F. and Guzowski J. F. (2007) A form of perforant path LTP can
occur without ERK1/2 phosphorylation or immediate early gene induction. Learn Mem 14(6), 433-45.
Steward O. and Levy W. B. (1982) Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci 2(3), 284-91.
Steward O., Wallace C. S., Lyford G. L. and Worley P. F. (1998) Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21(4), 741-51.
Swann J. W., Al-Noori S., Jiang M. and Lee C. L. (2000) Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 10(5), 617-25.
Tan Y. L., Zhou D. F., Cao L. Y., Zou Y. Z. and Zhang X. Y. (2005) Decreased BDNF in serum of patients with chronic schizophrenia on long-term treatment with antipsychotics. Neurosci Lett 382(1-2), 27-32.
Tancredi V., D'Arcangelo G., Mercanti D. and Calissano P. (1993) Nerve growth factor inhibits the expression of long-term potentiation in hippocampal slices. Neuroreport 4(2), 147-50.
Tao X., Finkbeiner S., Arnold D. B., Shaywitz A. J. and Greenberg M. E. (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20(4), 709-26.
Terlau H. and Seifert W. (1989) Influence of epidermal growth factor on long-term potentiation in the hippocampal slice. Brain Res 484(1-2), 352-6.
Terlau H. and Seifert W. (1990) Fibroblast Growth Factor Enhances Long-term Potentiation in the Hippocampal Slice. Eur J Neurosci 2(11), 973-977.
Tiedge H. and Brosius J. (1996) Translational machinery in dendrites of hippocampal neurons in culture. J Neurosci 16(22), 7171-81.
Timmusk T., Palm K., Metsis M., Reintam T., Paalme V., Saarma M. and Persson H. (1993) Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10(3), 475-89.
Tongiorgi E., Armellin M., Giulianini P. G., Bregola G., Zucchini S., Paradiso B., Steward O., Cattaneo A. and Simonato M. (2004) Brain-derived neurotrophic
101

References
factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci 24(30), 6842-52.
Tongiorgi E., Righi M. and Cattaneo A. (1997) Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17(24), 9492-505.
Toyooka K., Asama K., Watanabe Y., Muratake T., Takahashi M., Someya T. and Nawa H. (2002) Decreased levels of brain-derived neurotrophic factor in serum of chronic schizophrenic patients. Psychiatry Res 110(3), 249-57.
Trajkovska V., Marcussen A. B., Vinberg M., Hartvig P., Aznar S. and Knudsen G. M. (2007) Measurements of brain-derived neurotrophic factor: methodological aspects and demographical data. Brain Res Bull 73(1-3), 143-9.
Tsui C. C., Copeland N. G., Gilbert D. J., Jenkins N. A., Barnes C. and Worley P. F. (1996) Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci 16(8), 2463-78.
Vickers C. A., Dickson K. S. and Wyllie D. J. (2005) Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol 568(Pt 3), 803-13.
Walz R., Roesler R., Reinke A., Martins M. R., Quevedo J. and Izquierdo I. (2005) Short- and long-term memory are differentialy modulated by hippocampal nerve growth factor and fibroblast growth factor. Neurochem Res 30(2), 185-90.
Weickert C. S., Hyde T. M., Lipska B. K., Herman M. M., Weinberger D. R. and Kleinman J. E. (2003) Reduced brain-derived neurotrophic factor in prefrontal cortex of patients with schizophrenia. Mol Psychiatry 8(6), 592-610.
Weickert C. S., Ligons D. L., Romanczyk T., Ungaro G., Hyde T. M., Herman M. M., Weinberger D. R. and Kleinman J. E. (2005) Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol Psychiatry 10(7), 637-50.
Wetmore C., Ernfors P., Persson H. and Olson L. (1990) Localization of brain-derived neurotrophic factor mRNA to neurons in the brain by in situ hybridization. Exp Neurol 109(2), 141-52.
Willcutt E. G., Doyle A. E., Nigg J. T., Faraone S. V. and Pennington B. F. (2005) Validity of the executive function theory of attention-deficit/hyperactivity disorder: a meta-analytic review. Biol Psychiatry 57(11), 1336-46.
Williams J., Dragunow M., Lawlor P., Mason S., Abraham W. C., Leah J., Bravo R., Demmer J. and Tate W. (1995) Krox20 may play a key role in the stabilization of long-term potentiation. Brain Res Mol Brain Res 28(1), 87-93.
Woo N. H., Teng H. K., Siao C. J., Chiaruttini C., Pang P. T., Milner T. A., Hempstead B. L. and Lu B. (2005) Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci 8(8), 1069-77.
Yamamoto H. and Gurney M. E. (1990) Human platelets contain brain-derived neurotrophic factor. J Neurosci 10(11), 3469-78.
Ying S. W., Futter M., Rosenblum K., Webber M. J., Hunt S. P., Bliss T. V. and Bramham C. R. (2002) Brain-derived neurotrophic factor induces long-term
102

References
103
potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22(5), 1532-40.
Young K. M., Merson T. D., Sotthibundhu A., Coulson E. J. and Bartlett P. F. (2007) p75 neurotrophin receptor expression defines a population of BDNF-responsive neurogenic precursor cells. J Neurosci 27(19), 5146-55.
Zhang J., Geula C., Lu C., Koziel H., Hatcher L. M. and Roisen F. J. (2003) Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp Neurol 183(2), 469-81.
Zhou X. F., Song X. Y., Zhong J. H., Barati S., Zhou F. H. and Johnson S. M. (2004) Distribution and localization of pro-brain-derived neurotrophic factor-like immunoreactivity in the peripheral and central nervous system of the adult rat. J Neurochem 91(3), 704-15.
Ziegenhorn A. A., Schulte-Herbruggen O., Danker-Hopfe H., Malbranc M., Hartung H. D., Anders D., Lang U. E., Steinhagen-Thiessen E., Schaub R. T. and Hellweg R. (2007) Serum neurotrophins--a study on the time course and influencing factors in a large old age sample. Neurobiol Aging 28(9), 1436-45.
Zorner B., Wolfer D. P., Brandis D., Kretz O., Zacher C., Madani R., Grunwald I., Lipp H. P., Klein R., Henn F. A. and Gass P. (2003) Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than "depressive". Biol Psychiatry 54(10), 972-82.

List of papers
1. Leone E., Carlino D., De Vanna M., Tongiorgi E. - The ratio between BDNF
precursor and its mature form is decreased in serum of schizophrenic patients. –
Submitted to Journal of Neurochemistry.
2. Leone E., Vicario A., Baj G. – Business Plan “Neuroscrin” – selected as one of the
best ten projects of the university of Trieste “Start Cup 2006” competition.
3. Vicario A., Baj G., Leone E. and Tongiorgi E. (2006) – Evolutionary conserved
mechanisms of RNA trafficking in neurons and the regulation of spine morphology –
Caryologia Vol. 59 n. 4: 413-423.
4. Baj G., Leone E., Gan W.B., Chao M. V. and Tongiorgi E. - BDNF mRNA splice
variants represent a spatial code to regulate local plasticity of dendrites and spines in
developing and mature neurons – Submitted to PNAS.
104







![Blandino GIovanni MD CV January 2013 31[1] · p53 axis promotes aggressiveness in breast cancer", Università degli Studi di Trieste, Trieste, Italy 2010 (Opponent). Organization](https://static.fdocuments.us/doc/165x107/5b2dd5797f8b9a594c8cef3e/blandino-giovanni-md-cv-january-2013-311-p53-axis-promotes-aggressiveness.jpg)









