
UNIVERSIDADE DE LISBOArepositorio.ul.pt/bitstream/10451/2476/1/ulsd059472_Td_Ana_Vaz.pdf · À...
177
UNIVERSIDADE DE LISBOA FACULDADE DE FARMÁCIA IDENTIFICATION OF CELLULAR TARGETS FOR SPECIFIC THERAPIES IN NEURODEVELOPMENTAL DISORDERS Ana Rita Mendonça Vaz Doutoramento em Farmácia (Biologia Celular e Molecular) 2010
Transcript of UNIVERSIDADE DE LISBOArepositorio.ul.pt/bitstream/10451/2476/1/ulsd059472_Td_Ana_Vaz.pdf · À...

UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
IDENTIFICATION OF CELLULAR TARGETS FOR SPECIFIC
THERAPIES IN NEURODEVELOPMENTAL DISORDERS
Ana Rita Mendonça Vaz
Doutoramento em Farmácia
(Biologia Celular e Molecular)
2010


UNIVERSIDADE DE LISBOA
FACULDADE DE FARMÁCIA
IDENTIFICATION OF CELLULAR TARGETS FOR SPECIFIC
THERAPIES IN NEURODEVELOPMENTAL DISORDERS
Ana Rita Mendonça Vaz
Research advisor: Dora Maria Tuna de Oliveira Brites, PhD.
Co-advisor: Maria Alexandra de Oliveira Silva Braga Pedreira de Brito, PhD.
Doutoramento em Farmácia
(Biologia Celular e Molecular)
2010


IDENTIFICATION OF CELLULAR TARGETS FOR SPECIFIC
THERAPIES IN NEURODEVELOPMENTAL DISORDERS
IDENTIFICAÇÃO DE ALVOS TERAPÊUTICOS ESPECÍFICOS PARA O TRATAMENTO DE DOENÇAS DO
NEURODESENVOLVIMENTO
Dissertação apresentada à faculdade de Farmácia da Universidade de Lisboa para
obtenção do grau de Doutor em Farmácia (Biologia Celular e Molecular)
Ana Rita Mendonça Vaz
2010


Para a elaboração da presente tese de doutoramento foram usados integralmente
como capítulos, artigos científicos publicados, ou submetidos para publicação, em
revistas científicas internacionais indexadas. Estes trabalhos foram realizados em
colaboração com os seguintes autores: Sandra L. Silva, Maria Delgado-Esteban,
Andreia Barateiro, Adelaide Fernandes, Ana Sofia Falcão, Juan P. Bolaños, Angeles
Almeida, Maria Alexandra Brito e Dora Brites.
De acordo com o disposto no ponto 1 do artigo nº41 do Regulamento de Estudos Pós-
Graduados da Universidade de Lisboa, deliberação nº 93/2006, publicada em Diário
da República – II Série nº 153 – 5 de Julho de 2003, o Autor desta dissertação declara
que participou na concepção e execução do trabalho experimental, interpretação dos
resultados obtidos e redacção dos manuscritos.


Os estudos apresentados nesta dissertação foram realizados no grupo de
investigação “Neuron Glia Biology in Health & Disease”, Research Institute for
Medicines and Pharmaceutical Sciences (iMed.UL), Faculdade de Farmácia da
Universidade de Lisboa. Parte do trabalho foi também realizado no Departamento
de Bioquímica e Biologia Molecular da Universidade de Salamanca, Espanha, sob
a supervisão dos Professores Doutores Juan P. Bolaños e Angeles Almeida.
O trabalho foi subsidiado pelos projectos FCT-POCTI/SAU/MMO/55955/2004,
FCT-PTDC/SAU-NEU/64385/2006 concedidos à Professora Doutora Dora Brites
pela Fundação para a Ciência e Tecnologia (FCT), sendo que a Autora usufruiu de
uma bolsa de Doutoramento (SFRH/BD/30292/2006) concedida pela FCT, Lisboa,
Portugal,.


Agradecimentos __________________________________________________________________________
Agradecimentos
As minhas primeiras palavras de agradecimento vão para a Professsora Doutora Dora
Brites, orientadora deste trabalho. Agradeço-lhe por me ter recebido ainda enquanto
estudante de Licenciatura e me ter dado a a conhecer o mundo da investigação. Agradeço-
lhe também a oportunidade e o incentivo de fazer este Doutoramento, assim como todos os
conhecimentos que me transmitiu ao longo destes anos. Estou certa que o elevado nível de
exigência e rigor que estiveram sempre presentes na orientação científica deste trabalho
contribuíram de uma forma muito positiva para a elevada qualidade do mesmo. Do ponto de
vista científico, a sua capacidade de criação e raciocínio é bastante inspiradora, mesmo a
partir de ideias que ainda estejam a nascer nas nossas mentes, o que me faz sempre
acreditar no sucesso de novos projectos em que a Professora esteja envolvida. De um ponto
de vista mais pessoal, agradeço ainda a inteira disponibilidade para a orientação desta
Tese, assim como por me fazer acreditar que, se dermos o nosso melhor, podemos alcançar
o nível de excelência naquilo que fazemos, e assim contribuirmos activamente para nos
tornarmos pessoas especiais.
Agradeço também à Professora Doutora Alexandra Brito, minha co-orientadora. A
orientação científica que sempre me disponibilizou foi muito importante para a progressão
deste trabalho. Agradeço-lhe a disponiblidade para a orientação desta Tese, assim como a
constante motivação para fazer mais e melhor. Consigo aprendi que o “só mais um
esforçozinho” compensa quando queremos ser bem sucedidos. E quando alcançarmos esse
tão desejado sucesso, podemos acreditar que as coisas só têm tendência a melhorar!
Pessoalmente, agradeço-lhe toda a simpatia e preocupação que demonstrou comigo, assim
como o carinho que sempre me deu.
Me gustaría también agradecer a los Profesores Ángeles Almeida y Juan Bolaños del
Departamento de Bioquímica y Biología molecular de la Universidade de Salamanca por la
oportunidad que me ofrecieron de pasar parte significativa de mi doctorado en su
laboratorio. Durante esos periodos, me fue dada la oportunidad de aprender varias y muy
útiles metodologias experimentales. Nuestra colaboración se ha revelado muy provechosa
para el desarrollo de la presente tesis, una vez que los resultados ahí obtenidos constituyen
uno de los puntos clave aquí presentados.
À minha colega e mais que tudo, amiga, Sandra Guedes, deixo um agradecimento muito
especial. A tua amizade foi um dos achados mais preciosos desde que enveredei pelo
caminho da Ciência, quase que dava para a incluir no capítulo das conclusões! Sabes que
sempre contei com a tua força e sentido prático das coisas, tão importantes nas fases mais

Agradecimentos __________________________________________________________________________
difíceis. Esta fase final foi trabalhosa e houve momentos em que parecia que o caminho
estava continuamente a ser acrescentado mas ambas conseguimos lá chegar, e por isso
estamos de Parabéns! Ver-te concluir esta etapa ao mesmo tempo que eu vai ser uma fonte
adicional de satisfação. Para o futuro, desejo-te toda a felicidade, quer a nível profissional
como a nível pessoal. Não tenho dúvidas que serás bem sucedida nas duas pois para além
de inteligente, és muito justa e sensível e ainda por cima é fácil trabalhar e aprender contigo.
Quero deixar um OBRIGADA às minhas queridas colegas e amigas Adelaide Fernandes e
Sofia Falcão. Durante estes anos, vocês foram sempre os primeiros alvos das minhas
questões e dramas existenciais, tão característicos de quem procura o que ainda mais
ninguém encontrou. Adelaide, o teu conhecimento científico é uma fonte de inspiração, já
para não falar na tua capacidade de organização para teres sempre tempo para toda a
gente que te pede orientações sobre os próprios projectos e a quem tu nunca negas ajuda.
Sofia, a tua boa disposição e simpatia são contagiantes e as tuas orientações ao meu
trabalho foram sempre um grande contributo. Tem sido muito gratificante trabalhar e trocar
ideias convosco.
Quiero agradecer también a María Delgado-Esteban, que fue la responsable de
acompañarme en el laboratorio de la Universidad de Salamanca. Gracias por tu paciencia
com una recién estudiante de doctorado, acabada de llegar, siempre llena de preguntas y a
veces com menos respuestas. Contigo aprendí las bases de las regulaciones enzimáticas y
descubrí que se puede estudiar todo un mundo alrededor del metabolismo energético. De
esta colaboración nació también nuestra amistad. Creo que no podría haber tenido más
suerte con la persona con la que me tocó trabajar. Gracias por recibirme tan bien, incluso
fuera del laboratorio, y por hacerme sentir como si estuviera en casa en una ciudad
extranjera.
Me gustaría agradecer a todos los elementos del grupo de la Universidad de Salamanca, en
especial a Julia, Ángel y Mónica por la simpatia y amistad com que me acogieron y por el
esfuerzo constante en entender mi pseudo-español.
Agradeço também de uma forma muito carinhosa aos restantes elementos do grupo Neuron
Glia Biology in Health & Disease. Ao Professor Doutor Rui Silva agradeço a boa disposição
e a ajuda sempre presentes quando a ele recorri; consigo aprendi que às vezes o mais
importante é procurar qual é a pergunta certa! À Andreia um muito obrigada por seres
companheira de todas as horas; mesmo longe sei que posso contar sempre contigo. À Ema
e à Inês, minhas “pupilas” do coração, agradeço a amizade constante e os bons conselhos
que às vezes são tão precisos. À Filipa agradeço a espontaneidade que tantas vezes me fez

Agradecimentos __________________________________________________________________________
rir…Meninas, a todas vocês eu desejo as maiores felicidades para a continuação do vosso
Doutoramento e para a vossa vida posterior. Agradeço também à Cibelle e à Eduarda, que
mesmo tendo estado menos tempo connosco, contribuíram para as memórias felizes e
inesquecíveis que eu guardo destes anos.
A todos os colegas do Centro de Patogénese Molecular, obrigada pela vossa simpatia e
também partilharem das nossas venturas e aventuras do dia-a-dia.
Agradeço também a todos os amigos e familiares que me apoiaram na decisão de fazer o
Doutoramento, e que sempre me ouviram com entusiasmo e atenção a falar dos “meus
ratinhos”, ainda que por vezes não entendessem muito bem daquilo que eu falava…
Um especial agradecimento à minha Tia Nanda e ao meu Tio Tiago, por terem sempre
incentivado as minhas escolhas, provavelmente inspiradas neles, que foram as primeiras
pessoas mais próximas de cientistas que eu conheci. Eu sei que posso sempre contar com
vocês e vocês sabem que podem sempre contar comigo. A ti, Tiaguinho, agradeço o dom
que tens de me deixar sempre contente, mesmo nos momentos em ando mais desanimada.
E obrigada por me dizeres que também queres ser cientista como eu quando cresceres, faz-
me acreditar ainda mais naquilo que eu faço!
Aos meus Pais, Lídia e Francisco, deixo um agradecimento do tamanho do Mundo!
Obrigada pelo incentivo constante em querer estudar mais e procurar um futuro melhor,
especialmente porque neste momento isso ainda depende muito do vosso contributo. Bem
sei que às vezes não estive presente nem cheguei a horas mas nem por isso vocês
deixaram de me apoiar. E essa estabilidade em que me mantiveram a todos os níveis
contribuiu de forma decisiva para me tornar naquilo que eu sou, penso ou faço. Por isso,
tenho todo o prazer de partilhar esta Tese convosco, porque em verdade ela também é
vossa…
Ao Hugo deixo o meu agradecimento final. Aliás, a nós os dois. Porque a nossa vida estará
sempre ligada à Ciência e porque o gosto pela Ciência proporcionou que nos
conhecêssemos. Porque este ano foi atribulado e cheio de decisões difíceis e tu estiveste
sempre presente e deste-me força para continar. Porque todos os dias me fazes acreditar
que o que é necessário para estar contigo vale a pena…


Contents ________________________________________________________________________
xiii
Contents
I. Abbreviations ................................................................................................................ xix
Abstract ....................................................................................................................... xxiii
Resumo ........................................................................................................................ xxv
I. General Introduction ................................................................................................... 1 1. Redox status and cellular bioenergetics in central nervous system: regulation and
dysfunction ...................................................................................................................... 3
1.1. Free radicals, reactive species and antioxidants ............................................... 3
1.2. Pathways of glucose utilization .......................................................................... 7
1.3. Mitochondria: the powerhouse of the cell and the major source of ROS/RNS 10
1.4. Dysfunctional mitochondria ............................................................................. 12
2. Neuronal-glia actions and interplay in the brain ....................................................... 13
2.1. Glutathione shuttle ................................................................................................ 14
2.2. Glutamate shuttle ............................................................................................ 15
2.3. Lactate shuttle ................................................................................................. 17
2.4. Neuronal susceptibility to oxidative stress ....................................................... 18
2.4.1. Increased oxidant capacity in the brain ................................................. 18
2.4.2. Antioxidant capacity in the brain ............................................................ 20
2.5.Neuronal susceptibility bioenergetic crisis ........................................................ 21
3. Inflammation and cell death in central nervous system ........................................... 22
3.1. Cells involved in inflammation and CNS injury ................................................ 22
3.2. Inflammatory mediators and signalling pathways ............................................ 22
3.3. Neuronal susceptibility to inflammation ........................................................... 24
3.4. Death signalling pathways ............................................................................... 25
4. Bilirubin induced neurological damage and risk factors involved ............................ 27
4.1. Neonatal hyperbilirubinemia ............................................................................ 27

Contents ________________________________________________________________________
xiv
4.2. Prematurity as a risk factor of neonatal hyperbilirubinemia ............................. 28
4.3. Sepsis-associated neonatal hyperbilirubinemia .............................................. 29
4.4. Differential neuronal vulnerability among brain regions ................................... 31
4.5. Mechanisms underlying bilirubin-induced neurotoxicity .................................. 32
5. Promising molecules for modulation in hyperbilirubinemia ...................................... 34
5.1. Glycoursodeoxycholic acid (GUDCA) .............................................................. 34
5.2. N-ω-nitro-L-arginine methyl ester hydrochloride (L-NAME) ............................. 35
5.3. N-acetylcysteine (NAC) ................................................................................... 35
6. Global aims of the thesis ......................................................................................... 37
7. References .............................................................................................................. 38
II. Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in immature cortical neurons. Assessment of the protective effects of glycoursodeoxycholic acid ................................................................................................. 59
Abstract ....................................................................................................................... 61
1. Introduction .............................................................................................................. 62
2. Materials and Methods ............................................................................................ 63
2.1. Chemicals ........................................................................................................ 63
2.2. Neurons in primary culture .............................................................................. 64
2.3. Treatment of neurons ...................................................................................... 64
2.4. Determination of the mitochondrial respiratory chain complex activities and
citrate synthase ............................................................................................................. 64
2.5. Detection of superoxide anion radical (O2.-) ..................................................... 65
2.6. Determination of oxygen consumption ............................................................ 65
2.7. Ѱm measurements ........................................................................................... 65
2.8. Metabolite determinations ................................................................................ 66
2.9. Assessment of apoptotic cell death by flow citometry ..................................... 67
2.10. Analysis of apoptotic cell death by 4'-6-diamidino-2-phenylindole (DAPI)
nuclear staining ............................................................................................................. 67

Contents ________________________________________________________________________
xv
2.11. Caspase-3 and -9 activity assays .................................................................. 67
2.12. Statistical analysis ......................................................................................... 68
3. Results ..................................................................................................................... 68
3.1. UCB selectively impairs cytochrome c oxidase activity in immature neurons,
which is prevented by GUDCA...................................................................................... 68
3.2. UCB produces oxidative stress in immature neurons, which is prevented by
GUDCA ......................................................................................................................... 69
3.3. UCB impairs cellular oxygen consumption and collapses ΔѰm in immature
neurons and GUDCA exerts a preventive effect ........................................................... 70
3.4. UCB increases extracellular ATP content, glycolysis and F2,6P2 levels in
immature neurons, which are counteracted by GUDCA ............................................... 71
3.5. UCB triggers apoptotic cell death in immature neurons, which is prevented by
GUDCA ......................................................................................................................... 72
4. Discussion ............................................................................................................... 73
5. References .............................................................................................................. 78
III. Pro-inflammatory cytokines intensify the activation of .NO/NOS, JNK1/2 and caspase cascades in immature neurons exposed to elevated levels of unconjugated bilirubin ................................................................................................................................. 83
Abstract ....................................................................................................................... 85
1. Introduction .............................................................................................................. 86
2. Materials and Methods ............................................................................................ 88
2.1. Chemicals ........................................................................................................ 88
2.2. Neurons in primary culture ............................................................................... 88
2.3. Treatment of neurons ...................................................................................... 89
2.4. Quantification of nitrite levels ........................................................................... 89
2.5. Western blot assay .......................................................................................... 89
2.6. Caspase activity determination ........................................................................ 90
2.7. MTT reduction ................................................................................................. 90
2.8. Densitometry and statistical analysis ............................................................... 90

Contents ________________________________________________________________________
xvi
3. Results ..................................................................................................................... 91
3.1. UCB, alone or in combination with TNF-α+IL-1β, induces nNOS expression
and .NO production in immature neurons, which are counteracted by l-NAME ............ 91
3.2. Inhibition of nNOS by l-NAME prevents the cascade of apoptosis induced by
UCB or UCB+TNF-α+ IL-1β in immature neurons ........................................................ 91
3.3. Inhibition of nNOS by l-NAME decreases P-JNK1/2 in immature neurons
treated with UCB or UCB+TNF-α+IL-1β ....................................................................... 94
3.4. Inhibition of P-JNK1/2 by SP600125 prevents the cascade of apoptosis
induced by UCB or UCB+TNF-α+IL-1β in immature neurons ....................................... 96
3.5. Loss of neuronal functionality in immature cells exposed to UCB is increased
by UCB+TNF-α+IL-1β and prevented by inhibition of nNOS and JNK1/2 activation .... 96
4. Discussion ............................................................................................................... 98
5. References ............................................................................................................ 103
IV. Selective vulnerability of rat brain regions to unconjugated bilirubin ............... 109 Abstract ..................................................................................................................... 111
1. Introduction ............................................................................................................ 112
2. Materials and Methods .......................................................................................... 114
2.1. Chemicals ...................................................................................................... 114
2.2. Neurons in primary culture ............................................................................ 114
2.3. Treatment of neurons .................................................................................... 115
2.4. Quantification of nitrite levels ......................................................................... 115
2.5. Western blot assay ........................................................................................ 115
2.6. Determination of cGMP concentration: .......................................................... 116
2.7. Glutathione measurement ............................................................................. 116
2.8. Assessment of ROS formation ...................................................................... 116
2.9. Evaluation of cell death ................................................................................. 117
2.10. Neurite Extension and Ramification ............................................................ 117
2.11. Densitometry and statistical analysis ........................................................... 117
3. Results ................................................................................................................... 118

Contents ________________________________________________________________________
xvii
3.1. UCB-induced nNOS expression and production of nitrites and cGMP is
enhanced in immature hippocampal neurons as compared to cerebellar or cortical
neurons ....................................................................................................................... 118
3.2. UCB-induced oxidative stress is highest in immature hippocampal neurons,
probably as a result of the lowest levels of total glutathione ....................................... 118
3.3. UCB-induced neuronal death is higher in immature cells from hippocampus
than in those from cortex or cerebellum ...................................................................... 121
3.4. UCB-induced neuronal oxidative stress and cell death in immature neurons is
prevented by NAC ....................................................................................................... 123
3.5. UCB regulates DJ-1 protein expression in immature neurons, mainly in those
from hippocampus, which is reverted by NAC ............................................................ 123
3.6. UCB-induced reduction of neurite outgrowth and branching mainly in
immature neurons from hippocampus, is closely followed by those from cerebellar
and cortical regions, and is prevented by NAC ........................................................... 125
4. Discussion ............................................................................................................. 126
5. References ............................................................................................................ 131
V. Final considerations ................................................................................................ 137 1. Concluding remarks and perspectives ................................................................... 139
2. References ............................................................................................................ 145

xviii

Abbreviations ________________________________________________________________________
xix
Abbreviations 7-AAD 7-amino-actinomycin
AGUDC Ácido glico-ursodesoxicólico
Ala Alanine
Apaf 1 Protease activating factor 1
ATP Adenosine triphosphate
BCAA Branched-chain amino acid
BIND Bilirubin-induced neurologic dysfunction
BNC Bilirrubina não conjugada
CAT Catalase
cGMP cyclic Guanosine monophosphate
CHAPS Cholamidopropyldimethylammonio-1-propanesulfonate
CNS Central nervous system
CO Carbon monoxide
CO2 Carbon dioxide
CuZnSOD Cooper/zinc superoxide dismutase
Cys Cysteine
Cyt c Cytochrome c
DHR 123 Dihydrorhodamine 123
DNIB Disfunção neurológica induzida pela bilirrubina
ERK 1/2 Extracellular signal-regulated kinases 1 and 2
F2,6P2 Fructose-2,6-bisphosphate
FADH2 Reduced flavin adenine dinucleotide
FasR Fas receptor
FBS Fetal bovine serum
FCCP Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
G6P Glucose 6-phosphate
G6PD Glucose 6-phosphate dehydrogenase
Gln Glutamine
Glu Glutamate
Gly Glycine
GPx Glutathione peroxidase
GR Glutathione reductase
GSH Reduced glutathione
GSSG Oxidized glutathione
GST Glutathione S-transferase
GUDCA Glycoursodeoxycholic acid
H2O2 Hydrogen peroxide
HBSS Hanks’ balanced salt solution

Abbreviations ___________________________________________________________________
xx
HNE 4-Hydroxy-2-nonenal
HO Heme oxygenase
HSA Human serum albumin
IL-1 Interleukin-1
IL-1ra IL-1 receptor antagonist
IL-1β Interleukin-1β
ICE IL-1β-converting enzyme
IL-6 Interleukin-6
IM Inner mitochondrial membrane
IMS Intermembrane space
JNK 1/2 c-Jun N-terminal kinases 1 and 2
Leu Leucine
L-NAME N-ω-nitro-L-arginine methyl ester hydrochloride
L-NMMA N-ω-monomethyl-L-arginine
MAP-2 Microtubule-associated protein 2
MAPKs Mitogen-activated protein kinases
MEM Minimum essential medium
MnSOD Manganese superoxide dismutase
MPP+ 1-Methyl-4-phenylpyridinium ion
Mrp1 Multidrug resistance associated protein 1
NAC N-acetylcysteine
NAD Nicotinamide adenine dinucleotide
NADH Reduced nicotinamide adenine dinucleotide
NADPH Reduced nicotinamide adenine dinucleotide phosphate
NF-κB Nuclear factor κB
NH3 Ammonia
NMDA N-methyl-D-aspartate
NMDAR N-methyl-D-aspartate receptor .NO Nitric oxide
NOS Nitric oxide synthase
nNOS Neuronal isoform of NOS
mtNOS Mitochondrial isoform of NOS
iNOS Inducible isoform of NOS
eNOS Endothelial isoform of NOS
NOX NADPH oxidase enzymes
O2 Oxygen
O2.- Superoxide anion radical
O22- Peroxide anion
.OH Hydroxyl radical

Abbreviations ________________________________________________________________________
xxi
OM Outer mitochondrial membrane
ONOO- Peroxynitrite
PARP Poli (ADP-ribose) polymerase
PDH Pyruvate dehydrogenase
PFK1 6-phosphofructo-1-kinase
PFKFB 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
Pi Inorganic phosphate
Pgp P-glycoprotein
pNA p-nitroaniline
PPP Pentose phosphate pathway
Pyr Pyruvate
R. Radicals
RNS Reactive nitrogen species
ROOH Peroxides
ROS Reactive oxygen species
SAPKs Stress-activated protein kinases
SOD Superoxide dismutase
TNF-α Tumor necrosis factor-α
TACE TNF-α converting enzyme
TNFR TNF-α receptor
tBid truncated Bid
TCA Tricarboxylic acid cycle
TMRE Tetramethylrhodamine
TUDCA Tauroursodeoxycholic acid
UCB Unconjugated bilirubin
UDCA Ursodeoxycholic acid
VEGF Vascular endothelial growth factor
α-KG α-ketoglutarate
γ-GluCys γ-L-glutamyl-L-cysteinylglycine

xxii

Abstract __________________________________________________________________________
xxiii
Abstract The present dissertation is focused in neonatal hyperbilirubinemia, a very common
condition in the neonatal period, characterized by increased concentrations of unconjugated
bilirubin (UCB). High levels of UCB may lead to bilirubin-induced neurologic dysfunction
(BIND), particularly in premature infants, which may be a starting point to the appearance of
long-term neurodevelopment disabilities. In cultures isolated from rat brain, toxicity induced
by UCB is more pronounced in neuronal cells that in those from glia, and immature cells are
more prone to this injury. Among the UCB-induced cytotoxic effects, are the extracellular
accumulation of glutamate and the up-regulation of inflammatory pathways (mainly in glial
cells), permeabilization of the mitochondrial membrane (in isolated mitochondria), impairment
of neuritic development (in immature cortical and hippocampal neurons) and oxidative stress
(in differentiated neurons). Firstly, we intended to better understand the mechanisms of
neurotoxicity by UCB, mimicking a condition of prematurity, regarding oxidative stress,
mitochondrial dysfunction associated with bioenergetic alterations and cell death, as well as
to study the role of known modulators of oxidant species production in the prevention of
UCB-induced neuronal injury. The obtained results showed that rat immature cortical
neurons exposed to UCB undergo oxidative stress, mitochondrial dysfunction associated
with respiration failure and cell death, effects that are prevented in the presence of
glycoursodeoxycholic acid, a compound with antioxidant and anti-inflammatory properties. In
addition, since prematurity is often associated with sepsis, these studies evaluated the
additional effects of inflammation on hyperbilirubinemia. We demonstrated that UCB induced
nitrosative stress, c-Jun N-terminal kinases 1 and 2 signalling and cell death and that these
effects are intensified by pro-inflammatory cytokines tumor necrosis factor-α and interleukin-
1β, through the same cascade of mediators. Finally, it was investigated whether there is a
dissimilar brain regional susceptibility to UCB-induced oxidative damage and neurite
outgrowth and branching disruption in immature neurons, which might determine the
preferential UCB deposition and brain damage in specific brain areas characteristic of
kernicterus, such as cerebellum and hippocampus, and also the mechanisms that are
involved in the modulation of UCB-induced neurotoxicity. Rat hippocampal neurons were the
most susceptible to UCB-induced oxidative and nitrosative stress, as well as to UCB-induced
neuritic impairment and cell death. N-acetylcysteine, a precursor of glutathione synthesis,
was able to counteract the UCB-induced neurotoxicity. Taken together, these studies will
substantiate target-driven approaches to the prevention and treatment of BIND, and provide
fruitful opportunities for future investigations.

Abstract __________________________________________________________________________
xxiv
Keywords: Bilirubin-induced neurological dysfunction (BIND); BIND-associated
inflammation; oxidative and nitrosative stress; antioxidants; mitochondrial dysfunction;
caspase activation; brain regional vulnerability.

Resumo __________________________________________________________________________
xxv
Resumo A presente dissertação é dirigida para o estudo da hiperbilirrubinémia neonatal, uma
situação clínica frequente durante a primeira semana de vida, resultante da elevação das
concentrações da bilirrubina não conjugada (BNC). A disfunção neurológica induzida pela
bilirrubina (DNIB) poderá ser o ponto de partida para algumas doenças do
neurodesenvolvimento, especialmente nos bebés prematuros. Utilizando modelos de
culturas celulares obtidas a partir de cérebro de rato, verificou-se que a toxicidade induzida
pela BNC é mais pronunciada em neurónios do que em astrócitos, sendo as células mais
jovens particularmente susceptíveis. Do vasto leque de mecanismos moleculares envolvidos
na toxicidade induzida pela BNC, destacam-se a acumulação de glutamato extracelular e a
resposta inflamatória (nas células gliais), a diminuição do desenvolvimento neurítico (em
neurónios do córtex e do hipocampo) e o stresse oxidativo (em neurónios diferenciados).
Numa primeira fase, este trabalho teve como objectivo compreender os mecanismos
associados à lesão pela BNC, no que respeita ao stresse oxidativo, disfunção mitocondrial e
morte celular, assim como avaliar o efeito protector do ácido glico-ursodesoxicólico
(AGUDC), um composto com propriedades anti-oxidantes, em condições que mimetizam
uma situação de prematuridade. Para tal foram utilizadas culturas de neurónios corticais
com 3 dias, obtidos de cérebros de rato. Neste modelo, a exposição à BNC conduziu ao
stresse oxidativo, disfunção da respiração mitocondrial, e consequente morte celular, efeitos
que foram prevenidos na presença do AGUDC. De seguida, avaliaram-se os efeitos
adicionais da incubação concomitante da BNC com as citocinas pro-inflamatórias,
mimetizando uma reacção inflamatória associada à hiperbilirrubinémia. Utilizando o mesmo
modelo, observou-se que tanto o stresse nitrosativo, como a morte celular surgem
aumentados após esta a incubação concomitante da BNC com as citocinas pro-
inflamatórias, estando envolvidos os mesmos mediadores e vias sinalizadoras. Por fim,
investigou-se de que forma o padrão de deposição específico da BNC encontrado na
patologia de kernicterus é determinado pela diferente vulnerabilidade regional à lesão
oxidativa e ao desenvolvimento neurítico pela BNC. Para tal, isolaram-se neurónios não só
do córtex mas também do hipocampo e do cerebelo de rato. Os neurónios do hipocampo
mostraram ser mais susceptíveis ao stresse oxidativo e nitrosativo induzidos pela BNC,
assim como à disfunção do desenvolvimento neurítico e à morte celular. A incubação com o
precursor da síntese da glutationa N-acetilcisteína preveniu os efeitos tóxicos induzidos pela
BNC. Em conclusão, estes resultados contribuem para o melhor conhecimento dos
mecanismos moleculares subjacentes à DNIB no período neonatal, tendo as moléculas com
capacidade anti-oxidante um efeito notório na prevenção desta disfunção.

Resumo __________________________________________________________________________
xxvi
Palavras-chave: disfunção neurológica induzida pela bilirrubina (DNIB); DNIB associada
à sépsis; stresse oxidativo e nitrosativo; anti-oxidantes; disfunção mitocondrial; activação
das caspases; vulnerabilidade regional do encéfalo.

Chapter I
I. General Introduction


General Introduction _________________________________________________________________________
3
1. Redox status and cellular bioenergetics in central nervous system: regulation and dysfunction
1.1. Free radicals, reactive species and antioxidants Oxidative stress is classically defined as an imbalance between the levels of oxidants
and antioxidants and has been implicated in the cell death pathways of several disorders in
the central nervous system (CNS). Under normal circumstances, cells can regulate the
production of oxidants and antioxidants, resulting in redox equilibrium. Oxidative stress
occurs when cells are subjected to excess levels of reactive oxygen/nitrogen species
(ROS/RNS), or as a result of depletion in antioxidant defences Figure I.1. ROS result from
the body’s homeostatic response to the presence of molecular oxygen. Since it contains two
unpaired electrons, molecular oxygen is considered to be a diradical, accordingly with the
definition of a free radical - any chemical species containing one or more unpaired electrons
occupying an atomic or molecular orbital and can generate highly reactive species (Poli et
al., 2000).
Figure I.1 - Oxidative stress results from imbalance between the levels of reactive oxygen and nitrogen species (ROS/RNS) and antioxidants. Under normal circumstances, cells are able to balance the production of ROS/RNS and antioxidants, resulting in redox equilibrium. Oxidative stress occurs when cells are subjected to excess levels of ROS/RNS, or as a result of depletion in antioxidant defences.
ROS/RNS were originally considered to be exclusively detrimental to the cells but
nowadays they are recognized as key modulators in cellular functions, such as regulation of
redox cell signalling, gene modulation, neuromodulation, activation of signalling cascades,
differentiation, apoptosis and necrosis (Circu and Aw, 2010, Finkel, 2000, Yoneyama et al.,
ROS/RNSAntioxidants
ROS/RNS
Antioxidants
Equilibrium
Oxidative stress(Depleted Antioxidants)
ROS/RNS
Antioxidants
Oxidative stress(Excess ROS/RNS)

Chapter I __________________________________________________________________________
4
2010). Therefore, the classical concept of oxidative stress as “an imbalance between the
production of oxidants and the occurrence of cell antioxidant defences”, proposed by Sies H.
(Sies, 1997), is now being redefined as “a disruption of redox signalling and control that
recognizes the occurrence of compartmentalized cellular redox circuits”, as reviewed by
Packer and Cadenas (2007).
Among ROS group, the most common species are: superoxide anion radical (O2.-),
hydrogen peroxide (H2O2), peroxide anion (O22-) and hydroxyl radical (.OH). Among RNS, the
most important species are peroxynitrite (ONOO-) and nitric oxide (.NO). Mitochondria are
the main source of ROS, since generation of O2.- occurs during oxidative phosphorylation.
O2.- is easily converted into H2O2 by the action of superoxide dismutase (SOD). H2O2 can
originate .OH in the presence of iron (Fe2+) or copper (Cu+), by Fenton’s reaction. .OH is a
very potent inducer of lipid peroxidation and, along with peroxidation products, such as 4-
hydroxy-2-nonenal (HNE), is capable of impairing protein and acid nucleic functions, as well
as destroying cell membranes (Brito et al., 2007). .NO is a free radical generated from L-arginine, which is converted to L-citrulline in the
presence of O2, reduced nicotinamide adenine dinucleotide phosphate (NADPH) and
tetrahydrobiopterin, by a reaction catalysed by nitric oxide synthase (NOS) (Knowles et al.,
1989). In post-synaptic neurons, .NO is generated subsequently to activation of glutamate
receptor, mainly of the N-methyl-D-aspartate (NMDA) subtype. After this activation, Ca2+ is
transiently increased in the cytosol and forms a complex with calmodulin that binds to and
activates constitutive neuronal NOS (nNOS). Glial cells (astrocytes, microglia and
oligodendrocytes) synthesize .NO after the transcriptional expression of a Ca2+-independent
inducible NOS (iNOS). There is a third isoform of NOS, endothelial NOS (eNOS), that is
Ca2+-dependent (as nNOS) and is able to generate and release .NO from the brain
microvessels (Knowles and Moncada, 1994, Merrill et al., 1997). Different isoforms of NOS
are involved in distinct processes: nNOS is mainly involved in neuronal signalling, iNOS is
generally induced after an inflammatory stimulus and eNOS is involved in vasodilation
(Moncada and Bolaños, 2006). More recently, it was described a fourth isoform of NOS,
mitochondrial NOS (mtNOS), in rat liver mitochondria (Ghafourifar and Richter, 1997). The
mtNOS was identified as the splice variant α of the nNOS with the post-translational
modifications of myristilation and phosphorylation (Elfering et al., 2002). Although the
presence of mtNOS have been confirmed in several tissues, organs and cells, the NOS
isozyme that accounts for the formation of mtNOS is still a matter of debate. However, as
reviewed by Ghafourifar and Cadenas (2005), there is a growing notion that mtNOS is an
enzyme associated with the matrix face of the mitochondrial inner membrane, which

General Introduction _________________________________________________________________________
5
generates .NO in a Ca2+-dependent manner. It is also believed that .NO produced by mtNOS
regulates mitochondrial respiration. .NO can interact with O2
.-, generating ONOO-, which is very unstable and also a potent
inducer of lipid peroxidation. In addition, ONOO- participates in the nitration of tyrosine and in
oxidation of glutathione, processes that can impair several cellular functions (Moncada and
Bolaños, 2006). Besides being essential for neurotransmission, .NO accumulation leads to
excitotoxicity caused by over-activation of NMDA receptors (Dawson et al., 1991). However,
it should be taken into account that .NO is an important intercellular neuronal modulator and
plays a fundamental role not only in neuronal death but also in neuronal survival pathways.
Being an intercellular messenger, the rate and concentration of .NO are critical for its
modulatory function in the brain, as reviewed by Laranjinha and Ledo (2007).
In order to fight against oxidative injury, cells possess mechanisms to destroy or to expel
reactive species; these are called antioxidant defences, which can be divided enzymatic and
non-enzymatic systems. The most relevant antioxidant enzymes are SOD, catalase (CAT),
glutathione peroxidase (GPx) and glutathione S-transferase (GST). SOD, CAT and GPx
mainly have a preventive action, since they avoid oxidative damage by destroying or
inactivating ROS. GST acts by a repair mechanism, eliminating ROS-derived molecules,
such as hydroperoxides. Non-enzymatic systems are constituted by low molecular weight
compounds that act against peroxyl radicals. Some examples are: (i) α-tocopherol, that
inhibits lipid peroxidation by scavenging peroxyl radicals at the expense of a poorly reactive
radical generation, α- tocopheryl; (ii) ascorbic acid, that is able to remove the radical α-
tocopheryl, generating ascorbyl, a much less reactive radical; (iii) glutathione (γ-glutamyl-L-
cysteinylglycine), a tripeptide that serves as subtract to GPx and GST and also reacts
directly with radicals in non-enzymatic reactions, as represented in Figure I.2 (Brito et al.,
2007).
Glutathione is the most abundant cellular thiol present in mammalian cells. This
molecule constitutes one of the primary antioxidant defences of the cells, as it reacts directly
with radicals in nonenzymatic reactions and is also a donor of electrons in the reduction of
peroxides catalized by GPx (Dringen, 2000). The thiol group (SH) of cysteine serves as a
proton donor and is responsible for the biological activity of glutathione. Provision of this
amino acid is the rate-limiting factor in glutathione synthesis by the cells. In addition,
glutathione is essential for cell proliferation (Cotgreave and Gerdes, 1998) and regulation of
apoptosis (Ghibelli et al., 1998, Lu, 2009). In vivo, glutathione is synthesized by the action of
two enzymes: (i) γ-glutamylcysteine synthetase, which uses L-glutamate and cysteine to form
γ-glutamylcysteine; (ii) glutathione synthetase, which adds glycine to γ-glutamylcysteine,

Chapter I __________________________________________________________________________
6
originating the tripeptide glutathione. Both reactions require energy in the form of adenosine
triphosphate (ATP), being the first one the rate-limiting step in glutathione synthesis (Dringen
et al., 2000). Glutathione antioxidant action is extremely important in brain injury. In fact,
glutathione levels are reported to be markedly decreased in case of ischemia-reperfusion
lesion and inhibition of the enzymes involved in glutathione synthesis results in amplification
of brain damage (Mizui et al., 1992). In addition, GPx activity is considered determinant in the
recovery of the immature mouse brain subjected to traumatic brain injury (Tsuru-Aoyagi et
al., 2009) and several in vitro and in vivo studies support the neuroprotective effect of N-
acetylcysteine (NAC), an important precursor of cellular glutathione (Dringen, 2000,
Zachwieja et al., 2005) in lipid peroxidation and in antioxidant enzyme activities deficiencies
of rats’ brain (Nehru and Kanwar, 2004), as well as in hypoxia-induced oxidative stress in rat
cultured hippocampal neurons (Jayalakshmi et al., 2005).
Figure I.2 – Schematic representation of glutathione protective role in oxidative stress. Glutathione reacts directly with radicals (R.) in non-enzimatic reactions and is also a donor of electrons in the reduction of peroxides (ROOH), a reaction catalyzed by glutathione peroxidase (GPx). The resulting oxidized glutathione (GSSG) is recycled through the action of glutathione reductase (GR), a reaction dependent of reduced nicotinamide adenine dinucleotide phosphate (NADPH). Adapted from Brito et al. (2007).
Another compound that may have some antioxidant properties is bilirubin. The ability of
low nanomolar concentrations of bilirubin to overcome large amounts of oxidants by
efficiently scavenge peroxyl radicals was explained by a redox cycling mechanism, whereas
biliverdin reductase plays a key role. Through this catalytic cycle, and as schematically
represented in Figure I.3, bilirubin is oxidized to biliverdin by reactive species, neutralizing
Gly – Cys – Glu|
S|
Gly – Cys – Glu
NADPH
NADP
GR
ROOH
ROH+ H2O
2 R.
2 RH
GPx
2 GSH
GSSG
Gly - Cys - Glu

General Introduction _________________________________________________________________________
7
their toxicity, and then is regenerated by the action of biliverdin reductase, an enzyme
dependent of NADPH (Barañano et al., 2002, Stocker et al., 1987, Brito et al., 2006).
Figure I.3 – Amplification of the antioxidant properties of bilirubin by a redox cycling mechanism. Large amounts of oxidant species (R•) can be neutralized (RH) through bilirubin oxidation to biliverdin, which is rapidly reduced back to bilirubin by biliverdin reductase, a reaction dependent of nicotinamide adenine dinucleotide phosphate (NADPH). Adapted from Brito et al. (2006).
1.2. Pathways of glucose utilization
Maintenance of cellular activity within CNS requires large amounts of energy. Catabolic
pathways, in which organic nutrient molecules are converted into smaller and simpler end
products such as lactic acid, carbon dixode (CO2) and ammonia (NH3), release energy, some
of which is conserved in the formation of ATP and reduced electron carriers [reduced
nicotinamide adenine dinucleotide (NADH), NADPH, and reduced flavin adenine
dinucleotide (FADH2)]; the rest is lost as heat. In spite of fatty acids and aminoacids can be
bioenergetic precursors, glucose constitutes the main source of energy for most cells, being
the only one in the brain. Glucose is stored as high molecular weight polymers, such as
glycogen. However, glycogen stores are very limited in the brain, thus a permanent glucose
supply via the blood stream is necessary in order to maintain brain function. In the resting
brain, oxygen is mainly used for the oxidation of glucose. In fact, although brain represents
only ~2% of the total body weight, it contributes to more that 20% of the total consumption of
both oxygen and glucose. When energy demands increase, glucose is released from
glycogen and used to produce ATP either aerobically or anaerobically. In the first step of
glycolysis, glucose is activated for subsequent reactions by its phosphorylation to yield
glucose 6-phosphate (G6P), with ATP as the phosphoryl donor, in an irreversible reaction
catalyzed by hexokinase. G6P is then degraded during the sequential reactions of glycolysis,
NADPH
NADP2 R.
2 RH
Bilirubin
Biliverdin
Biliverdinreductase

Chapter I __________________________________________________________________________
8
where some of the free energy is conserved in the form of ATP and NADH. This process
occurs in the cytosol. The end product of glycolysis is pyruvate, which may have three
distinct metabolic fates, depending on tissue and environmental conditions (Nelson and Cox,
2005). Under normoxic conditions, pyruvate is converted into acetyl-coenzyme A by pyruvate
dehydrogenase (PDH) complex, a cluster of enzymes located in the mitochondria of
eukaryotic cells. The acetyl group is then oxidized to CO2 in the tricarboxylic acid cycle
(TCA), a process where energy of oxidation is temporarily held in the electron carriers FADH2
and NADH. The electrons resulting from these oxidations are passed to O2 through a chain
of carriers in the mitochondria (mitochondrial respiratory chain), in a process called oxidative
phosphorylation. The energy released by the flow of electrons through the mitochondrial
respiratory chain complexes is used to pump protons out of the inner mitochondria
membrane through complexes I (NADH dehydrogenase), II (succinate dehydrogenase), III
(ubiquinone: cytochrome c oxidoreductase) and IV (cytochrome c oxidase), coupling NADH
oxidation and passage of protons between mitochondrial matrix and intermembrane space.
This passage generates an electrochemical gradient across the inner mitochondrial
membrane, called proton-motive force, which drives protons back into the matrix, providing
the energy necessary for ATP synthesis, by the phosphorylation of ADP into ATP by F0F1-
ATPase (complex V, ATP synthase), a process denominated chemiosmotic theory. O2
serves as the ultimate electron acceptor and is reduced to water (Nicholls and Ferguson,
2002, Bolaños et al., 2010). Pathways of glucose utilization are schematically represented in
Figure I.4.
Under conditions of hypoxia or anoxia, or in case of impairment of the components of the
mitochondrial respiratory chain, NADH cannot be re-oxidized to nicotinamide adenine
dinucleotide (NAD), in spite of its requirement as an electron acceptor for the further
oxidation of pyruvate. Under these conditions, glycolytic rate increases and pyruvate is
reduced to lactate, accepting electrons from NADH and thereby regenerating the NAD
necessary for glycolysis to continue. Although this process is less efficient from
bioenergetics’ point of view, it may occur in a necessary level to provide the energetic needs
of the cells. This alternative is made at the expense of an increase rate in glucose
consumption (Nicholls and Ferguson, 2002).

General Introduction _________________________________________________________________________
9
Figure I.4 - Schematic representation of glucose utilization pathways. Glucose catabolism can be divided into three stages: (i) glycolysis, where glucose is metabolized in enzimatic sequential reactions. In aerobic conditions, the end product is pyruvate. During glycolysis, a small portion of free energy is conserved in the form of adenosine triphosphate (ATP) and the electron carrier reduced nicotinamide adenine dinucleotide (NADH); (ii) tricarboxylic acid cycle (TCA), a process where energy of oxidation is temporarily held in the electron carriers reduced flavin adenine dinucleotide (FADH2) and NADH and also conserved in the form of ATP; (iii) oxidative phosphorylation, where the electrons carried by NADH and FADH2 passes through a chain of carriers in the mitochondria, that constitute the respiratory chain. This passage generates an electrochemical gradient across the mitochondrial inner membrane, providing the energy necessary for ATP synthesis. An alternative pathway for glucose utilization is the pentose phosphate pathway, necessary for the maintenance of redox capacity of the cell, with the formation of reduced nicotinamide adenine dinucleotide phosphate (NADPH). Adapted from “Cellular respiration” from Department of Biology, University of Miami (2007).
In addition, G6P (the branching point of glucose metabolism) can be metabolized in the
pentose-phosphate pathway (PPP), an oxidative pathway where G6P is decarboxylated to
form ribose-5-phosphate, being NADPH the electron carrier that conserves the redox
potential. More important than participating in a bioenergetic metabolic route, NADPH is the
cofactor necessary for many reducing reactions, mainly those involved in fatty acids
biosynthesis and regeneration of reduced glutathione. The rate-limiting step of PPP is the
conversion of G6P into 6-phosphogluconate, catalyzed by glucose-6-phosphate
dehydrogenase (G6PD), an enzyme that is activated by oxidized glutathione (Eggleston and
Krebs, 1974) and in conditions of oxidative stress, in order to provide cytoprotection (Kletzien
et al., 1994). In addition, G6PD activation exerts its neuroprotective effects against .NO-
mediated apoptosis and glutathione depletion through up-regulation of PPP, which will
increase NADPH regeneration (García-Nogales et al., 2003, García-Nogales et al., 1999).
Therefore, NADPH plays a key role for the regeneration of reduced glutathione (GSH) from
GlycogenPentosephosphatepathway
NADPH
Acetyl-CoA
Lactate
TCAPyruvateGlucose
ATP ATP ATP
Mitochondrial respiratory chain
and oxidative phosphorylation
Glycolysis
Cytosol Mitochondria
Electronscarried via
NADH
Electrons carriedvia NADH and
FADH2

Chapter I __________________________________________________________________________
10
its oxidized form (GSSG), demonstrating that PPP is tightly connected with the maintenance
of cellular redox status (Figure I.5).
Figure I.5 - Branching point of glucose utilization. Glucose-6-phosphate (G6P), the metabolite resulting from glucose catalyzed by hexokinase, is metabolized either in glycolysis [first reaction catalyzed by glucose-6-phosphate isomerase (G6PI)] or in pentose phosphate pathway [first reaction catalyzed by glucose-6-phosphate dehydrogenase (G6PD)]. The main goal of glycolysis and subsequent metabolic pathways is energy production, whereas the main goal of pentose phosphate pathway is the maintenance of redox capacity of the cell, mainly due to production of reduced nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is an essential cofactor for glutathione reductase activity, which is responsible for regeneration of reduced glutathione (GSH) from oxidized form (GSSG).
1.3. Mitochondria: the powerhouse of the cell and the major source of ROS/RNS Mitochondria is the site where the oxidative phosphorylation machinery occurs.
However, during oxidative phosphorylation in mitochondria, electrons can directly react with
O2, generating ROS. As mentioned in section 1.2, O2 is the ultimate acceptor of electrons
that flow through mitochondrial respiratory chain complexes. However, electron leak to
oxygen through complexes I and III can generate O2.- (Figure I.6).
hexo
kina
se
ATP
Pyruvate, Lactate
Glutathione regeneration
Glucose
G6P
Energy production
Glycolysis
NADP+Glutathionereductase
GSH
GSSG
NADPH + H+
NADP+
Pentose phosphate pathwayG6PDG6PI

General Introduction _________________________________________________________________________
11
Figure I.6 - Respiratory chain is the major source of reactive species. The mitochondrial respiratory chain is embedded in the inner mitochondrial membrane (IM) and consists of complexes I–IV, coenzyme Q [ubiquinone (Q)] and ATP synthase (also denominated complex V). Cytochrome c is also a member of the chain, the only one present in the intermembrane space (IMS). These complexes are disposed in an electrochemical hierarchy based on their redox potentials. Electrons enter the chain through oxidation of either NADH at complex I or FADH2 at complex II and flow down the chain to complex IV to reduce O2 to H2O. However some O2 is reduced incompletely to superoxide anion (O2
-.) at the level of complexes I and III. In addition, mitochondria possess a NOS isoform (mtNOS), which is associated with the IM and generates nitric oxide (.NO) in a Ca2+-dependent manner. Mitochondrial .NO competes with O2 for binding to complex IV and regulates mitochondrial respiration. .NO produced by mtNOS reacts readily with O2
-. and produces the powerful oxidative species peroxynitrite (ONOO-). ONOO- produced inside mitochondria causes the release of cyto c and increases the peroxidation of mitochondrial membrane lipids. Outer mitochondrial membrane (OM). Adapted from Ghafourifar and Cadenas (2005).
The rate of O2
.- production is affected by mitochondrial metabolic state and increases
when the electron carriers harbor excess electrons, either from inhibition of oxidative
phosphorylation or from excessive calorie consumption (Nohl et al., 2005). In addition to O2.-
production, mitochondria also produces .NO, through the activity of mtNOS (Ghafourifar and
Cadenas, 2005, Ghafourifar and Richter, 1997). .NO is a physiological regulator of
mitochondrial respiration. In the arterioles, .NO promotes vasodilatation, increasing blood
flow and O2 delivery to the tissues (Clementi et al., 1999). However, .NO is capable of rapidly
and reversibly inhibit the mitochondrial respiratory chain by inhibition of complex IV, which

Chapter I __________________________________________________________________________
12
may be implicated in the cytotoxic effects in the CNS (Bolaños et al., 1994, Brown and
Cooper, 1994, Cleeter et al., 1994). When .NO is present at persistent higher concentrations,
it acts irreversibly at multiple sites, such as destruction of heme, compromising cellular
energy metabolism (Sharpe and Cooper, 1998). Additionally, inhibition of the mitochondrial
transport chain at the level of complex IV can further produce O2.- from O2 due to the
interruption of electron flow. O2.- can also react with .NO, generating the highly reactive
ONOO-. Damage to mitochondria by neurotoxins [such as 1-methyl-4-phenylpyridinium ion
(MPP+) and rotenone] generates more ROS from the electron transport chain and causes
oxidative damage that modifies proteins and other biomolecules (Szeto, 2006). Other
conditions can favor ROS production in the mitochondria, such as are high membrane
potentials, hyperoxia, excessive Ca2+ uptake and anoxia/reoxygenation (Kowaltowski, 2000).
1.4. Dysfunctional mitochondria Mitochondria plays a central position in the production of ATP and the decline of basal
metabolic rate and of physical performance in energy-requiring tasks is characteristic of
several neurological disorders. One example is mitocondrial dysfunction during the aging
process (Navarro and Boveris, 2007). An age-dependent impairment of mitochondrial
function includes: decreased electron transfer rates, increased permeability to H+ of the inner
membrane, and impairment of the driven ATP synthesis according to chemiosmotic theory.
As reviewed in Navarro & Boveris (2007), complexes I and IV activities are selectively
inhibited in isolated mitochondria from rat and mice liver, brain, heart, and kidney upon aging,
whereas complexes II and III are generally unaffected. Regarding enzyme activities of the
TCA cycle, only aconitase activity exhibited a significant decrease with age in isolated
mitochondria from kidneys of old mice and α-ketoglutarate dehydrogenase activity was
modestly decreased (Yarian et al., 2006). In the same study, the ratio of the
intramitochondrial redox indicator, NADPH/NADP+, was higher in young animals in
comparison to old ones, while the NADH/NAD+ ratio remained unchanged. Other metabolic
enzymes are reported to be selectively inhibited during the aging process, such as acyl
carnitine transferase, which catalyzes fatty acid transport to the mitochondrial matrix, thus
being essential for mitochondrial function (Liu et al., 2002). In addition, key glycolytic
enzymes activities, such as pyruvate kinase, α-enolase and triosephosphate isomerase, also
showed to be decreased in aging male monkey hearts (Yan et al., 2004).
Other neurological disorders present features of mitochondrial dysfunction, such as
hypoxia-ischemia. As reviewed by Vannucci et al. (2004), a cerebral hypoxic–ischemic event
rapidly depletes tissue energy reserves, promotes acidosis, glutamate excitotoxicity,
generation of ROS, with consequent inflammation and cell death (Vannucci and Hagberg,

General Introduction _________________________________________________________________________
13
2004). In addition, cultured neurons under conditions of hypoxia-ischemia demonstrated
specific loss of mitochondrial complex I activity, mitochondrial membrane collapse, ATP
depletion and consequent cell death (Almeida et al., 2002).
Mitochondrial dysfunction is also verified in sepsis. In fact, several studies have
implicated pro-inflammatory mediators in the impairment of metabolic function, namely at the
level of mitochondrial respiratory chain complexes and ATP production (Haden et al., 2007,
Suliman et al., 2004). Structural alterations of mitochondria were also found in intestinal
epithelial cells, hepatocytes and cardiomyocytes from septic animals, as reviewed by Wendel
and Heller (2010). Coupled with this less energetic efficiency, these neurological disorders
also present an increased production of free radicals, ROS and RNS in the mitochondria,
(Beckman and Ames, 1998, Sener et al., 2005, Vannucci and Hagberg, 2004, Wendel and
Heller, 2010). As a result, several mitochondrial proteins become nitrated, such as those
involved in TCA cycle, complex I, MnSOD, complex V, among others (Kanski et al., 2005),
which may cause inhibition of enzymatic activity. As a consequence, the mitochondrial
capacity to produce ATP is seriously compromised in this process.
2. Neuronal-glia actions and interplay in the brain Brain tissue encloses a complex network of different cells, each one with unique
structure and function. Neurons are the functioning unit of the CNS, with long processes
called dendrites and axons. Dendrites are multiple filaments that arise from the cell body,
often extending for hundreds of microns and branching multiple times, whereas axons are
single and usually ramified filaments that arise from the cell body. The interconnection of
these processes enables the reception, integration and transmission of information (Purves
et al., 2004). In contrast to neurons, glial cells do not fire action potentials, but instead
surround and enwrap neuronal cell bodies, axons and synapses throughout the CNS (Allen
and Barres, 2009). Astrocytes comprise about 85% of all glial cells, and contribute to the
maintenance of vascular, ionic, redox and metabolic homeostasis in the brain by providing
neurons with energy and substrates for neurotransmission, as well as glutathione precursors
(Allen and Barres, 2005, Dringen, 2000). Besides different brain cells have their own
particular functions and specialized machinery, bidirectional communication actually occurs
between neurons and astrocytes. This communication is essential in the maintenance of
several cellular processes, such as redox status regulation and metabolic pathways.

Chapter I __________________________________________________________________________
14
2.1. Glutathione shuttle
As mentioned in section 1.1, glutathione is synthesized by the action of two enzymes, at
the expense of ATP. Intracellular levels of glutathione are controlled by negative feedback of
γ-glutamylcysteine synthetase, thus, keeping glutathione homeostasis. These metabolic
steps occur in both neurons and astrocytes, however these two nerve cells use different
precursors for glutathione synthesis. In astrocytes, glutathione levels are limited by glutamate
content, and glutamine serves as a glutamate precursor when this aminoacid is not present.
In these cells, NAC and, most importantly, cystine serve as cysteine donors. However, since
neurons are not able to use cystine as cysteine donor, astrocytes supply the precursors
necessary for glutathione biosynthesis in neurons. Glutathione released by astrocytes is
hydrolyzed originating the dipeptide cysteine-glycine, which will be further hydrolyzed into
cysteine and glycine, taken up for neuronal usage (Dringen, 2000), as schematically
represented on Figure I.7.
Astrocytes also contain higher concentrations of glutathione, as well as greater activities
of enzymes involved in glutathione metabolism than neurons (Makar et al., 1994), indicating
that they are more resistant to ROS and that this ROS scavenging mechanism may function
to support neuronal survival. In fact, neurons co-cultured with astrocytes show increased
resistance to injury induced by .NO, H2O2 or O2.- than neurons cultured alone (Desagher et
al., 1996, Haskew-Layton et al., 2010, Lucius and Sievers, 1996) and differences in
glutathione content of neurons and astrocytes contribute to the increased susceptibility of
neurons to toxic agents that induce protein oxidation, such as unconjugated bilirubin (Brito et
al., 2008b).

General Introduction _________________________________________________________________________
15
Figure I.7 - Schematic view of interplay between astrocytes and neurons regarding glutathione metabolism. Neurons and astrocytes synthesize reduced glutathione (GSH) by the action of two enzymes: γ-L-glutamyl-L-cysteinylglycine (γGluCys) synthase, which uses glutamate (Glu) and cysteine (Cys) as substrates and GSH synthase, which combines γGluCys with Gly. While astrocytes are able to take up cystine through sodium independent channel and break it down to yield Cys. In contrast, neurons cannot use cystine as a Cys donor, therefore in these cells the rate limiting step of GSH synthesis is the usage of Cys and neurons rely on astrocytes for GSH synthesis. Reactive oxygen species (ROS) oxidize GSH to oxidized glutathione (GSSG), which is recycled back to GSH by the action of glutathione reductase (GR). Glutamine (Gln) and glycine (Gly). Adapted from Brito et al. (2007).
2.2. Glutamate shuttle
Glutamate toxicity plays an important role in neuronal cell death during brain injury (Yi
and Hazell, 2006). Therefore, control of extracellular glutamate levels is very important to the
prevention of neuronal excitotoxicity by excessive activation of glutamate receptors.
Astrocytes have an essential role in the maintenance of glutamate levels under the toxic
threshold, since they have Na+-dependent transporters which are responsible for the
clearance of glutamate from extracellular space, at the expense of ATP (Anderson and
Swanson, 2000). As schematically represented in Figure I.8, once taken up by astrocytes,
glutamate can be metabolized in different ways, of which glutamine formation and entry into
the TCA cycle are the most important. Glutamine formation is catalyzed by glutamine
synthetase, an enzyme present in astrocytes and in oligodendrocytes, but absent in neurons
(Suárez et al., 2002). Neuronal glutamate is also formed from α-ketoglutarate, a metabolite
produced in TCA cycle. Astrocytes take up leucine and transfer its amino group to
Neurons relie on astrocytes for GSH synthesis (Glu‐Cys‐Gly)

Chapter I __________________________________________________________________________
16
α-ketoglutarate by the action of branched-chain aminoacid transaminase, originating
α-ketoisocaproate. α-ketoisocaproate is then transferred to neurons and can originate
α-ketoglutarate by the reverse reaction (Daikhin and Yudkoff, 2000). Oxidative metabolism of
α-ketoglutarate produces more than 30 ATP, about 20-fold more than required for glutamate
uptake. In conditions of oxidative stress there is ATP depletion, which originates cessation of
glutamate uptake in astrocytes, together with its efflux. Accumulation of glutamate in the
synaptic cleft results in excitotoxicity phenomenon and neuronal death (Santos et al., 1996).
Therefore, astrocytes play an important role in the protection against oxidative stress-
induced excitotoxicity (de Arriba et al., 2006). In addition, astrocytes do use glutamate
released by neurons, for example in the synthesis of glutathione, as mentioned in 2.1, thus
removing excess of glutamate from the brain, which can be toxic when in elevated levels
(Dringen and Hamprecht, 1996).
Figure I.8 – Schematic view of interplay between astrocytes and neurons regarding glutamate metabolism. Astrocytes support neuronal glutamate metabolism. Glutamate (Glu) is released during neurotransmission and is taken up primarily by neighboring astrocytes through excitatory amino acid transporters. A portion of astrocytic Glu is converted to glutamine (Gln) by glutamine synthetase, which is abundant in astrocytes and absent in neurons. Gln is released from astrocytes and taken up by neurons through specific transporters. In neurons, Gln is deaminated into Glu by mitochondrial glutaminase. Neuronal Glu is also formed from α-ketoglutarate (α-KG). Astrocytes take up leucine (Leu), and the amino group of Leu is transferred to α-KG by branched-chain amino acid (BCAA) transaminase. Pyr, pyruvate; Ala, alanine; Pi, inorganic phosphate. Adapted from Chen and Swanson (2003).

General Introduction _________________________________________________________________________
17
2.3. Lactate shuttle
The coupling between synaptic activity and glucose utilization (neurometabolic coupling)
is a central physiological principle of brain function. Neurons and astrocytes are the two
major contributors for the massive consumption of oxygen and glucose in the brain. While
glycolysis occurs preferentially in astrocytes, most of the oxygen is consumed by neurons
(Jolivet et al., 2009). Under resting conditions, astrocytes metabolize ~85% of the glucose
consumed in lactate. As schematically represented in Figure I.9, glycogen, the main energy
store in the brain, is localized predominantly in astrocytes. Upon neuronal stimulation with
glutamate, both glucose uptake and lactate production are observed in surrounding
astrocytes (Pellerin and Magistretti, 1994). In addition to glucose, lactate (mainly provided by
astrocytes) can constitute a supplementary fuel for activated neurons. In fact, as reviewed by
Pellerin et al. (2007), a major glycolytic response in astrocytes upon activation, either by
direct application of glutamate or stimulation of glutamatergic pathways, represent an
important lactate source. Lactate accumulated in both extracellular and intracellular space in
astrocytes constitutes a pool readily available for neurons upon increased energy demands.
Upon neuronal activation, there is a rapid decrease in mitochondrial NADH in dendrites and
then an increase in TCA cycle activity, in order to replenish the mitochondrial NADH pool
(Kasischke et al., 2004). Moreover, there are additional reports that came to the conclusion
that lactate is the predominant oxidative substrate over glucose in cultured neurons (Itoh et
al., 2003, Bouzier-Sore et al., 2003).
In addition to glutamate/glutamine cycling between neurons and astrocytes referred in
2.2, neurons also rely on astrocytes for the supply of metabolic intermediates, particularly
oxaloacetate, formed by the condensation of pyruvate with CO2 (Haberg et al., 1998),
allowing the further synthesis of glutamate or γ-aminobutyric acid. Therefore, a transfer of
glucose-derived metabolites from glial cells to neurons is necessary for neuronal survival,
especially during severe hypoglycemia (Forsyth, 1996, Wender et al., 2000).

Chapter I __________________________________________________________________________
18
Figure I.9 - Neural activity triggers the release of the neurotransmitter glutamate (Glu) that is taken up into the astrocyte, and stimulates the breakdown of glycogen, the uptake of glucose, and glycolysis, to produce lactate in astrocytes. Astrocytic released glutamine (Gln) favors synaptic process, whereas astrocytic released lactate stimulates neuronal glucose uptake. Since neurons use more energy than they are able to produce by themselves, interplay with astrocytes constitutes an essential additional source of energy. Adapted from Magistretti (2006).
2.4. Neuronal susceptibility to oxidative stress
2.4.1. Increased oxidant capacity in the brain Mammalian brain cells are particularly susceptible to oxidative damage, since they
present higher oxidant capacities. The first reason is because large amounts of ATP are
required to maintain neuronal processes. As a consequence, in neuronal cells, a high O2 and
glucose consumption occurs, leading to a continuous production of ROS during oxidative
phosphorylation process. In fact, electrons leak to O2 through complexes I and III of the
respiratory chain, thus generating O2.-.
Brain cells are also more susceptible to oxidative stress because of the presence of
excitatory aminoacids. Oxidative stress damages neurons and induces the release of
glutamate. This aminoacid will bind to NMDA receptors on adjacent neurons, leading to an
increase in intracellular Ca2+ within them (Mailly et al., 1999). This increase in intracellular
Ca2+ concentrations can induce massive production of .NO, by activation of nNOS, a
Ca2+dependent enzyme, as mentioned in section 1.1. Rise in Ca2+ levels affects
mitochondrial function, contributing to the generation of O2.-. The excess of O2
.- may react
with .NO, generating ONOO-, which is responsible for inactivation of glutamine synthetase by
Glycolysis
Glycogen

General Introduction _________________________________________________________________________
19
tyrosine nitration (Görg et al., 2007). As a consequence of these events, it may occur an
increase in extracellular levels of glutamate, thus promoting excitotoxicity.
In addition, several neurotransmitters present in the brain, such as dopamine, serotonin
and norepinephrine are autoxidizable. By reacting with O2, they can generate O2.-, as well as
quinones/semiquinones that bind to thiol groups of reduced glutathione, causing its depletion
(Wrona and Dryhurst, 1998).
Another fact that accounts for brain increased susceptibility to oxidative stress is the
elevated concentrations of iron, mostly contained in ferritin in healthy brain (Burdo and
Connor, 2003). However, in damaged brain, iron accumulation is excessive relative to the
amount of ferritin and it will catalyze free radical reactions, namely Fenton’s reaction.
Neuronal membrane lipids are enriched in unsaturated fatty acids, which are thought to
be target molecules for free radical-induced peroxidation and neural cell damage, thus
playing a major role in the pathogenesis of many neurological diseases. HNE, one of the
main products of lipid peroxidation, especially induces neuronal cytotoxicity by increasing
Ca2+ levels, which will inactivate glutamate transporters and damage neurofilament proteins
(Mark et al., 1997). HNE also inactivates α-ketoglutarate dehydrogenase, a key enzyme in
TCA cycle (Sheu and Blass, 1999).
Brain metabolic pathways are also responsible for huge generation of H2O2, not only by
the action of SOD, as described in section 1.1, but also by other enzymes, being monoamine
oxidases A and B and flavoproteins located in the outer mitochondrial membranes of
neurons and glia particularly important for this process (Gal et al., 2005). Furthermore,
neuronal NADPH oxidase enzymes (NOX) become activated in response to oxidative stress
and may promote neuronal apoptosis. This process is extremely important during
development of the nervous system; however, if trophic support is lost in the developed
brain, NOX can become overactivated and leading to neuronal apoptosis (Sánchez-Carbente
et al., 2005, Tammariello et al., 2000).
Astrocytes and microglial cells can also contribute to oxidant environmental conditions,
when they become activated by inflammatory features, such as pro-inflammatory cytokines.
Activated astrocytes and, especially, activated microglia may produce O2.- and H2O2 and,
.NO, by activation of iNOS. Thus, activated glial cells are major players in oxidative stress
induced by inflammatory processes in the brain. Commonly, in studies with isolated cultures,
astrocytes appear less susceptible to ROS and RNS than neurons, since they have higher
glutathione levels and are more able to promote its synthesis under stress than neurons
(Halliwell, 2006).

Chapter I __________________________________________________________________________
20
2.4.2. Antioxidant capacity in the brain Efficiency of antioxidant defences of brain cells is low when compared to that of other
tissues. In fact, catalase levels are low in most brain regions, specifically located in
peroxisomes and are hardly able to counteract H2O2 produced in other cellular compartments
(Angermüller et al., 2009).
In order to battle against oxidative stress, all parts of the brain contain SODs with active-
site for manganese (MnSOD) in the mitochondrial matrix and for cooper/zinc (CuZnSOD) in
the mitochondrial intermembrane space and in the rest of the cell. Curiously, neurons
containing nNOS are reported to be relatively resistant to NMDA and .NO-mediated
neurotoxicity, by a mechanism involving MnSOD activation (Gonzalez-Zulueta et al., 1998).
Glutathione/GPx system is also present within all nervous cells. Since neuronal
concentrations of glutathione are lower than in glia, these cells might assist neurons by
supplying them with cysteinyl-glycine as a glutathione precursor. In fact, glutathione released
by astrocytes can be degraded by γ-glutamyl transpeptidase on their cell surface to produce
cysteinyl-glycine, which neurons then further cleave to release cysteine for uptake and use in
glutathione synthesis (Dringen et al., 2005).
In addition to glutathione, brain cells are enriched in low molecular compounds with
antioxidant activity, mainly ascorbate. In fact, neurons have specific transporters that
efficiently take up ascorbate and astrocytes take up dehydroascorbate and convert it to
ascorbate in intracellular space (Rice, 2000). However, in damaged brain, ascorbate can
stimulate the oxidation of Fe3+ and Cu2+ into Fe2+ and Cu+, respectively, thus potentiating
Fenton’s reaction and formation of .OH. Another low molecular compound that is present in
the brain is α-tocopherol, mostly derived from plasma high-density lipoprotein (Hayton and
Muller, 2004). Furthermore, brain contains elevated levels of histidine-containing dipeptides,
known for their antioxidant properties, namely, by chelating metal ions and binding cytotoxic
aldehydes produced during lipid peroxidation (Aruoma et al., 1989, De Marchis et al., 2000,
Decker et al., 2000).
Finally, bilirubin has antioxidant properties (Barañano et al., 2002, Stocker et al., 1987).
Heme oxygenase (HO) is a widespread enzyme in the brain, existing in both inducible
(HO-1) and constitutive (HO-2) isforms. HO catalyses the degradation of heme, with
generation of carbon monoxide (CO), which can act as neurotransmitter, and biliverdin that
will be further converted to bilirubin by biliverdin reductase. Although heme degradation
causes the release of Fe2+, which can potentiate .OH formation as abovementioned, HO is
involved in antioxidant mechanisms. HO-2 activation is able to prevent neuronal death in
cerebral ischemia (Doré et al., 2000, Doré et al., 1999) and HO-1 is rapidly upregulated by
oxidative and nitrosative stresses in some neurodegenerative diseases, as an attempt to

General Introduction _________________________________________________________________________
21
convert the highly damaging heme into the biliverdin and bilirubin (Calabrese et al., 2005).
However, bilirubin breakdown by ROS originates bilirubin oxidation products, which can
produce vasoconstricting compounds (Pyne-Geithman et al., 2005) and high levels of
bilirubin are neurotoxic, as it will be further discussed in section 4, due to its relevance for the
present thesis.
2.5. Neuronal susceptibility bioenergetic crisis
Astrocytes and neurons respond differently to .NO-induced inhibition of mitochondrial
respiration. In fact, whereas neurons suffer a rapid decline in ATP levels, a collapse in
mitochondrial membrane potential (ΔѰm) and apoptotic cell death, astrocytes utilize
glycolytically-generated ATP, thus maintaining their ΔѰm (Almeida et al., 2001). This
differential response in not exclusively to impaired mitochondrial respiration, since it also
occurs in case of over-activation of neuronal glutamate receptors, an event that inhibits
mitochondrial ATP synthesis or glucose uptake (Almeida and Bolaños, 2001, Porras et al.,
2004).
It was reported that one of the main reasons why neurons and astrocytes respond
differently to .NO-induced inhibition of respiration is the fact that they have very lower activity
levels of 6-phosphofructo-1-kinase (PFK1), a master regulator of glycolysis, in comparison to
astrocytes. The content of fructose-2,6-bisphosphate (F2,6P2), the powerful allosteric
activator of PFK1, is also lower in neurons. In addition, mitochondrial respiration inhibition
induced F2,6P2 in astrocytes, whereas has no effect on neuronal F2,6P2 (Almeida et al.,
2004). Recently, it was suggested that neurons are unable to increase glycolysis because
they almost does not possess 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
(PFKFB), which is the enzyme responsible to F2,6P2 formation (Herrero-Mendez et al.,
2009).
Glucose metabolism in neurons is directed mainly to the PPP, with the main goal of
reduced glutathione regeneration. In fact, the antioxidant function of the PPP in neurons was
demonstrated in response to pro-oxidant compounds exposure (García-Nogales et al., 2003,
Vaughn and Deshmukh, 2008) or glutamate receptor stimulation, mainly NMDA (Delgado-
Esteban et al., 2000). As described in section 1.1, over-activation of NMDA receptors favors .NO formation by activation of nNOS, a process implicated in the pathogenesis of several
CNS diseases (Suárez et al., 2002).

Chapter I __________________________________________________________________________
22
3. Inflammation and cell death in central nervous system
3.1. Cells involved in inflammation and CNS injury The inflammation in the CNS, also designated as neuroinflammation, represents an
essential response to peripheral inflammation and CNS injury or infection, necessary to the
maintenance of tissue survival, repair and recovery, and to conserve the energy of the
organism, by limiting the survival and proliferation of invading pathogens. The produced
damage may have different causes like infection, traumatism, ischemia, necrosis,
hemorrhage, among others. However, it is also recognized as a major contributor to acute
and chronic CNS disorders. As reviewed by Allan and Rothwell (2003), inflammatory
mediators, such as complement, adhesion molecules, cyclooxygenase enzymes and pro-
inflammatory cytokines, are increased in several neurological diseases, and intervention
studies in experimental animals suggest that several of these factors contribute directly to
neuronal injury.
In the past, studies on CNS injury have focused predominantly on neuronal death and
survival, since these cells largely determine CNS function and survival and cannot be
replaced once they are lost. However, the role of other cell types in CNS disease is
becoming increasingly apparent. Glial cells constitute the majority of the brain volume and
play an active role in normal physiology and pathology (Raivich et al., 1999). The primary
glial cells implicated in neuroinflammation are microglia. These are cells of the
monocyte/macrophage lineage, which are resident in the brain and are activated in response
to infection, inflammation and injury (Streit, 2002). They are important phagocytic cells and
release numerous inflammatory molecules, namely cytokines (Hanisch, 2002). Another glial
cell type important for neuroinflammation is astrocytes, which are the most abundant glial
cells, and play key physiological roles in supporting neurons, regulating ion and transmitter
concentrations and in electrical transmission, and are an important source of both
neuroprotective and inflammatory molecules (Allan and Rothwell, 2003). Oligodendrocytes,
the third type of glia, are crucial for myelination, but are also a source of specific
inflammatory molecules (Baumann and Pham-Dinh, 2001, Du and Dreyfus, 2002). Vascular
endothelial cells are also targets and sources of inflammatory mediators, being particularly
important in the adhesion of circulating cells of the immune system (Couraud, 1994).
3.2. Inflammatory mediators and signalling pathways Cytokines are among the major effectors of neuroinflammation. They can be involved in
either neuroprotection or neurodegeneration processes (Konsman et al., 2007). Specific pro-
inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) β,

General Introduction _________________________________________________________________________
23
have pleiotropic effects in the CNS, including their emerging role in neurodevelopment (Marx
et al., 2001) but are also described as mediators of neuronal apoptosis (Kajta et al., 2006).
TNF-α exerts its biological activity by binding to type 1 and type 2 receptors (TNFR1 and
TNFR2) and activating several signalling pathways. TNFR1 contains a common death
domain whereas TNFR2 does not. Thus, TNFR1 activation is involved in both cell survival
and cell death signalling, while TNFR2 mediates cell survival signals. However, it is
suggested that TNFR2 might potentiate death signal mediated by TNFR1 (Gupta, 2002).
Like TNF-α, IL-1β is a pro-inflammatory cytokine associated with several CNS
disorders. Activated microglial cells are the main source of this cytokine in the damaged
brain (Block and Hong, 2005). Released IL-1β directly affects neurons, astrocytes and
oligodendrocytes, promoting production of other cytokines and regulation of synaptic function
on hippocampal neurons (Bellinger et al., 1993). In the opposition of the dual role of TNF-α in
brain damage, IL-1β is mainly considered by its neurotoxic effects (Panegyres and Hughes,
1998, Yang et al., 1998).
The effects of cytokines depend on which cell type they act upon and whether it is a
direct or indirect effect (Allan and Rothwell, 2003). As reviewed by Allan and Rothwell (2003),
studies in vitro demonstrated that cytokines directly act on neurons, promoting changes in
Ca2+ entry, neurotransmitter release and synaptic plasticity, thus contributing to neuronal
viability in the injured brain. It is speculated that neuronal responses can be modified,
indirectly or directly, by cytokines. One example of this is in case of seizure activity, which is
enhanced by IL-1 administration (Vezzani et al., 1999). In addition, pro-inflammatory
cytokines such as IL-1 and TNF-α are reported to induce blood–brain barrier breakdown
(Blamire et al., 2000, Cardoso et al., 2010, Quagliarello et al., 1991), as well as to trigger the
release of toxic substances, such as .NO from the vascular endothelium (Bonmann et al.,
1997), allowing the entrance of leucocytes into the brain parenchyma, which will contribute to
neuronal injury.
In addition, several studies demonstrated the association between inflammation and
generation of ROS/RNS, leading to multiple organ dysfunction (Bian and Murad, 2001, Sener
et al., 2005). .NO is recognized as a mediator and regulator of inflammatory responses. It
was first reported that mouse macrophages produce nitrite and nitrate in response to
bacterial lipopolysaccharide (Stuehr and Marletta, 1985). However, although high levels of .NO produced in response to inflammatory stimuli can have deleterious effects, this molecule
is also important in cellular signalling, having an important role in the amelioration of the
pathogenesis of inflammation (Korhonen et al., 2005). Furthermore, .NO and induction of

Chapter I __________________________________________________________________________
24
NOS are involved in apoptosis induced by inflammatory mediators in neuronal cells (Hemmer
et al., 2001, Heneka et al., 1998, Thomas et al., 2008).
The mitogen-activated protein kinases (MAPKs) and the transcription factor nuclear
factor κB (NF- κB) are among the main effectors that participate in inflammatory signalling
pathways. MAPKs are divided into three major subfamilies, according to structural
differences between them: the p38 kinase, the c-Jun N-terminal kinases 1 and 2 (JNK1/2)
and the extracellular signal-regulated kinases 1 and 2 (ERK1/2), as reviewed by Roux and
Blenis (2004). In general, p38 and JNK1/2 are more responsive to environmental stress and
pro-inflammatory cytokines, being designated as stress-activated protein kinases (SAPKs),
while ERK1/2 are mostly activated in response to mitogens and growth factors (Kyriakis and
Avruch, 2001). NF- κB is an important transcription factor responsible for modulation of the
host immune and inflammatory response (O'Neill and Kaltschmidt, 1997). It will be given
preferential attention to JNK1/2, since their activation is discussed in the present thesis.
JNK1/2 become activated in response to toxic stimulus, such as ROS (Luo et al., 1998,
Marques et al., 2003) and pro-inflammatory cytokines TNF-α and IL-1, pointing these SAPKs
as strong effectors of neuronal apoptosis (Mielke and Herdegen, 2000, Tibbles and
Woodgett, 1999). The activation of JNK1/2 enzyme is related to toxicity in developing
neurons, since overexpression of activated c-Jun was shown to produce apoptosis and
suppression of this protein protected against neuronal death induced by deprivation of nerve
growth factor in sympathetic and hippocampal neurons (Estus et al., 1994, Ham et al., 1995,
Schlingensiepen et al., 1993). In addition, .NO-induced JNK phosphorylation is observed in
models of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s (Katsuki et al.,
2006, Marques et al., 2003).
3.3. Neuronal susceptibility to inflammation In spite of being essential to tissue survival, repair and recovery, extensive, prolonged or
unregulated inflammation is highly detrimental. During the last decades it has been observed
that not only cells of the immune system participate actively in the inflammation, but also
cells belonging to the CNS are a fundamental part of this process, especially glial cells, as
indicated in section 3.1. The neurons have a minor participation in inflammatory processes,
however they have the ability to express class I molecules, to produce several cytokines like
IFN-γ and also to induce apoptosis of T cells through the Fas receptor (FasR) interaction
(Chavarria and Alcocer-Varela, 2004).
TNF-α is the pro-inflammatory cytokine most characterized in neurologic diseases. This
cytokine directly affects every neural cell, by inducing the release of other cytokines in glial
cells (Allan and Rothwell, 2003), several chemokines (Croitoru-Lamoury et al., 2003) and

General Introduction _________________________________________________________________________
25
.NO (Madrigal et al., 2002). TNF-α has been demonstrated to induce neuronal apoptosis in
human brain cell cultures and animal models through TNFR1 signalling (Yang et al., 2002)
and further production of other inflammatory and/or neurotoxic molecules such as ONOO- by
induction of iNOS in glial cells (Combs et al., 2001). In addition, post-treatment with TNF-α
potentiates NMDA-mediated toxicity in organotypic hippocampal slice cultures (Wilde et al.,
2000). TNF-α produced by glial cells was also found to damage neural precursor cells and to
inhibit neurite elongation and branching during development and regeneration (Sheng et al.,
2005). Furthermore, it is reported that during aging, both TNF-α production and TNF-α-
induced apoptosis are increased (Gupta, 2002).
Other cytokine that is widely accepted for its role in neuronal injury is IL-1β (as referred
in section 3.2). In fact, increased expression of this cytokine in CNS is observed after a
variety of brain insults, and administration of exogenous IL-1β to animals undergoing
ischemic or excitotoxic challenges leads to a dramatic increase in the resulting cell death
(Allan and Rothwell, 2003). In addition, administration of the selective IL-1 receptor
antagonist (IL-1ra) clearly inhibits the extent of cell death induced by ischemic, traumatic or
excitotoxic injury in the mouse brain (Rothwell and Luheshi, 2000).
3.4. Death signalling pathways The principal mechanisms implicated in the death signalling cascades of the CNS cells
include the alteration of Ca2+ homeostasis, oxidative and nitrosative stress, the accumulation
of extracellular neurotransmitters, as glutamate and the activation of signalling cascades
(Neumar, 2000), as schematically represented in Figure I.10.
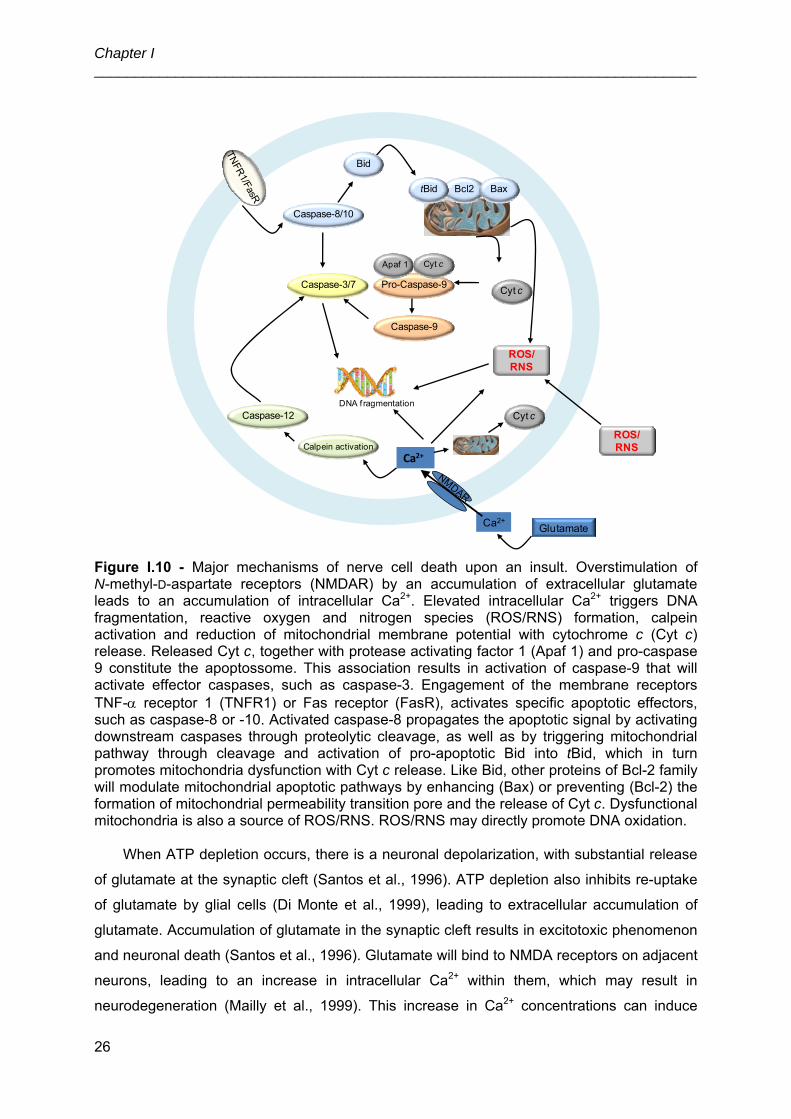
Chapter I __________________________________________________________________________
26
Figure I.10 - Major mechanisms of nerve cell death upon an insult. Overstimulation of N-methyl-D-aspartate receptors (NMDAR) by an accumulation of extracellular glutamate leads to an accumulation of intracellular Ca2+. Elevated intracellular Ca2+ triggers DNA fragmentation, reactive oxygen and nitrogen species (ROS/RNS) formation, calpein activation and reduction of mitochondrial membrane potential with cytochrome c (Cyt c) release. Released Cyt c, together with protease activating factor 1 (Apaf 1) and pro-caspase 9 constitute the apoptossome. This association results in activation of caspase-9 that will activate effector caspases, such as caspase-3. Engagement of the membrane receptors TNF-α receptor 1 (TNFR1) or Fas receptor (FasR), activates specific apoptotic effectors, such as caspase-8 or -10. Activated caspase-8 propagates the apoptotic signal by activating downstream caspases through proteolytic cleavage, as well as by triggering mitochondrial pathway through cleavage and activation of pro-apoptotic Bid into tBid, which in turn promotes mitochondria dysfunction with Cyt c release. Like Bid, other proteins of Bcl-2 family will modulate mitochondrial apoptotic pathways by enhancing (Bax) or preventing (Bcl-2) the formation of mitochondrial permeability transition pore and the release of Cyt c. Dysfunctional mitochondria is also a source of ROS/RNS. ROS/RNS may directly promote DNA oxidation.
When ATP depletion occurs, there is a neuronal depolarization, with substantial release
of glutamate at the synaptic cleft (Santos et al., 1996). ATP depletion also inhibits re-uptake
of glutamate by glial cells (Di Monte et al., 1999), leading to extracellular accumulation of
glutamate. Accumulation of glutamate in the synaptic cleft results in excitotoxic phenomenon
and neuronal death (Santos et al., 1996). Glutamate will bind to NMDA receptors on adjacent
neurons, leading to an increase in intracellular Ca2+ within them, which may result in
neurodegeneration (Mailly et al., 1999). This increase in Ca2+ concentrations can induce
ROS/RNS
Ca2+
Ca2+
DNA f ragmentation
Bid
Caspase-8/10
Bcl2tBid Bax
Cyt c
Pro-Caspase-9
Apaf 1 Cyt c
Caspase-3/7
Caspase-9
ROS/RNS
Caspase-12
Calpein activation
Glutamate
Cyt c

General Introduction _________________________________________________________________________
27
massive production of .NO, by activation of nNOS. Excessive elevation of intracellular Ca2+
also leads to the activation of hydrolytic enzymes and triggers mitochondrial permeability
transition and activation of many enzymes, including phospholipases and calpains. Calpain
activation is coupled to execution of caspase-independent apoptosis in cerebellar granule
neurons (Volbracht et al., 2005). Furthermore, cysteine proteases, including caspases are
also sensitive to the redox balance of the cell (Blomgren et al., 2007). In addition,
mitochondrial dysfunction induced by increased production of ROS/RNS is accompanied by
activation of caspase-3 and DNA fragmentation (Gilland et al., 1998, Puka-Sundvall et al.,
2000). This mitochondrial dysfunction may facilitate the release of proapoptotic factors from
the intermembrane space of the mitochondria to the cytosol, such as cytochrome c,
apoptosis-inducing factor 1 (Apaf 1), endonuclease G, SMAC/Diablo and HtrA2/Omi
(Ravagnan et al., 2002). Release of cytochrome c interacts with Apaf 1 and dATP/ATP to
form the apoptosome, leading to activation of pro-caspase-9 into the initiator caspase-9
(Acehan et al., 2002), which in turn cleaves and activates pro-caspase-3, the most abundant
effector caspase in the brain, and consequent activation of the apoptotic cell death
(Matapurkar and Lazebnik, 2006). The apoptotic pathway may also be triggered by the
engagement of the membrane receptors TNFR1 or FasR, often referred as “death
receptors”, since they are coupled to specific apoptotic effectors, such as caspase-8 or -10
(Walczak and Krammer, 2000). Activated caspase-8 propagates the apoptotic signal by
activating downstream caspases through proteolytic cleavage, as well as by triggering
mitochondrial pathway through cleavage and activation of pro-apoptotic Bid into tBid, which
in turn promotes mitochondria dysfunction and cytochrome c release (Adams, 2003).
The role of the inflammatory caspases (mainly caspase-1, also called IL-1 converting
enzyme) in apoptosis is not clear, however they may contribute to brain injury after ischemia
through their pro-inflammatory actions (Blomgren et al., 2007).
4. Bilirubin induced neurological damage and risk factors involved
4.1. Neonatal hyperbilirubinemia In fetal life, bilirubin production begins as early as 12 weeks’ gestation. In this period,
bilirubin clearance is made through its passage to maternal circulation. At birth, this placental
protection is suddenly lost and an accumulation of UCB takes place (Brito et al., 2006).
Neonatal hyperbilirubinemia occurs due to the three main reasons: (i) bilirubin
overproduction because in neonatal life there is a shortened red blood cell lifespan; (ii)
decreased bilirubin conjugation in the liver, due to immaturity of newborns’ hepatic

Chapter I __________________________________________________________________________
28
machinery; (iii) impaired bilirubin excretion because of the absence of bacterial flora (Porter
and Dennis, 2002).
Neonatal hyperbilirubinemia is a very common condition in the neonatal period, with total
serum bilirubin levels above to 5 mg/dl. This condition occurs in up to 60% of full term
newborns and 80% of preterms (Dennery et al., 2001). Commonly designated as neonatal
jaundice, this condition is characterized by accumulation of unconjugated bilirubin (UCB) in
the skin and mucous membranes, responsible for the yellow-orange coloration observed in
jaundiced babies (Stevenson et al., 2001).
Although most of newborn infants have mild to moderate elevated serum UCB levels
within the first days of life, a condition known as “physiologic jaundice”, higher levels of UCB,
known as “pathologic jaundice” cause nerve cell damage, a condition called UCB
encephalopathy, that may lead to adverse neurological outcomes (Hansen, 2002). In fact,
moderate degrees of hyperbilirubinemia may be a starting point to the appearance of long-
term neurodevelopment disabilities (Dalman and Cullberg, 1999, Soorani-Lunsing et al.,
2001). Neurologic dysfunctions reported to be related with elevated concentrations of UCB in
neonatal period include risk of otoxoxicity and hearing loss (de Vries et al., 1985), as well as
visual acuity or mild-to-moderate cerebral palsy (Sampath et al., 2005) in extremely low-birth-
weight infants. Changes in the auditory brainstem response were also found in rhesus
monkeys during the intravenous infusion of UCB (Ahlfors et al., 1986). In addition, high
levels of UCB can constitute the basis for chronic to permanent sequelae, or even death
(Ostrow et al., 2004, Shapiro, 2005). As reviewed by Hansen (2000), the term kernicterus
was first used by Schmorl in 1904 to describe the yellow staining of some brain regions,
notably basal ganglia and medulla oblongata, observed in postmortem analysis of brains of
term neonates. Statistically, around 70% of infants with kernicterus dye within seven days,
and the ~30% survivors commonly develop irreversible sequelae such as auditory
dysfunction, mental retardation and choreoathetoid cerebral palsy (Blanckaert and Fevery,
1990).
4.2. Prematurity as a risk factor of neonatal hyperbilirubinemia The risk of bilirubin-induced neurologic dysfunction is particularly enhanced in premature
newborns due to the higher rates of UCB production because of the shorter life span of their
red blood cells. This fact contributes to an increased UCB production, since this molecule
results from the degradation of heme proteins (Blanckaert and Fevery, 1990). In addition,
prematures present some metabolic deficiencies at the level of excretion pathways, that will
account for the decreased UCB clearance from the organism (Stevenson et al., 2001,

General Introduction _________________________________________________________________________
29
Watchko, 2006). Moreover, cerebral palsy was found in preterm infants with risk of
kernicterus, in spite of relatively low total serum bilirubin levels (Gkoltsiou et al., 2008).
Prematurity is frequently associated with hypoalbuminemia (Cartlidge and Rutter, 1986),
which will contribute to increased levels of free UCB, since bilirubin is released in circulation
reversibly bound to albumin, until reaching into the liver in order to be metabolized. If
concentrations of albumin are lower, a higher rate of free UCB easily crosses the blood brain
barrier, which is also immature and permeable and presents a reduced content in tight
junctions and pericytes, together with a more fragile brain vasculature in preterm newborns
(Ballabh et al., 2004). Furthermore, premature infants present a higher incidence of neonatal
pathophysiological processes such as hypoxia-ischemia insult and cerebral hemorrhage that
can contribute for their increased vulnerability to brain damage in comparison to the full term
infants (Volpe, 1997). Hypoxic-ischemic conditions may lead to development of acidosis
(O'Shea, 2002), contributing to an environment with lower pH, which favors cellular
deposition of UCB (Ostrow et al., 1994).
It should be noticed that in preterms is complicated to establish a threshold at which
UCB interferes with neurodevelopment outcome, since these infants are commonly clinically
ill, with other perinatal complications. Thus, UCB-induced neurotoxicity may be more
pronounced that in full term ones, even at relatively low levels of UCB (Oh et al., 2003). In
fact, low weight premature infants present decreased albumin concentration and a lower
affinity and/or capacity for UCB binding (Cashore, 1980, Kaplan and Hammerman, 2005).
4.3. Sepsis-associated neonatal hyperbilirubinemia Premature newborns are also more susceptible to some insults that are described as
risk factors for UCB-induced encephalopathy. In fact, prematurity is frequently associated
with sepsis, which is responsible for the alteration of blood brain barrier permeability through
the release of great amounts of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6
(Goldenberg and Andrews, 1996). In addition, a correlation between infection and the
increased risk of UCB-induced neurotoxicity is reported: (i) in an animal model of sepsis, it
was shown that serum concentration of both total and free bilirubin was increased, promoting
a net accumulation of UCB in the brain (Hansen, 1993); (ii) pro-inflammatory cytokines were
reported to increase blood-brain-barrier permeability, allowing UCB entrance in the brain
(Petty and Lo, 2002), and to exacerbate UCB-induced cytotoxicity in different cell lines, such
as in neuroblastoma, glioblastoma, umbilical vein endothelial, liver cell and mouse fibroblasts
(Ngai and Yeung, 1999), as well as in astrocytes (Fernandes et al., 2004).
The association between inflammation and generation of ROS/RNS described in section
3.2 should also be taken into account for the increased susceptibility in sepsis-associated

Chapter I __________________________________________________________________________
30
hyperbilirubinemia, since immature brain lacks antioxidant defences, such as catalase and
GPx, which may help to explain the differential susceptibility of the developing CNS to brain
injury (Chang et al., 2005). As discussed in section 3, although essential for survival in
response to tissue injury or infection, inflammatory response also causes neuronal damage,
through an increased production of pro-inflammatory cytokines, as well as .NO and low-
gestational-age newborns have a prominently increased risk of brain dysfunctions attributed
to cerebral-cortex damage, including excess of apoptosis and impairment of surviving
neurons (Leviton and Gressens, 2007). In addition, It has been suggested that infection
increases the risk for UCB encephalopathy (Dawodu et al., 1984) and presence of
inflammatory features, namely fever episodes and brain edema, were described during or
following moderate to severe hyperbilirubinemia (Kaplan and Hammerman, 2005). As
schematically represented in Figure I.11, inflammatory response of astrocytes and microglia
causes an increase of apoptotic neurons, which may produce neuropathological sequelae.
Interestingly, lipopolysaccharide exacerbates the release of TNF-α and IL-1β by cultured
astrocytes (Falcão et al., 2005). Because of the relevance for the present dissertation, this
issue will be further dissected into section 4.5.
.
Figure I.11 - Astrocytes and mostly microglia produce an inflammatory response in response to a toxic stimulus and may produce neuropathological sequelae.
astrocyte
Inflammatoryresponse
Apoptosis Degeneration
Developmental diseasemicroglia
Immatureneuron
Differentiatedneuron
Toxicstimulus

General Introduction _________________________________________________________________________
31
4.4. Differential neuronal vulnerability among brain regions Differential susceptibilities may be related with preferential deposition within brain
regions. In fact, UCB shows a specific deposition pattern when is able to cross the blood
brain barrier and enters into the brain. Kernicterus condition results from a specific pattern of
UCB deposition in the brain, mainly in basal ganglia, hippocampus lateral ventricular walls,
mid brain, pons, cerebellum and inferior cerebellar peduncles, and subthalamic nuclei,
together with brain edema (Ahdab-Barmada and Moossy, 1984, Perlman et al., 1997). The
tendency of bilirubin deposits in kernicterus localized in areas vulnerable to hypoxic ischemic
injury, such as the pyramidal cell layer of the hippocampus, raises the question of whether
hypoxic-ischemic injury is important to the development of the lesions of kernicterus
(Perlman et al., 1997). Purkinje cells in the cerebellum have also showed increased
susceptibility to bilirubin injury in Gunn rats, the well-established animal model for severe
hyperbilirubinemia (Conlee and Shapiro, 1997, Lin et al., 2005). Other studies have referred dissimilar vulnerability to toxic stimuli in different brain
regions show, such as ischemia and oxidative stress. Many factors have been proposed to
account for differential vulnerability of brain regions. This includes local differences in
synaptic input and in neurotransmitter released, differences in expression levels of specific
neurotransmitter receptors, differences in antioxidant defences, and differences in signaling
pathways (Xu et al., 2001). In vivo studies demonstrate that brief periods of global ischemia
cause selective neuronal loss, especially in the CA1 region of hippocampus (Papadopoulos
et al., 1997, Papadopoulos et al., 1998). Studies with rat primary cultures of neurons and
astrocytes isolated from cortex, striatum, or hippocampus revealed distinct profiles of
vulnerability when subjected to injury. While astrocytes from striatum showed increased
injury by oxygen and glucose deprivation, they were more resistant to H2O2 exposure or
glucose deprivation, since they presented higher levels of antioxidant defences, such as
increased glutathione levels and increased activities of GPx and SOD (Xu et al., 2001). In
addition, it was reported that antioxidant enzymes, such as xantine oxidase and catalase,
have maximum activity in cortex, followed by cerebellum and hippocampus in developing
mouse brain exposed to lead (Prasanthi et al., 2010) and glutathione peroxidase activity is
considered determinant in the recovery of the immature mouse brain subjected to traumatic
brain injury (Tsuru-Aoyagi et al., 2009).
In the neonatal brain, the basal ganglia are the most vulnerable region in term infants,
whereas in preterms the most susceptible one is the periventricular white matter region
(Barkovich et al., 1995). Neuronal damage after hypoxic-ischemic insult seems to affect
particularly hippocampal CA1 region, striatum and neocortical layers III, V, and VI in animal
models of both mature and immature brain (Guzzetta et al., 2000, Jiang et al., 2004). For

Chapter I __________________________________________________________________________
32
hypoxia-ischemia-induced regional vulnerability may account preferential distribution of
immature NMDA receptors, which corresponds to regions that preferentially express nNOS,
such as layers CA1 and CA3 of the hippocampus, pons and globus pallidus (Black et al.,
1995, Greenamyre et al., 1987, Mitani et al., 1998), regions described for preferential UCB
deposition (Ahdab-Barmada and Moossy, 1984, Hansen, 2000, Perlman et al., 1997). In
addition, regional differences in pro-oxidant and antioxidant defences (Candelario-Jalil et al.,
2001, Khan and Black, 2003), the Ca2+-induced mitochondrial permeability transition (Friberg
et al., 1999) and DNA damage (Cardozo-Pelaez et al., 2000) are observed between
hippocampus, cortex, and striatum. Among these possible mechanisms, it is suggested that
insufficient antioxidant defence during oxidative stress is a major contributor to regional
specificity in the immature brain after ischemia (Jiang et al., 2004).
4.5. Mechanisms underlying bilirubin-induced neurotoxicity Encephalopathy by UCB and kernicterus are the main complications of UCB-induced
neurotoxic effects on the newborn brain. However, accumulating evidence strongly suggests
that low concentrations of bilirubin have anti-oxidant properties (Stocker et al., 1987),
providing protection against injury resulting from oxidation (Doré et al., 2000, Doré et al.,
1999), as described in section 2.4.2. However, elevated levels of UCB in neonatal period
produce neurotoxic effects, as stated in section 4.1.
Several studies have been made in order to better understand the mechanisms
underlying UCB neurotoxicity. Experimentally, UCB participates in numerous toxic events
occurring in different study models. Pioneer studies of Ernster and Zetterstrom (1956)
showed that UCB inhibits respiration and uncouples oxidative phosphorylation in brain
homogenates or isolated mitochondria. The energy depletion was later on corroborated by
the reduced rates of glycolysis and decreased ATP levels observed in Gunn rats and in
newborn piglets (Hoffman et al., 1996, Park et al., 2001). However, discrepant findings were
reported in other studies that failed to document significant changes in brain glucose
metabolism or oxidative phosphorylation (Diamond and Schmid, 1967), whereas others
demonstrated that hyperbilirubinemia only disturbs brain energy metabolism in the presence
of additional factors that disrupt the blood brain barrier, such as hypoxia or hyperosmolarity
(Wennberg et al., 1991). In addition, UCB is reported to induce impairment in neurogenesis,
neuritogenesis and synaptogenesis of primary cultures from both cortical and hippocampal
neurons (Falcão et al., 2007b, Fernandes et al., 2009). Among the UCB-induced neurotoxic
effects are ionic imbalance (Brito et al., 2004), extracellular accumulation of glutamate and
release of pro-inflammatory cytokines TNF-α, IL-1β and IL-6 by both astrocytes (Falcão et
al., 2005, Falcão et al., 2006, Fernandes et al., 2004) and microglia (Gordo et al., 2006).

General Introduction _________________________________________________________________________
33
Inflammatory signalling pathways triggered by UCB includes the activation of MAPKs and
NF-κB (Fernandes et al., 2007a, Fernandes et al., 2006, Silva et al., 2010), as well as TNF-α
and IL-1β receptors signaling pathways (Fernandes et al., 2010).
Involvement of oxidative stress in the pathways of cellular demise by UCB was already
demonstrated in neocortical synaptosomes (Brito et al., 2004) and mature cultured neurons
(Brito et al., 2008a, Brito et al., 2008b). Particular attention has been given to the
involvement of .NO and induction of nNOS in UCB-induced neurotoxicity (Brito et al., 2010,
Mancuso et al., 2008), as well as in UCB-induced cytotoxicity in cultured oligodendrocytes,
pointed to be mediated by activation of iNOS and .NO production (Genc et al., 2003).
Interestingly, it has been recently reported that synaptic transmission failure observed in
auditory brainstem of Gunn rats occurs in neurons that are expressing high levels of nNOS,
whereas antagonism of this enzyme confers protection against hearing loss (Haustein et al.,
2010). These upstream events culminate in nerve cell death by both necrosis and apoptosis.
Indeed, neurons and astrocytes (Brites et al., 2009, Silva et al., 2002), oligodendrocytes
(Genc et al., 2003), endothelial cells (Akin et al., 2002) and microglia (Gordo et al., 2006,
Silva et al., 2010) show a concentration-dependent UCB-induced cell death by both
oncosis/necrosis and apoptotic processes. Other studies demonstrated that UCB interferes
with DNA and protein synthesis in Gunn rat model (Greenfield and Majumdar, 1974, Yamada
et al., 1977), as well as protein phosphorylation (Hansen et al., 1996). It is also established
that UCB directly interacts with mitochondria, influencing membrane lipid and protein
properties, redox status, and cytochrome c content (Rodrigues et al., 2002b, Rodrigues et
al., 2000). In addition, it was demonstrated that UCB induces apoptosis trough mitochondria-
caspase-3 pathway involving cytochrome c release, caspase-3 activation, and subsequent
poli (ADP-ribose) polymerase (PARP) cleavage in developing rat brain neurons (Rodrigues
et al., 2002a).
Comparison of nerve cell susceptibility to UCB showed that neurons and microglia are
more vulnerable to UCB-induced cell death and microglial cells present the most reactive
features, such as the highest levels of pro-inflammatory cytokines and glutamate release
(Brites et al., 2009). In addition, UCB-induced inflammatory response, extracellular
accumulation of glutamate and cell death were shown to be enhanced in both immature
neurons and astrocytes when compared to mature ones (Falcão et al., 2005, Falcão et al.,
2006), observations that provide a basis for the increased risk of hyperbilirubinemia in
premature neonates. Interestingly, the P-glycoprotein (Pgp) and the multidrug resistance
associated protein 1 (Mrp1), two ATP-dependent plasma membrane efflux pumps, were
pointed to be responsible for limiting UCB levels inside the nerve cell (Ostrow et al., 2004,
Watchko et al., 1998). In addition, these proteins are shown to increase along nerve cell

Chapter I __________________________________________________________________________
34
maturation (Falcão et al., 2007a, Tsai et al., 2002). Therefore, it seems likely that the limited
levels of Pgp and Mrp1 in the initial maturation cellular stages may have a role in the
increased vulnerability of immature nerve cells to UCB.
Finally, neonatal hyperbilirubinemia is considered a vulnerability factor for the
development of mental disorders (Dalman and Cullberg, 1999), such as schizophrenia
(Hayashida et al., 2009) and it is reported that structural abnormalities at cytoskeleton level
are produced by ROS generated by prolonged treatment with haloperidol, commonly used in
the treatment of schizophrenia (Benitez-King et al., 2010).
5. Promising molecules for modulation in hyperbilirubinemia Understanding the various molecular players involved in neurotoxicity induced by
hyperbilirubinemia or hyperbilirubinemia with associated inflammation will contribute to
identify adequate therapeutic targets. For the present thesis we focused on a better
understanding on the role of molecules involved in response to oxidative stress and whether
they represent important strategies to prevent neuronal injury in hyperbilirubinemia.
5.1. Glycoursodeoxycholic acid (GUDCA)
Ursodeoxycholic acid (UDCA), the 7β-hydroxy epimer of chenodeoxycholic acid, is an
endogenous bile acid that has been widely used for the treatment of hepatobiliary disorders
(Lazaridis et al., 2001) and is also considered an anti-apoptotic agent (Rodrigues and Steer,
2001). After oral administration, UDCA is conjugated with taurine and glycine in the liver,
originating tauroursodeoxycholic acid (TUDCA) and, mostly, glycoursodeoxycholic acid
(GUDCA), respectively (Lazaridis et al., 2001). Thus, GUDCA is the conjugate form of UDCA
with highest clinical relevance.
UDCA is able to suppress the production of pro-inflammatory cytokines by inactivation of
the NF-κB pathway in different cell types (Joo et al., 2004, Schoemaker et al., 2004, Shah et
al., 2006, Solá et al., 2003). In other studies UDCA or its conjugates were able to act as a
cytoprotective agent, by promoting the stabilization of the plasma and mitochondrial
membranes and preventing cellular apoptosis (Güldütuna et al., 1993, Solá et al., 2002).
Regarding neurotoxic effects of UCB, both UDCA and TUDCA are able to prevent form UCB-
induced neuronal apoptosis by inhibiting the Bax translocation to mitochondria, the
consequent mitochondrial depolarization, cytochrome c release, caspase-3 activation and
PARP cleavage (Rodrigues et al., 2000, Solá et al., 2002).
More recently, it was demonstrated that GUDCA prevents from cell death, as well as
from release of pro-inflammatory cytokines in astrocytes exposed to UCB. This
immunomodulatory effect is made at post-translational level, since it affects mainly the

General Introduction _________________________________________________________________________
35
activity of TNF-α- and IL-1β-converting enzymes (TACE and ICE, respectively), thus
preventing the maturation of this cytokines and their consequent release (Fernandes et al.,
2007b). Furthermore, GUDCA counteracts UCB-induced neuronal cell death and oxidative
stress, by inhibiting UCB-induced protein oxidation, lipid peroxidation and glutathione loss in
mature neurons (Brito et al., 2008a), as well as to induces a rapid and sustained decrease in
plasma UCB concentrations in Gunn rat model (Cuperus et al., 2009).
5.2. N-ω-nitro-L-arginine methyl ester hydrochloride (L-NAME)
L-NAME is an analog of L-arginine that inhibits NOS and subsequent .NO production in a
enantiomerically specific manner. L-NAME is structurally related to N-ω-monomethyl-L-
arginine (L-NMMA), which was previously used in studies of the cytotoxicity of activated
macrophages, before the discovery that .NO is involved in this process. L-NAME, unlike L-
NMMA, shows progressive and irreversible or only slowly reversible inhibition of brain NOS
following the initial binding as reviewed by Knowles and Moncada (1994). Analogues of
L-arginine are also muscarinic acetylcholine receptor antagonists (Buxton et al., 1993). As
mentioned in section 3.2, .NO and induction of NOS are involved in apoptotic pathways
induced by inflammatory mediators in neuronal cells. Therefore, .NO inhibitors represent
important strategies in the comprehension of cellular injury associated with inflammatory
processes. In fact, local elimination of nNOS in P7 rats resulted in a significant attenuation of
the damage after hypoxic-ischemic insult (Ferriero et al., 1995) and nNOS deficiency through
genetic targeting was also neuroprotective in neonatal mice (Ferriero et al., 1996). Other
experimental models of hypoxia-ischemia demonstrated that NOS inhibition reduced
apoptosis at the level of caspase-3 activation (Zhu et al., 2004) and conferred tissue
protection (Peeters-Scholte et al., 2002). The role of .NO in UCB-induced cytotoxicity was
demonstrated in primary cultures of oligodendrocytes (Genc et al., 2003) and, more recently,
in primary cultures of mature neurons concomitantly treated with UCB and L-NAME (Brito et
al., 2008a, Brito et al., 2010).
5.3. N-acetylcysteine (NAC)
As mentioned in section 1.1, NAC is a thiol compound that is converted to cysteine, an
important precursor of cellular glutathione (Dringen, 2000, Zachwieja et al., 2005).
Antioxidant effects of NAC embrace its action as a source of sulfydryl groups, promoting
glutathione biosynthesis and its supply for GPx. In addition, NAC reacts with ROS (Ocal et
al., 2004). There are several in vitro and in vivo studies supporting antioxidant effect of NAC.
Treatment with NAC was shown to confer neuroprotection in lead-induced lipid peroxidation

Chapter I __________________________________________________________________________
36
and inhibition of antioxidant enzyme activities in rats’ brain (Nehru and Kanwar, 2004). In
addition, supplementation of cultured hippocampal neurons subjected to hypoxia with NAC
resulted in a significant cytoprotection, decline in ROS generation, and higher antioxidant
levels similar to that of control cells (Jayalakshmi et al., 2005). NAC was also able to inhibit
DNA strand breaks induced by hypoxia. Moreover, NAC attenuated long-term depletion of
dopamine and lipid peroxidation in rat striatum subjected to hypothermia induced by
amphetamine (Wan et al., 2006).
Regarding NAC effects on hyperbilirubinemia models, treatment with NAC was able to
decrease lipid peroxidation in cerebral cortex, midbrain and cerebellum observed in
jaundiced rats (Karageorgos et al., 2006), as well as to protect against UCB-induced protein
oxidation, oxidative disruption and cell death in rat cultured mature cortical neurons (Brito et
al., 2008b).

General Introduction _________________________________________________________________________
37
6. Global aims of the thesis The main goal of the present work is to further dissect the cellular mechanisms of
neonatal neurotoxicity by hyperbilirubinemia. Oxidative stress, mitochondrial dysfunction and
consequent cell death will be the major features studied. Since prematurity and sepsis are
risk factors for hyperbilirubinemia, it will be used an experimental model that intends to mimic
conditions of a moderate to severe neonatal jaundice in prematures, alone or with
associatied inflammation. Regarding that bilirubin presents a specific deposition pattern in
the brain, regional susceptibility will also be evaluated in different areas. In addition, it will be
discussed the role of antioxidants or known modulators of oxidant species production in
prevention of neuronal injury in neonatal hyperbilirubinemia.
The major questions addressed in the present thesis are:
1. Does UCB interfere with mitochondrial function and energy metabolic pathways
through increased oxidative status in immature neurons? Can GUDCA counteract
such effects?
2. Does sepsis have an aggravating role in UCB-induced dysfunction in immature
neurons? Can .NO/NOS and JNK1/2 activation be considered signalling
determinants in this neuronal dysfunction?
3. Does UCB deposition specific pattern in the brain determine the differential
regional susceptibility to UCB-induced oxidative damage? Can DJ-1 protein
expression and glutathione content be considered potential modulators of this
neurotoxicity?
The studies developed to address these questions are described and discussed in the
three subsequent chapters.
In summary, they bring new insights into UCB effects on metabolic pathways, cellular
redox status and cell death in conditions mimicking a moderate to severe hyperbilirubinemia
in the early neonatal period. In addition, these results provide a basis for the commonly
indicated higher risk of UCB brain damage in a condition of inflammation. Furthermore, these
data provide specific features that may explain the differential susceptibility to UCB observed
in different brain areas. These advances may substantiate target-driven approaches to the
prevention and treatment of UCB-induced neurological damage, and provide fruitful
opportunities for future investigations.

Chapter I __________________________________________________________________________
38
7. References Cellular respiration (2007) In: http://fig.cox.miami.edu/Faculty/Dana/105F00_13.html, from
Department of Biology, University of Miami.
Acehan, D., Jiang, X., Morgan, D. G., Heuser, J. E., Wang, X. and Akey, C. W. (2002) Three-
dimensional structure of the apoptosome: implications for assembly, procaspase-9
binding, and activation. Mol Cell, 9, 423-432.
Adams, J. M. (2003) Ways of dying: multiple pathways to apoptosis. Genes Dev, 17, 2481-
2495.
Ahdab-Barmada, M. and Moossy, J. (1984) The neuropathology of kernicterus in the
premature neonate: diagnostic problems. J Neuropathol Exp Neurol, 43, 45-56.
Ahlfors, C. E., Bennett, S. H., Shoemaker, C. T., Ellis, W. G., Davis, S. L., Wennberg, R. P.
and Goetzman, B. W. (1986) Changes in the auditory brainstem response associated with
intravenous infusion of unconjugated bilirubin into infant rhesus monkeys. Pediatr Res, 20, 511-515.
Akin, E., Clower, B., Tibbs, R., Tang, J. and Zhang, J. (2002) Bilirubin produces apoptosis in
cultured bovine brain endothelial cells. Brain Res, 931, 168-175.
Allan, S. M. and Rothwell, N. J. (2003) Inflammation in central nervous system injury. Philos
Trans R Soc Lond B Biol Sci, 358, 1669-1677.
Allen, N. J. and Barres, B. A. (2005) Signaling between glia and neurons: focus on synaptic
plasticity. Curr Opin Neurobiol, 15, 542-548.
Allen, N. J. and Barres, B. A. (2009) Neuroscience: Glia - more than just brain glue. Nature,
457, 675-677.
Almeida, A., Almeida, J., Bolaños, J. P. and Moncada, S. (2001) Different responses of
astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte
protection. Proc Natl Acad Sci U S A, 98, 15294-15299.
Almeida, A. and Bolaños, J. P. (2001) A transient inhibition of mitochondrial ATP synthesis
by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J
Neurochem, 77, 676-690.
Almeida, A., Delgado-Esteban, M., Bolanos, J. P. and Medina, J. M. (2002) Oxygen and
glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones
but not in astrocytes in primary culture. J Neurochem, 81, 207-217.
Almeida, A., Moncada, S. and Bolaños, J. P. (2004) Nitric oxide switches on glycolysis
through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol, 6, 45-51.
Anderson, C. M. and Swanson, R. A. (2000) Astrocyte glutamate transport: review of
properties, regulation, and physiological functions. Glia, 32, 1-14.

General Introduction _________________________________________________________________________
39
Angermüller, S., Islinger, M. and Völkl, A. (2009) Peroxisomes and reactive oxygen species,
a lasting challenge. Histochem Cell Biol, 131, 459-463.
Aruoma, O. I., Laughton, M. J. and Halliwell, B. (1989) Carnosine, homocarnosine and
anserine: could they act as antioxidants in vivo? Biochem J, 264, 863-869.
Ballabh, P., Braun, A. and Nedergaard, M. (2004) The blood-brain barrier: an overview:
structure, regulation, and clinical implications. Neurobiol Dis, 16, 1-13.
Barañano, D. E., Rao, M., Ferris, C. D. and Snyder, S. H. (2002) Biliverdin reductase: a
major physiologic cytoprotectant. Proc Natl Acad Sci U S A, 99, 16093-16098.
Barkovich, A. J., Westmark, K., Partridge, C., Sola, A. and Ferriero, D. M. (1995) Perinatal
asphyxia: MR findings in the first 10 days. AJNR Am J Neuroradiol, 16, 427-438.
Baumann, N. and Pham-Dinh, D. (2001) Biology of oligodendrocyte and myelin in the
mammalian central nervous system. Physiol Rev, 81, 871-927.
Beckman, K. B. and Ames, B. N. (1998) The free radical theory of aging matures. Physiol
Rev, 78, 547-581.
Bellinger, F. P., Madamba, S. and Siggins, G. R. (1993) Interleukin 1 beta inhibits synaptic
strength and long-term potentiation in the rat CA1 hippocampus. Brain Res, 628, 227-234.
Benitez-King, G., Ortiz-Lopez, L., Jimenez-Rubio, G. and Ramirez-Rodriguez, G. (2010)
Haloperidol causes cytoskeletal collapse in N1E-115 cells through tau
hyperphosphorylation induced by oxidative stress: Implications for neurodevelopment. Eur
J Pharmacol, 644, 24-31.
Bian, K. and Murad, F. (2001) Diversity of endotoxin-induced nitrotyrosine formation in
macrophage-endothelium-rich organs. Free Radic Biol Med, 31, 421-429.
Black, S. M., Bedolli, M. A., Martinez, S., Bristow, J. D., Ferriero, D. M. and Soifer, S. J.
(1995) Expression of neuronal nitric oxide synthase corresponds to regions of selective
vulnerability to hypoxia-ischaemia in the developing rat brain. Neurobiol Dis, 2, 145-155.
Blamire, A. M., Anthony, D. C., Rajagopalan, B., Sibson, N. R., Perry, V. H. and Styles, P.
(2000) Interleukin-1beta -induced changes in blood-brain barrier permeability, apparent
diffusion coefficient, and cerebral blood volume in the rat brain: a magnetic resonance
study. J Neurosci, 20, 8153-8159.
Blanckaert, N. and Fevery, J. (1990) Physiology and patophysiology of bilirubin metabolism.
In: Hepatology. A textbook of liver disease., pp. 254-303. WB Saunders, Philadelphia.
Block, M. L. and Hong, J. S. (2005) Microglia and inflammation-mediated neurodegeneration:
multiple triggers with a common mechanism. Prog Neurobiol, 76, 77-98.
Blomgren, K., Leist, M. and Groc, L. (2007) Pathological apoptosis in the developing brain.
Apoptosis, 12, 993-1010.

Chapter I __________________________________________________________________________
40
Bolaños, J. P., Almeida, A. and Moncada, S. (2010) Glycolysis: a bioenergetic or a survival
pathway? Trends Biochem Sci, 35, 145-149.
Bolaños, J. P., Peuchen, S., Heales, S. J., Land, J. M. and Clark, J. B. (1994) Nitric oxide-
mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J
Neurochem, 63, 910-916.
Bonmann, E., Suschek, C., Spranger, M. and Kolb-Bachofen, V. (1997) The dominant role of
exogenous or endogenous interleukin-1 beta on expression and activity of inducible nitric
oxide synthase in rat microvascular brain endothelial cells. Neurosci Lett, 230, 109-112.
Bouzier-Sore, A. K., Voisin, P., Canioni, P., Magistretti, P. J. and Pellerin, L. (2003) Lactate is
a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb
Blood Flow Metab, 23, 1298-1306.
Brites, D., Fernandes, A., Falcão, A. S., Gordo, A. C., Silva, R. F. M. and Brito, M. A. (2009)
Biological risks for neurological abnormalities associated with hyperbilirubinemia. J
Perinatol, 29 Suppl 1, S8-13.
Brito, M. A., Brites, D. and Butterfield, D. A. (2004) A link between hyperbilirubinemia,
oxidative stress and injury to neocortical synaptosomes. Brain Res, 1026, 33-43.
Brito, M. A., Lima, S., Fernandes, A., Falcão, A. S., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008a) Bilirubin injury to neurons: contribution of oxidative stress and rescue by
glycoursodeoxycholic acid. Neurotoxicology, 29, 259-269.
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008b) Unconjugated bilirubin differentially affects the redox status of neuronal
and astroglial cells. Neurobiol Dis, 29, 30-40.
Brito, M. A., Rosa, A. I., Silva, R. F. M., Falcão, A. S., Fernandes, A. and Brites, D. (2007)
Oxidative stress and disruption of the nervous cell. In: Focus in Brain Research, pp. 1-33.
Nova Science Publishers, Inc., New York.
Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Cell response to hyperbilirrubinemia: a
journey along key molecular events. In: New Trends in Brain Research, pp. 1-38. Nova
Science Publishers, Inc., New York.
Brito, M. A., Vaz, A. R., Silva, S. L., Falcão, A. S., Fernandes, A., Silva, R. F. M. and Brites,
D. (2010) N-methyl-D-aspartate receptor and neuronal nitric oxide synthase activation
mediate bilirubin-induced neurotoxicity. Mol Med, 16, 372-380.
Brown, G. C. and Cooper, C. E. (1994) Nanomolar concentrations of nitric oxide reversibly
inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS
Lett, 356, 295-298.
Burdo, J. R. and Connor, J. R. (2003) Brain iron uptake and homeostatic mechanisms: an
overview. Biometals, 16, 63-75.

General Introduction _________________________________________________________________________
41
Buxton, I. L., Cheek, D. J., Eckman, D., Westfall, D. P., Sanders, K. M. and Keef, K. D.
(1993) NG-nitro L-arginine methyl ester and other alkyl esters of arginine are muscarinic
receptor antagonists. Circ Res, 72, 387-395.
Calabrese, V., Lodi, R., Tonon, C. et al. (2005) Oxidative stress, mitochondrial dysfunction
and cellular stress response in Friedreich's ataxia. J Neurol Sci, 233, 145-162.
Candelario-Jalil, E., Mhadu, N. H., Al-Dalain, S. M., Martinez, G. and Leon, O. S. (2001)
Time course of oxidative damage in different brain regions following transient cerebral
ischemia in gerbils. Neurosci Res, 41, 233-241.
Cardoso, F. L., Brites, D. and Brito, M. A. (2010) Looking at the blood-brain barrier:
molecular anatomy and possible investigation approaches. Brain Res Rev, 64, 328-363.
Cardozo-Pelaez, F., Brooks, P. J., Stedeford, T., Song, S. and Sanchez-Ramos, J. (2000)
DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free
Radic Biol Med, 28, 779-785.
Cartlidge, P. H. and Rutter, N. (1986) Serum albumin concentrations and oedema in the
newborn. Arch Dis Child, 61, 657-660.
Cashore, W. J. (1980) Free bilirubin concentrations and bilirubin-binding affinity in term and
preterm infants. J Pediatr, 96, 521-527.
Chang, E. F., Claus, C. P., Vreman, H. J., Wong, R. J. and Noble-Haeusslein, L. J. (2005)
Heme regulation in traumatic brain injury: relevance to the adult and developing brain. J
Cereb Blood Flow Metab, 25, 1401-1417.
Chavarria, A. and Alcocer-Varela, J. (2004) Is damage in central nervous system due to
inflammation? Autoimmun Rev, 3, 251-260.
Chen, Y. and Swanson, R. A. (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab,
23, 137-149.
Circu, M. L. and Aw, T. Y. (2010) Reactive oxygen species, cellular redox systems, and
apoptosis. Free Radic Biol Med, 48, 749-762.
Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. and Schapira, A. H. (1994)
Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial
respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett,
345, 50-54.
Clementi, E., Brown, G. C., Foxwell, N. and Moncada, S. (1999) On the mechanism by which
vascular endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci U S A,
96, 1559-1562.
Combs, C. K., Karlo, J. C., Kao, S. C. and Landreth, G. E. (2001) beta-Amyloid stimulation of
microglia and monocytes results in TNFalpha-dependent expression of inducible nitric
oxide synthase and neuronal apoptosis. J Neurosci, 21, 1179-1188.

Chapter I __________________________________________________________________________
42
Conlee, J. W. and Shapiro, S. M. (1997) Development of cerebellar hypoplasia in jaundiced
Gunn rats: a quantitative light microscopic analysis. Acta Neuropathol, 93, 450-460.
Cotgreave, I. A. and Gerdes, R. G. (1998) Recent trends in glutathione biochemistry--
glutathione-protein interactions: a molecular link between oxidative stress and cell
proliferation? Biochem Biophys Res Commun, 242, 1-9.
Couraud, P. O. (1994) Interactions between lymphocytes, macrophages, and central nervous
system cells. J Leukoc Biol, 56, 407-415.
Croitoru-Lamoury, J., Guillemin, G. J., Boussin, F. D. et al. (2003) Expression of chemokines
and their receptors in human and simian astrocytes: evidence for a central role of TNF
alpha and IFN gamma in CXCR4 and CCR5 modulation. Glia, 41, 354-370.
Cuperus, F. J., Hafkamp, A. M., Havinga, R., Vitek, L., Zelenka, J., Tiribelli, C., Ostrow, J. D.
and Verkade, H. J. (2009) Effective treatment of unconjugated hyperbilirubinemia with oral
bile salts in Gunn rats. Gastroenterology, 136, 673-682 e671.
Daikhin, Y. and Yudkoff, M. (2000) Compartmentation of brain glutamate metabolism in
neurons and glia. J Nutr, 130, 1026S-1031S.
Dalman, C. and Cullberg, J. (1999) Neonatal hyperbilirubinaemia--a vulnerability factor for
mental disorder? Acta Psychiatr Scand, 100, 469-471.
Dawodu, A. H., Owa, J. A. and Familusi, J. B. (1984) A prospective study of the role of
bacterial infection and G6PD deficiency in severe neonatal jaundice in Nigeria. Trop
Geogr Med, 36, 127-132.
Dawson, V. L., Dawson, T. M., London, E. D., Bredt, D. S. and Snyder, S. H. (1991) Nitric
oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U
S A, 88, 6368-6371.
de Arriba, S. G., Krugel, U., Regenthal, R. et al. (2006) Carbonyl stress and NMDA receptor
activation contribute to methylglyoxal neurotoxicity. Free Radic Biol Med, 40, 779-790.
De Marchis, S., Modena, C., Peretto, P., Migheli, A., Margolis, F. L. and Fasolo, A. (2000)
Carnosine-related dipeptides in neurons and glia. Biochemistry (Mosc), 65, 824-833.
de Vries, L. S., Lary, S. and Dubowitz, L. M. (1985) Relationship of serum bilirubin levels to
ototoxicity and deafness in high-risk low-birth-weight infants. Pediatrics, 76, 351-354.
Decker, E. A., Livisay, S. A. and Zhou, S. (2000) A re-evaluation of the antioxidant activity of
purified carnosine. Biochemistry (Mosc), 65, 766-770.
Delgado-Esteban, M., Almeida, A. and Bolaños, J. P. (2000) D-Glucose prevents glutathione
oxidation and mitochondrial damage after glutamate receptor stimulation in rat cortical
primary neurons. J Neurochem, 75, 1618-1624.
Dennery, P. A., Seidman, D. S. and Stevenson, D. K. (2001) Neonatal hyperbilirubinemia. N
Engl J Med, 344, 581-590.

General Introduction _________________________________________________________________________
43
Desagher, S., Glowinski, J. and Premont, J. (1996) Astrocytes protect neurons from
hydrogen peroxide toxicity. J Neurosci, 16, 2553-2562.
Di Monte, D. A., Tokar, I. and Langston, J. W. (1999) Impaired glutamate clearance as a
consequence of energy failure caused by MPP(+) in astrocytic cultures. Toxicol Appl
Pharmacol, 158, 296-302.
Diamond, I. and Schmid, R. (1967) Oxidative phosphorylation in experimental bilirubin
encephalopathy. Science, 155, 1288-1289.
Doré, S., Goto, S., Sampei, K. et al. (2000) Heme oxygenase-2 acts to prevent neuronal
death in brain cultures and following transient cerebral ischemia. Neuroscience, 99, 587-
592.
Doré, S., Takahashi, M., Ferris, C. D., Zakhary, R., Hester, L. D., Guastella, D. and Snyder,
S. H. (1999) Bilirubin, formed by activation of heme oxygenase-2, protects neurons
against oxidative stress injury. Proc Natl Acad Sci U S A, 96, 2445-2450.
Dringen, R. (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol, 62, 649-
671.
Dringen, R., Gutterer, J. M. and Hirrlinger, J. (2000) Glutathione metabolism in brain
metabolic interaction between astrocytes and neurons in the defence against reactive
oxygen species. Eur J Biochem, 267, 4912-4916.
Dringen, R. and Hamprecht, B. (1996) Glutathione content as an indicator for the presence of
metabolic pathways of amino acids in astroglial cultures. J Neurochem, 67, 1375-1382.
Dringen, R., Pawlowski, P. G. and Hirrlinger, J. (2005) Peroxide detoxification by brain cells.
J Neurosci Res, 79, 157-165.
Du, Y. and Dreyfus, C. F. (2002) Oligodendrocytes as providers of growth factors. J Neurosci
Res, 68, 647-654.
Eggleston, L. V. and Krebs, H. A. (1974) Regulation of the pentose phosphate cycle.
Biochem J, 138, 425-435.
Elfering, S. L., Sarkela, T. M. and Giulivi, C. (2002) Biochemistry of mitochondrial nitric-oxide
synthase. J Biol Chem, 277, 38079-38086.
Ernster, L. and Zetterstrom, R. (1956) Bilirubin, an uncoupler of oxidative phosphorylation in
isolated mitochondria. Nature, 178, 1335-1337.
Estus, S., Zaks, W. J., Freeman, R. S., Gruda, M., Bravo, R. and Johnson, E. M., Jr. (1994)
Altered gene expression in neurons during programmed cell death: identification of c-jun
as necessary for neuronal apoptosis. J Cell Biol, 127, 1717-1727.
Falcão, A. S., Bellarosa, C., Fernandes, A., Brito, M. A., Silva, R. F. M., Tiribelli, C. and
Brites, D. (2007a) Role of multidrug resistance-associated protein 1 expression in the in
vitro susceptibility of rat nerve cell to unconjugated bilirubin. Neuroscience, 144, 878-888.

Chapter I __________________________________________________________________________
44
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2005) Bilirubin-
induced inflammatory response, glutamate release, and cell death in rat cortical
astrocytes are enhanced in younger cells. Neurobiol Dis, 20, 199-206.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Bilirubin-
induced immunostimulant effects and toxicity vary with neural cell type and maturation
state. Acta Neuropathol, 112, 95-105.
Falcão, A. S., Silva, R. F. M., Pancadas, S., Fernandes, A., Brito, M. A. and Brites, D.
(2007b) Apoptosis and impairment of neurite network by short exposure of immature rat
cortical neurons to unconjugated bilirubin increase with cell differentiation and are
additionally enhanced by an inflammatory stimulus. J Neurosci Res, 85, 1229-1239.
Fernandes, A., Barateiro, A., Falcão, A. S., Silva, S. L., Vaz, A. R., Brito, M. A., Silva, R. F.
M. and Brites, D. (2010) Astrocyte reactivity to unconjugated bilirubin requires TNF-α and
IL-1β receptor signalling pathways. Glia, in press.
Fernandes, A., Falcão, A. S., Abranches, E., Bekman, E., Henrique, D., Lanier, L. M. and
Brites, D. (2009) Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and
synaptogenesis. Dev Neurobiol, 69, 568-582.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007a) MAPKs are
key players in mediating cytokine release and cell death induced by unconjugated bilirubin
in cultured rat cortical astrocytes. Eur J Neurosci, 25, 1058-1068.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Gordo, A. C., Gama, M. J., Brito, M. A. and
Brites, D. (2006) Inflammatory signalling pathways involved in astroglial activation by
unconjugated bilirubin. J Neurochem, 96, 1667-1679.
Fernandes, A., Silva, R. F. M., Falcão, A. S., Brito, M. A. and Brites, D. (2004) Cytokine
production, glutamate release and cell death in rat cultured astrocytes treated with
unconjugated bilirubin and LPS. J Neuroimmunol, 153, 64-75.
Fernandes, A., Vaz, A. R., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007b)
Glycoursodeoxycholic Acid and interleukin-10 modulate the reactivity of rat cortical
astrocytes to unconjugated bilirubin. J Neuropathol Exp Neurol, 66, 789-798.
Ferriero, D. M., Holtzman, D. M., Black, S. M. and Sheldon, R. A. (1996) Neonatal mice
lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury.
Neurobiol Dis, 3, 64-71.
Ferriero, D. M., Sheldon, R. A., Black, S. M. and Chuai, J. (1995) Selective destruction of
nitric oxide synthase neurons with quisqualate reduces damage after hypoxia-ischemia in
the neonatal rat. Pediatr Res, 38, 912-918.
Finkel, T. (2000) Redox-dependent signal transduction. FEBS Lett, 476, 52-54.

General Introduction _________________________________________________________________________
45
Forsyth, R. J. (1996) Astrocytes and the delivery of glucose from plasma to neurons.
Neurochem Int, 28, 231-241.
Friberg, H., Connern, C., Halestrap, A. P. and Wieloch, T. (1999) Differences in the activation
of the mitochondrial permeability transition among brain regions in the rat correlate with
selective vulnerability. J Neurochem, 72, 2488-2497.
Gal, S., Zheng, H., Fridkin, M. and Youdim, M. B. (2005) Novel multifunctional
neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative
diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-
induced striatal dopamine depletion. J Neurochem, 95, 79-88.
García-Nogales, P., Almeida, A. and Bolaños, J. P. (2003) Peroxynitrite protects neurons
against nitric oxide-mediated apoptosis. A key role for glucose-6-phosphate
dehydrogenase activity in neuroprotection. J Biol Chem, 278, 864-874.
García-Nogales, P., Almeida, A., Fernández, E., Medina, J. M. and Bolaños, J. P. (1999)
Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to
preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J
Neurochem, 72, 1750-1758.
Genc, S., Genc, K., Kumral, A., Baskin, H. and Ozkan, H. (2003) Bilirubin is cytotoxic to rat
oligodendrocytes in vitro. Brain Res, 985, 135-141.
Ghafourifar, P. and Cadenas, E. (2005) Mitochondrial nitric oxide synthase. Trends
Pharmacol Sci, 26, 190-195.
Ghafourifar, P. and Richter, C. (1997) Nitric oxide synthase activity in mitochondria. FEBS
Lett, 418, 291-296.
Ghibelli, L., Fanelli, C., Rotilio, G., Lafavia, E., Coppola, S., Colussi, C., Civitareale, P. and
Ciriolo, M. R. (1998) Rescue of cells from apoptosis by inhibition of active GSH extrusion.
FASEB J, 12, 479-486.
Gilland, E., Puka-Sundvall, M., Hillered, L. and Hagberg, H. (1998) Mitochondrial function
and energy metabolism after hypoxia-ischemia in the immature rat brain: involvement of
NMDA-receptors. J Cereb Blood Flow Metab, 18, 297-304.
Gkoltsiou, K., Tzoufi, M., Counsell, S., Rutherford, M. and Cowan, F. (2008) Serial brain MRI
and ultrasound findings: relation to gestational age, bilirubin level, neonatal neurologic
status and neurodevelopmental outcome in infants at risk of kernicterus. Early Hum Dev,
84, 829-838.
Goldenberg, R. L. and Andrews, W. W. (1996) Intrauterine infection and why preterm
prevention programs have failed. Am J Public Health, 86, 781-783.
Gonzalez-Zulueta, M., Ensz, L. M., Mukhina, G., Lebovitz, R. M., Zwacka, R. M., Engelhardt,
J. F., Oberley, L. W., Dawson, V. L. and Dawson, T. M. (1998) Manganese superoxide

Chapter I __________________________________________________________________________
46
dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J
Neurosci, 18, 2040-2055.
Gordo, A. C., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006)
Unconjugated bilirubin activates and damages microglia. J Neurosci Res, 84, 194-201.
Görg, B., Qvartskhava, N., Voss, P., Grune, T., Häussinger, D. and Schliess, F. (2007)
Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett,
581, 84-90.
Greenamyre, T., Penney, J. B., Young, A. B., Hudson, C., Silverstein, F. S. and Johnston, M.
V. (1987) Evidence for transient perinatal glutamatergic innervation of globus pallidus. J
Neurosci, 7, 1022-1030.
Greenfield, S. and Majumdar, A. P. (1974) Bilirubin encephalopathy: effect on protein
synthesis in the brain of the Gunn rat. J Neurol Sci, 22, 83-89.
Güldütuna, S., Zimmer, G., Imhof, M., Bhatti, S., You, T. and Leuschner, U. (1993) Molecular
aspects of membrane stabilization by ursodeoxycholate [see comment]. Gastroenterology,
104, 1736-1744.
Gupta, S. (2002) A decision between life and death during TNF-alpha-induced signaling. J
Clin Immunol, 22, 185-194.
Guzzetta, F., Deodato, F. and Rando, T. (2000) Brain ischemic lesions of the newborn.
Childs Nerv Syst, 16, 633-637.
Haberg, A., Qu, H., Haraldseth, O., Unsgard, G. and Sonnewald, U. (1998) In vivo injection
of [1-13C]glucose and [1,2-13C]acetate combined with ex vivo 13C nuclear magnetic
resonance spectroscopy: a novel approach to the study of middle cerebral artery
occlusion in the rat. J Cereb Blood Flow Metab, 18, 1223-1232.
Haden, D. W., Suliman, H. B., Carraway, M. S., Welty-Wolf, K. E., Ali, A. S., Shitara, H.,
Yonekawa, H. and Piantadosi, C. A. (2007) Mitochondrial biogenesis restores oxidative
metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med, 176, 768-
777.
Halliwell, B. (2006) Oxidative stress and neurodegeneration: where are we now? J
Neurochem, 97, 1634-1658.
Ham, J., Babij, C., Whitfield, J., Pfarr, C. M., Lallemand, D., Yaniv, M. and Rubin, L. L. (1995)
A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell
death. Neuron, 14, 927-939.
Hanisch, U. K. (2002) Microglia as a source and target of cytokines. Glia, 40, 140-155.
Hansen, T. W. (2000) Pioneers in the scientific study of neonatal jaundice and kernicterus.
Pediatrics, 106, E15.

General Introduction _________________________________________________________________________
47
Hansen, T. W., Mathiesen, S. B. and Walaas, S. I. (1996) Bilirubin has widespread inhibitory
effects on protein phosphorylation. Pediatr Res, 39, 1072-1077.
Hansen, T. W. R. (2002) Mechanisms of bilirubin toxicity: clinical implications. Clin Perinatol,
29, 765-778, viii.
Haskew-Layton, R. E., Payappilly, J. B., Smirnova, N. A. et al. (2010) Controlled enzymatic
production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an
Nrf2-independent pathway. Proc Natl Acad Sci U S A.
Haustein, M. D., Read, D. J., Steinert, J. R., Pilati, N., Dinsdale, D. and Forsythe, I. D. (2010)
Acute hyperbilirubinaemia induces presynaptic neurodegeneration at a central
glutamatergic synapse. J Physiol.
Hayashida, M., Miyaoka, T., Tsuchie, K. et al. (2009) Hyperbilirubinemia-related behavioral
and neuropathological changes in rats: a possible schizophrenia animal model. Prog
Neuropsychopharmacol Biol Psychiatry, 33, 581-588.
Hayton, S. M. and Muller, D. P. (2004) Vitamin E in neural and visual function. Ann N Y Acad
Sci, 1031, 263-270.
Hemmer, K., Fransen, L., Vanderstichele, H., Vanmechelen, E. and Heuschling, P. (2001) An
in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric
oxide and tumor necrosis factor-alpha. Neurochem Int, 38, 557-565.
Heneka, M. T., Loschmann, P. A., Gleichmann, M., Weller, M., Schulz, J. B., Wullner, U. and
Klockgether, T. (1998) Induction of nitric oxide synthase and nitric oxide-mediated
apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-
alpha/lipopolysaccharide. J Neurochem, 71, 88-94.
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S. and Bolaños, J.
P. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous
degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol, 11, 747-752.
Hoffman, D. J., Zanelli, S. A., Kubin, J., Mishra, O. P. and Delivoria-Papadopoulos, M. (1996)
The in vivo effect of bilirubin on the N-methyl-D-aspartate receptor/ion channel complex in
the brains of newborn piglets. Pediatr Res, 40, 804-808.
Itoh, Y., Esaki, T., Shimoji, K., Cook, M., Law, M. J., Kaufman, E. and Sokoloff, L. (2003)
Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro
and on glucose utilization by brain in vivo. Proc Natl Acad Sci U S A, 100, 4879-4884.
Jayalakshmi, K., Sairam, M., Singh, S. B., Sharma, S. K., Ilavazhagan, G. and Banerjee, P.
K. (2005) Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress
in primary hippocampal culture. Brain Res, 1046, 97-104.

Chapter I __________________________________________________________________________
48
Jiang, X., Mu, D., Manabat, C., Koshy, A. A., Christen, S., Tauber, M. G., Vexler, Z. S. and
Ferriero, D. M. (2004) Differential vulnerability of immature murine neurons to oxygen-
glucose deprivation. Exp Neurol, 190, 224-232.
Jolivet, R., Magistretti, P. J. and Weber, B. (2009) Deciphering neuron-glia
compartmentalization in cortical energy metabolism. Front Neuroenergetics, 1, 4.
Joo, S. S., Won, T. J. and Lee, D. I. (2004) Potential role of ursodeoxycholic acid in
suppression of nuclear factor kappa B in microglial cell line (BV-2). Arch Pharm Res, 27, 954-960.
Kajta, M., Trotter, A., Lason, W. and Beyer, C. (2006) Impact of 17beta-estradiol on cytokine-
mediated apoptotic effects in primary hippocampal and neocortical cell cultures. Brain
Res, 1116, 64-74.
Kanski, J., Behring, A., Pelling, J. and Schoneich, C. (2005) Proteomic identification of 3-
nitrotyrosine-containing rat cardiac proteins: effects of biological aging. Am J Physiol
Heart Circ Physiol, 288, H371-381.
Kaplan, M. and Hammerman, C. (2005) Understanding severe hyperbilirubinemia and
preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin Chim
Acta, 356, 9-21.
Karageorgos, N., Patsoukis, N., Chroni, E., Konstantinou, D., Assimakopoulos, S. F. and
Georgiou, C. (2006) Effect of N-acetylcysteine, allopurinol and vitamin E on jaundice-
induced brain oxidative stress in rats. Brain Res, 1111, 203-212.
Kasischke, K. A., Vishwasrao, H. D., Fisher, P. J., Zipfel, W. R. and Webb, W. W. (2004)
Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis.
Science, 305, 99-103.
Katsuki, H., Okawara, M., Shibata, H., Kume, T. and Akaike, A. (2006) Nitric oxide-producing
microglia mediate thrombin-induced degeneration of dopaminergic neurons in rat midbrain
slice culture. J Neurochem, 97, 1232-1242.
Khan, J. Y. and Black, S. M. (2003) Developmental changes in murine brain antioxidant
enzymes. Pediatr Res, 54, 77-82.
Kletzien, R. F., Harris, P. K. and Foellmi, L. A. (1994) Glucose-6-phosphate dehydrogenase:
a "housekeeping" enzyme subject to tissue-specific regulation by hormones, nutrients,
and oxidant stress. FASEB J, 8, 174-181.
Knowles, R. G. and Moncada, S. (1994) Nitric oxide synthases in mammals. Biochem J, 298 ( Pt 2), 249-258.
Knowles, R. G., Palacios, M., Palmer, R. M. and Moncada, S. (1989) Formation of nitric
oxide from L-arginine in the central nervous system: a transduction mechanism for
stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A, 86, 5159-5162.

General Introduction _________________________________________________________________________
49
Konsman, J. P., Drukarch, B. and Van Dam, A. M. (2007) (Peri)vascular production and
action of pro-inflammatory cytokines in brain pathology. Clin Sci (Lond), 112, 1-25.
Korhonen, R., Lahti, A., Kankaanranta, H. and Moilanen, E. (2005) Nitric oxide production
and signaling in inflammation. Curr Drug Targets Inflamm Allergy, 4, 471-479.
Kowaltowski, A. J. (2000) Alternative mitochondrial functions in cell physiopathology: beyond
ATP production. Braz J Med Biol Res, 33, 241-250.
Kyriakis, J. M. and Avruch, J. (2001) Mammalian mitogen-activated protein kinase signal
transduction pathways activated by stress and inflammation. Physiol Rev, 81, 807-869.
Laranjinha, J. and Ledo, A. (2007) Coordination of physiologic and toxic pathways in
hippocampus by nitric oxide and mitochondria. Front Biosci, 12, 1094-1106.
Lazaridis, K. N., Gores, G. J. and Lindor, K. D. (2001) Ursodeoxycholic acid 'mechanisms of
action and clinical use in hepatobiliary disorders'. J Hepatol, 35, 134-146.
Leviton, A. and Gressens, P. (2007) Neuronal damage accompanies perinatal white-matter
damage. Trends Neurosci, 30, 473-478.
Lin, S., Wei, X., Bales, K. R., Paul, A. B., Ma, Z., Yan, G., Paul, S. M. and Du, Y. (2005)
Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced
cerebellar hypoplasia in the Gunn rat. Eur J Neurosci, 22, 21-27.
Liu, J., Killilea, D. W. and Ames, B. N. (2002) Age-associated mitochondrial oxidative decay:
improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain
by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S
A, 99, 1876-1881.
Lu, S. C. (2009) Regulation of glutathione synthesis. Mol Aspects Med, 30, 42-59.
Lucius, R. and Sievers, J. (1996) Postnatal retinal ganglion cells in vitro: protection against
reactive oxygen species (ROS)-induced axonal degeneration by cocultured astrocytes.
Brain Res, 743, 56-62.
Luo, Y., Umegaki, H., Wang, X., Abe, R. and Roth, G. S. (1998) Dopamine induces
apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem, 273, 3756-3764.
Madrigal, J. L., Hurtado, O., Moro, M. A., Lizasoain, I., Lorenzo, P., Castrillo, A., Bosca, L.
and Leza, J. C. (2002) The increase in TNF-alpha levels is implicated in NF-kappaB
activation and inducible nitric oxide synthase expression in brain cortex after
immobilization stress. Neuropsychopharmacology, 26, 155-163.
Magistretti, P. J. (2006) Neuron-glia metabolic coupling and plasticity. J Exp Biol, 209, 2304-
2311.

Chapter I __________________________________________________________________________
50
Mailly, F., Marin, P., Israel, M., Glowinski, J. and Premont, J. (1999) Increase in external
glutamate and NMDA receptor activation contribute to H2O2-induced neuronal apoptosis.
J Neurochem, 73, 1181-1188.
Makar, T. K., Nedergaard, M., Preuss, A., Gelbard, A. S., Perumal, A. S. and Cooper, A. J.
(1994) Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of
glutathione metabolism in cultures of chick astrocytes and neurons: evidence that
astrocytes play an important role in antioxidative processes in the brain. J Neurochem, 62, 45-53.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P.,
Galeotti, T. and Pani, G. (2008) Bilirubin as an endogenous modulator of neurotrophin
redox signaling. J Neurosci Res, 86, 2235-2249.
Mark, R. J., Lovell, M. A., Markesbery, W. R., Uchida, K. and Mattson, M. P. (1997) A role for
4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion
homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem, 68, 255-
264.
Marques, C. A., Keil, U., Bonert, A., Steiner, B., Haass, C., Muller, W. E. and Eckert, A.
(2003) Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedish
amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J
Biol Chem, 278, 28294-28302.
Marx, C. E., Jarskog, L. F., Lauder, J. M., Lieberman, J. A. and Gilmore, J. H. (2001)
Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for
schizophrenia. Biol Psychiatry, 50, 743-749.
Matapurkar, A. and Lazebnik, Y. (2006) Requirement of cytochrome c for apoptosis in human
cells. Cell Death Differ, 13, 2062-2067.
Merrill, J. E., Murphy, S. P., Mitrovic, B., Mackenzie-Graham, A., Dopp, J. C., Ding, M.,
Griscavage, J., Ignarro, L. J. and Lowenstein, C. J. (1997) Inducible nitric oxide synthase
and nitric oxide production by oligodendrocytes. J Neurosci Res, 48, 372-384.
Mielke, K. and Herdegen, T. (2000) JNK and p38 stresskinases--degenerative effectors of
signal-transduction-cascades in the nervous system. Prog Neurobiol, 61, 45-60.
Mitani, A., Watanabe, M. and Kataoka, K. (1998) Functional change of NMDA receptors
related to enhancement of susceptibility to neurotoxicity in the developing pontine
nucleus. J Neurosci, 18, 7941-7952.
Mizui, T., Kinouchi, H. and Chan, P. H. (1992) Depletion of brain glutathione by buthionine
sulfoximine enhances cerebral ischemic injury in rats. Am J Physiol, 262, H313-317.
Moncada, S. and Bolaños, J. P. (2006) Nitric oxide, cell bioenergetics and
neurodegeneration. J Neurochem, 97, 1676-1689.

General Introduction _________________________________________________________________________
51
Navarro, A. and Boveris, A. (2007) The mitochondrial energy transduction system and the
aging process. Am J Physiol Cell Physiol, 292, C670-686.
Nehru, B. and Kanwar, S. S. (2004) N-acetylcysteine exposure on lead-induced lipid
peroxidative damage and oxidative defence system in brain regions of rats. Biol Trace
Elem Res, 101, 257-264.
Nelson, D. L. and Cox, M. M. (2005) Lehninger Principles of Biochemistry, 4th Ed. Worth
Publishers, New York.
Neumar, R. W. (2000) Molecular mechanisms of ischemic neuronal injury. Ann Emerg Med,
36, 483-506.
Ngai, K. C. and Yeung, C. Y. (1999) Additive effect of tumor necrosis factor-alpha and
endotoxin on bilirubin cytotoxicity. Pediatr Res, 45, 526-530.
Nicholls, D. G. and Ferguson, S. J. (2002) Bioenergetics, 3rd Ed. Academic Press, New York.
Nohl, H., Gille, L. and Staniek, K. (2005) Intracellular generation of reactive oxygen species
by mitochondria. Biochem Pharmacol, 69, 719-723.
O'Neill, L. A. and Kaltschmidt, C. (1997) NF-kappa B: a crucial transcription factor for glial
and neuronal cell function. Trends Neurosci, 20, 252-258.
O'Shea, T. M. (2002) Cerebral palsy in very preterm infants: new epidemiological insights.
Ment Retard Dev Disabil Res Rev, 8, 135-145.
Ocal, K., Avlan, D., Cinel, I., Unlu, A., Ozturk, C., Yaylak, F., Dirlik, M., Camdeviren, H. and
Aydin, S. (2004) The effect of N-acetylcysteine on oxidative stress in intestine and
bacterial translocation after thermal injury. Burns, 30, 778-784.
Oh, W., Tyson, J. E., Fanaroff, A. A. et al. (2003) Association between peak serum bilirubin
and neurodevelopmental outcomes in extremely low birth weight infants. Pediatrics, 112, 773-779.
Ostrow, J. D., Mukerjee, P. and Tiribelli, C. (1994) Structure and binding of unconjugated
bilirubin: relevance for physiological and pathophysiological function. J Lipid Res, 35, 1715-1737.
Ostrow, J. D., Pascolo, L., Brites, D. and Tiribelli, C. (2004) Molecular basis of bilirubin-
induced neurotoxicity. Trends Mol Med, 10, 65-70.
Packer, L. and Cadenas, E. (2007) Oxidants and antioxidants revisited. New concepts of
oxidative stress. Free Radic Res, 41, 951-952.
Panegyres, P. K. and Hughes, J. (1998) The neuroprotective effects of the recombinant
interleukin-1 receptor antagonist rhIL-1ra after excitotoxic stimulation with kainic acid and
its relationship to the amyloid precursor protein gene. J Neurol Sci, 154, 123-132.

Chapter I __________________________________________________________________________
52
Papadopoulos, M. C., Koumenis, I. L., Dugan, L. L. and Giffard, R. G. (1997) Vulnerability to
glucose deprivation injury correlates with glutathione levels in astrocytes. Brain Res, 748, 151-156.
Papadopoulos, M. C., Koumenis, I. L., Yuan, T. Y. and Giffard, R. G. (1998) Increasing
vulnerability of astrocytes to oxidative injury with age despite constant antioxidant
defences. Neuroscience, 82, 915-925.
Park, W. S., Chang, Y. S., Chung, S. H., Seo, D. W., Hong, S. H. and Lee, M. (2001) Effect
of hypothermia on bilirubin-induced alterations in brain cell membrane function and energy
metabolism in newborn piglets. Brain Res, 922, 276-281.
Peeters-Scholte, C., Koster, J., Veldhuis, W. et al. (2002) Neuroprotection by selective nitric
oxide synthase inhibition at 24 hours after perinatal hypoxia-ischemia. Stroke, 33, 2304-
2310.
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R. and
Magistretti, P. J. (2007) Activity-dependent regulation of energy metabolism by astrocytes:
an update. Glia, 55, 1251-1262.
Pellerin, L. and Magistretti, P. J. (1994) Glutamate uptake into astrocytes stimulates aerobic
glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad
Sci U S A, 91, 10625-10629.
Perlman, J. M., Rogers, B. B. and Burns, D. (1997) Kernicteric findings at autopsy in two sick
near term infants. Pediatrics, 99, 612-615.
Petty, M. A. and Lo, E. H. (2002) Junctional complexes of the blood-brain barrier:
permeability changes in neuroinflammation. Prog Neurobiol, 68, 311-323.
Poli, G., Cadenas, E. and Packer, L. (2000) Free Radicals in Brain Pathophysiology. Marcel
Dekker, Inc., New York.
Porras, O. H., Loaiza, A. and Barros, L. F. (2004) Glutamate mediates acute glucose
transport inhibition in hippocampal neurons. J Neurosci, 24, 9669-9673.
Porter, M. L. and Dennis, B. L. (2002) Hyperbilirubinemia in the term newborn. Am Fam
Physician, 65, 599-606.
Prasanthi, R. P., Devi, C. B., Basha, D. C., Reddy, N. S. and Reddy, G. R. (2010) Calcium
and zinc supplementation protects lead (Pb)-induced perturbations in antioxidant enzymes
and lipid peroxidation in developing mouse brain. Int J Dev Neurosci, 28, 161-167.
Puka-Sundvall, M., Wallin, C., Gilland, E., Hallin, U., Wang, X., Sandberg, M., Karlsson, J.,
Blomgren, K. and Hagberg, H. (2000) Impairment of mitochondrial respiration after
cerebral hypoxia-ischemia in immature rats: relationship to activation of caspase-3 and
neuronal injury. Brain Res Dev Brain Res, 125, 43-50.

General Introduction _________________________________________________________________________
53
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A., McNamara, J. O. and
Williams, S. M. (2004) Studying the Nervous Systems of Humans and Other Animals. In:
Neuroscience, 3rd Ed. Sinauer Associates Inc., Sunderland.
Pyne-Geithman, G. J., Morgan, C. J., Wagner, K., Dulaney, E. M., Carrozzella, J., Kanter, D.
S., Zuccarello, M. and Clark, J. F. (2005) Bilirubin production and oxidation in CSF of
patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow
Metab, 25, 1070-1077.
Quagliarello, V. J., Wispelwey, B., Long, W. J., Jr. and Scheld, W. M. (1991) Recombinant
human interleukin-1 induces meningitis and blood-brain barrier injury in the rat.
Characterization and comparison with tumor necrosis factor. J Clin Invest, 87, 1360-1366.
Raivich, G., Bohatschek, M., Kloss, C. U., Werner, A., Jones, L. L. and Kreutzberg, G. W.
(1999) Neuroglial activation repertoire in the injured brain: graded response, molecular
mechanisms and cues to physiological function. Brain Res Brain Res Rev, 30, 77-105.
Ravagnan, L., Roumier, T. and Kroemer, G. (2002) Mitochondria, the killer organelles and
their weapons. J Cell Physiol, 192, 131-137.
Rice, M. E. (2000) Ascorbate regulation and its neuroprotective role in the brain. Trends
Neurosci, 23, 209-216.
Rodrigues, C. M. and Steer, C. J. (2001) The therapeutic effects of ursodeoxycholic acid as
an anti-apoptotic agent. Expert Opin Investig Drugs, 10, 1243-1253.
Rodrigues, C. M. P., Solá, S. and Brites, D. (2002a) Bilirubin induces apoptosis via the
mitochondrial pathway in developing rat brain neurons. Hepatology, 35, 1186-1195.
Rodrigues, C. M. P., Solá, S., Brito, M. A., Brites, D. and Moura, J. J. (2002b) Bilirubin
directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat
mitochondria. J Hepatol, 36, 335-341.
Rodrigues, C. M. P., Solá, S., Silva, R. F. M. and Brites, D. (2000) Bilirubin and amyloid-β
peptide induce cytochrome c release through mitochondrial membrane permeabilization.
Mol Med, 6, 936-946.
Rothwell, N. J. and Luheshi, G. N. (2000) Interleukin 1 in the brain: biology, pathology and
therapeutic target. Trends Neurosci, 23, 618-625.
Roux, P. P. and Blenis, J. (2004) ERK and p38 MAPK-activated protein kinases: a family of
protein kinases with diverse biological functions. Microbiol Mol Biol Rev, 68, 320-344.
Sampath, V., Bowen, J. and Gibson, F. (2005) Risk factors for adverse neurodevelopment in
extremely low birth weight infants with normal neonatal cranial ultrasound. J Perinatol, 25, 210-215.

Chapter I __________________________________________________________________________
54
Sánchez-Carbente, M. R., Castro-Obregon, S., Covarrubias, L. and Narvaez, V. (2005)
Motoneuronal death during spinal cord development is mediated by oxidative stress. Cell
Death Differ, 12, 279-291.
Santos, M. S., Moreno, A. J. and Carvalho, A. P. (1996) Relationships between ATP
depletion, membrane potential, and the release of neurotransmitters in rat nerve
terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and
ischemia. Stroke, 27, 941-950.
Schlingensiepen, K. H., Schlingensiepen, R., Kunst, M., Klinger, I., Gerdes, W., Seifert, W.
and Brysch, W. (1993) Opposite functions of jun-B and c-jun in growth regulation and
neuronal differentiation. Dev Genet, 14, 305-312.
Schoemaker, M. H., Conde de la Rosa, L., Buist-Homan, M., Vrenken, T. E., Havinga, R.,
Poelstra, K., Haisma, H. J., Jansen, P. L. and Moshage, H. (2004) Tauroursodeoxycholic
acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival
pathways. Hepatology, 39, 1563-1573.
Sener, G., Toklu, H., Kapucu, C., Ercan, F., Erkanli, G., Kacmaz, A., Tilki, M. and Yegen, B.
C. (2005) Melatonin protects against oxidative organ injury in a rat model of sepsis. Surg
Today, 35, 52-59.
Shah, S. A., Volkov, Y., Arfin, Q., Abdel-Latif, M. M. and Kelleher, D. (2006) Ursodeoxycholic
acid inhibits interleukin 1 beta [corrected] and deoxycholic acid-induced activation of NF-
kappaB and AP-1 in human colon cancer cells. Int J Cancer, 118, 532-539.
Shapiro, S. M. (2005) Definition of the clinical spectrum of kernicterus and bilirubin-induced
neurologic dysfunction (BIND). J Perinatol, 25, 54-59.
Sharpe, M. A. and Cooper, C. E. (1998) Reactions of nitric oxide with mitochondrial
cytochrome c: a novel mechanism for the formation of nitroxyl anion and peroxynitrite.
Biochem J, 332 ( Pt 1), 9-19.
Sheng, W. S., Hu, S., Ni, H. T., Rowen, T. N., Lokensgard, J. R. and Peterson, P. K. (2005)
TNF-alpha-induced chemokine production and apoptosis in human neural precursor cells.
J Leukoc Biol, 78, 1233-1241.
Sheu, K. F. and Blass, J. P. (1999) The alpha-ketoglutarate dehydrogenase complex. Ann N
Y Acad Sci, 893, 61-78.
Sies, H. (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol, 82, 291-295.
Silva, R. F. M., Rodrigues, C. M. P. and Brites, D. (2002) Rat cultured neuronal and glial cells
respond differently to toxicity of unconjugated bilirubin. Pediatr Res, 51, 535-541.
Silva, S. L., Vaz, A. R., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F.
M. and Brites, D. (2010) Features of bilirubin-induced reactive microglia: From
phagocytosis to inflammation. Neurobiol Dis, 40, 663-675.

General Introduction _________________________________________________________________________
55
Solá, S., Brito, M. A., Brites, D., Moura, J. J. and Rodrigues, C. M. (2002) Membrane
structural changes support the involvement of mitochondria in the bile salt-induced
apoptosis of rat hepatocytes. Clin Sci (Lond), 103, 475-485.
Solá, S., Ma, X., Castro, R. E., Kren, B. T., Steer, C. J. and Rodrigues, C. M. P. (2003)
Ursodeoxycholic acid modulates E2F-1 and p53 expression through a caspase-
independent mechanism in transforming growth factor beta1-induced apoptosis of rat
hepatocytes. J Biol Chem, 278, 48831-48838.
Soorani-Lunsing, I., Woltil, H. A. and Hadders-Algra, M. (2001) Are moderate degrees of
hyperbilirubinemia in healthy term neonates really safe for the brain? Pediatr Res, 50, 701-705.
Stevenson, D. K., Dennery, P. A. and Hintz, S. R. (2001) Understanding newborn jaundice. J
Perinatol, 21 Suppl 1, S21-24; discussion S35-29.
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N. and Ames, B. N. (1987) Bilirubin
is an antioxidant of possible physiological importance. Science, 235, 1043-1046.
Streit, W. J. (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia,
40, 133-139.
Stuehr, D. J. and Marletta, M. A. (1985) Mammalian nitrate biosynthesis: mouse
macrophages produce nitrite and nitrate in response to Escherichia coli
lipopolysaccharide. Proc Natl Acad Sci U S A, 82, 7738-7742.
Suárez, I., Bodega, G. and Fernández, B. (2002) Glutamine synthetase in brain: effect of
ammonia. Neurochem Int, 41, 123-142.
Suliman, H. B., Welty-Wolf, K. E., Carraway, M., Tatro, L. and Piantadosi, C. A. (2004)
Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis.
Cardiovasc Res, 64, 279-288.
Szeto, H. H. (2006) Mitochondria-targeted peptide antioxidants: novel neuroprotective
agents. AAPS J, 8, E521-531.
Tammariello, S. P., Quinn, M. T. and Estus, S. (2000) NADPH oxidase contributes directly to
oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J
Neurosci, 20, RC53.
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A. and Gaestel, M. (2008)
MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing
neuroinflammation--relevance in a mouse model of Parkinson's disease. J Neurochem,
105, 2039-2052.
Tibbles, L. A. and Woodgett, J. R. (1999) The stress-activated protein kinase pathways. Cell
Mol Life Sci, 55, 1230-1254.

Chapter I __________________________________________________________________________
56
Tsai, C. E., Daood, M. J., Lane, R. H., Hansen, T. W., Gruetzmacher, E. M. and Watchko, J.
F. (2002) P-glycoprotein expression in mouse brain increases with maturation. Biol
Neonate, 81, 58-64.
Tsuru-Aoyagi, K., Potts, M. B., Trivedi, A. et al. (2009) Glutathione peroxidase activity
modulates recovery in the injured immature brain. Ann Neurol, 65, 540-549.
Vannucci, S. J. and Hagberg, H. (2004) Hypoxia-ischemia in the immature brain. J Exp Biol,
207, 3149-3154.
Vaughn, A. E. and Deshmukh, M. (2008) Glucose metabolism inhibits apoptosis in neurons
and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol, 10, 1477-1483.
Vezzani, A., Conti, M., De Luigi, A., Ravizza, T., Moneta, D., Marchesi, F. and De Simoni, M.
G. (1999) Interleukin-1beta immunoreactivity and microglia are enhanced in the rat
hippocampus by focal kainate application: functional evidence for enhancement of
electrographic seizures. J Neurosci, 19, 5054-5065.
Volbracht, C., Chua, B. T., Ng, C. P., Bahr, B. A., Hong, W. and Li, P. (2005) The critical role
of calpain versus caspase activation in excitotoxic injury induced by nitric oxide. J
Neurochem, 93, 1280-1292.
Volpe, J. J. (1997) Brain injury in the premature infant--from pathogenesis to prevention.
Brain Dev, 19, 519-534.
Walczak, H. and Krammer, P. H. (2000) The CD95 (APO-1/Fas) and the TRAIL (APO-2L)
apoptosis systems. Exp Cell Res, 256, 58-66.
Wan, F. J., Tung, C. S., Shiah, I. S. and Lin, H. C. (2006) Effects of alpha-phenyl-N-tert-butyl
nitrone and N-acetylcysteine on hydroxyl radical formation and dopamine depletion in the
rat striatum produced by d-amphetamine. Eur Neuropsychopharmacol, 16, 147-153.
Watchko, J. F. (2006) Hyperbilirubinemia and bilirubin toxicity in the late preterm infant. Clin
Perinatol, 33, 839-852; abstract ix.
Watchko, J. F., Daood, M. J. and Hansen, T. W. (1998) Brain bilirubin content is increased in
P-glycoprotein-deficient transgenic null mutant mice. Pediatr Res, 44, 763-766.
Wendel, M. and Heller, A. R. (2010) Mitochondrial function and dysfunction in sepsis. Wien
Med Wochenschr, 160, 118-123.
Wender, R., Brown, A. M., Fern, R., Swanson, R. A., Farrell, K. and Ransom, B. R. (2000)
Astrocytic glycogen influences axon function and survival during glucose deprivation in
central white matter. J Neurosci, 20, 6804-6810.
Wennberg, R. P., Johansson, B. B., Folbergrova, J. and Siesjo, B. K. (1991) Bilirubin-
induced changes in brain energy metabolism after osmotic opening of the blood-brain
barrier. Pediatr Res, 30, 473-478.

General Introduction _________________________________________________________________________
57
Wilde, G. J., Pringle, A. K., Sundstrom, L. E., Mann, D. A. and Iannotti, F. (2000) Attenuation
and augmentation of ischaemia-related neuronal death by tumour necrosis factor-alpha in
vitro. Eur J Neurosci, 12, 3863-3870.
Wrona, M. Z. and Dryhurst, G. (1998) Oxidation of serotonin by superoxide radical:
implications to neurodegenerative brain disorders. Chem Res Toxicol, 11, 639-650.
Xu, L., Sapolsky, R. M. and Giffard, R. G. (2001) Differential sensitivity of murine astrocytes
and neurons from different brain regions to injury. Exp Neurol, 169, 416-424.
Yamada, N., Sawasaki, Y. and Nakajima, H. (1977) Impairment of DNA synthesis in Gunn rat
cerebellum. Brain Res, 126, 295-307.
Yan, L., Ge, H., Li, H. et al. (2004) Gender-specific proteomic alterations in glycolytic and
mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol, 37, 921-929.
Yang, G. Y., Liu, X. H., Kadoya, C., Zhao, Y. J., Mao, Y., Davidson, B. L. and Betz, A. L.
(1998) Attenuation of ischemic inflammatory response in mouse brain using an adenoviral
vector to induce overexpression of interleukin-1 receptor antagonist. J Cereb Blood Flow
Metab, 18, 840-847.
Yang, L., Lindholm, K., Konishi, Y., Li, R. and Shen, Y. (2002) Target depletion of distinct
tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival
through different signal transduction pathways. J Neurosci, 22, 3025-3032.
Yarian, C. S., Toroser, D. and Sohal, R. S. (2006) Aconitase is the main functional target of
aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev, 127, 79-
84.
Yi, J. H. and Hazell, A. S. (2006) Excitotoxic mechanisms and the role of astrocytic glutamate
transporters in traumatic brain injury. Neurochem Int, 48, 394-403.
Yoneyama, M., Kawada, K., Gotoh, Y., Shiba, T. and Ogita, K. (2010) Endogenous reactive
oxygen species are essential for proliferation of neural stem/progenitor cells. Neurochem
Int, 56, 740-746.
Zachwieja, J., Zaniew, M., Bobkowski, W., Stefaniak, E., Warzywoda, A., Ostalska-Nowicka,
D., Dobrowolska-Zachwieja, A., Lewandowska-Stachowiak, M. and Siwinska, A. (2005)
Beneficial in vitro effect of N-acetyl-cysteine on oxidative stress and apoptosis. Pediatr
Nephrol, 20, 725-731.
Zhu, C., Wang, X., Qiu, L., Peeters-Scholte, C., Hagberg, H. and Blomgren, K. (2004)
Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor
in neonatal rat cerebral hypoxia-ischaemia. J Neurochem, 90, 462-471.


Chapter II
II. Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in immature cortical neurons. Assessment of
the protective effects of glycoursodeoxycholic acid
Ana Rita Vaz1, Maria Delgado-Esteban2, Maria Alexandra Brito1, Juan P. Bolaños3, Dora Brites1, Angeles Almeida2,3
1Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL),
Faculdade de Farmácia, University of Lisbon, Av. Professor Gama Pinto, Lisbon
1649-003, Portugal.
2Unidad de Investigación, Hospital Universitario de Salamanca, Instituto de Estudios
de Ciencias de la Salud de Castilla y León, Salamanca 37007, Spain.
3Departmento de Bioquímica y Biologia Molecular, University of Salamanca, Instituto
de Neurociencias de Castilla y León, Salamanca 37007, Spain.
Journal of Neurochemistry (2010) 112, 56–65.

Acknowledgements The skilful assistance of Mrs. Monica Resch is acknowledged. We are grateful to Dr.
Margarida Silva for her advice concerning ATP measurements. Supported by RENEVAS,
Fondo de Investigación Sanitaria (FIS06/0794) and Junta de Castilla y León (to A.A),
SAF2007-61492, CONSOLIDER RosasNet CSD2007-00020, SA066A07 and Red
Terapia Celular-ISCIII (to J.P.B.), grants POCI/SAU/MMO/55955/2004, PTDC/SAU-
NEU/64385/2006, POCI 2010 and FEDER (to D.B.) and BD/30292/2006 (to A.R.V.) from
Fundação para a Ciência e a Tecnologia, Lisbon, Portugal

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
61
Abstract
High levels of unconjugated bilirubin (UCB) may initiate encephalopathy in neonatal life,
mainly in premature infants. The molecular mechanisms of this bilirubin-induced neurologic
dysfunction (BIND) are not yet clarified and no neuroprotective strategy is currently
worldwide accepted. Here, we show that UCB, at conditions mimicking those of
hyperbilirubinemic newborns (50 µM UCB in the presence of 100 µM human serum albumin),
rapidly (within 1 h) inhibited cytochrome c oxidase activity and ascorbate-driven oxygen
consumption in 3 days in vitro rat cortical neurons. This was accompanied by a bioenergetic
and oxidative crisis, and apoptotic cell death, as judged by the collapse of the inner
mitochondrial membrane potential, increased glycolytic activity, superoxide anion radical
production and ATP release, as well as disruption of glutathione redox status. Furthermore,
the antioxidant compound glycoursodeoxycholic acid, fully abrogated UCB-induced
cytochrome c oxidase inhibition and significantly prevented oxidative stress, metabolic
alterations and cell demise. These results suggest that the neurotoxicity associated with
neonatal bilirubin-induced encephalopathy occur through a deregulation of energy
metabolism, and supports the notion that glycoursodeoxycholic acid may be useful in the
treatment of BIND.
Keywords: bilirubin neurotoxicity, glycolysis, glycoursodeoxycholic acid, mitochondrial
dysfunction, oxidative stress, respiratory chain.

Chapter II __________________________________________________________________________
62
1. Introduction As a consequence of the short half-life of fetal erythrocytes, and of the limited ability of
the neonate to conjugate and excrete bilirubin, newborn infants often show increased levels
of serum unconjugated bilirubin (UCB). This condition, known as the physiologic jaundice, is
usually resolved by the end of the first week of life without treatment (Ostrow et al., 2003,
Reiser, 2004). However, severe hyperbilirubinemia in neonates with prematurity and/or
systemic illnesses such as hemolytic disease, acidosis, and hypoxemia enhances their risk
for bilirubin-induced neurologic dysfunction (BIND) (McDonald et al., 1998, Ostrow et al.,
2004, Shapiro, 2005). In fact, in vitro experiments revealed that immature neurons have an
increased susceptibility to UCB (Falcão et al., 2006). Although the molecular mechanisms of
BIND remain to be fully clarified, it was shown to involve immunostimulation, accumulation of
extracellular glutamate, oxidative stress, apoptosis and loss of cell viability (Brites et al.,
2009, Brito et al., 2008b). This excitotoxic-like situation prompted us to hypothesize whether
UCB would exert the death of immature nerve cells through an impairment of energy
metabolism.
Many previous studies have examined the effects of UCB on cerebral energy status but
the results have been equivocal. In fact, initial reports of an inhibition of respiration and
uncoupling of oxidative phosphorylation, observed in brain homogenates or isolated
mitochondria, pointed to mitochondrial dysfunction as an important element of UCB toxicity
(Mustafa et al., 1969, Ernster and Zetterström, 1956, Day, 1954). The energy depletion was
later on corroborated by the reduced rates of glycolysis and decreased ATP levels observed
in Gunn rats and in newborn piglets (Katoh-Semba, 1976, McCandless and Abel, 1980,
Hoffman et al., 1996, Park et al., 2001). However, discrepant findings were reported in other
studies that failed to document significant changes in brain glucose metabolism or oxidative
phosphorylation (Diamond and Schmid, 1967, Brann et al., 1987), whereas others
demonstrated that hyperbilirubinemia only disturbs brain energy metabolism in the presence
of additional factors that disrupt the blood brain barrier, such as hypoxia or hyperosmolarity
(Ives et al., 1988, Ives et al., 1989, Wennberg et al., 1991). Thus, further studies are needed
to clarify the effects of UCB on brain energy and glucose metabolism in nerve cells,
particularly in poor differentiated neurons. This would provide a valuable contribute to the
current understanding of the neuropathological effects of UCB, as neuronal energy
metabolism is determinant for processes as neurotransmitter release, neurite outgrowth and
cell survival (Mattson and Liu, 2002), which are impaired by UCB. Moreover, energy
hypometabolism is one of the most consistent and earliest abnormalities seen in mild
cognitive impairment (Atamna and Frey, 2007), which is particularly relevant in premature
jaundiced infants.

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
63
Recent data indicate that ursodeoxycholic acid and its glycine-conjugated species are
able to prevent UCB-induced apoptosis and loss of cell viability, oxidative stress and
immunostimulation (Brito et al., 2008a, Fernandes et al., 2007) and induces a rapid and
sustained decrease in plasma UCB concentrations in Gunn rats, the well-established animal
model for severe hyperbilirubinemia (Cuperus et al., 2009). Thus, we decided to test the
neuroprotective effects of glycoursodeoxycholic acid (GUDCA) in our model of immature
neurons.
In this study we show that UCB at a UCB to HSA molar ratio that can be found in the
plasma of moderately jaundiced neonates (Brito et al., 2006), rapidly and selectively inhibits
the activity of cytochrome c oxidase, the terminal component of the mitochondrial respiratory
chain, in immature neurons; this led to an impairment in oxygen consumption, inner-
mitochondrial membrane potential (ΔѰm) collapse and apoptosis. These phenomena were
associated with an increase in cellular oxidized glutathione, production of superoxide anion
radical (O2.-) and a decrease in NADPH. Pretreatment of neurons with GUDCA prior to
exposure to UCB prevented inhibition of cytrochrome c oxidase activity together with
preservation of glutathione and NADPH status. These data indicate that cytochrome c
oxidase inhibition may be involved in the neurotoxicity associated with BIND and strongly
indicates the possible therapeutic potential of GUDCA in the treatment of this disorder.
2. Materials and Methods
2.1. Chemicals Neurobasal medium, B-27 supplement, Hanks’ balanced salt solution (HBSS-1), Hanks’
balanced salt solution without Ca2+ and Mg2+ (HBSS-2), gentamicin (50 mg/mL),
tetramethylrhodamine (TMRE), MitoSOX Red and trypsin (0.025%) were acquired from
Invitrogen (Carlsbad, CA). Human serum albumin (HSA) (fraction V, fatty acid free), carbonyl
cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), ubiquinone-5 (coenzyme Q1, CoQ1),
2,2,4-trimethyl-1,3-pentanediol (TMPD), sulfosalicyclic acid and 2-vinylpyridine were
purchased from Sigma Chemical Co (St Louis, MO). UCB was also from Sigma and purified
as previously described (McDonagh, 1979). Cytochrome c was obtained from Roche
Diagnostics (Heidelberg, Germany), and it was reduced with sodium ascorbate (Sigma) just
before use and passed through Sephadex G-25 M (PD-10 columns, Amersham Pharmacia
Biotech, Uppsala, Sweden) to remove excess ascorbate. GUDCA, as well as Caspases 3
and 9 substrates, Ac-DEVD-pNA and Ac-LEHD-pNA, respectively, were purchased from

Chapter II __________________________________________________________________________
64
Calbiochem (Darmstadt, Germany). Other substrates, inhibitors, enzymes, and coenzymes
were purchased from Sigma, Roche Diagnostics or Merck (Darmstadt, Germany).
2.2. Neurons in primary culture
Animal care followed the European Legislation on Protection of Animals Used for
Experimental and Scientific Purposes (EU directive L0065, 22/07/2003). Neurons were
isolated from fetuses of 16-17-day pregnant Wistar rats, as described previously (Silva et al.,
2002). The fetuses were collected in HBSS-1, the brain cortex was mechanically fragmented,
and the fragments transferred to a 0.025% (w/v) trypsin in HBSS-2 solution and incubated for
15 min at 37ºC. After trypsinization, cells were washed twice in HBSS-2 containing 10% (v/v)
FBS, and resuspended in Neurobasal medium supplemented with 0.5 mM L-glutamine, 25
μM L-glutamic acid, 2% B-27 supplement, and 0.12 mg/mL gentamicin. Finally, cells were
seeded on poly-D-lysine coated tissue culture plates at a density of 2.5 x 105 cells/cm2 and
maintained at 37ºC in a humidified atmosphere of 5% CO2. In this work we used neurons at 3
days in vitro (DIV).
2.3. Treatment of neurons
Neurons were incubated in Neurobasal medium without (control) or with 50 μM UCB
(from a 10 mM stock solution) in the presence of 100 μM HSA (UCB/HSA molar ratio of 0.5)
for 1 h at 37ºC. Stock UCB solutions were extemporarily prepared in 0.1 M NaOH under the
dark and the pH adjusted to 7.4 using 0.1 M HCl. When appropriate, neurons were pre-
incubated with GUDCA (50 µM) 1 h prior to UCB addition.
2.4. Determination of the mitochondrial respiratory chain complex activities and citrate
synthase Neurons plated on 60 cm2 Petri dishes were washed with ice-cold PBS and surviving
cells were collected by trypsinization, centrifuged and resuspended in 300 μL of 0.1 M
potassium phosphate buffer (pH 7.0). Cell suspensions (containing about 7-8 mg of
protein/mL) were frozen and thawed three times to ensure cell lysis. Enzyme activities were
determined in the cell lysates using an Uvikon XL spectrophotometer (Secomam, Domont,
France). NADH-CoQ1 reductase (complex I; EC 1.6.99.3) activity was measured as
described by Ragan et al. (1987). The activity of succinate-cytochrome c reductase (complex
II-III; EC 1.8.3.1) was determined following the method of King (1967). Cytochome c oxidase
(complex IV, EC 1.9.3.1) activity was assessed as described by Wharton and Tzagoloff
(1967). Citrate synthase (EC 4.1.3.7) activity was assessed as referred by Shepherd and
Garland (1969). Protein concentrations were determined by the method of Lowry et al.

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
65
(1951). All enzyme activities were expressed as nanomoles per minute per milligram of
protein, except for cytochrome c oxidase, which was expressed as the first-order rate
constant (k) per minute per milligram of protein (Almeida and Bolaños, 2001).
2.5. Detection of superoxide anion radical (O2.-)
After incubation in 9.6 cm2 wells, neurons were incubated in PBS containing MitoSox-
Red (2 μM) for 30 min, washed with PBS and fluorescence assessed by flow cytometry,
using MitoSox-Red method (Invitrogen), as previously described (Mukhopadhyay et al.,
2007). For the determination of mitochondrial superoxide by flow cytometry, the
measurements were carried out using FACScalibur flow cytometer (15 mW argon ion laser
tuned at 488 nm; CellQuest software, Becton Dickinson Biosciences) and the data collected
at 585/42 nm (FL2) and 670LP (FL3) channel. In this study, the data were presented in the
FL2 channel. Antimycin A at 20 µM was used for 15 minutes as a mitochondrial superoxide
generator.
2.6. Determination of oxygen consumption
Oxygen consumption was determined with a Clark-type electrode (Rank Brothers,
Cambridge, UK). Briefly, after the incubation period, neurons plated on 60 cm2 Petri dishes
were collected by trypsinization, centrifuged, rinsed once with buffered Hank’s solution and
resuspended in 500 μL of Hank’s solution (without glucose). Cell suspensions were kept on
ice until used for oxygen consumption (within 1 h). The rates of oxygen consumption were
calculated from the slopes (monitored for at least 15 min per trace), and expressed as
nanomoles of oxygen consumed per minute per 106 cells (Almeida et al., 1998). TMPD was
used together with ascorbate to assure the reduced form of cytochrome c.
2.7. ∆Ѱm measurements
For fluorescence measurements, neurons incubated in 9.6 cm2 wells were stained as
previously described (Almeida et al., 1999) with minor modifications. Briefly, neurons were
incubated in Hanks’ solution containing 1 μg/mL of tetramethylrhodamine (TMRE) for 30 min
at 37ºC. Excess dye was removed by washing cells twice with buffered Hanks’ solution and
covered with 1 mL of Hanks’solution. For each Petri dish, four fluorescence
microphotographs were taken with an inverted microscope with a fluorescein filter (excitation
filter 480-490 nm; emission filter 510-530 nm) and the intensity of fluorescence was
quantified using an image analyser system (NIH Image Program). The representative
selected area was always the same for all experimental conditions studied. The fluorescence
intensity corresponding to control cells was arbitrarily assigned a value of 100%

Chapter II __________________________________________________________________________
66
fluorescence. The 0% ΔѰm value was obtained when cells were loaded with TMRE in the
presence 10 μM of carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) as
previously referred (Almeida and Bolaños, 2001). The monotonic decrease of fluorescence
with FCCP assures that we are measuring mitochondrial membrane potential and not the
plasma membrane potential where a different pattern of fluorescence would be observed
(Farkas et al., 1989).
2.8. Metabolite determinations
Neurons (about 3x107) cultured in 60 cm2 Petri dishes and treated as abovementioned
were rapidly washed with ice-cold PBS, scrapped off with 0.3 M HClO4 and neutralized with 2
M KHCO3 to pH 6.5. The perchlorate precipitate was removed by centrifugation, and
fructose-6-phosphate (F6P) and fructose-1,6-bisphosphate (F1,6P2) concentrations were
measured in the supernatants as previously described (Almeida et al. 2004); intracellular
lactate concentrations were determined in the same neutralized extracts as previously
mentioned (Gutmann and Wahlefeld, 1974).
For the assessment of fructose-2,6-bisphosphate (F2,6P2), cell extracts were lysed in
0.1 M sodium hydroxide and centrifuged (20,000g for 20 min) as previously described
(Almeida et al., 2004). Briefly, an aliquot of the homogenate was used for protein
determination and the remaining sample was heated at 80°C (5 min), centrifuged (20,000g
for 20 min) and the resulting supernatant used for the determination of F2,6P2 concentrations
(Kawaguchi et al., 2001, Van Schaftingen et al., 1982).
For intracellular ATP evaluation, neurons were cultured in 2 cm2 wells and treated as
above. At the end of the incubation period, cells were rapidly washed with ice-cold PBS,
scrapped off with 2 x 0.5 mL of 0.3 M HClO4, and neutralized with 0.5 mL of 2 M KHCO3 at
pH 6.5. The perchlorate precipitate was removed by centrifugation, and ATP was determined
in the supernatants by chemiluminescence using a commercially available kit following the
manufacturer’s instructions. The released ATP was considered to be that found in the culture
medium and the quantification was an adaptation of the method previously described by
Silva et al. (1997). Briefly, after incubation period, supernatants were collected, placed on ice
and exposed to 2 M HClO4. A solution of 2 M KHCO3 was used to restore pH at 6.5. The
perchlorate precipitate was removed by centrifugation, and ATP levels were measured
fluorimetrically in the protein-free supernatants.
For glutathione measurement, neurons incubated in 9.6 cm2 wells were washed with ice-
cold PBS and immediately collected by scrapping off with 0.5 mL of 1% (w/v) sulfosalicyclic
acid. Cell lysates were centrifuged at 13,000 g for 5 min at 4ºC, and the supernatants used
for glutathione determinations on the same day. Total glutathione content (GSx, i.e. the

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
67
amount of GSH plus two times the amount of GSSG) and oxidized glutathione (GSSG) were
measured and calculated as previously described (Dringen and Hamprecht, 1996, García-
Nogales et al., 1999). GSX and GSSG concentrations were expressed as nanomoles per
milligram of protein.
NADPH concentrations were measured accordingly with García-Nogales et al. (1999). In
brief, neurons incubated in 2 cm2 wells were washed with ice-cold PBS, and collected in 200
μL of 0.5 M KOH in 50% (v/v) ethanol. Cell lysates were neutralized (pH 7.8) with 200 μL of
0.5 M triethanolamine/0.5 M potassium phosphate and centrifuged at 13,000 g for 2 min at
4ºC. A 50-μL aliquot of the supernatant was immediately used for NADPH determination
(Wulff, 1985), with the exception that NADH was oxidized by incubation of the samples with
0.5 mU/μL lactate dehydrogenase and 1 mM pyruvate (Klingerberg, 1985).
2.9. Assessment of apoptotic cell death by flow citometry
Allophycocyanin (APC)-conjugated annexin-V and 7-amino-actinomycin D (7-AAD)
(Apoptosis Assay Kit; Becton Dickinson Biosciences, San Jose, CA, USA) were used to
quantitatively determine the percentage of apoptotic cells by flow cytometry. After incubation
in 2 cm2 wells, neurons were stained with annexin-V-APC and 7-AAD, following the
manufacturer’s instructions, and they were analysed on a FACScalibur flow cytometer (15
mW argon ion laser tuned at 488 nm; CellQuest software, Becton Dickinson Biosciences).
The annexin V-APC-stained cells that were 7-AAD negative were considered apoptotic
(Almeida et al., 2004).
2.10. Analysis of apoptotic cell death by 4'-6-diamidino-2-phenylindole (DAPI) nuclear
staining Neurons were fixed with 4% (v/v, in PBS) paraformaldehyde for 30 min at room
temperature, rinsed with PBS and incubated with DAPI (30 µM, Sigma). After 10 min, cells
were washed three times with PBS and their nuclei examined under a fluorescence
microscope by an author blinded to the test. A total of ~200 cells per condition in three
different cultures were quantified, and the results were expressed as the percentage of
condensed or fragmented nuclei.
2.11. Caspase-3 and -9 activity assays
Activities of caspase-3 and -9 were measured by a colorimetric method (Calbiochem,
Darmstadt, Germany). Cells were harvested, washed with ice-cold PBS and lysed for 30
minutes on ice in the lysis buffer [50 mM HEPES (pH 7.4); 100 mM NaCl; 0.1% (w/v)
CHAPS; 1 mM DTT; 0.1 mM EDTA]. The lysate was centrifuged at 10,000 g for 10 min at

Chapter II __________________________________________________________________________
68
4ºC and the supernatants were collected and stored at -80ºC. Protein concentrations were
determined as aforementioned. The activity of caspases 3 and 9 was determined in cell
lysates by enzymatic cleavage of chromophore p-nitroanilide (pNA) from the substrate Ac-
YVAD-pNA, according to manufacturer’s instructions. The proteolytic reaction was carried
out in protease assay buffer buffer [50 mM HEPES (pH 7.4); 100 mM NaCl; 0.1% (w/v)
CHAPS; 10 mM DTT; 0.1 mM EDTA; 10% (v/v) glicerol], containing 2 mM substrate Ac-
DEVD-pNA for caspase-3 and Ac-LEHD-pNA for caspase-9. Following incubation of the
reaction mixtures for 2 h at 37ºC, the formation of pNA was measured at λ= 405 nm with a
reference filter of 620 nm.
2.12. Statistical analysis
Measurements from individual cultures were performed in triplicate. The results are
expressed as the mean ± SEM values for the number of culture preparations indicated in the
legends. Statistical analysis of the results was performed by one-way analysis of variance
followed by the least significant difference multiple range test, and p<0.05 was accepted as
statistically significant in all cases.
3. Results
3.1. UCB selectively impairs cytochrome c oxidase activity in immature neurons, which is prevented by GUDCA
To investigate the possible role of UCB on mitochondrial function in immature nerve
cells, 3 DIV cortical neurons were incubated with UCB at conditions mimicking neonatal
jaundice (50 µM UCB + 100 µM HSA) that had previously shown to induce oxidative injury
and cell death in mature neurons (Brites et al., 2009, Brito et al., 2008b). Cells were collected
1 h after treatment for the analysis of the activities of the mitochondrial respiratory chain
complexes. Activity of NADH-CoQ1 (Fig. II.1A) was unaffected and that of succinate-
cytochrome c reductase (Fig. II.1B), although slightly reduced, did not significantly change
after neuronal exposure to UCB. In contrast, cytochrome c oxidase activity (Fig. II.1C) was
inhibited by UCB in approximately 50% (p<0.01). Finally, since the activity of citrate synthase
(Fig. II.1D) was also unchanged, this indicates that no differences in mitochondrial
enrichment in our study model account for the decreased complex activity. Since GUDCA
has shown to have neuroprotective effects through the prevention of mitochondrial swelling
we have tested the beneficial effects of this bile acid on the UCB-induced alterations on the

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
69
mitochondrial respiratory chain. GUDCA revealed to be able to completely reverse the
inhibition of the cytochrome c oxidase activity (Fig. II.1C).
Figure II.1 - Unconjugated bilirubin (UCB) selectively impairs cytochrome c oxidase activity in immature neurons and glycoursodeoxycholic acid (GUDCA) exerts a preventive effect. Neurons at 3 days in vitro were incubated for 1 h with UCB (50 µM) plus human serum albumin (100 µM). When indicated, neurons were pre-treated with GUDCA (50 µM) for 1 h. After incubation, neurons were used for enzyme activity determinations, as indicated in Methods. UCB did not alter NADH-CoQ1 reductase - Complex I (A) as well as succinate-cytochrome c reductase - Complex II-III (B), but inhibited cytochrome c oxidase activity that was prevented by GUDCA (C). Additionally, no changes were noticed in citrate synthase activity (D). **p<0.01 vs. control; ##p<0.01 vs. UCB.
3.2. UCB produces oxidative stress in immature neurons, which is prevented by GUDCA
Next we explored the oxidative status of immature neurons exposed to UCB. We
observed that UCB markedly induced the production of reactive oxygen species (ROS),
namely O2.- (Fig. II.2A) and oxidized glutathione (Fig. II.2B), as well as decreased NADPH
0
50
100
150
200C
itrat
e sy
ntha
se a
ctiv
ity(n
mol
/min
/mg
prot
ein)
0.0
0.2
0.4
0.6
0.8
Com
plex
IV a
ctiv
ity(k
/min
/mg
prot
ein)
0
2
4
6
8
Com
plex
II a
ctiv
ity(n
mo/
min
/mg
prot
ein)
0
2
4
6
8C
ompl
ex I
activ
ity(n
mo/
min
/mg
prot
ein)
A B
**
##
C D

Chapter II __________________________________________________________________________
70
concentrations (Fig. II.2C). Moreover, pre-incubation of neurons with GUDCA efficiently
prevented all these oxidative events caused by UCB.
Figure II.2 - Unconjugated bilirubin (UCB) produces oxidative stress in immature neurons and glycoursodeoxycholic acid (GUDCA) exerts a preventive effect. Neurons at 3 days in vitro were treated as in Figure II.1. After incubation, neurons were used for metabolite assessments as indicated in Methods. UCB-induced oxidative stress through the increase in superoxide anion radical production, as indicated by a higher MitoSox fluoresence intensity (A), oxidized glutathione (B), as revealed by the increase in GSSG/GSx ratio, and the decrease in NADPH concentrations (C) that were prevented by GUDCA. **p<0.01 and *p<0.05 vs. control; #p<0.05 vs. UCB.
3.3. UCB impairs cellular oxygen consumption and collapses ΔѰm in immature
neurons and GUDCA exerts a preventive effect In view that the mitochondrial respiratory chain thresholds for oxygen consumption
(Davey et al., 1998), it could be speculated that the level of cytochrome c oxidase inhibition
caused by UCB might not be enough to impair the mitochondrial function. To elucidate this,
we first determined the rate of oxygen consumption in the dissociated neurons previously
incubated with UCB. As shown in Figure II.3A, UCB significantly reduced the rates of oxygen
consumption using glucose, succinate or ascorbate as substrates, effects that were
significantly prevented by GUDCA. The effects on O2 consumption are related to
mitochondria, since both succinate and ascorbate reproduced the same results as with
glucose. In addition, in all cases antimycin or potassium cyanide abolished O2 consumption
driven by succinate or ascorbate, respectively (data not shown). The inhibition of oxygen
consumption from ascorbate indicates that UCB-inhibition of cytochrome c oxidase, affects
cell respiration. To further test this possibility, we assessed the ΔѰm as an index of the
mitochondrial inner membrane integrity; as depicted in Figure II.3B, UCB caused the
collapse of ΔѰm, and GUDCA showed ability to restore mitochondrial integrity.
0
1
2
3
4
5
GS
SG
/GS
x x
100
0
50
100
150
NA
DP
H(p
mo/
mg
prot
ein)
0
10
20
Mito
Sox
Red
fluo
resc
ence
(arb
itrar
y un
its)
*#
A
*
#
B
**#
C
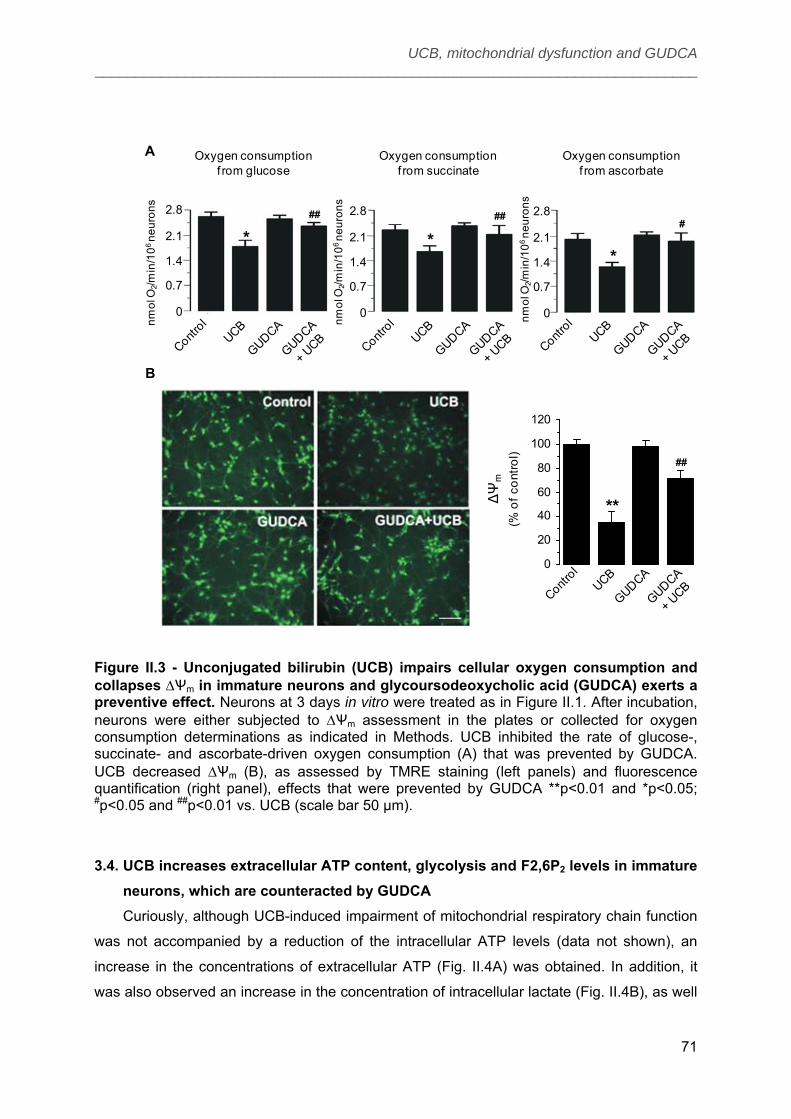
UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
71
Figure II.3 - Unconjugated bilirubin (UCB) impairs cellular oxygen consumption and collapses ΔѰm in immature neurons and glycoursodeoxycholic acid (GUDCA) exerts a preventive effect. Neurons at 3 days in vitro were treated as in Figure II.1. After incubation, neurons were either subjected to ΔѰm assessment in the plates or collected for oxygen consumption determinations as indicated in Methods. UCB inhibited the rate of glucose-, succinate- and ascorbate-driven oxygen consumption (A) that was prevented by GUDCA. UCB decreased ΔѰm (B), as assessed by TMRE staining (left panels) and fluorescence quantification (right panel), effects that were prevented by GUDCA **p<0.01 and *p<0.05; #p<0.05 and ##p<0.01 vs. UCB (scale bar 50 µm).
3.4. UCB increases extracellular ATP content, glycolysis and F2,6P2 levels in immature neurons, which are counteracted by GUDCA Curiously, although UCB-induced impairment of mitochondrial respiratory chain function
was not accompanied by a reduction of the intracellular ATP levels (data not shown), an
increase in the concentrations of extracellular ATP (Fig. II.4A) was obtained. In addition, it
was also observed an increase in the concentration of intracellular lactate (Fig. II.4B), as well
0
20
40
60
80
100
120
A
2.8
2.1
1.4
0.7
0
nmol
O2/m
in/1
06ne
uron
s
*##
Oxygen consumption f rom glucose
2.8
2.1
1.4
0.7
0
nmol
O2/m
in/1
06ne
uron
s
*## 2.8
2.1
1.4
0.7
0nmol
O2/m
in/1
06ne
uron
s
*
#
Oxygen consumption f rom succinate
Oxygen consumption f rom ascorbate
B
∆Ψ
m
(% o
f con
trol)
**
##

Chapter II __________________________________________________________________________
72
as in F1,6P2/F6P ratio (Fig. II.4C), suggesting an activation of glycolysis, which was further
supported by the concomitant elevated levels of F2,6P2 (Fig. II.4D). Noticeably, all these
effects were abolished by GUDCA.
Figure II.4 - Unconjugated bilirubin (UCB) increases extracellular ATP content, glycolysis and fructose-2,6-bisphosphate (F2,6P2) levels in immature neurons, and glycoursodeoxycholic acid (GUDCA) exerts a preventive effect. Neurons at 3 days in vitro were treated as in Figure II.1. After incubation, extracellular ATP was evaluated, and neurons were lysed for intracellular lactate, fructose-1,6-bisphosphate (F1,6P2), fructose-6-phosphate (F6P) and F2,6P2 measurements, as indicated in Methods. UCB triggered an increase in ATP release (A), intracellular lactate concentration (B), F1,6P2/F6P (C) and F2,6P2 concentration (D), which were prevented by GUDCA. **p<0.01 and *p<0.05 vs. control; ##p<0.01 and #p<0.05 vs. UCB.
3.5. UCB triggers apoptotic cell death in immature neurons, which is prevented by GUDCA Finally, we sought to investigate whether the mitochondrial impairment triggered by UCB
was associated with neurotoxicity. As shown in Figure II.5A, UCB enhanced the proportion of
annexin V+/7-AAD- neurons, as assessed by flow cytometry; it also triggered an increase in
the proportion of condensed or fragmented nuclei, as visualized with DAPI by fluorescence
0.00
0.05
0.10
0.15
0.0
0.5
1.0
1.5
2.0
F1,6
P2/
F6P
0
10
20
30
Ext
race
llula
r ATP
(nm
ol/m
g pr
otei
n)
0.0
1.0
2.0
3.0
4.0F2
,6P
2
(nm
ol/m
g pr
otei
n)
A B
**
##
C D
#
*
*
#
**
##
Intra
cellu
lar
Lact
ate
(µm
ol/m
g pr
otei
n)

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
73
microscopy (Fig. II.5B). Cell death by apoptosis was further corroborated by the increase in
the activation of caspase-3 (Fig. II.5C). Similar increase in the activation of caspase-9
indicates the involvement of mitochondria in this process. Such effects were again
completely counteracted by GUDCA. We can then speculate that the increased glycolytic
rate (Fig. II4) is a failed attempt to compensate the mitochondrial impairment. These results
support the notion that UCB causes nerve cell death by apoptosis, mainly in immature
neurons, and confirms that GUDCA efficiently protects cells against this type of neurotoxicity.
4. Discussion Here we show, for the first time, that brief exposure (1 h) of primary cortical immature
neurons to UCB in conditions that have relevance to the clinical manifestations of BIND (50
µM UCB + 100 µM HSA) inhibits mitochondrial respiratory chain, at the level of cytochrome c
oxidase complex. Mitochondrial dysfunction by UCB appears to involve .NO, accordingly with
previous studies (Brito et al., 2008a, Mancuso et al., 2008) reporting that neuronal oxidative
dysruption by UCB is abrogated by inhibition of neuronal NO synthase (nNOS). .NO is
capable of rapidly and reversibly inhibit the mitochondrial respiratory chain and may be
implicated in the cytotoxic effects in the CNS (Bolaños et al., 1994, Brown and Cooper, 1994,
Cleeter et al., 1994). Additionally, inhibition of the mitochondrial transport chain at the level of
complex IV can further produce O2.- from O2, a finding also observed in this study (Fig.
II.2A). Thus, inhibition of mitochondrial cytochrome c oxidase by .NO can lead to the
formation of both .NO and O2.- and thereby lead to the formation of ONOO- (Sharpe and
Cooper, 1998). Interestingly, we previously demonstrated that UCB induces protein oxidation
and lipid peroxidation, while diminishes the thiol antioxidant defences, events that were
correlated with the extent of cell death, and that GUDCA primarily acts as an antioxidant at
protecting neurons against UCB-induced oxidative stress (Brito et al., 2008a). The present
study extended our previous ones by showing that UCB decreases NADPH concentrations
(Fig. II.2C) in immature neurons, in adition to glutathione oxidation (Fig. II.2B) and O2.-
production, confirming oxidative stress by UCB.

Chapter II __________________________________________________________________________
74
Figure II.5 – Unconjugated bilirubin (UCB) triggers apoptotic death in immature neurons and glycoursodeoxycholic acid (GUDCA) exerts a preventive effect. Neurons at 3 days in vitro were treated as in Figure II.1. After incubation, neurons were subjected to assessment of apoptotic death by flow cytometry (annexin V+/7-AAD-), nuclear condensation or fragmentation in DAPI-stained cells, and caspase-3 and -9 activities, as indicated in Methods. UCB increased neuronal apoptosis as measured by the percentage of annexin V+/7-AAD- cells (A) (left panel shows a typical diagram; right panel represents the quantification) or fragmented or condensed nuclei (B) (left panel show a typical microphotograph of the DAPI-stained cells; right panel represents the quantification), which was prevented by GUDCA. Data was corroborated by the increase in the activation of caspase-3 and caspase-9 (C) pointing to the involvement of mitochondria. GUDCA was able to markedly prevent this effect. **p<0.01 and *p<0.05 vs. control; ##p<0.01 vs. UCB (scale bar 20 µm).
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Cas
pase
9 a
ctiv
ity (f
old
vs.c
ontro
l)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
Cas
pase
3 a
ctiv
ity (f
old
vs.c
ontro
l)
0
5
10
15
20
Con
dens
ed o
r fra
gmen
ted
nucl
ei(%
)
0
5
10
15
20
25
30
Apo
ptot
ic n
euro
ns(%
Ann
exin
+ /7A
AD
- )
A
B
*
##
C
**
##
##
**
##
**
Control
UCB

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
75
Moreover, pre-incubation of neurons with GUDCA efficiently prevented these oxidative
events, thus highlighting the antioxidant properties of the bile acid in this paradigm. In
addition, GUDCA abolished the inhibition of cytochrome c oxidase caused by UCB (Fig.
II.1C), indicating that the mitochondrial respiratory chain damage caused by UCB would be a
free radical-mediated process. To the best of our knowledge, this is the first evidence
reporting the ability of GUDCA to completely restore the activity of cytochrome c oxidase
when impaired.
Once a defective respiratory function also includes decreased oxygen consumption, we
explored the effects of UCB at such level, since contradictory results were previously found
by several authors using neither relevant physiological concentrations nor purified UCB
(Diamond and Schmid, 1967, Ernster and Zetterström, 1956, Mustafa et al., 1969). Since
glucose provides NAD-linked electrons through NADH-CoQ1 reductase, and succinate FAD-
linked ones through succinate dehydrogenase, the inhibition of cell respiration from these
substrates by UCB is compatible with, but does not demonstrate, inhibition at the terminal
complex, cytochrome c oxidase. However, the inhibition of oxygen consumption from
ascorbate, which directly supplies electrons to cytochrome c oxidase, confirms that the
inhibition at the level of this complex by UCB affects cell respiration. This was reinforced by
the collapse of ΔѰm produced by UCB indicating decreased mitochondrial membrane
potential and mitochondrial dysfunction, which was again prevented when neurons were pre-
treated with GUDCA before the exposure to UCB. Permeabilization of the mitochondrial
membrane by UCB and increased efflux of cytochrome c were previously observed in
isolated mitochondria from the brain and liver of adult male Wistar rats (Rodrigues et al.,
2002, Rodrigues et al., 2000). Such mitochondrial impairment by UCB was in the present
study accompanied by an increase in intracellular lactate concentrations and F1,6P2/F6P
ratio, suggesting an activation of glycolysis by stimulation of 6-phosphofructo-1-kinase
(Pfk1), i.e. a master regulator of this pathway (Uyeda, 1979); this notion was further
supported by the observed increased levels of F2,6P2, i.e. the positive effector of Pfk1 (Van
Schaftingen et al., 1982). Moreover, intracellular ATP content was unchanged, whereas
extracellular levels were increased. Altogether, these results suggest that UCB disrupts the
mitochondrial function in immature neurons leading to up-regulation of glycolysis, which
reflects the natural metabolic response of cells to cytochrome c oxidase deficiency (Almeida
et al., 2004, Bolaños et al., 1994) and may determine the unchanged intracellular ATP levels.
In fact, it is conceivable that the up-regulation of glycolysis in these still immature neuronal
cells may provide sufficient ATP as a self-protective attempt to support the bioenergetic
crisis, as previously demonstrated for astrocytes exposed to injurious stimuli (Bolaños et al.,
2004). In parallel, the release of ATP by neurons has been suggested to be determined by

Chapter II __________________________________________________________________________
76
the production of .NO and oxidative stress and to be associated with neuronal cell death by
apoptosis, in that ATP is critical to inducing several apoptotic events (Figueroa et al., 2006,
López et al., 2006). Noticeably, all these effects were prevented by GUDCA.
Another new finding on the mechanisms of UCB-induced oxidative stress is the
decrease of NADPH levels which may contribute to the altered glutathione redox status
(Dringen, 2000) observed in the presence of UCB. This result is consistent with the recently
reported notion that, in neurons, the activation of glycolysis leads to inhibition of the pentose-
phosphate pathway (PPP), causing glutathione oxidation (Herrero-Mendez et al., 2009).
Interestingly, through PPP, neurons maintain cytochrome c in a reduced status in order to
prevent its release and apoptotic death (Vaughn and Deshmukh, 2008). In good agreement
with this, GUDCA restored reduced glutathione levels in immature neurons, as it did with
differentiated cells (Brito et al., 2008a) and also re-established NADPH values reinforcing its
antioxidant capacity. An up-regulation of gamma-glutamyl cysteine synthetase, together with
the efficient scavenging of free radicals previously reported for the non conjugated form of
the bile acid (Lapenna et al., 2002, Rodriguez-Ortigosa et al., 2002, Serviddio et al., 2004)
shall contribute to the protective actions herewith found.
UCB induced mitochondrial dysfunction in 3 DIV neurons by selective inhibition of
cytochrome c oxidase activity, and decreased mitochondrial membrane potential, but
maintained intracellular ATP levels, ultimately leading to apoptotic cell death. The activation
of caspase-3, but mostly of caspase-9, points to the activation of the mitochondrial apoptotic
pathway in immature neurons after a short exposure to UCB in conditions mimicking an
acute neonatal jaundice. To the prevention of this neurotoxicity by GUDCA may account its
ability to re-establish the energy metabolism and redox status of the injured cell.
In conclusion, in this study we report evidence of UCB induced inhibition of cytochrome c
oxidase activity in immature rat neurons resulting in respiratory chain dysfunction, a
decrease in antioxidant defences and apoptosis, as schematically represented in Figure II.6.
The ability of GUDCA to ameliorate UCB induced mitochondrial respiratory chain dysfunction
and restore cellular antioxidant potential supports the efficacy of this compound as a
potential treatment for BIND.

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
77
Figure II.6 - Schematic representation of some important steps in neuronal injury by unconjugated bilirubin (UCB) and potential targets of glycoursodeoxycholic acid (GUDCA). UCB interaction with immature neurons causes the selective inhibition of complex IV (CIV) activity, impairs oxygen (O2) consumption, leads to a loss of mitochondrial membrane potential (ΔѰm) and superoxide anion radical production (O2
.-) production. UCB also induces the up-regulation of glycolysis and increased amounts of extracellular ATP, together with an inhibition of the pentose-phosphate pathway (PPP), as inferred by the decreased NADPH and increased oxidized glutathione (GSSG) levels. All these events shall contribute to the activation of caspases 9 and 3 and apoptotic cell death. GUDCA fully abrogated UCB-induced mitochondrial dysfunction, alterations in energy metabolism and disruption of the redox status, therefore counteraction immature nerve cell demise resulting from UCB exposure.

Chapter II __________________________________________________________________________
78
5. References
Almeida, A. and Bolaños, J. P. (2001) A transient inhibition of mitochondrial ATP synthesis
by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J
Neurochem, 77, 676-690.
Almeida, A., Bolaños, J. P. and Medina, J. M. (1999) Nitric oxide mediates glutamate-
induced mitochondrial depolarization in rat cortical neurons. Brain Res, 816, 580-586.
Almeida, A., Heales, S. J., Bolaños, J. P. and Medina, J. M. (1998) Glutamate neurotoxicity
is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione
depletion. Brain Res, 790, 209-216.
Almeida, A., Moncada, S. and Bolaños, J. P. (2004) Nitric oxide switches on glycolysis
through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol, 6,
45-51.
Atamna, H. and Frey, W. H., 2nd (2007) Mechanisms of mitochondrial dysfunction and
energy deficiency in Alzheimer's disease. Mitochondrion, 7, 297-310.
Bolaños, J. P., Cidad, P., García-Nogales, P., Delgado-Esteban, M., Fernandez, E. and
Almeida, A. (2004) Regulation of glucose metabolism by nitrosative stress in neural cells.
Mol Aspects Med, 25, 61-73.
Bolaños, J. P., Peuchen, S., Heales, S. J., Land, J. M. and Clark, J. B. (1994) Nitric oxide-
mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J
Neurochem, 63, 910-916.
Brann, B. S. t., Stonestreet, B. S., Oh, W. and Cashore, W. J. (1987) The in vivo effect of
bilirubin and sulfisoxazole on cerebral oxygen, glucose, and lactate metabolism in
newborn piglets. Pediatr Res, 22, 135-140.
Brites, D., Fernandes, A., Falcão, A. S., Gordo, A. C., Silva, R. F. M. and Brito, M. A. (2009)
Biological risks for neurological abnormalities associated with hyperbilirubinemia. J
Perinatol, 29 Suppl 1, S8-13.
Brito, M. A., Lima, S., Fernandes, A., Falcao, A. S., Silva, R. F., Butterfield, D. A. and Brites,
D. (2008a) Bilirubin injury to neurons: contribution of oxidative stress and rescue by
glycoursodeoxycholic acid. Neurotoxicology, 29, 259-269.
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008b) Unconjugated bilirubin differentially affects the redox status of neuronal
and astroglial cells. Neurobiol Dis, 29, 30-40.
Brown, G. C. and Cooper, C. E. (1994) Nanomolar concentrations of nitric oxide reversibly
inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS
Lett, 356, 295-298.

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
79
Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. and Schapira, A. H. (1994)
Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial
respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett,
345, 50-54.
Cuperus, F. J., Hafkamp, A. M., Havinga, R., Vitek, L., Zelenka, J., Tiribelli, C., Ostrow, J. D.
and Verkade, H. J. (2009) Effective treatment of unconjugated hyperbilirubinemia with oral
bile salts in Gunn rats. Gastroenterology, 136, 673-682 e671.
Davey, G. P., Peuchen, S. and Clark, J. B. (1998) Energy thresholds in brain mitochondria.
Potential involvement in neurodegeneration. J Biol Chem, 273, 12753-12757.
Day, R. L. (1954) Inhibition of brain respiration in vitro by bilirubin: reversal if inhibition by
various means. Am J Dis Child, 88, 504-506.
Diamond, I. and Schmid, R. (1967) Oxidative phosphorylation in experimental bilirubin
encephalopathy. Science, 155, 1288-1289.
Dringen, R. (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol, 62, 649-
671.
Dringen, R. and Hamprecht, B. (1996) Glutathione content as an indicator for the presence of
metabolic pathways of amino acids in astroglial cultures. J Neurochem, 67, 1375-1382.
Ernster, L. and Zetterström, R. (1956) Bilirubin, an uncoupler of oxidative phosphorylation in
isolated mitochondria. Nature, 178, 1335-1337.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Bilirubin-
induced immunostimulant effects and toxicity vary with neural cell type and maturation
state. Acta Neuropathol, 112, 95-105.
Farkas, D. L., Wei, M. D., Febbroriello, P., Carson, J. H. and Loew, L. M. (1989)
Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys J, 56,
1053-1069.
Fernandes, A., Vaz, A. R., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007)
Glycoursodeoxycholic Acid and interleukin-10 modulate the reactivity of rat cortical
astrocytes to unconjugated bilirubin. J Neuropathol Exp Neurol, 66, 789-798.
Figueroa, S., Oset-Gasque, M. J., Arce, C., Martinez-Honduvilla, C. J. and González, M. P.
(2006) Mitochondrial involvement in nitric oxide-induced cellular death in cortical neurons
in culture. J Neurosci Res, 83, 441-449.
García-Nogales, P., Almeida, A., Fernández, E., Medina, J. M. and Bolaños, J. P. (1999)
Induction of glucose-6-phosphate dehydrogenase by lipopolysaccharide contributes to
preventing nitric oxide-mediated glutathione depletion in cultured rat astrocytes. J
Neurochem, 72, 1750-1758.

Chapter II __________________________________________________________________________
80
Gutmann, I. and Wahlefeld, A. W. (1974) L-(+)-Lactate. Determination with Lactate
Dehydrogenase and NAD In: Methods of Enzymatic Analysis, Vol. 3, pp. 1464-1468.
Verlag Chemie GmbH, Weinheim.
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S. and Bolaños, J.
P. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous
degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol, 11, 747-752.
Hoffman, D. J., Zanelli, S. A., Kubin, J., Mishra, O. P. and Delivoria-Papadopoulos, M. (1996)
The in vivo effect of bilirubin on the N-methyl-D-aspartate receptor/ion channel complex in
the brains of newborn piglets. Pediatr Res, 40, 804-808.
Ives, N. K., Bolas, N. M. and Gardiner, R. M. (1989) The effects of bilirubin on brain energy
metabolism during hyperosmolar opening of the blood-brain barrier: an in vivo study using
31P nuclear magnetic resonance spectroscopy. Pediatr Res, 26, 356-361.
Ives, N. K., Cox, D. W., Gardiner, R. M. and Bachelard, H. S. (1988) The effects of bilirubin
on brain energy metabolism during normoxia and hypoxia: an in vitro study using 31P
nuclear magnetic resonance spectroscopy. Pediatr Res, 23, 569-573.
Katoh-Semba, R. (1976) Studies on cellular toxicity of bilirubin: effect on brain glycolysis in
the young rat. Brain Res, 113, 339-348.
Kawaguchi, T., Veech, R. L. and Uyeda, K. (2001) Regulation of energy metabolism in
macrophages during hypoxia. Roles of fructose 2,6-bisphosphate and ribose 1,5-
bisphosphate. J Biol Chem, 276, 28554-28561.
King, T. E. (1967) Preparation of succinate cytochrome c reductase and the cytochrome b-c1
particle, and reconstitution of succinate cytochrome c reductase. Methods Enzymology,
10, 216-225.
Klingerberg, M. (1985) NADH/NADPH. UV-methods. In: Methods of Enzymatic Analysis, Vol.
7, pp. 251-271. Verlag Chemie GmbH, Weinheim.
Lapenna, D., Ciofani, G., Festi, D., Neri, M., Pierdomenico, S. D., Giamberardino, M. A. and
Cuccurullo, F. (2002) Antioxidant properties of ursodeoxycholic acid. Biochem Pharmacol,
64, 1661-1667.
López, E., Arce, C., Oset-Gasque, M. J., Canadas, S. and González, M. P. (2006) Cadmium
induces reactive oxygen species generation and lipid peroxidation in cortical neurons in
culture. Free Radic Biol Med, 40, 940-951.
Lowry, O. H., Rosebrough, N. J., Farr, A. L. and Randall, R. J. (1951) Protein measurement
with the Folin phenol reagent. J Biol Chem, 193, 265-275.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P.,
Galeotti, T. and Pani, G. (2008) Bilirubin as an endogenous modulator of neurotrophin
redox signaling. J Neurosci Res, 86, 2235-2249.

UCB, mitochondrial dysfunction and GUDCA __________________________________________________________________________
81
Mattson, M. P. and Liu, D. (2002) Energetics and oxidative stress in synaptic plasticity and
neurodegenerative disorders. Neuromolecular Med, 2, 215-231.
McCandless, D. W. and Abel, M. S. (1980) The effect of unconjugated bilirubin on regional
cerebellar energy metabolism. Neurobehav Toxicol, 2, 81-84.
McDonagh, A. F. (1979) Bile pigments: Bilatrienes and 5,15-biladienes. In: The Porfirins, pp.
293-491. Academic Press, San Diego.
McDonald, J. W., Shapiro, S. M., Silverstein, F. S. and Johnston, M. V. (1998) Role of
glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat
model. Exp Neurol, 150, 21-29.
Mukhopadhyay, P., Rajesh, M., Hasko, G., Hawkins, B. J., Madesh, M. and Pacher, P.
(2007) Simultaneous detection of apoptosis and mitochondrial superoxide production in
live cells by flow cytometry and confocal microscopy. Nat Protoc, 2, 2295-2301.
Mustafa, M. G., Cowger, M. L. and King, T. E. (1969) Effects of bilirubin on mitochondrial
reactions. J Biol Chem, 244, 6403-6414.
Ostrow, J. D., Pascolo, L., Brites, D. and Tiribelli, C. (2004) Molecular basis of bilirubin-
induced neurotoxicity. Trends Mol Med, 10, 65-70.
Ostrow, J. D., Pascolo, L., Shapiro, S. M. and Tiribelli, C. (2003) New concepts in bilirubin
encephalopathy. Eur J Clin Invest, 33, 988-997.
Park, W. S., Chang, Y. S., Chung, S. H., Seo, D. W., Hong, S. H. and Lee, M. (2001) Effect
of hypothermia on bilirubin-induced alterations in brain cell membrane function and energy
metabolism in newborn piglets. Brain Res, 922, 276-281.
Ragan, C. I., Wilson, M. T., Darley-Usmar, V. M. and Lowe, P. N. (1987) Subfractionation of
mitochondria and isolation of the proteins of oxidative phosphorylation. In: Mitochondria: A
Practical Approach, pp. 79-112. IRL Press, London.
Reiser, D. J. (2004) Neonatal jaundice: physiologic variation or pathologic process. Crit Care
Nurs Clin North Am, 16, 257-269.
Rodrigues, C. M. P., Solá, S., Brito, M. A., Brites, D. and Moura, J. J. (2002) Bilirubin directly
disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat
mitochondria. J Hepatol, 36, 335-341.
Rodrigues, C. M. P., Stieers, C. L., Keene, C. D., Ma, X., Kren, B. T., Low, W. C. and Steer,
C. J. (2000) Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-
nitropropionic acid: evidence for a mitochondrial pathway independent of the permeability
transition. J Neurochem, 75, 2368-2379.
Rodriguez-Ortigosa, C. M., Cincu, R. N., Sanz, S., Ruiz, F., Quiroga, J. and Prieto, J. (2002)
Effect of ursodeoxycholic acid on methionine adenosyltransferase activity and hepatic
glutathione metabolism in rats. Gut, 50, 701-706.

Chapter II __________________________________________________________________________
82
Serviddio, G., Pereda, J., Pallardo, F. V. et al. (2004) Ursodeoxycholic acid protects against
secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress.
Hepatology, 39, 711-720.
Shapiro, S. M. (2005) Definition of the clinical spectrum of kernicterus and bilirubin-induced
neurologic dysfunction (BIND). J Perinatol, 25, 54-59.
Sharpe, M. A. and Cooper, C. E. (1998) Interaction of peroxynitrite with mitochondrial
cytochrome oxidase. Catalytic production of nitric oxide and irreversible inhibition of
enzyme activity. J Biol Chem, 273, 30961-30972.
Shepherd, D. and Garland, P. B. (1969) The kinetic properties of citrate synthase from rat
liver mitochondria. Biochem J, 114, 597-610.
Silva, M. F., Ruiter, J. P., Illst, L., Jakobs, C., Duran, M., de Almeida, I. T. and Wanders, R. J.
(1997) Valproate inhibits the mitochondrial pyruvate-driven oxidative phosphorylation in
vitro. J Inherit Metab Dis, 20, 397-400.
Silva, R. F. M., Rodrigues, C. M. P. and Brites, D. (2002) Rat cultured neuronal and glial cells
respond differently to toxicity of unconjugated bilirubin. Pediatr Res, 51, 535-541.
Uyeda, K. (1979) Phosphofructokinase. Adv Enzymol Relat Areas Mol Biol, 48, 193-244.
Van Schaftingen, E., Lederer, B., Bartrons, R. and Hers, H. G. (1982) A kinetic study of
pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers. Application
to a microassay of fructose 2,6-bisphosphate. Eur J Biochem, 129, 191-195.
Vaughn, A. E. and Deshmukh, M. (2008) Glucose metabolism inhibits apoptosis in neurons
and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol, 10, 1477-1483.
Wennberg, R. P., Johansson, B. B., Folbergrova, J. and Siesjo, B. K. (1991) Bilirubin-
induced changes in brain energy metabolism after osmotic opening of the blood-brain
barrier. Pediatr Res, 30, 473-478.
Wharton, D. C. and Tzagoloff, A. (1967) Cytochrome oxidase from beef heart mitochondria.
Methods Enzymology, 10, 245-250.
Wulff, K. (1985) NADH/NADPH. Luminometric method. In: Methods of Enzymatic Analysis,
Vol. 7, pp. 280-284. Verlag Chemie GmbH, Weinheim.

Chapter III
III. Pro-inflammatory cytokines intensify the activation of .NO/NOS,
JNK1/2 and caspase cascades in immature neurons exposed to elevated levels of unconjugated bilirubin
Ana Rita Vaz, Sandra Leitão Silva, Andreia Barateiro, Adelaide Fernandes, Ana Sofia Falcão, Maria A Brito, Dora Brites
Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculdade
de Farmácia, University of Lisbon, Av. Professor Gama Pinto, Lisbon 1649-003,
Portugal.
Experimental Neurology (submitted).

Acknowledgements This work was supported by grants PPCDT/SAU/MMO/55955/2004 and PTDC/SAU-
NEU/64385/2006 (to D.B.) and BD/30292/2006 (to A.R.V.) from Fundação para a Ciência
e a Tecnologia, Lisbon, Portugal.

BIND is increased by inflammation __________________________________________________________________________
85
Abstract
Hyperbilirubinemia may lead to encephalopathy in neonatal life, particularly in premature
infants. Although the mechanisms were never established, clinicians commonly consider
sepsis as a risk factor for bilirubin-induced neurological dysfunction (BIND). Our previous
studies showed that elevated levels of unconjugated bilirubin (UCB) have immunostimulant
effects, which are potentiated by LPS, and that immature neural cells are more vulnerable to
UCB. The present study was undertaken to explore the role of nitric oxide (.NO)/NO synthase
(NOS), c-Jun N-terminal kinase (JNK) 1/2 and caspase activation in BIND, as well as the
additional effects of inflammation, in immature neurons incubated from 1 h to 24 h, at 37ºC.
UCB, at conditions mimicking those of jaundiced newborns (UCB/serum albumin = 0.5),
induced .NO production, nNOS expression and JNK1/2 activation in 3 days in vitro neuron
cultures. As a consequence of these events, mitochondrial and extrinsic pathways of
apoptosis were initiated, ultimately leading to neuronal dysfunction. Co-incubation with TNF-
α+IL-1β intensified the activation of .NO/NOS, JNK1/2, caspase-8, caspase-9 and caspase-3
by UCB. Cleavage of Bid and truncated Bid (tBid) up-regulation, as well as increased
cytotoxic potential, was also observed. Interestingly, both L-NAME (NOS inhibitor) and
SP600125 (JNK1/2 inhibitor) reversed the effects produced by UCB either alone, or in
association to pro-inflammatory cytokines. Taken together, our data reveal not only that
activation of .NO/NOS, JNK1/2 and caspase cascades are important determinants of BIND
but also that the association of TNF-α+IL-1β have cumulative effects. These events provide a
reason for the risk of sepsis in BIND and point to potential targets for therapeutic
intervention.
Keywords: neuronal .NO/NOS system; JNK1/2 signalling pathway; Caspase activation; TNF-
α+IL-1β; Bilirubin-induced neurological dysfunction (BIND)
.

Chapter III __________________________________________________________________________
86
1. Introduction Elevated levels of unconjugated bilirubin (UCB) are responsible for the clinical
manifestation of jaundice, a common condition in the neonatal period. Although normal (or
slightly increased) levels of UCB provide protection against injury resulting from oxidation
(Doré et al., 2000), elevated UCB concentrations cause nerve cell damage, leading to
adverse neurological outcomes (Hansen, 2002), ranging from minimal damage to chronic
and permanent sequelae, or even death (Ostrow et al., 2004, Shapiro, 2005). The risk of
bilirubin-induced neurologic dysfunction (BIND) is particularly enhanced in premature
newborns due to the higher rates of UCB production and the immaturity of the excretion
pathways (Stevenson et al., 2001, Watchko, 2006).
Although the determinants of vulnerability to BIND are only partially understood, it is well
known that UCB triggers the accumulation of extracellular glutamate (Falcão et al., 2006),
oxidative stress (Brito et al., 2008a, Brito et al., 2010, Brito et al., 2008b), as well as the
release of the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α), interleukin-1β
(IL-1β) and IL-6 by both astrocytes (Fernandes et al., 2004) and microglia (Fernandes et al.,
2006). Inflammatory signalling pathway involves the activation of mitogen-activated protein
kinases (MAPKs) and nuclear factor (NF)-kB (Fernandes et al., 2006, Silva et al., 2010).
These upstream events culminate in nerve cell death by both necrosis and apoptosis (Silva
et al., 2002). UCB directly interacts with mitochondria influencing membrane lipid and protein
properties, redox status, and cytochrome c content (Rodrigues et al., 2002b). In addition, we
also demonstrated that UCB induces apoptosis trough mitochondria-caspase-3 pathway
involving cytochrome c release, caspase-3 activation, and subsequent PARP cleavage in
developing rat brain neurons (Rodrigues et al., 2002a). Recent findings evidenced that UCB
inhibits cytochrome c oxidase activity, and induces both ATP release and disruption of
glutathione redox status in immature neurons (Vaz et al., 2010).
Interestingly, immature nerve cells are more susceptible than differentiated ones to
UCB-induced toxicity and release higher levels of glutamate and TNF-α providing a basis for
the increased susceptibility of premature newborns to UCB deleterious effects (Falcão et al.,
2006). In addition, LPS showed to exacerbate the release of TNF-α and IL-1β by immature
astrocytes (Falcão et al., 2005).
It has been suggested that infection increases the risk for UCB encephalopathy
(Dawodu et al., 1984) and presence of inflammatory features, namely fever episodes and
brain edema, were described during or following moderate to severe hyperbilirubinemia
(Kaplan and Hammerman, 2005). The inflammatory reaction is essential for survival in
response to tissue injury or infection, but it can also cause neuronal damage, since cytokines
are not only involved in neuroprotection but also in neurodegeneration processes (Konsman

BIND is increased by inflammation __________________________________________________________________________
87
et al., 2007). Pro-inflammatory cytokines, such as TNF-α and IL-1β, besides having
pleiotropic effects in the central nervous system, including their emerging role in
neurodevelopment (Marx et al., 2001) have also been described as mediators of neuronal
apoptosis (Kajta et al., 2006, Takahashi et al., 2008). Several studies have shown that
inflammation is associated with the enhanced generation of reactive oxygen species (ROS),
and/or reactive nitrogen species (Bian and Murad, 2001, Sener et al., 2005, Brito et al.,
2007). One of the oxidant species, nitric oxide (.NO), although important in cellular signalling,
has an important role in the pathogenesis of inflammation (Korhonen et al., 2005). In fact, .NO and induction of nitric oxide synthase (NOS) are involved in apoptosis induced by
inflammatory mediators in neuronal cells (Hemmer et al., 2001, Heneka et al., 1998, Thomas
et al., 2008). To add that exposure to UCB leads to increased expression of neuronal NOS
(nNOS) and production of .NO, cyclic guanosine 3',5'-monophosphate (cGMP) and ROS,
along with protein oxidation and depletion of glutathione (Brito et al., 2008b, Fernandes et al.,
2010). Therefore, .NO inhibitors represent important strategies to prevent cellular injury
associated with jaundice and inflammatory processes. Among the upstream signals leading
to neuronal degeneration, one may include the stress-activated protein kinases (SAPKs). In
fact, c-Jun N-terminal kinases 1/2 (JNK1/2) become activated in response to toxic stimulus,
such as the reactive nitrogen species (Luo et al., 1998, Marques et al., 2003) and the pro-
inflammatory cytokines TNF-α and IL-1, pointing these SAPKs as strong effectors of
neuronal apoptosis (Tibbles and Woodgett, 1999, Mielke and Herdegen, 2000). JNK 1/2
directly mediates the UCB-stimulation of TNF-α by astrocytes as we showed by the use of
SP600125, a JNK1/2 inhibitor (Fernandes et al., 2007). This feature may be relevant if we
consider that astrocytic activation has also been reported in several neurodegenerative
disorders and that this transition may be accompanied by dysfunction of astrocytes leading to
incorrect glia-to-neuron cross-talk (Rossi and Volterra, 2009).
In this study, we investigated if .NO/NOS and JNK 1/2 activation were signalling
determinants for caspase-8 activation and mitochondrial death pathway in UCB-induced
dysfunction of immature neurons. Most of all, we explored if the combination of the pro-
inflammatory cytokines TNF-α and IL-1β aggravate the functional de-regulation of immature
neurons produced by UCB and whether use the same cascade of mediators.

Chapter III __________________________________________________________________________
88
2. Materials and Methods
2.1. Chemicals Neurobasal medium, B-27 supplement (50X), Hanks’ balanced salt solution (HBSS-1),
Hanks’ balanced salt solution without Ca2+ and Mg2+ (HBSS-2), gentamicin (50 mg/mL), and
trypsin (0.025%) were acquired from Invitrogen (Carlsbad, CA). Recombinant rat IL-1β and
TNF-α were form R&D Systems Inc. (Minneapolis, MN, USA). Human serum albumin (HSA)
(fraction V, fatty acid free), N-ω-nitro-L-arginine methyl ester hydrochloride (L-NAME),
primary monoclonal antibody mouse anti-β-actin, N-1-naphthylethylenediamine,
sulfanilamide, sodium nitrite and MTT [3-(4,5-dimethylthiazol, 2-yl)-2,5-diphenyltetrazolium
bromide] were purchased from Sigma Chemical Co (St Louis, MO). UCB was also from
Sigma and purified as previously described (McDonagh, 1979). Nitrocellulose membrane,
Hyperfilm ECL and horseradish peroxidase-labelled sheep anti-mouse IgG were from
Amersham Biosciences (Piscataway, NJ, USA). Cell lysis buffer and LumiGLO® were
acquired from Cell Signalling (Beverly, MA, USA). Antibodies directed to phosphorylated
JNK1/2 (P-JNK1/2) and Bid were from Santa Cruz Biotechnology (Santa Cruz, CA, USA)
while antibody against neuronal NOS (nNOS) was from Becton Dickinson Biosciences (San
José, CA, USA). Caspases 3, 8 and 9 substrates, Ac-DEVD-pNA, Ac-IETD-pNA and Ac-
LEHD-pNA, respectively, were purchased from Calbiochem (Darmstadt, Germany). A
concentrated solution (10 mM) of the JNK1/2 inhibitor SP600125 (Calbiochem) was prepared
in dimethylsulfoxide.
2.2. Neurons in primary culture
Animal care followed the recommendations of European Convention for the Protection of
Vertebrate Animals Used for Experimental and other Scientific Purposes (Council Directive
86/609/EEC) and National Law 1005/92 (rules for protection of experimental animals).
Neurons were isolated from fetuses of 16-17-day pregnant Wistar rats, as previously
described (Silva et al., 2002). The fetuses were collected in Hanks’ balanced salt solution
(HBSS), the brain cortices were mechanically fragmented, and the fragments transferred to a
0.5 g/L trypsin in Ca2+ and Mg2+ free HBSS medium and incubated for 15 min at 37ºC. After
trypsinization, cells were washed twice in Ca2+ and Mg2+ free HBSS medium containing 10%
fetal bovine serum, and resuspended in Neurobasal medium supplemented with 0.5 mM L-
glutamine, 25 μM L-glutamic acid, 2% B-27 supplement, and 0.12 mg/mL gentamicin. Finally,
cells were seeded on poly-D-lysine coated tissue culture plates at a density of 2 x 105

BIND is increased by inflammation __________________________________________________________________________
89
cells/cm2 and maintained at 37ºC in a humidified atmosphere of 5% CO2. In this work we
used neurons at 3 days in vitro (DIV).
2.3. Treatment of neurons Immature neurons were incubated in Neurobasal medium without (control) or with 50 μM
UCB (Sigma Chemical Co, St Louis, MO, USA) in the presence of 100 μM HSA from 1 h to
24 h, at 37ºC. For co-incubation studies, neurons were co-incubated with recombinant 50
ng/mL TNF-α plus 50 ng/mL IL-1β (R&D Systems Inc., Minneapolis, MN, USA). Stock UCB
solutions were extemporarily prepared in 0.1 M NaOH under the dark and the pH adjusted to
7.4 using 0.1 M HCl. In parallel studies, cells were co-incubated with 100 µM N-ω-nitro-L-
arginine methyl ester hydrochloride (L-NAME) (Sigma Chemcial Co), a non-selective NOS
inhibitor or treated with 0.2 µM SP600125 (Calbiochem, Darmstadt, Germany), an inhibitor of
JNK1/2, 1 h prior to UCB addition, alone or in combination with TNF-α+IL-1β.
2.4. Quantification of nitrite levels Nitric oxide levels were estimated by measuring the concentrations of nitrites (NO2
-),
which are the resulting .NO metabolites. Briefly, supernatants free from cellular debris were
mixed with Griess reagent [1 part 1% (w/v) sulfanilamide in 5% H3PO4, 1 part 0.1% (w/v) N-1-
naphthylethylenediamine (v/v)] in 96-well tissue culture plates for 10 min at room
temperature in the dark. The absorbance at 540 nm was determined using a microplate
reader (Bio-Rad Laboratories, Hercules, CA, USA).
2.5. Western blot assay The intracellular forms of nNOS, Bid and phosphorylated P-JNK1/2 were determined by
Western blot analysis as usual in our laboratory (Fernandes et al., 2006). Briefly, cells were
washed in ice-cold PBS and lysed in a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM
NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% (v/v) Triton X-100, 2.5 mM sodium pyrophosphate,
1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptine, 1 mM PMSF. The lysate was
sonicated for 20 s, centrifuged at 14.000 g for 10 min at 4ºC and the supernatants were
collected and stored at -80ºC. Protein concentrations were determined according to the
Bradford method (Bradford, 1976) using Bio-Rad’s Protein Assay reagent (Bio-Rad, CA,
USA). Equal amounts of protein were subjected to sodium dodecyl sulphate-polyacrylamide
gel electrophoresis and transferred to a nitrocellulose membrane. After blocking with 5% milk
solution, membranes were incubated with the primary antibody overnight at 4ºC [mouse anti-
nNOS (BD Biosciences, San José, CA, USA) diluted at 1:2.500, rabbit anti-Bid (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) diluted at 1:500, mouse anti-P-JNK1/2 (Santa Cruz
Biotechnology) diluted at 1:200 or anti-β-actin (Sigma Chemical Co) diluted at 1:10.000], and

Chapter III __________________________________________________________________________
90
finally with horseradish peroxidase-labelled secondary antibody. Protein bands were
detected by LumiGLO® and visualized by autoradiography with Hyperfilm ECL.
2.6. Caspase activity determination Activities of caspase-3, -8 and -9 were measured by a colorimetric method (Calbiochem,
Darmstadt, Germany) as usual in our laboratory (Vaz et al., 2010). Cells were harvested,
washed with ice-cold PBS and lysed for 30 minutes on ice in the lysis buffer [50 mM HEPES
(pH 7.4), 100 mM NaCl, 0.1% (w/v) cholamidopropyldimethylammonio-1-propanesulfonate
(CHAPS), 1 mM DTT, 0.1 mM EDTA]. The lysate was centrifuged at 10,000 g for 10 min at
4ºC and the supernatants were collected and stored at -80ºC. Protein concentrations were
determined as aforementioned. The activity of caspases 3, 8 and 9 was determined in cell
lysates by enzymatic cleavage of chromophore p-nitroaniline (pNA) from the substrate Ac-
YVAD-pNA, according to manufacturer’s instructions. The proteolytic reaction was carried
out in protease assay buffer [50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% (w/v) CHAPS, 10
mM DTT, 0.1 mM EDTA, 10% (v/v) glicerol], containing 2 mM substrate Ac-DEVD-pNA for
caspase-3, Ac-IETD-pNA for caspase-8 and Ac-LEHD-pNA for caspase-9. Following
incubation of the reaction mixtures for 2 h at 37ºC, the formation of pNA was measured at
405 nm with a reference filter of 620 nm.
2.7. MTT reduction Cellular reduction of [3-(4,5-dimethylthiazol, 2-yl)-2,5-diphenyltetrazolium bromide]
(MTT) was measured in nerve cells as previously described by us (Silva et al., 2002). Briefly,
a stock solution of MTT at 5 mg/mL was freshly prepared and after the incubation periods,
supernatants were removed and cells were incubated for 1 h, at 37°C, with 0.5 mL of MTT at
0.5 mg/mL. After incubation, medium was discarded and MTT formazan crystals were
dissolved by addition of 1 mL isopropanol/HCl 0.04 M and gentle shaking for 15 min, at room
temperature. After centrifugation, absorbance values at 570 nm were determined in a
Unicam UV2 spectrophotometer (Unicam Limited, UV2, Cambridge, UK). Results were
expressed as percentage of control, which was considered as 100%.
2.8. Densitometry and statistical analysis The relative intensities of protein and nucleic acid bands were analysed using the
Quantity One (version 4.6) program (Bio-Rad, CA, USA). Results of, at least, three different
experiments were expressed as mean ± S.E.M. Significant differences between two groups
were determined by the two-tailed t-test performed on the basis of equal and unequal
variance as appropriate. Comparison of more than two groups was done by ANOVA using
Instat 3.05 (GraphPad Software, San Diego, CA, USA) followed by multiple comparisons

BIND is increased by inflammation __________________________________________________________________________
91
Bonferroni post-hoc correction. Mean values were considered statistically significant when P
values were lower than 0.05.
3. Results 3.1. UCB, alone or in combination with TNF-α+IL-1β, induces nNOS expression and
.NO production in immature neurons, which are counteracted by L-NAME To investigate the possible role of UCB -induced neuronal damage in immature nerve
cells, 3 DIV cortical neurons were incubated with UCB at conditions mimicking moderate to
severe neonatal jaundice (UCB to HSA molar ratio of 0.5) that had previously shown to
promote oxidative injury and cell death in 8 DIVs neurons (Brito et al., 2008a). Cells were
collected after treatment during 1 h to 12 h for the analysis of nNOS expression and
production of nitrites. We also investigated the aggravating effects of pro-inflammatory
cytokines TNF-α and IL-1β on UCB-induced oxidative stress. In order to evaluate the
importance of .NO signalling in oxidative damage resulting from UCB or UCB+TNF-α+IL-1β,
we used L-NAME to inhibit nNOS activity. We observed that UCB induced nNOS expression,
together with a raise in nitrite production in immature neurons. Co-incubation with
TNF-α+IL-1β, in concentrations that have been described to induce neuronal loss (Patel and
Brewer, 2008, Zhang et al., 2008), aggravated UCB-induced nNOS expression and nitrite
production in these cells (Fig. III.1A and III.1C). In parallel studies, co-incubation with 100 µM
L-NAME led to at least ~70% and ~60% inhibition in nNOS expression and nitrite production
(p<0.05), respectively (Fig. III.1B and III.1D).
3.2. Inhibition of nNOS by L-NAME prevents the cascade of apoptosis induced by UCB or UCB+TNF-α+ IL-1β in immature neurons To deepen characterize the mechanisms of neuronal apoptosis upon exposure of
immature neurons to UCB and pro-inflammatory cytokines, we evaluated the activity of
initiator caspases from mitochondrial (intrinsic) and death receptor (extrinsic) pathways,
caspases-9 and -8, respectively, as well as of the effector caspase-3. As shown in Figures
III.2A and III.2C UCB led to the activation of caspases-3 and -9, indicating the stimulation of
the intrinsic pathway of apoptosis. Interestingly, co-incubation with TNF-α+IL-1β significantly
enhanced this activation.
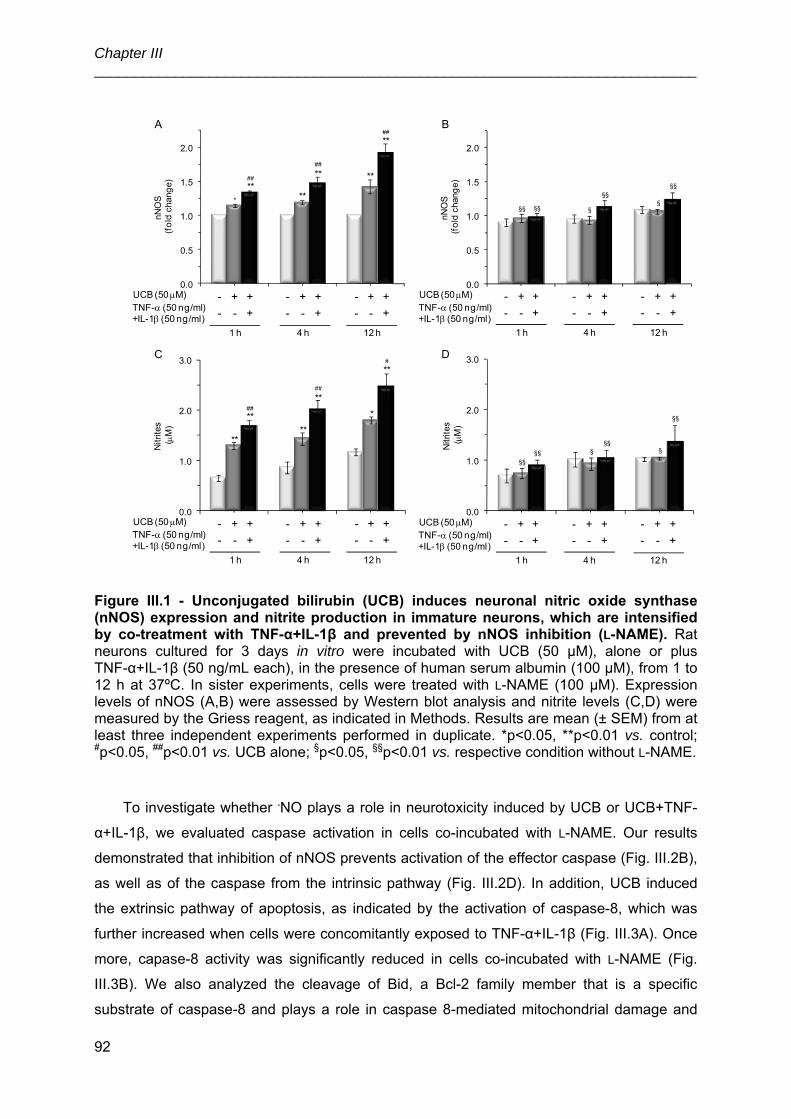
Chapter III __________________________________________________________________________
92
Figure III.1 - Unconjugated bilirubin (UCB) induces neuronal nitric oxide synthase (nNOS) expression and nitrite production in immature neurons, which are intensified by co-treatment with TNF-α+IL-1β and prevented by nNOS inhibition (L-NAME). Rat neurons cultured for 3 days in vitro were incubated with UCB (50 µM), alone or plus TNF-α+IL-1β (50 ng/mL each), in the presence of human serum albumin (100 μM), from 1 to 12 h at 37ºC. In sister experiments, cells were treated with L-NAME (100 μM). Expression levels of nNOS (A,B) were assessed by Western blot analysis and nitrite levels (C,D) were measured by the Griess reagent, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. control; #p<0.05, ##p<0.01 vs. UCB alone; §p<0.05, §§p<0.01 vs. respective condition without L-NAME.
To investigate whether .NO plays a role in neurotoxicity induced by UCB or UCB+TNF-
α+IL-1β, we evaluated caspase activation in cells co-incubated with L-NAME. Our results
demonstrated that inhibition of nNOS prevents activation of the effector caspase (Fig. III.2B),
as well as of the caspase from the intrinsic pathway (Fig. III.2D). In addition, UCB induced
the extrinsic pathway of apoptosis, as indicated by the activation of caspase-8, which was
further increased when cells were concomitantly exposed to TNF-α+IL-1β (Fig. III.3A). Once
more, capase-8 activity was significantly reduced in cells co-incubated with L-NAME (Fig.
III.3B). We also analyzed the cleavage of Bid, a Bcl-2 family member that is a specific
substrate of caspase-8 and plays a role in caspase 8-mediated mitochondrial damage and
0.0
0.5
1.0
1.5
2.0
nNO
S(f
old
chan
ge)
0.0
0.5
1.0
1.5
2.0nN
OS
(fol
d ch
ange
)
0.0
1.0
2.0
3.0
Nitr
ites
(μM
)
0.0
1.0
2.0
3.0
Nitr
ites
(μM
)
* **
**##**
##
**
##
**
§§
§§§
§§ §
§§
A B
****
*##**
##
**
#
**
§§§§
§
§§
§
§§
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
C D
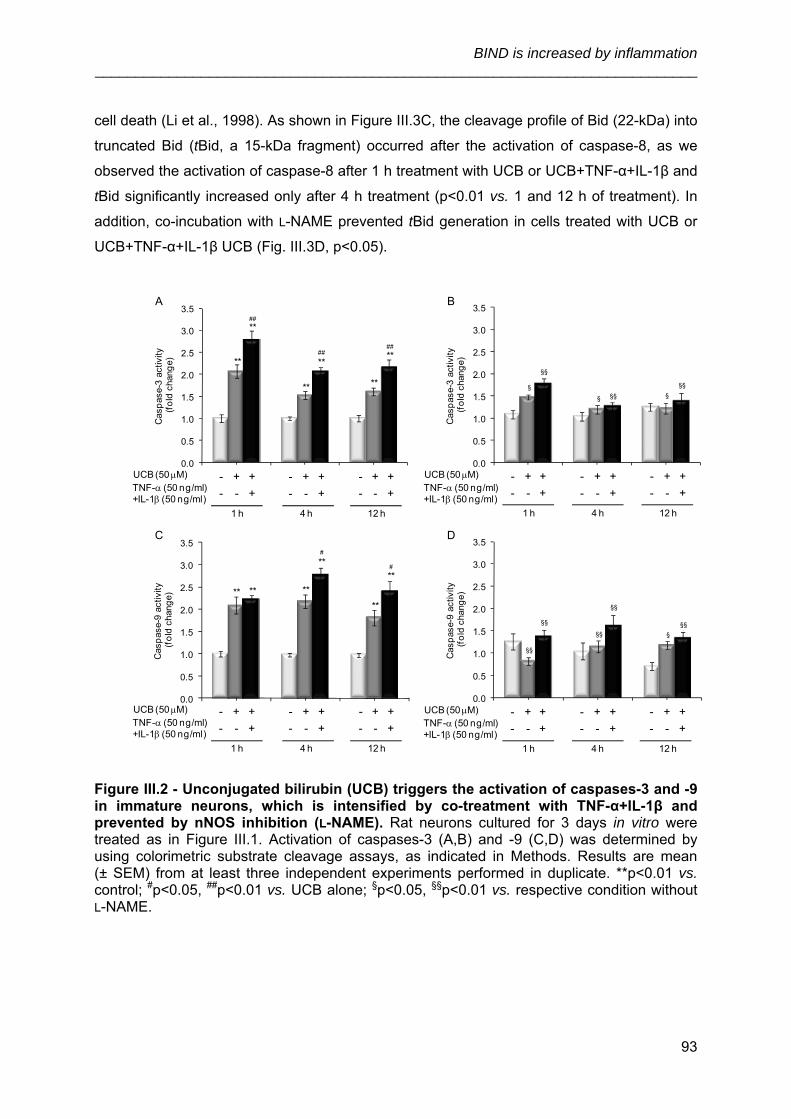
BIND is increased by inflammation __________________________________________________________________________
93
cell death (Li et al., 1998). As shown in Figure III.3C, the cleavage profile of Bid (22-kDa) into
truncated Bid (tBid, a 15-kDa fragment) occurred after the activation of caspase-8, as we
observed the activation of caspase-8 after 1 h treatment with UCB or UCB+TNF-α+IL-1β and
tBid significantly increased only after 4 h treatment (p<0.01 vs. 1 and 12 h of treatment). In
addition, co-incubation with L-NAME prevented tBid generation in cells treated with UCB or
UCB+TNF-α+IL-1β UCB (Fig. III.3D, p<0.05).
Figure III.2 - Unconjugated bilirubin (UCB) triggers the activation of caspases-3 and -9 in immature neurons, which is intensified by co-treatment with TNF-α+IL-1β and prevented by nNOS inhibition (L-NAME). Rat neurons cultured for 3 days in vitro were treated as in Figure III.1. Activation of caspases-3 (A,B) and -9 (C,D) was determined by using colorimetric substrate cleavage assays, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. **p<0.01 vs. control; #p<0.05, ##p<0.01 vs. UCB alone; §p<0.05, §§p<0.01 vs. respective condition without L-NAME.
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-9 a
ctiv
ity(f
old
chan
ge)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-9 a
ctiv
ity(f
old
chan
ge)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-3 a
ctiv
ity(f
old
chan
ge)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-3 a
ctiv
ity(f
old
chan
ge) **
** **
##**
##
**
##**
§§
§§ §§
§
§§
** ****
**
#
** #
**
§§
§§
§
§§
§§§§
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
A B
C D
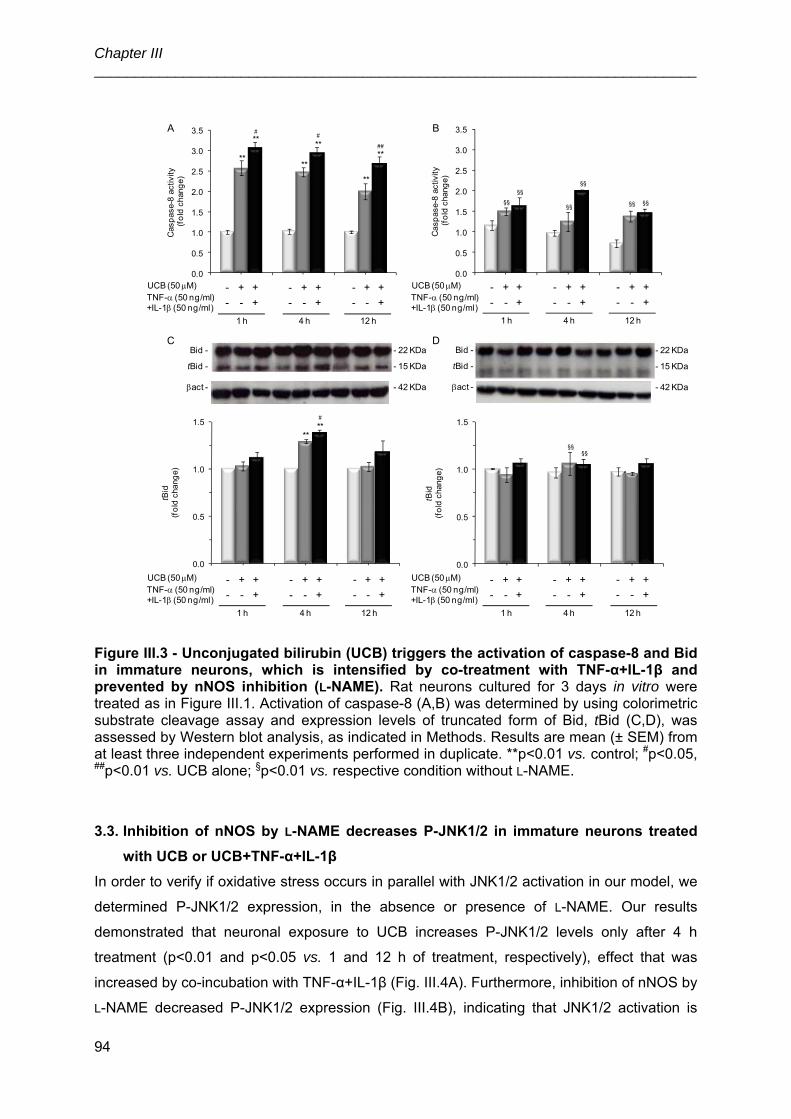
Chapter III __________________________________________________________________________
94
Figure III.3 - Unconjugated bilirubin (UCB) triggers the activation of caspase-8 and Bid in immature neurons, which is intensified by co-treatment with TNF-α+IL-1β and prevented by nNOS inhibition (L-NAME). Rat neurons cultured for 3 days in vitro were treated as in Figure III.1. Activation of caspase-8 (A,B) was determined by using colorimetric substrate cleavage assay and expression levels of truncated form of Bid, tBid (C,D), was assessed by Western blot analysis, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. **p<0.01 vs. control; #p<0.05, ##p<0.01 vs. UCB alone; §p<0.01 vs. respective condition without L-NAME.
3.3. Inhibition of nNOS by L-NAME decreases P-JNK1/2 in immature neurons treated with UCB or UCB+TNF-α+IL-1β
In order to verify if oxidative stress occurs in parallel with JNK1/2 activation in our model, we
determined P-JNK1/2 expression, in the absence or presence of L-NAME. Our results
demonstrated that neuronal exposure to UCB increases P-JNK1/2 levels only after 4 h
treatment (p<0.01 and p<0.05 vs. 1 and 12 h of treatment, respectively), effect that was
increased by co-incubation with TNF-α+IL-1β (Fig. III.4A). Furthermore, inhibition of nNOS by
L-NAME decreased P-JNK1/2 expression (Fig. III.4B), indicating that JNK1/2 activation is
0.0
0.5
1.0
1.5tB
id(f
old
chan
ge)
0.0
0.5
1.0
1.5
tBid
(fol
d ch
ange
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-8 a
ctiv
ity(f
old
chan
ge)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pase
-8 a
ctiv
ity(f
old
chan
ge)
****
**
#** #
** ##
**
§§§§
§§§§§§
§§
**
#
**
§§§§
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
- 22 KDa
- 15 KDa
Bid -
tBid -
βact - - 42 KDa
Bid -
tBid -
βact -
- 22 KDa
- 15 KDa
- 42 KDa
A B
C D

BIND is increased by inflammation __________________________________________________________________________
95
mediated by production of .NO. To better understand whether JNK1/2 activation takes part in
neuronal dysfunction, we tested the effects of SP600125, a specific inhibitor of JNK1/2, on
cells treated with UCB or UCB+TNF-α+IL-1β. Immature neurons were pre-treated with 0.2
µM of the inhibitor for 1 h, followed by 4 h stimulation with UCB alone or in combination with
TNF-α+IL-1β. As shown in Figures III.4C and III.4D, SP600125 efficiently prevented the
appearance of P-JNK1/2 induced by UCB or UCB+TNF-α+IL-1β (~90%, p<0.05).
Figure III.4 - Unconjugated bilirubin (UCB) leads to activation of c-Jun N-terminal kinases 1/2 (JNK1/2) in immature neurons, which is intensified by co-treatment with TNF-α+IL-1β and prevented by inhibition of nNOS (L-NAME) or JNK1/2 (SP600125). Rat neurons cultured for 3 days in vitro were treated as in Figure 1 (A,B). In another set of experiments, cells were treated for 1 h with 0.2 μM SP600125 (C,D), prior to UCB (50 µM) exposure, alone or in combination with TNF-α+IL-1β (50 ng/mL each), in the presence of human serum albumin (100 μM), for 4 h at 37ºC. Activation of JNK1/2 was determined by measuring the expression levels of phosphorylated forms of this enzyme (P-JNK1/2) by Western blot analysis, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. **p<0.01 vs. control; ##p<0.01 vs. UCB alone; §§p<0.01, §p<0.05 vs. respective condition without L-NAME; &&p<0.01 vs. respective condition without SP600125.
0.0
0.4
0.8
1.2
1.6
2.0
P-J
NK
1/2
(fol
d ch
ange
)
0.0
0.4
0.8
1.2
1.6
2.0
P-J
NK
1/2
(fol
d ch
ange
)
0.0
0.4
0.8
1.2
1.6
2.0
P-J
NK
1/2
(fol
d ch
ange
) **
##
**
§§
§
&&
- 55 KDa- 46 KDa
P-JNK1/2 -
βact - - 42 KDa
P-JNK1/2 -
βact -
- 55 KDa- 46 KDa
- 42 KDa
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
1 h
- + +- - +
4 h
- + +- - +
12 h
- + +- - +
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
- + +- - +SP600125 (0.2 µM)
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +
- - +
- + +
- - +
SP600125 (0.2 µM)
**
##
**
&&
- 55 KDa- 46 KDa
P-JNK1/2 -
βact - - 42 KDa
C D
A B

Chapter III __________________________________________________________________________
96
3.4. Inhibition of P-JNK1/2 by SP600125 prevents the cascade of apoptosis induced by UCB or UCB+TNF-α+IL-1β in immature neurons Having established that the appearance of P-JNK1/2 induced by UCB or UCB+TNF-
α+IL-1β is mediated, at least in part, by .NO production, we attempted to examine whether
JNK1/2 activation is also linked to the apoptotic events described previously. As shown in
Figure III.5, pre-treatment with SP600125 significantly reduced the activation of caspases-3,
-8 and -9, as well as activation of Bid into tBid in cells treated with UCB or UCB+TNF-α+IL-1β
(p<0.05), suggesting that, in our model, both intrinsic and extrinsic apoptotic pathways are
being initiated during JNK1/2 activation by bilirubin and cytokines.
3.5. Loss of neuronal functionality in immature cells exposed to UCB is increased by UCB+TNF-α+IL-1β and prevented by inhibition of nNOS and JNK1/2 activation To investigate whether induction of nNOS and JNK1/2 is implicated in neuronal
dysfunction, we assessed the effects of UCB, alone or in combination with TNF-α+IL-1β, on
the immature cell function in neurons treated with or without L-NAME or SP600125. Our
results demonstrated that the loss of functionality in immature neurons exposed to UCB for
24 h is increased by the pro-inflammatory cytokines TNF-α+IL-1β (Fig. III.6). In addition, this
neuronal dysfunction was prevented by co-incubation with L-NAME, as well as with
pretreatment with SP600125, suggesting both .NO and JNK1/2 as key elements in neuronal
dysfunction during hyperbilirubinemia with associated inflammation.

BIND is increased by inflammation __________________________________________________________________________
97
Figure III.5 - Inhibition of c-Jun N-terminal kinases 1/2 (JNK1/2) with SP600125 prevents the cascade of apoptosis induced either by unconjugated bilirubin (UCB) alone or by UCB co-treatment with TNF-α+IL-1β. Rat neurons cultured for 3 days in vitro were treated as in Figure III.4. Activation of caspases-3, -9 and -8 (A-C) was determined by using colorimetric substrate cleavage assays and expression levels of truncated form of Bid, tBid (D,E), was assessed by Western blot analysis, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. **p<0.01 vs. control; ##p<0.01, #p<0.05 vs. UCB alone; 0.05, &&p<0.01 vs. respective condition without SP600125.
0.0
0.5
1.0
1.5
tBid
(fol
d ch
ange
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pse-
8 ac
tivity
(fol
d ch
ange
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pse-
9 ac
tivity
(fol
d ch
ange
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Cas
pse-
3 ac
tivity
(fol
d ch
ange
)
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
- + +- - +SP600125 (0.2 µM)
**
##
**
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
- + +- - +SP600125 (0.2 µM)
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
- + +- - +SP600125 (0.2 µM)
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
- + +- - +SP600125 (0.2 µM)
**
#
**
**
#
** **
#
**
&&&
&&&&
&&
&&&&&
A B
C D
- 22 KDa
- 15 KDa
Bid -
tBid -
βact - - 42 KDa
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +
- - +
- + +
- - +
SP600125 (0.2 µM)
E

Chapter III __________________________________________________________________________
98
Figure III.6 - Loss of cell functionality in immature neurons exposed to unconjugated bilirubin (UCB) is intensified by co-treatment with TNF-α+IL-1β and prevented by inhibition of nNOS (L-NAME) or JNK1/2 (SP600125). Rat neurons cultured for 3 days in vitro were incubated with UCB (50 µM), alone or in combination with TNF-α+IL-1β (50 ng/mL each), in the presence of human serum albumin (100 μM), for 24 h at 37ºC. In one set of experiments, cells were treated with L-NAME (100 μM); in the other set, cells were pre-treated for 1 h with 0.2 μM SP600125. Cell functionality was evaluated by measuring 3-(4,5-dimethylthiazol, 2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction, as indicated in Methods. Results are mean (± SEM) from at least three independent experiments performed in duplicate. **p<0.01 vs. control; ##p<0.01 vs. UCB alone; §p<0.05, §§p<0.01 vs. respective condition without L-NAME; &&p<0.01, &p<0.05 vs. respective condition without SP600125.
4. Discussion Previously, we demonstrated that UCB induces neurotoxicity, which differ from neural
cell type and maturation state, being neurons and immature cells the most susceptible ones
(Falcão et al., 2006, Brito et al., 2008b). Moreover, co-incubation with LPS showed to
enhance the demise of astrocytes (Fernandes et al., 2004), namely in immature cells (Falcão
et al., 2005). Based on these data, we investigated whether pro-inflammatory cytokines are
able to strengthen the oxidative stress exerted by UCB in immature neurons, and if the effect
is reproduced on the extrinsic and intrinsic signalling apoptotic pathways, as well as at the
level of neuronal dysfunction.
In this paper, we demonstrate that UCB induces oxidative damage in immature rat
neurons, as indicated by the cell function impairment and apoptosis associated with .NO
production. Interestingly, we previously reported an inhibition of mitochondrial cytochrome c
0
25
50
75
100
MTT
(% o
f Con
trol)
UCB (50 μM)TNF-α (50 ng/ml)+IL-1β (50 ng/ml)
- + +- - +
SP600125 (0.2 µM)
L- NAME(100 µM)
- + +- - +
- + +- - +
**§
§§
**##
&&&

BIND is increased by inflammation __________________________________________________________________________
99
oxidase by UCB in the same in vitro model (Vaz et al., 2010), which can explain the
formation of .NO. In addition, we also observed that UCB decreases NADPH concentration
and increases glutathione oxidation and superoxide anion radical production, thus confirming
neuronal oxidative stress by UCB. Furthermore, UCB was shown to induce protein oxidation
and lipid peroxidation, and to diminish the antioxidant defences in mature neurons (8 DIV),
events that occur in parallel with necrotic cell death and could be due to low glutathione
stores (Brito et al., 2008a).
Here, we present evidence that mitochondrial-dependent and -independent apoptotic
pathways are induced in immature rat cultured neurons after UCB treatment. To the best of
our knowledge, this is the first evidence reporting UCB to induce neuronal extrinsic pathway
of apoptosis, in addition to that of mitochondria-mediated apoptosis observed in differentiated
neurons for UCB/HSA molar ratio of 3 (Rodrigues et al., 2002a). However, this molar ratio is
supposed to never occur in jaundiced babies, even during the worst pathological situations
and, therefore, we have repeated the experiments for the most suitable molar ratio of 0.5.
Interestingly, the same effect was significantly produced in immature neurons, after a short
exposure (1 h incubation) to UCB (Vaz et al., 2010). It is widely accepted that activated
caspase-8 propagates the apoptotic signal by activating downstream caspases through
proteolytic cleavage, as well as by triggering mitochondrial pathway through cleavage and
activation of pro-apoptotic Bid into tBid (Adams, 2003). In fact, in our model, activation of
caspase-8 begins early in time (at 1 h incubation), followed by tBid generation (at 4 h
incubation). This raises interesting possibilities as both caspase-8 and -9-initiated apoptotic
pathways converge independently to activate the execution phase of caspase-3 (Hengartner,
2000). In addition, we are tempted to propose that caspase-8/tBid-related route may play an
important role in UCB-induced neuronal cell death. The results herein obtained in immature
neurons are in line with our own previous observations showing that UCB induces intrinsic
signalling pathways by requiring Bax translocation to the mitochondria, mitochondrial
depolarization, release of cytochrome c and caspase-3 activation in isolated mitochondria
from the brain and liver of adult male Wistar rats (Rodrigues et al., 2002a, Rodrigues et al.,
2000). They also agree with other findings indicating activation of TNFR1 upon exposure of
astrocytes, to UCB reflecting the activation of the death receptor pathway (Fernandes et al.,
2006), and therefore the extrinsic cascade.
The present study also shows for the first time that pro-inflammatory cytokines TNF-α
and IL-1β intensify UCB-induced oxidative stress and apoptotic cell death by both
mitochondrial-dependent and –independent apoptotic pathways in immature neurons. These
observations are particularly important, since low-gestational-age newborns have a
prominently increased risk of brain dysfunction attributed to cerebral-cortex damage,

Chapter III __________________________________________________________________________
100
including excess of apoptosis and impairment of surviving neurons (Leviton and Gressens,
2007). With our data, we associate inflammation with the increased risk of UCB-induced
neurotoxicity. In fact, there are some reports in agreement with our finding: (i) in an animal
model of sepsis, it was shown that serum concentration of total and free bilirubin was
increased, promoting a net accumulation of UCB in the brain (Hansen et al., 1993); (ii) pro-
inflammatory cytokines were reported to increase blood-brain-barrier permeability (Petty and
Lo, 2002), allowing UCB entrance into the brain, and to exacerbate UCB-induced cytotoxicity
in different cell lines, such as in neuroblastoma (ATCC, HTB-10, SK-N-MC), glioblastoma
(ATCC, CRL 1690, T98G), umbilical vein endothelial (ATCC, CRL 1730, HUV-EC), liver cell
(ATCC, CCL 13) and mouse fibroblasts (L-929) (Ngai and Yeung, 1999, Yeung and Ngai,
2001). Thus, with our model, that mimics pro-inflammatory cytokine release from glial cells,
as we have previously reported for astrocytes and microglia exposed to UCB (Fernandes et
al., 2004, Fernandes et al., 2006), we can establish a relation between UCB-induced toxicity
and associated inflammation in immature neurons.
In this study we demonstrate that .NO production mediates, at least in part, neurotoxicity
in immature neurons exposed to UCB, alone or in combination with TNF-α+IL-1β. This
corroborates our recent observation in that oxidative stress and necrotic cell death induced
by UCB in mature neurons decreases by concomitant treatment with the NOS inhibitor, l-
NAME (Fernandes et al., 2010). Additionally, in P7 rats, local elimination of nNOS resulted in
a significant attenuation of the damage after hypoxic-ischemic insult (Ferriero et al., 1995)
and nNOS deficiency through genetic targeting was also neuroprotective in neonatal mice
(Ferriero et al., 1996). In other studies with hypoxia-ischemia models, NOS inhibition reduced
caspase-3 activation (Zhu et al., 2004) and conferred tissue protection (Peeters-Scholte et
al., 2002), further indicating that .NO production exerts cytotoxicity in the developing brain.
A very remarkable finding of our study is the JNK1/2 MAPK activation observed when
immature neurons are exposed to UCB, alone or in combination with TNF-α+IL-1β. The
activation of JNK1/2 is related to toxicity in developing neurons, since overexpression of
activated JNK1/2 was shown to produce apoptosis, as suppression of this protein protected
against neuronal death induced by deprivation of nerve growth factor in sympathetic and
hippocampal neurons (Estus et al., 1994, Ham et al., 1995, Schlingensiepen et al., 1993).
Interestingly, in our study, inhibition of .NO production led to a significant reduction in JKN1/2
activation. This observation is in good agreement with some reports on models of
neurodegenerative diseases, such as Alzheimer’s and Parkinson’s, where inhibition of .NO-
induced JNK 1/2 phosphorylation conferred protection against neuronal cell death (Katsuki et
al., 2006, Marques et al., 2003).

BIND is increased by inflammation __________________________________________________________________________
101
Here, we observed a reduction of either the intrinsic and extrinsic caspase cascades by
using the selective JNK1/2 inhibitor, SP600125. Due to the fact that members of the
antiapoptotic Bcl-2 family proteins are inactivated through JNK1/2 phosphorylation (Inoshita
et al., 2002, Maundrell et al., 1997), we may conclude that in our model JNK1/2 activation is
altering mitochondrial function. In agreement with our thoughts, there are several reports on
dopaminergic neuron models, where a correlation between SAPKs activation and
neurotoxicity was established: (i) phosphorylation of p38 by oxidative stress was linked to
activation of both caspases-8- and -9 (Choi et al., 2004); and (ii) repression of JNK1/2
activation by transfection of a dominant negative mutant SEK1(Lys 3 Arg) blocked dopamine-
induced apoptosis (Luo et al., 1998). In this last study, antioxidants, such as N-acetylcysteine
(NAC) and catalase, blocked dopamine-induced JNK1/2 activation and subsequent
apoptosis, confirming that dopamine-induced oxidative stress is involved in JNK1/2 pathway.
More recently, apoptotic cascade mediated by caspase-3 activation has been demonstrated
to be JNK1/2 dependent in different neuronal cell models (Cerezo-Guisado et al., 2007,
Sahara et al., 2008). Therefore, inhibition of SAPKs signalling might represent a therapeutic
target in acute brain insults, such as neonatal hyperbilirubinemia and associated
inflammation.
In summary, the data obtained in the present study contributes to a better understanding
of the mechanisms underlying neurotoxicity in conditions mimicking a moderate to severe
hyperbilirubinemia in the early neonatal period. Actually, and as schematically represented in
Figure III.7, they demonstrate that UCB-induced neuronal dysfunction in 3 DIV neurons
results from activation of both the intrinsic and extrinsic pathways of apoptosis, that injury is
linked to oxidative stress, and that .NO signalling and JNK1/2 activation are key players. The
association of pro-inflammatory cytokines, TNF-α+IL-1β, to the condition of
hyperbilirubinemia significantly increased the cytotoxic potential of UCB through the same
cascade of mediators. Most important, the results provide supportive evidence for the
commonly indicated higher risk of UCB brain damage in a condition of infection, thus
justifying that treatment should be carried out in these conditions at lower levels of UCB than
those for well-appearing jaundiced neonates. These advances may substantiate target-
driven approaches to the prevention and treatment of UCB-induced neurological damage,
and provide fruitful opportunities for future investigations.

Chapter III __________________________________________________________________________
102
Figure III.7 - Schematic representation of the cellular targets involved in unconjugated bilirubin (UCB) injury to immature cortical neurons, all of them further stimulated by the combination of UCB with pro-inflammatory cytokines (TNF-α and IL-1β), and modulating effects by specific inhibitors. Lighter grey and small arrows indicate the effects of UCB, while darkness grey and large arrows point to the combined effects of UCB plus TNF-α+IL-1β; lines with blocked ends indicate steps that are being modulated. UCB interaction with neurons leads to neuronal nitric oxide synthase (nNOS) increased expression, nitric oxide (.NO) production and activation of c-Jun N-terminal kinase (JNK1/2). As a consequence of these events, mitochondrial and extrinsic pathways of apoptosis are initiated, with activation of caspases-9 and -8, respectively. Activation of caspase-9 further activates the effector capase-3, leading to neuronal apoptosis. Activation of caspase-8 triggers the cleavage of the pro-apoptotic Bid into the truncated form (tBid) that is translocated to mitochondria, thus propagating the apoptotic signal by activation of caspase-3. Ultimately, these events lead to neuronal dysfunction, as assessed by
3-(4,5-dimethylthiazol, 2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. Co-incubation of UCB with TNF-α+IL-1β intensifies all the events activated by UCB alone. Inhibition of JNK1/2 activation by SP600125 conferred neuroprotection to immature neurons exposed either to UCB alone or UCB plus TNF-α+IL-1β, corroborating the involvement of this signaling pathway in the neuronal demise observed. Upstream inhibition of nNOS by N-ω-nitro-L-arginine methyl ester (L-NAME) prevents downstream events from occurring, thus pointing to .NO as a key mediator in UCB-induced neuronal dysfunction of immature neurons.
nNOS activation
JNK1/2 activation
NO production
Caspase-9Caspase-3
L- NAME
SP600125
Caspase-8
Bid
UCBTNF-α
IL-1β
UCB
tBid

BIND is increased by inflammation __________________________________________________________________________
103
5. References Adams, J. M. (2003) Ways of dying: multiple pathways to apoptosis. Genes Dev, 17, 2481-
2495.
Bian, K. and Murad, F. (2001) Diversity of endotoxin-induced nitrotyrosine formation in
macrophage-endothelium-rich organs. Free Radic Biol Med, 31, 421-429.
Bradford, M. M. (1976) A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72, 248-
254.
Brito, M. A., Vaz, A. R., Silva, S. L., Falcão, A. S., Fernandes, A., Silva, R. F. M. and Brites,
D. (2010) N-methyl-D-aspartate receptor and neuronal nitric oxide synthase activation
mediate bilirubin-induced neurotoxicity. Mol Med, 16, 372-380.
Brito, M. A., Lima, S., Fernandes, A., Falcão, A. S., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008a) Bilirubin injury to neurons: contribution of oxidative stress and rescue by
glycoursodeoxycholic acid. Neurotoxicology, 29, 259-269.
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008b) Unconjugated bilirubin differentially affects the redox status of neuronal
and astroglial cells. Neurobiol Dis, 29, 30-40.
Brito, M. A., Rosa, A. I., Silva, R. F. M., Falcão, A. S., Fernandes, A. and Brites, D. (2007)
Oxidative stress and disruption of the nervous cell. In: Focus in Brain Research, pp. 1-33.
Nova Science Publishers, Inc., New York.
Cerezo-Guisado, M. I., Alvarez-Barrientos, A., Argent, R., Garcia-Marin, L. J., Bragado, M. J.
and Lorenzo, M. J. (2007) c-Jun N-terminal protein kinase signalling pathway mediates
lovastatin-induced rat brain neuroblast apoptosis. Biochim Biophys Acta, 1771, 164-176.
Choi, W. S., Eom, D. S., Han, B. S. et al. (2004) Phosphorylation of p38 MAPK induced by
oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic
pathways in dopaminergic neurons. J Biol Chem, 279, 20451-20460.
Dawodu, A. H., Owa, J. A. and Familusi, J. B. (1984) A prospective study of the role of
bacterial infection and G6PD deficiency in severe neonatal jaundice in Nigeria. Trop
Geogr Med, 36, 127-132.
Doré, S., Goto, S., Sampei, K. et al. (2000) Heme oxygenase-2 acts to prevent neuronal
death in brain cultures and following transient cerebral ischemia. Neuroscience, 99, 587-
592.
Estus, S., Zaks, W. J., Freeman, R. S., Gruda, M., Bravo, R. and Johnson, E. M., Jr. (1994)
Altered gene expression in neurons during programmed cell death: identification of c-jun
as necessary for neuronal apoptosis. J Cell Biol, 127, 1717-1727.

Chapter III __________________________________________________________________________
104
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2005) Bilirubin-
induced inflammatory response, glutamate release, and cell death in rat cortical
astrocytes are enhanced in younger cells. Neurobiol Dis, 20, 199-206.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Bilirubin-
induced immunostimulant effects and toxicity vary with neural cell type and maturation
state. Acta Neuropathol, 112, 95-105.
Fernandes, A., Barateiro, A., Falcão, A. S., Silva, S. L., Vaz, A. R., Brito, M. A., Silva, R. F.
M. and Brites, D. (2010) Astrocyte reactivity to unconjugated bilirubin requires TNF-a and
IL-1b receptor signalling pathways. Glia, in press.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007) MAPKs are
key players in mediating cytokine release and cell death induced by unconjugated bilirubin
in cultured rat cortical astrocytes. Eur J Neurosci, 25, 1058-1068.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Gordo, A. C., Gama, M. J., Brito, M. A. and
Brites, D. (2006) Inflammatory signalling pathways involved in astroglial activation by
unconjugated bilirubin. J Neurochem, 96, 1667-1679.
Fernandes, A., Silva, R. F. M., Falcão, A. S., Brito, M. A. and Brites, D. (2004) Cytokine
production, glutamate release and cell death in rat cultured astrocytes treated with
unconjugated bilirubin and LPS. J Neuroimmunol, 153, 64-75.
Ferriero, D. M., Holtzman, D. M., Black, S. M. and Sheldon, R. A. (1996) Neonatal mice
lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury.
Neurobiol Dis, 3, 64-71.
Ferriero, D. M., Sheldon, R. A., Black, S. M. and Chuai, J. (1995) Selective destruction of
nitric oxide synthase neurons with quisqualate reduces damage after hypoxia-ischemia in
the neonatal rat. Pediatr Res, 38, 912-918.
Ham, J., Babij, C., Whitfield, J., Pfarr, C. M., Lallemand, D., Yaniv, M. and Rubin, L. L. (1995)
A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell
death. Neuron, 14, 927-939.
Hansen, T. W. R. (2002) Mechanisms of bilirubin toxicity: clinical implications. Clin Perinatol,
29, 765-778, viii.
Hansen, T. W. R., Maynard, E. C., Cashore, W. J. and Oh, W. (1993) Endotoxemia and brain
bilirubin in the rat. Biol Neonate, 63, 171-176.
Hemmer, K., Fransen, L., Vanderstichele, H., Vanmechelen, E. and Heuschling, P. (2001) An
in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric
oxide and tumor necrosis factor-alpha. Neurochem Int, 38, 557-565.
Heneka, M. T., Loschmann, P. A., Gleichmann, M., Weller, M., Schulz, J. B., Wullner, U. and
Klockgether, T. (1998) Induction of nitric oxide synthase and nitric oxide-mediated

BIND is increased by inflammation __________________________________________________________________________
105
apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-
alpha/lipopolysaccharide. J Neurochem, 71, 88-94.
Hengartner, M. O. (2000) The biochemistry of apoptosis. Nature, 407, 770-776.
Inoshita, S., Takeda, K., Hatai, T., Terada, Y., Sano, M., Hata, J., Umezawa, A. and Ichijo, H.
(2002) Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to
oxidative stress. J Biol Chem, 277, 43730-43734.
Kajta, M., Trotter, A., Lason, W. and Beyer, C. (2006) Impact of 17beta-estradiol on cytokine-
mediated apoptotic effects in primary hippocampal and neocortical cell cultures. Brain
Res, 1116, 64-74.
Kaplan, M. and Hammerman, C. (2005) Understanding severe hyperbilirubinemia and
preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin Chim
Acta, 356, 9-21.
Katsuki, H., Okawara, M., Shibata, H., Kume, T. and Akaike, A. (2006) Nitric oxide-producing
microglia mediate thrombin-induced degeneration of dopaminergic neurons in rat midbrain
slice culture. J Neurochem, 97, 1232-1242.
Konsman, J. P., Drukarch, B. and Van Dam, A. M. (2007) (Peri)vascular production and
action of pro-inflammatory cytokines in brain pathology. Clin Sci (Lond), 112, 1-25.
Korhonen, R., Lahti, A., Kankaanranta, H. and Moilanen, E. (2005) Nitric oxide production
and signaling in inflammation. Curr Drug Targets Inflamm Allergy, 4, 471-479.
Leviton, A. and Gressens, P. (2007) Neuronal damage accompanies perinatal white-matter
damage. Trends Neurosci, 30, 473-478.
Li, H., Zhu, H., Xu, C. J. and Yuan, J. (1998) Cleavage of BID by caspase 8 mediates the
mitochondrial damage in the Fas pathway of apoptosis. Cell, 94, 491-501.
Luo, Y., Umegaki, H., Wang, X., Abe, R. and Roth, G. S. (1998) Dopamine induces
apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem, 273,
3756-3764.
Marques, C. A., Keil, U., Bonert, A., Steiner, B., Haass, C., Muller, W. E. and Eckert, A.
(2003) Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedish
amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J
Biol Chem, 278, 28294-28302.
Marx, C. E., Jarskog, L. F., Lauder, J. M., Lieberman, J. A. and Gilmore, J. H. (2001)
Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for
schizophrenia. Biol Psychiatry, 50, 743-749.
Maundrell, K., Antonsson, B., Magnenat, E. et al. (1997) Bcl-2 undergoes phosphorylation by
c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the
constitutively active GTP-binding protein Rac1. J Biol Chem, 272, 25238-25242.

Chapter III __________________________________________________________________________
106
McDonagh, A. F. (1979) Bile pigments: Bilatrienes and 5,15-biladienes. In: The Porfirins, pp.
293-491. Academic Press, San Diego.
Mielke, K. and Herdegen, T. (2000) JNK and p38 stresskinases--degenerative effectors of
signal-transduction-cascades in the nervous system. Prog Neurobiol, 61, 45-60.
Ngai, K. C. and Yeung, C. Y. (1999) Additive effect of tumor necrosis factor-alpha and
endotoxin on bilirubin cytotoxicity. Pediatr Res, 45, 526-530.
Ostrow, J. D., Pascolo, L., Brites, D. and Tiribelli, C. (2004) Molecular basis of bilirubin-
induced neurotoxicity. Trends Mol Med, 10, 65-70.
Patel, J. R. and Brewer, G. J. (2008) Age-related changes to tumor necrosis factor receptors
affect neuron survival in the presence of beta-amyloid. J Neurosci Res, 86, 2303-2313.
Peeters-Scholte, C., Koster, J., Veldhuis, W. et al. (2002) Neuroprotection by selective nitric
oxide synthase inhibition at 24 hours after perinatal hypoxia-ischemia. Stroke, 33, 2304-
2310.
Petty, M. A. and Lo, E. H. (2002) Junctional complexes of the blood-brain barrier:
permeability changes in neuroinflammation. Prog Neurobiol, 68, 311-323.
Rodrigues, C. M. P., Solá, S. and Brites, D. (2002a) Bilirubin induces apoptosis via the
mitochondrial pathway in developing rat brain neurons. Hepatology, 35, 1186-1195.
Rodrigues, C. M. P., Solá, S., Brito, M. A., Brites, D. and Moura, J. J. (2002b) Bilirubin
directly disrupts membrane lipid polarity and fluidity, protein order, and redox status in rat
mitochondria. J Hepatol, 36, 335-341.
Rodrigues, C. M. P., Solá, S., Silva, R. F. M. and Brites, D. (2000) Bilirubin and amyloid-β
peptide induce cytochrome c release through mitochondrial membrane permeabilization.
Mol Med, 6, 936-946.
Rossi, D. and Volterra, A. (2009) Astrocytic dysfunction: insights on the role in
neurodegeneration. Brain Res Bull, 80, 224-232.
Sahara, N., Murayama, M., Lee, B., Park, J. M., Lagalwar, S., Binder, L. I. and Takashima, A.
(2008) Active c-jun N-terminal kinase induces caspase cleavage of tau and additional
phosphorylation by GSK-3beta is required for tau aggregation. Eur J Neurosci, 27, 2897-
2906.
Schlingensiepen, K. H., Schlingensiepen, R., Kunst, M., Klinger, I., Gerdes, W., Seifert, W.
and Brysch, W. (1993) Opposite functions of jun-B and c-jun in growth regulation and
neuronal differentiation. Dev Genet, 14, 305-312.
Sener, G., Toklu, H., Kapucu, C., Ercan, F., Erkanli, G., Kacmaz, A., Tilki, M. and Yegen, B.
C. (2005) Melatonin protects against oxidative organ injury in a rat model of sepsis. Surg
Today, 35, 52-59.

BIND is increased by inflammation __________________________________________________________________________
107
Shapiro, S. M. (2005) Definition of the clinical spectrum of kernicterus and bilirubin-induced
neurologic dysfunction (BIND). J Perinatol, 25, 54-59.
Silva, R. F. M., Rodrigues, C. M. P. and Brites, D. (2002) Rat cultured neuronal and glial cells
respond differently to toxicity of unconjugated bilirubin. Pediatr Res, 51, 535-541.
Silva, S. L., Vaz, A. R., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F.
M. and Brites, D. (2010) Features of bilirubin-induced reactive microglia: From
phagocytosis to inflammation. Neurobiol Dis.
Stevenson, D. K., Dennery, P. A. and Hintz, S. R. (2001) Understanding newborn jaundice. J
Perinatol, 21 Suppl 1, S21-24; discussion S35-29.
Takahashi, K., Funata, N., Ikuta, F. and Sato, S. (2008) Neuronal apoptosis and
inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-
2. J Neuroinflammation, 5, 11.
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A. and Gaestel, M. (2008)
MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing
neuroinflammation--relevance in a mouse model of Parkinson's disease. J Neurochem,
105, 2039-2052.
Tibbles, L. A. and Woodgett, J. R. (1999) The stress-activated protein kinase pathways. Cell
Mol Life Sci, 55, 1230-1254.
Vaz, A. R., Delgado-Esteban, M., Brito, M. A., Bolaños, J. P., Brites, D. and Almeida, A.
(2010) Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in
immature cortical neurons: assessment of the protective effects of glycoursodeoxycholic
acid. J Neurochem, 112, 56-65.
Watchko, J. F. (2006) Hyperbilirubinemia and bilirubin toxicity in the late preterm infant. Clin
Perinatol, 33, 839-852; abstract ix.
Yeung, C. Y. and Ngai, K. C. (2001) Cytokine- and endotoxin-enhanced bilirubin cytotoxicity.
J Perinatol, 21 Suppl 1, S56-58; discussion S59-62.
Zhang, R., Yamada, J., Hayashi, Y., Wu, Z., Koyama, S. and Nakanishi, H. (2008) Inhibition
of NMDA-induced outward currents by interleukin-1beta in hippocampal neurons.
Biochem Biophys Res Commun, 372, 816-820.
Zhu, C., Wang, X., Qiu, L., Peeters-Scholte, C., Hagberg, H. and Blomgren, K. (2004)
Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor
in neonatal rat cerebral hypoxia-ischaemia. J Neurochem, 90, 462-471.


Chapter IV
IV. Selective vulnerability of rat brain regions to unconjugated bilirubin
Ana Rita Vaz, Sandra Leitão Silva, Andreia Barateiro, Ana Sofia Falcão, Adelaide
Fernandes, Maria A Brito, Dora Brites
Research Institute for Medicines and Pharmaceutical Sciences (iMed.UL), Faculdade
de Farmácia, University of Lisbon, Av. Professor Gama Pinto, Lisbon 1649-003,
Portugal.
Molecular and Cellular Neuroscience (submitted).

Acknowledgements This work was supported by grants PTDC/SAU-NEU/64385/2006 (to D.B.) and
BD/30292/2006 (to A.R.V.) from Fundação para a Ciência e a Tecnologia, Lisbon,
Portugal.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
111
Abstract
Hippocampus is one of the brain regions most vulnerable to unconjugated bilirubin
(UCB) encephalopathy, although cerebellum also shows selective yellow staining in
kernicterus. We demonstrated that UCB induces oxidative stress in cortical neurons,
disruption of neuronal network dynamics, either in developing cortical or hippocampal
neurons, and that immature cortical neurons are more prone to UCB-induced injury.
Here, we studied features of oxidative stress and cell dysfunction induced by UCB in
immature rat neurons isolated from cortex, cerebellum and hippocampus. We also
explored whether oxidative damage and its regulation contribute to neuronal dysfunction
induced by hyperbilirubinemia, in terms of neurite extension and ramification, and cell
death. Our results show that UCB induces neuronal nitric oxide synthase expression, as
well as production of nitrites and cyclic guanosine monophosphate in immature neurons,
mainly in those from hippocampus. After exposure to UCB, hippocampal neurons
presented the highest content of reactive oxygen species, disruption of glutathione redox
status and cell death, when compared to those from cortex or cerebellum. In particular,
the results indicated that cells exposed to UCB undertake an adaptive response that
involves DJ-1, a multifunctional neuroprotective protein involved in cellular oxidation
status maintenance. However, longer neuronal exposure to UCB down-regulated DJ-1
expression, especially in hippocampal neurons. In addition, UCB induced impairment in
neurite outgrowth and branching, mainly in immature neurons from hippocampus.
Interestingly, pre-incubation with N-acetylcysteine, a precursor of glutathione synthesis,
conferred neuroprotection to UCB-induced oxidative stress and necrotic cell death, as
well as prevented DJ-1 down-regulation and neuritic impairment. Taken together, these
data point oxidative injury and disruption of neuritic network as hallmarks in hippocampal
susceptibility to UCB. Furthermore, it is suggested that local differences in glutathione
content may account to the different susceptibility found between brain regions exposed
to UCB.
Keywords: Cortex; cerebellum; DJ-1; hippocampus; immature neurons; oxidative and
nitrosative stress; neurite outgrowth and branching; unconjugated bilirubin.

Chapter IV __________________________________________________________________________
112
1. Introduction Hyperbilirubinemia, a very common condition in the neonatal period, characterized by
increased serum levels of unconjugated bilirubin (UCB) (Stevenson et al., 2001, Dennery et
al., 2001), is responsible for the clinical manifestation of jaundice. Although normal (or
slightly increased) levels of UCB provides protection against injury resulting from oxidation
(Doré et al., 2000), elevated UCB concentrations cause nerve cell damage, leading to
adverse neurological outcomes (Hansen, 2002), ranging from minor neurologic dysfunction
(Soorani-Lunsing et al., 2001) to chronic and permanent sequelae, or even death (Ostrow et
al., 2004, Shapiro, 2005). In addition, high levels of UCB are related with an augmented risk
for the appearance of long-term neurodevelopment disabilities (Dalman and Cullberg, 1999).
The risk of bilirubin-induced neurologic dysfunction is particularly enhanced in premature
newborns due to the higher rates of UCB production and the immaturity of the excretion
pathways (Stevenson et al., 2001, Watchko, 2006). Moreover, cerebral palsy is a common
condition in preterm infants at risk of kernicterus, in spite of the relatively low total serum
bilirubin levels (Gkoltsiou et al., 2008). However, little is known about mechanisms underlying
the increased vulnerability of selected regional neuronal populations in UCB-induced
neuronal damage. Hippocampus is one of the brain regions with preferential UCB
accumulation in severely jaundiced neonates who died with kernicterus, although cerebellum
and corpus striatum also showed selective yellow deposits (Ahdab-Barmada and Moossy,
1984, Hansen, 2000). This preferential deposition seems to be related with increased
vulnerability to hypoxic-ischemic injury (Perlman et al., 1997), as well as with the enhanced
vascularization and up-regulation of vascular endothelial growth factor (VEGF) that we have
recently observed in the hippocampus, cerebellum and striatum of a preterm infant with
sepsis who died with the diagnosis of kernicterus (personal communication Alexandra Brito,
2010).
In the last years, we demonstrated the involvement of oxidative stress in the
mechanisms underlying neuronal cell demise by clinically relevant concentrations of UCB
(Brito et al., 2004, Brito et al., 2008a, Brito et al., 2010). The UCB-induced dysfunction may
culminate in neuronal cell death (Silva et al., 2002, Falcão et al., 2006), which appears to be
mediated, at least in part, by perturbation of mitochondria (Rodrigues et al., 2002, Rodrigues
et al., 2000, Vaz et al., 2010). In addition, immature nerve cells were shown to be more
susceptible than more differentiated ones to UCB-induced toxicity (Falcão et al., 2006).
These findings provided a valuable contribute to the current understanding of the
neuropathological effects of UCB, as dysfunction and degeneration of neurons in several
neurological disorders are usually associated with reactive oxygen species (ROS) and/or
reactive nitrogen species (RNS) production (Mattson and Liu, 2002, Brito et al., 2007).

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
113
Recently, one of these oxidant species, nitric oxide (.NO), although important in cellular
signalling, showed to have a key role in the mechanisms of neurotoxicity by UCB, both in
mature (Brito et al., 2008a, Brito et al., 2010) and in immature neurons (personal
communication Ana Rita Vaz, 2010). RNS are also implicated in synapse injury (Sunico et
al., 2010) and early exposure to UCB provoked deleterious effects in neurogenesis,
neuritogenesis and synaptogenesis (Falcão et al., 2007, Fernandes et al., 2009), possibly
contributing to the development of mental illness in later life. Therefore, a better
understanding of the role of antioxidants and molecules involved in response to oxidative
stress represent important strategies to prevent neuronal injury by hyperbilirubinemia.
N-acetylcysteine (NAC) is a thiol compound that is converted to cysteine, an important
precursor of cellular glutathione (Zachwieja et al., 2005, Dringen, 2000). NAC is described by
its antioxidant effects in two ways: firstly, as a source of the cysteine aminoacid, it promotes
the biosynthesis of the tripeptide γ-l-glutamyl-l-cysteinylglycine, known as glutathione (GSH),
thus increasing GSH supply for glutathione peroxidise; secondly, as a source of sulfydryl
groups it promotes the reduction of ROS (Ocal et al., 2004). Several in vitro and in vivo
studies support the antioxidant effect of NAC. Treatment with NAC conferred neuroprotection
in lead-induced lipid peroxidation and in antioxidant enzyme activities deficiencies of rats’
brain (Nehru and Kanwar, 2004), as well as in hypoxia-induced oxidative stress in rat
cultured hippocampal neurons (Jayalakshmi et al., 2005). In addition, treatment with NAC
decreased lipid peroxidation in cerebral cortex, midbrain and cerebellum observed in
jaundiced rats (Karageorgos et al., 2006), and results from our group demonstrated that NAC
protects against UCB-induced protein oxidation in rat cultured cortical neurons (Brito et al.,
2008b).
DJ-1, also known as PARK7 [Parkinson disease (autosomal recessive, early onset) 7] is
a member of the peptidase C56 family of proteins, but is not known to exhibit proteolytic
activity. It functions as a redox-sensitive chaperone, like a sensor for oxidative stress,
protecting neurons against oxidative stress and cell death (Jin et al., 2005, Bonifati et al.,
2003). Recently, it was proposed that UCB-treated cells may undertake an adaptative
response that involves DJ-1 (Deganuto et al., 2010).
In this study, we investigated whether there is a dissimilar brain regional susceptibility to
UCB-induced oxidative damage and disruption of neurite outgrowth and branching in
immature neurons able to determine the selective pattern of UCB deposition and brain
damage in specific brain areas characteristic of kernicterus, such as cerebellum and
hippocampus. We also looked for potential mechanisms involved in UCB-induced
neurotoxicity modulation, namely DJ-1 protein expression and glutathione content.

Chapter IV __________________________________________________________________________
114
2. Materials and Methods
2.1. Chemicals Neurobasal medium, B-27 supplement (50X), Hanks’ balanced salt solution (HBSS),
Hanks’ balanced salt solution without Ca2+ and Mg2+ (Ca2+ and Mg2+ free HBSS), gentamicin
(50 mg/mL), and trypsin (2.5 g/L) were acquired from Invitrogen (Carlsbad, CA). Human
serum albumin (HSA) (fraction V, fatty acid free), NAC, dihydrorhodamine 123 (DHR 123),
3,8-diamino-5-(3-(diethyl-methylamino)propyl)-6-phenyl phenanthridinium diiodide,
sulfosalicyclic acid, and 2-vinylpyridine, Hoechst 33258 dye, 1-isobutyl-3-methylxanthine
(IBMX), primary monoclonal antibody mouse anti-β-actin, N-1-naphthylethylenediamine,
sulfanilamide and sodium nitrite were purchased from Sigma Chemical Co (St Louis, MO).
UCB was also from Sigma and purified as previously described (McDonagh 1979).
Nitrocellulose membrane, Hyperfilm ECL and horseradish peroxidase-labelled sheep anti-
mouse IgG were from Amersham Biosciences (Piscataway, NJ, USA). Cell lysis buffer,
LumiGLO® and antibody directed to caspase-3 were acquired from Cell Signalling (Beverly,
MA, USA).Cyclic guanosine monophosphate (cGMP) determination kit was from Enzo Life
Sciences (Plymouth Meeting, PA, USA). Antibodies directed to DJ-1 and microtubule
associated protein (MAP)-2 were from Chemicon (Temecula, CA, USA) while antibody
against neuronal nitric oxide synthase (nNOS) was from Becton Dickinson Biosciences (San
José, CA, USA).
2.2. Neurons in primary culture
Animal care followed the recommendations of European Convention for the Protection of
Vertebrate Animals Used for Experimental and other Scientific Purposes (Council Directive
86/609/EEC) and National Law 1005/92 (rules for protection of experimental animals).
Cortical, hippocampal and cerebellar neurons were isolated from fetuses of 16-17-day
pregnant Wistar rats, as previously described (Silva et al., 2002). Fetuses were collected in
HBSS medium, brain cortices, hippocampi and cerebella were mechanically fragmented, and
the fragments transferred to a 0.5 g/L trypsin in Ca2+ and Mg2+ free HBSS medium and
incubated for 15 min at 37ºC. After trypsinization, cells were washed twice in Ca2+ and Mg2+
free HBSS medium containing 10% fetal bovine serum, and resuspended in Neurobasal
medium supplemented with 0.5 mM L-glutamine, 25 μM L-glutamic acid, 2% B-27
supplement, and 0.12 mg/mL gentamicin. Finally, cells were seeded on poly-D-lysine coated
tissue culture plates at a density of 2 x 105 cells/cm2 and maintained at 37ºC in a humidified
atmosphere of 5% CO2. For hippocampal neurons, cells were first seeded in plating medium
(MEM with Earle’s salts supplemented with 10 mM HEPES, 10 mM sodium pyruvate, 1 mM

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
115
glutamine, 12.5 μM glutamate, 10% FBS and 0.6% glucose) and after 2 h, the media was
replaced with neuronal growth medium as abovementioned. In this work we used neurons at
3 days in vitro (DIV).
2.3. Treatment of neurons
Neurons from cortex, hippocampus and cerebellum were incubated in Neurobasal
medium without (control) or with 50 μM UCB in the presence of 100 μM HSA from 1 to 24 h,
at 37ºC. Stock UCB solutions were extemporarily prepared in 0.1 M NaOH under the dark
and the pH adjusted to 7.4 using 0.1 M HCl. In parallel studies, cells were incubated with
100 µM NAC, a precursor of glutathione synthesis, for 1 h prior to UCB addition.
2.4. Quantification of nitrite levels Nitric oxide levels were estimated by measuring the concentrations of nitrites (NO2
-),
which are the resulting .NO metabolites. Briefly, supernatants free from cellular debris were
mixed with Griess reagent [1 part 1% (w/v) sulfanilamide in 5% H3PO4, 1 part 0.1% (w/v) N-1-
naphthylethylenediamine (v/v)] in 96-well tissue culture plates for 10 min at room
temperature in the dark. The absorbance at 540 nm was determined using a microplate
reader (Bio-Rad Laboratories, Hercules, CA, USA).
2.5. Western blot assay The intracellular forms of nNOS, caspase-3 and DJ-1 were determined by Western blot
analysis as usual in our laboratory (Fernandes et al., 2006). Briefly, cells were washed in ice-
cold PBS and lysed in a buffer containing 20 mM Tris-HCl (pH 7.5); 150 mM NaCl; 1 mM
Na2EDTA; 1 mM EGTA; 1% (v/v) Triton X-100; 2.5 mM sodium pyrophosphate; 1 mM β-
glycerophosphate; 1 mM Na3VO4; 1 μg/mL leupeptine and 1 mM PMSF. The lysate was
sonicated for 20 s, centrifuged at 14000 g for 10 min at 4ºC and the supernatants were
collected and stored at -80ºC. Protein concentrations were determined according to the
Bradford method (Bradford, 1976) using Bio-Rad’s Protein Assay reagent (Bio-Rad, CA,
USA). Equal amounts of protein were subjected to sodium dodecyl sulphate-polyacrylamide
gel electrophoresis and transferred to a nitrocellulose membrane. After blocking with 5% milk
solution, membranes were incubated with the primary antibody overnight at 4ºC [mouse anti-
nNOS (1:2500), rabbit anti-caspase-3 (1: 1000), rabbit anti-DJ1 (1:200) or anti-β-actin
(1:5000)], and finally with horseradish peroxidase-labelled secondary antibody. Protein
bands were detected by LumiGLO® and visualized by autoradiography with Hyperfilm ECL.

Chapter IV __________________________________________________________________________
116
2.6. Determination of cGMP concentration:
For quantification of cGMP content, the phosphodiesterase inhibitor IBMX was included
in the incubation medium. Cell extracts collected from 9.6 cm2 wells were used for cGMP
determination using a commercially available kit from Enzo Life Sciences and measurements
were performed according to manufacturer’s instructions.
2.7. Glutathione measurement After incubation period, neurons were washed with ice-cold PBS and immediately
collected by scrapping off with 0.5 mL of 1% (w/v) sulfosalicyclic acid. Cell lysates were
centrifuged at 13,000 g for 5 min at 4ºC, and the supernatants used for glutathione
determinations. Total glutathione content (GSt, i.e. the amount of GSH plus two times the
amount of GSSG) and oxidized glutathione (GSSG) were measured and calculated as
previously described (Dringen and Hamprecht, 1996) and GSt and GSSG concentrations
were expressed as nanomoles per milligram of protein.
2.8. Assessment of ROS formation The nonfluorescent DHR 123 easily crosses cell membranes due to its lipophilicity and is
converted by ROS into rhodamine 123, a fluorescent compound that accumulates in
mitochondria and is considered as a sensitive indicator of ROS production in cell systems
(Gomes et al., 2005). To evaluate the production of ROS in neuronal cultures, cells were
seeded on glass coverslips placed in the 12-well culture plates. Cells were loaded, under
light protection, with 6 μM DHR 123 for 30 min at 37 °C, prior to cellular treatment. At the end
of the incubation period, cells were fixed with freshly prepared 4% paraformaldehyde in PBS,
and the nuclei immunostained with Hoechst 33258 dye. Cellular fluorescence was observed
using a fluorescence microscope (Axioskope®, Zeiss, Germany) and the intensity of the
fluorescence emission was quantified in at least six microscopic fields (×400) per sample
with an image analyzer software (ImageJ 1.29×, National Institutes of Health, USA) and
expressed as a percentage per total number of cells. Since UCB was referred as an
autofluorescent molecule (Özkan et al., 1995), a set of experiments was performed in
parallel, with no addition of DHR 123. The fact that no variations in the fluorescence intensity
were noticed in these control experiments guarantees that the rise in the fluorescence
intensity observed in the UCB-treated samples was due to ROS formation and not to UCB
interference.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
117
2.9. Evaluation of cell death Necrotic-like cell death was assessed by monitoring the cellular uptake of the fluorescent
dye propidium iodide (PI). PI readily enters and stains non-viable cells, but cannot cross the
membrane of viable cells. This dye binds to double-stranded DNA and emits red
fluorescence (630 nm; absorbance 493 nm). Unpermeabilized adherent cells cultured on
coverslips were incubated with a 75 μM PI solution for 15 min in the absence of light.
Subsequently, cells were fixed with freshly prepared 4% (w/v) paraformaldehyde in PBS and
the nuclei immunostained with Hoechst 33258 dye. Red-fluorescence and U.V. images of six
random microscopic fields (original magnification: 400×) were acquired per sample by using
a fluorescence microscope (Axioskope®, Zeiss, Germany) and the percentage of PI positive
cells was counted and expressed as a percentage per total number of cells.
2.10. Neurite Extension and Ramification For immunofluorescence detection of the cytoskeletal protein MAP-2, known to be
located mainly in dendrites and widely used as a neuritic marker (Hammond, 2001), cells
were fixed with freshly prepared 4% (w/v) paraformaldehyde in PBS and a standard indirect
immunocytochemical technique was carried out using a mouse anti-MAP-2 antibody (1:100)
as the primary antibody and a horse FITC-labeled anti-mouse antibody (1:227) as the
secondary antibody. Fluorescence was visualized by using a fluorescence microscope
(Axioskope®, Zeiss, Germany). Green-fluorescence images of ten random microscopic fields
were acquired per sample. Evaluation of neurite extension and number of nodes from
individual neurons were determined using HCA-Vision Neurite Analysis software (Australia).
2.11. Densitometry and statistical analysis The relative intensities of protein and nucleic acid bands were analysed using the
Quantity One (version 4.6) program (Bio-Rad, CA, USA). Results of, at least, three different
experiments were expressed as mean ± SEM. Significant differences between two groups
were determined by the two-tailed t-test performed on the basis of equal and unequal
variance as appropriate. Comparison of more than two groups was done by ANOVA using
Instat 3.05 (GraphPad Software, San Diego, CA, USA), followed by multiple comparisons
Bonferroni post-hoc correction. Mean values were considered statistically significant when P
values were lower than 0.05.

Chapter IV __________________________________________________________________________
118
3. Results
3.1. UCB-induced nNOS expression and production of nitrites and cGMP is enhanced in immature hippocampal neurons as compared to cerebellar or cortical neurons To investigate whether different brain regions present particular susceptibilities to UCB-
induced neuronal damage, we used 3 DIVs neurons isolated from cortex, hippocampus and
cerebellum. Neuronal cells were incubated with 50 μM UCB plus 100 μM HSA, to produce a
free UCB concentration of ~100 nM (Ostrow et al., 2003, Weisiger et al., 2001), that mimics
moderate to severe neonatal jaundice, a condition already shown to induce oxidative stress
and cell death in immature cortical neurons (Vaz et al., 2010). We started by investigating
whether UCB up-regulates nNOS expression, as well as production of nitrites and cGMP, the
resulting product of soluble guanylate cyclase stimulation by .NO (Knowles et al., 1989), in
neurons exposed to UCB for 1 to 24 h. As shown in Figure IV.1A, UCB rapidly (at 1 h)
induced nNOS expression in cortical and hippocampal neurons, but not in cerebellar
neurons. The effect was already pronounced after 4 h of treatment (p<0.01 vs. respective
controls) and decreased at 24 h. Furthermore, we observed a raise in the production of
nitrites (Fig. IV.1B), as well as cGMP (Fig. IV.1C), after 1 h of treatment with UCB, in neurons
from all brain regions (p<0.01 for nitrites and p<0.05 for cGMP vs. respective controls). To
note that hippocampal neurons, namely at 4 h of incubation with UCB, showed to be the
most sensitive regarding nNOS expression (1.4- vs. 1.2- and 1.0- fold in cortical and
cerebellar neurons p<0.01), nitrite production (3.8- vs. 2.2-fold in cortical and cerebellum
neurons, p<0.01) and cGMP content (1.7- vs. 1.3-fold, in cortical and cerebellar neurons,
p<0.01 and p<0.05, respectively).
3.2. UCB-induced oxidative stress is highest in immature hippocampal neurons, probably as a result of the lowest levels of total glutathione Having observed that RNS are produced by immature neurons exposed to UCB, we
considered relevant to look for alterations in the oxidative status of neuronal cells from the
three mentioned brain regions. Firstly, we determined whether UCB induced mitochondrial
ROS production by evaluation of fluorescence intensity of rhodamine 123.
As shown in Figure IV.2A-B, UCB induced oxidative stress more markedly after 4 h
treatment in immature hippocampal and cerebellar neurons (p<0.01 vs. respective controls)
by cortical cells (p<0.05 vs. respective control).

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
119
Figure IV.1 - Unconjugated bilirubin (UCB)-induced neuronal nitric oxide synthase (nNOS) expression and production of nitrites and cyclic GMP (cGMP) is enhanced in immature hippocampal neurons as compared to cerebellar or cortical neurons. Primary neuron cultures from rat cortex, hippocampus and cerebellum, at 3 days in vitro, were incubated with either no addition (control) or with UCB (50 µM), in the presence of human serum albumin (100 μM), from 1 to 24 h at 37ºC. It was determined nNOS expression by western blot analysis (A), nitrites by Griess reagent (B) and cyclic GMP by a colorimetric kit (C). Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $p<0.05, $$p<0.01 vs. cortical neurons; #p<0.05, ##p<0.01 vs. cerebellar neurons.
C
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
Control
UCB 50 μM
0
1
2
3
4
Nitr
ites
(μM
)
0.0
0.5
1.0
1.5
nNO
S (f
old
vs.c
ontro
l)
1h 4h 24hCortical neurons
Control
UCB 50 μM
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
A
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
Control
UCB 50 μM
B
***
** ****
$##
$$##
##
**
** **
**
**
$$##
$$##
##** **
* **
**
***$$#
*
*
0
5
10
15
20
cGM
P (p
mol
/mg
prot
ein)
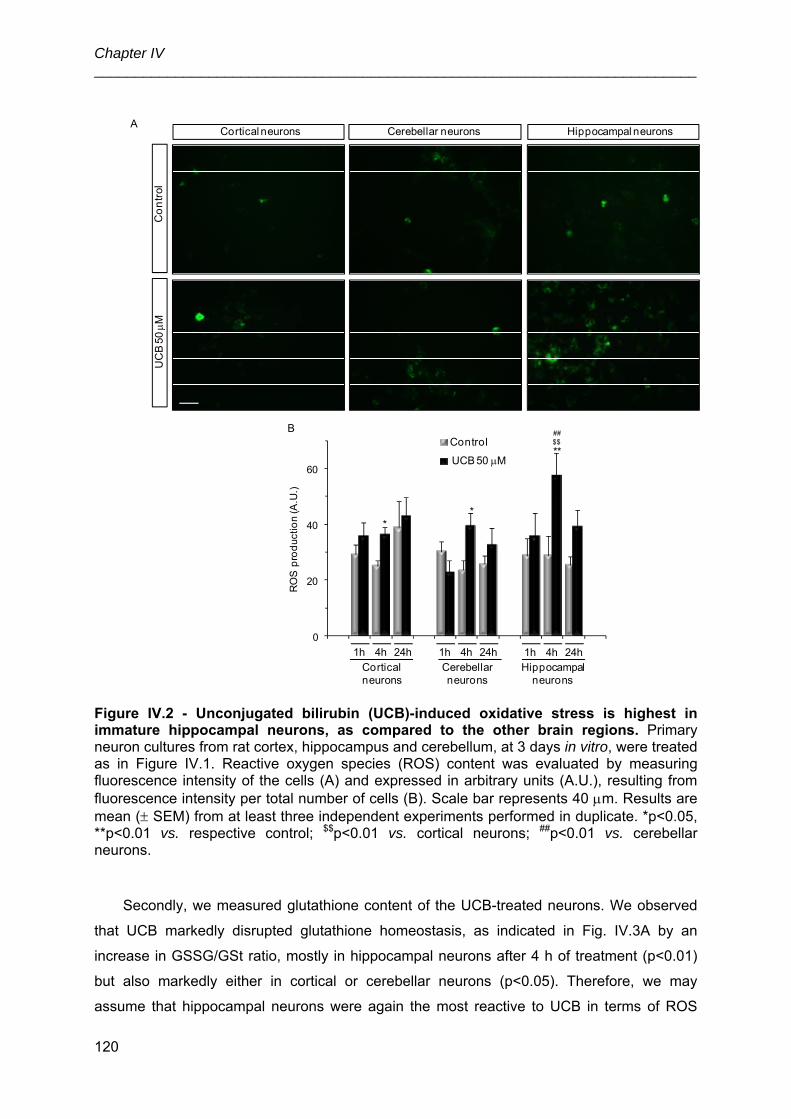
Chapter IV __________________________________________________________________________
120
Figure IV.2 - Unconjugated bilirubin (UCB)-induced oxidative stress is highest in immature hippocampal neurons, as compared to the other brain regions. Primary neuron cultures from rat cortex, hippocampus and cerebellum, at 3 days in vitro, were treated as in Figure IV.1. Reactive oxygen species (ROS) content was evaluated by measuring fluorescence intensity of the cells (A) and expressed in arbitrary units (A.U.), resulting from fluorescence intensity per total number of cells (B). Scale bar represents 40 μm. Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $$p<0.01 vs. cortical neurons; ##p<0.01 vs. cerebellar neurons.
Secondly, we measured glutathione content of the UCB-treated neurons. We observed
that UCB markedly disrupted glutathione homeostasis, as indicated in Fig. IV.3A by an
increase in GSSG/GSt ratio, mostly in hippocampal neurons after 4 h of treatment (p<0.01)
but also markedly either in cortical or cerebellar neurons (p<0.05). Therefore, we may
assume that hippocampal neurons were again the most reactive to UCB in terms of ROS
ACortical neurons Cerebellar neurons
Con
trol
UC
B 50
μM
Hippocampal neurons
0
20
40
60
RO
S p
rodu
ctio
n (A
.U.)
BControl
UCB 50 μM
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
*
**$$##
*

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
121
production (2- vs. 1.4- and 1.7- fold in cortical and cerebellar neurons, respectively, p<0.01),
and disruption of glutathione homeostasis (5.3- vs. 2.3- and 2.1-fold in cortical and cerebellar
neurons, respectively, p<0.01), probably because they show, as indicated in Figure IV.3B,
the lowest levels of total glutathione in the absence of UCB (p<0.01 vs. cortical neurons and
p<0.05 vs. cerebellar neurons), whereas no significant changes were produced by UCB (data
not shown).
Figure IV.3 - Unconjugated bilirubin (UCB)-induced disruption of glutathione metabolism is particularly evident in immature hippocampal neurons, whose vulnerability appears to be determined by the lowest total glutathione levels relatively to the other brain regions. Primary neuron cultures from rat cortex, hippocampus and cerebellum, at 3 days in vitro, were treated as in Figure IV.1. Total (GSt) and oxidized (GSSG) glutathione were determined by an enzymatic assay and expressed as GSSG/GSx ratio (A) and as nmol/mg protein in the case of GSt (B). Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $$p<0.01 vs. cortical neurons; #p<0.05, ##p<0.01 vs. cerebellar neurons.
3.3. UCB-induced neuronal death is higher in immature cells from hippocampus than in those from cortex or cerebellum We kept on determining whether UCB-induced oxidative stress courses in parallel with
cell death, by evaluating the necrotic-like cell death using PI uptake as an indicator of
membrane integrity and cell damage. We have also determined the possible involvement of
the apoptotic pathways by evaluating the relative levels of active form of caspase-3, an
effector caspase of apoptotic cascade (Fink and Cookson, 2005). As demonstrated in Figure
IV.4 A-B, UCB increased cellular uptake of PI in immature neurons, mainly after 4 h of
exposure (p<0.05 vs. respective control in cortical and cerebellar neurons and p<0.01 in
hippocampal ones). In Figure IV.4C, we verified an increase in the active form of caspase-3,
0
10
20
30
40
GS
t (nm
ol/m
g pr
otei
n)
Control
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
$$
#
$$
#
BA
0
10
20
30
40
GS
SG
/GS
t x
100
Control
UCB 50 μM
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
*
**$$
##
**
**
**
*
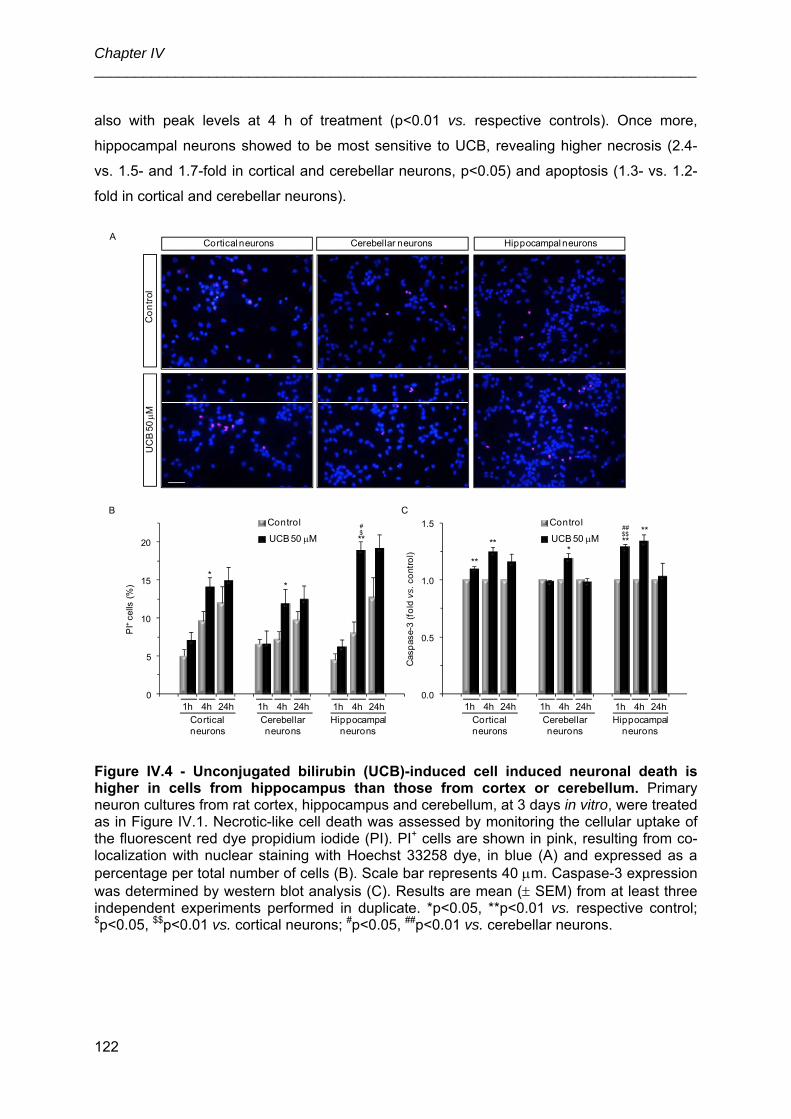
Chapter IV __________________________________________________________________________
122
also with peak levels at 4 h of treatment (p<0.01 vs. respective controls). Once more,
hippocampal neurons showed to be most sensitive to UCB, revealing higher necrosis (2.4-
vs. 1.5- and 1.7-fold in cortical and cerebellar neurons, p<0.05) and apoptosis (1.3- vs. 1.2-
fold in cortical and cerebellar neurons).
Figure IV.4 - Unconjugated bilirubin (UCB)-induced cell induced neuronal death is higher in cells from hippocampus than those from cortex or cerebellum. Primary neuron cultures from rat cortex, hippocampus and cerebellum, at 3 days in vitro, were treated as in Figure IV.1. Necrotic-like cell death was assessed by monitoring the cellular uptake of the fluorescent red dye propidium iodide (PI). PI+ cells are shown in pink, resulting from co-localization with nuclear staining with Hoechst 33258 dye, in blue (A) and expressed as a percentage per total number of cells (B). Scale bar represents 40 μm. Caspase-3 expression was determined by western blot analysis (C). Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $p<0.05, $$p<0.01 vs. cortical neurons; #p<0.05, ##p<0.01 vs. cerebellar neurons.
0.0
0.5
1.0
1.5
Cas
pase
-3 (f
old
vs.c
ontro
l)
ACortical neurons Cerebellar neurons
Con
trol
UC
B 50
μM
Hippocampalneurons
Control
UCB 50 μM
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
BControl
UCB 50 μM
1h 4h 24hCortical neurons
1h 4h 24hCerebellarneurons
Hippocampalneurons
1h 4h 24h
C
0
5
10
15
20
PI+
cells
(%)
**
****
*
$$##
**
#$**
**

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
123
3.4. UCB-induced neuronal oxidative stress and cell death in immature neurons is prevented by NAC To determine whether oxidative stress is related to UCB-induced neuronal death, we
evaluated ROS production, glutathione homeostasis and necrotic-like cell death in cells
treated with 100 µM NAC for 1 h, followed by 4 h incubation with UCB or not. NAC is a
cysteine donor, thus it promotes the synthesis of glutathione (Dringen, 2000, Zachwieja et
al., 2005). The concentration of this molecule was chosen based on our previous data
demonstrating that NAC prevents UCB-induced protein oxidation in 8 DIVs neurons (Brito et
al., 2008b). As stated in Table IV.1, pre-incubation with NAC significantly decreased ROS
production (p<0.05 vs. UCB-treated cortical neurons and p<0.01 vs. UCB-treated cerebellar
and hippocampal neurons), GSSG/GSt ratio (p<0.05 vs. UCB-treated cortical and cerebellar
neurons and p<0.01 vs. UCB-treated hippocampal neurons) and PI+ cells (p<0.05 vs. UCB-
treated cortical and hippocampal neurons and p<0.01 vs. UCB-treated cerebellar neurons).
Together, these results indicate that oxidative stress is involved in neuronal death by UCB.
3.5. UCB regulates DJ-1 protein expression in immature neurons, mainly in those from hippocampus, which is reverted by NAC There are several proteins that are differently expressed in response to toxic stimulus.
DJ-1, a protein involved in Parkinson ’s disease pathogenesis (Bonifati et al., 2003), is
particularly important as a redox-sensitive chaperone (Shendelman et al., 2004), protecting
neurons from oxidative stress and cell death (Lev et al., 2008, Lev et al., 2009), thus,
representing a putative protein for an adaptative response against UCB-induced oxidative
stress and neurotoxicity. Therefore, we evaluated DJ-1 protein expression in immature
neurons from the three regions, following incubation with UCB, in the absence or presence of
NAC. As observed in Figure IV.5A,C, 4 h treatment with UCB led to an increased expression
of DJ-1 in cortical and hippocampal neurons (respectively, p<0.05 and p<0.01 vs. respective
control), suggesting that DJ-1 protein expression is up-regulated, as an attempt to diminish
UCB-induced oxidative stress. However, after 24 h incubation, there is a significant decrease
of DJ1 expression in neurons from the three regions (p<0.05 vs. respective control for
cortical and cerebellar neurons and p<0.01 vs. respective control from hippocampal
neurons), suggesting that this effort to prevent oxidative stress is transient as it fails after
longer periods of neuronal exposure to UCB. Accordingly, with the abovementioned data,
hippocampal neurons are the most affected ones (36% reduction vs. 15% and 9% in cortical
and cerebellar neurons, respectively, p<0.01). In addition, this up or down-regulation of DJ-1
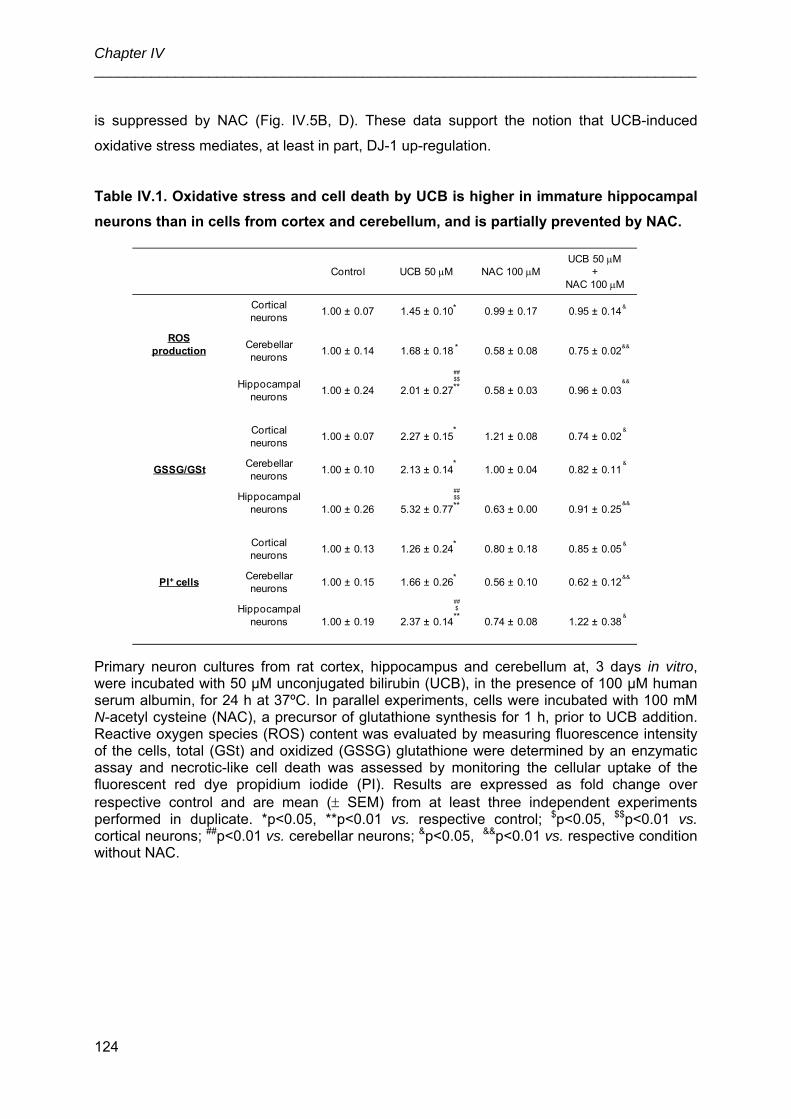
Chapter IV __________________________________________________________________________
124
is suppressed by NAC (Fig. IV.5B, D). These data support the notion that UCB-induced
oxidative stress mediates, at least in part, DJ-1 up-regulation.
Table IV.1. Oxidative stress and cell death by UCB is higher in immature hippocampal neurons than in cells from cortex and cerebellum, and is partially prevented by NAC.
Primary neuron cultures from rat cortex, hippocampus and cerebellum at, 3 days in vitro, were incubated with 50 µM unconjugated bilirubin (UCB), in the presence of 100 μM human serum albumin, for 24 h at 37ºC. In parallel experiments, cells were incubated with 100 mM N-acetyl cysteine (NAC), a precursor of glutathione synthesis for 1 h, prior to UCB addition. Reactive oxygen species (ROS) content was evaluated by measuring fluorescence intensity of the cells, total (GSt) and oxidized (GSSG) glutathione were determined by an enzymatic assay and necrotic-like cell death was assessed by monitoring the cellular uptake of the fluorescent red dye propidium iodide (PI). Results are expressed as fold change over respective control and are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $p<0.05, $$p<0.01 vs. cortical neurons; ##p<0.01 vs. cerebellar neurons; &p<0.05, &&p<0.01 vs. respective condition without NAC.
Control UCB 50 μM NAC 100 μMUCB 50 μM
+ NAC 100 μM
Cortical neurons 1.00 ± 0.07 1.45 ± 0.10 0.99 ± 0.17 0.95 ± 0.14
ROS production Cerebellar
neurons 1.00 ± 0.14 1.68 ± 0.18 0.58 ± 0.08 0.75 ± 0.02
Hippocampal neurons 1.00 ± 0.24 2.01 ± 0.27 0.58 ± 0.03 0.96 ± 0.03
Cortical neurons 1.00 ± 0.07 2.27 ± 0.15 1.21 ± 0.08 0.74 ± 0.02
GSSG/GSt Cerebellar neurons 1.00 ± 0.10 2.13 ± 0.14 1.00 ± 0.04 0.82 ± 0.11
Hippocampal neurons 1.00 ± 0.26 5.32 ± 0.77 0.63 ± 0.00 0.91 ± 0.25
Cortical neurons 1.00 ± 0.13 1.26 ± 0.24 0.80 ± 0.18 0.85 ± 0.05
PI+ cells Cerebellar neurons 1.00 ± 0.15 1.66 ± 0.26 0.56 ± 0.10 0.62 ± 0.12
Hippocampal neurons 1.00 ± 0.19 2.37 ± 0.14 0.74 ± 0.08 1.22 ± 0.38
&
&&
&&
&
&
&&
&
&
&&
*
*
**$$##
**$$##
*
*
**
##$
*
*

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
125
Figure IV.5 - Early up-regulation of DJ-1 protein expression by unconjugated bilirubin (UCB) is reverted for longer periods of incubation, and suppressed in the presence of N-acetyl cysteine (NAC), namely in immature neurons from hippocampus. Primary neuron cultures from rat cortex, hippocampus and cerebellum at, 3 days in vitro, were incubated with UCB (50 µM), in the presence of human serum albumin (100 μM), from 1 to 24 h at 37ºC (A). In parallel experiments, cells were incubated with 100 μM NAC, a precursor of glutathione synthesis for 1 h, prior to UCB addition (B). DJ-1 expression was determined by western blot analysis. Representative results for 24 h incubation with UCB are indicated in the absence (C) or in the presence (D) of NAC. Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $$p<0.01 vs. cortical neurons; ##p<0.01 vs. cerebellar neurons; &&p<0.01 vs. respective condition without NAC.
3.6. UCB-induced reduction of neurite outgrowth and branching mainly in immature neurons from hippocampus, is closely followed by those from cerebellar and cortical regions, and is prevented by NAC .NO showed to be involved in cell death, as well as in impairment of neurite ramification
and extension of immature cortical neurons after exposure to UCB, as we recently
demonstrated (personal communication Ana Rita Vaz, 2010; personal communication
Sandra L Silva, 2010, respectively). To investigate if oxidative stress affects neuritic
arborization, we assessed the neuronal network dynamics in cells non-treated or treated with
UCB in the presence or absence of NAC. We selected the longest time of incubation (24 h),
based on our previous reports showing UCB-induced neurite impairment in immature cortical
0.0
0.5
1.0
1.5
DJ-
1 (f
old
vs.c
ontro
l)
1h 4h 24hCortical neurons
Control
UCB 50 μM
1h 4h 24hCerebellar neurons
Hippocampal neurons
1h 4h 24h
##$$**
* *
*
##$$**
A B
0.0
0.5
1.0
1.5
DJ-
1 (f
old
vs.c
ontro
l)
1h 4h 24hCortical neurons
1h 4h 24hCerebellar neurons
Hippocampal neurons
1h 4h 24h
Control
UCB 50 μM
&& && &&
&&
- 22 KDaDJ-1 -
β-actin - - 42 KDa
UCB (50 μM) - - -+ + +Cortical neurons
Cerebellar neurons
Hippocampal neurons
- 22 KDaDJ-1 -
β-actin - - 42 KDa
UCB (50 μM) - - -+ + +Cortical neurons
Cerebellar neurons
Hippocampal neurons
C D
Without N-acetylcysteine With N-acetylcysteine

Chapter IV __________________________________________________________________________
126
neurons after 24 h treatment (Falcão et al., 2007) and because the decreased expression of
DJ-1 was found at 24 h incubation. As shown in Figure IV.6, UCB led to a decrease in
neurite extension and number of nodes in immature neurons from hippocampus and
cerebellum (p<0.01 vs. respective control) followed by those from cortex (p<0.05 vs.
respective control). In accordance with all the other results, neurite impairment by UCB was
higher in hippocampal neurons, both in total neurite output (42% reduction vs. 19% and 32%
in cortical and cerebellar neurons) and in number of branch points (38% reduction vs. 27%
and 29% in cortical and cerebellar neurons). Interestingly, pre-incubation with NAC
significantly prevented UCB-diminished neurite outgrowth (p<0.05), suggesting that
glutathione content may account to the resistance against UCB-induced neurite network
disruption. Therefore, different susceptibilities between brain regions may be partially due to
distinct levels of antioxidants.
4. Discussion In previous reports we demonstrated that UCB-induced neurotoxicity differs from nerve
cell type and maturation state, being cortical neurons more susceptible than cortical
astrocytes (Brito et al., 2008b, Falcão et al., 2006) and immature cells (3 DIVs) more
vulnerable than more differentiated (8 DIVs) ones (Falcão et al., 2005, Falcão et al., 2006).
Furthermore, oxidative stress and disruption of neuronal network dynamics were indicated as
pathological hallmarks in UCB-induced neurotoxicity (Brito et al., 2008a, Falcão et al., 2007,
Vaz et al., 2010, Fernandes et al., 2009). Based on these data, here we investigated for the
first time whether brain UCB specific pattern toxicity is determined by differential regional
susceptibility to UCB-induced oxidative stress and disruption of neurite arborization in
immature neurons, due to the increased vulnerability of premature babies. We also looked
for potential defence mechanisms as that of DJ-1 protein expression and glutathione content
as modulators of UCB-induced neurotoxicity.
In this study, we demonstrate that UCB leads to nitrosative and oxidative stress, as we
observed nNOS increased protein expression, production of nitrites and cGMP, as well as an
increase in the levels of ROS and of oxidized glutathione in immature neurons. Interestingly,
in spite of these biomarkers of oxidative stress are observed in immature neurons isolated
from all the three regions studied (cortex, cerebellum and hippocampus), they are mainly
detected in cells from hippocampus. For this differential vulnerability to UCB within brain
regions may account distinct responses at the cellular or biochemical processes, such as
antioxidant defences. In fact, total glutathione levels were markedly lower in immature
neurons from hippocampus, compared with those from cortex and cerebellum, as
demonstrated in Figure IV.3B.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
127
Figure IV.6 - Unconjugated bilirubin (UCB)-induced reduction of neurite outgrowth and branching in immature neurons from hippocampus is reduced at cerebellar and cortical levels, and is partly prevented by N-acetyl cysteine (NAC). Primary neuron cultures from rat cortex, hippocampus and cerebellum, at 3 days in vitro, were incubated with UCB (50 µM), in the presence of human serum albumin (100 μM), for 24 h at 37ºC. In parallel experiments, cells were incubated with 100 μM NAC, a precursor of glutathione synthesis for 1 h, prior to UCB addition. Neurite extension and number of nodes were evaluated by immunocytochemistry with MAP-2 labeling (A) as indicated in Methods. Scale bar 40 μM. Total neurite output (B) and number of branch points (C) were quantified in HCA-Vision Neurite Analysis software. Results are mean (± SEM) from at least three independent experiments performed in duplicate. *p<0.05, **p<0.01 vs. respective control; $$p<0.01 vs. cortical neurons; &p<0.05, &&p<0.01 vs. respective condition without NAC.
0
50
100
150
200
250
300
Tota
l neu
rite
outp
ut (m
icro
ns)
Control
UCB 50 μM
B
Cortical neurons
Cerebellarneurons
Hippocampalneurons
Cortical neurons
Cerebellarneurons
Hippocampalneurons
NAC 100 μM
Control
UCB 50 μM
C
Cortical neurons
Cerebellarneurons
Hippocampalneurons
Cortical neurons
Cerebellarneurons
Hippocampalneurons
NAC 100 μM
A Control UCB 50 μM NAC 100 μM UCB 50 μM + NAC 100 μM
Cor
tical
neu
rons
Cer
ebel
larn
euro
nsH
ippo
cam
paln
euro
ns
0
2
4
6
8
10
12
14
Num
ber o
f bra
nch
poin
ts
&
&& &&
&&
&
*
** $$
***
* **

Chapter IV __________________________________________________________________________
128
Supporting this concept, it was reported that antioxidant enzymes, such as xantine
oxidase and catalase, have maximum activity in cortex, followed by cerebellum and
hippocampus in developing mouse brain exposed to lead (Prasanthi et al., 2010).
Furthermore, primary astroglial cultures isolated from cortex, striatum, or hippocampus
revealed distinct profiles of vulnerability when subjected to injury. While astrocytes from
striatum showed increased injury by oxygen and glucose deprivation, they were more
resistant to an oxidative insult resultant from exposure to H2O2 since they have higher levels
of antioxidant defences, such as glutathione levels and glutathione peroxidase and
superoxide dismutase activities (Xu et al., 2001). Interestingly, glutathione peroxidase activity
was considered determinant in the recovery of the immature mouse brain subjected to
traumatic brain injury (Tsuru-Aoyagi et al., 2009). Taking these into account, together with
the preventive effects of NAC on UCB-induced neurotoxicity, we hypothesize that
hippocampal vulnerability to UCB is, at least in part, due to the lower content of the
antioxidant molecule glutathione.
For the differential susceptibilities to UCB neurotoxicity in the different brain areas may
also account the UCB specific affinities, after crossing the blood brain barrier. Hippocampus
is one of the brain regions stained by UCB in severely jaundiced neonates who died with
kernicterus (Ahdab-Barmada and Moossy, 1984, Hansen, 2000). The selective bilirubin
deposition in cases of kernicterus seems to take into account the areas more vulnerable to
hypoxic ischemic injury, such as the pyramidal cell layer of the hippocampus (Perlman et al.
1997), which raises the question of whether hypoxic-ischemic injury is important to the
development of the lesions by kernicterus. In addition, ischemic injury is also usually
associated with an increased production of the VEGF, which was recently detected in
hippocampal neurons from a kernicteric patient (personal communication Alexandra Brito,
2010). It is known that hippocampus is particularly affected by hypoxic-ischemic insult in
animal models of both mature and immature brain (Guzzetta et al., 2000, Jiang et al., 2004).
To this feature may account the distribution of immature NMDA receptors, which
corresponds to regions that preferentially express nNOS (Black et al., 1995, Greenamyre et
al., 1987, Mitani et al., 1998). Interestingly, hippocampal neurons were the main responders
to UCB in terms of nNOS protein expression and activation (increased concentrations of
nitrates and cGMP). Inhibition of mitochondrial cytochrome c oxidase observed in immature
neurons exposed to UCB (Vaz et al., 2010) is surely one of the causes. Accordingly, UCB
was shown to promote cytochrome c release in isolated mitochondria from the brain and liver
of adult male Wistar rats (Rodrigues et al., 2002, Rodrigues et al., 2000). Recently, our group
demonstrated that .NO signalling mediates, at least in part, UCB-induced neuronal injury,
both in mature (Brito et al., 2010) and in immature neurons (personal communication Ana

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
129
Rita Vaz, 2010), and also that inhibition of nNOS prevented glutathione impairment (Brito et
al., 2010). Additionally, neuronal damage mediated by nNOS was noticed in hypoxia-
ischemia (Ferriero et al., 1996, Ferriero et al., 1995) and its inhibition confered tissue
protection and reduction of caspase-3 activation (Peeters-Scholte et al., 2002, Zhu et al.,
2004). All these findings are reinforced by data demonstrating that .NO and NOS mediate
apoptosis during inflammation in neuronal cells (Hemmer et al., 2001, Heneka et al., 1998,
Thomas et al., 2008), indicating that .NO is a player in the developing brain cytotoxicity.
Besides antioxidant defences, regional differences in the Ca2+-induced mitochondrial
permeability transition (Friberg et al., 1999) and DNA damage (Cardozo-Pelaez et al., 2000)
were reported between hippocampus, cortex, and striatum, which are also signs of increased
pro-oxidant activity. Accordingly, our results demonstrated that intracellular production of
ROS from those specific brain regions is a result of UCB interaction. Protein oxidation, lipid
peroxidation, alterations in glutathione stores, decreased NADPH concentration and
superoxide anion radical production are also additional features produced by UCB in cortical
neurons (Brito et al., 2008b, Brito et al., 2010, Vaz et al., 2010).
Here, we evidenced that UCB-induced oxidative and nitrosative stress occur in parallel
with necrotic and apoptotic cell death, as demonstrated by increased levels of PI uptake and
caspase-3 activation, thus participating in the mechanisms of UCB-induced neurotoxicity.
Accordingly, experimental obstructive jaundice was shown to be correlated with oxidative
stress in rats' brain (Chroni et al., 2006). This surely accounts for the preventive effect of
NAC on UCB-induced oxidative stress and necrotic cell death. This is not without precedent
since NAC treatment was shown to be neuroprotective in lead-induced lipid peroxidation and
in antioxidant enzyme activities deficiencies of rats’ brain (Nehru and Kanwar, 2004), as well
as during hypoxia in cultures of rats' hippocampal neurons (Jayalakshmi et al., 2005).
Furthermore, results from our group demonstrated that NAC treatment protects from UCB-
induced protein oxidation (Brito et al., 2008b).
We extended our studies to the evaluation of the expression of DJ-1, a protein involved
in Parkinson’s disease pathogenesis (Bonifati et al., 2003), with particular relevance in
neuroprotection against oxidative stress and cell death (Lev et al., 2009, Lev et al., 2008).
Our data indicate that DJ-1 protein expression is transiently up-regulated suggesting an
intervening role to diminish acute UCB-induced oxidative in hippocampal neurons. Recently
DJ-1 was indicated as a protective factor against UCB-induced oxidative stress in human
neuroblastoma SH-SY5Y cell line (Deganuto et al., 2010). DJ-1 was also shown to negatively
regulate N-methyl-d-aspartate receptor (NMDAR) function and suppression of this protein led
to NMDAR-induced cell death (Chang et al., 2010). This is of particular interest, since UCB-

Chapter IV __________________________________________________________________________
130
induced neurotoxicity is mediated by glutamate receptors in cultured rat brain neurons
(Grojean et al., 2000, Grojean et al., 2001, Brito et al., 2010). Accumulation of extracellular
glutamate, secreted by UCB-exposed neurons (Brito et al., 2010, Falcão et al., 2006), and
glial cells, namely by microglia and immature astrocytes (Fernandes et al., 2004, Falcão et
al., 2005, Gordo et al., 2006) is a dominant cell response. Nevertheless, persistent
hyperbilirubinemia seems to down-regulated DJ-1, notoriously in hippocampus, what can
contribute for the deleterious effects of the condition. Most important, this effect was reverted
by NAC.
It was reported by our group that UCB impairs of neurite extension and ramification, in
immature cortical neurons, from which cells do not recover along the differentiation (Falcão
et al., 2007). UCB also showed to interfere with the development of hippocampal neurons,
reducing dendritic and axonal elongation and branching, axonal growth cones and number of
dendritic spines and synapses (Fernandes et al., 2009). This UCB-elicited impairment in
neuritic outgrowth was demonstrated to be mediated by .NO and overstimulation of NMDA
receptors and, thus, prevented by the use of inhibitors, even during nerve cell maturation
(personal communication Sandra L Silva, 2010). Interestingly, here we demonstrate that
hippocampus is the most affected in neurite impairment by UCB, what raises the possibility
that neonatal jaundice could have an impact on the infant’s learning and memory.
Observations by Weir and Millar revealed that adverse effects for learning are observed in
severe cases of hyperbilirubinemia history (Weir and Millar, 1997).
In conclusion, the data obtained show that UCB, in conditions mimicking a moderate
hyperbilirubinemia in the early neonatal period, induces oxidative stress in different brain
regions, particularly in the hippocampus. Furthermore, these results provide specific features
that may explain this differential susceptibility along different brain areas to UCB exposure,
namely local differences in glutathione content and in DJ-1 expression, important molecules
involved in the regulation of oxidative damage. Moreover, here we suggest that UCB
neuronal exposure will differently affect neurite outgrowth and ramification in distinct cell
populations, pointing hippocampal neurons as preferential target to short and long-term
UCB-induced neuronal dysfunction.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
131
5. References Ahdab-Barmada, M. and Moossy, J. (1984) The neuropathology of kernicterus in the
premature neonate: diagnostic problems. J Neuropathol Exp Neurol, 43, 45-56.
Black, S. M., Bedolli, M. A., Martinez, S., Bristow, J. D., Ferriero, D. M. and Soifer, S. J.
(1995) Expression of neuronal nitric oxide synthase corresponds to regions of selective
vulnerability to hypoxia-ischaemia in the developing rat brain. Neurobiol Dis, 2, 145-155.
Bonifati, V., Rizzu, P., van Baren, M. J. et al. (2003) Mutations in the DJ-1 gene associated
with autosomal recessive early-onset parkinsonism. Science, 299, 256-259.
Bradford, M. M. (1976) A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72, 248-
254.
Brito, M. A., Brites, D. and Butterfield, D. A. (2004) A link between hyperbilirubinemia,
oxidative stress and injury to neocortical synaptosomes. Brain Res, 1026, 33-43.
Brito, M. A., Lima, S., Fernandes, A., Falcão, A. S., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008a) Bilirubin injury to neurons: contribution of oxidative stress and rescue by
glycoursodeoxycholic acid. Neurotoxicology, 29, 259-269.
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008b) Unconjugated bilirubin differentially affects the redox status of neuronal
and astroglial cells. Neurobiol Dis, 29, 30-40.
Brito, M. A., Rosa, A. I., Silva, R. F. M., Falcão, A. S., Fernandes, A. and Brites, D. (2007)
Oxidative stress and disruption of the nervous cell. In: Focus in Brain Research, pp. 1-33.
Nova Science Publishers, Inc., New York.
Brito, M. A., Vaz, A. R., Silva, S. L., Falcão, A. S., Fernandes, A., Silva, R. F. M. and Brites,
D. (2010) N-methyl-D-aspartate receptor and neuronal nitric oxide synthase activation
mediate bilirubin-induced neurotoxicity. Mol Med, 16, 372-380.
Cardozo-Pelaez, F., Brooks, P. J., Stedeford, T., Song, S. and Sanchez-Ramos, J. (2000)
DNA damage, repair, and antioxidant systems in brain regions: a correlative study. Free
Radic Biol Med, 28, 779-785.
Chang, N., Li, L., Hu, R. et al. (2010) Differential regulation of NMDA receptor function by DJ-
1 and PINK1. Aging Cell.
Chroni, E., Patsoukis, N., Karageorgos, N., Konstantinou, D. and Georgiou, C. (2006) Brain
oxidative stress induced by obstructive jaundice in rats. J Neuropathol Exp Neurol, 65,
193-198.
Dalman, C. and Cullberg, J. (1999) Neonatal hyperbilirubinaemia--a vulnerability factor for
mental disorder? Acta Psychiatr Scand, 100, 469-471.

Chapter IV __________________________________________________________________________
132
Deganuto, M., Cesaratto, L., Bellarosa, C. et al. (2010) A proteomic approach to the bilirubin-
induced toxicity in neuronal cells reveals a protective function of DJ-1 protein. Proteomics,
10, 1645-1657.
Dennery, P. A., Seidman, D. S. and Stevenson, D. K. (2001) Neonatal hyperbilirubinemia. N
Engl J Med, 344, 581-590.
Doré, S., Goto, S., Sampei, K. et al. (2000) Heme oxygenase-2 acts to prevent neuronal
death in brain cultures and following transient cerebral ischemia. Neuroscience, 99, 587-
592.
Dringen, R. (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol, 62, 649-
671.
Dringen, R. and Hamprecht, B. (1996) Glutathione content as an indicator for the presence of
metabolic pathways of amino acids in astroglial cultures. J Neurochem, 67, 1375-1382.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2005) Bilirubin-
induced inflammatory response, glutamate release, and cell death in rat cortical
astrocytes are enhanced in younger cells. Neurobiol Dis, 20, 199-206.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Bilirubin-
induced immunostimulant effects and toxicity vary with neural cell type and maturation
state. Acta Neuropathol, 112, 95-105.
Falcão, A. S., Silva, R. F. M., Pancadas, S., Fernandes, A., Brito, M. A. and Brites, D. (2007)
Apoptosis and impairment of neurite network by short exposure of immature rat cortical
neurons to unconjugated bilirubin increase with cell differentiation and are additionally
enhanced by an inflammatory stimulus. J Neurosci Res, 85, 1229-1239.
Fernandes, A., Falcão, A. S., Abranches, E., Bekman, E., Henrique, D., Lanier, L. M. and
Brites, D. (2009) Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and
synaptogenesis. Dev Neurobiol, 69, 568-582.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Gordo, A. C., Gama, M. J., Brito, M. A. and
Brites, D. (2006) Inflammatory signalling pathways involved in astroglial activation by
unconjugated bilirubin. J Neurochem, 96, 1667-1679.
Fernandes, A., Silva, R. F. M., Falcão, A. S., Brito, M. A. and Brites, D. (2004) Cytokine
production, glutamate release and cell death in rat cultured astrocytes treated with
unconjugated bilirubin and LPS. J Neuroimmunol, 153, 64-75.
Ferriero, D. M., Holtzman, D. M., Black, S. M. and Sheldon, R. A. (1996) Neonatal mice
lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury.
Neurobiol Dis, 3, 64-71.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
133
Ferriero, D. M., Sheldon, R. A., Black, S. M. and Chuai, J. (1995) Selective destruction of
nitric oxide synthase neurons with quisqualate reduces damage after hypoxia-ischemia in
the neonatal rat. Pediatr Res, 38, 912-918.
Fink, S. L. and Cookson, B. T. (2005) Apoptosis, pyroptosis, and necrosis: mechanistic
description of dead and dying eukaryotic cells. Infect Immun, 73, 1907-1916.
Friberg, H., Connern, C., Halestrap, A. P. and Wieloch, T. (1999) Differences in the activation
of the mitochondrial permeability transition among brain regions in the rat correlate with
selective vulnerability. J Neurochem, 72, 2488-2497.
Gkoltsiou, K., Tzoufi, M., Counsell, S., Rutherford, M. and Cowan, F. (2008) Serial brain MRI
and ultrasound findings: relation to gestational age, bilirubin level, neonatal neurologic
status and neurodevelopmental outcome in infants at risk of kernicterus. Early Hum Dev,
84, 829-838.
Gomes, A., Fernandes, E. and Lima, J. L. (2005) Fluorescence probes used for detection of
reactive oxygen species. J Biochem Biophys Methods, 65, 45-80.
Gordo, A. C., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006)
Unconjugated bilirubin activates and damages microglia. J Neurosci Res, 84, 194-201.
Greenamyre, T., Penney, J. B., Young, A. B., Hudson, C., Silverstein, F. S. and Johnston, M.
V. (1987) Evidence for transient perinatal glutamatergic innervation of globus pallidus. J
Neurosci, 7, 1022-1030.
Grojean, S., Koziel, V., Vert, P. and Daval, J. L. (2000) Bilirubin induces apoptosis via
activation of NMDA receptors in developing rat brain neurons. Exp Neurol, 166, 334-341.
Grojean, S., Lievre, V., Koziel, V., Vert, P. and Daval, J. L. (2001) Bilirubin exerts additional
toxic effects in hypoxic cultured neurons from the developing rat brain by the recruitment
of glutamate neurotoxicity. Pediatr Res, 49, 507-513.
Guzzetta, F., Deodato, F. and Rando, T. (2000) Brain ischemic lesions of the newborn.
Childs Nerv Syst, 16, 633-637.
Hansen, T. W. (2000) Pioneers in the scientific study of neonatal jaundice and kernicterus.
Pediatrics, 106, E15.
Hansen, T. W. R. (2002) Mechanisms of bilirubin toxicity: clinical implications. Clin Perinatol,
29, 765-778, viii.
Hemmer, K., Fransen, L., Vanderstichele, H., Vanmechelen, E. and Heuschling, P. (2001) An
in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric
oxide and tumor necrosis factor-alpha. Neurochem Int, 38, 557-565.
Heneka, M. T., Loschmann, P. A., Gleichmann, M., Weller, M., Schulz, J. B., Wullner, U. and
Klockgether, T. (1998) Induction of nitric oxide synthase and nitric oxide-mediated

Chapter IV __________________________________________________________________________
134
apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-
alpha/lipopolysaccharide. J Neurochem, 71, 88-94.
Jayalakshmi, K., Sairam, M., Singh, S. B., Sharma, S. K., Ilavazhagan, G. and Banerjee, P.
K. (2005) Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress
in primary hippocampal culture. Brain Res, 1046, 97-104.
Jiang, X., Mu, D., Manabat, C., Koshy, A. A., Christen, S., Tauber, M. G., Vexler, Z. S. and
Ferriero, D. M. (2004) Differential vulnerability of immature murine neurons to oxygen-
glucose deprivation. Exp Neurol, 190, 224-232.
Jin, J., Meredith, G. E., Chen, L., Zhou, Y., Xu, J., Shie, F. S., Lockhart, P. and Zhang, J.
(2005) Quantitative proteomic analysis of mitochondrial proteins: relevance to Lewy body
formation and Parkinson's disease. Brain Res Mol Brain Res, 134, 119-138.
Karageorgos, N., Patsoukis, N., Chroni, E., Konstantinou, D., Assimakopoulos, S. F. and
Georgiou, C. (2006) Effect of N-acetylcysteine, allopurinol and vitamin E on jaundice-
induced brain oxidative stress in rats. Brain Res, 1111, 203-212.
Knowles, R. G., Palacios, M., Palmer, R. M. and Moncada, S. (1989) Formation of nitric
oxide from L-arginine in the central nervous system: a transduction mechanism for
stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A, 86, 5159-5162.
Lev, N., Ickowicz, D., Barhum, Y., Lev, S., Melamed, E. and Offen, D. (2009) DJ-1 protects
against dopamine toxicity. J Neural Transm, 116, 151-160.
Lev, N., Ickowicz, D., Melamed, E. and Offen, D. (2008) Oxidative insults induce DJ-1
upregulation and redistribution: implications for neuroprotection. Neurotoxicology, 29, 397-
405.
Mattson, M. P. and Liu, D. (2002) Energetics and oxidative stress in synaptic plasticity and
neurodegenerative disorders. Neuromolecular Med, 2, 215-231.
Mitani, A., Watanabe, M. and Kataoka, K. (1998) Functional change of NMDA receptors
related to enhancement of susceptibility to neurotoxicity in the developing pontine
nucleus. J Neurosci, 18, 7941-7952.
Nehru, B. and Kanwar, S. S. (2004) N-acetylcysteine exposure on lead-induced lipid
peroxidative damage and oxidative defence system in brain regions of rats. Biol Trace
Elem Res, 101, 257-264.
Ocal, K., Avlan, D., Cinel, I., Unlu, A., Ozturk, C., Yaylak, F., Dirlik, M., Camdeviren, H. and
Aydin, S. (2004) The effect of N-acetylcysteine on oxidative stress in intestine and
bacterial translocation after thermal injury. Burns, 30, 778-784.
Ostrow, J. D., Pascolo, L., Brites, D. and Tiribelli, C. (2004) Molecular basis of bilirubin-
induced neurotoxicity. Trends Mol Med, 10, 65-70.

Hallmarks in hippocampal susceptibility to UCB __________________________________________________________________________
135
Ostrow, J. D., Pascolo, L. and Tiribelli, C. (2003) Reassessment of the unbound
concentrations of unconjugated bilirubin in relation to neurotoxicity in vitro. Pediatr Res,
54, 926.
Özkan, H., Akkoc, N., Aydin, A., Kavukcu, Olgun, N., Irken, G., Akyol, F. and Cevik, N. T.
(1995) Relationship between serum unconjugated bilirubin levels and the
autofluorescence of white blood cells in neonatal jaundice. Biol Neonate, 68, 100-103.
Peeters-Scholte, C., Koster, J., Veldhuis, W. et al. (2002) Neuroprotection by selective nitric
oxide synthase inhibition at 24 hours after perinatal hypoxia-ischemia. Stroke, 33, 2304-
2310.
Perlman, J. M., Rogers, B. B. and Burns, D. (1997) Kernicteric findings at autopsy in two sick
near term infants. Pediatrics, 99, 612-615.
Prasanthi, R. P., Devi, C. B., Basha, D. C., Reddy, N. S. and Reddy, G. R. (2010) Calcium
and zinc supplementation protects lead (Pb)-induced perturbations in antioxidant enzymes
and lipid peroxidation in developing mouse brain. Int J Dev Neurosci, 28, 161-167.
Rodrigues, C. M. P., Solá, S. and Brites, D. (2002) Bilirubin induces apoptosis via the
mitochondrial pathway in developing rat brain neurons. Hepatology, 35, 1186-1195.
Rodrigues, C. M. P., Solá, S., Silva, R. F. M. and Brites, D. (2000) Bilirubin and amyloid-β
peptide induce cytochrome c release through mitochondrial membrane permeabilization.
Mol Med, 6, 936-946.
Shapiro, S. M. (2005) Definition of the clinical spectrum of kernicterus and bilirubin-induced
neurologic dysfunction (BIND). J Perinatol, 25, 54-59.
Shendelman, S., Jonason, A., Martinat, C., Leete, T. and Abeliovich, A. (2004) DJ-1 is a
redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation.
PLoS Biol, 2, e362.
Silva, R. F. M., Rodrigues, C. M. P. and Brites, D. (2002) Rat cultured neuronal and glial cells
respond differently to toxicity of unconjugated bilirubin. Pediatr Res, 51, 535-541.
Soorani-Lunsing, I., Woltil, H. A. and Hadders-Algra, M. (2001) Are moderate degrees of
hyperbilirubinemia in healthy term neonates really safe for the brain? Pediatr Res, 50,
701-705.
Stevenson, D. K., Dennery, P. A. and Hintz, S. R. (2001) Understanding newborn jaundice. J
Perinatol, 21 Suppl 1, S21-24; discussion S35-29.
Sunico, C. R., Gonzalez-Forero, D., Dominguez, G., Garcia-Verdugo, J. M. and Moreno-
Lopez, B. (2010) Nitric oxide induces pathological synapse loss by a protein kinase G-,
Rho kinase-dependent mechanism preceded by myosin light chain phosphorylation. J
Neurosci, 30, 973-984.

Chapter IV __________________________________________________________________________
136
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A. and Gaestel, M. (2008)
MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing
neuroinflammation--relevance in a mouse model of Parkinson's disease. J Neurochem,
105, 2039-2052.
Tsuru-Aoyagi, K., Potts, M. B., Trivedi, A. et al. (2009) Glutathione peroxidase activity
modulates recovery in the injured immature brain. Ann Neurol, 65, 540-549.
Vaz, A. R., Delgado-Esteban, M., Brito, M. A., Bolaños, J. P., Brites, D. and Almeida, A.
(2010) Bilirubin selectively inhibits cytochrome c oxidase activity and induces apoptosis in
immature cortical neurons: assessment of the protective effects of glycoursodeoxycholic
acid. J Neurochem, 112, 56-65.
Watchko, J. F. (2006) Hyperbilirubinemia and bilirubin toxicity in the late preterm infant. Clin
Perinatol, 33, 839-852; abstract ix.
Weir, C. and Millar, W. S. (1997) The effects of neonatal jaundice and respiratory
complications on learning and habituation in 5- to 11-month-old infants. J Child Psychol
Psychiatry, 38, 199-206.
Weisiger, R. A., Ostrow, J. D., Koehler, R. K., Webster, C. C., Mukerjee, P., Pascolo, L. and
Tiribelli, C. (2001) Affinity of human serum albumin for bilirubin varies with albumin
concentration and buffer composition: results of a novel ultrafiltration method. J Biol
Chem, 276, 29953-29960.
Xu, L., Sapolsky, R. M. and Giffard, R. G. (2001) Differential sensitivity of murine astrocytes
and neurons from different brain regions to injury. Exp Neurol, 169, 416-424.
Zachwieja, J., Zaniew, M., Bobkowski, W., Stefaniak, E., Warzywoda, A., Ostalska-Nowicka,
D., Dobrowolska-Zachwieja, A., Lewandowska-Stachowiak, M. and Siwinska, A. (2005)
Beneficial in vitro effect of N-acetyl-cysteine on oxidative stress and apoptosis. Pediatr
Nephrol, 20, 725-731.
Zhu, C., Wang, X., Qiu, L., Peeters-Scholte, C., Hagberg, H. and Blomgren, K. (2004)
Nitrosylation precedes caspase-3 activation and translocation of apoptosis-inducing factor
in neonatal rat cerebral hypoxia-ischaemia. J Neurochem, 90, 462-471.

Chapter V
V. Final considerations


Final considerations __________________________________________________________________________
139
1. Concluding remarks and perspectives The present thesis was designed to bring new insights into the cellular mechanisms
underlying neonatal hyperbilirubinemia neurotoxicity, and particular attention was given to
the role of oxidative stress in immature neurons exposed to unconjugated bilirubin (UCB). It
is widely accepted that hyperbilirubinemia in preterm infants is more prevalent, more severe
and represents an increased risk for the development of neurologic dysfunction in later life
(Stevenson et al., 2001, Watchko, 2006). Therefore, it is important to better understand the
essential mechanisms that contributes to the increased vulnerability of prematures to UCB
encephalopathy.
Cultured neuronal cells are powerful experimental models that we have adopted to
evaluate the deleterious effects of UCB. We have decided to use rat primary cultures of
neurons at 3 days in vitro (DIV), to mimic a condition of prematurity. This model was chosen
based on previous results demonstrating that immature cells are more susceptible to UCB-
induced neurotoxic effects than more differentiated ones (Falcão et al., 2005, Falcão et al.,
2006). In addition, this work focused on primary cultures of neurons, since they showed to be
more prone to oxidative injury and mitochondrial dysfunction and consequent cell death than
astrocytes, for which differences in glutathione content appear to have an important role
(Almeida et al., 2002, Almeida et al., 1998, Bolaños et al., 1996, Brito et al., 2008b).
Using this model, in the first experimental part of this thesis (chapter II), it is
demonstrated that UCB neuronal exposure rapidly inhibits cytochrome c oxidase (complex
IV) activity and ascorbate-driven oxygen consumption in 3 DIV rat cortical neurons. This
impairment of mitochondrial respiration was accompanied by a bioenergetic crisis, as judged
by the collapse of the inner-mitochondrial membrane potential, increased glycolytic activity,
and adenosine triphosphate (ATP) release. In addition, it was observed a disruption of
oxidized status of the cells, with increased superoxide anion radical production, disruption of
glutathione metabolism and impairment of reduced nicotinamide adenine dinucleotide
phosphate (NADPH) production. These events coursed in parallel with apoptotic cell death,
as determined by increased activities of mitochondrial-dependent caspases-9 and -3, as well
as increased annexin V+ neurons and condensed or fragmented nuclei. The mitochondrial
dysfunction observed by UCB may involve nitric oxide (.NO), which is capable of rapidly and
reversibly inhibit the mitochondrial respiratory chain (Bolaños et al., 1994, Brown and
Cooper, 1994, Cleeter et al., 1994). In fact, recent studies reported that neuronal oxidative
disruption by UCB is counteracted by inhibition of neuronal NO synthase (nNOS) (Brito et al.,
2008a, Mancuso et al., 2008). In the present work, the mitochondrial impairment by UCB
accompanied by the up-regulation of glycolysis suggests an attempt of immature neuronal
cells to support the bioenergetic crisis, as previously demonstrated for astrocytes exposed to

Chapter V __________________________________________________________________________
140
toxic stimulus (Bolaños et al., 2004). Nevertheless, excess of neuronal released ATP
determines .NO production and may be associated with neuronal apoptosis (Figueroa et al.,
2006, López et al., 2006). Finally, since NADPH levels are necessary to restore reduced
glutathione (Dringen, 2000), UCB-induced up-regulation of glycolysis may lead to inhibition of
the pentose-phosphate pathway (PPP), causing decrease of NADPH levels and,
consequently, glutathione oxidation, a notion recently reported for neuronal cultures
(Herrero-Mendez et al., 2009). In this study, neuroprotective effects of glycoursodeoxycholic
acid (GUDCA) were tested, since this bile acid was recently demonstrated to inhibit neuronal
cell death and oxidative stress (Brito et al., 2008a), as well as astrocytic inflammatory
response and consequent apoptotic and necrotic cell death (Fernandes et al., 2007b). In
summary, treatment of neurons with GUDCA prior to exposure to UCB prevented inhibition of
cytrochrome c oxidase activity, preserved cellular redox status and maintained cellular
viability. Taken together, these data suggest that cytochrome c oxidase inhibition is involved
in the neurotoxicity associated with UCB-induced neurological dysfunction and strongly
indicates the possible therapeutic potential of GUDCA in the treatment of neonatal jaundice.
In the second part of the work presented (chapter III), it is discussed the role of
neuroinflammation, a risk factor that is often associated with neonatal hyperbilirubinemia and
that is responsible for the alteration of blood brain barrier permeability through the release of
great amounts of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α+,
interleukin-1β (IL-1β) and interleukin-6 (IL-6) (Goldenberg and Andrews, 1996). In fact,
infection increases the risk for UCB encephalopathy (Dawodu et al., 1984) and presence of
inflammatory features, namely fever episodes and brain edema, were already described
during or following moderate to severe hyperbilirubinemia (Kaplan and Hammerman, 2005).
Interestingly, immature neuronal and astroglial cells showed higher levels of released
glutamate and TNF-α induced by exposure to UCB than more differentiated ones (Falcão et
al., 2006). Moreover, LPS showed to exacerbate the release of TNF-α and IL-1β by
immature astrocytes (Falcão et al., 2005), which provides a basis for the increased risk of
hyperbilirubinemia in the presence of sepsis. The results presented here reveal neuronal
dysfunction in 3 DIV rat cortical neurons by exposure to UCB, resulting from activation of
both the intrinsic and extrinsic pathways of apoptosis and loss of cellular functionality. Most
important, the results provide supportive evidence for the commonly indicated higher risk of
UCB brain damage in a condition of inflammation, since the association of pro-inflammatory
cytokines, TNF-α+IL-1β, to the condition of hyperbilirubinemia significantly increased the
cytotoxic potential of UCB through the same cascade of mediators. In this study, .NO
signalling and JNK1/2 activation are pointed to be key players in neurotoxicity induced by
hyperbilirubinemia and associated inflammation. In fact, several studies highlighted the

Final considerations __________________________________________________________________________
141
association between inflammation and the generation of reactive oxygen species and/or
reactive nitrogen species (ROS/RNS) (Bian and Murad, 2001, Brito et al., 2007, Sener et al.,
2005) and one of them, .NO, is important in the pathogenesis of inflammation (Korhonen et
al., 2005). .NO and induction of NOS are involved in apoptosis induced by inflammatory
mediators in neuronal cells (Hemmer et al., 2001, Heneka et al., 1998, Thomas et al., 2008).
Interestingly, in recent studies with mature neurons (rat primary cultures used at 8 DIV), UCB
exposure increased the expression of nNOS and production of .NO, cyclic guanosine 3',5'-
monophosphate (cGMP) and ROS, along with protein oxidation and depletion of glutathione
(Brito et al., 2008b, Brito et al., 2010). Furthermore, stress-activated protein kinases, such as
c-Jun N-terminal kinases 1/2 (JNK1/2), become activated in response to toxic stimulus, such
as the RNS (Luo et al., 1998, Marques et al., 2003), and the pro-inflammatory cytokines
TNF-α and IL-1, pointing these kinases as strong effectors of neuronal apoptosis (Mielke and
Herdegen, 2000, Tibbles and Woodgett, 1999). More recently, JNK 1/2 showed to directly
mediate the UCB-stimulation of TNF-α by astrocytes (Fernandes et al., 2007a). This feature
may be relevant if we consider that astrocytic activation has also been reported in several
neurodegenerative disorders and that this transition may be accompanied by dysfunction of
astrocytes leading to incorrect glia-to-neuron cross-talk (Rossi and Volterra, 2009). In
conclusion, these results provide evidence for the commonly indicated higher risk of UCB
brain damage in an inflammatory-associated condition.
In the third part of this work (chapter IV), it is discussed the different regional
vulnerability to UCB-induced neurotoxicity. For this purpose, rat primary neurons were
isolated from cortex, hippocampus and cerebellum and cultured for 3 DIV. Hippocampal and
cerebellar neurons were chosen based on preferential deposition pattern in the brain in
kernicteric conditions (Ahdab-Barmada and Moossy, 1984, Hansen, 2000). Here it is
demonstrated that neurons from hippocampus are more susceptible to UCB-induced
oxidative and nitrosative stress than those from cerebellum and cortex. For this differential
vulnerability to UCB within brain regions may account distinct antioxidant defences, since the
lowest levels of total glutathione were found in immature neurons from hippocampus. In
agreement with this concept, antioxidant enzymes, such as xantine oxidase, catalase and
glutathione peroxidase, showed minimum activity in hippocampus in different models, such
as developing mouse brain and astrocytic cultures (Prasanthi et al., 2010, Tsuru-Aoyagi et
al., 2009, Xu et al., 2001). Considering these facts, together with the results achieved with
the precursor of glutathione, N-acetylcysteine (NAC), where there was a protection from UCB
neurotoxicity, it is conceivable that hippocampal preferential vulnerability to UCB is, at least
in part, due to the lower glutathione content. Another important finding is the increased
vulnerability of hippocampal neurons to UCB-induced neurite arborization impairment.

Chapter V __________________________________________________________________________
142
Interestingly, in a very recent study with the same cellular model, inhibition of nNOS
abrogated the deleterious effects produced by UCB in network dynamics (personal
communication Sandra L Silva, 2010), result that were maintained along cell maturation,
indicating that short and long-term UCB toxic effects are prevented through inhibition of .NO
release. Since hippocampus is crucial for the linkage of short-term memory to the learning
process and the storage of spatial information (de Hoz et al., 2003, O'Neill et al., 2010,
Scoville and Milner, 1957), disassembly of neuritic development in neonatal
hyperbilirubinemia may account for a reduced learning memory ability and memory loss.
Cytoskeleton disassembly, loss of dendrites and axons and impairment of
neurotransmission are responsible for synaptic connectivity disruption, which are contributors
to the development of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s
diseases (Benitez-King et al., 2010, Evans et al., 2008). Since oxidative stress is a hallmark
of these pathologies (Halliwell, 2006, Lin et al., 2005), a better understanding of the neuronal
models comprising both oxidative stress and neuronal network dynamics is extremely
important in the comprehension of neurodegenerative diseases pathogenesis. In this study it
was also evaluated the expression of DJ-1, a protein involved in Parkinson’s disease
pathogenesis (Bonifati et al., 2003). However, in our experimental model, the effort to
prevent oxidative stress by increasing DJ-1 levels fails after long periods of UCB neuronal
exposure in neurons from all the three regions. Since down-regulation of DJ-1 is suppressed
in the presence of NAC, we consider that UCB-induced oxidative stress participates in DJ-1
regulation. In conclusion, the data obtained in the present study show that the condition of
neonatal hyperbilirubinemia induces oxidative stress in different brain regions, particularly in
the hippocampus. Furthermore, these results provide specific features that may explain the
differential susceptibility throughout different brain areas to UCB exposure, such as local
differences in glutathione content and in DJ-1 expression, important molecules involved in
the regulation of oxidative damage.
The major findings of this thesis are summarized in Figure V.1. Although we were able to
answer the questions that constitute the starting point of this work, others were raised and
remain to be clarified.

Final considerations __________________________________________________________________________
143
Figure V.1 – Integrative schematic representation of the major findings achieved in the present work, using immature neurons (3 DIV) isolated from brain cortex of fetuses from 16 days (E16) pregnant Wistar rats as experimental model. In the first part of this work (A), 3 DIV cortical neurons (Cx) exposed to unconjugated bilirubin (UCB), in conditions mimicking neonatal hyperbilirubinemia in the prematures, suffered oxidative stress, mitochondrial dysfunction associated with bioenergetic crisis and cell death, effects that were prevented in the presence of glycoursodeoxycholic acid (GUDCA). In the second part (B), it is concluded that UCB-induced nitrosative stress, c-Jun N-terminal kinases 1 and 2 (JNK1/2) and cell death are intensified by pro-inflammatory cytokines as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), used to mimic infection, in 3 DIV cortical neurons. In addition, both L-NAME (nitric oxide synthase inhibitor) and SP600125 (JNK1/2 inhibitor) reversed the effects produced by UCB either alone, or in association to pro-inflammatory cytokines. In the third part (C), neurons were isolated not only from cortex but also from cerebellum (Cb) and hippocampus (Hc). Hc neurons were the most susceptible to UCB-induced oxidative and nitrosative stress, as well as to UCB-induced neuritic impairment and cell death. N-acetylcysteine (NAC), a precursor of glutathione synthesis, was able to counteract the UCB-induced neurotoxicity.
Since hippocampal neurons were shown to be more vulnerable to UCB-induced
neurotoxicity than those from cortex and cerebellum, and that antioxidant defences will
appear to account for this differential vulnerability, it would be interesting to study whether
treatment of neuronal cultures with molecules that showed here to have antioxidant capacity,
such as GUDCA and NAC, are able to prevent from long term oxidative stress and energy
dysfunction in immature cultured neurons exposed to UCB, either alone or in association with
pro-inflammatory cytokines. This will contribute to elucidate the molecular events underlying
the relation between neonatal hyperbilirubinemia, alone or with associated inflammation, and
the development of neurological disorders in latter life. It would also be important to
determine if glutathione is the only molecule presented in lower levels in neurons from
hippocampus or if other antioxidant systems, like superoxide dismutase and catalase are
also compromised in these cells.
E16 rat brain3 DIV neurons
CxHc Cb
UCBOxidative stressMitochondrial dysfunction
Bioenergetic crisisCell death
(A)
UCB Nitrosative stressJNK1/2 signalling
Cell deathUCB + TNF-α + IL-1β
(B)
UCBOxidative/Nitrosative
stressNeuritic impairment
Cell death
(C)
GUDCA
L-NAME; SP600125
NAC

Chapter V __________________________________________________________________________
144
Other interesting issue that deserves to be addressed is whether activated astrocytes
and microglia exert a preventive or toxic effect on immature neuronal cells. Neuronal-glial
interactions constitute a major feature in the maintenance of neuronal homeostasis regarding
vascular, ionic, redox and metabolic function in the brain. In fact, astrocytes provide neurons
with energy and substrates for neurotransmission, as well as glutathione precursors (Allen
and Barres, 2005, Dringen et al., 2000). In addition, microglia is one of the brain’s major
sources of ROS and RNS and elevated concentrations of .NO (Bishop and Anderson, 2005),
and may be implicated in neurodegenerative diseases and neural injury as previously
discussed. Interestingly it has been demonstrated that UCB is able to activate microglia by
the acquisition of a phagocytic and inflammatory phenotype, which includes the release of
pro-inflammatory cytokines and release of glutamate (Gordo et al., 2006, Silva et al., 2010).
Interestingly, studies performed in our laboratory (personal communication Sandra L Silva,
2010) suggest that microglia exposure to UCB increases .NO production, effect that seems to
be further augmented when microglia is incubated with UCB-treated neuron conditioned
medium. In addition, stimulation of hippocampal slice cultures by UCB was recently reported
to increase nitrites release into the culture medium, as well as intracellular production of
glutamate, which is completed abrogated in microglia-depleted slice cultures. Since .NO
signalling by UCB is one important finding of the studies discussed in this thesis, these
observations suggest that microglial reactivity may be important for UCB-induced
neurotoxicity. Therefore, it would be interesting to evaluate whether microglia plays a role in
mitochondrial dysfunction and consequent cell death observed in chapter II, by using other
experimental models, such as neuron-microglia co-cultures or in models with depleted or
non-depleted microglia slices.
These advances may substantiate target-driven approaches to the prevention and
treatment of UCB-induced neurological damage, and provide fruitful opportunities for future
investigations.

Final considerations __________________________________________________________________________
145
2. References
Ahdab-Barmada, M. and Moossy, J. (1984) The neuropathology of kernicterus in the
premature neonate: diagnostic problems. J Neuropathol Exp Neurol, 43, 45-56.
Allen, N. J. and Barres, B. A. (2005) Signaling between glia and neurons: focus on synaptic
plasticity. Curr Opin Neurobiol, 15, 542-548.
Almeida, A., Delgado-Esteban, M., Bolanos, J. P. and Medina, J. M. (2002) Oxygen and
glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones
but not in astrocytes in primary culture. J Neurochem, 81, 207-217.
Almeida, A., Heales, S. J., Bolanos, J. P. and Medina, J. M. (1998) Glutamate neurotoxicity
is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione
depletion. Brain Res, 790, 209-216.
Benitez-King, G., Ortiz-Lopez, L., Jimenez-Rubio, G. and Ramirez-Rodriguez, G. (2010)
Haloperidol causes cytoskeletal collapse in N1E-115 cells through tau
hyperphosphorylation induced by oxidative stress: Implications for neurodevelopment. Eur
J Pharmacol, 644, 24-31.
Bian, K. and Murad, F. (2001) Diversity of endotoxin-induced nitrotyrosine formation in
macrophage-endothelium-rich organs. Free Radic Biol Med, 31, 421-429.
Bishop, A. and Anderson, J. E. (2005) NO signaling in the CNS: from the physiological to the
pathological. Toxicology, 208, 193-205.
Bolaños, J. P., Cidad, P., García-Nogales, P., Delgado-Esteban, M., Fernández, E. and
Almeida, A. (2004) Regulation of glucose metabolism by nitrosative stress in neural cells.
Mol Aspects Med, 25, 61-73.
Bolaños, J. P., Heales, S. J., Peuchen, S., Barker, J. E., Land, J. M. and Clark, J. B. (1996)
Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for
glutathione. Free Radic Biol Med, 21, 995-1001.
Bolaños, J. P., Peuchen, S., Heales, S. J., Land, J. M. and Clark, J. B. (1994) Nitric oxide-
mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J
Neurochem, 63, 910-916.
Bonifati, V., Rizzu, P., van Baren, M. J. et al. (2003) Mutations in the DJ-1 gene associated
with autosomal recessive early-onset parkinsonism. Science, 299, 256-259.
Brito, M. A., Lima, S., Fernandes, A., Falcão, A. S., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008a) Bilirubin injury to neurons: contribution of oxidative stress and rescue by
glycoursodeoxycholic acid. Neurotoxicology, 29, 259-269.

Chapter V __________________________________________________________________________
146
Brito, M. A., Rosa, A. I., Falcão, A. S., Fernandes, A., Silva, R. F. M., Butterfield, D. A. and
Brites, D. (2008b) Unconjugated bilirubin differentially affects the redox status of neuronal
and astroglial cells. Neurobiol Dis, 29, 30-40.
Brito, M. A., Rosa, A. I., Silva, R. F. M., Falcão, A. S., Fernandes, A. and Brites, D. (2007)
Oxidative stress and disruption of the nervous cell. In: Focus in Brain Research, pp. 1-33.
Nova Science Publishers, Inc., New York.
Brito, M. A., Vaz, A. R., Silva, S. L., Falcão, A. S., Fernandes, A., Silva, R. F. M. and Brites,
D. (2010) N-methyl-D-aspartate receptor and neuronal nitric oxide synthase activation
mediate bilirubin-induced neurotoxicity. Mol Med, 16, 372-380.
Brown, G. C. and Cooper, C. E. (1994) Nanomolar concentrations of nitric oxide reversibly
inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS
Lett, 356, 295-298.
Cleeter, M. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. and Schapira, A. H. (1994)
Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial
respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett,
345, 50-54.
Dawodu, A. H., Owa, J. A. and Familusi, J. B. (1984) A prospective study of the role of
bacterial infection and G6PD deficiency in severe neonatal jaundice in Nigeria. Trop
Geogr Med, 36, 127-132.
de Hoz, L., Knox, J. and Morris, R. G. (2003) Longitudinal axis of the hippocampus: both
septal and temporal poles of the hippocampus support water maze spatial learning
depending on the training protocol. Hippocampus, 13, 587-603.
Dringen, R. (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol, 62, 649-
671.
Dringen, R., Gutterer, J. M. and Hirrlinger, J. (2000) Glutathione metabolism in brain
metabolic interaction between astrocytes and neurons in the defence against reactive
oxygen species. Eur J Biochem, 267, 4912-4916.
Evans, N. A., Facci, L., Owen, D. E., Soden, P. E., Burbidge, S. A., Prinjha, R. K.,
Richardson, J. C. and Skaper, S. D. (2008) Abeta(1-42) reduces synapse number and
inhibits neurite outgrowth in primary cortical and hippocampal neurons: a quantitative
analysis. J Neurosci Methods, 175, 96-103.
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2005) Bilirubin-
induced inflammatory response, glutamate release, and cell death in rat cortical
astrocytes are enhanced in younger cells. Neurobiol Dis, 20, 199-206.

Final considerations __________________________________________________________________________
147
Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006) Bilirubin-
induced immunostimulant effects and toxicity vary with neural cell type and maturation
state. Acta Neuropathol, 112, 95-105.
Fernandes, A., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007a) MAPKs are
key players in mediating cytokine release and cell death induced by unconjugated bilirubin
in cultured rat cortical astrocytes. Eur J Neurosci, 25, 1058-1068.
Fernandes, A., Vaz, A. R., Falcão, A. S., Silva, R. F. M., Brito, M. A. and Brites, D. (2007b)
Glycoursodeoxycholic Acid and interleukin-10 modulate the reactivity of rat cortical
astrocytes to unconjugated bilirubin. J Neuropathol Exp Neurol, 66, 789-798.
Figueroa, S., Oset-Gasque, M. J., Arce, C., Martinez-Honduvilla, C. J. and González, M. P.
(2006) Mitochondrial involvement in nitric oxide-induced cellular death in cortical neurons
in culture. J Neurosci Res, 83, 441-449.
Goldenberg, R. L. and Andrews, W. W. (1996) Intrauterine infection and why preterm
prevention programs have failed. Am J Public Health, 86, 781-783.
Gordo, A. C., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F. M. and Brites, D. (2006)
Unconjugated bilirubin activates and damages microglia. J Neurosci Res, 84, 194-201.
Halliwell, B. (2006) Oxidative stress and neurodegeneration: where are we now? J
Neurochem, 97, 1634-1658.
Hansen, T. W. (2000) Pioneers in the scientific study of neonatal jaundice and kernicterus.
Pediatrics, 106, E15.
Hemmer, K., Fransen, L., Vanderstichele, H., Vanmechelen, E. and Heuschling, P. (2001) An
in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric
oxide and tumor necrosis factor-alpha. Neurochem Int, 38, 557-565.
Heneka, M. T., Loschmann, P. A., Gleichmann, M., Weller, M., Schulz, J. B., Wullner, U. and
Klockgether, T. (1998) Induction of nitric oxide synthase and nitric oxide-mediated
apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-
alpha/lipopolysaccharide. J Neurochem, 71, 88-94.
Herrero-Mendez, A., Almeida, A., Fernández, E., Maestre, C., Moncada, S. and Bolaños, J.
P. (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous
degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol, 11, 747-752.
Kaplan, M. and Hammerman, C. (2005) Understanding severe hyperbilirubinemia and
preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin Chim
Acta, 356, 9-21.
Korhonen, R., Lahti, A., Kankaanranta, H. and Moilanen, E. (2005) Nitric oxide production
and signaling in inflammation. Curr Drug Targets Inflamm Allergy, 4, 471-479.

Chapter V __________________________________________________________________________
148
Lin, S., Wei, X., Bales, K. R., Paul, A. B., Ma, Z., Yan, G., Paul, S. M. and Du, Y. (2005)
Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced
cerebellar hypoplasia in the Gunn rat. Eur J Neurosci, 22, 21-27.
López, E., Arce, C., Oset-Gasque, M. J., Canadas, S. and González, M. P. (2006) Cadmium
induces reactive oxygen species generation and lipid peroxidation in cortical neurons in
culture. Free Radic Biol Med, 40, 940-951.
Luo, Y., Umegaki, H., Wang, X., Abe, R. and Roth, G. S. (1998) Dopamine induces
apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem, 273, 3756-3764.
Mancuso, C., Capone, C., Ranieri, S. C., Fusco, S., Calabrese, V., Eboli, M. L., Preziosi, P.,
Galeotti, T. and Pani, G. (2008) Bilirubin as an endogenous modulator of neurotrophin
redox signaling. J Neurosci Res, 86, 2235-2249.
Marques, C. A., Keil, U., Bonert, A., Steiner, B., Haass, C., Muller, W. E. and Eckert, A.
(2003) Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedish
amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J
Biol Chem, 278, 28294-28302.
Mielke, K. and Herdegen, T. (2000) JNK and p38 stresskinases--degenerative effectors of
signal-transduction-cascades in the nervous system. Prog Neurobiol, 61, 45-60.
O'Neill, J., Pleydell-Bouverie, B., Dupret, D. and Csicsvari, J. (2010) Play it again:
reactivation of waking experience and memory. Trends Neurosci, 33, 220-229.
Prasanthi, R. P., Devi, C. B., Basha, D. C., Reddy, N. S. and Reddy, G. R. (2010) Calcium
and zinc supplementation protects lead (Pb)-induced perturbations in antioxidant enzymes
and lipid peroxidation in developing mouse brain. Int J Dev Neurosci, 28, 161-167.
Rossi, D. and Volterra, A. (2009) Astrocytic dysfunction: insights on the role in
neurodegeneration. Brain Res Bull, 80, 224-232.
Scoville, W. B. and Milner, B. (1957) Loss of recent memory after bilateral hippocampal
lesions. J Neurol Neurosurg Psychiatry, 20, 11-21.
Sener, G., Toklu, H., Kapucu, C., Ercan, F., Erkanli, G., Kacmaz, A., Tilki, M. and Yegen, B.
C. (2005) Melatonin protects against oxidative organ injury in a rat model of sepsis. Surg
Today, 35, 52-59.
Silva, S. L., Vaz, A. R., Barateiro, A., Falcão, A. S., Fernandes, A., Brito, M. A., Silva, R. F.
M. and Brites, D. (2010) Features of bilirubin-induced reactive microglia: From
phagocytosis to inflammation. Neurobiol Dis.
Stevenson, D. K., Dennery, P. A. and Hintz, S. R. (2001) Understanding newborn jaundice. J
Perinatol, 21 Suppl 1, S21-24; discussion S35-29.

Final considerations __________________________________________________________________________
149
Thomas, T., Timmer, M., Cesnulevicius, K., Hitti, E., Kotlyarov, A. and Gaestel, M. (2008)
MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing
neuroinflammation-relevance in a mouse model of Parkinson's disease. J Neurochem,
105, 2039-2052.
Tibbles, L. A. and Woodgett, J. R. (1999) The stress-activated protein kinase pathways. Cell
Mol Life Sci, 55, 1230-1254.
Tsuru-Aoyagi, K., Potts, M. B., Trivedi, A. et al. (2009) Glutathione peroxidase activity
modulates recovery in the injured immature brain. Ann Neurol, 65, 540-549.
Watchko, J. F. (2006) Hyperbilirubinemia and bilirubin toxicity in the late preterm infant. Clin
Perinatol, 33, 839-852; abstract ix.
Xu, L., Sapolsky, R. M. and Giffard, R. G. (2001) Differential sensitivity of murine astrocytes
and neurons from different brain regions to injury. Exp Neurol, 169, 416-424.


















