UNIVERSIDAD DE VIGO TESIS DOCTORAL
140
UNIVERSIDAD DE VIGO TESIS DOCTORAL Realizada en el Departamento de Química Física Grupo de Química Cuántica ESTUDIO QTAIM DE NITRILOS Y COMPUESTOS RELACIONADOS José Luis López Fernández Memoria para optar al grado de Doctor por la Universidad de Vigo Septiembre 2015
Transcript of UNIVERSIDAD DE VIGO TESIS DOCTORAL

UNIVERSIDAD DE VIGO
TESIS DOCTORAL
Realizada en el Departamento de Química
Física Grupo de Química Cuántica
ESTUDIO QTAIM DE NITRILOS
Y
COMPUESTOS RELACIONADOS
José Luis López Fernández
Memoria para optar al grado de
Doctor por la Universidad de Vigo
Septiembre 2015


“Science may set limits to knowledge, but should not set limits to imagination”
Bertrand Russell

AGRADECIMIENTOS
Mi más sincero y profundo agradecimiento al Prof. Dr. Ricardo A.
Mosquera Castro, gracias a esfuerzo y dedicación fue posible terminar el
trabajo de esta tesis.
El Dr. Ricardo A. Mosquera me ha enseñado, corregido, orientado,
apoyado de manera que ha sobrepasado con mucho las expectativas que
uno desearía tener en un director de tesis.
Sin lugar a dudas una de las mejoras cosas que me ha deparado la vida
es haber conocido a Ricardo, mi primer contacto con él hace años tuvo
lugar cuando dirigió mi tesina de grado, con el tiempo lo más valioso que
he conseguido, no es el terminar este trabajo, sino tener un amigo de un
valor tanto profesional como personal inestimable. Mi agradecimiento
eterno para Ricardo.
Mi agradecimiento también al Departamento de Química-Física de la
Universidad de Vigo por poner a mi disposición los equipos informáticos
y programas de computación usados en la elaboración de la tesis.


DEDICATORIA
Quisiera dedicar esta tesis a todos aquellos que han dedicado parte del
tiempo de su vida a está fascinante parte del conocimiento humano
como es La Química Teórica, tanto profesional como si lo han hecho por
simple curiosidad o afición, creo que el tiempo es uno de los bienes más
preciados que tiene un ser humano, mi recuerdo para todos ellos.
Por supuesto también aquí quiero mencionar expresamente a mis
directores de tesis: Prof. Dr. Ricardo A. Mosquera y Prof. Dra. Aña María
Graña.
Y finalmente también va dedicada a aquellos que han “padecido” un
poco mi afición por la Química, mis hijos Elena y Luis Alberto.


TABLA DE CONTENIDOS
RESUMEN .............................................................................................. 1
1. INTRODUCCIÓN ............................................................................. 17
2. OBJETIVOS ..................................................................................... 25
3. DISCUSIÓN GENERAL ................................................................... 27
3.1 Methodology ............................................................................. 29
3.1.1 Density Functional Theory (DFT) ................................................... 29
3.1.2 An Overview on the Quantum Theory of Atoms in Molecules (QTAIM)... 39
3.1.3 Approximate Transferability........................................................... 43
3.1.4 On the limitations of the Resonance Model…………………………………52
3.2 Discusión general de resultados ....................................................... 59
4. TRABAJOS DE INVESTIGACION ................................................... 62
4.1 Aproximate Transferability in Alkanenitriles ......................................... 64
4.2 A Charge Denstiy Analysis on the Proximity Effect in Dicyanoalkanes ...... 74
4.3 Electron Density Analysis on the Protonation of Nitriles ......................... 82
4.4 Electron Density Analysis on the Alpha Acidity of Nitriles ....................... 88
4.5 QTAIM Study of Rearregement Reactions in Nitrogenated Compounds .. 100
5. CONCLUSIONES ........................................................................... 120
6. BIBLIOGRAFIA............................................................................... 126


Estudio QTAIM de nitrilos y compuestos relacionados
1
RESUMEN
Esta Tesis se origina dentro de una investigación más general sobre la
transferibilidad de grupos funcionales que en aquellos momentos
desarrollaba el Grupo de Química Cuántica de la Universidad de Vigo [1-
4]. A lo largo de su desarrollo los objetivos iniciales fueron modificados
para considerar también problemas de reactividad. En concreto se
examinaron las propiedades ácido-base de los nitrilos, considerando su
N-protonación y la abstracción de hidrógenos enlazados a la posición al
grupo nitrilo. Ambos estudios, que analizaban únicamente los estados
inicial y final de dichos procesos, se relacionaban con un nuevo objetivo
más general del grupo, el estudio de las limitaciones del modelo de
resonancia para describir la evolución electrónica en procesos químicos
simples [5-11]. Por último, recientemente se añadió un estudio sobre la
evolución de la densidad electrónica en algunas transposiciones que
tienen lugar con compuestos que guardan alguna relación con el grupo
nitrilo. De manera general, puede decirse que el presente trabajo se
centra en el estudio de las propiedades y comportamiento de nitrilos y
compuestos afines mediante el uso de la teoría cuántica de átomos en
moléculas (QTAIM) [12,13]. Sus objetivos concretos son: i) Definir grupos
atómicos aproximadamente transferibles en los alcanonitrilos; ii) Analizar
como la proximidad entre dos grupos CN afecta a la transferibilidad
atómica; iii) Obtener afinidades protónicas y acideces de diversos nitrilos,

Estudio QTAIM de nitrilos y compuestos relacionados
2
relacionando sus valores con la estructura electrónica de los compuestos;
iv) Describir los efectos electrónicos que acompañan a los principales
procesos ácido-base que los nitrilos pueden experimentar; y v) Detallar
como evoluciona la densidad electrónica en etapas de procesos químicos
(transposiciones de Curtius, Beckmann y Hofmann) en que intervienen
compuestos nitrogenados estructuralmente semejantes a los nitrilos.
Formalmente, el trabajo se ha divido en seis secciones: Introducción,
Objetivos, Discusión General, Resultados y discusión (que contienen los
trabajos de investigación publicados o en proceso de publicación),
Conclusiones y Bibliografía.
En el primer capítulo se hace una breve descripción y encuadre del
trabajo dentro del marco de la teoría QTAIM, justificando el tipo de
estudio llevado a cabo y reseñando brevemente trabajos análogos que
fueron realizados en otras series de compuestos tales como aldehídos,
cetonas, éteres, etc. Dichos trabajos se utilizan como punto de
comparación con el de esta Tesis.
En el segundo capítulo se definen de manera explicita los objetivos
perseguidos. De manera genérica se alcanzarán a partir de analizar
propiedades atómicas y de enlace, definidas en el contexto de la QTAIM,
calculadas para nitrilos y compuestos afines.
El tercer capítulo combina la metodología empleada y una discusión
general de resultados, obligada por la normativa vigente en la
Universidad de Vigo en el caso de las Tesis presentadas como compendio

Estudio QTAIM de nitrilos y compuestos relacionados
3
de trabajos de investigación. Para realizar este trabajo se han utilizado
varios tratamientos basados en la Mecánica Cuántica, tales como
cálculos Hartree-Fock (HF) y DFT/B3LYP (Density Functional Theory,
funcional B3LYP) y, principalmente, la teoría QTAIM (Quantum Theory of
atoms in molecules).
Aunque las funciones de onda contienen toda la información extraíble de
un sistema, su forma, en el caso molecular, suele ser demasiado
complicada para proporcionar de manera directa una imagen sencilla de
la molécula. Basta pensar que, incluso al nivel HF restringido (RHF) (sin
tener en cuenta la correlación electrónica), la parte electrónica de la
función de onda molecular obtenida con un método de combinación
lineal de orbitales atómicos (CLOA) o con su variante habitual, la
combinación de funciones base, es normalmente un determinante de
tantas funciones espín orbital como electrones. La función
polielectrónica resultante depende de las coordenadas de posición y
espín de todos los electrones y presenta numerosos parámetros. Resulta,
por tanto, fundamental, disponer de alguna magnitud o cantidad que
permita obtener información fácilmente visualizable de la función de
onda y refleje sus características fundamentales. Las funciones de
densidad son una vía tradicional para conseguir este objetivo. En este
trabajo, se utilizará la función densidad electrónica monodimensional e
independiente del espín, ρ(r). Esto es, la misma función del espacio
tridimensional que puede obtenerse por vía experimental a través de un

Estudio QTAIM de nitrilos y compuestos relacionados
4
estudio de difracción de rayos X. En nuestro caso, sin embargo, dicha
función se obtiene por vía computacional integrando una función de
onda HF o a partir de los orbitales Kohn-Sham de un cálculo DFT.
Las diversas herramientas desarrolladas en el contexto de la QTAIM son
las que se usarán para analizar la función ρ(r) suministrada por los
cálculos HF o DFT. Por todo ello, en esta parte debe hacerse una
referencia a los métodos de obtención de la densidad electrónica y al
utilizado para su interpretación. Considerando que el método HF se
estudia suficientemente en los actuales programas de grado y postgrado,
sólo se presenta aquí una descripción general de la DFT (sección 3.1).
Respecto a la teoría QTAIM se ha optado por presentar únicamente una
breve introducción (sección 3.2), ya que existen excelentes monografías
[12-16] que recogen con detalle los extremos de esta teoría desarrollada
por Richard F. W. Bader [12-14] para analizar la densidad electrónica de
sistemas moleculares. Es importante, no obstante, recordar aquí que
esta teoría particiona el espacio físico real, a diferencia de otras
metodologías basadas en el espacio de configuración (espacio orbital) y
que trabaja sobre un observable físico, ρ(r), y no sobre entidades
matemáticas (orbitales moleculares) [17,18].
Otras dos secciones del tercer capítulo recogen aspectos generales de
dos temas fundamentales para esta Tesis y que son aplicaciones
habituales de la teoría QTAIM: transferibilidad aproximada (sección 3.3) y
revisión crítica de las predicciones del modelo de resonancia (sección

Estudio QTAIM de nitrilos y compuestos relacionados
5
3.4). Debe señalarse que se ha demostrado que la transferibilidad
completa es un límite inalcanzable (como consecuencia de los teoremas
de Hohenberg y Kohn) [19,20], por lo que sólo es posible hablar de
transferibilidad aproximada. Por otro lado, las predicciones del modelo
de resonancia no son compatibles en numerosos casos con la evolución
de la densidad electrónica que muestran los análisis QTAIM.
En la cuarta sección se recopilan los artículos de investigación publicados
(los tres primeros [21-23]) o en fase de publicación (los dos últimos
[24,25]) que se han elaborado como consecuencia de este trabajo.
En un primer estudio se analizaron las propiedades atómicas y de enlace
de una serie de doce alcanonitrilos lineales en conformación
antiperiplanar [21]. El objetivo es analizar la transferabilidad de los
grupos CN, CH2 y CH3. La geometría de todos ellos fue optimizada con el
nivel de cálculo RHF/6-31G(d,p), obteniéndose posteriormente una
función de onda con una base que adicionaba funciones difusas sobre
todos los átomos: RHF/6-31++G(d,p). Se establece una clasificación para
los grupos anteriores en virtud de su transferabilidad aproximada. Debe
destacarse que el trabajo indica que no se observa transferabilidad de la
energía atómica, E(Ω). Por el contrario esta propiedad exhibe una
dependencia con el tamaño molecular. En el momento de la publicación
de este trabajo, este comportamiento había sido observado también en
otras series homólogas y se conocía como efecto Z [1-4,26,27]. No
obstante, en un estudio realizado paralelamente a esta Tesis [28],

Estudio QTAIM de nitrilos y compuestos relacionados
6
nuestro grupo demostró que este problema tenía su origen en la forma
en que el programa AIMPAC calcula la energía atómica, aplicando la
relación virial, γ, sobre la integración atómica de la densidad de energía
cinética electrónica, K(r) [28,29]. Cuando se analiza la energía cinética
electrónica atómica, K(Ω), se observa, en cambio, una transferibilidad
semejante a la observada a partir de poblaciones electrónicas atómicas.
Como cabría esperar por los resultados ya conocidos de otras series de
compuestos el grupo metilo presenta un comportamiento específico
para moléculas pequeñas. Los grupos metileno se han clasificado de
acuerdo con su distancia al grupo CN. En este primer artículo también se
muestra que, tanto energías electrónicas moleculares calculadas, como
calores de formación experimentales, muestran un excelente ajuste a un
modelo de contribuciones de grupo en el que sólo se consideran los
grupos ciano (CN), CH3 y el número de grupos metileno. Este intrigante
comportamiento, que fue denominado “transferibilidad compensatoria”
por Bader [30], tiene un origen que puede remontarse a los estudios
sobre alcanos llevados a cabo por el propio Bader [31,32] y fue ya
también observado en otras series de moléculas funcionalizadas por
nuestro grupo [2,4,26].
En un segundo artículo se estudia el resultado de introducir en las
moléculas grupos funcionales adicionales al grupo CN original [22]. El
objetivo es estudiar las influencias mutuas entre grupos funcionales
(denominadas efecto de proximidad por Kehiaian [33]). Se realiza así un

Estudio QTAIM de nitrilos y compuestos relacionados
7
estudio QTAIM de una serie de dicianoalcanos. En este caso se estudian
las propiedades atómicas y de enlace de 21 dicianoalcanos. En este
artículo se ha calculado también la entropía normalizada de Shannon
para la distribución electrónica, Sh(Ω) [34-36]. Las densidades
electrónicas analizadas fueron obtenidas con el nivel RHF/6-31++G(d,p)
aplicado sobre geometrías totalmente optimizadas con el nivel RHF/6-
31G(d,p). De nuevo se establece una clasificación de los grupos CN y CH2
al tiempo que se hace una comparativa con los resultados de trabajos
análogos. Se encuentra que los grupos ciano son estadísticamente
equivalentes cuando entre ellos hay una separación de al menos 14
grupos metileno. Los efectos del grupo CN sobre los grupos metileno son
casi independientes de la posición, en este aspecto se ha visto que los
hidrógenos son más sensibles que los átomos de carbono. También se ha
encontrado un comportamiento específico en un grupo metileno cuando
su número en la molécula es menor de 19. Debe señalarse, que a
diferencia de lo indicado en el trabajo anterior, en este artículo ya se
utiliza la propiedad K(Ω), en lugar de E(Ω), a la hora de estudiar
transferibilidades aproximadas. Asimismo, en el estudio se tuvieron en
cuenta dos tipos de confórmeros: aquellos con conformación
completamente antiperiplanar, t, y los que presentan un ángulo diedro
central de aproximadamente 60:, g.
Un tercer estudio se centra en la reactividad de los nitrilos frente a la
protonación [23]. A través de un análisis QTAIM se testearon las

Estudio QTAIM de nitrilos y compuestos relacionados
8
predicciones del modelo de resonancia (RM) para una serie de 15 nitrilos.
En este caso se incluyeron también compuestos con conjugación π. Las
densidades electrónicas fueron obtenidas con dos niveles de cálculo
distintos: B3LYP/6-31++G** y HF/6-31**G**, sin que ello diese lugar a
encontrar diferencias significativas. Se observa que las afinidades
protónicas (PA) calculadas concuerdan en todos los casos con buena
precisión con las experimentales (salvo un caso la diferencia es siempre
inferior a 10 kJ mol-1). Como conclusión principal debe destacarse que
tras la protonación del cianocompuesto, el protón mantiene una elevada
carga positiva. De hecho, se concluye que las estructuras de Lewis del
tipo +H-N≡C-R son más adecuadas que las del tipo H-N≡C+-R y H-N+≡C-R
para describir la distribución electrónica de las especies protonadas.
Además, el estudio de las propiedades de enlace pone de manifiesto que
en el enlace N≡C aumenta la densidad electrónica π y se reduce la
densidad electrónica ς como consecuencia de la protonación. Asimismo,
durante la protonación de un cianocompuesto la densidad electrónica
molecular evoluciona de forma análoga a la observada en las O-
protonaciones y N-protonaciones de otros compuestos [6,37-40],
observándose transferencias de densidad electrónica entre átomos
vecinos. Así, comparando la evolución de la densidad electrónica en las
protonaciones de HCN y de sus derivados alquílicos, se observa que la
población electrónica del átomo de carbono del grupo ciano, N(C), es
siginificativamente mayor en los segundos, debido a la transferencia

Estudio QTAIM de nitrilos y compuestos relacionados
9
desde otros átomos, particularmente desde los hidrógenos del grupo
alquilo que, una vez más, actúan como fuentes (en este caso) o
sumideros de densidad electrónica en el proceso químico tal como había
sido propuesto inicialmente por Stutchbury y Cooper [41]. También
resulta significativo que cuando el cianocompuesto contiene un sistema
con conjugación π, la protonación da lugar a una importante reducción
de la densidad electrónica π de dicho sistema, mientras que la densidad
electrónica ς se mantiene prácticamente inalterada. Por último, se
destaca que los procesos de protonación dan lugar a variaciones de
poblaciones electrónicas atómicas y energías atómicas que guardan una
buena correlación.
En el cuarto artículo se analiza la acidez de la posición al grupo ciano
[24], propiedad frecuentemente utilizada en síntesis orgánica [42]. Para
ello se considera una serie de 24 nitrilos sustituidos CNCHR1R2 con
diferentes grupos dadores y receptores de densidad electrónica. Se
comparan las densidades para cada compuesto neutro con la del
obtenido por su desprotonación en la posición que da lugar al anión
[CNCR1R2]-. Todas las densidades electrónicas se obtienen con
optimizaciones geométricas completas al nivel B3LYP/6-311++G(2d,2p)
6d. Se analiza: i) la estabilización del anión -desprotonado frente a la
desprotonación en otras posiciones; ii) El efecto de la sustitución sobre la
distribución electrónica y su relación con la diferencia de energía entre
las especies neutra y protonada y iii) la fiabilidad de las predicciones del

Estudio QTAIM de nitrilos y compuestos relacionados
10
modelo de resonancia, tanto desde un punto de vista energético como
en términos de cargas atómicas.
En primer lugar es notoria la prioridad energética de la desprotonación
sobre las restantes. Esto se demuestra estudiando las diversas
desprotonaciones de dos cianuros de alquilo de cadena larga
(CN(CH2)9CH3 y CN(CH2)10CH3). La diferencia observada supera en todos
los casos los 100 kJ mol-1.
En principio, la presencia de sustituyentes que retiran densidad
electrónica reduce notablemente la energía de desprotonación. Así en la
serie CNCH3, (CN)2CH2, (CN)3CH, dichas energías presentan,
respectivamente, valores de 1549, 1376 y 1229 kJ mol-1. Asimismo, la
combinación del grupo ciano con otros aceptores de densidad
electrónica por efecto mesómero (-NO2, -COOCH3) da lugar a notables
reducciones de la energía implicada en el proceso (1402 y 1334 kJ mol-1,
respectivamente). Sin embargo, el efecto contrario no es tan claro
cuando se incluye un dador de densidad electrónica por resonancia (-OH,
-NH2), que no incrementan desprotE más allá de 3 kJ mol-1 con respecto al
caso del CNCH3. la longitud y topología de la cadena alquílica unida al
grupo -CH2 tampoco dan lugar a cambios significativos. Así, los valores
de desprotE no difieren en más de 10 kJ mol-1 de los hallados para CNCH3
cuando el grupo CH3 se reemplaza por etilo, isopropilo, alquilos de
cadena lineal larga o, incluso, un grupo CH2=CH-. Un poco más intenso es
el efecto observado con derivados fluorados (1527 y 1503 kJ mol-1 para

Estudio QTAIM de nitrilos y compuestos relacionados
11
FCH2CN y F2CHCN, respectivamente). En cambio, la incorporación de
grupos hidrocarbonados que dan lugar a conjugación π con el grupo CN
(CH2=CH-CH2- y C6H5-CH2-) vuelve a provocar notables descensos de
desprotE (1458 y 1444 kJ mol-1, respectivamente). También, debe
destacarse que los grupos bencilo que incluyen sustituyentes receptores
por resonancia (NO2) reducen más desprotE. Por el contrario, si el
sustituyente incluído en el grupo bencilo es dador por resonancia (NH2),
la reducción observada para desprotE es menor. Incluso, se observan los
efectos debidos a la posición del sustituyente en el grupo bencilo que
predice el modelo de resonancia.
Por último, y en contraste con lo encontrado en los estudios de
protonación [23], se observa que, en general el modelo de resonancia
proporciona predicciones compatibles con las variaciones de población
electrónica atómica observadas en nuestro estudio QTAIM, desprotN(Ω).
Así, los incrementos de población electrónica observados en la especie
aniónica se reparten con mayor intensidad entre aquellos átomos sobre
los que el modelo de resonancia deslocaliza la carga negativa.
En el quinto artículo de esta Tesis se estudia la evolución de la densidad
electrónica en varios procesos de transposición que tienen lugar en
compuestos nitrogenados [25]. En primer lugar se considera la migración
de un átomo de hidrógeno para formar un isocianato con liberación de
N2 a partir de una acilazida (transposición de Curtius [43]). Además se
analizan dos migraciones de un grupo metilo: i) la formación de un catión

Estudio QTAIM de nitrilos y compuestos relacionados
12
nitrilio (R-C+=N-R ↔ R-CN+-R’) a partir de la protonación de una oxima
en la etapa inicial de la transposición de Beckmann [44]; y ii) la evolución
de un anión haloamida hasta el correspondiente isocianato (etapa de la
transposición de Hofmann [45]). El estudio utiliza densidades
electrónicas B3LYP/6-311++G(d,p). En los tres casos se llevaron a cabo
cálculos IRC (intrinsic reaction coordinate), así como optimizaciones de
reactivos y productos con el mismo nivel de cálculo.
El estudio de la transposición de Curtius confirma el carácter concertado
establecido en trabajos recientes para el mecanismo de este proceso [46-
49]. En el estado de transición el átomo de hidrógeno (átomo migrante)
está simultáneamente unido a los átomos de C (enlace C-H en el
reactivo) y N (enlace N-H en el producto), según indica la existencia de
dos puntos críticos de enlace. Si bien, la distancia de enlace C-H es más
próxima en el estado de transición a la del reactivo que la del enlace C-N
a la que muestra en el producto. En el estado de transición la molécula
de N2 está prácticamente formada desde un punto de vista geométrico.
Respecto a las poblaciones electrónicas atómicas, llama la atención la
carga inicialmente negativa del átomo de nitrógeno central en la unidad
N-NN, que de acuerdo con la estructura de Lewis habitualmente
empleada para describir a la azida debería presentar carga positiva. A lo
largo de la reacción la carga del átomo de oxígeno se mantiene
prácticamente constante, mientras se observa una transmisión de

Estudio QTAIM de nitrilos y compuestos relacionados
13
densidad electrónica desde H y C al átomo de N que terminará formando
parte del isocianato.
Nuestro estudio indica que la etapa seleccionada de la transposición de
Hofmann es un proceso elemental. El punto crítico del enlace C-N surge
al desaparecer el correspondiente al enlace C-C que se rompe en el
proceso. Esto no tiene lugar hasta después de la formación del estado de
transición y en todo ese intervalo de la reacción el enlace haloamida
continúa establecido. Se observa una reducción constante de la
población electrónica del C sp2 (coherente con la evolución desde un
enlace C-C a un C=N) y del C sp3 que, de manera semejante, reemplaza
un enlace C-C por un C-N. Es posible plantear que en el proceso
concertado tienen lugar dos transferencias electrónicas principales: de C
sp2 a Br y de C sp3 a N.
La etapa seleccionada de la reacción de Beckmann es también elemental.
El reactivo presenta una estructura compatible con un enlace C=N
mientras que la estructura del producto es compatible con la existencia
de un enlace C≡N. Sin embargo, el cálculo de las cargas atómicas sobre el
grupo C≡N del reactivo presenta valores que no coinciden con la forma
resonante que presenta este enlace triple y presentan mayor
coincidencia con una forma resonante similar a la encontrada para
cianocompuestos protonados : H+-N≡C-R. El enlace entre el grupo metilo
que migra y el nitrógeno aparece después del estado de transición
cuando se rompe su enlace al C sp2 y se rompe el enlace N-O. Al principio

Estudio QTAIM de nitrilos y compuestos relacionados
14
de la reacción, antes del estado de transición, se produce transferencia
de carga del C sp2 al nitrógeno, ya que la carga del C que migra apenas
varía y después del estado de transición, la carga se transfieres desde
estos dos carbonos al nitrógeno.
Finalmente figuran las conclusiones extraídas del trabajo realizado, así
como las referencias bibliográficas. Como principales conclusiones
resaltamos las siguientes.
En los cianoalcanos lineales se han encontrado valores transferibles para
todas las propiedades atómicas y de enlace calculadas para el grupo CN.
Se exceptúa la energía atómica, que muestra una dependencia del
tamaño molecular, cuantificado por la suma de los números atómicos.
Este efecto, encontrado en otras series homólogas, es un artificio debido
a los diferentes valores del cociente virial. Por el contrario, la energía
cinética electrónica atómica si presenta valores transferibles. Los valores
de estas propiedades permiten considerar como cuasi-transferibles y
específicos de la serie de cianoalcanos a: i) Los átomos C y N del grupo
CN; ii) CH2 en α respecto al grupo CN; y iii) CH2 en β respecto al grupo CN.
El grupo CH3 terminal, el CH2 previo al grupo metilo terminal y el resto de
los grupos metileno de la cadena presentan un comportamiento similar
al de los n-alcanos. Además, los siguientes átomos presentan un
comportamiento específico: C en posición α en los nitrilos de metilo y
etilo; C en posición β en los nitrilos de etilo y propilo; así como C y N del
grupo CN en los cianuros de hidrógeno y metilo.

Estudio QTAIM de nitrilos y compuestos relacionados
15
En los dicianoalcanos se observa una mayor sensibilidad a la
transferencia en las propiedades atómicas que en las propiedades de
enlace. Utilizando parámetros estadísticos se han caracterizado 12
grupos metilenos diferentes. La influencia mutua entre grupos CN (efecto
de proximidad) puede considerarse despreciable cuando estos grupos
están separados por más de 14 grupos metileno.
En los nitrilos protonados, el protón presenta una carga positiva grande
lo que indica que está más acorde con la estructura de Lewis H+N≡CR
que con las estructuras: HN+≡CR, HNC+R. La densidad
electrónica de los nitrilos se modifica con la protonación de manera
análoga a la observado para las protonaciones de O y N de otras series de
compuestos orgánicos.
La desprotonación de un metileno al grupo CN está significativamente
favorecida cuando el grupo CN se encuentra en conjugación π con grupos
de carácter atrayente de densidad electrónica por efecto mesómero. El
efecto de los grupos dadores de densidad electrónica, incluso por efecto
mesómero, no altera significativamente la energía del proceso. La
variación de las poblaciones atómicas en este proceso no presenta
contradicciones con el modelo de resonancia semejantes a las
observadas en proceceos de protonación o adición de hidruros.
La evolución de la densidad electrónica en etapas seleccionadas de las
transposiciones de Curtius, Beckmann y Hofmann indica un carácter
concertado para los tres procesos. Los nuevos enlaces formados durante

Estudio QTAIM de nitrilos y compuestos relacionados
16
la reacción solo aparecen cuando los originales se rompen, después del
estado de transición. La transferencia de carga durante estas reacciones
tiene lugar implicando esencialmente al átomo que migra y a los átomos
unidos a él al principio y al final de la reacción.
.

Estudio QTAIM de nitrilos y compuestos relacionados
17
1.- INTRODUCCIÓN
El grupo CN está considerado como uno de los principales grupos
funcionales. El uso de nitrilos en química orgánica preparativa comenzó a
adquirir importancia en la segunda mitad del siglo XIX. Sus características
propiedades reactivas lo convirtieron en un compuesto de uso común en
síntesis orgánica e inorgánica. En concordancia con su utilidad, su
estructura geométrica y electrónica ha sido objeto de una enorme
cantidad de estudios teóricos y experimentales [50]. Muchos de estos
estudios se centraron en la estructura del primer miembro de la serie de
los nitrilos, HCN, que se ha convertido en uno de los sistemas de
referencia más utilizados para testear métodos teóricos y niveles de
cálculo [51,52]. Los nitrilos, especialmente HCN, son también
importantes moléculas interestelares que han sido detectadas por
radioastronomía en varias fuentes [53].
En el estudio que se presenta en esta Tesis Doctoral juegan un papel
fundamental conceptos químicos como: similaridad, grupo funcional,
transferabilidad, efecto de proximidad, modelo de resonancia, etc. Los
conceptos de similaridad, grupo funcional y transferabilidad atómica han
jugado un papel muy importante en el desarrollo de la Química [54]. Sin
embargo, debe resaltarse que, normalmente, estos conceptos, han sido
empleados de manera intuitiva, sin tener en cuenta definiciones precisas
ni ningún tipo de cuantificación. Aceptando que las propiedades de la

Estudio QTAIM de nitrilos y compuestos relacionados
18
materia son una manifestación de su estructura interna, la similaridad
entre substancias debe originarse en distribuciones de carga similares. En
1980 Carbó et al. [55] propusieron el primer índice de similaridad
mecano cuántico basado en las distribuciones de densidad electrónica de
las moléculas. Este índice marcó un punto de arranque para muchos
otros que han surgido posteriormente con objeto de abordar el
problema de evaluar la similaridad entre moléculas, entre otros
relacionados, cabe citar los índices de Cioslowski [56,57].
La densidad electrónica, ya sea obtenida a partir de cálculos
computacionales, como se hace en este trabajo, o bien
experimentalmente por métodos como la difracción de rayos X, puede
someterse a un análisis topológico. Aunque para esta tarea existen varios
métodos, entre ellos ha adquirido un notable grado de aceptación la
teoría cuántica de átomos en moléculas (QTAIM) desarrollada por Bader
[12,13]. Este será el método que utilizaremos fundamentalmente en
nuestro trabajo, aunque también se ha considerado el estudio topológico
de distintos campos escalares relacionados con ρ, como es el caso de su
laplaciana.
La introducción de la QTAIM hizo posible la partición de una molécula
de manera precisa y rigurosa (sin utilizar hipótesis ajenas a los principios
fundamentales) [58,59], en subsistemas discretos que verifican los
teoremas de la Mecánica Cuántica. Se puede demostrar de manera
precisa que esta división se realiza mediante superficies de flujo cero
para el gradiente de la densidad electrónica,ρ(r) [60-62]. Estas

Estudio QTAIM de nitrilos y compuestos relacionados
19
superficies dividen el espacio en regiones, Ω, que se identifican con los
átomos de la molécula. Las propiedades atómicas se obtienen, entonces,
por integración de la correspondiente densidad de la propiedad sobre
esa región.
La teoría QTAIM ofrece la herramienta teórica para definir un grupo
funcional como un átomo (o grupo de átomos) que en una serie de
moléculas mantiene una similitud importante [61]. La similaridad de los
átomos a lo largo de una serie puede ser cuantificada por medio de un
índice de similaridad, como los introducidos por Cioslowski et al. [56,57]
o bien estimada comparando los valores de las propiedades atómicas en
varias moléculas [1-4]. Así, QTAIM proporciona una vía para establecer el
concepto de grupo funcional de manera cuantitativa. Se ha demostrado
que la transferabilidad perfecta de las propiedades es un límite
inalcanzable [19,20]. Por ello, se utiliza el término “transferabilidad
aproximada”. Este se aplica cuando las variaciones observadas en una
serie de moléculas son menores que los errores experimentales o se
aproximan a la precisión atribuida a los métodos numéricos.
La transferabilidad de átomos y grupos de átomos presenta una
aplicación práctica ampliamente utilizada en Química: predecir las
propiedades de una molécula a partir de las propiedades de sus
fragmentos constituyentes, esto es debido a la ligera variación que
presentan muchas propiedades atómicas a lo largo de series homólogas
de moléculas.

Estudio QTAIM de nitrilos y compuestos relacionados
20
Nosotros estamos interesados en el estudio de las propiedades de
grupos funcionales obtenidos por combinación de los átomos de la teoría
QTAIM. Especialmente en comparar sus propiedades con objeto de
establecer límites para entornos moleculares en los que una propiedad o
grupo funcional pueda ser considerado aproximadamente transferible.
Estudios realizados en este departamento, de aplicación de la teoría
QTAIM, han permitido hacer una clasificación de los átomos de aldehídos
y cetonas [1,2], éteres [3,4] y otros compuestos [26,27] en grupos casi
transferibles. Para lograr estos resultados, se hizo uso de relaciones
empíricas encontradas y que no habían sido publicadas hasta ese
momento, tales como la relación entre varias propiedades atómicas y el
nivel de precisión con el que se determinan las superficies de flujo cero
[1]. Cabe añadir, que, pese a la presencia de diferentes grupos metileno
cuasi-transferibles, aldehídos, cetonas [2] y éteres [4] presentan un buen
ajuste lineal para la energía total HF y para el calor de formación
experimental con el número de grupos metileno presentes en la
molécula, de manera que para estas magnitudes la reproducción de
resultados para las moléculas de la serie nunca presentan discrepancias
en E mayores que 2.5 kJ mol-1 en éteres y 1.5 kJ mol-1 en aldehídos y
cetonas.
El término “efecto proximidad” fue acuñado hace más de 30 años [33] y
está relacionado con el desarrollo de modelos moleculares para
disoluciones de no electrolitos [63-64]. Estos modelos particionan una
molécula en bloques (“building blocks”) que se supone que son

Estudio QTAIM de nitrilos y compuestos relacionados
21
independientes, transferibles y que están caracterizados por un conjunto
de parámetros empleados para calcular diversas propiedades de mezclas
de no electrolitos. El efecto de proximidad hace referencia a una de las
principales deficiencias de los modelos de contribuciones de grupos: la
interacción intramolecular entre dos o más grupos funcionales. Esta
afecta a sus propiedades así como a las de los grupos situados en su
entorno invalidando así la tranferabilidad [33]. Estas variaciones que
sufren las propiedades de los átomos debido a la presencia de otro grupo
funcional han sido usadas repetidamente en discusiones cualitativas
sobre el comportamiento de mezclas de compuestos polifuncionales
[65,66]. Se han propuesto varias soluciones para tratar este problema
desde variaciones empíricas de los parámetros de grupo dependiendo
de primeros y segundos vecinos [33] hasta correcciones cuantitativas
basadas en los análisis de población de Mulliken con la finalidad de
adaptar grupos definidos para compuestos monofuncionales a moléculas
polifuncionales [63]. La adecuación de todos estos tratamientos está
relacionada con la siguiente pregunta: ¿Son equivalentes (en una buena
aproximación) los cambios sufridos por la distribución electrónica de un
átomo en una molécula a la suma de los efectos producidos por estos
grupos funcionales en compuestos monofuncionales? o, por el contrario:
¿el efecto de proximidad involucra efectos cooperativos importantes
entre grupos funcionales?
La aplicación de QTAIM a las densidades electrónicas HF/6-31++G**
muestra que los átomos de oxígeno de las moléculas RO-(CH2)-OR’ son

Estudio QTAIM de nitrilos y compuestos relacionados
22
significativamente diferentes de los correspondientes a los monoéteres
cuando n<4 [67]. Esto es, se confirma la existencia de un efecto de
proximidad cuando los oxígenos están separados por menos de cinco
enlaces. En esta tesis se estudia el efecto proximidad en α,ω-
dicianoalcanos usando un particionamiento QTAIM. Estos compuestos
han sido empleados para formar complejos con enlaces de hidrógeno y
compuestos de inclusión con urea [68], de interés en Química
Supramolecular [69]. Concretamente se estudia el efecto proximidad en
los grupos –CN y –CH2- usando criterios estadísticos para establecer los
límites de transferabilidad. Estos criterios se basan en las máximas
desviaciones presentadas por las propiedades de grupos que son
claramente transferibles en moléculas grandes para las que las
propiedades atómicas y de enlace son equivalentes sin lugar a duda. Este
procedimiento da lugar a un mayor número de grupos específicos que los
obtenidos en el trabajo sobre cianoalcanos lineales. También se estudia
en esta tesis si el efecto proximidad está compuesto por contribuciones
aditivas de los grupos funcionales aislados.
Generalmente se ha aceptado la aplicación del modelo de resonancia
(RM) para explicar la estructura y reactividad de compuestos orgánicos
[70,71] siendo una herramienta muy útil en Química. No obstante, el
análisis topológico de las densidades electrónicas realizados con la teoría
QTAIM para diversos procesos ha mostrado una evolución de la densidad
electrónica que no concuerda con las predicciones del modelo RM. Estos
desacuerdos aparecen incluso para procesos tan simples como

Estudio QTAIM de nitrilos y compuestos relacionados
23
rotaciones internas [72,73], protonaciones [6,8,37-40] o adiciones de
hidruro [10]. Asimismo los resultados QTAIM son inconsistentes con las
estructuras de Lewis tradicionalmente aceptadas para algunos
compuestos cargados, tales como sales de diazonio [74] o éteres
protonados [37,38]. La publicación del primer estudio sobre los
desacuerdos entre RM y QTAIM fue seguida por una controversia acerca
de la adecuadabilidad de QTAIM para este tipo de estudios [75-77].
Actualmente la controversia parece que se ha inclinado en favor de la
aplicabilidad de QTAIM. Además, muchas de las conclusiones cualitativas
obtenidas a partir de estudios QTAIM sobre protonación y adiciones de
hidruro han sido confirmadas por estudios que emplean otros métodos
de analísis de las densidades electrónicas [8,10] tales como el
particionamiento de Hirshfeld [78,79].
Las estructuras de Lewis H-N=C+-R se han usado tradicionalmente para
describir los nitrilos protonados en varios mecanismos de reacción. Estas
estructuras son, en el marco del modelo RM, el resultado de tranformar
un par electrónico π del triple enlace N≡C en un enlace N-H.
Alternativamente, el proceso de protonación puede ser entendido como
la formación de un enlace dativo entre N y el protón usando un par
solitario (par no enlazante) del N, proceso representado por la fórmula
H+-X, que esta acompañado por una redistribución electrónica que afecta
a toda la molécula. Los hidrógenos actúan como una fuente muy efectiva
de densidad electrónica en está redistribución, tal y como confirman los
estudios sobre la basicidad de NH3 y una serie de metilaminas realizados

Estudio QTAIM de nitrilos y compuestos relacionados
24
por Stuchbury y Cooper [41]. En esta Tesis se ha llevado a cabo un
estudio QTAIM sobre la protonación de varios ciano compuestos en fase
gas que permite analizar esta cuestión. También permite estudiar si el
triple enlace modifica las tendencias que se han observado hasta este
momento en otros compuestos. Las moléculas estudiadas en este trabajo
incluyen ciano alcanos lineales y ramificados así como compuestos en los
que la función ciano está conjugada con sistemas π deslocalizados, por lo
tanto ha sido posibles establecer tendencias según el tamaño de las
cadenas alquílicas lineales, el cambio conformacional, cadenas alquílicas
ramificadas, electronegatividad de sustituyentes y deslocalización π.
Además, la reacción de protonación se puede tomar como modelo de
estudio para analizar las tendencias que muestran ciertos compuestos en
su reactividad con sistemas electrofílicos. También la protonación a
menudo resulta ser un primer paso en muchos mecanismos de reacción,
por estos motivos se han calculado las afinidades protónicas de 15
nitrilos con objeto de estudiar la evolución de la densidad electrónica
durante la misma. La evolución de la densidad electrónica también ha
servido para testear el modelo de resonancia (RM), como ha sucedido en
otros estudios anteriores [5-11,37-40]. Las tendencias mostradas por el
proceso de protonación no son compatibles con las predichas por el
modelo de resonancia para procesos de protonación en fase gas.

Estudio QTAIM de nitrilos y compuestos relacionados
25
2.- OBJETIVOS
De manera general esta tesis aborda analizar las propiedades
electrónicas de los nitrilos y de compuestos directamente
relacionados con ellos, sea por motivos de reactividad o de semejanza
estructural. En concreto se persiguen los siguientes objetivos:
1. Determinar en qué condiciones y de qué manera se pueden
considerar grupos aproximadamente transferibles en la serie de
nitrilos de alquilo. En este análisis se considera tanto el grupo
nitrilo (-CN) como los grupos metilo y metileno del resto alquílico.
2. Analizar como el efecto de proximidad entre grupos nitrilos puede
modificar las reglas de transferibilidad aproximada que se
obtengan como respuesta al objetivo anterior. Es decir, estudiar la
influencia mutua entre dos grupos nitrilo separados por un resto
alquílico, así como el efecto sufrido por los grupos metileno
intermedios, considerando la serie homóloga de los
dicianoalcanos, CN(CH2)nCN.
3. Obtener computacionalmente afinidades protónicas y acideces de
diversos nitrilos que presenten diferencias estructurales
significativas.

Estudio QTAIM de nitrilos y compuestos relacionados
26
4. Describir los efectos electrónicos que acompañan a los principales
procesos ácido-base que experimentan los nitrilos: a) protonación
y b) abstracción de hidrógenos en posiciones α. La descripción
obtenida se comparará con la prevista según el modelo de
resonancia. Esta comparación permitirá evaluar la viabilidad del
modelo de resonancia para describir estos procesos.
5. Describir como evoluciona la densidad electrónica en ciertos
procesos que involucran a compuestos nitrogenados que guardan
cierta semejanza estructural con los nitrilos. Concretamente: a) la
formación de un isocianato y liberación de nitrógeno a partir de
una acilazida en la transposición de Curtius; b) la etapa inicial de la
transposición de Beckmann, que proporciona un catión nitrilio (al
que se asignan formas resonante R-C+=N-R y R-CN+-R’) a partir de
la protonación de una oxima; y c) la etapa de la transposición de
Hofmann que considera la evolución desde un anión haloamida
hasta el correspondiente isocianato.

Estudio QTAIM de nitrilos y compuestos relacionados
27
3. DISCUSIÓN GENERAL

Estudio QTAIM de nitrilos y compuestos relacionados
28

Estudio QTAIM de nitrilos y compuestos relacionados
29
"Las teorías son redes: solo quién lance cogerá". Novalis (citado por Kart R. Popper en
la Lógica de la investigación científica)
3.1. METHODOLOGY
This chapter overviews the two methods which were most extensively
used throughout this Thesis: Density Functional Theory (DFT) and the
Quantum Theory of Atoms in Molecules (QTAIM). The first one has been
our usual tool to obtain electron densities, which were subsequentely
analyzed by means of the second one in order to get insight about the
chemical problems here addressed (presented in chapter 2 and discussed
in section 3.2). We have also included two sections devoted to a couple
of important issues we are directly involved in this Thesis: approximate
transferability and the limitations of the resonance model. As a lot of
work has been done previously on both we believe it is worth to make a
short review and introduce some general considerations on them before
presenting the results here obtained.
3.1.1. DENSITY FUNCTIONAL THEORY (DFT)
In computational chemistry, density functional theory (DFT) usually
stands for the Kohn–Sham implementation. Certainly, the initial
approaches to DFT can be traced back to the statistical method,
independently proposed by Thomas [80] and Fermi [81]. In this method,
the electron density of polyelectronic atoms is treated locally as a Fermi

Estudio QTAIM de nitrilos y compuestos relacionados
30
gas in which the free-electron relations apply. Nevertheless, it is the
Kohn–Sham implementation which has gained ground, mainly due to its
similarity with the self-consistentfield Hartree–Fock method.
The Kohn–Sham formulation of DFT relies on the fact that the electron
density of the ground state of a system, can be computed as the density
of a system of independent particles, moving in an effective one-particle
potential, whose precise formal construction forms part of the method.
Once this effective potential has been determined, the Kohn–Sham
method solves self consistently the nonlinear Kohn–Sham equations
which contain an unknown exchange-correlation functional [82-84]. The
exchange-correlation functional contains the description of the electron–
electron interactions within the system. The theoretical foundation for
the Kohn–Sham method is the Hohenberg–Kohn theorems [85].
The first Hohenberg-Kohn theorem, as published in 1964, states that
there is a unique relation of the external potential Vext(r) (arising from the
positive charges of the nuclei) within an N electron system and its
(ground state) electron density ρ(r). Since the complete ground state
energy E0 is a unique functional of the electron density distribution ρ(r),
so must be its individual parts 1.
rVrVrTrE ext
int 1
In this expression we find a system-dependent part, Vext[ρ(r)], which
changes from one system to another. In contrast, two parts are universal,
in the sense that the form of the functional is independent of the actual
system determined by N, RA and ZA.

Estudio QTAIM de nitrilos y compuestos relacionados
31
The system independent parts define the Hohenberg-Kohn functional 2.
rVrTrFHK
int 2
The second Hohenberg-Kohn theorem is nothing else than the variational
principle formulated for densities. Given any electron density distribution
ρ*(r) associated to an N electron system with external potential Vext, one
can state 3, with the equal sign only valid if ρ*(r) = ρ(r).
rVrVrTrEE ext
**** int0 3
Up till now, both the exact ground state density, ρ*(r), as well as the
Hohenberg-Kohn functional, FHK, are still unknown, so one cannot make
use of the Hohenberg theorems to calculate the molecular properties.
In the FHK both known and unknown parts can be identified 4 with
potential energy term, Vee, giving by 5, where J(ρ) is the classical
interaction of two charge densities and ENCL(ρ) contains all non-classical
parts. Thus, the complete energy functional can be written 6, where the
two first terms are known and the latter are unkonwn.
rVrTrF eeHK
4
rErdrdrr
rrrErJrV NCLNCLee
21
21
21
2
1
5
rErTrVrJrE NCLext
6
The basic problem is the unknown functional for the kinetic energy. A
solution to this problem was given by Kohn and Sham in the paper

Estudio QTAIM de nitrilos y compuestos relacionados
32
published in 1965 [82], where they suggested to formaly split this
functional into two parts 7.
rTrTrT cS
7
where the first part Ts[ρ], the kinetic energy of the non interacting
electrons, will be expressed in a one particle approach similar to Hartree-
Fock, thus being well known, and the second, the correction to the
kinetic energy deriving from the interacting nature of the electrons (still
unknown part) contains the difference between the real functional T[ρ]
and the one particle term Ts[ρ], as well as the other remaining parts of
the total energy functional, which are still unknown, in a approximative
way. Thus one can write 9.
rErTrTrVrJrE NCLCSext
8
rErTrVrJrE XCSext
9
The so-called exchange-correlation functional EXC[ρ] (summation of
TS[ρ(r)] and ENCL[ρ(r)]) remains unknown and the rest are well defined
terms.
No reference is made in the proof to the Hartree–Fock level of
approximation. That is, the approximations made in DFT enter at the
level of the Hamiltonian, when an approximate form for the functional is
chosen. Such a Halmitonian can be expressed as a sum of one-electron
operators, has eigenfunctions that are Slater determinants of the
individual one-electron eigenfunctions, and has eigenvalues that are
simply the sum of the one-electron eigenvalues. Due to the similarity,

Estudio QTAIM de nitrilos y compuestos relacionados
33
one can solve the Kohn-Sham equations using the same algorithms as in
the Hartree-Fock theory, including the usage of basis functions and the
self consistent field (SCF) approach.
Within an orbital expression the energy functional 9 may then rewritten
as 10.
rErdr
r
Rr
ZrE XC
N
i
ii
N
i
i
A Ai
Aii
ii
1
1
12
1
1
2
2
1
2
10
where N is the number of electrons and the density for a Slater
monodeterminantal wave function 11.
N
i
i rr1
2
11
If we undertake in the usual fashion to find the orbitals φ that minimize E
in 10 we find that they satisfy the pseudoeigenvalue equations 12.
rrf iii
KS ˆ
12
where the Kohn-Sham (KS) one-electron operator is defined as 13
where the exchange-correlation potential, defined by 14, is a so-called
functional derivative, and it is best described as the one-electron
operator for which the expectation value of the KS Slater determinant is
Exc.
A A
AXC
iKS
Rr
ZrVrd
r
rf
1
12
12
2
2
2ˆ
13
r
rErV xc
XC
1
14

Estudio QTAIM de nitrilos y compuestos relacionados
34
Because the E of 10 that we are minimizing is exact, the orbitals φ must
provide the exact density. KS orbitals are determined expressing them
within a basis set of functions and the individual orbital coefficients are
determined by solution of a secular equation entirely analogous to that
employed for HF theory. Noneless, the KS orbitals are not the same as
the HF orbitals, and they lack of the physical interpretation of the HF one
electron molecular orbitals, but in some cases [86,87].
Therefore, the accuracy of a DFT calculation depends upon the quality of
the exchange–correlation (XC) functional. Exc is an unknown object that
includes all non-trivial many–body effects required to make KS theory
exact. The past two decades have seen remarkable progress in the
development and validation of XC density functionals [88-91]. There are
two main strategies for developing new functional, namely the
nonempirical and the semiempirical approach. The nonempirical
approach, favored in physics, is to construct functional subject to several
exact constraints. The typical nonempirical is the “Jacobs ladder” scheme
[92-95] advanced by Perdew and co-workers. This strategy can be viewed
as a ladder with five rungs, from the local density approximation (LDA) up
to the “divine” exact exchange and exact correlation functional. In this
approximation Exc[ρ(r)] is taken to be the exchange and correlation
energy of a homogeneous electron gas with density ρ=ρ(r). Although
there exists an exact expression for the exchange energy in this model,
the exact value of the correlation energy is known only in the limit of
very high densities. The semiempirical way to construct Exc[ρ(r)], which

Estudio QTAIM de nitrilos y compuestos relacionados
35
has been very successful in chemistry, is to choose a flexible
mathematical functional form depending on one or more parameters
and then to fit these parameters to molecular thermochemical data. This
approach is only empirical in part because the functional form is guided
by theory.
The fisrt generation of functionals were the local spin density
approximation (LSDA), in which density functionals depend only on the
up- and down-spin (ς = α, β) local spin densities ρς. Although LSDA gives
accurate predictions for solid-state physics, it is not a useful model for
chemistry due to its severe overbinding of chemical bonds and
underestimation of barrier heights. The second generation of density
functionals is called the generalized gradient approximation (GGA), in
which functionals depend on the ρς and their gradients ρς. GGA
functionals have been shown to give more accurate predictions for
thermochemistry than LSDA ones, but they still underestimate barrier
heights. In third-generation functionals, two additional variables, the spin
kinetic energy densities, τς(r), are included in the functional form; such
functionals are called meta-GGAs. LSDAs, GGAs, and meta-GGAs are
“local” functionals because the electronic energy density at a single
spatial point depends only on the behavior of the electronic density and
kinetic energy at and near that point [96-98]; local functionals can be
mixed with nonlocal Hartree–Fock (HF) exchange as justified by the
adiabatic connection theory [99]. Functionals containing HF exchange are
usually called hybrid functionals, and they are often more accurate than

Estudio QTAIM de nitrilos y compuestos relacionados
36
local functionals for maingroup thermochemistry. HF exchange would be
exact if the Kohn–Sham orbitals were the accurate ones determined by
the exact XC functional, which is unknown. We do, however, know some
properties of the exact XC functional, for example, it is nonlocal [100],
and these properties can serve as constraints during functional
development. In the last six years, the development of new functional
forms for meta-GGAs and hybrid meta-GGAs and their validation against
diverse databases have yielded powerful new density functionals with
broad applicability to many areas of chemistry. There has also been
much interest in including noncovalent interactions in DFT [93,101-107].
The B3LYP [99,108,109] functional, which is a hybrid GGA, is largely
responsible for DFT becoming one of the most popular tools in
computational chemistry. However, B3LYP is unable to describe van der
Waals complexes bound by medium-range interactions, such as the
interactions in methane dimers and benzene dimers. Kohn et al. [110]
point out that “the commonly used LDA and GGA, designed for
nonuniform electron gases, fail to capture the essence of vdW energies”.
Mourik and Gdanitz [111] confirmed this point by showing that the local
density approximation and some well-established GGA functional are
incapable of accounting for dispersion effects in a quantitative way. This
inability of B3LYP (and most of other popular functionals) to describe
accurately medium-range XC energy limits their applicability for
biological systems where medium-range dispersion-like interactions play
vital roles. For biological systems it is essential to describe London

Estudio QTAIM de nitrilos y compuestos relacionados
37
dispersion forces (van der Waals attraction) accurately along with
electrostatic and hydrogen bond interaction. For those studies, those in
which we study stacking interaction, we use new improved functionals
like the MPW1B95 [112]. This functional belongs to the generation of
functionals called meta-GGAs, because it incorporates electron spin
density, density gradient, kinetic energy density, and Hartree-Fock (HF)
exchange. Spin density, density gradient, and kinetic energy density are
local properties of the density, although the latter two are sometimes
called semilocal whereas HF exchange is nonlocal. The inclusion of HF
exchange is a permanent feature of accurate exchange-correlation
functional. The one-parameter hybrid Fock-Kohn-Sham operator can be
written as 15, [113,114]
CorGCESEHFEH FFFX
FX
FF ˆˆˆ100
1ˆ100
ˆˆ
15
where FH is the Hartree operator (i.e., the nonexchange part of the HF
exchange operator), FHFE is the HF exchange operator, X is the percentage
of HF exchange, FSE is the Dirac-Slater local density functional for
exchange [115,116], FGCE is the gradient correction for the exchange
functional, and FCor is the total correlation functional including both local
and gradient-corrected parts and (where applicable) a dependence on
kinetic energy density. In this functional, Adamo and Barone´s mPW
exchange functional [117] is used for FGCE and Becke95 [109] functional
for FCor (meta-GGA). For the MPW1B95 model, X is optimized to minimize
the root-mean-square error for the AE6[118] representative atomization
energy database. This functional is suitable for general applications in

Estudio QTAIM de nitrilos y compuestos relacionados
38
thermochemistry and gives good performance for hydrogen bonding and
weak interaction calculations [112].

Estudio QTAIM de nitrilos y compuestos relacionados
39
“Es gibt nichts mehr praktisches al seine gute Theorie” (No hay nada más práctico que
una bunea teoría). Clausius
3.1.2. AN OVERVIEW ON THE QUANTUM THEORY OF ATOMS IN
MOLECULES (QTAIM)
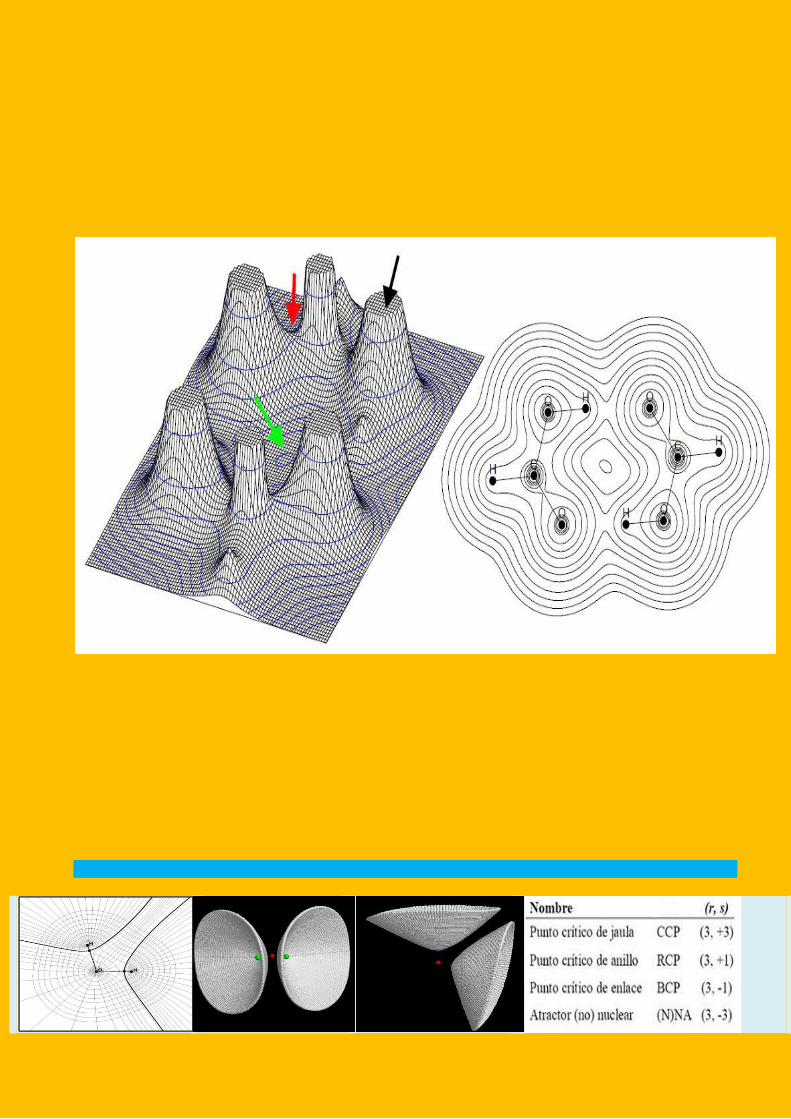
QTAIM can be viewed as a topological analysis of the electron density
function, ρ(r),.As in any topological analysis, the localization of singular
points plays a basic role. In this case we have singular points in the real
space spanned by the 3 coordinates representing the position of any
electron, r. Looking at the relief map of ρ(r) in the plane of pyrrole (figure
1) that contains all its nuclei, we can observe ten local maxima, also
called electron density attractors, whose coordinates correspond very
approximately to those of the ten nuclei in the molecule. Along every
bond there is a saddle point with two negative eigenvalues of the
Hessian matrix of ρ(r). These points are also called bond critical points or
BCPs. Finally, inside the ring, we observe another saddle point, whose
Hessian matrix presents two positive eigenvalues. It is called a ring critical
point (or RCP) and it is characteristic of cyclic structures. Finally in
molecules like cubane, where there is a cage structure, we observe the
presence of a relative minimum, one per cage, which is named cage
critical point (CCP) and is surrounded by ring critical points, six in this
case. It this context, a recent paper by Castillo et al. has proved that in a

Estudio QTAIM de nitrilos y compuestos relacionados
40
highly twisted system (1,12-difluorobenzo[c]phenanthrene) a cage
structure can be delimited by only two ring surfaces [119].
Figure 1. Relief plot of the electron density of pyrrole.
N 1 H 2
C 3
C 4
C 5
C 6
H 7
H 8
H 9
H 10
Figure 2. ρ(r) plot in the main plane of pyrrole.

Estudio QTAIM de nitrilos y compuestos relacionados
41
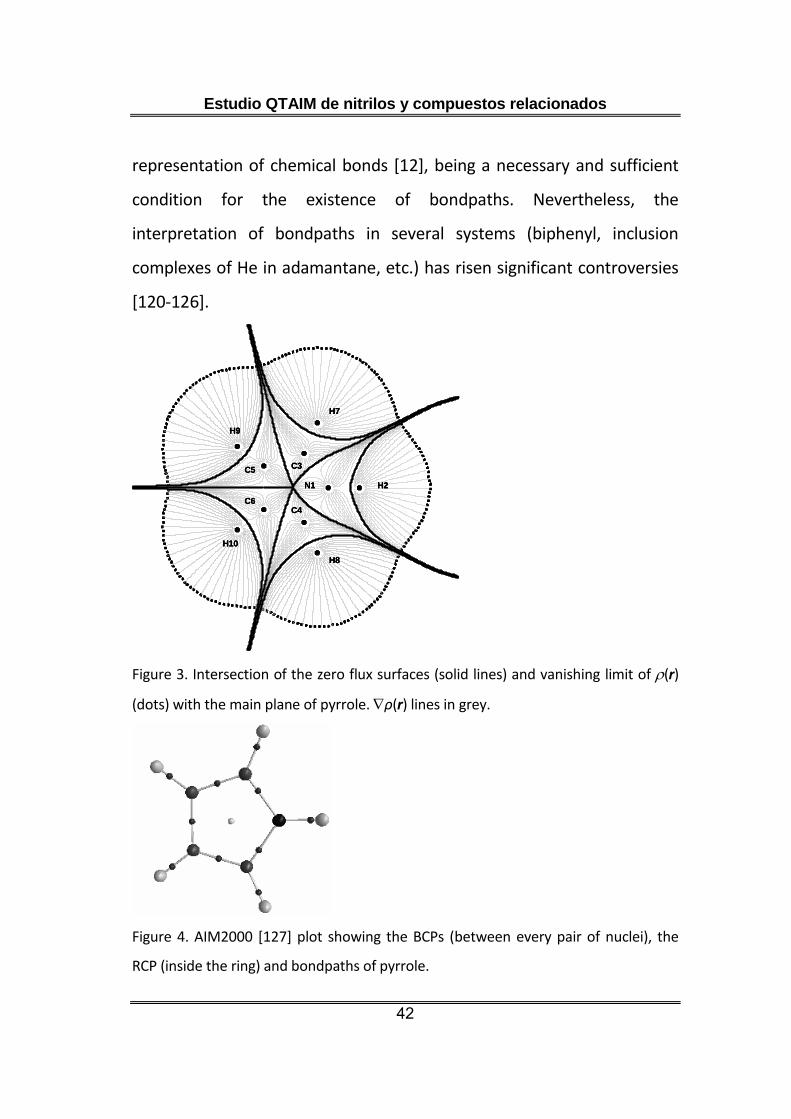
The topological analysis also looks at the gradient paths of the electron
density, which are shown for the main plane of pyrrole in figure 2. We
observe they form a vector field where every group of field lines ends at
a different nucleus. These groups of lines are delimited by surfaces given
by what is known as the zero flux condition 16, which is a mathematical
condition rigorously derived [58] from Schwinger’s principle of stationary
action [59]. These surfaces are represented in the pyrrole plane as lines
that intersect a certain vanishing limit of the molecular electron density
defining the atomic basins, that are disjoints regions of the space (figure
3). In this context, an atom can be defined as the joint of a nucleus and
its electron basin. The integration of the proper density function within
the atomic basin provides the atomic properties, like the atomic electron
population 17, the atomic electron kinetic energy 18 or the atomic
volume 19.
0 rnr
16;
rdrN
17;
rdrrrK rr
2
,24
1
18;
rdv
19
Turning back to critical points, it has to be said that the conduction of the
eigenvector associated to the positive eigenvalue of every BCP gives rise
to the atomic interaction lines or bondpaths (figure 4). According to
Bader's original formulation of QTAIM, bondpaths are the physical
Estudio QTAIM de nitrilos y compuestos relacionados
42
representation of chemical bonds [12], being a necessary and sufficient
condition for the existence of bondpaths. Nevertheless, the
interpretation of bondpaths in several systems (biphenyl, inclusion
complexes of He in adamantane, etc.) has risen significant controversies
[120-126].
N1 H2
C3
C4
C5
C6
H7
H8
H9
H10
N1 H2
C3
C4
C5
C6
H7
H8
H9
H10
N1 H2
C3
C4
C5
C6
H7
H8
H9
H10
Figure 3. Intersection of the zero flux surfaces (solid lines) and vanishing limit of (r)
(dots) with the main plane of pyrrole. ρ(r) lines in grey.
Figure 4. AIM2000 [127] plot showing the BCPs (between every pair of nuclei), the
RCP (inside the ring) and bondpaths of pyrrole.
Estudio QTAIM de nitrilos y compuestos relacionados
43
3.1.3.- APPROXIMATE TRANSFERABILITY
9.747
9.748
9.749
9.750
9.751
9.752
9.753
1 4 7 10n
N(F
) [a
u]
9.335
9.336
9.337
9.338
9.339
9.340
9.341
1 4 7 10n
N(O
) [a
u]
F
C
(CH2) n
CH3
H H
H
C
O
(CH2) n
CH3
N(O) = 9.3387(2) au N(F) = 9.7503(2) au
|L(O)| < 6·10-4 au |L(F)| < 5·10-4 au
9.747
9.748
9.749
9.750
9.751
9.752
9.753
1 4 7 10n
N(F
) [a
u]
9.747
9.748
9.749
9.750
9.751
9.752
9.753
1 4 7 10n
N(F
) [a
u]
9.335
9.336
9.337
9.338
9.339
9.340
9.341
1 4 7 10n
N(O
) [a
u]
9.335
9.336
9.337
9.338
9.339
9.340
9.341
1 4 7 10n
N(O
) [a
u]
F
C
(CH2) n
CH3
H H
F
C
(CH2) n
CH3
H H
H
C
O
(CH2) n
CH3
N(O) = 9.3387(2) au N(F) = 9.7503(2) au
|L(O)| < 6·10-4 au |L(F)| < 5·10-4 au
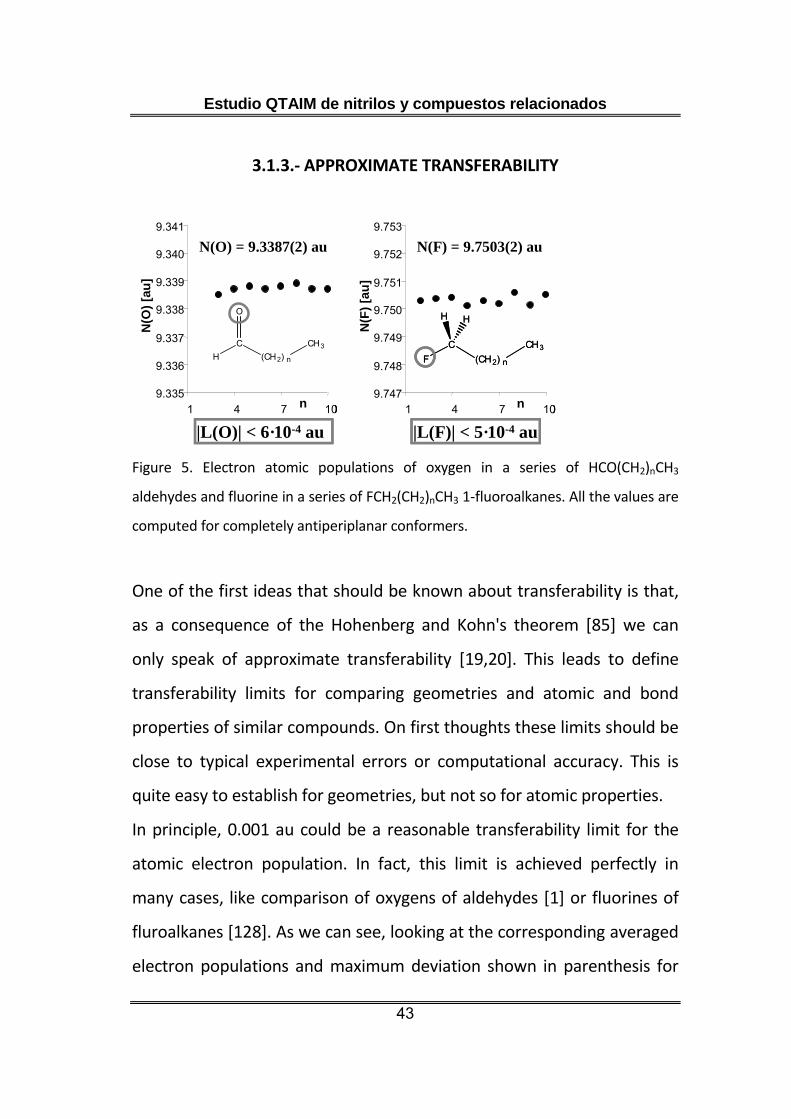
Figure 5. Electron atomic populations of oxygen in a series of HCO(CH2)nCH3
aldehydes and fluorine in a series of FCH2(CH2)nCH3 1-fluoroalkanes. All the values are
computed for completely antiperiplanar conformers.
One of the first ideas that should be known about transferability is that,
as a consequence of the Hohenberg and Kohn's theorem [85] we can
only speak of approximate transferability [19,20]. This leads to define
transferability limits for comparing geometries and atomic and bond
properties of similar compounds. On first thoughts these limits should be
close to typical experimental errors or computational accuracy. This is
quite easy to establish for geometries, but not so for atomic properties.
In principle, 0.001 au could be a reasonable transferability limit for the
atomic electron population. In fact, this limit is achieved perfectly in
many cases, like comparison of oxygens of aldehydes [1] or fluorines of
fluroalkanes [128]. As we can see, looking at the corresponding averaged
electron populations and maximum deviation shown in parenthesis for
Estudio QTAIM de nitrilos y compuestos relacionados
44
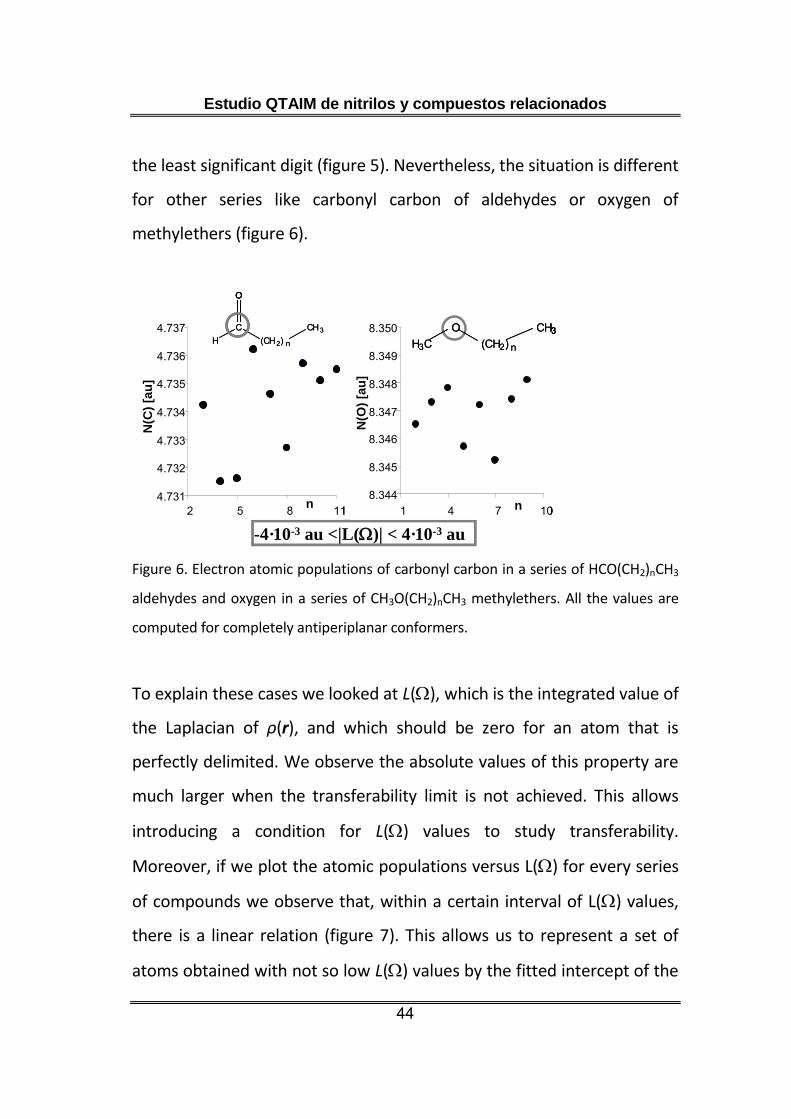
the least significant digit (figure 5). Nevertheless, the situation is different
for other series like carbonyl carbon of aldehydes or oxygen of
methylethers (figure 6).
-4·10-3 au <|L()| < 4·10-3 au
4.731
4.732
4.733
4.734
4.735
4.736
4.737
2 5 8 11n
N(C
) [a
u]
H
C
O
(CH2) n
CH3
8.344
8.345
8.346
8.347
8.348
8.349
8.350
1 4 7 10n
N(O
) [a
u]
H3C
O
(CH2) n
CH3
-4·10-3 au <|L()| < 4·10-3 au
4.731
4.732
4.733
4.734
4.735
4.736
4.737
2 5 8 11n
N(C
) [a
u]
H
C
O
(CH2) n
CH3
4.731
4.732
4.733
4.734
4.735
4.736
4.737
2 5 8 11n
N(C
) [a
u]
4.731
4.732
4.733
4.734
4.735
4.736
4.737
2 5 8 11n
N(C
) [a
u]
H
C
O
(CH2) n
CH3
H
C
O
(CH2) n
CH3
8.344
8.345
8.346
8.347
8.348
8.349
8.350
1 4 7 10n
N(O
) [a
u]
H3C
O
(CH2) n
CH3
8.344
8.345
8.346
8.347
8.348
8.349
8.350
1 4 7 10n
N(O
) [a
u]
8.344
8.345
8.346
8.347
8.348
8.349
8.350
1 4 7 10n
N(O
) [a
u]
H3C
O
(CH2) n
CH3
H3C
O
(CH2) n
CH3
Figure 6. Electron atomic populations of carbonyl carbon in a series of HCO(CH2)nCH3
aldehydes and oxygen in a series of CH3O(CH2)nCH3 methylethers. All the values are
computed for completely antiperiplanar conformers.
To explain these cases we looked at L(), which is the integrated value of
the Laplacian of ρ(r), and which should be zero for an atom that is
perfectly delimited. We observe the absolute values of this property are
much larger when the transferability limit is not achieved. This allows
introducing a condition for L() values to study transferability.
Moreover, if we plot the atomic populations versus L() for every series
of compounds we observe that, within a certain interval of L() values,
there is a linear relation (figure 7). This allows us to represent a set of
atoms obtained with not so low L() values by the fitted intercept of the

Estudio QTAIM de nitrilos y compuestos relacionados
45
electron population, written as N0(), which is the electron population
obtained with no integration error.
4.731
4.732
4.733
4.734
4.735
4.736
4.737
-3.0 -1.0 1.0 3.0 5.0L(C) [au]
N(C
) [
au
]
8.344
8.345
8.346
8.347
8.348
8.349
8.350
-4.0 -2.0 0.0 2.0 4.0L(C) [au]
N(O
) [
au
]
Nº(C) = 4.734 au Nº(O) = 8.346 au
4.731
4.732
4.733
4.734
4.735
4.736
4.737
-3.0 -1.0 1.0 3.0 5.0L(C) [au]
N(C
) [
au
]
8.344
8.345
8.346
8.347
8.348
8.349
8.350
-4.0 -2.0 0.0 2.0 4.0L(C) [au]
N(O
) [
au
]
Nº(C) = 4.734 au Nº(O) = 8.346 au
Figure 7. N() vs. L() plot for the carbonyl carbon in a series of HCO(CH2)nCH3
aldehydes and oxygen in a series of CH3O(CH2)nCH3 methylethers. All the values are
computed for completely antiperiplanar conformers.
Atomic energy was a more problematic quantity for establishing
transferability limits. Looking again at the atomic energy of the oxygen
atom, E(O), of a homologous series of aldehydes [1], we observe that so
similar atoms like the oxygen of dodecanal and nonanal differed by
almost 30 kJ mol-1. The situation was completely similar for all the
ketones studied, with the oxygen atom becoming more destabilized
when the size of the molecule increases, and giving rise to unexpected
differences of the oxygen energy in position isomers, like 5-undecanone
and 6-undecanone (figure 8).
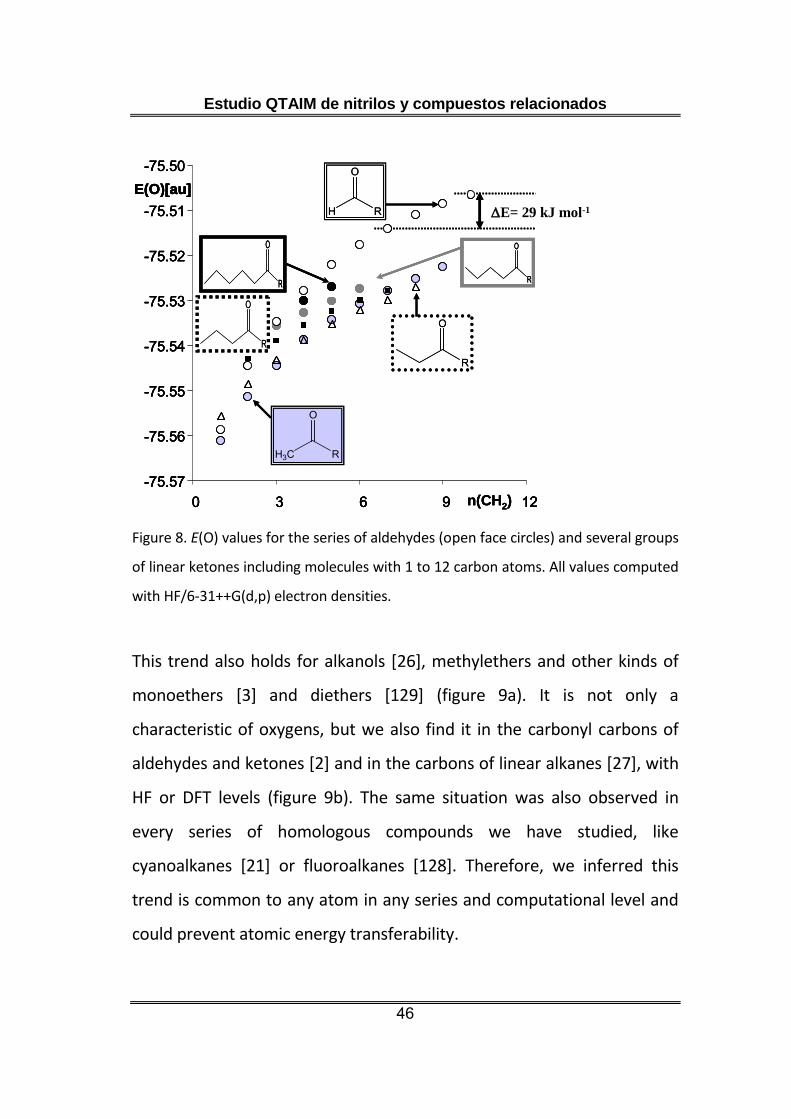
Estudio QTAIM de nitrilos y compuestos relacionados
46
H
O
R
H3C
O
R
O
R
O
R
n(CH2)
-75.57
-75.56
-75.55
-75.54
-75.53
-75.52
-75.51
-75.50
0 3 6 9 12
E(O)[au]
O
R
O
R
E= 29 kJ mol-1H
O
R
H3C
O
R
O
R
O
R
n(CH2)
-75.57
-75.56
-75.55
-75.54
-75.53
-75.52
-75.51
-75.50
0 3 6 9 12
E(O)[au]
n(CH2)
-75.57
-75.56
-75.55
-75.54
-75.53
-75.52
-75.51
-75.50
0 3 6 9 12
E(O)[au]
-75.57
-75.56
-75.55
-75.54
-75.53
-75.52
-75.51
-75.50
0 3 6 9 12
E(O)[au]
O
R
O
R
O
R
O
R
E= 29 kJ mol-1
Figure 8. E(O) values for the series of aldehydes (open face circles) and several groups
of linear ketones including molecules with 1 to 12 carbon atoms. All values computed
with HF/6-31++G(d,p) electron densities.
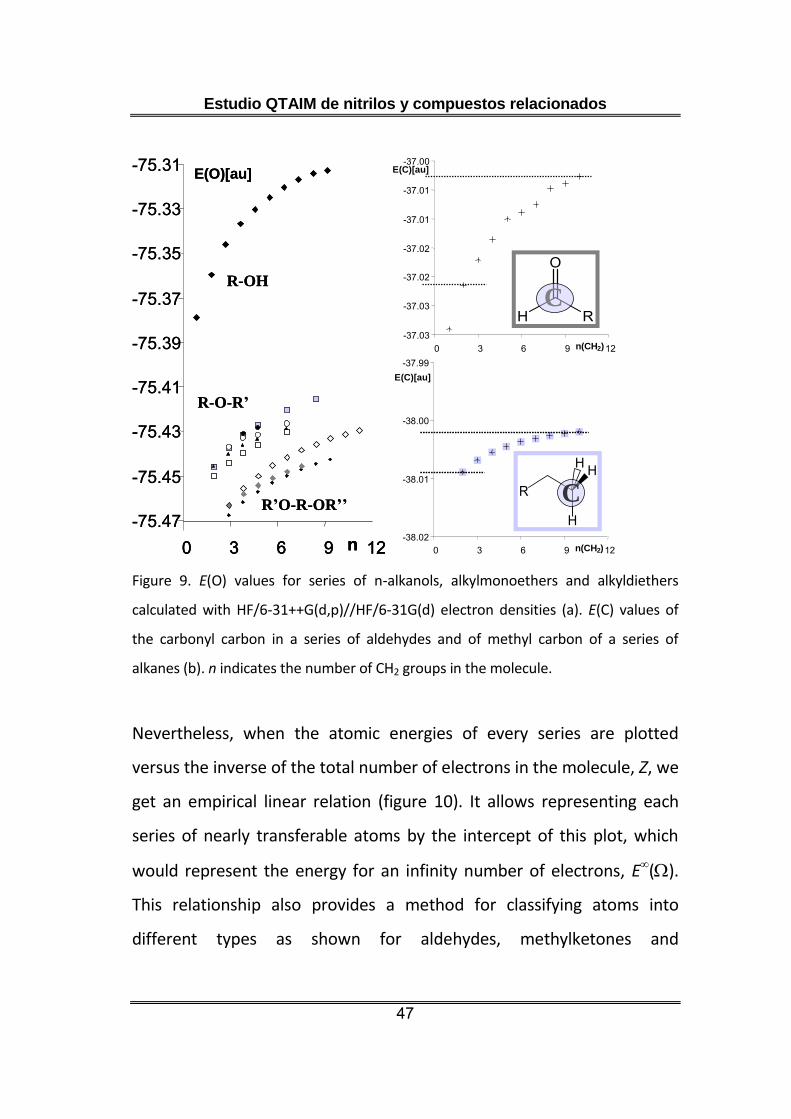
This trend also holds for alkanols [26], methylethers and other kinds of
monoethers [3] and diethers [129] (figure 9a). It is not only a
characteristic of oxygens, but we also find it in the carbonyl carbons of
aldehydes and ketones [2] and in the carbons of linear alkanes [27], with
HF or DFT levels (figure 9b). The same situation was also observed in
every series of homologous compounds we have studied, like
cyanoalkanes [21] or fluoroalkanes [128]. Therefore, we inferred this
trend is common to any atom in any series and computational level and
could prevent atomic energy transferability.
Estudio QTAIM de nitrilos y compuestos relacionados
47
R-OH
R-O-R’
R’O-R-OR’’-75.47
-75.45
-75.43
-75.41
-75.39
-75.37
-75.35
-75.33
-75.31
0 3 6 9 12n
E(O)[au]
R-OH
R-O-R’
R’O-R-OR’’-75.47
-75.45
-75.43
-75.41
-75.39
-75.37
-75.35
-75.33
-75.31
0 3 6 9 12n0 3 6 9 12n
E(O)[au]
-38.02
-38.01
-38.00
-37.99
0 3 6 9 12n(CH2)
E(C)[au]
R
HH
H
C
-37.03
-37.03
-37.02
-37.02
-37.01
-37.01
-37.00
0 3 6 9 12n(CH2)
E(C)[au]
H
O
RC
-38.02
-38.01
-38.00
-37.99
0 3 6 9 12n(CH2)
E(C)[au]
R
HH
H
C
-38.02
-38.01
-38.00
-37.99
0 3 6 9 12n(CH2)
E(C)[au]
R
HH
H
R
HH
H
C
-37.03
-37.03
-37.02
-37.02
-37.01
-37.01
-37.00
0 3 6 9 12n(CH2)
E(C)[au]
H
O
RC
-37.03
-37.03
-37.02
-37.02
-37.01
-37.01
-37.00
0 3 6 9 12n(CH2)
E(C)[au]
H
O
RC
Figure 9. E(O) values for series of n-alkanols, alkylmonoethers and alkyldiethers
calculated with HF/6-31++G(d,p)//HF/6-31G(d) electron densities (a). E(C) values of
the carbonyl carbon in a series of aldehydes and of methyl carbon of a series of
alkanes (b). n indicates the number of CH2 groups in the molecule.
Nevertheless, when the atomic energies of every series are plotted
versus the inverse of the total number of electrons in the molecule, Z, we
get an empirical linear relation (figure 10). It allows representing each
series of nearly transferable atoms by the intercept of this plot, which
would represent the energy for an infinity number of electrons, E().
This relationship also provides a method for classifying atoms into
different types as shown for aldehydes, methylketones and
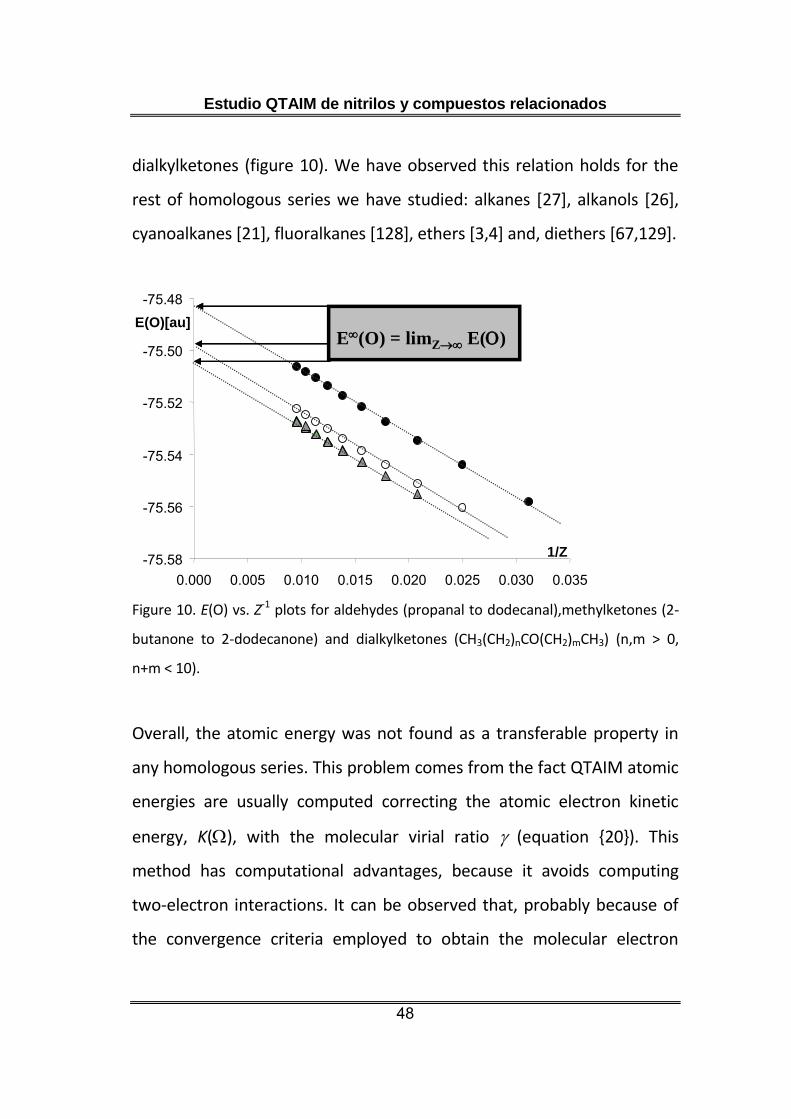
Estudio QTAIM de nitrilos y compuestos relacionados
48
dialkylketones (figure 10). We have observed this relation holds for the
rest of homologous series we have studied: alkanes [27], alkanols [26],
cyanoalkanes [21], fluoralkanes [128], ethers [3,4] and, diethers [67,129].
-75.58
-75.56
-75.54
-75.52
-75.50
-75.48
0.000 0.005 0.010 0.015 0.020 0.025 0.030 0.035
1/Z
E(O)[au]
E(O) = limZ E()
-75.58
-75.56
-75.54
-75.52
-75.50
-75.48
0.000 0.005 0.010 0.015 0.020 0.025 0.030 0.035
1/Z
E(O)[au]
-75.58
-75.56
-75.54
-75.52
-75.50
-75.48
0.000 0.005 0.010 0.015 0.020 0.025 0.030 0.035
1/Z
E(O)[au]
E(O) = limZ E()
Figure 10. E(O) vs. Z-1 plots for aldehydes (propanal to dodecanal),methylketones (2-
butanone to 2-dodecanone) and dialkylketones (CH3(CH2)nCO(CH2)mCH3) (n,m > 0,
n+m < 10).
Overall, the atomic energy was not found as a transferable property in
any homologous series. This problem comes from the fact QTAIM atomic
energies are usually computed correcting the atomic electron kinetic
energy, K(), with the molecular virial ratio (equation 20). This
method has computational advantages, because it avoids computing
two-electron interactions. It can be observed that, probably because of
the convergence criteria employed to obtain the molecular electron
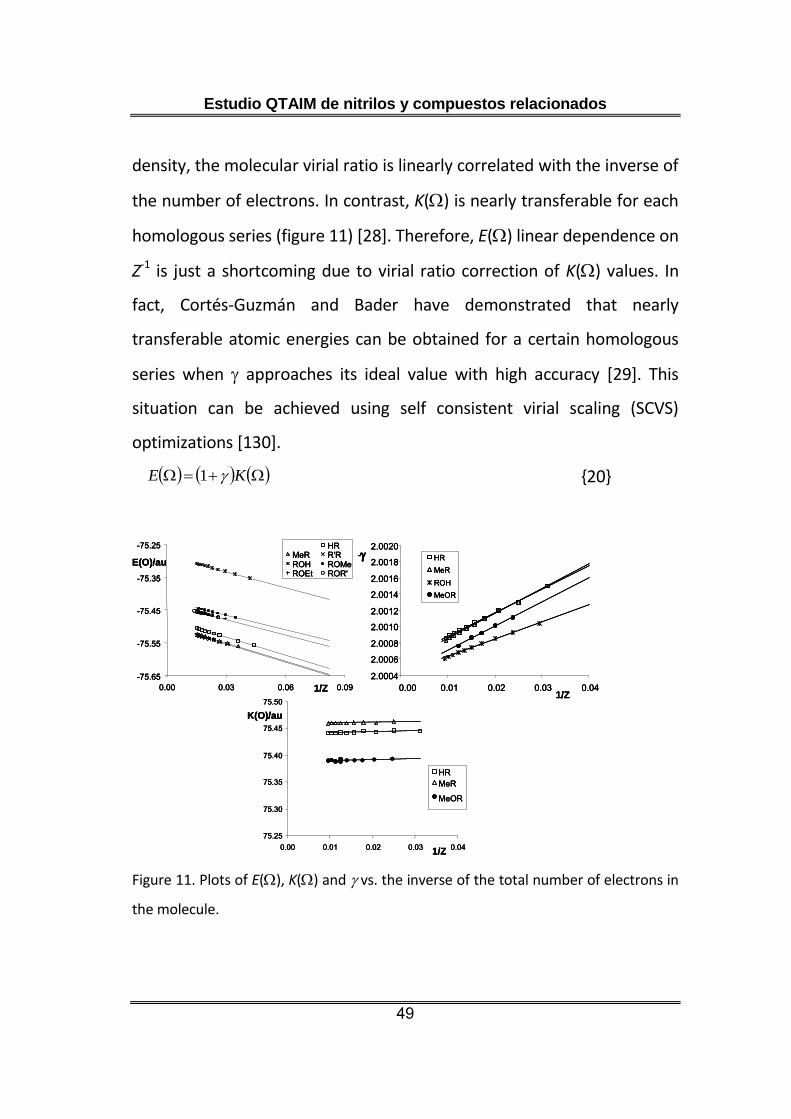
Estudio QTAIM de nitrilos y compuestos relacionados
49
density, the molecular virial ratio is linearly correlated with the inverse of
the number of electrons. In contrast, K() is nearly transferable for each
homologous series (figure 11) [28]. Therefore, E() linear dependence on
Z-1 is just a shortcoming due to virial ratio correction of K() values. In
fact, Cortés-Guzmán and Bader have demonstrated that nearly
transferable atomic energies can be obtained for a certain homologous
series when approaches its ideal value with high accuracy [29]. This
situation can be achieved using self consistent virial scaling (SCVS)
optimizations [130].
KE 1 20
-75.65
-75.55
-75.45
-75.35
-75.25
0.00 0.03 0.06 0.091/Z
E(O)/au
HRMeR R'RROH ROMeROEt ROR'
75.25
75.30
75.35
75.40
75.45
75.50
0.00 0.01 0.02 0.03 0.041/Z
K(O)/au
HR
MeR
MeOR
2.0004
2.0006
2.0008
2.0010
2.0012
2.0014
2.0016
2.0018
2.0020
0.00 0.01 0.02 0.03 0.041/Z
- HR
MeR
ROH
MeOR
-75.65
-75.55
-75.45
-75.35
-75.25
0.00 0.03 0.06 0.091/Z
E(O)/au
HRMeR R'RROH ROMeROEt ROR'
-75.65
-75.55
-75.45
-75.35
-75.25
0.00 0.03 0.06 0.091/Z
E(O)/au
HRMeR R'RROH ROMeROEt ROR'
75.25
75.30
75.35
75.40
75.45
75.50
0.00 0.01 0.02 0.03 0.041/Z
K(O)/au
HR
MeR
MeOR
75.25
75.30
75.35
75.40
75.45
75.50
0.00 0.01 0.02 0.03 0.041/Z
K(O)/au
HR
MeR
MeOR
2.0004
2.0006
2.0008
2.0010
2.0012
2.0014
2.0016
2.0018
2.0020
0.00 0.01 0.02 0.03 0.041/Z
- HR
MeR
ROH
MeOR
2.0004
2.0006
2.0008
2.0010
2.0012
2.0014
2.0016
2.0018
2.0020
0.00 0.01 0.02 0.03 0.041/Z
- HR
MeR
ROH
MeOR
Figure 11. Plots of E(), K() and vs. the inverse of the total number of electrons in
the molecule.

Estudio QTAIM de nitrilos y compuestos relacionados
50
No significant problems were found for obtaining accurate transferability
limits for other atomic properties like the first moment of the electron
density, dipole moment, volume, or Shannon entropy, as well as for bond
properties.
F
CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3
C
C
C
C C
5.240
5.245
5.250
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
C
FF
CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3
CC
CC
CC
CC CC
5.240
5.245
5.250
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.240
5.245
5.250
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.240
5.245
5.250
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
C
Figure 11. N(C) vs. L(C) plots for nearly transferable carbons in 1-fluoroalkanes in
completely antiperiplanar conformer.
Once the transferability limits were established we investigated how
transferability is affected by diverse factors. One of the simplest is the
conformational change. If we take a series of functionalized linear
alkanes, like 1-fluoralkanes, in completely antiperiplanar arrangement,
we observe (using the above commented N() vs. L() plots shown in

Estudio QTAIM de nitrilos y compuestos relacionados
51
figure 11), that carbons form a nearly transferable group, the same is
true for , , and atoms, whereas all the rest of the carbons (excluding
the terminal groups) belong to a common nearly transferable group
[128]. They are called by us normal or C carbons, and present the same
properties obtained for the inner methylenes of unfuctionalized n-
alkanes.
C
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
F
C
C
C
CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3
C
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
F
C
C
C
C
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
F
C
C
C
C
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
F
C
C
C
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.235
5.240
5.245
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
5.790
5.795
5.800
5.805
5.810
5.815
5.820
-0.004 -0.002 0.000 0.002 0.004
L(C) [au]
N(C
) [a
u]
C
C
C
C
F
C
C
C
C
C
C
CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3CH3-CH2-CH2-(CH2)n-CH2-CH2-CH3
Figure 12. N(C) vs. L(C) plots for nearly transferable carbons in 1-fluoroalkanes in
gauche conformers.
Rotating from anti to gauche conformer, we observe that the population
of and carbons is different, but they continue as different types. In
contrast, C cannot be distinguished from and carbons, which are not
specific groups in gauche conformers (figure 12) [128]. In summary, , ,

Estudio QTAIM de nitrilos y compuestos relacionados
52
and carbons of an alkyl chain are significantly influenced by a
functional group in antiperiplanar arrangement, but this influence
reduces to and in gauche. This trend also holds for other series and
computational levels [131].
3.1.4. ON THE LIMITATIONS OF THE RESONANCE MODEL
The resonance model (RM) is still one of the most often employed tools
for explaining the mechanism of chemical processes or predicting their
products. The study and application of this model consumes a significant
amount of time to chemistry students. Nevertheless diverse evidences
found by several research groups point to its inadequacy to describe the
evolution of the electron density in various simple chemical processes.
Even, RM cannot explain some experimental facts like the evolution of
pKas along certain series of organic compounds. Conformational
equilibria, protonations or hydride additions are examples of simple
processes where the resonance model leads to explanations that
contradict those obtained using modern quantum chemical methods for
the electron density analysis. Among them, the quantum theory of atoms
in molecules (QTAIM) [12,13] can be considered as a very reliable one, as
it is based exclusively on the basic principles of Physics without
introducing any other hypothesis.

Estudio QTAIM de nitrilos y compuestos relacionados
53
Several QTAIM studies have contradicted well known and
generally accepted conclusions of the RM. To the best of our knowledge,
Wiberg and Laidig’s work on the origin of ester and amide resonance
[72], can be reported as the first serious difference between QTAIM
results and RM explanations. This work shows that in diverse R-CO-XR’
compounds, comprising formamide(XR’=NH2), formic acid (XR’=OH),
methyl formate (XR’=OCH3), etc., the atomic electron population of
nitrogen/oxygen, N(X), is smaller in the transition states for the C-X
rotation than in the corresponding planar conformers. In contrast, N(O)
remains nearly constant along this process. According to the RM, the
non-planar geometry of transition states for C-X rotation breaks the
electron delocalization along the O=C-X unit (usually represented by
resonant forms: O=C-X ↔ O--C=X+) present in the planar conformer.
Clearly, RM predicts that, at transition states and with regard to
conformers, N(X) should be larger, as the O--C-X+ form should have a
negligible weight, and N(O) should be smaller. These results, obtained
initially at the HF level, were confirmed later at the MP2 level [73].
Slightly later, Slee and MacDougall observed that the comparison of
QTAIM atomic electron populations, N(Ω), of allyl ions and the
corresponding neutral compounds is not in line with the evolution of
electron density expected with the resonance model [132]. In this
context, it should be mentioned that the publication of the first of
Wiberg’s papers on “ester/amide resonance” led to a long argument
about the reliability of QTAIM atomic populations [75], ended by clear

Estudio QTAIM de nitrilos y compuestos relacionados
54
demonstrations of unreliability of the basic postulates against QTAIM
charges [76,77]. Moreover, the same kind of contradiction between RM
predictions and QTAIM relative charges observed for simple amides and
esters was also obtained for thioformamide [133].
In the same vein, Glaser and Chao obtained that the electron
density distribution of diazonium ions are inconsistent with the
commonly used Lewis structure R-N+≡N and would be better
represented by a combination of two unconnected structures: R+···N≡N
and R+···N-≡N+ [74,134]. Also, the acidity sequence followed by dimethyl
sulfide, sulfoxide and sulfone, cannot be explained by the RM, which
reverses the order. In contrast, QTAIM atomic populations explain the
real sequence and provide no evidence for the delocalization of the
charge from the anionic carbon in the rest of the anion [135].
In the second half of the 90’s our group started a systematic study
on protonation processes of oxygenated compounds employing QTAIM
as basic tool for analyzing the evolution of the molecular electron density
(computed at diverse computational levels: HF, B3LYP, MP2 and
sometimes QCISD) along the protonation. This study comprised
carbonylic compounds [1,2]. linear [3,4] and cyclic ethers [38,136]. The
general conclusion obtained was that the positive charge was mainly
concentrated on the proton while the oxygen formally attached to it
does not reduce its electron population, as postulated by classic
protonation scheme shown in figure 13. On the contrary, N(O) increases
upon protonation, gaining electron density from the remaining hydrogen

Estudio QTAIM de nitrilos y compuestos relacionados
55
atoms in the molecule, as had been previously proposed by Stutchbury
and Cooper [41].
R1
O
R2 R1
O
R2
H+
+ H+
Figure 13. Classic mechanism of protonation for carbonyl compounds
+ H+
+406
-25
+310
-105 -105
-83 -39
-112
-112
-47 -76 a
b
Figure 14. Evolution of atomic electron population, ΔN(Ω), upon protonation of
propanone. All values in au multiplied by 103.
Later on, our work was extended to other systems of practical
interest, as uracil and cytosine [5,6,39], and to compounds without
oxygen, like nitriles [23] or indole [40]. RM was only able to predict the
stability sequence of protonated forms and explain the changes exhibited
by most of the bond properties upon protonation. Even, both the O- and
N-protonated forms of uracil and cytosine are found to be better
described by RO-H+ and RN-H+ forms than by the classical RO+-H and RN+-

Estudio QTAIM de nitrilos y compuestos relacionados
56
H structures. Again, according to the QTAIM analysis the electron charge
gained by the proton is mainly provided by the other hydrogens of the
molecule. The study of several model systems, like vinylketone, methyl
formiate and N-methyl formamide [6] led us to explain the previously
reported stability sequence of uracil [5] and cytosine protonated forms,
as well as the evolution of atomic electron populations. Thus, we
developed an alternative model, not based on the resonance concept but
mainly on electrostatic interactions [6,39], which we think can be applied
to any protonation. This model is based upon the following points: i) the
donation of electron population is easier when the atomic number is
smaller; ii) the closer the distance to the proton is, the easier the electron
donation will be; iii) the donation of electron population between
bonded atoms follows the direction of the bond. The orientation of the
bond with regard to the proton can make the electron transference
easier (if the electron density approaches the proton), or more difficult (if
the electron density moves away from the proton) (see, respectively,
hydrogens labeled “a” and “b” in figure 14); and iv) π-transferences are
generally easier than ς ones, when both are possible.
At this point, we should highlight that other modern methods for
electron density analysis, like the Hirshfeld scheme [78], implemented
for several computational levels [79], which was employed to analyze
several simple oxygenated compounds [8], provide different absolute
values for the evolution of atomic electron populations, ΔN(Ω), but the
same qualitative description, contradicting RM expectations.

Estudio QTAIM de nitrilos y compuestos relacionados
57
As protonation can be considered as a model for electrophilic
attacks, our group also studied how activant and deactivant substituents
modify the evolution of electron density in this process. QTAIM analysis
carried out the protonation of a set of aniline derivatives, indicates that
most of the electron density gained by the proton is provided neither by
the nitrogen atom nor by activant substituens like OH [7]. In a similar
way, the acidity of phenol derivatives can be rationalized on the basis of
atomic QTAIM properties, but not on the RM predictions [9].
On the other hand, the evolution of molecular electron density upon
hydride addition, (simple model for nucleophilic attacks), computed both
with QTAIM or Hirshfeld methods, has been shown to display general
trends that are also not in line with RM predictions summarized in the
scheme shown in figure 15 [10]. Thus, we observe that most of the
electron density provided by the hydride is not taken by the oxygen. In
fact ΔN(O) never reaches 0.2 au, whereas for the carbon attached to
hydride ΔN(C) always exceeds 0.4 au and ΣΔN(H) goes from 0.44 au to
0.53 au in the compounds hitherto studied. When the study is repeated
using other anionic nucleophiles (CN-, OH-) the results do not change
substantially.
R1
O
R2 R1 R2
H- +
O H-
Figure 15. Classic mechanism for hydride addition to carbonyl compounds.

Estudio QTAIM de nitrilos y compuestos relacionados
58
Among the discrepancies observed between RM predictions and
relative atomic charges, we highlight the specific behavior of
heteroatoms, X, reducing the extent of electron reorganization with
regard to that displayed when they are replaced by carbons [6]. In fact C-
X bonds were found to act as barriers to ς-electron reorganization,
precluding (or reducing substantially) the transference of ς electron
density throughout them [6]. We thought of interest to show if the
discrepancies previously described for pyrimidinic and puric bases
[5,6,39], affect in general to all heterocycles. In particular, it was shown
that RM predictions are not in line with conclusions derived from the
QTAIM analysis carried out for the protonation (in some cases, also other
processes) of diverse heterocycles: indoles [40], 1,3-azoles [137], and
anthocyanidins [138].

Estudio QTAIM de nitrilos y compuestos relacionados
59
3.2. DISCUSIÓN GENERAL DE RESULTADOS
De acuerdo con la normativa vigente, se presenta en esta sección una
discusión que unifica los resultados presentados en los artículos
contenidos en la sección 4. En esta Tesis se ha abordado el estudio de la
estructura y reactividad de nitrilos y compuestos relacionados utilizando
la Teoría Cuántica de Átomos en Moléculas (QTAIM).
Hemos considerado de interés, comenzar caracterizando la estructura
electrónica de los nitrilos más simples: los cianoalcanos. Para ello, se
analizaron las propiedades atómicas y de enlace de una serie de doce
alcanonitrilos lineales en conformación antiperiplanar [21]. Se estudió en
que condiciones se podía considerar como cuasi-transferibles a los
grupos CN, CH2 y CH3.
Nuestro siguiente reto consistía en averiguar como se distorsiona está
imagen de la estructura electrónica considerando dos posibles
alteraciones:
i) La presencia de un sustituyente en la molécula. De entre todos
los posibles hemos considerado únicamente la introducción de
otro grupo ciano en la molécula.
ii) Que tenga lugar un proceso reactivo. Hemos considerado los
dos principales procesos ácido-base que pueden experimentar
los cianocompuestos: protonación y liberación de un protón
desde un carbono al grupo funcional.

Estudio QTAIM de nitrilos y compuestos relacionados
60
El primer estudio nos ha permitido establecer que el efecto de
proximidad en los dicianoalcanos es importante y se mantiene como
estadísticamente significativo sobre la distribución electrónica hasta que
ambos grupos se separan por más de 14 grupos CH2 [22].
Con el segundo grupo de estudios hemos podido comprobar que: a) las
distorsiones introducidas por la protonación afectan al total de la
molécula, de manera particular a los átomos de hidrógeno [23]; b) los
protones mantienen una elevada carga positiva en el nitrilo, de manera
que la especie protonada está mejor descrita por una estructura de Lewis
del tipo +H-N≡C-R que por las tipo H-N≡C+-R o H-N+≡C-R [23]; c) la
desprotonación está significativamente favorecida sobre las restantes
en todos los cianocompuestos estudiados; d) Sólo los sustituyentes que
retiran densidad electrónica por efecto resonante reducen notablemente
la energía de desprotonación; y e) en general el modelo de resonancia
proporciona predicciones compatibles con las variaciones de población
electrónica atómica en la desprotonación de un nitrilo [24].
Por último, se estudia la evolución dinámica de la densidad electrónica
en tres procesos de transposición que tienen lugar en compuestos
nitrogenados, concluyéndose que en todos ellos la reacción tiene lugar
en una sola etapa y de forma concertada [25]. La reacción transcurre de
forma similar en los tres casos: el enlace se forma después del estado de
transición cuando ya se ha roto el enlace original. La transferencia de
carga se produce esencialmente entre los átomos implicados en la
transposición: el átomo que migra y los enlazados a él en el reactivo y el

Estudio QTAIM de nitrilos y compuestos relacionados
61
producto.

Estudio QTAIM de nitrilos y compuestos relacionados
62
4. TRABAJOS DE INVESTIGACIÓN
4.1. Approximate Transferability in Alkanenitriles, J. L. López, M. Mandado, A. M. Graña, R. A. Mosquera, Int. J. Quantum Chem. 86 (2002) 190. 4.2. A Charge Density Analysis on the Proximity Effect in Dicyanoalkanes, J. L. López, M. Mandado, M. J. González Moa, R. A. Mosquera Chem. Phys. Lett. 422 (2006) 558. 4.3. Electron Density Analysis on the Protonation of Nitriles, J. L. López, A. M. Graña, R. A. Mosquera J. Phys. Chem. A 113 (2009) 2652. 4.4. Electron Density Analysis on the Alpha Acidity of Nitriles, J. L. López, A. M. Graña, R. A. Mosquera Chem. Soc. Adv. Para ser sometido (2015). 4.5. Electron Density Evolution in Rearregements on Nitrogenated Compounds, J. L. López, R. A. Mosquera, A. M. Graña Eur. J. Org. Chem. Para ser sometido (2015).

Estudio QTAIM de nitrilos y compuestos relacionados
63

Approximate Transferabilityin Alkanenitriles
JOS LUIS LÓPEZ, MARCOS MANDADO, ANA M. GRAÑA,RICARDO A. MOSQUERADepartamento de Química Física, Universidade de Vigo, Lagoas-Marcosende, E-36200 Vigo,Galicia, Spain
Received 4 October 2000; revised 19 March 2001; accepted 15 May 2001
ABSTRACT: The atomic and bond properties of a series of alkanenitriles werecalculated in order to analyze the transferability of the CN, methyl, and methylene groups.The calculations were carried out using the atoms in molecules (AIM) theory onRHF/6-31++G∗∗//RHF/6-31G∗∗ wave functions obtained for compounds CN–R(R ranging from H to C11H23). Linear correlations between L() and N() were used toestablish N(CH2) and N(CH3) nearly transferable values. Average values and maximumdifferences to the mean value of several properties were used for classifying the CN group.It shows a transferable behavior along the CN–R series for R > Et. The methyl grouppresents specific properties when R < Pr. The methylene groups can be classifiedconsidering both their position with respect to the end of the chain and the position withrespect to the CN group. The atomic energy displays a dependence on the molecular size.Although this behavior does not allow to consider this property as transferable, both theab initio total electronic molecular energies and the experimental heats of formation can befitted, by linear regression analysis, as a function of the number of methylene groups.© 2002 John Wiley & Sons, Inc. Int J Quantum Chem 86: 190–198, 2002
Key words: AIM; transferability; energy additivity; functional group; nitriles
Correspondence to: R. A. Mosquera; e-mail: [email protected].
Contract grant sponsor: Government of Galicia.Contract grant number: PGIDT-99X130102B.Contract grant sponsor: CICyT.Contract grant number: PD98-1085.
International Journal of Quantum Chemistry, Vol. 86, 190–198 (2002)© 2002 John Wiley & Sons, Inc.
Estudio QTAIM de nitrilos y compuestos relacionados
64

APPROXIMATE TRANSFERABILITY IN ALKANENITRILES
Introduction
T he cyano group, CN, is known as one of thefundamental functional groups in chemistry.
The use of nitriles in preparative organic chemistrybegan to acquire importance in the second half ofthe nineteenth century. Their useful characteristicreactions converted them in very common reagentsboth in organic and inorganic synthesis. In accor-dance with their utility, its electron structure hasbeen the subject of a tremendous amount of exper-imental and theoretical studies [1]. Many of themconcentrated on the structure of the first member ofthe nitrile series, HCN, which became part of themost usual benchmark systems to test theoreticalmethods and calculation levels [2, 3]. Nitriles, es-pecially HCN, are also important interstellar mole-cules that had been detected by radioastronomy invarious sources [4].
The concepts of similarity, functional group, andatomic transferability have played a very importantrole in the historical development of chemistry [5].However, these concepts were usually employed inan intuitive way, without including accurate defi-nitions and quantifications. Accepting that proper-ties of matter are caused by the internal structure,the similarity between substances must originate insimilar charge distributions. In 1980 Carbó et al. [6]proposed the first quantum mechanical similarityindex based on the electron density distributions ofmolecules. This index was a starting point for manyothers that also faced the problem of measuring thesimilarity between complete molecules. The devel-opment of Bader’s atoms in molecules (AIM) theory[7, 8] made possible to split a molecule, in a uniqueand accurate way [9], into discrete subsystems thatverify the theorems of quantum mechanics. It can beaccurately deduced that this division is performedby the zero-flux surfaces for the gradient of thecharge density, ∇ρ(r) [10 – 12]. These surfaces dividethe spaces into regions, , that are readily identi-fied with the constituent atoms of the molecule. Theproperties of the atom are obtained by the integra-tion of a corresponding property density over thatregion.
AIM provides the theoretical tool to define afunctional group as an atom (or groups of atoms)that present in a series of molecules keeps an impor-tant similarity along the series [13]. The similarityof the atoms along a molecule series can be quan-tified by a similarity index, as those introduced byCioslowski et al. [14, 15], or described by comparing
the values of the atomic properties for several mole-cules [16 – 23]. Although absolute transferability ofthe properties has proved to be an unattainable limit[24, 25], the concept of transferability for atoms andgroups of atoms is still widely invoked in chem-istry to predict the properties of a molecule fromthe properties of its constituent fragments. This is,at least, partially due to the very slight variationexperienced by many atomic properties along ho-mologous series of molecules. Variations that arebelow the limits of the experimental error or withinthe accuracy of numerical methods, allow us tospeak of approximately transferable groups.
We are interested in the study of the proper-ties of the functional groups obtained by combiningthe atoms of the AIM theory. Especially in com-paring their properties in order to establish limitsfor the molecular environments in which a certainproperty of a functional group can be consideredas approximately transferable. In our recent stud-ies the application of the AIM theory has allowedthe classification of the atoms of aldehydes, ketones[21, 22], other carbonyl compounds [26, 27], andethers [23, 28, 29] into nearly transferable groups.To achieve these results, we have found and madeuse of empirical relationships that had not beenreported previously, such as the relationship be-tween several atomic properties and the level ofaccuracy with which the zero-flux surfaces weredetermined, or the dependency of the energy prop-erties on the size of the molecule. Because of thisdependency, the energy properties cannot be con-sidered transferable. Nevertheles, for alkyl linearaldehydes, ketones [21], and ethers [23], we havefound that linear fittings for both the HF total en-ergy, E, and the experimental heat of formation, tothe number of methylene groups, n, in the molecule,provide excellent reproductions of those magni-tudes for every molecule in the series (maximumdiscrepancies for E do not overpass 2.5 kJ mol−1 inethers, and 1.5 kJ mol−1 in aldehydes and ketones).
Methodology
Atomic and bond properties were computed us-ing the AIMPAC [30] series of programs on RHF/6-31++G∗∗//RHF/6-31G∗∗ [31, 32] wave functionsobtained with the Gaussian 94 program [33]. Thecalculations were performed on 12 molecules of lin-ear alkanenitriles, R–CN, with R ranging from H toC11H23 (Table I). For all the compounds we havestudied the conformation corresponding to the an-
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 191
Estudio QTAIM de nitrilos y compuestos relacionados
65
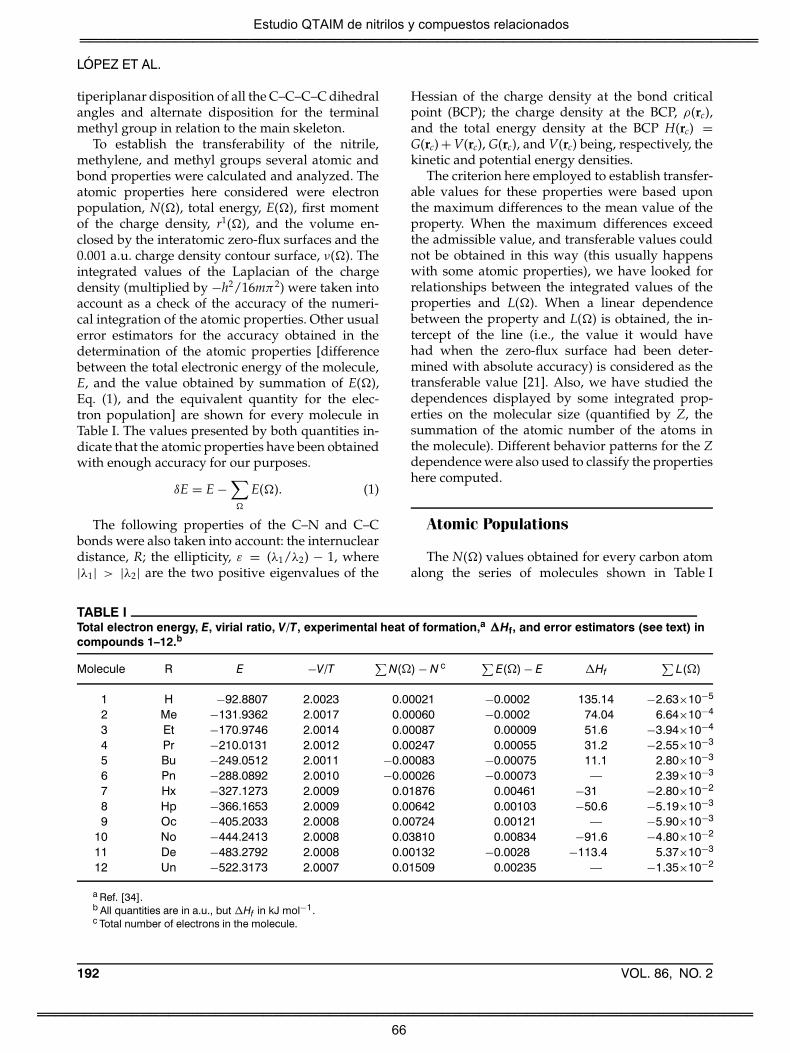
LÓPEZ ET AL.
tiperiplanar disposition of all the C–C–C–C dihedralangles and alternate disposition for the terminalmethyl group in relation to the main skeleton.
To establish the transferability of the nitrile,methylene, and methyl groups several atomic andbond properties were calculated and analyzed. Theatomic properties here considered were electronpopulation, N(), total energy, E(), first momentof the charge density, r1(), and the volume en-closed by the interatomic zero-flux surfaces and the0.001 a.u. charge density contour surface, ν(). Theintegrated values of the Laplacian of the chargedensity (multiplied by −h2/16mπ2) were taken intoaccount as a check of the accuracy of the numeri-cal integration of the atomic properties. Other usualerror estimators for the accuracy obtained in thedetermination of the atomic properties [differencebetween the total electronic energy of the molecule,E, and the value obtained by summation of E(),Eq. (1), and the equivalent quantity for the elec-tron population] are shown for every molecule inTable I. The values presented by both quantities in-dicate that the atomic properties have been obtainedwith enough accuracy for our purposes.
δE = E −∑
E(). (1)
The following properties of the C–N and C–Cbonds were also taken into account: the internucleardistance, R; the ellipticity, ε = (λ1/λ2) − 1, where|λ1| > |λ2| are the two positive eigenvalues of the
Hessian of the charge density at the bond criticalpoint (BCP); the charge density at the BCP, ρ(rc),and the total energy density at the BCP H(rc) =G(rc) + V(rc), G(rc), and V(rc) being, respectively, thekinetic and potential energy densities.
The criterion here employed to establish transfer-able values for these properties were based uponthe maximum differences to the mean value of theproperty. When the maximum differences exceedthe admissible value, and transferable values couldnot be obtained in this way (this usually happenswith some atomic properties), we have looked forrelationships between the integrated values of theproperties and L(). When a linear dependencebetween the property and L() is obtained, the in-tercept of the line (i.e., the value it would havehad when the zero-flux surface had been deter-mined with absolute accuracy) is considered as thetransferable value [21]. Also, we have studied thedependences displayed by some integrated prop-erties on the molecular size (quantified by Z, thesummation of the atomic number of the atoms inthe molecule). Different behavior patterns for the Zdependence were also used to classify the propertieshere computed.
Atomic Populations
The N() values obtained for every carbon atomalong the series of molecules shown in Table I
TABLE ITotal electron energy, E, virial ratio, V/T, experimental heat of formation,a Hf, and error estimators (see text) incompounds 1–12.b
Molecule R E −V/T∑
N() − N c ∑E() − E Hf
∑L()
1 H −92.8807 2.0023 0.00021 −0.0002 135.14 −2.63×10−5
2 Me −131.9362 2.0017 0.00060 −0.0002 74.04 6.64×10−4
3 Et −170.9746 2.0014 0.00087 0.00009 51.6 −3.94×10−4
4 Pr −210.0131 2.0012 0.00247 0.00055 31.2 −2.55×10−3
5 Bu −249.0512 2.0011 −0.00083 −0.00075 11.1 2.80×10−3
6 Pn −288.0892 2.0010 −0.00026 −0.00073 — 2.39×10−3
7 Hx −327.1273 2.0009 0.01876 0.00461 −31 −2.80×10−2
8 Hp −366.1653 2.0009 0.00642 0.00103 −50.6 −5.19×10−3
9 Oc −405.2033 2.0008 0.00724 0.00121 — −5.90×10−3
10 No −444.2413 2.0008 0.03810 0.00834 −91.6 −4.80×10−2
11 De −483.2792 2.0008 0.00132 −0.0028 −113.4 5.37×10−3
12 Un −522.3173 2.0007 0.01509 0.00235 — −1.35×10−2
a Ref. [34].b All quantities are in a.u., but Hf in kJ mol−1.c Total number of electrons in the molecule.
192 VOL. 86, NO. 2
Estudio QTAIM de nitrilos y compuestos relacionados
66

APPROXIMATE TRANSFERABILITY IN ALKANENITRILES
TABLE IITransferable and specific values of the atomicproperties for different atoms according to theirposition with respect to CN (α, β, or further away, n)and to their position with respect to CH3(terminal = T, α to the terminal = P, and furtheraway = n).a
N() r1() v()
CN 13.378 5.536 21.58Cα 5.700 5.511 4.94Cβ 5.790 5.643 4.82Cn 5.806 5.668 4.74CP 5.794 5.654 4.85CT 5.778 5.690 5.73
CαP 5.687 5.491 5.03CαT 5.657 5.489 5.90CβP 5.778 5.629 4.92CβT 5.755 5.649 5.81
a N() and r1() in a.u. and v() in cm3 mol−1.
have been classified considering maximum differ-ences of 5 × 10−3 a.u. as the limit for approximatetransferability in this quantity. Table II shows theN(C) transferable values obtained by averaging allthe atomic populations inside a group displayingsmaller differences than 5 × 10−3 a.u. Accordingto this criterion, 6 groups with different behaviorpatterns have been found: (i) The CN group forwhich the atomic population shows no dependencyon the value of L() nor on the value of Z, so thetransferable value can be directly obtained as the av-erage value when molecules 1 and 2 are excluded. Ifmolecule 3 is also considered as a specific case, themaximum differences are not reduced. (ii) C in α tothe CN group (henceforth, Cα) for which a transfer-able value of the population is also obtained as amean value excluding molecules 2 and 3. For mole-cule 2, Cα belongs to a terminal methyl group; andfor molecule 3, Cα belongs to a methylene previ-ous to the terminal methyl. Both terminal methyl(CT), and methylene bonded to a methyl (CP) areconsidered as different behavior patterns, as waspreviously found in n-alkanes [17, 20], aldehydes,and ketones [22]. Once again, maximum differencesdo not decrease if molecule 4 is excluded. (iii) Cin β to the CN group (henceforth, Cβ) for whichthe transferable value can be obtained as an aver-age of molecules 5–12. (iv) The remaining C (Cn),excluding CT and CP, for which N(C) is found todepend linearly on L(C) (Fig. 1). The r2 value ex-
FIGURE 1. Plot of the electron population of the Cn
atoms vs. L(Cn).
ceeds 0.90. The intercept of the straight line is takenas the transferable value of the property. (v) C in α tothe terminal methyl in the chain (CP) for which a lin-ear dependence on L(C) is also obtained (r2 = 0.92).(vi) C at the end of the chain, that is, those belong-ing to the terminal methyl group (CT) for which thetransferable value is obtained as a mean value whenmolecules 2 and 3 are excluded. Summing up, tak-ing into account the specific cases: those atoms that(at the same time) are α and T, β and T, α and P, andβ and P, a total of 10 different groups can be found.As a general rule, electron population increases asthe distances to the terminal methyl and to the CNgroup increase. Thus, the populations for Cα and Cβ
are larger as the distance to the methyl group in-creases, the populations for CT and CP increase withthe distance to the CN group, and the largest popu-lations are found for the “standard” carbons in themiddle of the chain, Cn. N() values obtained forCT, CP, and Cn (Table II) in this series do not differby more than 3 × 10−3 a.u. from the correspondingN() in hydrocarbons. The values obtained in thesame level of calculation for n-dodecane are 5.779,5.792, and 5.803 a.u. for CT, CP, and Cn, respectively.
Energies
The results obtained for the total electronic en-ergy, E, of the molecules here studied (Table III)indicate, as was previously found for several ho-mologous series (alkanes, aldehydes and ketones,and ethers), that E values can be very well fit-ted by expression 2 (Fig. 2), where n indicates thenumber of methylene groups in the molecule andE(CH2) and E0 represent, according to a group con-tribution scheme, respectively, the energy of themethylene and the addition of the energies of the
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 193
Estudio QTAIM de nitrilos y compuestos relacionados
67

LÓPEZ ET AL.
TAB
LEII
IE
ner
gie
s(a
.u.)
for
the
ato
ms
of
the
CN
gro
up
,an
dfo
rth
ed
iffer
entm
eth
ylen
ean
dm
eth
ylg
rou
ps
inm
ole
cule
s1–
12.
ZN
CC
H2α
CH
2β
CH
2γ
CH
2δ
CH
2ε
CH
2ξ
CH
2η
CH
2θ
CH
2ι
CH
2P
CH
3
114
−55.
2782
−37.
0713
222
−55.
2691
−37.
1658
−39.
5015
330
−55.
2540
−37.
1781
−38.
9189
−39.
6236
438
−55.
2456
−37.
1720
−38.
9302
−39.
0156
−39.
6492
546
−55.
2392
−37.
1677
−38.
9258
−39.
0309
−39.
0414
−39.
6470
654
−55.
2355
−37.
1650
−38.
9219
−39.
0261
−39.
0563
−39.
0361
−39.
6490
762
−55.
2322
−37.
1629
−38.
9200
−39.
0225
−39.
0503
−39.
0506
−39.
0363
−39.
6479
870
−55.
2296
−37.
1609
−38.
9185
−39.
0212
−39.
0500
−39.
0483
−39.
0532
−39.
0352
−39.
6474
978
−55.
2275
−37.
1599
−38.
9158
−39.
0199
−39.
0487
−39.
0466
−39.
0507
−39.
0518
−39.
0349
−39.
6463
1086
−55.
2265
−37.
1593
−38.
9153
−39.
0179
−39.
0451
−39.
0446
−39.
0480
−39.
0486
−39.
0500
−39.
0316
−39.
6461
1194
−55.
2252
−37.
1583
−38.
9138
−39.
0173
−39.
0468
−39.
0459
−39.
0486
−39.
0484
−39.
0498
−39.
0504
−39.
0328
−39.
6448
1210
2−5
5.22
39−3
7.15
76−3
8.91
26−3
9.01
61−3
9.04
51−3
9.04
34−3
9.04
71−3
9.04
72−3
9.04
81−3
9.04
84−3
9.04
98−3
9.03
11−3
9.64
46
FIGURE 2. RHF/6-31++G∗∗//RHF/6-31G∗∗ energiesvs. number of CH2 groups, n, for molecules 4–12.
CN and CH3 groups.
E = nE∗(CH2) + E0. (2)
Table IV shows how this fitting is significantlyimproved when molecules 1–3 are excluded. Nev-ertheless, the exclusion of more molecules does notintroduce any meaningful improvement. Even stan-dard deviations, σE0 and σ [E∗(CH2)], for the fittingparameters are enlarged when molecule 4 is also ex-cluded. This confirms nitriles 4–12 can be treated asan homologous series suitable for any kind of groupcontribution treatment of their energies. Also, whena similar fitting to the number of methylene groupsis tested for the experimental standard heat of for-mation (Table I), the results also indicate a very goodadditivity for this magnitude (Table IV), though inthis case no significant improvement is obtained byexluding molecules 2 and 3.
In spite of this linear behavior, neither CH3, CN,nor CH2 groups present a common energy alongthe series. In fact, the CH2 group energies, obtainedby adding their atomic energies, can differ by morethan 329 kJ (CH2
α of molecule 12) from the E∗(CH2)value. It has to be pointed out that most of these dif-ferences overpass the limits of the total integrationerror for the energy, δE [Eq. (1)], which reachs a max-imum value of 21.8 kJ mol−1 (molecule 10). Thesedifferences between the AIM-calculated group ener-gies, E(CH2), and E∗(CH2) are shown in Figure 3. Itcan be said that they are the result of two combinedeffects: (a) The position of the CH2 group with refer-ence to the CN functional group and to the methylgroup, positions used above to define nearly trans-ferable methylenes for nonenergetic properties andclassify the methylenes (Table II). (b) The effect ofthe total size of the molecule, quantified by the sum-mation of its nuclear charges, Z, and displayed inFigure 3 for every group of Table III, and previously
194 VOL. 86, NO. 2
Estudio QTAIM de nitrilos y compuestos relacionados
68
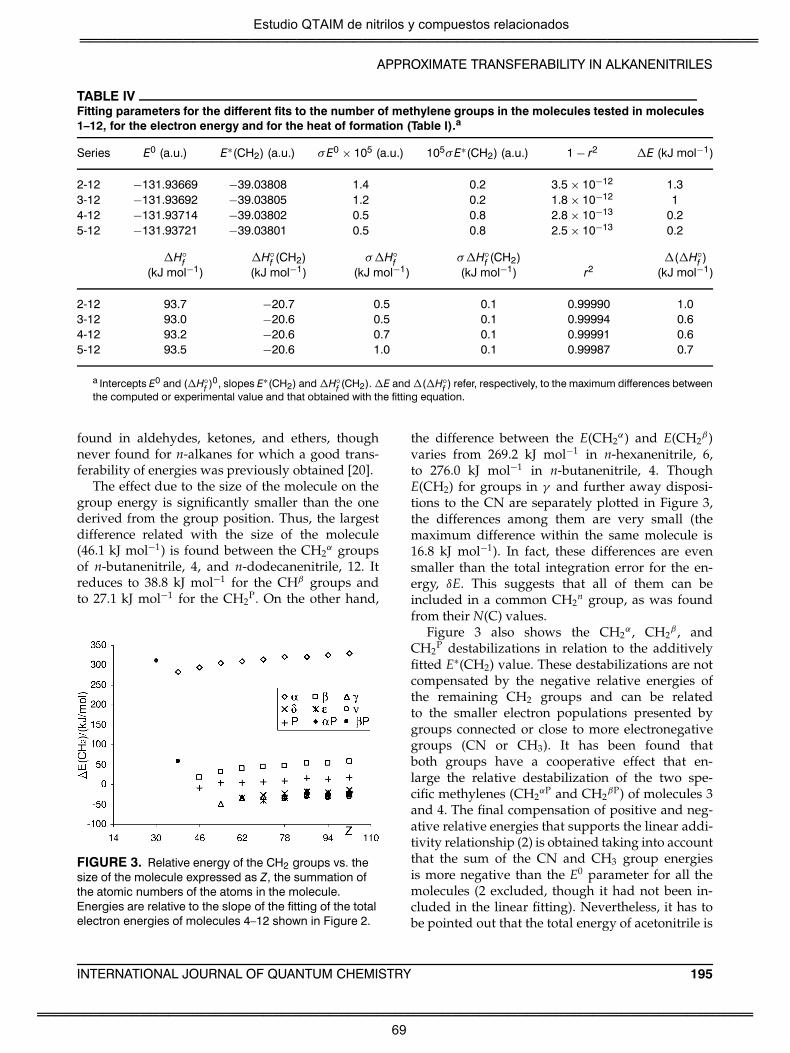
APPROXIMATE TRANSFERABILITY IN ALKANENITRILES
TABLE IVFitting parameters for the different fits to the number of methylene groups in the molecules tested in molecules1–12, for the electron energy and for the heat of formation (Table I).a
Series E0 (a.u.) E∗(CH2) (a.u.) σE0 × 105 (a.u.) 105σE∗(CH2) (a.u.) 1 − r2 E (kJ mol−1)
2-12 −131.93669 −39.03808 1.4 0.2 3.5 × 10−12 1.33-12 −131.93692 −39.03805 1.2 0.2 1.8 × 10−12 14-12 −131.93714 −39.03802 0.5 0.8 2.8 × 10−13 0.25-12 −131.93721 −39.03801 0.5 0.8 2.5 × 10−13 0.2
Hf H
f (CH2) σHf σH
f (CH2) (Hf )
(kJ mol−1) (kJ mol−1) (kJ mol−1) (kJ mol−1) r2 (kJ mol−1)
2-12 93.7 −20.7 0.5 0.1 0.99990 1.03-12 93.0 −20.6 0.5 0.1 0.99994 0.64-12 93.2 −20.6 0.7 0.1 0.99991 0.65-12 93.5 −20.6 1.0 0.1 0.99987 0.7
a Intercepts E0 and (Hf )0, slopes E∗(CH2) and H
f (CH2). E and (Hf ) refer, respectively, to the maximum differences between
the computed or experimental value and that obtained with the fitting equation.
found in aldehydes, ketones, and ethers, thoughnever found for n-alkanes for which a good trans-ferability of energies was previously obtained [20].
The effect due to the size of the molecule on thegroup energy is significantly smaller than the onederived from the group position. Thus, the largestdifference related with the size of the molecule(46.1 kJ mol−1) is found between the CH2
α groupsof n-butanenitrile, 4, and n-dodecanenitrile, 12. Itreduces to 38.8 kJ mol−1 for the CHβ groups andto 27.1 kJ mol−1 for the CH2
P. On the other hand,
FIGURE 3. Relative energy of the CH2 groups vs. thesize of the molecule expressed as Z, the summation ofthe atomic numbers of the atoms in the molecule.Energies are relative to the slope of the fitting of the totalelectron energies of molecules 4–12 shown in Figure 2.
the difference between the E(CH2α) and E(CH2
β)varies from 269.2 kJ mol−1 in n-hexanenitrile, 6,to 276.0 kJ mol−1 in n-butanenitrile, 4. ThoughE(CH2) for groups in γ and further away disposi-tions to the CN are separately plotted in Figure 3,the differences among them are very small (themaximum difference within the same molecule is16.8 kJ mol−1). In fact, these differences are evensmaller than the total integration error for the en-ergy, δE. This suggests that all of them can beincluded in a common CH2
n group, as was foundfrom their N(C) values.
Figure 3 also shows the CH2α, CH2
β , andCH2
P destabilizations in relation to the additivelyfitted E∗(CH2) value. These destabilizations are notcompensated by the negative relative energies ofthe remaining CH2 groups and can be relatedto the smaller electron populations presented bygroups connected or close to more electronegativegroups (CN or CH3). It has been found thatboth groups have a cooperative effect that en-large the relative destabilization of the two spe-cific methylenes (CH2
αP and CH2βP) of molecules 3
and 4. The final compensation of positive and neg-ative relative energies that supports the linear addi-tivity relationship (2) is obtained taking into accountthat the sum of the CN and CH3 group energiesis more negative than the E0 parameter for all themolecules (2 excluded, though it had not been in-cluded in the linear fitting). Nevertheless, it has tobe pointed out that the total energy of acetonitrile is
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 195
Estudio QTAIM de nitrilos y compuestos relacionados
69

LÓPEZ ET AL.
in very good agreement with the intercept of Eq. (2),as was found between the intercept of this equationfor ketones and dimethylketone and for aldehydesand ethanal [21, 22].
The methyl group is destabilized as the molec-ular size increase along the series. Though this de-stabilization is really smaller (12.2 kJ mol−1 be-tween molecules 4 and 12) than that found formethylenes, it is still significantly larger thanany difference between CH3 in n-alkanes. In fact,our calculated values for E(CH3) in n-eicosane(−39.63347 a.u.), n-dodecane (−39.63394 a.u.), andn-pentane (−39.63429 a.u.) differ by less than2.2 kJ mol−1 and cannot be presented as an exam-ple of the Z dependence of the group energies. Onthe other hand, this destabilization was found as26.8 kJ mol−1 in the CH3
α of methoxyethers, whencomparing these group energies for methoxybutaneand methoxydecane. These facts seem to indicatethat the presence of an electronegative substituentin the molecule is crucial to observe this Z effect.When the values of E(CH3) of nitriles are comparedwith those of a n-alkane (n-dodecane), we concludethat this group is more stabilized in nitriles.
Finally, the CN group is stabilized by the elec-trons supplied by the alkyl chain with respect to theHCN molecule, but this stabilization is reduced asthe length of the chain increases. This results in thebehavior displayed in Figure 4. It can be observedthat the energy of the nitrogen atom is less and lessnegative as Z increases. On the other hand, the en-ergy of the C atom is first stabilized by increasingthe chain (methyl and ethyl groups) and then expe-riences a continuous destabilization.
FIGURE 4. Relative energies of the C and N atoms andthe CN group vs. the size of the molecule expressedas Z, the summation of the atomic numbers of the atomsin the molecule. Energies are relative to thecorresponding values in molecule 12.
First Moment of the Electron Charge
For this property no appreciable relationshipwith Z was found. Thus, we have establishedtransferable values by using mean values and de-pendences on L(). The patterns of behavior areexactly the same found for the electron populationand the procedure to obtain transferable values isalso the same. The highest values for methyl car-bons whose charge is, therefore, further (in average)from the nucleus (Table II). The calculated values forn-dodecane are 5.693, 5.647, and 5.661 for CT, CP,and Cn, respectively. The differences with the valuesin Table I are within 7 × 10−3 only slightly higherthan those admissible to define a group.
Atomic Volume
Patterns of behavior and transferable values forvolumes were obtained in the same way, that is, byusing dependences on L() for Cn and CP and byusing mean values in the other cases. For the atomicvolume the admissible maximum difference was es-tablished in 5 × 10−2 cm3 mol−1, which comparessatisfactorily with the usual limit of the experimen-tal accuracy for molecular volumes. Once more, wecan compare the results with those obtained for CT,CP, and Cn atoms in n-dodecane (5.74, 4.82, and4.71 cm3 mol−1, respectively). Once again, the dif-ferences allow us to establish that CT, CP, and Cn
atoms are similar to those in hydrocarbons.
Geometrical Features
The optimized geometries of the completely an-tiperiplanar conformer of compounds 1–12 are verysimilar. Table V shows transferable values for bonddistances and bond angles obtained as mean val-ues for the series. Mean values in every case arecalculated excluding the smallest molecule contain-ing the bond or the angle. It can be concluded thatmolecules in the series show common geometricalfeatures, with an angle N–C–C slightly smaller than180 as it has been shown previously [35]. Oncemore, the values observed for geometric parame-ters including atoms distant from the CN group arein good agreement with the same parameters in n-alkanes (maximum differences in bond distances are7 × 10−4 Å and less than 0.01 in bond angles). Thebond distances allow us to distinguish six different
196 VOL. 86, NO. 2
Estudio QTAIM de nitrilos y compuestos relacionados
70
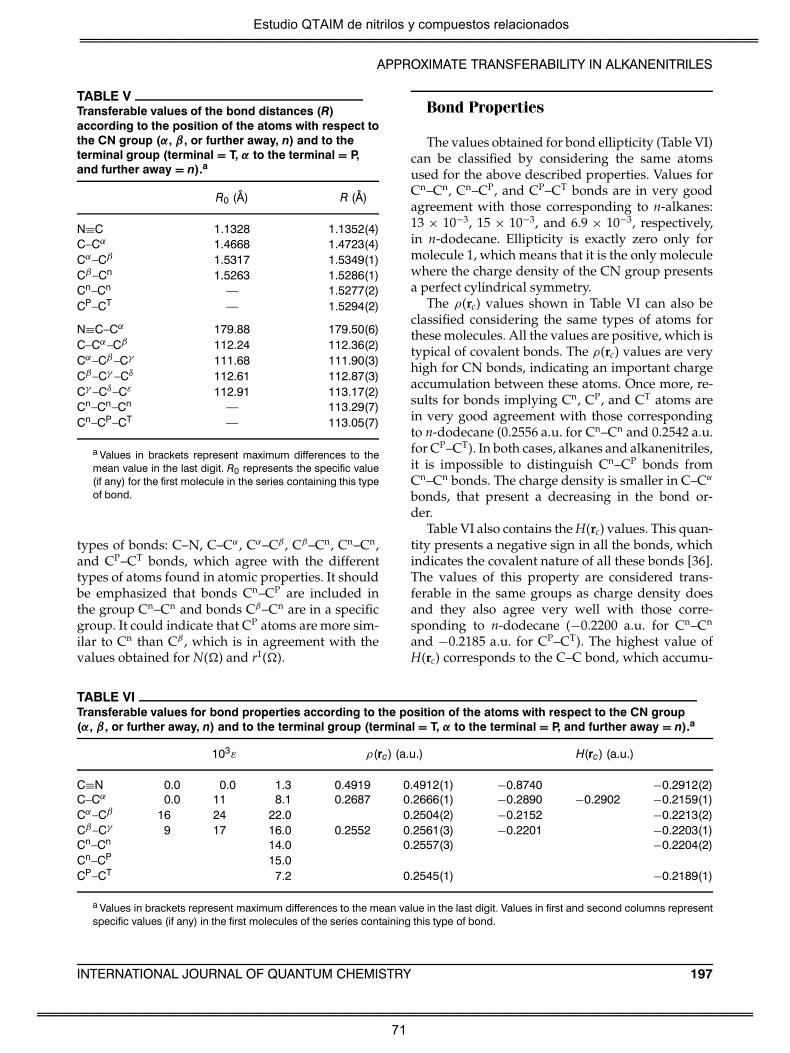
APPROXIMATE TRANSFERABILITY IN ALKANENITRILES
TABLE VTransferable values of the bond distances (R)according to the position of the atoms with respect tothe CN group (α, β, or further away, n) and to theterminal group (terminal = T, α to the terminal = P,and further away = n).a
R0 (Å) R (Å)
N≡C 1.1328 1.1352(4)C–Cα 1.4668 1.4723(4)Cα–Cβ 1.5317 1.5349(1)Cβ–Cn 1.5263 1.5286(1)Cn–Cn — 1.5277(2)CP–CT — 1.5294(2)
N≡C–Cα 179.88 179.50(6)C–Cα–Cβ 112.24 112.36(2)Cα–Cβ–Cγ 111.68 111.90(3)Cβ–Cγ –Cδ 112.61 112.87(3)Cγ –Cδ–Cε 112.91 113.17(2)Cn–Cn–Cn — 113.29(7)Cn–CP–CT — 113.05(7)
a Values in brackets represent maximum differences to themean value in the last digit. R0 represents the specific value(if any) for the first molecule in the series containing this typeof bond.
types of bonds: C–N, C–Cα, Cα–Cβ , Cβ–Cn, Cn–Cn,and CP–CT bonds, which agree with the differenttypes of atoms found in atomic properties. It shouldbe emphasized that bonds Cn–CP are included inthe group Cn–Cn and bonds Cβ–Cn are in a specificgroup. It could indicate that CP atoms are more sim-ilar to Cn than Cβ , which is in agreement with thevalues obtained for N() and r1().
Bond Properties
The values obtained for bond ellipticity (Table VI)can be classified by considering the same atomsused for the above described properties. Values forCn–Cn, Cn–CP, and CP–CT bonds are in very goodagreement with those corresponding to n-alkanes:13 × 10−3, 15 × 10−3, and 6.9 × 10−3, respectively,in n-dodecane. Ellipticity is exactly zero only formolecule 1, which means that it is the only moleculewhere the charge density of the CN group presentsa perfect cylindrical symmetry.
The ρ(rc) values shown in Table VI can also beclassified considering the same types of atoms forthese molecules. All the values are positive, which istypical of covalent bonds. The ρ(rc) values are veryhigh for CN bonds, indicating an important chargeaccumulation between these atoms. Once more, re-sults for bonds implying Cn, CP, and CT atoms arein very good agreement with those correspondingto n-dodecane (0.2556 a.u. for Cn–Cn and 0.2542 a.u.for CP–CT). In both cases, alkanes and alkanenitriles,it is impossible to distinguish Cn–CP bonds fromCn–Cn bonds. The charge density is smaller in C–Cα
bonds, that present a decreasing in the bond or-der.
Table VI also contains the H(rc) values. This quan-tity presents a negative sign in all the bonds, whichindicates the covalent nature of all these bonds [36].The values of this property are considered trans-ferable in the same groups as charge density doesand they also agree very well with those corre-sponding to n-dodecane (−0.2200 a.u. for Cn–Cn
and −0.2185 a.u. for CP–CT). The highest value ofH(rc) corresponds to the C–C bond, which accumu-
TABLE VITransferable values for bond properties according to the position of the atoms with respect to the CN group(α, β, or further away, n) and to the terminal group (terminal = T, α to the terminal = P, and further away = n).a
103ε ρ(rc) (a.u.) H(rc) (a.u.)
C≡N 0.0 0.0 1.3 0.4919 0.4912(1) −0.8740 −0.2912(2)C–Cα 0.0 11 8.1 0.2687 0.2666(1) −0.2890 −0.2902 −0.2159(1)Cα–Cβ 16 24 22.0 0.2504(2) −0.2152 −0.2213(2)Cβ–Cγ 9 17 16.0 0.2552 0.2561(3) −0.2201 −0.2203(1)Cn–Cn 14.0 0.2557(3) −0.2204(2)Cn–CP 15.0CP–CT 7.2 0.2545(1) −0.2189(1)
a Values in brackets represent maximum differences to the mean value in the last digit. Values in first and second columns representspecific values (if any) in the first molecules of the series containing this type of bond.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 197
Estudio QTAIM de nitrilos y compuestos relacionados
71

LÓPEZ ET AL.
lates the greatest charge density in the internuclearzone, that is, the bond that has the highest values forρ(rc) and ε.
Conclusions
We have found transferable values for all theatomic and bond properties calculated, but the en-ergy, which displays variation with molecular size,cannot be considered as a transferable property. Thecomparison of the different values for the proper-ties, and the different patterns of variation with Zin the case of the energy, allow us to consider dif-ferent types of transferable atoms in alkanenitriles:(i) C and N atoms of the CN group; (ii) C in α to theCN group; (iii) C in β to the CN group; (iv) carbonof the terminal methyl group; (v) C previous to theterminal methyl group; and (vi) remaining C in thechain, whose behavior is similar to the C in hydro-carbons. On the other hand, a total of eight specificheavy atoms are present in the smallest linear alka-nenitriles: (i) C α of molecules 2 and 3; (ii) C β inmolecule 3 and 4; and (iii) C and N of the CN groupin molecules 1 and 2.
ACKNOWLEDGMENTS
Financial support by the autonomous govern-ment of Galicia (Project PGIDT-99X130102B), andCICyT, Spain (PD98-1085) is gratefully acknowl-edged. We are also indebted to Prof. R. F. W. Baderfor providing us a copy of AIMPAC suite of pro-grams and to CESGA for computer time. One of us(M.M.) thanks Universidade de Vigo for a postgrad-uated fellowship.
References
1. Shorter, J. In Patai, S.; Rapport, Z., Eds. The Chemistry ofTriple-Bonded Functional Groups; Wiley: Chichester, 1994;Chapter 5.
2. Botschwina, P.; Horn, M.; Matuschewski, M.; Schick, E.;Sebals, P. J Mol Struct (Theochem) 1997, 400, 119.
3. Allen, F. H.; Garner, S. E. In Patai, S.; Rapport, Z., Eds.The Chemistry of Triple-Bonded Functional Groups; Wiley:Chichester, 1994; Chapter 2.
4. Irvine, W. M. Space Sci Rev 1999, 90, 203.5. Rouvray, D. H. Topics Curr Chem 1995, 173, 1.6. Carbó, R.; Leyda, L.; Arnau, M. Int J Quantum Chem 1980,
17, 1185.7. Bader, R. F. W. Atoms in Molecules—A Quantum Theory;
International Series of Monographs on Chemistry, No. 22;Oxford University Press: Oxford, 1990.
8. Bader, R. F. W. Chem Rev 1991, 91, 893.
9. Bader, R. F. W. Can J Chem 1998, 77, 86.
10. Srebenik, S.; Bader, R. F. W. J Chem Phys 1975, 63, 3945.
11. Bader, R. F. W. Pure Appl Chem 1988, 60, 145.
12. Schwinger, J. Phys Rev 1951, 82, 914.
13. Bader, R. F. W.; Popelier, P. L. A.; Keith, T. A. Angew ChemInt Ed Engl 1994, 33, 620.
14. Cioslowski, J.; Nanayakkara, A. J Am Chem Soc 1993, 115,11213.
15. Cioslowski, J.; Stefanov, B. B.; Constans, P. J Comput Chem1996, 17, 1352.
16. Wiberg, K. B.; Bader, R. F. W.; Lau, C. D. J Am Chem Soc1987, 109, 1001.
17. Bader, R. F. W.; Carroll, M. T.; Cheeseman, J. R.; Chang, C.J Am Chem Soc 1987, 109, 7968.
18. Bader, R. F. W.; Larouche, A.; Gatti, C.; Carroll, M. T.; Mac-Dougall, P. J.; Wiberg, K. B. J Chem Phys 1987, 87, 1142.
19. Bader, R. F. W.; Keith, T. A.; Gough, K. M.; Laidig, K. E. MolPhys 1992, 75, 1167.
20. Bader, R. F. W.; Bayles, D. J Phys Chem A 2000, 104, 5579.
21. Graña, A. M.; Mosquera, R. A. J Chem Phys 1999, 110, 6606.
22. Graña, A. M.; Mosquera, R. A. J Chem Phys 2000, 113, 1492.
23. Vila, A.; Carballo, E.; Mosquera, R. A. Can J Chem 2000, 78,1535.
24. Bader, R. F. W.; Becker, P. Chem Phys Lett 1988, 148, 452.
25. Riess, J.; Münch, W. Theoret Chim Acta 1981, 58, 295.
26. Graña, A. M.; Mosquera, R. A. Chem Phys 1999, 243, 17.
27. Graña, A. M.; Mosquera, R. A. J Comput Chem 1999, 20,1444.
28. Vila, A.; Mosquera, R. A. J Phys Chem 2001, 115, 1264.
29. Vila, A.; Mosquera, R. A.; Hermida-Ramón, J. M. J MolStruct (Theochem) 2001, 541, 149.
30. Bader, R. F. W.; co-workers, Eds. AIMPAC: A suite of pro-grams for the Theory of Atoms in Molecules; McMasterUniversity, Hamilton, Ontario, Canada, L8S 4M1. [email protected].
31. Clark, T.; Chandrasekar, J.; Spitznagel, G. W.; Schleyer, P. R.J Comput Chem 1983, 4, 294.
32. Hariharan, P. C.; Pople, J. A. Theor Chim Acta 1973, 28, 213.
33. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.;Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T. A.;Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman,J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challa-combe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.;Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.;Fox, D. J.; Binkley, J. S.; DeFrees, D. J.; Baker, J.; Stewart,J. J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian94; Gaussian: Pittsburgh, PA, 1995.
34. NIST Chemistry Webbook, NIST Standard Reference Data-base Number 69, February 2000 Release, National Insti-tute of Standards and Technology, Gaithersburg, MD 20899(http://webbook.nist.gov).
35. Ishii, K.; Nakayama, H.; Koyama, K. Yokoyama, Y.; Ohashi,Y. Bull Chem Soc Jpn 1997, 70, 2085.
36. Cremer, D.; Kraka, E. Croat Chem Acta 1984, 57, 1259.
198 VOL. 86, NO. 2
Estudio QTAIM de nitrilos y compuestos relacionados
72

Estudio QTAIM de nitrilos y compuestos relacionados
73

A charge density analysis on the proximity effect in dicyanoalkanes
Jose Luis Lopez, Marcos Mandado, Marıa J. Gonzalez Moa, Ricardo A. Mosquera *
Departamento de Quımica Fısica, Facultade de Quımica, Universidade de Vigo, Lagoas-Marcosende, ES36310-Vigo, Galicia, Spain
Received 10 January 2006; in final form 5 March 2006Available online 10 March 2006
Abstract
QTAIM atomic and bond properties of 21 linear alkyl dicyanoalkanes of formula NC(CH2)nCN (n = 0–20), and three larger mole-cules: C32H66, NC(CH2)30CH3, and NC(CH2)30CN, indicate that cyano groups can be considered statistically equivalent to those of alarge cyanoalkane when they are separated by at least 14 methylene groups. When n < 19 there is at least one methylene group in thedicyanoalkane that differs significantly from those of NC(CH2)30CH3 or NC(CH2)30CN. Every cyano group produces an effect onthe methylenes that is nearly independent of the position of the other one, hydrogens being more sensitive than carbons. 2006 Elsevier B.V. All rights reserved.
1. Introduction
The term ‘proximity effect’ [1] was coined more than 20years ago and it is related to the development of molecularmodels for non electrolytes solutions [2]. These models dis-sect a molecule into building blocks that are assumed to beindependent, transferable, and characterised by a set ofparameters employed to compute diverse properties ofnon electrolyte mixtures. The proximity effect makes refer-ence to one of the main shortcomings of group contribu-tions models: the intramolecular interaction between two(or more) functional groups that affects their propertiesand those of the groups placed in their surroundings, inval-idating group transferability [3]. Thus, variations under-gone by the properties of atoms because of the presenceof another functional group has been invoked in qualitativediscussions on the behaviour of several mixtures of poly-functional compounds [4–6]. Several treatments have beenproposed to deal with this effect, ranging from empiricalvariations of the group parameters depending on their firstand second neighbouring groups [1], to quantitative correc-tions based upon Mulliken population analysis to adaptgroups defined for monofunctional compounds to poly-
functional molecules [3]. The suitability of these diversetreatments can be related to one question: are the changesundergone by the electron distribution of a certain atom ina molecule with two functional groups equivalent (in agood approximation) to the summation of the effects pro-duced by these functional groups in monofunctionalisedcompounds? or, on the contrary, does the proximity effectinvolve important cooperative effects between both func-tional groups?
The application of the Quantum Theory of Atoms in Mol-ecules (QTAIM) [7,8] on HF/6-31++G** electron densitiesproved that the oxygen atoms of RAOA(CH2)nAOAR 0
molecules are significantly different from those of the corre-sponding monoethers when n < 4 [9], confirming the pres-ence of the proximity effect when the oxygens are separatedby less than five bonds. QTAIM was also employed to ana-lyse the specificity of methylene groups placed between thetwo oxygens of diethers [10].
This work revisits the proximity effect using the QTAIMpartitioning but focusing on a,x-dicyanoalkanes. Thesecompounds have been recently employed to form hydro-gen-bonded complexes and inclusion compounds with urea[11], that are of practical interest in supramolecular chem-istry [12]. Atomic and bond properties of cyanoalkaneswere analysed in a previous QTAIM study [13], concludingthe approximate transferability of the ACN and ACH3
0009-2614/$ - see front matter 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.cplett.2006.03.019
* Corresponding author. Fax: +34 968 812 321.E-mail address: [email protected] (R.A. Mosquera).
www.elsevier.com/locate/cplett
Chemical Physics Letters 422 (2006) 558–564
Estudio QTAIM de nitrilos y compuestos relacionados
74

groups for CH3A(CH2)nACN molecules when n > 2, andthat of ACH2A groups separated by three bonds fromACN and by two bonds from ACH3. This conclusion dif-fers from those obtained for homologous series that con-tain oxygen atoms: aldehydes and ketones [14], ethers[15], and alkanols [16]. In fact, ACH2A groups in thesecompounds are significantly modified by the functionalgroup when they are separated up to four bonds. Neverthe-less, it is a consequence of extending the Hohenberg andKohn theorem [17] to open systems [18] that perfect trans-ferability is an unreachable limit [19]. Therefore, we onlycan speak about approximate transferability that is foundwithin a certain transferability limit. It fact, most of theapproximate transferability studies hitherto performedwith the QTAIM have employed diverse subjective limits oftransferability and some of them, even, present alternativeconclusions depending on the specific criteria considered.However, Lorenzo et al. [20] revisited the approximatetransferability in n-alkanes by using statistical criteria forestablishing the limits of transferability. They concludedthat the ACH2A groups separated by three or four bondsfrom the ACH3 are specific groups in n-alkanes.
This Letter investigates the proximity effect on the ACNand ACH2A groups in a,x-dicyanoalkanes using statisticalcriteria for establishing the limits of transferability. Thesecriteria are based on the maximum deviations displayedby the properties of clearly transferable groups in largemolecules (see Section 2 for details), for which the atomicand bond properties can be considered equivalentundoubtedly. This procedure provides a larger number ofspecific groups than those obtained in previous works[13–16]. We also aim to explore if the proximity effect ismade up by additive contributions of isolated functionalgroups.
2. Computational details
HF/6-31++G** charge densities for full optimisedgeometries at the HF/6-31G** level were obtained forthe completely antiperiplanar conformers of the 21CNA(CH2)nACN molecules verifying 0 6 n 6 20, hereaf-ter denoted by their n value. Three larger molecules werealso studied. They included one n-alkane, C32H66 (A), onecyanoalkane, NC(CH2)30CH3 (C), and one dicyanoal-kane, NC(CH2)30CN (D). These molecules were fully opti-mised from the completely antiperiplanar conformation, t,and from that obtained after rotating the central dihedralangle to 60, g. All of these calculations were carried outusing the GAUSSIAN-98 program [21] setting the criterionfor SCF convergence to 1012 au. Although DFT calcula-tions would not increase the computational cost substan-tially, HF calculations were used in order to compare ourresults with those previously obtained for cyanoalkanes[13] and diethers [9,10] at the same level. Moreover, theHF method was proved to provide similar results, forstudies of transferability, to those obtained with DFT cor-related methods [22]. The topological QTAIM charge
density analysis was performed with the AIMPAC pack-age of programs [23].
This work is mainly concerned with atomic propertiessuch as the atomic electron population, N(X), the atomickinetic energy, K(X), and the normalized Shannon entropyof the electron distribution, Sh(X), as well as with bondproperties such as the bond distance, R, and the electrondensity at the bond critical points (BCP), q(rc). The prop-erties above were previously proved to be the very usefulin QTAIM studies of group transferability [13–16,20,22,24]. Moreover, the use of K(X) instead of the totalatomic energy, E(X), is required for transferability studies[25], unless the charge densities used satisfy the virial theo-rem to a high approximation, like those obtained in selfconsistent virial scaling (SCVS) calculations in the calcula-tion of E(X) [26].
The summations of QTAIM N(X) and E(X) valuesobtained in this work reproduce the total electron popula-tion, N, and the HF molecular energy, E, with a maximumdifference of 0.004 au and 4.0 kcal mol1 respectively. NoQTAIM atom was integrated with absolute values of theL(X) function [7] larger than 3.0 · 103 au. This accuracylevel was obtained at a larger computational cost for theg conformers than for the t ones. Thus, PROMEGA algo-rithm with a large number of gaussian quadrature rays wasrequired for the former, whereas PROAIM with standardintegration conditions was enough for the latter.
N(X) and L(X) values obtained for nearly transferableatoms display very good linear relationships, as previouslyfound in several studies on approximate transferability fordiverse series of compounds [9,10,13–16,20,24,27–29] andby Aicken and Popelier looking for an improvement inthe accuracy of computed atomic properties [30]. Also here,as in all the reported cases, the slopes of these N(X) vs.L(X) fitting lines approach 1 which indicates that L(X)mimics approximately the error made in the calculationof N(X). Therefore, the values of N(X) shown in this workwere obtained by correcting those computed by numericalintegration, Ncomp(X), with the corresponding value of theL(X) function through Eq. (1).
NðXÞ ¼ N compðXÞ þ LðXÞ ð1ÞLimits of transferability for atomic and bond properties
used throughout this work (Table 1) were establishedaccording to a statistic criterion: the maximum deviationwith respect to the mean value of groups that could be con-sidered equivalent ‘a priori’ in t conformers. Here weassume this equivalence for the cyano groups of C andD, the methyl groups of n-alkane A and cyanoalkane C,and the methylenes of the central backbone of the threelarge molecules that are separated from the ACN andACH3 groups by at least 9 and 3 methylene groups respec-tively. To obtain the limits of transferability for N atomswe have also considered the ACN groups of dicyanoalk-anes 16–20.
The effect on a given atomic property, A, of atom, X, ofa methylene due to a group in k or l positions can be
J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564 559
Estudio QTAIM de nitrilos y compuestos relacionados
75
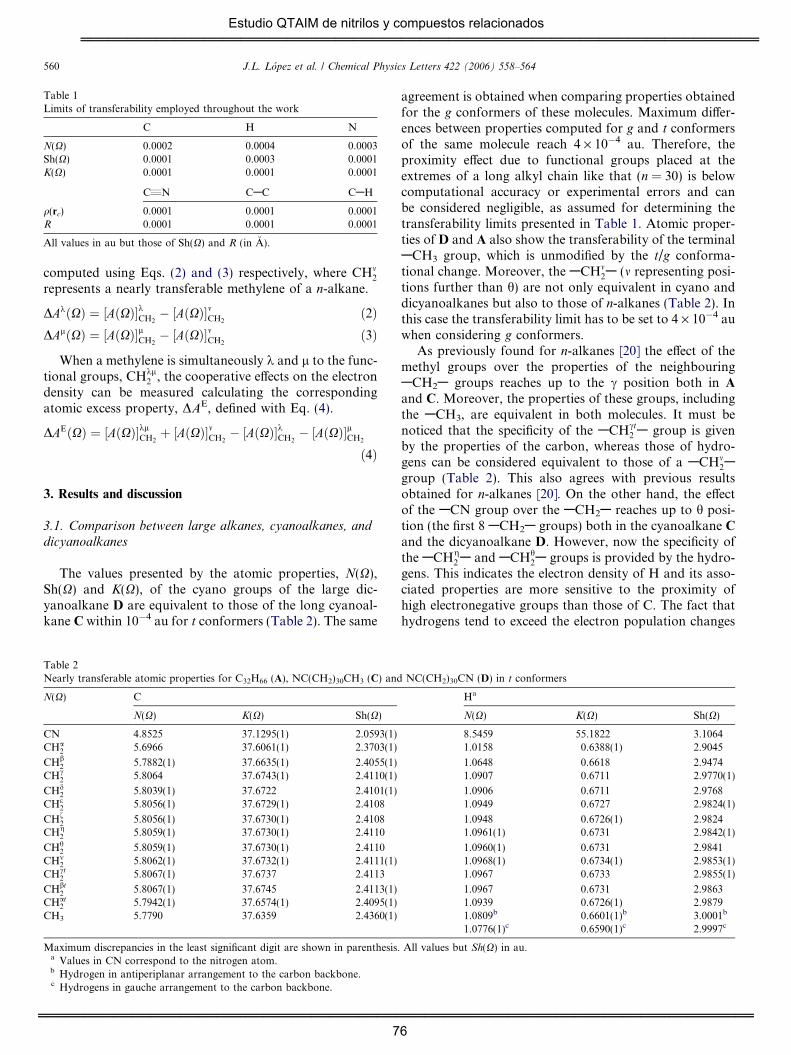
computed using Eqs. (2) and (3) respectively, where CHm2
represents a nearly transferable methylene of a n-alkane.
DAkðXÞ ¼ ½AðXÞkCH2 ½AðXÞmCH2
ð2ÞDAlðXÞ ¼ ½AðXÞlCH2
½AðXÞmCH2ð3Þ
When a methylene is simultaneously k and l to the func-tional groups, CHkl
2 , the cooperative effects on the electrondensity can be measured calculating the correspondingatomic excess property, DAE, defined with Eq. (4).
DAEðXÞ ¼ ½AðXÞklCH2þ ½AðXÞmCH2
½AðXÞkCH2 ½AðXÞlCH2
ð4Þ
3. Results and discussion
3.1. Comparison between large alkanes, cyanoalkanes, and
dicyanoalkanes
The values presented by the atomic properties, N(X),Sh(X) and K(X), of the cyano groups of the large dic-yanoalkane D are equivalent to those of the long cyanoal-kane C within 104 au for t conformers (Table 2). The same
agreement is obtained when comparing properties obtainedfor the g conformers of these molecules. Maximum differ-ences between properties computed for g and t conformersof the same molecule reach 4 · 104 au. Therefore, theproximity effect due to functional groups placed at theextremes of a long alkyl chain like that (n = 30) is belowcomputational accuracy or experimental errors and canbe considered negligible, as assumed for determining thetransferability limits presented in Table 1. Atomic proper-ties of D and A also show the transferability of the terminalACH3 group, which is unmodified by the t/g conforma-tional change. Moreover, the ACHm
2A (m representing posi-tions further than h) are not only equivalent in cyano anddicyanoalkanes but also to those of n-alkanes (Table 2). Inthis case the transferability limit has to be set to 4 · 104 auwhen considering g conformers.
As previously found for n-alkanes [20] the effect of themethyl groups over the properties of the neighbouringACH2A groups reaches up to the c position both in A
and C. Moreover, the properties of these groups, includingthe ACH3, are equivalent in both molecules. It must benoticed that the specificity of the ACHct
2 A group is givenby the properties of the carbon, whereas those of hydro-gens can be considered equivalent to those of a ACHm
2Agroup (Table 2). This also agrees with previous resultsobtained for n-alkanes [20]. On the other hand, the effectof the ACN group over the ACH2A reaches up to h posi-tion (the first 8 ACH2A groups) both in the cyanoalkane C
and the dicyanoalkane D. However, now the specificity ofthe ACHg
2 A and ACHh2A groups is provided by the hydro-
gens. This indicates the electron density of H and its asso-ciated properties are more sensitive to the proximity ofhigh electronegative groups than those of C. The fact thathydrogens tend to exceed the electron population changes
Table 2Nearly transferable atomic properties for C32H66 (A), NC(CH2)30CH3 (C) and NC(CH2)30CN (D) in t conformers
N(X) C Ha
N(X) K(X) Sh(X) N(X) K(X) Sh(X)
CN 4.8525 37.1295(1) 2.0593(1) 8.5459 55.1822 3.1064CHa
2 5.6966 37.6061(1) 2.3703(1) 1.0158 0.6388(1) 2.9045
CHb2 5.7882(1) 37.6635(1) 2.4055(1) 1.0648 0.6618 2.9474
CHc2 5.8064 37.6743(1) 2.4110(1) 1.0907 0.6711 2.9770(1)
CHd2 5.8039(1) 37.6722 2.4101(1) 1.0906 0.6711 2.9768
CHe2 5.8056(1) 37.6729(1) 2.4108 1.0949 0.6727 2.9824(1)
CHf2 5.8056(1) 37.6730(1) 2.4108 1.0948 0.6726(1) 2.9824
CHg2 5.8059(1) 37.6730(1) 2.4110 1.0961(1) 0.6731 2.9842(1)
CHh2 5.8059(1) 37.6730(1) 2.4110 1.0960(1) 0.6731 2.9841
CHm2 5.8062(1) 37.6732(1) 2.4111(1) 1.0968(1) 0.6734(1) 2.9853(1)
CHct2 5.8067(1) 37.6737 2.4113 1.0967 0.6733 2.9855(1)
CHbt2 5.8067(1) 37.6745 2.4113(1) 1.0967 0.6731 2.9863
CHat2 5.7942(1) 37.6574(1) 2.4095(1) 1.0939 0.6726(1) 2.9879
CH3 5.7790 37.6359 2.4360(1) 1.0809b 0.6601(1)b 3.0001b
1.0776(1)c 0.6590(1)c 2.9997c
Maximum discrepancies in the least significant digit are shown in parenthesis. All values but Sh(X) in au.a Values in CN correspond to the nitrogen atom.b Hydrogen in antiperiplanar arrangement to the carbon backbone.c Hydrogens in gauche arrangement to the carbon backbone.
Table 1Limits of transferability employed throughout the work
C H N
N(X) 0.0002 0.0004 0.0003Sh(X) 0.0001 0.0003 0.0001K(X) 0.0001 0.0001 0.0001
C„N CAC CAH
q(rc) 0.0001 0.0001 0.0001R 0.0001 0.0001 0.0001
All values in au but those of Sh(X) and R (in A).
560 J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564
Estudio QTAIM de nitrilos y compuestos relacionados
76
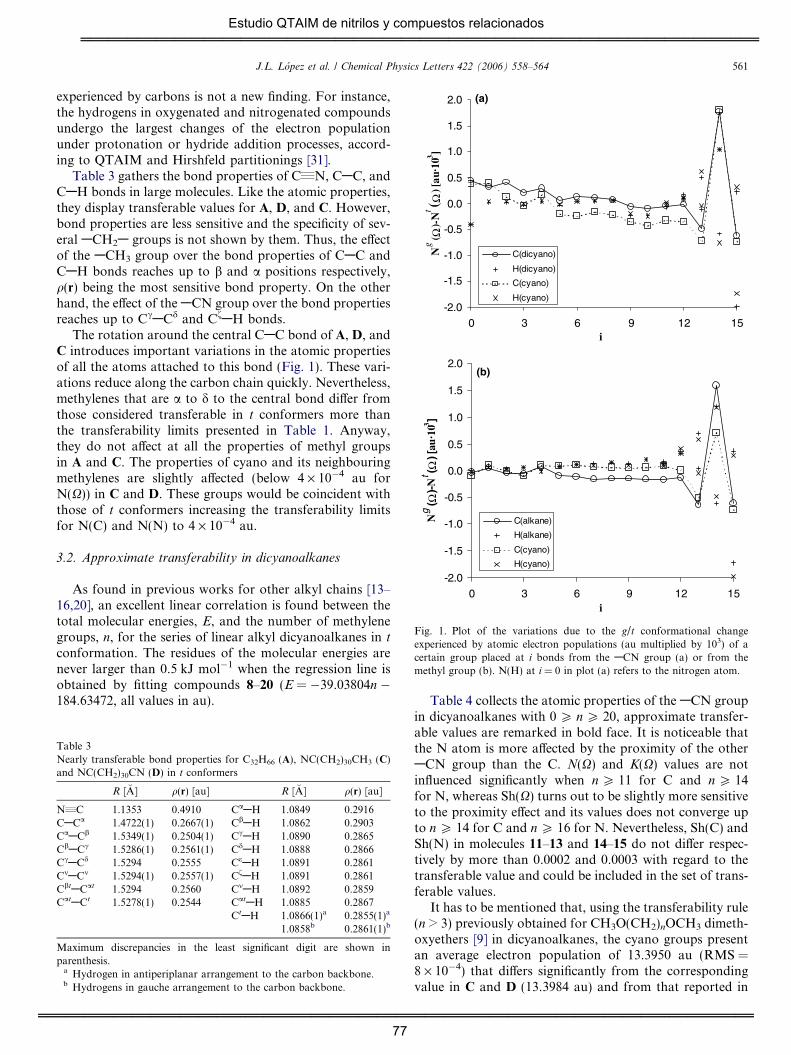
experienced by carbons is not a new finding. For instance,the hydrogens in oxygenated and nitrogenated compoundsundergo the largest changes of the electron populationunder protonation or hydride addition processes, accord-ing to QTAIM and Hirshfeld partitionings [31].
Table 3 gathers the bond properties of C„N, CAC, andCAH bonds in large molecules. Like the atomic properties,they display transferable values for A, D, and C. However,bond properties are less sensitive and the specificity of sev-eral ACH2A groups is not shown by them. Thus, the effectof the ACH3 group over the bond properties of CAC andCAH bonds reaches up to b and a positions respectively,q(r) being the most sensitive bond property. On the otherhand, the effect of the ACN group over the bond propertiesreaches up to CcACd and CfAH bonds.
The rotation around the central CAC bond of A, D, andC introduces important variations in the atomic propertiesof all the atoms attached to this bond (Fig. 1). These vari-ations reduce along the carbon chain quickly. Nevertheless,methylenes that are a to d to the central bond differ fromthose considered transferable in t conformers more thanthe transferability limits presented in Table 1. Anyway,they do not affect at all the properties of methyl groupsin A and C. The properties of cyano and its neighbouringmethylenes are slightly affected (below 4 · 104 au forN(X)) in C and D. These groups would be coincident withthose of t conformers increasing the transferability limitsfor N(C) and N(N) to 4 · 104 au.
3.2. Approximate transferability in dicyanoalkanes
As found in previous works for other alkyl chains [13–16,20], an excellent linear correlation is found between thetotal molecular energies, E, and the number of methylenegroups, n, for the series of linear alkyl dicyanoalkanes in t
conformation. The residues of the molecular energies arenever larger than 0.5 kJ mol1 when the regression line isobtained by fitting compounds 8–20 (E = 39.03804n 184.63472, all values in au). Table 4 collects the atomic properties of the ACN group
in dicyanoalkanes with 0 P n P 20, approximate transfer-able values are remarked in bold face. It is noticeable thatthe N atom is more affected by the proximity of the otherACN group than the C. N(X) and K(X) values are notinfluenced significantly when n P 11 for C and n P 14for N, whereas Sh(X) turns out to be slightly more sensitiveto the proximity effect and its values does not converge upto n P 14 for C and n P 16 for N. Nevertheless, Sh(C) andSh(N) in molecules 11–13 and 14–15 do not differ respec-tively by more than 0.0002 and 0.0003 with regard to thetransferable value and could be included in the set of trans-ferable values.
It has to be mentioned that, using the transferability rule(n > 3) previously obtained for CH3O(CH2)nOCH3 dimeth-oxyethers [9] in dicyanoalkanes, the cyano groups presentan average electron population of 13.3950 au (RMS =8 · 104) that differs significantly from the correspondingvalue in C and D (13.3984 au) and from that reported in
Table 3Nearly transferable bond properties for C32H66 (A), NC(CH2)30CH3 (C)and NC(CH2)30CN (D) in t conformers
R [A] q(r) [au] R [A] q(r) [au]
N„C 1.1353 0.4910 CaAH 1.0849 0.2916CACa 1.4722(1) 0.2667(1) CbAH 1.0862 0.2903CaACb 1.5349(1) 0.2504(1) CcAH 1.0890 0.2865CbACc 1.5286(1) 0.2561(1) CdAH 1.0888 0.2866CcACd 1.5294 0.2555 CeAH 1.0891 0.2861CmACm 1.5294(1) 0.2557(1) CfAH 1.0891 0.2861CbtACat 1.5294 0.2560 CmAH 1.0892 0.2859CatACt 1.5278(1) 0.2544 CatAH 1.0885 0.2867
CtAH 1.0866(1)a 0.2855(1)a
1.0858b 0.2861(1)b
Maximum discrepancies in the least significant digit are shown inparenthesis.
a Hydrogen in antiperiplanar arrangement to the carbon backbone.b Hydrogens in gauche arrangement to the carbon backbone.
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
0 3 6 9 12 15i
Ng
( Ω)-
Nt ( Ω
) [au
·103 ]
C(dicyano)
H(dicyano)
C(cyano)
H(cyano)
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
0 3 6 9 12 15i
Ng
( Ω)-
Nt ( Ω
) [au
·103
]
C(alkane)
H(alkane)
C(cyano)
H(cyano)
(a)
(b)
Fig. 1. Plot of the variations due to the g/t conformational changeexperienced by atomic electron populations (au multiplied by 103) of acertain group placed at i bonds from the ACN group (a) or from themethyl group (b). N(H) at i = 0 in plot (a) refers to the nitrogen atom.
J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564 561
Estudio QTAIM de nitrilos y compuestos relacionados
77
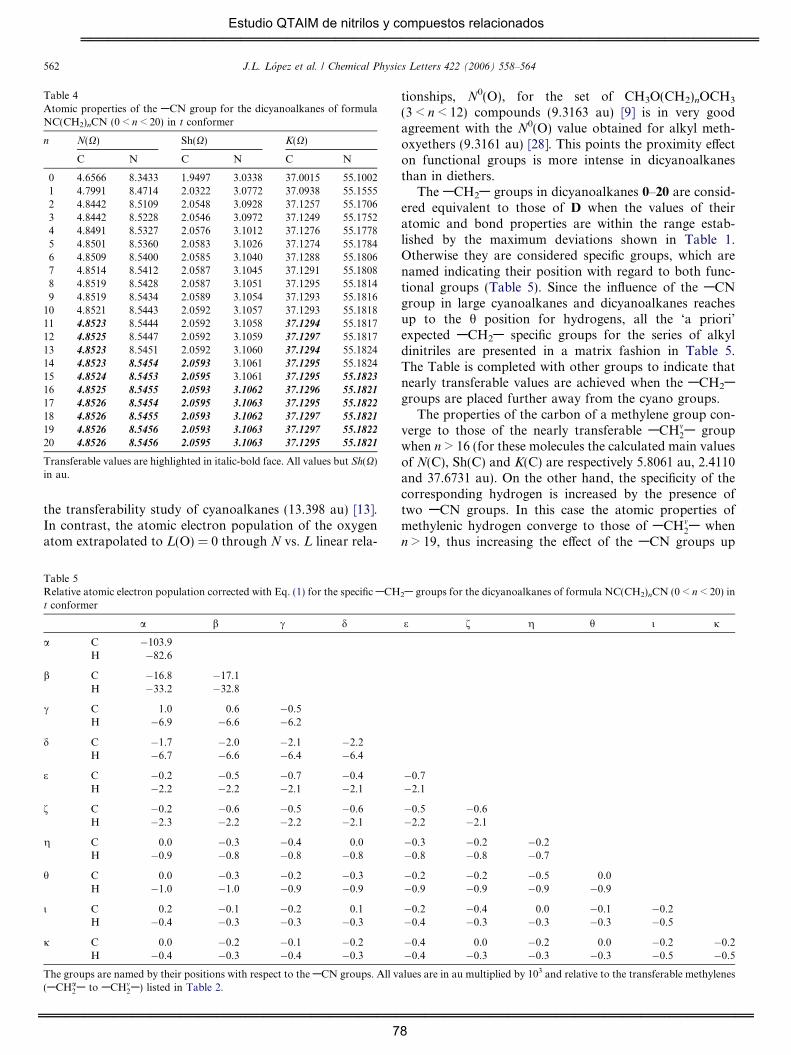
the transferability study of cyanoalkanes (13.398 au) [13].In contrast, the atomic electron population of the oxygenatom extrapolated to L(O) = 0 through N vs. L linear rela-
tionships, N0(O), for the set of CH3O(CH2)nOCH3
(3 < n < 12) compounds (9.3163 au) [9] is in very goodagreement with the N0(O) value obtained for alkyl meth-oxyethers (9.3161 au) [28]. This points the proximity effecton functional groups is more intense in dicyanoalkanesthan in diethers.
The ACH2A groups in dicyanoalkanes 0–20 are consid-ered equivalent to those of D when the values of theiratomic and bond properties are within the range estab-lished by the maximum deviations shown in Table 1.Otherwise they are considered specific groups, which arenamed indicating their position with regard to both func-tional groups (Table 5). Since the influence of the ACNgroup in large cyanoalkanes and dicyanoalkanes reachesup to the h position for hydrogens, all the ‘a priori’expected ACH2A specific groups for the series of alkyldinitriles are presented in a matrix fashion in Table 5.The Table is completed with other groups to indicate thatnearly transferable values are achieved when the ACH2Agroups are placed further away from the cyano groups.
The properties of the carbon of a methylene group con-verge to those of the nearly transferable ACHm
2A groupwhen n > 16 (for these molecules the calculated main valuesof N(C), Sh(C) and K(C) are respectively 5.8061 au, 2.4110and 37.6731 au). On the other hand, the specificity of thecorresponding hydrogen is increased by the presence oftwo ACN groups. In this case the atomic properties ofmethylenic hydrogen converge to those of ACHm
2A whenn > 19, thus increasing the effect of the ACN groups up
Table 4Atomic properties of the ACN group for the dicyanoalkanes of formulaNC(CH2)nCN (0 < n < 20) in t conformer
n N(X) Sh(X) K(X)
C N C N C N
0 4.6566 8.3433 1.9497 3.0338 37.0015 55.10021 4.7991 8.4714 2.0322 3.0772 37.0938 55.15552 4.8442 8.5109 2.0548 3.0928 37.1257 55.17063 4.8442 8.5228 2.0546 3.0972 37.1249 55.17524 4.8491 8.5327 2.0576 3.1012 37.1276 55.17785 4.8501 8.5360 2.0583 3.1026 37.1274 55.17846 4.8509 8.5400 2.0585 3.1040 37.1288 55.18067 4.8514 8.5412 2.0587 3.1045 37.1291 55.18088 4.8519 8.5428 2.0587 3.1051 37.1295 55.18149 4.8519 8.5434 2.0589 3.1054 37.1293 55.1816
10 4.8521 8.5443 2.0592 3.1057 37.1293 55.181811 4.8523 8.5444 2.0592 3.1058 37.1294 55.181712 4.8525 8.5447 2.0592 3.1059 37.1297 55.181713 4.8523 8.5451 2.0592 3.1060 37.1294 55.182414 4.8523 8.5454 2.0593 3.1061 37.1295 55.182415 4.8524 8.5453 2.0595 3.1061 37.1295 55.1823
16 4.8525 8.5455 2.0593 3.1062 37.1296 55.1821
17 4.8526 8.5454 2.0595 3.1063 37.1295 55.1822
18 4.8526 8.5455 2.0593 3.1062 37.1297 55.1821
19 4.8526 8.5456 2.0593 3.1063 37.1297 55.1822
20 4.8526 8.5456 2.0595 3.1063 37.1295 55.1821
Transferable values are highlighted in italic-bold face. All values but Sh(X)in au.
Table 5Relative atomic electron population corrected with Eq. (1) for the specific ACH2A groups for the dicyanoalkanes of formula NC(CH2)nCN (0 < n < 20) int conformer
a b c d e f g h i j
a C 103.9H 82.6
b C 16.8 17.1H 33.2 32.8
c C 1.0 0.6 0.5H 6.9 6.6 6.2
d C 1.7 2.0 2.1 2.2H 6.7 6.6 6.4 6.4
e C 0.2 0.5 0.7 0.4 0.7H 2.2 2.2 2.1 2.1 2.1
f C 0.2 0.6 0.5 0.6 0.5 0.6H 2.3 2.2 2.2 2.1 2.2 2.1
g C 0.0 0.3 0.4 0.0 0.3 0.2 0.2H 0.9 0.8 0.8 0.8 0.8 0.8 0.7
h C 0.0 0.3 0.2 0.3 0.2 0.2 0.5 0.0H 1.0 1.0 0.9 0.9 0.9 0.9 0.9 0.9
i C 0.2 0.1 0.2 0.1 0.2 0.4 0.0 0.1 0.2H 0.4 0.3 0.3 0.3 0.4 0.3 0.3 0.3 0.5
j C 0.0 0.2 0.1 0.2 0.4 0.0 0.2 0.0 0.2 0.2H 0.4 0.3 0.4 0.3 0.4 0.3 0.3 0.3 0.5 0.5
The groups are named by their positions with respect to the ACN groups. All values are in au multiplied by 103 and relative to the transferable methylenes(ACHa
2A to ACHm2A) listed in Table 2.
562 J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564
Estudio QTAIM de nitrilos y compuestos relacionados
78
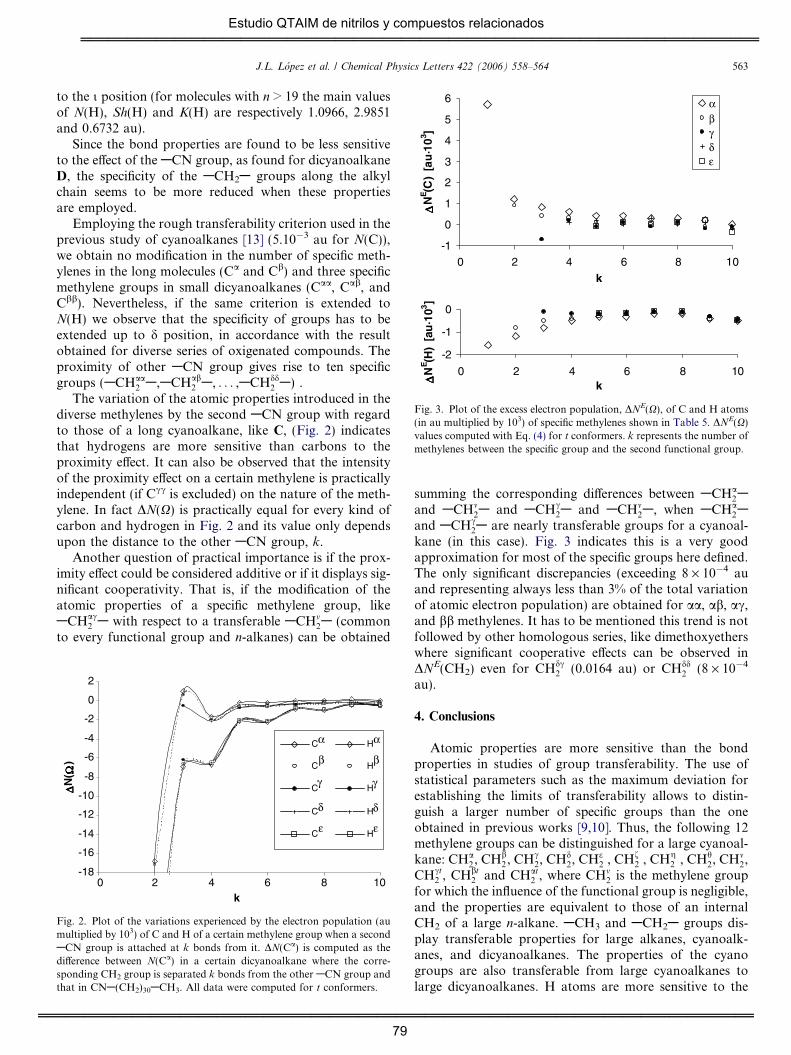
to the i position (for molecules with n > 19 the main valuesof N(H), Sh(H) and K(H) are respectively 1.0966, 2.9851and 0.6732 au).
Since the bond properties are found to be less sensitiveto the effect of the ACN group, as found for dicyanoalkaneD, the specificity of the ACH2A groups along the alkylchain seems to be more reduced when these propertiesare employed.
Employing the rough transferability criterion used in theprevious study of cyanoalkanes [13] (5.103 au for N(C)),we obtain no modification in the number of specific meth-ylenes in the long molecules (Ca and Cb) and three specificmethylene groups in small dicyanoalkanes (Caa, Cab, andCbb). Nevertheless, if the same criterion is extended toN(H) we observe that the specificity of groups has to beextended up to d position, in accordance with the resultobtained for diverse series of oxigenated compounds. Theproximity of other ACN group gives rise to ten specificgroups (ACHaa
2 A,ACHab2 A, . . . ,ACHdd
2 A) .The variation of the atomic properties introduced in the
diverse methylenes by the second ACN group with regardto those of a long cyanoalkane, like C, (Fig. 2) indicatesthat hydrogens are more sensitive than carbons to theproximity effect. It can also be observed that the intensityof the proximity effect on a certain methylene is practicallyindependent (if Ccc is excluded) on the nature of the meth-ylene. In fact DN(X) is practically equal for every kind ofcarbon and hydrogen in Fig. 2 and its value only dependsupon the distance to the other ACN group, k.
Another question of practical importance is if the prox-imity effect could be considered additive or if it displays sig-nificant cooperativity. That is, if the modification of theatomic properties of a specific methylene group, likeACHac
2 A with respect to a transferable ACHm2A (common
to every functional group and n-alkanes) can be obtained
summing the corresponding differences between ACHa2A
and ACHm2A and ACHc
2A and ACHm2A, when ACHa
2Aand ACHc
2A are nearly transferable groups for a cyanoal-kane (in this case). Fig. 3 indicates this is a very goodapproximation for most of the specific groups here defined.The only significant discrepancies (exceeding 8 · 104 auand representing always less than 3% of the total variationof atomic electron population) are obtained for aa, ab, ac,and bb methylenes. It has to be mentioned this trend is notfollowed by other homologous series, like dimethoxyetherswhere significant cooperative effects can be observed inDNE(CH2) even for CHdc
2 (0.0164 au) or CHdd2 (8 · 104
au).
4. Conclusions
Atomic properties are more sensitive than the bondproperties in studies of group transferability. The use ofstatistical parameters such as the maximum deviation forestablishing the limits of transferability allows to distin-guish a larger number of specific groups than the oneobtained in previous works [9,10]. Thus, the following 12methylene groups can be distinguished for a large cyanoal-kane: CHa
2, CHb2, CHc
2, CHd2, CHe
2 , CHf2 , CHg
2 , CHh2, CHm
2,CHct
2 , CHbt2 and CHat
2 , where CHm2 is the methylene group
for which the influence of the functional group is negligible,and the properties are equivalent to those of an internalCH2 of a large n-alkane. ACH3 and ACH2A groups dis-play transferable properties for large alkanes, cyanoalk-anes, and dicyanoalkanes. The properties of the cyanogroups are also transferable from large cyanoalkanes tolarge dicyanoalkanes. H atoms are more sensitive to the
-18
-16
-14
-12
-10
-8
-6
-4
-2
0
2
0 2 4 6 8 10k
ΔΔ ΔΔ(
NΩΩ ΩΩ
)
C H
C H
C H
C H
C H
α α
β β
γ γ
δ δ
ε ε
Fig. 2. Plot of the variations experienced by the electron population (aumultiplied by 103) of C and H of a certain methylene group when a secondACN group is attached at k bonds from it. DN(Ca) is computed as thedifference between N(Ca) in a certain dicyanoalkane where the corre-sponding CH2 group is separated k bonds from the other ACN group andthat in CNA(CH2)30ACH3. All data were computed for t conformers.
-1
0
1
2
3
4
5
6
0 2 4 6 8 10
k
ΔΔ ΔΔN
E01·
ua[ )C(
3 ]
αβγδε
-2
-1
0
0 2 4 6 8 10kΔΔ ΔΔ
NE
01·ua[ )
H(3 ]
Fig. 3. Plot of the excess electron population, DNE(X), of C and H atoms(in au multiplied by 103) of specific methylenes shown in Table 5. DNE(X)values computed with Eq. (4) for t conformers. k represents the number ofmethylenes between the specific group and the second functional group.
J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564 563
Estudio QTAIM de nitrilos y compuestos relacionados
79

presence of ACN than C, however they are less sensitive tothe presence of a ACH3.
The mutual influence between two ACN groups (prox-imity effect) in alkyl dinitriles is negligible when n > 14.The specificity is mainly due to the N atom whose atomicproperties converge to a transferable value later than thoseof C. The presence of two ACN increases the specificity ofthe ACH2A groups, their atomic properties does not con-verge to those of a large dicyanoalkane or cyanoalkaneuntil the i position due to the hydrogens, whose atomicproperties converge to a transferable value later than thoseof C. The effects observed in a specific methylene of a dic-yanoalkane can be considered as the summation of thosedue to independent CN groups if we exclude the methyl-enes of molecules CN(CH2)nCN with n < 4, where cooper-ative effects for the atomic electron population are between5.7 · 103 and 8 · 104 au. This trend cannot be extendedto other homologous series.
The effect of the conformation change was studied con-sidering the rotation around the CAC central bond ofC32H66, NC(CH2)30CH3, and NC(CH2)30CN. We havefound significant differences for methylene groups thatare a to d to that bond. Nevertheless the conformationaleffect does not change the conclusions here presented ifthe transferability limits for N(C) and N(N) are increasedto 4 · 104 au.
Acknowledgements
We thank Luis Alberto Lopez for his encouraging sup-port and ‘Centro de Supercomputacion de Galicia’ (CES-GA) for access to their computational facilities.
References
[1] H.V. Kehiaian, Fluid Phase Equilibria 13 (1983) 243.[2] S.I. Sandler, Models for Thermodynamic and Phase Equilibria
Calculations, Marcel Dekker, New York, 1994.[3] S.-T. Lin, S.I. Sandler, J. Phys. Chem. A 104 (2000) 7099.
[4] D. Gonzalez-Salgado, C.A. Tovar, C.A. Cerdeirina, E. Carballo, L.Romanı, Fluid Phase Equilibria 199 (2002) 121.
[5] S. Delcros, J.R. Quint, J.P.E. Grolier, H.V. Kehiaian, Fluid PhaseEquilibria 113 (1995) 1.
[6] H.S. Wu, S.I. Sandler, AIChE Journal 35 (1989) 168.[7] R.F.W. Bader, Atoms in Molecules – A Quantum Theory Interna-
tional Series of Monographs on Chemistry, vol. 22, Oxford Univer-sity Press, Oxford, 1990.
[8] R.F.W. Bader, Chem. Rev. 91 (1991) 893.[9] A. Vila, R.A. Mosquera, Chem. Phys. Lett. 345 (2001) 445.
[10] A. Vila, E. Carballo, R.A. Mosquera, J. Mol. Struct. (THEOCHEM)617 (2002) 219.
[11] A.E. Aliev, K.D.M. Harris, P.H. Champkin, J. Phys. Chem. B 109(2005) 23342.
[12] K.D.M. Harris, in: J.L. Atwood, J.W. Steed (Eds.), Encyclopedia ofSupramolecular Chemistry, vol. 2, Marcel Dekker, New York, 2004,pp. 1538–1549.
[13] J.L. Lopez, M. Mandado, A.M. Grana, R.A. Mosquera, Int. J.Quantum Chem. 86 (2002) 190.
[14] A.M. Grana, R.A. Mosquera, J. Chem. Phys. 113 (2000) 1492.[15] A. Vila, R.A. Mosquera, J. Chem. Phys. 115 (2001) 1264.[16] M. Mandado, A.M. Grana, R.A. Mosquera, J. Mol. Struct.
(THEOCHEM) 584 (2002) 221.[17] L. Lorenzo, R.A. Mosquera, Chem. Phys. Lett. 356 (2002) 305.[18] P. Hohenberg, B. Kohn, Phys. Rev. B 136 (1964) 864.[19] J. Riess, W. Munch, Theor. Chim. Acta 58 (1981) 295.[20] R.F.W. Bader, P. Becker, Chem. Phys. Lett. 148 (1988) 452.[21] M.J. Frisch et al., GAUSSIAN 98, Revision A.7., Gaussian Inc.,
Pittsburgh, PA, 1998.[22] M. Mandado, R.A. Mosquera, A.M. Grana, Chem. Phys. Lett. 355
(2002) 529.[23] AIMPAC: A suite of programs for the Theory of Atoms in
Molecules; R.F.W. Bader and coworkers, Eds. McMaster University,Hamilton, Ontario, Canada, L8S 4M1. Available from:<[email protected]>.
[24] M. Mandado, A.M. Grana, R.A. Mosquera, J. Mol. Struct.(THEOCHEM) 572 (2001) 223.
[25] M. Mandado, A. Vila, A.M. Grana, R.A. Mosquera, J. Cioslowski,Chem. Phys. Lett. 371 (2003) 739.
[26] F. Cortes-Guzman, R.F.W. Bader, Chem. Phys. Lett. 379 (2003) 183.[27] A.M. Grana, R.A. Mosquera, J. Chem. Phys. 110 (1999) 6606.[28] A. Vila, E. Carballo, R.A. Mosquera, Can. J. Chem. 78 (2000) 1535.[29] P.B. Quinonez, A. Vila, A.M. Grana, R.A. Mosquera, Chem. Phys.
287 (2003) 227.[30] F.M. Aicken, P.L.A. Popelier, Can. J. Chem. 78 (2000) 415.[31] M. Mandado, C. Van Alsenoy, R.A. Mosquera, J. Phys. Chem. 108
(2004) 7050.
564 J.L. Lopez et al. / Chemical Physics Letters 422 (2006) 558–564
Estudio QTAIM de nitrilos y compuestos relacionados
80

Estudio QTAIM de nitrilos y compuestos relacionados
81

Electron Density Analysis on the Protonation of Nitriles
Jose Luis Lopez, Ana M. Grana, and Ricardo A. Mosquera*Departamento Quımica Fısica, UniVersidade de Vigo, Lagoas-Marcosende, 36310-Vigo, Galicia (Spain)
ReceiVed: December 14, 2008; ReVised Manuscript ReceiVed: January 9, 2009
The applicability of the resonance model to explain the evolution of electron density was tested for a set of15 nitriles whose protonation processes were studied by means of the quantum theory of atoms in molecules(QTAIM). The electron densities were obtained at the B3LYP/6-31++G**//B3LYP/6-31++G** and HF/6-31++G**//HF/6-31++G** levels. QTAIM atomic and bond properties do not follow the trends that shouldbe expected according to the resonance model and our results are more in line with a H+-NtC-R Lewisstructure than with the H-N+tC-R and H-NdC+-R ones. Also, reasonable agreement between experimentaland calculated PA values as well as good correlations between variations in atomic energies and populationsas a result of protonation were found.
Introduction
The applicability of the resonance model (RM) to explainthe structure and reactivity of organic compounds has beengenerally accepted1,2 and has proved to be a very useful tool inchemistry. Nevertheless, topological analysis of electron densi-ties carried out with the quantum theory atoms in molecules(QTAIM)3,4 for diverse processes have reported evolutions ofthe electron density that are not in line with the predictionsprovided by the RM. These disagreements appear even for sosimple processes as internal rotations,5,6 protonations,7-9 orhydride additions.10 Also, QTAIM results are inconsistent withthe Lewis structures traditionally accepted for some chargedcompounds, like diazonium salts11 or protonated ethers.12-14 Thepublication of the first study reporting on the disagreementsbetween RM and QTAIM was followed by a controversy aboutthe suitability of QTAIM for this kind of studies.15-17 Nowadays,this controversy seems to be solved clearly in favor of QTAIMapplicability.16,17 Moreover, most of the qualitative conclusionsobtained from QTAIM studies on protonation and hydrideaddition are confirmed by other electron density analysis,9,10 likeHirshfeld partitioning.18,19
H-NdC+-R Lewis structures have been traditionally em-ployed for describing protonated nitriles in diverse reactionmechanisms. These structures are, in the context of the RM,the result of transforming one π electron pair of the NtC triplebond into the N-H bond. Alternatively, the protonation processcould be understood as the formation of a dative bond betweenN and proton using the nitrogen lone pair, a process representedby the H-N+tC-R resonance form. These are basically thesame schemes used for explaining protonations at other elec-tronegative sites, which have been recently found in controversywith the QTAIM studies carried out for the N-protonation ofindole,20 O-protonation of simple carbonyl systems,9 and N/O-protonations on diverse pyrimidinic bases.7,8,21 All of thesestudies point to H+-X-R structures (XdO or N and the X-Rbond being single or double). QTAIM results for these systemsalso indicate that the formation of the H+-X bond is ac-companied by an electron density redistribution affecting thewhole molecule. Hydrogens act very effectively as a source ofelectron density for this redistribution, as reported by Stuchbury
and Cooper studying the basicity of NH3 and the series ofmethylamines.22
In this work, we have carried out a QTAIM study on theprotonation of several cyanocompounds. This allows to studyif the triple bonding modifies the trends hitherto observed forother compounds. The molecules here studied include both linearand branched cyanoalkanes as well as compounds where thecyano function is conjugated with π delocalized systems. Thus,we have been able to establish trends for the size of linear alkylchains (1-6), conformational change (4, 7), alkyl chainramification (2, 3, 8, 9), electronegativity of the substitutents(2, 10), and π-delocalization (11-15) (Table 1).
Computational Details
QTAIM allows the partitioning of a molecule into disjointsubsystems without resorting to hypothesis alien to quantummechanics.3,4 With a few exceptions,23 each of these subsystemsconsists of a nucleus, which acts as an attractor for thetrajectories of the gradient of the electron density vector field,3F(r), and its associated atomic basin, throughout thesetrajectories spread. An atom, Ω, is defined as the union of theattractor and its associated basin, and it is surrounded by zeroflux surfaces for 3F(r). The integration of the proper densityfunctions within these limits provides diverse atomic propertiessuch as the electron population, N(Ω), or the total atomicelectron energy, E(Ω). In this article, we have considered the σand π components of the atomic electron population, Nσ(Ω)and Nπ(Ω), respectively.
QTAIM also recovers main elements of molecular structurein terms of the critical points, rc, of the electron density, F(r).Prominent among them are the bond critical points (BCPs),which are located roughly in between every pair of bondedatoms. Although the relationship between the presence of a BCPand the existence of a chemical bonding has become acontroversial and it is still a debated point of the theory,24-29
the electron density at a certain BCP is regarded as an indicatorfor bond strength.
All the neutral (1 to 15) and protonated (1+ to 15+) specieshere considered (Table 1) were fully optimized at the HF/6-31++G(d,p) and B3LYP/6-31++G(d,p) levels using the pro-gram GAMESS.32 The optimization was performed using theself-consistent virial scaling (SCVS) method introduced by Lehd* To whom correspondence should be addressed.
J. Phys. Chem. A 2009, 113, 2652–26572652
10.1021/jp811023x CCC: $40.75 2009 American Chemical SocietyPublished on Web 02/23/2009
Estudio QTAIM de nitrilos y compuestos relacionados
82
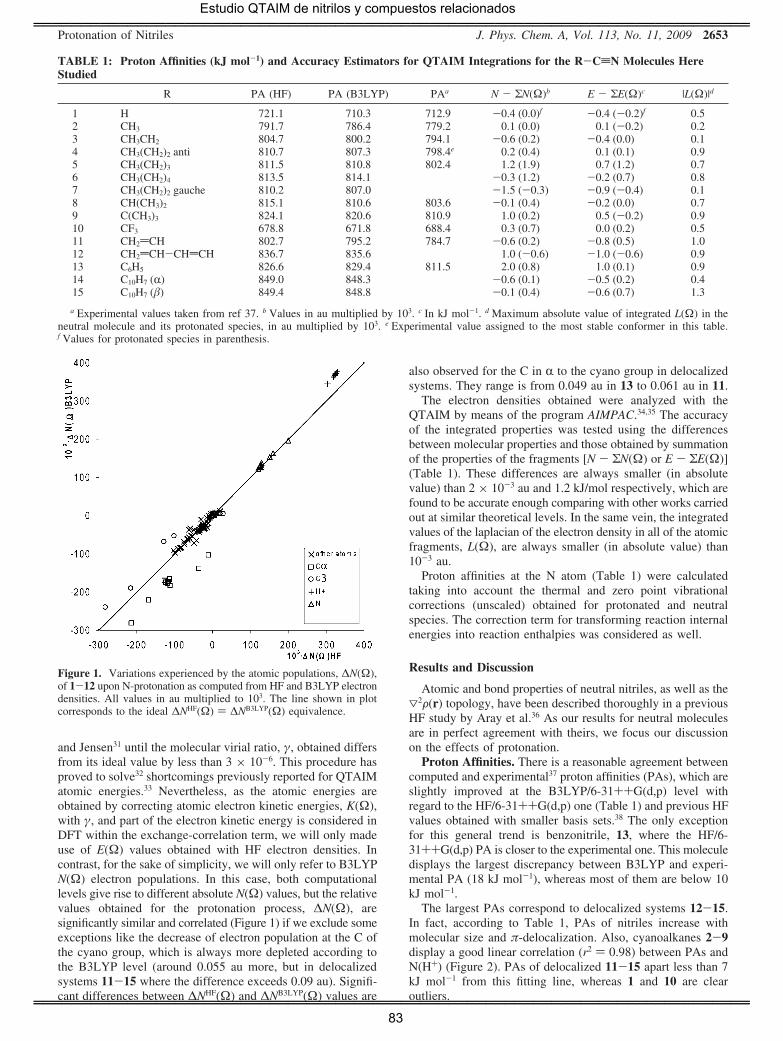
and Jensen31 until the molecular virial ratio, γ, obtained differsfrom its ideal value by less than 3 × 10-6. This procedure hasproved to solve32 shortcomings previously reported for QTAIMatomic energies.33 Nevertheless, as the atomic energies areobtained by correcting atomic electron kinetic energies, K(Ω),with γ, and part of the electron kinetic energy is considered inDFT within the exchange-correlation term, we will only madeuse of E(Ω) values obtained with HF electron densities. Incontrast, for the sake of simplicity, we will only refer to B3LYPN(Ω) electron populations. In this case, both computationallevels give rise to different absolute N(Ω) values, but the relativevalues obtained for the protonation process, ∆N(Ω), aresignificantly similar and correlated (Figure 1) if we exclude someexceptions like the decrease of electron population at the C ofthe cyano group, which is always more depleted according tothe B3LYP level (around 0.055 au more, but in delocalizedsystems 11-15 where the difference exceeds 0.09 au). Signifi-cant differences between ∆NHF(Ω) and ∆NB3LYP(Ω) values are
also observed for the C in R to the cyano group in delocalizedsystems. They range is from 0.049 au in 13 to 0.061 au in 11.
The electron densities obtained were analyzed with theQTAIM by means of the program AIMPAC.34,35 The accuracyof the integrated properties was tested using the differencesbetween molecular properties and those obtained by summationof the properties of the fragments [N - ΣN(Ω) or E - ΣE(Ω)](Table 1). These differences are always smaller (in absolutevalue) than 2 × 10-3 au and 1.2 kJ/mol respectively, which arefound to be accurate enough comparing with other works carriedout at similar theoretical levels. In the same vein, the integratedvalues of the laplacian of the electron density in all of the atomicfragments, L(Ω), are always smaller (in absolute value) than10-3 au.
Proton affinities at the N atom (Table 1) were calculatedtaking into account the thermal and zero point vibrationalcorrections (unscaled) obtained for protonated and neutralspecies. The correction term for transforming reaction internalenergies into reaction enthalpies was considered as well.
Results and Discussion
Atomic and bond properties of neutral nitriles, as well as the32F(r) topology, have been described thoroughly in a previousHF study by Aray et al.36 As our results for neutral moleculesare in perfect agreement with theirs, we focus our discussionon the effects of protonation.
Proton Affinities. There is a reasonable agreement betweencomputed and experimental37 proton affinities (PAs), which areslightly improved at the B3LYP/6-31++G(d,p) level withregard to the HF/6-31++G(d,p) one (Table 1) and previous HFvalues obtained with smaller basis sets.38 The only exceptionfor this general trend is benzonitrile, 13, where the HF/6-31++G(d,p) PA is closer to the experimental one. This moleculedisplays the largest discrepancy between B3LYP and experi-mental PA (18 kJ mol-1), whereas most of them are below 10kJ mol-1.
The largest PAs correspond to delocalized systems 12-15.In fact, according to Table 1, PAs of nitriles increase withmolecular size and π-delocalization. Also, cyanoalkanes 2-9display a good linear correlation (r2 ) 0.98) between PAs andN(H+) (Figure 2). PAs of delocalized 11-15 apart less than 7kJ mol-1 from this fitting line, whereas 1 and 10 are clearoutliers.
TABLE 1: Proton Affinities (kJ mol-1) and Accuracy Estimators for QTAIM Integrations for the R-CtN Molecules HereStudied
R PA (HF) PA (B3LYP) PAa N - ΣN(Ω)b E - ΣE(Ω)c |L(Ω)|d
1 H 721.1 710.3 712.9 -0.4 (0.0)f -0.4 (-0.2)f 0.52 CH3 791.7 786.4 779.2 0.1 (0.0) 0.1 (-0.2) 0.23 CH3CH2 804.7 800.2 794.1 -0.6 (0.2) -0.4 (0.0) 0.14 CH3(CH2)2 anti 810.7 807.3 798.4e 0.2 (0.4) 0.1 (0.1) 0.95 CH3(CH2)3 811.5 810.8 802.4 1.2 (1.9) 0.7 (1.2) 0.76 CH3(CH2)4 813.5 814.1 -0.3 (1.2) -0.2 (0.7) 0.87 CH3(CH2)2 gauche 810.2 807.0 -1.5 (-0.3) -0.9 (-0.4) 0.18 CH(CH3)2 815.1 810.6 803.6 -0.1 (0.4) -0.2 (0.0) 0.79 C(CH3)3 824.1 820.6 810.9 1.0 (0.2) 0.5 (-0.2) 0.910 CF3 678.8 671.8 688.4 0.3 (0.7) 0.0 (0.2) 0.511 CH2dCH 802.7 795.2 784.7 -0.6 (0.2) -0.8 (0.5) 1.012 CH2dCH-CHdCH 836.7 835.6 1.0 (-0.6) -1.0 (-0.6) 0.913 C6H5 826.6 829.4 811.5 2.0 (0.8) 1.0 (0.1) 0.914 C10H7 (R) 849.0 848.3 -0.6 (0.1) -0.5 (0.2) 0.415 C10H7 () 849.4 848.8 -0.1 (0.4) -0.6 (0.7) 1.3
a Experimental values taken from ref 37. b Values in au multiplied by 103. c In kJ mol-1. d Maximum absolute value of integrated L(Ω) in theneutral molecule and its protonated species, in au multiplied by 103. e Experimental value assigned to the most stable conformer in this table.f Values for protonated species in parenthesis.
Figure 1. Variations experienced by the atomic populations, ∆N(Ω),of 1-12 upon N-protonation as computed from HF and B3LYP electrondensities. All values in au multiplied to 103. The line shown in plotcorresponds to the ideal ∆NHF(Ω) ) ∆NB3LYP(Ω) equivalence.
Protonation of Nitriles J. Phys. Chem. A, Vol. 113, No. 11, 2009 2653
Estudio QTAIM de nitrilos y compuestos relacionados
83
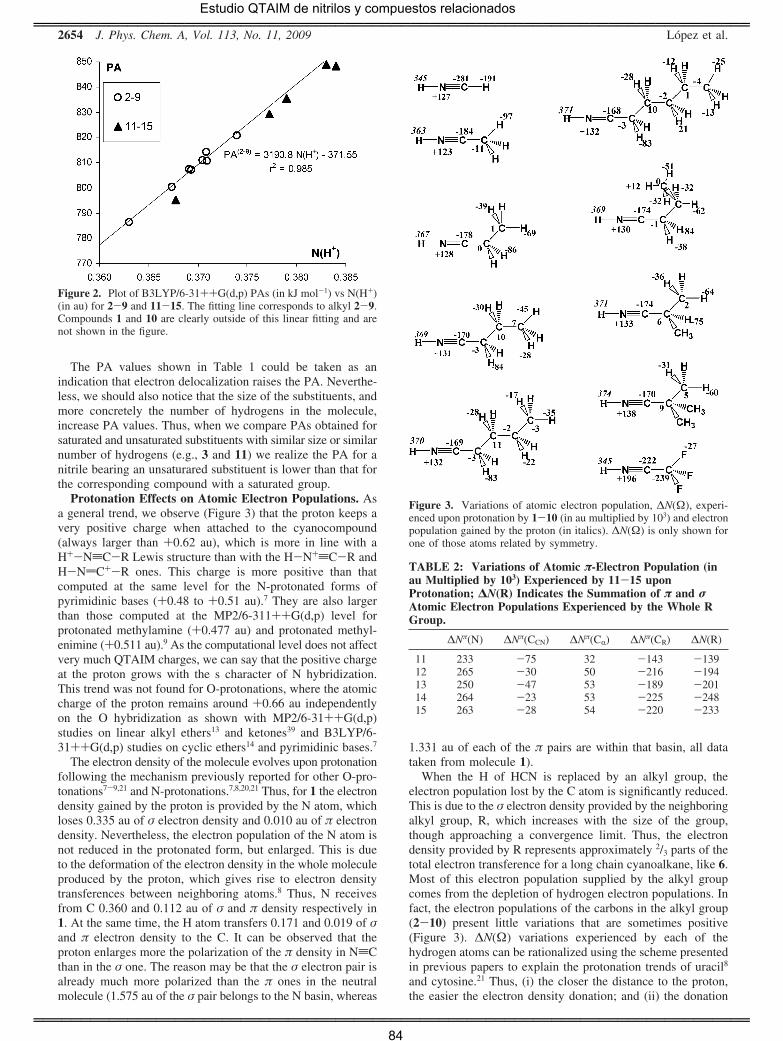
The PA values shown in Table 1 could be taken as anindication that electron delocalization raises the PA. Neverthe-less, we should also notice that the size of the substituents, andmore concretely the number of hydrogens in the molecule,increase PA values. Thus, when we compare PAs obtained forsaturated and unsaturated substituents with similar size or similarnumber of hydrogens (e.g., 3 and 11) we realize the PA for anitrile bearing an unsaturared substituent is lower than that forthe corresponding compound with a saturated group.
Protonation Effects on Atomic Electron Populations. Asa general trend, we observe (Figure 3) that the proton keeps avery positive charge when attached to the cyanocompound(always larger than +0.62 au), which is more in line with aH+-NtC-R Lewis structure than with the H-N+tC-R andH-NdC+-R ones. This charge is more positive than thatcomputed at the same level for the N-protonated forms ofpyrimidinic bases (+0.48 to +0.51 au).7 They are also largerthan those computed at the MP2/6-311++G(d,p) level forprotonated methylamine (+0.477 au) and protonated methyl-enimine (+0.511 au).9 As the computational level does not affectvery much QTAIM charges, we can say that the positive chargeat the proton grows with the s character of N hybridization.This trend was not found for O-protonations, where the atomiccharge of the proton remains around +0.66 au independentlyon the O hybridization as shown with MP2/6-31++G(d,p)studies on linear alkyl ethers13 and ketones39 and B3LYP/6-31++G(d,p) studies on cyclic ethers14 and pyrimidinic bases.7
The electron density of the molecule evolves upon protonationfollowing the mechanism previously reported for other O-pro-tonations7-9,21 and N-protonations.7,8,20,21 Thus, for 1 the electrondensity gained by the proton is provided by the N atom, whichloses 0.335 au of σ electron density and 0.010 au of π electrondensity. Nevertheless, the electron population of the N atom isnot reduced in the protonated form, but enlarged. This is dueto the deformation of the electron density in the whole moleculeproduced by the proton, which gives rise to electron densitytransferences between neighboring atoms.8 Thus, N receivesfrom C 0.360 and 0.112 au of σ and π density respectively in1. At the same time, the H atom transfers 0.171 and 0.019 of σand π electron density to the C. It can be observed that theproton enlarges more the polarization of the π density in NtCthan in the σ one. The reason may be that the σ electron pair isalready much more polarized than the π ones in the neutralmolecule (1.575 au of the σ pair belongs to the N basin, whereas
1.331 au of each of the π pairs are within that basin, all datataken from molecule 1).
When the H of HCN is replaced by an alkyl group, theelectron population lost by the C atom is significantly reduced.This is due to the σ electron density provided by the neighboringalkyl group, R, which increases with the size of the group,though approaching a convergence limit. Thus, the electrondensity provided by R represents approximately 2/3 parts of thetotal electron transference for a long chain cyanoalkane, like 6.Most of this electron population supplied by the alkyl groupcomes from the depletion of hydrogen electron populations. Infact, the electron populations of the carbons in the alkyl group(2-10) present little variations that are sometimes positive(Figure 3). ∆N(Ω) variations experienced by each of thehydrogen atoms can be rationalized using the scheme presentedin previous papers to explain the protonation trends of uracil8
and cytosine.21 Thus, (i) the closer the distance to the proton,the easier the electron density donation; and (ii) the donation
Figure 2. Plot of B3LYP/6-31++G(d,p) PAs (in kJ mol-1) vs N(H+)(in au) for 2-9 and 11-15. The fitting line corresponds to alkyl 2-9.Compounds 1 and 10 are clearly outside of this linear fitting and arenot shown in the figure.
Figure 3. Variations of atomic electron population, ∆N(Ω), experi-enced upon protonation by 1-10 (in au multiplied by 103) and electronpopulation gained by the proton (in italics). ∆N(Ω) is only shown forone of those atoms related by symmetry.
TABLE 2: Variations of Atomic π-Electron Population (inau Multiplied by 103) Experienced by 11-15 uponProtonation; ∆N(R) Indicates the Summation of π and σAtomic Electron Populations Experienced by the Whole RGroup.
∆Nπ(N) ∆Nπ(CCN) ∆Nπ(CR) ∆Nπ(CR) ∆N(R)
11 233 -75 32 -143 -13912 265 -30 50 -216 -19413 250 -47 53 -189 -20114 264 -23 53 -225 -24815 263 -28 54 -220 -233
2654 J. Phys. Chem. A, Vol. 113, No. 11, 2009 Lopez et al.
Estudio QTAIM de nitrilos y compuestos relacionados
84
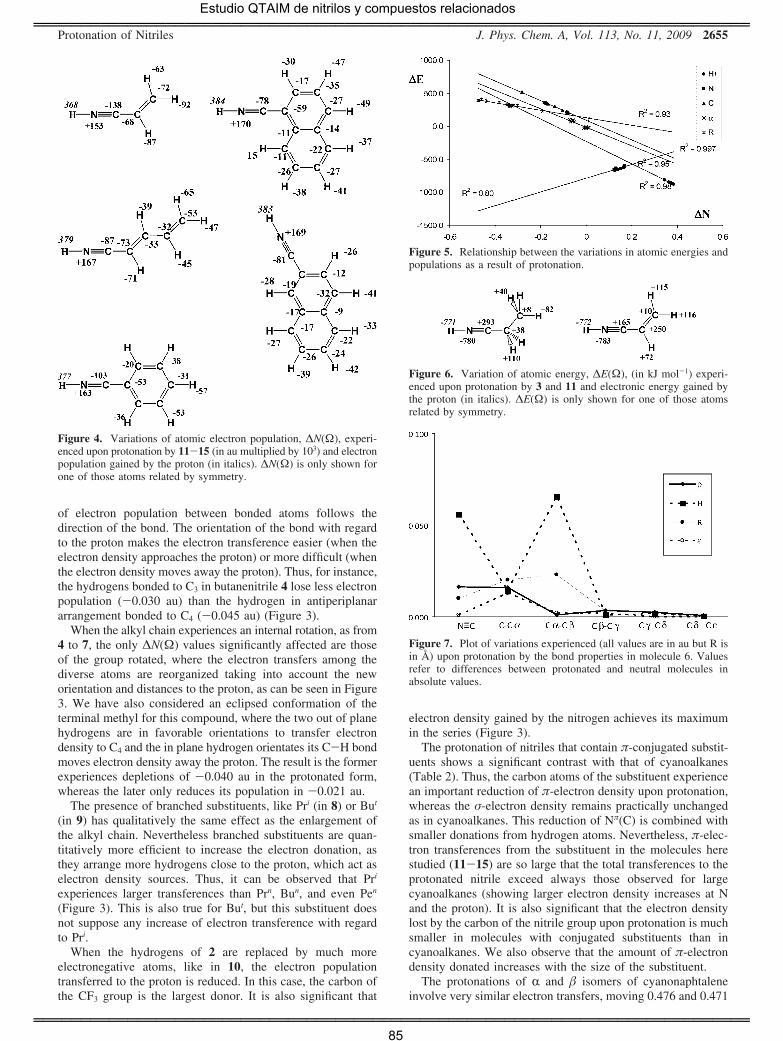
of electron population between bonded atoms follows thedirection of the bond. The orientation of the bond with regardto the proton makes the electron transference easier (when theelectron density approaches the proton) or more difficult (whenthe electron density moves away the proton). Thus, for instance,the hydrogens bonded to C3 in butanenitrile 4 lose less electronpopulation (-0.030 au) than the hydrogen in antiperiplanararrangement bonded to C4 (-0.045 au) (Figure 3).
When the alkyl chain experiences an internal rotation, as from4 to 7, the only ∆N(Ω) values significantly affected are thoseof the group rotated, where the electron transfers among thediverse atoms are reorganized taking into account the neworientation and distances to the proton, as can be seen in Figure3. We have also considered an eclipsed conformation of theterminal methyl for this compound, where the two out of planehydrogens are in favorable orientations to transfer electrondensity to C4 and the in plane hydrogen orientates its C-H bondmoves electron density away the proton. The result is the formerexperiences depletions of -0.040 au in the protonated form,whereas the later only reduces its population in -0.021 au.
The presence of branched substituents, like Pri (in 8) or But
(in 9) has qualitatively the same effect as the enlargement ofthe alkyl chain. Nevertheless branched substituents are quan-titatively more efficient to increase the electron donation, asthey arrange more hydrogens close to the proton, which act aselectron density sources. Thus, it can be observed that Pri
experiences larger transferences than Prn, Bun, and even Pen
(Figure 3). This is also true for But, but this substituent doesnot suppose any increase of electron transference with regardto Pri.
When the hydrogens of 2 are replaced by much moreelectronegative atoms, like in 10, the electron populationtransferred to the proton is reduced. In this case, the carbon ofthe CF3 group is the largest donor. It is also significant that
electron density gained by the nitrogen achieves its maximumin the series (Figure 3).
The protonation of nitriles that contain π-conjugated substit-uents shows a significant contrast with that of cyanoalkanes(Table 2). Thus, the carbon atoms of the substituent experiencean important reduction of π-electron density upon protonation,whereas the σ-electron density remains practically unchangedas in cyanoalkanes. This reduction of Nπ(C) is combined withsmaller donations from hydrogen atoms. Nevertheless, π-elec-tron transferences from the substituent in the molecules herestudied (11-15) are so large that the total transferences to theprotonated nitrile exceed always those observed for largecyanoalkanes (showing larger electron density increases at Nand the proton). It is also significant that the electron densitylost by the carbon of the nitrile group upon protonation is muchsmaller in molecules with conjugated substituents than incyanoalkanes. We also observe that the amount of π-electrondensity donated increases with the size of the substituent.
The protonations of R and isomers of cyanonaphtaleneinvolve very similar electron transfers, moving 0.476 and 0.471
Figure 4. Variations of atomic electron population, ∆N(Ω), experi-enced upon protonation by 11-15 (in au multiplied by 103) and electronpopulation gained by the proton (in italics). ∆N(Ω) is only shown forone of those atoms related by symmetry.
Figure 5. Relationship between the variations in atomic energies andpopulations as a result of protonation.
Figure 6. Variation of atomic energy, ∆E(Ω), (in kJ mol-1) experi-enced upon protonation by 3 and 11 and electronic energy gained bythe proton (in italics). ∆E(Ω) is only shown for one of those atomsrelated by symmetry.
Figure 7. Plot of variations experienced (all values are in au but R isin Å) upon protonation by the bond properties in molecule 6. Valuesrefer to differences between protonated and neutral molecules inabsolute values.
Protonation of Nitriles J. Phys. Chem. A, Vol. 113, No. 11, 2009 2655
Estudio QTAIM de nitrilos y compuestos relacionados
85
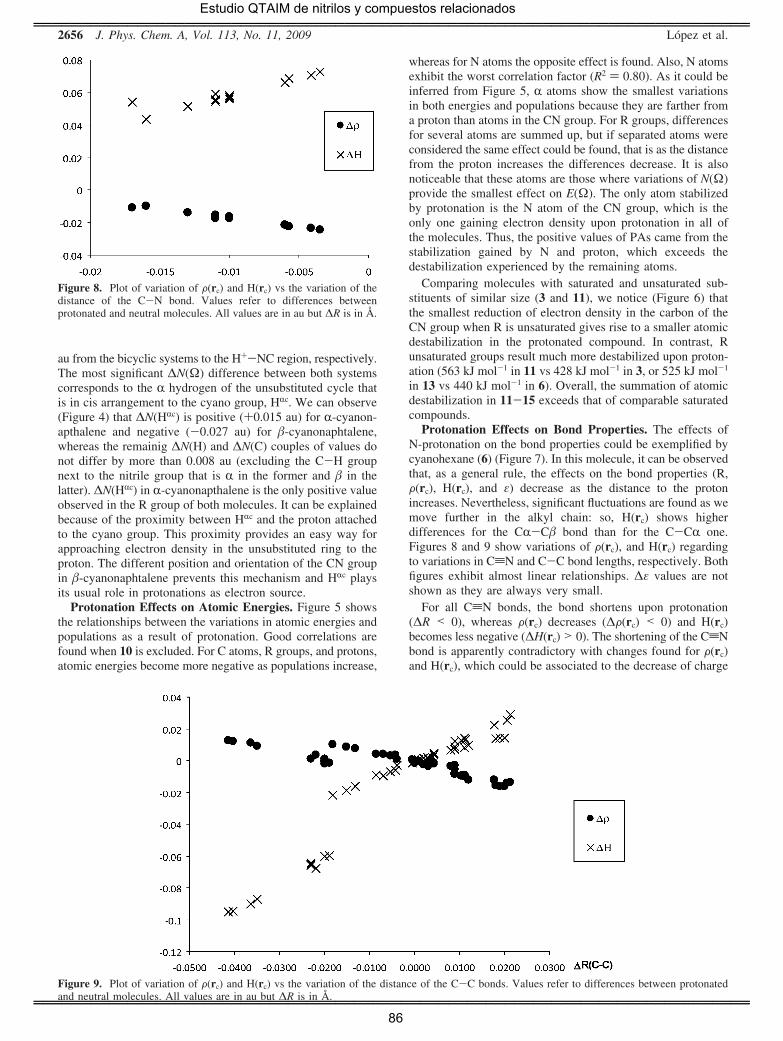
au from the bicyclic systems to the H+-NC region, respectively.The most significant ∆N(Ω) difference between both systemscorresponds to the R hydrogen of the unsubstituted cycle thatis in cis arrangement to the cyano group, HRc. We can observe(Figure 4) that ∆N(HRc) is positive (+0.015 au) for R-cyanon-apthalene and negative (-0.027 au) for -cyanonaphtalene,whereas the remainig ∆N(H) and ∆N(C) couples of values donot differ by more than 0.008 au (excluding the C-H groupnext to the nitrile group that is R in the former and in thelatter). ∆N(HRc) in R-cyanonapthalene is the only positive valueobserved in the R group of both molecules. It can be explainedbecause of the proximity between HRc and the proton attachedto the cyano group. This proximity provides an easy way forapproaching electron density in the unsubstituted ring to theproton. The different position and orientation of the CN groupin -cyanonaphtalene prevents this mechanism and HRc playsits usual role in protonations as electron source.
Protonation Effects on Atomic Energies. Figure 5 showsthe relationships between the variations in atomic energies andpopulations as a result of protonation. Good correlations arefound when 10 is excluded. For C atoms, R groups, and protons,atomic energies become more negative as populations increase,
whereas for N atoms the opposite effect is found. Also, N atomsexhibit the worst correlation factor (R2 ) 0.80). As it could beinferred from Figure 5, R atoms show the smallest variationsin both energies and populations because they are farther froma proton than atoms in the CN group. For R groups, differencesfor several atoms are summed up, but if separated atoms wereconsidered the same effect could be found, that is as the distancefrom the proton increases the differences decrease. It is alsonoticeable that these atoms are those where variations of N(Ω)provide the smallest effect on E(Ω). The only atom stabilizedby protonation is the N atom of the CN group, which is theonly one gaining electron density upon protonation in all ofthe molecules. Thus, the positive values of PAs came from thestabilization gained by N and proton, which exceeds thedestabilization experienced by the remaining atoms.
Comparing molecules with saturated and unsaturated sub-stituents of similar size (3 and 11), we notice (Figure 6) thatthe smallest reduction of electron density in the carbon of theCN group when R is unsaturated gives rise to a smaller atomicdestabilization in the protonated compound. In contrast, Runsaturated groups result much more destabilized upon proton-ation (563 kJ mol-1 in 11 vs 428 kJ mol-1 in 3, or 525 kJ mol-1
in 13 vs 440 kJ mol-1 in 6). Overall, the summation of atomicdestabilization in 11-15 exceeds that of comparable saturatedcompounds.
Protonation Effects on Bond Properties. The effects ofN-protonation on the bond properties could be exemplified bycyanohexane (6) (Figure 7). In this molecule, it can be observedthat, as a general rule, the effects on the bond properties (R,F(rc), H(rc), and ε) decrease as the distance to the protonincreases. Nevertheless, significant fluctuations are found as wemove further in the alkyl chain: so, H(rc) shows higherdifferences for the CR-C bond than for the C-CR one.Figures 8 and 9 show variations of F(rc), and H(rc) regardingto variations in CtN and C-C bond lengths, respectively. Bothfigures exhibit almost linear relationships. ∆ε values are notshown as they are always very small.
For all CtN bonds, the bond shortens upon protonation(∆R < 0), whereas F(rc) decreases (∆F(rc) < 0) and H(rc)becomes less negative (∆H(rc) > 0). The shortening of the CtNbond is apparently contradictory with changes found for F(rc)and H(rc), which could be associated to the decrease of charge
Figure 8. Plot of variation of F(rc) and H(rc) vs the variation of thedistance of the C-N bond. Values refer to differences betweenprotonated and neutral molecules. All values are in au but ∆R is in Å.
Figure 9. Plot of variation of F(rc) and H(rc) vs the variation of the distance of the C-C bonds. Values refer to differences between protonatedand neutral molecules. All values are in au but ∆R is in Å.
2656 J. Phys. Chem. A, Vol. 113, No. 11, 2009 Lopez et al.
Estudio QTAIM de nitrilos y compuestos relacionados
86

density in the bond critical point and so to the weakening ofthe bond. However, it should be taken into account that theproperties in the bond critical point only reflect what happensto σ density as the π density is out of the plane of the bond.Then, the shortening of the bond length and the changes in bondproperties upon protonation are compatible with the increaseof the π density and the decrease of the σ density in the CtNbond, which confirms the important π character of the CtNbond in protonated compounds and so the predominance of theresonance form containing a triple bond (H+-NtC-R).
From Figure 9 it could be inferred that when the bond lengthremains constant all of the properties of the BCP remain alsounchanged. This happens for C-C bonds placed further awaywithin the R group. The most negative values of ∆R correspondto the most negative ones of ∆H(rc) and to the most positiveones of ∆F(rc). So, when the bond shortens, bond propertiesreflect a strengthening of the bond, whereas the opposite happenswhen the bond lengthens.
Conclusions
After the protonation of cyanocompounds, the proton keepsa very positive charge, which is more in line with aH+-NtC-R Lewis structure than with the H-N+tC-R andH-NdC+-R ones. This is also confirmed by the increase ofthe π density and the decrease of the σ density in the CtNbond obtained from results of its bond properties.
The electron density of HCN evolves upon protonationfollowing the mechanism previously reported for other O-pro-tonations7-9,21 and N-protonations7,8,20,21 due to the deformationof the electron density in the whole molecule produced by theproton, which gives rise to electron density transferencesbetween neighboring atoms. When the H of HCN is replacedby an alkyl group the electron population lost by the C atom issignificantly reduced, due to the σ-electron density providedby the neighboring alkyl group, R. When the alkyl chainexperiences an internal rotation, the only ∆N(Ω) valuessignificantly affected are those of the group rotated. For nitrileswith π-conjugated substituents, an important reduction ofπ-electron density appears upon protonation, whereas the σ-elec-tron density remains practically unchanged as in cyanoalkanes.
Also, we have found a reasonable agreement betweenexperimental and calculated PA values as well as goodcorrelations between variations in atomic energies and popula-tions as a result of protonation.
Acknowledgment. We are indebted to “Centro de Super-computacion de Galicia” (CESGA) for access to their compu-tational facilities and to “Xunta de Galicia” and Spanish MECfor financial support through, respectively, projects C217122P-64100 and CTQ2006-15500/BQU.
References and Notes(1) Carey, F. A.; Sundberg, R. J. AdVanced Organic Chemistry; Kluwer
Academic: New York, 2001.(2) Wheland, G. W. Resonance in Organic Chemistry; Wiley: New
York, 1955.(3) Bader, R. F. W. Atoms in Molecules: A Quantum Theory; Oxford
University Press: New York, 1990.(4) Bader, R. F. W. Chem. ReV. 1991, 91, 893.(5) Wiberg, K. B.; Laidig, K. E. J. Am. Chem. Soc. 1987, 109, 5935.(6) Wiberg, K. B.; Breneman, C. M. J. Am. Chem. Soc. 1992, 114,
831.(7) Gonzalez Moa, M. J.; Mosquera, R. A. J. Phys. Chem. A 2003,
107, 5361.(8) Gonzalez Moa, M. J.; Mosquera, R. A. J. Phys. Chem. A 2005,
109, 3682.(9) Mandado, M.; Van Alsenoy, C.; Mosquera, R. A. J. Phys. Chem.
A 2004, 108, 7050.(10) Mandado, M.; Van Alsenoy, C.; Mosquera, R. A. Chem. Phys. Lett.
2005, 405, 10.(11) Glaser, R.; Choy, G. S. K. J. Am. Chem. Soc. 1993, 115, 2340.(12) Vila, A.; Mosquera, R. A. J. Phys. Chem. A 2000, 104, 12006.(13) Vila, A.; Mosquera, R. A. Chem. Phys. Lett. 2000, 332, 474.(14) Vila, A.; Mosquera, R. A. Tetrahedron 2001, 57, 9415.(15) Perrin, C. J. Am. Chem. Soc. 1991, 113, 2865.(16) Laidig, K. E. J. Am. Chem. Soc. 1992, 114, 7912.(17) Gatti, C.; Fantucci, P. J. Phys. Chem. 1993, 97, 11677.(18) Hirshfeld, F. L. Theor. Chim. Acta 1977, 44, 129.(19) De Proft, F.; Van Alsenoy, C.; Peeters, A.; Langenaker, W.;
Geerlings, P. J. Comput. Chem. 2002, 23, 1198.(20) Otero, N.; Gonzalez.Moa, M. J.; Mandado, M.; Mosquera, R. A.
Chem. Phys. Lett. 2006, 428, 249.(21) Gonzalez Moa, M. J.; Mandado, M.; Mosquera, R. A. Chem. Phys.
Lett. 2006, 428, 255.(22) Stutchbury, N. C. J.; Cooper, D. L. J. Chem. Phys. 1983, 79, 4967.(23) Alcoba, D. R.; Lain, L.; Torre, A.; Bochicchio, R. C. Chem. Phys.
Lett. 2005, 407, 379.(24) Cioslowski, J.; Mixon, S. T. Can. J. Chem. 1992, 70, 443.(25) Bader, R. F. W. J. Phys. Chem. A 1998, 102, 7314.(26) Haaland, A.; Shorokhov, D. J.; Tverdova, N. V. Chem.sEur. J.
2004, 10, 4416.(27) Poater, J.; Sola, M.; Bickelhaupt, F. M. Chem.sEur. J. 2006, 12,
2889.(28) Bader, R. F. W. Chem.sEur. J. 2006, 12, 2896.(29) Poater, J.; Sola, M.; Bickelhaupt, F. M. Chem.sEur. J. 2006, 12,
2902.(30) GAMESS: Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert,
S. T.; Gordon, M. S.; Jensen, J. H.; Koseki, S.; Matsunaga, N.; Nguyen,K. A.; Su, S. J.; Windus, T. L.; Dupuis, M.; Montgomery, J. A. J. Comput.Chem. 1993, 14, 1347–1363.
(31) Lehd, M.; Jensen, F. J. Comput. Chem. 1991, 12, 1089.(32) Cortes-Guzman, F.; Bader, R. F. W. Chem. Phys. Lett. 2003, 379,
183.(33) Mandado, M.; Vila, A.; Grana, A. M.; Mosquera, R. A.; Cioslowski,
J. Chem. Phys. Lett. 2003, 371, 739.(34) Bader, R. F. W. et al. AIMPAC: A Suite of Programs for the AIM
Theory; McMaster University: Hamilton, Ontario, Canada, L8S 4M1.Contact [email protected].
(35) Biegler-Konig, F. W.; Bader, R. F. W.; Nguyen-Dang, T. T.J. Comput. Chem. 1982, 3, 371.
(36) Aray, Y.; Murgich, J.; Luna, M. A. J. Am. Chem. Soc. 1991, 113,7135.
(37) Hunter, E. P.; Lias, S. G. J. Phys. Chem. Ref. Data 1998, 27, 413.(38) Mariott, S.; Topsom, R. D.; Lebrilla, C. B.; Koppel, I.; Mishima,
M.; Taft, R. W. J. Mol. Struct. 1986, 137, 133.(39) Grana, A. M.; Mosquera, R. A. Chem. Phys. 1999, 243, 17.
JP811023X
Protonation of Nitriles J. Phys. Chem. A, Vol. 113, No. 11, 2009 2657
Estudio QTAIM de nitrilos y compuestos relacionados
87

Estudio QTAIM de nitrilos y compuestos relacionados
88
ELECTRON DENSITY ANALYSIS ON THE ALPHA ACIDITY OF NITRILES
José Luis López, Ana M. Graña, Ricardo A. Mosquera*
Dpto. Química Física, Universidade de Vigo,
Lagoas-Marcosende, 36310-Vigo, Galicia (Spain).
Abstract
24 substituded cyanocompounds and the corresponding anions obtained upon H+-
abstraction from diverse positions were subjected to an electron density analysis with
the Quantum Theory of Atoms in Molecules (QTAIM). All the electron densities were
obtained at the B3LYP/6-31++G(2d,2p) level on completely optimized geometries. -
H+ abstraction is found as the most favored one (by at least 100 kJ mol
-1 in all the
tested compounds). The presence of additional resonance electron attractors reduces
significantly the -deprotonation energy, whereas this magnitude is quite insensitive to
the inclusion of resonance electron donors. The electron density rearrangement
accompanying the deprotonation is basically in line with the predictions of the
resonance model. Thus a significant part of the electron density gained by expelling
the proton is transferred to cyano N and to other groups where significant resonance
structures delocalize the negative charge.

Estudio QTAIM de nitrilos y compuestos relacionados
89
Introduction
Carbanions stabilized by mesomeric electron acceptor groups are a class of
compounds with a certain practical interest. In fact, they have been employed widely in
organic synthesis.1 Even, some important biochemical intermediates display this
chemical moiety.2 From a theoretical point of view, they are convenient systems for
testing the reliability of electron delocalization models. According to the resonance
model (RM), the negative charge of the C atom is expected to be delocalized on the
mesomeric electron acceptor, e.g. O in a carbonyl group or N in a cyano substituent. In
this context, we acknowledge the very good services provided by RM in Chemistry.3,4
Even, different possibilities to supply resonance structures with more reliable and
quantitative weighting coefficients obtained making use of modern tools of electron
density topological analysis can be explored.5 Nevertheless, since the publication of
the seminal paper by Wiberg and Laidig on the electronic origin of the esther and
amide resonance,6 it is not possible to deny that a large amount of inconsistencies
between RM predictions and computed evolutions of electron densities have been
reported.7-26
Most of these discrepancies where obtained studying protonation
processes or in nucleophilic addition reactions. In contrast, deprotonations seem to
have been much less explored.
This paper aims to get insight into the stabilization of carbanions by cyano
groups that is on the basis of the significant acidity displayed by the hydrogens of
methylenes (and other groups) that are α to CN units. In order to achieve this objective
we have performed an electron density analysis of neutral and deprotonated anionic
species of a series of substituted N≡CHRR’ cyanocompounds (R’=H in most of them)

Estudio QTAIM de nitrilos y compuestos relacionados
90
(Table 1). This analysis was carried out with the Quantum Theory Atoms in Molecules
(QTAIM).27,28
As an starting point, we remember the N≡C-RR’ anions are considered to be
stabilized by delocalization of the negative charge on the N atom. This is represented
by –N≡CRR’ resonance Lewis structures. In polysubstituted nitriles, delocalizations
can be extended to other atoms of R and R’ groups where similar resonance structures
could be written. Scheme 1 shows an example of them for compound N (containing an
additional π-acceptor substituent: NO2. The opposite effect, should be expected when
the additional substituent is a π-donor like -NH2 or –OH.
C NC
N
H
O
O
-C NC
N
H
O
O
--+
-N +
C N-C
N
H
O
O
-+
Scheme 1
Computational details
QTAIM allows the partitioning of a molecule into disjoint subsystems without
resorting to hypothesis alien to Quantum Mechanics.27,28
With a few exceptions,29
each
of these subsystems consists of a nucleus, which acts as an attractor for the trajectories
of the gradient of the electron density vector field, (r), and its associated atomic
basin, throughout these trajectories spread. An atom, , is defined as the union of the
attractor and its associated basin, and is surrounded by zero flux surfaces for (r).

Estudio QTAIM de nitrilos y compuestos relacionados
91
The integration of the proper density functions within these limits provides diverse
atomic properties such as the electron population, N(), or the total atomic electron
energy, E().
QTAIM also recovers main elements of molecular structure in terms of the
critical points, rc, of the electron density, (r). Prominent among them are the bond
critical points (BCPs), which are located roughly in between every pair of bonded
atoms. Although the relationship between the presence of a BCP and the existence of a
chemical bonding has become a controversial and it is still a debated point of the
theory,30-35
the electron density at a certain BCP is regarded as an indicator for bond
strength.
All the neutral (1-24) and deprotonated (1a-24a) species here considered
(Table 1) were fully optimized at the B3LYP/6-31++G(2d,2p) levels using the
Gaussian-09 program.36
Exclunding the long chain linear cyanoalkanes (14 and 15),
initial geometries were optimized for all expected conformers. The completely
antiperiplanar conformation was the only initial geometry optimized for 14 and 15.
The electron densities obtained were analyzed with the QTAIM by means of the
program AIMPAC.37
The accuracy of the integrated properties was tested using the
differences between molecular properties and those obtained by summation of the
properties of the fragments [N-N() or E-E()] (Table 1). These differences are
always smaller (in absolute value) than 2 10-3
au and 1.2 kJ/mol, respectively, which
are found to be accurate enough comparing with other works carried out at similar
theoretical levels. In the same vein, the integrated values of the Laplacian of the

Estudio QTAIM de nitrilos y compuestos relacionados
92
electron density in all the atomic fragments, L(), are always smaller (in absolute
value) than 10-3
au.
Deprotonation energies, dpE, (Table 1) were calculated taken into account the
thermal and zero point vibrational corrections (unscaled) obtained for deprotonated
and neutral species. All the optimized structures were real minima as they do not
display any imaginary frequency. When more than one local minima is present in the
neutral or anionic form o certain compound dpE is computed as the different between
the lowest energy conformer found for each species.
Results and Discussion
Atomic and bond properties of neutral nitriles, as well as the 2(r) topology, have been described thoroughly in a previous HF study by Aray et al.
38 As our results for neutral molecules are in perfect agreement with theirs, we focus our discussion on the effects of deprotonation.
Deprotonation energies.
Table 1 lists the dpE energies obtained for -deprotonation of the 24 cyanocompounds here studied. For the sake of simplicity, in what follows compound 1 (cyanomethane) will be our reference, and deprotonation energies will be commented as relative values to that computed for 1 (dpE). First we notice that, in spite of
large structural changes, deprotonation energies do not span in a wide range. It is also
noticeable that positive values are scarce. Especially when we take into account that
the most positive value corresponds to LiCH2CN (compound 8), whose neutral
optimized structure is significantly different from those of the remaining species, with
the Li atom attached to the CN group and not to the methylene, denoting its ionic
character. Thus, one [CH2CN]- is already formed in neutral 8. As a consequence
abstracting a proton from it demands the largest amount of energy and this compound
can be excluded from the series because of this singular bonding structure.

Estudio QTAIM de nitrilos y compuestos relacionados
93
Table 1. Deprotonation energies, ΔdpE (in kJ mol-1
) and accuracy estimators for
QTAIM integrations for the RR’CH-CN molecules here studiede.
R R’ ΔdpE ΔdpE N-N()b
E-E()c
|L()|d
1 H H 1549.3 0 -0.4 (0.0)f
-0.4 (-0.2)f
0.5
2 H CN 1375.6 -173.7 0.1 (0.0) 0.1 (-0.2) 0.2
3 CN CN 1228.9 -320.4 -0.6 (0.2) -0.4 (0.0) 0.1
4 CH3-O-CO H 1402.2 -147.1 0.2 (0.4) 0.1 (0.1) 0.9
5 NO2 H 1334.4 -214.9 1.2 (1.9) 0.7 (1.2) 0.7
6 OH H 1547.3 -2 -0.3 (1.2) -0.2 (0.7) 0.8
7 NH2 H 1552.2 2.9 -1.5 (-0.3) -0.9 (-0.4) 0.1
8 Li H 1660.7 111.4 -0.1 (0.4) -0.2 (0.0) 0.7
9 SiH3 H 1472.0 -77.3 1.0 (0.2) 0.5 (-0.2) 0.9
10 F H 1526.6 -22.7 0.3 (0.7) 0.0 (0.2) 0.5
11 F F 1503.4 -45.9 -0.6 (0.2) -0.8 (0.5) 1.0
12 CH3 H 1558.8 9.5 1.0 (-0.6) -1.0 (-0.6) 0.9
13 CH3 CH3 1556.6 7.3 2.0 (0.8) 1.0 (0.1) 0.9
14 CH3(CH2)8 H 1548.0 -1.3 -0.6(0.1) -0.5(0.2) 0.4
15 CH3(CH2)9 H 1548.2 -1.1 -0.1(0.4) -0.6(0.7) 1.3
16a CH2=CH-CN - 1548.4 -0.9 -0.3 (1.2) -0.2 (0.7) 0.8
17 CH2=CH H 1458.0 -91.3 -1.5 (-0.3) -0.9 (-0.4) 0.1
18 C6H5 H 1444.2 -105.1 -0.1 (0.4) -0.2 (0.0) 0.7
19 p-NO2C6H4 H 1342.6 -206.7 0.1 (0.0) 0.1 (-0.2) 0.2
20 p-NH2C6H4 H 1469.5 -79.8 -0.6 (0.2) -0.4 (0.0) 0.1
21 m-NO2C6H4 H 1388.3 -161 -1.5 (-0.3) -0.9 (-0.4) 0.1
22 m-NH2C6H4 H 1453.5 -95.8 -0.6 (0.2) -0.8 (0.5) 1.0
23 o-NO2C6H4 H 1363.0 -186.3 -0.3 (1.2) -0.2 (0.7) 0.8
24 o-NH2C6H4 H 1455.5 -93.8 0.2 (0.4) 0.1 (0.1) 0.9 aThis compound, CH2=CH-CN, does not follow the general RR’CH-CN formula.
bValues in au multiplied by 10
3.
cin kJ mol
-1.
dMaximum absolute value of integrated L() in the neutral molecule and its protonated
species, in au multiplied by 103.
eValues for protonated species in parenthesis.
The other positive dpE values do not exceed 10 kJ mol-1
(7, 12 and 13). 12 and 13
correspond to other short alkyl chains (cyanoethane and cyano-iso-propane). The small
difference between them (Table 1) leads us to think that chain ramifications are not
Estudio QTAIM de nitrilos y compuestos relacionados
94
significant to this problem. The effect of chain size is even smaller for larger alkyl
groups (14 and 15), becoming negligible.
7 should be compared with the other resonance electron donor containing compound (+R) here considered: 6. Both values are really close (slightly positive one and slightly negative the other). Thus we conclude that the inclusion of +R substituents does not really modify dpE.
-20
0
20
40
60
80
100
120
140
160
180
0 1 2 3 4 5 6 7 8 9 10 11
14 14(t)
15 15(t)
Figure 1. Relative values (in kJ mol-1
) of deprotonation energies (dpE) vs. H+-
abstraction position in compounds 14 and 15.
In contrast, significantly negative dpE values are displayed by those compounds (2-5) that include additional (CN being also one of them) resonance electron withdrawers (-R). According to the RM these compounds allow a larger delocalization of the formal negative charge formed at the C (scheme 1). A similar mechanism can be
considered for π-conjugated substituents, such as vinyl (17) of phenyl (parent, 18, or
susbtitued, 19-24) groups. In fact all these compounds (2-5, 17-24) reduce the
deprotonation energy by more than 90 kJ mol-1
with regard to the reference (1).
We have also checked that -deprotonations are preferred over other possible processes for H+-abstractions. To this end we computed all possible H-abstractions along the alkyl chain of 14 and 15 (Figure 1). Other deprotonations cost at least 100 kJ mol
-1 more than the alpha one. Moreover the only significant difference between both
points is due to the displacement (by one position) of the terminal and previous to
terminal methyl or methylene group.

Estudio QTAIM de nitrilos y compuestos relacionados
95
Proton abstraction effects on atomic electron populations and bond properties in
cyanomethane.
As in the previous section, we will refer to compound 1 as our basic model to
describe the electron density change due to -deprotonation. The variations
experienced by its atomic electron populations, N(Ω), in the process are shown in
Figure 2. All the atoms increase their electron population after expelling the proton,
which means sharing 0.948 au. Whereas a little more than one half of the electron
density is kept within the CH2 unit, the electron density taken by the cyano group is
important, and there is an important transference of electron density to its N atom.
C NC
H
H 217
151
343
118
118
Figure 2. Variations of atomic electron populations in 1 upon -deprotonation,
dpN(Ω), (in au multiplied by 103).
C NC
H
H
-26.50.042 -83
+33.40.296 +87
Figure 3. Variations of most significant BCP properties in 1 upon -deprotonation:
Relative electron density values (in au and bolface) are multiplied by 103, absolute
values of ellipticities (in italics) and relative values of the total electronic energy
density (multiplied by 103).

Estudio QTAIM de nitrilos y compuestos relacionados
96
In the same vein the evolutions of BCPs properties is in line with the
predictions of the RM model. Thus, we notice (Figure 3) the C-C bond is reinforced
while the C-N linkage gets weaker. At the same time the first bond shrinks by 0.08 Å,
while the later lengths by 0.03 Å. More meaningful, both bond ellipticities that are
perfectly null in the neutral compound become 0.296 and 0.042 in the anion. Finally
the values of the total energy density function become more negative for C-C in the
anion and less negative for the C-N likange, pointing to a reinforcement of covalent
character in the former and to its depletion towards a significant polarization in the
latter.
Conclusions
QTAIM analysis of the electron densities of 24 substituded cyanocompounds
and the corresponding anions obtained upon H+-abstraction allowed us to establish the
following conclusions: -deprotonation is at least favored by 100 kJ mol-1
with regard
to other deprotonation processes. While resonance electron attractors reduce
significantly the energy involved in the process, the effect of resonance electron donors
is nearly negligible. Deprotonation involves a significant variation of atomic electron
populations. Whereas hydrogen atoms are involved in this rearrangement, the role they
play is not so important as that in protonation. In contrast, those atoms where the
resonance model predicts significant delocalizations of the negative charge gain an
important part of the electron density left by the hydrogen.
Acknowledgements We are indebted to “Centro de Supercomputación de Galicia”
(CESGA) for access to their computational facilities.

Estudio QTAIM de nitrilos y compuestos relacionados
97
References
(1) Trost, B. M., Ed. Comprehensive Organic Synthesis, Vol. 3, Carbon-Carbon u-
Bond Formation; Pergamon Press: Oxford, 1991.
(2) Richard, J. P. In The Chemistry of Enols; Rappoport, Z., Ed.; Wiley: New York,
1990.
(3) Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry; Kluwer Academic:
New York, 2001.
(4) Wheland, G.W. Resonance in Organic Chemistry; Wiley: New York, 1955.
(5) Ferro-Costas, D., Mosquera, R. A. Phys. Chem. Chem. Phys. 2015, 17, 7424.
(6) Wiberg, K.B.; Laidig, K.E. J. Am. Chem. Soc. 1987, 109, 5935-5943.
(7) Wiberg, K.B.; Breneman, C.M. J. Am. Chem. Soc. 1992, 114, 831.
(8) Slee, T.S.; MacDougall, P.J. Can. J. Chem. 1988, 66, 2961.
(9) Laidig, K.E. J. Am. Chem. Soc. 1992, 114, 7912.
(10)Laidig, K.E.; Cameron, L.M. J. Am. Chem. Soc. 1996, 118, 1737.
(11)Glaser, R. J. Phys. Chem. 1989, 93, 7993.
(12)Glaser, R.; Choy, G.S.-C. J. Am. Chem. Soc. 1993, 115, 2340.
(13)Speers, P.; Laidig, K.E.; Streitwieser, J. Am. Chem. Soc. 1994, 116, 9257.
(14)Vila, A.; Mosquera, R.A. Tetrahedron 2001, 57, 9415.
(15)González Moa, M.J.; Mosquera, R.A. J. Phys. Chem. A 2003, 107, 5361.
(16)González Moa, M.J.; Mosquera, R.A. J. Phys. Chem. A 2005, 109, 3682.
(17)González Moa, M.J.; Mandado, M.; Mosquera, R.A. Chem. Phys. Lett. 2006, 428,
255.
(18)López, J.L.; Graña, A.M.; Mosquera, R.A. J. Phys. Chem. A 2009, 113, 2652-
2657.
(19)Mandado, M.; Van Alsenoy, C.; Mosquera, R.A. J. Phys. Chem. A 2004, 108,
7050.
(20)Graña, A.M.; Hermida-Ramón, J.M.; Mosquera, R.A. Chem. Phys. Lett. 2005,
412, 106.
(21)Mandado, M.; Mosquera, R.A.; Graña, A.M. Chem. Phys. Lett. 2004, 386, 454.

Estudio QTAIM de nitrilos y compuestos relacionados
98
(22)Mandado, M.; Van Alsenoy, C.; Mosquera, R.A. Chem. Phys. Lett. 2005, 405, 10.
(23)Mandado, M.; Van Alsenoy, C.; Mosquera, R.A. J. Phys. Chem. A 2005, 109,
8624.
(24)Otero, N.; González Moa, M. J.; Mandado, M.; Mosquera, R. A Chem. Phys. Lett.
2006, 428, 249.
(25)Otero, N.; Estévez, L.; Mandado, M.; Mosquera, R. A. Eur. J. Org. Chem. 2012,
12, 2403.
(26)Estévez, L.; Mosquera, R. A. Chem. Phys. Lett. 2008, 451, 121.
(27)Bader, R. F. W. Atoms in Molecules: A Quantum Theory Oxford University Press:
New York, 1990.
(28)Bader, R. F. W. Chem. Rev. 1991, 91, 893.
(29) Alcoba, D. R.; Lain, L.; Torre, A.; Bochicchio, R. C. Chem. Phys. Lett. 2005, 407,
379.
(30) Cioslowski, J.; Mixon, S. T. Can. J. Chem. 1992, 70, 443.
(31) Bader, R. F. W. J. Phys. Chem. A 1998, 102, 7314.
(32) Haaland, A.; Shorokhov, D. J.; Tverdova, N. V. Chem. Eur. J. 2004, 10, 4416.
(33) Poater, J.; Solà, M.; Bickelhaupt, F. M. Chem. Eur. J. 2006, 12, 2889.
(34) Bader, R. F. W. Chem. Eur. J. 2006,12, 2896.
(35) Poater, J.; Solà, M.; Bickelhaupt, F. M. Chem. Eur. J. 2006, 12, 2902.
(36) G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R.
Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M.
Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L.
Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T.
Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E.
Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N.
Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S.
S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B.
Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev,
A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V.
G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,

Estudio QTAIM de nitrilos y compuestos relacionados
99
Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc.,
Wallingford CT, 2009.
(37) Bader, R. F. W. et al. AIMPAC: A suite of programs for the AIM theory; Mc
Master University, Hamilton, Ontario, Canada L8S 4M1. Contact
(38) Aray, Y.; Murgich, J.; Luna, M. A. J. Am. Chem. Soc. 1991, 113, 7135.
(39) Stutchbury, N. C. J.; Cooper, D. L. J. Chem. Phys. 1983, 79, 4967.

Estudio QTAIM de nitrilos y compuestos relacionados
100
QTAIM STUDY OF REARRANGEMENT REACTIONS IN NITROGENATED
COMPOUNDS
José Luis López, Ricardo A. Mosquera, Ana M. Graña*
Dpto. Química Física, Universidade de Vigo,
Lagoas-Marcosende, 36310-Vigo, Galicia (Spain)
ABSTRACT
The variations of the geometries, the electron density and atomic charges along the
reaction path of rearrangement reactions were studied by B3LYP/6-311++G**
methods. The reaction paths were studied for the Curtius rearrangement of
formaldehyde oxime, the step of rearrangement of the Hofmann reaction of
acetamide and the step of rearrangement of the Beckmann reaction of propanone
oxime. The atomic and bond properties for the minima, the transition states and
selected points along the reaction coordinated were analysed, including atomic and
bond properties obtained from QTAIM analysis of the electron density. The results
show similar patterns for all the three reactions: the new bonds are built when the old
ones break, after the reactions go through the transition state. Regarding to the
electron transfer during the reaction, a similar behaviour is found for the Curtius and
the Beckmann rearrangements: only the atoms involved in the migration (the atom
that moves and the two atoms bonded to it in the reagent and the product) exhibit
large variation of charge along the path, i.e., the electron charge transfer happen
among them. For the Hofmann reaction the Br atom is also involved in the charge
transfer, because is the atom which moves away as an anion. The study of the
reagent, transition state and product structures reveals that the usual resonance forms
representing the geometries do not correspond to the obtained results for bond
lengths and atomic charges.
KEYWORDS: QTAIM; Density functional calculations; Electron density analysis;
Reaction mechanisms

Estudio QTAIM de nitrilos y compuestos relacionados
101
INTRODUCTION
Rearrangement reactions are an important group of reactions in which an atom or a
bond moves from a site in the reagent to a different site in the product. These
reactions may occur in a concerted manner or through a step-wise mechanism.
In this paper, we have studied three different rearrangement reactions: Curtius
rearrangement of formaldehyde oxime, the step of rearrangement of the Hofmann
reaction of acetamide and the step of rearrangement of the Beckmann reaction of
propanone oxime. From the mechanism of these reactions we have selected the step
which involves the migration of one atom in order to study how the geometries and
the different atomic and bond properties, specifically atomic charges, change along
the reaction path.
The Curtius rearrangement[1]
allows us to obtain isocyanates from acyl-azides:
Scheme 1
The mechanism for this reaction remained unknown for years. The main point in
discussion was if the reaction takes place in one stage or if it occurs in several steps
with acylnitrenes (RC(O)N:) as intermediates.
Different theoretical studies [2]
[3-6]
[4]
[5]
[6]
[7]
[8]
on aryl and acyl azides show that the
syn conformers with respect to the C-N bond are more stable than the anti ones and
that for syn conformers Curtius rearrangement occurs in one stage. The barriers for

Estudio QTAIM de nitrilos y compuestos relacionados
102
syn- anti isomerization achieve 7-9 kcal mol-1
whereas the barriers for the
transformation of syn compounds into isocyanates are considerably lower than the
barriers corresponding to the rearrangement of anti compounds into isocyanates. For
this reason the Curtius reaction occurs by a concerted mechanism as shown in
Scheme I.
The Hofmann rearrangement[9]
allows us to obtain amines from amides following a
multi-stage mechanism shown in Scheme 2. One of the steps in the mechanism
involves the migration of a methyl group in a similar way that in the Curtius reaction.
This step appears in scheme into a box. To the best of our knowledge the only
theoretical study[10]
of this reaction was performed into a study about the mechanism
of the synthesis of oxazolidines, which includes a Hofmann rearrangement as the
most important step of the reaction. This study does not include the evolution of the
reaction and the obtained value for the energy barrier (ΔG at 298 K is 123.4 kJ mol-1
,
from DFT calculations with chloroform as a solvent) is not comparable to our results
as the size and structure of the molecules are very different.
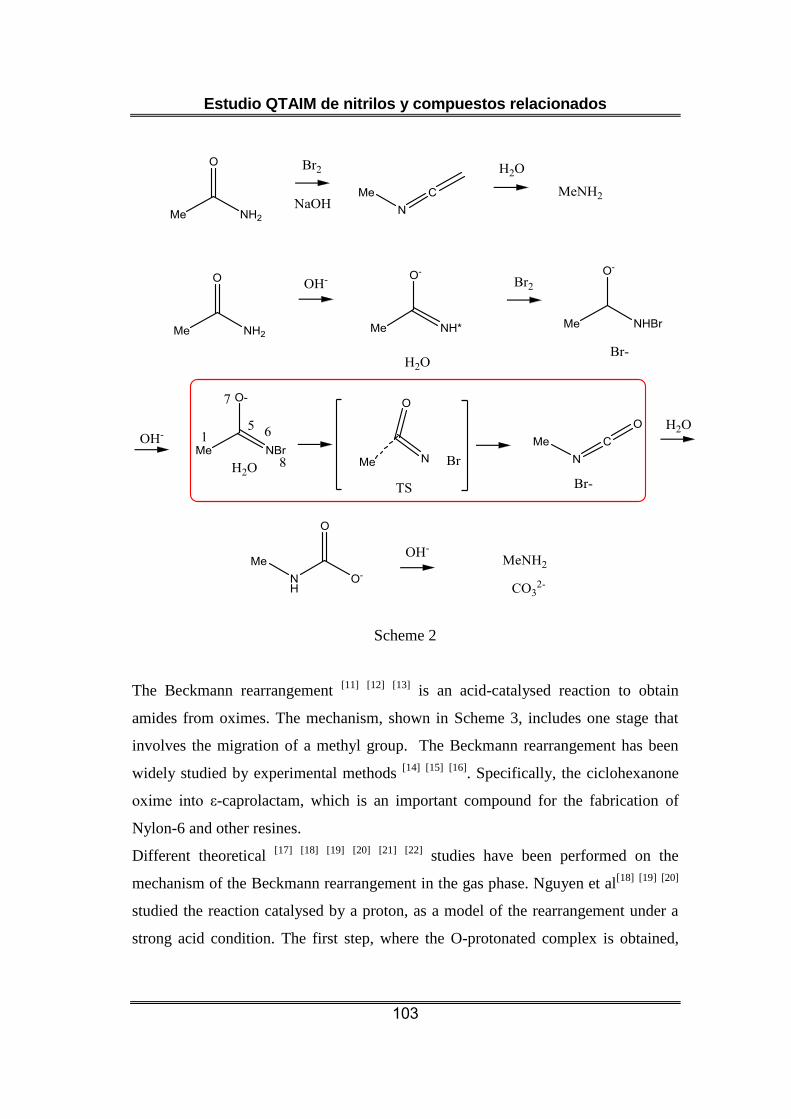
Estudio QTAIM de nitrilos y compuestos relacionados
103
Scheme 2
The Beckmann rearrangement [11]
[12]
[13]
is an acid-catalysed reaction to obtain
amides from oximes. The mechanism, shown in Scheme 3, includes one stage that
involves the migration of a methyl group. The Beckmann rearrangement has been
widely studied by experimental methods [14]
[15]
[16]
. Specifically, the ciclohexanone
oxime into ε-caprolactam, which is an important compound for the fabrication of
Nylon-6 and other resines.
Different theoretical [17]
[18]
[19]
[20]
[21]
[22]
studies have been performed on the
mechanism of the Beckmann rearrangement in the gas phase. Nguyen et al[18]
[19]
[20]
studied the reaction catalysed by a proton, as a model of the rearrangement under a
strong acid condition. The first step, where the O-protonated complex is obtained,

Estudio QTAIM de nitrilos y compuestos relacionados
104
was found to be the rate-limiting step. An important number of studies have been
performed [23]
[17]
by using different heterogeneous catalyst for this reaction.
Scheme 3
COMPUTATIONAL METHODS
All calculations were performed by using B3LYP method in Gaussian09 [24]
with 6-
311++G** basis set. For the three reactions, intrinsic coordinate (IRC) calculations
were carried out. All the optimized structures were characterized as minima in the
frequencies calculation The wavefunctions were obtained for reagents, products,
transition states and selected points of the path. On these wavefunctions we
performed QTAIM[25]
[26]
topological electron density analysis by using the
AIMPAC package[27]
of programs in order to obtain bond and atomic properties.
The value of the electron density at the bond critical points, (rc ) are employed as an
indicator for bond strength. Electron populations, N(Ω), were calculated by
numerical integration of the respective density function. The absolute values
achieved for the integrated values of the laplacian of the electron density in all the
atomic fragments, L(Ω), were smaller than 1.0∙10-3
au. The differences between total
electron population and that obtained by summation of properties of the fragments
[N-ΣN(Ω)], were always smaller (in absolute value) than 2.0∙10-3
au.
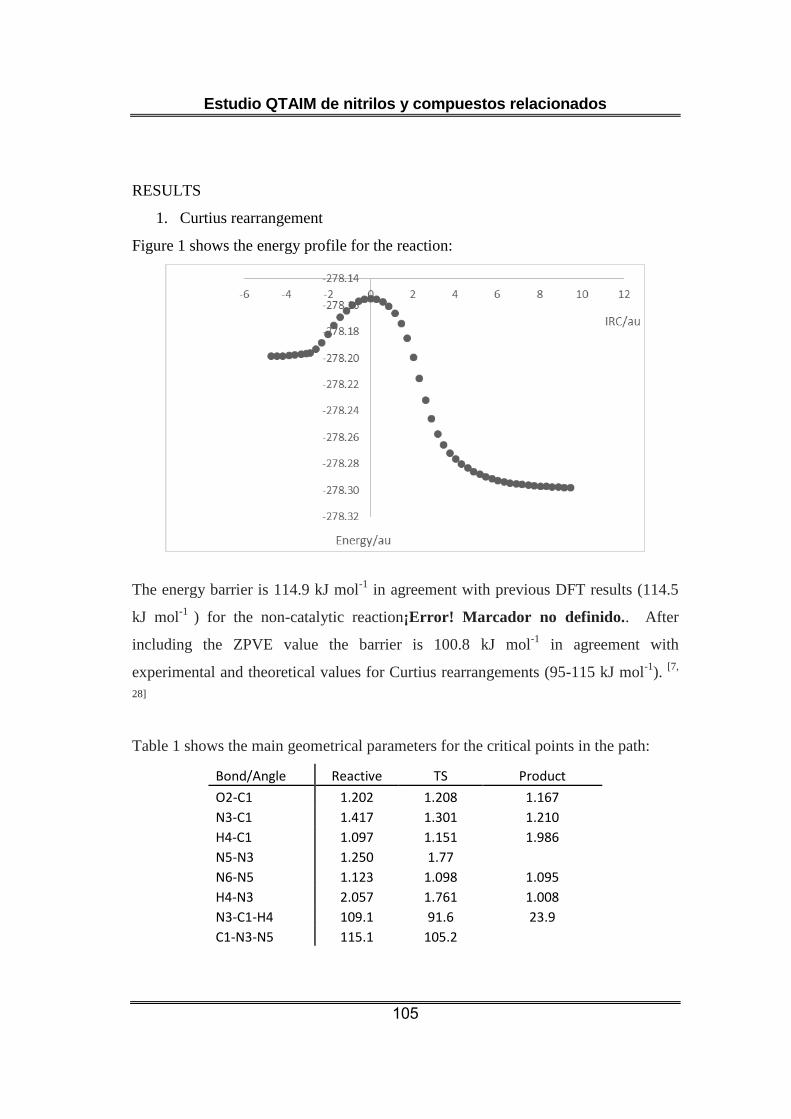
Estudio QTAIM de nitrilos y compuestos relacionados
105
RESULTS
1. Curtius rearrangement
Figure 1 shows the energy profile for the reaction:
The energy barrier is 114.9 kJ mol-1
in agreement with previous DFT results (114.5
kJ mol-1
) for the non-catalytic reaction¡Error! Marcador no definido.. After
including the ZPVE value the barrier is 100.8 kJ mol-1
in agreement with
experimental and theoretical values for Curtius rearrangements (95-115 kJ mol-1
). [7,
28]
Table 1 shows the main geometrical parameters for the critical points in the path:
Bond/Angle Reactive TS Product
O2-C1 1.202 1.208 1.167
N3-C1 1.417 1.301 1.210
H4-C1 1.097 1.151 1.986
N5-N3 1.250 1.77
N6-N5 1.123 1.098 1.095
H4-N3 2.057 1.761 1.008
N3-C1-H4 109.1 91.6 23.9
C1-N3-N5 115.1 105.2

Estudio QTAIM de nitrilos y compuestos relacionados
106
Table 1. Main geometrical parameters. Distances in Å and angles in degrees.
The geometric parameters are in good agreement with the those obtained from both
previous theoretical and experimental studies [29]
[30]
[31]
[32]
. For the reagent,
different bond distances are found from N5 to N3 and N6. The N5-N6 distance is
closer to the distance in N2 while the N3-N5 length is larger. From these values it
seems that the resonance form in Scheme 1 showing two double bonds could not
represent the structure of the molecule. It is confirmed when the values of the atomic
charges for N5 and N6 are considered (-0.099 and 0.186 au respectively). They are
small values for the charges which do not agree with the partial charges in the
proposed resonance form.
In the transition state the H4 atom is bonded to C1 and N3, taking into account the
existence of a bond critical point in both cases. The bond to C1 is very close to that
in the reactive, increasing 0.054 Å the reagent value. The bond to N3 is also closer to
that in the reagent but the difference is 0.293 Å. The N5-N6 distance is in the
transition state very similar to that in N2 molecule. The geometry for the transition
state, with small angles involving C1, causes the high energy barrier of the reaction.
Figure 2 exhibits the variations of the main bonds involved in the reaction: C1-H4,
N3-H4 and N3-N5 considering the length of the bonds and the value of the electron
density in the critical point.

Estudio QTAIM de nitrilos y compuestos relacionados
107
Figure 2
The C1-H4 distance increases along the reaction path showing the most important
variation in the central part of the path, after the transition state. The electron density
decreases as the distance increases and a critical point for the electron density of the
bond, is found after the transition state until a value about 0.87 Å. The N3-H4
distance increases during the reaction and a critical point for the electron density
appears after the transition state (for distances shorter than 1.24 Å). There is a point
where he distances from H4 to C1 and N3 arise the same value. In that point there is
a bond to C1 (considering the existence of a critical point for the electron density)
but there is not a bond to N3. The N3-N5 bond becomes larger and a critical point for
ρ, is found along the whole path, but the values are smaller than 0.1 au for lengths
larger than 1.91 Å.
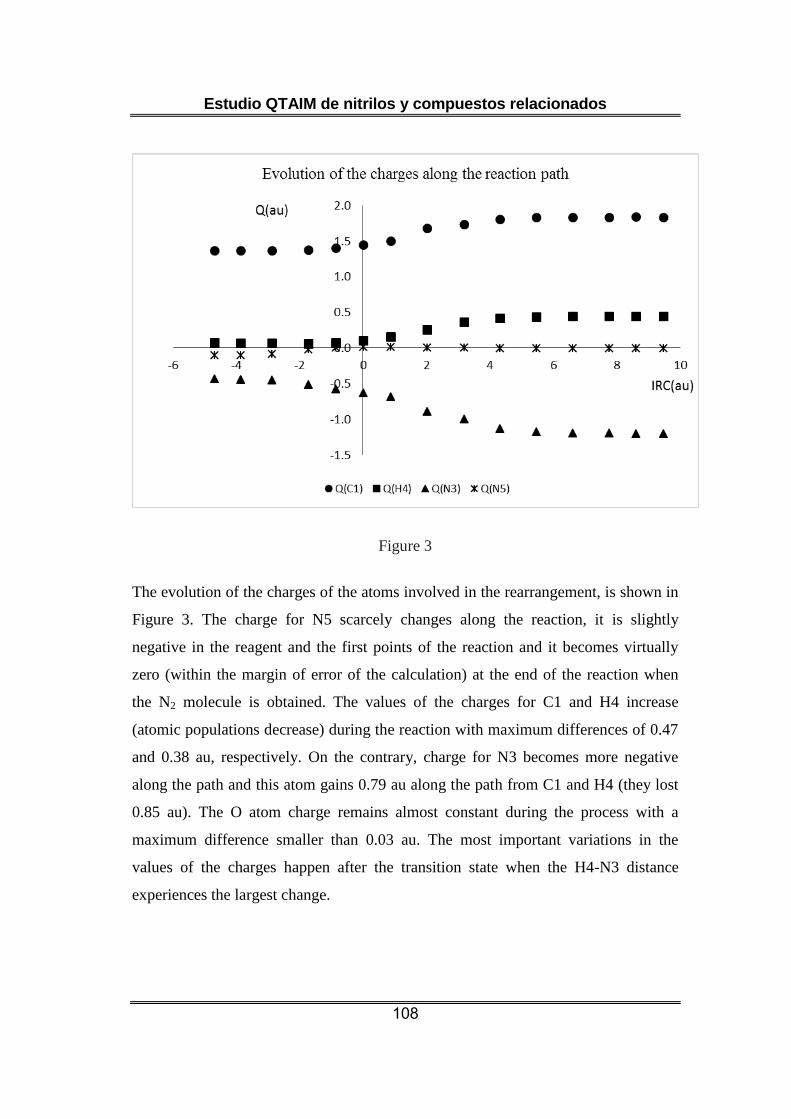
Estudio QTAIM de nitrilos y compuestos relacionados
108
Figure 3
The evolution of the charges of the atoms involved in the rearrangement, is shown in
Figure 3. The charge for N5 scarcely changes along the reaction, it is slightly
negative in the reagent and the first points of the reaction and it becomes virtually
zero (within the margin of error of the calculation) at the end of the reaction when
the N2 molecule is obtained. The values of the charges for C1 and H4 increase
(atomic populations decrease) during the reaction with maximum differences of 0.47
and 0.38 au, respectively. On the contrary, charge for N3 becomes more negative
along the path and this atom gains 0.79 au along the path from C1 and H4 (they lost
0.85 au). The O atom charge remains almost constant during the process with a
maximum difference smaller than 0.03 au. The most important variations in the
values of the charges happen after the transition state when the H4-N3 distance
experiences the largest change.
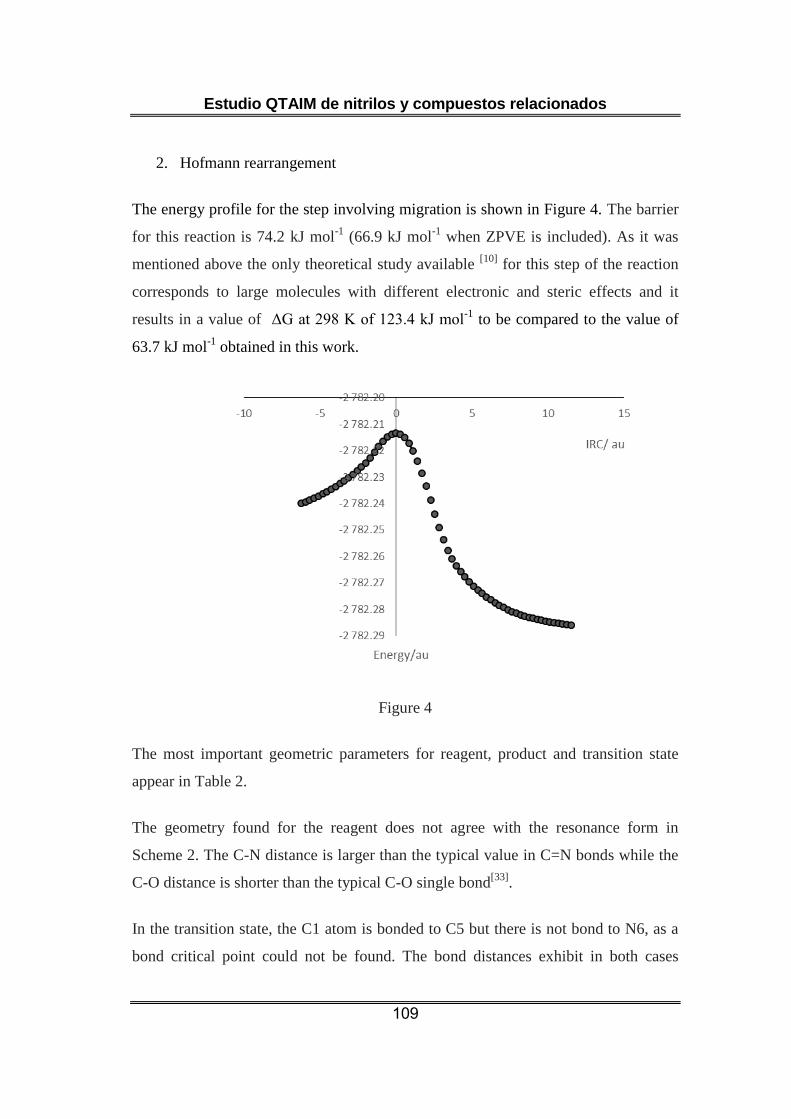
Estudio QTAIM de nitrilos y compuestos relacionados
109
2. Hofmann rearrangement
The energy profile for the step involving migration is shown in Figure 4. The barrier
for this reaction is 74.2 kJ mol-1
(66.9 kJ mol-1
when ZPVE is included). As it was
mentioned above the only theoretical study available [10]
for this step of the reaction
corresponds to large molecules with different electronic and steric effects and it
results in a value of ΔG at 298 K of 123.4 kJ mol-1
to be compared to the value of
63.7 kJ mol-1
obtained in this work.
Figure 4
The most important geometric parameters for reagent, product and transition state
appear in Table 2.
The geometry found for the reagent does not agree with the resonance form in
Scheme 2. The C-N distance is larger than the typical value in C=N bonds while the
C-O distance is shorter than the typical C-O single bond[33]
.
In the transition state, the C1 atom is bonded to C5 but there is not bond to N6, as a
bond critical point could not be found. The bond distances exhibit in both cases

Estudio QTAIM de nitrilos y compuestos relacionados
110
intermediate vales between reagent and product, but they are closer to the value in
the reagent. The distance between O atom and C5 as well as the distance between N6
and C5 show smaller differences but they are in the transition state closer to those in
the product of the reaction.
Bond/Angle Reactant TS Product
C5-C1 1.550 1.844 2.540
N6-C1 2.336 2.021 1,456
O7-C5 1.246 1.199 1.186
N6-C5 1.328 1.247 1.188
Br8-N6 1.985 2.591
C1-C5-N6 108.2 79.1 17.9
N6-C5-O7 133.7 162.4 174.3
Table 2. Main geometrical parameters. Distances in Å and angles in degrees.
The evolution of the main distances between atoms and the electron density in the
bond critical point, is shown in Figure 5.
The distance between Br and C5 increases almost linearly from a standard value for
bonded atoms. The bond critical point is not found when the distance is higher than
2.27 Å. In the transition state, the C1 atom is bonded to the C5 but there is no bond
to N6 atom. The bond critical point for C1-N6 appears when the one for C1-C5
disappears, so in the point where both distances equals, there is a bond between C1
and C5 but there is not a bond between C1 and N6.
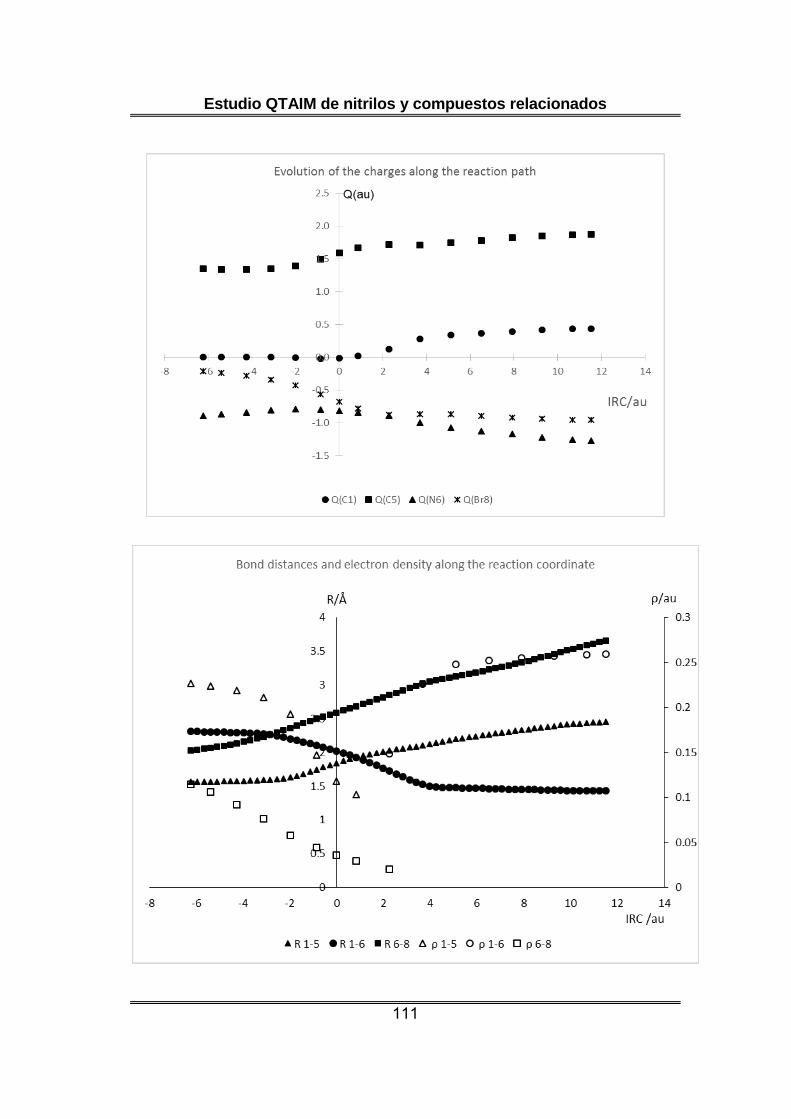
Estudio QTAIM de nitrilos y compuestos relacionados
111

Estudio QTAIM de nitrilos y compuestos relacionados
112
Figure 5
Figure 6
Figure 6 shows the variation of the atomic charges for the most important atoms in
the molecules. The charges for oxygen and hydrogen atoms remain almost constant
during the reaction with maximum differences smaller than 0.1 au for oxygen and
0.05 au for hydrogen atoms. The value for Q(C1) changes after the transition state:
from the reagent to the transition state changes less than 0.02 au and from the
transition state to the product the variation is 0.39 au. A similar pattern is found for
the evolution of the charge of N6, with a difference between the value in the
transition state and the value in the product of -0.51 au. It suggest a charge transfer
between these atoms in this stage of the reaction. The results show a continuous
decrease of the charge of C5 from the reagent to the product with an intermediate
values in the transition state. The Br atomic charge becomes gradually more negative
although the change is slightly more important before the transition state. After the
transition state C5 charge decreases 0.24 au (becomes more positive) and Br charge
increases 0.28 au (becomes more negative). So, the electron transfer happen from C5
to Br through N6 and from C1 to N6. The negative atomic charge of the oxygen atom
decreases during the reaction less than 0.1 au showing values between -1.22 and -
1.13 au from reagent to product. So, the differences in the charge of this atom in
reagent, transition state and product structures confirm the results for the C-O bond
length and show that the resonance forms in Scheme 2 are not representative of the
structures of the molecules.
3. Beckmann rearrangement
Figure 7 shows the value of the energy along the reaction path for this reaction. The
value of the barrier for this step of the reaction is 23.7 kJ mol-1
(12.5 kJ mol-1
taking
into account the ZPVE correction). It is lower than that found from DFT
calculations[18]
for formaldehyde oxime (44 kJ mol-1
) but higher to those found Chu
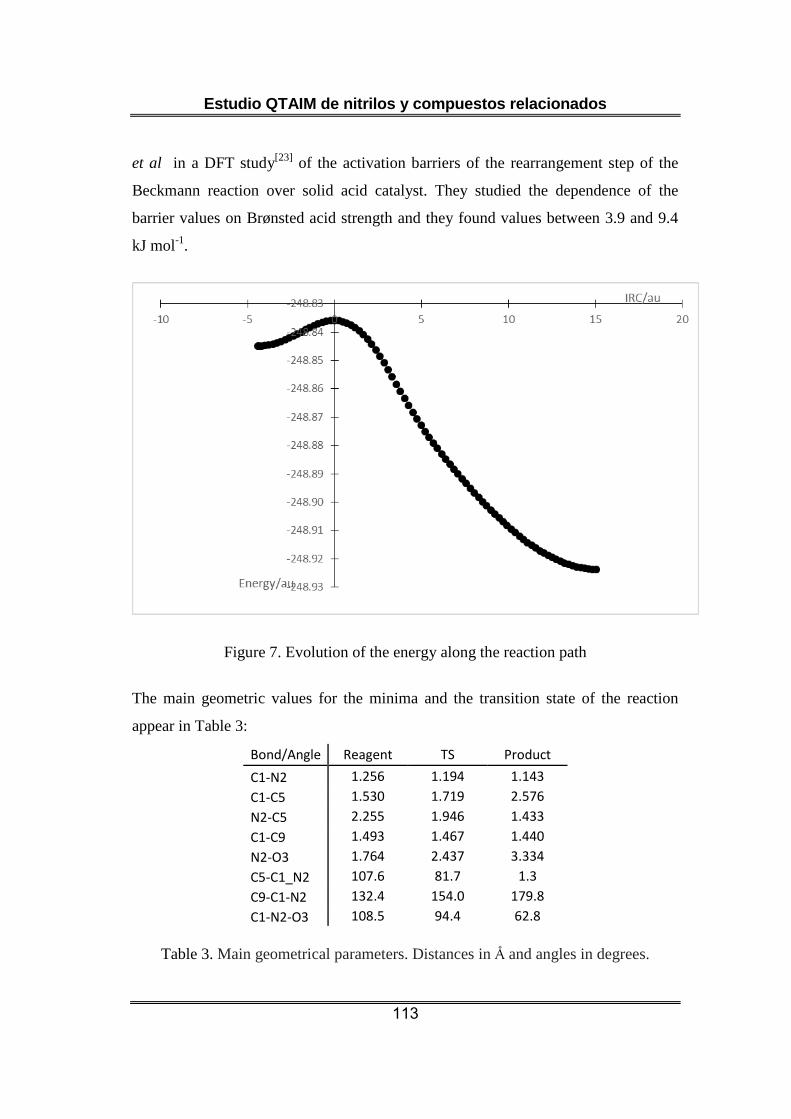
Estudio QTAIM de nitrilos y compuestos relacionados
113
et al in a DFT study[23]
of the activation barriers of the rearrangement step of the
Beckmann reaction over solid acid catalyst. They studied the dependence of the
barrier values on Brønsted acid strength and they found values between 3.9 and 9.4
kJ mol-1
.
Figure 7. Evolution of the energy along the reaction path
The main geometric values for the minima and the transition state of the reaction
appear in Table 3:
Bond/Angle Reagent TS Product
C1-N2 1.256 1.194 1.143
C1-C5 1.530 1.719 2.576
N2-C5 2.255 1.946 1.433
C1-C9 1.493 1.467 1.440
N2-O3 1.764 2.437 3.334
C5-C1_N2 107.6 81.7 1.3
C9-C1-N2 132.4 154.0 179.8
C1-N2-O3 108.5 94.4 62.8
Table 3. Main geometrical parameters. Distances in Å and angles in degrees.

Estudio QTAIM de nitrilos y compuestos relacionados
114
The reagent exhibits a CN bond with a C=N typical value, whereas in the product the
CN bond is closer to the experimental (1.157 Å) or to the DFT calculated (1.149 Å)
value of CN in acetonitrile. So, it could be inferred that the most important resonance
structure in the product should be that with a CN triple bond. However, when the
values of the atomic charges are analysed for this structure they are not compatible
with the expected positive charge on the N atom. So, the value for Q(C1) is +1.05 au
and the value for Q(N2) is -1.29 au. Although there is a hydrogen bond between the
oxygen of the water molecule and one of the H atoms bonded to C9, the charge for
the H2O molecule is 0.01 au and the charge for C9 is 0.09 au. However, the charge of
all the hydrogen atoms bonded to C5 and C9 is larger than 0.1 au and the total charge
of the methyl group bonded to N atom is 0.72 au. These results agree with those
obtained for the resonance forms and atomic charges in CN-protonated molecules [34]
with structures more compatible with a H+-N≡C-R Lewis structure than with the H-
N+≡C-R and H-N=C
+-R ones.
The main parameters of the geometry of the transition state show values intermediate
between the values in the reagent and the product, except for C1-N2 bond and C5-
C1-N2 angle with values closer to those in the reactive.
Figure 8 shows the evolution of the main bond distances during the reaction as well
as the value for the electron density in the bond critical point. The bond length
between C1 and C5 decreases whereas the distance between N2 andC5 increases by a
similar amount. The bond critical point for N2-C5 bond appears when the ones for
C1-C5 and N2-O3 disappear, after the transition state for the reaction. At the point
where the C1-C5 and N2-C5 arises the same value C5 is bonded to C1 but it is not
bonded to N2. The N2-C3 bond is always a weak bond with values of (rc) in the
bond critical point less than 0.12 au. The lowest values of (rc) for C1-C5 and N2-C5
in Figure 8 are larger than the highest value for N2-C3.
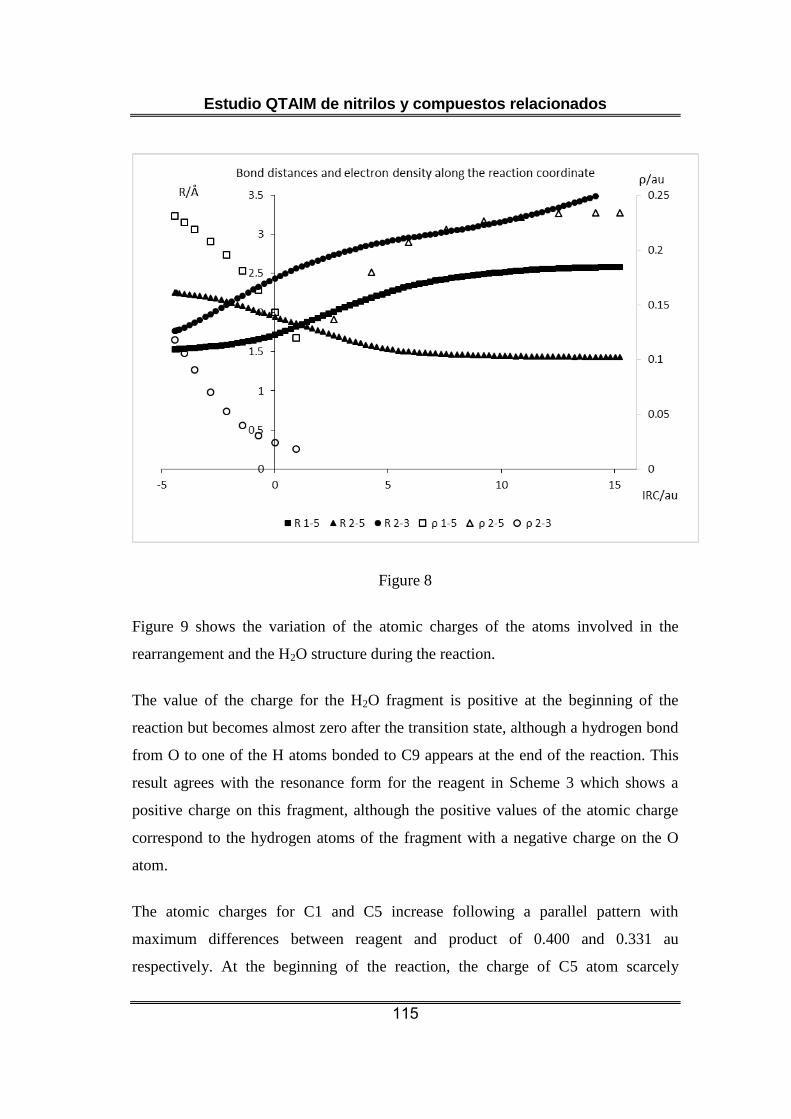
Estudio QTAIM de nitrilos y compuestos relacionados
115
Figure 8
Figure 9 shows the variation of the atomic charges of the atoms involved in the
rearrangement and the H2O structure during the reaction.
The value of the charge for the H2O fragment is positive at the beginning of the
reaction but becomes almost zero after the transition state, although a hydrogen bond
from O to one of the H atoms bonded to C9 appears at the end of the reaction. This
result agrees with the resonance form for the reagent in Scheme 3 which shows a
positive charge on this fragment, although the positive values of the atomic charge
correspond to the hydrogen atoms of the fragment with a negative charge on the O
atom.
The atomic charges for C1 and C5 increase following a parallel pattern with
maximum differences between reagent and product of 0.400 and 0.331 au
respectively. At the beginning of the reaction, the charge of C5 atom scarcely
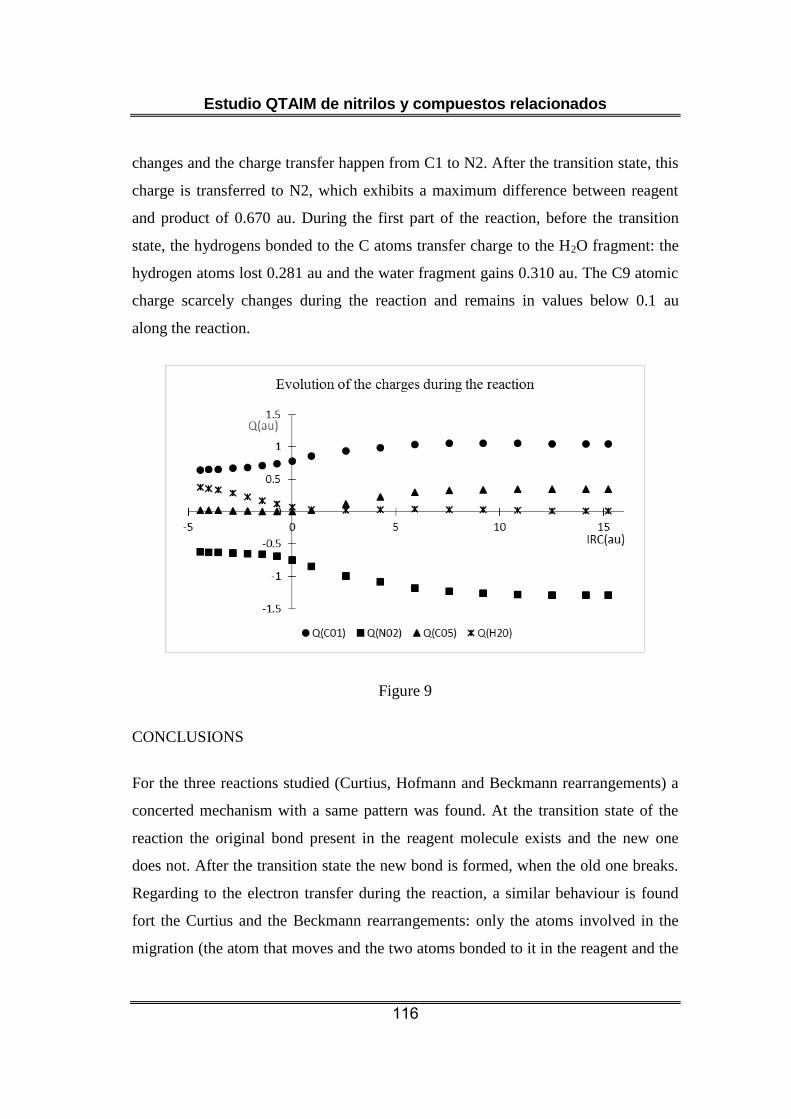
Estudio QTAIM de nitrilos y compuestos relacionados
116
changes and the charge transfer happen from C1 to N2. After the transition state, this
charge is transferred to N2, which exhibits a maximum difference between reagent
and product of 0.670 au. During the first part of the reaction, before the transition
state, the hydrogens bonded to the C atoms transfer charge to the H2O fragment: the
hydrogen atoms lost 0.281 au and the water fragment gains 0.310 au. The C9 atomic
charge scarcely changes during the reaction and remains in values below 0.1 au
along the reaction.
Figure 9
CONCLUSIONS
For the three reactions studied (Curtius, Hofmann and Beckmann rearrangements) a
concerted mechanism with a same pattern was found. At the transition state of the
reaction the original bond present in the reagent molecule exists and the new one
does not. After the transition state the new bond is formed, when the old one breaks.
Regarding to the electron transfer during the reaction, a similar behaviour is found
fort the Curtius and the Beckmann rearrangements: only the atoms involved in the
migration (the atom that moves and the two atoms bonded to it in the reagent and the

Estudio QTAIM de nitrilos y compuestos relacionados
117
product) exhibit large variation of charge along the path, i.e., the electron charge
transfer happen among them. For the Hofmann reaction the Br atom is also involved
in the charge transfer, because is the atom which moves away as an anion. The study
of the reagent, transition state and product structures reveals that the usual resonance
forms representing the geometries do not correspond to the obtained results for bond
lengths and atomic charges.
ACNOWLEDGEMENTS
We thank CESGA for acces to their computational facilities.
REFRENCES
[1] T. Curtius, Berichte der deutschen chemischen Gesellschaft 1890, 23, 3023-
3033.
[2] N. P. Gritsan, M. S. Platz, W. T. Borden, Computational Methods in
Photochemistry 2005, 13, 235-356.
[3] M. V. Zabalov, R. P. Tiger, Russian Chemical Bulletin 2012, 61, 1694-1704.
[4] J. Liu, S. Mandel, C. M. Hadad, M. S. Platz, Journal of Organic Chemistry
2004, 69, 8583-8593.
[5] M. V. Zabalov, R. P. Tiger, Russian Chemical Bulletin 2005, 54, 2270-2280.
[6] M. V. Zabalov, R. P. Tiger, Russian Chemical Bulletin 2007, 56, 7-13.
[7] V. Tarwade, O. Dmitrenko, R. D. Bach, J. M. Fox, Journal of Organic
Chemistry 2008, 73, 8189-8197.
[8] R. Kakkar, S. Zaidi, R. Grover, International Journal of Quantum Chemistry
2009, 109, 1058-1069.
[9] E. S. Wallis, J. F. Lane, in Organic Reactions, John Wiley & Sons, Inc.,
2004.

Estudio QTAIM de nitrilos y compuestos relacionados
118
[10] A. Duan, L. Peng, D. Peng, F. L. Gu, Journal of Organic Chemistry 2013, 78,
12585-12592.
[11] E. Beckmann, Berichte der deutschen chemischen Gesellschaft 1886, 19,
988-993.
[12] A. H. Blatt, Chemical Reviews 1933, 12, 215-260.
[13] W. Z. Heldt, Journal of Organic Chemistry 1961, 26, 1695-1702.
[14] V. R. R. Marthala, Y. Jiang, J. Huang, W. Wang, R. Gläser, M. Hunger,
Journal of the American Chemical Society 2006, 128, 14812-14813.
[15] B. M. Reddy, G. K. Reddy, K. N. Rao, L. Katta, 2009, 306, 62-68.
[16] J. C. Jochims, S. Hehl, S. Herzberger, Synthesis-Stuttgart 1990, 1128-1133.
[17] J. Sirijaraensre, T. N. Truong, J. Limtrakul, The Journal of Physical
Chemistry B 2005, 109, 12099-12106.
[18] M. T. Nguyen, G. Raspoet, L. G. Vanquickenborne, Journal of the American
Chemical Society 1997, 119, 2552-2562.
[19] M. T. Nguyen, G. Raspoet, L. G. Vanquickenborne, Journal of the Chemical
Society-Perkin Transactions 2 1997, 821-825.
[20] G. Raspoet, M. T. Nguyen, L. G. Vanquickenborne, Bulletin Des Societes
Chimiques Belges 1997, 106, 691-697.
[21] J. Sirijaraensre, J. Limtrakul, Physical Chemistry Chemical Physics 2009, 11,
578-585.
[22] N. An, B.-X. Tian, H.-J. Pi, L. A. Eriksson, W.-P. Deng, Journal of Organic
Chemistry 2013, 78, 4297-4302.
[23] Y. Chu, P. Ji, X. Yi, S. Li, P. Wu, A. Zheng, F. Deng, Catalysis Science &
Technology 2015, 5, 3675-3681.
[24] G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman,
G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato,
X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M.
Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y.
Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F.
Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R.
Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J.

Estudio QTAIM de nitrilos y compuestos relacionados
119
Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V.
Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J.
Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G.
Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,
Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc.,
Wallingford CT, 2009.
[25] R. W. F. Bader, Atoms in Molecules: A Quantum Theory Oxford University
Press: New York, 1990.
[26] R. F. W. Bader, Chemical Reviews 1991, 91, 893-928.
[27] F. W. Bieglerkonig, R. F. W. Bader, T. H. Tang, Journal of Computational
Chemistry 1982, 3, 317-328.
[28] C. Wentrup, H. Bornemann, European Journal of Organic Chemistry 2005,
4521-4524.
[29] K. Yokoyama, S. Takane, T. Fueno, Bulletin of the Chemical Society of
Japan 1991, 64, 2230-2235.
[30] A. M. Mebel, A. Luna, M. C. Lin, K. Morokuma, Journal of Chemical
Physics 1996, 105, 6439-6454.
[31] W. A. Shapley, G. B. Bacskay, Journal of Physical Chemistry A 1999, 103,
6624-6631.
[32] A. L. L. East, W. D. Allen, Journal of Chemical Physics 1993, 99, 4638-
4650.
[33] P. J. a. M. Linstrom, W. G., Eds., NIST Chemistry WebBook, NIST
Standard Reference Database Number 69, National Institute of Standards
and Technology, Gaithersburg MD, 20899, http://webbook.nist.gov.
[34] J. Luis Lopez, A. M. Grana, R. A. Mosquera, Journal of Physical Chemistry
a 2009, 113, 2652-2657.

Estudio QTAIM de nitrilos y compuestos relacionados
120
5. CONCLUSIONES
El análisis QTAIM de la densidad electrónica de varias series de
cianocompuestos considerados como inertes o sometidos a procesos
ácido-base, así como el estudio de la evolución de la densidad electrónica
en varias transposiciones que tienen asiento en compuestos
nitrogenados, han permitido establecer las siguientes conclusiones:
El grupo CN de los cianoalcanos lineales presenta valores
transferibles para todas sus propiedades atómicas y de enlace si
se exceptúa la energía atómica, que muestra una dependencia
del tamaño molecular. Este efecto, encontrado en otras series
homólogas, es un artificio debido a los diferentes valores del
cociente virial. Por el contrario, la energía cinética electrónica
atómica si presenta valores transferibles.
Se han encontrado varios tipos de átomos transferibles, propios
de la serie de cianoalcanos lineales:
i. Los átomos C y N del grupo CN.
ii. C en α respecto al grupo CN.
iii. C en β respecto al grupo CN.

Estudio QTAIM de nitrilos y compuestos relacionados
121
Los siguientes átomos presentan un comportamiento específico
en los cianoalcanos lineales:
i. El C en posición α en los nitrilos de metilo y etilo.
ii. El C en posición β en los nitrilos de etilo y propilo.
iii. El C y el N del grupo CN en los cianuros de hidrógeno y
metilo.
El resto de los carbonos de la cadena presentan un
comportamiento similar a los correspondientes de los n-alcanos,
sean estos: internos, terminales o previos al C terminal.
El número de grupos CH2 que se puede distinguir en un
cianoalcano crece cuando se tienen en cuenta simultáneamente
propiedades atómicas y de enlace de todos los átomos
(incluyendo a los hidrógenos) y el conjunto de datos se trata con
criterios estadísticos rigurosos. Así, se pueden considerar hasta
12 grupos CH2 diferentes en los cianoalcanos lineales de cadena
larga. Estos son los metilenos situados a hasta 8 enlaces del
grupo CN, el CH2 interno y los CH2 α, β y γ al metilo terminal. Los
cuatro últimos grupos son comunes con la serie de n- alcanos.

Estudio QTAIM de nitrilos y compuestos relacionados
122
La influencia mutua entre grupos CN (efecto de proximidad) sólo
puede considerarse despreciable cuando están separados por
más de 14 grupos CH2. No obstante, los efectos debidos a la
presencia de dos grupos CN pueden considerarse aditivos salvo
en compuestos CN(CH2)nCN con n<4.
La rotación del enlace central de un dicianoalcano modifica
sensiblemente las propiedades de los grupos CH2 situados entre
posiciones α y δ al enlace.
En los cianocompuestos N-protonados, el protón conserva una
elevada carga positiva. Por ello, lo representa más
adecuadamente la estructura de Lewis H+N≡CR que las
estructuras: HN+≡CR o HNC+R tradicionalmente
empleadas. También favorece la prioridad de dicha estructura el
hecho de que tras la protonación, en el enlace C≡N, aumenta la
densidad π y disminuye la ς.
La N-protonación de un nitrilo no conjugado origina
transferencias electrónicas que afectan a toda la molécula, tal
como sucede en las O- y N-protonaciones de otros compuestos

Estudio QTAIM de nitrilos y compuestos relacionados
123
alifáticos. Sin embargo, en las protonaciones de nitrilos
conjugados las variaciones de población electrónica son
fundamentalmente de tipo π.
La desprotonación está significativamente favorecida frente a
las restantes en todos los cianocompuestos estudiados. Cuando
la molécula presenta sustituyentes adicionales que retiran
densidad electrónica por efecto resonante la energía de
desprotonación se reduce significativamente. No se observa un
efecto opuesto cuando la molécula incorpora dadores por
efecto resonante. Por el contrario, las energías de
desprotonación son muy semejantes a las del cianometano.
Tampoco tiene efectos significativos: el tamaño y la ramificación
del grupo alquilo o la inclusión de átomos electronegativos.
En general el modelo de resonancia proporciona predicciones
compatibles con las variaciones de población electrónica
atómica que acompañan a la desprotonación de un nitrilo. Así,
se observa un importante aumento de la población electrónica
del N del grupo ciano.

Estudio QTAIM de nitrilos y compuestos relacionados
124
Las etapas de migración de las reacciones de transposición de
Curtius, Hofmann y Beckmann son elementales.
Estas reacciones transcurren siguiendo un patrón similar: el
nuevo enlace se forma después del estado de transición cuando
ya se ha roto el enlace original. En ningún punto intermedio de
la reacción coexisten los enlaces originales y los que se forman
durante la misma.
La transferencia de carga a lo largo de las reacciones de
transposición se produce esencialmente entre los átomos
implicados en la transposición: el átomo que migra y los
enlazados a él en el reactivo y el producto. En la transposición
de Hofmann también está implicado en la transferencia de carga
el Br que se separa como anión.

Estudio QTAIM de nitrilos y compuestos relacionados
125

Estudio QTAIM de nitrilos y compuestos relacionados
126
6. BIBLIOGRAFÍA
1. A. M. Graña, R. A. Mosquera, J. Chem. Phys. 110 (1999) 6606. 2. A. M. Graña, R. A. Mosquera, J. Chem. Phys. 113 (2000) 1492. 3. A. Vila, E. Carballo, R. A. Mosquera, Can. J. Chem. 78 (2000) 1535. 4. A. Vila, R. A. Mosquera, J. Chem. Phys. 115 (2001) 1564. 5. M. J. González Moa, R. A. Mosquera, J. Phys. Chem. A 107 (2003) 5361. 6. M. J. González Moa, R. A. Mosquera, J. Phys. Chem. A 109 (2005) 3682. 7. A. M. Graña, J. M. Hermida-Ramón, R. A. Mosquera, Chem. Phys. Lett. 412 (2005) 106. 8. M. Mandado, C. Van Alsenoy, R. A. Mosquera, J. Phys. Chem. A 108 (2004) 7050. 9. M. Mandado, R. A. Mosquera, A. M. Graña, Chem. Phys. Lett. 386 (2004) 454. 10. M. Mandado, C. Van Alsenoy, R. A. Mosquera, Chem. Phys. Lett. 405 (2005) 10. 11. A. Vila, R. A. Mosquera, J. Phys. Chem. A 109 (2005) 6985. 12. R. F. W. Bader, Atoms in Molecules a Quantum Theory, Oxford University Press, 1990. 13. R. F. W. Bader, Chem. Rev. 91 (1991) 893. 14. R. F. W. Bader, Acc. Chem. Res. 18 (1985) 9. 15. P. L. A. Popelier, Atoms in Molecules: An Introduction, Prentice & Hall, 2000. 16. C. Gatti, P. Macchi, Modern Charge-Density Analysis, Springer Verlag, 2012. 17. R. F. W. Bader, Can. J. Chem. 77 (1999) 86. 18. R. F. W. Bader, Int. J. Quantum Chem. 94 (2003) 173. 19. R. F. W. Bader, P. Becker, Chem. Phys. Lett. 148 (1988) 452. 20. J. Riess, W. Münch, Theor. Chmi. Acta 58 (1981) 295. 21. J. L. López, M. Mandado, A. M. Graña, R. A. Mosquera Int. J. Quantum Chem. 86 (2002) 190. 22. J. L. López, M. Mandado, M. J. González Moa, R. A. Mosquera Chem. Phys. Lett. 422 (2006) 558. 23. J. L. López, A. M. Graña, R. A. Mosquera J. Phys. Chem. A 113 (2009) 2652. 24. J. L. López, A. M. Graña, R. A. Mosquera Roy. Soc. Adv. Para ser sometido (2015). 25. J. L. López, R. A. Mosquera, A. M. Graña Eur. J. Org. Chem. Par ser sometido (2015). 26. M. Mandado, A. M. Graña, R. A. Mosquera J. Mol. Struct. (THEOCHEM) 584 (2002) 221. 27. L. Lorenzo, R. A. Mosquera Chem. Phys. Lett. 356 (2002) 305. 28. M Mandado, A. Vila, A. M. Graña, R. A. Mosquera, J. Cioslowski Chem. Phys. Lett. 371 (2003) 739. 29. F. Cortés-Guzmán, R. F. W. Bader Chem. Phys. Lett. 379 (2003) 183. 30. R. F. W. Bader, D. Bayles, J Phys. Chem. A 104 (2000) 5579. 31. R. F. W. Bader, Ting-Hua Tang, Y. Tal, F. W. Biegler-König J. Am. Chem. Soc. 104 (1982) 940. 32. R. F. W. Bader, Ting-Hua Tang, Y. Tal, F. W. Biegler-König J. Am. Chem. Soc. 104 (1982) 946. 33. H. V. Kehiaian Fluid Phase Equilibria 13 (1983) 243. 34. C. E. Shannon The Bell System Technical Journal 27 (1948) 379. 35. M. Hô, V. H. Smith Jr., D. F. Weaver, C. Gatti, R. P. Sagar, R. O. Esquivel J. Chem. Phys. 108 (1998) 5469.

Estudio QTAIM de nitrilos y compuestos relacionados
127
36. M. Hô, B. J. Clark, V. H. Smith Jr., D. F. Weaver, C. Gatti, R. P. Sagar, R.O. Esquivel J. Chem. Phys. 112 (2000) 7572. 37. A. Vila, R. A. Mosquera Chem. Phys. Lett. 332 (2000) 474. 38. A. Vila, R. A. Mosquera Tetrahedron 57 (2001) 9415. 39. M. J. González Moa, M. Mandado, R. A. Mosquera Chem. Phys. Lett. 428 (2006) 255. 40. N. Otero, M. J. González Moa, M. Mandado, R. A. Mosquera Chem. Phys. Lett. 428 (2006) 249. 41. N. C. J. Stutchbury, D. L. Cooper J. Chem. Phys. 79 (1983) 4967. 42. S. Arseniyadis, K. S. Kyler, D.S. Watt, Org. React. 31 (1984) 1. 43. T. Curtius Chem. Ber. 23 (1890) 3023. 44. E. Beckmann, Chem. Ber. 19 (1886) 988. 45. A. W. v. Hofman, Chem. Ber. 14 (1881) 2725. 46. J. Liu, S. Mandel, C. M. Hadad, M. S. Platz, J. Org. Chem. 69 (2004) 8583. 47. M. V. Zabalov, R. P. Tiger, Russ. Chem. Bull. (Int. Ed.) 54 (2005) 2270. 48. M. V. Zabalov, R. P. Tiger, Russ. Chem. Bull. (Int. Ed.) 56 (2007) 7. 49. V. Tarwade, O. Dmitrenko, R. D. Bach, J. M. Fox, J. Org. Chem. 73 (2008) 8189. 50. J. Shorter, en S. Patai, Z. Rapport (eds.) The Chemistry o f Triple-Bonded Functional Groups, Wiley, 1994, capítulo 5. 51. P. Botschwina, M. Horn, M. Matuschewski, E. Schick, P. Sebals, J. Mol. Struct. (THEOCHEM) 400 (1997) 119. 52. F. H. Allen, S. E. Garner, en S. Patai, Z. Rapport (eds.) The Chemistry o f Triple-Bonded Functional Groups, Wiley, 1994, capítulo 2. 53. W. M. Irvine, Space Sci. Rev. 90 (1999) 203. 54. D. H. Rouvray Topics. Curr. Chem. 173 (1995) 1. 55. R. Carbó, L. Leyda, M. Arnau Int. J. Quantum Chem. 17 (1980) 1185. 56. J. Cioslowski, A. Nanayakkara J. Am. Chem. Soc. 115 (1993) 11213. 57. J. Cioslowski, B. B.Stefanov, P. J. Constans J. Comput. Chem. 17 (1996) 1352. 58. R. F. W. Bader Pure Appl. Chem. 60 (1988) 145. 59. J. Schwinger Phys. Rev. 82 (1951) 914. 60. R. F. W. Bader Can. J. Chem. 77 (1998) 86. 61. R. F. W. Bader, P. L. A. Popelier, T. A. Keith Angew. Chem. Int. Ed. Engl. 33 (1994) 620. 62. S. Srebenik, R. F. W. Bader J. Chem. Phys. 63 (1975) 3945. 63. S. I. Sandler, Models for Thermodynamic and Phase Equilibria Calculations, Marcel Dekker, 1994. 64. S.-T. Lin, S. I. Sandler, J. Phys. Chem. A 104 (2000) 7099. 65. D. González-Salgado, C. A. Tovar, C. A. Cerdeiriña, E. Carballo, L. Romaní, Fluid Phase Equilibria 199 (2002) 121. 66. S. Delcros, J. R. Quint, J. P. E. Grolier, H. V. Kehiaian Fluid Phase Equilibria 113 (1995) 1. 67. A. Vila, E. Carballo, R. A. Mosquera, J. Mol. Struct. (THEOCHEM) 617 (2002) 219. 68. A. E. Aliev, K. D. M. Harris, P. H. Champkin J. Phys. Chem. B 109 (2005) 23342. 69. K. D. M. Harris, en: J. L. Atwood, J. W. Steed (eds.) Encyclopedia of Supramolecular Chemistry, vol. 2, Marcel Dekker, 2004, pp. 1538–1549. 70. F. A. Carey, R. J.Sundberg Advanced Organic Chemistry, Kluwer Academic, 2001. 71. G.W. Wheland Resonance in Organic Chemistry, Wiley, 1955. 72. K. B. Wiberg, K. E. Laidig J. Am. Chem. Soc.109 (1987) 5935. 73. K. B. Wiberg, C. M. Breneman, J. Am. Chem. Soc. 114 (1992) 831.

Estudio QTAIM de nitrilos y compuestos relacionados
128
74. R. Glaser, G. S. K. Choy J. Am. Chem. Soc. 115 (1993) 2340. 75. C. L. Perrin J. Am. Chem. Soc. 113 (1991) 2865. 76. K. E. Laidig J. Am. Chem. Soc. 114 (1992) 7912. 77. C. Gatti, P. Fantucci J. Phys. Chem. 97 (1993) 11677. 78. F. L. Hirshfeld, Theor. Chim. Acta 44 (1977) 129. 79. F. De Proft, C. Van Alsenoy, A. Peeters, W. Langenaker, P. J. Geerlings, J. Comput. Chem. 23 (2002) 1198. 80. L. H. Thomas Proc. Camb. Phil. Soc. 23 (1926) 542. 81. E. Fermi, Zeits. für Phys. 48 (1928) 73. 82. W. Kohn, L. Sham Phys. Rev. A 140 (1965) 1133. 83. R.G. Parr, W. Yang Density Functional Theory of Atoms and Molecules, Oxford University Press, 1989. 84. E. S. Kryachko, E. V. Ludeña, Energy Density Functiona Theory of Atoms in Molecules, Kluwer Academic Publishres, 1990. 85. P. Hohenberg, W. Kohn Phys. Rev. B 136 (1964) 864. 86. J. F. Janak, Phys. Rev. B 18 (1978) 7165. 87. A. Strowasser, R. Hoffman J. Am. Chem. Soc. 121 (1999) 3414. 88. G. E. Scuseria, V. N. Staroverov en: Theory and Application of Computational Chemistry: The First 40 Years. C. E. Dykstra, G. Frenking, K. S. Kim, G. E. Scuseria, (eds.) Elsevier, 2005; p. 669. 89. Y. Zhao, N. E. Schultz, D. G. Truhlar J. Chem. Theory Comput. 2 (2006) 364. 90. Y. Zhao, D. G. Truhlar Theor. Chem. Acc. 120 (2008) 215. 91. Y. Zhao, D. G. Truhlar Acc. Chem. Res. 41 (2008) 157. 92. J. Tao, J. P. Perdew. V. N. Staroverov, G. E. Scuseria Phys. Rev. Lett. 91 (2003) 146401. 93. X. Xu, W. A. Goddard, III, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 2673. 94. J. P. Perdew, K. Schmidt en Density Functional Theory and Its Applications to Materials, V. Doren, C. Van Alsenoy, P. Geerlings.(eds.) American Institute of Physics 2001. 95. A. E. MattssonScience 298 (2002) 759. 96. R. Van Leeuwen, E. Baerends J. Phys. Rev. A 49 (1994) 2421. 97. A. D. Becke, J. Chem. Phys. 109 (1998) 2092. 98. P. Mori-Sanchez, A. J. Cohen, W. Yang J. Chem. Phys. 124 (2006) 91102. 99. A. D. Becke J. Chem. Phys. 98 (1993) 5648. 100. J. Perdew, A. Ruzsinszky, J. Tao, V. N. Staroverov, G. E. Scuseria, G. I. Csonka J. Chem. Phys. 123 (2005) 62201. 101. Q. Wu, P. W. Ayers, W. Yang, J. Chem. Phys. 119 (2003) 2978. 102. M. Dion, H. Rydberg, E. Schroder, D. C. Langreth, B. I. Lundqvist Phys. Rev. Lett. 92 (2004) 246401. 103. T. Sato, T. Tsuneda, K.; Hirao, J. Chem. Phys. 123 (2005) 4307. 104. E. R. Johnson, A. D. Becke, J. Chem. Phys. 124 (2006) 174104 105. S. Grimme, J. Comput. Chem. 27 (2006) 1787. 106. T. Sato, T. Tsuneda, K. Hirao, J. Chem. Phys. 126 (2007) 234114. 107. P. Jurecka, J. Cerny, P. Hobza, D. R. Salahub J. Comput. Chem. 28 (2007) 555. 108. C. Lee, W. Yang, R. G. Parr, R. G. Phys. Rev. B 37 (1988) 785. 109. A. D. Becke Phys. Rev. A 38 (1988) 3098. 110. W. Kohn, Y. Meir, D. E. Makarov Phys. Rev. Lett. 80 (1998) 4153. 111. T. V. Mourik, R. J. Gdanitz, J. Chem. Phys. 116 (2002) 9620.

Estudio QTAIM de nitrilos y compuestos relacionados
129
112. Y. Zhao, D. G. Truhlar J. Phys. Chem. A 108 (2004) 6908. 113. A. D. Becke, J. Chem. Phys. 104 (1996) 1040. 114. B. J. Lynch, P. L. Fast, M. Harris, D. G. Truhlar J. Phys. Chem. A 104 (2000) 4811. 115. P. A. M. Dirac Proc. Cambridge Philos. Soc. 26 (1930) 376. 116. J. C. Slater Quantum Theory of Matter, McGraw-Hill, 1968. 117. C. Adamo, V. Barone, J. Chem. Phys. 108 (1998) 664. 118. B. J. Lynch, D. G. Truhlar J. Phys. Chem. A 107 (2003) 8996. 119. N. Castillo, C. F. Matta, R. Boyd J. Chem. Phys. Lett. 409 (2005) 265. 120. J. Poater, M. Solà, F. M. Bickelhaupt, Chem. Eur. J. 12 (2006) 2889. 121. R. F. W. Bader Chem. Eur. J. 12 (2006) 2896. 122. A. Haaland, D. J. Shorokhov, N. V. Tverdova, Chem. Eur. J. 10 (2004) 4416; Chem. Eur. J. 10 (2004) 6210 (Corrigendum). 123. R. F. W. Bader, F. De-Cai, J. Chem. Theory Comput. 1 (2005) 403. 124. J. Cioslowski, S. T. Mixon, Can. J. Chem. 70 (1992) 443. 125. J. Cioslowski, S. T. Mixon, J. Am. Chem. Soc. 114 (1992) 4382. 126. R. F. W. Bader J. Phys. Chem. A 102 (1998) 7314. 127. F. W. Biegler-König, J. Schönbohm, D. Bayles J. Comp. Chem. 22 (2001) 545. 128. P. B. Quiñónez, A. Vila, A. M. Graña, R. A. Mosquera Chem. Phys. 287 (2003) 227. 129. A. Vila, R. A. Mosquera Chem. Phys. Lett. 345 (2001) 445. 130. M. Lehd, F. Jensen J. Comput. Chem. 12 (1991) 1089. 131. M. Mandado, A. M. Graña, R. A. Mosquera Chem. Phys. Lett. 355 (2002) 529. 132. T. S. Slee, P. J. MacDougall Can. J. Chem. 66 (1988) 2961. 133. K. E. Laidig, L. M. Cameron J. Am. Chem. Soc. 118 (1996) 1737. 134. R. Glaser J. Phys. Chem. 93 (1989) 7993. 135. P. Speers, K. E. Laidig, A. Streitwieser J. Am. Chem. Soc. 116 (1994) 9261. 136. A. Vila, R. A. Mosquera Chem. Phys. Lett. 371 (2003) 540. 137. N. Otero, L. Estévez, M. Mandado, R. A. Mosquera, Eur. J. Org. Chem. 12 (2012) 2403. 138. L. Estévez, R. A. Mosquera, Chem. Phys. Lett. 451 (2008) 121.

130