
Understanding the Molecular Mechanisms of Post-Exercise ...
163
Understanding the Molecular Mechanisms of Post-Exercise Muscle Inflammation and Repair by Luke Vella BAppSc(Ex&SpSc)(Hons) Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy Deakin University December 2016
Transcript of Understanding the Molecular Mechanisms of Post-Exercise ...

Understanding the Molecular Mechanisms of Post-Exercise Muscle
Inflammation and Repair
by
Luke Vella
BAppSc(Ex&SpSc)(Hons)
Submitted in fulfilment of the requirements for the degree of
Doctor of Philosophy
Deakin University
December 2016


lswan
Redacted stamp


lswan
Redacted stamp


Acknowledgements
I dedicate this thesis to my parents, Steve and Anne-Maree Vella, you are my support, my inspiration,
my family.
To my supervisory team, Aaron Russell, David Cameron-Smith and Rod Snow. Thank you for your
constant support, hard work, care and diligence. Your patience and understanding of my career
outside of my PhD has been enormously appreciated. Your passion and knowledge in your
respective fields is something that I will always admire and aspire to.
To my co-authors, James Markworth, Jonathan Peake, Rao Maddipati, Gøran Paulsen, Truls Raastad,
Marissa Caldow, Petra Graves, Amy Larsen, Paul Della Gatta and Daniella Tassoni. You have all
contributed significantly to my research and to this thesis. Thank you for your time, dedication and
support over the past 8 years.
To Dr. Andrew Garnham. It was a pleasure working with you through the exercise trials. Your
expertise is unrivalled and I hope our paths will cross again in the future.
To Dr. Sean Williams and Professor John Reynolds. Thank you for all of your support with my
statistical analysis, I cannot express how lost I would have been without your guidance. Your
patience and willingness to help me through the process was a saving grace.
To Laura. My best friend, my never ending support, my inspiration and soon-to-be, my wife. I could
write another thesis on how much I treasure your love and support. Thank you for being all of those
things and so much more. You have kept me sane and grounded throughout this entire process.
Finally to my family. My brother Mark, my Mum Anne-Maree and my Dad Steve. You have provided
me with every opportunity that a person could possibly wish for. You are the first people I want to
talk to when things go well, and the first people I turn too when things go wrong. Your love,
generosity, selflessness and modesty is something that I aspire to every day of my life.

List of publications
Vella L, Markworth JF, Paulen G, Raastad T, Peake JM, Snow RJ, Cameron-Smith D, Russell
AP: Ibuprofen ingestion does not affect markers of post-exercise muscle inflammation.
Frontiers in Physiology, March 2016, 7:86.
Vella L, Markworth JF, Peake JM, Snow RJ, Cameron-Smith D, Russell AP: Ibuprofen
supplementation and its effects on NF-κB activation in skeletal muscle following resistance
exercise. Physiological reports, September 2014, 2:10.
Vella L, Caldow MK, Larsen AE, Tassoni D, Della Gatta PA, Gran P, Russell AP, Cameron-Smith
D: Resistance exercise increases NF-κB activity in human skeletal muscle. American Journal
of Physiology. Regulatory, Integrative and Comparative Physiology, March 2012, 302:6.

Table of Contents
LIST OF FIGURES .......................................................................................................................... I
LIST OF TABLES .......................................................................................................................... III
ABBREVIATIONS ........................................................................................................................IV
CHAPTER 1 – LITERATURE REVIEW ............................................................................................ 1
1.1 Introduction: ....................................................................................................................... 2
1.2 Exercise-induced inflammation: ......................................................................................... 3
1.3 Anti-inflammatory treatments and muscle recovery: ........................................................ 6
1.3.1. Prostaglandin response to exercise. ............................................................................ 7
1.3.2. The effect of NSAIDs on muscle inflammation and recovery. ..................................... 7
1.4 Resolution of exercise-induced inflammation: .................................................................. 9
1.5 Transcriptional regulation of exercise-induced inflammation: ........................................ 12
1.5.1 Nuclear Factor-kappa B: ........................................................................................ 13
1.5.2 NF-κB and inflammation: ................................................................................... 15
1.5.3 NF- κB and skeletal muscle myogenesis: ........................................................... 15
1.5.4 NF- κB and exercise: ........................................................................................... 17
1.6 Summary: .......................................................................................................................... 18
1.7 General hypothesis: .......................................................................................................... 19
1.8 Specific Aims: .................................................................................................................... 21
1.9 Hypothesis: ....................................................................................................................... 21
CHAPTER 2: .............................................................................................................................. 22
2.1 Abstract: ............................................................................................................................. 23
2.2 Introduction: ...................................................................................................................... 24
2.3 Methods: ............................................................................................................................ 27
2.3.1 Participants: ................................................................................................................ 27
2.3.2 Ethics approval: ........................................................................................................... 27

2.3.3 Familiarization: ............................................................................................................ 28
2.3.4 Experimental Procedures: ........................................................................................... 28
2.3.5 Standardized meals: .................................................................................................... 28
2.3.6 NSAID Administration: ................................................................................................ 29
2.3.7 Sample collection: ....................................................................................................... 29
2.3.8 Immunohistochemistry: .............................................................................................. 30
2.3.9 Biochemical assays: ..................................................................................................... 31
2.3.10 Muscle soreness assessment: ................................................................................... 31
2.3.11 Statistics: ................................................................................................................... 32
2.4 Results: ............................................................................................................................... 33
2.4.1 Muscle Soreness: ......................................................................................................... 33
2.4.2 Immunohistochemistry: .............................................................................................. 33
2.4.3 Serum proteins: ........................................................................................................... 35
2.4.4 Correlation analysis: .................................................................................................... 36
2.5 Discussion: ......................................................................................................................... 38
2.6 Conclusion: ......................................................................................................................... 40
2.7 Acknowledgements:........................................................................................................... 41
2.8 Disclosures: ........................................................................................................................ 41
CHAPTER 3: .............................................................................................................................. 42
3.1 Abstract: ............................................................................................................................. 43
3.2 Introduction: ...................................................................................................................... 44
3.3 Methods: ............................................................................................................................ 47
3.3.1 Subjects: ...................................................................................................................... 47
3.3.2 Ethics approval: ........................................................................................................... 47
3.3.3 Familiarization: ............................................................................................................ 47
3.3.4 Experimental design: ................................................................................................... 48

3.3.5 Muscle biopsy procedure: ........................................................................................... 48
3.3.6 Liquid chromatography-mass spectrometry: .............................................................. 48
3.3.7 Statistics: ..................................................................................................................... 49
3.4 Results: ............................................................................................................................... 50
3.4.1 Lipidomic analysis:....................................................................................................... 50
3.4.1.1 Cyclooxygenase pathway: .................................................................................... 50
3.4.1.2 Lipoxygenase pathway: ........................................................................................ 52
3.4.1.3 Epoxygenase pathway: ......................................................................................... 55
3.5 Discussion: ......................................................................................................................... 57
3.6 Conclusion: ......................................................................................................................... 61
3.7 Acknowledgments: ............................................................................................................ 61
3.8 Disclosures: ........................................................................................................................ 61
CHAPTER 4: .............................................................................................................................. 63
4.1 Abstract: ............................................................................................................................. 64
4.2 Introduction: ...................................................................................................................... 65
4.3 Methods: ............................................................................................................................ 68
4.3.1 Participants: ................................................................................................................ 68
4.3.2 Ethics approval: ........................................................................................................... 68
4.3.3 Familiarization and strength testing: .......................................................................... 69
4.3.4 Experimental procedures: ........................................................................................... 69
4.3.5 NSAID administration: ................................................................................................. 70
4.3.6 Sample collection: ....................................................................................................... 70
4.3.7 Protein extraction and quantification: ........................................................................ 71
4.3.8 RNA extraction and RT-PCR: ........................................................................................ 71
4.3.9 Nuclear extraction and Transcription Factor (TF) Assay: ............................................ 72
4.3.10 Statistics: ................................................................................................................... 73
4.4 Results: ............................................................................................................................... 74

4.4.1 Protein expression: ..................................................................................................... 74
4.4.2 Electrophoretic mobility shift assay: ........................................................................... 76
4.4.3 RT-PCR: ........................................................................................................................ 77
4.5 Discussion: ......................................................................................................................... 79
4.6 Conclusion: ......................................................................................................................... 81
CHAPTER 5 – GENERAL DISCUSSION AND FUTURE DIRECTIONS: ............................................ 83
5.1 – Introduction: ................................................................................................................... 83
5.2 – Summary of results: ........................................................................................................ 84
5.3 – Future directions: ............................................................................................................ 85
5.4 – Concluding remarks: ....................................................................................................... 87
REFERENCE LIST: ...................................................................................................................... 89
Appendix A: ............................................................................................................................ 122
Appendix B: ............................................................................................................................ 129
Appendix C: ............................................................................................................................ 145

I
LIST OF FIGURES
Figure 1.1: A schematic of the process of acute exercise-induced muscle damage,
inflammation and repair.
Figure 1.2: NF-κB - a central regulator of stress signaling pathways.
Figure 1.3: Classical and alternative NF-κB signaling pathways.
Figure 2.1: Subjective rating for delayed onset muscle soreness (DOMS). This figure has
been adapted from Vella et al. (2014).
Figure 2.2: Immunohistochemistry data for muscle cells staining positive for WBC cell
surface markers.
Figure 2.3: Immunohistochemical analysis of skeletal muscle samples following a bout of
resistance exercise.
Figure 2.4: Blood derived proteins creatine kinase and myoglobin acute resistance exercise
Figure 3.1: Metabolites of the cyclooxygenase pathway derived from arachidonic acid.
Figure 3.2: Metabolites of the lipoxygenase pathway derived from arachidonic acid.

II
Figure 3.3: Metabolites of the lipoxygenase pathway derived from docosahexaenoic acid.
Figure 3.4: Metabolites of the epoxygenase pathway derived from arachidonic acid.
Figure 3.5: Metabolites of the epoxygenase pathway derived from linoleic acid.
Figure 4.1: Protein expression of NF-κB subunits p65, p50, p52 and cREL.
Figure 4.2: NF-κB subunits p65 (A) and p50 (B) binding to nuclear protein.
Figure 4.3: RT-PCR analysis of NF-κB target genes IL-6 (A), IL-8 (B), MCP-1 (C), TNF-α (D) and
COX-2 (E) in skeletal muscle cDNA.

III
LIST OF TABLES
TABLE 2.1 SUBJECT CHARACTERISTICS AND STRENGTH TESTING DATA.
TABLE 2.2 SPEARMAN RANK CORRELATION ANALYSIS.
TABLE 4.1 SUBJECT CHARACTERISTICS AND STRENGTH TESTING DATA.
TABLE 4.2 RT-PCR PRIMER SEQUENCES

IV
ABBREVIATIONS
1RM 1 repetition maximum
AA Arachidonic acid
ATP Adenosine triphosphate
BMI Body mass index
BSA Bovine serum albumin
CK Creatine kinase
COX Cyclooxygenase
CYP Cytochrome P450
DHA Docosahexaenoic acid
DiHETrE Vicinal diol dihydroxyeicosatrienoic acid
DOMS Delayed onset muscle soreness
EMSA Electrophoretic mobility shift assay
EPA Eicosapentaenoic acid
EpETrE Epoxyeicosatrienoic acid
HDoHE Monohydroxylated- docosahexaenoate
HETE Hydroxyeicosatetraenoic acids
IBU Ibuprofen
IGF-BP1 Insulin-like growth factor binding protein 1
IGF-BP2 Insulin-like growth factor binding protein 2
IGF-II Insulin-like growth factor II
IKK Inhibitor of κB kinase
IL-6 Interleukin-6
IL-8 Interleukin-8
iMVC Isokinetic maximal voluntary contraction
IκB Inhibitor of κB
LC-MS Liquid chromatography-mass spectrometry
LOX Lipoxygenase
LSD Least significant difference
LT Leukotriene
LX Lipoxin
MAPK Mean arterial pressure kinase
MCP-1 Monocyte chemoattractant protein-1
MHC Myosin heavy chain
MPO Myeloperoxidase
MYO Myoglobin
n-3 Omega-3
n-6 Omega-6

V
NF-κB Nuclear factor-kappa B
NSAID Non-steroidal anti-inflammatory drug
PCR Polymerase chain reaction
PD Protectin
PDGF Plasma-derived growth factor
PG Prostaglandin
PLA Placebo
PMN Polymorphonuclear neutrophils
PUFA Polyunsaturated fatty acid
Rv Resolvin
SEM Standard error of the mean
SPM Specialized pro-resolving mediator
TGF-β Transforming growth factor-β
TMB Tetramethylhylbenzidine
TNF-ɑ Tumour necrosis factor-ɑ
TX Thromboxane
VEGF Vascular endothelial growth factor
YY1 Ying Yang1

1
CHAPTER 1 – LITERATURE REVIEW

2
1.1 Introduction:
Skeletal muscle is a remarkably heterogeneous tissue with a vast capacity to adapt and
respond to its external environment [1-4]. It is therefore not surprising that the intense
muscular contractions common to resistance exercise results in a marked increase in muscle
size, commonly termed hypertrophy [5, 6]. However, following most bouts of intense exercise,
there is often considerable muscle damage, inflammation and muscle soreness. Human studies
exploring the physiological mechanisms of exercise-induced muscle damage date back to the
beginning of the 20th century [7]. Early works with muscle preparations and animal models
demonstrated that physical exercise causes damage to muscle cells including the mechanical
disruption of sarcomeres and adjacent cellular membranes, and disturbed excitation
contraction coupling and calcium signalling. [8-15]. Pioneering work using human skeletal
muscle biopsy samples reported muscle damage at the ultrastructural level and changes in
circulating leucocytes [16-22]. These studies identified a role for inflammatory white blood
cells in post-exercise muscle recovery. Modern molecular biology has superimposed additional
layers of complexity by, identifying functional roles for protein and lipid derivatives including
cytokines, acute phase proteins and eicosanoids, in exercise-induced inflammation. Whilst
precise cellular and molecular signalling pathways remain poorly understood, post-exercise
muscle recovery occurs according to a well-described temporal scheme: (1) tissue injury via
mechanical and metabolic stress, (2) release of vasoactive and chemotactic substances from
damaged tissue, (3) migration and adhesion of systemic leucocytes to the injury site, and (4)
tissue repair and adaptation (Figure 1). Research efforts over the last few decades have sought
to understand the physiological significance of acute post-exercise inflammation and its
functional role in muscle recovery and adaptation. Developing an understanding of the
molecular mechanisms that regulate this phenomenon may permit the development of
therapeutic treatment strategies to manipulate selective events of muscle inflammation and
optimise the cellular processes of muscle repair.

3
Figure 1.1: A schematic of the process of acute exercise-induced muscle damage, inflammation and
repair. This figure was adapted from Pillon et al. and demonstrates the interaction between skeletal
muscle and both local and systemic immune cells [23].
1.2 Exercise-induced inflammation:
It appears that cellular events occurring early in exercise-induced muscle damage engage a
biologically active and tightly regulated acute inflammatory process that is of critical
importance to post-exercise muscle recovery. While the complexity of inflammation is
currently acknowledged herein, the fundamental objective of any inflammatory program is to
repair injury and restore tissue function. Historically, the paradigm that exercise-induced
muscle damage is linked to an acute inflammatory response has been driven by reports from
various animal reports and in-vitro study designs, and has been extensively reviewed [9, 12, 13,
24-30]. Because of the various physiological and anatomical differences between species, and
the extent of muscle damage that can be caused in in-vitro research models, findings from
these studies may or may not be applicable to voluntary physical activity in humans.
Localized inflammation is initiated by damage to the extracellular matrix, increasing the
permeability of the sarcolemmal membrane, causing a rapid extravasation of fluid and blood-
borne polymorphonuclear neutrophils (PMNs) into the damaged tissue [31-33]. It is well
accepted that neutrophils play a critical role in acute inflammation, removing necrotic tissue
and cellular debris via a process termed phagocytosis [31, 34]. Phagocytosis triggers a

4
respiratory burst and subsequent release of cytotoxic granulocyte products, reactive oxygen
species and inflammatory cytokines, which cause further tissue damage and exacerbate the
inflammatory response [35-38]. Neutrophil infiltration appears to peak between 3-24 h post-
exercise. Interestingly this time point is associated with a rise in histological and functional
markers of muscle damage. This suggests that neutrophil function may lead to secondary
tissue injury, a phenomenon termed delayed onset muscle soreness (DOMS) [8, 39-43]. Peak
accumulation of PMNs is overlapped in time by the accumulation of monocyte/macrophages.
These are highly versatile cell types whose function depends largely on the extracellular
environment [44, 45]. Two sub-types of macrophages exist within skeletal muscle, and perform
multiple and largely disparate functions within the contraction-induced inflammatory cascade
[25, 34, 46-48]. Classically activated, or M1 macrophages, appear to peak between 12 and 24 h
post exercise [49] and are most prominent within necrotic muscle fibres [50, 51]. The
distribution of M1 cells is consistent with their capability of phagocytosis [52]. This precedes a
shift in macrophage phenotype to an alternatively activated, or M2 macrophage. This shift in
phenotype coincides with a change from the proliferative stage to the early differentiation
stage of myogenesis, and substantiates a long suspected concept that M2 macrophages
regulate key pathways in muscle regeneration [25, 33, 47, 53-60]. These findings rationalize
the concept that cellular events occurring early in the acute inflammatory cascade, trigger a
series of cellular processes that are a key regulatory component of muscle regeneration and
tissue repair.
More recent human research identified two key concepts that have challenged the paradigm
of exercise-induced muscle damage, DOMS, and muscle regeneration. Firstly, a tissue may be
influenced by pro-inflammatory signaling molecules, including cytokines and eicosanoids, in
the absence of inflammatory cell invasion. Subsequently, humans can experience symptoms of
exercise-induced muscle damage without presenting with cellular signs of inflammation [61-
65]. Radionuclide imaging techniques have confirmed large changes in circulating leucocytes
(primarily neutrophils) immediately following exercise [63, 66]. However, these techniques
give no information about the infiltration of leucocytes into skeletal muscle tissue.
Investigations of exercise-induced muscle inflammation that explore the infiltration of
leucocytes in to skeletal muscle tissue are relatively scarce, and conflicting results have often

5
been attributed to variations in exercise protocols, muscle sampling procedures, individual
responsiveness to exercise stress. Additionally, the use of non-specific nomenclature to
identify and describe muscle damage and inflammation [17, 19, 67-74] has also created some
confusion in the field. The main criticism of these studies is the risk of the muscle biopsy
procedure triggering local inflammatory responses that could confound the effects of the
exercise protocol. Muscle biopsy procedures have been shown to cause an increase in the
activity of mean arterial pressure kinase (MAPK) [75] and the expression of leucocyte specific
antigens [62], while impairing adenosine triphosphate (ATP) and glycogen re-synthesis [76].
Alternatively, indirect proxy measures of muscle damage and inflammation including
subjective DOMS, swelling and serum measures of blood-borne proteins have been used,
however these markers do not accurately reflect the extent of cellular muscle damage and
show poor correlation with each other [20, 77-82]. In order to determine a functional role for
acute inflammation in exercise-induced muscle damage, further research including a non-
exercise control group, and measuring direct markers of cellular inflammation are required.
The presence of a post-exercise inflammatory response remains well accepted, however its
relationship with myofibrillar disruption, cellular damage and muscle soreness remains
speculative [19, 65, 83, 84]. A review from Paulsen et al. [85] suggested that when intracellular
damage exceeds a specific threshold, leucocytes infiltrate the damaged tissue and this
manifests itself as a reduction in muscle force generating capacity. A reduction in muscle force
generating capacity of greater than 20% appears necessary to initiate leucocyte accumulation
in skeletal muscle tissue, however results remain inconsistent [19, 84, 86-92]. Studies reporting
a reduction of muscle force generating capacity greater than 50%, consistently resulted in an
increase in intramuscular leucocyte infiltration [69, 70, 84, 93-96].
Immunohistochemical staining of leucocyte specific antigens has become the gold standard for
the temporal profiling of the appearance of leucocyte populations in human muscle following
exercise. While animal and in-vitro studies present a highly predictable sequence of cellular
events in acute inflammation, these findings have yet to be replicated in human in-vivo
exercise clinical trials. Pioneering works from Paulsen et al. [84] attempted to profile the time

6
course appearance of leucocyte populations by staining muscle cells for CD16 (primarily a
neutrophil marker) and CD68 (primarily a monocyte/macrophage marker). This study showed
increased numbers of both CD16 and CD68 localised to the endomysium and perimysium as
soon as 0.5 h post-exercise and persisting until 4 d post-exercise [84]. Further, this study
established a strong correlation between the individual changes in muscle function and the
accumulation of radiolabelled leucocytes, providing further justification for a role of exercise-
induced inflammation in delaying the restoration of muscle function post-exercise. The
appearance of monocyte/macrophage populations has been a consistent histological finding
and appears common in both early stage [86, 88, 89, 92, 93, 97] and late stage inflammation
[32, 70, 98]. Among those publications exclusively examining neutrophil infiltration only two
studies have identified an increase in both early stage [68, 84] and late stage inflammation [84,
98]. Further research profiling the time-course of leucocyte recruitment and infiltration in
human subjects is required to understand the complexity of the exercise-induced inflammation
response.
1.3 Anti-inflammatory treatments and muscle recovery:
Non-steroidal anti-inflammatory drugs (NSAIDs) are a commonly used as a treatment strategy
in exercise and sports medicine to assist with recovery from exercise-induced inflammation,
particularly following soft-tissue injury [99]. NSAIDs inhibit the cyclooxygenase (COX-1 and -2)
enzymes and consequently the formation prostanoids derived from mobilised free intracellular
arachidonic acid (AA) [100]. The COX pathway converts AA to prostaglandin G2 (PGG2), and
subsequently catalyses the reduction of PGG2 to form PGH2 [101]. Various synthase enzymes
convert PGH2 to form five primary prostanoids that play a diverse role in acute inflammation,
including; thromboxane A2, PGD2, PGF2ɑ, PGE2 and PGI2. These prostaglandin species are often
ascribed to the cardinal signs of acute inflammation through their effects on local blood flow,
vascular permeability, leucocyte infiltration, and triggering sensations of pain [102, 103].
However, more recent information from both animal [104-106] and human research [87, 107,
108], suggests a role for prostaglandin species in skeletal muscle regeneration.

7
1.3.1. Prostaglandin response to exercise.
Research in to the prostaglandin response to muscle loading and exercise-induced muscle
damage has produced equivocal findings that appear to be influenced by the nature of the
exercise stimulus. Early studies implementing a prolonged submaximal exercise protocol
demonstrated heightened levels of various circulating prostaglandin species including PGE2
[109-112], PGF2ɑ [102, 109], PGI2 [113-116] and thromboxane [113, 116]. Fewer studies have
investigated the prostaglandin response to muscle damaging resistance exercise protocols. The
limited studies available demonstrated an increase in circulating PGE2 and 13,-14-dihydro-15-
keto-PGF2ɑ following both stretch-shortening cycling exercise [117] and traditional resistance
exercise [118-120]. No such effect was observed in isolated eccentric knee extension [121, 122]
and elbow extension exercise [123-125], or downhill running protocols [126]. Similarly, few
studies have attempted to profile intramuscular prostaglandin responses to exercise-induced
muscle damage. Skeletal muscle cells express both COX-1 and -2. Few studies have reported an
increase in skeletal muscle COX mRNA expression and enzymatic activity during exercise
recovery [127-130], while others have found no change [87, 131]. Eccentric resistance exercise
triggers an increase in intramuscular PGF2ɑ at 24 h post-exercise [132], while a more traditional
bout of resistance exercise increases PGF2ɑ in muscle microdialysates at 5-6 and 8-9 h post-
exercise, with no increase in PGE2 [133]. Likewise, an incremental cycling based intervention
triggered an increase in PGE2 concentration during exercise, measured using microdialysis,
however an intermittent plantar flexion protocol elicited no such response [134, 135]. Further,
unilateral arm exercise including 70 maximal eccentric contractions, achieved no change in
PGE2 levels in tissue dialysate [93]. The reason for such conflicting findings remains unclear,
however may relate to differences in the exercise protocol, specifically in the intensity of
muscle load, or the volume of active muscle mass in the respective exercise trials.
1.3.2. The effect of NSAIDs on muscle inflammation and recovery.
Administration of non-selective NSAIDs (e.g. ibuprofen) effectively blocks the exercise-induced
rise in prostaglandins in both skeletal muscle [132, 136, 137] and peripheral blood [102].
Therefore, NSAID treatment has been used as a research model to explore the physiological
role of prostaglandin species in exercise-induced inflammation, and the biological link between
acute inflammation and skeletal muscle regeneration. Research into the effects of NSAIDs on

8
exercise-induced muscle damage and inflammation has produced largely controversial
findings. Many studies have explored the short term effects of NSAID treatment on indirect
markers of muscle damage and inflammation and produced disparate findings for an effect on
muscle swelling [93, 138, 139], circulating creatine kinase [42, 119-121, 140-146], restoration
of muscle function [42, 93, 122, 138, 140, 142, 144, 146-150] and subjective muscle soreness
[42, 71, 87, 93, 107, 119, 138-158]. While these markers often represent the symptoms of
exercise-induced muscle damage, many of these parameters have a relatively weak association
with structural muscle damage or, more specifically, cellular inflammation. In animal models of
acute muscle damage, treatment with NSAIDs blunts the infiltration of leucocytes into muscle
tissue [104, 159], however this finding has not been replicated in human subjects [147, 160].
Oral consumption of both ibuprofen, and acetaminophen, an analgesic also known as
paracetamol, had no effect on skeletal muscle macrophage infiltration at 24 h following an
eccentric exercise protocol [160]. Similarly treatment with naproxen, another non-selective
NSAID, had no effect on the infiltration of leucocyte common antigen positive cells following a
unilateral, isotonic resistance exercise protocol [147]. Interestingly, Paulsen et al., (2010)
suggested a blunting effect of the COX-2 specific celecoxib following maximum eccentric
muscle contractions [93]. This research identified a tendency for higher monocyte/macrophage
numbers in subjects within the placebo group, and within those subjects who were identified
as ‘high-responders’ to the exercise protocol based on the number of inflammatory leucocytes
[93]. Although this is not a particularly robust finding, it has led to the hypothesis that NSAIDs
may influence leucocyte infiltration in skeletal muscle when a sufficiently strong and early
inflammatory reaction is present [93]. This hypothesis would suggest that the intensity of the
exercise stimulus and the consequent muscle-damage response would be largely influential in
determining the effect of a pharmacologically based anti-inflammatory intervention.
Experimental evidence also indicates that NSAID administration can influence the regenerative
capacity of skeletal muscle following exercise-induced muscle damage. The non-selective COX
inhibitor ibuprofen, blunted skeletal muscle protein synthesis [107], while local intramuscular
infusion of indomethacin inhibited satellite cell proliferation following maximum eccentric
exercise protocols [87]. Similarly, oral administration of indomethacin attenuated the exercise-
induced increase in satellite cell number following an endurance running protocol. Further,

9
treatment with ibuprofen inhibited early translational signaling responses involved in post-
exercise muscle hypertrophy following a bout of traditional resistance exercise [161]. These
findings support earlier in-vitro research models that implicate a role for prostaglandin species
in stimulating skeletal muscle adaptive pathways [162, 163]. However, follow up studies using
selective COX-2 specific NSAID celecoxib showed no effect on the exercise-induced increase in
skeletal muscle protein turnover [130] or satellite cell number [93] following an eccentric
exercise protocol. These findings present two potential alternatives. Firstly, despite the well-
established role for the inducible COX-2 enzyme in the animal muscle reparative response to
acute damage [104-106, 111, 164], the constitutively active COX-1 may play an important
function in the human adaptive response to exercise. Alternatively, NSAID administration may
have the capacity to influence alternative signalling pathways within the exercise-induced
inflammation-tissue repair paradigm. Markworth et al. employed a targeted lipidomics
approach to characterize temporal changes in the human peripheral blood fatty acyl lipid
mediator profile in response to acute resistance exercise [102]. This research demonstrated
that ibuprofen administration has the capacity to influence the appearance of a series of lipid-
derived mediators that have the potential to engage a biologically active inflammatory
resolution programme that may provide a crucial physiological link between inflammatory and
adaptive signaling pathways.
1.4 Resolution of exercise-induced inflammation:
Historically, the label of inflammation has been ascribed to a distinct group of symptoms
including heat, redness, swelling, pain and a loss of function [165]. The resolution of
inflammation was previously thought to be a passive process whereby chemotactic gradients
were thought to dissipate and the exodus of inflammatory leucocytes through lymphatic
drainage enabled muscle tissue to return to homeostatic function. However, the discovery of a
novel class of lipid-derived mediators has established the resolution of inflammation as an
active biochemical and metabolic process. The profiling of these bioactive lipids was pioneered
by Serhan et al. [166-174] and represents an area of critical importance in understanding the
biological significance of the inherent self-resolving nature of acute skeletal muscle
inflammation.

10
Lipid mediators are biosynthesized endogenously from numerous polyunsaturated fatty acid
(PUFA) precursors including essential omega-3 fatty acids (n-3) eicosapentaenoic (EPA) and
docosahexaenoic acid (DHA), and major omega-6 (n-6) arachidonic acid (AA). Under basal
conditions PUFA substrates remain esterfied within cell membrane phospholipids. In response
to injury or inflammatory insult hundreds of distinct lipid species can be synthesized via 3
major enzymatic pathways: (1) cyclooxygenase, (2) lipoxygenase, and (3) epoxygenase which is
catalysed by cytochrome P450 (CYP) [101]. The development of mass spectrometry (MS)-based
lipidomic profiling has identified and quantified large numbers of pro-inflammatory, pro-
resolution and anti-inflammatory mediators, and enabled the sequential profiling of lipid
species in a variety of acute inflammatory models. The majority of research has focussed on
classic prostaglandin and leuokotriene species which are pro-inflammatory in nature and
derived from omega-6 AA. These lipids play a diverse role in stimulating acute inflammation by
controlling local blood flow, cytokine production, cell membrane permeability and leucocyte
infiltration [102]. More recently, a secondary class of eicosanoids also generated from AA,
termed lipoxins [172, 175, 176], as well as newly identified EPA (E-Series) and DHA (D-Series)
derived resolvins, protectins [166, 169-171, 177, 178], play dual anti-inflammatory and pro-
resolution functions in acute inflammation. These endogenous mediators possess potent
immunoregulatory actions via several pathways including the inhibition of pro-inflammatory
cytokines and the chemotaxis neutrophils, and stimulating the recruitment of monocytes and
the nonphlogistic phagocytosis of apoptotic neutrophils [175, 177-182]. Evidence from in-vitro
models suggests that the phagocytosis of apoptotic neutrophils can trigger a phenotypic shift
in macrophages from the classical M1 macrophage, to the alternative M2 macrophage [181].
This process may suppress the release of pro-inflammatory agents and increase the
macrophage release of anti-inflammatory mediators such as transforming growth factor-β
(TGF-β) and vascular endothelial growth factor (VEGF) [44, 183].
Lipoxins, resolvins and protectins are formed during inflammatory transcellular interactions,
involving the sequential actions of two or more LOX and/or COX enzymes. During inflammation
cell-cell interactions between platelets, leucocytes and resident tissue cells facilitates the
transcellular biosynthesis of unique lipid derivatives [101]. For example, LX biosynthesis
involves cellular interactions between 5-LOX expressing PMNs with 12-LOX expressing platelets

11
or 15-LOX expressing M2 monocytes [184]. The complex signalling network that exists within
damaged skeletal musculature remains poorly understood, and it appears from other models
of acute inflammation that the temporal regulation of lipids and their role in inflammation is
likely to be cell-type and stimulus specific.
Our understanding of pro-resolving lipid mediator circuits has been limited to in-vitro assays
utilizing blood borne immune cell populations and pharmacologically induced models of acute
inflammation. The first study to explore the time-course appearance of lipid mediators
implemented a TNF-ɑ-stimulated model of acute inflammation in the murine air pouch [185].
Within this model, early formation of LTB4 and PGE2 at the onset of inflammation was
succeeded by a class-switching of eicosanoids to LXA4 which was concurrent with spontaneous
inflammatory resolution. During this process, interactions between inflammatory and host
tissue cells enabled the biosynthesis of resolution mediators [185]. Human PMN exposed to
PGE2 switched eicosanoid biosynthesis from predominately LTB4 to a LXA4 product, establishing
a pathway termed lipid mediator class switching. Alternatively, in a model of zymosan-A
stimulated murine peritonitis, the onset of inflammation was characterized by a concomitant
increase in LTB4 and LXA4 followed by a late appearance of PGE2 at the onset of resolution
[186]. The discrepancies in the temporal patterns of eicosanoid generation likely reflect
differences in the nature of the experimental models (microbial-initiated vs wound model)
and/or the stimulus specific signalling pathways (zymosan A vs TNF-ɑ).
In the first study to explore the appearance of lipid derived resolution mediators in an in-vivo
exercise model, maximal physical exertion on a cycle ergometer caused a rapid increase in the
urinary excretion of LXA4 immediately post-exercise [187]. These findings led the author to
suggest that the enhanced LX biosynthesis may represent a protective pro-resolving
mechanism programmed in to the early stages of acute inflammation to act as a self-regulatory
defence against exercise-induced cellular stress [187]. More recently Markworth et al. [102]
undertook an unbiased metabololipidomic profiling of human peripheral blood serum samples
obtained during the first 24 h of muscle recovery following an acute bout of unaccustomed
resistance exercise. This study showed an increase in key prostaglandin, leukotriene, lipoxin

12
and E-series resolvin species during the early stages of acute post-exercise inflammation (0-3
h). Subsequently D-series resolvins and protectins, along with 15-LOX derivatives and some
prostaglandin metabolites (6-keto-PGF1ɑ and 13,14dh-15kPGE2) remained elevated within the
circulation following 24 h of recovery [102]. Collectively these results indicate that pro-
resolving lipid mediator biosynthesis is increased in humans in response to an exercise stress
and the concept of an active inflammatory resolution model must be considered. Distinct
classes of pro-resolution lipid mediators appear to exhibit specific temporal patterns
throughout the time-course of post-exercise muscle recovery. Therefore, future research
needs to establish the profile of resolution mediators, and the transcriptional mechanisms that
regulate this pathway, in skeletal muscle tissue in models of exercise-induced inflammation.
1.5 Transcriptional regulation of exercise-induced inflammation:
To understand the link between inflammatory and regenerative processes requires the
identification of molecular signalling pathways coupling cellular stress with tissue adaptation.
NF-κB is a pleiotropic transcription factor that acts as a central regulator of stress signaling
pathways by mediating a wide array of cellular responses including immune cell recruitment,
cellular survival, proliferation and differentiation (Figure 2) [188-195]. The regulation of NF-κB
is implicated in many of the physiological processes that occur in response to tissue injury,
specifically: (1) the onset of inflammation, (2) the resolution of inflammation and (3) skeletal
muscle myogenesis.

13
Figure 1.2: NF-κB acts as a central regulator of stress signaling pathways and is involved in a diverse
number of stress signaling pathways. The function of NF-κB within cellular physiology depends on the
inciting stimulus and the tissue type.
1.5.1 Nuclear Factor-kappa B:
In mammalian tissue the Rel/NF-κB family of eukaryotic transcription factors is composed of
five structurally related subunits, RelA (p65), cREL, RelB, NF-κB1 (p50) and NF-κB2 (p52) [191,
192]. These proteins bind to form hetero- and homodimers that produce diverse combinations
of dimeric complexes [196, 197]. Each dimer possesses different DNA binding specificities, thus
different complexes can induce distinct signaling pathways specific to the activating stimulus
and cell type [196, 197]. Under basal conditions NF-κB remains sequestered within the
cytoplasm in an inactive complex bound to the inhibitor protein IκB [190, 193, 198]. There are
seven mammalian IκBs; IκBα, IκBβ, IκBε and IκBγ, BCL-3 and precursor proteins p100 and p105
that interact with specific NF-κB dimers. Upon stimulation the IκB kinase (IKK) complex
regulates the degradation of IκB proteins via phosphorylation and subsequent
polyubiquitination. The IKK complex is composed of two catalytic subunits IKK-α and IKKβ and a
regulatory subunit IKK-γ [192]. The activation and subsequent nuclear translocation of NF-κB
dimeric complexes occurs via two well described mechanisms; the classic and alternative NF-κB
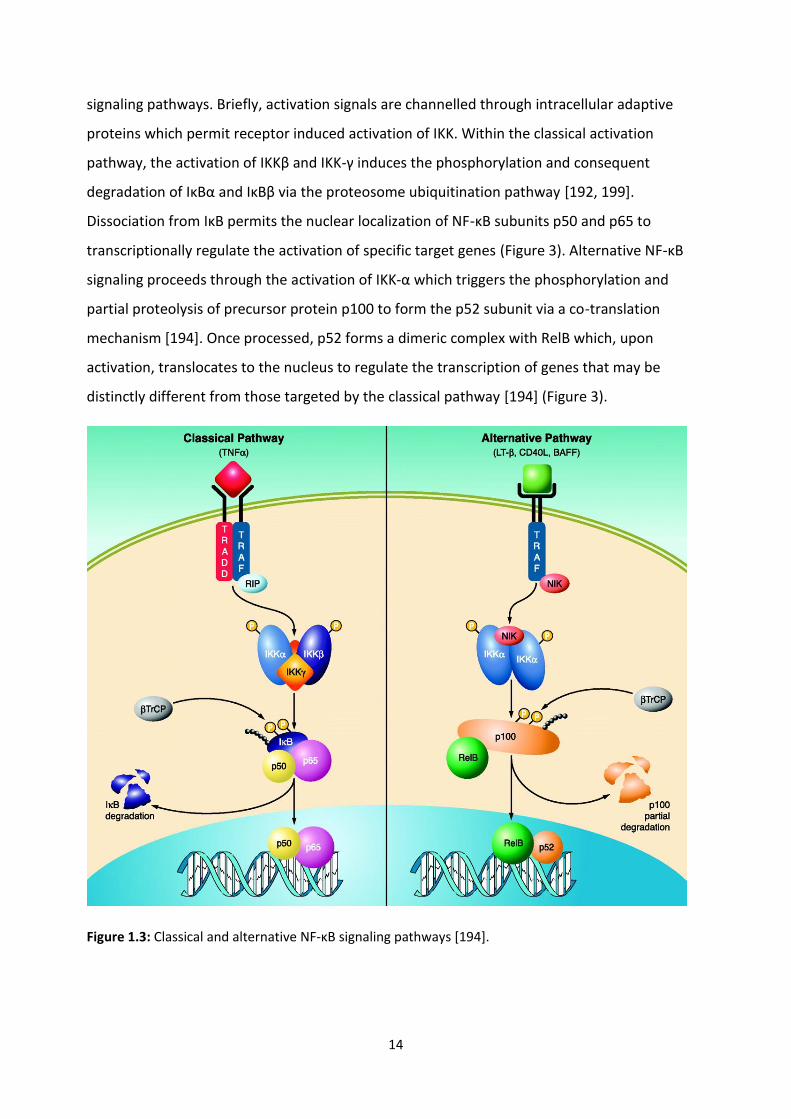
14
signaling pathways. Briefly, activation signals are channelled through intracellular adaptive
proteins which permit receptor induced activation of IKK. Within the classical activation
pathway, the activation of IKKβ and IKK-γ induces the phosphorylation and consequent
degradation of IκBα and IκBβ via the proteosome ubiquitination pathway [192, 199].
Dissociation from IκB permits the nuclear localization of NF-κB subunits p50 and p65 to
transcriptionally regulate the activation of specific target genes (Figure 3). Alternative NF-κB
signaling proceeds through the activation of IKK-α which triggers the phosphorylation and
partial proteolysis of precursor protein p100 to form the p52 subunit via a co-translation
mechanism [194]. Once processed, p52 forms a dimeric complex with RelB which, upon
activation, translocates to the nucleus to regulate the transcription of genes that may be
distinctly different from those targeted by the classical pathway [194] (Figure 3).
Figure 1.3: Classical and alternative NF-κB signaling pathways [194].

15
1.5.2 NF-κB and inflammation:
NF-κB regulates the expression of various genes that play a pivotal role in both acute and
chronic inflammatory conditions. In response to a diverse range of pro-inflammatory stimuli
including; cytokines, oxidative and thermal stress, ischemia and apoptotic mediators, NF-κB
induces pro-inflammatory genes including cytokines [200], chemokines [192], vascular and
intracellular adhesion molecules [201, 202], and acute phase proteins [203]. NF-κB also plays a
role in the promoting the cessation of an inflammatory program via the inhibition of neutrophil
apoptosis [204]. Dysregulation of apoptosis inevitably leads to the disintegration of
extravasated neutrophils. This in turn exposes surrounding tissue to large quantities of
histotoxic products causing further damage and prolonging the inflammatory response [183].
Not surprisingly chronic up regulation of NF-κB is implicated in the pathogenesis of several
inflammatory myopathies [205, 206] establishing NF-κB as a prospective target for
manipulation in the treatment of chronic inflammatory disorders [207]. However recent
research has identified a role for NF-κB in the active resolution program of acute inflammation
[208-210]. Homodimers of the alternative NF-κB activation pathway (p50 subunit) which lack a
transactivation domain, are associated with the expression of anti-inflammatory genes, the
inhibition of pro-inflammatory stimuli and the induction of apoptosis [210-212]. In a model of
rodent carageenin-induced pleurisy NF-κB activation was biphasic in nature [210]. Early
activation of the classical NF-κB pathway coincided with the onset of inflammation and the
expression of pro-inflammatory cytokines, while a secondary activation of the alternative NF-
κB pathway comprising p50 homodimers corresponded with the resolution of inflammation
[210].
1.5.3 NF- κB and skeletal muscle myogenesis:
The role of NF-κB in this process remains enigmatic and appears to be specific to the duration
of NF-κB activation, the inducing stimuli and the dimeric complex activated. Accumulating
evidence suggests that NF-κB may be essential in both inhibiting and promoting skeletal
muscle myogenesis. Evidence for a direct role of NF-κB in the inhibition of satellite cell
differentiation is well documented. Inhibition of NF-κB either via the administration of known
suppressors such as curcumin [213], or genetic manipulation [214], accelerates myogenesis, as
well as myotube formation. Likewise, pharmacological activation of NF-κB inhibits myogenesis

16
in response to severe muscle trauma and in skeletal muscle myopathies [193, 207, 215].
Further, chronic inflammation activates skeletal muscle degradation pathways involved in the
pathology of chronic disease. Chronic activation of NF-κB is observed in several diseased states
and the activation of downstream inflammatory targets are linked to skeletal muscle wasting
and suppressed myogenesis [207, 216-218]. Mechanistically, NF-κB can act through multiple
pathways to block muscle growth and repair. Recent investigations have indicated that NF-κB
inhibits myogenesis via the up regulation of transcriptional factor Ying Yang1 (YY1) which acts
as a repressor of late stage differentiation genes MyoD and MHC [193]. Alternative pathways
that have been explored include the NF-κB regulation of cyclinD1. CyclinD1 is a cyclin-
dependent kinase complex that can cause myoblasts to undergo irreversible growth arrest
[214]. Further, activation of NF-κB by a supraphysiological dose of TNF-α has deleterious
effects on satellite cell signaling [200]. Contrary to these initial findings, numerous in vitro
reports have established a positive association between NF-κB activation and skeletal muscle
myogenesis. Pharmacological inhibition of NF-κB impairs myogenesis in L6 rodent myoblasts
[219, 220]. Both lactacystin [220] and three dimensional clinorotation [219] attenuated
signaling through the classical NF-κB pathway and concomitantly inhibited the expression of
muscle-specific proteins and myoblast differentiation. Whilst our understanding of precise
signaling pathways remains poor, several different mechanisms have been proposed. Briefly,
several mitogenic precursors stimulate or are induced by NF-κB. Growth factors such as PDGF
and VEGF appear to function via an autocrine loop, acting both upstream and downstream of
NF-κB [192, 221]. Further, IGF-II signaling promotes NF-κB DNA binding during C2C12
differentiation via IKKα activation, a key component of the alternative NF-κB signaling pathway
[192]. Correspondingly, NF-κB activity increases the transcription of IGF-BP1 [222, 223] and
IGF-BP2 [224] which is required for terminal myogenic differentiation in the presence of IGF-II.
A number of reports have further identified promyogenic factor p38 MAPK as an activator of
NF-κB signaling through the classical signaling pathway [225-227]. Baeza-Raja et al. [225-227]
established a p38-dependent increase in NF-κB DNA binding activity during myogenic
differentiation in C2C12 cells. It was proposed that NF-κB activation is required for IL-6
production, a cytokine implicated in myogenesis and satellite cell signaling [225]. It is also
plausible to consider a role for TNF-α in this pathway. This cytokine is a known transcriptional
target of NF-κB and at high levels acts as a potent inhibitor of myogenesis in skeletal muscle
myopathies. However, recent in-vitro research discovered low levels of TNF-α promotes

17
myogenesis via signaling through the myogenic activity of p38 MAPK [228], establishing a
conceivable link between NF-κB and TNF-α in skeletal muscle satellite cell activation [229]. This
premise is validated by the role of TNF- α as a positive regulator of myogenesis in cardiotoxin-
injured muscle [230]. Additionally NF-κB induces hypertrophic growth in cardiomyocytes in-
vitro [231]. The discrepancies that exists within the literature suggest that the effects NF-κB
and TNF-α signaling exert on myogenic pathways is likely to be specific to the activating
stimulus, the timing and duration of activation and the precise NF-κB pathway that is activated.
1.5.4 NF- κB and exercise:
Acute exercise triggers a transient rise in various components of the NF-κB signaling pathway
in various cell types [232-236]. However, a precise functional role for NF-κB in contraction-
induced muscle damage remains speculative. Key components of the NF-κB signaling pathway
are up regulated in human peripheral blood lymphocytes and rodent skeletal muscle tissue
immediately post exercise, implicating a potential role for NF-κB in acute muscle damage [232,
234, 235]. Further, this rise appears to peak between 1 and 5 h post exercise, a time point that
coincides with the recruitment and activation of inflammatory neutrophils [232, 235]. Protein
expression of p50/p65 complex and NF-κB DNA binding remains activated at 24 h post-exercise
[232, 234], however a functional role for NF-κB at this time point was not established.
Interestingly Ho et al. [233] observed no change in NF-κB DNA binding activity at 5 h post
exercise in rodent skeletal muscle tissue whilst Ji et al. [234] employed a similar research
model, and identified an increase in NF-κB DNA binding at 4 and 24 h. Collectively these two
experiments may provide an insight into the regulatory patterns of NF-κB following exercise.
Models of TNF-α induced activation of NF-κB and ischemic reperfusion injury, have identified a
biphasic activation pattern of NF-κB in response to cellular stress [216, 237]. The first phase is
transient in nature and appears to peak between 30 min and 3 h post exercise. Activation of a
secondary phase differs between studies, with TNF-α triggering an increase in NF-κB
transactivation function between 24 and 36 h post exposure [216] and ischemic reperfusion
injury causing an increase NF-κB DNA binding between 6 and 16 h [237]. The discrepancy in the
literature is likely to reflect a difference in the stress induced stimulus. This also highlights a
need for further research to examine the time course activation of both the classical and
alternative NF-κB signaling pathways following exercise-induced muscle damage.

18
Understanding the complexity of this pathway and the potential interactions between classical
and alternative NF-κB pathways will provide new insights into the mechanistic link between
exercise-induced inflammation and cellular regeneration pathways.
Remarkably, a paucity of research exists examining the effect of exercise on NF-κB signaling in
human skeletal muscle tissue. Durham et al. [238] conducted an initial trial examining the
effect of an acute resistance exercise bout on the activation of the NF-κB pathway. This
research established a 44% decrease in NF-κB DNA binding activity at 10 min post exercise and
no change at 1 h post [238]. Contrastingly, Tantiwong et al. [236] used a moderate intensity
cycling protocol and observed no change in DNA binding activity immediately post exercise and
a 90% increase at 210 min post. It is likely that the observed disparities between the two trials
may be caused by the differences in exercise protocol or the difference in the time course of
muscle biopsy sampling. Further research from Vella et al. [239] demonstrated a significant
increase in NF-κB (p65) in response to traditional resistance exercise at 2 h post-exercise.
These findings led to the novel exploration for a potential target of NF-κB (p65) in response to
a resistance exercise stimulus and identified NF-κB as a transcriptional regulator of pro-
inflammatory myokines MCP-1, IL-6 and IL-8. EMSA data revealed that NF-κB binding to the
promoter region on all three respective genes increased significantly at 2 h post exercise and
returned to basal levels by 4 h. While these findings establish NF-κB as a key regulator of post-
exercise inflammatory pathways, the regulatory circuitry of the NF-κB pathway, and the
heterogeneity of NF-κB target genes, suggests that our understanding of NF-κB signaling
following exercise is incomplete. Future research needs to consider the activation of both
classical and alternative NF-κB pathways beyond the 4 h time point examined in human
research, and further explore the role of NF-κB in post-exercise inflammatory resolution and
skeletal muscle myogenesis.
1.6 Summary:
The human response to resistance exercise presents a highly complex paradigm. Simplistically,
the outcome of post-exercise inflammation can be regarded as a battle between those
mechanisms that augment the inflammatory response and cause further injury, against those

19
that tend to promote the resolution of inflammation and skeletal muscle repair. The skeletal
muscle response to exercise-induced muscle damage occurs in a well described schematic of
events; degeneration, inflammation and, regeneration. While processes of degeneration and
inflammation can exacerbate symptoms of muscle injury and promote sensations of pain, a
considerable body of literature has established a clear link between signaling pathways in
inflammation and skeletal muscle adaptation. Developing an understanding of the molecular
mechanisms regulating this physiological link is vital in establishing therapeutic treatment
strategies to optimise muscle recovery from exercise-induced tissue injury.
The acute post-exercise inflammatory response is self-resolving in nature and recent works
have identified a series of novel lipid derivatives that play a role in regulating a biologically
active inflammatory resolution program. While this concept remains largely unexplored in
humans post-exercise, initial research demonstrates these lipids are increased in human serum
samples following exercise and may provide a novel mechanism in understanding the link
between inflammation and tissue adaptation [102]. Further, NF-κB is a transcription factor
shown to couple cellular stress with an adaptive or maladaptive response. It has been
implicated in post-exercise inflammation and also demonstrated to be involved in an in-vitro
model of acute inflammatory resolution. The concept that events occurring early in acute
inflammation engage an active cellular resolution program that is intimately necessary for the
activation tissue adaptive pathways, presents a novel and unexplored link between cellular
stress and tissue regeneration that may provide insight into the mechanisms that regulate
post-exercise muscle recovery.
1.7 General hypothesis:
The overall hypothesis of this thesis is that an acute inflammatory resolution pathway exists in
post-exercise inflammation. A biologically active inflammatory resolution pathway provides a
potential mechanism through which cellular events occurring early within an acute
inflammation engage a complex cascade that ultimately leads to the restoration of tissue
homeostasis and skeletal muscle adaptation. NF-κB presents a transcription factor involved in
processes of acute inflammation, inflammatory resolution and myogenesis. Interactions

20
between classical and alternative NF-κB signaling pathways may provide a transcriptional
target that links these cellular processes and plays an integral role in recovery from exercise-
induced muscle damage. This hypothesis will be tested by investigating the specific aims
outlined below.

21
1.8 Specific Aims:
1)
a) To investigate if oral administration of ibuprofen influences skeletal muscle leucocyte
infiltration during the first 24 h after muscle-damaging resistance exercises.
b) To determine if individual changes in leucocyte infiltration are correlated to markers of
muscle damage, including circulating muscle proteins CK and myoglobin, and subjective
markers of muscle soreness.
2) To determine the effect of acute resistance exercise on a novel family of lipid derived
eicosanoids and docosanoids that play a key role in driving a biologically active
inflammatory resolution program.
3) To examine the effect of an acute bout of resistance exercise on regulating members of the
classical and alternative NF-κB signaling pathways in skeletal muscle of young untrained
men.
1.9 Hypothesis:
1. The ingestion of ibuprofen following acute resistance exercise will not influence the
infiltration of leucocytes, however it will attenuate sensations of muscle soreness.
2. Acute resistance exercise will trigger an increase in a novel series of lipid-derived
inflammatory mediators that are involved in regulating an active inflammatory
resolution pathway.
3. Acute resistance exercise will trigger an increase in key parameters of the NF- κB
signaling pathways. The activation of NF-κB will be biphasic in nature, involving the
activation of the classical NF-κB pathway at the onset of inflammation, and the
alternative NF-κB pathway coincident with the resolution of acute inflammation.

22
CHAPTER 2:
Ibuprofen ingestion does not affect markers of post-exercise muscle inflammation.
Vella L, Markworth JF, Paulsen G, Raastad T, Peake JM, Snow RJ, Cameron-Smith D, Russell
AP. Ibuprofen ingestion does not affect markers of post-exercise muscle inflammation.
Frontiers in Exercise Physiology, 2016 Mar 29;7:86. doi: 10.3389/fphys.2016.00086
(Appendix A).

23
2.1 Abstract:
Purpose: Chapter 2 investigated if oral ingestion of ibuprofen influenced leucocyte recruitment
and infiltration following an acute bout of traditional resistance exercise. Methods: Sixteen
male subjects were divided into two groups that received the maximum over-the-counter dose
of ibuprofen (1200 mg d-1) or a similarly administered placebo following lower body resistance
exercise. Muscle biopsies were taken from m.vastus lateralis and blood serum samples were
obtained before and immediately after exercise, and at 3 h and 24 h after exercise. Muscle
cross-sections were stained with antibodies against neutrophils (CD66b and MPO) and
macrophages (CD68). Muscle damage was assessed via creatine kinase and myoglobin in blood
serum samples, and muscle soreness was rated on a ten-point pain scale. Results: The
resistance exercise protocol stimulated a significant increase in the number of CD66b+ and
MPO+ cells when measured at 3 h post exercise. Serum creatine kinase, myoglobin and
subjective muscle soreness all increased post-exercise. Muscle leucocyte infiltration, creatine
kinase, myoglobin and subjective muscle soreness were unaffected by ibuprofen treatment
when compared to placebo. There was also no association between increases in inflammatory
leucocytes and any other marker of cellular muscle damage. Conclusion: Ibuprofen
administration had no effect on the accumulation of neutrophils, markers of muscle damage or
muscle soreness during the first 24 h of post-exercise muscle recovery.

24
2.2 Introduction:
Unaccustomed resistance exercise often results in tissue damage and inflammation, leading to
delayed onset muscle soreness (DOMS) and a consequent reduction in force production [31,
240]. Animal models and in vitro studies have identified that local and systemic inflammation
exerts a regulatory influence during the different phases of muscle recovery, including
myofibrillar disruption, cellular necrosis, satellite cell activation, maturation and subsequent
regeneration and adaptation [17, 24]. Thus, post-exercise inflammation is intimately necessary
and a key feature of the normal process of tissue regeneration and adaptation following acute
muscle damage. However, excessive inflammation is considered a potential cause of prolonged
post-exercise muscle soreness and may have a negative effect on muscle recovery [24, 25];
consequently strategies to reduce or counteract inflammation are commonly implemented to
aid in improving muscle recovery after exercise [103].
Non-steroidal anti-inflammatory drugs (NSAIDs) are commonly used as a treatment strategy in
exercise and sports medicine to assist with recovery from exercise-induced inflammation,
particularly following soft-tissue injury. NSAIDs inhibit the cyclooxygenase (COX-1 and 2)
enzymes and consequently the formation of prostanoids (prostaglandins, prostacyclins and
thromboxanes) that play a diverse roll in acute inflammation [102]. Prostanoids stimulate an
acute inflammatory process by controlling local blood flow, vascular permeability, leucocyte
infiltration, and triggering sensations of pain [102, 103]. In animal models of acute muscle
damage, treatment with NSAIDs blunts the infiltration of leucocytes into muscle tissue [104,
159] and causes a reduction in creatine kinase (CK) [106]. Consequently, NSAIDs can also
inhibit myofiber regeneration, satellite cell proliferation and differentiation, and overload-
induced muscle hypertrophy [104-106]. These findings provide preliminary evidence that
NSAIDs compromise the physiological link between processes of acute muscle damage,
inflammation and cellular regeneration.
Research into the effects of NSAIDs on exercise-induced muscle damage and inflammation has
produced equivocal findings. In exercise models, NSAID administration has been shown to
attenuate post-exercise DOMS in some [93, 140, 241], but not all studies [87, 107, 151, 152].

25
While a precise cellular mechanism for an analgesic effect of NSAIDs remains unclear, it has
been largely ascribed to their effect on prostaglandin synthesis and the capacity of NSAIDs to
interfere with aspects of inflammatory cell function [160, 242]. Previous research has
demonstrated that oral consumption of both ibuprofen, a non-selective NSAID and
acetaminophen, an analgesic also known as paracetamol, had no effect on macrophage
infiltration at 24 h following an eccentric exercise protocol [160]. Similarly treatment with
naproxen, another non-selective NSAID, had no effect on the infiltration of leucocyte common
antigen positive cells following a unilateral, isotonic resistance exercise protocol [147].
Interestingly, Paulsen et al., (2010) suggested a blunting effect of the COX-2 specific celecoxib
following maximum eccentric muscle contractions [93]. This research identified a tendency for
higher monocyte/macrophage numbers in subjects within the placebo group, and those
subjects who were identified as ‘high-responders’ to the exercise protocol based on the
number of inflammatory leucocytes [93]. Although this is not a particularly robust finding, it
has led to the hypothesis that NSAIDs may influence leucocyte infiltration in skeletal muscle
when a sufficiently strong and early inflammatory reaction is present [93]. This hypothesis
would suggest that the intensity of the exercise stimulus and the consequent muscle-damage
response would be largely influential in determining the effect of a pharmacologically based
anti-inflammatory intervention.
Conflicting findings also exist with regard to the effect of NSAIDs on the regenerative capacity
of skeletal muscle following exercise-induced muscle damage. The non-selective COX inhibitor
ibuprofen blunted skeletal muscle protein synthesis [107] while local intramuscular infusion of
indomethacin [243] and the oral administration of the COX-2 selective NSAID celecoxib [93,
130] had no such effect. Similarly, treatment with indomethacin inhibited post-exercise
satellite cell proliferation [87]. Recent work from our group demonstrated that treatment with
ibuprofen inhibited early translational signalling responses involved in post-exercise muscle
hypertrophy [161]. A clear mechanistic pathway for NSAIDs to influence the physiological link
between post-exercise inflammation and skeletal muscle regeneration remains elusive.

26
These discrepancies in the research to date are likely due to differences in the exercise
protocol (concentric vs. eccentric muscle contractions), the training status of subjects, the
timing of muscle biopsies, and the type of NSAID, the dosage administered, and method of
administration. Further research is required to determine whether NSAID administration
affects the infiltration of leucocyte populations following exercise-induced muscle damage and
how this influences post-exercise adaptive pathways. The aim of the present study was to
investigate if oral administration of the NSAID ibuprofen influenced skeletal muscle leucocyte
infiltration during the first 24 h after muscle-damaging resistance exercises. A further aim was
to explore how any changes in leucocyte infiltration were related to markers of muscle
damage, including circulating muscle proteins CK and myoglobin, and subjective markers of
muscle soreness. It was hypothesized that the ingestion of ibuprofen following acute
resistance exercise would not influence the infiltration of leucocytes, however it would
attenuate sensations of DOMS.

27
2.3 Methods:
2.3.1 Participants:
Sixteen healthy male subjects were recruited to participate in the study (Table 1) [102].
Exclusion criteria included participation in a lower body resistance exercise program within the
last 6 months to ensure a muscle damage response from the exercise stimulus, and/or
previous chronic treatments with anti-inflammatory medication. Participants also completed a
medical screening to identify any potential risk factors for them to perform strenuous physical
activity.
Table 2.1: Subject characteristics and strength testing data. Values are mean values ± SEM. No
significant differences were observed between the two groups.
Characteristics Strength (1RM)
Age (y) Height
(m)
Body mass
(kg)
BMI Squat (kg) Leg Press
(kg)
Leg
Extension
(kg)
PLA 23.9
± 1.3
1.89
± 0.1
86.9
± 4.5
24.5
± 1.2
94.9
± 5.4
237
± 17
236
± 18
IBU 23.0
± 0.5
1.89
± 0.1
89.1
± 4.4
24.8
± 0.8
91.9
± 6.0
240
± 15
196
± 22
PLA, placebo; IBU, Ibuprofen; BMI, body mass index
2.3.2 Ethics approval:
All procedures involved in this study were approved by the Deakin University Human Research
Ethics Committee (DUHREC 2010-019) and muscle sampling procedures were performed in
accordance with the Helsinki declaration. Each participant was provided with written and oral
details of the nature and requirements of the study and provided written consent to
participate.

28
2.3.3 Familiarization:
Each participant completed a familiarization and strength testing session at least 7 days prior
to completing the exercise protocol. Briefly, subjects performed repetition maximum testing
for the Smith machine-assisted squat, the leg press and the leg extension to determine their
experimental exercise load (80% of a 1 repetition maximum (1RM)). The maximum weight the
subject could lift for 5-8 reps was determined, and these data were entered into the Brzycki
equation to predict 1RM [1 RM = 100 X load rep/(102.78 – 2.78 X reps completed] [244].
Subjects were asked to abstain from any further activity until the completion of the trial.
2.3.4 Experimental Procedures:
The participants reported to the laboratory on the morning of the trial in an overnight fasted
state. They were asked to abstain from caffeine, tobacco and alcohol for the 24 h preceding
the trial. Participants rested in a supine position for 30 min, following which the first muscle
biopsy sample was taken. Each participant then completed a 10-min warm-up protocol
comprised of 5 min of low intensity cycling on a stationary bike, and one low-intensity set of
each exercise at a weight of each subject’s own choice within the range of 30-50% 1RM. The
resistance exercise session consisted of 3 sets of 8-10 repetitions performed on a Smith
machine assisted squat, a 45-degree leg press and a leg extension at 80% of a predicted 1RM.
The exercises were performed as a circuit with 1 min rest permitted between exercises and 3
min rest between sets. This protocol has been used previously and has been a sufficient
stimulus to activate inflammatory signalling pathways [239] and was implemented to replicate
a commonly used exercise routine. After exercise, the subjects rested while subsequent muscle
biopsy samples were collected. After the 3 h biopsy, participants were provided a standardized
meal and could go home. The following morning, participants reported to the laboratory in an
over-night fasted state for a 24 h muscle biopsy and blood sample.
2.3.5 Standardized meals:
Standardized meals were provided to participants on the night before the trial, (carbohydrate
57%, fat 22%, protein 21%), immediately following the 3 h muscle biopsy (carbohydrate 71%,
fat 13%, protein 16%), and in the evening (carbohydrate 64%, fat 27%, protein 18%) on the day

29
of the exercise trial. All participants received the same meal and individualised energy content
based on body mass was not provided. Participants were permitted to drink water ad libitum
and were asked to report if they could not finish their allocated meals. This nutrition plan was
included to ensure that each participant received the same relative percentage of macro- and
micro-nutrients.
2.3.6 NSAID Administration:
Prior to exercise, participants were randomly assigned in a double-blind method, to consume
either the maximum recommended dose of ibuprofen (IBU, n = 8) or a placebo control (gelatin
capsules identical in appearance containing powdered sugar in place of ibuprofen) (PLA, n = 8).
The IBU group consumed three doses of 400 mg of ibuprofen throughout the trial day. The first
dose was administered immediately prior to the first muscle biopsy sample. Participants were
instructed to consume the following two doses at 6 h and 12 h following the exercise protocol.
Follow-up phone calls from the research team ensured compliance. To ensure this dosing
structure was appropriate to maintain biologically active levels of ibuprofen, ibuprofen
concentration was measured in blood serum samples and these data are reported elsewhere
[102].
2.3.7 Sample collection:
Venous blood samples were drawn prior to exercise, within 5 min post exercise (here-after
referred to as 0 h post-exercise), and at 1, 2, 3 and 24 h post-exercise. Blood samples were
drawn through an indwelling catheter into VACUETTE serum tubes (Greiner,Bio-One,
Stonehouse UK). Whole blood was allowed to clot at room temperature. Samples were then
centrifuged at 1,000 g for 10 min. The serum layer was collected and stored at -80°C for
subsequent analysis.
Muscle biopsy samples were obtained from the vastus lateralis muscle under local anaesthesia
(Xylocaine 1%) using a percutaneous needle biopsy technique modified to include suction
[245]. To minimize interference from the biopsy procedure, samples prior to exercise and at 24

30
h post exercise were taken from the same leg, while samples taken at 0 and 3 h post-exercise
were taken from the contralateral leg. Samples obtained within the same leg were taken at
least 5cm from the previous incision site. A section of excised tissue was immersed in Tissue-
Tek and stored at -80°C until further analysis. We have previously reported that this technique
is effective for minimizing the risk of any inflammation arising from the biopsy procedure
confounding exercise-induced inflammation [239]. This research showed no effect of the
biopsy protocol on mRNA expression of acute phase inflammatory cytokines, however did not
directly investigate a potential effect on leucocyte infiltration or markers of cellular muscle
damage. The absence of a non-exercise control group is a limitation of the present study.
2.3.8 Immunohistochemistry:
Cross sections (8 μm) of muscle tissue were cut using a microtome at -20 ⁰C (CM3050, Leica,
Germany) and mounted on microscope slides (Superfrost Plus, Thermo Scientific, MA, USA),
air-dried and stored at -80 °C. Each subject’s muscle sections from all time points were
mounted on the same microscope slide. Serial muscle cross-sections were stained with three
different leucocyte antibodies, MPO (#0398, DakoCytomation, Glostrup, Denmark; dilution
1:2000), CD66b (#CLB-B13.9, Sanquin Reagents, Amsterdam, The Netherlands; dilution 1:500)
and CD68 (#EBM-11, DakoCytomation, Glostrup, Denmark; dilution 1:300). Furthermore, the
sections were also stained with dystrophin antibody (ab15277, Abcam, Cambridge, UK) for
visualizing borders of muscle fibers
.
Sections were fixed in 4 % paraformaldehyde solution in a staining jar for 5 min at room
temperature and rinsed twice for 10 min in phosphate buffered saline (PBS) containing 0.5 %
Tween 20 (Sigma-Aldrich). The microscope slides were moved into a humidified chamber and
non-specific binding sites were blocked with 1% bovine serum albumin (BSA) on the section for
45 min at room temperature. Sections were incubated overnight with leucocyte and
dystrophin antibodies diluted in 1% BSA at 4°C. After overnight incubation, the slides were
washed three times for 10 min in PBS-Tween in a staining jar. Slides were moved back to the
humidified chamber and sections were incubated for 45 min with secondary antibodies diluted
1:200 in 1% BSA at room temperature. Alexa Fluor® 594 F(ab’)2 fragment of goat anti-rabbit

31
and Alexa Fluor®488 anti-mouse IgG (Invitrogen, Eugene, Oregon, USA) were used as a
secondary antibodies. The fluorochrome-stained sections were washed three times for 10 min
in PBS-Tween. After the last washing in PBS-Tween, the sections were mounted on ProLong®
Gold Antifade reagent with DAPI (Invitrogen, Eugene, Orgeon, USA).
Muscle sections were visualized using a high-resolution camera (DP72, Olympus, Japan)
mounted on a microscope (BX61, Olympus, Japan) with a fluorescence light source (X-Cite
120PCQ, EXFO, Canada). The number of MPO, CD66b and CD68 positive cells (as well as the
total number of muscle fibres from the area included) was counted. Data was calculated as
percentage of MPO, CD66b or CD68 positively stained cells per 100 skeletal muscle fibres.
Areas of sections that contained freeze damage or were folded due to the cutting procedure
were not included in the analysis.
2.3.9 Biochemical assays:
Serum creatine kinase activity in pre- and post-exercise blood samples was analyzed using an
enzymatic assay (CK-NAC kit, CDT14010, Thermo-Fisher Scientific Clinical Diagnostics, Sydney,
Australia) and an automated clinical analyser (Cobas Mira, Roche Diagnostics, Germany).
Serum myoglobin concentration was also measured in these samples using an immunoassay
(Roche Diagnostics, Germany) and an automated clinical analyzer (Cobas E411, Roche
Diagnostics, Germany). The intra-assay coefficient of variation was 10.4% for creatine kinase
and 1.7% for myoglobin.
2.3.10 Muscle soreness assessment:
Upon arrival at the laboratory and prior to the first muscle biopsy procedure subjects were
asked to rate their subjective muscle soreness on a 0-10 visual analogue scale. The subjects
were instructed to contract, stretch and palpate the quadriceps muscle to assess general
muscle soreness. In both instances 0 was considered to represent no pain and correspondingly
a rating of 10 was considered to represent intense pain.

32
2.3.11 Statistics:
Data are expressed as means ± SEM. Prior to analysis the data displaying a lack of normality
were log transformed to stabilise variance. Data were analysed using a two-way ANOVA with
repeated measures for time. The sphericity adjustment was checked, and if required, a
Greenhouse-Geisser epsilon correction was applied. Where no significant effect for treatment
was observed, we explored pair-wise comparisons between individual time-points using the
Least Significant Difference (LSD) of means. Bivariate relationships were examined with a
Spearman rank correlation test. Confidence intervals were calculated at 90% using a
bootstrapping technique for non-normally distributed data. Statistical analyses were
performed using GenStat for Windows 16th Edition (VSN International, Hemel Hempstead, UK).
Statistical significance was set at P < 0.05.

33
2.4 Results:
2.4.1 Muscle Soreness:
Muscle soreness showed a main effect for time (p < 0.01), suggesting that the exercise protocol
was sufficient to induce a DOMS response. No effect of the ibuprofen treatment was observed
(Figure 2.1).
Figure 2.1: Subjective rating for delayed onset muscle soreness (DOMS) (B). Values depicted are
mean values ± SEM. * denotes statistical significance from pre-exercise values (p<0.05). White bars
= PLA group; black bars = IBU group. This figure has been adapted from Vella et al. (2014) [246].
2.4.2 Immunohistochemistry:
The number of MPO+ cells per 100 myofibres analysed showed a main effect for time (P <
0.01). No main effect for treatment (P = 0.250) or time x treatment (P = 0.709) was observed
(Figure 2.2A). LSD pairwise comparisons indicated a significant increase in MPO+ cells at all
sampling time points post-exercise. The greatest increase in MPO+ cells was observed at 3 h
post-exercise (Figure 2.2B). Similarly, the number of CD66b+ cells per 100 myofibres showed a
main effect for time (P < 0.05), with no main effect for treatment (P = 0.149) or time x
treatment interaction (P = 0.907) (Figure 2.2C). LSD pairwise comparisons indicated a
significant increase in the number of CD66b+ cells at 3 h post exercise (Figure 2D). The number
of CD68+ cells per 100 myofibres analysed showed no effect for time (P = 0.061), treatment (P
= 0.530) or time x treatment interaction (P = 0.688) (Figure 2.2D). MPO+, CD66b+, and CD68+
cells were observed within the endomysium and the perimysium, with no cellular infiltration
occurring at any time point (Figure 2.2E). Representative images for each cell surface marker
are presented in Figure 3.

34
Figure 2.2: Immunohistochemistry data for muscle cells staining positive for myeloperoxidase
(MPO), showing group specific data (A) and collapsed data (B), CD66b showing group specific data
(C) and collapsed data (D), and CD68 showing group data only (E). Data represent the mean number
of positively stained cells per 100 fibres analysed ± SEM. ** denotes statistical significance from
pre-exercise values (p<0.01). * denotes statistical significance from pre-exercise values (p<0.05). ^
denotes statistical significance from 0 h post-exercise (p<0.05). White bars =PLA group; black bars =
IBU group.

35
Figure 2.3: Immunohistochemical analysis of skeletal muscle samples following a bout of resistance
exercise. Sections were probed with antibodies raised against leucocyte cell surface markers (green;
Figure 3A– MPO, Figure 3B – CD66b, Figure 3C – CD68) and dystrophin (red), while DAPI was used
to stain nuclei (blue). Scale bars = 50µm.
2.4.3 Serum proteins:
Serum CK activity demonstrated a main effect for time (P < 0.01) with no effect for treatment
(P = 0.843) or time x treatment interaction (P = 0.494) (Figure 2.4A). LSD comparisons revealed
that serum CK increased from pre-exercise values at 2 h post-exercise and peaked at 24 h post-
exercise (Figure 2.4B). Similarly, serum myoglobin concentration showed a main effect for time
(P < 0.01) with no effect for treatment (P = 0.710) or time x treatment interaction (P = 0.666)
(Figure 2.4C). LSD comparisons demonstrated that the biggest increase in serum myoglobin
concentrations occurred at 1 h post-exercise and remained significantly elevated up to 3 h post
exercise (Figure 2.4D).

36
Figure 2.4: Blood derived proteins creatine kinase (A and B) and myoglobin (C and D). Values
depicted are mean values ± SEM. Figures A and C represent data divided into two treatment
groups; figures B and D represent collapsed data. *denotes statistical significance from pre-exercise
values (p<0.05). ** denotes statistical significance from pre-exercise values (p<0.01). ^ denotes
statistical significance from 1, 2 and 3 h post exercise (p<0.05). White bars = PLA; black bars = IBU.
2.4.4 Correlation analysis:
In general, large inter-individual differences were seen in the appearance of all of the leucocyte
cell types. Results from the Spearman rank correlation test showed no correlation between the
histochemical appearance of inflammatory leucocytes, biological markers of muscle damage
and subjective markers of muscle soreness (Table 2). Anecdotally, three subjects were
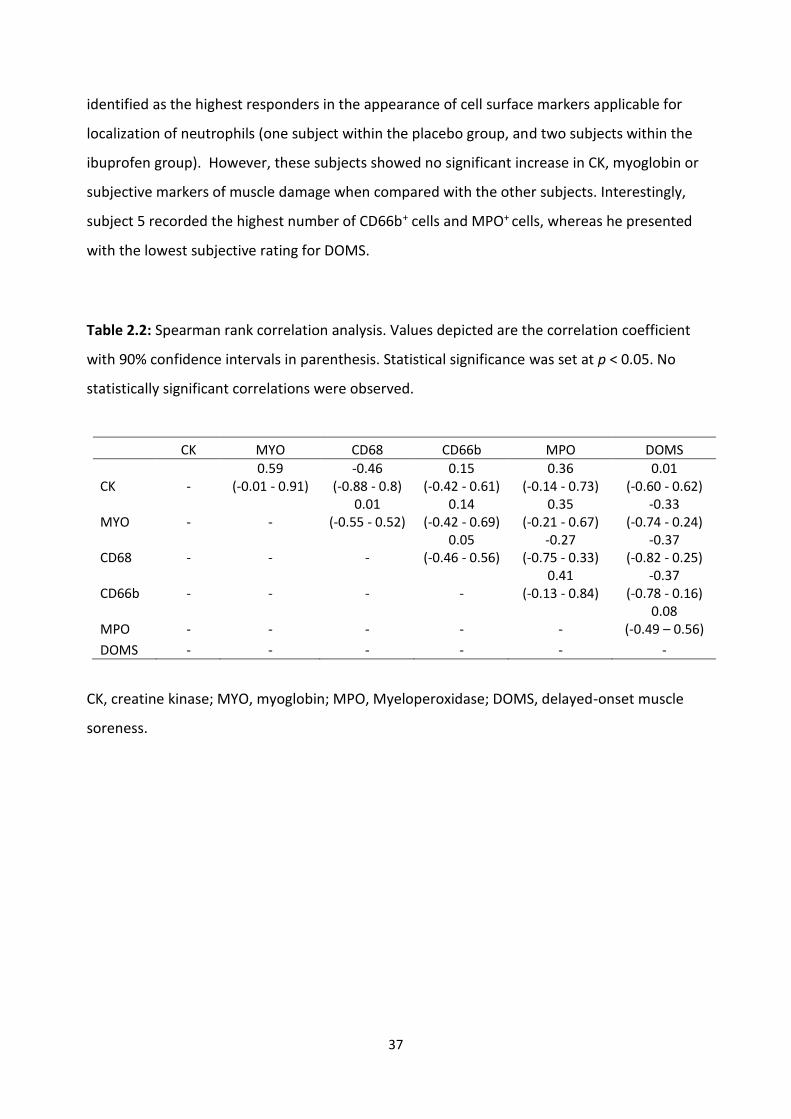
37
identified as the highest responders in the appearance of cell surface markers applicable for
localization of neutrophils (one subject within the placebo group, and two subjects within the
ibuprofen group). However, these subjects showed no significant increase in CK, myoglobin or
subjective markers of muscle damage when compared with the other subjects. Interestingly,
subject 5 recorded the highest number of CD66b+ cells and MPO+ cells, whereas he presented
with the lowest subjective rating for DOMS.
Table 2.2: Spearman rank correlation analysis. Values depicted are the correlation coefficient
with 90% confidence intervals in parenthesis. Statistical significance was set at p < 0.05. No
statistically significant correlations were observed.
CK MYO CD68 CD66b MPO DOMS
CK - 0.59
(-0.01 - 0.91) -0.46
(-0.88 - 0.8) 0.15
(-0.42 - 0.61) 0.36
(-0.14 - 0.73) 0.01
(-0.60 - 0.62)
MYO - - 0.01
(-0.55 - 0.52) 0.14
(-0.42 - 0.69) 0.35
(-0.21 - 0.67) -0.33
(-0.74 - 0.24)
CD68 - - - 0.05
(-0.46 - 0.56) -0.27
(-0.75 - 0.33) -0.37
(-0.82 - 0.25)
CD66b - - - - 0.41
(-0.13 - 0.84) -0.37
(-0.78 - 0.16)
MPO - - - - - 0.08
(-0.49 – 0.56)
DOMS - - - - - -
CK, creatine kinase; MYO, myoglobin; MPO, Myeloperoxidase; DOMS, delayed-onset muscle
soreness.

38
2.5 Discussion:
The main finding of this study was that ibuprofen, a non-selective COX inhibitor, had no effect
on the histological detection of leukocytes following an acute bout of traditional resistance
exercise. Serum CK and myoglobin, and muscle soreness also did not change in response to
ibuprofen. Furthermore, no correlation was observed between the accumulation of
inflammatory leucocytes, increases in CK or myoglobin, and sensations of DOMS. These
observations suggest that the intramuscular infiltration of inflammatory white blood cells, or
an increase in serum levels of intramuscular proteins, are not predictive of post-exercise
muscle soreness.
The current study focused on the acute local inflammatory response to exercise-induced
muscle damage. Our protocol was effective in inducing muscle soreness as shown in Figure 2.1.
An increase in the histological appearance of CD66b+ and MPO+ cells occurred at 3 h post
exercise, while no effect of the NSAID treatment was observed. CD66b is a highly specific cell
surface marker for detecting human neutrophils, and this is the first paper to show a small but
significant increase in the intramuscular number of CD66b+ cells following acute resistance
exercise. Paulsen et al. (2013) were unable to identify any change in the number of CD66b+
cells in the elbow flexors at 1 h, 2, 4 or 7 days following 70 maximal eccentric contractions [97].
The discrepancy between the two trials may be the result of a combination of methodological
differences, such as the timing of muscle biopsy samples, the mode of exercise and the muscle
group sampled. A small but significant increase in the number of MPO+ cells at all sampled time
points was also observed, with a peak in expression at 3 h post exercise. MPO has typically
been used to detect changes in neutrophils, and an increased number of MPO+ cells have been
reported following high-force eccentric muscle contractions [98, 247]. However, MPO is also
expressed on the lysosomes of monocytes and has been detected on basophils and
eosinophils, perhaps providing a reason why there were a higher number of MPO+ cells when
compared to CD66b+ cells detected in the present study [97].
No change was identified in the number of CD68+ cells. CD68 cell counts are widely used as a
marker of monocyte/macrophage infiltration. However, CD68 may also be expressed on other

39
cell types including satellite cells and fibroblasts [97]. Other researchers have been able to
identify a change in CD68+ cells following exercise-induced muscle damage [98, 160, 247]. Each
of these studies have used a high-force maximal eccentric muscle contraction protocol, which
may suggest that the detection of CD68+ cells is dependent on the extent of skeletal muscle
damage.
Previous research exploring the effect on NSAIDs on post-exercise inflammation has produced
equivocal findings. Earlier research from in-vivo rodent trials demonstrated a blunted
inflammatory response to eccentric muscle contractions following the administration of both
selective and non-selective NSAIDs [104, 105, 248, 249]. However, these findings are yet to be
supported in human trials [140, 147, 160]. Petersen et al. (2003) found no effect of oral
ibuprofen administration on the appearance of CD68+ cells 24 h following 100 eccentric muscle
contractions [160]. Similarly, Tokmakidis showed no effect of ibuprofen on circulating white
blood cells 24 h following an eccentric exercise protocol [140]. Paulsen et al. (2010) proposed
the concept of an inflammatory threshold, suggesting that NSAIDs may only influence post-
exercise inflammation in response to a sufficiently strong inflammatory stimulus [93]. It was
suggested that the absence of an intramuscular prostaglandin response to exercise, specifically
PGE2, could explain the lack of an NSAID effect on leucocyte recruitment [93, 132].
Interestingly, our group recently reported an increase in serum PGE2 in samples that were
obtained during the same exercise trials as those currently reported [102]. This change in
serum PGE2 was blunted by the administration of ibuprofen and suggests that in the presence
of a blunted prostaglandin response to exercise, NSAID treatment had no effect on leucocyte
recruitment within skeletal muscle. Although this finding does not refute the possibility of an
inflammatory threshold, based on our previous findings [102], it is reasonable to suggest that
the presence of an exercise-induced serum prostaglandin response is not the required
underlying mechanism for neutrophil infiltration of muscle during the early hours of post-
exercise recovery.
In accordance with previous studies, resistance exercise resulted in an increase in serum CK
and myoglobin, and subjective muscle soreness [250]. Although large inter-individual variations

40
in the human response to exercise can make it difficult to establish causal relationships, there
was no relationship between the appearance of leucocytes and blood-derived markers of
muscle damage or sensations of muscle soreness. Interestingly, the subject with the highest
number of CD66b+ and MPO+ cells presented with the lowest subjective rating of DOMS. These
findings provide support for the assumption that leucocyte recruitment and inflammation do
not consistently cause DOMS.
From animal studies, it appears that cellular events in post-exercise inflammation, occur
concomitantly with the onset of DOMS [24, 240]. The mechanistic link has been proposed to be
a leucocyte-mediated release of cytotoxic substances and reactive oxygen species as a bi-
product of phagocytosis [251]. Furthermore, the prostaglandin response to exercise has been
shown to occur concurrently to the onset of DOMS, and is known to influence both the
recruitment of inflammatory leucocytes and neural afferents to pain [102]. However, in line
with the current findings, the accumulation of inflammatory leucocytes in human skeletal
muscle tissue during exercise recovery appears to be both spatially and temporally out of
phase with the development of DOMS [93]. Malm et al. [252] explored the concept that the
activation of local leucocytes present in the epimysium may be involved in the regulation of
DOMS. This hypothesis has since received support from both rodent [253] and human trials
[61, 254] and presents a promising area for future research.
2.6 Conclusion:
This is the first study to demonstrate that ibuprofen administration has no effect on the
histological appearance of inflammatory white blood cells following an acute bout of
traditional resistance exercise. Ibuprofen administration had no effect on blood markers of
muscle damage or subjective muscle soreness, and no significant correlations between
leucocyte numbers and post-exercise muscle damage or soreness was observed. Future
research needs to explore the possibility of an inflammatory threshold above which NSAID
administration influences post-exercise inflammation. The underlying cause of post-exercise
muscle soreness also remains largely unknown. Future investigations are needed to determine
the role that inflammation plays in exercise-induced DOMS.

41
2.7 Acknowledgements:
The authors would like to acknowledge to assistance of Associate Professor John Reynolds for
his contributions in the statistical analysis of the dataset. We further thank Dr. Andrew
Garnham (Deakin University) for all muscle biopsy procedures, and the participants for
volunteering their time to be involved in the trial.
2.8 Disclosures:
This research was conducted in the absence of any commercial or financial relationships that
could be construed as a potential conflict of interest.

42
CHAPTER 3:
Intramuscular inflammatory lipid profile response to an acute bout of resistance exercise in
humans.
Vella L, Markworth JF, Maddipati KR, Cameron-Smith D, Russell AP. Intramuscular
inflammatory lipid profile in response to an acute bout of resistance exercise in humans.
Submitted for review to Frontiers in Physiology: Exercise Physiology on 20/11/2016.

43
3.1 Abstract:
Purpose: Classic eicosanoid species including prostaglandins (PG) and leukotrienes (LT) play
essential roles in initiating and regulating acute inflammation. Recent identification of novel
classes of bioactive lipid mediators derived from long-chain (LC) polyunsaturated fatty acids
(PUFA), collectively termed specialized resolving mediators (SPM) has led to the proposal of an
active biochemical pathway for resolution of the acute inflammatory response. Chapter 3
investigated the human intramuscular lipid mediator profile at rest and in response to an acute
bout of unaccustomed resistance exercise. Methods: Fourteen male subjects completed a bout
of lower body resistance exercise. Muscle biopsy samples were taken from the m.vastus
lateralis before and at 2, 4 and 24 h post-exercise. Intramuscular lipid mediator composition
was analysed via liquid chromatography–mass spectrometry (LC-MS)-based targeted
lipidomics. Results: A total of 129 unique lipid mediators were reliably identified in human
skeletal muscle tissue. Post-exercise recovery was characterized by increased levels of COX-
derived thromboxane (TXB2 and 12(S)-HHTrE) and prostaglandin metabolites (2,3-dinor PGE1
and 15d-D12,14-PGJ3), LOX derived leukotrienes (LTB4 and 20-COOH LTB4),
hydroxyeicosatetraenoic acids (5-HETE, 12-HETE, tetranor 12-HETE and 15-HETE),
monohydroxylated- docosahexaenoate (HDoHE) species (4-HDoHE, 7-HDoHE and 8-HDoHE),
and CYP derived epoxyeicosatrienoic acids (5,6-EpETrE) / vicinal diol dihydroxyeicosatrienoic
acids (11,12-DiHETrE and 14,15-DiHETrE). Conclusion: This research utilised a targeted
lipidomics approach to profile the human skeletal muscle lipid mediator response to an acute
bout of resistance exercise. The profile suggests that the activation of classical pro-
inflammatory eicosanoids such as PG and LT species, as well as novel lipid intermediates in
SPM biosynthesis derived from both LOX and CYP pathways, form a highly complex self-
resolving model of acute inflammation following an acute bout of resistance exercise.

44
3.2 Introduction:
Skeletal muscle is a remarkably heterogeneous tissue with the capacity to adapt and respond
to external stress. It is well established that intense resistance exercise can lead to
improvements in muscle strength through changes in muscle fiber type, myofibrillar
hypertrophy and neuromuscular mechanisms [1]. However, unaccustomed exercise, especially
when comprising a large eccentric component, can cause skeletal muscle injury and initiate an
acute inflammatory response [24, 31, 240]. Experimental models targeted at manipulating the
post-exercise inflammatory response have identified that exercise-induced inflammation is a
key regulatory feature in the normal process of tissue regeneration and adaptation following
acute muscle damage [103, 161, 255]. This suggests that molecular signaling events occurring
early during acute inflammation play an active role in promoting the restoration of normal
tissue function and promote skeletal muscle adaptation following an exercise stimulus.
The humoral and local muscular changes that occur during exercise-induced inflammation
closely resemble that of an acute phase response to cellular stress. As observed in chapter 2, a
resistance exercise stimulus triggers the production of pro-inflammatory signaling molecules,
establishing a chemotactic gradient and the diapedesis as well as the infiltration of
inflammatory leukocytes [85, 246]. These chemoattractants consist of lipid-derived mediators
such as leukotrienes (LTs) and prostaglandins (PGs), as well as protein mediators, including
cytokines and chemokines [168]. The usual outcome of an acute inflammatory response is its
successful resolution and repair of damaged tissue [174]. Traditionally, the resolution of
inflammation was thought to be a passive process involving the dilution and catabolism of pro-
inflammatory mediators leading to exodus of leukocytes from the site of muscle damage.
However, with the discovery of novel classes of lipid-derived mediators, the resolution of acute
inflammation has evolved to be seen as an active and finely controlled biochemical and
metabolic process that may provide a critical link between cellular stress and tissue
regeneration/adaptation [185, 186].
Lipid mediators are biosynthesized endogenously from essential omega-6 (n-6) and omega-3
(n-3) polyunsaturated fatty acids (PUFA) and are involved in a wide range of physiological and

45
pathophysiological processes [186]. The majority of research has focused on classic
prostaglandins (synthesized via cyclooxygenase (COX) enzymes, COX-1 and COX-2) and
leukotrienes (synthesized via the lipoxygenase (LOX) enzyme, 5-LOX), which are derived from
the n-6 PUFA arachidonic acid (AA). These lipids play a diverse role in stimulating acute
inflammation by controlling local blood flow, vascular permeability, cytokine production,
leucocyte chemotaxis and pain [102]. More recently, a second class of eicosanoids also
generated from AA, termed lipoxins (LX) [172, 175, 176], as well as newly identified
eicosapentaenoic (EPA) (E-Series) and docosahexaenoic acid (DHA) (D-Series) derived resolvins
(Rv), protectins (PD) [166, 169-171, 177, 178], and maresins (MaR) [168, 173] have been shown
to play pro-resolution functions following acute inflammation. These novel lipid mediators,
collectively termed specialized pro-resolving mediators (SPMs), act to block acute
inflammatory signals by inhibiting pro-inflammatory cytokine production and subsequent
neutrophil chemotaxis [173, 182]. They simultaneously promote the nonphlogistic infiltration
of blood monocytes/macrophages and stimulate tissue macrophages to phagocytize and clear
apoptotic neutrophils whilst promoting wound healing [175, 181].
SPMs are formed during inflammatory transcellular interactions, involving the sequential
actions of two or more cell types expressing the required LOX and/or COX enzymes in a
compartmentalized manner. During the time-course of inflammation, cell-cell interactions
between platelets, leucocytes, the vasculature and resident tissue cells facilitates the
transcellular biosynthesis of unique SPMs [101]. The temporal regulation of these lipids is
therefore specific to the tissue type and the inciting inflammatory stimulus[185, 186]. For
example, LX biosynthesis involves cellular interactions between 5-LOX expressing neutrophils
with 12-LOX expressing platelets or 15-LOX expressing M2 monocytes [184]. Recent findings
from Markworth et al. (2013) demonstrated that pro-resolving lipid mediators, including
lipoxins, resolvins and protectins, are elevated in human blood serum samples following an
acute bout of resistance exercise [102]. The present study used the same targeted lipidomics
approach to characterize the local time-course changes of eicosanoid and docosanoid species
in human skeletal muscle tissue following an acute bout of resistance exercise. The primary
aim was to identify which species of lipids are present within skeletal muscle tissue, and hence
may be locally generated and acting, following an acute bout of resistance exercise. It was

46
hypothesized that there would be a rapid increase in pro-inflammatory prostaglandin and
leukotriene biosynthesis, followed by the activation of SPMs at the onset of inflammation
resolution. Identification of the lipid mediator profile of skeletal muscle and ability of exercise
stress to modulate intramuscular bioactive lipids will help to contribute to the understanding
of a biologically active inflammatory resolution pathway that may be essential to muscle
recovery and adaptation following an inflammatory event.

47
3.3 Methods:
3.3.1 Subjects:
As previously described [256], fourteen healthy men aged 18-25 years were recruited to
participate in the acute exercise study. A subset of 12 male participants, for which sufficient
muscle biopsy tissue remained, were included in the analysis performed here. Exclusion criteria
included participation in regular resistance exercise within one year prior to commencing the
study, and/or the consumption of any nutritional or purported muscle building supplements.
Each participant also completed a medical history questionnaire to identify any potential risk
factors that would prevent the subjects from completing strenuous exercise.
3.3.2 Ethics approval:
Each participant was provided with a written and oral explanation of the nature of the study
and potential risks of the experimental procedures before providing written consent to
participate. All procedures involved in the study were formally approved by the Deakin
University Human Research Ethics Committee (DUHREC 2004-017) and muscle biopsy
procedures were performed in order with Helsinki declaration.
3.3.3 Familiarization:
For the 24 h preceding exercise, subjects consumed a standardised diet (20% fat, 14% protein,
and 66% carbohydrate) [257]. At least seven days prior to the trial day, each subject completed
a familiarization session on the Cybex NORM dynamometer (Cybex International Inc. UK). The
session involved performing isokinetic maximal voluntary contractions (iMVC) during
concentric and eccentric knee extension exercise. Maximal force production measured as peak
torque (N.m) was determined at 60°/s over 12 maximal concentric and eccentric contractions.
Subjects were provided with verbal encouragement throughout the test to ensure maximal
effort.

48
3.3.4 Experimental design:
On the morning of the trial, subjects reported to the laboratory in an overnight fasted state
having abstained from alcohol, caffeine and tobacco for the previous 24 h. Participants rested
in a supine position for 30 min, following which a resting muscle biopsy sample was collected.
Each participant then completed an acute bout of concentric and eccentric isokinetic unilateral
knee extension exercise on the Cybex NORM dynamometer. Subjects completed three sets of
12 maximal voluntary repetitions at a constant speed of 60°/s with 2 mins of rest between
each set. In a previous research project, this exercise stimulus was shown to trigger an increase
in pro-inflammatory gene expression, STAT-3 phosphorylation and nuclear translocation, and
STAT-3 target gene expression [257]. Subjects were instructed to contract maximally during
each repetition and were provided with verbal encouragement throughout each set. Further
muscle biopsy samples were obtained from the exercised leg at 2 and 4 h after completion of
the exercise protocol. The following morning, subjects reported to the laboratory again in an
overnight fasted state for a final follow up 24 h post-exercise muscle biopsy sample.
3.3.5 Muscle biopsy procedure:
Muscle biopsy samples were obtained from vastus lateralis under local anaesthesia (Xylocaine
1%) using a percutaneous needle biopsy technique modified to include suction [245]. A section
of excised tissue was rapidly snap frozen in liquid nitrogen and stored at -80°C for further
analysis. Repeat muscle biopsy samples were collected from the same leg through separate
incisions separated byat least 2 cm from the previous biopsy site to minimize the risk of any
localized inflammation arising from the biopsy procedure confounding exercise-induced
inflammation.
3.3.6 Liquid chromatography-mass spectrometry:
Muscle biopsy samples were weighed and homogenized in 1 ml phosphate buffered saline (50
mM phosphate containing 0.9% sodium chloride, pH 7.4) using Zirconium beads on a high-
frequency oscillator (Precellys homogenizer, Bertig Instruments). The homogenates were
centrifuged at 6,000g for 10 min and the supernatant was collected for the extraction fatty acyl
lipid mediators using C18 solid phase extraction cartridges as described earlier [258-260]. Fatty

49
acyl lipid mediator extracts were subjected to LC-MS analysis essentially as described before
[258]. Under the LC-MS conditions employed, the detection limit for most of the lipid
mediators was 1 pg on the column and the quantitation limit was 5 pg on the column with a
signal/noise ratio >3. Tissue weights from each sample (range: 14-70 mg, average: 43 mg,
inter-quartile range: 32-56 mg) were used for normalization of the LC-MS data and the data are
reported as ng per gram of tissue.
3.3.7 Statistics:
Statistical analysis was performed using IBM SPSS Statistics Version 22 (IBM United Kingdom
Limited, Hampshire, England). Data were analyzed using a one-way repeated measures
ANOVA. The sphericity adjustment was checked, and if required, a Greenhouse-Geisser epsilon
correction was applied. In the instance, a metabolite verged achieving a main effect for time (P
< 0.1), a post-hoc analysis was run using a Bonferonni correction. Data are presented as mean
± standard error of the mean (SEM). Statistical significance was set at P < 0.05. This research
was an exploratory study and as such statistical power calculations based off a predicted mean
and standard deviation scores were unable to be completed. Sample size was determined by
the availability of existing muscle sample from a previous clinical trial exploring the effect of an
acute resistance exercise protocol on markers of post-exercise muscle damage [257].

50
3.4 Results:
3.4.1 Lipidomic analysis:
Lipid mediator profiles of human skeletal muscle tissue were generated via LC-MS. A total 129
unique lipid mediators were reliably detected (Supplemental Table 1, displayed in Appendix D).
However, many of these lipids were very lowly expressed or below the limits of quantification
of the assay (signal/noise ratio >3) in particular sample sets. Therefore, only lipid species that
were quantified in all post-exercise samples were statistically analysed. A main effect for time
was observed in a range of lipid-derived inflammatory mediators and their downstream
metabolites during post-exercise recovery. Peak concentration was generally identified at 2 h
post-exercise, however we were often unable to detect the specific time points at which these
changes became significant.
3.4.1.1 Cyclooxygenase pathway:
COX enzymes catalyse the first step in the conversion of AA to prostaglandins (PGE2, PGF2ɑ,
PGD2 and PGI2) and thromboxane (TXA2). TXA2 is a highly unstable and non-enzymatically
decomposes to TXB2 and 12(S)-HHTrE. Both metabolites serve as surrogate marker of TXA2
biosynthesis. Both of these inactive metabolites were detected within skeletal muscle tissue
and were significantly increased post-resistance exercise (P < 0.05) with the greatest
concentrations identified at 2 h post exercise (Figure 3.1). There were no significant changes
detected in any of the primary prostaglandins derived from AA. PGE2 and PGF2ɑ were the most
abundant prostaglandins in skeletal muscle tissue, detected at concentrations of 1.13 ng/g and
0.68 ng/g respectively at rest. Post-exercise, both PGE2 and PGF2ɑ were elevated 2 h post
exercise, however, the increase did not reach statistical significance (PGE2: P = 0.075, PGF2ɑ: P =
0.078) (Figure 3.1). PGD2 levels were very low and often below detectable limits, while PGI2
(measured as the stable non-enzymatic hydrolysis product 6-keto-PGF1α) was not detected.
Within muscle tissue, PGF2α and PGE2 were present at considerably higher concentrations
compared to their inactive circulating secondary (15-keto-PGE2/PGF2α) and tertiary (13,14-
dihydro-15-keto-PGE2/PGF2α) enzymatic degradation products. However, downstream
metabolites of other prostaglandin species were identified in muscle. A bioactive downstream
metabolite of the EPA-derived PGD3, 15-Deoxy-Δ12,14-prostaglandin J3, was detected at
concentrations of 2.93 ng/g in resting muscle and increased significantly during the acute

51
stages of post-exercise recovery with a peak response observed at 2 h post (P < 0.05) (Figure
3.1).
Figure 3.1: Metabolites of the cyclooxygenase pathway derived from arachidonic acid. Values depicted
are mean values ± SEM. *denotes a main effect for the exercise intervention (p < 0.05).

52
3.4.1.2 Lipoxygenase pathway:
Lipoxygenase isoform expression has been characterized in various cell types involved in acute
inflammation including neutrophils (5-LOX), platelets (12-LOX) and monocytes (15-LOX) [261].
5-LOX metabolises AA to 5-hydroperoxyeicosatetranoic acid (5-HpETE), which is either reduced
to 5-hydroxyeicosatetraenoic acid (5-HETE) or further metabolised to the potent neutrophil
chemoattractant leukotriene B4 (LTB4). In resting skeletal muscle tissue, LTB4 was below the
level of detection in the majority of samples analysed and when detected it was generally
present only at very low concentrations (≤1ng/g). However, LTB4 was consistently detected at
much more abundant levels within muscle tissue (~3ng/g) during post-exercise recovery,
specifically at the 2 h time point, however this failed to reach statistical significance (Figure
3.2). 5-HETE also verged on a time-dependent increase (P = 0.085). Post-hoc analysis verged on
a significant increase in intramuscular 5-HETE to concentrations of 8.99 ng/g at 2 h post-
exercise when compared to resting levels of 3.38 ng/g) (P = 0.058) (Figure 3.2). Further, 20-
COOH-LTB4, a downstream metabolite of LTB4 showed a significant increase, with the highest
response occurring at 2 h post exercise (P < 0.05) (Figure 3.2). In addition to AA, the 5-LOX
enzyme also has the capacity to oxidize the omega-3 fatty acids DHA and EPA.
Monohydroxylated-DHA (HDoHE) products of the 5-LOX pathway including 4-
hydroxydocosahexanoic acid (4-HDoHE) and 7-hydroxydocosahexanoic acid (7-HDoHE) were
detected in resting muscle at concentrations of 2.23 ng/g and 0.98 ng/g respectively. Both 4-
HDoHE and 7-HDoHE showed a significant increase with peak muscle concentrations of 3.76
ng/g and 1.54 ng/g respectively observed at 2 h post-exercise (P < 0.05) (Figure 3.3). The 5-LOX
product of EPA, 5-HEPE was similarly detected within skeletal muscle tissue but at
concentrations of 0.52 ng/g, approximately 10 times lower than the corresponding AA product
5-HETE. Intramuscular 5-HEPE was not significantly influenced by resistance exercise.
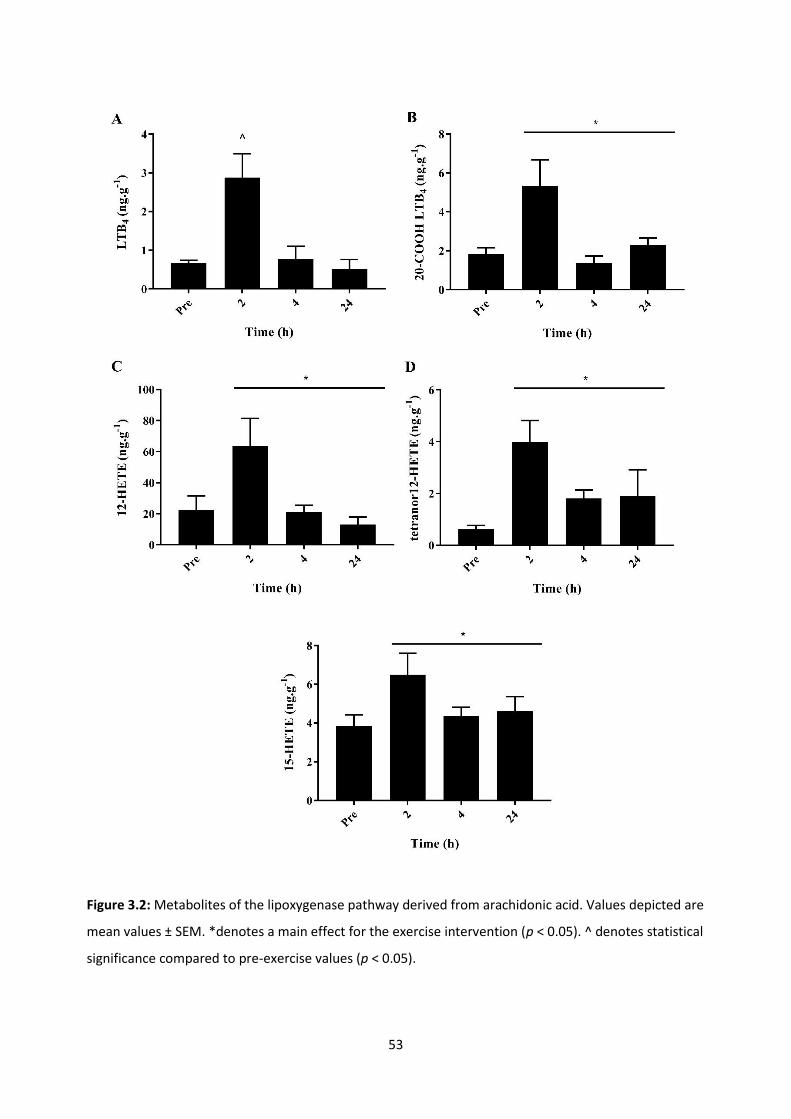
53
Figure 3.2: Metabolites of the lipoxygenase pathway derived from arachidonic acid. Values depicted are
mean values ± SEM. *denotes a main effect for the exercise intervention (p < 0.05). ^ denotes statistical
significance compared to pre-exercise values (p < 0.05).

54
Figure 3.3: Metabolites of the lipoxygenase pathway derived from docosahexaenoic acid. Values
depicted are mean values ± SEM. *denotes a main effect for the exercise intervention (p < 0.05).
The 12-LOX enzyme is expressed primarily in platelets and metabolizes AA to form 12-
hydroxyeicosatetraenoic acid (12-HETE), which can stimulate leucocyte chemotaxis and
platelet aggregation [262-264]. Additionally, in one key pathway of LX biosynthesis, 5-LOX
derived LTA4 is converted to LXA4 and LXB4 via the sequential action of platelet 12-LOX. 12-
HETE was by far the most abundant monohydroxylated-FA product detected in resting muscle,
present at concentrations of ~20 ng/g. We found 12-HETE increased with time (P < 0.05) with
peak concentrations of 63.81 ng/g observed at 2 h post-exercise (Figure 3.2). Similarly, muscle
tetranor 12-HETE, a downstream metabolite of 12-HETE increased significantly post-exercise (P
< 0.05), with the greatest response at 2 h post-exercise (Figure 3.2). The 15-LOX enzyme is
highly expressed in eosinophils, epithelial cells and alternatively activated M2 macrophages
and acts to convert AA to 15-hydroxyeicosatetranoic acid (15-HETE) [265]. While 15-HETE did
not achieve a main effect for time overall, post-hoc analysis showed a significant increase at 2
h post-exercise (6.50 ng/g) when compared to pre-exercise levels (3.84 ng/g) (P = 0.013)
(Figure 3.2).
LOX metabolites of the n-3 PUFA DHA have been established to be key pathway markers and
intermediates in the biosynthesis of SPMs. The 12-LOX enzyme converts the 22-carbon DHA to
14-hydroperxydocosahexanoic acid (14-HpDoHE) which is reduced to 14-HDoHE or

55
metabolised to MaR1/2 via further action of 12-LOX. Thus, 14-HDoHE is considered a key MaR
pathway activation marker. Similarly, 15-LOX converts DHA substrate to 17-H(p)DoHE, a key
intermediary step in the biosynthesis of both the D-series resolvins and protectins. We
detected 14-HDoHE in resting muscle at concentrations of 0.68 ng/g whereas 17-HDoHE was
not found to be present at detectable levels. There was no significant effect of exercise on 14-
HDoHE or 17-HDoHE. Nevertheless, peak intramuscular concentrations of 1.60 ng/g 14-HDoHE
were observed at the 2 h post-exercise time-point. MRM transitions corresponding to mature
SPMs including lipoxins (LXA4 and LXB4), E-series resolvins (RvE1 & RvE3), D-series resolvins
(RvD1, RvD2 and RvD5), protectins (PD1 and 10S,17S-DiHDoHE) and maresins (MaR1) were
additionally monitored by our LC-MS/MS assay. However, mature SPMs were not found to be
present at detectable levels locally within muscle tissue biopsies at rest or throughout 24 h
post-exercise recovery.
3.4.1.3 Epoxygenase pathway:
The cytochrome P-450 (CYP) enzymes metabolize n-6 PUFA AA to a family of
epoxyeicosatrienoic acid (EpETrE) regioisomers. Once formed, these EpETrEs are rapidly
metabolized by the soluble epoxide hydrolase (sEH) enzyme to form corresponding vicinal
diols, dihydroxyeicosatrienoic acids (DiHETrEs). 5,6-EpETrE was the only primary enzymatic
epoxy AA-metabolite that showed an effect for the exercise intervention, with concentrations
increasing 3-fold to ~15ng/g at 2h post-exercise (P < 0.05) (Figure 3.4). Whilst 8,9-EpETrE,
11,12-EpETrE, 14,15-EpETrE were also found in relative abundance in skeletal muscle tissue at
rest (~3-10 ng/g), they did not display a significant effect of exercise. Alternatively,
downstream products of the sEH enzyme, 11,12-DiHETrE and 14,15-DiHETrE were present at
lower absolute concentrations in resting muscle (~1 ng/g), but significantly increased in
response to exercise with a peak response at 2 h of recovery (P < 0.05) (Figure 3.4). 5,6-
DiHETrE and 8,9-DiHETrE regioisomers were also detected at low levels in resting muscle,
however no effect of the exercise intervention was observed.
CYP epoxidase enzymes also have the capacity to metabolise the n-6 linoleic acid to bioactive
products. Linoleic acid derived 9,10-DiHoME, is a leukotoxin generated by neutrophils during
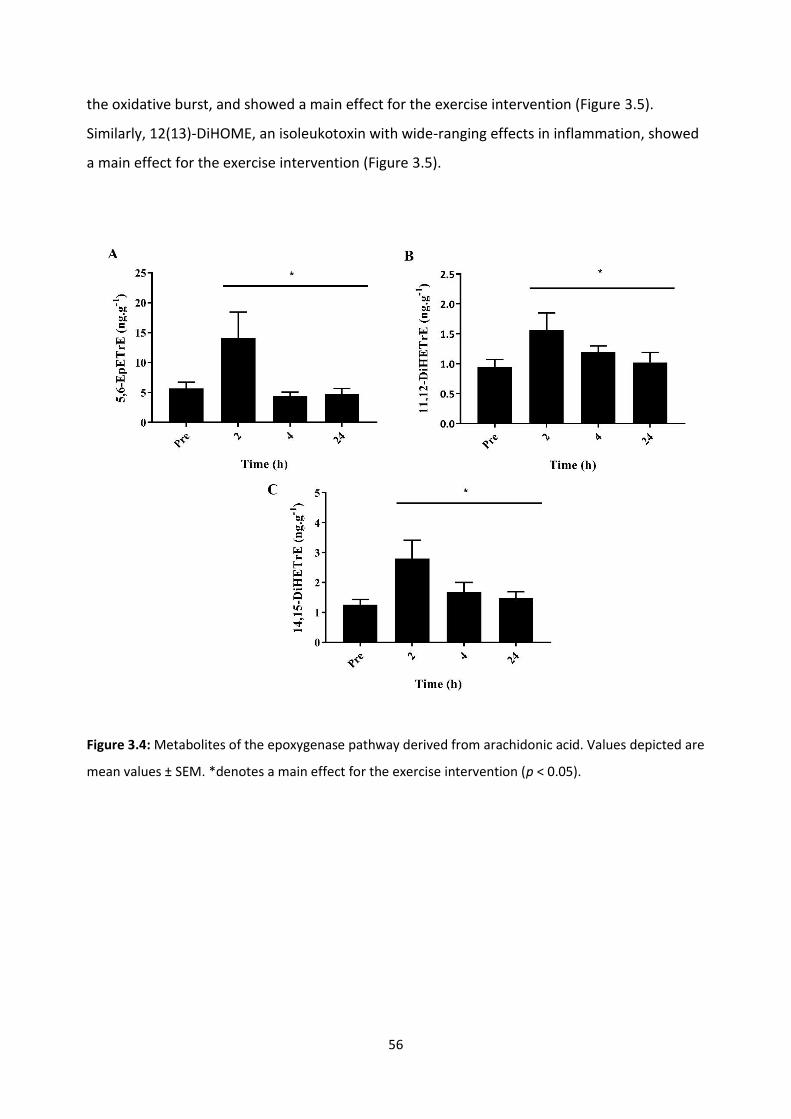
56
the oxidative burst, and showed a main effect for the exercise intervention (Figure 3.5).
Similarly, 12(13)-DiHOME, an isoleukotoxin with wide-ranging effects in inflammation, showed
a main effect for the exercise intervention (Figure 3.5).
Figure 3.4: Metabolites of the epoxygenase pathway derived from arachidonic acid. Values depicted are
mean values ± SEM. *denotes a main effect for the exercise intervention (p < 0.05).
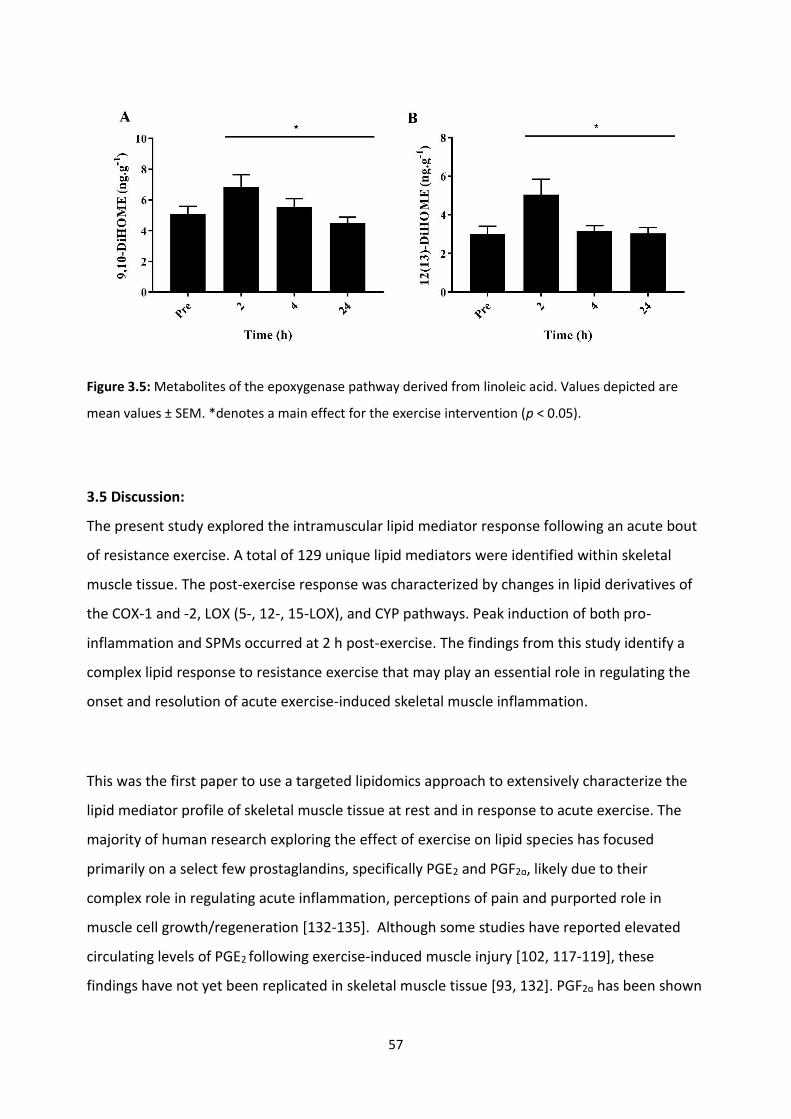
57
Figure 3.5: Metabolites of the epoxygenase pathway derived from linoleic acid. Values depicted are
mean values ± SEM. *denotes a main effect for the exercise intervention (p < 0.05).
3.5 Discussion:
The present study explored the intramuscular lipid mediator response following an acute bout
of resistance exercise. A total of 129 unique lipid mediators were identified within skeletal
muscle tissue. The post-exercise response was characterized by changes in lipid derivatives of
the COX-1 and -2, LOX (5-, 12-, 15-LOX), and CYP pathways. Peak induction of both pro-
inflammation and SPMs occurred at 2 h post-exercise. The findings from this study identify a
complex lipid response to resistance exercise that may play an essential role in regulating the
onset and resolution of acute exercise-induced skeletal muscle inflammation.
This was the first paper to use a targeted lipidomics approach to extensively characterize the
lipid mediator profile of skeletal muscle tissue at rest and in response to acute exercise. The
majority of human research exploring the effect of exercise on lipid species has focused
primarily on a select few prostaglandins, specifically PGE2 and PGF2ɑ, likely due to their
complex role in regulating acute inflammation, perceptions of pain and purported role in
muscle cell growth/regeneration [132-135]. Although some studies have reported elevated
circulating levels of PGE2 following exercise-induced muscle injury [102, 117-119], these
findings have not yet been replicated in skeletal muscle tissue [93, 132]. PGF2ɑ has been shown

58
to increase locally within skeletal muscle tissue following both eccentric [132] and isotonic
resistance exercise protocols [133] and plays a significant role in in-vitro myofibre hypertrophy
[266] and post-exercise muscle protein synthesis [107]. Further, recent research from our
group found a significant increase in serum levels of the circulating PGF2ɑ metabolite 15-keto-
PGF2ɑ early (1 h) following resistance exercise in humans [102]. While the increase in muscle
PGE2 or PGF2ɑ failed to reach statistical significance, they were the most abundant AA derived
PGs detected within muscle and both markers showed a strong trend for an effect of the
exercise intervention (P = 0.075 and P = 0.078 respectively). Further, increase in TXA2
(measured as TXB2 and 12(S)-HHTrE) post-exercise with peak responses at the 2 h time-point
may reflect a pro-aggregatory, pro-thrombotic circulatory effect that occurs in response to
both maximal aerobic [267] and resistance exercise protocols [102].
The other major metabolic pathway leading to the formation of lipid species involved in the
regulation of inflammation is the lipoxygenase pathway. Increases in the 5-LOX derived LTB4
has been reported in blood serum samples following acute resistance exercise [102] and high
speed running [268]. LTB4 is a potent neutrophil chemoattractant and a powerful stimulator of
vasoconstriction and blood vessel permeability [269]. Expression of 5-LOX is essentially limited
to bone-marrow derived cells including inflammatory neutrophils and
monocytes/macrophages [270]. It is therefore not surprising that in the present study LTB4 was
very lowly expressed in skeletal muscle tissue prior to exercise. At 2 h post-exercise, LTB4 was
detectable in the majority of subjects, and a downstream derivative, 20-COOH LTB4,
demonstrated a main effect for the exercise intervention. A less well-described branch of the
5-LOX pathway involves the metabolism of n-3 PUFA DHA to form mono-hydroxylated fatty
acids 4-HDoHE and 7-HDoHE. These fatty acids were found in abundance in skeletal muscle
tissue and increased during post-exercise recovery, however no such change was observed
previously in blood serum samples [102].
In addition to 5-LOX, metabolites of the human platelet type 12-LOX enzyme 12-HETE and its
downstream derivate tetranor 12-HETE were also elevated within muscle during post-exercise
recovery. Both metabolites are pro-inflammatory in nature and have been shown to act

59
transcellularly to modify the responsiveness of neutrophils to other chemotactic factors [271].
We previously observed a similar increase in 12-HETE and downstream tetranor 12-HETE in
human blood serum samples during recovery from resistance exercise [102]. Interestingly 12-
LOX expressing platelets have been implicated in the transcellular biosynthesis of pro-
resolution LX mediators through interactions with 5-LOX expressing PMNs. This pathway
involves leucocyte-platelet interactions during which the 5-LOX derived leukotriene
intermediate LTA4 is taken up by 12-LOX expressing platelets for subsequent conversion to
LXA4 [272, 273]. Further, the 15-LOX pathway has been implicated as a second the endogenous
route of LX biosynthesis species. 15-LOX is highly expressed in alternatively activated
macrophages and epithelial cells. The secretion of 15-LOX product 15-HETE can be taken up by
5-LOX expressing cells and converted to LXA4 and LXB4 [274]. In this study we demonstrated a
significant increase in 15-HETE at 2 h post-exercise, a finding which has been replicated in
human blood serum samples [102]. Our finding of simultaneous induction the primary
products of both the 5-LOX/12-LOX and 15-LOX/5-LOX pathways locally within skeletal muscle
during post-exercise recovery would presumably be a permissive environment for local LX
biosynthesis to occur. However, LXA4 or LXB4 were unable to be detected locally within muscle
biopsy homogenates. Nevertheless, these local changes within the exercised musculature
suggests that skeletal muscle tissue may potentially contribute to the exercise-induced
systemic lipoxin response previously reported [102].
The CYP pathway is a third, perhaps less well-known, branch of the AA metabolic pathway. The
increase in epoxyeicosatrienoic acid regioisomer 5-6-EpETrE and dihydroxyeicosatrienoic acids
11,12-DiHETrE and 14,15-DiHETrE in skeletal muscle supports observations made in serum
samples when measured during the early stages of post-exercise inflammation [102]. The
physiological function of these derivatives remains unexplored in skeletal muscle tissue.
However, in vascular smooth muscle and endothelium cells they play anti-inflammatory roles
through in the inhibition of prostaglandin and cytokine induced inflammatory responses [275].
CYP enzymes also metabolise linoleic acid via epoxidation to form epoxyoctadecmonoeic acids
(EpOMEs). EpOMEs are rapidly hydrolysed by the sEH enzyme to form DiHOMEs [276]. EpOMEs
and DiHOMEs are leukotoxins that play a role in the suppression of neutrophil respiratory burst
activity, vasodilation and cellular apoptosis [277, 278]. A cycling based intervention comprising

60
of a 75km time trial had no effect on 9-10,DiHOME 1.5 h and 21 h post-exercise in plasma
samples of competitive road cyclists [277]. Alternatively, a bout of acute resistance exercise
triggered an increase in 9(10)-EpOME and 9-10,DiHOME in serum samples [102]. Results from
the present study showed an increase in 9-10,DiHOME and 12(13)-DiHOME suggesting that
discrepancies in the previous literature may be due to the differences in the type of exercise
performed and the training status of the subjects used in each study.
Collectively, these findings demonstrate an increase in lipid derived mediators of the COX, LOX
and CYP pathways during post-exercise muscle recovery. The concept of a biologically active
inflammatory resolution programme governed by lipid derivatives was first proposed in a TNF-
ɑ-stimulated model of acute inflammation in the murine air pouch [185]. Within this model,
early formation of LTB4 and PGE2 at the onset of inflammation was succeeded by a class-
switching of eicosanoids to LXA4. During this process, interactions between inflammatory and
host tissue cells enabled the biosynthesis of resolution mediators [185]. Alternatively, in a
model of zymosan-A stimulated murine peritonitis, the onset of inflammation was
characterized by a concomitant increase in LTB4 and LXA4 followed by a late appearance of
PGE2 at the onset of resolution [186]. These findings demonstrate that the temporal regulation
of lipids and their role in inflammation is likely cell-type and stimulus specific. Recent work
from our group profiled the human lipid response to acute resistance exercise in serum
samples [102]. This study showed an increase in key prostaglandin, leukotriene, lipoxin and
resolvin species during the early stages of acute inflammation (1-3 h), followed by an increase
in 15-LOX derivatives and some prostaglandin metabolites (6-keto-PGF1ɑ and 13,14dh-
15kPGE2) at 24 h post- exercise [102]. One of the limitations of the present study was the lack
of statistical power preventing post-hoc analysis of the observed effect of the exercise
intervention. However, for all intramuscular lipid derivatives peak expression occurred at 2 h
post-exercise, which appears consistent with findings from serum samples [102]. Interestingly
lipid species that require transcellular interactions, including lipoxins, resolvins and protectins,
were either undetected, or very lowly expressed in skeletal muscle tissue. These lipids were
detectable previously in abundance in human serum samples during post exercise recovery
and are known to play a vital role in the active resolution of acute inflammation [102].

61
3.6 Conclusion:
This is the first study to profile the lipid mediator response to acute resistance exercise in
skeletal muscle tissue. An increase in lipids derived from the COX, LOX and CYP pathways was
observed during the early stages of post-exercise muscle recovery. Peak induction of AA
derived classical pro-inflammatory markers PGE2, PGF2ɑ, and both upstream and downstream
markers of LTB4 occurred at 2 h post-exercise. Further various derivatives of the 5-LOX (4-
HDoHE and 7-HDoHE), 12-LOX (12-HETE and tetranor 12-HETE) and 15-LOX (15-HETE)
pathways were identified in abundance in skeletal muscle tissue post-exercise and may
resemble transient cellular intermediates for the formation of pro-resolution lipoxin and
resolvin species. In alternative models of acute inflammation, these lipids are involved in
coordinating a biologically active inflammatory resolution programme that has been
mechanistically linked to processes tissue healing. Future research exploring the physiological
significance and function of these intramuscular lipids in both skeletal muscle and
serum/plasma samples post-exercise, will be useful in further characterizing the significance of
the intramuscular inflammatory response in post-exercise muscle recovery.
3.7 Acknowledgments:
The authors would like to acknowledge to assistance of Associate Professor John Reynolds for
his contributions in the statistical analysis of the dataset. We further thank Dr. Andrew
Garnham (Deakin University) for all muscle biopsy procedures, and the participants for
volunteering their time to be involved in the trial.
3.8 Disclosures:
This research was conducted in the absence of any commercial or financial relationships that
could be construed as a potential conflict of interest.

62

63
CHAPTER 4:
Ibuprofen supplementation and its effects on NF-κB activation in skeletal muscle
following resistance exercise.
Vella L, Markworth JF, Peake JM, Snow RJ, Cameron-Smith D, Russell AP. Ibuprofen
supplementation and its effects on NF-κB activation in skeletal muscle following resistance
exercise. Physiological Reports, 2014 Oct 24; 2:10. doi: 10.14814/phy2.12172 (Appendix B).

64
4.1 Abstract:
Purpose: Resistance exercise triggers a subclinical inflammatory response that plays a pivotal
role in skeletal muscle regeneration. Nuclear factor-κB (NF-κB) is a key stress signaling
transcription factor that regulates acute and chronic states of inflammation. The classical NF-
κB pathway regulates the early activation of post-exercise inflammation; however there
remains scope for this complex transcription factor to play a more detailed role in the muscle
regeneration process. Methods: Sixteen volunteers completed a single bout of lower body
resistance exercise with the ingestion of three 400 mg doses of ibuprofen or a placebo control.
Muscle biopsy samples were obtained prior to exercise and at 0, 3 and 24 h post-exercise.
Samples were analysed for key markers of NF-κB activity. Results: Phosphorylated p65 protein
expression and p65 inflammatory target genes were elevated immediately post-exercise.
These responses were independent of the two treatments. These changes did not translate to
an increase in p65 DNA binding activity. NF-κB p50 protein expression and NF-κB p50 binding
activity were lower than pre-exercise at 0 and 3 h post-exercise, but were elevated at 24 h
post-exercise. Conclusion: These findings provide novel evidence that two distinct NF-κB
pathways are active in skeletal muscle after resistance exercise. The initial wave of activity
involving p65 resembles the classical NF-κB pathway and is associated with the onset of an
acute inflammatory response. The second wave of NF-κB activity comprises the p50 subunit,
which has been previously shown to resolve acute inflammation in rodent inflammatory
models. The current study showed no effect of the ibuprofen treatment on markers of the NF-
κB pathway, however examination of the within group effects of the exercise protocol suggests
that this pathway warrants further research.

65
4.2 Introduction:
Unaccustomed resistance exercise causes skeletal muscle damage that impairs muscle function
and promotes sensations of pain. The precise signaling mechanisms that initiate muscle repair
following exercise-induced damage remain a topic of ongoing debate (previously reviewed by
Paulsen et al. [279]). The onset of muscle damage triggers a complex interplay of intracellular
events that involve myofibre injury, acute inflammation and cellular repair. At a symptomatic
level, inflammation in skeletal muscle is characterised by swelling and soreness [279].
Consequently post-exercise inflammation is often ascribed as a cause of delayed-onset muscle
soreness (DOMS). DOMS typically occurs 2448 hours post-exercise, and is accompanied by a
secondary reduction in muscle force-generating capacity. Treatment of DOMS has focused on
reducing inflammation; however this may be detrimental to processes of cellular repair [103].
Attenuating exercise-induced inflammation using non-steroidal anti-inflammatory drugs
reduces skeletal muscle protein synthesis [280] and impairs satellite cell proliferation [87]. To
develop more efficacious treatments for DOMS, a better understanding of the mechanisms
that govern inflammation and tissue repair in skeletal muscle after exercise is required.
The nuclear factor-kappa B (NF-κB) transcription factor acts as a central regulator of
inflammatory signaling pathways. The NF-B family consists of five subunits, including NF-κB1
(p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB and cREL that share an amino terminus Rel
homology domain. The Rel domain permits DNA binding, nuclear localization, dimerization and
interaction with its own inhibitory protein IκB [190, 191, 194]. Under basal conditions RelA
(p65), cREL and RelB subunits remain sequestered within the cytoplasm bound to an inhibitory
IB protein. The IB family that regulates NF-B includes the subunits IκBβ, IκBα, IκBγ IκBε, and
Bcl-3. Unlike the Rel proteins, subunits p50 and p52 are synthesized as large precursor proteins
(p105 and 100, respectively) that require proteolytic processing to permit nuclear localization.
These subunits lack a REL domain, to initiate gene transcription, and hence are primarily
considered as repressors of gene transcription [194, 281].

66
Upon stimulation, the IκB kinase complex (IKK) controls the degradation of IκB and its
precursor proteins, thereby enabling the NF-κB REL subunits to control gene transcription. The
IKK complex consists of two catalytic kinases IKKα and IKKβ, and a regulatory IKKγ subunit.
IKKβ activates the classical NF-κB signaling pathway through the phosphorylation and
subsequent degradation of the inhibitor IκBα [192, 199]. The classical pathway typically
comprises p65:p50 heterodimers, and is essential for the activation of acute inflammation by
controlling the transcription on inflammatory cytokines and acute phase proteins [192, 199].
More recently an alternative NF-κB pathway has been described, which is dependent on IKKα
[282]. Alternative signaling involves the activation of a secondary NF-κB inducing kinase (NIK),
and has been linked to the activation of both p52 and p50 subunits. The functional significance
of IKKα-dependent gene expression in acute inflammation is not yet well established.
However, preliminary research in rat muscle tissue suggests p50 homodimers may play a
crucial role in the resolution of acute inflammation [283, 284]. Transgenic rodents expressing
enhanced p50-p50 DNA binding showed reduced expression of inflammatory cytokines and
impaired leucocyte recruitment during inflammatory resolution [285]. Further, p50-p50
complexes bind to distinct κB response elements in COX-2 promoter regions, and have shown
to regulate COX-2 expression through association with transcriptional co-activators of the
C/EBP family [286, 287]. These findings implicate that a biphasic NF-κB activation patterns may
represent a key regulatory pathway in the expression of COX derived inflammatory resolution
mediators, as identified in chapter 3.
Despite the importance of inflammation in tissue regeneration post-exercise, very little is
known about how NF-κB activity is regulated in skeletal muscle after acute exercise. A transient
increase in various components of the classical NF-κB signaling pathway is observed in rat
muscle post-exercise [233, 234, 288-290]. In contrast, a decrease in NF-κB DNA binding at 0
hours post-exercise with a return to near baseline 1 hour post-exercise was observed in human
muscle following a traditional resistance exercise model [238]. Recent work from our
laboratory, using a similar resistance exercise model, demonstrated an increase in NF-κB
binding to the promoter region of key inflammatory cytokines at 2 hours post-exercise, with a

67
return to baseline levels at 4 hours post-exercise [239]. These findings suggest that a transient
NF-κB response contributes to acute post-exercise inflammation.
To enhance our understanding of the cellular mechanisms that regulate both the onset and
resolution of post-exercise inflammation, the current study aimed to investigate changes in the
activity of the subunits that comprise the classical and alternative NF-κB signaling pathways. It
was hypothesized that the regulation of NF-κB post-exercise would involve two distinct waves
of activation. Specifically, we hypothesized that the classical NF-κB pathway (involving p65-p50
heterodimers) would be activated soon after exercise during the early phases of inflammation,
while the alternative pathway (comprising p50 homodimers) would be activated at later time
points after exercise that correspond with the resolution of acute inflammation [284, 285]. It
was further hypothesized that the administration of ibuprofen would blunt both waves of NF-
κB activation, providing a potential mechanism through which anti-inflammatory medication
might attenuate exercise-induced inflammation in skeletal muscle.

68
4.3 Methods:
4.3.1 Participants:
As previously described in chapter 2, sixteen healthy male subjects were recruited to
participate in the study (characteristics shown in Table 1) [102]. All participants completed a
medical history questionnaire that was used to identify and exclude participants with a
diagnosed condition or illness that prevented them from completing strenuous exercise.
Exclusion criteria included participation in a lower body resistance exercise program within the
last 6 months to ensure a muscle damage response from the exercise stimulus, and/or chronic
treatment with anti-inflammatory drugs. Current use of prescription medication or nutritional
supplements also excluded subjects from participating.
Table 4.1: Subject characteristics and strength testing data. Values are mean values ± SEM. No
significant differences were observed between the two groups.
Characteristics Strength (1RM)
Age (y) Height (m) Body mass
(kg) BMI Squat (kg) Leg Press (kg)
Leg
Extension
(kg)
PLA 23.9
± 1.3
1.89
± 0.1
86.9
± 4.5
24.5
± 1.2
94.9
± 5.4
237
± 17
236
± 18
IBU 23.0
± 0.5
1.89
± 0.1
89.1
± 4.4
24.8
± 0.8
91.9
± 6.0
240
± 15
196
± 22
4.3.2 Ethics approval:
Prior to participation each subject received written and oral information regarding the nature
of the experiment before providing written consent to participate. All procedures involved in
this study were approved by the Deakin University Human Research Ethics Committee
(DUHREC 2010-019). All muscle sampling procedures were performed in accordance with the
Helsinki declaration.

69
4.3.3 Familiarization and strength testing:
Each participant completed a familiarization session at least 7 days prior to completing the
exercise trial. Participants performed repetition maximum testing to determine the
experimental exercise load (80% of 1 repetition maximum (1RM)). The maximal weight each
subject could lift was determined for the Smith machine-assisted squat, the leg press, and the
leg extension. These data were substituted into the validated Brzycki equation to predict 1RM
for each participant [244, 291]. The participants were required to abstain from any further
exercise until completion of the trial.
4.3.4 Experimental procedures:
The participants reported to the laboratory in an over-night fasted state, having abstained
from caffeine, tobacco and alcohol for the preceding 24 h. Following 30 min of supine resting a
pre-exercise muscle biopsy was taken. Participants then completed a 10 min warm up
consisting of low intensity cycling on a bicycle ergometer, and one low resistance set for each
exercise at approximately 30-50% of the participants 1RM. Each participant then completed a
single bout of intense resistance exercise. This session consisted of 3 sets of 8-10 repetitions of
a bilateral Smith machine assisted squat, 45-degree leg press and leg extension. These
exercises were all performed at 80% 1RM. The exercises were performed sequentially as a
circuit, with 1 min rest between each exercise, and 3 min rest between sets. We have
previously used this exercise protocol, and demonstrated that it activates inflammatory
signaling pathways [239]. Following the completion of the exercise bout, the participants
rested in a supine position while muscle biopsy samples were collected. The participants
returned to the laboratory the following morning in an over-night fasted state for a final
muscle biopsy sample. Standardized meals were provided to participants the night preceding
the trial (carbohydrate 57%, fat 22%, protein 21%), in the laboratory immediately following the
3 h muscle biopsy (carbohydrate 71%, fat 13%, protein 16%), and an evening meal
(carbohydrate 64%, fat 27%, protein 18%).

70
4.3.5 NSAID administration:
Prior to the exercise bout, the participants were randomly assigned to either the ibuprofen
(NSAID) group (n = 8) or the placebo group (PLA) (n = 8). Participants in the NSAID group
consumed the maximum recommended over-the-counter dose of 1200 mg of ibuprofen as
three doses of 400 mg throughout the trial day. The first dose was administered upon arriving
to the laboratory on the first morning of the trial, immediately prior to the first muscle biopsy
sample. Participants were instructed to consume two additional doses at 2:00pm and 8:00 pm
the same evening. This dosing structure was prescribed to optimise levels of circulating
ibuprofen to biologically active levels throughout the course of the trial day. This protocol has
previously been validated by our research group in this same group of study participants [102].
Alternatively, the placebo consumed a gelatin capsule containing powdered sugar, identical in
appearance to the ibuprofen capsule.
4.3.6 Sample collection:
Muscle biopsy samples were obtained under local anaesthesia (Xylocaine 1%) from the vastus
lateralis muscle of either leg using the percutaneous needle biopsy technique modified to
include suction [245]. Samples were obtained prior to exercise, immediately following exercise
and at 3 h and 24 h post-exercise. The muscle biopsy sample obtained immediately post-
exercise was taken within 1-2 min of the completion of the exercise protocol, and will herein
after being referred to as 0 h post-exercise. The muscle biopsy procedure has been shown to
trigger a local inflammatory response [62]. To minimize interference from the biopsy
procedure, samples prior to exercise and at 24 h post-exercise were taken from the same leg,
while muscle samples obtained at 0 and 3 h post-exercise were taken from the opposite leg.
Samples within the same leg were taken at least 5 cm from the previous site. We have
previously reported that this technique is effective for minimizing cytokine gene expression
and NF-B activity in response to muscle biopsies [239]. Excised tissue was immediately
immersed in liquid nitrogen and stored at 80°C until further analysis.

71
4.3.7 Protein extraction and quantification:
Muscle samples were homogenized in ice cold RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM
NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA supplemented with protease and
phosphatase inhibitors including 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 1 mM
Na3VO4 & 1mM NaF). The homogenate was agitated for 1 h at 4°C and centrifuged at 13,000
g at 4°C for 15 min. The resultant supernatant was removed and stored at 80°C until further
analysis. Total protein concentration was determined using a BCA protein assay kit according
to the manufacturer’s instruction (Pierce, Rockford, IL). Protein samples (50 µg) were
denatured in loading buffer and separated by a 10% SDS-PAGE and transferred to a PVDF
membrane. Membranes were blocked for 90 min at room temperature in 5% BSA/Tris buffered
saline with 0.1% Tween 20 (TBST). Primary antibodies [phosphorylated NF-κB p65 Ser536, total
NF-κB p65, NF-κB p100/p52, NF-κB p105/p50, NF-κB cREL and β-actin (all obtained from Cell
Signaling Technologies, Arundel, QLD)] were diluted to 1:1,000 in 5% BSA/TBST, applied and
incubated overnight at 4°C with gentle agitation. Membranes were washed for 30 min in TBST
and probed with HRP conjugated secondary antibodies diluted to 1:2,000 in 5%BSA/TBST, and
incubated for 1 h at room temperature. Proteins were visualised by using Western Lighting
enhanced chemiluminescence (Perkin Elmer Lifesciences, Boston, MA). Signals were captured
using a Kodak Digital Image Station 2000M (model: 440CF; Eastman Kodak, Rochester, NY) and
quantified by densitometry band analysis using Kodak Molecular Imaging Software (Version
4.0.5, © 1994-2005, Eastman Kodak). Phosphorylated NF-κB p65 protein was normalized to
total p65 protein (Supplementary Figure 4.1A). NF-κB p50 protein expression was normalized
to its precursor protein p105 (Supplementary Figure 4.1B). NF-κB p52 and cREL protein was
normalized to β-actin (Supplementary Figure 4.1C).
4.3.8 RNA extraction and RT-PCR:
Total cellular RNA was extracted as previously described [257] using the ToTALLY RNA Kit
(Ambion, Austin, TX). RNA quality and concentration were determined using the Agilent 2100
Biolanalyzer (Agilent Technologies, Palo Alto, CA). First strand cDNA was generated from 0.5 µg
total RNA using the AMV RT kit (Promega, Madison, WI). RT-PCR was performed in duplicate
using the Biorad CFX384 system (Biorad, Hercules, CA), containing 5XHOT FirePol® EvaGreen
Mix (Integrated Science, Sydney, NSW), forward primer, reverse primer, sterile nuclease free
72
water and cDNA (0.125 ng/μL). Data were analysed using a comparative critical threshold (Ct)
method, where the amount of the specified target gene normalised to the amount of
endogenous control, relative to the control value is given by 2-ΔΔCt. The endogenous control
used in this experiment was GAPDH. The efficacy of GAPDH as an endogenous control was
examined using the equation 2-ΔCt. Primers were designed using Primer Express software
package version 3.0 (Applied Biosystems). Gene sequences were obtained from GenBank
(Table 2). Primer sequence specificity was confirmed using BLAST. A melting point dissociation
curve was generated by the PCR instrument for all PCR products to confirm the presence of a
single amplified product.
Table 4.2: Primer sequences were designed using Primer Express Software v 3.0 (Applied
Biosystems) using sequences accessed through Genbank and checked for specificity using
nucleotide-nucleotide BLAST search.
Gene Accession No. Primer Sequence
GAPDH NM_21130 Forward CAT CCA TGA CAA CTT TGG TAT CGT
Reverse CAG TCT TCT GGG TGG CAG TGA
MCP-1 NM_002982 Forward TCC CAA AGA AGC TGT GAT CTT CA
Reverse CAG ATT CTT GGG TGG AGT GA
IL-6 NM_000600 Forward GCG AAA GGA TGA AAG TGA CCA T
Reverse AGA CAA GCC CAG CAA TGA AAA
IL-8 NM_000584 Forward CTG GCC GTC GCT CTC TGG
Reverse TTA GCA CTC CTT GGC AAA ACT
TNF-α X_02910 Forward GGA GAA GGG TGA CCG ACT CA
Reverse TGC CCA GAC TCG GCA AAG
COX-2 U_20548 Forward GAA TCA TTC ACC AGG CAA ATT G
Reverse TGG AAG CCT GTG ATA CTT TCT GTA CT
4.3.9 Nuclear extraction and Transcription Factor (TF) Assay:
Nuclear and cytoplasmic proteins were extracted from 20 mg of muscle tissue using a NE-PER
Nuclear and Cytoplasmic Extractions Reagents (Pierce, Rockford, IL), according to the
manufacturer’s instructions. Western blot analysis probed for GAPDH, α-tubulin and Lamin A
was performed to ensure the nuclear extract was not contaminated by cytoplasmic proteins

73
(Supplementary Figure 2). A Transcription Factor Assay detecting specific transcription factor
DNA binding activity was performed according to manufacturer’s instructions (Cayman
Chemical, Ann Arbour, MI). Briefly, 10 µg of nuclear protein were loaded into a well containing
an immobilized NF-B consensus sequence (5’GGGACTTTCC-3’) and incubated overnight at
4°C. Primary antibodies for NF-B subunits p65 and p50 were loaded in to each well and
incubated at room temperature for 1 h. Each well was flushed using a diluted wash buffer for
30 min. Following a secondary 30 min wash, samples were incubated with secondary HRP-
conjugated antibody for a further 1 h at room temperature. To quantify transcription factor
binding, a developing solution containing a 3,3’,5,5’-Tetramethhylbenzidine (TMB) solution was
added to each well and incubated for 45 min at room temperature. DNA binding was then
quantified using photospectrometry with absorbance measurements taken at 450 nm using a
Multiscan RC plate reader (Labsystems, Finland) and Gen5 Data Analysis Software (BioTek,
Winooski, VT).
4.3.10 Statistics:
Data are expressed as means ± SEM. Prior to analysis the data displaying a lack of normality
were log transformed to stabilise variance. Data were analysed using a two-way ANOVA with
repeated measures for time. The sphericity adjustment was checked, and if required, a
Greenhouse-Geisser epsilon to the residual degrees of freedom was applied. A data point was
considered to be a statistical outlier where a z-score exceeded a pre-determined threshold of ±
4.00. Where appropriate we explored within-group pair-wise comparisons between individual
time-points using the Least Significant Differences of means at P < 0.05 to determine
statistically significant changes [292, 293]. Statistical analyses were performed using GenStat
for Windows 16th Edition (VSN International, Hemel Hempstead, UK).

74
4.4 Results:
4.4.1 Protein expression:
Expression of NF-κB phosphorylated p65 (Ser 536) increased over time (time effect p=0.006),
but this response was overall not different between the groups (interaction effect p=0.253,
treatment effect p= 0.176). Nevertheless, LSD pairwise comparisons indicated that the main
increase from baseline was in the placebo group at 0 h post-exercise. Within the placebo group
phosphorylated p65 protein expression remained elevated at 3 h post-exercise and returned to
baseline by 24 h (Figure 4.1A). No significant changes were observed in the ibuprofen group by
LSD comparisons. There was no effect of time or treatment for NF-κB total p65 (Figure
Supplementary Figure 4.1A). There was a trend toward a significant time treatment
interaction (p=0.057) for the protein expression of the p50 subunit, although a main effect for
time (p=0.376) or treatment (p=0.865) was not achieved. Further analysis revealed a statistical
outlier in the ibuprofen group at the 24 h time point. When this subject was removed from the
analysis, this trend was weaker (p=0.115) (Figure 4.1B). LSD comparisons indicated an increase
in p50 protein expression was observed at 24h post exercise in the placebo group only (Figure
4.1B). There were no main or interaction effects for the NF-κB p50 precursor protein p105
(Supplementary Figure 4.1B) or for p52 and cREL subunits (Figure 4.1C and Figure 4.1D).

75
Figure 4.1: Protein expression of NF-κB subunits p65, p50, p52 and cREL. Representative Western blots
for p-p65 (A), p50/p105 (B), p52 (C) and cREL (D) measured in muscle biopsy samples. Data are mean
arbitrary units ± SEM. * denotes statistical significance from pre-exercise values in the placebo group; ^
denotes statistical significance from 24 h post-exercise in the placebo group (p<0.05). White bars =
placebo group; black bars =ibuprofen group.
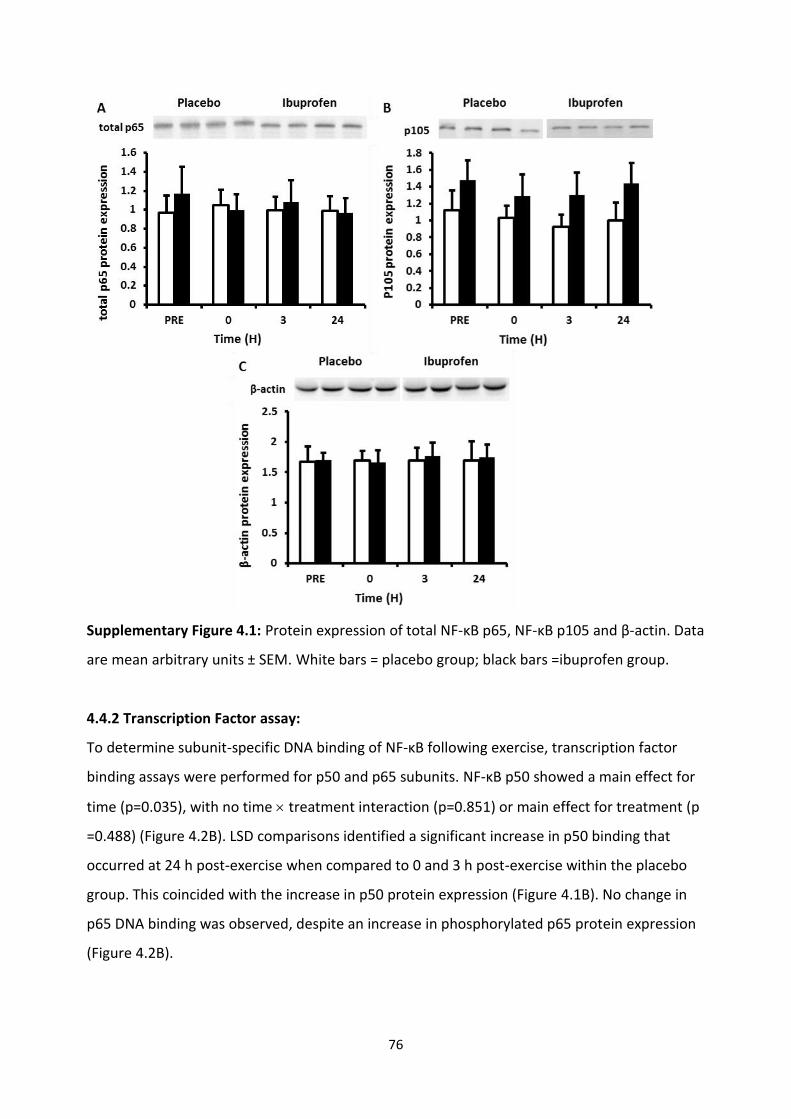
76
Supplementary Figure 4.1: Protein expression of total NF-κB p65, NF-κB p105 and β-actin. Data
are mean arbitrary units ± SEM. White bars = placebo group; black bars =ibuprofen group.
4.4.2 Transcription Factor assay:
To determine subunit-specific DNA binding of NF-κB following exercise, transcription factor
binding assays were performed for p50 and p65 subunits. NF-κB p50 showed a main effect for
time (p=0.035), with no time treatment interaction (p=0.851) or main effect for treatment (p
=0.488) (Figure 4.2B). LSD comparisons identified a significant increase in p50 binding that
occurred at 24 h post-exercise when compared to 0 and 3 h post-exercise within the placebo
group. This coincided with the increase in p50 protein expression (Figure 4.1B). No change in
p65 DNA binding was observed, despite an increase in phosphorylated p65 protein expression
(Figure 4.2B).

77
Figure 4.2: NF-κB subunits p65 (A) and p50 (B) binding to nuclear protein. Data are mean arbitrary units
± SEM. # denotes statistical significance from 0 h post-exercise in the placebo group; $ denotes
statistical significance from 3 h post-exercise in the placebo group. White bars = placebo group; black
bars =ibuprofen group.
4.4.3 RT-PCR:
The mRNA levels of IL-6 (Figure 4.3A), IL-8 (Figure 4.3B) and MCP-1 (Figure 4.3C) demonstrated
a main effect for time (p<0.01), with the highest elevation in expression levels at 3 h post-
exercise after significant increases at 0 h post-exercise. TNF-α mRNA remained unchanged
after exercise (Figure 4.3D). COX-2 mRNA showed a main effect for time (p<0.01), increasing at
0, 3 and 24 h post-exercise (Figure 4.3E). There were no significant interaction effects or main
effects for treatment for the inflammatory cytokines or COX-2 mRNA expression. Consistently,
LSD pairwise comparisons revealed similar changes over time from baseline within both
groups, demonstrating no effect of the ibuprofen supplementation protocol on exercise-
induced cytokine expression.

78
Figure 4.3: RT-PCR analysis of NF-κB target genes IL-6 (A), IL-8 (B), MCP-1 (C), TNF-α (D) and COX-2 (E) in
skeletal muscle cDNA. Data are mean arbitrary units ± SEM. * denotes statistical significance from pre-
exercise in the same treatment group; # denotes statistical significance from 0 h post-exercise in the
same treatment group; ^ denotes statistical significance from 24 h post-exercise in the same treatment
group (p<0.05). White bars = placebo group; black bars =ibuprofen group.

79
4.5 Discussion:
The current study aimed to explore the regulation of the different NF-κB subunits following a
single bout of lower body resistance exercise, and investigate the mechanism through which
ibuprofen treatment may influence post-exercise inflammation. The results of this study
provide preliminary evidence that an alternative NF-κB signaling pathway comprising
predominately p50 subunits is activated 24 h post-exercise. This pathway appears distinctly
different to the activation of NF-κB p65 subunit that occurs during the early stages of acute
post-exercise inflammation.
Phosphorylated NF-κB p65 protein expression was significantly elevated in the placebo group
when measured at 0 and 3 h post-exercise, and returned to baseline levels at 24 h post-
exercise. These findings support those of previous research showing a post-exercise increase in
key components of the classical NF-κB signaling pathway in skeletal muscle, including
phosphorylated NF-κB p65 protein [234, 239], phosphorylated IκBα protein [233, 239] and NF-
κB p65 DNA binding activity [236, 294]. In the present study, the increase in phosphorylated
NF-κB p65 protein expression was not associated with an increase in p65 transcription factor
binding activity at 0, 3 and 24 h post-exercise. However, NF-κB regulated cytokine genes
including MCP-1, IL-6 and IL-8 were increased at 3 h post-exercise. Previous work from our
group showed a transient increase in NF-κB p65 binding to the promoter region of genes
coding for inflammatory cytokines at 2 h after traditional resistance exercise that returned to
basal levels at 4 h [239]. This research performed a series of electrophoretic mobility shift
assays that looked specifically NF-κB binding to genes coding MCP-1, IL-6 and IL-8 and thus
differences in the methodology, particularly the timing of muscle biopsy samples and the
different experimental methods used to quantify DNA binding, may explain conflicting results.
Research models using an eccentric resistance exercise model could demonstrate an increase
in p65 DNA binding to nuclear protein at 3 h post-exercise [294, 295]. By contrast, research
from Durham et al showed a decrease in NF-κB DNA binding activity immediately post-
exercise, which returned to baseline levels at 1 h post-exercise [238]. Collectively, these
findings suggest that the activation of NF-κB DNA binding occurs transiently within the first few
hours of exercise. Changes in NF-κB activity in skeletal muscle may depend on the mode of
exercise and training status of participants.

80
This research also provides novel evidence for a potential role of NF-κB p50 activation as a
component of a biologically active inflammation-resolution program as outlined in chapter 3.
In our initial analysis of p50 protein expression, there was a trend towards a time group
interaction effect (p=0.057) but this trend was weaker (p=0.115) after removing a statistical
outlier. Despite this weaker interaction, within-group analysis by LSD comparisons revealed
that p50 protein expression was elevated in the placebo group at 24 h post-exercise.
Coincident with this response, p50 transcription factor binding activity was highest at 24 h
post-exercise. The biological significance of this delayed increase in NF-B p50 signaling during
the latter stages of post-exercise recovery remains uncertain. However, in vitro work suggests
that it may play a role in regulating the active resolution of acute inflammation in skeletal
muscle [282, 283, 296]. In a model of carageenin-induced pleurisy in rats, Lawrence et al [283]
provided preliminary evidence for a complex interplay between distinct NF-κB signaling
pathways that control an acute and transient inflammatory response. They reported that the
preliminary phase of NF-κB activation that occurred at 6 h was characteristic of the classical
NF-κB pathway, and was associated with the onset of acute inflammation. The secondary
phase was typical of the alternative NF-κB pathway, comprising p50 homodimers. Importantly,
inhibiting this wave of activity caused a prolonged inflammatory response, delaying the
resolution of key inflammatory mediators [283]. Findings from the present study support the
concept of two functionally distinct waves of NF-κB activity following traditional resistance
exercise. A primary wave consistent with classical NF-κB signaling pathways was observed
during the early stages of post-exercise muscle recovery and the onset of inflammation. A
secondary rise in NF-κB p50 expression occurred at 24 h post-exercise, at a time-point that
could be indicative of inflammation resolution. Based on findings from chapter 3, a biologically
active inflammation resolution program may exist followed exercise-induced muscle damage.
Future work is warranted to determine how this secondary wave of NF-κB pathway activity
influences post-exercise inflammation and skeletal muscle recovery.
Supplementation with the NSAID ibuprofen was used to investigate whether manipulating the
inflammatory response to exercise through the cyclooxygenase-prostaglandin pathway alters

81
post-exercise NF-κB signaling. Previous reports indicate that NF-κB can function upstream of
COX-2 to control transcription of this gene [192, 297]. Alternatively, prostaglandin activity may
also affect NF-κB [297-299]. In-vitro studies demonstrate that the effect of prostaglandins on
NF-κB activity is specific to the class of prostaglandin. Prostaglandin E2 activates the classical
NF-κB pathway, whereas prostaglandin A2, and prostaglandin J2 and its downstream analogues
can inhibit NF-κB activation in response to pro-inflammatory stimuli [298-301]. Our group
recently reported changes in prostaglandins in blood serum samples after traditional resistance
exercise [102]. Interestingly, PGE2, PGA2 and PGD2 all peaked in expression at 2 h post-exercise.
No significant time group interaction effects for the change in p65 phosphorylation was
observed; however, LSD comparisons suggested that phosphorylated p65 expression only
seemed to increase in the placebo group. Similarly, the observed changes in p50 protein
expression and DNA binding were only identified within the placebo group. These findings are
supported by data from the study by Lawrence et al [283], which demonstrated no change in
the secondary wave of NF-B DNA binding activity following the administration of an
alternative COX inhibitor, NS398, in rats with pleurisy. While these findings do not offer any
conclusive evidence that ibuprofen treatment inhibits the NF-κB pathway, it does provide
justification to further explore this pathway as a potential mechanism through which ibuprofen
treatment inhibits post-exercise inflammation.
4.6 Conclusion:
This research provides new insights into the regulation of NF-κB following a bout of acute
resistance exercise. The primary finding from this study is that NF-κB activation follows a
biphasic activation pattern that is subunit-specific. The first wave involves the classical NF-κB
p65 subunit, and corresponds with the onset of an acute inflammatory response and an
increase in inflammatory cytokine gene expression. The second wave is consistent with a
previously identified secondary wave of NF-κB activity that involves the alternative p50
subunit. Research in alternative models of inflammation suggests that this second wave of
activity may be associated with an active inflammatory resolution program. Further research
into the role of p50 signaling in skeletal muscle represents a key area for future research in
order to better understand the mechanisms that regulate post-exercise inflammation. Analysis
of the LSD to examine pair-wise within-group differences suggested that the observed changes

82
in both NF-κB p65 and p50 were detected only within the placebo group. The complex
interplay between the NF-κB and the COX/prostaglandin pathway in exercise-induced muscle
damage remains poorly understood, and should be a focus for future research.

83
CHAPTER 5 – GENERAL DISCUSSION AND FUTURE DIRECTIONS:
5.1 – Introduction:
The human response to resistance exercise presents a highly complex paradigm. Intense
muscular contractions have the capacity to trigger large increases in muscle size and strength.
However, following most bouts of intense exercise, there is often considerable muscle damage,
inflammation and muscle soreness. Whilst recognising the complexity of the skeletal muscle
response to exercise-induced muscle damage, the time-course of cellular events in post-
exercise muscle recovery appear to follow a well described schematic of events; (1) tissue
injury via mechanical and metabolic stress, (2) release of vasoactive and chemotactic
substances from damaged tissue, (3) migration and adhesion of systemic leucocytes to the
injury site, and (4) tissue repair and adaptation. The accumulation of inflammatory leucocytes
coincides with a secondary rise in histological and functional markers of muscle damage
including DOMS and a reduction in force generating capacity [24, 31, 39, 42, 302, 303]. These
findings have led researchers to propose that exercise-induced inflammation is a potential
cause of secondary muscle damage, and led to the implementation of anti-inflammatory
recovery strategies. However, with the past few years has been a growing recognition that
interventions targeted at the inhibition of key pro-inflammatory mediator pathways, have also
had deleterious effects of tissue adaptation and skeletal muscle repair [103, 106, 107, 161,
255]. These findings rationalize the concept that cellular events occurring within acute
inflammation form a key regulatory component of muscle regeneration and tissue repair.
The overall aim of my PhD was to explore the possibility of an active post-exercise
inflammatory resolution programme that may play a key role in establishing a biological link
between skeletal muscle damage and tissue repair. Specifically, chapter 2 involved a clinical
trial looking at the effect of ant-inflammatory medication on key markers of inflammation of
muscle damage. This project was part of a wider trial that examined the effect of anti-
inflammatory medication of muscle hypertrophy signaling pathways. The aim of chapter 3 was
to explore the human lipid profile in response to an acute resistance exercise. This paper
identified various lipids involved in regulating the bioactive resolution of acute inflammation.
These findings permitted the investigation of potential pathways that regulate both the

84
activation and resolution of acute inflammation. Chapter 4 examined the effect of an acute
bout of resistance exercise on the classical and alternative NF-κB signaling pathways in skeletal
muscle of young untrained men.
5.2 – Summary of results:
The aim for chapter 2 was to investigate the effect of oral ibuprofen administration on
leucocyte infiltration and indirect markers of muscle damage, including circulating muscle
proteins CK and myoglobin, and subjective muscle soreness. This was the first study to
demonstrate that ibuprofen administration has no effect on the histological appearance of
inflammatory white blood cells following an acute bout of traditional resistance exercise. We
also found no effect of ibuprofen administration on blood markers of muscle damage or
subjective muscle soreness, and no significant correlations between leucocyte numbers and
post-exercise muscle damage or soreness.
Chapter 3 sought to explore the possibly of a biologically active inflammatory resolution
pathway that may provide a physiological link between processes of acute exercise-induced
inflammation and skeletal muscle adaptation. Post-exercise recovery was characterized by
increased levels of COX-1 derived TXB2 and 12(S)-HHTrE, COX-2 derived prostaglandin
metabolites 2,3-dinor PGE1 and 15d-D12,14-PGJ3, LOX derivatives LTB4, 20-COOH LTB4, 5-HETE,
12-HETE, tetranor 12-HETE and 15-HETE, HDoHE species 4-HDoHE, 7-HDoHE, and CYP derived
5,6-EpETrE, 11,12-DiHETrE and 14,15-DiHETrE. This lipid profile suggests that novel lipid
intermediates involved in the formation of specialized pro-resolution mediators form a highly
complex self-resolving model of acute inflammation following a bout of resistance exercise.
The aim of chapter 4 was to explore the regulation of NF-κB as a potential regulator of an
active post-exercise inflammatory resolution programme. The primary finding from this study
is that NF-B activation follows a biphasic activation pattern that is subunit-specific. The first
wave involves the classical NF-B p65 subunit, and corresponds with the onset of an acute
inflammatory response and an increase in inflammatory cytokine gene expression. The second
wave is consistent with a previously identified secondary wave of NF-B activity that involves
the alternative p50 subunit. These findings are consistent with those from alternative models

85
of acute inflammation and identifies NF-κB as a critically important regulator of post-exercise
inflammation.
5.3 – Future directions:
This thesis represents a broad approach to exploring the possibility of a bioactive inflammatory
resolution programme following exercise-induced muscle damage. The important outcomes
from this research are; (1) leucocyte infiltration occurs following an acute resistance exercise
stimulus, however does not appear to be associated with exercise-induced muscle soreness,
(2) pro-resolution inflammatory mediators derived from omega-3 and omega-6 fatty acids are
increased following a bout of resistance exercise, (3) the biphasic regulation of NF-κB, a key
regulator of stress signaling pathways, coincides with the onset and resolution of acute
inflammation. Collectively, these findings provide preliminary evidence for an active
inflammatory resolution programme that may provide a biological link between processes of
acute muscle damage, inflammation and soft-tissue regeneration.
In chapter 2 I identified that the administration of a non-selective NSAID had no effect on the
observed increase in white blood cell infiltration or indirect measures of muscle damage.
Paulsen et al. (2010) proposed the concept of an inflammatory threshold, suggesting that
NSAIDs may only influence post-exercise inflammation in response to a sufficiently strong
inflammatory stimulus [93]. It was suggested that the absence of an intramuscular
prostaglandin response to exercise, specifically PGE2, could explain the lack of an NSAID effect
on leucocyte recruitment [93, 132]. Interestingly, in a follow up study within the same sample,
NSAID administration blunted early translational pathways involved in skeletal muscle
hypertrophy [161]. These findings present two potential paradigms. Firstly, despite the well-
established role for the inducible COX-2 enzyme in the animal muscle reparative response to
acute damage [104-106, 111, 164], the constitutively active COX-1 may play an important
function in the human adaptive response to exercise. Alternatively, NSAID administration may
have the capacity to influence alternative signalling pathways within the exercise-induced
inflammation-tissue repair paradigm.

86
These findings led our research team to explore the regulation of a novel family of n-3 and n-6
derived lipid mediators known to play a role in acute inflammatory resolution. In addition to
their role in regulating inflammation, lipid mediators have been implicated in processes of soft
tissue regeneration and skeletal muscle adaptation [304]. Firstly, Markworth et al. [102]
demonstrated elevated levels of pro-resolving lipoxins, resolvins and protectins in human
blood serum samples during the early hours of post-exercise muscle recovery. Treatment with
ibuprofen inhibited the lipid mediator response to exercise-induced cellular stress. In chapter 3
I identified a similar increase in lipids derived from the COX, LOX and CYP pathways in skeletal
muscle tissue following acute resistance exercise. In each research trial a unique lipid profile
was identified in blood serum and skeletal muscle tissue samples. Future research exploring
the functions of these lipids in exercise-induced muscle damage is necessary in understanding
the physiological significance of acute inflammation in post-exercise skeletal muscle
regeneration. At this stage the mechanism of action for lipid derived resolution mediators is
not clear and requires further investigation. This can be explored further in-vivo using rodent
models of muscle damage. Utilizing an eccentrically loaded muscle contraction, or an
unloading/reloading muscle damaging protocol, lipoxin and resolvin species can be artificially
over-expressed, or pharmacologically inhibited [171, 305, 306]. This research needs to consider
the implications of modifying lipid mediator synthesis on the onset and resolution of acute
inflammation, and what impact this has on downstream markers of skeletal muscle tissue
regeneration. Further human trials need to consider the effect of dietary interventions on key
markers of post-exercise muscle damage, inflammation and repair. Supplementation with n-3
EPA and DPA has a highly divergent effect on the human lipid mediator profile, specifically
resolvin and maresin species [307]. This avenue of research would explore the concept of
recovery interventions designed to promote resolving mediators and biochemical pathways
that are homeostatic and modulatory in their actions, to optimize the outcome of an acute
inflammatory program.
Chapter 4 identified NF-κB as a potential transcriptional regulator involved in both the onset
and resolution of acute post-exercise inflammation. The primary finding from this study is that
NF-κB regulation follows a biphasic activation pattern that is subunit-specific. The first wave
involves the classical NF-κB p65 subunit, and corresponds with the onset of an acute

87
inflammatory response and an increase in inflammatory cytokine gene expression. The second
wave is consistent with a previously identified secondary wave of NF-κB activity that involves
the alternative p50 subunit. While research in alternative models of acute inflammation
suggest this second wave of increased NF-κB activity may be associated with an active
inflammatory resolution program, no research has explored the physiological significance of
this pathway in acute post-exercise inflammation. Recent work from our group utilized a series
of electrophoretic mobility shift assays (EMSA) to demonstrate an increase in NF-κB p65
binding to the promoter region of genes coding for inflammatory cytokines in the early stages
of exercise-induced inflammation [239]. This research model may provide an avenue to explore
whether the secondary increase in key markers of the alternative NF-κB signaling pathway are
involved in regulating a biologically active inflammatory resolution program. Potential targets
of NF-κB p50 homodimers include the inducible COX-2 enzyme, transcription factor p53 and
the pro-apoptotic Bcl2 homolog Bax [192, 210].
An inherent limitation of muscle biopsy studies is the test-retest reliability of muscle biopsies
obtained from different sites along the vastus lateralis. The muscle biopsy protocol used
throughout this thesis was used to minimize interference from the biopsy procedure on
markers of acute inflammation [239]. However, previous research has demonstrated the
regional differences in muscle hypertrophy/atrophy exist across an individual muscle length
[308]. Further evidence suggests that intramuscular differences in fibre type have been
observed across individual muscle biopsy samples from the vastus lateralis [309]. The absence
of a non-exercise control group, and the inherent lack of test-retest reliability of the muscle
biopsy protocol are acknowledged as accepted limitations of the current research.
5.4 – Concluding remarks:
This thesis presents preliminary evidence for a biologically active inflammatory resolution
program that may provide a crucial link between processes of exercise-induced muscle
damage, and tissue regeneration and adaptation. This concept would challenge the current use
of anti-inflammatory therapies to promote post-exercise muscle recovery. Most of the anti-
inflammatory interventions studied to date, arise from our need to control the cardinal signs of

88
inflammation by inhibiting key pro-inflammatory mediator pathways. Experimental models
targeted at inhibiting the post-exercise inflammatory response have identified that exercise-
induced inflammation is a key regulatory feature in the restoration of tissue structure and
function.
The overall aim of this thesis was to explore the regulation of a family of novel lipid derivatives
that have been shown to play dual anti-inflammatory and pro-resolution functions in acute
inflammation. In chapter 2 we demonstrated that the administration of ibuprofen had no
effect on key inflammatory and muscle damage markers. These findings challenge the
suitability of NSAID administration as an effective treatment strategy for reducing acute
inflammation and muscle soreness following resistance exercise. Further research from within
our laboratory [161], amongst others [87, 107, 130], have demonstrated a detrimental effect
of anti-inflammatory interventions on post-exercise early translational signalling responses via
the inhibition of eicosanoids derived from the COX pathway. Taken together, these findings
suggest that inhibiting eicosanoid formation during acute post-exercise inflammation may
directly affect muscle hypertrophy pathways without directly impacting on cellular
inflammation. In chapter 3 we explored the regulation of lipid species during acute post-
exercise muscle recovery. We identified a post-exercise increase in eicosanoids and
docosanoids species derived from COX, LOX and CYP pathways, presenting the possibility of a
highly complex self-resolving model of acute inflammation following an acute bout of
resistance exercise. For chapter 4, an in-depth analysis of the regulation of NF-κB,
demonstrated a bi-phasic regulation pattern that coincides with the onset and resolution of
acute inflammation. This thesis shows for the first time that events occurring early in acute
post-exercise inflammation may engage an active and coordinated inflammatory resolution
programme, governed by the production of specialized pro-resolution lipid mediators. Further
work is required to elucidate the specific functions of each of these lipid variables and explore
the efficacy of interventions designed to promote resolving mediators and biochemical
pathways that will be homeostatic and modulatory in their actions.

89
REFERENCE LIST:

90
1. Flück, M., Functional, structural and molecular plasticity of mammalian skeletal muscle
in response to exercise stimuli. The Journal Of Experimental Biology, 2006. 209(Pt 12): p.
2239-2248.
2. Harridge, S.D.R., Plasticity of human skeletal muscle: gene expression to in vivo function.
Experimental Physiology, 2007. 92(5): p. 783-797.
3. Rasmussen, B.B. and S.M. Phillips, Contractile and nutritional regulation of human
muscle growth. Exercise And Sport Sciences Reviews, 2003. 31(3): p. 127-131.
4. Schiaffino, S. and G. Salviati, Molecular diversity of myofibrillar proteins: isoforms
analysis at the protein and mRNA level. Methods In Cell Biology, 1997. 52: p. 349-369.
5. Seynnes, O.R., M. de Boer, and M.V. Narici, Early skeletal muscle hypertrophy and
architectural changes in response to high-intensity resistance training. Journal of
Applied Physiology, 2007. 102(1): p. 368-373.
6. Vierck, J., et al., Satellite cell regulation following myotrauma caused by resistance
exercise. Cell Biology International, 2000. 24(5): p. 263-272.
7. Hough, T., Ergographic studies in muscular fatigue and soreness. Journal. Boston
Society Of Medical Sciences, 1900. 5(3): p. 81-92.
8. Faulkner, J.A., D.A. Jones, and J.M. Round, Injury to skeletal muscles of mice by forced
lengthening during contractions. Quarterly Journal Of Experimental Physiology
(Cambridge, England), 1989. 74(5): p. 661-670.
9. Armstrong, R.B., R.W. Ogilvie, and J.A. Schwane, Eccentric exercise-induced injury to rat
skeletal muscle. Journal Of Applied Physiology: Respiratory, Environmental And Exercise
Physiology, 1983. 54(1): p. 80-93.
10. Kuipers, H., et al., Muscle degeneration after exercise in rats. International Journal Of
Sports Medicine, 1983. 4(1): p. 45-51.
11. Lieber, R.L., L.E. Thornell, and J. Fridén, Muscle cytoskeletal disruption occurs within the
first 15 min of cyclic eccentric contraction. Journal Of Applied Physiology (Bethesda,
Md.: 1985), 1996. 80(1): p. 278-284.

91
12. Lieber, R.L., T.M. Woodburn, and J. Fridén, Muscle damage induced by eccentric
contractions of 25% strain. Journal Of Applied Physiology (Bethesda, Md.: 1985), 1991.
70(6): p. 2498-2507.
13. McNeil, P.L. and R. Khakee, Disruptions of muscle fiber plasma membranes. Role in
exercise-induced damage. The American Journal Of Pathology, 1992. 140(5): p. 1097-
1109.
14. Salminen, A., K. Hongisto, and V. Vihko, Lysosomal changes related to exercise injuries
and training-induced protection in mouse skeletal muscle. Acta Physiologica
Scandinavica, 1984. 120(1): p. 15-19.
15. Salminen, A. and V. Vihko, Exercise myopathy: selectively enhanced proteolytic capacity
in rat skeletal muscle after prolonged running. Experimental And Molecular Pathology,
1983. 38(1): p. 61-68.
16. Fridén, J. and R.L. Lieber, Segmental muscle fiber lesions after repetitive eccentric
contractions. Cell And Tissue Research, 1998. 293(1): p. 165-171.
17. Fridén, J., M. Sjöström, and B. Ekblom, A morphological study of delayed muscle
soreness. Experientia, 1981. 37(5): p. 506-507.
18. Fridén, J., U. Kjörell, and L.E. Thornell, Delayed muscle soreness and cytoskeletal
alterations: an immunocytological study in man. International Journal Of Sports
Medicine, 1984. 5(1): p. 15-18.
19. Friden, J., M. Sjoestroem, and B. Ekblom, Myofibrillar damage following intense
eccentric exercise in man. International Journal of Sports Medicine, 1983. 4(3): p. 170-
176.
20. Newham, D.J., The consequences of eccentric contractions and their relationship to
delayed onset muscle pain. European Journal Of Applied Physiology And Occupational
Physiology, 1988. 57(3): p. 353-359.
21. Newham, D.J., D.A. Jones, and R.H.T. Edwards, Large delayed plasma creatine kinase
changes after stepping exercise. Muscle & Nerve, 1983(6): p. 380-385.

92
22. Newham, D.J., et al., Muscle fatigue and pain after eccentric contractions at long and
short length. Clinical Science (London, England: 1979), 1988. 74(5): p. 553-557.
23. Pillon, N.J., et al., Cross-talk between skeletal muscle and immune cells: muscle-derived
mediators and metabolic implications. American Journal of Physiology - Endocrinology
and Metabolism, 2013. 304(5): p. E453-E465.
24. Armstrong, R.B., G.L. Warren, and J.A. Warren, Mechanisms of exercise-induced muscle
fibre injury. Sports Medicine, 1991. 12(3): p. 184-207.
25. Smith, L.L., Acute inflammation: the underlying mechanism in delayed onset muscle
soreness? Medicine & Science in Sports & Exercise, 1991. 23(5): p. 542-551.
26. Appell, H.J., J.M.C. Soares, and J.A.R. Duarte, Exercise, muscle damage and fatigue.
Sports Medicine, 1992. 13(2): p. 108-115.
27. Lieber, R.L. and J. Fridén, Mechanisms of muscle injury gleaned from animal models.
American Journal Of Physical Medicine & Rehabilitation / Association Of Academic
Physiatrists, 2002. 81(11 Suppl): p. S70-S79.
28. Michna, H., Ultrastructural features of skeletal muscle in mice after physical exercise: its
relation to the pathogenesis of leucocyte invasion. Acta Anatomica, 1989. 134(4): p.
276-282.
29. Smith, C., et al., The inflammatory response to skeletal muscle injury: illuminating
complexities. Sports Medicine (Auckland, N.Z.), 2008. 38(11): p. 947-969.
30. Tidball, J.G., Inflammatory processes in muscle injury and repair. American Journal Of
Physiology. Regulatory, Integrative And Comparative Physiology, 2005. 288(2): p. R345-
R353.
31. Tidball, J.G., Inflammatory cell response to acute muscle injury. Medicine & Science in
Sports & Exercise, 1995. 27(7): p. 1022-1032.
32. Stauber, W.T., et al., Extracellular matrix disruption and pain after eccentric muscle
action. Journal of Applied Physiology, 1990. 69(3): p. 868-874.

93
33. Tidball, J.G. and S.A. Villalta, Regulatory interactions between muscle and the immune
system during muscle regeneration. American Journal Of Physiology. Regulatory,
Integrative And Comparative Physiology, 2010. 298(5): p. R1173-R1187.
34. Butterfield, T.A., T.M. Best, and M.A. Merrick, The Dual Roles of Neutrophils and
Macrophages in Inflammation: A Critical Balance Between Tissue Damage and Repair.
Journal of Athletic Training, 2006. 41(4): p. 457-465.
35. Evans, W.J. and J.G. Cannon, The metabolic effects of exercise-induced muscle damage.
Exercise & Sport Sciences Reviews, 1991. 19: p. 99-125.
36. Frenette, J., et al., Muscle impairment occurs rapidly and precedes inflammatory cell
accumulation after mechanical loading. American Journal of Physiology: Regulatory,
Integrative & Comparative Physiology, 2002. 51(2): p. R351.
37. Nguyen, H.X. and J.G. Tidball, Null mutation of gp91phox reduces muscle membrane
lysis during muscle inflammation in mice. The Journal Of Physiology, 2003. 553(Pt 3): p.
833-841.
38. Toumi, H., S. F’guyer, and T.M. Best, The role of neutrophils in injury and repair
following muscle stretch. Journal of Anatomy, 2006. 208(4): p. 459-470.
39. Brickson, S., et al., Oxidant production and immune response after stretch injury in
skeletal muscle. Medicine & Science in Sports & Exercise, 2001. 33(12): p. 2010-2015.
40. Hurme, T., et al., Healing of skeletal muscle injury: an ultrastructural and
immunohistochemical study. Medicine & Science in Sports & Exercise, 1991. 23(7): p.
801-810.
41. Newham, D.J., et al., Ultrastructural changes after concentric and eccentric contractions
of human muscle. Journal of Neurological Sciences, 1983. 61(1): p. 109-122.
42. Pizza, F.X., et al., Neutrophils contribute to muscle injury and impair its resolution after
lengthening contractions in mice. The Journal Of Physiology, 2005. 562(Pt 3): p. 899-
913.
43. St Pierre Schneider, B., et al., CD11b+ neutrophils predominate over RAM11+
macrophages in stretch-injured muscle. Muscle & Nerve, 2002. 25(6): p. 837-844.

94
44. Erwig, L.P., et al., Initial cytokine exposure determines function of macrophages and
renders them unresponsive to other cytokines. Journal Of Immunology (Baltimore, Md.:
1950), 1998. 161(4): p. 1983-1988.
45. MacIntyre, D.L., W.D. Reid, and D.C. McKenzie, Delayed muscle soreness. The
inflammatory response to muscle injury and its clinical implications. Sports Medicine
(Auckland, N.Z.), 1995. 20(1): p. 24-40.
46. Honda, H., H. Kimura, and A. Rostami, Demonstration and phenotypic characterization
of resident macrophages in rat skeletal muscle. Immunology, 1990. 70(2): p. 272-277.
47. St. Pierre, B.A. and J.G. Tidball, Differential response of macrophage subpopulations to
soleus muscle reloading after rat hindlimb suspension. Journal of Applied Physiology,
1994. 77(1): p. 290-297.
48. Arnold, L., et al., Inflammatory monocytes recruited after skeletal muscle injury switch
into antiinflammatory macrophages to support myogenesis. The Journal Of
Experimental Medicine, 2007. 204(5): p. 1057-1069.
49. McLennan, I.S., Degenerating and regenerating skeletal muscles contain several
subpopulations of macrophages with distinct spatial and temporal distributions. Journal
of Anatomy, 1996. 188 ( Pt 1): p. 17-28.
50. Krippendorf, B.B., Riley, D. A., Distinguishing unloading versus reloading-induced
changes in rat soleus muscle. Muscle & Nerve, 1993. 16: p. 99-108.
51. McLennan, I.S., Resident macrophages (ED2- and ED3-positive) do not phagocytose
degenerating rat skeletal muscle fibres. Cell And Tissue Research, 1993. 272(1): p. 193-
196.
52. Honda, H., H. Kimura, and A. Rostami, Isolation and characterization of macrophages
from rat embryonic muscle culture. Journal Of Leukocyte Biology, 1992. 52(5): p. 537-
544.
53. Cantini, M., et al., Macrophage-secreted myogenic factors: a promising tool for greatly
enhancing the proliferative capacity of myoblasts in vitro and in vivo. Neurological
Sciences, 2002. 23(4): p. 189.

95
54. Chazaud, B., et al., Satellite cells attract monocytes and use macrophages as a support
to escape apoptosis and enhance muscle growth. Journal of Cell Biology, 2003. 163(5):
p. 1133-1143.
55. Gierer, P., et al., Selective cyclooxygenase-2 inhibition reverses microcirculatory and
inflammatory sequelae of closed soft tissue trauma in an animal model. Journal of Bone
& Joint Surgery, American Volume, 2005. 87(1): p. 153-160.
56. Massimino, M.L., et al., ED2+ macrophages increase selectively myoblast proliferation in
muscle cultures. Biochemical And Biophysical Research Communications, 1997. 235(3):
p. 754-759.
57. Merly, F., et al., Macrophages enhance muscle satellite cell proliferation and delay their
differentiation. Muscle & Nerve, 1999. 22(6): p. 724-732.
58. Sonnet, C., et al., Human macrophages rescue myoblasts and myotubes from apoptosis
through a set of adhesion molecular systems. Journal Of Cell Science, 2006. 119(Pt 12):
p. 2497-2507.
59. Summan, M., et al., Macrophages and skeletal muscle regeneration: a clodronate-
containing liposome depletion study. American Journal Of Physiology. Regulatory,
Integrative And Comparative Physiology, 2006. 290(6): p. R1488-R1495.
60. Tidball, J.G. and M. Wehling-Henricks, Macrophages promote muscle membrane repair
and muscle fibre growth and regeneration during modified muscle loading in mice in
vivo. The Journal Of Physiology, 2007. 578(Pt 1): p. 327-336.
61. Crameri, R.M., et al., Myofibre damage in human skeletal muscle: effects of electrical
stimulation versus voluntary contraction. The Journal Of Physiology, 2007. 583(Pt 1): p.
365-380.
62. Malm, C., et al., Immunological changes in human skeletal muscle and blood after
eccentric exercise and multiple biopsies. The Journal Of Physiology, 2000. 529 Pt 1: p.
243-262.

96
63. Raastad, T., et al., Temporal relation between leukocyte accumulation in muscles and
halted recovery 10-20 h after strength exercise. Journal Of Applied Physiology
(Bethesda, Md.: 1985), 2003. 95(6): p. 2503-2509.
64. Yu, J.-G., D.O. Fürst, and L.-E. Thornell, The mode of myofibril remodelling in human
skeletal muscle affected by DOMS induced by eccentric contractions. Histochemistry
And Cell Biology, 2003. 119(5): p. 383-393.
65. Yu, J.-G., C. Malm, and L.-E. Thornell, Eccentric contractions leading to DOMS do not
cause loss of desmin nor fibre necrosis in human muscle. Histochemistry and Cell
Biology, 2002. 118(1): p. 29-34.
66. MacIntyre, D.L., et al., Presence of WBC, decreased strength, and delayed soreness in
muscle after eccentric exercise. Journal Of Applied Physiology (Bethesda, Md.: 1985),
1996. 80(3): p. 1006-1013.
67. Crenshaw, A.G., et al., Increased technetium uptake is not equivalent to muscle necrosis:
scintigraphic, morphological and intramuscular pressure analyses of sore muscles after
exercise. Acta Physiologica Scandinavica, 1993. 148(2): p. 187-198.
68. Fielding, R.A., et al., Acute phase response in exercise. III. Neutrophil and IL-1 beta
accumulation in skeletal muscle. The American Journal Of Physiology, 1993. 265(1 Pt 2):
p. R166-R172.
69. Hellsten, Y., et al., Xanthine oxidase in human skeletal muscle following eccentric
exercise: a role in inflammation. The Journal Of Physiology, 1997. 498 ( Pt 1): p. 239-
248.
70. Jones, D.A., et al., Experimental human muscle damage: morphological changes in
relation to other indices of damage. The Journal Of Physiology, 1986. 375: p. 435-448.
71. Kuipers, H., et al., Influence of a prostaglandin-inhibiting drug on muscle soreness after
eccentric work. International Journal Of Sports Medicine, 1985. 6(6): p. 336-339.
72. Nurenberg, P., et al., MR imaging-guided muscle biopsy for correlation of increased
signal intensity with ultrastructural change and delayed-onset muscle soreness after
exercise. Radiology, 1992. 184(3): p. 865-869.

97
73. O'Reilly, K.P., et al., Eccentric exercise-induced muscle damage impairs muscle glycogen
repletion. Journal Of Applied Physiology (Bethesda, Md.: 1985), 1987. 63(1): p. 252-256.
74. Round, J.M., D.A. Jones, and G. Cambridge, Cellular infiltrates in human skeletal muscle:
exercise induced damage as a model for inflammatory muscle disease? Journal Of The
Neurological Sciences, 1987. 82(1-3): p. 1-11.
75. Aronson, D., et al., Extracellular-regulated protein kinase cascades are activated in
response to injury in human skeletal muscle. The American Journal Of Physiology, 1998.
275(2 Pt 1): p. C555-C561.
76. Constantin-Teodosiu, D., et al., The effect of repeated muscle biopsy sampling on ATP
and glycogen resynthesis following exercise in man. European Journal Of Applied
Physiology And Occupational Physiology, 1996. 73(1-2): p. 186-190.
77. Chleboun, G.S., J.N. Howell, and R.R. Conatser, Relationship between muscle swelling
and stiffness after eccentric exercise. Medicine & Science in Sports & Exercise, 1998.
30(4): p. 529-535.
78. Cleak, M.J. and R.G. Eston, Muscle soreness, swelling, stiffness and strength loss after
intense eccentric exercise. British Journal Of Sports Medicine, 1992. 26(4): p. 267-272.
79. Fridén, J. and R.L. Lieber, Serum creatine kinase level is a poor predictor of muscle
function after injury. Scandinavian Journal Of Medicine & Science In Sports, 2001. 11(2):
p. 126-127.
80. Nosaka, K. and P.M. Clarkson, Variability in serum creatine kinase response after
eccentric exercise of the elbow flexors. International Journal Of Sports Medicine, 1996.
17(2): p. 120-127.
81. Nosaka, K., M. Newton, and P. Sacco, Delayed-onset muscle soreness does not reflect
the magnitude of eccentric exercise-induced muscle damage. Scandinavian Journal of
Medicine & Science in Sports, 2002. 12(6): p. 337.
82. Rodenburg, J., P. Bar, and R. De Baer, Relations between muscle soreness and
biochemical and functional outcomes of eccentric exercise. Journal of Applied
Physiology, 1993. 74(6): p. 2976-2983.

98
83. Féasson, L., et al., Molecular adaptations of neuromuscular disease-associated proteins
in response to eccentric exercise in human skeletal muscle. The Journal Of Physiology,
2002. 543(Pt 1): p. 297-306.
84. Paulsen, G., et al., Time course of leukocyte accumulation in human muscle after
eccentric exercise. Medicine And Science In Sports And Exercise, 2010. 42(1): p. 75-85.
85. Paulsen, G., et al., Leucocytes, cytokines and satellite cells: what role do they play in
muscle damage and regeneration following eccentric exercise? Exercise Immunology
Review, 2012. 18: p. 42-97.
86. Hubal, M.J., et al., Inflammatory gene changes associated with the repeated-bout
effect. American Journal of Physiology: Regulatory, Integrative & Comparative
Physiology, 2008. 63(5): p. R1628-R1637.
87. Mikkelsen, U.R., et al., Local NSAID infusion inhibits satellite cell proliferation in human
skeletal muscle after eccentric exercise. Journal Of Applied Physiology (Bethesda, Md.:
1985), 2009. 107(5): p. 1600-1611.
88. Beaton, L.J., et al., Contraction-induced muscle damage is unaffected by vitamin E
supplementation. Medicine And Science In Sports And Exercise, 2002. 34(5): p. 798-805.
89. Beaton, L.J., M.A. Tarnopolsky, and S.M. Phillips, Contraction-induced muscle damage in
humans following calcium channel blocker administration. The Journal Of Physiology,
2002. 544(Pt 3): p. 849-859.
90. Crenshaw, A.G., et al., Knee extension torque and intramuscular pressure of the vastus
lateralis muscle during eccentric and concentric activities. European Journal Of Applied
Physiology And Occupational Physiology, 1995. 70(1): p. 13-19.
91. Crenshaw, A.G., L.E. Thornell, and J. Fridén, Intramuscular pressure, torque and swelling
for the exercise-induced sore vastus lateralis muscle. Acta Physiologica Scandinavica,
1994. 152(3): p. 265-277.
92. Stupka, N., et al., Cellular adaptation to repeated eccentric exercise-induced muscle
damage. Journal Of Applied Physiology (Bethesda, Md.: 1985), 2001. 91(4): p. 1669-
1678.

99
93. Paulsen, G., et al., A COX-2 inhibitor reduces muscle soreness, but does not influence
recovery and adaptation after eccentric exercise. Scandinavian Journal of Medicine &
Science in Sports, 2010. 20(1): p. 1-13.
94. Child, R., et al., Changes in indices of antioxidant status, lipid peroxidation and
inflammation in human skeletal muscle after eccentric muscle actions. Clinical Science
(London, England: 1979), 1999. 96(1): p. 105-115.
95. Hikida, R.S., et al., Muscle fiber necrosis associated with human marathon runners.
Journal Of The Neurological Sciences, 1983. 59(2): p. 185-203.
96. Lauritzen, F., et al., Gross ultrastructural changes and necrotic fiber segments in elbow
flexor muscles after maximal voluntary eccentric action in humans. Journal of Applied
Physiology, 2009. 107(6): p. 1923-1934.
97. Paulsen, G., et al., Inflammatory markers CD11b, CD16, CD66b, CD68, myeloperoxidase
and neutrophil elastase in eccentric exercised human skeletal muscles. Histochemistry
And Cell Biology, 2013. 139(5): p. 691-715.
98. Mahoney, D.J., et al., Gene expression profiling in human skeletal muscle during
recovery from eccentric exercise. American Journal Of Physiology. Regulatory,
Integrative And Comparative Physiology, 2008. 294(6): p. R1901-R1910.
99. Elnachef, N., et al., Changing perceptions and practices regarding aspirin, nonsteroidal
anti-inflammatory drugs, and cyclooxygenase-2 selective nonsteroidal anti-
inflammatory drugs among US primary care providers. Alimentary Pharmacology &
Therapeutics, 2008. 28(10): p. 1249-1258.
100. Green, G.A., Understanding NSAIDs: from aspirin to COX-2. Clinical Cornerstone, 2001.
3(5): p. 50-60.
101. Markworth, J.F., K.R. Maddipati, and D. Cameron-Smith, Emerging roles of pro-resolving
lipid mediators in immunological and adaptive responses to exercise-induced muscle
injury. Exercise Immunology Review, 2016. 22: p. 110-134.

100
102. Markworth, J.F., et al., Human inflammatory and resolving lipid mediator responses to
resistance exercise and ibuprofen treatment. American Journal Of Physiology.
Regulatory, Integrative And Comparative Physiology, 2013. 305(11): p. R1281-R1296.
103. Urso, M.L., Anti-inflammatory interventions and skeletal muscle injury: benefit or
detriment? Journal Of Applied Physiology (Bethesda, Md.: 1985), 2013. 115(6): p. 920-
928.
104. Bondesen, B.A., et al., The COX-2 pathway is essential during early stages of skeletal
muscle regeneration. American Journal of Physiology: Cell Physiology, 2004. 56(2): p.
C475-C483.
105. Bondesen, B.A., S.T. Mills, and G.K. Paviath, The COX-2 pathway regulates growth of
atrophied muscle via multiple mechanisms. American Journal of Physiology: Cell
Physiology, 2006. 59(6): p. C1651-C1659.
106. Mishra, D.K., et al., Anti-inflammatory medication after muscle injury. A treatment
resulting in short-term improvement but subsequent loss of muscle function. The
Journal Of Bone And Joint Surgery. American Volume, 1995. 77(10): p. 1510-1519.
107. Trappe, T.A., et al., Effect of ibuprofen and acetaminophen on postexercise muscle
protein synthesis. American Journal Of Physiology. Endocrinology And Metabolism,
2002. 282(3): p. E551-E556.
108. Mackey, A.L., et al., The influence of anti-inflammatory medication on exercise-induced
myogenic precursor cell responses in humans. Journal of Applied Physiology, 2007.
103(2): p. 425-431.
109. Demers, L.M., et al., Effect of prolonged exercise on plasma prostaglandin levels.
Prostaglandins And Medicine, 1981. 6(4): p. 413-418.
110. Laustiola, K., et al., Exercise-induced increase in plasma arachidonic acid and
thromboxane B2 in healthy men: effect of beta-adrenergic blockade. Journal of
Cardiovascular Pharmacology, 1984. 6(3): p. 449-454.

101
111. Nowak, J. and A. Wennmalm, Effect of exercise on human arterial and regional venous
plasma concentrations of prostaglandin E. Prostaglandins And Medicine, 1978. 1(6): p.
489-497.
112. Venkatraman, J.T., X. Feng, and D. Pendergast, Effects of dietary fat and endurance
exercise on plasma cortisol, prostaglandin E2, interferon-gamma and lipid peroxides in
runners. Journal Of The American College Of Nutrition, 2001. 20(5): p. 529-536.
113. Carter, J.W., et al., The effect of exercise on bleeding time and local production of
prostacyclin and thromboxane. European Journal Of Applied Physiology And
Occupational Physiology, 1989. 59(5): p. 355-359.
114. Feng, D.L., et al., Upright posture and maximal exercise increase platelet aggregability
and prostacyclin production in healthy male subjects. British Journal Of Sports
Medicine, 1999. 33(6): p. 401-404.
115. Mourits-Andersen, T., et al., Plasma 6-keto-PGF1 alpha, thromboxane B2 and PGE2 in
type 1 (insulin-dependent) diabetic patients during exercise. Diabetologia, 1987. 30(7):
p. 460-463.
116. Vinikka, L., J. Vuori, and O. Ylikorkala, Lipid peroxides, prostacyclin, and thromboxane A2
in runners during acute exercise. Medicine & Science in Sports & Exercise, 1984. 16(3):
p. 275-277.
117. Dousset, E., et al., Bimodal recovery pattern in human skeletal muscle induced by
exhaustive stretch-shortening cycle exercise. Medicine And Science In Sports And
Exercise, 2007. 39(3): p. 453-460.
118. Uchida, M.C., et al., Effect of bench press exercise intensity on muscle soreness and
inflammatory mediators. Journal of Sports Sciences, 2009. 27(5): p. 499-507.
119. Tartibian, B., B.H. Maleki, and A. Abbasi, Omega-3 Fatty Acids Supplementation
Attenuates Inflammatory Markers After Eccentric Exercise in Untrained Men. Clinical
Journal of Sport Medicine, 2011. 21(2): p. 131-137.

102
120. Boatwright, D., R. Byrd, and M. Mangum, Relationship between serum prostaglandin
formation, creatine kinase activity, and ratings of perceived soreness. Research in
Sports Medicine: An International Journal, 1991. 2(2): p. 85-88.
121. Croisier, J.L., et al., Myocellular Enzyme Leakage, Polymorphonuclear Neutrophil
Activation and Delayed Onset Muscle Soreness Induced by Isokinetic Eccentric Exercise.
Archives of Physiology and Biochemistry, 1996. 104(3): p. 322-329.
122. Croisier, J.L., et al., Piroxicam fails to reduce myocellular enzyme leakage and delayed
onset muscle soreness induced by isokinetic eccentric exercise. Mediators Of
Inflammation, 1996. 5(3): p. 230-234.
123. Black, C.D., et al., Ginger (Zingiber officinale) reduces muscle pain caused by eccentric
exercise. Journal of Pain, 2010. 11(9): p. 894-903.
124. Braun, W.A., et al., The effects of chondroitin sulfate supplementation on indices of
muscle damage induced by eccentric arm exercise. The Journal Of Sports Medicine And
Physical Fitness, 2005. 45(4): p. 553-560.
125. Hirose, L., et al., Changes in inflammatory mediators following eccentric exercise of the
elbow flexors. Exercise Immunology Review, 2004. 10: p. 75-90.
126. Buford, T.W., et al., Protease supplementation improves muscle function after eccentric
exercise. Medicine & Science in Sports & Exercise, 2009. 41(10): p. 1908-1914.
127. Carroll, C.C., et al., The influence of acute resistance exercise on cyclooxygenase-1 and -
2 activity and protein levels in human skeletal muscle. American Journal Of Physiology.
Regulatory, Integrative And Comparative Physiology, 2013. 305(1): p. R24-R30.
128. Testa, M., et al., Expression and activity of cyclooxygenase isoforms in skeletal muscles
and myocardium of humans and rodents. Journal Of Applied Physiology (Bethesda, Md.:
1985), 2007. 103(4): p. 1412-1418.
129. Weinheimer, E.M., et al., Resistance exercise and cyclooxygenase (COX) expression in
human skeletal muscle: implications for COX-inhibiting drugs and protein synthesis.
American Journal Of Physiology. Regulatory, Integrative And Comparative Physiology,
2007. 292(6): p. R2241-R2248.

103
130. Burd, N.A., et al., Effect of a cyclooxygenase-2 inhibitor on postexercise muscle protein
synthesis in humans. American Journal of Physiology: Endocrinology & Metabolism,
2010. 61(2): p. E354-E361.
131. Trappe, T.A., et al., Influence of acetaminophen and ibuprofen on skeletal muscle
adaptations to resistance exercise in older adults. American Journal Of Physiology.
Regulatory, Integrative And Comparative Physiology, 2011. 300(3): p. R655-R662.
132. Trappe, T.A., et al., Skeletal muscle PGF(2)(alpha) and PGE(2) in response to eccentric
resistance exercise: influence of ibuprofen acetaminophen. The Journal Of Clinical
Endocrinology And Metabolism, 2001. 86(10): p. 5067-5070.
133. Trappe, T., et al., Effects of age and resistance exercise on skeletal muscle interstitial
prostaglandin F(2alpha). Prostaglandins, Leukotrienes, And Essential Fatty Acids, 2006.
74(3): p. 175-181.
134. Karamouzis, M., et al., The response of muscle interstitial prostaglandin E(2)(PGE(2)),
prostacyclin I(2)(PGI(2)) and thromboxane A(2)(TXA(2)) levels during incremental
dynamic exercise in humans determined by in vivo microdialysis. Prostaglandins,
Leukotrienes, And Essential Fatty Acids, 2001. 64(4-5): p. 259-263.
135. Karamouzis, M., et al., In situ microdialysis of intramuscular prostaglandin and
thromboxane in contracting skeletal muscle in humans. Acta Physiologica Scandinavica,
2001. 171(1): p. 71-76.
136. Boushel, R., et al., Combined inhibition of nitric oxide and prostaglandins reduces
human skeletal muscle blood flow during exercise. The Journal Of Physiology, 2002.
543(Pt 2): p. 691-698.
137. Burian, A., et al., An exploratory microdialysis study investigating the effect of repeated
application of a diclofenac epolamine medicated plaster on prostaglandin
concentrations in skeletal muscle after standardized physical exercise. British Journal Of
Clinical Pharmacology, 2013. 76(6): p. 880-887.
138. Dudley, G.A., et al., Efficacy of naproxen sodium for exercise-induced dysfunction muscle
injury and soreness. Clinical Journal Of Sport Medicine: Official Journal Of The Canadian
Academy Of Sport Medicine, 1997. 7(1): p. 3-10.

104
139. Reynolds, J.F., et al., Non-steroidal anti-inflammatory drugs fail to enhance healing of
acute hamstring injuries treated with physiotherapy. South African Medical Journal,
1995. 85(6): p. 517-522.
140. Tokmakidis, S.P., et al., The effects of ibuprofen on delayed muscle soreness and
muscular performance after eccentric exercise. Journal Of Strength And Conditioning
Research / National Strength & Conditioning Association, 2003. 17(1): p. 53-59.
141. Donnelly, A.W., R.J. Maughan, and P.H. Whiting, Effects of ibuprofen on exercise-
induced muscle soreness and indices of muscle damage. British Journal of Sports
Medicine, 1990. 24(3): p. 191-195.
142. Hasson, S.M., et al., Effect of ibuprofen use on muscle soreness, damage, and
performance: a preliminary investigation. Medicine & Science in Sports & Exercise,
1993. 25(1): p. 9-17.
143. Hill, D.W. and J.D. Richardson, Effectiveness of 10% trolamine salicylate cream on
muscular soreness induced by a reproducible program of weight training. The Journal Of
Orthopaedic And Sports Physical Therapy, 1989. 11(1): p. 19-23.
144. Lecomte, J.M., V.J. Lacroix, and D.L. Montgomery, A randomized controlled trial of the
effect of naproxen on delayed onset muscle soreness and muscle strength. Clinical
Journal Of Sport Medicine: Official Journal Of The Canadian Academy Of Sport
Medicine, 1998. 8(2): p. 82-87.
145. O'Grady, M., et al., Diclofenac sodium (Voltaren) reduced exercise-induced injury in
human skeletal muscle. Medicine & Science in Sports & Exercise, 2000. 32(7): p. 1191-
1196.
146. Rahnama, N., F. Rahmani-Nia, and K. Ebrahim, The isolated and combined effects of
selected physical activity and ibuprofen on delayed-onset muscle soreness. Journal Of
Sports Sciences, 2005. 23(8): p. 843-850.
147. Bourgeois, J., et al., Naproxen does not alter indices of muscle damage in resistance-
exercise trained men. Medicine & Science in Sports & Exercise, 1999. 31(1): p. 4-9.

105
148. Baldwin, A.C., S.W. Stevenson, and G.A. Dudley, Nonsteroidal anti-inflammatory
therapy after eccentric exercise in healthy older individuals. The Journals Of
Gerontology. Series A, Biological Sciences And Medical Sciences, 2001. 56(8): p. M510-
M513.
149. Cannavino, C.R., et al., Efficacy of transdermal ketoprofen for delayed onset muscle
soreness. Clinical Journal Of Sport Medicine: Official Journal Of The Canadian Academy
Of Sport Medicine, 2003. 13(4): p. 200-208.
150. Sayers, S.P., et al., Effect of ketoprofen on muscle function and sEMG activity after
eccentric exercise. Medicine And Science In Sports And Exercise, 2001. 33(5): p. 702-
710.
151. Hyldahl, R.D., et al., Effects of Ibuprofen Topical Gel on Muscle Soreness. Medicine &
Science in Sports & Exercise, 2010. 42(3): p. 614-621.
152. Krentz, J.R., et al., The effects of ibuprofen on muscle hypertrophy, strength, and
soreness during resistance training. Applied Physiology, Nutrition & Metabolism, 2008.
33(3): p. 470-475.
153. Loram, L.C.M., D.; Fuller, A., Rofecoxib and tramadol do not attenuate delayed-onset
muscle soreness or ischaemic pain in human volunteers. Canadian Journal of Physiology
and Pharmacology, 2005. 83(12): p. 1137-1145.
154. Barlas, P., et al., Managing delayed-onset muscle soreness: lack of effect of selected oral
systemic analgesics. Archives Of Physical Medicine And Rehabilitation, 2000. 81(7): p.
966-972.
155. Grossman, J.M., et al., Effect of ibuprofen use on delayed onset muscle soreness of the
elbow flexors. Journal of Sport Rehabilitation, 1995. 4(4): p. 253-263.
156. Nieman, D.C., et al., Ibuprofen use, endotoxemia, inflammation, and plasma cytokines
during ultramarathon competition. Brain, Behavior, and Immunity, 2006. 20(6): p. 578-
584.

106
157. Semark, A., et al., The effect of a prophylactic dose of flurbiprofen on muscle soreness
and sprinting performance in trained subjects. Journal Of Sports Sciences, 1999. 17(3):
p. 197-203.
158. Stone, M.B., et al., Preliminary comparison of bromelain and Ibuprofen for delayed
onset muscle soreness management. Clinical Journal Of Sport Medicine: Official Journal
Of The Canadian Academy Of Sport Medicine, 2002. 12(6): p. 373-378.
159. Lapointe, B.M., P. Fremont, and C.H. Cote, Influence of nonsteroidal anti-inflammatory
drug treatment duration and time of onset on recovery from exercise-induced muscle
damage in rats. Archives of Physical Medicine & Rehabilitation, 2003. 84(5): p. 651-655.
160. Peterson, J.M., et al., Ibuprofen and acetaminophen: effect on muscle inflammation
after eccentric exercise. Medicine & Science in Sports & Exercise, 2003. 35(6): p. 892-
896.
161. Markworth, J.F., et al., Ibuprofen treatment blunts early translational signaling
responses in human skeletal muscle following resistance exercise. Journal Of Applied
Physiology (Bethesda, Md.: 1985), 2014. 117(1): p. 20-28.
162. Markworth, J.F. and D. Cameron-Smith, Prostaglandin F2α stimulates PI3K/ERK/mTOR
signaling and skeletal myotube hypertrophy. American Journal Of Physiology. Cell
Physiology, 2011. 300(3): p. C671-C682.
163. Rodemann, H.P. and A.L. Goldberg, Arachidonic acid, prostaglandin E2 and F2 alpha
influence rates of protein turnover in skeletal and cardiac muscle. The Journal Of
Biological Chemistry, 1982. 257(4): p. 1632-1638.
164. Shen, W., et al., Inhibited skeletal muscle healing in cyclooxygenase-2 gene-deficient
mice: the role of PGE2 and PGF2alpha. Journal Of Applied Physiology (Bethesda, Md.:
1985), 2006. 101(4): p. 1215-1221.
165. Scott, A., et al., What do we mean by the term "inflammation: a contemporary basic
science update for sports medicine. British Journal of Sports Medicine, 2004. 38(3): p.
372-380.

107
166. Serhan, C.N., Resolvins and protectins: novel lipid mediators in anti-inflammation and
resolution. Scandinavian Journal of Food & Nutrition, 2006. 50: p. 68-78.
167. Serhan, C.N., et al., Resolution of inflammation: state of the art, definitions and terms.
FASEB Journal: Official Publication Of The Federation Of American Societies For
Experimental Biology, 2007. 21(2): p. 325-332.
168. Serhan, C.N., N. Chiang, and T.E. Van Dyke, Resolving inflammation: dual anti-
inflammatory and pro-resolution lipid mediators. Nature Reviews. Immunology, 2008.
8(5): p. 349-361.
169. Serhan, C.N., et al., Novel functional sets of lipid-derived mediators with
antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-
nonsteroidal antiinflammatory drugs and transcellular processing. The Journal Of
Experimental Medicine, 2000. 192(8): p. 1197-1204.
170. Serhan, C.N., et al., Protectins and maresins: New pro-resolving families of mediators in
acute inflammation and resolution bioactive metabolome. BBA - Molecular & Cell
Biology of Lipids, 2015. 1851(4): p. 397-413.
171. Serhan, C.N., et al., Resolvins: a family of bioactive products of omega-3 fatty acid
transformation circuits initiated by aspirin treatment that counter proinflammation
signals. The Journal Of Experimental Medicine, 2002. 196(8): p. 1025-1037.
172. Serhan, C.N., et al., Design of lipoxin A4 stable analogs that block transmigration and
adhesion of human neutrophils. Biochemistry, 1995. 34(44): p. 14609-14615.
173. Serhan, C.N. and N.A. Petasis, Resolvins and protectins in inflammation resolution.
Chemical Reviews, 2011. 111(10): p. 5922-5943.
174. Serhan, C.N. and J. Savill, Resolution of inflammation: the beginning programs the end.
Nature Immunology, 2005. 6(12): p. 1191-1197.
175. Maddox, J.F. and C.N. Serhan, Lipoxin A4 and B4 are potent stimuli for human monocyte
migration and adhesion: selective inactivation by dehydrogenation and reduction. The
Journal Of Experimental Medicine, 1996. 183(1): p. 137-146.

108
176. Ryan, A. and C. Godson, Lipoxins: regulators of resolution. Current Opinion In
Pharmacology, 2010. 10(2): p. 166-172.
177. Duvall, M.G. and B.D. Levy, DHA- and EPA-derived resolvins, protectins, and maresins in
airway inflammation. European Journal Of Pharmacology, 2015.
178. Hong, S., et al., Novel docosatrienes and 17S-resolvins generated from docosahexaenoic
acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. The
Journal Of Biological Chemistry, 2003. 278(17): p. 14677-14687.
179. Bannenberg, G. and C.N. Serhan, Specialized pro-resolving lipid mediators in the
inflammatory response: An update. Biochimica Et Biophysica Acta, 2010. 1801(12): p.
1260-1273.
180. Fadok, V.A., et al., Differential effects of apoptotic versus lysed cells on macrophage
production of cytokines: role of proteases. Journal Of Immunology (Baltimore, Md.:
1950), 2001. 166(11): p. 6847-6854.
181. Godson, C., et al., Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of
apoptotic neutrophils by monocyte-derived macrophages. Journal Of Immunology
(Baltimore, Md.: 1950), 2000. 164(4): p. 1663-1667.
182. Schwab, J.M., et al., Resolvin E1 and protectin D1 activate inflammation-resolution
programmes. Nature, 2007. 447(7146): p. 869-874.
183. Haslett, C., Granulocyte apoptosis and its role in the resolution and control of lung
inflammation. American Journal Of Respiratory And Critical Care Medicine, 1999. 160(5
Pt 2): p. S5-S11.
184. Serhan, C.N., M. Hamberg, and B. Samuelsson, Lipoxins: novel series of biologically
active compounds formed from arachidonic acid in human leukocytes. Proceedings Of
The National Academy Of Sciences Of The United States Of America, 1984. 81(17): p.
5335-5339.
185. Levy, B.D., et al., Lipid mediator class switching during acute inflammation: signals in
resolution. Nature Immunology, 2001. 2(7): p. 612-619.

109
186. Bannenberg, G.L., et al., Molecular circuits of resolution: formation and actions of
resolvins and protectins. Journal Of Immunology (Baltimore, Md.: 1950), 2005. 174(7):
p. 4345-4355.
187. Gangemi, S., et al., Physical exercise increases urinary excretion of lipoxin A4 and related
compounds. Journal Of Applied Physiology (Bethesda, Md.: 1985), 2003. 94(6): p. 2237-
2240.
188. Baldwin, A.S., Jr., The NF-kappa B and I kappa B proteins: new discoveries and insights.
Annual Review Of Immunology, 1996. 14: p. 649-683.
189. Cai, D., et al., IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell,
2004. 119(2): p. 285-298.
190. Delhalle, S., et al., A beginner's guide to NF-kappaB signaling pathways. Annals Of The
New York Academy Of Sciences, 2004. 1030: p. 1-13.
191. Mourkioti, F. and N. Rosenthal, NF-κB signaling in skeletal muscle: prospects for
intervention in muscle diseases. Journal of Molecular Medicine, 2008. 86(7): p. 747-759.
192. Pahl, H.L., Activators and target genes of Rel/NF-kappaB transcription factors.
Oncogene, 1999. 18(49): p. 6853-6866.
193. Wang, H., et al., NF-kappaB regulation of YY1 inhibits skeletal myogenesis through
transcriptional silencing of myofibrillar genes. Molecular And Cellular Biology, 2007.
27(12): p. 4374-4387.
194. Bakkar, N. and D.C. Guttridge, NF-kappaB signaling: a tale of two pathways in skeletal
myogenesis. Physiological Reviews, 2010. 90(2): p. 495-511.
195. Ghosh, S. and M. Karin, Missing pieces in the NF-kappaB puzzle. Cell, 2002. 109 Suppl:
p. S81-S96.
196. Chen, F.E. and G. Ghosh, Regulation of DNA binding by Rel/NF-kappaB transcription
factors: structural views. Oncogene, 1999. 18(49): p. 6845-6852.
197. Shishodia, S. and B.B. Aggarwal, Nuclear factor-kappaB activation: a question of life or
death. Journal Of Biochemistry And Molecular Biology, 2002. 35(1): p. 28-40.

110
198. Li, H., S. Malhotra, and A. Kumar, Nuclear factor-kappa B signaling in skeletal muscle
atrophy. Journal Of Molecular Medicine (Berlin, Germany), 2008. 86(10): p. 1113-1126.
199. Zandi, E., et al., The IkappaB kinase complex (IKK) contains two kinase subunits,
IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB
activation. Cell, 1997. 91(2): p. 243-252.
200. Langen, R.C., et al., Inflammatory cytokines inhibit myogenic differentiation through
activation of nuclear factor-kappaB. The FASEB Journal: Official Publication Of The
Federation Of American Societies For Experimental Biology, 2001. 15(7): p. 1169-1180.
201. Layne, M.D., et al., Characterization of the mouse aortic carboxypeptidase-like protein
promoter reveals activity in differentiated and dedifferentiated vascular smooth muscle
cells. Circulation Research, 2002. 90(6): p. 728-736.
202. van de Stolpe, A., et al., 12-O-tetradecanoylphorbol-13-acetate- and tumor necrosis
factor alpha-mediated induction of intercellular adhesion molecule-1 is inhibited by
dexamethasone. Functional analysis of the human intercellular adhesion molecular-1
promoter. The Journal Of Biological Chemistry, 1994. 269(8): p. 6185-6192.
203. Jozci, A.C., et al., Molecular Characteristics of Aged Muscle Reflect an Altered Ability to
Respond to Exercise. International Journal of Sport Nutrition & Exercise Metabolism,
2001. 11(4): p. S9.
204. Ward, C., et al., NF-kappaB activation is a critical regulator of human granulocyte
apoptosis in vitro. The Journal Of Biological Chemistry, 1999. 274(7): p. 4309-4318.
205. Monici, M.C., et al., Activation of nuclear factor-kappaB in inflammatory myopathies
and Duchenne muscular dystrophy. Neurology, 2003. 60(6): p. 993-997.
206. Yang, C.C., et al., Immunolocalization of transcription factor NF-kappaB in inclusion-
body myositis muscle and at normal human neuromuscular junctions. Neuroscience
Letters, 1998. 254(2): p. 77-80.
207. Mourkioti, F., et al., Targeted ablation of IKK2 improves skeletal muscle strength,
maintains mass, and promotes regeneration. The Journal Of Clinical Investigation, 2006.
116(11): p. 2945-2954.

111
208. Fong, C.H.Y., et al., An antiinflammatory role for IKKbeta through the inhibition of
"classical" macrophage activation. The Journal Of Experimental Medicine, 2008. 205(6):
p. 1269-1276.
209. Greten, F.R., et al., NF-kappaB is a negative regulator of IL-1beta secretion as revealed
by genetic and pharmacological inhibition of IKKbeta. Cell, 2007. 130(5): p. 918-931.
210. Lawrence, T., et al., Possible new role for NF-κB in the resolution of inflammation.
Nature Medicine, 2001. 7(12): p. 1291.
211. Lawrence, T. and C. Fong, The resolution of inflammation: anti-inflammatory roles for
NF-kappaB. The International Journal Of Biochemistry & Cell Biology, 2010. 42(4): p.
519-523.
212. Ziegler-Heitbrock, H.W., I. Petersmann, and M. Frankenberger, p50 (NF-kappa B1) is
upregulated in LPS tolerant P388D1 murine macrophages. Immunobiology, 1997. 198(1-
3): p. 73-80.
213. Thaloor, D., et al., Systemic administration of the NF-kappaB inhibitor curcumin
stimulates muscle regeneration after traumatic injury. The American Journal Of
Physiology, 1999. 277(2 Pt 1): p. C320-C329.
214. Guttridge, D.C., et al., NF-kappaB controls cell growth and differentiation through
transcriptional regulation of cyclin D1. Molecular And Cellular Biology, 1999. 19(8): p.
5785-5799.
215. Acharyya, S., et al., Interplay of IKK/NF-kappaB signaling in macrophages and myofibers
promotes muscle degeneration in Duchenne muscular dystrophy. The Journal Of Clinical
Investigation, 2007. 117(4): p. 889-901.
216. Ladner, K.J., M.A. Caligiuri, and D.C. Guttridge, Tumor Necrosis Factor-regulated
Biphasic Activation of NF-κB Is Required for Cytokine-induced Loss of Skeletal Muscle
Gene Products. Journal of Biological Chemistry, 2003. 278(4): p. 2294.
217. Dogra, C., et al., Tumor Necrosis Factor-like Weak Inducer of Apoptosis Inhibits Skeletal
Myogenesis through Sustained Activation of Nuclear Factor-κB and Degradation of
MyoD Protein. Journal of Biological Chemistry, 2006. 281(15): p. 10327-10336.

112
218. Szalay, K., Z. Rázga, and E. Duda, TNF inhibits myogenesis and downregulates the
expression of myogenic regulatory factors myoD and myogenin. European Journal Of
Cell Biology, 1997. 74(4): p. 391-398.
219. Hirasaka, K., et al., Clinorotation prevents differentiation of rat myoblastic L6 cells in
association with reduced NF-kappa B signaling. Biochimica Et Biophysica Acta, 2005.
1743(1-2): p. 130-140.
220. Kim, S.S., et al., Inhibitors of the proteasome block the myogenic differentiation of rat L6
myoblasts. FEBS Letters, 1998. 433(1-2): p. 47-50.
221. Olashaw, N.E., et al., Induction of NF-kappa B-like activity by platelet-derived growth
factor in mouse fibroblasts. Molecular Biology Of The Cell, 1992. 3(10): p. 1131-1139.
222. Canicio, J., et al., Nuclear factor kappa B-inducing kinase and Ikappa B kinase-alpha
signal skeletal muscle cell differentiation. The Journal Of Biological Chemistry, 2001.
276(23): p. 20228-20233.
223. Frost, R.A., G.J. Nystrom, and C.H. Lang, Stimulation of insulin-like growth factor binding
protein-1 synthesis by interleukin-1beta: requirement of the mitogen-activated protein
kinase pathway. Endocrinology, 2000. 141(9): p. 3156-3164.
224. Kaliman, P., et al., Insulin-like growth factor-II, phosphatidylinositol 3-kinase, nuclear
factor-kappaB and inducible nitric-oxide synthase define a common myogenic signaling
pathway. The Journal Of Biological Chemistry, 1999. 274(25): p. 17437-17444.
225. Baeza-Raja, B. and P. Muñoz-Cánoves, p38 MAPK-induced nuclear factor-kappaB
activity is required for skeletal muscle differentiation: role of interleukin-6. Molecular
Biology Of The Cell, 2004. 15(4): p. 2013-2026.
226. De Alvaro, C., et al., Nuclear exclusion of forkhead box O and Elk1 and activation of
nuclear factor-kappaB are required for C2C12-RasV12C40 myoblast differentiation.
Endocrinology, 2008. 149(2): p. 793-801.
227. Madrid, L.V., et al., Akt stimulates the transactivation potential of the RelA/p65 Subunit
of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-

113
activated protein kinase p38. The Journal Of Biological Chemistry, 2001. 276(22): p.
18934-18940.
228. Chen, S.-E., B. Jin, and Y.-P. Li, TNF-alpha regulates myogenesis and muscle
regeneration by activating p38 MAPK. American Journal Of Physiology. Cell Physiology,
2007. 292(5): p. C1660-C1671.
229. Collins, R.A. and M.D. Grounds, The role of tumor necrosis factor-alpha (TNF-alpha) in
skeletal muscle regeneration. Studies in TNF-alpha(-/-) and TNF-alpha(-/-)/LT-alpha(-/-)
mice. The Journal Of Histochemistry And Cytochemistry: Official Journal Of The
Histochemistry Society, 2001. 49(8): p. 989-1001.
230. Chen, S.-E., et al., Role of TNF-{alpha} signaling in regeneration of cardiotoxin-injured
muscle. American Journal Of Physiology. Cell Physiology, 2005. 289(5): p. C1179-C1187.
231. Purcell, N.H., et al., Activation of NF-kappa B is required for hypertrophic growth of
primary rat neonatal ventricular cardiomyocytes. Proceedings Of The National Academy
Of Sciences Of The United States Of America, 2001. 98(12): p. 6668-6673.
232. García-López, D., et al., Effects of eccentric exercise on NF-kappaB activation in blood
mononuclear cells. Medicine And Science In Sports And Exercise, 2007. 39(4): p. 653-
664.
233. Ho, R.C., et al., Regulation of IkappaB kinase and NF-kappaB in contracting adult rat
skeletal muscle. American Journal Of Physiology. Cell Physiology, 2005. 289(4): p. C794-
C801.
234. Ji, L.L., et al., Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in
rat skeletal muscle. The FASEB Journal: Official Publication Of The Federation Of
American Societies For Experimental Biology, 2004. 18(13): p. 1499-1506.
235. Jiménez-Jiménez, R., et al., Eccentric training impairs NF-kappaB activation and over-
expression of inflammation-related genes induced by acute eccentric exercise in the
elderly. Mechanisms Of Ageing And Development, 2008. 129(6): p. 313-321.

114
236. Tantiwong, P., et al., NF-κB activity in muscle from obese and type 2 diabetic subjects
under basal and exercise-stimulated conditions. American Journal Of Physiology.
Endocrinology And Metabolism, 2010. 299(5): p. E794-E801.
237. Lille, S.T., et al., Inhibition of the initial wave of NF-kappaB activity in rat muscle reduces
ischemia/reperfusion injury. Muscle & Nerve, 2001. 24(4): p. 534-541.
238. Durham, W.J., et al., Fatiguing exercise reduces DNA binding activity of NF-κB in skeletal
muscle nuclei. Journal of Applied Physiology, 2004. 97(5): p. 1740-1745.
239. Vella, L., et al., Resistance exercise increases NF-κB activity in human skeletal muscle.
American Journal Of Physiology. Regulatory, Integrative And Comparative Physiology,
2012. 302(6): p. R667-R673.
240. Faulkner, J.A., S.V. Brooks, and J.A. Opiteck, Injury to skeletal muscle fibers during
contractions; conditions of occurrence and prevention. Physical Therapy, 1993. 73(12):
p. 911-921.
241. Baldwin, A.C., S.W. Stevenson, and G.A. Dudley, Nonsteroidal anti-inflammatory
therapy after eccentric exercise in healthy older individuals. Journals of Gerontology
Series A: Biological Sciences & Medical Sciences, 2001. 56(8): p. M510-m513.
242. Hersh, E.V., P.A. Moore, and G.L. Ross, Over-the-counter analgesics and antipyretics: a
critical assessment. Clinical Therapeutics, 2000. 22(5): p. 500-548.
243. Mikkelsen, U.R., et al., Local NSAID infusion does not affect protein synthesis and gene
expression in human muscle after eccentric exercise. Scandinavian Journal of Medicine
& Science in Sports, 2011. 21(5): p. 630-644.
244. Whisenant, M.J., et al., Validation of submaximal prediction equations for the 1
repetition maximum bench press test on a group of collegiate football players. Journal
Of Strength And Conditioning Research / National Strength & Conditioning Association,
2003. 17(2): p. 221-227.
245. Buford, T.W., M.B. Cooke, and D.S. Willoughby, Resistance exercise-induced changes of
inflammatory gene expression within human skeletal muscle. European Journal Of
Applied Physiology, 2009. 107(4): p. 463-471.

115
246. Vella, L., et al., Ibuprofen supplementation and its effects on NF-κB activation in skeletal
muscle following resistance exercise. Physiological Reports, 2014. 2(10).
247. MacNeil, L.G., et al., 17β-estradiol attenuates exercise-induced neutrophil infiltration in
men. American Journal Of Physiology. Regulatory, Integrative And Comparative
Physiology, 2011. 300(6): p. R1443-R1451.
248. Lapointe, B.M., P. Frémont, and C.H. Côté, Adaptation to lengthening contractions is
independent of voluntary muscle recruitment but relies on inflammation. American
Journal Of Physiology. Regulatory, Integrative And Comparative Physiology, 2002.
282(1): p. R323-R329.
249. Lapointe, B.M., J. Frenette, and C.H. Côté, Lengthening contraction-induced
inflammation is linked to secondary damage but devoid of neutrophil invasion. Journal
Of Applied Physiology (Bethesda, Md.: 1985), 2002. 92(5): p. 1995-2004.
250. Nosaka, K., P. Sacco, and K. Mawatari, Effects of Amino Acid Supplementation on Muscle
Soreness and Damage. International Journal of Sport Nutrition & Exercise Metabolism,
2006. 16(6): p. 620-635.
251. Connolly, D.A.J., S.P. Sayers, and M.P. McHugh, Treatment and prevention of delayed
onset muscle soreness. Journal of Strength & Conditioning Research (Allen Press
Publishing Services Inc.), 2003. 17(1): p. 197-208.
252. Malm, C., et al., Leukocytes, cytokines, growth factors and hormones in human skeletal
muscle and blood after uphill or downhill running. The Journal Of Physiology, 2004.
556(Pt 3): p. 983-1000.
253. Gibson, W., et al., Increased pain from muscle fascia following eccentric exercise: animal
and human findings. Experimental Brain Research, 2009. 194(2): p. 299-308.
254. Raastad, T., et al., Changes in Calpain Activity, Muscle Structure, and Function after
Eccentric Exercise. Medicine & Science in Sports & Exercise, 2010. 42(1): p. 86-95.
255. Roberts, L.A., et al., Post-exercise cold water immersion attenuates acute anabolic
signalling and long-term adaptations in muscle to strength training. The Journal Of
Physiology, 2015. 593(18): p. 4285-4301.

116
256. Farnfield, M.M., et al., Whey protein ingestion activates mTOR-dependent signalling
after resistance exercise in young men: a double-blinded randomized controlled trial.
Nutrients, 2009. 1(2): p. 263-275.
257. Trenerry, M.K., et al., STAT3 signaling is activated in human skeletal muscle following
acute resistance exercise. J Appl Physiol, 2007. 102(4): p. 1483-9.
258. Markworth, J.F., et al., Human inflammatory and resolving lipid mediator responses to
resistance exercise and ibuprofen treatment. Am J Physiol Regul Integr Comp Physiol,
2013. 305(11): p. R1281-96.
259. Maddipati, K.R., et al., Eicosanomic profiling reveals dominance of the epoxygenase
pathway in human amniotic fluid at term in spontaneous labor. FASEB J, 2014. 28(11):
p. 4835-46.
260. Maddipati, K.R., et al., Clinical chorioamnionitis at term: the amniotic fluid fatty acyl
lipidome. Journal of Lipid Research, 2016. 57(10): p. 1906-1916.
261. Haeggström, J.Z. and C.D. Funk, Lipoxygenase and Leukotriene Pathways: Biochemistry,
Biology, and Roles in Disease. Chemical Reviews, 2011. 111(10): p. 5866-5898.
262. Dobrian, A.D., et al., Functional and pathological roles of the 12- and 15-lipoxygenases.
Progress In Lipid Research, 2011. 50(1): p. 115-131.
263. Yeung, J. and M. Holinstat, 12-lipoxygenase: a potential target for novel anti-platelet
therapeutics. Cardiovascular & Hematological Agents In Medicinal Chemistry, 2011.
9(3): p. 154-164.
264. Yin, B., et al., Arachidonate 12-lipoxygenase may serve as a potential marker and
therapeutic target for prostate cancer stem cells. International Journal Of Oncology,
2011. 38(4): p. 1041-1046.
265. Wuest, S.J.A., et al., Expression and regulation of 12/15-lipoxygenases in human
primary macrophages. Atherosclerosis, 2012. 225(1): p. 121-127.
266. Markworth, J.F. and D. Cameron-Smith, Prostagiandin F2α stimulates PI3K/ERK/mTOR
signaling and skeletal myotube hypertrophy. American Journal of Physiology: Cell
Physiology, 2011. 69(3): p. C671-C682.

117
267. Laustiola, K., et al., Exercise-induced increase in plasma arachidonic acid and
thromboxane B2 in healthy men: effect of beta-adrenergic blockade. Journal Of
Cardiovascular Pharmacology, 1984. 6(3): p. 449-454.
268. Hilberg, T., et al., Transcription in response to physical stress--clues to the molecular
mechanisms of exercise-induced asthma. FASEB Journal: Official Publication Of The
Federation Of American Societies For Experimental Biology, 2005. 19(11): p. 1492-1494.
269. Massoumi, R. and A. Sjölander, The role of leukotriene receptor signaling in
inflammation and cancer. Thescientificworldjournal, 2007. 7: p. 1413-1421.
270. Rouzer, C.A., et al., Cloning and expression of human leukocyte 5-lipoxygenase.
Advances In Prostaglandin, Thromboxane, And Leukotriene Research, 1989. 19: p. 474-
477.
271. Reynaud, D. and C.R. Pace-Asciak, 12-HETE and 12-HPETE potently stimulate
intracellular release of calcium in intact human neutrophils. Prostaglandins,
Leukotrienes, And Essential Fatty Acids, 1997. 56(1): p. 9-12.
272. Romano, M., et al., Lipoxin synthase activity of human platelet 12-lipoxygenase. The
Biochemical Journal, 1993. 296 ( Pt 1): p. 127-133.
273. Serhan, C.N. and K.A. Sheppard, Lipoxin formation during human neutrophil-platelet
interactions. Evidence for the transformation of leukotriene A4 by platelet 12-
lipoxygenase in vitro. The Journal Of Clinical Investigation, 1990. 85(3): p. 772-780.
274. Serhan, C.N., On the relationship between leukotriene and lipoxin production by human
neutrophils: evidence for differential metabolism of 15-HETE and 5-HETE. Biochimica Et
Biophysica Acta, 1989. 1004(2): p. 158-168.
275. Spector, A.A., et al., Epoxyeicosatrienoic acids (EETs): metabolism and biochemical
function. Progress In Lipid Research, 2004. 43(1): p. 55-90.
276. Konkel, A. and W.-H. Schunck, Role of cytochrome P450 enzymes in the bioactivation of
polyunsaturated fatty acids. Biochimica Et Biophysica Acta, 2011. 1814(1): p. 210-222.

118
277. Nieman, D.C., et al., Influence of pistachios on performance and exercise-induced
inflammation, oxidative stress, immune dysfunction, and metabolite shifts in cyclists: a
randomized, crossover trial. Plos One, 2014. 9(11): p. e113725-e113725.
278. Thompson, D.A. and B.D. Hammock, Dihydroxyoctadecamonoenoate esters inhibit the
neutrophil respiratory burst. Journal of Biosciences, 2007. 32(2): p. 279-291.
279. Paulsen, G., et al., Leucocytes, cytokines and satellite cells: what role do they play in
muscle damage and regeneration following eccentric exercise? Exercise Immunology
Review, 2012. 18: p. 42-97.
280. Trappe, T.A., et al., Effect of ibuprofen and acetaminophen on postexercise muscle
protein synthesis. American Journal of Physiology: Endocrinology & Metabolism, 2002.
45(3): p. E551.
281. Bonizzi, G. and M. Karin, The two NF-kappaB activation pathways and their role in
innate and adaptive immunity. Trends In Immunology, 2004. 25(6): p. 280-288.
282. Senftleben, U., et al., Activation by IKKalpha of a second, evolutionary conserved, NF-
kappa B signaling pathway. Science (New York, N.Y.), 2001. 293(5534): p. 1495-1499.
283. Lawrence, T., et al., Possible new role for NF-kappaB in the resolution of inflammation.
Nature Medicine, 2001. 7(12): p. 1291-1297.
284. Bohuslav, J., et al., Regulation of an essential innate immune response by the p50
subunit of NF-kappaB. The Journal Of Clinical Investigation, 1998. 102(9): p. 1645-1652.
285. Ishikawa, H., et al., Chronic inflammation and susceptibility to bacterial infections in
mice lacking the polypeptide (p)105 precursor (NF-kappaB1) but expressing p50. The
Journal Of Experimental Medicine, 1998. 187(7): p. 985-996.
286. Thomas, B., et al., Critical role of C/EBPdelta and C/EBPbeta factors in the stimulation of
the cyclooxygenase-2 gene transcription by interleukin-1beta in articular chondrocytes.
European Journal Of Biochemistry / FEBS, 2000. 267(23): p. 6798-6809.
287. Wadleigh, D.J., et al., Transcriptional activation of the cyclooxygenase-2 gene in
endotoxin-treated RAW 264.7 macrophages. The Journal Of Biological Chemistry, 2000.
275(9): p. 6259-6266.

119
288. Hollander, J., et al., Superoxide dismutase gene expression is activated by a single bout
of exercise in rat skeletal muscle. Pflügers Archiv: European Journal Of Physiology, 2001.
442(3): p. 426-434.
289. Kramer, H.F. and L.J. Goodyear, Exercise, MAPK, and NF-kappaB signaling in skeletal
muscle. Journal Of Applied Physiology (Bethesda, Md.: 1985), 2007. 103(1): p. 388-395.
290. Spangenburg, E.E., et al., Exercise increases SOCS-3 expression in rat skeletal muscle:
potential relationship to IL-6 expression. The Journal Of Physiology, 2006. 572(Pt 3): p.
839-848.
291. Mayhew, J.L., et al., Muscular endurance repetitions to predict bench press strength in
men of different training levels. The Journal Of Sports Medicine And Physical Fitness,
1995. 35(2): p. 108-113.
292. Mead, R., The design of experiments: Statistical principles for practical applications. The
design of experiments: Statistical principles for practical applications. 1988, New York,
NY US: Cambridge University Press.
293. Saville, D.J., Multiple comparison procedures: The practical solution. American
Statistician, 1990. 44(2): p. 174.
294. Hyldahl, R.D., et al., Activation of nuclear factor-κB following muscle eccentric
contractions in humans is localized primarily to skeletal muscle-residing pericytes.
FASEB Journal: Official Publication Of The Federation Of American Societies For
Experimental Biology, 2011. 25(9): p. 2956-2966.
295. Xin, L., et al., A contralateral repeated bout effect attenuates induction of NF-κB DNA-
binding following eccentric exercise. Journal Of Applied Physiology (Bethesda, Md.:
1985), 2013.
296. Lawrence, T., et al., IKKalpha limits macrophage NF-kappaB activation and contributes
to the resolution of inflammation. Nature, 2005. 434(7037): p. 1138-1143.
297. Poligone, B. and A.S. Baldwin, Positive and negative regulation of NF-kappaB by COX-2:
roles of different prostaglandins. The Journal Of Biological Chemistry, 2001. 276(42): p.
38658-38664.

120
298. Rossi, A., et al., Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors
of IkappaB kinase. Nature, 2000. 403(6765): p. 103-108.
299. Straus, D.S., et al., 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the
NF-kappa B signaling pathway. Proceedings Of The National Academy Of Sciences Of
The United States Of America, 2000. 97(9): p. 4844-4849.
300. Castrillo, A., et al., Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-
deoxy-Delta(12,14)-prostaglandin J(2) in activated murine macrophages. Molecular And
Cellular Biology, 2000. 20(5): p. 1692-1698.
301. Lawrence, T., D.A. Willoughby, and D.W. Gilroy, Anti-inflammatory lipid mediators and
insights into the resolution of inflammation. Nature Reviews. Immunology, 2002. 2(10):
p. 787-795.
302. Brickson, S., et al., M1/70 attenuates blood-borne neutrophil oxidants, activation, and
myofiber damage following stretch injury. Journal Of Applied Physiology (Bethesda,
Md.: 1985), 2003. 95(3): p. 969-976.
303. Tidball, J.G., Interactions between muscle and the immune system during modified
musculoskeletal loading. Clinical Orthopedics, 2002. 403(suppl): p. S100-S109.
304. Veliça, P. and C.M. Bunce, Prostaglandins in muscle regeneration. Journal Of Muscle
Research And Cell Motility, 2008. 29(6-8): p. 163-167.
305. Arita, M., et al., Stereochemical assignment, antiinflammatory properties, and receptor
for the omega-3 lipid mediator resolvin E1. Journal of Experimental Medicine, 2005.
201(5): p. 713-722.
306. Bandeira-Melo, C., et al., Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4
accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats:
relationship with concurrent eosinophilia. Journal of Immunology (Baltimore), 2000.
164(2): p. 1029-1036.
307. Markworth, J.F., et al., Divergent shifts in lipid mediator profile following
supplementation with n-3 docosapentaenoic acid and eicosapentaenoic acid. FASEB

121
Journal: Official Publication Of The Federation Of American Societies For Experimental
Biology, 2016.
308. Miokovic, T., et al., Heterogeneous atrophy occurs within individual lower limb muscles
during 60 days of bed rest. Journal Of Applied Physiology (Bethesda, Md.: 1985), 2012.
113(10): p. 1545-1559.
309. Lexell, J., K. Henriksson-Larsén, and M. Sjöström, Distribution of different fibre types in
human skeletal muscles. 2. A study of cross-sections of whole m. vastus lateralis. Acta
Physiologica Scandinavica, 1983. 117(1): p. 115-122.

122
Appendix A:

ORIGINAL RESEARCHpublished: 29 March 2016
doi: 10.3389/fphys.2016.00086
Frontiers in Physiology | www.frontiersin.org 1 March 2016 | Volume 7 | Article 86
Edited by:
Vincent Pialoux,
University Lyon 1, France
Reviewed by:
Gianluca Vernillo,
University of Milan, Italy
Scott Forbes,
Okanagan College, Canada
*Correspondence:
Luke Vella
Specialty section:
This article was submitted to
Exercise Physiology,
a section of the journal
Frontiers in Physiology
Received: 12 December 2015
Accepted: 22 February 2016
Published: 29 March 2016
Citation:
Vella L, Markworth JF, Paulsen G,
Raastad T, Peake JM, Snow RJ,
Cameron-Smith D and Russell AP
(2016) Ibuprofen Ingestion Does Not
Affect Markers of Post-exercise
Muscle Inflammation.
Front. Physiol. 7:86.
doi: 10.3389/fphys.2016.00086
Ibuprofen Ingestion Does Not AffectMarkers of Post-exercise MuscleInflammationLuke Vella 1*, James F. Markworth 2, Gøran Paulsen 3, Truls Raastad 3, Jonathan M. Peake 4,
Rod J. Snow 1, David Cameron-Smith 2 and Aaron P. Russell 1
1Centre for Physical Activity and Nutrition Research, School of Exercise and Nutrition Science, Deakin University, Burwood,
VIC, Australia, 2 Liggins Institute, University of Auckland, Auckland, New Zealand, 3Department of Physical Performance,
Norwegian School of Sport Science, Oslo, Norway, 4 School of Biomedical Sciences and Institute of Health and Biomedical
Innovation, Queensland University of Technology, Brisbane, QLD, Australia
Purpose: We investigated if oral ingestion of ibuprofen influenced leucocyte recruitment
and infiltration following an acute bout of traditional resistance exercise.
Methods: Sixteen male subjects were divided into two groups that received the
maximum over-the-counter dose of ibuprofen (1200mg d−1) or a similarly administered
placebo following lower body resistance exercise. Muscle biopsies were taken from
m.vastus lateralis and blood serum samples were obtained before and immediately after
exercise, and at 3 and 24 h after exercise. Muscle cross-sections were stained with
antibodies against neutrophils (CD66b and MPO) and macrophages (CD68). Muscle
damage was assessed via creatine kinase and myoglobin in blood serum samples, and
muscle soreness was rated on a ten-point pain scale.
Results: The resistance exercise protocol stimulated a significant increase in the number
of CD66b+ and MPO+ cells when measured 3 h post exercise. Serum creatine kinase,
myoglobin and subjective muscle soreness all increased post-exercise. Muscle leucocyte
infiltration, creatine kinase, myoglobin and subjective muscle soreness were unaffected
by ibuprofen treatment when compared to placebo. There was also no association
between increases in inflammatory leucocytes and any other marker of cellular muscle
damage.
Conclusion: Ibuprofen administration had no effect on the accumulation of neutrophils,
markers of muscle damage or muscle soreness during the first 24 h of post-exercise
muscle recovery.
Keywords: exercise recovery, NSAID treatment, inflammation, resistance exercise, leucocyte
INTRODUCTION
Unaccustomed resistance exercise often results in tissue damage and inflammation, leadingto delayed onset muscle soreness (DOMS) and a consequent reduction in force production(Faulkner et al., 1993; Tidball, 1995). Animal models and in vitro studies have identified that localand systemic inflammation exerts a regulatory influence during the different phases of musclerecovery, including myofibrillar disruption, cellular necrosis, satellite cell activation, maturationand subsequent regeneration, and adaptation (Fridén et al., 1981; Armstrong et al., 1991). Thus,

Vella et al. Ibuprofen and Post-exercise Inflammation
post-exercise inflammation is intimately necessary and a keyfeature of the normal process of tissue regeneration andadaptation following acute muscle damage. However, excessiveinflammation is considered a potential cause of prolongedpost-exercise muscle soreness and may have a negative effecton muscle recovery (Armstrong et al., 1991; Smith, 1991);consequently strategies to reduce or counteract inflammation arecommonly implemented to aid in improving muscle recoveryafter exercise (Urso, 2013).
Non-steroidal anti-inflammatory drugs (NSAIDs) arecommonly used as a treatment strategy in exercise andsports medicine to assist with recovery from exercise-inducedinflammation, particularly following soft-tissue injury. NSAIDsinhibit the cyclooxygenase (COX-1 and 2) enzymes andconsequently the formation of prostanoids (prostaglandins,prostacyclins, and thromboxanes) that play a diverse role inacute inflammation (Markworth et al., 2013). Prostanoidsstimulate an acute inflammatory process by controlling localblood flow, vascular permeability, leucocyte infiltration, andtriggering sensations of pain (Markworth et al., 2013; Urso,2013). In animal models of acute muscle damage, treatmentwith NSAIDs blunts the infiltration of leucocytes into muscletissue (Lapointe et al., 2003; Bondesen et al., 2004) and causesa reduction in creatine kinase (CK) (Mishra et al., 1995).Consequently, NSAIDs can also inhibit myofiber regeneration,satellite cell proliferation and differentiation, and overload-induced muscle hypertrophy (Mishra et al., 1995; Bondesenet al., 2004, 2006). These findings provide preliminary evidencethat NSAIDs compromise the physiological link betweenprocesses of acute muscle damage, inflammation and cellularregeneration.
Research into the effects of NSAIDs on exercise-inducedmuscle damage and inflammation has produced equivocalfindings. In exercise models, NSAID administration has beenshown to attenuate post-exercise DOMS in some (Baldwin et al.,2001; Tokmakidis et al., 2003; Paulsen et al., 2010), but notall studies (Trappe et al., 2002; Krentz et al., 2008; Mikkelsenet al., 2009; Hyldahl et al., 2010). While a precise cellularmechanism for an analgesic effect of NSAIDs remains unclear,it has been largely ascribed to their effect on prostaglandinsynthesis and the capacity of NSAIDs to interfere with aspectsof inflammatory cell function (Hersh et al., 2000; Petersonet al., 2003). Previous research has demonstrated that oralconsumption of both ibuprofen, a non-selective NSAID andacetaminophen, an analgesic also known as paracetamol, had noeffect on macrophage infiltration at 24 h following an eccentricexercise protocol (Peterson et al., 2003). Similarly treatment withnaproxen, another non-selective NSAID, had no effect on theinfiltration of leucocyte common antigen positive cells followinga unilateral, isotonic resistance exercise protocol (Bourgeois et al.,1999). Interestingly, Paulsen et al. (2010) suggested a bluntingeffect of the COX-2 specific celecoxib following maximumeccentric muscle contractions (Paulsen et al., 2010). This researchidentified a tendency for higher monocyte/macrophage numbersin subjects within the placebo group, and those subjects who wereidentified as “high-responders” to the exercise protocol based onthe number of inflammatory leucocytes (Paulsen et al., 2010).
Although this is not a particularly robust finding, it has led to thehypothesis that NSAIDs may influence leucocyte infiltration inskeletal muscle when a sufficiently strong and early inflammatoryreaction is present (Paulsen et al., 2010). This hypothesis wouldsuggest that the intensity of the exercise stimulus and theconsequent muscle-damage response would be largely influentialin determining the effect of a pharmacologically based anti-inflammatory intervention.
Conflicting findings also exist with regard to the effectof NSAIDs on the regenerative capacity of skeletal musclefollowing exercise-induced muscle damage. The non-selectiveCOX inhibitor ibuprofen blunted skeletal muscle proteinsynthesis (Trappe et al., 2002) while local intramuscularinfusion of indomethacin (Mikkelsen et al., 2011) and theoral administration of the COX-2 selective NSAID celecoxib(Burd et al., 2010; Paulsen et al., 2010) had no such effect.Similarly, treatment with indomethacin inhibited post-exercisesatellite cell proliferation (Mikkelsen et al., 2009). Recent workfrom our group demonstrated that treatment with ibuprofeninhibited early translational signaling responses involved in post-exercise muscle hypertrophy (Markworth et al., 2014). A clearmechanistic pathway for NSAIDs to influence the physiologicallink between post-exercise inflammation and skeletal muscleregeneration remains elusive.
These discrepancies in the research to date are likely dueto differences in the exercise protocol (concentric vs. eccentricmuscle contractions), the training status of subjects, the timingof muscle biopsies, and the type of NSAID, the dosageadministered, and method of administration. Further research isrequired to determine whether NSAID administration affects theinfiltration of leucocyte populations following exercise-inducedmuscle damage and how this influences post-exercise adaptivepathways. The aim of the present study was to investigate iforal administration of the NSAID ibuprofen influenced skeletalmuscle leucocyte infiltration during the first 24 h after muscle-damaging resistance exercises. We also aimed to explore howany changes in leucocyte infiltration were related to markersof muscle damage, including circulating muscle proteins CKand myoglobin, and subjective markers of muscle soreness.We hypothesized that the ingestion of ibuprofen followingacute resistance exercise would not influence the infiltration ofleucocytes, however it would attenuate sensations of DOMS.
MATERIALS AND METHODS
ParticipantsAs described previously, 16 healthy male subjects were recruitedto participate in the study (Table 1; Markworth et al., 2013). Itis important to note that the purpose of the original study byMarkworth et al. (2013) was to profile the human eicosanoidresponse to acute exercise in blood plasma samples, as opposedto the aim of the present study which was to determine theeffect of oral ingestion of ibuprofen on leucocyte recruitmentand infiltration following an acute bout of traditional resistanceexercise.While the subjects and experimental design are the samein both studies the aims and dependent variables reported aredifferent. Exclusion criteria included participation in a lower
Frontiers in Physiology | www.frontiersin.org 2 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
TABLE 1 | Subject characteristics and strength testing data.
Characteristics Strength (1 RM)
Age (y) Height (m) Body mass (kg) BMI Squat (kg) Leg press (kg) Leg extension (kg)
PLA 23.9 ± 1.3 1.89 ± 0.1 86.9 ± 4.5 24.5 ± 1.2 94.9 ± 5.4 237 ± 17 236 ± 18
IBU 23 ± 0.5 1.89 ± 0.1 89.1 ± 4.4 24.8 ± 0.8 91.9 ± 6.0 240 ± 15 196 ± 22
Values are mean values ± SEM. No significant differences were observed between the two groups. PLA, placebo; IBU, Ibuprofen; BMI, body mass index.
body resistance exercise program within the last 6 months toensure a muscle damage response from the exercise stimulus,and/or previous chronic treatments with anti-inflammatorymedication. Participants also completed a medical screening toidentify any potential risk factors for them to perform strenuousphysical activity.
Ethics ApprovalAll procedures involved in this study were approved bythe Deakin University Human Research Ethics Committee(DUHREC 2010-019) and muscle sampling procedures wereperformed in accordance with the Helsinki declaration. Eachparticipant was provided with written and oral details of thenature and requirements of the study and provided writtenconsent to participate.
FamiliarizationEach participant completed a familiarization and strength testingsession at least 7 days prior to completing the exercise protocol.Details of the familiarization session have been describedpreviously (Markworth et al., 2013). Briefly, subjects performedrepetition maximum testing for the Smith machine-assistedsquat, the leg press and the leg extension to determine theirexperimental exercise load [80% of a 1 repetition maximum (1RM)]. The maximum weight the subject could lift for 5–8 repswas determined, and these data were entered into the Brzyckiequation to predict 1 RM (Whisenant et al., 2003). Subjects wereasked to abstain from any further activity until the completion ofthe trial.
Experimental ProceduresThe participants reported to the laboratory on the morningof the trial in an overnight fasted state. They were asked toabstain from caffeine, tobacco and alcohol for the 24 h precedingthe trial. Participants rested in a supine position for 30min,following which the first muscle biopsy sample was taken.Each participant then completed a 10min warm-up protocolcomprised of 5min of low intensity cycling on a stationarybike, and one low-intensity set of each exercise at a weight ofeach subject’s own choice within the range of 30–50% 1 RM.The resistance exercise session consisted of three sets of 8–10repetitions performed on a Smith machine assisted squat, a 45◦
leg press and a leg extension at 80% of a predicted 1 RM. Theexercises were performed as a circuit with 1min rest permittedbetween exercises and 3min rest between sets. This protocolhas been used previously and has been a sufficient stimulus toactivate inflammatory signaling pathways (Vella et al., 2012) and
was implemented to replicate a commonly used exercise routine.After exercise, the subjects rested while subsequentmuscle biopsysamples were collected. After the 3 h biopsy, participants wereprovided a standardized meal and were allowed to go home.The following morning, participants reported to the laboratoryin an over-night fasted state for a 24 h muscle biopsy and bloodsample.
Standardized MealsStandardized meals were provided to participants on the nightbefore the trial, (carbohydrate 57%, fat 22%, protein 21%),immediately following the 3 h muscle biopsy (carbohydrate 71%,fat 13%, protein 18%), and in the evening (carbohydrate 64%, fat27%, protein 18%) on the day of the exercise trial. Participantswere permitted to drinkwater ad libitum andwere asked to reportif they could not finish their allocated meals. This nutrition planwas included to ensure that each participant received the samerelative percentage of macro- and micro-nutrients.
NSAID AdministrationPrior to exercise, participants were randomly assigned ina double-blind method, to consume either the maximumrecommended dose of ibuprofen (IBU, n = 8) or a placebocontrol (gelatin capsules identical in appearance containingpowdered sugar in place of ibuprofen) (PLA, n = 8). The IBUgroup consumed three doses of 400mg of ibuprofen throughoutthe trial day. The first dose was administered immediately priorto the first muscle biopsy sample. Participants were instructedto consume the following two doses at 6 and 12 h followingthe exercise protocol. Follow-up phone calls from the researchteam ensured compliance. To ensure this dosing structure wasappropriate to maintain biologically active levels of ibuprofen,ibuprofen concentration was measured in blood serum samplesand these data are reported elsewhere (Markworth et al.,2013).
Sample CollectionVenous blood samples were drawn prior to exercise, within 5minpost exercise (here-after referred to as 0 h post-exercise), and at 1,2, 3, and 24 h post-exercise. Blood samples were drawn throughan indwelling catheter into VACUETTE serum tubes (Greiner,Bio-One, Stonehouse UK). Whole blood was allowed to clot atroom temperature. Samples were then centrifuged at 1000 × gfor 10min. The serum layer was collected and stored at −80◦Cfor subsequent analysis.
Muscle biopsy samples were obtained from the vastuslateralis muscle under local anesthesia (Xylocaine 1%) using
Frontiers in Physiology | www.frontiersin.org 3 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
a percutaneous needle biopsy technique modified to includesuction (Buford et al., 2009). To minimize interference fromthe biopsy procedure, samples prior to exercise and at 24 h postexercise were taken from the same leg, while samples taken at0 and 3 h post-exercise were taken from the contralateral leg.Samples obtained within the same leg were taken at least 5 cmfrom the previous incision site. We have previously reportedthat this technique is effective for minimizing the risk of anyinflammation arising from the biopsy procedure confoundingexercise-induced inflammation (Vella et al., 2012). Tissue-Tekimmersed tissue was rapidly frozen in isopentane cooled liquidnitrogen before storage at−80◦C.
ImmunohistochemistryCross sections (8µm) of muscle tissue were cut using amicrotome at −20◦C (CM3050, Leica, Germany) and mountedon microscope slides (Superfrost Plus, Thermo Scientific,MA, USA), air-dried and stored at −80◦C. Each subject’smuscle sections from all time points were mounted on thesame microscope slide. Serial muscle cross-sections werestained with three different leucocyte antibodies, MPO (#0398,DakoCytomation, Glostrup, Denmark; dilution 1:2000), CD66b(#CLB-B13.9, Sanquin Reagents, Amsterdam, The Netherlands;dilution 1:500), and CD68 (#EBM-11, DakoCytomation,Glostrup, Denmark; dilution 1:300). Furthermore, the sectionswere also stained with dystrophin antibody (ab15277, Abcam,Cambridge, UK) for visualizing borders of muscle fibers.
Sections were fixed in 4% paraformaldehyde solution in astaining jar for 5min at room temperature and rinsed twicefor 10min in phosphate buffered saline (PBS) containing 0.5%Tween 20 (Sigma-Aldrich). The microscope slides were movedinto a humidified chamber and non-specific binding sites wereblocked with 1% bovine serum albumin (BSA) on the section for45min at room temperature. Sections were incubated overnightwith leucocyte and dystrophin antibodies diluted in 1% BSAat 4◦C. After overnight incubation, the slides were washedthree times for 10min in PBS-Tween in a staining jar. Slideswere moved back to the humidified chamber and sectionswere incubated for 45min with secondary antibodies diluted1:200 in 1% BSA at room temperature. Alexa Fluor R© 594F(ab′)2 fragment of goat anti-rabbit and Alexa Fluor R©488 anti-mouse IgG (Invitrogen, Eugene, Oregon, USA) were used as asecondary antibodies. The fluorochrome-stained sections werewashed three times for 10min in PBS-Tween. After the lastwashing in PBS-Tween, the sections weremounted on ProLong R©
Gold Antifade reagent with DAPI (Invitrogen, Eugene, Orgeon,USA).
Muscle sections were visualized using a high-resolutioncamera (DP72, Olympus, Japan) mounted on a microscope(BX61, Olympus, Japan) with a fluorescence light source (X-Cite 120PCQ, EXFO, Canada). The number of MPO, CD66b,and CD68 positive cells (as well as the total number of musclefibers from the area included) was counted. Data was calculatedas percentage of MPO, CD66b, or CD68 positively stained cellsper 100 skeletal muscle fibers. Areas of sections that containedfreeze damage or were folded due to the cutting procedure werenot included in the analysis.
Biochemical AssaysSerum creatine kinase activity in pre- and post-exercise bloodsamples was analyzed using an enzymatic assay (CK-NACkit, CDT14010, Thermo-Fisher Scientific Clinical Diagnostics,Sydney, Australia) and an automated clinical analyser (CobasMira, Roche Diagnostics, Germany). Serum myoglobinconcentration was also measured in these samples using animmunoassay (Roche Diagnostics, Germany) and an automatedclinical analyzer (Cobas E411, Roche Diagnostics, Germany).The intra-assay coefficient of variation was 10.4% for creatinekinase and 1.7% for myoglobin.
Muscle Soreness AssessmentUpon arrival at the laboratory and prior to the first musclebiopsy procedure, as well as 24 h following exercise prior tothe fourth and final muscle biopsy, subjects were asked to ratetheir subjective muscle soreness on a 0–10 visual analog scale.The subjects were instructed to contract, stretch, and palpatethe quadriceps muscle to assess general muscle soreness. Inboth instances 0 was considered to represent no pain andcorrespondingly a rating of 10 was considered to representintense pain.
StatisticsData are expressed as means ± SEM. Prior to analysis thedata displaying a lack of normality were log transformed tostabilize variance. Data were analyzed using a two-way ANOVAwith repeated measures for time. The sphericity adjustmentwas checked, and if required, a Greenhouse-Geisser epsiloncorrection was applied. Where no significant effect for treatmentwas observed, we explored pair-wise comparisons betweenindividual time-points using the Least Significant Difference(LSD) of means. Bivariate relationships were examined witha Spearman rank correlation test. These statistical analyseswere performed using GenStat for Windows 16th Edition (VSNInternational, Hemel Hempstead, UK). Confidence intervalswere calculated at 90% using a bootstrapping technique for non-normally distributed data using SPSS Statistics Version 22 (IBM,Portsmouth Hampshire, UK). Statistical significance was set atP < 0.05.
RESULTS
Muscle SorenessMuscle soreness showed a main effect for time (p < 0.01),suggesting that the exercise protocol was sufficient to induce amuscle stress response. No effect of ibuprofen treatment wasobserved (Figure 1).
ImmunohistochemistryThe number of MPO+ cells per 100 myofibers analyzed showed amain effect for time (P < 0.01). No main effect for treatment(P = 0.250) or time × treatment (P = 0.709) was observed(Figure 2A). LSD pairwise comparisons indicated a significantincrease in MPO+ cells at all sampling time points post-exercise.The greatest increase in MPO+ cells was observed at 3 h post-exercise (Figure 2B). Similarly, the number of CD66b+ cells per
Frontiers in Physiology | www.frontiersin.org 4 March 2016 | Volume 7 | Article 86
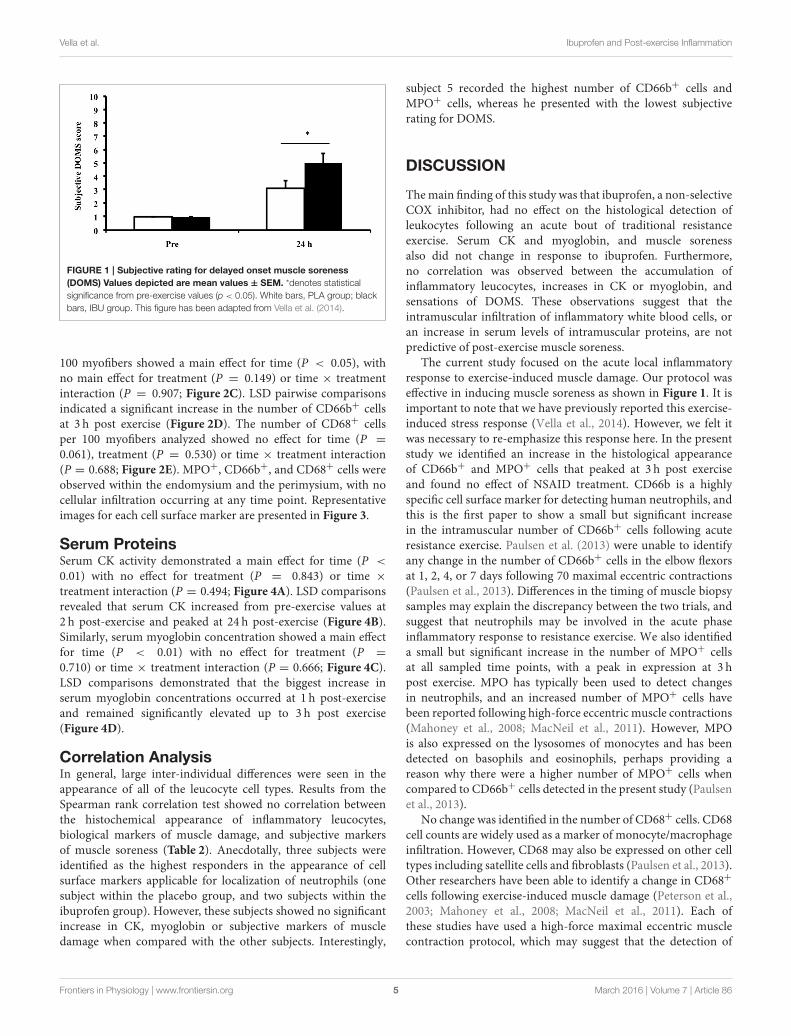
Vella et al. Ibuprofen and Post-exercise Inflammation
FIGURE 1 | Subjective rating for delayed onset muscle soreness
(DOMS) Values depicted are mean values ± SEM. *denotes statistical
significance from pre-exercise values (p < 0.05). White bars, PLA group; black
bars, IBU group. This figure has been adapted from Vella et al. (2014).
100 myofibers showed a main effect for time (P < 0.05), withno main effect for treatment (P = 0.149) or time × treatmentinteraction (P = 0.907; Figure 2C). LSD pairwise comparisonsindicated a significant increase in the number of CD66b+ cellsat 3 h post exercise (Figure 2D). The number of CD68+ cellsper 100 myofibers analyzed showed no effect for time (P =
0.061), treatment (P = 0.530) or time × treatment interaction(P = 0.688; Figure 2E). MPO+, CD66b+, and CD68+ cells wereobserved within the endomysium and the perimysium, with nocellular infiltration occurring at any time point. Representativeimages for each cell surface marker are presented in Figure 3.
Serum ProteinsSerum CK activity demonstrated a main effect for time (P <
0.01) with no effect for treatment (P = 0.843) or time ×
treatment interaction (P = 0.494; Figure 4A). LSD comparisonsrevealed that serum CK increased from pre-exercise values at2 h post-exercise and peaked at 24 h post-exercise (Figure 4B).Similarly, serum myoglobin concentration showed a main effectfor time (P < 0.01) with no effect for treatment (P =
0.710) or time × treatment interaction (P = 0.666; Figure 4C).LSD comparisons demonstrated that the biggest increase inserum myoglobin concentrations occurred at 1 h post-exerciseand remained significantly elevated up to 3 h post exercise(Figure 4D).
Correlation AnalysisIn general, large inter-individual differences were seen in theappearance of all of the leucocyte cell types. Results from theSpearman rank correlation test showed no correlation betweenthe histochemical appearance of inflammatory leucocytes,biological markers of muscle damage, and subjective markersof muscle soreness (Table 2). Anecdotally, three subjects wereidentified as the highest responders in the appearance of cellsurface markers applicable for localization of neutrophils (onesubject within the placebo group, and two subjects within theibuprofen group). However, these subjects showed no significantincrease in CK, myoglobin or subjective markers of muscledamage when compared with the other subjects. Interestingly,
subject 5 recorded the highest number of CD66b+ cells andMPO+ cells, whereas he presented with the lowest subjectiverating for DOMS.
DISCUSSION
Themain finding of this study was that ibuprofen, a non-selectiveCOX inhibitor, had no effect on the histological detection ofleukocytes following an acute bout of traditional resistanceexercise. Serum CK and myoglobin, and muscle sorenessalso did not change in response to ibuprofen. Furthermore,no correlation was observed between the accumulation ofinflammatory leucocytes, increases in CK or myoglobin, andsensations of DOMS. These observations suggest that theintramuscular infiltration of inflammatory white blood cells, oran increase in serum levels of intramuscular proteins, are notpredictive of post-exercise muscle soreness.
The current study focused on the acute local inflammatoryresponse to exercise-induced muscle damage. Our protocol waseffective in inducing muscle soreness as shown in Figure 1. It isimportant to note that we have previously reported this exercise-induced stress response (Vella et al., 2014). However, we felt itwas necessary to re-emphasize this response here. In the presentstudy we identified an increase in the histological appearanceof CD66b+ and MPO+ cells that peaked at 3 h post exerciseand found no effect of NSAID treatment. CD66b is a highlyspecific cell surface marker for detecting human neutrophils, andthis is the first paper to show a small but significant increasein the intramuscular number of CD66b+ cells following acuteresistance exercise. Paulsen et al. (2013) were unable to identifyany change in the number of CD66b+ cells in the elbow flexorsat 1, 2, 4, or 7 days following 70 maximal eccentric contractions(Paulsen et al., 2013). Differences in the timing of muscle biopsysamples may explain the discrepancy between the two trials, andsuggest that neutrophils may be involved in the acute phaseinflammatory response to resistance exercise. We also identifieda small but significant increase in the number of MPO+ cellsat all sampled time points, with a peak in expression at 3 hpost exercise. MPO has typically been used to detect changesin neutrophils, and an increased number of MPO+ cells havebeen reported following high-force eccentric muscle contractions(Mahoney et al., 2008; MacNeil et al., 2011). However, MPOis also expressed on the lysosomes of monocytes and has beendetected on basophils and eosinophils, perhaps providing areason why there were a higher number of MPO+ cells whencompared to CD66b+ cells detected in the present study (Paulsenet al., 2013).
No change was identified in the number of CD68+ cells. CD68cell counts are widely used as a marker of monocyte/macrophageinfiltration. However, CD68 may also be expressed on other celltypes including satellite cells and fibroblasts (Paulsen et al., 2013).Other researchers have been able to identify a change in CD68+
cells following exercise-induced muscle damage (Peterson et al.,2003; Mahoney et al., 2008; MacNeil et al., 2011). Each ofthese studies have used a high-force maximal eccentric musclecontraction protocol, which may suggest that the detection of
Frontiers in Physiology | www.frontiersin.org 5 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
FIGURE 2 | Immunohistochemistry data for muscle cells staining positive for myeloperoxidase (MPO), showing group specific data (A) and collapsed
data (B), CD66b showing group specific data (C), and collapsed data (D), and CD68 showing group data only (E). Data represent the mean number of
positively stained cells per 100 fibers analyzed ± SEM. **denotes statistical significance from pre-exercise values (p < 0.01). *denotes statistical significance from
pre-exercise values (p < 0.05). ∧denotes statistical significance from 0h post-exercise (p < 0.05). White bars, PLA group; black bars, IBU group.
CD68+ cells is dependent on the extent of skeletal muscledamage.
Previous research exploring the effect on NSAIDs onpost-exercise inflammation has produced equivocal findings.Earlier research from in-vivo rodent trials demonstrated ablunted inflammatory response to eccentric muscle contractionsfollowing the administration of both selective and non-selectiveNSAIDs (Lapointe et al., 2002a,b; Bondesen et al., 2004, 2006).However, these findings are yet to be supported in humantrials (Bourgeois et al., 1999; Peterson et al., 2003; Tokmakidiset al., 2003). Peterson et al. (2003) found no effect of oralibuprofen administration on the appearance of CD68+ cells 24 h
following 100 eccentric muscle contractions (Peterson et al.,2003). Similarly, Tokmakidis showed no effect of ibuprofenon circulating white blood cells 24 h following an eccentricexercise protocol (Tokmakidis et al., 2003). Paulsen et al. (2010)proposed the concept of an inflammatory threshold, suggestingthat NSAIDs may only influence post-exercise inflammation inresponse to a sufficiently strong inflammatory stimulus (Paulsenet al., 2010). It was suggested that the absence of an intramuscularprostaglandin response to exercise, specifically PGE2, couldexplain the lack of an NSAID effect on leucocyte recruitment(Trappe et al., 2001; Paulsen et al., 2010). Interestingly, ourgroup recently reported an increase in serum PGE2 in samples
Frontiers in Physiology | www.frontiersin.org 6 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
that were obtained during the same exercise trials as thosecurrently reported (Markworth et al., 2013). This change inserum PGE2 was blunted by the administration of ibuprofen
FIGURE 3 | Immunohistochemical analysis of skeletal muscle samples
following about of resistance exercise. Sections were probed with
antibodies raised against leucocyte cell surface markers (green; A—MPO,
B—CD66b, C—CD68) and dystrophin (red), while DAPI was used to stain
nuclei (blue). Scale bars = 50µm.
and suggests that in the presence of a blunted prostaglandinresponse to exercise, NSAID treatment had no effect on leucocyterecruitment within skeletal muscle. Although this finding doesnot refute the possibility of an inflammatory threshold, basedon our previous findings (Markworth et al., 2013), it isreasonable to suggest that the presence of an exercise-inducedserum prostaglandin response is not the required underlyingmechanism for neutrophil infiltration of muscle during the earlyhours of post-exercise recovery.
In accordance with previous studies, resistance exerciseresulted in an increase in serum CK and myoglobin, andsubjective muscle soreness (Nosaka et al., 2006). Although largeinter-individual variations in the human response to exercisecan make it difficult to establish causal relationships, therewas no relationship between the appearance of leucocytesand blood-derived markers of muscle damage or sensationsof muscle soreness. Interestingly, the subject with the highestnumber of CD66b+ and MPO+ cells presented with the lowestsubjective rating of DOMS. These findings provide support forthe assumption that leucocyte recruitment and inflammation donot consistently cause DOMS.
From animal studies it appears that cellular events in post-exercise inflammation, occur concomitantly with the onset ofDOMS (Armstrong et al., 1991; Faulkner et al., 1993). Themechanistic link has been proposed to be a leucocyte-mediated
FIGURE 4 | Blood derived proteins creatine kinase (A,B) and myoglobin (C,D). Values depicted are mean values ± SEM. (A,C) represent data divided into two
treatment groups; (B,D) represent collapsed data. *denotes statistical significance from pre-exercise values (p < 0.05). **denotes statistical significance from
pre-exercise values (p < 0.01). ∧denotes statistical significance from 1, 2, and 3 h post exercise (p < 0.05). White bars, PLA; black bars, IBU.
Frontiers in Physiology | www.frontiersin.org 7 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
TABLE 2 | Spearman rank correlation analysis.
CK MYO CD68+ CD66b+ MPO+ DOMS
CK – 0.59 (−0.01 − 0.91) −0.46 (−0.88 − 0.8) 0.15 (−0.42 − 0.61) 0.36 (−0.14 − 0.73) 0.01 (−0.60 − 0.62)
MYO – – 0.01 (−0.55 − 0.52) 0.14 (−0.42 − 0.69) 0.35 (−0.21 − 0.67) −0.33 (−0.74 − 0.24)
CD68+ – – – 0.05 (−0.46 − 0.56) −0.27 (−0.75 − 0.33) −0.37 (−0.82 − 0.25)
CD66b+ – – – – 0.41 (−0.13 − 0.84) −0.37 (−0.78 − 0.16)
MPO+ – – – – – 0.08 (−0.49 − 0.56)
DOMS – – – – – –
Values depicted are the correlation coefficient with 90% confidence intervals in parenthesis. Statistical significance was set at p < 0.05. No statistically significant correlations were
observed. CK, creatine kinase; MYO, myoglobin; MPO, Myeloperoxidase; DOMS, delayed-onset muscle soreness.
release of cytotoxic substances and reactive oxygen speciesas a bi-product of phagocytosis (Connolly et al., 2003).Furthermore, the prostaglandin response to exercise has beenshown to occur concurrently to the onset of DOMS, andis known to influence both the recruitment of inflammatoryleucocytes and neural afferents to pain (Markworth et al., 2013).However, in line with the current findings, the accumulationof inflammatory leucocytes in human skeletal muscle tissueduring exercise recovery appears to be both spatially andtemporally out of phase with the development of DOMS(Paulsen et al., 2010). Malm et al. (2004) explored theconcept that the activation of local leucocytes present in theepimysium may be involved in the regulation of DOMS.This hypothesis has since received support from both rodent(Gibson et al., 2009) and human trials (Crameri et al., 2007;Raastad et al., 2010) and presents a promising area for futureresearch.
CONCLUSION
This is the first study to demonstrate that ibuprofenadministration has no effect on the histological appearanceof inflammatory white blood cells following an acute boutof traditional resistance exercise. We also found no effectof ibuprofen administration on blood markers of muscledamage or subjective muscle soreness, and no significantcorrelations between leucocyte numbers and post-exercisemuscle damage or soreness. Future research needs to explorethe possibility of an inflammatory threshold above whichNSAID administration influences post-exercise inflammation.The underlying cause of post-exercise muscle soreness also
remains largely unknown. Future investigations are needed todetermine the role that inflammation plays in exercise-inducedDOMS.
AUTHOR CONTRIBUTIONS
LV—Research design, sample acquisition and analysis, dataanalysis, drafting and formatting manuscript. JM—Researchdesign, data analysis, revising manuscript, final approval ofversion to be published. GP—Data collection and analysis,revising manuscript, final approval of version to be published.TR—Data collection and analysis, interpretation of data, revising
manuscript, final approval of version to be published. JP—Research design, data analysis, interpretation of data, revisingmanuscript, final approval of version to be published. RS—Research design, interpretation of data, revising manuscript,final approval of version to be published. DC—Research design,interpretation of data, revising manuscript, final approval ofversion to be published. AR—Data analysis, interpretation ofdata, revising manuscript, final approval of version to bepublished.
ACKNOWLEDGMENTS
The authors would like to acknowledge to assistance of AssociateProfessor John Reynolds of Deakin University, and Dr. SeanWilliams of University of Bath, for their contributions to thestatistical analysis of the dataset. We further thank Dr. AndrewGarnham of Deakin University for all muscle biopsy procedures,and the participants for volunteering their time to be involved inthe trial.
REFERENCES
Armstrong, R. B.,Warren, G. L., andWarren, J. A. (1991). Mechanisms of exercise-
induced muscle fibre injury. Sports Med. 12, 184–207. doi: 10.2165/00007256-
199112030-00004
Baldwin, A. C., Stevenson, S. W., and Dudley, G. A. (2001). Nonsteroidal
anti-inflammatory therapy after eccentric exercise in healthy older
individuals. J. Gerontol. A Biol. Sci. Med. Sci. 56, M510–M513. doi:
10.1093/gerona/56.8.M510
Bondesen, B. A., Mills, S. T., Kegley, K. M., and Pavlath, G. K. (2004). The COX-2
pathway is essential during early stages of skeletal muscle regeneration. Am. J.
Physiol. Cell Physiol. 56, C475–C483. doi: 10.1152/ajpcell.00088.2004
Bondesen, B. A., Mills, S. T., and Paviath, G. K. (2006). The COX-2 pathway
regulates growth of atrophied muscle via multiple mechanisms. Am. J. Physiol.
Cell Physiol. 59, C1651–C1659. doi: 10.1152/ajpcell.00518.2005
Bourgeois, J., MacDougall, D., MacDonald, J., and Tarnopolsky, M. (1999).
Naproxen does not alter indices of muscle damage in resistance-exercise
trainedmen.Med. Sci. Sports Exerc. 31, 4–9. doi: 10.1097/00005768-199901000-
00002
Buford, T. W., Cooke, M. B., and Willoughby, D. S. (2009). Resistance exercise-
induced changes of inflammatory gene expression within human skeletal
muscle. Eur. J. Appl. Physiol. 107, 463–471. doi: 10.1007/s00421-009-1145-z
Burd, N. A., Dickinson, J. M., LeMoine, J. K., Carroll, C. C., Sullivan, B. E., Haus, J.
M., et al. (2010). Effect of a cyclooxygenase-2 inhibitor on postexercise muscle
Frontiers in Physiology | www.frontiersin.org 8 March 2016 | Volume 7 | Article 86

Vella et al. Ibuprofen and Post-exercise Inflammation
protein synthesis in humans. Am. J. Physiol. Endocrinol. Metab. 61, E354–E361.
doi: 10.1152/ajpendo.00423.2009
Connolly, D. A. J., Sayers, S. P., and McHugh, M. P. (2003). Treatment and
prevention of delayed onset muscle soreness. J. Strength Cond. Res. 17, 197–208.
doi: 10.1519/00124278-200302000-00030
Crameri, R. M., Aagaard, P., Qvortrup, K., Langberg, H., Olesen, J., and
Kjaer, M. (2007). Myofibre damage in human skeletal muscle: effects of
electrical stimulation versus voluntary contraction. J. Physiol. 583, 365–380. doi:
10.1113/jphysiol.2007.128827
Faulkner, J. A., Brooks, S. V., and Opiteck, J. A. (1993). Injury to skeletal muscle
fibers during contractions; conditions of occurrence and prevention. Phys.
Ther. 73, 911–921.
Fridén, J., Sjöström, M., and Ekblom, B. (1981). A morphological study of delayed
muscle soreness. Experientia 37, 506–507. doi: 10.1007/BF01986165
Gibson,W., Arendt-Nielsen, L., Taguchi, T., Mizumura, K., andGraven-Nielsen, T.
(2009). Increased pain from muscle fascia following eccentric exercise: animal
and human findings. Exp. Brain Res. 194, 299–308. doi: 10.1007/s00221-008-
1699-8
Hersh, E. V., Moore, P. A., and Ross, G. L. (2000). Over-the-counter analgesics and
antipyretics: a critical assessment. Clin. Ther. 22, 500–548. doi: 10.1016/S0149-
2918(00)80043-0
Hyldahl, R. D., Keadle, J., Rourier, P. A., Pearl, D., and Clarkson, P. M. (2010).
Effects of ibuprofen topical gel on muscle soreness. Med. Sci. Sports Exerc. 42,
614–621. doi: 10.1249/MSS.0b013e3181b95db2
Krentz, J. R., Quest, B., Farthing, J. P., Quest, D. W., and Chilibeck, P. D. (2008).
The effects of ibuprofen on muscle hypertrophy, strength, and soreness during
resistance training. Appl. Physiol. Nutr. Metab. 33, 470–475. doi: 10.1139/H08-
019
Lapointe, B. M., Frémont, P., and Côté, C. H. (2003). Influence of nonsteroidal
anti-inflammatory drug treatment duration and time of onset on recovery from
exercise-induced muscle damage in rats. Arch. Phys. Med. Rehabil. 84, 651–655.
doi: 10.1016/S0003-9993(02)04899-2
Lapointe, B. M., Frémont, P., and Côté, C. H. (2002a). Adaptation to lengthening
contractions is independent of voluntary muscle recruitment but relies on
inflammation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R323–R329.
doi: 10.1152/ajpregu.00339.2001
Lapointe, B. M., Frenette, J., and Côté, C. H. (2002b). Lengthening contraction-
induced inflammation is linked to secondary damage but devoid of neutrophil
invasion. J. Appl. Physiol. 92, 1995–2004. doi: 10.1152/japplphysiol.00803.2001
MacNeil, L. G., Baker, S. K., Stevic, I., and Tarnopolsky, M. A. (2011).
17β-estradiol attenuates exercise-induced neutrophil infiltration in men.
Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1443–R1451. doi:
10.1152/ajpregu.00689.2009
Mahoney, D. J., Safdar, A., Parise, G., Melov, S., Fu, M., MacNeil, L., et al.
(2008). Gene expression profiling in human skeletal muscle during recovery
from eccentric exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294,
R1901–R1910. doi: 10.1152/ajpregu.00847.2007
Malm, C., Sjödin, T. L. B., Sjöberg, B., Lenkei, R., Renström, P., Lundberg, I. E., et al.
(2004). Leukocytes, cytokines, growth factors and hormones in human skeletal
muscle and blood after uphill or downhill running. J. Physiol. 556, 983–1000.
doi: 10.1113/jphysiol.2003.056598
Markworth, J. F., Vella, L. D., Figueiredo, V. C., and Cameron-Smith, D. (2014).
Ibuprofen treatment blunts early translational signaling responses in human
skeletal muscle following resistance exercise. J. Appl. Physiol. 117, 20–28. doi:
10.1152/japplphysiol.01299.2013
Markworth, J. F., Vella, L., Lingard, B. S., Tull, D. L., Rupasinghe, T.W., Sinclair, A.
J., et al. (2013). Human inflammatory and resolving lipid mediator responses to
resistance exercise and ibuprofen treatment.Am. J. Physiol. Regul. Integr. Comp.
Physiol. 305, R1281–R1296. doi: 10.1152/ajpregu.00128.2013
Mikkelsen, U. R., Langberg, H., Helmark, I. C., Skovgaard, D., Andersen, L. L.,
Kjaer, M., et al. (2009). Local NSAID infusion inhibits satellite cell proliferation
in human skeletal muscle after eccentric exercise. J. Appl. Physiol. 107,
1600–1611. doi: 10.1152/japplphysiol.00707.2009
Mikkelsen, U. R., Schjerling, P., Helmark, I. C., Reitelseder, S., Holm, L.,
Skovgaard, D., et al. (2011). Local NSAID infusion does not affect protein
synthesis and gene expression in human muscle after eccentric exercise.
Scand. J. Med. Sci. Sports 21, 630–644. doi: 10.1111/j.1600-0838.2010.
01170.x
Mishra, D. K., Fridén, J., Schmitz, M. C., and Lieber, R. L. (1995). Anti-
inflammatory medication after muscle injury. A treatment resulting in short-
term improvement but subsequent loss of muscle function. J. Bone Joint Surg.
Am. 77, 1510–1519.
Nosaka, K., Sacco, P., and Mawatari, K. (2006). Effects of amino acid
supplementation on muscle soreness and damage. Int. J. Sport Nutr. Exerc.
Metab. 16, 620–635.
Paulsen, G., Egner, I. M., Drange, M., Langberg, H., Benestad, H. B., Fjeld, J. G.,
et al. (2010). A COX-2 inhibitor reducesmuscle soreness, but does not influence
recovery and adaptation after eccentric exercise. Scand. J. Med. Sci. Sports 20,
1–13. doi: 10.1111/j.1600-0838.2009.00947.x
Paulsen, G., Egner, I., Raastad, T., Reinholt, F., Owe, S., Lauritzen, F., et al. (2013).
Inflammatory markers CD11b, CD16, CD66b, CD68, myeloperoxidase and
neutrophil elastase in eccentric exercised human skeletal muscles. Histochem.
Cell Biol. 139, 691–715. doi: 10.1007/s00418-012-1061-x
Peterson, J. M., Trappe, T. A., Mylona, E., White, F., Lambert, C. P., Evans,
W. J., et al. (2003). Ibuprofen and acetaminophen: effect on muscle
inflammation after eccentric exercise. Med. Sci. Sports Exerc. 35, 892–896. doi:
10.1249/01.MSS.0000069917.51742.98
Raastad, T., Owe, S. G., Paulsen, G., Enns, D., Overgaard, K., Crameri,
R., et al. (2010). Changes in calpain activity, muscle structure, and
function after eccentric exercise. Med. Sci. Sports Exerc. 42, 86–95. doi:
10.1249/MSS.0b013e3181ac7afa
Smith, L. L. (1991). Acute inflammation: the underlying mechanism in
delayed onset muscle soreness. Med. Sci. Sports Exerc. 23, 542–551. doi:
10.1249/00005768-199105000-00006
Tidball, J. G. (1995). Inflammatory cell response to acute muscle injury.
Med. Sci. Sports Exerc. 27, 1022–1032. doi: 10.1249/00005768-199507000-
00011
Tokmakidis, S. P., Kokkinidis, E. A., Smilios, I., and Douda, H. (2003). The effects
of ibuprofen on delayed muscle soreness and muscular performance after
eccentric exercise. J. Strength Condition. Res. 17, 53–59. doi: 10.1519/00124278-
200302000-00009
Trappe, T. A., Fluckey, J. D., White, F., Lambert, C. P., and Evans, W. J. (2001).
Skeletal muscle PGF(2)(alpha) and PGE(2) in response to eccentric resistance
exercise: influence of ibuprofen acetaminophen. J. Clin. Endocrinol. Metab. 86,
5067–5070. doi: 10.1210/jcem.86.10.7928
Trappe, T. A., White, F., Lambert, C. P., Cesar, D., Hellerstein, M., and Evans,
W. J. (2002). Effect of ibuprofen and acetaminophen on postexercise muscle
protein synthesis. Am. J. Physiol. Endocrinol. Metab. 282, E551–E556. doi:
10.1152/ajpendo.00352.2001
Urso, M. L. (2013). Anti-inflammatory interventions and skeletal muscle
injury: benefit or detriment? J. Appl. Physiol. 115, 920–928. doi:
10.1152/japplphysiol.00036.2013
Vella, L., Caldow, M. K., Larsen, A. E., Tassoni, D., Della Gatta, P. A., Gran, P.,
et al. (2012). Resistance exercise increases NF-κB activity in human skeletal
muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R667–R673. doi:
10.1152/ajpregu.00336.2011
Vella, L., Markworth, J. F., Peake, J. M., Snow, R. J., Cameron-Smith, D., and
Russell, A. P. (2014). Ibuprofen supplementation and its effects on NF-
κB activation in skeletal muscle following resistance exercise. Physiol. Rep.
2:e12172. doi: 10.14814/phy2.12172
Whisenant, M. J., Panton, L. B., East, W. B., and Broeder, C. E. (2003). Validation
of submaximal prediction equations for the 1 repetition maximum bench press
test on a group of collegiate football players. J. Strength Condition. Res. 17,
221–227. doi: 10.1519/00124278-200305000-00002
Conflict of Interest Statement: The authors declare that the research was
conducted in the absence of any commercial or financial relationships that could
be construed as a potential conflict of interest.
Copyright © 2016 Vella, Markworth, Paulsen, Raastad, Peake, Snow, Cameron-
Smith and Russell. This is an open-access article distributed under the terms
of the Creative Commons Attribution License (CC BY). The use, distribution or
reproduction in other forums is permitted, provided the original author(s) or licensor
are credited and that the original publication in this journal is cited, in accordance
with accepted academic practice. No use, distribution or reproduction is permitted
which does not comply with these terms.
Frontiers in Physiology | www.frontiersin.org 9 March 2016 | Volume 7 | Article 86

131
Appendix B:

ORIGINAL RESEARCH
Ibuprofen supplementation and its effects on NF-jBactivation in skeletal muscle following resistance exerciseLuke Vella1, James F. Markworth2, Jonathan M. Peake3, Rod J. Snow1, David Cameron-Smith2 &Aaron P. Russell1
1 Centre for Physical Activity and Nutrition, School of Exercise and Nutrition Science, Deakin University, Burwood, Vic., Australia
2 Liggins Institute, University of Auckland, Auckland, New Zealand
3 School of Biomedical Sciences and Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, Qld, Australia
Keywords
Exercise recovery, inflammation, NF-jB,
NSAID treatment, resistance exercise.
Correspondence
Luke Vella, Centre for Physical Activity and
Nutrition, School of Exercise and Nutrition
Science, Deakin University, 221 Burwood
Highway, Burwood, Victoria 3125, Australia.
Tel: (03) 9244 6100
Fax: (03) 9786 0141
E-mail: [email protected]
Funding Information
No funding information provided.
Received: 10 September 2014; Accepted: 10
September 2014
doi: 10.14814/phy2.12172
Physiol Rep, 2 (10), 2014, e12172,
doi: 10.14814/phy2.12172
Abstract
Resistance exercise triggers a subclinical inflammatory response that plays a
pivotal role in skeletal muscle regeneration. Nuclear factor-jB (NF-jB) is a
stress signalling transcription factor that regulates acute and chronic states of
inflammation. The classical NF-jB pathway regulates the early activation of
post-exercise inflammation; however there remains scope for this complex
transcription factor to play a more detailed role in post-exercise muscle recov-
ery. Sixteen volunteers completed a bout of lower body resistance exercise
with the ingestion of three 400 mg doses of ibuprofen or a placebo control.
Muscle biopsy samples were obtained prior to exercise and at 0, 3 and 24 h
post-exercise and analysed for key markers of NF-jB activity. Phosphorylated
p65 protein expression and p65 inflammatory target genes were elevated
immediately post-exercise independent of the two treatments. These changes
did not translate to an increase in p65 DNA binding activity. NF-jB p50 pro-
tein expression and NF-jB p50 binding activity were lower than pre-exercise
at 0 and 3 h post-exercise, but were elevated at 24 h post-exercise. These find-
ings provide novel evidence that two distinct NF-jB pathways are active in
skeletal muscle after resistance exercise. The initial wave of activity involving
p65 resembles the classical pathway and is associated with the onset of an
acute inflammatory response. The second wave of NF-jB activity comprises
the p50 subunit, which has been previously shown to resolve an acute inflam-
matory program. The current study showed no effect of the ibuprofen treat-
ment on markers of the NF-jB pathway, however examination of the within
group effects of the exercise protocol suggests that this pathway warrants
further research.
Introduction
Unaccustomed resistance exercise causes skeletal muscle
damage that impairs muscle function and promotes sen-
sations of pain. The precise signalling mechanisms that
initiate muscle repair following exercise-induced damage
remain a topic of ongoing debate [previously reviewed by
Paulsen et al. (2012)]. The onset of muscle damage trig-
gers a complex interplay of intracellular events that
involve myofibre injury, acute inflammation and cellular
repair. At a symptomatic level, inflammation in skeletal
muscle is characterised by swelling and soreness (Paulsen
et al. 2012). Consequently post-exercise inflammation is
often ascribed as a cause of delayed-onset muscle soreness
(DOMS). DOMS typically occurs 24–48 h post-exercise,
and is accompanied by a secondary reduction in muscle
force-generating capacity. Treatment of DOMS has
focused on reducing inflammation; however this may be
detrimental to processes of cellular repair (Urso 2013).
Attenuating exercise-induced inflammation using non-ste-
roidal anti-inflammatory drugs reduces skeletal muscle
protein synthesis (Trappe et al. 2002) and impairs satellite
cell proliferation (Mikkelsen et al. 2009). To develop
more efficacious treatments for DOMS, a better
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
This is an open access article under the terms of the Creative Commons Attribution License,
which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
2014 | Vol. 2 | Iss. 10 | e12172Page 1
Physiological Reports ISSN 2051-817X

understanding of the mechanisms that govern inflamma-
tion and tissue repair in skeletal muscle after exercise is
required.
The nuclear factor-kappa B (NF-jB) transcription
factor acts as a central regulator of inflammatory signal-
ling pathways. The NF-jB family consists of five subunits,
including NF-jB1 (p105/p50), NF-jB2 (p100/p52), RelA
(p65), RelB and cREL that share an amino terminus Rel
homology domain. The Rel domain permits DNA bind-
ing, nuclear localization, dimerization and interaction
with its own inhibitory protein IjB (Delhalle et al. 2004;
Mourkioti and Rosenthal 2008; Bakkar and Guttridge
2010). Under basal conditions RelA (p65), cREL and RelB
subunits remain sequestered within the cytoplasm bound
to an inhibitory IjB protein. The IjB family that regu-
lates NF-jB includes the subunits IjBb, IjBa, IjBc IjBe,and Bcl-3. Unlike the Rel proteins, subunits p50 and p52
are synthesized as large precursor proteins (p105 and 100,
respectively) that require proteolytic processing to permit
nuclear localization. These subunits lack a REL domain,
to initiate gene transcription, and hence are primarily
considered as repressors of gene transcription (Bonizzi
and Karin 2004; Bakkar and Guttridge 2010).
Upon stimulation, the IjB kinase complex (IKK) con-
trols the degradation of IjB and its precursor proteins,
thereby enabling the NF-jB REL subunits to control gene
transcription. The IKK complex consists of two catalytic
kinases IKKa and IKKb, and a regulatory IKKc subunit.
IKKb activates the classical NF-jB signalling pathway
through the phosphorylation and subsequent degradation
of the inhibitor IjBa (Zandi et al. 1997; Pahl 1999). The
classical pathway typically comprises p65:p50 heterodi-
mers, and is essential for the activation of acute inflam-
mation by controlling the transcription on inflammatory
cytokines and acute phase proteins (Zandi et al. 1997;
Pahl 1999). More recently an alternative NF-jB pathway
has been described, which is dependent on IKKa (Senftle-
ben et al. 2001). Alternative signalling involves the activa-
tion of a secondary NF-jB inducing kinase (NIK), and
has been linked to the activation of both p52 and p50
subunits. The functional significance of IKKa-dependentgene expression in acute inflammation is not yet well
established. However, preliminary research in rat muscle
tissue suggests p50 homodimers may play a crucial role
in the resolution of acute inflammation (Bohuslav et al.
1998; Lawrence et al. 2001).
Despite the importance of inflammation in tissue
regeneration post-exercise, very little is known about how
NF-jB activity is regulated in skeletal muscle after acute
exercise. A transient increase in various components of
the classical NF-jB signalling pathway is observed in rat
muscle post-exercise (Hollander et al. 2001; Ji et al. 2004;
Ho et al. 2005; Spangenburg et al. 2006; Kramer and
Goodyear 2007). In contrast, a decrease in NF-jB DNA
binding at 0 h post-exercise with a return to near baseline
1 h post-exercise was observed in human muscle follow-
ing a traditional resistance exercise model (Durham et al.
2004). Recent work from our laboratory, using a similar
resistance exercise model, demonstrated an increase in
NF-jB binding to the promoter region of key inflamma-
tory cytokines at 2 h post-exercise, with a return to base-
line levels at 4 h post-exercise (Vella et al. 2012). These
findings suggest that a transient NF-jB response contrib-
utes to acute post-exercise inflammation.
To enhance our understanding of the cellular mecha-
nisms that regulate both the onset and resolution of post-
exercise inflammation, the current study aimed to investi-
gate changes in the activity of the subunits that comprise
the classical and alternative NF-jB signalling pathways.
We hypothesized that the regulation of NF-jB post-exer-
cise would involve two distinct waves of activation. Spe-
cifically, we hypothesized that the classical NF-jBpathway, involving p65 and p50 dimers would be acti-
vated soon after exercise during the early phases of
inflammation, while the alternative pathway, comprising
mainly the p50 subunit would be activated at later time
points after exercise that correspond with the resolution
of acute inflammation (Bohuslav et al. 1998; Ishikawa
et al. 1998). We also hypothesized that the administration
of ibuprofen would blunt both waves of NF-jB activa-
tion, providing a potential mechanism through which
anti-inflammatory medication might attenuate exercise-
induced inflammation in skeletal muscle.
Methods
Participants
As previously described, sixteen healthy male subjects
were recruited to participate in the study (characteristics
shown in Table 1) (Markworth et al. 2013). All partici-
pants completed a medical history questionnaire that was
used to identify and exclude participants with a diagnosed
condition or illness that prevented them from completing
strenuous exercise. Exclusion criteria included participa-
tion in a lower body resistance exercise program within
the last 6 months to ensure a muscle damage response
from the exercise stimulus, and/or chronic treatment with
anti-inflammatory drugs. Current use of prescription
medication or nutritional supplements also excluded sub-
jects from participating.
Ethics approval
Prior to participation each subject received written and
oral information regarding the nature of the experiment
2014 | Vol. 2 | Iss. 10 | e12172Page 2
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
NF-jB and Resistance Exercise L. Vella et al.

before providing written consent to participate. All proce-
dures involved in this study were approved by the Deakin
University Human Research Ethics Committee (DUHREC
2010-019). All muscle sampling procedures were per-
formed in accordance with the Helsinki declaration.
Familiarization and strength testing
Each participant completed a familiarization session at
least 7 days prior to completing the exercise trial. Partici-
pants performed repetition maximum testing to deter-
mine the experimental exercise load (80% of 1 repetition
maximum (1RM)). The maximal weight each subject
could lift was determined for the Smith machine-assisted
squat, the leg press, and the leg extension. These data
were substituted into the validated Brzycki equation to
predict 1RM for each participant (Mayhew et al. 1995;
Whisenant et al. 2003). The participants were required to
abstain from any further exercise until completion of the
trial.
Experimental Procedures
The participants reported to the laboratory in an over-
night fasted state, having abstained from caffeine, tobacco
and alcohol for the preceding 24 h. Following 30 min of
supine resting a pre-exercise muscle biopsy was taken.
Participants then completed a 10 min warm up consisting
of low intensity cycling on a bicycle ergometer, and one
low resistance set for each exercise at approximately 30–50% of the participants 1RM. Each participant then com-
pleted a single bout of intense resistance exercise. This
session consisted of three sets of 8–10 repetitions of a
bilateral Smith machine assisted squat, 45° leg press and
leg extension. These exercises were all performed at 80%
1RM. The exercises were performed sequentially as a cir-
cuit, with 1 min rest between each exercise, and 3 min
rest between sets. We have previously used this exercise
protocol and demonstrated that it activates inflammatory
signalling pathways (Vella et al. 2012). Following the
completion of the exercise bout, the participants rested in
a supine position while muscle biopsy samples were col-
lected. The participants returned to the laboratory the fol-
lowing morning in an over-night fasted state for a final
muscle biopsy sample. Standardized meals were provided
to participants the night preceding the trial (carbohydrate
57%, fat 22%, protein 21%), in the laboratory immedi-
ately following the exercise bout (carbohydrate 71%, fat
13%, protein 16%), as additional snacks throughout the
day and an evening meal (carbohydrate 64%, fat 27%,
protein 18%).
NSAID administration
Prior to the exercise bout, the participants were randomly
assigned to either the ibuprofen (NSAID) group (n = 8)
or the placebo group (PLA) (n = 8). Participants in the
NSAID group consumed the maximum recommended
over-the-counter dose of 1200 mg of ibuprofen as three
doses of 400 mg throughout the trial day. The first dose
was administered upon arriving to the laboratory on the
first morning of the trial, immediately prior to the first
muscle biopsy sample. Participants were instructed to
consume two additional doses at 2:00 pm and 8:00 pm
the same evening. This dosing structure was prescribed to
optimise levels of circulating ibuprofen to biologically
active levels throughout the course of the trial day. This
protocol has previously been validated by our research
group in this same group of study participants and the
same NSAID administration protocol (Markworth et al.
2013). Alternatively the placebo consumed a gelatin cap-
sule containing powdered sugar, identical in appearance
to the ibuprofen capsule.
Sample collection
Muscle biopsy samples were obtained under local anaes-
thesia (Xylocaine 1%) from the vastus lateralis muscle of
either leg using the percutaneous needle biopsy technique
modified to include suction (Buford et al. 2009). Samples
were obtained prior to exercise, immediately following
exercise and at 3 and 24 h post-exercise. The muscle
biopsy sample obtained immediately post-exercise was
taken within 1–2 min of the completion of the exercise
protocol, and will herein after be referred to as 0 h post-
exercise. The muscle biopsy procedure has been shown to
Table 1. Subject characteristics and strength testing data.
Characteristics Strength (1RM)
Age (y) Height (m) Body mass (kg) BMI Squat(kg) Leg press (kg) Leg extension (kg)
PLA 23.9 � 1.3 1.89 � 0.1 86.9 � 4.5 24.5 � 1.2 94.9 � 5.4 237 � 17 236 � 18
IBU 23.0 � 0.5 1.89 � 0.1 89.1 � 4.4 24.8 � 0.8 91.9 � 6.0 240 � 15 196 � 22
Values are mean values � SEM. No significant differences were observed between the two groups.
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf ofthe American Physiological Society and The Physiological Society.
2014 | Vol. 2 | Iss. 10 | e12172Page 3
L. Vella et al. NF-jB and Resistance Exercise

trigger a local inflammatory response (Malm et al. 2000).
To minimize interference from the biopsy procedure,
samples prior to exercise and at 24 h post-exercise were
taken from the same leg, while muscle samples obtained
at 0 and 3 h post-exercise were taken from the opposite
leg. Samples within the same leg were taken at least 5 cm
from the previous site. We have previously reported that
this technique is effective for minimizing cytokine gene
expression and NF-jB activity in response to muscle
biopsies (Vella et al. 2012). Excised tissue was immedi-
ately immersed in liquid nitrogen and stored at �80°Cuntil further analysis.
Subjective assessment of DOMS and rangeof movement
Subjective assessment of muscle soreness (DOMS) and
range of movement were recorded prior to exercise and
at 24 h post exercise. Subjects were asked to rate their
levels of muscle soreness and range of movement on a
0–10 scale. In both instances 0 was considered as the best
possible result and a rating of 10 was considered to be
the worst result.
Protein extraction and quantification
Muscle samples were homogenized in ice cold RIPA buf-
fer (50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl,
0.25% deoxycholic acid, 1% NP-40, 1 mmol/L EDTA
supplemented with protease and phosphatase inhibitors
including 1 mmol/L PMSF, 1 lg/mL aprotinin, 1 lg/mL
leupeptin, 1 mmol/L Na3VO4 & 1 mmol/L NaF). The
homogenate was agitated for 1 h at 4°C and centrifuged
at 13,000 9 g at 4°C for 15 min. The resultant superna-
tant was removed and stored at �80°C until further
analysis. Total protein concentration was determined
using a BCA protein assay kit according to the manufac-
turer’s instruction (Pierce, Rockford, IL). Protein samples
(50 lg) were denatured in loading buffer and separated
by a 10% SDS-PAGE and transferred to a PVDF mem-
brane. Membranes were blocked for 90 min at room
temperature in 5% BSA/Tris buffered saline with 0.1%
Tween 20 (TBST). Primary antibodies [phosphorylated
NF-jB p65 Ser536, total NF-jB p65, NF-jB p100/p52,
NF-jB p105/p50, NF-jB cREL and b-actin (all obtained
from Cell Signaling Technologies, Arundel, QLD)] were
diluted to 1:1000 in 5% BSA/TBST, applied and incu-
bated overnight at 4°C with gentle agitation. Membranes
were washed for 30 min in TBST and probed with HRP
conjugated secondary antibodies diluted to 1:2000 in 5%
BSA/TBST, and incubated for 1 h at room temperature.
Proteins were visualised by using Western Lighting
enhanced chemiluminescence (Perkin Elmer Lifesciences,
Boston, MA). Signals were captured using a Kodak Digi-
tal Image Station 2000M (model: 440CF; Eastman Kodak,
Rochester, NY) and quantified by densitometry band
analysis using Kodak Molecular Imaging Software (Ver-
sion 4.0.5, © 1994–2005, Eastman Kodak). Phosphory-
lated NF-jB p65 protein was normalized to total p65
protein (Fig. 2A). NF-jB p50 protein expression was
normalized to its precursor protein p105 (Fig. 2B). NF-
jB p52 and cREL protein was normalized to b-actin(Fig. 2C).
RNA extraction and RT-PCR
Total cellular RNA was extracted as previously described
(Trenerry et al. 2007) using the ToTALLY RNA Kit (Am-
bion, Austin, TX). RNA quality and concentration were
determined using the Agilent 2100 Biolanalyzer (Agilent
Technologies, Palo Alto, CA). First strand cDNA was gen-
erated from 0.5 lg total RNA using the AMV RT kit
(Promega, Madison, WI). RT-PCR was performed in
duplicate using the Biorad CFX384 system (Biorad, Her-
cules, CA), containing 5XHOT FirePol� EvaGreen Mix
(Integrated Science, Sydney, NSW), forward primer,
reverse primer, sterile nuclease free water and cDNA
(0.125 ng/lL). Data were analysed using a comparative
critical threshold (Ct) method, where the amount of the
specified target gene normalised to the amount of endog-
enous control, relative to the control value is given by
2�DDCt . The endogenous control used in this experiment
was GAPDH. The efficacy of GAPDH as an endogenous
control was examined using the equation 2�DCt . Primers
were designed using Primer Express software package ver-
sion 3.0 (Applied Biosystems, Mulgrave, Vic., Australia).
Gene sequences were obtained from GenBank (Table 2).
Primer sequence specificity was confirmed using BLAST.
A melting point dissociation curve was generated by the
PCR instrument for all PCR products to confirm the
presence of a single amplified product.
Nuclear extraction and Transcription Factor(TF) Assay
Nuclear and cytoplasmic proteins were extracted from
20 mg of muscle tissue using a NE-PER Nuclear and
Cytoplasmic Extractions Reagents (Pierce, Rockford, IL),
according to the manufacturer’s instructions. Western
blot analysis probed for GAPDH, a-tubulin and Lamin A
was performed to ensure the nuclear extract was not
contaminated by cytoplasmic proteins. A Transcription
Factor Assay detecting specific transcription factor
DNA binding activity was performed according to
manufacturer’s instructions (Cayman Chemical, Ann
Arbour, MI). Briefly, 10 lg of nuclear protein were
2014 | Vol. 2 | Iss. 10 | e12172Page 4
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
NF-jB and Resistance Exercise L. Vella et al.

loaded into a well containing an immobilized NF-jB con-
sensus sequence (50GGGACTTTCC-30) and incubated
overnight at 4°C. Primary antibodies for NF-jB subunits
p65 and p50 were loaded in to each well and incubated at
room temperature for 1 h. Each well was flushed using a
diluted wash buffer for 30 min. Following a secondary
30 min wash, samples were incubated with secondary
HRP-conjugated antibody for a further 1 h at room tem-
perature. To quantify transcription factor binding, a
developing solution containing a 3,30,5,50-Tetrame-
thhylbenzidine (TMB) solution was added to each
well and incubated for 45 min at room temperature.
DNA binding was then quantified using photospectrome-
try with absorbance measurements taken at 450 nm
using a Multiscan RC plate reader (Labsystems, Finland)
and Gen5 Data Analysis Software (BioTek, Winooski,
VT).
Statistics
Data are expressed as means � SEM. Prior to analysis the
data displaying a lack of normality were log transformed
to stabilise variance. Data were analysed using a two-way
ANOVA with repeated measures for time. The sphericity
adjustment was checked, and if required, a Greenhouse-
Geisser epsilon to the residual degrees of freedom was
applied. A data point was considered to be a statistical
outlier where a z-score exceeded a pre-determined thresh-
old of � 4.00. Where appropriate we explored within-
group pair-wise comparisons between individual time-
points using the Least Significant Differences of means at
P < 0.05 to determine statistically significant changes
(Mead 1988; Saville 1990). Statistical analyses were per-
formed using GenStat for Windows 16th Edition (VSN
International, Hemel Hempstead, UK).
Results
Subjective DOMS analysis showed a main effect for time
(time effect P < 0.01) with no effect for treatment. The
average post-exercise DOMS score was 4.81 � 0.5 and
the post-exercise ROM score was 3.3 � 0.5. This data
demonstrates that the exercise protocol induced a moder-
ate level of muscle soreness and this effect was irrespective
of ibuprofen treatment.
Expression of NF-jB phosphorylated p65 (Ser 536)
increased over time (time effect P = 0.006), but this
response was overall not different between the groups
(interaction effect P = 0.253, treatment effect P = 0.176).
Nevertheless, LSD pairwise comparisons indicated that
the main increase from baseline was in the placebo
group at 0 h post-exercise. Within the placebo group
phosphorylated p65 protein expression remained elevated
at 3 h post-exercise and returned to baseline by 24 h
(Fig. 1A). No significant changes were observed in the
ibuprofen group by LSD comparisons. There was no
effect of time or treatment for NF-jB total p65
(Fig. 2A).
There was a trend toward a significant time 9 treat-
ment interaction (P = 0.057) for the protein expression
of the p50 subunit, although a main effect for time
(P = 0.376) or treatment (P = 0.865) was not achieved.
Our analysis revealed a statistical outlier in the ibuprofen
group at the 24 h time point. When this subject was
removed from the analysis, this trend was weaker
(P = 0.115) (Fig. 1B). LSD comparisons indicated an
increase in p50 protein expression was observed at 24 h
post exercise in the placebo group only (Fig. 1B). There
were no main or interaction effects for the NF-jB p50
precursor protein p105 (Fig. 2B) or for p52 and cREL
subunits (Fig. 1C and D).
To determine subunit-specific DNA binding of NF-jBfollowing exercise, transcription factor binding assays
were performed for p50 and p65 subunits. NF-jB p50
showed a main effect for time (P = 0.035), with no
time 9 treatment interaction (P = 0.851) or main effect
for treatment (P = 0.488) (Fig. 3B). LSD comparisons
identified a significant increase in p50 binding that
occurred at 24 h post-exercise when compared to 0 and
3 h post-exercise within the placebo group. This coin-
cided with the increase in p50 protein expression
(Fig. 1B). No change in p65 DNA binding was observed,
despite an increase in phosphorylated p65 protein expres-
sion (Fig. 3B).
We sought to determine whether any increase in
NF-jB signalling coincided with an increase in down-
stream inflammatory cytokines, and if the administration
of ibuprofen influenced post-exercise cytokine expression.
The mRNA levels of IL-6 (Fig. 4A), IL-8 (Fig. 4B) and
MCP-1 (Fig. 4C) demonstrated a main effect for time
(P < 0.01), with the highest elevation in expression levels
at 3 h post-exercise after significant increases at 0 h post-
exercise. TNF-a mRNA remained unchanged after exer-
cise (Fig. 4D). COX-2 mRNA showed a main effect for
time (P < 0.01), increasing at 0, 3 and 24 h post-exercise
(Fig. 4E). There were no significant interaction effects or
main effects for treatment for the inflammatory cytokines
or COX-2 mRNA expression. Consistently, LSD pairwise
comparisons revealed similar changes over time from
baseline within both groups.
Discussion
The current study aimed to explore the regulation of
the different NF-jB subunits following a single bout of
lower body resistance exercise, and investigate the mech-
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf ofthe American Physiological Society and The Physiological Society.
2014 | Vol. 2 | Iss. 10 | e12172Page 5
L. Vella et al. NF-jB and Resistance Exercise
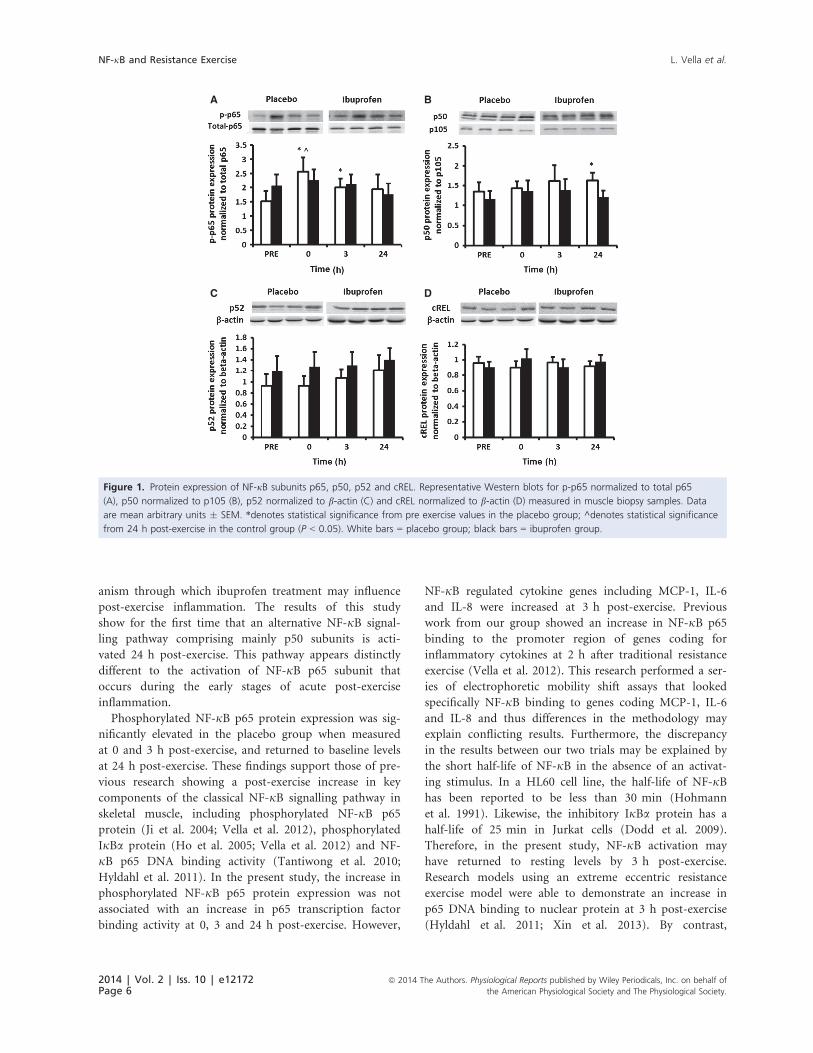
anism through which ibuprofen treatment may influence
post-exercise inflammation. The results of this study
show for the first time that an alternative NF-jB signal-
ling pathway comprising mainly p50 subunits is acti-
vated 24 h post-exercise. This pathway appears distinctly
different to the activation of NF-jB p65 subunit that
occurs during the early stages of acute post-exercise
inflammation.
Phosphorylated NF-jB p65 protein expression was sig-
nificantly elevated in the placebo group when measured
at 0 and 3 h post-exercise, and returned to baseline levels
at 24 h post-exercise. These findings support those of pre-
vious research showing a post-exercise increase in key
components of the classical NF-jB signalling pathway in
skeletal muscle, including phosphorylated NF-jB p65
protein (Ji et al. 2004; Vella et al. 2012), phosphorylated
IjBa protein (Ho et al. 2005; Vella et al. 2012) and NF-
jB p65 DNA binding activity (Tantiwong et al. 2010;
Hyldahl et al. 2011). In the present study, the increase in
phosphorylated NF-jB p65 protein expression was not
associated with an increase in p65 transcription factor
binding activity at 0, 3 and 24 h post-exercise. However,
NF-jB regulated cytokine genes including MCP-1, IL-6
and IL-8 were increased at 3 h post-exercise. Previous
work from our group showed an increase in NF-jB p65
binding to the promoter region of genes coding for
inflammatory cytokines at 2 h after traditional resistance
exercise (Vella et al. 2012). This research performed a ser-
ies of electrophoretic mobility shift assays that looked
specifically NF-jB binding to genes coding MCP-1, IL-6
and IL-8 and thus differences in the methodology may
explain conflicting results. Furthermore, the discrepancy
in the results between our two trials may be explained by
the short half-life of NF-jB in the absence of an activat-
ing stimulus. In a HL60 cell line, the half-life of NF-jBhas been reported to be less than 30 min (Hohmann
et al. 1991). Likewise, the inhibitory IjBa protein has a
half-life of 25 min in Jurkat cells (Dodd et al. 2009).
Therefore, in the present study, NF-jB activation may
have returned to resting levels by 3 h post-exercise.
Research models using an extreme eccentric resistance
exercise model were able to demonstrate an increase in
p65 DNA binding to nuclear protein at 3 h post-exercise
(Hyldahl et al. 2011; Xin et al. 2013). By contrast,
A B
C D
Figure 1. Protein expression of NF-jB subunits p65, p50, p52 and cREL. Representative Western blots for p-p65 normalized to total p65
(A), p50 normalized to p105 (B), p52 normalized to b-actin (C) and cREL normalized to b-actin (D) measured in muscle biopsy samples. Data
are mean arbitrary units � SEM. *denotes statistical significance from pre exercise values in the placebo group; ^denotes statistical significance
from 24 h post-exercise in the control group (P < 0.05). White bars = placebo group; black bars = ibuprofen group.
2014 | Vol. 2 | Iss. 10 | e12172Page 6
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
NF-jB and Resistance Exercise L. Vella et al.
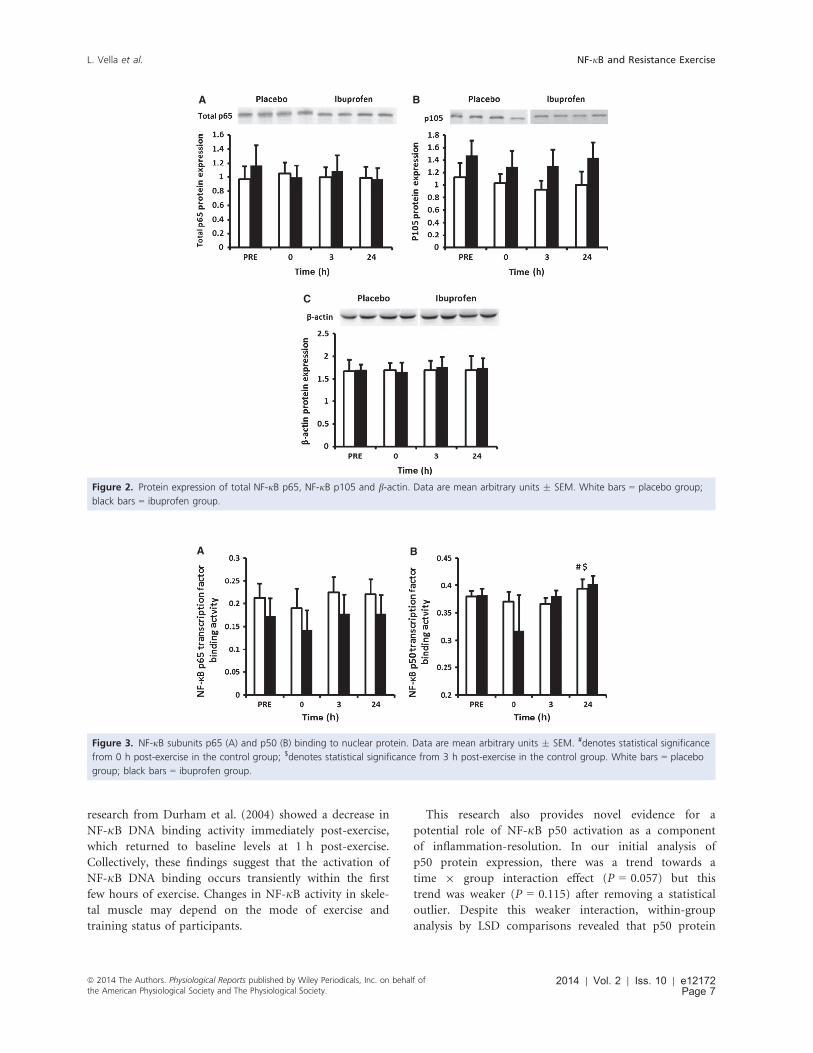
research from Durham et al. (2004) showed a decrease in
NF-jB DNA binding activity immediately post-exercise,
which returned to baseline levels at 1 h post-exercise.
Collectively, these findings suggest that the activation of
NF-jB DNA binding occurs transiently within the first
few hours of exercise. Changes in NF-jB activity in skele-
tal muscle may depend on the mode of exercise and
training status of participants.
This research also provides novel evidence for a
potential role of NF-jB p50 activation as a component
of inflammation-resolution. In our initial analysis of
p50 protein expression, there was a trend towards a
time 9 group interaction effect (P = 0.057) but this
trend was weaker (P = 0.115) after removing a statistical
outlier. Despite this weaker interaction, within-group
analysis by LSD comparisons revealed that p50 protein
A B
C
Figure 2. Protein expression of total NF-jB p65, NF-jB p105 and b-actin. Data are mean arbitrary units � SEM. White bars = placebo group;
black bars = ibuprofen group.
A B
Figure 3. NF-jB subunits p65 (A) and p50 (B) binding to nuclear protein. Data are mean arbitrary units � SEM. #denotes statistical significance
from 0 h post-exercise in the control group; $denotes statistical significance from 3 h post-exercise in the control group. White bars = placebo
group; black bars = ibuprofen group.
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf ofthe American Physiological Society and The Physiological Society.
2014 | Vol. 2 | Iss. 10 | e12172Page 7
L. Vella et al. NF-jB and Resistance Exercise

expression was elevated in the placebo group at 24 h
post-exercise. Coincident with this response, p50 tran-
scription factor binding activity was highest at 24 h
post-exercise. The biological significance of this delayed
increase in NF-jB p50 signalling during the latter stages
of post-exercise recovery remains uncertain. However, in
vitro work suggests that it may play a role in regulating
the active resolution of acute inflammation in skeletal
muscle (Lawrence et al. 2001, 2005; Senftleben et al.
2001). In a model of carageenin-induced pleurisy in
rats, Lawrence et al. (2001) provided preliminary evi-
dence for a complex interplay between distinct NF-jBsignalling pathways that control an acute and transient
inflammatory response. They reported that the prelimin-
ary phase of NF-jB activation that occurred at 6 h was
characteristic of the classical NF-jB pathway, and was
associated with the onset of acute inflammation. The
secondary phase was typical of the alternative NF-jBpathway, comprising p50 homodimers. Importantly,
inhibiting this wave of activity caused a prolonged
inflammatory response (Lawrence et al. 2001). Findings
from the present study support the concept of two
functionally distinct waves of NF-jB activity following
traditional resistance exercise. Future work is warranted
to determine how this secondary wave of NF-jB path-
way activity influences post-exercise inflammation and
skeletal muscle recovery.
We used ibuprofen supplementation to investigate
whether manipulating the inflammatory response to exer-
cise through the cyclooxygenase-prostaglandin pathway
alters post-exercise NF-jB signalling. Previous reports
indicate that NF-jB can function upstream of COX-2 to
control transcription of this gene (Pahl 1999; Poligone
and Baldwin 2001). Alternatively, prostaglandin activity
may also affect NF-jB (Rossi et al. 2000; Straus et al.
2000; Poligone and Baldwin 2001). In-vitro studies dem-
onstrate that the effect of prostaglandins on NF-jB activ-
ity is specific to the class of prostaglandin. Prostaglandin
E2 activates the classical NF-jB pathway, whereas prosta-
glandin A2, and prostaglandin J2 and its downstream
analogues can inhibit NF-jB activation in response to
pro-inflammatory stimuli (Castrillo et al. 2000; Rossi
et al. 2000; Straus et al. 2000; Lawrence et al. 2002). Our
group recently reported changes in prostaglandins in
blood serum samples after traditional resistance exercise
(Markworth et al. 2013). Interestingly, PGE2, PGA2 and
PGD2 all peaked in expression at 2 h post-exercise. We
did not find any significant time 9 group interaction
effects for the change in p65 phosphorylation; however,
LSD comparisons suggested that phosphorylated p65
expression only seemed to increase in the placebo group.
Similarly the observed changes in p50 protein expression
and DNA binding were only identified within the placebo
group. These findings are supported by data from the
study by Lawrence et al. (2001), which demonstrated no
change in the secondary wave of NF-jB DNA binding
activity following the administration of an alternative
COX inhibitor, NS398, in rats with pleurisy. While these
findings do not offer any conclusive evidence that ibupro-
fen treatment inhibits the NF-jB pathway, it does provide
justification to further explore this pathway as a potential
mechanism through which ibuprofen treatment inhibits
post-exercise inflammation.
There were several limitations to the present study that
need to be considered. Firstly, this analysis was run as
part of a larger study investigating the effects of ibuprofen
administration on multiple components of the post-exer-
cise inflammatory and hypertrophy response (Markworth
et al. 2013, 2014). Therefore, limited muscle sample
remained to complete further analyses. Future research
Table 2. Primer sequences were designed using Primer Express Software v 3.0 (Applied Biosystems) using sequences accessed through Gen-
bank and checked for specificity using nucleotide-nucleotide BLAST search.
Gene Accession No. Primer sequence
GAPDH NM_21130 Forward: CAT CCA TGA CAA CTT TGG TAT CGT
Reverse: CAG TCT TCT GGG TGG CAG TGA
MCP-1 NM_002982 Forward: TCC CAA AGA AGC TGT GAT CTT CA
Reverse: CAG ATT CTT GGG TGG AGT GA
IL-6 NM_000600 Forward: GCG AAA GGA TGA AAG TGA CCA T
Reverse: AGA CAA GCC CAG CAA TGA AAA
IL-8 NM_000584 Forward: CTG GCC GTC GCT CTC TGG
Reverse: TTA GCA CTC CTT GGC AAA ACT
TNF-a X_02910 Forward: GGA GAA GGG TGA CCG ACT CA
Reverse: TGC CCA GAC TCG GCA AAG
COX-2 U_20548 Forward: GAA TCA TTC ACC AGG CAA ATT G
Reverse: TGG AAG CCT GTG ATA CTT TCT GTA CT
2014 | Vol. 2 | Iss. 10 | e12172Page 8
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
NF-jB and Resistance Exercise L. Vella et al.

should consider the post-exercise regulation of upstream
markers including IKKa and IKKb.
Conclusion
This research provides new insights into the regulation of
NF-jB following a bout of acute resistance exercise. The
primary finding from this study is that NF-jB activation
follows a biphasic activation pattern that is subunit-spe-
cific. The first wave involves the classical NF-jB p65 sub-
unit, and corresponds with the onset of an acute
inflammatory response and an increase in inflammatory
cytokine gene expression. The second wave is consistent
with a previously identified secondary wave of NF-jB
activity that involves the alternative p50 subunit. Research
in alternative models of inflammation suggests that this
second wave of activity may be associated with an active
inflammatory resolution program. Further research into
the role of p50 signalling in skeletal muscle represents a
key area for future research in order to better understand
the mechanisms that regulate post-exercise inflammation.
Analysis of the LSD to examine pair-wise within-group
differences suggested that the observed changes in both
NF-jB p65 and p50 were detected only within the pla-
cebo group. The complex interplay between the NF-jBand the COX/prostaglandin pathway in exercise-induced
muscle damage remains poorly understood, and should
be a focus for future research.
A B
C D
E
Figure 4. RT-PCR analysis of NF-jB target genes IL-6 (A), IL-8 (B), MCP-1 (C), TNF-a (D) and COX-2 (E) in skeletal muscle cDNA. Data are mean
arbitrary units � SEM. *denotes statistical significance from pre exercise in the same treatment group; #denotes statistical significance from 0 h
post-exercise in the same treatment group; ^denotes statistical significance from 24 h post-exercise in the same treatment group (P < 0.05).
White bars = placebo group; black bars = ibuprofen group.
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf ofthe American Physiological Society and The Physiological Society.
2014 | Vol. 2 | Iss. 10 | e12172Page 9
L. Vella et al. NF-jB and Resistance Exercise

Acknowledgments
The authors would like to acknowledge to assistance of
Associate Professor John Reynolds for his contributions
in the statistical analysis of the dataset.
Conflict of Interest
There were no conflicts of interest relevant to this paper.
References
Bakkar, N., and D. C. Guttridge. 2010. NF-kappaB signaling: a
tale of two pathways in skeletal myogenesis. Physiol. Rev.
90:495–511.
Bohuslav, J., V. V. Kravchenko, G. C. Parry, J. H. Erlich,
S. Gerondakis, N. Mackman, et al. 1998. Regulation of an
essential innate immune response by the p50 subunit of
NF-kappaB. J. Clin. Investig. 102:1645–1652.
Bonizzi, G., and M. Karin. 2004. The two NF-kappaB
activation pathways and their role in innate and adaptive
immunity. Trends Immunol. 25:280–288.
Buford, T. W., M. B. Cooke, and D. S. Willoughby. 2009.
Resistance exercise-induced changes of inflammatory gene
expression within human skeletal muscle. Eur. J. Appl.
Physiol. 107:463–471.
Castrillo, A., M. J. D�ıaz-Guerra, S. Hortelano, P. Mart�ın-Sanz,
and L. Bosc�a. 2000. Inhibition of IkappaB kinase and
IkappaB phosphorylation by 15-deoxy-Delta
(12,14)-prostaglandin J(2) in activated murine macrophages.
Mol. Cell. Biol. 20:1692–1698.
Delhalle, S., R. Blasius, M. Dicato, and M. Diederich. 2004.
A beginner’s guide to NF-kappaB signaling pathways. Ann.
N. Y. Acad. Sci. 1030:1–13.
Dodd, S. L., B. Hain, S. M. Senf, and A. R. Judge. 2009.
Hsp27 inhibits IKKb-induced NF-jB activity and skeletal
muscle atrophy. FASEB J. 23:3415–3423.
Durham, W. J., L. Yi-Ping, E. Gerken, M. Farid, S. Arbogast,
R. R. Wolfe, et al. 2004. Fatiguing exercise reduces DNA
binding activity of NF-jB in skeletal muscle nuclei. J. Appl.
Physiol. 97:1740–1745.
Ho, R. C., M. F. Hirshman, Y. Li, D. Cai, J. R. Farmer,
W. G. Aschenbach, et al. 2005. Regulation of IkappaB kinase
and NF-kappaB in contracting adult rat skeletal muscle.
Am. J. Physiol. Cell Physiol. 289:C794–C801.
Hohmann, H. P., R. Remy, C. Scheidereit, and A. P. van
Loon. 1991. Maintenance of NF-kappa B activity is
dependent on protein synthesis and the continuous presence
of external stimuli. Mol. Cell. Biol. 11:259–266.
Hollander, J., R. Fiebig, M. Gore, T. Ookawara, H. Ohno, and
L. L. Ji. 2001. Superoxide dismutase gene expression is
activated by a single bout of exercise in rat skeletal muscle.
Pfl€ugers Arch. 442:426–434.
Hyldahl, R. D., L. Xin, M. J. Hubal, S. Moeckel-Cole,
S. Chipkin, and P. M. Clarkson. 2011. Activation of nuclear
factor-jB following muscle eccentric contractions in humans
is localized primarily to skeletal muscle-residing pericytes.
FASEB J. 25:2956–2966.
Ishikawa, H., E. Claudio, D. Dambach, C. Ravent�os-Su�arez,
C. Ryan, and R. Bravo. 1998. Chronic inflammation and
susceptibility to bacterial infections in mice lacking the
polypeptide (p)105 precursor (NF-kappaB1) but expressing
p50. J. Exp. Med. 187:985–996.
Ji, L. L., M. C. Gomez-Cabrera, N. Steinhafel, and J. Vina.
2004. Acute exercise activates nuclear factor (NF)-kappaB
signaling pathway in rat skeletal muscle. FASEB J. 18:
1499–1506.
Kramer, H. F., and L. J. Goodyear. 2007. Exercise, MAPK, and
NF-kappaB signaling in skeletal muscle. J. Appl. Physiol.
103:388–395.
Lawrence, T., D. W. Gilroy, P. R. Colville-Nash, and
D. A. Willoughby. 2001. Possible new role for NF-kappaB in
the resolution of inflammation. Nat. Med. 7:1291–1297.
Lawrence, T., D. A. Willoughby, and D. W. Gilroy. 2002.
Anti-inflammatory lipid mediators and insights into the
resolution of inflammation. Nat. Rev. Immunol. 2:787–795.
Lawrence, T., M. Bebien, G. Y. Liu, V. Nizet, and M. Karin.
2005. IKKalpha limits macrophage NF-kappaB activation
and contributes to the resolution of inflammation. Nature
434:1138–1143.
Malm, C., P. Nyberg, M. Engstrom, B. Sjodin, R. Lenkei,
B. Ekblom, et al. 2000. Immunological changes in human
skeletal muscle and blood after eccentric exercise and
multiple biopsies. J. Physiol. 529(Pt 1):243–262.
Markworth, J. F., L. Vella, B. S. Lingard, D. L. Tull,
T. W. Rupasinghe, A. J. Sinclair, et al. 2013. Human
inflammatory and resolving lipid mediator responses to
resistance exercise and ibuprofen treatment. Am. J. Physiol.
Regul. Integr. Comp. Physiol. 305:R1281–R1296.
Markworth, J. F., L. D. Vella, V. C. Figueiredo, and
D. Cameron-Smith. 2014. Ibuprofen treatment blunts early
translational signaling responses in human skeletal muscle
following resistance exercise. J. Appl. Physiol. 117:20–28.
Mayhew, J. L., J. L. Prinster, J. S. Ware, D. L. Zimmer,
J. R. Arabas, and M. G. Bemben. 1995. Muscular endurance
repetitions to predict bench press strength in men of
different training levels. J. Sports Med. Phys. Fitness 35:
108–113.
Mead, R.. 1988. The design of experiments: statistical
principles for practical applications. Cambridge University
Press, New York, NY.
Mikkelsen, U. R., H. Langberg, I. C. Helmark,
D. Skovgaard, L. L. Andersen, M. Kjaer, et al. 2009. Local
NSAID infusion inhibits satellite cell proliferation in
human skeletal muscle after eccentric exercise. J. Appl.
Physiol. 107:1600–1611.
Mourkioti, F., and N. Rosenthal. 2008. NF-jB signaling in
skeletal muscle: prospects for intervention in muscle
diseases. J. Mol. Med. 86:747–759.
2014 | Vol. 2 | Iss. 10 | e12172Page 10
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf of
the American Physiological Society and The Physiological Society.
NF-jB and Resistance Exercise L. Vella et al.

Pahl, H. L. 1999. Activators and target genes of Rel/
NF-kappaB transcription factors. Oncogene 18:6853–6866.
Paulsen, G., U. R. Mikkelsen, T. Raastad, and J. M. Peake.
2012. Leucocytes, cytokines and satellite cells: what role do
they play in muscle damage and regeneration following
eccentric exercise? Exerc. Immunol. Rev. 18:42–97.
Poligone, B., and A. S. Baldwin. 2001. Positive and negative
regulation of NF-kappaB by COX-2: roles of different
prostaglandins. J. Biol. Chem. 276:38658–38664.
Rossi, A., P. Kapahi, G. Natoli, T. Takahashi, Y. Chen,
M. Karin, et al. 2000. Anti-inflammatory cyclopentenone
prostaglandins are direct inhibitors of IkappaB kinase.
Nature 403:103–108.
Saville, D. J. 1990. Multiple comparison procedures: the
practical solution. Am. Stat. 44:174.
Senftleben, U., Y. Cao, G. Xiao, F. R. Greten, G. Kr€ahn,
G. Bonizzi, et al. 2001. Activation by IKKalpha of a second,
evolutionary conserved, NF-kappa B signaling pathway.
Science 293:1495–1499.
Spangenburg, E. E., D. A. Brown, M. S. Johnson, and
R. L. Moore. 2006. Exercise increases SOCS-3 expression in
rat skeletal muscle: potential relationship to IL-6 expression.
J. Physiol. 572:839–848.
Straus, D. S., G. Pascual, M. Li, J. S. Welch, M. Ricote,
C. H. Hsiang, et al. 2000. 15-deoxy-delta
12,14-prostaglandin J2 inhibits multiple steps in the
NF-kappa B signaling pathway. Proc. Natl Acad. Sci. USA
97:4844–4849.
Tantiwong, P., K. Shanmugasundaram, A. Monroy, S. Ghosh,
M. Li, R. A. DeFronzo, et al. 2010. NF-jB activity in muscle
from obese and type 2 diabetic subjects under basal and
exercise-stimulated conditions. Am. J. Physiol. Endocrinol.
Metab. 299:E794–E801.
Trappe, T. A., F. White, C. P. Lambert, D. Cesar,
M. Hellerstein, and W. J. Evans. 2002. Effect of ibuprofen
and acetaminophen on postexercise muscle protein
synthesis. Am. J. Physiol. Endocrinol. Metab. 45:E551.
Trenerry, M. K., K. A. Carey, A. C. Ward, and
D. Cameron-Smith. 2007. STAT3 signaling is activated in
human skeletal muscle following acute resistance exercise.
J. Appl. Physiol. 102:1483–1489.
Urso, M. L.. 2013. Anti-inflammatory interventions and
skeletal muscle injury: benefit or detriment? J. Appl. Physiol.
115: 920–928.
Vella, L., M. K. Caldow, A. E. Larsen, D. Tassoni, P. A. Della
Gatta, P. Gran, et al. 2012. Resistance exercise increases
NF-jB activity in human skeletal muscle. Am. J. Physiol.
Regul. Integr. Comp. Physiol. 302:R667–R673.
Whisenant, M. J., L. B. Panton, W. B. East, and
C. E. Broeder. 2003. Validation of submaximal prediction
equations for the 1 repetition maximum bench press test on
a group of collegiate football players. J. Strength Cond. Res.
17:221–227.
Xin, L., R. D. Hyldahl, S. R. Chipkin, and P. M. Clarkson.
2013. A contralateral repeated bout effect attenuates
induction of NF-?B DNA-binding following eccentric
exercise. J. Appl. Physiol. 116:1473–1478.
Zandi, E., D. M. Rothwarf, M. Delhase, M. Hayakawa, and
M. Karin. 1997. The IkappaB kinase complex (IKK)
contains two kinase subunits, IKKalpha and IKKbeta,
necessary for IkappaB phosphorylation and NF-kappaB
activation. Cell 91:243–252.
ª 2014 The Authors. Physiological Reports published by Wiley Periodicals, Inc. on behalf ofthe American Physiological Society and The Physiological Society.
2014 | Vol. 2 | Iss. 10 | e12172Page 11
L. Vella et al. NF-jB and Resistance Exercise

145
Appendix C:
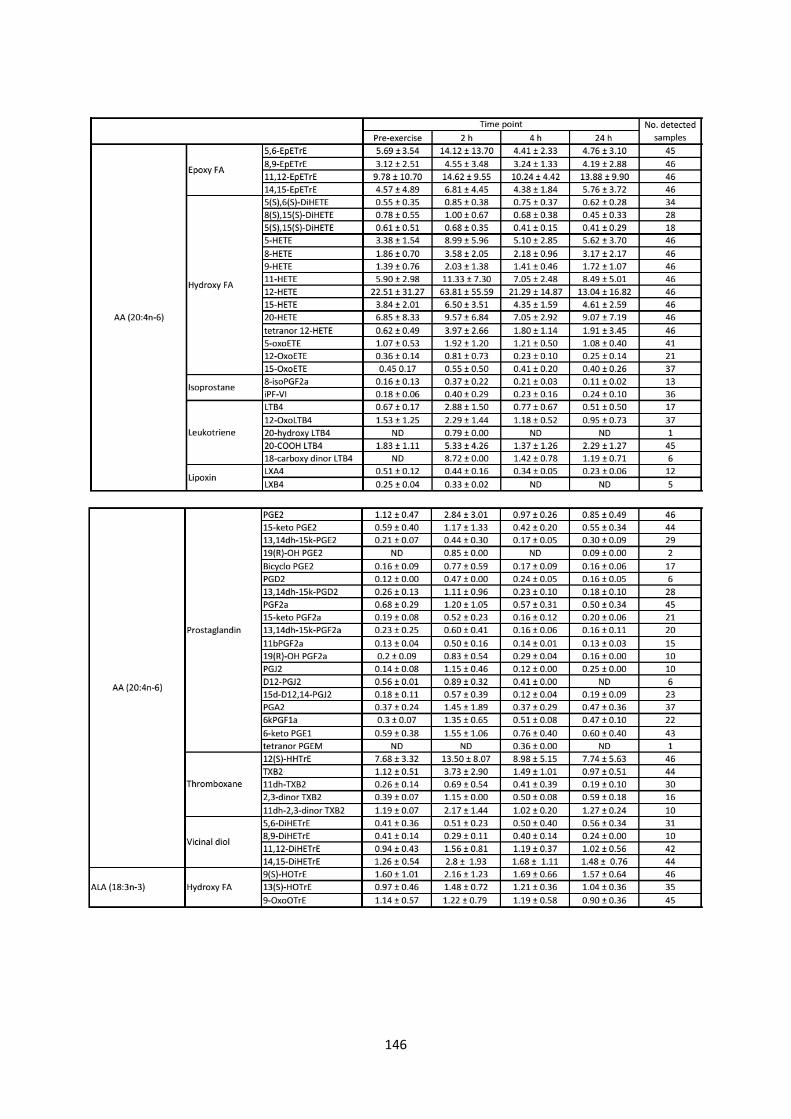
146

147


















