
Forecasting two-photon absorption based on one-photon properties

Chemical Physics Letters 390 (2004) 433–439
www.elsevier.com/locate/cplett
Two-photon absorption properties of star-shapedmolecules containing peripheral diarylthienylamines
Sean Liu a, Kuen Shen Lin a, Victor M. Churikov a, Yi Zhen Su b,Jiann T’suen Lin b, Tzer-Hsiang Huang c, Chia Chen Hsu d,*
a Department of Physics, National Chung Cheng University, Ming Hsiung, Chia-Yi 621, Taiwanb Institute of Chemistry, Academia Sinica, Taipei 11529, Taiwan
c Department of Electronic Engineering, Wu Feng Institute of Technology, Ming Hsiung, Chia-Yi 621, Taiwand Department of Physics and Graduate Institute of Opto-Mechatronics, National Chung Cheng University, Ming Hsiung, Chia-Yi 621, Taiwan
Received 24 September 2003; in final form 5 March 2004
Available online 6 May 2004
Abstract
Two-photon absorption (TPA) properties of several novel donor–acceptor–donor (D–A–D) quadrupolar and octupolar star-
shaped molecules containing peripheral diarylthienylamines were investigated and are discussed in this Letter. The effects of donor–
acceptor strength, conjugation length, and molecular symmetry on the effective TPA cross-section of these novel molecules were
studied. It was found that incorporation of a triazine acceptor center in octupolar molecules can effectively enhance TPA response.
This provides a strategy for designing novel octupolar molecules with a large TPA response but without red-shifting their linear
absorption wavelengths.
� 2004 Published by Elsevier B.V.
1. Introduction
Research on two-photon absorption (TPA) prop-
erties of organic chromophores has become important
recently due to applications in optical power limiting
[1–4], three-dimensional optical data storage [5–7],
two-photon fluorescence microscopy [8–10], micro ornano structure fabrication [11,12], upconversion
[13,14], etc. These applications use two key aspects of
TPA: (1) the electronic transition from ground to
excited states takes place when a pair of photons with
photon energy that is about half of the transition
energy is absorbed, and (2) absorption is proportional
to the square of the incident intensity [15]. The former
aspect of TPA provides improved penetration into a
* Corresponding author. Present address: Department of Economics,
National Central University, Chung Li 32054, Taiwan. Fax: +886-3-
4222876.
E-mail address: [email protected] (C.C. Hsu).
0009-2614/$ - see front matter � 2004 Published by Elsevier B.V.
doi:10.1016/j.cplett.2004.03.050
sample, and the latter aspect allows a high degree of
spatial selectivity in three dimensions when chromo-
phores are excited by the use of a tightly focused laser
beam [16].
Unfortunately, TPA responses of most known or-
ganic chromophores are too small to allow the im-
plementation of the above-mentioned applications. Itis thus necessary to design and synthesize organic
molecules with highly efficient TPA. Knowledge of the
relationship between molecular structure and TPA
cross-section r2 can provide a guideline for this design
and synthesis. Previous research results showed that
some quadrupolar molecules with conjugated p-elec-tron donor–acceptor–donor (D–A–D) or acceptor–
donor–acceptor (A–D–A) structure possess very larger2, due mainly to their centrosymmetric charge re-
distribution [16–18].
Recently, TPA properties of molecules with octu-
polar and multi-branched structures have attracted
considerable attention. Prasad et al. [19] showed that
multi-branched structures exhibit large cooperative

434 S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439
enhancement of TPA in comparison to its one-bran-
ched counterpart. Cho et al. [20–22] investigated the
TPA properties of 1,3,5-tricyano-2,4,6-tris(styryl)ben-
zene derivatives with octupolar structure. They found
that some of these molecules have very large r2, andthe maximum value of r2 increases as the donor
strength and conjugation length increase. A linear
relationship was observed between r2 and the first
hyperpolarizability b. This r2–b relationship served as
a useful synthetic strategy for the design of novel TPA
dyes with octupolar structure [20–22].
In this Letter, we report the TPA properties of several
novel D–A–D quadrupolar and octupolar star-shapedmolecules containing peripheral diarylthienylamines.
The effects of donor–acceptor strength, conjugation
length, and molecular symmetry on TPA properties
were studied and are discussed here. Fig. 1 shows the
molecular structures of the molecules studied in this
work.
Due to theweak absorption of the incident radiation, it
is very difficult to determine r2 from measurement of adirect transmission. Two-photon absorption induced
(a)
(c)
(e)
P2SS2Ph
S
S S
S
S
S
NPh2
NPh2
Ph2N
Ph2N
S
NPh2
SNPh2SPh2N
NN N
S
S
SNPh(Tol)
NPh(Tol)
(Tol)PhN TRZ-Ph-Tol
P3SPh
Fig. 1. Structures of the star-shaped molecules studied in this work: (a) P2SS
(e) TRZ-Ph-Tol are octupolar, and (f) Rhodamine B is the reference sample
fluorescence (TPF) measurement is an alternative ap-
proach to the determination of r2. In theory, the time-
averaged TPF flux density hF ðtÞi measured by an ideal
detecting systemwith uniform spectrum-response is given
by [23]
F ðtÞh i � ð1=2Þgj/Cr2
8n P ðtÞh i2
pk
¼Z 1
0
h~F ðt;xÞidx; ð1Þ
where j is the fluorescence collection factor for the ideal
detecting system. Ideally, j should remain constant for
all samples when the same experimental configuration is
employed. g ¼ hI20 ðtÞi=hI0ðtÞi2is the normalized inten-
sity–intensity autocorrelation of the laser used. / is the
fluorescence quantum efficiency. C, n, and k are the
sample’s concentration, refractive index, and excitationwavelength, respectively. hP ðtÞi is the time-averaged
excitation power. h~F ðt;xÞi is the time-averaged TPF flux
density emitted from molecules at frequency x.In most experiments, a detecting system with a non-
uniform spectrum-response is used. Therefore, the time-
(b)
(d)
(f)
P2SS2PhNp
SS S
S
S NPhNp
NPhNp
NpPhN
S
NpPhN
S
S
S
SS
S
S
S
S
NPh2
Ph2N
NPh2
Ph2N
NPh2
NPh2
COH
O
O N+−CH2CH3CH3CH
2−N
CH2CH
3CH
2CH
3
P3SS2Ph
Rhodamine B
2Ph and (b) P2SS2PhNp are quadrupolar, (c) P3SPh, (d) P3SS2Ph and
.

eoctupolarmolecules(P3SPh,P3SS2Ph,TRZ-Ph-Tol)andthereference
molecule
RhodamineB
SS2PhNp
(c)P3SPh
(d)P3SS2Ph
(e)TRZ-Ph-Tol
(f)RhodamineB
373
432
424
545
438
547
552
572
0.493
0.603
0.678
0.107
740
862
825
813
120
170
2300
160
0.082
0.075
0.059
0.7
12
7.5
5.3
5.3
ence’and‘two-photonabsorption’.Stokes’shiftDEisdefined
asE(k
ð1Þ
max)–E(D
OPF
max).
fphotons.
ismeasured.
S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439 435
averaged TPF flux density hF 0ðtÞi measured in an ex-
periment should be expressed as
F 0ðtÞ� �
� ð1=2Þgj0/Cr2
8n P ðtÞh i2
pk
¼Z 1
0
h~F ðt;xÞiRðxÞdx; ð2Þ
where j0 is the fluorescence collection factor for the real
experimental system and RðxÞ is the spectrum-responsefunction of the detecting system. A spectrum-response
correction factor f between the ideal and real experi-
mental systems is defined as
f � jj0 ¼
hF ðtÞihF 0ðtÞi ¼
R10h~F ðt;xÞidxR1
0h~F ðt;xÞiRðxÞdx
; ð3Þ
where f can be different for different samples because
they may have different h~F ðt;xÞi distributions. From
Eq. (2), we determine r2 of the sample studied bycomparing hF 0ðtÞi of the sample with that of the [24,25].
If hF 0ðtÞi of both the sample studied and the reference
are measured in identical experimental conditions,
8n=pk, g, and hP ðtÞi2 in Eq. (2) can be cancelled, leading
to
r2s ¼/rCrnrfshF 0ðtÞis/sCsnsfrhF 0ðtÞir
r2r; ð4Þ
where the subscripts s and r refer to the sample studied
and the reference, respectively. Once we know the valuesof all the physical parameters on the right hand side of
Eq. (4), the TPA cross-section r2s of the sample studied
can be determined.
Table
1
Spectroscopic
properties
ofthequadrupolarmolecules(P2SS2Ph,P2SS2PhNp),th
Compound
(a)P2SS2Ph
(b)P2
Linearabsorptionpeak,kð
1Þ
max(nm)
437
440
OPFspectralpeak,kO
PF
max(nm)
548
553
Stokes’shiftDE(eV)a
0.575
0.576
TPA
cross-sectionpeak,kð
2Þ
max(nm)
807
804
Effectivemaxim
alTPA
cross-section,b
r2;eff;m
ax(�
10�50cm
4s/#)
340
360
Fluorescence
quantum
efficiency,/
0.054
0.052
Excitationphotonfluxdensity
c
(�1027photoncm
�2s�
1)
5.3
5.3
aOPFandTPA
stand,respectively,for‘one-photonabsorptioninducedfluoresc
bThedata
listed
pertain
tothemaxim
alcross-section.#
standsforthenumber
ocExcitationphotonfluxdensity
isgiven
forthewavelength
atwhichther2;eff;m
ax
2. The experiment
Synthesis of P3SPh: The compound was synthesizedfrom 5-(N,N-diphenylamino)-2-(tributylstannyl)thioph-
ene, 1,3,5-tribromobenzene, and PdCl2(PPh3)2 follow-
ing the procedure described in an earlier report [26].
The pale yellow products were isolated with a 65%
yield.
Synthesis of P3SS2Ph: Orange powdery P3SS2Ph was
synthesized (37% yield) by the same procedure used for
P3SPh except that N5,N 5,N500 ,N500-tetraphenyl-50-tribu-tylstannyl-[2,20;30,200]terthiophene-5, 500-diamine [27] was
used instead of 5-(N,N-diphenylamino)-2-(tributylstan-
nyl)thiophene. The compounds P2SS2Ph and P2SS2-
PhNp were synthesized as described in [27].
Synthesis of TRZ-Ph-Tol: Yellow-orange powdery
TRZ-Ph-Tol was synthesized (89% yield) from 2,4,6-
tris-(4-bromo-phenyl)-[1,3,5]triazine, 5-(N,N-(m-tolyl)
phenylamino)-2-(tributylstannyl)thiophene [27], andPdCl2(PPh3)2 by a procedure similar to P3SPh. All the
sample molecular structures were identified by the NMR
technique.

436 S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439
The molecules from the samples studied were dis-
solved in tetrahydrofuran (THF) solvent, whereas the
reference Rhodamine B molecules were dissolved in
methanol. The concentration of the studied and refer-
ence samples was about 1:0� 1016 and 7:5� 1014 cm�3,respectively. All the sample solutions were passed
through a 0.22 lm filter to eliminate the indissoluble
particles, and the concentrations of all the sample so-
lutions remained unchanged after filtration.
An optical parametric oscillation (OPO) laser (Con-
tinuum Mirage 500) with a 10 ns pulse width and a 10
Hz repetition rate was used as the light source in the
experiment. The excitation light was focused into thecenter of a cylindrical glass sample cell, and the TPF was
collected by a photo-multiplier tube (PMT, Hamamatsu
R928) in the direction perpendicular to the propagation
direction of the excitation beam. For the results shown
in Fig. 3 and r2;eff ;max in Table 1, a broad band-pass
filter instead of a monochromator was used in front of
the PMT so that all the TPF signals from the sample in
the wavelength range 400–700 nm were collected. InEq. (3) the denominator
R10h~F ðt;xÞiRðxÞdx was ob-
tained directly from the experiment, and the numerator
0.0
0.5
1.0
0
0
1
0.0
0.5
1.0
1.5
0
0
1
1
0.0
0.5
1.0
1.5
2.0
0
0
1
2
4
6
8
300 400 500 600 700 800
300 400 500 600 700 800
2
4
6
300 400 500 600 700 800
2
4
6
P3SPh
ab
sorb
ance
(A
.U.)
TRZ-Ph-Tol
P2SS2Ph
wavelengt
Fig. 2. Linear absorption spectra (solid lines) and two-photon fluorescence (T
were detected with a monochromator/PMT system with a spectral resolution
R10h~F ðt;xÞidx was determined by removing the con-
tribution of the spectrum-response function RðxÞ of thedetecting system from the TPF spectra of all the samples
in Fig. 2. The spectrum-response function RðxÞ of the
detecting system was acquired from the manufacturers’data sheets.
The TPA cross-sections r2s of the samples were de-
termined from Eq. (4) based on the values of r2r and
/r(/r ¼ 0:7) of Rhodamine B reported in the literature
[23,28] and the refractive indices ns ¼ 1:407 and
nr ¼ 1:328. Here, we assume that TPF and one-photon
absorption induced fluorescence (OPF) have the identi-
cal fluorescence quantum efficiency, which remainsconstant in the excitation wavelength range used in this
work. Support for this assumption is given below.
3. Results and discussions
Linear absorption spectra of all samples studied are
shown, along with the TPF spectra caused by the831 nm radiation, in Fig. 2. The TPF spectra of all the
samples, except that of P3SS2Ph, remain unchanged as
.0
.5
.0
.0
.5
.0
.5
.0
.5
.0
2
4
6
2
4
6
300 400 500 600 700 800
300 400 500 600 700 800
300 400 500 600 700 800
2
4
6
8
P2SS2PhNp
P3SS2Ph
Rhodamine B
inten
sity (A.U
.)
h (nm)
PF) spectra (dotted solid lines) of the molecules studied. TPF spectra
of 10 nm.
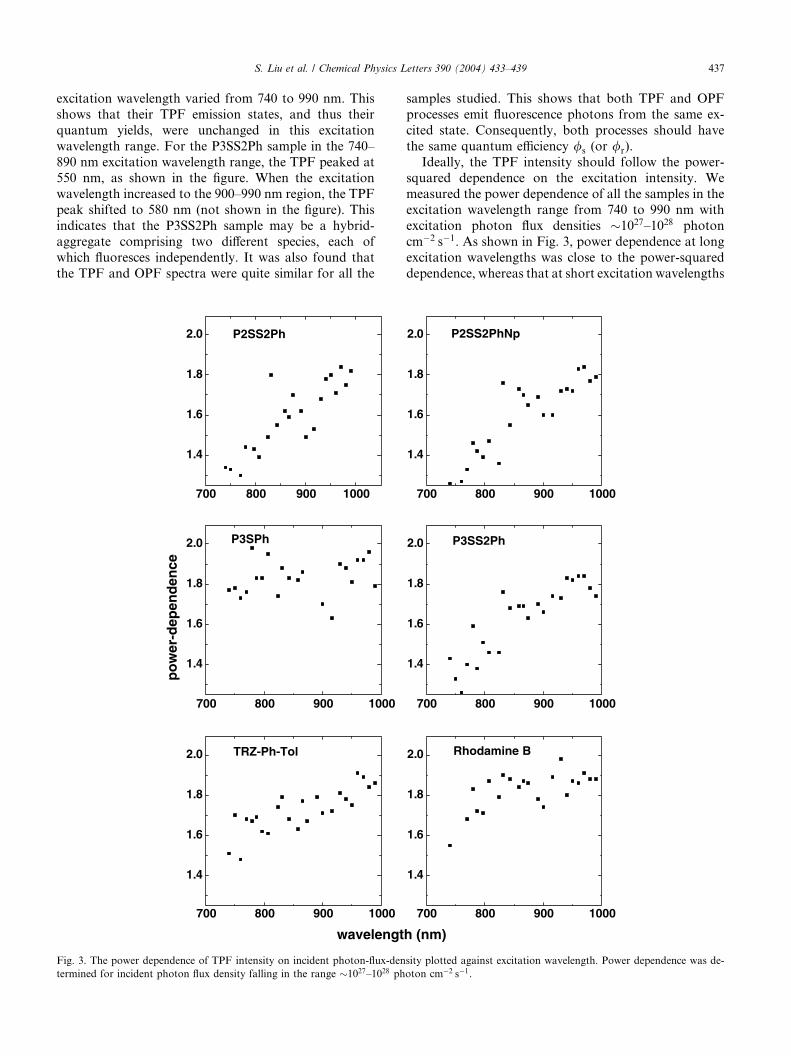
S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439 437
excitation wavelength varied from 740 to 990 nm. This
shows that their TPF emission states, and thus their
quantum yields, were unchanged in this excitation
wavelength range. For the P3SS2Ph sample in the 740–
890 nm excitation wavelength range, the TPF peaked at550 nm, as shown in the figure. When the excitation
wavelength increased to the 900–990 nm region, the TPF
peak shifted to 580 nm (not shown in the figure). This
indicates that the P3SS2Ph sample may be a hybrid-
aggregate comprising two different species, each of
which fluoresces independently. It was also found that
the TPF and OPF spectra were quite similar for all the
700 800 900 1000
1.4
1.6
1.8
2.0
700 800 900 1000
700 800 900 1000
1.4
1.6
1.8
2.0
1.4
1.6
1.8
2.0
P2SS2Ph
TRZ-Ph-Tol
wavelengt
po
wer
-dep
end
ence
P3SPh
Fig. 3. The power dependence of TPF intensity on incident photon-flux-den
termined for incident photon flux density falling in the range �1027–1028 ph
samples studied. This shows that both TPF and OPF
processes emit fluorescence photons from the same ex-
cited state. Consequently, both processes should have
the same quantum efficiency /s (or /r).
Ideally, the TPF intensity should follow the power-squared dependence on the excitation intensity. We
measured the power dependence of all the samples in the
excitation wavelength range from 740 to 990 nm with
excitation photon flux densities �1027–1028 photon
cm�2 s�1. As shown in Fig. 3, power dependence at long
excitation wavelengths was close to the power-squared
dependence, whereas that at short excitation wavelengths
700 800 900 1000
700 800 900 1000
700 800 900 1000
1.4
1.6
1.8
2.0
1.4
1.6
1.8
2.0
1.4
1.6
1.8
2.0 Rhodamine B
P3SS2Ph
P2SS2PhNp
h (nm)
sity plotted against excitation wavelength. Power dependence was de-
oton cm�2 s�1.

438 S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439
deviated more from the power-squared dependence. It is
possible that higher excited states were reached with
shorter excitation wavelength (higher photon energy).
As a result, extra non-fluorescent energy dissipation
processes, such as excited-state absorption, intersystemcrossing, and stimulated emission, involved. These dis-
sipation processes competed with TPF (i.e., use up in-
cident photon energy without producing TPF) and
hence resulted in deviation from the power-squared
dependence.
Thus, the r2 values determined in this work cannot
fully represent the true TPA property of the molecules
studied. Nonetheless, it is useful to introduce the effec-tive TPA cross-section r2;eff to qualitatively describe the
TPA response of the samples. r2;eff depends critically on
the excitation conditions used and the energy dissipation
processes involved. It will be equal to r2 only when no
non-TPF energy dissipation processes are involved. As
suggested in Fig. 3, the power dependence of the P3SPh
molecule does not follow the general trend of a decrease
in power dependence with the decrease of the excitationwavelength. This may be because the transition energy
of P3SPh molecules is higher than twice the excitation
photon energy used in Fig. 3. Therefore, the above-
mentioned non-TPF energy dissipation processes are
insignificant under short wavelength excitations shown
in Fig. 3.
The results we obtained for all samples are summa-
rized in Table 1. The fluorescence quantum efficiencies /listed in the table were determined by the OPF tech-
nique. The errors of the r2;eff ;max values are approxi-
mately 20%.
We first look at the effect of conjugation length and
electron donation. As shown in Table 1, the effective
maximal TPA cross-section r2;eff ;max and Stokes’ shift
DE of the octupolar P3SS2Ph were larger than those of
octupolar P3SPh, which has a less extended conjugationlength than that of P3SS2Ph. The change of peripheral
electron donor from NPh2 in the quadrupolar P2SS2Ph
to NPhNp in the quadrupolar P2SS2PhNp resulted in a
small increase in r2;eff ;max. Since the difference is within
the 20% experimental uncertainty range, we cannot as-
certain the trend of the r2;eff ;max increase solely from the
present measurement.
Next, let us examine the effect of changing the electronacceptor center from benzene to triazine on TPA re-
sponse. Comparing the octupolar star-shaped molecules
P3SS2Ph (with a benzene at the center) and TRZ-Ph-Tol
(with a triazine at the center), we found that the latter had
a DE that was slightly larger, a r2;eff ;max that was more
than ten times larger, and a kð1Þmax that was not red-shifted,
as is usually the case. Since the molecule TRZ-Ph-Tol has
a strong central acceptor, triazine, this comparison hintsat a plausible strategy for designing novel octupolar
molecules with a large TPA response but without red-
shifting their linear absorption wavelength.
As shown in Table 1, the peak wavelength positions
of the effective TPA cross-section kð2Þmax of the star-
shaped octupolar molecules are generally twice their
linear absorption peak positions kð1Þmax. This indicates
that in these non-centrosymmetric structures both one-and two-photon transitions to the same electronic
state are allowed [20]. On the contrary, the kð2Þmax of the
star-shaped quadrupolar molecules are relatively
smaller than twice their linear absorption peaks kð1Þmax.
This means the final state of TPA in these quadru-
polar molecules is higher than the zero-vibration level
of the first excited electronic state, which is the final
state of one-photon absorption, because of their cen-trosymmetric structures [17].
4. Summary
Using a tunable wavelength OPO laser, we investi-
gated the TPA properties of several novel D–A–D
quadrupolar and octupolar star-shaped molecules con-
taining peripheral diarylthienylamines. Experimental
results showed that the use of a triazine electron–acceptorcenter in star-shaped octupolar molecules provides an
effective way to enhance TPA efficiency without in-
creasing the absorption peak wavelength. The locations
of the one- and the two-photon states were found to be
related to inversion symmetry: in non-centrosymmetric
octupolar molecules both one- and two-photon transi-
tions to the same electronic state were allowed, but in
centrosymmetric quadrupolar molecules the two-photonstate was above the lowest one-photon state.
References
[1] A.A. Said, C. Wamsley, D.J. Hagan, E.W. Van Stryland, B.A.
Reinhardt, P. Roderer, A.G. Dillard, Chem. Phys. Lett. 228
(1994) 646.
[2] G.S. He, R. Gvishi, P.N. Prasad, B. Reindardt, Opt. Commun.
117 (1995) 133.
[3] J.W. Perry, K. Mansour, I.-Y.S. Lee, X.-L. Wu, P.V. Bedworth,
C.-T. Chen, D. Ng, S.R. Marder, P. Miles, T. Wada, M. Tian, H.
Sasabe, Science 273 (1996) 1533.
[4] J.E. Ehrlich, X.L. Wu, I.-Y.S. Lee, Z.-H. Hu, H. R€ockel, S.R.
Marder, J.W. Perry, Opt. Lett. 22 (1997) 1843.
[5] D.A. Parthenopoulos, P.M. Rentzepis, Science 245 (1989) 843.
[6] J.H. Strikler, W.W. Webb, Opt. Lett. 16 (1991) 1780.
[7] B.H. Cumpston, S.P. Ananthavel, S. Barlow, D.L. Dyer, J.E.
Ehrlich, L.L. Erskine, A.A. Heikal, S.M. Kuebler, I.-Y.S. Lee, D.
McCord-Maughon, J. Qin, H. R€ockel, M. Rumi, X.-L. Wu, S.R.
Marder, J.W. Perry, Nature 398 (1999) 51.
[8] J.D. Bhawalkar, J. Swiatkiewicz, P.N. Prasad, S.J. Pan, A. Shin,
J.K. Samarabandu, P.C. Cheng, B.A. Reinhardt, Polymer 38
(1997) 4551.
[9] D.W. Piston, Trends Cell Biol. 9 (1999) 66.
[10] W. Denk, J.H. Strickler, W.W. Webb, Science 248 (1990) 73.
[11] I. Wang, M. Bouriau, P.L. Baldeck, C. Martineau, C. Andraud,
Opt. Lett. 27 (2002) 1348.

S. Liu et al. / Chemical Physics Letters 390 (2004) 433–439 439
[12] S. Kawata, H.-B. Sun, T. Tanaka, K. Takada, Nature 412 (2001)
697.
[13] A. Mukherjee, Appl. Phys. Lett. 62 (1993) 3423.
[14] J.D. Bhawalkar, G.S. He, C.-K. Park, C.F. Zhao, G. Ruland,
P.N. Prasad, Opt. Commun. 124 (1996) 33.
[15] Z. Sekkat, H. Ishitobi, S. Kawata, Opt. Commun. 222 (2003)
269.
[16] M. Albota, D. Beljonne, J.-L. Bredas, J.E. Ehrlich, J.-Y. Fu, A.A.
Heikal, S.E. Hess, T. Kogej, M.D. Levin, S.R. Marder, D.
McCord-Maughon, J.W. Perry, H. R€ockel, M. Rumi, G. Subr-
amaniam, W.W. Webb, X.-L. Wu, C. Wu, Science 281 (1998)
1653.
[17] W.H. Lee, M. Cho, S.-J. Jeon, B.R. Cho, J. Phys. Chem. A 104
(2000) 11033.
[18] B.A. Reinhardt, L.L. Brott, S.J. Clarson, A.G. Dillard, J.C. Bhatt,
R. Kannan, L. Yuan, G.S. He, P.N. Prasad, Chem. Mater. 10
(1998) 1863.
[19] S.-J. Chung, K.-S. Kim, T.-C. Lin, G.S. He, J. Swiatkiewicz, P.N.
Prasad, J. Phys. Chem. B 103 (1999) 10741.
[20] B.R. Cho, K.H. Son, S.H. Lee, Y.-S. Song, Y.-K. Lee, S.-J. Jeon,
J.H. Choi, H. Lee, M. Cho, J. Am. Chem. Soc. 123 (2001) 10039.
[21] B.R. Cho, M.J. Piao, K.H. Son, S.H. Lee, S.J. Yoon, S.-J. Jeon,
M. Cho, Chem. Eur. J. 8 (2002) 3907.
[22] W.-H. Lee, H. Lee, J.-A. Kim, J.-H. Choi, M. Cho, S.J. Jeon, B.R.
Cho, J. Am. Chem. Soc. 123 (2001) 10658.
[23] C. Xu, W.W. Webb, J. Opt. Soc. Am. B 13 (1996) 481.
[24] D.A. Oulianov, I.V. Tomov, A.S. Dvornikov, P.M. Rentzepis,
Opt. Commun. 191 (2001) 235.
[25] A. Fischer, C. Cremer, E.H.K. Stelzer, Appl. Opt. 34 (1995) 1989.
[26] I.-Y. Wu, J.T. Lin, Y.-T. Tao, E. Balasubramaniam, E. Adv.
Mater. 12 (2000) 668.
[27] Y.Z. Su, J.T. Lin, Y.-T. Tao, C.-W. Ko, S.-C. Lin, S.-S. Sun,
Chem. Mater. 14 (2002) 1884.
[28] J.N. Demas, G.A. Crosby, J. Phys. Chem. 75 (1971) 991.


















