
Tuning peptide self-assembly by an in-tether chiral...
9
MATERIALS SCIENCE Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC). Tuning peptide self-assembly by an in-tether chiral center Kuan Hu, 1,2 * Yixiang Jiang, 1 * Wei Xiong, 3 Hu Li, 4 Pei-Yu Zhang, 5 Feng Yin, 1 Qianling Zhang, 6 Hao Geng, 1 Fan Jiang, 1† Zhou Li, 2† Xinwei Wang, 3† Zigang Li 1† The self-assembly of peptides into ordered nanostructures is important for understanding both peptide molec- ular interactions and nanotechnological applications. However, because of the complexity and various self- assembling pathways of peptide molecules, design of self-assembling helical peptides with high controllability and tunability is challenging. We report a new self-assembling mode that uses in-tether chiral center-induced helical peptides as a platform for tunable peptide self-assembly with good controllability. It was found that self-assembling behavior was governed by in-tether substitutional groups, where chirality determined the forma- tion of helical structures and aromaticity provided the driving force for self-assembly. Both factors were essential for peptide self-assembly to occur. Experiments and theoretical calculations indicate long-range crystal-like pack- ing in the self-assembly, which was stabilized by a synergy of interpeptide p-p and p-sulfur interactions and hydrogen bond networks. In addition, the self-assembled peptide nanomaterials were demonstrated to be promising candidate materials for applications in biocompatible electrochemical supercapacitors. INTRODUCTION Molecular self-assembly of short peptides into well-ordered nanostruc- tures is a cornerstone of advanced bionanomaterial fabrication (1–3). It has been used in a variety of applications, including in biomedical (4, 5), optical engineering (6), and electronic nanotechnology applications (7). Peptide self-assembly is complicated, because peptides display signifi- cant variation in their sequences as well as secondary structures (8). As- semblies of amphiphilic peptides (9), b sheets (10), collagens (11), and diphenylalanine peptides and their analogs (12, 13) have been studied previously. This list could be extended to include specially designed planar cyclic-D, L-peptides (14) and foldectures (15), which are self-assembled molecular architectures of b-peptide foldamers. However, these peptide self-assemblies are highly sequence-dependent and susceptible to sub- tle changes in primary structure. Therefore, the de novo design of tun- able and controllable peptide self-assembly remains challenging. Helices are the basic units of protein secondary structures, and more than 60% of protein-protein interactions involve a helices at their interface (16). In this context, efforts have been made toward engineer- ing helical peptide self-assemblies. Recently, self-assemblies of specially designed short linear a-helical peptides were demonstrated to form hol- low vesicles (17) or two-dimensional nanostructures (18). Gazit et al. reported a constrained single heptad repeat peptide that could form functional superhelical assemblies (19). However, linear peptides are highly flexible, and their secondary structures can be significantly inter- fered with by subtle perturbations (20). Recently, a head-to-tail cyclized helical short peptide was reported to self-assemble through the incor- poration of hydrophobic residues (21). However, head-to-tail cyclization of helical peptides is sequence-dependent, thus limiting the residue scope for assembly. By contrast, side-chain–to–side-chain cross-linked helical peptides have more conformational stability than their linear analogs (22, 23). Therefore, we hypothesized that constrained helical peptides are an ide- al platform for tunable peptide self-assembly that could minimize environmental perturbations and allow further modifications for fine- tuning the self-assembly for various purposes. We recently demon- strated a precisely positioned in-tether chiral center could induce the backbone peptides into a helical conformation with a preferable abso- lute configuration generating chirality-induced helical (CIH) peptides, as shown in Fig. 1A (24–26). We hypothesized that tuning the on-tether substitution group would be a de novo avenue of helical peptide as- sembly. We envisioned an aromatic substituent that provided p-p intermolecular interactions would serve as a driving force for pep- tides to self-assemble. Here, as proof of concept, we demonstrate that CIH peptides assem- ble into different nanostructures. As shown in Fig. 1A, self-assembling behavior was dominated by the peptide helicity and in-tether func- tional groups (X). Meanwhile, detailed nanostructures were formed in a sequence-dependent manner, providing significant chemical space for structural and functional tuning. Aromatic phenyl and naphthyl sub- stituents with suitable chirality (R epimers for the helical peptides) facili- tated self-assembly. Their nonhelical epimers (S epimers) with aromatic substituents or helical peptides with nonaromatic substituents could not assemble. De novo assembly was demonstrated by the assembly behavior of CIH peptides with different backbone sequences. Notably, the self- assembled bionanomaterials are promising for use in biocompatible electrochemical supercapacitors. RESULTS Exploration of stapled peptide self-assembly Cyclic pentapeptides of Ac-CAAAS 5 (X)-NH 2 (BDCPs; Fig. 1A) were chosen as model peptides to investigate self-assembling behavior. These BDCP peptides did not contain amino acid residues with significant hydrophobic interaction or hydrogen bonding features, but varied in 1 Key Laboratory of Chemical Genomics, School of Chemical Biology and Biotech- nology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. 2 Beijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences, Beijing 100083, China. 3 School of Advanced Materials, Peking University Shenzhen Graduate School, Shenzhen 518055, China. 4 Key Laboratory for Biomechanics and Mechanobiology of the Ministry of Education, School of Biological Science and Medical Engineering, Beihang University, Beijing 100083, China. 5 XtalPi Inc., One Broadway, 9th floor, Cambridge, MA 02142, USA. 6 Shenzhen Key Laboratory of Func- tional Polymer, College of Chemistry and Environmental Engineering, Shenzhen Uni- versity, Shenzhen 518060, China. *These authors contributed equally to this work. †Corresponding author. Email: [email protected] (Zigang L.); wangxw@pkusz. edu.cn (X.W.); [email protected] (Zhou L.); [email protected] (F.J.) SCIENCE ADVANCES | RESEARCH ARTICLE Hu et al., Sci. Adv. 2018; 4 : eaar5907 11 May 2018 1 of 8 on July 14, 2018 http://advances.sciencemag.org/ Downloaded from
Transcript of Tuning peptide self-assembly by an in-tether chiral...

SC I ENCE ADVANCES | R E S EARCH ART I C L E
MATER IALS SC I ENCE
1Key Laboratory of Chemical Genomics, School of Chemical Biology and Biotech-nology, Peking University Shenzhen Graduate School, Shenzhen 518055, China.2Beijing Institute of Nanoenergy and Nanosystems, Chinese Academy of Sciences,Beijing 100083, China. 3School of Advanced Materials, Peking University ShenzhenGraduate School, Shenzhen 518055, China. 4Key Laboratory for Biomechanics andMechanobiology of the Ministry of Education, School of Biological Science andMedical Engineering, Beihang University, Beijing 100083, China. 5XtalPi Inc., OneBroadway, 9th floor, Cambridge, MA 02142, USA. 6Shenzhen Key Laboratory of Func-tional Polymer, College of Chemistry and Environmental Engineering, Shenzhen Uni-versity, Shenzhen 518060, China.*These authors contributed equally to this work.†Corresponding author. Email: [email protected] (Zigang L.); [email protected] (X.W.); [email protected] (Zhou L.); [email protected] (F.J.)
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
Copyright © 2018
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim to
originalU.S. Government
Works. Distributed
under a Creative
Commons Attribution
NonCommercial
License 4.0 (CC BY-NC).
Dow
n
Tuning peptide self-assembly by an in-tetherchiral centerKuan Hu,1,2* Yixiang Jiang,1* Wei Xiong,3 Hu Li,4 Pei-Yu Zhang,5 Feng Yin,1 Qianling Zhang,6
Hao Geng,1 Fan Jiang,1† Zhou Li,2† Xinwei Wang,3† Zigang Li1†
The self-assembly of peptides into ordered nanostructures is important for understanding both peptide molec-ular interactions and nanotechnological applications. However, because of the complexity and various self-assembling pathways of peptide molecules, design of self-assembling helical peptides with high controllabilityand tunability is challenging. We report a new self-assembling mode that uses in-tether chiral center-inducedhelical peptides as a platform for tunable peptide self-assembly with good controllability. It was found thatself-assembling behavior was governed by in-tether substitutional groups, where chirality determined the forma-tion of helical structures and aromaticity provided the driving force for self-assembly. Both factors were essentialfor peptide self-assembly to occur. Experiments and theoretical calculations indicate long-range crystal-like pack-ing in the self-assembly, which was stabilized by a synergy of interpeptide p-p and p-sulfur interactions and hydrogenbond networks. In addition, the self-assembled peptide nanomaterials were demonstrated to be promising candidatematerials for applications in biocompatible electrochemical supercapacitors.
loa
on July 14, 2018http://advances.sciencemag.org/
ded from
INTRODUCTIONMolecular self-assembly of short peptides into well-ordered nanostruc-tures is a cornerstone of advanced bionanomaterial fabrication (1–3). Ithas been used in a variety of applications, including in biomedical (4, 5),optical engineering (6), and electronic nanotechnology applications (7).Peptide self-assembly is complicated, because peptides display signifi-cant variation in their sequences as well as secondary structures (8). As-semblies of amphiphilic peptides (9), b sheets (10), collagens (11), anddiphenylalanine peptides and their analogs (12, 13) have been studiedpreviously. This list could be extended to include specially designed planarcyclic-D, L-peptides (14) and foldectures (15), which are self-assembledmolecular architectures of b-peptide foldamers. However, these peptideself-assemblies are highly sequence-dependent and susceptible to sub-tle changes in primary structure. Therefore, the de novo design of tun-able and controllable peptide self-assembly remains challenging.
Helices are the basic units of protein secondary structures, andmorethan 60% of protein-protein interactions involve a helices at theirinterface (16). In this context, efforts have been made toward engineer-ing helical peptide self-assemblies. Recently, self-assemblies of speciallydesigned short linear a-helical peptides were demonstrated to formhol-low vesicles (17) or two-dimensional nanostructures (18). Gazit et al.reported a constrained single heptad repeat peptide that could formfunctional superhelical assemblies (19). However, linear peptides arehighly flexible, and their secondary structures can be significantly inter-fered with by subtle perturbations (20). Recently, a head-to-tail cyclizedhelical short peptide was reported to self-assemble through the incor-
poration of hydrophobic residues (21). However, head-to-tail cyclizationof helical peptides is sequence-dependent, thus limiting the residue scopefor assembly.
By contrast, side-chain–to–side-chain cross-linked helical peptideshave more conformational stability than their linear analogs (22, 23).Therefore, we hypothesized that constrained helical peptides are an ide-al platform for tunable peptide self-assembly that could minimizeenvironmental perturbations and allow further modifications for fine-tuning the self-assembly for various purposes. We recently demon-strated a precisely positioned in-tether chiral center could induce thebackbone peptides into a helical conformation with a preferable abso-lute configuration generating chirality-induced helical (CIH) peptides,as shown in Fig. 1A (24–26).We hypothesized that tuning the on-tethersubstitution group would be a de novo avenue of helical peptide as-sembly. We envisioned an aromatic substituent that provided p-pintermolecular interactions would serve as a driving force for pep-tides to self-assemble.
Here, as proof of concept, we demonstrate that CIH peptides assem-ble into different nanostructures. As shown in Fig. 1A, self-assemblingbehavior was dominated by the peptide helicity and in-tether func-tional groups (X).Meanwhile, detailed nanostructures were formed in asequence-dependent manner, providing significant chemical space forstructural and functional tuning. Aromatic phenyl and naphthyl sub-stituents with suitable chirality (R epimers for the helical peptides) facili-tated self-assembly. Their nonhelical epimers (S epimers) with aromaticsubstituents or helical peptides with nonaromatic substituents could notassemble. De novo assembly was demonstrated by the assembly behaviorof CIH peptides with different backbone sequences. Notably, the self-assembled bionanomaterials are promising for use in biocompatibleelectrochemical supercapacitors.
RESULTSExploration of stapled peptide self-assemblyCyclic pentapeptides of Ac-CAAAS5(X)-NH2 (BDCPs; Fig. 1A) werechosen asmodel peptides to investigate self-assembling behavior. TheseBDCP peptides did not contain amino acid residues with significanthydrophobic interaction or hydrogen bonding features, but varied in
1 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from
terms of in-tether chiral center substituents. As shown in our previouswork, the helicity of the BDCP peptides was determined by the chiralconfiguration of the in-tether chiral center, where the R configurationwas required for ana-helical structure (24). Here, BDCPpeptides withthree different substituents, whereX =methyl, phenyl, or naphthyl, weresynthesized in two different configurations: R and S. The resulting sixpurified BDCP peptides consisting of three R/S epimer pairs were char-acterized using circular dichroism (CD; fig. S1). It was confirmed that onlythe R-configured BDCP peptides were helical in water, whereas theS-configured epimers were all nonhelical, which is consistent with ourpreviously reportedwork (24, 25). Figure 1B presents the simulated struc-ture of BDCP-1-R, which was first reported in our previous work (24).
Characterization of self-assembly morphologySelf-assembly experiments were performed by placing each type ofBDCP peptide (listed in Fig. 1C) -R separately in water at room tem-perature and then sonicating for 10 min to assist in self-assembly (27).Different peptide concentrations were tested to find the optimal condi-tion for self-assembly. Using BDCP-1-R as an example, the concentra-tion ranged from 0.25 to 8 mg/ml. The peptide became fully dissolvedwhen the concentration was below 1 mg/ml, whereas suspended solidswere visible when the concentration exceeded 2 mg/ml. These sus-pended solids were self-assembled BDCP-1 peptides. Under an opticalmicroscope, they appeared needle-shaped and colorless. Further char-acterization by scanning electron microscopy (SEM) was performedto inspect the morphology of the BDCP-1-R assemblies. Images 1 to4 in Fig. 1D present the microscopic view of the assemblies, whichshow that these solid materials were tetragonal-shaped nanobelts ornanotubes tens of micrometers long and hundreds of nanometers toseveralmicrometers wide. Image 5 is amacroscopic view of the assem-blies and reveals the assemblies had a large aspect ratio that leads tointerweaving of the assemblies.
Conversely, the nonhelical S-configured epimer BDCP-1-S was notable to self-assemble, evenwhen awider range of peptide concentrationswas tested. A similar phenomenon was observed for the naphthyl-substituted BDCP peptide epimers (BDCP-2-R/S), where the helicalBDCP-2-R peptide was able to self-assemble into nanostructures (Fig.1D, image 6), but its nonhelical epimer BDCP-2-S was not. These results
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
indicate that the helical structure was necessary for undergoing self-assembly. Thehelix constrained the peptide into a compact unit inwater,because its peptide chain wasmuch less flexible than those in the random-coil structures of the nonhelical epimers. Additional tests with uncyclizedlinear peptides (that is, BDLP) withmolecular structures that were evenmore flexible further support these findings. None of the substitutedBDLP peptides were able to self-assemble (Fig. 1C).
However, having only an a helix may not be sufficient for self-assembly, and certain functional groups with ample intermolecular in-teractions are needed to drive assembly. The in-tether substituents ofphenyl and naphthyl were critically important for the successful self-assembly of BDCP-1-R and BDCP-2-R, because both aromatic groupswere hydrophobic and facilitated adequate intermolecular p-p interac-tions. These p-p interactions are directional anddrive the aromatic ringsto stack on one another and, therefore, enable the formation of self-assemblies. If the peptide did not contain an aromatic group, then itcould not self-assemble despite its helical content due to a lack of inter-molecularp-p interactions to serve as the driving force for self-assembly,as shown by the BDCP-0-R/S pair in Fig. 1C.
After demonstrating the crucial role of both the helical conformationsand aromatic substituent groups in self-assembly, the self-assemblednanostructures of the BDCP-1-R peptide were characterized in detail.As shown in Fig. 2A, the left part of the BDCP-1-R peptides clearly hada tubular nanostructure (indicated by the yellow box); however, theright part did not have an obvious tubular shape. This phenomenonmay be attributed to the diffusion-limited crystal growth, which was al-so reported by Lee et al. in their short b-peptide foldamer self-assemblysystem (15). On the basis of this mechanism, without any agitation, masstransport on the most rapidly growing face of the crystal is diffusion-limited, resulting in site-dependent growth rate on this crystal face.Diffusion of solute molecules to the outside apexes is easier than that tothe central region. During the crystal growth, the concentration of thesolution at the center of the crystal growth face decreases until it is nolonger supersaturated, leaving only the growth of edges to give tubularstructure (15). We found that the above explanation can explain thecoexistence of nanobelt and nanotubes in our system. The associatedelectron diffraction (Fig. 2B) reveals a pattern of rings, which suggeststhat the peptide nanostructures are polycrystalline. Powder x-ray
Fig. 1. Study of BDCP peptides self-assembly. (A) Molecular structure of cyclic pentapeptides of Ac-CAAAS5(X)-NH2 (BDCPs). (B) Simulated structure of helical BDCP-1-Rpeptide in water. Figure was adapted from our previous article (24). (C) Table of peptides tested for self-assembly in H2O, where “√” and “−” indicate self-assembly wasachieved or not achieved, respectively. [a] S and R represents the absolute configuration of the in-tether chiral center in the peptide epimer, and L denotes the peptideis an uncyclized linear peptide (structural details in the Supplementary Materials). (D) SEM images of self-assembled BDCP-1-R at various magnifications (images 1 to 5)and BDCP-2-R (image 6). Image 4 is a high-magnification SEM image of the region inside the yellow box in image 1, indicating rectangular cross section. Scale bars,1 mm (upper left), 3 mm (upper middle), 500 nm (upper right), 200 nm (lower left), 15 mm (lower middle), and 5 mm (lower right).
2 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from
diffraction (XRD) also revealed sharp diffraction peaks for the nano-materials (Fig. 2C), indicating long-range ordered crystallites. The dif-fraction peakswere located at 3.4, 4.44, 5.04, 8.5, 17, and 24.6 Å in latticespacing, where two were consistent with the transmission electron mi-croscopy (TEM) electron diffraction rings. Atomic force microscopy(AFM) further confirmed the tetragonal shape of the nanostructures,and the average tube height was ~150 nm (Fig. 2D), which is close tothe dimension observed by SEM. We also found peptide nanodots inthe solution. As shown in Fig. 2D (image 3), these nanodots were about10 to 20 nm in diameter. A previous report (28) suggests that the ele-mentary building blocks for self-assembled peptide nanotubes arenanodots, and the assembling process from dots to tubes is reversible.Perhaps, BDCP-1-R assembled in a similar process involving the forma-tion of nanodots. We also detected the reverse, peptide nanostructuredisassembly, upon simply diluting the aqueous solution.
Study of the self-assembly behavior of the assembliesBDCP-1-R nanomaterials were further characterized by Fourier trans-form infrared spectroscopy (FTIR) and Raman spectroscopy. FTIRprovided information on the chemical nature and secondary structuresof the assemblies (29). The spectrum for the assemblies (Fig. 3A) con-tains intense bands for amide I at 1654 cm−1 and amide II at 1530 and1547 cm−1, indicating an a-helical structure (30). This observation wasfurther corroborated by the amide III band at 1308 cm−1.
Moreover, the N-H stretching frequencies at 3319 and 3266 cm−1
correspond to a hydrogen bond network (Fig. 3A). The Raman spec-trum contained a peak at 1652 cm−1, indicating a helical structure forindividual peptides and is consistent with the FTIR spectra (Fig. 3B).Therefore, we demonstrated that helical BDCP-1-R is the buildingblock for nanostructure assembly.
To probe the molecular interactions in the nanostructures, ultra-violet (UV)–vis absorption and fluorescence spectroscopy were per-formed (31). UV-vis spectra (Fig. 3C) contained a broad absorptionand increased intensity for the BDCP-1-R assemblies compared to theBDCP-1-R monomers, indicating strong p-p interactions between thestacked aromatic groups (32). The absorption of the BDCP-1-R mono-mers wasmainly due to the n-p* transitions of the phenyl group, and thephenyl p electrons could interactwith a neighboring phenyl group through
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
p-p interactions in the assembled state. Fluorescence spectroscopy wasused to measure changes in photoluminescence excitation (PLE) andemission after assembly (33). Figure 3D demonstrates that the photo-luminescence (PL) and PLE spectra of BDCP-1-R were very different be-tween the peptide nanomaterials (solid lines) andmonomers (dash lines).
The red shift in the maximum emission wavelength (red solid line)indicates significant electron delocalization, suggesting strong p-p inter-actions in the assembly (31). Fluorescence imaging was performed onthe BDCP-1-R assemblies using standard UV-blue, blue-green, andgreen-red filters to detect emissions (Fig. 3E).When a suitable excitationwavelength was used, the BDCP-1-R assemblies emitted blue, green, orred fluorescence. The broad range exhibited in the PL spectra by thepeptide assemblies indicates they are promising candidates for use inoptical and imaging technologies.
Modeling of crystal packing in the self-assemblyThe observed sharp XRD peaks indicate long-range ordered packing.We performed crystal structure prediction of the BDCP-1-R peptideby searching peptide conformational and crystal packing degrees offreedom, followed by molecular mechanics and subsequent densityfunctional theory calculations. One low-energy structure fitting the pre-dicted XRD pattern was identified (fig. S2). In this structure (Fig. 4A),each peptide had a backbone and side-chain conformation similar tothat in water (Fig. 1B) in agreement with the a-helical structure sug-gested by our FTIR and Raman spectra (Fig. 3, A and B).
The predicted crystallite structure had alternating polar and nonpolarlayers. The nonpolar regions were formed by packing of side-chaintethers in different peptides and driven by aromatic p-p interactions be-tween in-tether phenyl groups and p-sulfur interactions between thephenyl and thioether groups (Fig. 4B). Aromatic side-chain interactionsare frequent in protein crystal structures and lead to strong stabilization(34). Aromatic-sulfur interactions have also been observed in the crystalstructures of many molecules and can help to stabilize peptide helicalfolding (35). The simulation results explainwhyBDCP-0peptides lackingaromatic groups cannot self-assemble (Fig. 1C). The p-p interactions ofphenyl groups can also explain the observed red shift and broadening offluorescence and UV-vis spectra upon self-assembly (Fig. 3, C and D).The peptide backbones also packed together to form polar layers by
Fig. 2. Characterization of BDCP-1-R peptide nanomaterials. TEM image (A), electron diffraction pattern (B), and XRD pattern (C) of BDCP-1-R peptide nanomaterials.The labels contained a star “*” indicate the diffraction peaks of BDLP-1-R peptide nanomaterials. (D) AFM images of the peptide nanomaterials (1) and peptide nanodots (3).Three-dimensional views of (1) and (3) are shown in (2) and (4), respectively. Sectional height plots along the labeled arrows in (1) and (3) are shown on the right.
3 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from
interpeptide hydrogen bond networks (Fig. 4C). Unlike previously re-ported superhelical assemblieswith only head-to-tail hydrogen bonding(19), the assembly here had zipper-like interlocked patterns with onemolecule forming hydrogen bonds simultaneously with four othermolecules. In each polar layer, all a-helical peptides were aligned with
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
theirmacrodipoles in similar directions. Thus, the hydrogen bondswerestrengthened by cooperative long-range electrostatics and polarizationeffects (36). In addition, two nearby polar layers (intermitted by onenonpolar layer) have macrodipoles in opposite directions, which elim-inate the dipole moment of the whole crystallite.
Fig. 4. Elucidation of BDCP-1-R nanostructure packing model. (A) Predicted crystallite packing of BDCP-1-R molecules. (B) Nonpolar region formed by the aromatic-aromatic and aromatic-sulfur interactions of the side-chain tethers. (C) Polar region formed by hydrogen bonding of the a-helical backbones. Carbon atoms in differentmolecules are shown in different colors. In all the structures, N/O/S atoms are in blue/red/yellow, respectively, and nonpolar hydrogen atoms are omitted for clarity.
Fig. 3. Characterization of self-assembly behavior in BDCP-1-R nanomaterials. (A) FTIR spectrum of the amide bond bands and hydrogen bond region (inset) and(B) Raman spectrum of BDCP-1-R peptide nanomaterials. (C) UV-vis spectra of BDCP-1-R monomers (black line) and peptide nanomaterials (blue dashed line). a.u.,arbitrary units. (D) Fluorescence spectra of BDCP-1-R monomers (dashed lines) and peptide nanomaterials (solid lines) dissolved or dispersed in water (1 mg/ml, 20°C).Peptide nanomaterial peaks are broader and red-shifted. (E) Optical image of BDCP-1-R peptide nanomaterials and associated fluorescence images with visible lumi-nescence using UV-blue, blue-green, and green-red filters.
4 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
http://advances.sD
ownloaded from
Extending sequence scope of the self-assemblyTo examine the de novonature of this assembly process, the assembly ofother sequences, including Ac-CAGAS5(Ph)-NH2 (BDCP-3-S/R) andAc-CAVAS5(Ph)-NH2 (BDCP-4-S/R), was examined under the sameconditions (Fig. 5A). As expected, both BDCP-3-R and BDCP-4-Rassembled whereas their S epimers did not. CD (Fig. 5C) indicatedBDCP-3-S and BDCP-4-S were nonhelical, whereas their R isomersadopted helical conformations in solution. The SEM images (Fig. 5B)revealed BDCP-3-R and BDCP-4-R formed flower- or ribbon-shapedstructures. The FTIR also confirmed that BDCP-3-R and BDCP-4-Radopted an a helix structure when assembled. Therefore, the aboveresults suggest that our strategy may allow de novo assembly of peptides,where the resulting detailed nanostructures are sequence-dependent.This provides a broad chemical space for developingmaterials with dif-ferent nanostructures for different purposes.
Application of CIH peptide nanomaterialsin electrochemical supercapacitorsConstraint peptides are known for their superior chemical and thermalstability compared to their linear counterparts (22). The nanomaterialsassembled from constrained helical peptides are therefore highlypromising for use in bionanotechnology applications because theirproperties could be tuned using sequence variation (12). One possibleapplication is in biocompatible supercapacitors (37–41), where the useof biocompatible active materials could extend their applications towearable, implantable, and biodegradable devices (42). Here, we dem-onstrated the BDCP nanomaterials were excellent candidate materialsfor supercapacitors and, therefore, are promising for use in biocom-patible energy devices.
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
on July 14, 2018ciencem
ag.org/
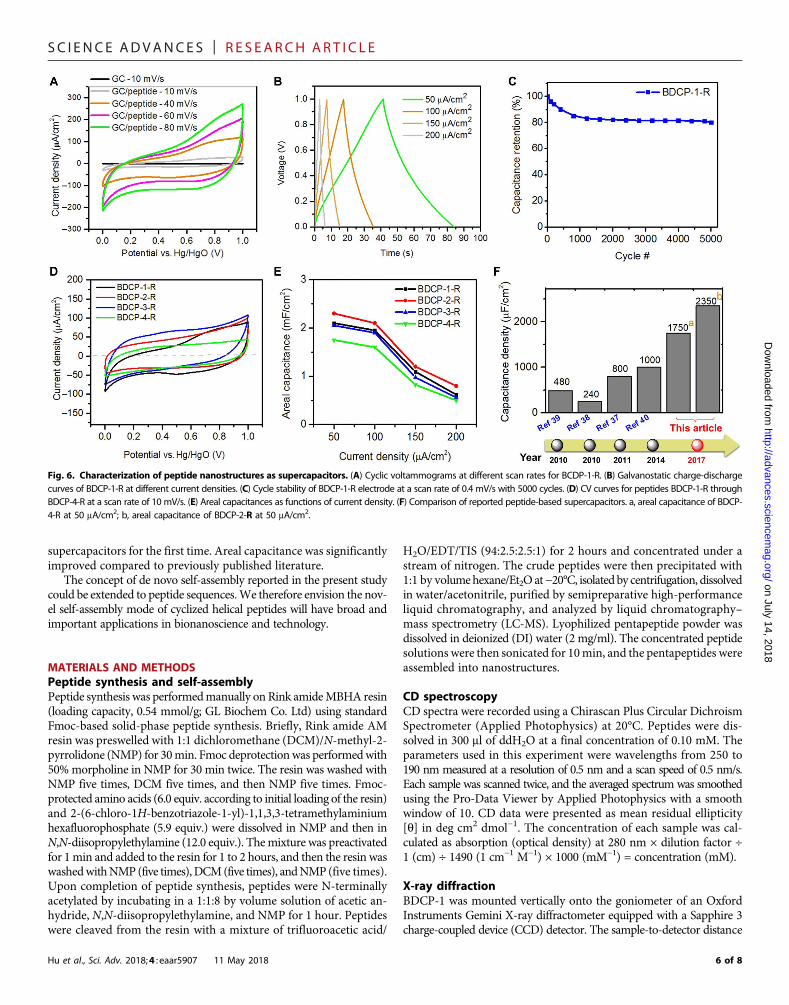
The supercapacitor behavior of the BDCP-1-R nanomaterials wasinvestigated by loading the assemblies onto a glassy carbon (GC) work-ing electrode (1mg/cm2). The peptide nanomaterialmorphology on theelectrode was characterized using optical microscopy and SEM. Theimage in fig. S3A is a macroscopic view of the peptide-modified elec-trode. The images in fig. S3 (B to D) show the microscopic details ofthe peptide assemblies, where peptide hollow tubes and solid beltswere both observed, consistent with the characterized morphology.Cyclic voltammetry (CV) measurements were obtained using a three-electrode cell in 0.05MKH2PO4/0.5MKCl at room temperature (fig.S4A). Figure 6A presents the CV curves for BDCP-1-R peptide as-semblies at different scan rates ranging from 10 to 80 mV/s with avoltage range of 0 to 1 V (versus Hg/HgO). The curves exhibited typicalcapacitor shapes. A control experiment using a bare GC electrodeconfirmed that the capacitance could largely be attributed to the pep-tide nanomaterials. Typical capacitive charge-discharge curves of thepeptide-loaded electrode are shown in Fig. 6B, with galvanostatic currentdensities ranging from 50 to 200 mA/cm2. The capacitance retention wasfound to be ~80% after 5000 cycles, as shown in Fig. 6C, demonstratingthe excellent electrochemical stability of the peptide nanomaterials.
Because peptide morphology can influence energy storage capacity,we also studied the relationships between peptide sequence, morphol-ogy, and energy storage capacity. We performed CVmeasurements forpeptides BDCP-2-R to BDCP-4-R (fig. S4, B to D). These peptides haddifferent areal capacitances that trended 2 > 1 > 3 > 4 (Fig. 6D). Thegalvanostatic charging/discharging ofBDCP-2-R toBDCP-4-Rwas alsoperformed (data shown in fig. S4, E to G), and the areal capitances werecalculated and plotted versus current density in Fig. 6E. In particular,peptide BDCP-2-Rhad an areal capacitance of 2.35mF/cm2 at a currentdensity of 50 mA/cm2. We also investigated the influence of the elec-trolyte on capacitance and found the capacitance could be furtherincreased by using 0.1 M H2SO4 instead of 0.05 M KH2PO4 and 0.5 MKCl (fig. S5A) (37). We observed the morphology of peptide assemblieson the electrode after CV measurement. We found that the peptidenanostrucutures revealed little change compared to that before CVmeasurement (Fig. 5, B to D). These results suggest that the peptidenanomaterials are very stable in different electrolytes. Figure 6Hcompares the areal capacitance of the reported peptide-based super-capacitors and presents the clear improvement in areal capacitance inthis work.
DISCUSSIONIn summary, we demonstrated cyclized helical peptides can self-assembleinto well-ordered nanostructures. This is a novel self-assembly modefor peptides. The self-assembly was controlled by an in-tether substitu-ent group. Whereas the absolute configuration of the chiral centerdetermined thehelicity, the aromaticity of the chiral centerwas the drivingforce for self-assembly. As a proof of principle, self-assembled BDCP-1-Rpeptide nanostructures were carefully characterized to confirm thebuilding blocks of the BDCP-1-R peptides in the nanostructures.Theoretical calculations suggest the existence of interpeptide p-p andp-sulfur interactions and hydrogen bond networks in the BDCP-1-Rself-assembly.
The self-assembled peptide nanomaterials also performed excel-lently as biocompatible active materials in electrochemical superca-pacitors, which are promising for use in wearable, implantable, andbiodegradable devices. We systematically studied the energy storagecapacity of the peptide nanomaterials for use in electrochemical
Fig. 5. CIH peptide assembly is tolerant of amino acid mutations. (A) Molecularstructures of BDCP-3(R/S) and BDCP-4(R/S). Side chains of middle amino acids arelabeled in blue. (B) SEM images of self-assembled BDCP-3-R and BDCP-4-R. (C) CDspectra of BDCP-3/4-R/S in water (pH 7, 20°C, and 100 mM). (D) FTIR spectra of BDCP-3-R and BDCP-4-R. Wavenumbers of peaks in pink circles are given.
5 of 8
SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14,http://advances.sciencem
ag.org/D
ownloaded from
supercapacitors for the first time. Areal capacitance was significantlyimproved compared to previously published literature.
The concept of de novo self-assembly reported in the present studycould be extended to peptide sequences.We therefore envision the nov-el self-assembly mode of cyclized helical peptides will have broad andimportant applications in bionanoscience and technology.
2018
MATERIALS AND METHODSPeptide synthesis and self-assemblyPeptide synthesis was performedmanually on Rink amideMBHA resin(loading capacity, 0.54 mmol/g; GL Biochem Co. Ltd) using standardFmoc-based solid-phase peptide synthesis. Briefly, Rink amide AMresin was preswelled with 1:1 dichloromethane (DCM)/N-methyl-2-pyrrolidone (NMP) for 30min. Fmoc deprotectionwas performedwith50%morpholine in NMP for 30 min twice. The resin was washed withNMP five times, DCM five times, and then NMP five times. Fmoc-protected amino acids (6.0 equiv. according to initial loading of the resin)and 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminiumhexafluorophosphate (5.9 equiv.) were dissolved in NMP and then inN,N-diisopropylethylamine (12.0 equiv.). Themixture was preactivatedfor 1min and added to the resin for 1 to 2 hours, and then the resin waswashedwithNMP (five times),DCM(five times), andNMP(five times).Upon completion of peptide synthesis, peptides were N-terminallyacetylated by incubating in a 1:1:8 by volume solution of acetic an-hydride, N,N-diisopropylethylamine, and NMP for 1 hour. Peptideswere cleaved from the resin with a mixture of trifluoroacetic acid/
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
H2O/EDT/TIS (94:2.5:2.5:1) for 2 hours and concentrated under astream of nitrogen. The crude peptides were then precipitated with1:1 by volumehexane/Et2O at−20°C, isolated by centrifugation, dissolvedin water/acetonitrile, purified by semipreparative high-performanceliquid chromatography, and analyzed by liquid chromatography–mass spectrometry (LC-MS). Lyophilized pentapeptide powder wasdissolved in deionized (DI) water (2 mg/ml). The concentrated peptidesolutions were then sonicated for 10min, and the pentapeptides wereassembled into nanostructures.
CD spectroscopyCD spectra were recorded using a Chirascan Plus Circular DichroismSpectrometer (Applied Photophysics) at 20°C. Peptides were dis-solved in 300 ml of ddH2O at a final concentration of 0.10 mM. Theparameters used in this experiment were wavelengths from 250 to190 nm measured at a resolution of 0.5 nm and a scan speed of 0.5 nm/s.Each sample was scanned twice, and the averaged spectrum was smoothedusing the Pro-Data Viewer by Applied Photophysics with a smoothwindow of 10. CD data were presented as mean residual ellipticity[q] in deg cm2 dmol−1. The concentration of each sample was cal-culated as absorption (optical density) at 280 nm × dilution factor ÷1 (cm) ÷ 1490 (1 cm−1 M−1) × 1000 (mM−1) = concentration (mM).
X-ray diffractionBDCP-1 was mounted vertically onto the goniometer of an OxfordInstruments Gemini X-ray diffractometer equipped with a Sapphire 3charge-coupled device (CCD) detector. The sample-to-detector distance
Fig. 6. Characterization of peptide nanostructures as supercapacitors. (A) Cyclic voltammograms at different scan rates for BCDP-1-R. (B) Galvanostatic charge-dischargecurves of BDCP-1-R at different current densities. (C) Cycle stability of BDCP-1-R electrode at a scan rate of 0.4 mV/s with 5000 cycles. (D) CV curves for peptides BDCP-1-R throughBDCP-4-R at a scan rate of 10 mV/s. (E) Areal capacitances as functions of current density. (F) Comparison of reported peptide-based supercapacitors. a, areal capacitance of BDCP-4-R at 50 mA/cm2; b, areal capacitance of BDCP-2-R at 50 mA/cm2.
6 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from
was 45mm.CLEARER softwarewas used to reduce the two-dimensionaldata to a one-dimensional intensity profile. The peptide structures of thesamples were studied using a PAN analytical X’Pert X-ray diffractometerwith CuKa radiation. The sample powders were scanned in the range of2q = 2° to 60° with a step size of 0.02°.
FTIR spectroscopyTo characterize interactions within the samples, qualitative analysis wasperformed by analyzing the infrared absorption spectrum of each sam-ple. KBr pellets were prepared beforemeasurement. A Bruker Vertex 70FTIR spectrometer was used for FTIR analysis with awavenumber rang-ing from 4000 to 400 cm−1.
High-resolution TEMSamples (10 ml) of peptide assemblies were sonicated in a bath sonicatorfor 5 s. The samples were placed on Formvar/carbon mesh 400 coppergrids (Electron Microscopy Sciences) for 20 s and then removed usingnitrocellulose paper. The Cu grids were left to dry at room temperature.TEM images were recorded using a high-resolution transmission elec-tron microscope (JEM-100F; 200 kV).
Scanning electron microscopyPentapeptide samples were placed on silicon slides and left to dry atroom temperature. Samples were then viewed using a scanning electronmicroscope (ZEISS Supra 55, Oxford X-Max 20; 20 kV).
Atomic force microscopyPentapeptide solutions were prepared at a concentration of 5 mg/ml.A droplet of solution was placed on a siliconized glass for more than30min to allow volatilization of the solvent. Large areas were imagedat different magnifications using an optical microscope (Axio Imager,Carl Zeiss). AFM analysis was carried out using a Bruker AFMmulti-mode (MultiMode 8, Bruker). The probes used for AFMwere antimony-doped (n) silicon cantilevers with a spring constant of 40 N/m (Bruker)and resonance frequency of 300 kHz. Observation was performed usingtapping mode with a scan rate of 0.9 Hz. After acquiring the AFMimages, pentapeptide data were analyzed using theNanoScope Analysisprogram.
Characterization of PLMeasurements of PL and PLE were performed using a Horiba JobinYvon FL3-11 spectrofluorometer. PL/PLE measurements were takenof peptide nanofibers and monomers on a standard cuvette. Fluores-cence measurements of peptide nanofiber film were acquired on a glasscoverslip.
Raman spectroscopyRaman data were acquired using a Horiba Jobin-Yvon LabRamAramisRaman spectrometer. A 532-nmAr-ion laser was used as the excitationsource, and the substrate plate was stainless steel with no Raman signal,guaranteeing a fluorescence-free background. Wavenumbers rangingfrom 200 to 3000 cm−1 were scanned, and each spectrum was acquiredfor 60 s. The analysis was executed for more than three points for eachsample for reproducibility and reliability.
UV-vis spectroscopic analysisUV-vis spectra were obtained using anOtsuka ElectronicsMCPD 7000UV-visible spectrophotometer with fiber optics and an ultra-high sen-sitive CCD array cooled by a Peltier device. The sample room was a
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
custom-made dark box in which versatile measurements, includingtransmission measurements of LB films, could be performed. Trans-mitted light was collected through an integral sphere. For visible spec-trometric measurements, a quartz plate was used, which was purchasedfrom the same provider as the germanium substrate.
Computational modeling of peptide crystal structureCrystal structure prediction was performed in the cloud platform ofXtalPi Inc., which integrates conformation analysis, force-field param-eterization, crystal structure searching, clustering, and ranking. Thecrystal packings were sampled in the five most frequent space groups(P21, P212121, P1, C2, and P21212) according to the Cambridge struc-tural database statistics for all chiral entries. The searching space includedlattice parameters, molecular positions and orientations, and rotationsalong some single bonds. Initially, the lattice energies were evaluatedusing a system-specific force field. Finally, the geometries and energieswere reoptimized using the Perdew-Burke-Ernzerhof functional withdispersion correction using VASP software.
Preparation of GC/peptide electrode andelectrochemical measurementsAGCelectrode (3mm indiameter)was polished to bemirror-like using0.05 mm of alumina medium and then coated with a layer of active ma-terials to serve as a working electrode. The active material ink was pre-pared as follows: 2 mg of peptide was dispersed in 1 ml of DI water.Nafion [80 ml, obtained as a 5 wt % (weight %) solution; ElectrochemInc.] was added, and the mixture was sonicated for 30 min to form ahomogeneous ink. The resulting ink (5 ml) was drop-cast onto theGC electrode and subsequently air-dried to obtain a mass loadingof 1mg/cm2. The electrode was tested on a CHI 660e electrochemicalworkstation (Chenhua) in three-electrode electrochemical cells using0.05MKH2PO4/0.5MKCl or 0.1MH2SO4. Pt wire andHg/HgOwereused as the counter and reference electrodes, respectively. For the GC/peptide electrode, the areal capacitance (F/cm2) was calculated from thecharging/discharging curves using the equation Cv = IDt/SDV, where Irefers to the discharging current (A), S is the geometrical area of theglass carbon (cm2),Dt is the discharging time (s), andDV is the potentialwindow (V).
SUPPLEMENTARY MATERIALSSupplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaar5907/DC1Synthesis of BDCP peptidesLC-MS, mass, and 1H and 13C spectra for nonnatural amino acids or peptidesBDCP-2-S/RBDCP-3-S/RBDCP-4-S/RNuclear magnetic resonance data of unnatural amino acids1H of Fmoc-S5(2-Me)-OH13C of Fmoc-S5(2-Me)-OHHigh-resolution mass spectrum1H of Fmoc-S5(2-Ph)-OH13C of Fmoc-S5(2-Ph)-OHHigh-resolution mass spectrum1H of Fmoc-S5(2-naphnal)-OHfig. S1. CD spectra of BDCP-0/1/2/-R or S in water, respectively.fig. S2. Simulated powder XRD pattern from the predicted crystal structure of BDCP-1-R.fig. S3. Morphology of the BDCP-1-R nanostructure on GC electrode.fig. S4. Electrochemical performance of the BDCP-2/3/4-R peptide assemblies.fig. S5. Electrochemical performance of BDCP-1-R nanostructures in different electrolytes.table S1. Relationship between peptide structures and self-assemblies.cif file
7 of 8

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from
REFERENCES AND NOTES1. T. G. Barclay, K. Constantopoulos, J. Matisons, Nanotubes self-assembled from
amphiphilic molecules via helical intermediates. Chem. Rev. 114, 10217–10291 (2014).2. Z. Luo, S. Zhang, Designer nanomaterials using chiral self-assembling peptide systems
and their emerging benefit for society. Chem. Soc. Rev. 41, 4736–4754 (2012).3. J. Wang, K. Liu, R. Xing, X. Yan, Peptide self-assembly: Thermodynamics and kinetics.
Chem. Rev. 45, 5589–5604 (2016).4. R. G. Ellis-Behnke, Y.-X. Liang, S.-W. You, D. K. C. Tay, S. Zhang, K.-F. So, G. E. Schneider,
Nano neuro knitting: Peptide nanofiber scaffold for brain repair and axon regenerationwith functional return of vision. Proc. Natl. Acad. Sci. U.S.A. 103, 5054–5059 (2006).
5. A. L. Boyle, D. N. Woolfson, De novo designed peptides for biological applications.Chem. Soc. Rev. 40, 4295–4306 (2011).
6. J. H. Kim, M. Lee, J. S. Lee, C. B. Park, Self-assembled light-harvesting peptide nanotubesfor mimicking natural photosynthesis. Angew. Chem. Int. Ed. Engl. 51, 517–520 (2012).
7. K. Tao, P. Makam, R. Aizen, E. Gazit, Self-assembling peptide semiconductors. Science 358,eaam9756 (2017).
8. I. W. Hamley, Peptide nanotubes. Angew. Chem. Int. Ed. Engl. 53, 6866–6881 (2014).9. S. E. Paramonov, H.-W. Jun, J. D. Hartgerink, Self-assembly of peptide–amphiphile
nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc.128, 7291–7298 (2006).
10. A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly,S. E. Radford, Responsive gels formed by the spontaneous self-assembly of peptides intopolymeric b-sheet tapes. Nature 386, 259–262 (1997).
11. I. C. Tanrikulu, A. Forticaux, S. Jin, R. T. Raines, Peptide tessellation yields micrometre-scalecollagen triple helices. Nat. Chem. 8, 1008–1014 (2016).
12. L. Adler-Abramovich, E. Gazit, The physical properties of supramolecular peptide assemblies:From building block association to technological applications. Chem. Soc. Rev. 43,6881–6893 (2014).
13. B. Sun, Q. Li, H. Riegler, S. Eickelmann, L. Dai, Y. Yang, R. Perez-Garcia, Y. Jia, G. Chen,J. Fei, K. Holmberg, J. Li, Self-assembly of ultralong aligned dipeptide single crystals.ACS Nano 11, 10489–10494 (2017).
14. R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe, S. Perrier, Design and properties offunctional nanotubes from the self-assembly of cyclic peptide templates. Chem. Soc. Rev.41, 6023–6041 (2012).
15. J. Kim, S. Kwon, S. H. Kim, C.-K. Lee, J.-H. Lee, S. J. Cho, H.-S. Lee, H. Ihee, Microtubes withrectangular cross-section by self-assembly of a short b-peptide foldamer. J. Am. Chem. Soc.134, 20573–20576 (2012).
16. V. Azzarito, K. Long, N. S. Murphy, A. J. Wilson, Inhibition of a-helix-mediatedprotein–protein interactions using designed molecules. Nat. Chem. 5, 161–173(2013).
17. X. Chen, Y. He, Y. Kim, M. Lee, Reversible, short a-peptide assembly for controlled captureand selective release of enantiomers. J. Am. Chem. Soc. 138, 5773–5776 (2016).
18. J. Lee, I. R. Choe, N.-K. Kim, W.-J. Kim, H.-S. Jang, Y.-S. Lee, K. T. Nam, Water-floating giantnanosheets from helical peptide pentamers. ACS Nano 10, 8263–8270 (2016).
19. S. Mondal, L. Adler-Abramovich, A. Lampel, Y. Bram, S. Lipstman, E. Gazit, Formation offunctional super-helical assemblies by constrained single heptad repeat. Nat. Commun. 6,8615 (2015).
20. H. J. Dyson, P. E. Wright, Defining solution conformations of small linear peptides.Annu. Rev. Biophys. Biophys. Chem. 20, 519–538 (1991).
21. S. Sim, Y. Kim, T. Kim, S. Lim, M. Lee, Directional assembly of a-helical peptides inducedby cyclization. J. Am. Chem. Soc. 134, 20270–20272 (2012).
22. Y. H. Lau, P. de Andrade, Y. Wu, D. R. Spring, Peptide stapling techniques based ondifferent macrocyclisation chemistries. Chem. Soc. Rev. 44, 91–102 (2015).
23. J. C. Phelan, N. J. Skelton, A. C. Braisted, R. S. McDowell, A general method for constrainingshort peptides to an a-helical conformation. J. Am. Chem. Soc. 119, 455–460 (1997).
24. K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y.-D. Wu,Z. Li, An in-tether chiral center modulates the helicity, cell permeability, and targetbinding affinity of a peptide. Angew. Chem. Int. Ed. Engl. 55, 8013–8017 (2016).
25. Y. Jiang, K. Hu, X. Shi, Q. Tang, Z. Wang, X. Ye, Z. Li, Switching substitution groups on thein-tether chiral centre influences backbone peptides’ permeability and target bindingaffinity. Org. Biomol. Chem. 15, 541–544 (2017).
26. K. Hu, C. Sun, D. Yang, Y. Wu, C. Shi, L. Chen, T. Liao, J. Guo, Y. Liu, Z. Li, A preciselypositioned chiral center in an i, i + 7 tether modulates the helicity of the backbonepeptide. Chem. Commun. 53, 6728–6731 (2017).
Hu et al., Sci. Adv. 2018;4 : eaar5907 11 May 2018
27. A. C. Santos, P. Pattekari, S. Jesus, F. Veiga, Y. Lvov, A. J. Ribeiro, Sonication-assistedlayer-by-layer assembly for low solubility drug nanoformulation. ACS Appl. Mater.Interfaces 7, 11972–11983 (2015).
28. N. Amdursky, M. Molotskii, E. Gazit, G. Rosenman, Elementary building blocks ofself-assembled peptide nanotubes. J. Am. Chem. Soc. 132, 15632–15636 (2010).
29. D. A. Middleton, J. Madine, V. Castelletto, I. W. Hamley, Insights into the moleculararchitecture of a peptide nanotube using FTIR and solid-state NMR spectroscopicmeasurements on an aligned sample. Angew. Chem. Int. Ed. Engl. 52, 10537–10540 (2013).
30. J. D. Hartgerink, J. R. Granja, R. A. Milligan, M. R. Ghadiri, Self-assembling peptidenanotubes. J. Am. Chem. Soc. 118, 43–50 (1996).
31. M. Pellach, S. Mondal, L. J. W. Shimon, L. Adler-Abramovich, L. Buzhansky, E. Gazit,Molecular engineering of self-assembling diphenylalanine analogues results in theformation of distinctive microstructures. Chem. Mater. 28, 4341–4348 (2016).
32. H. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, X. Lu, R. Collins, S. Y. Liem,A. Song, P. L. A. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch, R. V. Ulijn, An investigation ofthe conductivity of peptide nanotube networks prepared by enzyme-triggeredself-assembly. Nanoscale 2, 960–966 (2010).
33. N. C. Burgess, T. H. Sharp, F. Thomas, C. W. Wood, A. R. Thomson, N. R. Zaccai, R. L. Brady,L. C. Serpell, D. N. Woolfson, Modular design of self-assembling peptide-basednanotubes. J. Am. Chem. Soc. 137, 10554–10562 (2015).
34. S. K. Burley, G. A. Petsko, Aromatic-aromatic interaction: A mechanism of proteinstructure stabilization. Science 229, 23–28 (1985).
35. R. J. Zauhar, C. L. Colbert, R. S. Morgan, W. J. Welsh, Evidence for a strong sulfur–aromaticinteraction derived from crystallographic data. Biopolymers 53, 233–248 (2000).
36. Y.-D. Wu, Y.-L. Zhao, A theoretical study on the origin of cooperativity in the formation of310- and a-helices. J. Am. Chem. Soc. 123, 5313–5319 (2001).
37. P. Beker, G. Rosenman, Bioinspired nanostructural peptide materials for supercapacitorelectrodes. J. Mater. Res. 25, 1661–1666 (2010).
38. P. Beker, I. Koren, N. Amdursky, E. Gazit, G. Rosenman, Bioinspired peptide nanotubes assupercapacitor electrodes. J. Mater. Sci. 45, 6374–6378 (2010).
39. L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky,G. Rosenman, E. Gazit, Self-assembled arrays of peptide nanotubes by vapour deposition.Nat. Nanotechnol. 4, 849–854 (2009).
40. J. Zhang, X. Wu, Z. Gan, X. Zhu, Y. Jin, Unidirectionally aligned diphenylalanine nanotube/microtube arrays with excellent supercapacitive performance. Nano Res. 7, 929–937 (2014).
41. A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo,P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu,C. J. Bettinger, R. V. Ulijn, Polymeric peptide pigments with sequence-encodedproperties. Science 356, 1064–1068 (2017).
42. V. Nguyen, R. Zhu, K. Jenkins, R. Yang, Self-assembly of diphenylalanine peptide withcontrolled polarization for power generation. Nat. Commun. 7, 13566 (2016).
AcknowledgmentsFunding: We acknowledge financial support from the Natural Science Foundation of China(grants 21778009, 81701818, and 51673029), Ministry of Science and Technology ofChina (2015DFA31590), and the Shenzhen Science and Technology Innovation Committee(JCYJ20170412150719814 and GJHS20170310093122365). Author contributions: K.H. and Z.G.L.conceived and designed the experiments. K.H., Y.J., and F.Y. synthesized and characterizedthe peptide nanomaterial. P.-Y.Z., H.G., and F.J. performed the computational modeling. K.H.,Y.J., W.X., and H.L. performed the electrochemical experiments. K.H., Y.J., W.X., F.J., X.W.,Q.L.Z., Z.G.L., and Z.L. wrote the manuscript. All authors participated in discussions on the dataand critiques of the manuscript. Competing interests: The authors declare that they have nocompeting interests. Data and materials availability: All data needed to evaluate the conclusionsin the paper are present in the paper and/or the Supplementary Materials. Additional data relatedto this paper may be requested from the authors.
Submitted 27 November 2017Accepted 28 March 2018Published 11 May 201810.1126/sciadv.aar5907
Citation: K. Hu, Y. Jiang, W. Xiong, H. Li, P.-Y. Zhang, F. Yin, Q. Zhang, H. Geng, F. Jiang, Z. Li,X. Wang, Z. Li, Tuning peptide self-assembly by an in-tether chiral center. Sci. Adv. 4, eaar5907(2018).
8 of 8

Tuning peptide self-assembly by an in-tether chiral center
Wang and Zigang LiKuan Hu, Yixiang Jiang, Wei Xiong, Hu Li, Pei-Yu Zhang, Feng Yin, Qianling Zhang, Hao Geng, Fan Jiang, Zhou Li, Xinwei
DOI: 10.1126/sciadv.aar5907 (5), eaar5907.4Sci Adv
ARTICLE TOOLS http://advances.sciencemag.org/content/4/5/eaar5907
MATERIALSSUPPLEMENTARY http://advances.sciencemag.org/content/suppl/2018/05/04/4.5.eaar5907.DC1
REFERENCES
http://advances.sciencemag.org/content/4/5/eaar5907#BIBLThis article cites 42 articles, 4 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS.is aScience Advances Association for the Advancement of Science. No claim to original U.S. Government Works. The title
York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive licensee American (ISSN 2375-2548) is published by the American Association for the Advancement of Science, 1200 NewScience Advances
on July 14, 2018http://advances.sciencem
ag.org/D
ownloaded from


















