
To every complex question there is a simple answer ... · Struktur- og vibrationsegenskaber hos...
64
To every complex question there is a simple answer − and it is wrong. (Menckens Lov. H. L. Mencken, 1880-1956) 12 Hydrogenbindinger til alkylradikaler Hydrogenbindinger – randbetingelser og kriterier Beregningsmetoder; forventninger Protondonorer med ladning Intramolekylære hydrogenbindinger Ammoniumioner. Forgrening Alkoholer og ethere, protoniserede carbonylgrupper Intermolekylære hydrogenbindinger – addukter Donors protonaffinitet, acceptors ioniseringsenergi Struktur- og vibrationsegenskaber hos hydrogenbundne addukter Ioniske vekselvirkninger Molekyler er ikke punkter. Neutrale uden H-bindinger Kationer uden H-bindinger; ladning; delokalisering Bindingsenergi Deformering Elektrostatik eller kovalens – VB og MO beskrivelse Ladningsoverførsel. Relationer til ΔPA og Δr Fravær af symmetri; H + eller H . overførsel Acceptorer med åbne og lukkede skaller Alkaner; vand og formaldehyd; ternære addukter Hydrogenbindinger i ikke-ladede systemer Elektrofilt aktiverede neutrale protondonorer
Transcript of To every complex question there is a simple answer ... · Struktur- og vibrationsegenskaber hos...

To every complex question there is a simple answer− and it is wrong.
(Menckens Lov. H. L. Mencken, 1880-1956)
12 Hydrogenbindinger til alkylradikaler
Hydrogenbindinger – randbetingelser og kriterierBeregningsmetoder; forventningerProtondonorer med ladning
Intramolekylære hydrogenbindingerAmmoniumioner. ForgreningAlkoholer og ethere, protoniserede carbonylgrupper
Intermolekylære hydrogenbindinger – addukterDonors protonaffinitet, acceptors ioniseringsenergiStruktur- og vibrationsegenskaber hos hydrogenbundne addukter
Ioniske vekselvirkningerMolekyler er ikke punkter. Neutrale uden H-bindingerKationer uden H-bindinger; ladning; delokalisering
BindingsenergiDeformering
Elektrostatik eller kovalens – VB og MO beskrivelseLadningsoverførsel. Relationer til ΔPA og ΔrFravær af symmetri; H+ eller H. overførsel
Acceptorer med åbne og lukkede skallerAlkaner; vand og formaldehyd; ternære addukter
Hydrogenbindinger i ikke-ladede systemerElektrofilt aktiverede neutrale protondonorer

Hydrogenbindinger 12.284
Forsidecitatets kølige konstatering af at én-parameterforklaringer på mange-parameter problemstillinger nødvendigvis er utilstrækkelige kaldes ofteMenckens lov og gives den anførte formulering. Den oprindelige form synesdog at være "There is always an easy solution to every human problem −neat, plausible and wrong" (skal have stået in New York Evening Mail, 1917,siden i samlingen Prejudices, Second Series, 1920).
Den del af kapitlets resultater der foreligger på publiceret form findes i S.Hammerum og C. B. Nielsen, Intramolecular hydrogen bonding and hydrogenatom abstraction in gas-phase amine radical cations . J. Phys. Chem. A, 109, 12046-53 (2005); S. Hammerum, Alkyl radicals as hydrogen bond acceptors: computa-tional evidence. J. Am. Chem. Soc., 131, 8627-35 (2009) (supporting informationheri indeholder en stor del af de beregningsresultater som dette kapiteltrækker på); S. G. Olesen og S. Hammerum, Redshift or adduct stabilization − acomputational study of hydrogen bonding in adducts of protonated carboxylic acids .Europ. J. Mass Spectrom., 15, 239-48 (2009) (tilgængelig på http://www-mf.kiku.dk/art/carbox.pdf); S. G. Olesen og S. Hammerum, Hydrogen bondingto alkanes: computational evidence. J. Phys. Chem. A, 113, 7940-44 (2009); og S.G. Olesen, T. L. Gausco, G. H. Weddle, S. Hammerum og M. A. Johnson,Vibrational predissociation spectra of the Ar tagged [CH4 H3O+] binary complex:spectroscopic signature of hydrogen bonding to an alkane. Mol. Phys., 108, 1191-97(2010).
December 2017, Steen Hammerum, [email protected](mindre rettelser, august 2018)

Hydrogenbindinger 12.285
Sammenfatning
Protondonorer med ladning danner moderate til gode inter- og intra-molekylære hydrogenbindinger til alkylradikaler. Dette fremgår af denbetydelige stabilisering og de bindingslængdeændringer og IR rødskiftsom hydrogenbundne radikaladdukter udviser. Eksempelvis er tert-butylradikalet i gasfase lige så god en hydrogenbindingsacceptor somvand og formaldehyd overfor fx H3O+
eller CH3OH2+.. Disse vekselvirkningerundersøges her ved hjælp af beregningermed sammensatte kvantekemiske abinitio metoder.
De hydrogenbundne radikaladdukters stabilisering skyldes dels denelektrostatiske tiltrækning som betinges af at den ene komponent bæreren ladning, dels hydrogenbindinger, som i første række afhænger af denpotentielle H-donors syrestyrke og af alkylradikalets ioniseringsenergi.Dette sidste forhold afspejler at for alkylradikalers hydrogenbindingerspiller ladningsoverførsel til H-donormolekylet en særligt stor rolle.
Den H-overførsel hydrogenbindingen tager forskud på kan involvereenten H+ eller H., men de ændringer af struktur, energi og spektro-skopiske egenskaber der ledsager dannelsen afhydrogenbindinger til alkylradikaler er de samme; deindledende vekselvirkninger bestemmes ikke af denpotentielle H-flytnings art.
Hydrogenbindinger mellem alkylradikaler og neutrale H-donorer erforholdsvis svage; de styrkes imidlertid betydeligt ved komplex-dannelse med Lewis-syrer.
Kapitlet er struktureret således at forsidens overskrifter falder i treafsnit, en beskrivelse af forudsætninger og resultater, en diskussion afresultaternes sammenhæng og systematik, og en omtale af nært-beslægtede forhold.

Hydrogenbindinger 12.286
Hydrogenbindinger til alkylradikaler
Den intra- og intermolekylære vekselvirkning vi kalder 'hydrogen-binding' blev først beskrevet omkring 1920, af Latimer og Rodebush [1]og Huggins [2]. Rigtig kendt blev hydrogenbindingen dog først nogetsenere, gennem en oversigtsartikel af Lassettre (1937) [3] og, især, ved atvære emnet for et helt kapitel i Paulings The Nature of the Chemical Bond(1939) [4]. Betegnelsen er imidlertid noget kamæleonagtig, og dersavnes tilsyneladende enighed om hvordan fænomenet opstår ogafgrænses [5]. Lærebøgerne har gerne meget at sige om hydrogen-bindingers konsekvenser − men meget lidt om hydrogenbindingersoprindelse. De er der bare. Miseren har sammenhæng med atbetegnelsen 'hydrogenbinding' udtrykker det samlede resultat af enrække forskellige vekselvirkninger, og med det forhold at sammebetegnelse skal dække fænomener med et meget bredt virknings-spektrum, herunder nogle så svage at de næppe er virkelige [5,6].Blandt andet derfor har terminologien været emne for et af IUPACsprojekter [7].
Uden randbetingelser. I fravær af en kontant definition (fordi en sådanikke findes) er et udsagn om at hydrogenbindinger er til stede i et givetsystem ikke nødvendigvis særlig indholdsrigt, og, værre, fraværet kangøre det vanskeligt at argumentere konkret for at hydrogenbindingerikke skulle være til stede; det er især på dette punkt at IUPACs forsøg påafgrænsning af fænomenet [7] er utilstrækkeligt. Det kan imidlertidvære hensigtsmæssigt at se bort fra helt svage vekselvirkninger (uansetdisses betegnelse) og undgå diskussion af skarpe definitioner og rand-betingelser, og i stedet undersøge og eventuelt sandsynliggøre tilstede-værelsen af hydrogenbindinger ved først og fremmest at beskæftige sigmed de konkrete fysiske egenskaber som typiske hydrogenbundnesystemer almindeligvis besidder.
Kort sammenfattet gør hydrogenbindinger sig i reglen gældende derhvor protonoverførsel i princippet er mulig; de forekommer når et labilt(dvs surt) H kan vekselvirke med et område med (relativt) høj elektron-tæthed. Den typiske hydrogenbinding involverer således ofte et lonepair på et elektronegativt atom [5], men også π-systemer skal kunnevære hydrogenbindingsacceptorer. Som det vil fremgå af det efter-følgende gælder dette tillige alkylradikaler.

Hydrogenbindinger 12.287
Objektive kriterier. Hydrogenbindinger giver sig til kende gennemderes indvirkning på molekylernes struktur og termokemi, og på defysiske og spektroskopiske egenskaber. Er vinkler og bindingslængderændret? Gør en usædvanlig stabilisering sig gældende? Er infrarødfrekvenser og intensiteter eller NMR chemical shifts ændret på karak-teristisk vis? Alt sammen set i forhold til nærtbeslægtede systemer udenhydrogenbindinger.
Disse egenskaber hos hydrogenbundne systemer kan hensigtsmæssigtundersøges ved beregning (se fx [8-18]). Her er det imidlertid ikketilstrækkeligt at undersøge hvorvidt en særlig stabilisering gør siggældende. Beregninger kan nok vise at ét system har lavere energi endet andet, men ikke nødvendigvis hvorfor; mange forskellige veksel-
virkninger kan spille ind. Derimod udgør en samtidigstabilisering og en bindingslængdeforøgelse og etmarkant fald i IR bølgetal for strækning af bindingentil det involverede H en god begrundelse for at antageat hydrogenbindinger er til stede.
Beregningsmetoder. I denne undersøgelse er de energimæssige forhold(dannelsesvarmer og addukters stabilisering) belyst med sammensatteab initio metoder, G3 og G3//B3LYP [19]; struktur og IR spektroskopiskeegenskaber er bestemt med B3LYP/6-31+G(d,p) beregninger. Tilsammenligning og kontrol er yderligere benyttet andre DFT-metodersåvel som overalt MP2/6-31+G(d,p) beregninger; hverken DFT- ellerMP2-beregninger har udvist problemer med spinforurening.
Generelt stemmer de forskellige DFT-metoders resultater godt overens,såvel hvad angår struktur som vibrationsfrekvenser, og der er ret godoverensstemmelse mellem DFT- og MP2-resultaterne, omend det medhydrogenbindinger forbundne IR rødskift er noget mindre når detbestemmes ved MP2-beregninger end ved brug af DFT-metoder.Derimod synes struktur og frekvenser bestemt ved simple Hartree-Fockberegninger på hydrogenbundne systemer ikke at være pålidelige [20].
Beregning af addukters elektroniske energi kan lide af basissætsuperpositionsfejl (BSSE). Det er uklart om der i ioniske systemer børsøges korrigeret herfor [21], men den korrektion man kan foretage vhacounterpoise-metoden [22] viser sig ved både G3- og G3(MP2)-beregninger på de ioniske addukter at være på små 10% af den samledestabilisering, med en sådan regelmæssighed at det kunne være udtryk

Hydrogenbindinger 12.288
for et alment fænomen, eller for en systematisk fejl. For hydrogen-bundne addukter uden ladning kan korrektionen derimod være nogetstørre. De her samlede beregningsresultater er ikke søgt korrigeret forBSSE; counterpoise-korrektionerne er anført for sig i de fleste tabeller.Der er ligeledes ikke søgt korrigeret for de særlige BSSE-problemer vedberegninger vedrørende delvis kovalent bundne addukter [23].
Undersøgelse af bindingsforholdene med Baders atoms-in-moleculesmetode (AIM) [24] og af orbital-vekselvirkninger med Reed ogWeinholds natural bond orbital metode (NBO) [25] er, som de øvrigeberegninger, udført med Gaussian 03 og Gaussian 09. Såvel AIM somNBO undersøgelserne har taget udgangspunkt i B3LYP/6-31+G(d,p)beregningsresultater.
Forventninger. At alkylradikaler skulle kunne være gode hydrogen-bindingsacceptorer har ikke været almindeligt anerkendt (tværtimod[8]). Det vides dog at F-atomers reaktion med methan i kolde ædel-gasser fører til et svagt bundet addukt af CH3
. og HF [26], hvilketstemmer godt overens med et (mindre) antal beregningsmæssige under-søgelsers resultater [8,27,28], der også omfatter andre neutrale H-donorer. Den beregnede bindingsstyrke i disse systemer ligger nogetunder det der svarer til hydrogenbindingen mellem to vandmolekyler,dvs få kJ mol−1.
Imidlertid er det egentlig ret naturligt at forvente hydrogenbindinger tilalkylradikaler. Ganske som en sædvanlig hydrogenbinding i en visforstand tager forskud på en H+-overførsel [6,29] kunne man forventeen analog vekselvirkning, en radikal-hydrogenbinding, i de systemerhvor reversibel H-atomflytning let finder sted. Dermed ville hydrogen-bindinger til alkylradikaler særlig være at forvente når H-donor bærerladning, idet hydrogenatomflytning mellem alkylgrupper og hetero-atomer i organiske kationradikaler har ret lav barriere [30,31]. Detteviser sig at holde stik, og det viser sig tillige at hydrogenbindinger tilalkylradikaler manifesterer sig helt på samme måde som'konventionelle' hydrogenbindinger [32].
Protondonorer med ladning
I lige-elektron systemer forekommer særligt stærke hydrogenbindingernår protondonor har en positiv ladning [33], af Gilli [34] kaldet charge-assisted hydrogen bonds. Det var da også hos ioner, protoniserede

Hydrogenbindinger 12.289
aminoalkylradikaler, at en systematisk undersøgelse [31] først kunnepåvise at intramolekylære N−H...C hydrogenbindinger til radikal-carbon gør sig gældende og bl.a. øver afgørende indflydelse på dekonformationsmæssige forhold.
Der er enkelte tidlige beslægtede resultater i litteraturen: Holmes [35] karakteriserede som den første det "elektrostatisk bundne" gasfase-addukt af H3O+ og CH3
.. Gil [36] beskrev kort tid senere dette som et"hydrogenbundet komplex", og Audier [37] fremhævede at såvel H3O+
som CH3OH2+ danner moderate bindinger til CH3
.. En usædvanligstabilisering af to protoniserede hydroxyalkylradikalers gauche-konfor-mere blev af Bouchoux og Choret [30] tilskrevet intramolekylærhydrogenbinding mellem OH og radikal-carbon, ligesom Semialjac [38]for de distoniske isomere af valeramids kationradikal antog atforskellen mellem trans- og gauche-formernes elektroniske energier(DFT-beregninger) skyldtes intramolekylære hydrogenbindinger tilcarbon. Mayer [39] havde noget tidligere foreslået at ionisering afdimethylamins van der Waals dimer kan føre til dannelse af ioner medN−H...C hydrogenbindinger.
Disse observationer foregreb at moderate eller endog stærke hydrogen-bindinger mellem gode protondonorer og alkylradikaler bevirker enofte betydelig stabilisering og indvirker afgørende på addukternesstruktur og på deres IR-spektroskopiske egenskaber. Denne hidtilupåagtede vekselvirkning kan være af betydning for alkylradikalerssolvatisering, den indvirker givetvis på deres reaktioner under surebetingelser og med gode protondonorer, og den øver betydelig ind-flydelse på de konformationelle forhold ved intramolekylære hydrogen-atomafrivninger. Dertil kommer at den ladningsoverførsel der udgørdet kovalente bidrag til hydrogenbindingen spiller en særligt iøjne-faldende rolle når alkylradikaler er H-acceptorer, og dermed bidragerdisse resultater til at fremhæve betydningen af denne vekselvirkning,der udgør en vigtig del af moderate og stærke hydrogenbindinger.
Undersøgelsen omfatter ikke simple uorganiske hydrogenbindings-acceptorer med åbne skaller, som fx .OH; det fremgår af adskilligearbejder, fx [40], at disse ikke opfører sig exceptionelt.

Hydrogenbindinger 12.290
Intramolekylære hydrogenbindinger
Ammoniumioner. En systematisk beregningsmæssig undersøgelse [31]af protoniserede γ- og δ-aminoalkylradikaler (distoniske isomere afalifatiske amin kationradikaler) viste, at disse er særlig stabile på dengauche-form der bringer aminogruppen tættest muligt på radikal-carbon. Eksemplet i Tabel 12.1 illustrerer at denne konformer har egen-skaber der er ret karakteristiske for et molekyle med intramolekylærehydrogenbindinger, til forskel fra all-trans-formen. I gauche,gauche-formen er det involverede hydrogenatoms binding til nitrogen 0.026 Ålængere end i trans-formen, bølgetallet for N−H strækningsvibrationener faldet mere end 400 cm−1 og absorptionsintensiteten samtidigbetydeligt forøget, og dannelsesvarmen er 23 kJ mol−1 lavere (Tabel12.1); netop de forskelle som normalt ville tilskrives systemer medmoderate hydrogenbindinger [5]. Foruden denne konformer, der hvadstruktur angår ligner cyclohexan i stolform, findes en hydrogenbundettwist-boat-form, hvor ændringerne i forhold til trans-formen er mindreog hydrogenbindingen dermed svagere (Tabel 12.1).
Tabel 12.1. Beregnede egenskaber for det protoniserede 4-amino-1-methylbutyl-radikal, i trans- og gauche,gauche-konformationer.
rN−H υN−H ΔHf
1.028 33891 1.028 3480 701
1.028 3482
1.054 29542 1.026 3433 678
(chair) 1.026 3499
1.043 31483 1.026 3438 689
(twist-boat) 1.026 3500
G3//B3LYP beregninger; r i Å, υ i cm−1, 298 K ΔHf i kJ mol−1.

Hydrogenbindinger 12.291
Tiltrækningen mellem C og N kan illustreres yderligere ved sammen-ligning med 2-hexylradikalets gauche,gauche-stolform. Den beregnedeCHC vinkel er her 116o og afstanden mellem C-2 og C-6 er 3.28 Å, hvordet hydrogenbundne ammoniopentylradikal, 2, har en CHN vinkel på142o og en afstand mellem N og C-4 på 2.94 Å. Den betydeligtformindskede afstand skyldes den stabiliserende N−H ...C vekselvirk-ning, og den mere åbne vinkel at vekselvirkningen styrkes af linearitet.
Forgrening. Forskellen mellem N−H bindingslængder og IR egenskaberhos all-trans-formen og den relevante gauche,gauche-form viser for enrække protoniserede 4-aminobutylradikaler (Tabel 12.2) at substitutionpå N ikke indvirker meget på den intramolekylære hydrogenbinding;den synes nogenlunde lige udpræget hvad enten de protoniserederadikaler er afledt af primære, sekundære eller tertiære aminer.Substitution på radikal-C gør derimod hydrogenbindingerne stærkere.For protoniserede aminoalkylradikaler med primært, sekundært ogtertiært radikal-carbon er der således tydelige forskelle mellem N−H ...Cvekselvirkningernes konsekvenser (Tabel 12.2); forøget substitution påcarbon medfører at N−H bindingen bliver længere og viser større IRrødskift, og at stabiliseringsenergien vokser.
Tabel 12.2. Beregnede forskelle mellem protoniserede 4aminobutylradikalers alltrans og gauche,gauche konformationers N−H længder, strækningsvibrationer og dannelsesvarme.
ΔrN−H ΔυN−H rN−C Estab
4a R,R=H,H 0.020 401 2.95 194b R,R=H,CH3 0.019 357 2.98 18 44c R,R=CH3,CH3 0.018 349 2.99 18
5a R,R=H,H 0.020 401 2.95 195b R,R=H,CH3 0.028 480 2.94 23 55c R,R=CH3,CH3 0.033 570 2.92 29
Estab = ΔHf(trans) − ΔHf(gauche,gauche); G3//B3LYP beregninger; r i Å, υ i cm−1,298 K ΔHf i kJ mol−1.

Hydrogenbindinger 12.292
Protoniserede alkoholer og ethere. Endnu mere udpræget stabilise-rende intramolekylære vekselvirkninger gør sig gældende mellem OHog radikal-carbon i protoniserede γ- og δ-hydroxy (alkoxy) alkyl-radikaler, dvs γ- og δ-distoniske isomere af alkoholers (etheres) kation-radikaler (Tabel 12.3 og 12.4). At hydrogenbindingerne i protoniseredeoxybutylradikaler er noget stærkere end i aminobutylradikaler finderdirekte udtryk i at IR-rødskift og bindingslængdeændringerne er større,og i den større forskel mellem trans- og gauche,gauche-formernesdannelsesvarme. Resultaterne i Tabel 12.3 illustrerer dernæst at intra-molekylære hydrogenbindinger også optræder som 1,5-vekselvirk-ninger, men at disse er noget svagere end 1,6-vekselvirkninger.
Tabel 12.3. Beregnede egenskaber for protoniserede 4-methoxybutyl- og 3-methoxy-propylradikaler, i trans- og gauche-former.
rO−H υO−H rO−C ΔHf
6 0.980 3643 − 6757 1.029 2673 2.75 636
6 78 0.979 3644 − 696 8 9 9 0.996 3318 2.80 681
G3//B3LYP beregninger; r i Å, υ i cm−1, 298 K ΔHf i kJ mol−1.
Hydrogenbindinger i protoniserede hydroxyalkylradikaler er ligeledesstærkere når radikal-carbon bærer substituenter (Tabel 12.4). Proton-iserede alkoxyalkylradikaler udviser intramolekylære hydrogen-bindinger af nogenlunde samme styrke som protoniserede hydroxy-alkylradikaler (ikke vist).

Hydrogenbindinger 12.293
Tabel 12.4. Beregnede forskelle mellem protoniserede 4-hydroxyalkylradikalers all-trans- og gauche,gauche-konformeres O−H afstande, strækningsvibrationer og dannelsesvarme.
ΔrO−H ΔυO−H rO−C Estaba
10a R,R=H,H 0.063 1153 2.72 4010b R,R=H,CH3 0.086 1494 2.69 51 1010c R,R=CH3,CH3 0.118 1835 2.65 60
Estab = ΔHf(trans) − ΔHf(gauche,gauche); G3//B3LYP beregninger; r i Å, υ i cm−1,298 K ΔHf i kJ mol−1.
Protoniserede carbonylgrupper. O-protoniserede oxoalkylradikaler(distoniske isomere af aldehyders, carboxylsyrers og estres kation-radikaler) kan ligeledes udvise bemærkelsesværdige intramolekylærehydrogenbindinger, hvilket ses af de hydrogenbundne formers O−Hbindingslængder og IR egenskaber (Tabel 12.5). For γ-oxoalkylradikalerer sammenligning af dannelsesvarmen for konformere med og udenmulighed for hydrogenbinding mindre nyttig, eftersom de der ikkedanner hydrogenbinding tilsyneladende stabiliseres ved strækning afbindingen mellem C-2 og C-3 til omkring 1.7 Å; dette kunne havesammenhæng med at det protoniserede γ-oxoalkylradikal er et interme-diat i McLafferty-omlejringen, som netop indebærer at denne bindingbrydes.
Bindingslængderne og IR-egenskaberne viser også her at intra-molekylære hydrogenbindinger der indgår i en 5-leddet ring (1,5-vekselvirkninger) er noget svagere end de der indgår i en 6-leddet, og atde i en 7-leddet til gengæld synes noget stærkere (Tabel 12.5). Dettekunne skyldes at i en 7-leddet cyklisk struktur kan OHC vinklenkomme nærmere 180o, hvilket normalt [5] er fordelagtigt.

Hydrogenbindinger 12.294
Tabel 12.5. Beregnede O−H bindingslængder, O−C afstande og O−H stræknings-vibrationer for protoniserede oxoalkylradikaler i gauche-konformationer (metode og enheder som i Tabel 12.1).
rO−Ha υO−H
a rO−Hb υO−H
b rO−C
11a X,R=H,H 1.033 2640 − − 2.7611b X,R=H,CH3 1.056 2277 − − 2.7111c X,R=OH,H 1.017 2906 0.984 3598 2.8011d X,R=OH,CH3 1.032 2654 0.983 3602 2.7611e X,R=OCH3,H 1.012 2991 − − 2.81
12c 0.990 3455 0.983 3597 2.82
13d 1.041 2481 0.983 3608 2.76
(a) Hydrogen-bundet H; (b) Frit H; (c) 1,5-vekselvirkning; (d) 1,7-vekselvirkning
11 12 13

Tabel 12.6. Beregnet stabiliseringa af alkylradikalers hydrogenbundne addukter, D−H+...A.
Donor (PA) CH3. C2H5
. C3H7. iso-C3H7
. sec-C4H9. tert-C4H9
.
(522) (611) (686) (675) (725) (708)
H2O (689) 58 (4) 85 (5) 87 (6) 104 (8) 105 (8) 120 (10)
CH2=O (712) 51 (4) 76 (5) 80 (6) 95 (7) 100 [7] 112 [10]
HCN (713) 43 (3) 66 (5) 70 (5) 85 (7) 88 [7] 102 [8]
HCOOH (743) 43 (4) 66 (5) 69 (6) 82 (8) 87 [8] 97 (10)
CH3OH (755) 45 (4) 68 (5) 71 (6) 84 (7) 90 [8] 100 (9)
CH3CHO (772) 43 (3) 65 (5) 68 (6) 81 (7) 86 [8] 95 [9]
C2H5OH (777) 43 (3) 64 (5) 66 (6) 78 (7)
HCOOCH3 (783) 38 (3) 58 (5)
CH3CN (784) 34 (3) 53 (4) 55 [5] 68 [6]
CH3COOH (788) 36 (4) 56 (5) 59 [6] 70 [7] 83 [9]
(CH3)2O (793) 41 (4) 61 (5) 64 [6] 76 [7] 80 [7] 91 [9]
C2H5CN (796) 33 (3) 52 (4)
C2H5OCH3 (810) 36 (4) 56 [5]
(CH3)2CO (815) 33 (3) 53 (5) 55 [5] 66 [7]
HCONH2 (833) 33 (3) 52 (5) 53 [5] 64 [6]
NH3 (856) 28 (2) 44 (3) 46 (3) 55 (4) 59 [5] 66 (6)
CH3CONH2 (869) 29 (3) 45 [5]
CH2=NH (870) 26 (2) 41 (3) 43 (4) 52 (4) 56 [5] 63 [6]
CH3NH2 (902) 24 (3) 38 (3) 40 (4) 49 (4) 53 [5] 59 [6]
CH3N=CH2 (903) 24 (2) 38 (3) 39 [4] 48 [4]
CH3CH=NH (909) 21 (2) 35 (3) 36 [3] 44 [4]
(CH3)2NH (932) 23 (3) 36 (4) 38 [4] 47 [5]
(a) Stabilisering = ΣΔHf(komponenter) − ΔHf(addukt); alle værdier beregnet med G3//B3LYP ogangivet i kJ mol−1 (298 K); G3 counterpoise korrektioner i parentes, G3(MP2) ditto i kantetparentes; øvrige data i [32], supporting information.

Hydrogenbindinger 12.296
Intermolekylære hydrogenbindinger – Addukter
Hydrogenbindinger mellem molekyler er i almindelighed noget stær-kere end hydrogenbindinger indenfor samme molekyle, antagelig fordimolekylerne i et addukt nogenlunde frit kan orientere sig således atD−H...A bindingsvinklen kan nærme sig 180o [5]. Dette gælder ogsåintermolekylære hydrogenbindinger til alkylradikaler, hvor adduktermed gode, ladede protondonorer kan være stabiliserede med over 100kJ mol−1 (Tabel 12.6). Addukternes interatomare afstande og deberegnede IR-bølgetal og intensiteter bekræfter entydigt at ioniskeprotondonorer som ammoniumioner, oxoniumioner og molekyler medprotoniserede carbonylgrupper danner gode intermolekylærehydrogenbindinger til radikal-carbon
Der er god overensstemmelse mellem resultaterne i Tabel 12.6 ogresultater fra beregninger med mere krævende metoder. Den addukt-stabilisering for [H3O+...CH3
.] og [NH4+...CH3
.] der fremgår afberegninger med CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ ligger tætpå G3-resultaterne (Tabel 12.7). Tilsvarende beregninger er ikke udførtfor hovedparten af de øvrige addukter i Tabel 12.6; det er nok tvivlsomtom resultaterne rent faktisk ville komme 'virkelighedens' termokemimeget nærmere end G3-beregningers.
Tabel 12.7. Stabilisering (elektronisk energi, kJ mol−1) af simple addukter beregnetmed forskellige metoder.
[H3O+...CH3.] G3 62 [NH4
+...CH3.] G3 34
G3//B3LYP 61 G3//B3LYP 34CCSD(T)a 65 CCSD(T)a 37
(a) CCSD(T)/aug-cc-pVTZ//MP2/aug-cc-pVTZ.
Det er vanskeligt at afgøre præcis hvor stor en del hydrogen-bindingernes bidrag udgør af den samlede stabilisering (Tabel 12.6).Addukter af neutrale molekyler og ioner af enhver art bindes altidsammen af elektrostatiske vekselvirkninger, også i fuldstændigt fraværaf hydrogenbindinger, og det er ikke umiddelbart muligt at sondremellem de forskellige bidrags størrelse; dette forhold diskuteresnærmere nedenfor.
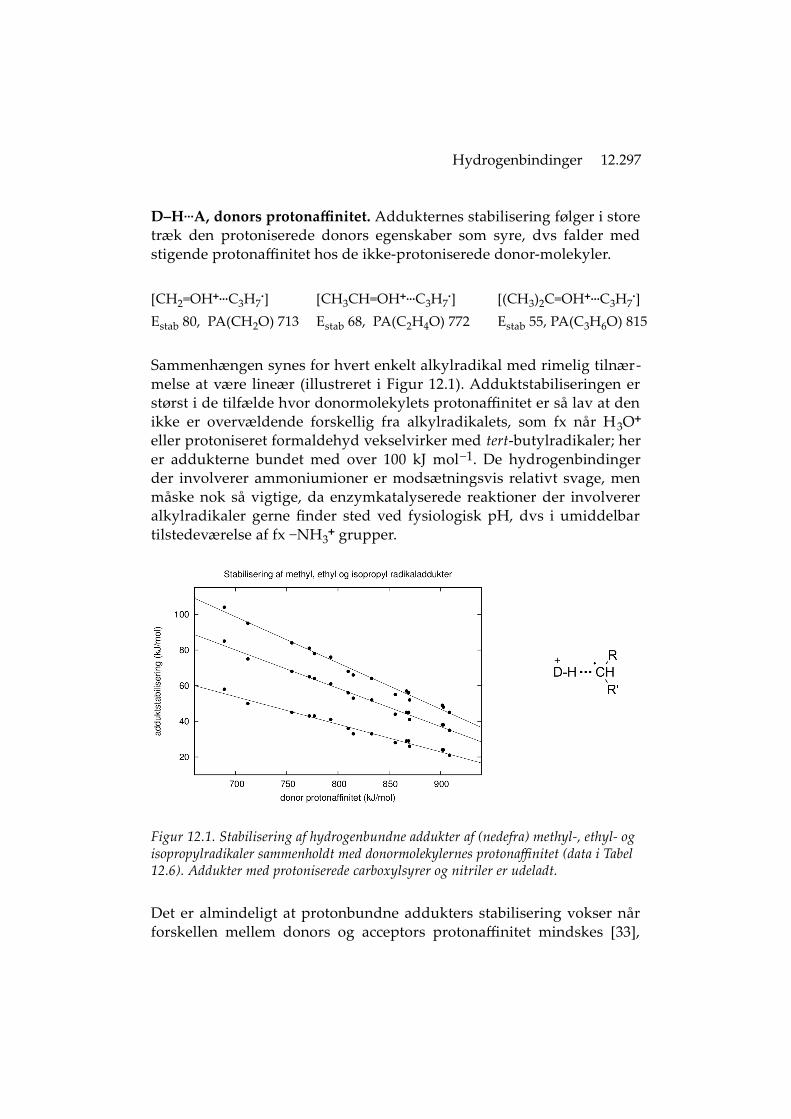
Hydrogenbindinger 12.297
D‒H...A, donors protonaffinitet. Addukternes stabilisering følger i storetræk den protoniserede donors egenskaber som syre, dvs falder medstigende protonaffinitet hos de ikke-protoniserede donor-molekyler.
[CH2=OH+...C3H7.] [CH3CH=OH+...C3H7
.] [(CH3)2C=OH+...C3H7.]
Estab 80, PA(CH2O) 713 Estab 68, PA(C2H4O) 772 Estab 55, PA(C3H6O) 815
Sammenhængen synes for hvert enkelt alkylradikal med rimelig tilnær-melse at være lineær (illustreret i Figur 12.1). Adduktstabiliseringen erstørst i de tilfælde hvor donormolekylets protonaffinitet er så lav at denikke er overvældende forskellig fra alkylradikalets, som fx når H3O+
eller protoniseret formaldehyd vekselvirker med tert-butylradikaler; herer addukterne bundet med over 100 kJ mol−1. De hydrogenbindingerder involverer ammoniumioner er modsætningsvis relativt svage, menmåske nok så vigtige, da enzymkatalyserede reaktioner der involvereralkylradikaler gerne finder sted ved fysiologisk pH, dvs i umiddelbartilstedeværelse af fx −NH3
+ grupper.
Figur 12.1. Stabilisering af hydrogenbundne addukter af (nedefra) methyl-, ethyl- og isopropylradikaler sammenholdt med donormolekylernes protonaffinitet (data i Tabel 12.6). Addukter med protoniserede carboxylsyrer og nitriler er udeladt.
Det er almindeligt at protonbundne addukters stabilisering vokser nårforskellen mellem donors og acceptors protonaffinitet mindskes [33],
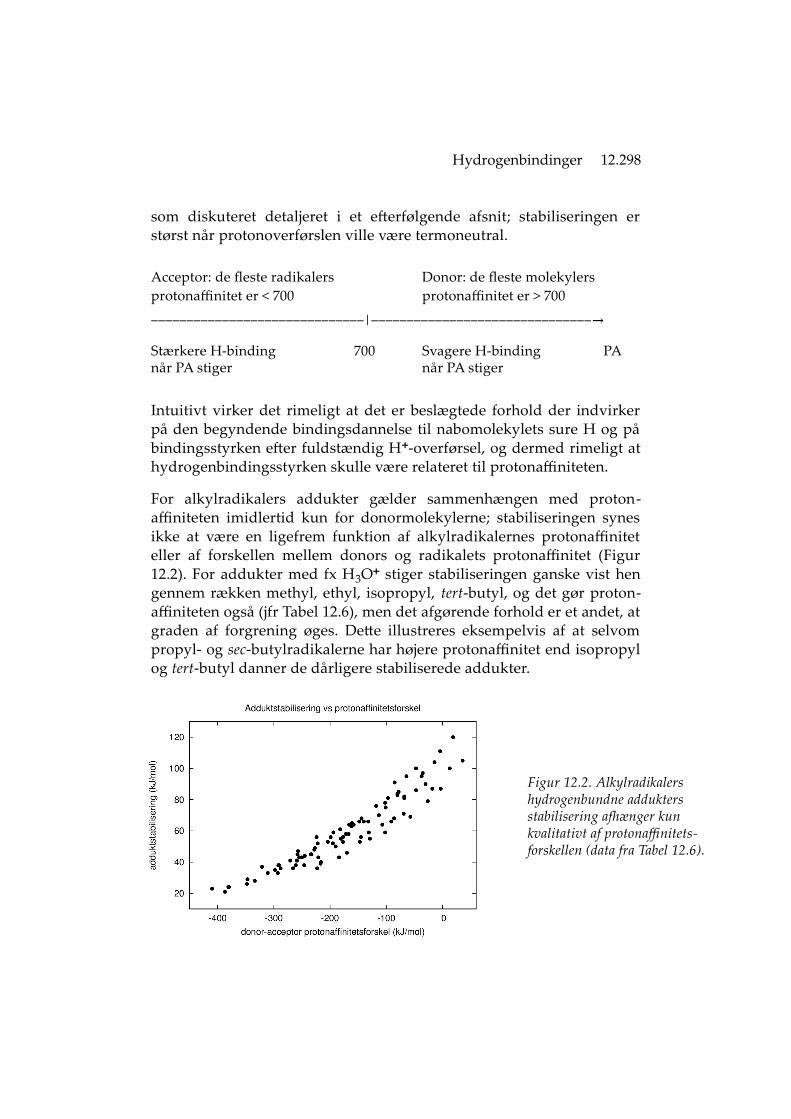
Hydrogenbindinger 12.298
som diskuteret detaljeret i et efterfølgende afsnit; stabiliseringen erstørst når protonoverførslen ville være termoneutral.
Acceptor: de fleste radikalers Donor: de fleste molekylersprotonaffinitet er < 700 protonaffinitet er > 700
−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−|−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−−→
Stærkere H-binding 700 Svagere H-binding PAnår PA stiger når PA stiger
Intuitivt virker det rimeligt at det er beslægtede forhold der indvirkerpå den begyndende bindingsdannelse til nabomolekylets sure H og påbindingsstyrken efter fuldstændig H+-overførsel, og dermed rimeligt athydrogenbindingsstyrken skulle være relateret til protonaffiniteten.
For alkylradikalers addukter gælder sammenhængen med proton-affiniteten imidlertid kun for donormolekylerne; stabiliseringen synesikke at være en ligefrem funktion af alkylradikalernes protonaffiniteteller af forskellen mellem donors og radikalets protonaffinitet (Figur12.2). For addukter med fx H3O+ stiger stabiliseringen ganske vist hengennem rækken methyl, ethyl, isopropyl, tert-butyl, og det gør proton-affiniteten også (jfr Tabel 12.6), men det afgørende forhold er et andet, atgraden af forgrening øges. Dette illustreres eksempelvis af at selvompropyl- og sec-butylradikalerne har højere protonaffinitet end isopropylog tert-butyl danner de dårligere stabiliserede addukter.
Figur 12.2. Alkylradikalers hydrogenbundne addukters stabilisering afhænger kun kvalitativt af protonaffinitets-forskellen (data fra Tabel 12.6).

Hydrogenbindinger 12.299
D‒H...A, acceptors ioniseringsenergi. Addukternes stabilisering synesderimod tydeligt korreleret med alkylradikalernes ioniseringsenergi,højere for tertiære radikaler end for sekundære, højere for sekundæreend for primære, og for primære højere end for methyl (Figur 12.3). Denvarierer på nogenlunde samme måde indenfor de enkelte grupper.
Figur 12.3. Stabilise-ring af alkylradikalers addukter (Tabel 12.6) med (nedefra) NH4
+, CH3OH2+ og H3O + sammenholdt med radikalernes vertikale ioniseringsenergi; de indtegnede rette linier skal blot fremhæve tendensen.
At acceptors ioniseringsenergi kunne være af betydning for hydrogen-bindingers styrke falder godt i tråd med en række tidlige overvejelser,der med teoretisk udgangspunkt inddrog H-acceptormolekylets ionise-ringsenergi som en medbestemmende faktor [41-47]. Det aspekt har i endel år været tillagt mindre betydning, uden dog at være helt glemt, menden sammenhæng som resultaterne i Figur 12.3 peger på kan tages tilindtægt for at et væsentligt bidrag til hydrogenbindingsstyrken i alkyl-radikalers addukter skyldes vekselvirkninger beslægtet med ladnings-overførsel; også dette forhold diskuteres nærmere nedenfor.
Korrelationen mellem addukternes stabilisering og både donorernesprotonaffinitet og alkylradikalernes vertikale ioniseringsenergi viser sigat kunne udtrykkes simpelt, med ligning (1), der indenfor få kJ mol −1
gengiver stabiliseringsenergien tilstrækkelig godt til at vise at disseegenskaber er af stor betydning for vekselvirkningen (se Figur 12.4øverst):
Estab/kJ mol−1 = 394 − 0.22 PA(donor) − 0.19 IE(radikal) (1)
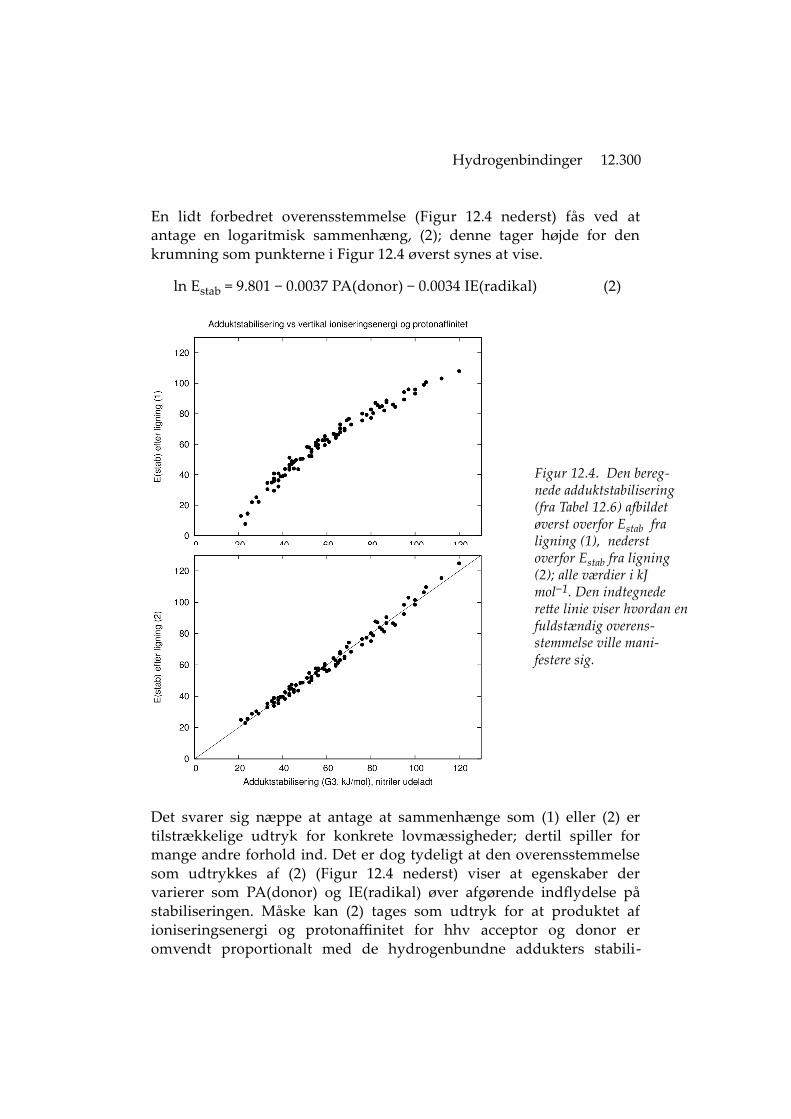
Hydrogenbindinger 12.300
En lidt forbedret overensstemmelse (Figur 12.4 nederst) fås ved atantage en logaritmisk sammenhæng, (2); denne tager højde for denkrumning som punkterne i Figur 12.4 øverst synes at vise.
ln Estab = 9.801 − 0.0037 PA(donor) − 0.0034 IE(radikal) (2)
Figur 12.4. Den bereg-nede adduktstabilisering (fra Tabel 12.6) afbildet øverst overfor Estab fra ligning (1), nederst overfor Estab fra ligning (2); alle værdier i kJ mol−1. Den indtegnede rette linie viser hvordan enfuldstændig overens-stemmelse ville mani-festere sig.
Det svarer sig næppe at antage at sammenhænge som (1) eller (2) ertilstrækkelige udtryk for konkrete lovmæssigheder; dertil spiller formange andre forhold ind. Det er dog tydeligt at den overensstemmelsesom udtrykkes af (2) (Figur 12.4 nederst) viser at egenskaber dervarierer som PA(donor) og IE(radikal) øver afgørende indflydelse påstabiliseringen. Måske kan (2) tages som udtryk for at produktet afioniseringsenergi og protonaffinitet for hhv acceptor og donor eromvendt proportionalt med de hydrogenbundne addukters stabili-

Hydrogenbindinger 12.301
sering, en mulig sammenhæng som ligger i forlængelse af et forslagsom Allen fremførte allerede i 1975 [46] (dog med en anden angivelse afdonormolekylets baseegenskaber). Denne opfattelse har ikke sidenværet meget diskuteret i litteraturen, men omtales nærmere i etefterfølgende afsnit. Adduktstabilisering er imidlertid ikke det sammesom hydrogenbindingsstyrke, omend denne indgår. Sondringen er envigtig del at bindingsovervejelserne.
Lidt overraskende synes korrelationen at holde selv når proton-affinitetsforskellene er så store som 300 kJ mol−1 eller mere, somtilfældet er når hydrogenbindingen går mellem fx methylradikalet ogammoniumionen. Dette afspejler dog måske mest at vekselvirkningen idisse tilfælde er relativt svag. Adduktstabiliseringen for CH3
. medprimære, sekundære eller tertiære ammoniumioner (Tabel 12.6) variererikke meget i styrke, uanset at aminernes protonaffinitet er op til 100 kJmol−1 forskellige.

Hydrogenbindinger 12.302
Struktur- og vibrationsegenskaber hos hydrogenbundne addukter
Hydrogenbindingen i et [D−H...A] addukt giver sig direkte til kendegennem sin indvirkning på D−H bindingslængden og derigennem på IRstrækningsvibrationerne; de resulterende rødskift og intensitetsfor-øgelser betragtes gerne som hydrogenbindingens signatur [5,20,48].Som diskuteret nedenfor synes der i nærtbeslægtede systemer at væreen systematisk relation mellem hydrogenbindingens styrke og rød-skiftets størrelse. Også intensitetsforøgelsen følger i almindelighedhydrogenbindingsstyrken [49].
Alkylradikalers hydrogenbundne addukter udviser i overensstemmelsehermed forøget donor O−H eller N−H bindingslængde. Eksempelvis seshos det relativt svagt bundne [NH4
+...CH3.] addukt en forøgelse på 3%; i
det stærkere bundne [CH3OH2+...tert-C4H9
.] addukt er forøgelsen på17%. I store træk følger bindingslængdens ændring den beregnedestabilisering og det rødskift som donor−H strækningsvibrationerneudviser. For moderat bundne addukter drejer det sig om ændringer påmåske 5-600 cm−1, for stærkt bundne om over 2000 cm−1 (eksem-plificeret i Tabel 12.8-12.10). Som indikation på at hydrogenbindinger ertil stede udgør ændringen af strækningsbølgetallet et meget følsomtmål, måske endog lovlig følsomt.
Tabel 12.8. Beregnede O−H IR rødskift og O−H bindingslængdeændringer for hydrogen-bundne addukter af alkylradikaler og protoniseret methanol, [CH3OH2+...alkyl .].
ΔrOH ΔνOH rO−C Estab
CH3. 0.056 1062 2.83 45
C2H5. 0.089 1537 2.75 68
C3H7. 0.102 1717 2.73 71 CH3OH2
+ rOH 0.978iso-C3H7
. 0.120 1895 2.71 84 νOH 3742, 3648C4H9
. 0.118 1921 2.70 76 CH3OH2+...CH3
. rOH 0.977, 1.033iso-C4H9
. 0.113 1860 2.71 77 νOH 2649, 3711sec-C4H9
. 0.123 1946 2.70 90tert-C4H9
. 0.145 2125 2.69 100
Afstande (Å) og bølgetal (cm-1) fra B3LYP/6-31+G(d,p) beregninger; Estab (kJ mol1) bestemt ud fra G3//B3LYP beregningsresultater.

Hydrogenbindinger 12.303
I de tilfælde hvor bølgetallet for den involverede OH eller NHstrækningsvibration ligger omkring 3000 cm−1 eller under 1900 cm−1
viser beregningerne ofte stærk kobling til andre vibrationer. Det er dogsædvanligvis muligt at bestemme det 'rigtige' bølgetal ved hvad mankunne kalde 'virtuel isotopmærkning', dvs ved at beregne IR-egen-skaberne for hensigtsmæssigt deuterium-substituerede analoger, enfremgangsmåde som også andre har benyttet [50]. Eksempelvis koblerden bindende N−H strækningsvibration i [NH4
+...CH3.] med strækning
af de øvrige N−H bindinger; rødskiftet kan imidlertid bestemmes ifravær af forstyrrende kobling ud fra de beregnede IR-egenskaber for[ND3H+...CH3
.] og [NHD3+...CH3
.].
Tabel 12.9. Beregnede IR rødskift og bindingslængdeændringer for hydrogenbundne addukter af methylradikaler og forbindelser med protoniserede carbonylgrupper.
ΔrOH ΔνOH rO−C Estab
CH2=OH+ 0.078 1498 2.77 53CH3CH=OH+ 0.053 1042 2.83 43(CH3)2C=OH+ 0.039 790 2.90 33HC(OH)2
+ 0.053 1034 2.83 43CH3C(OH)2
+ 0.038 790 2.88 36
Afstande (Å) og bølgetal (cm−1) fra B3LYP/6-31+G(d,p) beregninger, Estab fra Tabel12.6; sammenligning mellem adduktets OH-grupper for protoniserede carboxylsyrer,ellers sammenligning mellem addukt og de tilsvarende adskilte komponenter.
De beregnede O−H og N−H afstande afhænger lidt af beregnings-metoden; ændringerne i forhold til de ikke-hydrogenbundne kompo-nenter er mindre når MP2-metoden anvendes end ved DFT-beregninger.Imidlertid er afstanden mellem de involverede tunge atomer ret nærden samme uanset beregningsmetode, i god overensstemmelse med atden beregnede stabilisering også er nogenlunde den samme. Forskel-lene afspejler at beregningsmetoderne placerer det bindende hydrogen-atom lidt forskelligt mellem C og O (N); dermed påvirkes også deberegnede bølgetal for donor−H strækningsvibrationerne, som i over-ensstemmelse med Badgers regel [51] er lavere når bindingerne erlængere.

Hydrogenbindinger 12.304
Tabel 12.10. Beregnede IR rødskift og bindingslængdeændringer for hydrogenbundneaddukter af ethylradikaler og protoniserede aminer og iminer, [XNH+...C2H5
.].
ΔrNH ΔνNH rN−C Estab
NH4+ 0.047 810 3.00 44
CH3NH3+ 0.036 636 3.05 38
(CH3)2NH2+ 0.030 539 3.09 36
CH2=NH2+ 0.040 730 3.01 41
CH3CH=NH2+ 0.030 572 3.07 35
CH3NH=CH2+ 0.032 586 3.06 38
Enheder og metoder som i Tabel 12.9
O−H og N−H strækningsvibrationernes intensitet forøges betydeligt nårH-atomet indgår i hydrogenbindinger [49]; for addukter af alkyl-radikaler vokser de beregnede absorptionsintensiteter i reglen medmere end en faktor 10 (eksempler i Tabel 12.11 og Figur 12.5). Detteletter tilordningen for såvidt angår de vibrationer der involvererbindingshydrogenatomet.
Tabel 12.11. Beregnede IR bølgetal (cm−1) og absorptionsintensiteter (km mol−1) for hydrogenbundent og frit H i simple addukters −OH2+ og −NH2+ grupper.
νhb inthb νfri intfri
HC(OH)2+...CH3
. 2608 2322 3642 241HC(OH)2
+...C2H5. 2078 2816 3653 227
CH3OH2+...CH3. 2649 1927 3711 230
CH3OH2+...iso-C3H7
. 1830 2701 3725 176(CH3)2NH2
+...CH3. 3070 662 3462 68
(CH3)2NH2+...tert-C4H9
. 2754 1434 3467 56
B3LYP/6-31+G(d,p) beregningsresultater
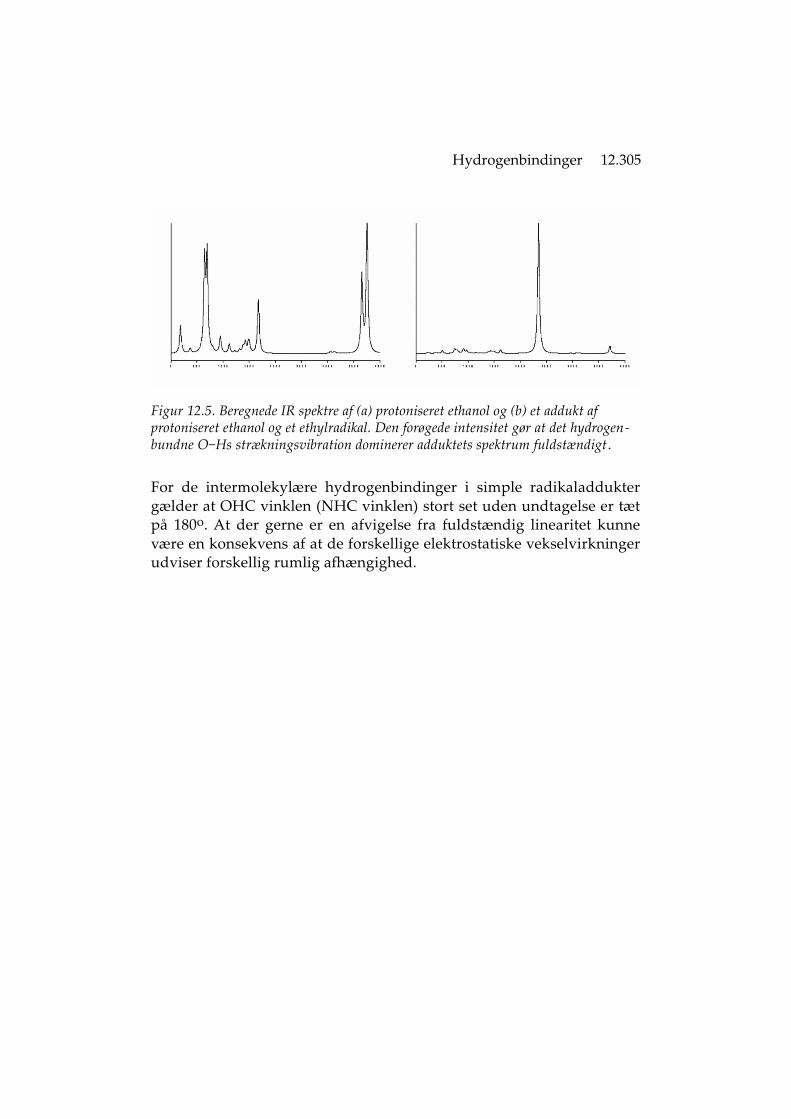
Hydrogenbindinger 12.305
Figur 12.5. Beregnede IR spektre af (a) protoniseret ethanol og (b) et addukt af protoniseret ethanol og et ethylradikal. Den forøgede intensitet gør at det hydrogen-bundne O−Hs strækningsvibration dominerer adduktets spektrum fuldstændigt .
For de intermolekylære hydrogenbindinger i simple radikaladduktergælder at OHC vinklen (NHC vinklen) stort set uden undtagelse er tætpå 180o. At der gerne er en afvigelse fra fuldstændig linearitet kunnevære en konsekvens af at de forskellige elektrostatiske vekselvirkningerudviser forskellig rumlig afhængighed.

Hydrogenbindinger 12.306
Ioniske vekselvirkninger
Ladede hydrogenbundne addukters stabilisering indeholder et betyde-ligt bidrag fra den elektrostatiske vekselvirkning der nødvendigvis gørsig gældende når en ion befinder sig i umiddelbar nærhed af et polari-serbart molekyle. Alkylradikalers dipolmomenter er tilstrækkeligt småtil at det er rimeligt at se bort fra ion-dipol vekselvirkninger, men det erderimod nødvendigt at tage hensyn til det bidrag til stabiliseringen derstammer fra højere multipoler og fra inducerede multipoler. For atskønne over hvor stort et bidrag der kunne skyldes den inducerededipol kan vekselvirkningen mellem en punktladning og et polariserbartatom beregnes, V = −695α/r4 (i kJ mol−1), hvor α er atomets polari-sabilitet og r er afstanden mellem atom og punktladning. Anvendt på[H3O+...CH3
.] adduktet giver dette udtryk en beregnet elektrostatiskvekselvirkning på 29 kJ mol−1, idet methylradikalet har nogenlundesamme polarisabilitet som methan, 2.6 Å3, og r tages som C−O af-standen. Til sammenligning er adduktets samlede stabilisering 58 kJmol−1. For [NH4
+...CH3.] fås 20 kJ mol−1 (samlet stabilisering 28 kJ
mol−1).
Molekyler er ikke punkter. Når afstanden er så lille som i disseaddukter udgør de således beregnede værdier dog kun en grovtilnærmelse; de færreste ioner og molekyler er punktformede, og veksel-virkning med en delokaliseret ladning adskiller sig fra vekselvirkningmed en punktladning. Dertil kommer at højere-ordens effekter kanspille en betydelig rolle. Bl.a. Kebarle [52] og senere Dopfer [53] harpåpeget at også ion-quadrupol vekselvirkninger bidrager til stabilise-ringen af addukter med ladning, i god overensstemmelse med at højere-ordens vekselvirkninger generelt vejer tungere når ion og molekylebefinder sig tæt på hinanden [54,55].
Neutrale uden H-bindinger. Det er imidlertid problematisk at finde etsystem der giver mulighed for at anslå størrelsen af den elektrostatiskevekselvirkning mellem fx H3O+ og et polariserbart, neutralt molekyleder ikke danner hydrogenbinding. Selv til Ar og N2 danner H3O+
hydrogenbindinger, at dømme efter de målte og beregnede IR rødskift[56], og det synes ikke muligt ad denne vej at få et godt mål for den rentelektrostatiske vekselvirkning mellem neutrale molekyler og ladedeprotondonorer med en vis udstrækning.

Hydrogenbindinger 12.307
Kationer uden H-bindinger. Et vist indtryk af disse forhold kanderimod fås ved at betragte addukter med ioner der ikke kan dannehydrogenbinding, fx metalioner. Med G3-beregninger findes at et[Na+ CH3
.] addukt er 40 kJ mol−1 lavere i energi end de adskiltekomponenter; for [Na+ CH4] fås 31 kJ mol−1, i god overensstemmelsemed den eksperimentelle værdi på 30 kJ mol−1 [57] (Tabel 12.12). Dissetal svarer nogenlunde til den ovenfor anslåede ion-induceret dipolvekselvirkning, og kunne tyde på at måske omkring halvdelen af denberegnede stabilisering for [H3O+...CH3
.] adduktet blot skyldes at denene komponent bærer ladning.
Tabel 12.12. Addukter af simple ioner og carbonhydrider.
CH3. CH4 tert-C4H9
. iso-C4H10Estab rC−X Estab rC−X Estab rC−X Estab rC−X
H3O+ 58 2.79 35 2.94 120 2.67 51 3.16Na+ 40 2.67 31 2.59 76 2.56 52 2.71K+ 22 3.13 15 3.14 50 3.09 29 3.31NH4
+ 28 3.07 16 3.27 66 2.93 25 3.48
Stabilisering bestemt med G3//B3LYP (kJ mol−1, 298 K); tung-atom afstande (Å)bestemt med B3LYP/6-31+G(d,p).
K+ og NH4+. For addukter med svagere hydrogenbindinger udgør deioniske vekselvirkninger en forholdsmæssigt endnu større andel. K+ ogNH4
+ er nogenlunde lige store, og deres addukter med neutrale mole-kyler burde udvise nogenlunde de samme ioniske vekselvirkninger.Eksperimentelle undersøgelser har da også vist at K+ og NH4
+ adduktermed polære og upolære neutrale molekyler ikke er særlig forskelligtstabiliseret [58], hvilket indebærer at hydrogenbindinger i NH4
+-addukter kun kan stå for en mindre del af vekselvirkningen.
Dersom K+-addukternes stabilisering er et udtryk for de ioniske veksel-virkninger kan denne trækkes fra NH4
+-addukternes; forskellen skulleså udgøre hydrogenbindingsstyrken. Ud fra resultaterne i Tabel 12.12fås at hydrogenbindinger bidrager til stabiliseringen af addukternemellem NH4
+ og hhv CH3. og tert-C4H9
. med omkring 5 og 15 kJ mol−1,

Hydrogenbindinger 12.308
hvor vekselvirkningen mellem to vandmolekyler til sammenligningudgør omkring 20 kJ mol−1 [59]. Dette overslag er dog nok på den laveside når man tager den beregnede stabilisering af δdistoniske aminioners gaucheformer i betragtning (ovenfor, Tabel 12.1).
Addukter af neutrale molekyler og hhv Na+ og H3O+ kan sammenlignes på tilsvarende vis, med nogenlunde tilsvarende resultater.
Ladning. Hydrogenbindinger i ioniske systemer er gerne betydeligtstærkere end hydrogenbindinger mellem neutrale molekyler [60,61]. Envigtig årsag hertil er at komponenternes indbyrdes afstand formindskesaf den ioniske tiltrækning, og alene af den grund vil hydrogen-bindingerne kunne spille en større rolle.
Eftersom en betydelig del af de tiltrækkende vekselvirkninger i ioniskeaddukter alene skyldes at en af komponenterne bærer en ladning er detret påfaldende at adduktstabiliseringen alligevel hyppigt fremstillessom var den et direkte udtryk for hydrogenbindingernes styrke, uansetat de ioniske vekselvirkninger ikke er en del af hydrogenbindingen ogkun i begrænset omfang er en forudsætning. Der er med Steiners ord tittale om "ionic interactions with a moderate hydrogen bond on top" [6]. Da deioniske vekselvirkninger varierer fra addukt til addukt kan denmanglende sondring være en medvirkende årsag til vanskelighederne[33] med at få overordnet sammenhæng ud af resultaternes systematik.
Terminologien udgør i tilgift et problem, idet der gør sig to former forelektrostatiske vekselvirkninger gældende, de der optræder i kraft af atadduktets ene komponent bærer en ladning (her benævnt ioniskevekselvirkninger), og de der fortrinsvis skyldes lokale dipol-veksel-virkninger omkring det hydrogenatom der er involveret i hydrogen-bindingen. Imidlertid er det ioniske bidrag til stabiliseringen af etkonkret alkylradikals addukter nok med rimelig tilnærmelse en faststørrelse, til og med mindre end for de fleste andre hydrogenbindings-acceptorer, på grund af fraværet af en kendelig permanent dipol.Desuden er alkylradikalets orientering i forhold til ladningen nogen-lunde uændret fra addukt til addukt, og afstanden varierer tilsyne-ladende nogenlunde monotont med den samlede stabilisering. Dermedvil de ioniske vekselvirkninger ikke maskere eventuelle tydeligtsystematiske træk ved hydrogenbindingsstyrkens variation, som fxsammenhængen med ioniseringsenergi eller protonaffinitet; de viltværtimod kunne have en forstærkende virkning.

Hydrogenbindinger 12.309
Delokalisering? Hydrogenbundne addukter af protoniserede nitrilerog andre carboxylsyrederivater er lidt dårligere stabiliseret endforventet ud fra donorernes protonaffinitet. Dette gælder særligprotoniserede nitriler (se Tabel 12.6 og Figur 12.1), hvilket kunneafspejle at de ioniske vekselvirkninger her er noget svækket på grund afdelokalisering af ladningen. Mulige årsager til at også det kovalentebidrag til hydrogenbindingen kunne være lavere for protoniseredenitriler omtales nedenfor.
For protoniserede carboxylsyrer gør sig noget måske beslægtetgældende. Disse kan på deres mest stabile form danne to forskelligeaddukter med alkylradikaler (Figur 12.6); i det bedre stabiliseredeaddukt indgår E-hydrogenatomet i hydrogenbindingen (a i Figur 12.6),hvorimod den stærkeste hydrogenbinding involverer det mindre sureZ-hydrogenatom (b i Figur 6). Dette ses for fx [HC(OH)2
+...C2H5.] ved at
(a) er bedre stabiliseret men har kortere O−H binding og mindrerødskift. Med lukket-skals hydrogenbindingsacceptorer ses helt tilsva-rende forskelle [62,63], ligeledes i tydelig modstrid med den forventedeoverensstemmelse mellem adduktstabilisering og protonaffinitets-forskel (se nedenfor). Dette afspejler antagelig at de to OH-grupper erforskellige i henseende til såvel elektrostatik som elektronacceptor-egenskaber. Rablen et al. [12] har konstateret at der også kan væreforskel på styrken af de hydrogenbindinger som forskellige konformereaf neutrale carboxylsyrer kan danne.
ΔrOH 0.091 Å ΔrOH 0.101 ÅΔνOH 1575 cm−1 ΔνOH 1801 cm−1 Estab 66 kJ mol−1 Estab 63 kJ mol−1
PA 743 kJ mol−1 PA 760 kJ mol−1
Figur 12.6. To addukter af protoniseret myresyre og et ethylradikal. Form (a) er bedrestabiliseret, mens form (b) har stærkere hydrogenbinding. ΔrOH og ΔνOH er angiveti forhold til protoniseret myresyre (E,Z), PA er for hhv Z- og E-myresyre.

Hydrogenbindinger 12.310
Den stabilisering som ledsager adduktdannelse med protoniseredecarboxylsyrer er desuden kendeligt mindre end den man ser med andredonorer, hvilket eksemplificeres af indgangene for HCOOH og CH3OHi Tabel 12.6 − samme stabilisering på trods af ~30 kJ mol−1 proton-affinitetsforskel − og for CH3COOH og CH3OCH3 − nogenlunde sammeprotonaffinitet, men tydeligt forskellig stabilisering.

Hydrogenbindinger 12.311
Bindingsenergi
Hydrogenbindingers styrke kan ikke måles for sig, da de altid ledsagesaf andre vekselvirkninger, i særdeleshed når der er tale om ladedeprotondonorer. Af samme grund kan hydrogenbindingers styrke hellerikke beregnes, om end de for svage bindinger nok kan anslås [64-67].
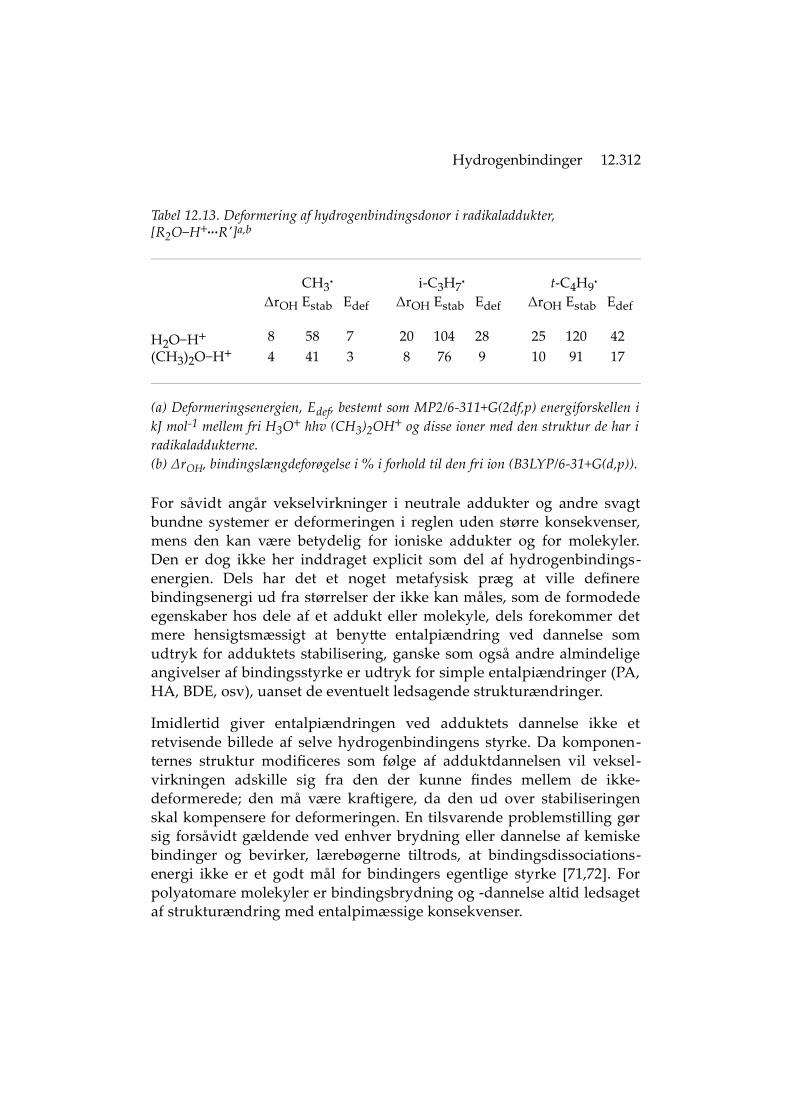
For addukter med to komponenter bestemmes stabiliseringen ved attrække systemets samlede energi fra summen af komponenternes, dvsudtrykkes som entalpiændringen når adduktet frembringes ud frakomponenterne (kaldet Estab i det foranstående). Når adduktet bærerladning, fx [D−H+...A], er Estab den samlede konsekvens af hydrogen-bindingsvekselvirkningerne og de ioniske vekselvirkninger. Imidlertidhar D−H+ og A i adduktet ikke samme struktur (og dermed ikke sammeenergi) som de har som frie molekyler (før dannelse af addukt)[15,23,68-70]. D−H+ og A deformeres når de føres sammen, stiger dervedi energi, og den 'virkelige' bindingsenergi bliver dermed højere,eftersom den afspejler komponenternes egentlige energi i adduktet.
Deformering. I hydrogenbindingsvekselvirkningerne indgår bidrag frasåvel hydrogenbindingen, EHB, som fra den energi der er medgået til atdeformere adduktets komponenter, Edef. Deformeringen (eng. fragmentrelaxation) består i det væsentlige i at D−H bindingen er blevetforlænget; stærkere hydrogenbinding indebærer forøget strækning.Derved kan deformeringsenergien blive anseelig (op imod en trediedelaf Estab). Alkylradikalets struktur er derimod ikke ændret i særlig grad;tert-butylradikalets energi vokser således mindre end 3 kJ mol -1 vedadduktdannelse med H3O+ eller (CH3)2OH+ (Tabel 12.13).
Entalpiændringer og strukturændringer ved adduktdannelse kanberegnes eller i det mindste anslås, men vurderingen af hydrogen-bindingens styrke afhænger af hvordan (eller om) deformeringen ind-regnes. Begrebsmæssigt består adduktstabiliseringen af tre elementer:
Estab = EHB + Eion ‒ Edef
En sådan opdeling er dog ikke så nyttig i praksis, da de tre elementerikke er uafhængige; ændring af ét har umiddelbare konsekvenser for deto øvrige, hvilket gør simple årsag-virkning undersøgelser vanskelige.Eksempelvis vil forstærkede ioniske vekselvirkninger bevirke for-mindsket afstand mellem adduktets komponenter, og dervedbegunstige hydrogenbindingen, hvilket igen forøger deformeringen.
Hydrogenbindinger 12.312
Tabel 12.13. Deformering af hydrogenbindingsdonor i radikaladdukter, [R2O−H+...R']a,b
CH3. i-C3H7
. t-C4H9.
ΔrOH Estab Edef ΔrOH Estab Edef ΔrOH Estab Edef
H2O−H+ 8 58 7 20 104 28 25 120 42(CH3)2O−H+ 4 41 3 8 76 9 10 91 17
(a) Deformeringsenergien, Edef, bestemt som MP2/6-311+G(2df,p) energiforskellen ikJ mol-1 mellem fri H3O+ hhv (CH3)2OH+ og disse ioner med den struktur de har iradikaladdukterne.(b) ΔrOH, bindingslængdeforøgelse i % i forhold til den fri ion (B3LYP/6-31+G(d,p)).
For såvidt angår vekselvirkninger i neutrale addukter og andre svagtbundne systemer er deformeringen i reglen uden større konsekvenser,mens den kan være betydelig for ioniske addukter og for molekyler.Den er dog ikke her inddraget explicit som del af hydrogenbindings-energien. Dels har det et noget metafysisk præg at ville definerebindingsenergi ud fra størrelser der ikke kan måles, som de formodedeegenskaber hos dele af et addukt eller molekyle, dels forekommer detmere hensigtsmæssigt at benytte entalpiændring ved dannelse somudtryk for adduktets stabilisering, ganske som også andre almindeligeangivelser af bindingsstyrke er udtryk for simple entalpiændringer (PA,HA, BDE, osv), uanset de eventuelt ledsagende strukturændringer.
Imidlertid giver entalpiændringen ved adduktets dannelse ikke etretvisende billede af selve hydrogenbindingens styrke. Da komponen-ternes struktur modificeres som følge af adduktdannelsen vil veksel-virkningen adskille sig fra den der kunne findes mellem de ikke-deformerede; den må være kraftigere, da den ud over stabiliseringenskal kompensere for deformeringen. En tilsvarende problemstilling gørsig forsåvidt gældende ved enhver brydning eller dannelse af kemiskebindinger og bevirker, lærebøgerne tiltrods, at bindingsdissociations-energi ikke er et godt mål for bindingers egentlige styrke [71,72]. Forpolyatomare molekyler er bindingsbrydning og -dannelse altid ledsagetaf strukturændring med entalpimæssige konsekvenser.

Hydrogenbindinger 12.313
Deformering ved strækning af donor−H bindingen indebærer forbedretmulighed for ladningsoverførsel. Herved kan entalpiændringerne vedladningsoverførsel tildels opveje deformeringen, og deres bidrag til densamlede stabilisering fremstår derfor mindre tydeligt.
IR rødskif. Den ændring af vibrationsegenskaber der ledsager addukt-dannelse er et konkret udtryk for sammenhængen mellem hydrogen-bindingens styrke og D−H bindingens svækkelse. IR frekvensændringener en direkte konsekvens af deformeringen og dermed af hydrogen-bindingens tilstedeværelse og styrke. Det kan måske synes lidtparadoksalt, men det bedste tilgængelige mål for hydrogenbindingensstyrke fås således ikke ved at betragte hydrogenbindingen, menderimod svækkelsen af den binding som hydrogenbindingen tildelserstatter. Den bagved liggende ændring af donor−H bindingslængdenkan også bestemmes direkte, ved måling eller ved beregning, ikke kungennem den ændring af vibrationsegenskaberne den forårsager.
Figur 12.7. Bliver hydrogenbindingen stærkere bliver O−H bindingen længere og svagere, derfor IR rødskift.
Det blev ret tidligt konstateret at hydrogenbindingsdannelse bevirkedeændring af donors IR egenskaber og at der tilsyneladende var enrelation mellem rødskiftets størrelse og hydrogenbindingens styrke[73,74]. Den oprindeligt foreslåede proportionalitet synes dog kun atgælde for nærtbeslægtede forbindelser [75]. Da rødskiftet direkteafspejler egenskaber hos det hydrogenbundne system (bindingslængde-ændringer) antages rødskiftets størrelse at afspejle relativ hydrogen-bindingsstyrke. I litteraturen er de mulige sammenhænge mellemhydrogenbindingens styrke, adduktstabilisering, IR-egenskaber, ogdonor-acceptor protonaffinitetsforskellen stadigt tilbagevendendeemner [76], ind imellem suppleret med korrelation med beregnedestørrelser (se afsnittet om NBO og AIM beregninger).
Komponenternes protonaffinitet. Hydrogenbindingens styrke i kat-ioniske addukter, [D–H+...A], er afhængig af hvor stærkt H+ er bundetog hvor stærkt det kunne blive bundet, dvs af hvor sur D−H+ er (hvilketgerne angives med den komplementære størrelse, Ds protonaffinitet),og af hvor tilgængelige de ikke-bindende elektroner på A er (udtryktved As protonaffinitet eller ioniseringsenergi).

Hydrogenbindinger 12.314
Det virker rimeligt at forvente at vekselvirkningen vil være forholdsvissvag dersom protonaffinitetsforskellen er stor; er acceptors tilbøjelighedtil at danne binding til H+ betydeligt mindre end donors vil ogsåacceptors binding til et 'fælles' H nok være svagere. Hvis donor ogacceptor kemisk set er beslægtede, fx begge alkoholer, kan man forventeat modifikationer der påvirker syre-baseegenskaberne også i nogengrad vil indvirke på hydrogenbindingens styrke, omend ændring afdonors egenskaber måske vejer tungere end ændring af acceptors [77].Er donor og acceptor derimod ret forskellige er det vanskeligt atforudsige hvilken betydning deres syre-base egenskaber vil have forvekselvirkningen. For umage donor-acceptor par kan det eksempelvisikke forventes at samme forskel i protonaffinitet vil give anledning tilsamme adduktstabilisering [78-80].
Figur 12.8. Større protonaffinitetsforskel indebærer mindre adduktstabilisering.
PA og ∆PA. En direkte sammenhæng mellem hydrogenbindingsstyrkeog protonaffinitetsforskel synes oprindelig at være forslået af Taft [81]og underbygget eksperimentelt af Kebarle [60]. Litteraturens forslag tilhvordan afhængigheden konkret ytrer sig er imidlertid noget forskel-lige. Kebarles oprindelige beskrivelse (der siden er blevet fulgt op afmange) indebar nogenlunde lineær afhængighed [33,82-86]; dette harfundet støtte i beregningsmæssige undersøgelser [61,87-89]. Der er dogingen særlig grund til at sammenhængen skulle være lineær, og det harværet foreslået at donors og acceptors protonaffinitet skulle vægtesforskelligt, eller at der var tale om eksponentiel afhængighed [78,90]. Demange eksempler i litteraturen har imidlertid hovedsaglig involveretbeslægtede forbindelser. Herved kan andre effekter nemt skjule sig,dersom deres bidrag er nogenlunde konstant eller ændrer sig monotont.

Hydrogenbindinger 12.315
En yderligere komplikation optræder når hydro-genbindingsdannelse ikke involverer sammeatom som det der angribes ved protonoverførsel.Radom [16] har eksempelvis vist at enaminerhelst danner hydrogenbindinger til N menprotoniseres på C; derfor er molekylets proton-affinitet her antagelig uden større betydning forhydrogenbindingsstyrken.
Disse resultater understreger at hydrogenbindingsdannelse kan styresaf andre forhold end de der bestemmer protonaffinitetsforskellen, dvsreaktionsvarmen ved den potentielle H+-overførsel. Denne er udtryk foregenskaber hos de involverede molekyler i fravær af hydrogenbinding,egenskaber der nok kan indvirke på hydrogenbindingens styrke, mensom næppe alene bestemmer den.
Hydrogenbindingsstyrke. Det er imidlertid påfaldende så tydeligt Estaber korreleret med såvel rødskift som med ∆PA (eller PA og IE). Dettemere end antyder at de ioniske vekselvirkninger bidrager nogenlundekonstant til adduktstabiliseringen, eller ændrer sig monotont ogsystematisk, antagelig relateret til den neutrale komponents størrelse.For addukter hvor denne er et simpelt alkylradikal virker formod-ningen om monotont varierende ioniske vekselvirkninger ikke urimelig,omend forventninger til hydrogenbindingens styrke baseret på stabili-sering eller protonaffinitetsforskelle som omtalt ikke altid stemmeroverens med adduktets IR rødskift [62].
∆PA og (næsten)symmetriske hydrogenbindinger. De fleste veldoku-menterede eksempler på systematiske træk ved stabiliseringen afhydrogenbundne addukter gælder næsten-symmetriske systemer, fx[ROH...H+...HOR'], hvor forhold af samme art bestemmer donors ogacceptors bidrag til hydrogenbindingsstyrken. Den stærkeste hydrogen-binding optræder når protonaffinitetsforskellen er mindst, men detindebærer ikke at hydrogenbindingsstyrken er den samme i alle tilfældehvor ∆PA = 0; den varierer med bl.a. heteroatomets(ernes) elektronegativitet og er eksempelvis forskellig for protoniserede alkoholdimere og protoniserede amin dimere [33,79,80,87].
Fravær af symmetri: OH...C vs. O...HC. I næsten-symmetriske sammen-hænge er det naturligt at forvente at adduktets struktur umiddelbartafspejler de termokemiske forhold, dvs at det bindende H-atom er

Hydrogenbindinger 12.316
knyttet nærmest til den mest basiske komponent [91]. Er donor ogacceptor ikke kemisk set beslægtede er forventninger som denneimidlertid ikke velbegrundede, og heller ikke altid opfyldt. Bian [77] hareksempelvis vist at H-donors indflydelse på adduktegenskaberne ofteer større end acceptors.
I H3O+ addukter af sec-butyl- og tert-butylradikalerne sker ifølgestrukturberegningerne ikke fuldstændig protonoverførsel fra O til C,uanset at begge alkylradikalers protonaffinitet er noget højere endvands (se Tabel 12.6); det bindende H forbliver tættest knyttet tiloxygen. Hvis ikke en robust beregningsmæssig artefakt kunne detteafspejle at hydrogenbindingen i fx [H3O+...sec-C4H9
.] er så megetstærkere end hydrogenbindingen i [H2O...C4H10
+.] at det opvejerprotonaffinitetsforskellen (Figur 12.9). Scheiner [63], Fridgen [92] ogJohnson [93] har gjort tilsvarende observationer for protoniseredeaddukter i en række tilfælde hvor komponenternes dipolmomenter erbetydeligt forskellige, i tilsyneladende modstrid med Radoms genera-lisering [16,91]. Hvor vidt fremskreden protonoverførsel er mellemadduktets komponenter anslås lidt forskelligt med forskelligeberegningsmetoder (Figur 12.9); også eksperimentelle bestemmelser kanvære tvetydige i denne henseende [94].
B3LYP MP2
rO−H 1.233 Å 1.113 ÅrH−C 1.420 Å 1.547 Å
Figur 12.9. H+-placeringen i [H3O+...sec-C4H9.] adduktet afspejler ikke proton-
affiniteterne; PA(H2O) 689 kJ mol−1, PA(sec-C4H9.) 725 kJ mol−1. Afstande beregnet
med hhv B3LYP/6-31+G(d,p) og MP2(full)/6-31+G(d,p).
Fravær af symmetri: NH...C vs. N...HC. Når donor og acceptor er retforskellige kan det være tydeligt at deres bidrag til hydrogenbindingenikke bestemmes af de samme egenskaber. Hvad angår vekselvirkningermed alkylradikaler finder dette blandt andet udtryk i forskellen mellemhydrogenbindingerne hos reaktanterne og hos produkterne ved termo-neutrale H-overførselsreaktioner (Figur 12.10).
Omdannelsen af N-methylbutylamins kationradikal til den δ-distoniskeisomer (Figur 12.10) er reversibel og med god tilnærmelse termoneutral[31]. Begge ioner er i deres gauche-konformation stabiliseret af intra-

Hydrogenbindinger 12.317
molekylære hydrogenbindinger, men disse er betydeligt stærkere i dendistoniske isomer. Det fremgår af stabiliseringen (i forhold til trans-formerne), af bindingslængdeændringerne og af IR rødskiftet at NH ...Chydrogenbindingen er en del stærkere end CH...N hydrogenbindingen(Figur 12.10). Årsagen er antagelig at ladningsoverførselsbidraget erhelt forskelligt i de to ioner, idet C−H bindingens σ*-orbital er enringere elektronacceptor end N−H bindingens [95,96], samtidig med atden distoniske ions C-radikal er en betydeligt bedre elektrondonor endden ioniserede amins N-radikal.
H+ eller H. vekselvirkning. Det kan være uklart om den H-overførselsom hydrogenbindingen tager forskud på i hydrogenbundne addukteraf ioniske protondonorer og alkylradikaler ville indebære flytning af H+
eller af H.. Betragtes fx [H3O+...C2H5.] og [CH3OH2
+...C2H5.] ville H+-
overførsel fra H3O+ til ethylradikalet være 81 kJ mol−1 mere favorabelend H.-overførsel, mens det modsatte gør sig gældende for CH3OH2
+
(forskel 79 kJ mol−1) (Figur 12.11).
De to ethyl-addukter opfører sig imidlertid ens i henseende til hydro-genbinding, dvs betydelig stabilisering, bindingslængdeændringer, IR-rødskift og -intensitetsforøgelse. Dette er et udtryk for at de indledendevekselvirkninger manifesterer sig på samme måde, uanset om denpotentielle H-overførsel ville involvere H+ eller H.; en sondring er ikkevelbegrundet. Yderligere illustration fås ved sammenligning af alkyl-radikalers intramolekylære hydrogenbindinger med binære og ternæreradikal-addukters hydrogenbindinger. De giver sig til kende på sammemåde, men den intramolekylære H-overførsel (der ofte er reversibel)beskrives bedst som flytning af et hydrogenatom mens den i adduktergerne ses som flytning af H+.
ΔrC−H 0.007 Å ΔrN−H 0.017 ÅΔνC−H 76 cm−1 ΔνN−H 331 cm−1
rC−N 3.12 Å rC−N 2.99 ÅEstab ~ 0 Estab ~ 17 kJ mol−1
Figur 12.10. NH...C. hydrogenbindinger er stærkere end CH...N. H-flytningen i N-methylbutylamin kationradikalet er omtrent termoneutral og har en beregnet kritisk energi på 32 kJ mol−1 [31]; Estab er angivet i forhold til de tilsvarende all-trans former.

Hydrogenbindinger 12.318
Figur 12.11. Termokemi ved H-flytning fra oxoniumioner til ethylradikalet (kJ mol1).
Derved fremhæves igen at de forhold der styrer den indledende veksel-virkning, hydrogenbindingen, ikke nødvendigvis er de samme som deder bestemmer de termokemiske konsekvenser af den fuldstændige H-overførsel [16].
Ioniseringsenergi. Som beskrevet ovenfor indgår acceptors ioniserings-energi frem for protonaffinitet når det gælder alkylradikaladduktersstabilisering. Ioniseringsenergien udtrykker et potentielt bidrag tilstabiliseringen men siger umiddelbart intet om hvor favorabelfuldbyrdet H+-flytning vil være. Protonaffinitet siger modsætningsvisnoget om entalpiændringer ved H+-flytning og dermed formodentligogså noget om villighed til at indgå i de forudgående vekselvirkninger.
Som påpeget af Beauchamp og siden også andre [97,98], og især under-streget af Maksić og Vianello [99], er de to egenskaber imidlertid nærtforbundne, da molekylets protonaffinitet kan udtrykkes som forskellenmellem dets ioniseringsenergi og det tilsvarende kationradikals hydro-genatomaffinitet,
PA(M) = HA(M+.) – IE(M) + konstant,
dvs som funktion af to én-elektronsegenskaber, hvilket ligger bag detbidrag som ladningsoverførsel yder til hydrogenbindingsstyrken(diskuteres i det følgende).

Hydrogenbindinger 12.319
Hydrogenbindingen med bred pensel; elektrostatik og kovalens
Hydrogenbindingen er det samlede resultat af en række forskelligevekselvirkninger, der optræder når et molekyle med et surt H bringessammen med et molekyle med tilgængelig elektrontæthed (lone pair,uparret elektron, π-system). De vigtigste stabiliserende vekselvirk-ninger skyldes lokal elektrostatisk tiltrækning og begyndende kovalentbindingsdannelse (ladningsoverførsel); dette sidste element spiller ensærligt stor rolle for hydrogenbindinger i ladede systemer.
Det er mest almindeligt at inddrage hydrogenbindingen ved beskrivelseaf systemer hvor fuldstændig H-overførsel ikke finder sted (eller harfundet sted), men alle H+-overførselsreaktioner indledes med hydrogen-bindingsvekselvirkninger (se fx [6,29]); det viser sig frugtbart at antageat vekselvirkninger af denne art også kan indlede overførsel afhydrogenatomer.
Synet på hydrogenbindingen har varieret en del. I mange år blev den,efter Pauling [4], betragtet som resultatet af hovedsaglig elektrostatiskevekselvirkninger. I halvtresserne viste bl.a. Tsubomura [41] og Coulson[100,101] at hydrogenbindingernes indvirken på IR-egenskaberne, rød-skift og intensitetsforøgelse, gør det nødvendigt at inddrage et kovalentelement i beskrivelsen, dog uden at dette blev nøjere konkretiseret. Enrække forfattere argumenterede senere for at hydrogenbindingen burdeses som en art ladningsoverførselsvekselvirkning, og for at hydrogen-bindingens styrke skulle være direkte afhængig af acceptormolekyletsioniseringsenergi [41-47], en opfattelse der iflg Coulson [101] går tilbagetil Sokolov [102]. Den dannede dog på det tidspunkt ikke skole, menladningsoverførsel som en del af hydrogenbindingsbeskrivelsen har i desenere år vundet frem igen [103] (se dog [104]).
Den fremherskende opfattelse er i dag, at såvel elektrostatiske somkovalente elementer indgår, hvor disse sidste overvejende beror påvekselvirkning mellem acceptoratomets ikke-bindende elektron(er) ogdonor−H bindingens σ*-orbital, ladningsoverførsel [25,68,96,97,103-105,106]. Der kan være ret stor forskel på hvor den enkelte forfatterlægger vægten, og beskrivelsen varierer desuden alt efter den samledebindingsstyrke. Helt svage hydrogenbindinger skal have fortrinsviselektrostatisk oprindelse [107,108], mens meget stærke hydrogen-bindinger ind imellem beskrives som 4-elektron 3-center kovalentebindinger [34,106]. For moderate og stærke hydrogenbindinger er både

Hydrogenbindinger 12.320
elektrostatik og ladningsoverførsel vigtige [6,9,25,79,107], men der erikke én enkelt 'forklaring' eller oprindelse; flere former for veksel-virkning spiller ind, i varierende forhold. Dette er imidlertid ikke etsærkende for hydrogenbindinger − det samme gælder 'almindelige'kemiske bindinger.
VB og MO modeller. At flere vekselvirkninger bidrager afspejles ilitteraturens principielle overvejelser om hydrogenbindingen. I denvalence bond beskrivelse som Coulson [100] og Tsubomura [43] tidligtbenyttede tilskrives de vigtigste bidrag til hydrogenbindinger i neutraleog ladede systemer disse former:
D−H :A D− H+ :A D− H−A+ (a)D: H+ :A D: H. .A+ +D. H. :A (b)D: H+ .A D: H. A+ +D. H. .A (c)
Figur 12.12. De vigtigste elementer i en valence bond beskrivelse af hydrogen-bundne addukter (efter (a) Coulson [100] og (b) Humbel [109]); (c) er Humbels beskrivelse dersom A er et radikal.
Valence bond modeller for hydrogenbindinger ses i dag kun sjældentanvendt explicit (fx [106,109]); den mest almindelige begrebsmæssigeramme bygger på en opdeling (energy decomposition) som Morokuma[17] udformede på grundlag af ab initio MO teori. De vigtigsteelementer heri er elektrostatisk vekselvirkning, elektronfrastødning,polarisering og ladningsoverførsel. I svage hydrogenbindinger udensynderlig ladningsoverførsel beror bindingen mest på vekselvirkningmellem lokale multipoler. Dette gør sig især gældende når afstandenmellem hydrogenbindingens tunge atomer er forholdsvis stor, eftersomden stabilisering som ladningsoverførsel bevirker i særlig gradafhænger af denne afstand [25,68,107].
Ladningsoverførsel. Alkylradikalers hydrogenbindinger til ladedeprotondonorer er stærkere end til neutrale (omtalt nærmere i etefterfølgende afsnit); den elektrostatiske vekselvirkning forstærkertiltrækningen mellem donor og acceptormolekylerne og formindskerafstanden sammenlignet med upolære acceptormolekylers veksel-virkning med uladede donorer, hvorved muligheden for ladnings-overførsel forbedres.

Hydrogenbindinger 12.321
Med Reed og Weinholds natural bond orbital (NBO) metode [25] kanhydrogenbundne addukters bindingsforhold og især ladningsover-førselsbidragets del af den samlede vekselvirkning undersøges direkte[25,68,96] (se afsnittet om AIM og NBO nedenfor). Herved synes i særliggrad protondonors O−H (N−H) σ*-orbitals egenskaber som elektron-acceptor at være af betydning. Ladningsoverførsel til den antibindendeorbital* bevirker som omtalt ovenfor svækkelse og dermed forlængelseaf O−H (N−H) bindingen, hvoraf bl.a. IR rødskiftet følger (se dog [110]).Som elektronacceptor er O−H bindingers σ*-orbital bedre end N−Hbindingers [97], og oxoniumioners O−H og ammoniumioners N−Hbedre end de uladede forbindelsers. Det er mindre klart om ellerhvordan acceptoregenskaberne varierer med O−H (eller N−H)bindingernes hybridisering, om fx protoniserede nitrilers N−H bindingsσ*-orbital (sp-hybridisering) er en bedre eller dårligere acceptor endprotoniserede aminers (sp3-hybridisering). Det forhold at protoniseredenitrilers addukter med alkylradikaler ikke er helt så stabiliserede somforventet ud fra nitrilernes protonaffinitet er måske en konsekvens af atladningsoverførsel fra alkylradikalet til en sp σ* er mindre favorabel.
Figur 12.13. Donor-H hybridisering, sp og sp3; acceptor lone pair hybridisering, sp og sp3.
Hvad angår acceptor er der for alkylradikaler ikke nogen tilsvarendeproblemstilling; disse binder ved hjælp af en nogenlunde ren p-orbital.For lone-pair H-acceptorer kan hybridiseringen variere fra sp3 til sp, ogdet stod tidligt klart [111] at denne forskel indvirkede på hydrogen-bindingsstyrken. Aminer danner stærkere hydrogenbindinger endnitriler, hvilket skulle vise at bindingsstyrken var relateret til graden afs-karakter; med højere s-bidrag fulgte svagere hydrogenbinding; alkyl-radikalers gode hydrogenbindingsegenskaber kunne således bl.a. væreknyttet til at acceptororbitalen er uden s-bidrag. Der er en tydeligsammenhæng med Aue og Bowers' påvisning af relationerne mellem*Denne del af vekselvirkningen benævnes somme tider 'hyperkonjugation' (sefx [97]), men det er et udtryk der har set ret mange forskellige anvendelser,og det umiddelbare informationsindhold er blevet betænkeligt lavt. Den derskriver kender den tilsigtede betydning, men kan ikke altid have envelbegrundet formodning om hvad den der læser vil opfatte.

Hydrogenbindinger 12.322
basestyrke og lone pair hybridisering [113], som peger på at til højere s-bidrag svarer dårligere baseegenskaber. Korzhenevskaya [112] har medandre begrundelser gjort tilsvarende synspunkter gældende.
At adduktstabiliseringen vokser med faldende ioniseringsenergi hosalkylradikalet er et konkret udtryk for at bindingen til det 'fælles' Hberor direkte på radikalets evne til at bidrage med en enkelt elektron.Dette bidrag, ladningsoverførslen, fremstår særlig tydeligt for alkyl-radikaler, fordi det her ikke maskeres af syre-baseegenskaberne; alkyl-radikaler har nemlig på én gang lav ioniseringsenergi og lav proton-affinitet, hvilket er usædvanligt i mættede systemer. Det er særliginteressant at ladningsoverførslens betydning fremgår direkte, ikke somen tolkning af teoretiske overvejelser, og udtrykker konkrete fysiskeegenskaber hvis indflydelse træder tydeligt frem. Den komplementæresammenhæng, at hydrogenbindingsstyrken påvirker ioniserings-energien, kendes også [114].
Afhængigheden af H-acceptors ioniseringsenergi står i tilsyneladendekontrast til at hydrogenbindingers styrke i reglen relateres til proton-affiniteten hos såvel donor som acceptor. Denne forskel er imidlertidikke udtryk for at bindingsforholdene i alkylradikalers addukteradskiller sig grundlæggende fra forholdene i andre hydrogenbundneaddukter. Protonaffiniteten udtrykker molekylets villighed til at bidragemed et elektronpar, ioniseringsenergien villigheden til at bidrage meden enkelt elektron, og disse er som omtalt beslægtede størrelser [98,113].
Maksić og Vianello [99] har endog argumenteret for at én-elektrons-overførsel kan være et vigtigt element ved syre-basereaktioner, oginddrager tillige forskellen mellem vertikal og adiabatisk ioniserings-energi ved beskrivelsen af basers protonaffinitet. Her videreudvikler deAue og Bowers' [113] syn på relationen mellem protonaffinitet,ioniseringsenergi og hybridisering. Det er værd at bemærke, at deresbeskrivelse af protonoverførsel er nært beslægtet med Humbels VB-model (Figur 12.12b) for hydrogenbindinger, og understøtter atacceptors ioniseringsenergi er en relevant størrelse.
Efter Humbel kan et system hvor protonacceptor er et frit radikalbeskrives som i Figur 12.12c, hvor elektronafgivelse fra acceptorillustreres af den midterste resonansform. Det er meget vel muligt atresonansformer som den højre i Figur 12.12c bør tillægges mindre vægtpå grund af antallet af uparrede elektroner, men den antyder dog, at

Hydrogenbindinger 12.323
også donors ioniseringsenergi kunne spille en rolle forhydrogenbindingsstyrken. Dermed kunne den dårligere stabilisering afprotoniserede nitrilers hydrogenbundne addukter være udtryk for atnitrilers ioniseringsenergi er så høj at denne resonansform især her ikkebidrager synderligt.
Opdeling i vekselvirkningsbidrag. Morokuma-analyse kan føre til denslutning at ladningsoverførsel kun spiller en ret begrænset rolle, typiskmindre end 10% af den samlede vekselvirkning [17], hvilket en over-gang blev anvendt som begrundelse for at beskrive hydrogenbindingensom et hovedsaglig klassisk elektrostatisk fænomen. Tilsvarendeopdelinger foretaget med et NBO udgangspunkt [25,68,115] kommerimidlertid til ganske andre resultater, at ladningsoverførsel og dermeddet kovalente bidrag til hydrogenbindingen kan udgøre mere endhalvdelen af den samlede vekselvirkning. Opdeling foretaget med enrække nyere teoretiske metoder har bidraget til beskrivelsen af veksel-virkningernes forskellige dele; de fleste tillægger ligeledes ladnings-overførsel stor betydning [103].
En mulig syntese af disse ret forskellige (og uforligelige, efter uforson-ligheden at dømme; se fx [104,115]) betragtningsmåder skyldes MartínPendás [18], som har påpeget at deformeringsenergien kan inddragespå forskellig vis, at det ikke er den samme samlede vekselvirkning somde forskellige opdelinger beskriver, og dermed at alle på en måde kanhave nogenlunde ret samtidig. Det springende punkt er hvorvidthydrogenbindingen bedst ses som en mindre ændring for donor ogacceptor (svag binding, ubetydelig deformering, mest elektrostatik)eller som en stærk vekselvirkning som modificerer adduktets kompo-nenter (rimelig binding, kendelig deformering, betydeligt kovalentbidrag), hvor fx Morokumas beskrivelse forudsætter ringe deformering.Måske er det hovedsaglig en strid om ord, der synes at have situdspring i de forventninger den enkelte forfatter har til hydrogen-bindingens styrke, hvilket igen viser tilbage til fænomenets uklareafgrænsning (se kapitlets indledning).

Hydrogenbindinger 12.324
Hydrogenbindinger til acceptorer med åbne og lukkede skaller
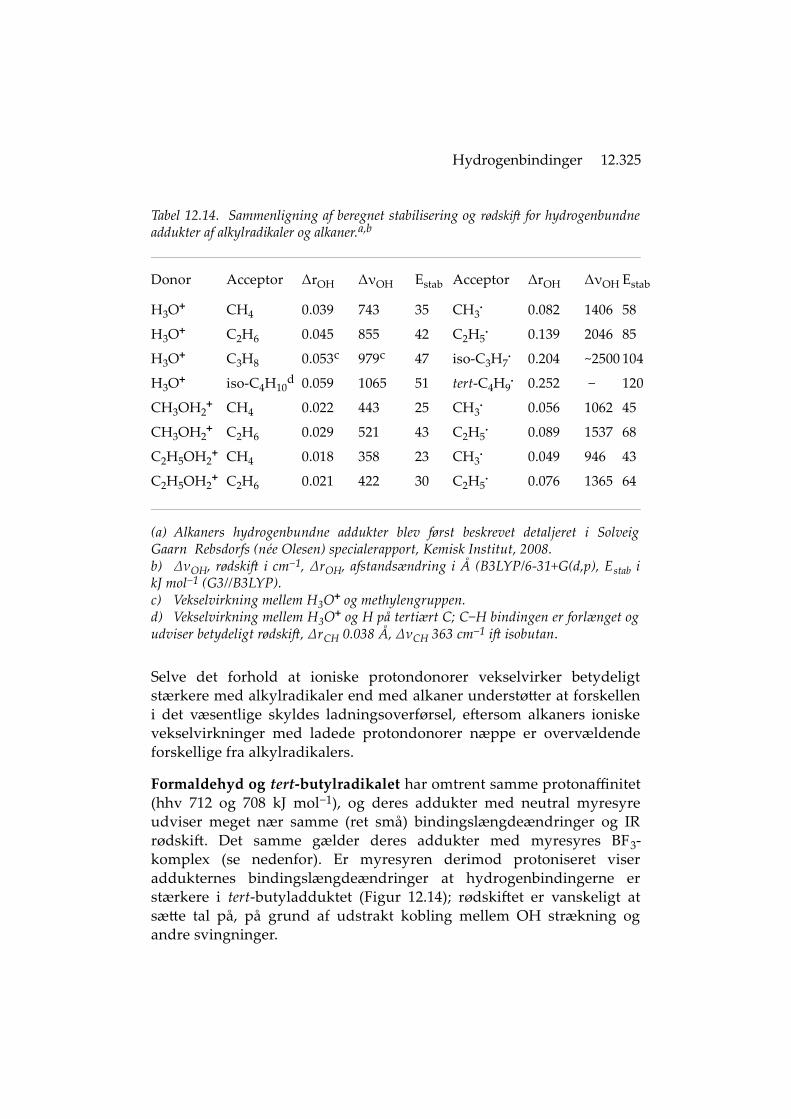
Ioniske protondonorers vekselvirkning med alkaner. Vekselvirk-ningerne mellem alkaner og stærke gasfasesyrer som H3O+ viser sig,måske uventet, at have betydelig indvirkning på addukternes IR-egen-skaber [116]. De kan resultere i beregnede rødskift for O−H stræknings-vibrationer på op til 1000 cm−1 (Tabel 12.14), hvilket er mere endobserveret med fx N2 eller Ar som acceptorer [56]. Interessant nokvekselvirker et enkelt H på H3O+ eller CH3OH2
+ med flere alkan-hydrogenatomer samtidig (dog undtaget alkaner med tertiære carbon-atomer som isobutan, hvor vekselvirkningen kun omfatter det H dersidder ved forgreningen). Sammenlignet med alkylradikalernesaddukter er alkan-addukternes stabilisering dog en del mindre. De er tilgengæld til at verificere eksperimentelt, ved IR fotodissociations-målinger på argon-addukter [116]. Her observeres et rødskift på mereend 600 cm‒1 for den bindende O–H, hvilket bekræfter at en hydrogen-binding af betragtelig styrke gør sig gældende.
Alkan-addukternes beregnede struktur indicerer at C−H σ bindingen erH-acceptor, idet den sure O−H binding er rettet (næsten) mod midten afC−H bindingen, ikke mod H-atomet. Dette er i overensstemmelse medOlahs sigma-basicitetsbetragtninger [117] og med NBO beregnings-resultaterne, der viser vekselvirkning mellem den bindende C−H σorbital og O−H bindingens σ*. Såvel CH som OH strækningsvibra-tionerne udviser rødskift, og CH rødskiftet er betragteligt (af en C−Hbindings at være) [116].
Enkelte eksempler på dannelse af hydrogenbindinger mellem ioniskeprotondonorer og alkaner er tidligere blevet beskrevet af Field [118],Ahlberg [119] og Zeegers-Huyskens [50]; vore egne resultater [116] erblevet suppleret af Isaevs AIM undersøgelser [120]. Det viser sig atvekselvirkningerne generelt gør sig gældende når protondonor ikke erhelt så stærk at regelret protonisering af alkanen finder sted. Grænsensynes at gå omkring H2F+ (som protoniserer alkaner, hvorimod H3F2
+
ikke er stærk nok [119]). Hydrogenbindingen til alkaner kan ses som etforstadium til en dissociativ protonisering, da alkaners protonisering ireglen følges af tab af H2 [121].
Hydrogenbindinger 12.325
Tabel 12.14. Sammenligning af beregnet stabilisering og rødskift for hydrogenbundneaddukter af alkylradikaler og alkaner.a,b
Donor Acceptor ΔrOH ΔνOH Estab Acceptor ΔrOH ΔνOH Estab
H3O+ CH4 0.039 743 35 CH3. 0.082 1406 58
H3O+ C2H6 0.045 855 42 C2H5. 0.139 2046 85
H3O+ C3H8 0.053c 979c 47 iso-C3H7. 0.204 ~2500 104
H3O+ iso-C4H10d 0.059 1065 51 tert-C4H9
. 0.252 − 120
CH3OH2+ CH4 0.022 443 25 CH3
. 0.056 1062 45
CH3OH2+ C2H6 0.029 521 43 C2H5
. 0.089 1537 68
C2H5OH2+ CH4 0.018 358 23 CH3
. 0.049 946 43
C2H5OH2+ C2H6 0.021 422 30 C2H5
. 0.076 1365 64
(a) Alkaners hydrogenbundne addukter blev først beskrevet detaljeret i SolveigGaarn Rebsdorfs (née Olesen) specialerapport, Kemisk Institut, 2008.b) ΔνOH, rødskift i cm−1, ΔrOH, afstandsændring i Å (B3LYP/6-31+G(d,p), Estab ikJ mol−1 (G3//B3LYP).c) Vekselvirkning mellem H3O+ og methylengruppen.d) Vekselvirkning mellem H3O+ og H på tertiært C; C−H bindingen er forlænget ogudviser betydeligt rødskift, ΔrCH 0.038 Å, ΔνCH 363 cm−1 ift isobutan.
Selve det forhold at ioniske protondonorer vekselvirker betydeligtstærkere med alkylradikaler end med alkaner understøtter at forskelleni det væsentlige skyldes ladningsoverførsel, eftersom alkaners ioniskevekselvirkninger med ladede protondonorer næppe er overvældendeforskellige fra alkylradikalers.
Formaldehyd og tert-butylradikalet har omtrent samme protonaffinitet(hhv 712 og 708 kJ mol−1), og deres addukter med neutral myresyreudviser meget nær samme (ret små) bindingslængdeændringer og IRrødskift. Det samme gælder deres addukter med myresyres BF3-komplex (se nedenfor). Er myresyren derimod protoniseret viseraddukternes bindingslængdeændringer at hydrogenbindingerne erstærkere i tert-butyladduktet (Figur 12.14); rødskiftet er vanskeligt atsætte tal på, på grund af udstrakt kobling mellem OH strækning ogandre svingninger.

Hydrogenbindinger 12.326
[HC(OH)2+...C(CH3)3
.] [HC(OH)2+...O=CH2]
ΔrOH 0.206 Å, Estab 97 kJ mol1 ΔrOH 0.188 Å, Estab 115 kJ mol1
Figur 12.14. tert-Butylradikalet danner stærkere hydrogenbindinger til protoniseret myresyre end formaldehyd gør, men adduktet er svagere stabiliseret.
Med methanol og protoniseret methanol gør noget tilsvarende siggældende; formaldehyd og tert-butylradikalet er begge kun svagtbundet i deres addukter med methanol, men med protoniseretmethanol danner tert-butylradikalet stærkere hydrogenbindinger endformaldehyd. Hos vand og propylradikalet, som har omtrent sammeprotonaffinitet, 689 og 686 kJ mol−1, ses ændringer af helt samme artoverfor methanol og protoniseret methanol.
Benyttes bindingslængdeændringerne som mål for hydrogenbinding-ernes styrke er formaldehyd ikke helt så god en acceptor som tert-butylradikalet, og vand og propylradikalet nogenlunde lige gode. Denstabilisering som adduktdannelse indebærer er betydeligt større forvand og formaldehyd (Figur 12.14), eftersom de ioniske veksel-virkninger er betydeligt kraftigere når det neutrale molekyle har etrimeligt dipolmoment. Derved understreges endnu en gang hensigts-mæssigheden af at sondre mellem de egenskaber der skyldes ioniskevekselvirkninger og de der skyldes hydrogenbindinger. Desuden udgørdisse resultater en yderligere påvisning af at alkylradikalers acceptor-egenskaber forstærkes overfor ladede donormolekyler.
Hydrogenbindinger i ternære addukter. Hydrogenbindinger antræffesogså i addukter med flere end to komponenter, hvilket giver yderligeremuligheder for at sammenligne hydrogenbindinger til alkylradikaler ogandre acceptorer. Et illustrativt eksempel er [H2O...H3O+...tert-C4H9
.]adduktet, hvori H3O+ danner hydrogenbinding til såvel vand som tert-butyl. Bindingslængdeændringerne viser tydeligt at hydrogenbindingentil alkylradikalet er stærkere end den til vandmolekylet; denpågældende O−H binding er længere og O−H strækningsvibrationen ermere rødskiftet og har større intensitet (Figur 12.15 og Tabel 12.15).

Hydrogenbindinger 12.327
Figur 12.15. [H2O...H3O+...tert-C4H9.]; (a) beregnet IR spektrum der tydeligt viser
forskel i rødskift (Tabel 12.15); (b) AIM elektrontæthed i OOC-planet, der illustrerer at hydrogenbindingsforholdene ikke er drastisk forskellige.
Helt tilsvarende resultater fås for de ternære addukter af NH4+ med
vand og et tert-butylradikal, og med protoniseret methanol og desamme to acceptorer. Under disse omstændigheder er isopropyl-radikalet derimod ikke bundet stærkere end H2O.
Tabel 12.15. Hydrogenbindinger i ternære addukter.
νOH(C) νOH(O) rOH(C) rOH(O) rCO (rCN)
H2O...H3O+...tert-C4H9. 2114 2419 1.071 1.050 2.74
2419 2500 1.047 1.043 2.73H2O...CH3OH2
+...tert-C4H9. 2375 2744 1.047 1.028 2.78
2598 2837 1.034 1.022 2.75H2O...NH4
+...tert-C4H9. 2703 3012 1.068 1.051 3.00
2961 3174 1.051 1.041 2.99H2O...H3O+...iso-C3H7
. 2324 2316 1.054 1.058 2.772684 2390 1.030 1.051 2.78
ν i cm−1, r i Å, B3LYP/6-31+G(d,p) resultater over MP2(full)/6-31+G(d,p).
NBO-analyse af bindingsforholdene i de ternære addukter bekræfter athydrogenbindingerne til C og til heteroatomet begge er stærke og afnogenlunde samme styrke. Den AIM-beregnede elektrontæthed(eksempel i Figur 12.15) er i overensstemmelse hermed.
Hydrogenbindinger 12.328
Neutrale protondonorers hydrogenbindinger til alkylradikaler
Hydrogenbindinger mellem alkylradikaler og ikke-protoniserededonormolekyler er relativt svage (Tabel 12.16), som oprindeligt påpegetaf Alkorta et al. [8].
Tabel 12.16. Beregnede O−H bindingslængder og strækningsvibrationer for hydrogen-bundne addukter af alkylradikaler og uladede protondonorer.
rO−Ha υO−H
a rO-Cb ΔHf
c Estabd
CH3OH...CH3. 0.970 (0.005) 3722 (119) 3.31 −60 2 (2)
0.966 (0.002) 3858 (43) 3.39CH3OH...C(CH3)3
. 0.973 (0.008) 3658 (183) 3.22 −160 13 (6)0.971 (0.007) 3748 (162) 3.14
HCOOH...CH3. 0.984 (0.010) 3513 (223) 3.19 −243 5 (3)
0.977 (0.004) 3688 (99) 3.30HCOOH...C(CH3)3
. 0.992 (0.018) 3347 (389) 3.07 −350 24 [7]e
0.988 (0.015) 3452 (335) 3.03CH3COOH...CH3
. 0.981 (0.009) 3568 (189) 3.23 −296 6 (3)0.976 (0.004) 3712 (84) 3.32
CH3COOH...C(CH3)3. 0.987 (0.015) 3426 (331) 3.12 −397e 22e [7]e
0.986 (0.014) 3492 (304) 3.04
(a) O−H bindingslængde (Å) og IR strækningsbølgetal (cm−1) for det hydrogen-bundne addukt (B3LYP/6-31+G(d,p) beregningsresultater over MP2/6-31+G(d,p));forskellen fra det ikke-hydrogenbundne donormolekyle i parentes. (b) OHC vinkel tæt på 180o.(c) Adduktets 298 K dannelsesvarme i kJ mol−1 (G3//B3LYP).(d) Estab, forskellen mellem dannelsesvarmerne (G3//B3LYP) for det hydrogen-bundne addukt og de frie komponenter; BSSE korrektion i parentes.(e) G3(MP2)//B3LYP.
Således er bindingen mellem tert-butylradikalet og neutral myresyreeller eddikesyre omtrent lige så stærk (eller svag) som bindingenmellem methylradikalet og ammoniumioner. Sammenlignes stabili-sering, bindingslængdeændringer og rødskift for addukter af CH3
. ogtert-C4H9
. (Tabel 12.16) fremgår det dog tydeligt at der er hydrogen-

Hydrogenbindinger 12.329
bindinger til tert-butylradikalet, omend svage. Hvad methylradikaletangår er effekten ubetydelig (en AIM analyse bekræfter dogtilstedeværelsen af et bond critical point også her).
De intramolekylære vekselvirkninger i neutrale systemer med mulighedfor hydrogenbinding mellem O−H/N−H og et radikal-carbon er endnusvagere. Der er således ingen væsentlig ændring af bindingslængdereller IR-egenskaber i 4-hydroxyalkyl- eller 4-aminoalkylradikalersgauche-former, sammenlignet med trans-formerne eller med de tilsva-rende alkoholer eller aminer. Som omtalt ovenfor ændres dettebetydeligt dersom H-donor protoniseres, dels på grund af de ændredeioniske vekselvirkninger, dels fordi acceptoregenskaberne hos OH ogNH σ* orbitalerne ændres.
Elektrofilt aktiverede neutrale protondonorer. Sammenhængenmellem adduktstabilisering og protondonors syrestyrke indicerer ataddukter af Lewis-syrer og neutrale protondonorer skulle kunne dannestærkere hydrogenbindinger til alkylradikaler end de neutrale proton-donorer gør selv. Dette bekræftes af de beregnede IR-egenskaber forradikal-addukter af neutrale protondonorer og BF3, AlF3 og beslægtedeLewis-syrer.
HCOOH νOH 3736 cm−1 HCOOH...C(CH3)3. νOH 3347 cm−1
BF3:HCOOH νOH 3715 cm−1 BF3:HCOOH...C(CH3)3. νOH 2974 cm−1
Disse resultater falder godt i tråd med nyere resultater fra den præpa-rative radikalkemi, hvor Lewis-syre aktiverede protondonorer (H2O,CH3OH) reagerer med sekundære og tertiære tertiære alkylradikaler,med reduktion af disse til følge ved overførsel af H . fra vand og fra OH imethanol til carbon [122-124]. Dette er en konsekvens af at oxygen-atomets koordination til Lewis-syren modificerer de termokemiskeforhold således at overførsel af H. fra oxygen lettes (OH-bindingensbindingsdissociationsenergi falder betydeligt).
Figur 12.16. Ti(III)-aktiveret reduktion af tert-butylradikalet (efter [124]).

Hydrogenbindinger 12.330
Figur 12.17. Reduktion af isopropylradikalet med methanol-triethylboran (efter [125]).
Tabel 12.17. Lewis-syre-forstærkede hydrogenbindinger mellem tert-butylradikaler ogdonorer uden ladning.a
ΔrOH ΔνOH rO−C Estab
AlF3:HCOOH...C(CH3)3. 0.039b 812b 2.90 41
BF3:HCOOH...C(CH3)3. 0.036b 741b 2.92 38
AlF3:H2O...C(CH3)3. 0.035 744 2.91 39 (10)
BF3:H2O...C(CH3)3. 0.031 641 2.96 33 (10)
AlH3:H2O...C(CH3)3. 0.026 559 2.98 32 (7)
BH3:H2O...C(CH3)3. 0.024 523 2.99 31 (7)
a) Stabiliseringsenergi bestemt ved G3MP2//B3LYP beregninger (kJ mol−1), counterpoise korrektion i parentes; struktur og IR-egenskaber bestemt med B3LYP/6-31+G(d,p) beregninger.b) Forskelle angivet i forhold til AlF3:HCOOH hhv BF3:HCOOH

Hydrogenbindinger 12.331
AIM og NBO undersøgelser
Atoms in Molecules. Under betegnelsen Atoms in Molecules har Baderudviklet en beskrivelse af molekylers elektronstruktur som bygger påelektrontæthedens topologi [24]. Metoden har fundet en vis anvendelseved beregningsmæssige undersøgelser af hydrogenbindinger, og Kochog Popelier [125] har med udgangspunkt i Baders beskrivelse opstilleten række kriterier der efter deres mening bør være opfyldt for systemermed hydrogenbindinger. De to vigtigste er at hydrogenbindinger ligesom alle andre bindinger bør have et såkaldt bond critical point (bcp), ogat størrelsen af den Laplace-transformerede ladningstæthed ved dettepunkt skal ligge indenfor bestemte grænser. Den simple fysiske tolk-ning heraf er ikke klar.
Det viser sig at disse kriterier er opfyldt for alkylradikalers hydrogen-bundne addukter. For de stærkest bundne viser AIM-analysen endog atbindingen efter Koch og Popeliers kriterier er så stærk at den snareremå betegnes som kovalent.
Koch og Popeliers kriterier er empiriske; de sammenfatter en rækkeforhold der sædvanligvis gør sig gældende når hydrogenbindinger erinvolveret. Som AIM-resultater iøvrigt kan de være overordentlignyttige i afgrænsningsøjemed, men giver kun undtagelsesvis kvanti-tative udsagn og forudsigelser der lader sig sammenholde med andetend andre AIM-resultater. AIM metoden udsættes ind imellem foralvorlig kritik (se fx [126]), men denne skyldes nok i et vist mål detanstrøg af vækkelse der kan præge AIM-proselytters publikationer.
Flere forfattere har foreslået at den beregnede ladningstæthed vedhydrogenbindingens bond critical point skulle hænge sammen medbindingsstyrken [14,125,127]. Det forekommer at være i god overens-stemmelse med vore resultater (se Figur 12.18, som indeholder enafbildning af ladningstæthed ved bcp overfor den samlede stabiliseringfor alle undersøgte ethylradikal-addukter; en tilsvarende rigtig godkorrelation findes for hydrogenbundne addukter af ammoniumioner ogalkoholer/ethere). Som ved relationen mellem protonaffinitetsforskelleog stabilisering er disse korrelationer kun helt tilfredsstillende vedbeskrivelse af nærtbeslægtede forbindelsers egenskaber [128], menemnet er ikke her gjort til genstand for yderligere undersøgelser.
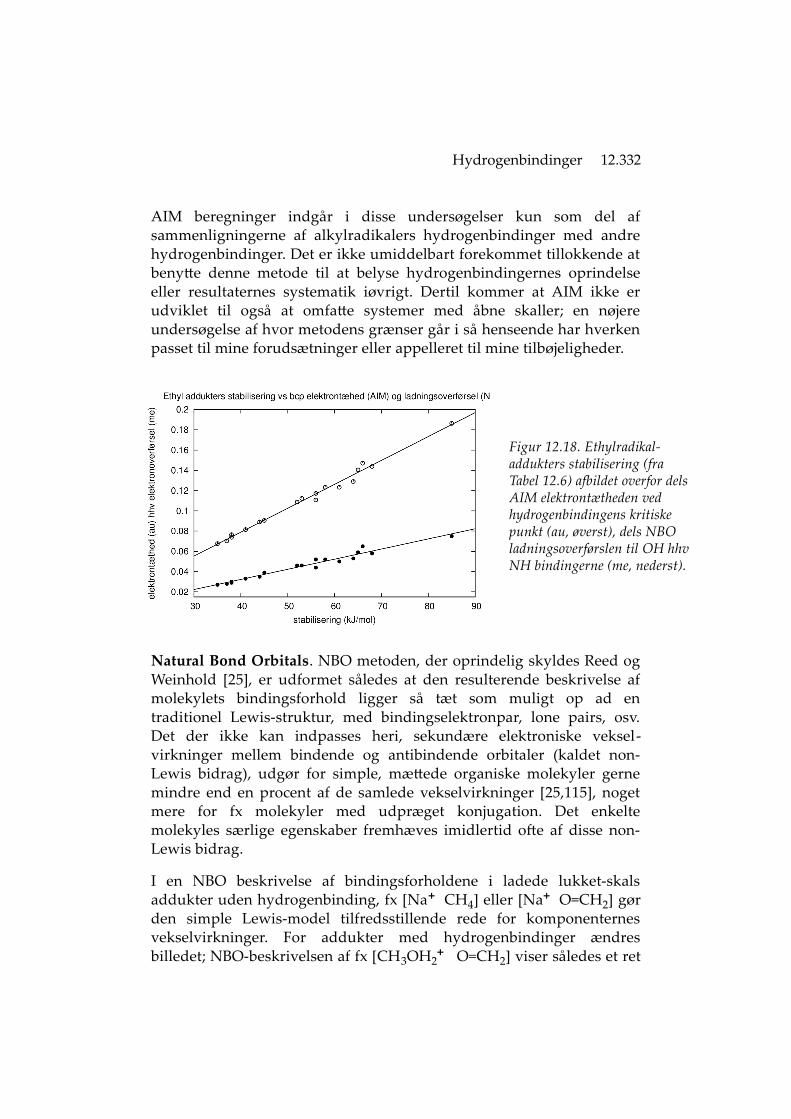
Hydrogenbindinger 12.332
AIM beregninger indgår i disse undersøgelser kun som del afsammenligningerne af alkylradikalers hydrogenbindinger med andrehydrogenbindinger. Det er ikke umiddelbart forekommet tillokkende atbenytte denne metode til at belyse hydrogenbindingernes oprindelseeller resultaternes systematik iøvrigt. Dertil kommer at AIM ikke erudviklet til også at omfatte systemer med åbne skaller; en nøjereundersøgelse af hvor metodens grænser går i så henseende har hverkenpasset til mine forudsætninger eller appelleret til mine tilbøjeligheder.
Figur 12.18. Ethylradikal-addukters stabilisering (fra Tabel 12.6) afbildet overfor dels AIM elektrontætheden ved hydrogenbindingens kritiske punkt (au, øverst), dels NBO ladningsoverførslen til OH hhv NH bindingerne (me, nederst).
Natural Bond Orbitals. NBO metoden, der oprindelig skyldes Reed ogWeinhold [25], er udformet således at den resulterende beskrivelse afmolekylets bindingsforhold ligger så tæt som muligt op ad entraditionel Lewis-struktur, med bindingselektronpar, lone pairs, osv.Det der ikke kan indpasses heri, sekundære elektroniske veksel-virkninger mellem bindende og antibindende orbitaler (kaldet non-Lewis bidrag), udgør for simple, mættede organiske molekyler gernemindre end en procent af de samlede vekselvirkninger [25,115], nogetmere for fx molekyler med udpræget konjugation. Det enkeltemolekyles særlige egenskaber fremhæves imidlertid ofte af disse non-Lewis bidrag.
I en NBO beskrivelse af bindingsforholdene i ladede lukket-skalsaddukter uden hydrogenbinding, fx [Na+ CH4] eller [Na+ O=CH2] gørden simple Lewis-model tilfredsstillende rede for komponenternesvekselvirkninger. For addukter med hydrogenbindinger ændresbilledet; NBO-beskrivelsen af fx [CH3OH2
+ O=CH2] viser således et ret

Hydrogenbindinger 12.333
stort bidrag fra en enkelt non-Lewis vekselvirkning, nemlig ladnings-overførsel fra et formaldehyd lone pair til en OH bindings σ*-orbital.
Vekselvirkninger af denne art er ligeledes særdeles fremtrædende foralkylradikalers hydrogenbundne addukter. Her viser NBO-beskrivelsenat et betydeligt bidrag til adduktets binding skyldes ladningsoverførselfra alkylradikalets enkeltbesatte orbital til donors O−H eller N−H σ*-orbital. En NBO analyse af bindingsforholdene i molekyler elleraddukter med uparrede elektroner kan imidlertid vanskeliggøres afforskellig bindingstilordning for α- og β-elektronerne [115,129]; dette ersærlig et problem når ladningsoverførselsbidraget er stort. Det er muligtat afgrænse NBO-beregningerne således at en eventuel uoverens-stemmelse forsvinder (ved explicit at angive eletronparrenes placering),men det er uklart i hvilket omfang dette indskrænker nytten af sammen-ligninger med andre NBO-resultater.
NBO-beskrivelsen fremhæver bestemte vigtige vekselvirkninger, menmetodens nytte begrænses af at den ikke umiddelbart kan anvendeskvantitativt når hydrogenbindingerne er moderate eller stærkere. Dettegælder såvel sammenligning med andre vekselvirkninger indenforadduktet som sammenligning forskellige addukter imellem. Som detillustreres i Figur 18 er der imidlertid et gennemgående fællestræk, atbetydelig adduktstabilisering ledsages af betydelig NBO elektron-overførsel, i god overensstemmelse med at hydrogenbindingsveksel-virkninger har et afgørende ladningsoverførselselement. Ikke allebetragter dog metoden som hensigtsmæssig og pålidelig [104].

334
Litteraturhenvisninger til Kapitel 12
1 W. M. Latimer og W. H. Rodebush, Polarity and ionization from the standpoint of the Lewistheory of valence. J. Am. Chem. Soc., 42, 1419-33 (1920).
2 (a) M. L. Huggins, Phys. Rev., 18, 333 (1921); henvisning ses ofte men efterkontrol er vanskelig.(b) M. L. Huggins, The electronic structure of atoms. J. Phys. Chem., 26, 601-25 (1922).(c) 50 år senere sammenfattede Huggins selv sit tidlige arbejde med hydrogen-bindinger, M. L. Huggins, 50 years of hydrogen bond theory. Angew. Chem. Int. Ed., 10, 147-53 (1971).
3 E. N. Lassettre, The hydrogen bond and association. Chem. Rev., 20, 259-303 (1937).4 L. Pauling, The Nature of the Chemical Bond. Cornell University Press, Ithaca 1939.5 G. A. Jeffrey, An Introduction to Hydrogen Bonding. Oxford University Press, New York
1997. Enhver ny læser burde begynde her.6 T. Steiner, The hydrogen bond in the solid state. Angew. Chem. Int. Ed., 41, 48-76 (2002).7 E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R.
H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci, og D. J. Nesbitt, Defining the hydrogen bond: An account (IUPAC Technical Report). Pure Appl. Chem., 83, 1619–36 (2011).
8 I. Alkorta, I. Rozas og J. Elguero, Radicals as hydrogen bond acceptors. Ber. Bunsenges., 102, 429-36 (1998).
9 (a) S. Scheiner, Ab initio studies of hydrogen bonds: the water dimer paradigm. Annu. Rev. Phys. Chem., 45, 23-56 (1994).(b) S. Scheiner, Hydrogen Bonding, a Theoretical Perspective. Oxford University Press, New York 1997.
10 M. S. Gordon og J. H. Jensen, Understanding the hydrogen bond using quantum chemistry. Acc. Chem. Res., 29, 536-43 (1996).
11 M. Koné, B. Illien, J. Graton og C. Laurence, B3LYP and MP2 calculations of the enthalpies of hydrogen-bonded complexes of methanol with neutral bases and anions: comparison with experimental data. J. Phys. Chem. A, 109, 11907-13 (2005).
12 P. R. Rablen, J. W. Lockman og W. L. Jorgensen, Ab initio study of hydrogen-bonded complexes of small organic molecules with water. J. Phys. Chem. A, 102, 3782-97 (1998).
13 S. D. Wetmore, R. Schofield, D. M. Smith og L. Radom, A theoretical investigation of the effects of electronegative substitution on the strength of C−H···N hydrogen bonds. J. Phys. Chem. A, 105, 8718-26 (2001).
14 S. J. Grabowski, Ab initio calculations on conventional and unconventional hydrogen bonds‒study of the hydrogen bond strength. J. Phys. Chem. A, 105, 10739-46 (2001).
15 S. J. Grabowski, What is the covalency of hydrogen bonding? Chem. Rev., 111, 2597-2625 (2011).
16 B. Chan, J. E. Del Bene, J. Elguero og L. Radom, On the relationship between the preferred site of hydrogen bonding and protonation. J. Phys. Chem. A, 109, 5509-17 (2005).
17 (a) H. Umeyama og K. Morokuma, The origin of hydrogen bonding. An energy decomposition study. J. Am. Chem. Soc., 99, 1316-32 (1977).

335
(b) K. Morokuma, Why do molecules interact? The origin of electron donor-acceptor complexes, hydrogen bonding, and proton affinity. Acc. Chem. Res., 10, 294-300 (1977).
18 A. Martín Pendás, M. A. Blanco og E. Francisco, The nature of the hydrogen bond: A synthesis from the interacting quantum atoms picture. J. Chem. Phys., 125, 184112 (2006).
19 (a) L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov og J. A. Pople, Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys., 109, 7764-76 (1998).(b) A. G. Baboul, L. A. Curtiss, P. C. Redfern og K. Raghavachari, Gaussian-3 theory using density functional geometries and zero-point energies. J. Chem. Phys., 110, 7650-57 (1999).(c) L. A. Curtiss, P. C. Redfern, V. Rassolov, G. Kedziora og J. A. Pople, Extension of Gaussian-3 theory to molecules containing third-row atoms K, Ca, Ga–Kr . J. Chem. Phys., 114, 9287-95 (2001).
20 J. E. Del Bene og I. Shavitt, i Molecular Interactions. Red. S. Scheiner, Wiley, New York 1997; kap 5, pp 157-79.
21 (a) M. Masamura, The effect of basis set superposition error on the convergence of interaction energies. Theor. Chem. Acc., 106, 301-13 (2001).(b) C. J. Cramer, Essentials of Computational Chemistry. Wiley, Chichester 2002.
22 F. Jensen, Introduction to Computational Chemistry. Wiley, Chichester 1999.23 S. S. Xantheas, On the importance of the fragment relaxation energy terms in the estimation
of the basis set superposition error correction to the intermolecular interaction energy . J. Chem. Phys., 104, 8821-24 (1996).
24 R. A. W. Bader, Atoms in Molecules. Clarendon Press, Oxford 1990.25 A. E. Reed, L. A. Curtiss og F. Weinhold, Intermolecular interactions from a natural bond
orbital, donor-acceptor viewpoint. Chem. Rev., 88, 899-926 (1988).26 (a) M. E. Jacox, The reaction of F atoms with methane in an argon matrix. Chem. Phys., 42,
133-48 (1979).(b) E. Y. Misochko, V. A. Benderskii, A. U. Goldschleger, A. V. Akimov og A. F. Shestakov, Formation of the CH3-HF complex in reaction of thermal F atoms with CH4 in solid Ar. J. Am. Chem. Soc., 117, 11997-98 (1995).(c) J. M. Merritt, S. Rudić og R. E. Miller, Infrared laser spectroscopy of CH3...HF in helium nanodroplets: the exit-channel complex of the F + CH4 reaction. J. Chem. Phys., 124, 084301 (2006).(d) S. Rudić, J. M. Meritt og R. E. Miller, Study of the CH3...H2O radical complex stabilizedin helium nanodroplets. Phys. Chem. Chem. Phys., 11, 5345-52 (2009).
27 Y. Chen, E. Tschuikow-Roux og A. Rauk, Intermediate complexes and transition structuresfor the reactions CH3 + HX CH4 + X (X=Cl,Br): application of G1 theory. J. Phys. Chem.,95, 9832-36 (1991).
28 (a) H. Tachikawa, A direct ab initio dynamics study on the finite temperature effects on the hyperfine coupling constant of a weakly bonded complex. J. Phys. Chem. A, 102, 7065-69 (1998).(b) M. Igarashi, T. Ishibashi og H. Tachikawa, A direct ab initio molecular dynamics study of the finite temperature effects on the hyperfine coupling constant of methyl radical–water complexes. J. Mol. Struct. Theochem, 594, 61-69 (2002).

336
(c) B.-Q. Wang, Z.-R. Li, D. Wu, X.-Y. Hao, R.-J. Li og C.-C. Sun, Single-electron hydrogenbonds in the methyl radical complexes H3C HF and H⋯ 3C HCCH: an ab initio study. ⋯Chem. Phys. Lett., 375, 91-95 (2003).(d) K. Tang og F. Q. Shi, Comparative analysis of blue-shifted hydrogen bond versus conventional hydrogen bond in methyl radical complexes. Int. J. Quantum. Chem., 107, 665-69 (2007).(e) B. Raghavendra og E. Arunan, Unpaired and σ bond electrons as H, Cl, and Li bond acceptors: An anomalous one-electron blue-shifting chlorine bond. J. Phys. Chem. A, 111, 9699-9706 (2007).(f) X. An, H. Lin, Q. Li, B. Gong og J. Cheng, Influence of substitution, hybridization, and solvent on the properties of C−HO single-electron hydrogen bond in CH3−H2O complex. J. Phys. Chem A, 112, 5258-63 (2008).(g) Q. Li, X. An, F. Luan, W. Li, B. Gong, J. Cheng og J. Sun, Cooperativity between two types of hydrogen bond in H(3)C-HCN-HCN and H(3)C-HNC-HNC complexes. J. Chem. Phys., 128, 154102 (2008).
29 (a) F. Fuster, B. Silvi, S. Berski og Z. Latajka, Topological aspects of protonation and hydrogen bonding: the dihydrogen bond case. J. Mol. Struct., 555, 75-84 (2000).(b) M. Barroso, L. G. Arnaut og S. J. Formosinho, Absolute rate calculations: atom and proton transfers in hydrogen-bonded systems. ChemPhysChem, 6, 363-71 (2005).(c) H. B. Bürgi og J. D. Dunitz, From crystal statics to chemical dynamics. Acc. Chem. Res.,16, 153-61 (1983). (d) P. Gilli, V. Bertolasi, V. Feretti og G. Gilli, Covalent nature of the strong homonuclear hydrogen bond. Study of the O‒H...O system by crystal structure correlation methods. J. Am. Chem. Soc., 116, 909-15 (1994).
30 G. Bouchoux og N. Choret, Intramolecular hydrogen migrations in ionized aliphatic alcohols. Barton type and related rearrangements. Int. J. Mass Spectrom., 201, 161-77 (2000).
31 S. Hammerum og C. B. Nielsen, Intramolecular hydrogen bonding and hydrogen atom abstraction in gas-phase aliphatic amine radical cations. J. Phys. Chem. A, 109, 12046-53 (2005).
32 S. Hammerum, Alkyl radicals as hydrogen bond acceptors – computational evidence . J. Am. Chem. Soc., 131, 8627-35 (2009).
33 M. Meot-Ner (Mautner), The ionic hydrogen bond. Chem. Rev., 105, 213-84 (2005); Chem.Rev. 112, PR22-PR103 (2012).
34 G. Gilli og P. Gilli, Towards an unified hydrogen-bond theory. J. Mol. Struct., 552, 1-15 (2000).
35 (a) J. Cao, W. Sun og J. L. Holmes, The ions CH6O+. and CH7O+. Int. J. Mass Spectrom., 217, 179-84 (2002).(b) X. Wang, W. Sun og J. L. Holmes, Water-assisted interconversions and dissociations of the acetaldehyde ion and its isomers. An experimental and theoretical study. J. Phys. Chem. A, 110, 8409-17 (2006).
36 A. Gil, M. Sodupe og J. Bertran, Unusual hydrogen bonds in [AH3–H3O]+ radical cations (A = C, Si, Ge, Sn and Pb): Single-electron hydrogen bond, proton-hydride hydrogen bond andformation of [H2AOH2]+–H2 complexes. Chem. Phys. Lett., 395, 27-32 (2004).

337
37 (a) H. Nedev, G. van der Rest, P. Mourgues og H. E. Audier, Reactions of CH3CHO+. andof CH3CHO+. with water upon FT-ICR conditions. Europ. J. Mass Spectrom., 9, 319-26 (2003).(b) H. Nedev, G. van der Rest, P. Mourgues og H. E. Audier, Hydrogen radical abstraction by small ionized molecules, distonic ions and ionized carbenes in the gas phase . Int.J. Mass Spectrom., 231, 197-205 (2004).
38 M. Semialjac, J. Loos, D. Schröder og H. Schwarz, Dissociation behavior of ionized valeramide: Part II. Theoretical exploration of the potential-energy surface. Int. J. Mass Spectrom., 214, 129-54 (2002).
39 (a) P. M. Mayer, J. W. Keister, T. Baer, M. Evans, C. Y. Ng og C.-W. Hsu, The dimethylamine dimer ion is an ion-radical complex: A combined TPEPICO, variational RRKM, and ab initio MO study of the fragmentation of ionized dimers of dimethylamine . J. Phys. Chem. A, 101, 1270-76 (1997).(b) P. M. Mayer, Ion rearrangement at the beginning of cluster formation: isomerization pathways and dissociation kinetics for the ionized dimethylamine dimer. Int. J. Mass Spectrom., 218, 1-10 (2002).
40 (a) S. Aloisio og J. S. Francisco, Radical-water complexes in Earth's atmosphere. Acc. Chem. Res., 33, 825-30 (2000).(b) J. C. Hansen og J. S. Francisco, Radical-molecule complexes: changing our perspective onthe molecular mechanisms of radical-molecule reactions and their impact on atmospheric chemistry. ChemPhysChem, 3, 833-40 (2002).(c) H. Hernández-Soto, F. Weinhold og J. S. Francisco, Radical hydrogen bonding: origin of the stability of radical-molecule complexes. J. Chem. Phys., 127, 164102 (2007).
41 H. Tsubomura, Investigations into the hydrogen bonding between n-butanol and some ketones by infra-red spectroscopy. Bull. Chem. Soc. Jap., 27, 445-50 (1954).
42 P. G. Puranik og V. Kumar, The charge transfer theory of the hydrogen bond. Proc. Ind. Acad. Sci., 58, 29-37 (1963).
43 (a) H. Ratajczak, Charge-transfer properties of the hydrogen bond. I. Theory of the enhancement of dipole moment of hydrogen-bonded systems. J. Phys. Chem., 76, 3000-04 (1972). (b) H. Ratajczak og W. J. Orville-Thomas, Charge-transfer properties of hydrogen bonds. III. Charge-transfer theory and the relation between the energy and the enhancement of dipole moment of hydrogen-bonded complexes. J. Chem. Phys., 58, 911-19 (1973).(c) H. Ratajczak og W. J. Orville-Thomas, Charge-transfer properties of the hydrogen bond. Part 6 . Charge-transfer theory and dipole moments of hydrogen-bonded complexes . J. Mol. Struct., 26, 387-91 (1975).
44 (a) H. Ratajczak, W. J. Orville-Thomas og C. N. R. Rao, Charge transfer theory of hydrogen bonds: relations between vibrational spectra and energy of hydrogen bonds. Chem. Phys., 17, 197-216 (1976).(b) C. N. R. Rao, P. C. Dwivedi, H. Ratajczak og W. J. Orville-Thomas, Relation between O—H stretching frequency and hydrogen bond energy: re-examination of the Badger–Bauer rule. J. Chem. Soc. Faraday 2, 1974, 955-66.
45 P. A. Kollman, A theory of hydrogen bond directionality. J. Am. Chem. Soc., 94, 1837-42 (1972).

338
46 (a) L. C. Allen, A model for the hydrogen bond. Proc. Nat. Acad. Sci. USA, 72, 4701-05 (1975).(b) L. C. Allen, Simple model of hydrogen bonding. J. Am. Chem. Soc., 97, 6921-40 (1975).
47 (a) S. Furutaka, H. Kondo og S. Ikawa, Infrared spectroscopic study of water–aromatic hydrocarbon mixtures at high temperatures and pressures. Bull. Chem. Soc. Japan, 74, 1775-88 (2001). Betydeligt element af autoplagiat; stort set samme indhold findes i (b) S. Furutaka og S. Ikawa, J. Chem. Phys., 117, 751-55 (2002), og (c) S. Furutaka og S. Ikawa, J. Chem. Phys., 108, 5159-60 (1998).
48 (a) A. Allerhand og P. v. R. Schleyer, A survey of C-H groups as proton donors in hydrogen bonding. J. Am. Chem. Soc., 85, 1715-23 (1963).(b) C. G. Pimentel og A. L. McClellan, The Hydrogen Bond. Freeman, San Francisco 1960.(c) F. Fuster og B. Silvi, Does the topological approach characterize the hydrogen bond?Theor. Chem. Acc., 104, 13-21 (2000).
49 (a) D. J. Swanton, G. B. Bacskay og N. S. Hush, An ab initio SCF calculation of the dipole-moment derivatives and infrared-absorption intensities of the water-dimer molecule. Chem. Phys., 82, 303-15 (1983).(b) A. V. Iogansen, Direct proportionality of the hydrogen bonding energy and the intensi-fication of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta, 55, 1585-1612 (1999).
50 E. S. Kryachko og T. Zeegers-Huyskens, Theoretical study of the CH4.(H2O)2 and
CH4.H5O2
+ complexes. Three-hydrogen-atoms interaction. J. Phys. Chem. A, 107, 7546-51 (2003).
51 (a) R. M. Badger, A relation between internuclear distances and bond force constants. J. Chem. Phys. 2, 128-131 (1934).(b) R. M. Badger, The relation between the internuclear distances and force constants of molecules and its application to polyatomic molecules. J. Chem. Phys. 3, 710-714 (1935).
52 (a) J. Sunner, K. Nishizawa og P. Kebarle, Ion-solvent molecule interactions in the gas phase. The potassium ion and benzene. J. Phys. Chem., 85, 1814-20 (1981).(b) M. Hanna, Bonding in donor-acceptor complexes. I. Electrostatic contributions to the ground-state properties of benzene-halogen complexes. J. Am. Chem. Soc., 90, 285-91 (1968).
53 N. Solcà og O. Dopfer, Hydrogen-bonded networks in ethanol proton wires: IR spectra of (EtOH)qH+-Ln clusters (L = Ar/N2, q < or = 4, n < or = 5). J. Phys. Chem. A, 109, 6174-86 (2005).
54 (a) W. Qian og S. Krimm, Limitations of the molecular multipole expansion treatment of electrostatic interactions for C-H...O and O-H...O hydrogen bonds and application of a general charge density approach. J. Phys. Chem. A, 109, 5608-18 (2005).(b) A. J. Stone, The Theory of Intramolecular Forces. Oxford University Press, Oxford, 1996.
55 I. Corral, O. Mo, M. Yáñez og L. Radom, Why are the Ca2+ and K+ binding energies of formaldehyde and ammonia reversed with respect to their proton affinities? J. Phys. Chem. A, 109, 6735-42 (2005).
56 N. I. Hammer, E. G. Diken, J. R. Roscoli, M. A. Johnson, E. M. Myshakin, K. D. Jordan, A. B. McCoy, X. Huang, J. M. Bowman og S. Carter, The vibrational predissociation

339
spectra of the H5O2+RGn (RG = Ar,Ne) clusters: correlation of the solvent perturbations in the free OH and shared proton transitions of the Zundel ion. J. Chem. Phys., 122, 244301 (2005).
57 A. W. Castleman, K. I. Peterson, B. L. Upschulte og F. J. Schilling, Energetics and structure of Na+ cluster ions. Int. J. Mass Spectrom. Ion Proc., 47, 203-06 (1983).
58 J. F. Liebman, M. J. Romm, M. Meot-Ner (Mautner), S. M. Cybulski og S. Scheiner, Isotropy in ionic interactions. 2. How spherical is the ammonium ion? Comparison of the gas-phase clustering energies and condensed-phase thermochemistry of K+ and NH4+. J. Phys. Chem., 95, 1112-19 (1991).
59 (a) D. J. Goebbert og P. G. Wenthold, Water dimer proton affinity from the kinetic method: dissociation energy of the water dimer. Europ. J. Mass Spectrom., 10, 837-45 (2004).(b) W. Klopper, J. G. C. M. van Duijneveldt-van de Rijdt og F. B. van Duijneveldt, Computational determination of equilibrium geometry and dissociation energy of the water dimer. Phys. Chem. Chem. Phys., 2, 2227-34 (2000).
60 (a) P. Kebarle, Ion thermochemistry and solvation from gas phase ion equilibria. Ann. Rev. Phys. Chem., 28, 445-76 (1977).(b) W. R. Davidson, J. Sunner og P. Kebarle, Hydrogen bonding of water to onium ions. Hydration of substituted pyridinium ions and related systems. J. Am. Chem. Soc., 101, 1675-80 (1979).
61 P. J. Desmeules og L. C. Allen, Strong, positive-ion hydrogen bonds: the binary complexes formed from NH3, OH2, FH, PH3, SH2, and ClH. J. Chem. Phys., 72, 4731-48 (1980).
62 S. G. Olesen og S. Hammerum, Redshift or adduct stabilization – a computational study of hydrogen bonding in adducts of protonated carboxylic acids. Europ. J. Mass Spectrom., 15, 239-48 (2009) (tilgængelig på http://www-mf.kiku.dk/art/carbox.pdf).
63 E. A. Hillenbrand og S. Scheiner, Analysis of the principles governing proton-transfer reactions. Carboxyl group. J. Am. Chem. Soc., 108, 7178-86 (1986).
64 K. Wendler, J. Thar, S. Zahn og B. Kirchner, Estimating the hydrogen bond energy. J. Phys.Chem. A, 114, 9529-36 (2010).
65 M. Reiher, D. Sellmann og B. A. Hess, Stabilization of diazene in Fe(II)-sulfur model complexes relevant for nitrogenase activity. I. A new approach to the evaluation of intramolecular hydrogen bond energies. Theor. Chem. Acc., 106, 379-92 (2001).
66 A. T. Ayoub, J. Tuszynski og M. Klobukowski, Estimating hydrogen bond energies: comparison of methods. Theor. Chem. Acc., 133, 1520 (2014).
67 M. Freindorf, E. Kraka og D. Cremer, A comprehensive analysis of hydrogen bond interactions based on local vibrational modes. Int. J. Quant. Chem., 112, 3174-87 (2012).
68 E. D. Glendening og A. Streitwieser, Natural energy decomposition analysis: An energy partitioning procedure for molecular interactions with application to weak hydrogen bonding, strong ionic, and moderate donor–acceptor interactions. J. Chem. Phys., 100, 2900-09 (1994).
69 S. J. Grabowski, Theoretical studies of strong hydrogen bonds. Ann. Rep. Progr. Chem., sect C, 102, 131-65 (2006).
70 J. P. Bowen, J. B. Sorensen og K. N. Kirschner, Calculating interaction energies using first principle theories: consideration of basis set superposition error and fragment relaxation . J. Chem. Ed., 84, 1225-29 (2007).
71 D. J. Grant, M. H. Matus, J. R. Switzer, D. A. Dixon, J. S. Francisco og K. O. Christe, Bond dissociation energies in second-row compounds. J. Phys. Chem. A, 112, 3145-56 (2008).

340
72 M. von Hopffgarten og G. Frenking, Energy decomposition analysis. WIREs Comput. Mol. Sci., 2, 43-62 (2012).
73 (a) G. E. Hilbert, O. R. Wulf, S. B. Hendricks og U. Liddel, The hydrogen bond between oxygen atoms in some organic compounds. J. Am. Chem. Soc., 58, 548-55 (1936).(b) S. B. Hendricks, O. R. Wulf, G. E. Hilbert og U. Liddel, Hydrogen bond formation between hydroxyl groups and nitrogen atoms in some organic compounds. J. Am. Chem. Soc.,58, 1991-96 (1936).
74 R. M. Badger og S. H. Bauer, Spectroscopic studies of the hydrogen bond. II. The shift of the O–H vibrational frequency in the formation of the hydrogen bond. J. Chem. Phys., 5, 839-51 (1937).
75 (a) E. L. Lippincott og R. Schroeder, One-dimensional model of the hydrogen bond. J. Chem. Phys., 23, 1099-1106 (1955).(b) R. Schroeder og E. L. Lippincott, Potential function model of hydrogen bonds. J. Phys. Chem., 61, 921-28 (1957).
76 (a) B. S. Ault, E. Steinback og G. C. Pimentel, Matrix isolation studies of hydrogen bonding. Vibrational correlation diagram. J. Phys. Chem., 79, 615-20 (1975).(b) A. J. Barnes, Molecular complexes of the hydrogen halides studied by matrix isolation infrared spectroscopy. J. Mol. Struct., 100, 259-80 (1983).(c) T. Zeegers-Huyskens, Correlation between the frequency shift of the stretching vibration of hydrogen bond complexes and the proton affinity. J. Mol. Liq., 67, 33-47 (1995).(d) O. Dopfer, R. V. Olkhov, D. Roth og J. P. Maier, Intermolecular interaction in proton-bound dimers: infrared photodissociation spectra of Rg–HOCO+ (Rg=He, Ne, Ar) complexes. Chem. Phys. Lett., 296, 585-91 (1998).(e) T. D. Fridgen, L. MacAleese, P. Maitre, T. B. McMahon, P. Boissel og J. Lemaire, Infrared spectra of homogeneous and heterogeneous proton-bound dimers in the gas phase. Phys. Chem. Chem. Phys., 7, 2747-55 (2005).(f) J. R. Roscioli, L. R. McCunn og M. A. Johnson, Quantum structure of the inter-molecular proton bond. Science, 316, 249-54 (2007).(g) L. Tao, J. Han og F.-M. Tao, Correlations and predictions of carboxylic acid pKa values using intermolecular structure and properties of hydrogen-bonded complexes. J. Phys. Chem. A, 112, 775-82 (2008).
77 L. Bian, Proton donor is more important than proton acceptor in hydrogen bond formation: a universal equation for calculation of hydrogen bond strength. J. Phys. Chem. A, 107, 11517-24 (2003).
78 L. C. Remer og J. H. Jensen, Toward a general theory of hydrogen bonding: the short, strong hydrogen bond [HOH···OH]-.. J. Phys. Chem. A, 104, 9266-75 (2000).
79 P. Gilli og G. Gilli, Hydrogen bond models and theories: The dual hydrogen bond model and its consequences. J. Mol. Struct., 972, 2-10 (2010).
80 (a) B. Chan, J. E. Del Bene og Leo Radom, Proton-bound homodimers: How are the binding energies related to proton affinities? J. Am. Chem. Soc., 129, 12197-99 (2007).(b) B. Chan, J. E. Del Bene og Leo Radom, What factors determine whether a proton-bound homodimer has a symmetric or an asymmetric hydrogen bond? Mol. Phys., 107, 1095-1105 (2009).

341
(c) B. Chan, J. E. Del Bene og Leo Radom, Proton-bound homodimers involving second-rowatoms. Theor. Chem. Acc., 131, 1088-96 (2012).
81 E. M. Arnett, B. Chawla, L. Bell, M. Taagepera, W. J. Hehre og R. W. Taft, Solvation and hydrogen bonding of pyridinium ions. J. Am. Chem. Soc., 99, 5729-39 (1977).
82 P. Gilli, V. Bertolasi, V. Feretti og G. Gilli, Predicting hydrogen-bond strengths from acid-base molecular properties. The pKA slide rule: toward the solution to a long-lasting problem. Acc. Chem. Res., 42, 33-44 (2009).
83 J. W. Larson og T. B. McMahon, Formation, thermochemistry, and relative stabilities of proton-bound dimers of oxygen n-donor bases from ion cyclotron resonance solvent-exchange equilibrium measurements. J. Am. Chem. Soc., 104, 6255-61 (1982).
84 (a) M. Meot-Ner (Mautner), The ionic hydrogen bond and ion solvation. 1. NH+...O, NH+...N, and OH+...O bonds. Correlations with proton affinity. Deviations due to structural effects. J. Am. Chem. Soc., 106, 1257-64 (1984).(b) M. Meot-Ner (Mautner) og L.W. Sieck, The ionic hydrogen bond and ion solvation. 5. OH...O bonds. Gas-phase solvation and clustering of alkoxide and carboxylate anions . J. Am. Chem. Soc., 108, 7525 (1986).(c) C. V. Speller og M. Meot-Ner (Mautner), The ionic hydrogen bond and ion solvation. 3. Bonds involving cyanides. Correlations with proton affinities. J. Phys. Chem., 89, 5217-22 (1985).(d) M. Meot-Ner (Mautner) og L. W. Sieck, The ionic hydrogen bond and ion solvation. 4. SH...O and NH...S bonds. Correlation with proton affinity. Mutual effects of weak and strong ligands in mixed clusters. J. Phys. Chem., 89, 5222-25 (1985).
85 J.-F. Gal og P.-C. Maria, Correlation analysis of acidity and basicity: from the solution to the gas phase. Prog. Phys. Org. Chem., 17, 159-238 (1990).
86 J. Graton, M. Berthelot og C. Laurence, Hydrogen-bond basicity pKHB scale of secondary amines. J. Chem. Soc., Perkin 2, 2001, 2130-35.
87 J. Chen, M. A. McAllister, J. K. Lee og K. N. Houk, Short, strong hydrogen bonds in the gas phase and in solution: theoretical exploration of pKa matching and environmental effects on the strengths of hydrogen bonds and their potential roles in enzymatic catalysis. J. Org. Chem., 63, 4611-19 (1998).
88 P. M. Mayer, Structures and binding energies of proton-bound pairs of HCN and CH3CN with NH3, H2O, HF, CH3NH2, CH3OH, and CH3F. J. Phys. Chem. A, 103, 5905-09 (1999).
89 T. Lankau og C.-H. Yu, Correlated proton motion in hydrogen bonded systems: tuning proton affinities. Phys. Chem. Chem. Phys., 9, 299-310 (2007).
90 T. Zeegers-Huyskens, Enthalpies of hydrogen bonds and proton affinities. J. Mol. Struct., 135, 93-103 (1986).(a) M. T. Nguyen, N. Leroux og T. Zeegers-Huyskens, Ab initio calculations on the hydrogen bond interaction between diacetamide and ammonia. J. Mol. Struct., 404, 75-82 (1997).(b) T. Zeegers-Huyskens, Are the enol-enolate hydrogen bonds at matched PA really symmetrical? J. Org. Chem., 64, 4946-48 (1999).(c) A. K. Chandra og T. Zeegers-Huyskens, Theoretical study of the symmetry of the (OH...O)- hydrogen bonds in vinyl alcohol-vinyl alcoholate systems. J. Org. Chem., 68, 3618-25 (2003).

342
(d) A. K. Chandra, M. T. Nguyen, T. Zeegers-Huyskens, Theoretical study of the interaction between thymine and water. Protonation and deprotonation enthalpies and comparison with uracil. J. Phys. Chem. A, 102, 6010-16 (1998). Se dog også A. S. Özen, F.De Proft, V. Aviyente og P. Geerlings, Interpretation of hydrogen bonding in the weak and strong regions using conceptual DFT descriptors. J. Phys. Chem. A, 110, 5860-68 (2006).(e) T. Zeegers-Huyskens, Energies of OH···X- hydrogen bonds in the gas phase and proton affinities. Chem. Phys. Lett., 129, 172-75 (1986).(f) T. Zeegers-Huyskens, Importance of the proton affinities in hydrogen bond studies. A new exponential expression. J. Mol. Struct., 177, 125-41 (1988).(g) A. K. Chandra, M. T. Nguyen, T. Uchimaru og T. Zeegers-Huyskens, Protonation and deprotonation enthalpies of guanine and adenine and implications for the structure and energy of their complexes with water: comparison with uracil, thymine, and cytosine . J. Phys. Chem. A, 103, 8853-60 (1999).
91 (a) W. J. Bouma og L. Radom, Is the carboxylic acid-fluoride bond really the strongest type ofhydrogen bond? Chem. Phys. Lett., 64, 216-18 (1979).(b) A. Pross og L. Radom, Effect of substituents on the structure of reaction complexes. J. Am. Chem. Soc., 103, 6049-53 (1981).
92 (a) T. D. Fridgen, Structures of heterogeneous proton-bond dimers with a high dipole momentmonomer: covalent vs electrostatic interactions. J. Phys. Chem. A, 110, 6122-28 (2006).(b) M. B. Burt og T. D. Fridgen, Heterogeneous proton-bound dimers with a high dipole moment monomer: How could we experimentally observe these anomalous ionic hydrogen bonds? J. Phys. Chem. A, 111, 10738-44 (2007).
93 G. H. Gardenier, J. R. Roscioli og M. A. Johnson, Intermolecular proton binding in the presence of a large electric dipole: Ar-tagged vibrational predissociation spectroscopy of the CH3CN x H+ x OH2 and CH3CN x D+ x OD2 complexes. J. Phys. Chem. A, 112, 12022-6 (2008).
94 G. Sedo og K. L. Leopold, Proton transfer in a molecular complex: assessments from both thedonor and acceptor points of view. J. Phys. Chem. A, 115, 1787-94 (2011).
95 A. E. Reed, F. Weinhold, L. A. Curtiss og D. J. Pochatko, Natural bond orbital analysis of molecular interactions: theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO, and CO2 with HF, H2O, and NH3. J. Chem. Phys., 84, 5687-5705 (1986).
96 (a) I. V. Alabugin, M. Manoharan, S. Peabody og F. Weinhold, Electronic basis of improper hydrogen bonding: a subtle balance of hyperconjugation and rehybridization. J. Am. Chem. Soc., 125, 5973-87 (2003).(b) I. V. Alabugin, K. M. Gilmore og P. W. Peterson, Hyperkonjugation. WIREs Comput. Mol. Sci., 1, 109-41 (2011).
97 (a) J. L. Beauchamp, Ion cyclotron resonance spectroscopy. Annu. Rev. Phys. Chem., 22, 527-61 (1971).(b) C. R. Moylan og J. I. Brauman, Gas phase acid-base chemistry. Annu. Rev. Phys. Chem., 34, 187-215 (1983).(c) W.-Z. Liu og F. G. Bordwell, Gas-phase and solution-phase homolytic bond dissociation energies of N-H+ bonds in the conjugate acids of nitrogen bases. J. Org. Chem., 61, 4778-83 (1996).
98 (a) S. Campbell, E. M. Marzluff, M. T. Rodgers, J. L. Beauchamp, M. E. Rempe, K. F. Schwinck og D. L. Lichtenberger, Proton affinities and photoelectron spectra of phenyl-

343
alanine and N-methyl- and N,N-dimethylphenylalanine. Correlation of lone-pair ionization energies with proton affinities, and implications for N-methylation as a method for site specific protonation of peptides. J. Am. Chem. Soc., 116, 5257-64 (1994).(b) J. Catalan, O. Mo, P. Perez og M. Yañez, Prediction of proton affinities and protonation sites using a multivariate linear correlation. J. Chem. Soc. Perkin 2, 1982, 1409-18.(c) E. Uggerud, Properties and reactions of protonated molecules in the gas phase. Experimentand theory. Mass Spectrom. Rev., 11, 389-430 (1992).(d) S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin og W. G. Mallard, Gas-phase ion and neutral thermochemistry. J. Phys. Chem. Ref. Data, 17 suppl. vol. 1 (1988).
99 (a) Z. B. Maksić og R. Vianello, Quest for the origin of basicity: initial vs final state effect inneutral nitrogen bases. J. Phys. Chem. A, 106, 419-30 (2002).(b) Z. B. Maksić, B. Kovačević og R. Vianello, Advances in determining the absolute protonaffinities of organic molecules in the gas phase and their interpretation: a theoretical account. Chem. Rev., 112, 5240-70 (2012).(c) C. Deakyne, Proton affinities and gas-phase basicities: theoretical methods and structural effects. Int. J. Mass Spectrom., 227, 601-16 (2003).
100 (a) C. A. Coulson og U. Danielsson, Ionic and covalent contributions to the hydrogen bond. 1. Ark. Fys., 8, 239-44 (1954).(b) C. A. Coulson og U. Danielsson, Ionic and covalent contributions to the hydrogen bond. 2. Ark. Fys., 8, 245-55 (1954).(c) C. A. Coulson, The hydrogen bond – A review of the present position. Sci. Appl. Ind., 10, 149-59 (1957).
101 C. A. Coulson, The hydrogen bond. I Hydrogen Bonding. Red. D. Hadzi; Pergamon Press, London 1959; pp 339-55.
102 (a) N. D. Sokolov, On the quantum theory of the hydrogen bond. I Hydrogen Bonding. Red. D. Hadzi; Pergamon Press, London 1959; pp 385-92.(b) N. D. Sokolov, Dokl. Akad. Nauk. SSSR, 58, 611 (1947) (henvisning vanskelig at verificere).
103 (a) P. R. Horn, Y. Mao og M. Head-Gordon, Defining the contributions of permanent electrostatics, Pauli repulsion, and dispersion in density functional theory calculations of intermolecular interaction energies. J. Chem. Phys., 144, 114107 (2016). (b) P. R. Horn, E. J. Sundstrom, T. A. Baker og M. Head-Gordon, Unrestricted absolutely localized molecular orbitals for energy decomposition analysis: theory and applications to intermolecular interactions involving radicals. J. Chem. Phys., 138, 134119 (2013).(c) K. U. Lao og J. M. Herbert, Energy decomposition analysis with a stable charge-transfer term for interpreting intermolecular ionteractions. J. Chem. Theory Comput., 12, 2569-82 (2016).(d) D. Cappelletti, E. Ronca, L. Belpassi, F. Tarantelli og F. Pirani, Revealing charge-transfer effects in gas-phase water chemistry. Acc. Chem. Res., 45, 1571-80 (2012).(e) R. Z. Khaliullin, A. T. Bell og M. Head-Gordon, Electron donation in the water—water hydrogen bond. Chem. Eur. J., 15, 851-55 (2009).

344
(f) E. Ramos-Cordoba, D. S. Lambrecht og M. Head-Gordon, Charge-transfer and the hydrogen bond: Spectroscopic and structural implications from electronic structure calculations. Faraday Discuss., 150, 345-62 (2011).(g) M. J. S. Phipps, T. Fox, C. S. Tautermann og C.-K. Skylaris, Energy decomposition analysis approaches and their evaluation on typical protein-drug interaction patterns . Chem. Soc. Rev., 44, 3177-3211 (2015).(h) E. D. Glendening, Natural energy decomposition analysis: Extension to density functional methods and analysis of cooperative effects in water clusters . J. Phys. Chem. A, 109, 11936-40 (2005).
104 (a) A. J. Stone, Natural bond orbitals and the nature of the hydrogen bond. J. Phys. Chem. A,121, 1531-34 (2017). Et noget polemisk arbejde der har foranlediget svar og gentagelse.(b) F. Weinhold og E. D. Glendening, Comment on "Natural bond orbitals and the nature ofthe hydrogen bond". J. Phys. Chem. A, 122, 724-32 (2017). (c) A. J. Stone og K. Szalewicz, Reply to comment "Natural bond orbitals and the nature of the hydrogen bond". J. Phys. Chem. A, 122, 733-36 (2017).
105 (a) C. F. Guerra og F. M. Bickelhaupt, Charge transfer and environment effects responsible for characteristics of DNA base pairing. Angew. Chem. Int. Ed., 38, 2942-45 (1999).(b) C. F. Guerra, F. M. Bickelhaupt, J. G. Snijders og E. J. Baerends, The nature of the hydrogen bond in DNA base pairs: the role of charge transfer and resonance assistance. Chem.Eur. J., 5, 3581-94 (1999).(c) C. F. Guerra og F. M. Bickelhaupt, Orbital interactions in strong og weak hydrogen bonds essential for DNA replication. Angew. Chem. Int. Ed., 41, 2092-95 (2002).
106 R. D. Harcourt, Four-electron three-center bonding: one-electron and concerted two-electron delocalizations into bonding and antibonding molecular orbitals. J. Phys. Chem. A, 103, 4293-97 (1999).
107 P. A. Kollman og L. C. Allen, Theory of the hydrogen bond. Chem. Rev., 72, 283-303 (1972).
108 G. R. Desiraju, Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res., 35, 565-73 (2002).
109 (a) S. Humbel, Short strong hydrogen bonds: a valence bond analysis. J. Phys. Chem. A, 106,5517-20 (2002).(b) S. Humbel, N. Hoffmann, I. Côte og J. Bouquart, Substituent effects on two-center three-electron bonds and hydrogen bonds involving unsaturated organic functional groups andan ammonia radical cation - the resonance contribution. Chem. Eur. J., 6, 1592-1600 (2000).
110 S. A. C. McDowell og A. D. Buckingham, On the correlation between bond-length change and vibrational frequency shift in hydrogen-bonded complexes: a computational study of Y...HCl dimers (Y = N2, CO, BF). J. Am. Chem. Soc., 127, 15515-20 (2005).
111 F. B. van Duijneveldt, Effect of lone-pair hybridization on the stability of weak hydrogen bonds. J. Chem. Phys., 49, 1424-29 (1968).
112 N. G. Korzhenevskaya, V. I. Rybachenko og V. V. Kovalenko, Peculiarities of the effect of the electronic nature of substituents on the basic properties of aliphatic amines . Russ. Chem. Bull., Int. Ed., 52, 893-99 (2003); Izvest. Akad. Nauk., Ser. Khim., 2003, 848-53.

345
113 (a) D. H. Aue, H. M. Webb og M. T. Bowers, Proton affinities, ionization potentials, and hydrogen affinities of nitrogen and oxygen bases. Hybridization effects . J. Am. Chem. Soc., 97, 4137-9 (1975).(b) D. H. Aue og M. T. Bowers, Stabilities of positive ions from equilibrium gas-phase basicity measurements. I Gas Phase Ion Chemistry, bd. 2, pp 2-52. Red. M. T. Bowers. Academic Press, New York 1979.(c) D. H. Aue, H. M. Webb, W. R. Davidson, M. Vidal, M. T. Bowers, H. Goldwhite, L. E. Vertal, J. E. Douglas, P. A. Kollman og G. L. Kenyon, Proton affinities and photo-electron spectra of three-membered-ring heterocycles. J. Am. Chem. Soc., 102, 5151-57 (1980).
114 S. X. Tian og J. Yang, Effects of intramolecular hydrogen bonding on the ionization energies of proline. Angew. Chem. Int. Ed., 45, 2969-72 (2006).
115 E. D. Glendening, C. R. Landis og F. Weinhold, Natural orbital bond methods. WIREs Comput. Mol. Sci., 2, 1-42 (2012)
116 (a) S. G. Olesen og S. Hammerum, Hydrogen bonding to alkanes: computational evidence. J. Phys. Chem. A, 113, 7940-44 (2009).(b) S. G. Olesen, T. L. Gausco, G. H. Weddle, S. Hammerum og M. A. Johnson, Vibrational predissociation spectra of the Ar tagged [CH4 H3O+] binary complex: spectroscopic signature of hydrogen bonding to an alkane. Mol. Phys., 108, 1191-97 (2010).
117 (a) G. A. Olah, Y. Halpern, J. Shen og Y. K. Mo, Electrophilic reactions at single bonds. III. Hydrogen-deuterium exchange and protolysis (deuterolysis) of alkanes with superacids. Mechanism of acid-catalyzed hydrocarbon transformation reactions involving the electron pair donor ability of single bonds (shared electron pairs) via three-center bond formation . J. Am. Chem. Soc., 93, 1251-56 (1971).(b) G. A. Olah, Carbocations and electrophilic reactions. Angew. Chem. Int. Ed., 12, 173-212 (1973).(c) A. Goeppert og J. Sommer, H/D exchange, protolysis and oxidation of C3-C5 alkanes in HF-SbF5. σ-basicity vs. reactivity of C-H bonds. New J. Chem., 26, 1335-39 (2002).
118 S. L. Bennett og F. H. Field, Reversible reactions of gaseous ions. V. Methane-water system atlow temperatures. J. Am. Chem. Soc., 94, 5188-95 (1972).
119 P. Ahlberg, A. Karlsson, A. Goeppert, S. O. N. Lill, P. Dinér og J. Sommer, Solvated CH5
+ in liquid superacid. Chem. Eur. J., 7, 1936-43 (2001).120 A. N. Isaev, Intermolecular charge transfer as evidence for unusual O—H...C(sp3) hydrogen
bond. Comput. Theor. Chem., 1090, 180-92 (2016).121 (a) P. Dinér, Superacid-promoted ionization of alkanes without carbonium ion formation: a
density functional study. J. Phys. Chem. A, 116, 9979-84 (2012).(b) P. Vogel, Carbocation Chemistry. Elsevier, Amsterdam 1985.
122 (a) D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros og J. L. Wood, Deoxygenation of alcohols employing water as the hydrogen atom source. J. Am. Chem. Soc., 127, 12513-15 (2005).(b) D. Pozzi, E. M. Scanlan og P. Renaud, A mild radical procedure for the reduction of B-alkylcatecholboranes to alkanes. J. Am. Chem. Soc., 127, 14204-5 (2005).
123 (a) W. Tantawy og H. Zipse, Hydroxylic solvents as hydrogen atom donors in radical reactions. Eur. J. Org. Chem., 2007, 5817-20.

346
(b) G. Povie, M. Marzorati, P. Bigler og P. Renaud, Role of equilibrium associations on the hydrogen atom transfer from the triethylborane—methanol complex. J. Org. Chem., 78, 1553-58 (2013).
124 (a) J. M. Cuerva, A. G. Campaña, J. Justicia, A. Rosales, J. L. Oller-López, R. Robles, D. J. Cárdenas, E. Buñuel og J. E. Oltra, Water: The ideal hydrogen-atom source in free-radical chemistry mediated by TiIII and other single-electron-transfer metals? Angew. Chem. Int. Ed., 45, 5522-26 (2006).(b) M. Paradas, A. G. Campaña, T. Jiménez, R. Robles, J. E. Oltra, E. Buñuel, J. Justicia, D. J. Cárdenas og J. M. Cuerva, Understanding the exceptional hydrogen-atom donor characteristics of water in Ti(III)-mediated free-radical chemistry. J. Am. Chem. Soc., 132, 12748-56 (2010).(c) A. Gansäuer, M. Behlendorf, A. Cangönül, C. Kube, J. M. Cuerva, J. Friedrich og M. van Gastel, H2O activation for hydrogen-atom transfer: correct structures and revised mechanisms. Angew. Chem. Int. Ed., 51, 3266-70 (2012).
125 U. Koch og P. L. A. Popelier, Characterization of C-H-O hydrogen bonds on the basis of the charge density. J. Phys. Chem., 99, 9747-54 (1995).
126 J. Poater, M. Solà og F. M. Bickelhaupt, A model of the chemical bond must be rooted in quantum mechanics, provide insight, and possess predictive power. Chem. Eur. J., 12, 2902-5 (2006).
127 (a) S. J. Grabowski, W. A. Sokalski og J. Leszczynski, Is a π···H+···π complex hydrogen bonded? J. Phys. Chem. A, 108, 1806-12 (2004).(b) R. Parthasarathi, V. Subramanian og N. Sathyamurthy, Hydrogen bonding without borders: an atoms-in-molecules perspective. J. Phys. Chem. A, 110, 3349-51 (2006).(c) M. Torrent-Sucarrat og J. M. Anglada, On the gas phase hydrogen bond complexes between formic acid and hydroperoxyl radical. A theoretical study. J. Phys. Chem. A, 110, 9718-26 (2006).(d) B. Bankiewicz, P. Matczak og M. Palusiak, Electron density characteristics in bond critical point (QTAIM) versus interaction energy components (SAPT): the case of charge-assisted hydrogen bonding. J. Phys. Chem. A, 116, 452-59 (2012).(e) A. Ebrahimi, S. M. H. Khorassani og H. Delarami, Estimation of individual binding energies in some dimers involving multiple hydrogen bonds using topological properties of electron charge density. Chem. Phys., 365, 18-23 (2009).(f) O. Mó, M. Yañez og J. Elguero, Cooperative (nonpairwise) effects in water trimers: An abinitio molecular orbital study. J. Chem. Phys., 97, 6628-38 (1992).
128 E. Espinosa, M. Souhassou, H. Lachekar og C. Lecomte, Topological analysis of the electron density in hydrogen bonds. Acta Cryst., B55, 563-72 (1999).
129 J. E. Carpenter og F. Weinhold, Analysis of the geometry of the hydroxymethyl radical by the “Different hybrids for different spins” natural orbital procedure. J. Mol. Struct. Theochem., 169, 41-62 (1988).


















