
TLR2 Signaling and Th2 Responses Drive Tannerella forsythia-Induced Periodontal Bone · PDF...
10
of May 12, 2018. This information is current as Bone Loss -Induced Periodontal Tannerella forsythia TLR2 Signaling and Th2 Responses Drive Achsah D. Keegan, Sarah L. Gaffen and Ashu Sharma Srinivas R. Myneni, Rajendra P. Settem, Terry D. Connell, http://www.jimmunol.org/content/187/1/501 doi: 10.4049/jimmunol.1100683 2011; 2011; 187:501-509; Prepublished online 1 June J Immunol Material Supplementary 3.DC1 http://www.jimmunol.org/content/suppl/2011/06/01/jimmunol.110068 References http://www.jimmunol.org/content/187/1/501.full#ref-list-1 , 24 of which you can access for free at: cites 62 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2011 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on May 12, 2018 http://www.jimmunol.org/ Downloaded from by guest on May 12, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of TLR2 Signaling and Th2 Responses Drive Tannerella forsythia-Induced Periodontal Bone · PDF...

of May 12, 2018.This information is current as
Bone Loss-Induced PeriodontalTannerella forsythia
TLR2 Signaling and Th2 Responses Drive
Achsah D. Keegan, Sarah L. Gaffen and Ashu SharmaSrinivas R. Myneni, Rajendra P. Settem, Terry D. Connell,
http://www.jimmunol.org/content/187/1/501doi: 10.4049/jimmunol.11006832011;
2011; 187:501-509; Prepublished online 1 JuneJ Immunol
MaterialSupplementary
3.DC1http://www.jimmunol.org/content/suppl/2011/06/01/jimmunol.110068
Referenceshttp://www.jimmunol.org/content/187/1/501.full#ref-list-1
, 24 of which you can access for free at: cites 62 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
TLR2 Signaling and Th2 Responses Drive Tannerellaforsythia-Induced Periodontal Bone Loss
Srinivas R. Myneni,* Rajendra P. Settem,* Terry D. Connell,†,‡ Achsah D. Keegan,x
Sarah L. Gaffen,{ and Ashu Sharma*
Periodontal disease (PD) is a chronic inflammation of the tooth-supporting soft tissue and alveolar bone due to infection by a select
group of Gram-negative microbes, which leads to tooth loss if untreated. Because mice deficient in CD4+ cells are resistant to
infection-induced alveolar bone loss, Th cells have been implicated in bone-destructive processes during PD. However, the extent
to which different Th cell subtypes play roles in pathogenesis or host protection remains to be defined and is likely to vary
depending on the dominant microorganism involved. By far, Porphyromonas gingivalis is the best-studied periodontal microbe in
PD. Although the Gram-negative anaerobe Tannerella forsythia is also a vital contributor to periodontal bone loss, almost nothing
is known about immune responses to this organism. Previous studies from our laboratory revealed that T. forsythia induces
periodontal bone loss in mice and that this bone loss depends on the bacterially expressed BspA protein. In this study, we showed
that T. forsythia activates murine APCs primarily through TLR2-dependent signaling via BspA. Furthermore, T. forsythia in-
fection causes a pronounced Th2 bias, evidenced by T cell expression of IL-5, but not IFN-g or IL-17, in draining lymph nodes.
Consistently, deficiencies in TLR2 or STAT6 result in resistance to T. forsythia-induced alveolar bone loss. Thus, TLR2 signaling
and Th2 cells play pathogenic roles in T. forsythia-induced alveolar bone destruction. The Journal of Immunology, 2011, 187:
501–509.
Periodontal disease (PD) is an inflammatory disease of thetooth-supporting tissue (periodontium) that frequentlyleads to tooth loss (1) and is the most common cause of
inflammation-induced bone loss in humans. PD is caused bya select group of anaerobic Gram-negative bacteria that colonizesubgingival spaces as biofilms. Periodontal bone destruction re-sults mainly from the effects of the immune response to thesebiofilm bacteria (2, 3). The pathogenesis of PD is mediated bya polymicrobial consortium consisting of the Gram-negativepathogens Porphyromonas gingivalis, Treponema denticola, andTannerella forsythia, collectively known as the red complex (4–6).Although T. forsythia has been increasingly implicated in thedevelopment and severity of PD, it remains a highly understudiedpathogen (7, 8). Its etiological role in PD has been recognizedrelatively recently, and the virulence mechanisms of T. forsythiaare only just beginning to be defined (7). Unlike P. gingivalis, T.
forsythia is a fastidious microbe with stringent growth require-ments. We were the first, to our knowledge, to document the vir-ulence potential of T. forsythia in a murine model of PD (9). In sodoing, we found that the alveolar bone loss is dependent on thebacterially expressed virulence protein BspA (9). However, theimmune response to T. forsythia remains almost entirely undefined.Alveolar bone loss in response to oral infection by P. gingivalis
is dependent on the host response. For example, SCID mice ormice specifically deficient in CD4+ T cells are resistant to alveolarbone loss due to P. gingivalis infection (10–12). Moreover, Th1responses are associated with P. gingivalis-stimulated alveolarbone loss in mice (13). In humans, the role of T cells in PDpathobiology is complex, and data support a role for T cells inboth protection and pathogenesis (6, 14–16). The existence of twodistinct effector CD4 T cell subsets has been recognized since1986 (17), with the description of Th1 (IFN-g–producing) andTh2 (IL-4-, IL-5- and IL-13–producing) cells. The Th17 lineage ofIL-17–producing T cells was recognized in 2005 (18). In P. gin-givalis infection, both Th1 and Th2 cytokines are found in theperiodontal lesion (6, 14–16). Prior to the discovery of Th17 cells,it was suggested that Th1 cells are characteristic of stable lesions,whereas Th2 cells are associated with disease (14). However, IL-17 was also documented in periodontitis patients with severedisease (19–22), and a significant number of CD4+ T cells isolatedfrom gingival tissue of periodontitis patients express IL-17 (23). InP. gingivalis murine infections, IL-17 signaling is host protectiveby virtue of limiting infection via neutrophil mobilization (24).The Th effector responses to T. forsythia are still poorly defined
and may be different from responses to P. gingivalis. This couldarise from differences in the nature of the pathogen-associatedmolecular patterns expressed on different periodontal pathogens,which ultimately shape the adaptive response. In support of thisnotion, T. forsythia, unlike P. gingivalis, is unable to block neu-trophil recruitment during infection in a mouse model (25), andrecent studies demonstrated that T. forsythia, but not P. gingivalis,preferentially activates TLR2. For instance, although P. gingivalis
*Department of Oral Biology, School of Dental Medicine, University at Buffalo,Buffalo, NY 14214; †Department of Microbiology and Immunology, University atBuffalo, Buffalo, NY 14214; ‡Witebsky Center for Microbial Pathogenesis and Im-munology, University at Buffalo, Buffalo, NY 14214; xDepartment of Microbiologyand Immunology, Center for Vascular and Inflammatory Diseases, University ofMaryland School of Medicine, Baltimore, MD 21201; and {Division of Rheumatol-ogy and Clinical Immunology, Department of Medicine, University of Pittsburgh,Pittsburgh, PA 15261
Received for publication March 8, 2011. Accepted for publication April 28, 2011.
This work was supported by U.S. Public Health R01 Grants DE1074905 (to A.S.and S.L.G.), DE14749 (to A.S.), DE13833 (to T.D.C.), and AI059775 and AI038985(to A.D.K.).
Address correspondence and reprint requests to Dr. Ashu Sharma, Department ofOral Biology, School of Dental Medicine, University at Buffalo, 311 Foster Hall,3435 Main Street, Buffalo, NY 14214. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: ABC, alveolar bone crest; BMDC, bone marrow-derived dendritic cell; CEJ, cemento-enamel junction; cLN, cervical lymph node;DC, dendritic cell; eLPS, Escherichia coli LPS; PD, periodontal disease; TRAP,tartrate-resistant acid phosphatase; WT, wild-type.
Copyright� 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1100683
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

through its fimbriae and LPS predominantly activate TLR2 (26),whole bacteria (27) and both minor and major fimbrial proteinsactivate TLR4/CD14/MD2 (28, 29). Thus, the role of CD4 ef-fector responses in PD bone loss remains poorly defined (14, 30),particularly specific responses to T. forsythia.TLRs can play protective or destructive roles, depending on the
nature of the invading pathogen and its associated virulence de-terminants (31). T. forsythia, as well as its virulence factor BspA,induce proinflammatory cytokine and chemokine secretion throughTLR2 (32–34). Signaling by TLR2 in APCs and expression ofspecific cytokines were suggested to favor Th2 responses (35–38).Moreover, the Th2-specific transcription factor STAT6 was linkedto susceptibility to PD in mice (39). Accordingly, it was compellingto determine the role of TLR2 and STAT6-mediated responses in T.forsythia-induced bone destruction. We predicted that T. forsythiawould favor a Th2 inflammatory response by activating TLR2. Wefurther hypothesized that the Th2 response would exert a de-structive role, based on our prior observation that T. forsythia causesalveolar bone loss in mice (9). As shown in this study, we found thatTLR22/2 or STAT62/2 mice indeed showed markedly decreasedsusceptibility to T. forsythia-induced PD bone loss, associated withdecreased Th2 responses. These results imply a critical involvementof a bone-destructive Th2 response following oral infection withT. forsythia, mediated via TLR2 signaling.
Materials and MethodsMice
Specific-pathogen free BALB/cJ mice (wild-type [WT]) and STAT62/2
mice (BALB/cJ background) were from The Jackson Laboratory (BarHarbor, ME). TLR22/2 mice on a BALB/cJ background used in this studyrepresent progeny obtained after backcrossing the tlr2 deletion (tlr22/2)originally on C57BL/6 to BALB/cJ background for six generations (T.D.Connell, unpublished observations). These mice were healthy and showedno signs of gross abnormalities. Mice were maintained in HEPA-filteredcages with autoclaved food, water, and bedding. All procedures wereperformed in accordance with protocols approved by the University atBuffalo Institutional Animal Care and Use Committee.
Bacterial strains and culture conditions
T. forsythia was cultured in T. forsythia broth or on T. forsythia-agar plates(1.5% agar in T. forsythia broth) under anaerobic conditions, as describedpreviously (40).
Oral infection and alveolar bone loss assessment
Animals within groups were matched by age and sex (6–7 wk at the start ofthe experiment; n = 8–10 per group) and quarantined for 1 wk prior to theexperiment. Mice were infected with T. forsythia, as previously described,with the following modifications (9): mice were treated with kanamycin (1mg/ml) for 7 d ad libitum, followed by a 3-d antibiotic-free period. Thiswas followed by infection with live bacteria (T. forsythia ATCC 43037)via oral gavage. Infection was given as 100-ml bacterial suspensions (109
CFU/ml) in 2% carboxymethyl cellulose three times at 48-h intervals for 2wk. The control (sham-infected) mice received antibiotic pretreatment and100 ml 2% carboxymethyl cellulose. Mice were sacrificed after 6 wk, andserum was collected by cardiac puncture. Jaws were autoclaved, defleshed,immersed overnight in 3% H2O2, and stained with 1% methylene blue.Horizontal bone loss was assessed morphometrically by measuring thedistance between the cemento-enamel junction (CEJ) and the alveolarbone crest (ABC). Measurements at 14 buccal sites per mouse (7 on theleft and right maxillary molars) were made under a dissecting microscope(Brook-Anco, Rochester, NY) fitted with an Aquinto imaging measure-ment system (a4i America). Random, blinded bone measurements weretaken by two independent evaluators. Data were analyzed on GraphPadPrism 5 software (GraphPad, San Diego, CA).
ELISA
T. forsythia-specific ELISAs were performed as described (9). Briefly, 96-well Immuno-Maxisorp plates (Nalgene Nunc International, Rochester,NY) were coated with formalin-fixed T. forsythia (108 cells/well). Serawere added in 2-fold serial dilutions, and T. forsythia-specific IgG was
detected using HRP-conjugated goat anti-mouse IgG (Bethyl Laboratories,Montgomery, TX). Specific serum IgG isotype Ab was detected by additionof biotinylated isotype-specific secondary Ab (rat anti-mouse IgG1 orIgG2a; Southern Biotechnology, Birmingham, AL) followed by streptavidin-conjugated HRP (Southern Biotechnology). ELISA wells were color de-veloped with TMB Microwell enzyme substrate (Kirkgaards and Perry,Gaithersburg, MD). After stopping the enzyme reaction by adding 0.1 NH2SO4, plates were read at 495 nm. The titer was defined as the log2 of thehighest dilution with a signal that was 0.1 OD units above background.
Isolation of dendritic cells and macrophages and stimulation
Bone marrow-derived dendritic cells (BMDCs) were generated as described(41). Briefly, femurs and tibias were flushed with PBS. Erythrocytes werelysed using M-Lyse buffer (R&D Systems, Minneapolis, MN), and cells weresuspended in RPMI 1640 supplemented with 10% heat-inactivated FCS,penicillin (50 U/ml), streptomycin (50 mg/ml), L-glutamine (2 mM), 2-ME(50 mM), sodium pyruvate (1 mM), sodium bicarbonate (1.5 mg/ml), andHEPES (25 mM). BMDCs were cultured in 24-well plates at 106 cells/ml/well. GM-CSF (R&D Systems) was added at 20 ng/ml. Media + GM-CSFwere replaced on days 2, 4, and 6. Cells were restimulated on day 7. Thisprotocol yielded BMDCs with dendritic morphology and .85% purity byCD11c+ staining. Peritoneal macrophages were prepared as described (42).
Cytokine ELISA
Cytokines were measured in triplicate by ELISA kits from eBiosciences.Detection limits were 15 pg/ml for IFN-g, 4 pg/ml for IL-10, 10 pg/ml forIL-12 p40, and 8 pg/ml for IL-17.
Intracellular staining and FACS analysis
The following Abs and isotype controls were purchased from eBiosciences:PE-conjugated anti–IFN-g and rat IgG2a; FITC-conjugated anti–IL-4;PECy5-labeled anti-CD4; anti-CD80 (16-10A1) and anti-CD83 (Michel-17); rat IgG1, IgG2a, and IgG2b; PE-conjugated anti-CD11c (N418);hamster IgG; PE-conjugated anti–IL-17A (eBio17B7); and rat IgG2a. Forintracellular staining, cell suspensions from cervical lymph nodes (cLNs)were stimulated with anti-CD3 and anti-CD28 Abs (eBioscience) for 48 h,followed by PMA (50 ng/ml), ionomycin (1 mg/ml), and brefeldin A (10mg/ml) for 6 h. Cells were washed, incubated with FcR block (1 mg/ml;eBiosciences), and stained for CD4. Cells were fixed with 2% para-formaldehyde, permeabilized with 0.1% saponin (Sigma), and stained forIL-4, IL-17, or IFN-g. Cells were analyzed on a FACSCalibur (BD Bio-sciences) with FCS Express software (DeNovo Software).
Histological staining
Murine maxillary and mandibular bones (n = 4) were fixed in 10%phosphate-buffered formalin and decalcified in 10% EDTA. Samples wereembedded in paraffin, and sections at 4 mm were prepared and stained fortartrate-resistant acid phosphatase (TRAP; Sigma). Slides were digitallyscanned with a ScanScope CS system (Aperio) to minimize color fadingand viewed with ImageScope viewing software (Aperio). The right max-illary and mandibular interdental areas (average of 10 higher-power fields/slide) of the crestal alveolar bone from the first molar to third molar wereused to quantify osteoclasts.
CD45 staining
The right and left halves of the maxillary and mandibular bones wereremoved, fixed, and embedded in paraffin, and 4-mm sections were cut andmounted. They were deparaffinized in xylene and hydrated in gradedethanol. After Ag retrieval by incubating at 90˚C for 10 min with BDRetrievagen A (BD Pharmingen), specimens were sequentially incubatedin blocking solution containing 0.1% Triton X-100 in 0.1 M PBS for 1 hat room temperature and then monoclonal rat anti-mouse CD45 (BDPharmingen) (1:30 dilution in PBS containing 0.1% Triton X-100) for 1 hat room temperature. The slides were incubated with a biotinylated goatanti-rat secondary Ab, followed by an avidin–biotin complex developedwith 3,3-diaminobenzidine (Vector Labs, Burlingame, CA); the counter-stain was hematoxylin. After each step, slides were rinsed in PBST (threetimes for 10 min). Slides were analyzed with a ScanScope CS system andviewed with ImageScope viewing software (both from Aperio). The in-terdental areas from the first to third molar were used to quantify in-flammatory cells.
Data analysis
Data were analyzed on GraphPad Prism software (GraphPad). Comparisonsbetween groups were made using a Student t test (between two groups) or
502 TLR2/Th2 RESPONSES DRIVE T. FORSYTHIA-INDUCED BONE LOSS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
ANOVA (multiple group comparisons), as appropriate. Statistical signifi-cance was defined as p , 0.05.
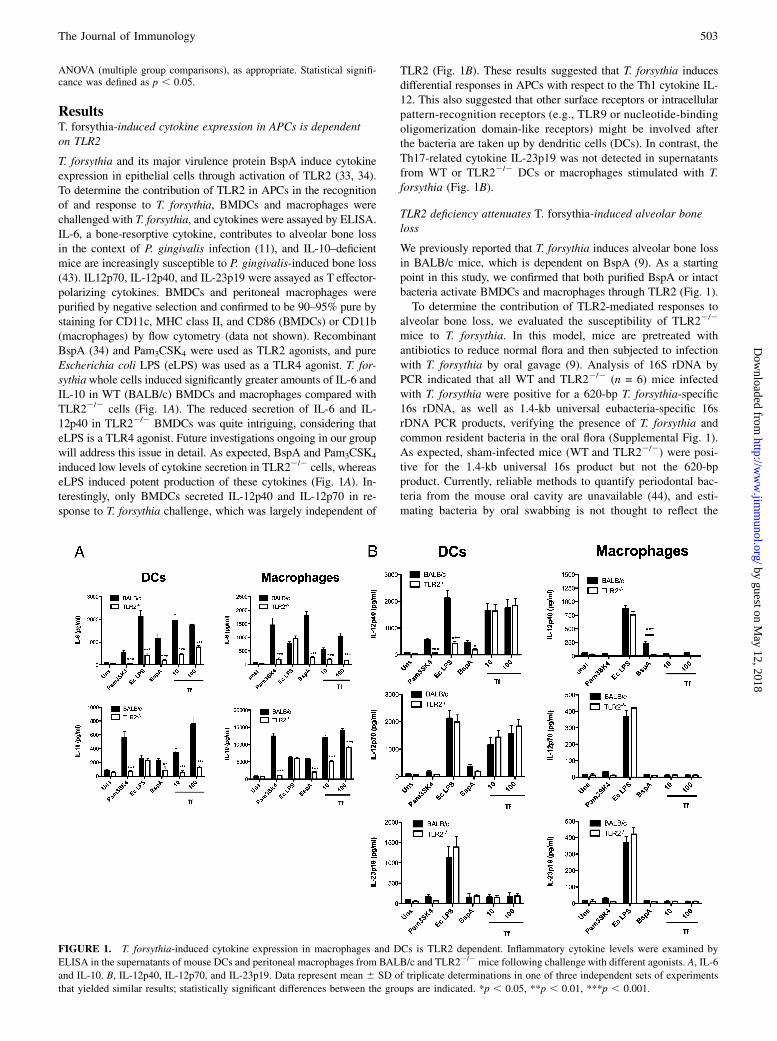
ResultsT. forsythia-induced cytokine expression in APCs is dependenton TLR2
T. forsythia and its major virulence protein BspA induce cytokineexpression in epithelial cells through activation of TLR2 (33, 34).To determine the contribution of TLR2 in APCs in the recognitionof and response to T. forsythia, BMDCs and macrophages werechallenged with T. forsythia, and cytokines were assayed by ELISA.IL-6, a bone-resorptive cytokine, contributes to alveolar bone lossin the context of P. gingivalis infection (11), and IL-10–deficientmice are increasingly susceptible to P. gingivalis-induced bone loss(43). IL12p70, IL-12p40, and IL-23p19 were assayed as T effector-polarizing cytokines. BMDCs and peritoneal macrophages werepurified by negative selection and confirmed to be 90–95% pure bystaining for CD11c, MHC class II, and CD86 (BMDCs) or CD11b(macrophages) by flow cytometry (data not shown). RecombinantBspA (34) and Pam3CSK4 were used as TLR2 agonists, and pureEscherichia coli LPS (eLPS) was used as a TLR4 agonist. T. for-sythiawhole cells induced significantly greater amounts of IL-6 andIL-10 in WT (BALB/c) BMDCs and macrophages compared withTLR22/2 cells (Fig. 1A). The reduced secretion of IL-6 and IL-12p40 in TLR22/2 BMDCs was quite intriguing, considering thateLPS is a TLR4 agonist. Future investigations ongoing in our groupwill address this issue in detail. As expected, BspA and Pam3CSK4
induced low levels of cytokine secretion in TLR22/2 cells, whereaseLPS induced potent production of these cytokines (Fig. 1A). In-terestingly, only BMDCs secreted IL-12p40 and IL-12p70 in re-sponse to T. forsythia challenge, which was largely independent of
TLR2 (Fig. 1B). These results suggested that T. forsythia inducesdifferential responses in APCs with respect to the Th1 cytokine IL-12. This also suggested that other surface receptors or intracellularpattern-recognition receptors (e.g., TLR9 or nucleotide-bindingoligomerization domain-like receptors) might be involved afterthe bacteria are taken up by dendritic cells (DCs). In contrast, theTh17-related cytokine IL-23p19 was not detected in supernatantsfrom WT or TLR22/2 DCs or macrophages stimulated with T.forsythia (Fig. 1B).
TLR2 deficiency attenuates T. forsythia-induced alveolar boneloss
We previously reported that T. forsythia induces alveolar bone lossin BALB/c mice, which is dependent on BspA (9). As a startingpoint in this study, we confirmed that both purified BspA or intactbacteria activate BMDCs and macrophages through TLR2 (Fig. 1).To determine the contribution of TLR2-mediated responses to
alveolar bone loss, we evaluated the susceptibility of TLR22/2
mice to T. forsythia. In this model, mice are pretreated withantibiotics to reduce normal flora and then subjected to infectionwith T. forsythia by oral gavage (9). Analysis of 16S rDNA byPCR indicated that all WT and TLR22/2 (n = 6) mice infectedwith T. forsythia were positive for a 620-bp T. forsythia-specific16s rDNA, as well as 1.4-kb universal eubacteria-specific 16srDNA PCR products, verifying the presence of T. forsythia andcommon resident bacteria in the oral flora (Supplemental Fig. 1).As expected, sham-infected mice (WT and TLR22/2) were posi-tive for the 1.4-kb universal 16s product but not the 620-bpproduct. Currently, reliable methods to quantify periodontal bac-teria from the mouse oral cavity are unavailable (44), and esti-mating bacteria by oral swabbing is not thought to reflect the
FIGURE 1. T. forsythia-induced cytokine expression in macrophages and DCs is TLR2 dependent. Inflammatory cytokine levels were examined by
ELISA in the supernatants of mouse DCs and peritoneal macrophages from BALB/c and TLR22/2 mice following challenge with different agonists. A, IL-6
and IL-10. B, IL-12p40, IL-12p70, and IL-23p19. Data represent mean 6 SD of triplicate determinations in one of three independent sets of experiments
that yielded similar results; statistically significant differences between the groups are indicated. *p , 0.05, **p , 0.01, ***p , 0.001.
The Journal of Immunology 503
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

bacterial load. Periodontal bacteria, and particularly T. forsythia,invade buccal and gingival epithelial cells and form biofilms (45–47), thereby avoiding detection. As another confirmation of in-fection, T. forsythia-specific serum IgG titers in T. forsythia-infected mice were increased over sham-infected mice. The netAb response, defined as the titer of sham-infected mice subtractedfrom T. forsythia-infected mice, increased in both WTand TLR22/2
mice (Fig. 2A). Although there was a low titer, even in sham-infected mice, these Abs presumably represent cross-reactivenonspecific Abs against normal resident bacteria. TLR2-deficientmice elicited lower levels of T. forsythia-specific Abs comparedwith WT mice following infection (Fig. 2A). Thus, both WT andTLR22/2 mice were productively infected with T. forsythia.Conceivably, TLR2 deficiency directly impairs Ab productionagainst T. forsythia by affecting cellular pathways associated withAb maturation. Alternatively, this occurs indirectly by reducing T.forsythia proliferation and survival, resulting in suboptimal hu-moral responses. In support of the latter, it was suggested that P.gingivalis survival is impaired in TLR22/2 mice (48). The lack ofsuitable methods to quantify bacteria precluded us from de-termining whether TLR2 deficiency indeed causes lower T. for-sythia loads.After 6 wk, alveolar bone loss was measured as the distance
between the CEJ and ABC at 14 buccal sites per mouse (horizontalbone loss, see Fig. 2B). As expected, significant alveolar bone losswas observed in T. forsythia-infected WT mice compared with
controls (Fig. 2B, 2C). Strikingly, TLR22/2 mice infected with T.forsythia showed bone loss at fewer sites compared with sham-infected TLR22/2 mice. Although the baseline ABC–CEJ dis-tances showed a trend for higher CEJ–ABC distances in TLR22/2
mice compared with WT mice, this did not reach statistical sig-nificance. Nevertheless, the average net alveolar bone loss inducedby T. forsythia (measured as total ABC–CEJ for sham-treatedmice subtracted from T. forsythia-infected mice) in TLR22/2
mice was significantly lower than was the average net bone lossobserved for WT mice (Fig. 2C). These results implied that TLR2signaling stimulates bone loss following T. forsythia infection,which is ameliorated in TLR22/2 mice.
Stat6 deletion attenuates T. forsythia-induced alveolar boneloss
TLR2 was suggested to stimulate primarily Th2 responses (35–38),although there are exceptions to this finding. To test the hypothesisthat Th2 responses are responsible for inflammatory alveolar bonedestruction, mice deficient in STAT6 were assessed. Notably, stat6gene expression has been linked to alveolar bone loss suscepti-bility in response to P. gingivalis infection, although its role in T.forsythia infection has never been evaluated (39). One week afterthe final infection, all mice tested positive for the 620-bp T. for-sythia rDNA band by PCR (Supplemental Fig. 2). Similarly, thepresence of increased T. forsythia-specific IgG titers in sera ofanimals confirmed that all animals were productively infected
FIGURE 2. TLR2 promotes T. forsythia-induced alveolar bone loss. A, Oral infection with T. forsythia elicits serum IgG response in WT and TLR22/2
mice. After 6 wk of infection (sham or T. forsythia), sera from WT and TLR22/2 mice were analyzed for T. forsythia-specific IgG by ELISA. Net Ab
response in each group (WT or TLR22/2) was determined by subtracting the Ab titers of sham-infected mice from that of T. forsythia-infected mice. Data
represent means and standard deviations for each group (n = 8–10). *p , 0.05, unpaired t test. B and C, TLR22/2 mice exhibit reduced net alveolar bone
loss in response to T. forsythia infection. WT and TLR22/2 mice (n = 8–10) were infected with T. forsythia or were sham infected. Alveolar bone de-
struction was assessed after 6 wk by measuring the distance from the ABC to the CEJ at 14 maxillary buccal sites per mouse (R1–R7, right jaw; L1–L7, left
jaw). B, Representative maxillary phenotypes of male BALB/c and TLR22/2 mice. Maxillary jaws were stained with methylene blue, and images were
acquired with a Nikon SMZ 1000 microscope (original magnification33). C, Average alveolar bone loss at 14 buccal sites for BALB/c and TLR22/2 mice.
Net bone loss shows ABC–CEJ distance of T. forsythia-infected sites minus the mean ABC–CEJ distance of sham-treated sites. Data were analyzed by
unpaired t test; SDs are shown. *p , 0.05, **p , 0.01, ***p , 0.001, T. forsythia infection versus sham infection.
504 TLR2/Th2 RESPONSES DRIVE T. FORSYTHIA-INDUCED BONE LOSS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
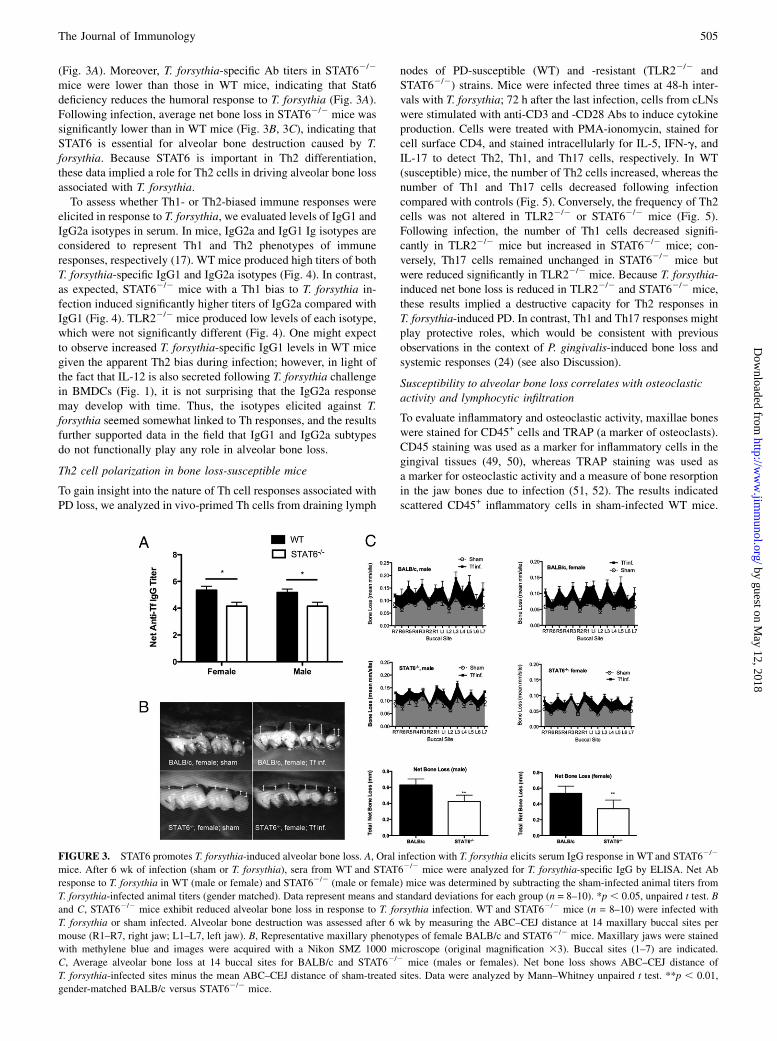
(Fig. 3A). Moreover, T. forsythia-specific Ab titers in STAT62/2
mice were lower than those in WT mice, indicating that Stat6deficiency reduces the humoral response to T. forsythia (Fig. 3A).Following infection, average net bone loss in STAT62/2 mice wassignificantly lower than in WT mice (Fig. 3B, 3C), indicating thatSTAT6 is essential for alveolar bone destruction caused by T.forsythia. Because STAT6 is important in Th2 differentiation,these data implied a role for Th2 cells in driving alveolar bone lossassociated with T. forsythia.To assess whether Th1- or Th2-biased immune responses were
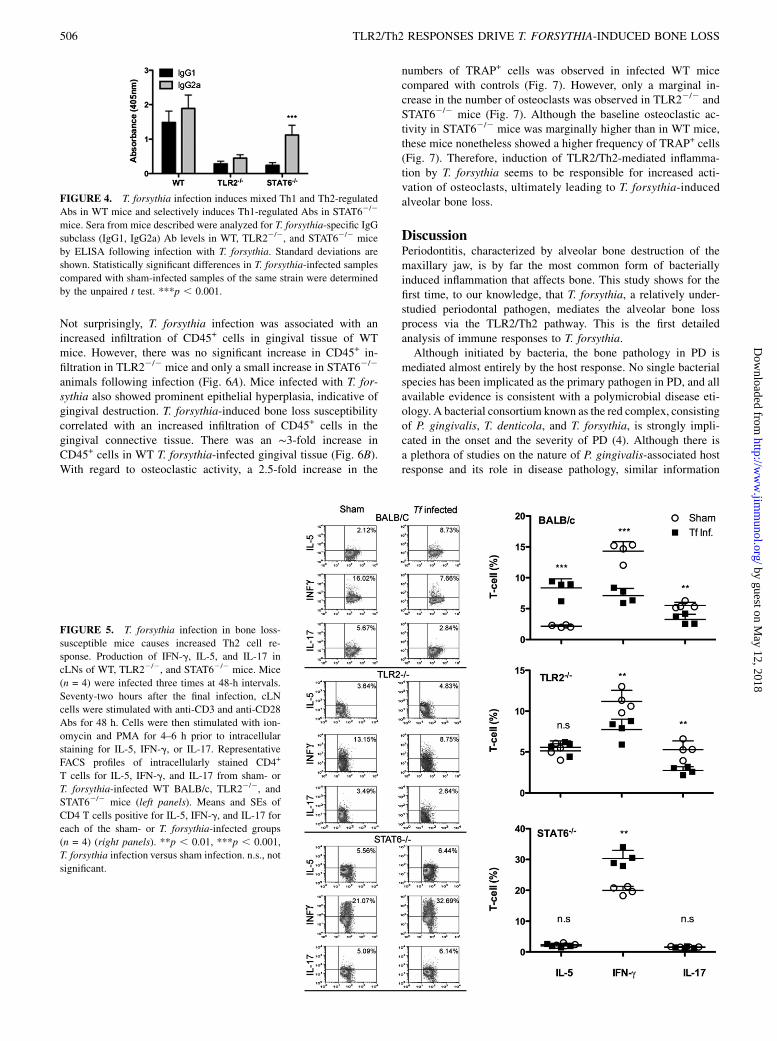
elicited in response to T. forsythia, we evaluated levels of IgG1 andIgG2a isotypes in serum. In mice, IgG2a and IgG1 Ig isotypes areconsidered to represent Th1 and Th2 phenotypes of immuneresponses, respectively (17). WT mice produced high titers of bothT. forsythia-specific IgG1 and IgG2a isotypes (Fig. 4). In contrast,as expected, STAT62/2 mice with a Th1 bias to T. forsythia in-fection induced significantly higher titers of IgG2a compared withIgG1 (Fig. 4). TLR22/2 mice produced low levels of each isotype,which were not significantly different (Fig. 4). One might expectto observe increased T. forsythia-specific IgG1 levels in WT micegiven the apparent Th2 bias during infection; however, in light ofthe fact that IL-12 is also secreted following T. forsythia challengein BMDCs (Fig. 1), it is not surprising that the IgG2a responsemay develop with time. Thus, the isotypes elicited against T.forsythia seemed somewhat linked to Th responses, and the resultsfurther supported data in the field that IgG1 and IgG2a subtypesdo not functionally play any role in alveolar bone loss.
Th2 cell polarization in bone loss-susceptible mice
To gain insight into the nature of Th cell responses associated withPD loss, we analyzed in vivo-primed Th cells from draining lymph
nodes of PD-susceptible (WT) and -resistant (TLR22/2 andSTAT62/2) strains. Mice were infected three times at 48-h inter-vals with T. forsythia; 72 h after the last infection, cells from cLNswere stimulated with anti-CD3 and -CD28 Abs to induce cytokineproduction. Cells were treated with PMA-ionomycin, stained forcell surface CD4, and stained intracellularly for IL-5, IFN-g, andIL-17 to detect Th2, Th1, and Th17 cells, respectively. In WT(susceptible) mice, the number of Th2 cells increased, whereas thenumber of Th1 and Th17 cells decreased following infectioncompared with controls (Fig. 5). Conversely, the frequency of Th2cells was not altered in TLR22/2 or STAT62/2 mice (Fig. 5).Following infection, the number of Th1 cells decreased signifi-cantly in TLR22/2 mice but increased in STAT62/2 mice; con-versely, Th17 cells remained unchanged in STAT62/2 mice butwere reduced significantly in TLR22/2 mice. Because T. forsythia-induced net bone loss is reduced in TLR22/2 and STAT62/2 mice,these results implied a destructive capacity for Th2 responses inT. forsythia-induced PD. In contrast, Th1 and Th17 responses mightplay protective roles, which would be consistent with previousobservations in the context of P. gingivalis-induced bone loss andsystemic responses (24) (see also Discussion).
Susceptibility to alveolar bone loss correlates with osteoclasticactivity and lymphocytic infiltration
To evaluate inflammatory and osteoclastic activity, maxillae boneswere stained for CD45+ cells and TRAP (a marker of osteoclasts).CD45 staining was used as a marker for inflammatory cells in thegingival tissues (49, 50), whereas TRAP staining was used asa marker for osteoclastic activity and a measure of bone resorptionin the jaw bones due to infection (51, 52). The results indicatedscattered CD45+ inflammatory cells in sham-infected WT mice.
FIGURE 3. STAT6 promotes T. forsythia-induced alveolar bone loss. A, Oral infection with T. forsythia elicits serum IgG response in WT and STAT62/2
mice. After 6 wk of infection (sham or T. forsythia), sera from WT and STAT62/2 mice were analyzed for T. forsythia-specific IgG by ELISA. Net Ab
response to T. forsythia in WT (male or female) and STAT62/2 (male or female) mice was determined by subtracting the sham-infected animal titers from
T. forsythia-infected animal titers (gender matched). Data represent means and standard deviations for each group (n = 8–10). *p , 0.05, unpaired t test. B
and C, STAT62/2 mice exhibit reduced alveolar bone loss in response to T. forsythia infection. WT and STAT62/2 mice (n = 8–10) were infected with
T. forsythia or sham infected. Alveolar bone destruction was assessed after 6 wk by measuring the ABC–CEJ distance at 14 maxillary buccal sites per
mouse (R1–R7, right jaw; L1–L7, left jaw). B, Representative maxillary phenotypes of female BALB/c and STAT62/2 mice. Maxillary jaws were stained
with methylene blue and images were acquired with a Nikon SMZ 1000 microscope (original magnification 33). Buccal sites (1–7) are indicated.
C, Average alveolar bone loss at 14 buccal sites for BALB/c and STAT62/2 mice (males or females). Net bone loss shows ABC–CEJ distance of
T. forsythia-infected sites minus the mean ABC–CEJ distance of sham-treated sites. Data were analyzed by Mann–Whitney unpaired t test. **p , 0.01,
gender-matched BALB/c versus STAT62/2 mice.
The Journal of Immunology 505
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Not surprisingly, T. forsythia infection was associated with anincreased infiltration of CD45+ cells in gingival tissue of WTmice. However, there was no significant increase in CD45+ in-filtration in TLR22/2 mice and only a small increase in STAT62/2
animals following infection (Fig. 6A). Mice infected with T. for-sythia also showed prominent epithelial hyperplasia, indicative ofgingival destruction. T. forsythia-induced bone loss susceptibilitycorrelated with an increased infiltration of CD45+ cells in thegingival connective tissue. There was an ∼3-fold increase inCD45+ cells in WT T. forsythia-infected gingival tissue (Fig. 6B).With regard to osteoclastic activity, a 2.5-fold increase in the
numbers of TRAP+ cells was observed in infected WT micecompared with controls (Fig. 7). However, only a marginal in-crease in the number of osteoclasts was observed in TLR22/2 andSTAT62/2 mice (Fig. 7). Although the baseline osteoclastic ac-tivity in STAT62/2 mice was marginally higher than in WT mice,these mice nonetheless showed a higher frequency of TRAP+ cells(Fig. 7). Therefore, induction of TLR2/Th2-mediated inflamma-tion by T. forsythia seems to be responsible for increased acti-vation of osteoclasts, ultimately leading to T. forsythia-inducedalveolar bone loss.
DiscussionPeriodontitis, characterized by alveolar bone destruction of themaxillary jaw, is by far the most common form of bacteriallyinduced inflammation that affects bone. This study shows for thefirst time, to our knowledge, that T. forsythia, a relatively under-studied periodontal pathogen, mediates the alveolar bone lossprocess via the TLR2/Th2 pathway. This is the first detailedanalysis of immune responses to T. forsythia.Although initiated by bacteria, the bone pathology in PD is
mediated almost entirely by the host response. No single bacterialspecies has been implicated as the primary pathogen in PD, and allavailable evidence is consistent with a polymicrobial disease eti-ology. A bacterial consortium known as the red complex, consistingof P. gingivalis, T. denticola, and T. forsythia, is strongly impli-cated in the onset and the severity of PD (4). Although there isa plethora of studies on the nature of P. gingivalis-associated hostresponse and its role in disease pathology, similar information
FIGURE 5. T. forsythia infection in bone loss-
susceptible mice causes increased Th2 cell re-
sponse. Production of IFN-g, IL-5, and IL-17 in
cLNs of WT, TLR22/2, and STAT62/2 mice. Mice
(n = 4) were infected three times at 48-h intervals.
Seventy-two hours after the final infection, cLN
cells were stimulated with anti-CD3 and anti-CD28
Abs for 48 h. Cells were then stimulated with ion-
omycin and PMA for 4–6 h prior to intracellular
staining for IL-5, IFN-g, or IL-17. Representative
FACS profiles of intracellularly stained CD4+
T cells for IL-5, IFN-g, and IL-17 from sham- or
T. forsythia-infected WT BALB/c, TLR22/2, and
STAT62/2 mice (left panels). Means and SEs of
CD4 T cells positive for IL-5, IFN-g, and IL-17 for
each of the sham- or T. forsythia-infected groups
(n = 4) (right panels). **p , 0.01, ***p , 0.001,
T. forsythia infection versus sham infection. n.s., not
significant.
FIGURE 4. T. forsythia infection induces mixed Th1 and Th2-regulated
Abs in WT mice and selectively induces Th1-regulated Abs in STAT62/2
mice. Sera from mice described were analyzed for T. forsythia-specific IgG
subclass (IgG1, IgG2a) Ab levels in WT, TLR22/2, and STAT62/2 mice
by ELISA following infection with T. forsythia. Standard deviations are
shown. Statistically significant differences in T. forsythia-infected samples
compared with sham-infected samples of the same strain were determined
by the unpaired t test. ***p , 0.001.
506 TLR2/Th2 RESPONSES DRIVE T. FORSYTHIA-INDUCED BONE LOSS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

regarding T. denticola or T. forsythia is evidently lacking (5).Previous studies have largely been restricted to P. gingivalis asa model periodontal pathogen. Although informative, such studiesare limited to the nuances of this organism.Although initiated by bacteria, bone loss is mediated by the
immune response. A destructive role for T and B cells in PD boneloss is well documented (10–12). In contrast, impaired neutrophiland macrophage recruitment due to deficiencies in P-selectin andICAM-1 result in increased susceptibility to P. gingivalis-inducedalveolar bone loss (53). Thus, although lymphocytes can drivebone loss, a robust innate immune response controls infection and,hence, limits overall bone destruction. Despite the potential bone-destructive capacity of IL-17 and Th17 cells seen in the context ofrheumatoid arthritis (54), a deficiency in IL-17 signaling in PDresults in increased susceptibility to P. gingivalis-induced alveolarbone loss. It was also demonstrated that TLR2-mediated in-flammatory responses are critical to P. gingivalis-induced alveolarbone loss through promoting pathogen survival and burden in thehost (48). Taken together, these studies demonstrated that bothinnate and adaptive arms of immunity play critical roles in PD.However, the contribution of individual T cell responses (i.e., Th1,Th2, or Th17) needs to be explored further in PD (6, 55).The mechanisms by which T. forsythia induces inflammation and
alveolar bone loss are poorly understood. We previously demon-strated that T. forsythia expresses a TLR2-activating molecule,BspA (34), which is required for inducing alveolar bone loss inmice (9). Because both BspA and intact T. forsythia signal throughTLR2 (33, 34), the focus of this study was to evaluate the role ofTLR2-mediated responses in alveolar bone loss. Moreover, becauseTLR2-mediated responses were shown to favor Th2 development(31, 35, 36, 38), we predicted that Th2 responses would be elicitedand dictate the alveolar bone loss associated with T. forsythia in-
fection. Indeed, our results showed that TLR2 plays a significantrole in stimulating DCs and macrophages to elicit cytokineresponses via BspA (Fig. 1). DCs or macrophages stimulated withbacteria or BspA did not express IL-23, suggesting that Th17 cellsare not induced in response to T. forsythia. Interestingly, DCs, butnot macrophages, produced IL-12 (Th1 cytokine) in response tobacteria or BspA. It is possible that engagement of other pathways,such as through C-type lectin receptors, intracellular nucleotide-binding oligomerization domain-like receptors, or TLR9, might beoperative in DCs. Indeed, we showed that T. forsythia DNA isa strong inducer of TLR9 (56). Consistently, Mycobacterium tu-berculosis induces strong IL-12 expression in DCs through theTLR9 pathway but weak IL-12 and strong IL-10 expression inmacrophages through engagement of TLR2 (57). TLR2 activationinhibits IL-12 by dampening the Th1 IFN-g amplification loop inDCs and promotes induction of Th2 and Th17 responses (38).Furthermore, IL-10, produced primarily via TLR2, inhibits IP-10and IL-12p35; therefore, it is considered a Th1-suppressing cyto-kine. Although IL-10 is primarily an inhibitor of Th1, recentstudies indicated that it also suppresses Th2 and Th17 cell re-sponses (58). The failure of T. forsythia to induce IL-23 in DCs ormacrophages (Fig. 1B) indicated that Th17 responses are likely notinduced in this setting, which was confirmed by our findings in T.forsythia-infected mice (Fig. 5). Also consistent with our hypoth-esis, the net bone loss caused by T. forsythia in TLR22/2 mice wasattenuated. This finding indicated that TLR2 activation plays a de-structive role. This finding is similar to what has been observed forP. gingivalis infections, where TLR22/2 mice are resistant to P.gingivalis-induced alveolar bone loss (48). It is likely that T. for-sythia, which depends on the availability of host factors, such aspeptides, heme, and sialic acid for growth, exploits TLR2-mediatedinflammation for its growth and survival.
FIGURE 6. Alveolar bone loss correlates with
lymphocytic infiltration. A, CD45 staining for in-
flammatory infiltrate cells in gingival tissue 3 wk
following infection of WT, TLR22/2, and STAT62/2
mice (original magnification 3400). B, Inflam-
matory cells were quantified as number of CD45+
cells/mm2. *p , 0.05, ***p , 0.001, T. forsythia
infection versus sham infection.
FIGURE 7. Alveolar bone loss correlates with os-
teoclastic activity. A, Representative histological sec-
tions showing TRAP+ cells (arrows) from T. forsythia-
and sham-infected WT, TLR22/2, and STAT62/2
mice (original magnification 3400). B, Average num-
ber of TRAP+ cells in 10 high-power magnification
fields/slide (n = 4 mice/group). *p, 0.05, ***p, 0.001,
T. forsythia infection versus sham infection.
The Journal of Immunology 507
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

To explore the possibility of polarization of Th2 responsesdownstream of TLR2, STAT62/2mice were assessed. In STAT62/2
mice T. forsythia-induced alveolar bone loss was significantly re-duced (Fig. 3). Consistent with our findings, the stat6 gene wasshown to be associated with increased susceptibility in P. gingi-valis-induced PD (39). We observed increased osteoclastic activityfollowing infection only in alveolar bone loss-susceptible WTmice. Consistently, WT mice presented increased inflammatorycells in the connective tissue surrounding alveolar bone followinginfection, which correlated with orthoclastic activity. These resultsstrongly suggested that the TLR2–Th2 inflammatory axis playsa significant role in T. forsythia-induced alveolar bone loss.Our observations suggested that STAT6 does not play a bone-
protective role during T. forsythia-induced alveolar bone loss.However, STAT6 is considered important for dampening the ac-tivity of osteoclasts via IL-4 and IL-13 in vitro (59, 60), so othermechanisms are likely to be involved whereby Th2 mediates boneloss. In that respect, transgenic expression of IL-4 in mice wasshown to suppress osteoblast activity, leading to general osteo-porosis (61). Thus, it is possible that Th2 cytokines suppress localosteoblast activity in the jaw. Another possibility is that thisoccurs via increased levels of RANKL expression, driving oste-oclast differentiation. However, RANKL expression is thought tobe limited mainly to Th1 (62) and Th17 (63) cells, although thismay vary depending on the infection stimulus, and it was notassessed in our study. Alternatively, it is tempting to speculatethat Th2 responses induce RANKL-mediated osteoclastogenesisthrough effects on B cells, which also express RANKL and candrive osteoclastogenesis in inflamed periodontium (64, 65).In summary, we presented evidence for the roles of TLR2
signaling and Th2 differentiation in mediating T. forsythia-inducedalveolar bone destruction. We showed that T. forsythia-inducedTLR2 activation resulted in alveolar bone destruction. Further-more, Th2 development downstream of TLR2 activation was as-sociated with alveolar bone destruction caused by T. forsythia.
AcknowledgmentsWe thank Raymond J. Kelleher and Rajesh Rao for assistance with flow
cytometry and Moon-Il Cho and Shuying Yang for advice on processing
jaw bones for histological staining.
DisclosuresThe authors have no financial conflicts of interest.
References1. Zambon, J. J., S. Grossi, R. Dunford, V. I. Harazsthy, H. Preus, and R. J. Genco.
1994. Epidemiology of subgingival bacterial pathogens in periodontal diseases.In Molecular Pathogenesis of Periodontal Disease. R. J. Genco, S. Hamada,J. R. Lehrer, J. R. McGhee, and S. Mergenhagen, eds. American Society forMicrobiology, Washington, D.C., p. 3–12.
2. Darveau, R. P. 2010. Periodontitis: a polymicrobial disruption of host homeo-stasis. Nat. Rev. Microbiol. 8: 481–490.
3. Graves, D. T., J. Li, and D. L. Cochran. 2011. Inflammation and uncoupling asmechanisms of periodontal bone loss. J. Dent. Res. 90: 143–153.
4. Socransky, S. S., A. D. Haffajee, M. A. Cugini, C. Smith, and R. L. Kent Jr. 1998.Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25: 134–144.
5. Holt, S. C., and J. L. Ebersole. 2005. Porphyromonas gingivalis, Treponemadenticola, and Tannerella forsythia: the “red complex”, a prototype polybacterialpathogenic consortium in periodontitis. Periodontol. 2000 38: 72–122.
6. Gaffen, S. L., and G. Hajishengallis. 2008. A new inflammatory cytokine on theblock: re-thinking periodontal disease and the Th1/Th2 paradigm in the contextof Th17 cells and IL-17. J. Dent. Res. 87: 817–828.
7. Sharma, A. 2010. Virulence mechanisms of Tannerella forsythia. Periodontol.2000 54: 106–116.
8. Tanner, A. C., and J. Izard. 2006. Tannerella forsythia, a periodontal pathogenentering the genomic era. Periodontol. 2000 42: 88–113.
9. Sharma, A., S. Inagaki, K. Honma, C. Sfintescu, P. J. Baker, and R. T. Evans.2005. Tannerella forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J. Dent. Res. 84: 462–467.
10. Baker, P. J. 2000. The role of immune responses in bone loss during periodontaldisease. Microbes Infect. 2: 1181–1192.
11. Baker, P. J., M. Dixon, R. T. Evans, L. Dufour, E. Johnson, and D. C. Roopenian.1999. CD4(+) T cells and the proinflammatory cytokines gamma interferon andinterleukin-6 contribute to alveolar bone loss in mice. Infect. Immun. 67: 2804–2809.
12. Baker, P. J., R. T. Evans, and D. C. Roopenian. 1994. Oral infection with Por-phyromonas gingivalis and induced alveolar bone loss in immunocompetent andsevere combined immunodeficient mice. Arch. Oral Biol. 39: 1035–1040.
13. Stashenko, P., R. B. Goncalves, B. Lipkin, A. Ficarelli, H. Sasaki, andA. Campos-Neto. 2007. Th1 immune response promotes severe bone resorptioncaused by Porphyromonas gingivalis. Am. J. Pathol. 170: 203–213.
14. Gemmell, E., K. Yamazaki, and G. J. Seymour. 2007. The role of T cells inperiodontal disease: homeostasis and autoimmunity. Periodontol. 2000 43: 14–40.
15. Teng, Y. T. 2006. Protective and destructive immunity in the periodontium: Part1–innate and humoral immunity and the periodontium. J. Dent. Res. 85: 198–208.
16. Teng, Y. T., H. Nguyen, X. Gao, Y. Y. Kong, R. M. Gorczynski, B. Singh,R. P. Ellen, and J. M. Penninger. 2000. Functional human T-cell immunity andosteoprotegerin ligand control alveolar bone destruction in periodontal infection.J. Clin. Invest. 106: R59–R67.
17. Mosmann, T. R., and R. L. Coffman. 1989. TH1 and TH2 cells: different patternsof lymphokine secretion lead to different functional properties. Annu. Rev.Immunol. 7: 145–173.
18. Korn, T., E. Bettelli, M. Oukka, and V. K. Kuchroo. 2009. IL-17 and Th17 Cells.Annu. Rev. Immunol. 27: 485–517.
19. Honda, T., Y. Aoki, N. Takahashi, T. Maekawa, T. Nakajima, H. Ito, K. Tabeta,T. Okui, K. Kajita, H. Domon, and K. Yamazaki. 2008. Elevated expression ofIL-17 and IL-12 genes in chronic inflammatory periodontal disease. Clin. Chim.Acta 395: 137–141.
20. Johnson, R. B., N. Wood, and F. G. Serio. 2004. Interleukin-11 and IL-17 and thepathogenesis of periodontal disease. J. Periodontol. 75: 37–43.
21. Schenkein, H. A., T. E. Koertge, C. N. Brooks, R. Sabatini, D. E. Purkall, andJ. G. Tew. 2010. IL-17 in sera from patients with aggressive periodontitis. J.Dent. Res. 89: 943–947.
22. Vernal, R., N. Dutzan, A. Chaparro, J. Puente, M. Antonieta Valenzuela, andJ. Gamonal. 2005. Levels of interleukin-17 in gingival crevicular fluid and insupernatants of cellular cultures of gingival tissue from patients with chronicperiodontitis. J. Clin. Periodontol. 32: 383–389.
23. Ito, H., T. Honda, H. Domon, T. Oda, T. Okui, R. Amanuma, T. Nakajima, andK. Yamazaki. 2005. Gene expression analysis of the CD4+ T-cell clones derivedfrom gingival tissues of periodontitis patients. Oral Microbiol. Immunol. 20:382–386.
24. Yu, J. J., M. J. Ruddy, G. C. Wong, C. Sfintescu, P. J. Baker, J. B. Smith,R. T. Evans, and S. L. Gaffen. 2007. An essential role for IL-17 in preventingpathogen-initiated bone destruction: recruitment of neutrophils to inflamed bonerequires IL-17 receptor-dependent signals. Blood 109: 3794–3802.
25. Gosling, P. T., E. Gemmell, C. L. Carter, P. S. Bird, and G. J. Seymour. 2005.Immunohistological analysis of Tannerella forsythia-induced lesions in a murinemodel. Oral Microbiol. Immunol. 20: 25–30.
26. Eskan, M. A., G. Hajishengallis, and D. F. Kinane. 2007. Differential activationof human gingival epithelial cells and monocytes by Porphyromonas gingivalisfimbriae. Infect. Immun. 75: 892–898.
27. Zhou, Q., T. Desta, M. Fenton, D. T. Graves, and S. Amar. 2005. Cytokineprofiling of macrophages exposed to Porphyromonas gingivalis, its lipopoly-saccharide, or its FimA protein. Infect. Immun. 73: 935–943.
28. Davey, M., X. Liu, T. Ukai, V. Jain, C. Gudino, F. C. Gibson, III, D. Golenbock,A. Visintin, and C. A. Genco. 2008. Bacterial fimbriae stimulate proin-flammatory activation in the endothelium through distinct TLRs. J. Immunol.180: 2187–2195.
29. Gibson, F. C., III, T. Ukai, and C. A. Genco. 2008. Engagement of specific innateimmune signaling pathways during Porphyromonas gingivalis induced chronicinflammation and atherosclerosis. Front. Biosci. 13: 2041–2059.
30. Gaffen, S. L., J. M. Kramer, J. J. Yu, and F. Shen. 2006. The IL-17 cytokinefamily. Vitam. Horm. 74: 255–282.
31. Netea, M. G., J. W. M. Van der Meer, R. P. Sutmuller, G. J. Adema, and B.-J. Kullberg. 2005. From the Th1/Th2 paradigm towards a Toll-like receptor/T-helper bias. Antimicrob. Agents Chemother. 49: 3991–3996.
32. Hasebe, A., A. Yoshimura, T. Into, H. Kataoka, S. Tanaka, S. Arakawa,H. Ishikura, D. T. Golenbock, T. Sugaya, N. Tsuchida, et al. 2004. Biologicalactivities of Bacteroides forsythus lipoproteins and their possible pathologicalroles in periodontal disease. Infect. Immun. 72: 1318–1325.
33. Kikkert, R., M. L. Laine, L. A. Aarden, and A. J. van Winkelhoff. 2007. Acti-vation of toll-like receptors 2 and 4 by gram-negative periodontal bacteria. OralMicrobiol. Immunol. 22: 145–151.
34. Onishi, S., K. Honma, S. Liang, P. Stathopoulou, D. Kinane, G. Hajishengallis,and A. Sharma. 2008. Toll-like receptor 2-mediated interleukin-8 expression ingingival epithelial cells by the Tannerella forsythia leucine-rich repeat proteinBspA. Infect. Immun. 76: 198–205.
35. Dillon, S., A. Agrawal, T. Van Dyke, G. Landreth, L. McCauley, A. Koh,C. Maliszewski, S. Akira, and B. Pulendran. 2004. A Toll-like receptor 2 ligandstimulates Th2 responses in vivo, via induction of extracellular signal-regulatedkinase mitogen-activated protein kinase and c-Fos in dendritic cells. J. Immunol.172: 4733–4743.
36. Re, F., and J. L. Strominger. 2004. IL-10 released by concomitant TLR2 stim-ulation blocks the induction of a subset of Th1 cytokines that are specifically
508 TLR2/Th2 RESPONSES DRIVE T. FORSYTHIA-INDUCED BONE LOSS
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

induced by TLR4 or TLR3 in human dendritic cells. J. Immunol. 173: 7548–7555.
37. Redecke, V., H. Hacker, S. K. Datta, A. Fermin, P. M. Pitha, D. H. Broide, andE. Raz. 2004. Cutting edge: activation of Toll-like receptor 2 induces a Th2immune response and promotes experimental asthma. J. Immunol. 172: 2739–2743.
38. Wenink, M. H., K. C. Santegoets, J. C. Broen, L. van Bon, S. Abdollahi-Roodsaz, C. Popa, R. Huijbens, T. Remijn, E. Lubberts, P. L. van Riel, et al.2009. TLR2 promotes Th2/Th17 responses via TLR4 and TLR7/8 by abrogatingthe type I IFN amplification loop. J. Immunol. 183: 6960–6970.
39. Hart, G. T., D. J. Shaffer, S. Akilesh, A. C. Brown, L. Moran, D. C. Roopenian,and P. J. Baker. 2004. Quantitative gene expression profiling implicates genes forsusceptibility and resistance to alveolar bone loss. Infect. Immun. 72: 4471–4479.
40. Sharma, A., H. T. Sojar, I. Glurich, K. Honma, H. K. Kuramitsu, and R. J. Genco.1998. Cloning, expression, and sequencing of a cell surface antigen containinga leucine-rich repeat motif from Bacteroides forsythus ATCC 43037. Infect.Immun. 66: 5703–5710.
41. Inaba, W. J., R. M. Swiggard, N. Steinman, N. Romani, G. Schular, and C.Brinster. 2009. Isolation of dendritic cells. Curr. Protoc. Immunol. Chapter 3:Unit 3.7.
42. Zheng, X., R. Goncalves, and D. M. Mosser. 2008. The isolation and charac-terization of murine macrophages. Curr. Protoc. Immunol. Chapter 14: Unit 14.1.
43. Sasaki, H., Y. Okamatsu, T. Kawai, R. Kent, M. Taubman, and P. Stashenko.2004. The interleukin-10 knockout mouse is highly susceptible to Porphyr-omonas gingivalis-induced alveolar bone loss. J. Periodontal Res. 39: 432–441.
44. Graves, D. T., D. Fine, Y. T. Teng, T. E. Van Dyke, and G. Hajishengallis. 2008.The use of rodent models to investigate host-bacteria interactions related toperiodontal diseases. J. Clin. Periodontol. 35: 89–105.
45. Honma, K., E. Mishima, and A. Sharma. 2011. Role of Tannerella forsythiaNanH sialidase in epithelial cell attachment. Infect. Immun. 79: 393–401.
46. Inagaki, S., S. Onishi, H. K. Kuramitsu, and A. Sharma. 2006. Porphyromonasgingivalis vesicles enhance attachment, and the leucine-rich repeat BspA proteinis required for invasion of epithelial cells by “Tannerella forsythia”. Infect.Immun. 74: 5023–5028.
47. Rudney, J. D., R. Chen, and G. J. Sedgewick. 2005. Actinobacillus actino-mycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis arecomponents of a polymicrobial intracellular flora within human buccal cells. J.Dent. Res. 84: 59–63.
48. Burns, E., G. Bachrach, L. Shapira, and G. Nussbaum. 2006. Cutting Edge:TLR2 is required for the innate response to Porphyromonas gingivalis: activationleads to bacterial persistence and TLR2 deficiency attenuates induced alveolarbone resorption. J. Immunol. 177: 8296–8300.
49. Seguier, S., G. Godeau, and N. Brousse. 2000. Collagen fibers and inflammatorycells in healthy and diseased human gingival tissues: a comparative and quan-titative study by immunohistochemistry and automated image analysis. J.Periodontol. 71: 1079–1085.
50. Bage, T., A. Kats, B. S. Lopez, G. Morgan, G. Nilsson, I. Burt, M. Korotkova,L. Corbett, A. J. Knox, L. Pino, et al. 2011. Expression of prostaglandin esynthases in periodontitis immunolocalization and cellular regulation. Am. J.Pathol. 178: 1676–1688.
51. Kawai, T., R. Eisen-Lev, M. Seki, J. W. Eastcott, M. E. Wilson, andM. A. Taubman. 2000. Requirement of B7 costimulation for Th1-mediated in-
flammatory bone resorption in experimental periodontal disease. J. Immunol.164: 2102–2109.
52. Liu, R., H. S. Bal, T. Desta, N. Krothapalli, M. Alyassi, Q. Luan, andD. T. Graves. 2006. Diabetes enhances periodontal bone loss through enhancedresorption and diminished bone formation. J. Dent. Res. 85: 510–514.
53. Baker, P. J., L. DuFour, M. Dixon, and D. C. Roopenian. 2000. Adhesionmolecule deficiencies increase Porphyromonas gingivalis-induced alveolar boneloss in mice. Infect. Immun. 68: 3103–3107.
54. Onishi, R. M., and S. L. Gaffen. 2010. Interleukin-17 and its target genes:mechanisms of interleukin-17 function in disease. Immunology 129: 311–321.
55. Gemmell, E., K. Yamazaki, and G. J. Seymour. 2002. Destructive periodontitislesions are determined by the nature of the lymphocytic response. Crit. Rev. OralBiol. Med. 13: 17–34.
56. Sahingur, S. E., X. J. Xia, S. Alamgir, K. Honma, A. Sharma, andH. A. Schenkein. 2010. DNA from Porphyromonas gingivalis and Tannerellaforsythia induce cytokine production in human monocytic cell lines. Mol. OralMicrobiol. 25: 123–135.
57. Pompei, L., S. Jang, B. Zamlynny, S. Ravikumar, A. McBride, S. P. Hickman,and P. Salgame. 2007. Disparity in IL-12 release in dendritic cells and macro-phages in response toMycobacterium tuberculosis is due to use of distinct TLRs.J. Immunol. 178: 5192–5199.
58. Saraiva, M., and A. O’Garra. 2010. The regulation of IL-10 production by im-mune cells. Nat. Rev. Immunol. 10: 170–181.
59. Moreno, J. L., M. Kaczmarek, A. D. Keegan, and M. Tondravi. 2003. IL-4suppresses osteoclast development and mature osteoclast function bya STAT6-dependent mechanism: irreversible inhibition of the differentiationprogram activated by RANKL. Blood 102: 1078–1086.
60. Palmqvist, P., P. Lundberg, E. Persson, A. Johansson, I. Lundgren, A. Lie,H. H. Conaway, and U. H. Lerner. 2006. Inhibition of hormone and cytokine-stimulated osteoclastogenesis and bone resorption by interleukin-4 andinterleukin-13 is associated with increased osteoprotegerin and decreased RANKLand RANK in a STAT6-dependent pathway. J. Biol. Chem. 281: 2414–2429.
61. Lewis, D. B., H. D. Liggitt, E. L. Effmann, S. T. Motley, S. L. Teitelbaum,K. J. Jepsen, S. A. Goldstein, J. Bonadio, J. Carpenter, and R. M. Perlmutter.1993. Osteoporosis induced in mice by overproduction of interleukin 4. Proc.Natl. Acad. Sci. USA 90: 11618–11622.
62. Kotake, S., Y. Nanke, M. Mogi, M. Kawamoto, T. Furuya, T. Yago,T. Kobashigawa, A. Togari, and N. Kamatani. 2005. IFN-gamma-producinghuman T cells directly induce osteoclastogenesis from human monocytes viathe expression of RANKL. Eur. J. Immunol. 35: 3353–3363.
63. Sato, K., A. Suematsu, K. Okamoto, A. Yamaguchi, Y. Morishita, Y. Kadono,S. Tanaka, T. Kodama, S. Akira, Y. Iwakura, et al. 2006. Th17 functions as anosteoclastogenic helper T cell subset that links T cell activation and bone de-struction. J. Exp. Med. 203: 2673–2682.
64. Lin, X., X. Han, T. Kawai, and M. A. Taubman. 2010. Antibody to RANKLAmeliorates T cell-mediated Periodontal Bone Resorption. Infect. Immun. 79:911–917.
65. Han, X., X. Lin, A. R. Seliger, J. Eastcott, T. Kawai, and M. A. Taubman. 2009.Expression of receptor activator of nuclear factor-kappaB ligand by B cells inresponse to oral bacteria. Oral Microbiol. Immunol. 24: 190–196.
The Journal of Immunology 509
by guest on May 12, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















