
This bioinformatics lesson is brought to you by the letter 'D'
57
Today's bioinformatics lesson is brought to you by the letter 'D' by Keith Bradnam Image from flickr.com/91619273@N00/ Today ' s bloinformaties lesson is brought to you by the letter 101 Image fromflickr.com/91619273©NO0/
-
Upload
keith-bradnam -
Category
Education
-
view
2.118 -
download
1
Transcript of This bioinformatics lesson is brought to you by the letter 'D'

Today's bioinformatics lesson is brought to you by the letter 'D'
by
Keith Bradnam
Image from flickr.com/91619273@N00/
Today's bloinformaties lessonis brought to you by the letter 101
Image from flickr.com/91619273©NO0/

Dis for Default parametersis for Default parameters

Dis also for Danger!is also for Danger!


about 0,91-6-1? proeloots please frtalt us at 415,1v.ostet:co/?7caiwetalatlal7s ori your pateliase of an ostz-Rt toaster/ To leatt? %re
bralleltaclones a b e t efectaado coMPla ere rostadota OSIZ.R*lPapa apreovet ozrls sabre los procluctos ae OSTER', visite/7os pot tavot eo
ivit*ostet:coto.

X

XNobody reads a toaster manual!Nobody reads a toaster manual!


But everyone would read a manual for thisBut everyone would read a manual for this
, - - r -----------
opts....tos

Bioinformatics programs are not toasters!
ANL
Bloinformatics programs are not toasters!-EL

Read the manual!Read the manual!

At least, read *some* of the manualAt least, read *some* of the manual

TIEBOW BowtieAn ultrafest memory-efficient short reed aligner
OHNS HOPKINSU N I V E R S I T Y
Bowtle is an ultrafast, memory-efficient short read aligner. It aligns short DNA sequences (reads) tothe human genome at a rate of over 25 million 35-bp reads per hour. Bowtie indexes the genome witha Burrows-Wheeler index to keep its memory footprint small: typically about 2.2 GB for the humangenome (2.9 GB for paired-end). OSI certified
Recent news
" Lighter releasedO Lighter is an extremely fast and memory-efficient program for
correcting sequencing errors in DNA sequencing data. For details onhow error correction can help improve the speed and accuracy ofdownstream analysis tools, see the paper in Genome Biology.Source and software available at GitHub.
" 1.1.1 - 101112014O Fixed a compiling linkage problem related with Mac OS X Mavericks.O Improved performance for cases where the reference contains many
stretches of Ns.O Some minor automatic tests updates.1.1.0 - 7/19/2014
O Added support for large and small indexes, removing 4-billion-nucleotide barrier. Bowtie can now be used with reference genomesof any size
O No longer releasing 32-bit binaries. Simplified manual and Makefileaccordingly.
O Phased out CygWin support.O Improved efficiency of index files loading.O Fixed a bug that made bowtic-inspecz fail in some situations.O (This release was briefly given version number 2.0.0, but we
changed it to 1.1.0 to avoid confusion with Bowtie 2.)1.0.1 release - 3/1412014
bowie-bio_sourceforge.ne:
Site MapHomeNews archiveGetting startedManualTools that use Bowtie
Latest Release
Bowtie 1.1.1 1 0 / 1 / 1 4Please cite. Langmead 8, Trapnell Co Pop M, Salzberg
Ultraast anc memory-efficient alignment of shotDNA sequences to the numan genome. Genome Eltol10:1125.For release updates, subscribe to the mailing list.
related ToolsBowtie 2: Fast, accurate read alignmentCrossbow: Genotyping, cloud computingTophat: RNA-Seq splice junction mapperCufflinks: Isoform assembly, quantitationMyrna: Cloud, differential gene expressionLighter: Fast error correctionOther tools using Bowtie
Pre-built indexes
Consider using Illumina's iGenomescollection. Each iGenomes archive containspre-built Bowtie and Bowtie 2 indexes.
H. sapiens, NCBI GRCh38 2 . 7 GB

How to use bowtie
bowtie [options]* <ebwt> {-1 <m1> -2 <m2> | --12 <r> | <s>} [<hit>]
How to use bowtie
bowtie [options]* <ebwt> 1-1 <ml> -2 <m2> 1 --12 <r> 1 <s>1 [<hit>]

How to use bowtie
bowtie [options]* <ebwt> {-1 <m1> -2 <m2> | --12 <r> | <s>} [<hit>]
How to use bowtie
bowtie [options]* <ebwt> 1-1 <ml> -2 <m2> 1 --12 <r> 1 <s>1 [<hit>]

bowtie [options]* <ebwt> {-1 <m1> -2 <m2> | --12 <r> | <s>} [<hit>]
Bowtie has a lot of options!Bowtie has a lot of options!

The query input files (specified either as <m1> and <m2>, or as <s>) are FASTQ files (usually having extension • fq or , fastg).This is the default. See also: --solexa-quais and --integer-quals.
The query input files (specified either as <mi> and <m2>, or as <s>) are FASTA files (usually having extension fa, .mfa, fnaor similar). All quality values are assumed to be 40 on the Phred quality scale.
-r T h e query input files (specified either as <rni> and <m2>, or as <s>) are Raw files: one sequence per line, without quality valuesor names. All quality values are assumed to be 40 on the Phred quality scale.
-c T h e query sequences are given on command line. I.e. <ml>, <m2> and <singles> are comma-separated lists of reads ratherthan lists of read files.
-C/--color A l i g n in colorspace. Read characters are interpreted as colors. The index specified must be a colorspace index (i.e. built withbowtie-build -C, or bowtie will print an error message and quit. See Colorspace alignment for more details.
-Qt--quals <files> Comma-separated list of files containing quality values for corresponding unpaired CSFASTA reads. Use in combination with -cand -t. --integer-quais is set automatically when -Q/--guals is specified.
--Q1 <files> Comma-sepa ra ted list of files containing quality values for corresponding CSFASTA #1. mates. Use in combination with -C, -f,and -1. --integer-quals is set automatically when --Q1 is specified.
--Q2 <files> Comma-sepa ra ted list of files containing quality values for corresponding CSFASTA #2 mates. Use in combination with -C, -f,and -2. --integer-quals is set automatically when --Q2 is specified.
-s/--skip <int> S k i p (i.e. do not align) the first <int> reads or pairs in the input.
-u/--qupto <int> O n l y align the first <int> reads or read pairs from the input (after the -s/--skip reads or pairs have been skipped). Default:no limit.
-51--trim5 <int> T r i m <int> bases from high-quality (left) end of each read before alignment (default: 0).
-3/--trim3 <int> T r i m <int> bases from low-quality (right) end of each read before alignment (default: 0).
--phred33-quals I n p u t qualities are ASCII chars equal to the Phred quality plus 33. Default: on.
--phred64-quals I n p u t qualities are ASCII chars equal to the Phred quality plus 64. Default: off.
--solexa-quals C o n v e r t input qualities from Solexa (which can be negative) to Phred (which can't). This is usually the right option for use with(unconverted) reads emitted by GA Pipeline versions prior to 1.3. Default: off.
--solexa1.3-quals S a m e as --phred64-quals. This is usually the right option for use with (unconverted) reads emitted by GA Pipeline version 1.3or later. Default: off.
--integer-quals Quality values are represented in the read input file as space-separated ASCII integers, e.g., 40 40 30 40-, rather than ASCIIcharacters, e.g., I n t e g e r s are treated as being on the Phred quality scale unless --s01.exa-quals is also specified.

-k <int>
-m <int>
-M <int>
--best
Report up to <int> valid alignments per read or pair (default: 1). Validity of alignments is determined by the alignment policy (combinedeffects of -n, -v, -1, and -e). If more than one valid alignment exists and the --best and --strata options are specified, then only thosealignments belonging to the best alignment 'stratum" will be reported. Bowtie is designed to be very fast for small -k but bowtie canbecome significantly slower as -k increases. If you would like to use Bowtie for larger values of consider building an index with adenser suffix-array sample, i.e. specify a smaller -ot—offrate when invoking bowtie-build for the relevant index (see the Performancetuning section for details).
-a/--all Report all valid alignments per read or pair (default: off). Validity of alignments is determined by the alignment policy (combined effects of-n, -v, -1, and -e). If more than one valid alignment exists and the --best and --strata options are specified, then only thosealignments belonging to the best alignment "stratum" will be reported. Bowtie is designed to be very fast for small -k but bowtie canbecome significantly slower if -a/--all is specified. If you would like to use Bowtie with -a, consider building an index with a denser suffix-array sample, i.e. specify a smaller -oi—offrate when invoking bowtie-build for the relevant index (see the Performance tuning sectionfor details).Suppress all alignments for a particular read or pair if more than <int> reportable alignments exist for it. Reportable alignments are thosethat would be reported given the -n, -v, -1, -e, -k, -a, --best, and --strata options. Default: no limit. Bowtie is designed to be very fastfor small -m but bowtie can become significantly slower for larger values of -in. If you would like to use Bowtie for larger values of -k,consider building an index with a denser suffix-array sample, i.e. specify a smaller -0/--offrate when invoking bowtie-build for therelevant index (see the Performance tuning section for details).Behaves like -ra except that if a read has more than <int> reportable alignments, one is reported at random. In default output mode, theselected alignment's 7th column is set to <int>-1-1 to indicate the read has at least <int>+1 valid alignments. In -S/--sam mode, theselected alignment is given a MAPQ (mapping quality) of 0 and the xm:i field is set to <int>4-1. This option requires --best; if specifiedwithout --best, --best is enabled automatically.
Make Bowtie guarantee that reported singleton alignments are "best" in terms of stratum (i.e. number of mismatches, or mismatches inthe seed in the case of -r_ mode) and in terms of the quality values at the mismatched position(s). Stratum always trumps quality; e.g. a1-mismatch alignment where the mismatched position has Phred quality 40 is preferred over a 2-mismatch alignment where themismatched positions both have Phred quality 10. When --best is not specified, Bowtie may report alignments that are sub-optimal interms of stratum and/or quality (though an effort is made to report the best alignment). --bes7_ mode also removes all strand bias. Notethat --best does not affect which alignments are considered "valid" by bowtie, only which valid alignments are reported by boTertie. When--best is specified and multiple hits are allowed (via -k or -a), the alignments for a given read are guaranteed to appear in best-to-worstorder in bewtie's output. bowtie is somewhat slower when --best is specified.
--strata I f many valid alignments exist and are reportable (e.g. are not disallowed via the -k option) and they fall into more than one alignment"stratum", report only those alignments that fall into the best stratum. By default, Bowtie reports all reportable alignments regardless ofwhether they fall into multiple strata. When --strata is specified, --best must also be specified.

-v <int> R e p o r t alignments with at most <int> mismatches. -0 and -1 options are ignored and quality values have no effect on whatalignments are valid. -v is mutually exclusive with -n.
-n/--seedmms <int> Maximum number of mismatches permitted in the "seed", i.e. the first L base pairs of the read (where L is set with -1/--seedien). This may be 0, 1, 2 or 3 and the default is 2. This option is mutually exclusive with the -v option.
-ef--magerr <int> Maximum permitted total of quality values at all mismatched read positions throughout the entire alignment, not just in the"seed". The default is 70. Like Maq, Dow-tie rounds quality values to the nearest 10 and saturates at 30; rounding can bedisabled with --nomaground.
-1/--seedien <int>
--nomaground
-I/--minins <int>
-X/--maxins <int>
--nofw/--norc
The "seed length"; i.e., the number of bases on the high-quality end of the read to which the -n ceiling applies. The lowestpermitted setting is 5 and the default is 28. bowtie is faster for larger values ofMaq accepts quality values in the Phred quality scale, but internally rounds values to the nearest 10, with a maximum of 30. Bydefault, bowtie also rounds this way. --nomagrounci prevents this rounding in bowtie.
The minimum insert size for valid paired-end alignments. E.g. if -I 60 is specified and a paired-end alignment consists of two20-bp alignments in the appropriate orientation with a 20-bp gap between them, that alignment is considered valid (as long as-x is also satisfied). A 19-bp gap would not be valid in that case. If trimming options -3 or -!,; are also used, the constraintis applied with respect to the untrimmed mates. Default: O.The maximum insert size for valid paired-end alignments. E.g. if -x 100 is specified and a paired-end alignment consists of two20-bp alignments in the proper orientation with a 60-bp gap between them, that alignment is considered valid (as long as -I isalso satisfied). A 61-bp gap would not be valid in that case. If trimming options -3 or -5 are also used, the -x constraint isapplied with respect to the untrimmed mates, not the trimmed mates. Default: 250.The upstream/downstream mate orientations for a valid paired-end alignment against the forward reference strand. E.g., if --fr is specified and there is a candidate paired-end alignment where matel appears upstream of the reverse complement ofmate2 and the insert length constraints are met, that alignment is valid. Also, if mate2 appears upstream of the reversecomplement of matel and all other constraints are met, that too is valid. --rf likewise requires that an upstream matel bereverse-complemented and a downstream mate2 be forward-oriented. --ff requires both an upstream matel and adownstream mate2 to be forward-oriented. Default: --fr when -C (colorspace alignment) is not specified, --ff when -C isspecified.If --now is specified, bowtie will not attempt to align against the forward reference strand. If --nort is specified, bowtie willnot attempt to align against the reverse-complement reference strand. For paired-end reads using --fr or --rf modes, --nofIsTand --norc apply to the forward and reverse-complement pair orientations. I.e. specifying --nofw and --±r will only find readsin the R/F orientation where mate 2 occurs upstream of mate 1 with respect to the forward reference strand.
--maxbts T h e maximum number of backtracks permitted when aligning a read in 2 or -n 3 mode (default: 125 without --best, 800with --best). A "backtrack" is the introduction of a speculative substitution into the alignment. Without this limit, the default

Print the amount of wall-clock time taken by each phase.
-V--offbase <int> When outputting alignments in Bowtie format, consider the first base of a reference sequence to have offset <int>. This optionhas no effect in -si—sala mode, since SAM mandates 1-based offsets. Default: O.
--quiet P r i n t nothing besides alignments.
--refout
--al <filename>
--un <filename>
--max <filename>
--suppress <cols>
--fullref
Write alignments to a set of files named refXXXXX.map, where xxxXX is the 0-padded index of the reference sequence alignedto. This can be a useful way to break up work for downstream analyses when dealing with, for example, large numbers of readsaligned to the assembled human genome. If <hits> is also specified, it will be ignored.
--refidx W h e n a reference sequence is referred to in a reported alignment, refer to it by 0-based index (its offset into the list ofreferences that were indexed) rather than by name.Write all reads for which at least one alignment was reported to a file with name <filename>. Written reads will appear as theydid in the input, without any of the trimming or translation of quality values that may have taken place within bowtie. Paired-end reads will be written to two parallel files with _1 and inserted in the filename, e.g., if <f ilename> is aligned. fq, the #1and It2 mates that align at least once will be written to aligned_l.fq and aligned_2. fa_ respectively.Write all reads that could not be aligned to a file with name <filename>. Written reads will appear as they did in the input,without any of the trimming or translation of quality values that may have taken place within Bowtie. Paired-end reads will bewritten to two parallel files with _1 and _2 inserted in the filename, e.g., if <filename> is unaligned. fq, the #1 and #2 matesthat fail to align will be written to unaligned_l fo and unaligned_2 q respectively. Unless --max is also specified, reads witha number of valid alignments exceeding the limit set with the -m option are also written to <filenane>.Write all reads with a number of valid alignments exceeding the limit set with the -m option to a file with name <filename>.Written reads will appear as they did in the input, without any of the trimming or translation of quality values that may havetaken place within •zowtie. Paired-end reads will be written to two parallel files with _1 and _2 inserted in the filename, e.g., if<filename> is max. fa, the # 1 and # 2 mates that exceed the -m limit will be written to max_1.fq and max_2.fq respectively.These reads are not written to the file specified with --lart.Suppress columns of output in the default output mode. E.g. if --suppress 1, 5, 6 is specified, the read name, read sequence,and read quality fields will be omitted. See Default Bowtie output for field descriptions. This option is ignored if the outputmode is -S/--sarr..
Print the full refernce sequence name, including whitespace, in alignment output. By default bowtie prints everything up to butnot including the first whitespace.

Colorspace
--snpphred <int>
--snpfrac <dec>
--col-cseq
--col-equal
--col-keepends
SAM
-S/--sam
When decoding colorspace alignments, use <int> as the SNP penalty. This should be set to the user's best guess of the true ratioof SNPs per base in the subject genome, converted to the Phred quality scale. E.g., if the user expects about 1 SNP every 1,000positions, --snpphred should be set to 30 (which is also the default). To specify the fraction directly, use --snpfrac.
When decoding colorspace alignments, use <dot> as the estimated ratio of SNPs per base. For best decoding results, this shouldbe set to the user's best guess of the true ratio. bowtie internally converts the ratio to a Ph red quality, and behaves as if thatquality had been set via the --zinpphred option. Default: 0.001.
If reads are in colorspace and the default output mode is active, --col-cseq causes the reads' color sequence to appear in theread-sequence column (column 5) instead of the decoded nucleotide sequence. See the Decoding colorspace alignments sectionfor details about decoding. This option is ignored in -s/--sam mode.If reads are in colorspace and the default output mode is active, --col-cguai causes the reads original (color) quality sequenceto appear in the quality column (column 6) instead of the decoded qualities. See the Colorspace alignment section for detailsabout decoding. This option is ignored in -S1--sarri mode.
When decoding colorpsace alignments, bowtie trims off a nucleotide and quality from the left and right edges of the alignment.This is because those nucleotides are supported by only one color, in contrast to the middle nucleotides which are supported bytwo. Specify --col-keepends to keep the extreme-end nucleotides and qualities.
Print alignments in SAM format. See the SAM output section of the manual for details. To suppress all SAM headers, use --sam-nohead in addition to -S/--sam. To suppress just the headers (e.g. if the alignment is against a very large number of referencesequences), use --sam-nosq in addition to -S/--sam. bowtie does not write BAM files directly, but SAM output can be converted toBAM on the fly by piping •DowtielS output to samtools view. -Si—sarn is not compatible with --refout.
--mapo <int> I f an alignment is non-repetitive (according to -m, --strata and other options) set the MAPQ (mapping quality) field to this value.See the SAM Spec for details about the MAK, field Default: 255.
--sam-hohead S u p p r e s s header lines (starting with @) when output is -S/--sarr.. This must be specified in addition to -S/--sam. --sam-nohead isignored unless -s/--sarr. is also specified.
--sam-hosq S u p p r e s s 1S0 header lines when output is --Si—sam. This must be specified in addition to -S/--sam. --sam-hosq is ignored unless-sj--sam is also specified.
--sam-RG <text> A d d <text> (usually of the form TAG:VAL, e.g. ID:IL-1LANE2) as a field on the 2:RG header line. Specify --sam-RG multiple times toset multiple fields. See the SAM Spec for details about what fields are legal. Note that, if any @RG fields are set using this option,the ID and SM fields must both be among them to make the gRG line legal according to the SAM Spec. --sari-RG is ignored unless -

Performance
-of—offrate <int>
-pi—threads <int>
--mm
--shmem
Other
Override the offrate of the index with <int>. If <int> is greater than the offrate used to build the index, then some rowmarkings are discarded when the index is read into memory. This reduces the memory footprint of the aligner but requiresmore time to calculate text offsets. <int> must be greater than the value used to build the index.Launch <in':> parallel search threads (default: 1). Threads will run on separate processors/cores and synchronize when parsingreads and outputting alignments. Searching for alignments is highly parallel, and speedup is fairly close to linear. This option isonly available if b,owtie is linked with the othreads library (i.e. if BOVIIE_PTHREADS=0 is not specified at build time).Use memory-mapped I/O to load the Index, rather than normal C file I/O. Memory-mapping the index allows many concurrentbowtio processes on the same computer to share the same memory image of the index (i.e. you pay the memory overheadjust once). This facilitates memory-efficient parallelization of bowtie In situations where using -p is not possible.Use shared memory to load the index, rather than normal C file I/O. Using shared memory allows many concurrent bowtieprocesses on the same computer to share the same memory image of the index (i.e. you pay the memory overhead just once).This facilitates memory-efficient parallelization of bowtie in situations where using -p is not desirable. Unlike --mm, --shneminstalls the index into shared memory permanently, or until the user deletes the shared memory chunks manually. See youroperating system documentation for details on how to manually list and remove shared memory chunks (on Linux and Mac OSX, these commands are ipcs and ipcm). You may also need to increase your OS's maximum shared-memory chunk size toaccomodate larger indexes; see your OS documentation.
--seed <int> U s e <int> as the seed for pseudo-random number generator.
--verbose P r i n t verbose output (for debugging).
--version P r i n t version information and quit.
-hi—help P r i n t usage information and quit.

flickr.com/photos/dannyjacksonflickrcomiphotosidannyjackson
4• -

"I'll just use the default parameters!""I'll just use the default parameters!"

"What could go wrong?""What could go wrong?"

First, some terminology…
Read 1 Read 2
'Insert'
inner-mate pair distance
DNA/RNA Fragmentadapter adapteradapter
First, some terminology...
DNA/RNA Fragment adapter
Read 1inner-mate pair distance
'Insert'
Read 2

We can plot the distributionof inner mate pair distancesWe can plot the distributionof inner mate pair distances

Reads mapped to Transcriptome with Bowtie 2
200 4 0 0 6 0 0 8 0 0Inner size between mapped read pairs

Notice anything unusual?
Reads mapped to Transcriptome with Bowtie 2
Notice anything unusual?
200 4 0 0 6 0 0 8 0 0Inner size between mapped read pairs

Bowtie 2 has an -X option for 'max fragment length'
The default value is 500 bp
= 100 + 100 + 300
What happens if we increase -X to 2000 bp?
Bowtie 2 has an -X optionfor 'max fragment length'
The default value is 500 bp
= 100 + 100 + 300
What happens if weincrease -X to 2000 bp?

New data!
c7,
0 2 0 0
Reads mapped to Transcriptome with Bowtie 2
1
New data!
11111r1n1Ithimin
Inner size between mapped read pairs400 6 0 0 8 0 0

Most programs will have some options that you should consider changing
Most programs will have some optionsthat you should consider changing

Some options from TopHat
TopHat command-line option Meaning Default
value
--num-threadsHow many CPU threads to use when running TopHat 1
--min-intron-length Minimum intron length 70
-r / --mate-inner-distExpected (mean) inner distance between mate pairs 50
--mate-std-devStandard deviation for the distribution on inner distances 20
Some options from Top Hat1WTopHatcommand-line option Meaning Default
value
--num-threads How many CPU threads touse when running TopHat 1

You nearly always can run with more processors/threads than the default (1)You nearly always can run with more
processors/threads than the default (1)

Some options from TopHat
TopHat command-line option Meaning Default
value
--num-threadsHow many CPU threads to use when running TopHat 1
--min-intron-length Minimum intron length 70
-r / --mate-inner-distExpected (mean) inner distance between mate pairs 50
--mate-std-devStandard deviation for the distribution on inner distances 20
Some options from Top Hat1WTopHatcommand-line option Meaning Default
value
--num-threads How many CPU threads touse when running TopHat 1
--min-intron-length Minimum intron length 7 0

This might not be suitable for non-vertebratesThis might not be suitable for non vertebrates

Dis for Documentationis for Documentation

You should document your efforts!You should document your efforts!

You should document as you goYou should document as you go

1iortt, peke),,,v,
4Z-c; (>t_t'
1 7 : 11tresP\ L
LA-r,,oc -nrt
t Lek
(20 tf\-1-*) 1 re-4,
(3,31 - or-
1?-1., tokos • ,,,,, 4
Rool1,11--
12 ProN
rc
RIcA. 046-AccAlitipow)
Pr5T Plow e
opoy.•)&!).
tr Aer -od %Pt,'
• •••••-
t'vsa ( ( A t %44.--5 0-F 6 Co% c -( tcei L0pAr COV Pv-0 t ) ‘_
5i e t-f)triz. a c et 0 ) Loe_ev-T,t,vo S
otir•I'L r e e?
(c• ctfte,4411,rev 6-1 esTmok•as,
kJ, So &IS IFir,at- 05.771 - efive,:t •••1_
0 V (t,d2 (sty
LI 5
IP(112,'A
lActi - r r e r
Cc. 5 4 , e c e 14(,,r5 ,Fe rv 'A4 t r3L.PSAe e V O I N 4
o r- ti-t1/4 etec.vt,
6,11,2) eot%Ps. C E-4 • a t . t ,LAJo-o evw'_1__ s v l y, - 1 c / 9 4 , e r Ttrei-4/. _(3 e o s - 1 - r . v c-rf Pi410 /4,5
ok.1 t% ' t / e n AY) 6"-',00•4)6 •, O f f t ) s s .
.SV.Cte,(v-11 re,i YVIte% kV%) ,,,,,e1r,C rot ; C : )r p u ) 4 61 Cke,4tteV c•At r a t tocr
CT') kJ s Ilk); 0 1 ; , 4 - S P -h 0460w b l ' " "re. 4,ki, 6- 1 S t . e12-",r,POT cve,e3e4crey
"egg, 40 LS-TAN oPcaf t le - r t , *ger o-yreleve_ A t , 1 7 . 7 L t 4- ,5 F a t
1 1 0
PI21 rA, ke, i 2 eleta
I:3.0ex 4 itiv a , - Pp v . / )•% ) 1 a rien tor-
g, v A.) e s - $ • T4P••••• _ 3
covy L a _ Tsres
\) ti7t- eltri—1 TO r e
(Drr n)t-i PM-N4C Arc- CA-13
rrsArtf.. tg-S 6,„tid—toAtt-
e1244-r(0;:t tn.)
•••, trttrteT—.1.t.,,IA1
(..• Oren
t
irtra
t4..c..ec4
IAI/oe4 ( I N T cyt _tu.s.+-•
ST ft:,S
i42the4.1st 2_7\
IRO Ft- ca*.$
nit.P0-6

Lab books are good…
tiortt, peke),,,v,17-6, Nos •••,,,. 4
PsIcA. 0* s6 .._111Q•-kce.iitoow)
RIN'YrIl-1--- - —
T c p- efIc•I:rt _ 3
4 4, elk, -r 4rer-:-1)
re-v6-1 esTA.Was,U_Si)S IFINit( OS:71 — C plAlt " 1 n r • t e . . . n c ,
1:Y4'k.. r e P IONCti(C•Nitt. C I E tot,
L
wkoD°'6'.% L•
54,e c e 14e.m-, r a 1E4-- iter,'A‘t r
1 )PsAe e
(vo f;) wtE ct.c1,..J)116041 1,pt„,„
1
V )1. N„.
41.-ettneT—.1„.„;1,1
ab books are good...
L• • 11 • • 1 • - •
•nrt (20• - • - • • • • — •
- • • • • • • • -opoysn&i)
4._ (3,3-, 0 r- r
FpaL ( f A ( 0 5 - F 6 col 6,1 c -L /tors E 0 P v - 0 s p . ) _
P v i z , 5 i e r •-t• 1 2 0 et 0 ) Weaft-Tt 0,1 S
, A r c , 6 ( el2-•
ittg, LSTANOr:-
L/ ( r , 1 7 2 - 1 ) ,oex ' L k - P p 1 . 1 1 ) ) ) 1 a 1 f l I
1/1/1,-1_1w4,12,'A
•5\) eltri
OPco2 at1/4.)P'1't F a t * -
or-
"••••••-)a. Q r44-19—
v1,6 A *5 z•-
*ger E-yrn,J, 6_1PQ trAl / 2 e
C er 7 / rvs-1,- I t 4tA>11 )*-, it-N4C
1244--r(or's esn
IAI/to'N NI (INT cytV I - , E WA '
nii.P0-6

…but electronic lab books are more helpful
few,":-)
1iortt, peke),,,v,17-1- Nos • et, 4
RIcA. e*s6-kce.tiovw)
ROVNI-11-1----- —
51 NA
_
L
1:3"kk- r e x? IONCti
(C•Nist. ClEtoc.,
PsAe e
v— ef4c- -;rt t
(Les-$ Tcp
t - r 4rer-z1)
01"14
i1/4J itsTA.LIVai1241- C
5-0Z c e k.,041,,,,r,
1 )
(vo ,;) e ct.cro,) _
f i c tert-t'4(-1 r
416041 4 , 1
1
V)I
d_et41
-but electronic lab books are more helpful
LLA-1--,oc. •nrt tve7). 1 re-4,
(3,310ife --or-
( / / k . / 4 L t e l l " - S
e P p A r SP i t
Pit$ rc
• ••••-opoysn&i)
F C014:140 *C-C 0 ev—o 1)\_%--t vs")5i e or-(3,11-0.'" a ° ec.,- ) t w o s
ittg. LST phekjeOr-
,/71-111kAest-I:100v4 lt-Ae 'Lk- p p 1 . 1 1 ) ) ) 1 1 4 t (1 :1 "•5
co2 at1/4.) ci17-7,E, Fat
•g, cv,rOrtor>y
or- 904---,
aver to,Vreteve 6-1
Pi2trA,Ike,t, /2e/ta
mc.A
It71-
-1 b C LI 7 / rr.,1-1-t 4a>11n),-, P 11-N4c Agz Awk-o_p_Q r44-19-
e12-44--r
rat orN N.) eiNtl cyt
nit.P0-6

MicrosoftWord

XJftord
w

Tools like Microsoft Word might not be future proofTools like Microsoft Wordmight not be future proof

Consider using plain text filesConsider using plain text files

I.e. something that can be read using 'less'La something that can be read using 'less'

I like to write README files in Markdown format for everything
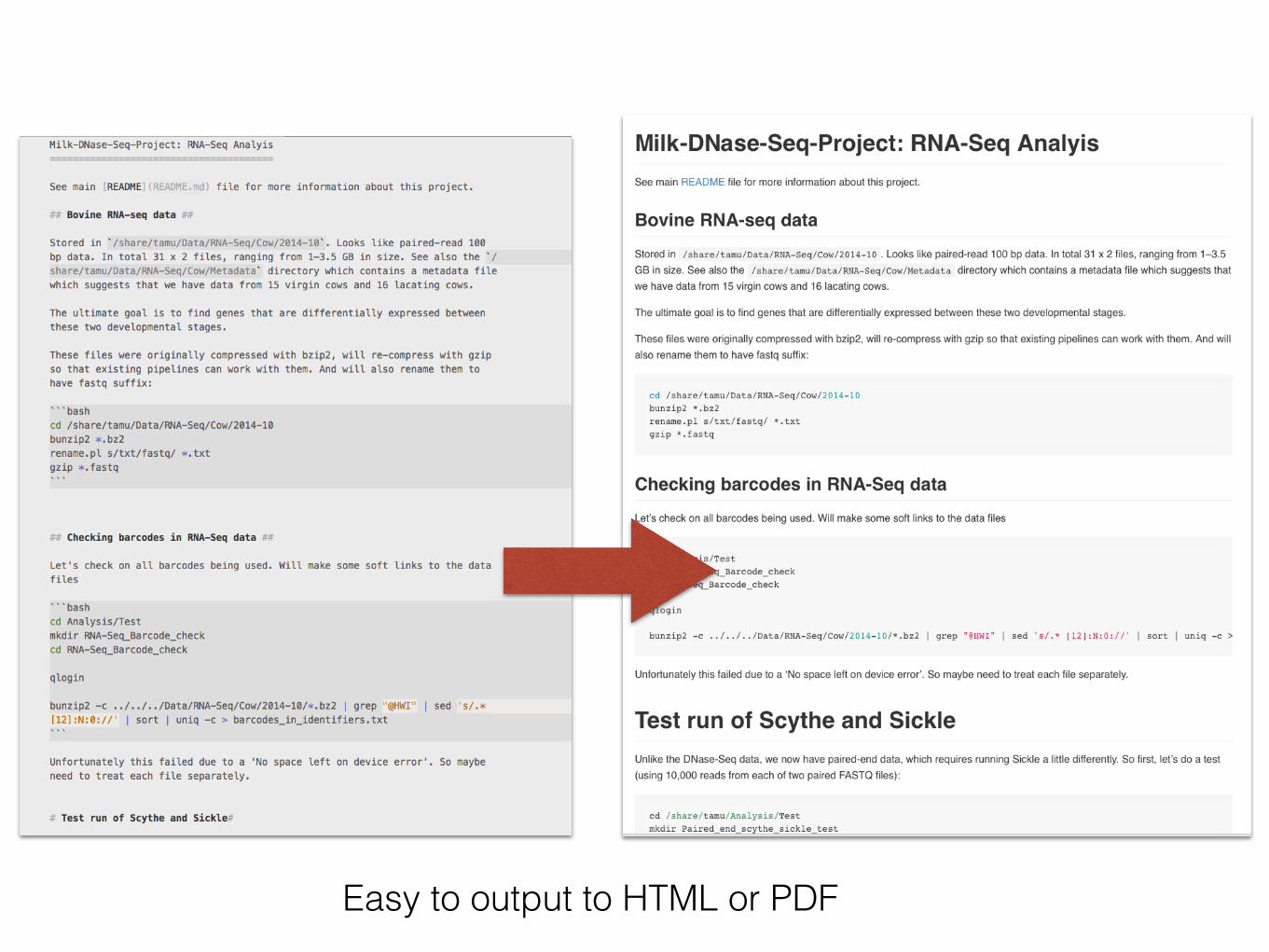
Milk-DNase-Seq-Project: RNA-Seq Analyis--- - - -
See main ,README rl'ADME.md) f i le for more information about this project.
4* Bovine RNA-seq data ##
Stored in 'ishare/tamu/Data/RNA-Seq/Cow/2014-10'. Looks like paired-read 100bp data. In total 31 x 2 files, ranging from 1-3.5 GB in size. See also the ' /share/tamu/Data/RNA-Seq/Cow/Metadata' directory which contains a metadata fi lewhich suggests that we have data from 15 virgin cows and 16 lacating cows.
The ultimate goal is to find genes that are differentially expressed betweenthese two developmental stages.
These files were originally compressed with bzip2, will re-compress with gzipso that existing pipelines can work with them. And will also rename them tohave fastq suffix:—bashcd ishare/tamu/Data/RNA-Seq/Cow/2014-10bunzip2 *.bz2rename.pl sitxt/fastq/ *.txtgzip *.fastq
ttt Checking barcodes in RNA-Seq data ##
Let's check on all barcodes being used. Will make some soft links to the datafiles
"'bashcd Analysis/Testmkdir RNA-Seq_Barcode_checkcd RNA-Seq_Barcode_check
qlogin
bunzip2 -c ../../../Data/RNA-Seq/Cow/2014-10/*.bz2 1 grep @HWI" 1 sed 's/.*[12]:N:0://' I sort I uniq -c > barcodes_in_identifiers.txt
Unfortunately this failed due to a 'No space left on device error'. So maybeneed to treat each fi le separately.
# Test run of Scythe and Sickle#
I like to write README files in Markdown format for everything
Easy to output to HTML or PDF
Milk-DNase-Seq-Project: RNA-Seq Analyis--- - - -
See main ,README rl'ADME.md) f i le for more information about this project.
4* Bovine RNA-seq data ##
Stored in 'ishare/tamu/Data/RNA-Seq/Cow/2014-10'. Looks like paired-read 100bp data. In total 31 x 2 files, ranging from 1-3.5 GB in size. See also the ' /share/tamu/Data/RNA-Seq/Cow/Metadata' directory which contains a metadata fi lewhich suggests that we have data from 15 virgin cows and 16 lacating cows.
The ultimate goal is to find genes that are differentially expressed betweenthese two developmental stages.
These files were originally compressed with bzip2, will re-compress with gzipso that existing pipelines can work with them. And will also rename them tohave fastq suffix:—bashcd ishare/tamu/Data/RNA-5eq/Cow/2014-10bunzip2 *.bz2rename.pl sitxt/fastq/ *.txtgzip *.fastq
ttt Checking barcodes in RNA-Seq data ## 1is/TestLet's check on all barcodes being used. Will make some soft links to the data Barcode check
files q Barcode_check"'bashcd Analysis/Testmkdir RNA-Seq_Barcode_checkcd RNA-Seq_Barcode_check
qlogin
bunzip2 -c ../../../Data/RNA-Seq/Cow/2014-10/*.bz2 1 grep @HWI" 1 sed 's/.*[12]:N:0://' I sort I uniq -c > barcodes_in_identifiers.txt
Unfortunately this failed due to a 'No space left on device error'. So maybeneed to treat each fi le separately.
# Test run of Scythe and Sickle#
Milk-DNase-Seq-Project: RNA-Seq AnalyisSee main README file for more information about this project.
Bovine RNA-seci dataStored in /share/tamu/Data/RNA-Seq/Cow/2014-10 Looks like paired-read 100 bp data. In total 31 x 2 files, ranging from 1-3.5GB in size. See also the ishareitamo/Data/RNA-Seq/Cow/Metadata directory which contains a metadata file which suggests thatwe have data from 15 virgin cows and 16 lacating cows.The ultimate goal is to find genes that are differentially expressed between these two developmental stages.These files were originally compressed with bzip2, will re-compress with gzip so that existing pipelines can work with them. And willalso rename them to have fastq suffix:
cd /share/tamu/Data/RNA-Seq/Cow/2014-10bunzip2 *.b22rename.pl s/txt/fastq/ *.txtgzip *.fastq
Checking barcodes in RNA-Seq dataet's check on all barcodes being used. Will make some soft links to the data files
ogin
bunzip2 -c ../../../Data/RNA-Seq/Cow/2014-10/*.bz2 1 grep "MI " 1 sed 's/. . f121:N:0://' 1 sort 1 unlq -c >
Unfortunately this failed due to a No space left on device error'. So maybe need to treat each file separately.
Test run of Scythe and SickleUnlike the DNase-Seg data. we now have paired-end data, which requires running Sickle a little differently. So first, let's do a test(using 10.000 reads from each of two paired FAST() files):
cd ishare/tamu/Analysis/Testmkdir Paired_end_seythe_sickle test
Easy to output to HIM_ or PDF

http://korflab.ucdavis.edu/bootcamp.md
http://korflab.ucdavis.edu/bootcamp.html
Markdown is easy to read, and converts to useful HTML (with hyperlinks and formatting)
http://kortlabiucdavis.edu/bootcamp.mdhttp://kortlabiucdavis.edu/bootcampihtml
Markdown is easy to read, and converts touseful HIM_ (with hyperlinks and formatting)

Title: Command-line BootcampAuthors: Ke i t h BradnamDate: 2015-06-14Address: Genome Center, UC Davis, Davis, CA, 95616
# Command-line Bootcamp### Keith Bradnam### UC Davis Genome Center#10 Version 1.0 - - - June 2015<br><br><br>
><a rel="license" href="http://creativecommons.org/licenses/by-nc-sa/4.0/"><imgait="Creative Commons License" style-"border-width:0"src="https://i.creativecommons.org/l/by-nc-sa/4.0188x31.png" /></a><br />This work islicensed under a <a rel-"license" href-"http://creativecommons.org/licenses/by-nc-sa/4.0/">Creative Commons Attribution-NonCommercial-ShareAlike 4.0 InternationalLicense</a>. Please send feedback, questions, money, o r abuse to <krbradnamquedavis.edu>
Introduction [Introduction]This 'bootcamp' i s intended to provide the reader with a basic overview of essentialUnix/Linux commands that w i l l allow them to navigate a f i l e system and move, copy, ed i tf i les. I t w i l l also introduce a brief overview of some 'power' commands i n Unix.
## Why Unix? [Why Unix]The [Unix operating system][Unix] has been around since 1969. Back thenthing as a graphical user interface. You typed everything. I t may seem akeyboard to issue commands today, but i t ' s much easier to automate keybomouse tasks. There are several variants of Unix (including [Linux][Linux o u gdifferences do not matter much for most basic functions.
[Unix]: http://en.wikipedia.org/wiki/Unix[Linux]: http://en.wikipedia.org/wiki/Linux
Increasingly, the raw output of biological research exists as _in si l ico_ data, usuallyin the form of large text f i les . Unix i s particularly suited to working with such f i lesand has several powerful (and flexible) commands that can process your data for you. Thereal strength of learning Unix i s that most of these commands can be combined in analmost unlimited fashion. So i f you can learn just f ive Unix commands, you w i l l be ableto do a lo t more than just f ive things.
Of Typeset Conventions [Typeset]Command-line examples that you are meant to type into a terminal window w i l l be shown_
Command-line BootcampKeith BradnamUC Davis Genome CenterVersion 1.0 — June 2015
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Please sendfeedback, questions, money, or abuse to krbradnamgucdavis.edu
IntroductionThis 'bootcamp is intended to provide the reader with a basic overview of essentialUnix/Linux commands that will allow them to navigate a file system and move, copy,edit files. It will also introduce a brief overview of some 'power' commands in Unix.
Why Unix?The Unix operating system has been around since 1969. Back then there was nosuch thing as a graphical user interface. You typed everything. it may seem archaic touse a keyboard to issue commands today, but its much easier to automate keyboard

0 This repository Search Explore Gist Blog Help k b r a d n a m 0 0
Korf Lab / Milk-DNase-Seq-Project
i2/ branch: master • Milk-DNase-Seq-Project/ README_RNA-SecLanalysisand
0 Watch
cd Analysis/Testmkdir RNA—Seq_Barcode_checkcd RNA—Seq_Barcode_check
qlogin
*Star 0 V F o r k 0
i=
koradnam 3 days ago New analysis using R to run DEseq21 _c::,tribiAtOr
317 lines (213 sloc) 12.729 kb R a w Blame History m
Milk-DNase-Seq-Project: RNA-Seq AnalyisSee main README file for more information about this prolect.
Bovine RNA-seq dataStored in /she reitamu/Data/RNA-Seq/Cow/2014-1.0 Looks like paired-read 100 bp data. In total 31 x 2 files, rangingfrom 1-3.5 GB in size. See also the isharettamuiData/RNA-Seq/CowiMetadata directory which contains a metadatafile which suggests that we have data from 15 virgin cows and 16 lacating cows.The ultimate goal is to find genes that are differentially expressed between these two developmental stages.These files were originally compressed with bzip2, will re-compress with gzip so that existing pipelines can work withthem. And will also rename them to have faste suffix:
cd ishare/tamu/Data/RNA—Seq/Cow/2014-10bunzip2 *.bz2rename.pl sitxt/fastq/ *.txtgzip *.fastq
Checking barcodes in RNA-Sell dataLet's check on all barcodes being used. Will make some soft links to the data files
1

Sites like GitHub use Markdown
This repository Search 1 Explore Gist Blog Help kbradnam 0 01
Korf Lab / Milk-DNase-Seq-Project
i2/ branch: master • Milk-DNase-Seq-Project/ README_RNA-SecLanalysisand
0 Watch *Star 0 V F o r k 0
i=
kbradnam 3 days ago New analysis using R to run DEseq21 b 10 r
317 lines (213 sloc) 12.729 kb Raw Blame History I l m
Milk-DNase-Seq-Project: RNA-Seq AnalyisSee main README file for more information about this prolect.
Sites like GitHub use Markdownbunzip2 *.bz2rename.pl sitxt/fastq/ *.txtgzip *.fastq
Checking barcodes in RNA-Sell dataLet's check on all barcodes being used. Will make some soft links to the data files
cd Analysis/Testmkdir RNA-Seq_Barcode_checkcd RNA-Seg_Barcode_check
qlogin

Reproducible science is important!Reproducible science is important!

Reviewers increasingly want more details regarding bioinformatics methods
Reviewers increasingly want moredetails regarding bloinformatics methods

Make it easy to for others to follow your workMake it easy to for others to follow your work

The endThe end


















