
Thesis AS 07 - ETH Z
244
Research Collection Doctoral Thesis Synthesis of chemical libraries based on a Bicyclo 3.1.0 hexane scaffold and studies on the synthesis of Bicyclo 3.1.0 hexane- based S-Adenosylhomocysteine and Pentostatin analogues Author(s): Schlegel, Anna Maria Publication Date: 2012 Permanent Link: https://doi.org/10.3929/ethz-a-7355965 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of Thesis AS 07 - ETH Z

Research Collection
Doctoral Thesis
Synthesis of chemical libraries based on a Bicyclo 3.1.0 hexanescaffold and studies on the synthesis of Bicyclo 3.1.0 hexane-based S-Adenosylhomocysteine and Pentostatin analogues
Author(s): Schlegel, Anna Maria
Publication Date: 2012
Permanent Link: https://doi.org/10.3929/ethz-a-7355965
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Diss. ETH No.20158
Synthesis of Chemical Libraries based on a Bicyclo[3.1.0]hexane Scaffold and Studies on the Synthesis
of Bicyclo[3.1.0]hexane-based S-Adenosylhomocysteine and Pentostatin Analogues
A dissertation submitted to
ETH Zurich
for the degree of
Doctor of Sciences
presented by
Anna Maria Schlegel
Dipl. Natw. ETH Zurich
November 9, 1977
citizen of Buchs/SG, Switzerland
accepted on the recommendation of
Prof. Dr. Karl-Heinz Altmann, examiner Prof. Dr. Dario Neri, co-examiner
2011

Acknowledgments
In the first place I would like to thank Prof. Karl-Heinz Altmann for the opportunity to
accomplish my PhD thesis in his research group.
I am grateful to Prof. Dario Neri for being my co-examiner. Besides, I was allowed to use the
SpectraMaxmicroplate reader for protease activity tests in his lab, and I would also like to
thank all the people from his lab who showed me how to use it.
I would like to thank my collaborators: Dr. Doriano Fabbro and Dr. Daniel D’Orazio, Dr.
Thomas Keller, Prof. Stewart Cole and Dr. Ruben Hartkoorn, Prof. Jürg Getsch und Dr.
Andrea Chicca, Dr. Sebastian Sonntag and Dr. Paul Monaghan, Dr. Bernhard Pfeiffer, and
special thanks to Kurt Hauenstein and Tizian Herzog.
I would like to thank the whole Altmann group, especially Andrea Jantsch, Sascha Kopp,
Christian Kuzniewski, Raphael Schiess, Philipp Gersbach, Oliver Horlacher and Ana Simao,
as well as Sylvia Peleg. Heaps of gratitude go especially to Raphael (and lots of admiration).
In particular, I want to express my gratitude to my family and friends (especially Christoph
Schmid, Jeannine Lingeri, Nina Maag, Sofia Deloudi, Mike Günther, Besnik Kasumaj, and
Naomi Kirchgraber) for emotional and financial support.

Table of Contents
Abstract........................................... ...................................................................................... I
Zusammenfassung ................................... ........................................................................... V
Abbreviations, Acronyms and Symbols ............... ............................................................ IX
1 Introduction ...................................... .......................................................................... 1
1.1 Lead Finding in Drug Discovery .......................................................................... 1
1.2 Natural Products Containing the Bicyclo[3.1.0]hexane Scaffold .......................... 2
1.3 Conformational Characteristics of the Bicyclo[3.1.0]hexane Scaffold .................. 4
1.4 The Bicyclo[3.1.0]hexane Scaffold as a Rigid Sugar Analogue ........................... 8
1.4.1 Conformational Characteristics of the Sugar Part in Nucleosides............. 8
1.4.2 Discrimination between Sugar Conformations by Nucleoside-
processing Enzymes ..............................................................................11
1.4.2.1 HIV Reverse Transcriptase ....................................................................... 11
1.4.2.2 Other DNA Polymerases ........................................................................... 12
1.4.2.3 Adenosine Deaminase .............................................................................. 14
1.4.2.4 Cytidine Deaminase .................................................................................. 17
1.4.2.5 DNA Methyltransferases ........................................................................... 18
1.4.3 Receptors Discriminating between Sugar Pucker: Adenosine and
P2Y-Receptors .......................................................................................19
1.4.3.1 Adenosine Receptors ................................................................................ 19
1.4.3.2 P2Y Receptors .......................................................................................... 21
1.5 Synthesis of Bicyclo[3.1.0]hexane Systems .......................................................22
1.5.1 Natural Products .....................................................................................22
1.5.2 Sugar Analogues ....................................................................................25
1.5.3 Bicyclo[3.1.0]hexane-based Nucleoside Analogues ................................28
1.5.3.1 Northern (N)-Type Analogues ................................................................... 29
1.5.3.2 Southern (S)-Type Analogues ................................................................... 38
1.6 Conclusions .......................................................................................................42

2 Aims and Scope .................................... ....................................................................43
2.1 Chemical Library Synthesis ................................................................................43
2.2 Synthesis of Bicyclo[3.1.0]hexane-based Analogues of Pentostatin and S-
Adenosylhomocysteine ......................................................................................46
2.3 Alternative Synthesis of (+)-4 .............................................................................49
2.4 Biological Screening of the Bicyclo[3.1.0]hexane-based Chemical Library .........50
3 Results and Discussions ........................... ...............................................................51
3.1 Chemical Library Synthesis ................................................................................51
3.1.1 Synthesis of Advanced Intermediates 3, 4 and 5 ...................................51
3.1.2 Synthesis of Library Members with General Structure 1 ..........................58
3.1.2.1 Target Structures Incorporating Heterocyclic Amines ............................... 58
3.1.2.2 N-(2-((4-Methoxyphenyl)amino)ethyl)amides ............................................ 62
3.1.3 Synthesis of Library Members with General Structure 2 ..........................66
3.1.3.1 N-linked Heterocycles ............................................................................... 66
3.1.3.2 C-linked Heterocycles ............................................................................... 71
3.1.4 Final Library ............................................................................................75
3.2 Synthesis of Bicyclo[3.1.0]hexane-based 5-Alkynyl-deoxypyrimidine
Nucleoside Analogue 243 ..................................................................................76
3.3 Miscellaneous Derivatives ..................................................................................77
3.3.1 Tetrazole 250 ..........................................................................................77
3.3.2 Iodides 238 and 239 ...............................................................................78
3.3.3 C3-O-Alkyl Derivatives ............................................................................79
3.4 Structural Studies ...............................................................................................81
3.4.1 Conformational Analysis of 1aa and 1ag by Solution NMR and
Simulated Annealing (SA) ......................................................................81
3.4.2 Conformational Analysis by X-ray Crystallography ..................................85
3.5 Synthesis of Bicyclo[3.1.0]hexane-based Analogues of Pentostatin and S-
Adenosylhomocysteine ......................................................................................88
3.5.1 Bicyclo[3.1.0]hexane-based Pentostatin 6 ..............................................88
3.5.2 Bicyclo[3.1.0]hexane-based S-Adenosylhomocysteine 7 ........................92

3.6 Biological Evaluation of the Bicyclo[3.1.0]hexane-based Chemical Library ....... 100
3.6.1 Reporter Gene Assay Screening for Inhibitory Effects on Selected
Cellular Signaling Pathways ................................................................. 100
3.6.2 In Vitro Protease Inhibition .................................................................... 105
3.6.2.1 Thrombin Inhibition Assay ....................................................................... 105
3.6.2.2 Urokinase Inhibition Assay ...................................................................... 107
3.6.2.3 Cathepsin B Inhibition Assay ................................................................... 108
3.6.3 Antibiotic Activity against Mycobacteria................................................. 109
3.6.3.1 Compounds of General Structure 1 and 2 .............................................. 109
3.6.3.2 Antibiotic Activity against Mycobacterium Tuberculosis of 5-Alkynyl-
deoxypyrimidine Nucleoside Analogue 243 ............................................ 110
3.6.4 Cytotoxicity ........................................................................................... 111
3.7 Biological Evaluation of the Bicyclo[3.1.0]hexane-based S-
Adenosylhomocysteine Analogue ..................................................................... 114
3.7.1 Inhibition of AdoMet-dependant Methyltransferases by S-
Adenosylhomocysteine analogue 7 ...................................................... 114
4 Conclusions and Outlook ........................... ............................................................ 116
5 Experimental Section .............................. ................................................................ 118
5.1 General Methods .............................................................................................. 118
5.2 Experimental Procedures and Analytical Data .................................................. 120
5.2.1 Synthesis of Bicyclo[3.1.0]hexane-based Chemical Library .................. 120
5.2.1.1 Synthesis of Lactone 161 ........................................................................ 120
5.2.1.2 Synthesis of Advanced Intermediates 3, 4, and 5 ................................... 120
5.2.1.3 Synthesis of library members of general structure 1 ............................... 128
5.2.1.4 Synthesis of library members of general structure 2 ............................... 161
5.2.1.5 Crystallographic Data .............................................................................. 174
5.2.2 Biological Evaluation of Bicyclo[3.1.0]hexane-based Chemical
Library .................................................................................................. 186
5.2.3 Synthesis of Miscellaneous Derivatives and Bicyclo[3.1.0]hexane-
based 5-Alkynyl-deoxypyrimidine Nucleoside Analogue 243 ................ 187

5.2.4 Synthesis of Bicyclo[3.1.0]hexane based Pentostatin- and (S)-
Adenosylhomocysteine Analogues ....................................................... 194
5.2.4.1 Preparation of lactone (-)-44 (enantiomeric series)................................. 194
5.2.4.2 Preparation of Advanced Intermediate (+)-4 ........................................... 197
5.2.4.3 Attempted Preparation of Pentostatin Analogue 6 .................................. 202
5.2.4.4 Preparation of S-Adenosylhomocysteine Analogue 7 ............................. 205
6 Curriculum Vitae................................... ................................................................... 218
7 Bibliography ...................................... ...................................................................... 220

I
Abstract
The bicyclo[3.1.0]hexane scaffold possesses unique conformational characteristics and as
such constitutes an attractive starting point for the exploration of new types of lead structures
for drug discovery. In contrast to a simple cyclohexane ring, the bicyclo[3.1.0]hexane system
(Fig. 1) exhibits a pronounced preference for a boat-type conformation, which is the result of
the staggered arrangement of substituents/hydrogens attached to C1, C2 and C5, C4 as well
as favorable orbital interactions. Collectively, these effects outplay the adverse 1,4-
interactions of substituents/hydrogens attached to C6 and C3.
Figure 1. The boat-type conformation of bicyclo[3.1.0]hexane.
The bicyclo[3.1.0]hexane structural unit has been used as a (deoxy)ribose surrogate in
different nucleosides and nucleotides, in order to probe the conformational preferences of
various nucleoside- and nucleotide-processing enzymes, such as HIV-RT, adenosine
deaminase or DNA methyltransferases, and GPCR’s such as the adenosine or P2Yx
receptors.
One goal of this research project was to extend the exploitation of the bicyclo[3.1.0]hexane
scaffold’s special conformational properties to structures other than nucleosides and
biological targets other then nucleoside/nuceotide-interacting enzymes or receptors. To this
end, a small screening library of bicyclo[3.1.0]hexane-based molecules of the general
structures 1 and 2 was to be synthesized (Fig. 2).
Figure 2. Bicyclo[3.1.0]hexane-based library members.
II
The substituents attached to the bicyclo[3.1.0]hexane scaffold in 1 and 2 comprise various
pharmacophoric groups that were chosen based on their prevalence of occurrence in known
drugs; the heterocycles directly attached to the bridgehead carbon atom in 2 frequently occur
in biologically active nucleoside analogues or natural products. Due to the exceptional
conformational rigidity of the bicyclo[3.1.0]hexane scaffold, these groups are present in well
defined spatial orientations.
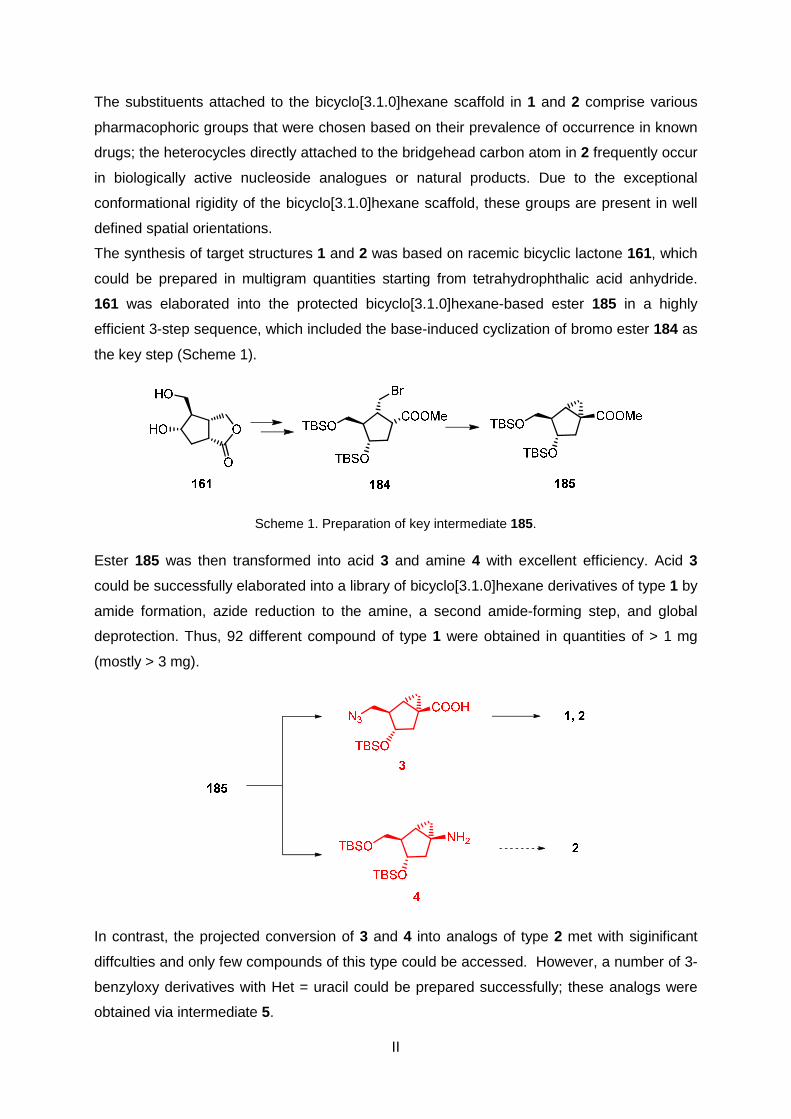
The synthesis of target structures 1 and 2 was based on racemic bicyclic lactone 161, which
could be prepared in multigram quantities starting from tetrahydrophthalic acid anhydride.
161 was elaborated into the protected bicyclo[3.1.0]hexane-based ester 185 in a highly
efficient 3-step sequence, which included the base-induced cyclization of bromo ester 184 as
the key step (Scheme 1).
Scheme 1. Preparation of key intermediate 185.
Ester 185 was then transformed into acid 3 and amine 4 with excellent efficiency. Acid 3
could be successfully elaborated into a library of bicyclo[3.1.0]hexane derivatives of type 1 by
amide formation, azide reduction to the amine, a second amide-forming step, and global
deprotection. Thus, 92 different compound of type 1 were obtained in quantities of > 1 mg
(mostly > 3 mg).
In contrast, the projected conversion of 3 and 4 into analogs of type 2 met with siginificant
diffculties and only few compounds of this type could be accessed. However, a number of 3-
benzyloxy derivatives with Het = uracil could be prepared successfully; these analogs were
obtained via intermediate 5.

III
Structual studies by x-ray crystallography and solution NMR spectroscopy confirmed the
purported boat-like conformation of the bicylic ring system in these derivatives.
The biological evaluation of the library was performed in three protease inhibition assays (for
thrombin, urokinase, and cathepsin B) and a number of reporter gene assays that reflect the
activity of different cellular signalling pathways and networks. While no activity was
detectable in any of the protease inhibition assays, 4 compounds were found to display sub-
µM inhibitory activity in reporter gene assays that measure AP-1-mediated transcription in
response to activation of the EGF or TNFα receptor, respectively.
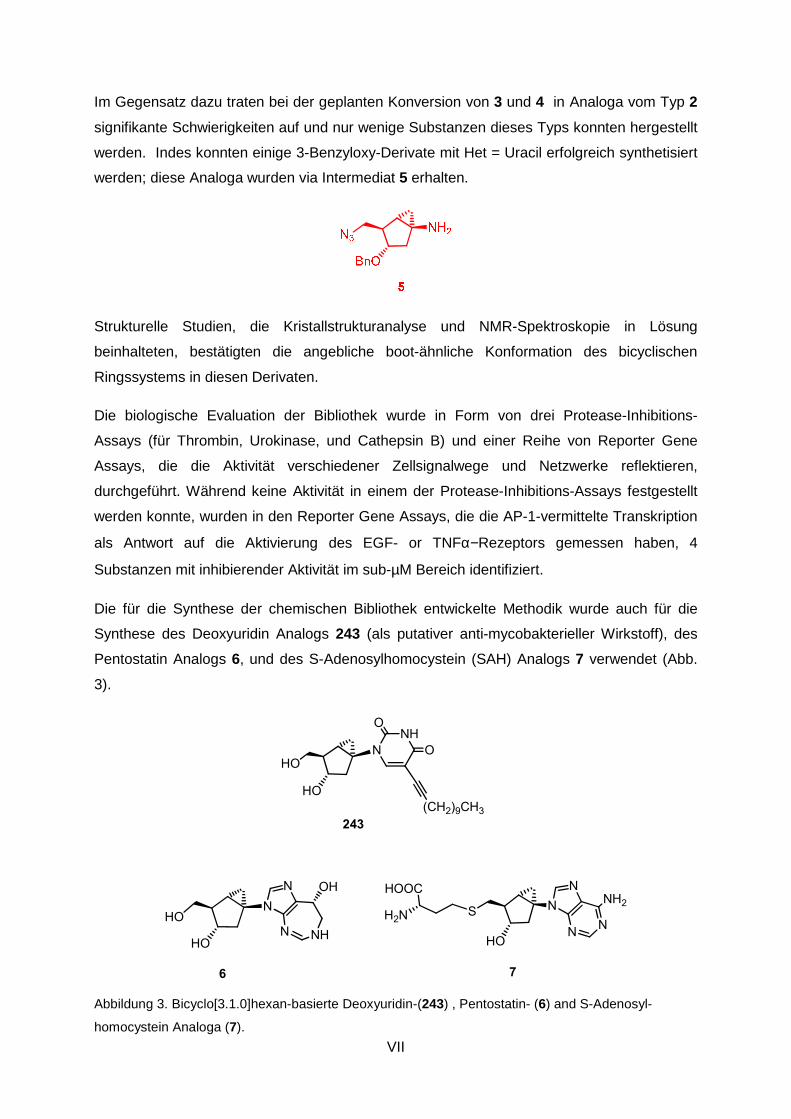
The methodology developed for the synthesis of the above chemical library was also to be
exploited for the synthesis of deoxyuridine analog 243 (as a putative anti-mycobacterial
agent), pentostatin analog 6, and S-adenosylhomocysteine (SAH) analog 7 (Fig. 3).
N
N
N NHHO
HO
6
HO
S N
N
NN
NH2
H2N
7
OH HOOC
HO
HON
NHO
O
243
(CH2)9CH3
Figure 3. Bicyclo[3.1.0]hexane-based deoxyuridine-, pentostatin- (6) and S-adenosylhomocysteine
analogues (7).
The synthesis of racemic 243 was successfully accomplished from amine 4, but the
compound was found to be inactive against M. tuberculosis.
Analogs 6 and 7 were to be prepared as single enantiomers via the dextrorotatory
enantiomer of 4 (i. e. (+)-4). The latter was obtained through fractionate crystallization of
racemic acid (±)-175 with (-)-ephedrine; (±)-175 is an early precursor in the synthesis of
racemic 4.

IV
(±)-175
COOMe
COOH
TBSO
TBSON
NOTBS
NH2
163
N3
Amine (+)-4 could transformed into the imidazole derivative 163 in low yield; however, all
attempts at the elaboration of this intermediate into target structure 6 failed. The synthesis of
bicyclo[3.1.0]hexane-based SAH analogue 7 was successfully accomplished via
intermediates 168 and 287 (Scheme 2).
Scheme 2. Synthesis of SAH analogue 7.
While NMR-spectroscopic analysis of 7 clearly suggested the presence of a single isomer,
HPLC analysis of the material gave two distinct peaks in a 1:1 ratio. Preparative separation
of these peaks and re-analysis of the pure fractions again produced an analytical
chromatogram with two peaks. This finding indicates that 7 (under the conditions of the
HPLC experiments) exists as an equilibrium mixture of two species; the nature of this
equilibrium was not elucidated.
SAH analogue 7 did not show any inhibition of Dengue methyltransferase.

V
Zusammenfassung
Das Bicyclo[3.1.0]hexan-Gerüst besitzt einzigartige konformationelle Eigenschaften und stellt
als solches einen attraktiven Ansatzpunkt für die Erschliessung neuer Leitstrukturen für die
Wirkstoffentwicklung dar. Im Gegensatz zu einem einfachen Cyclohexanring besitzt das
Bicyclo[3.1.0]hexan-System eine ausgeprägte Präferenz für eine boot-ähnliche
Konformation, welche aus der gestaffelten Anordnung von Substituenten/H-Atomen an C1,
C2 und C5, C4, sowie vorteilhaften Orbitalwechselwirkungen resultiert. Diese Effekte
zusammen machen die ungünstige 1,4-Wechselwirkung von Substituenten/H-Atomen an C6
und C3 wett.
Abbildung 1. Die boot-ähnliche Konformation von Bicyclo[3.1.0]hexan.
Die strukturelle Einheit des Bicyclo[3.1.0]hexans wurde bisher als (Deoxy)ribose-Surrogat in
verschiedenen Nucleosiden and Nucleotiden verwendet, um die konformationellen
Präferenzen verschiedener Nukleosid- und Nukleotid-prozessierender Enzyme, wie HIV-RT,
Adenosin Deaminase oder DNA-Methyltransferasen, und GPCRs wie die Adenosin- oder
P2Yx -Rezeptoren, zu untersuchen.
Ein Ziel dieses Forschungsprojekts war es, die Nutzung der speziellen konformationellen
Eigenschaften des Bicyclo[3.1.0]hexan-Gerüsts auf andere Strukturen ausser Nukleosiden
und andere biologische Angriffsziele ausser Nucleosid-/Nucleotid-interagierende Enzyme
oder Rezeptoren auszuweiten. Zu diesem Zweck wurde eine kleine Screening-Bibliothek
Bicyclo[3.1.0]hexan-basierter Moleküle mit den allgemeinen Strukturen 1 und 2 synthetisiert
(Abbildung 2).
Abbildung 2. Bicyclo[3.1.0]hexan-basierte Bibliothekssubstanzen.

VI
Die an das Bicyclo[3.1.0]hexan-Gerüst gebundenen Substituenten der Zielstrukturen 1 und 2
umfassen verschiedenste pharmakophore Gruppen, die nach der Häufigkeit ihres
Vorkommens in bekannten Wirkstoffen ausgewählt wurden; die direkt an das
Brückenkopfatom gebundenen Heterozyklen in 2 kommen häufig in biologisch aktiven
Nukleosid-Analoga oder Naturprodukten vor. Dank der aussergewöhnlichen
konformationellen Starrheit des Bicyclo[3.1.0]hexan-Gerüsts liegen diese Gruppen in einer
definierten räumlichen Anordnung vor.
Die Synthese der Zielstrukturen 1 und 2 basierte auf dem racemischen bicyclischen Lacton
161, das von Tetrahydrophthalsäure ausgehend in mehreren Gramm hergestellt wurde. 161
wurde in einer hocheffizienten 3-Schritt-Sequenz in den geschützten bicyclo[3.1.0]hexan-
basierten Ester 185 überführt, mit der basisch induzierten Cyclisierung des Bromoesters 184
als Schlüsselschritt (Schema 1).
Schema 1. Synthese des Intermediats 185.
Der Ester 185 wurde dann mit exzellenter Effizienz in die Säure 3 und das Amin 4 überführt.
Die Säure 3 konnte durch eine Amidkopplung, eine Azidreduktion zum Amin, eine zweite
Amidkopplung, und globale Entschützung erfolgreich in eine Bibliothek von
Bicyclo[3.1.0]hexan-Derivaten vom Typ 1 überführt werden. Auf diese Weise wurden 92
verschiedene Substanzen vom Typ 1 in Mengen von > 1 mg (> 3 mg für den Grossteil)
erhalten.
VII
Im Gegensatz dazu traten bei der geplanten Konversion von 3 und 4 in Analoga vom Typ 2
signifikante Schwierigkeiten auf und nur wenige Substanzen dieses Typs konnten hergestellt
werden. Indes konnten einige 3-Benzyloxy-Derivate mit Het = Uracil erfolgreich synthetisiert
werden; diese Analoga wurden via Intermediat 5 erhalten.
Strukturelle Studien, die Kristallstrukturanalyse und NMR-Spektroskopie in Lösung
beinhalteten, bestätigten die angebliche boot-ähnliche Konformation des bicyclischen
Ringssystems in diesen Derivaten.
Die biologische Evaluation der Bibliothek wurde in Form von drei Protease-Inhibitions-
Assays (für Thrombin, Urokinase, und Cathepsin B) und einer Reihe von Reporter Gene
Assays, die die Aktivität verschiedener Zellsignalwege und Netzwerke reflektieren,
durchgeführt. Während keine Aktivität in einem der Protease-Inhibitions-Assays festgestellt
werden konnte, wurden in den Reporter Gene Assays, die die AP-1-vermittelte Transkription
als Antwort auf die Aktivierung des EGF- or TNFα−Rezeptors gemessen haben, 4
Substanzen mit inhibierender Aktivität im sub-µM Bereich identifiziert.
Die für die Synthese der chemischen Bibliothek entwickelte Methodik wurde auch für die
Synthese des Deoxyuridin Analogs 243 (als putativer anti-mycobakterieller Wirkstoff), des
Pentostatin Analogs 6, und des S-Adenosylhomocystein (SAH) Analogs 7 verwendet (Abb.
3).
N
N
N NHHO
HO
6
HO
S N
N
NN
NH2
H2N
7
OH HOOC
HO
HON
NHO
O
243
(CH2)9CH3
Abbildung 3. Bicyclo[3.1.0]hexan-basierte Deoxyuridin-(243) , Pentostatin- (6) and S-Adenosyl-
homocystein Analoga (7).

VIII
Die Synthese von racemischem 243 wurde von 4 ausgehend erfolgreich durchgeführt, aber
die Substanz war inaktiv gegen M. tuberculosis.
Analoga 6 und 7 sollten enantiomerenrein über das dextrorotatorische Enantiomer von 4 (d.
h. (+)-4) hergestellt werden. Letzteres wurde durch eine fraktionierte Kristallisation der
racemischen Säure (±)-175 mit (-)-Ephedrin hergestellt; (±)-175 ist ein früher Vorläufer in der
Synthese von racemischem 4.
(±)-175
COOMe
COOH
TBSO
TBSON
NOTBS
NH2
163
N3
Amin (+)-4 konnte in niedrigen Ausbeuten in das Imidazol-Derivat 163 überführt werden; alle
Versuche, dieses Intermediat zur Endverbindung 6 umzusetzen schlugen hingegen fehl. Die
Synthese des Bicyclo[3.1.0]hexane-basierten SAH Analogs 7 wurde erfolgreich via die
Intermediate 168 and 287 ausgeführt (Schema 2).
Schema 2. Synthese des SAH Analogs 7.
Während die Analyse mittels NMR-Spektroskopie klar auf das Vorliegen eines einzelnen
Isomers hinwies, ergab die HPLC Analyse zwei individuelle Peaks mit einem Verhältnis von
1:1. Präparative Trennung dieser Peaks und erneute Analyse der reinen Fraktionen
produzierte erneut ein analytisches Chromatogramm mit zwei Peaks. Diese Ergebnis deutet
darauf hin, dass 7 (unter den Bedingungen des HPLC-Experiments) in einer
Gleichgewichtsmischung zweier Spezies vorliegt; die Natur dieses Gleichgewichts wurde
nicht aufgeklärt.
SAH Analog 7 zeigte keine Inhibition der Dengue Methyltransferase.

IX
Abbreviations, Acronyms and Symbols
))) microwave irradiation
A
AcOH acetic acid
AgNO3 silver nitrate
AP 1 activating protein 1
ARE antoxidant responsive element
B
BH3·Me2S borane dimethylsulfide complex
Bu2NH dibutylamine
BnBr benzylbromide
BnOH benzyl alcohol
C
cAMP cyclic adenosine monophopsphate
CAN cerium ammonium nitrate
cat. catalyst
CMV cytomegalovirus
CRE cAMP response element
CSA camphor sulfonic acid
D
δ delta; chemical shift in ppm (NMR)
d doublet (NMR)
DEAD diethyl azodicarboxylate
DIPEA diisopropylethylamine
DMF dimethylformamide
DPPA diphenylphosphoryl azide
E
ee enantiomeric excess
EGF epidermal growth factor
EDAC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid
ESI electron spay ionisation
Et2O diethylether

X
Et3N triethylamine
EtOH ethanol
F
FADD Fas-Associated protein with Death Domain
FRET fluorescence resonance energy transfer
H
h hours
HATU 2-(1H-7-azabenzotriazol-1-yl)--1,1,3,3-tetramethyl-
uronium hexafluorophosphate methanaminium
HOBt hydroxybenzotriazole
HPLC high performance liquid chromatography
HR-MS high resolution mass spectrometry
I
i-PrOH isopropanol
Im2CO carbonyldiimidazole
Im imidazole
M
Me3BH trimethylborane
min minutes
MsCl mesyl chloride
MTBSA
N
NFκB nuclear factor κB
n-BuLi butyl lithium
P
PCC pyridinium chlorochromate
Pd/C 10% palladium on carbon
Pd/CaCO3 5% palladium on calcium carbonate (Lindlar cat.)
Pd(PPh3)4 tetrakis-triphenyl palladium
PhCOOH benzoic acid
PPh3 triphenylphosphine
PPh2N3 diphenylphosphorylazide

XI
py pyridine
(PhO)2PON3 diphenylphosphorylazide
R
rt room temperature
S
SBE SMAD binding element
T
TBAI tetrabutylammonium iodide
TBAF tetrabutylammonium fluoride
TBS tert-butyldimethylsilyl
TBDMSTf tert-butyldimethylsilyl triflate
tert-BuOK potassium tert-butoxide
tert-BuOH tert-butanol
TGFβ transforming growth factor
THF tetrahydrofuran
TIPDSCl2 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane
TMSBr trimethylsilylbromide
TMSN3 trimethylsilyl azide
TNFα tumor necrosis factor alpha
TosMIC tosylmethyl isocyanide
TRADD Tumor necrosis factor receptor type 1-associated
DEATH domain protein
TsN3 p-toluenesulfonylazide
U
UV ultraviolet

Introduction
1
1 Introduction
1.1 Lead Finding in Drug Discovery
Lead finding marks a decisive phase in the drug discovery process and requires the co-
operation of several disciplines, in order to identify a structural starting point for an extensive
chemical optimization effort that is directed at the discovery of a candidate drug. The aim is
to find those substances in the vast chemical space of all possible compounds that possess
a biological profile which could be potentially useful for medicinal purposes. The disciplines
involved in lead finding include synthetic and computational chemistry, biology,
pharmacology and biopharmacy.
There are different approaches currently employed in the drug discovery process for lead
finding. One important starting point is the high-throughput screening1 (HTS) of compound
collections, in the meaning of a systematic assessment of compound effects in biochemical,
cellular, or biophysical assays. The compound collections screened can be obtained by
means of combinatorial chemistry/parallel synthesis, they can be bioextracts from plants or
microbes, or pure natural products. However, the high-throughput screening in the first place
only generates hits, i.e. non-reactive compounds of verified structure and purity with a
confirmed minimal in vitro potency. These hits have to be transformed into leads in the next
phase of the drug discovery process.2 The structure and purity of compounds reaching the
minimum biological activity threshold set for a given screen have to be confirmed; likewise,
the activity, the physico-chemical and ADME (absorption, distribution, metabolism and
excretion) properties are assessed and the possibilities for further chemical derivatization
have to be investigated. Extensive hit-to-lead chemistry can then identify the core structure
required for a specific activity of interest (inhibition of a certain target).
An alternative screening approach is fragment-based screening, but again this method does
not directly generate lead compounds.3 With this method, small (MW < 300) fragments that
possess low biological activity are screened and subsequently developed into higher potency
lead structures by combining them.
Existing drugs or clinical development compounds can be used as lead structures by
extending their targets or by their further optimization for a chosen target. Natural substrates
or known receptor ligands can serve as lead structures. In general, the exploitation of
biological information won by biochemical or cellular assays, in animal experiments, by the
detection of side effects in human clinical trials or by basic biological research can be used to
restrict oneself to certain chemical structures.

Introduction
2
There are also in silico approaches such as virtual screening4 or structure-based rational
drug design.5
High-throughput screening is the presently the most widely applicable technology to deliver
chemical starting points for drug discovery. This method enables the screening of up to 100
000 compounds a day, but it has not led to the expected abundance of suitable lead
compounds; of 58 drugs introduced in the market between 1991 and 2008 only 19 were
HTS-derived.6 Although the compounds in large libraries are by now successfully subjected
to prediction tools for physicochemical properties7 ensuring that hits with drug-like properties
are generated, the issue of covering a significant portion of chemical diversity space is not
addressed satisfactorily.1
Instead of screening huge compound libraries, the directed synthesis of smaller focused
libraries which are subsequently assessed biologically is therefore also considered an
important means of lead finding.8 These libraries are designed around a specific core
structure or a certain scaffold.
However, it is difficult to choose the scaffold that is to serve as a common structural feature
of a small chemical library. In this context, the bicyclo[3.1.0]hexane system represents a
promising scaffold that has not yet been explored widely outside the nucleoside analogue
research area. With its special conformational characteristics, it embodies an excellent
starting point for the exploration of new types of structures, opening up the possibility to
predefine the spatial orientation of pharmacophoric groups attached to it.
1.2 Natural Products Containing the Bicyclo[3.1.0]h exane Scaffold
The bicyclo[3.1.0]hexane structural unit can be found in a number of natural products;
however, overall, its occurrence in nature is relatively rare. Two prominent examples of
bicyclo[3.1.0]hexane-containing natural products are (-)-α- and (+)-β-thujone (8a and 8b,
respectively), monoterpenes that have been isolated from Artemisia absinthium or several
other plants and that act on GABA and 5HT3 receptors in the brain (Fig. 1).9

Introduction
3
Figure 1. Natural products containing the bicyclo[3.1.0]hexane scaffold.
Chlorajaponilide A (11b), very recently isolated from Chloranthus japonicus, is a
sesquiterpenoid that contains two bicyclo[3.1.0]hexane units and possesses anti-HIV-1
activity.10 From the same plant, which mainly grows in East Asia, other sesquiterpenoids with
biological activities have been isolated, such as the chlorahololides (11c, 11d);11 all of these
compounds share a lindenane scaffold as the common core structure. Chloranthalactone A
(11a) is possibly a biosynthetic precursor of 11b-d.12 Syntheses of the heptacyclic cores of
chlorahololides (11c and 11d) have been recently accomplished.13
Laurinterol (9) has been isolated from seaweeds of the genus Laurencia and constitutes an
unusual natural product with its bicyclo[3.1.0]hexane unit and its bromo functionality.14 From
Jatropha curcas L., a plant widely distributed in Asian and African countries, Jatropha factor
C1 (10a) and the related compounds 10b and 10c have been isolated.15 Compounds 10a-c
are probably biosynthetically derived from the diterpene 12-deoxy-16-hydroxyphorbol; it is
unknown, however, which reactions lead to the formation of the bicyclo[3.1.0]hexane
structural unit in these compounds.16
Introduction
4
1.3 Conformational Characteristics of the Bicyclo[3 .1.0]hexane
Scaffold
The most specific conformational feature of the bicyclo[3.1.0]hexane moiety is its exclusive
occurrence as a boat conformer. As a consequence, the cyclopentane ring embedded in the
bicyclo[3.1.0]hexane scaffold is permanently locked in a single envelope conformation; in
contrast, cyclopentane itself, while also residing in an envelope conformation, normally
undergoes a rapid pseudorotation process during which its five carbon atoms take turns in
adopting the endo position, interconverting with half-chair forms that are of equally lower
energy compared to a planar conformation.17 It has to be noted that, for simplicity, the typical
conformation of a bicyclo[3.1.0]hexane system is referred to here as “boat” conformation,
although it is more planar than the corresponding form of cyclohexane (and, hence, is also
called pseudoboat conformation). The conformational preference of the bicyclo[3.1.0]hexane
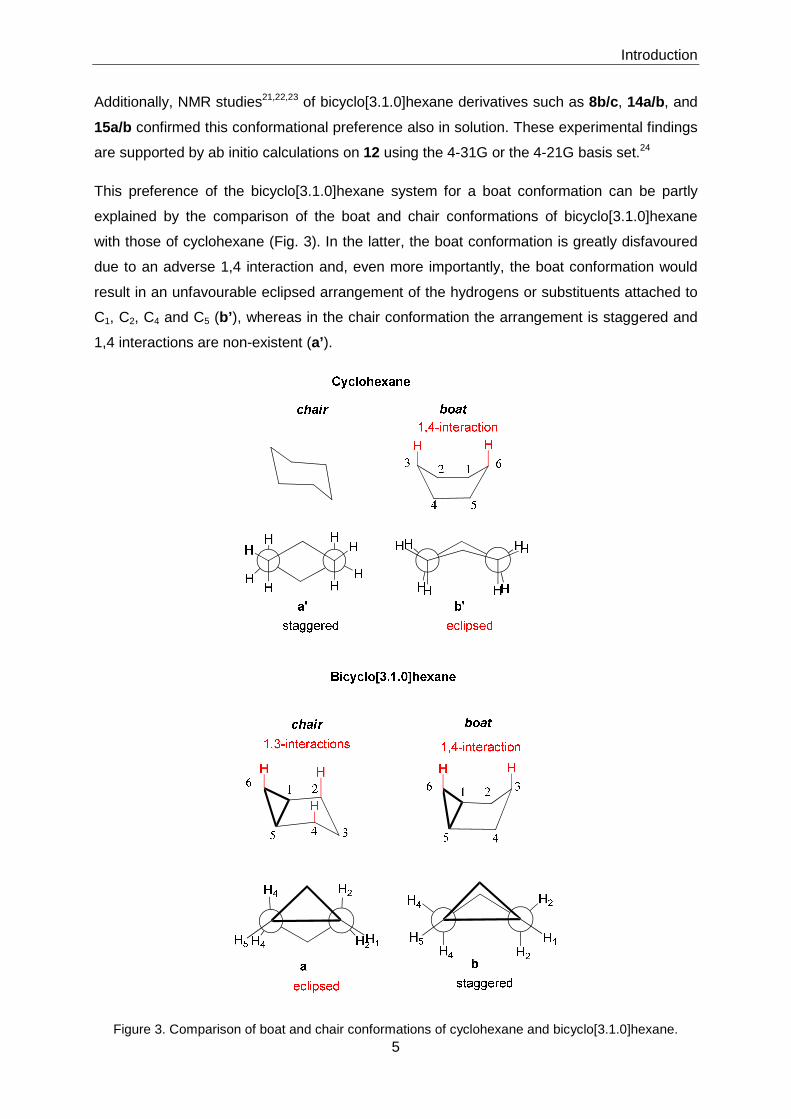
system for a boat conformation was first established experimentally by microwave18 and
combined electron-diffraction microwave studies19 of bicyclo[3.1.0]hexane (12) itself as well
as X-ray crystallographic analysis of bicyclo[3.1.0]hexane-based p-bromosulfonamide 13
(Fig. 2).20
Figure 2. Bicyclo[3.1.0]hexane and derivatives used in molecular geometry studies.
Introduction
5
Additionally, NMR studies21,22,23 of bicyclo[3.1.0]hexane derivatives such as 8b/c , 14a/b, and
15a/b confirmed this conformational preference also in solution. These experimental findings
are supported by ab initio calculations on 12 using the 4-31G or the 4-21G basis set.24
This preference of the bicyclo[3.1.0]hexane system for a boat conformation can be partly
explained by the comparison of the boat and chair conformations of bicyclo[3.1.0]hexane
with those of cyclohexane (Fig. 3). In the latter, the boat conformation is greatly disfavoured
due to an adverse 1,4 interaction and, even more importantly, the boat conformation would
result in an unfavourable eclipsed arrangement of the hydrogens or substituents attached to
C1, C2, C4 and C5 (b’ ), whereas in the chair conformation the arrangement is staggered and
1,4 interactions are non-existent (a’).
Figure 3. Comparison of boat and chair conformations of cyclohexane and bicyclo[3.1.0]hexane.

Introduction
6
In contrast, the Newman projections of bicyclo[3.1.0]hexane reveal an adverse eclipsed
arrangement of hydrogen atoms or substituents in the case of the chair conformer (a). In the
case of the boat conformer, the corresponding hydrogen atoms/substituents arrange
staggered and gauche, respectively, and this favorable arrangement outplays the 1,4
interaction between the C3 and C6 hydrogens or substituents, respectively. The argument of
replacing two 1,3 interactions by a single 1,4 interaction has also been suggested to account
for a favourable boat conformation.23 In contrast, a combined electron-diffraction microwave
study19 of 12 additionally suggested an unusually short C1-C5 bond, which would render the
1,4 interaction in the boat bicyclo[3.1.0]hexane conformer more severe; however, this finding
is neither supported by the ab inito results, nor by later studies that combine experimental
data with quantum mechanical calculations, such as MM2 and MM3 force field studies, like
the one carried out in 1992 by Allinger and co-workers on 12.25
The lengths of the C1-C2, C2-C3 and the C1-C6 bonds in bicyclo[3.1.0]hexane (12) have
been determined by Kato and co-workers by ab initio calculations using the 3.21G, 3-21G*,
and 6-31G* basis sets to be all around 1.52 Å.26 A bond length in the same range has been
obtained for the C1-C5 bond.26 The torsion angle between the planes spanned by C4, C5,
C1, C2 and C4, C3, C2 was determined to be 150° ( β), the one between planes spanned by
C4, C5, C1, C2 and C5, C6, C1 to be 110° ( α). The bond angle between the bonds C1- C2
and C2- C3 was found to be 105° ( γ, Fig. 4). These results were in good agreement with
observed values18 and with earlier calculations as well as with the molecular mechanics
study.25
Figure 4. Bond lengths, torsion angles (α, β) and bond angle (γ) in bicyclo[3.1.0]hexane.
Most importantly, Kato’s work showed that stereoelectronic effects greatly add to the purely
steric analysis, such that a stabilizing orbital interaction exists between the lowest
unoccupied molecular orbital (LUMO) of the cyclopropane ring, a Walsh-type orbital, and the
back lobes of the pseudoequatorial C-H2 and C-H4 highest occupied molecular orbitals
(HOMOs). As can be seen in Figure 5, the stabilizing interaction can only take place in the
boat conformation, where the cyclopropane LUMO is in line with the C-H2 and C-H4
pseudoequatorial bonds, which is not the case for the chair conformer.26

Introduction
7
boatchair
H4
H4
H5
H2
H2
H1
1 23
45
6
Bicyclo[3.1.0]hexane
in-phase orbital interaction
of cyclopropane LUMO with
C-H4 and C-H2 HOMOs
H4
H4H5
no stabilizing orbital interaction
H2
H2H1
1 2
345
6
ba
Figure 5. Orbital interactions in the boat (b) and chair (a) conformation of bicyclo[3.1.0]hexane.26 In
case b), there is an in-phase orbital interaction existing which is geometrically not possible in case a).
All the above steric and stereoelectronic arguments in favour of the boat conformer should
also hold for substituted bicyclo[3.1.0]hexane derivatives. As indicated above, several X-ray
crystal structures of bicyclo[3.1.0]hexane derivatives, e.g. 16, 17, 18 27 indeed prove this
point.30, 28 No other conformations for the bicyclo[3.1.0]hexane scaffold have been found in
existing compounds (based on a Cambridge Structural Database search).
Accordingly, the rigid conformation of the bicyclo[3.1.0]hexane scaffold leads to a significant
degree of structural preorganization of substituents attached to this scaffold, and this, in turn,
enables the defined spatial arrangement of pharmacophores, a feature that was to be
exploited in this thesis. So far, exploitation of the bicyclo[3.1.0]hexane scaffold’s unique
conformational properties has been largely limited to its use as a rigid sugar analogue. The
bicyclo[3.1.0]hexane scaffold’s use as a means to impart rigidity on nucleoside analogues is
shortly discussed in the next chapter.

Introduction
8
1.4 The Bicyclo[3.1.0]hexane Scaffold as a Rigid Su gar Analogue
1.4.1 Conformational Characteristics of the Sugar P art in Nucleosides
Nucleosides are N-glycosides consisting of a nucleobase (adenine, guanine, cytosine, uracil
or thymine) bound to a 5-or 6-carbon sugar via a glycosidic linkage. Most common examples
of nucleosides include cytidine, uridine, adenosine, guanosine, thymidine and inosine,
consisting of either a ribose or a deoxyribose and a base moiety (Fig. 6).
Figure 6. Common nucleosides and nucleotides.
When an enzyme or receptor interacts with its nucleoside substrate, the efficiency of the
enzymatic reaction depends on all parts of the nucleoside (ribose and base) being properly
arranged to fit the active site. The critical conformational parameter of the ribose (or the 2’-
deoxyribose) moiety that ensures that these interactions are optimal is the sugar pucker,
which is defined by the pseudorotational phase angle P.29 P can adopt values between 0° and
360° and is a function of the five endocyclic torsi on angles of the tetrahydrofuran ring (Fig. 7).
A range of P between 342° and 18° ( 2E -->3T2-->3E) by convention corresponds to a northern
conformation of the sugar in nucleosides/nucleotides. A southern conformation is defined by
a range of P between 162° and 198° ( 2E -->2T3-->3E). The radius of the pseudorotational
cycle is equal to the maximum out-of-plane pucker. In principle, the whole variety of
conformations designated in the pseudorotational cycle can be adopted by nucleosides. The
preference for any of these specific conformations in solution is largely determined by the
interplay of gauche and anomeric effects. The anomeric effect30 is characterized by the
orbital interaction between the lone pair of oxygen O4’ and the highest p-orbital component
of the anti-bonding orbital of the C1’-N1/N9 glycosyl bond, and it is strongest when the
conformation of the nucleoside is northern. Gauche effects existing between O4’-C4’-C3’-O3’
and O4’-C1’-C2’-O2’ fragments exert a stronger influence than the anomeric effect, steering
the conformation for 2’-deoxyribonucleosides towards south, whereas for ribonucleosides,

Introduction
9
these effects result in a two-state 3’-endo 2’-endo equilibrium of an approximate 1:1
ratio.31
Rapid interconversion between these two extreme forms of sugar pucker via the
pseudorotational cycle takes place constantly, as the energetic barrier is very low (in the
range of 4 kcal/mol).32
E0
North
South
2E
1E
3E
0E90°270°
180°
0°
3T2
2T3
1T2
1T0
OBHOH2CBHOH2C
O
O
BHOH2C
O
BHOH2C
3E
2E
3'-endo
2'-endo
r = max
Figure 7. The pseudorotational cycle.
Although the energy difference between these two conformations is small, experimental
findings lead to the conclusion that the majority of nucleoside/nucleotide-interacting enzymes
accept the furanose ring only in one of the two conformations.32 These enzymes seem to
have strict conformational preferences for substrate binding, as in many cases only one of
the two conformers is present in the active site of the enzyme in structures of enzyme-
substrate complexes.33 Thus, methods have been searched for to elucidate those
conformational preferences for different enzymes.
Experimental methods to achieve that goal include a rigidification of the nucleoside in one of
the two conformations, southern or northern. In this context, the bicyclo[3.1.0]hexane scaffold
offers the opportunity of a fixation of either of the two main conformations of a nucleoside by
substituting the furanose ring part of the nucleoside with a bicyclo[3.1.0]hexane moiety (Fig.
8). Depending on the location of the fused cyclopropane ring supplanting the ribofuranoside
oxygen, either a northern or a southern nucleoside conformation can be achieved, in which
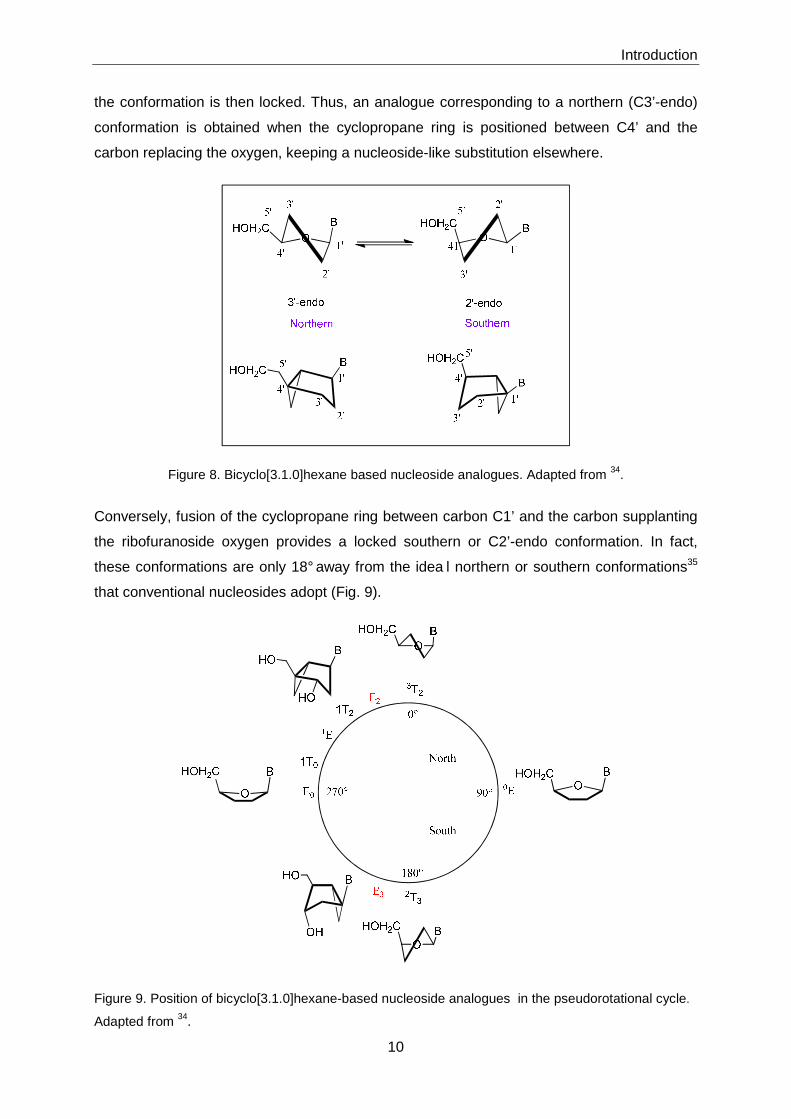
Introduction
10
the conformation is then locked. Thus, an analogue corresponding to a northern (C3’-endo)
conformation is obtained when the cyclopropane ring is positioned between C4’ and the
carbon replacing the oxygen, keeping a nucleoside-like substitution elsewhere.
Figure 8. Bicyclo[3.1.0]hexane based nucleoside analogues. Adapted from 34.
Conversely, fusion of the cyclopropane ring between carbon C1’ and the carbon supplanting
the ribofuranoside oxygen provides a locked southern or C2’-endo conformation. In fact,
these conformations are only 18° away from the idea l northern or southern conformations35
that conventional nucleosides adopt (Fig. 9).
Figure 9. Position of bicyclo[3.1.0]hexane-based nucleoside analogues in the pseudorotational cycle.
Adapted from 34.

Introduction
11
Furthermore, the replacement of the furanose oxygen by carbon also provides higher
chemical and enzymatic stability of the resulting analogues, due to the absence of a
glycosidic bond. While this is also true for simple carbocyclic nucleosides where the furanose
ring is replaced by a cyclopentane unit, these latter analogues adopt an aberrant 1‘-exo
conformation, due to the steric bulk of the nucleobase, which prefers to adopt an equatorial
orientation. Consequently, such analogues are not suitable to probe conformational
preferences of nucleoside- or nucleotide-targeting, as the orientation of the hydroxyl groups
and/or base is not similar to the original nucleoside/nucleotide conformation.
1.4.2 Discrimination between Sugar Conformations by Nucleoside-
processing Enzymes
1.4.2.1 HIV Reverse Transcriptase
Dideoxynucleoside analogues targeting viral reverse transcriptase (RT), a RNA-dependant
DNA polymerase, were the first drugs enabling an effective treatment of HIV-1 infections.36
After phosphorylation by viral and host kinases, these compounds are incorporated into the
viral DNA and subsequently act as chain terminators, thereby stopping viral transcription.
The nucleoside reverse transcriptase inhibitors (NRTI’s) such as AZT or stavudine (Fig. 10)
achieve a certain selectivity, since DNA production by reverse transcriptase is very different
from human DNA replication.37
Figure 10. Selected nucleoside analogue HIV RT inhibitors (NRTI’s) and carbocyclic analogues.

Introduction
12
For HIV-1 RT, there exists evidence that the conformation of the sugar moiety preferred by
the reverse transcriptase is similar to the northern conformation.38 When the 5’-triphosphate
of AZT (19cb) was compared with its conformationally restricted analogues 19aa and 19ba
(Fig. 10), in terms of anti-RT activity, it was found that the southern analogue 19ba was
completely inactive against HIV-1 RT, whereas the northern triphosphorylated analogue
19aa inhibited RT as effectively as the (flexible) AZT nucleotide.33a However, 19a as such
shows no anti-HIV effect, meaning that it is not recognized by cellular kinases. In contrast,
the northern-type analogue of the NRTI stavudine (20b) shows anti-HIV activity; the double
bond in the carbocyclic scaffold of 20a changes the conformation of the five-membered ring
from envelope to planar, a change that made the molecule recognizable to cellular kinases.39
These findings suggest that a southern conformation is advantageous for the interaction with
cellular kinases, whereas RT interacts preferentially with the northern conformers. AZT itself,
which is flexible, has a preference for the southern conformation, due to a gauche effect
between the 3’-azido group and the ribose O4’-oxygen. This preference is reflected in the
fact that phosphorylation of AZT by thymidine kinase (tk1) is 500-fold more efficient than the
phosphorylation of stavudine or northern analogues.40 However, once triphosporylated the
compound would bind to RT in a northern conformation.
1.4.2.2 Other DNA Polymerases
Northern-methanocarbathymidine (N-MCT, 21a, Fig. 11) is another conformationally
restricted thymidine analogue41 with high activity against Herpes simplex viruses of type I
(HSV-1) and type II (HSV-2). It exhibits its antiviral activity by interaction with type I as well
as type II kinase (HSV-1-tk, and HSV-2-tk, respectively), both key enzymes in the herpes
simplex virus life cycle.
Figure 11. Conformationally restricted thymidine analogues.
Once phosphorylated to the triphosphate level, N-MCT triphosphate inhibits viral DNA
synthesis by inhibition of DNA polymerase. In addition, N-MCT has shown good in vitro

Introduction
13
activity against Kaposi’s sarcoma-associated herpesvirus (KSHV), displaying greater potency
than the reference compounds ganciclovir and cidofovir.42
When examining the processing of this compound in the cell, similarly opposed
conformational penchants towards kinases and DNA polymerase had been expected to be
observed as in the context of anti-HIV drugs acting on reverse transcriptase. The supposedly
general preference of cellular kinases for a southern conformation and the opposing
preference of DNA polymerase for a northern sugar conformation could indeed be confirmed
with the synthesis of southern-type analogue 21b. Comparing 21a and 21b, S-MCT 21b was
readily triphosphorylated, while much lower levels of the triphosphate of N-MCT 21a were
formed. Despite the high levels of S-MCT-5’-triphosphate, only triphosphorylated 21a was
incorporated into DNA and therefore displayed anti-HSV activity, while S-MCT-5’-
triphosphate was completely ineffective. However, examining the 3 phosphorylation steps in
detail, the first step in the processing of S-MCT and N-MCT, carried out by viral thymidine
kinases (HSV-1-tk or HSV-2-tk), strikingly seemed to prefer a northern conformation of the
drug, being more effective on 21a than on 21b. The second and third phosphorylation steps,
conducted by cellular thymidine kinases (tk1), clearly preferred a southern conformation,
while DNA polymerase only recognized and incorporated tri-phosphorylated northern
analogues.43 A possible explanation for the different behavior of viral HSV-tk and cellular
kinases against S-type nucleoside analogues can be found in the orientation of the
nucleobase; the latter is described by the glycosyl torsion angle χ (= torsion angle of the C1’-
C5’-N1-C2 sequence, Fig. 12) whose value determines the syn or anti disposition of the base
relative to the sugar moiety.
Figure 12. syn-anti base conformation.
For southern thymidine analogue 21b the preferred base conformation is syn (χ = 60°),
stabilized by a hydrogen bond between the C-7’-hydroxyl group and the C-2 carbonyl of the
pyrimidine (Fig. 12).50 Northern analogues preferentially adopt an anti conformation of the
base (χ = -150°). 44 This finding is in agreement with the trend observed in conventional

Introduction
14
nucleosides, where the nucleobase conformation tends to be syn for a southern sugar
pucker and anti for a northern sugar pucker,45 with the difference of a higher syn-anti energy
barrier for bicyclo[3.1.0]hexane based analogues such as 21b, a consequence of the fusion
of the cyclopropane ring immediately adjacent to the C-N bond.44b
Viral thymidine kinase HSV-tk prefers a southern sugar (or pseudosugar-) conformation but
with the nucleobase adopting an anti conformation.46 The significance of base conformation
for the recognition of bicyclo[3.1.0]hexane-based nucleoside analogs by HSV-tk was
confirmed with thymidine analogue 21c, which mimicks a south-like conformation, but
displays the nucleobase in an anti orientation. 21c was phosphorylated by viral HSV-tk at
least twice as effectively as 21b.47
1.4.2.3 Adenosine Deaminase
Adenosine deaminase (ADA) is a key enzyme in purine metabolism, catalyzing the
deamination of adenosine (22a) and 2’-deoxy-adenosine (22b), to the corresponding
inosines 24, presumably via the tetrahedral intermediates 23a or 23b, respectively
(Scheme 1).
Scheme 1. Adenosine deamination reaction catalyzed by ADA.
ADA regulates both intra- and extracellular adenosine concentrations. By regulating
extracellular adenosine levels, it modulates signal transduction through adenosine receptors,
e.g. the A2a receptor, and is considered an important factor in the control of inflammation.
ADA is ubiquitious in almost all human tissues, and anomalous levels of ADA have been
detected in a range of diseases such as AIDS (acquired immunodeficiency syndrome),
anemia, lymphomas, and leukemias.48 The inhibition of ADA could also be useful in the
therapy of viral infections and of some types of lymphoproliferative disorders.49 Furthermore,
ADA inhibitors can modulate the immune response in B-or T-cell malignancies,50 as
adenosine plays an essential role in the differentiation and maturation of the immune system.
The use of extracellular ADA inhibitors may offer cardiovascular protection in hypertension.51

Introduction
15
ADA inhibitors are also needed in order to prevent the deamination, which would lead to their
inactivation, of antileukemic and antiviral agents containing adenine bases.52
The most potent ADA inhibitors known are the natural product coformycin (25a) and its 2’-
deoxy analogue pentostatin (25b) (Fig. 13), with Ki values of 1 x 10-11 M and 2.5 x 10-12 M,
respectively.53 Their high activity is assumed to be due to nearly irreversible binding of these
compounds to ADA, by mimicking the transition state for the deamination reaction
(Scheme 1).
Figure 13. Structures of coformycin and pentostatin.
Pentostatin is a FDA-approved drug, for the treatment of hairy cell leukemia.54
Coformycin, as a highly potent in vitro inhibitor, was clinically tested but exhibited
unacceptable toxicities.55
Mammalian adenosine deaminase preferentially interacts with its natural substrate
adenosine in a C3’-endo sugar conformation and, likewise, inhibitors of ADA have been
found to bind to the enzyme in a C3’-endo conformation exclusively.56 Interestingly, ADA
from Plasmodium falciparum, the parasite causing malaria, appears to interact with
adenosine-type compounds adopting a C2’-endo sugar pucker.57 For the parasite, ADA is
essential since it provides the fundamental building blocks for its sole purine salvage
pathway.58 Unlike the human host’s ADA, however, Plasmodium ADA also acts as an
essential enzyme in the recycling of methylthiopurines from the polyamine biosynthetic
pathway.59 In this role, Plasmodium ADA catalyzes the deamination of 5’-
methylthioadenosine to 5’-methylthioinosine (Scheme 2), although less efficiently than it
catalyzes the deamination of adenosine. Human and other mammalian ADAs do not accept
5’-methylthioadenosine as a substrate.60
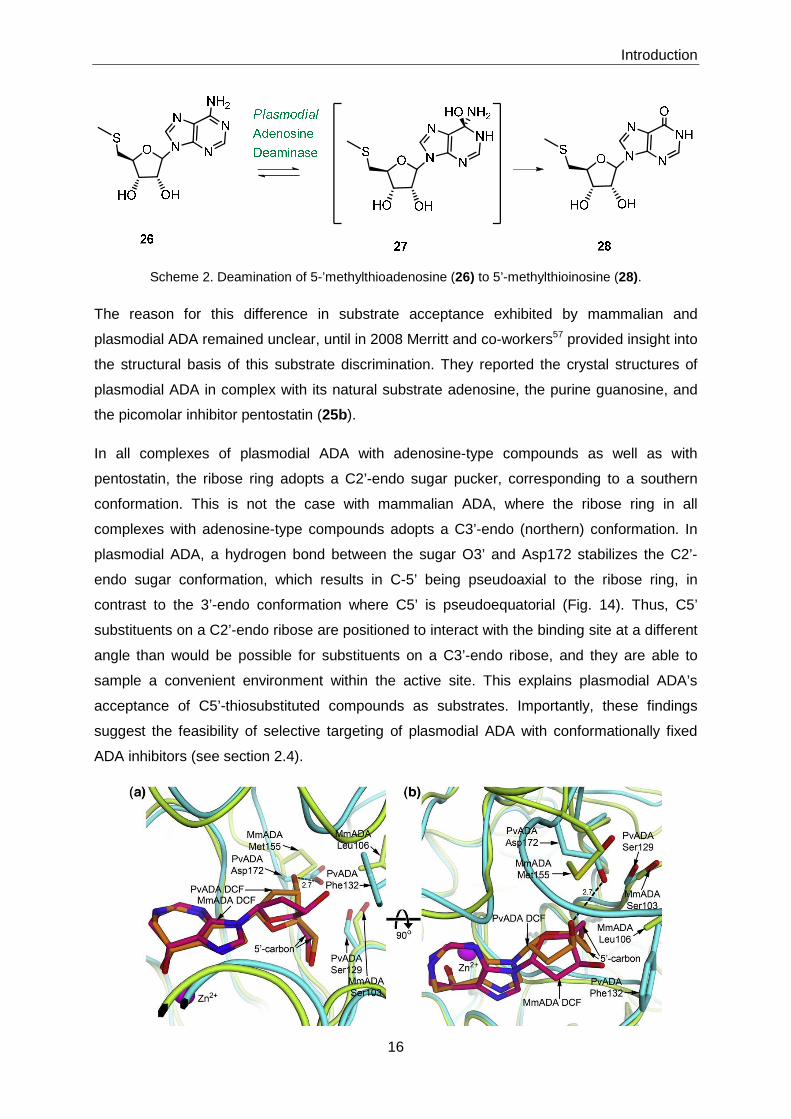
Introduction
16
Scheme 2. Deamination of 5-’methylthioadenosine (26) to 5’-methylthioinosine (28).
The reason for this difference in substrate acceptance exhibited by mammalian and
plasmodial ADA remained unclear, until in 2008 Merritt and co-workers57 provided insight into
the structural basis of this substrate discrimination. They reported the crystal structures of
plasmodial ADA in complex with its natural substrate adenosine, the purine guanosine, and
the picomolar inhibitor pentostatin (25b).
In all complexes of plasmodial ADA with adenosine-type compounds as well as with
pentostatin, the ribose ring adopts a C2’-endo sugar pucker, corresponding to a southern
conformation. This is not the case with mammalian ADA, where the ribose ring in all
complexes with adenosine-type compounds adopts a C3’-endo (northern) conformation. In
plasmodial ADA, a hydrogen bond between the sugar O3’ and Asp172 stabilizes the C2’-
endo sugar conformation, which results in C-5’ being pseudoaxial to the ribose ring, in
contrast to the 3’-endo conformation where C5’ is pseudoequatorial (Fig. 14). Thus, C5’
substituents on a C2’-endo ribose are positioned to interact with the binding site at a different
angle than would be possible for substituents on a C3’-endo ribose, and they are able to
sample a convenient environment within the active site. This explains plasmodial ADA’s
acceptance of C5’-thiosubstituted compounds as substrates. Importantly, these findings
suggest the feasibility of selective targeting of plasmodial ADA with conformationally fixed
ADA inhibitors (see section 2.4).

Introduction
17
Figure 14. (a and b) Alternate sugar pucker of substrate/inhibitor induced by the plasmodial ADA Asp172: mammalian ADA Met155 sequence difference. Plasmodial ADA is cyan and its bound pentostatin in orange, while mammalian ADA is green and its bound pentostatin in pink. Plasmodial ADA Asp172 hydrogen-bonds with the ribose 3′-hydroxyl group, an interaction that mammalian Met155 is incapable of making. This causes the plasmodial ADA-bound inhibitor to adopt a C2′-endo sugar pucker, while the mammalian ADA-bound inhibitor adopts a C3′-endo pucker. The result is that the 5′-carbon of the two riboses are oriented significantly differently with respect to the ribose ring, although the 5′-hydroxyl groups occupy nearly the same location and are less than 0.4 Å apart.57
1.4.2.4 Cytidine Deaminase
Cytidine deaminase (CDA) is a zinc-dependent enzyme that catalyzes the deamination of
cytidine to uridine, analogously to ADA deaminating adenosine to inosine61 (Scheme 1).
Therapeutically, inhibition of CDA is of specific interest to improve the activity of readily
deaminated antitumor nucleosides such as cytosine arabinoside or 2’-deoxycytidine.62
Marquez and co-workers63 synthesized the two bicyclo[3.1.0]hexane based analogues of the
known CDA inhibitor 29, with fixed northern (30, Fig. 15) and southern (31) conformations.
For comparative purposes they also investigated the flexible cyclopentane-based derivative
32 - to examine CDA’s preference for a certain sugar pucker. Since 29 inhibits CDA via a
mechanism independent from the zinc-promoted hydration mechanism for which the sugar
O4’ oxygen is critical, analogues of 29 were considered suitable compounds to study the
conformational preferences of CDA. Both the southern (31) and the northern (30) analogues
were found to be competitive inhibitors of CDA; the southern conformer 31 was found to be
2-fold more potent than the northern conformer. This finding confirmed the assumed
preference of CDA for a southern conformation, first derived from the result that all crystal
structures of inhibitors bound to CDA had shown the conformation of the sugar to be in the
southern hemisphere.64 This finding stood also in sharp contrast to the similar enzyme ADA,
which prefers a northern conformation of its substrates and is inhibited by nucleoside
analogues with a 3’-endo conformation (see section 1.5.3).

Introduction
18
Figure 15. Bicyclo[3.1.0]hexane based analogues of (29).
However, the flexible analogue 32 was also a competitive inhibitor of CDA. 32 was
approximately 100-fold more potent compared to the southern diazepinone 31, and thus also
more potent than northern conformer 30, with the original nucleoside 29 still being the most
active. A likely explanation for this smaller than expected potency of the bicyclo[3.1.0]-
hexane based southern diazepinone analogue is that its glycosyl torsion angle (χ = -149°) is
clearly in the syn region as X-ray structures have shown, leading to an unfavourable
orientation in space of the base moiety, although the pseudosugar conformation itself would
enhance affinity. Supporting this assumption is the finding that the crystal structure of CDA in
complex with parent riboside 29 shows that its glycosyl torsion angle is clearly in the anti
range.44b
1.4.2.5 DNA Methyltransferases
DNA methyltransferases (DNMTs) are enzymes transferring methyl groups to DNA, using
S-adenosyl-methionine (AdoMet, 33, Fig. 16) as the methyl donor molecule. AdoMet is
converted to S-adenosylhomocysteine (SAH, 34) during the reaction.
Changes in the pattern of DNA methylation are associated with cancer, as increased or
decreased methylation has been identified in all types of cancer cells so far.65 Generally,
genomic tumor DNA is characterized by definite methylation changes that are also termed
epimutations. As such epimutations appear rarely in healthy tissue, the epigenetic approach
to cancer therapy may have high tumor specificity.66

Introduction
19
Figure 16. Structures of AdoMet (33) and SAH (34).
With structural modification of S-adenosylhomocysteine, the product inhibitor of the
methylation reaction, Lim and co-workers recently achieved selective inhibition of Dengue
virus methyltransferase.67 The development of selective inhibitors of viral methyltransferases
in general could also be of high therapeutic interest.
DNA (cytosine C5)-methyltransferase as one important representative of the DNMT enzyme
family catalyses the methylation of carbon-5 of cytosine in the DNA. The enzyme links
covalently to C-6 of cytosine, along with a protonation at N-3, thus forming an activated
enamine intermediate; C-5 is rendered nucleophilic and attacks the methyl sulfonium center
of AdoMet. Evidence for this mechanism stems from X-ray crystallographic studies of DNA
(cytosine C5)-methyltransferase linked to methylated 5-fluorocytosine in DNA. The cytosine
residue targeted for methylation was “flipped” out of the helix during the transfer reaction.68
The removal of the target cytosine base, i.e. introduction of an abasic site, enhanced binding
of DNA (cytosine C5)-methyltransferase to DNA and the conformation of the sugar-
phosphate backbone at the abasic site in the resultant complexes was the same as that of
the sugar attached to a “flipped” cytosine. Conformationally constrained sugar analogues
based on bicyclo[3.1.0]hexane templates were placed in DNA duplexes as abasic target sites
in the DNA (cytosine C5)-methyltransferase recognition sequence. Biochemical studies
demonstrated that the binding affinity of the enzyme for abasic sites increases when the
abasic target sugar is constrained to the southern conformation and decreases when it is
constrained to the northern conformation.69
1.4.3 Receptors Discriminating between Sugar Pucker : Adenosine and
P2Y-Receptors
1.4.3.1 Adenosine Receptors
The nucleoside adenosine plays an important role in many aspects of cellular metabolism. It
influences different mammalian organ systems, specifically the cardiac, nervous, and

Introduction
20
immune system.70 Biochemical and pharmacological studies have validated that most of
adenosine’s physiological actions are mediated via cell surface receptors, of which there are
four subtypes: A1, A2a, A2b, and A3-receptors (AR’s). They are G-protein-coupled receptors of
which the A1 adenosine receptors are the best studied within the AR family; agonists and
antagonists for this receptor have therapeutic potential as anti-arrhythmic, neuroprotective,
and analgesic agents.71
A3 AR agonists and antagonists are proposed for the treatment of cancer and inflammatory
diseases.72
Jacobson and co-workers examined the conformational preferences of the A1, A2A, and A3
receptors using the conformationally restricted adenosine analogues 35 and 36 (Fig. 17) and
some N6-modified derivatives.72, 73 All three receptors displayed a higher binding affinity for
northern adenosine analogues (with an affinity ratio northern to southern > 150 for the A3-
receptor).
N
N
N
N
NH2
HO
HO OH
N
N
N
N
NH2
HO
HO OH
35 36
Figure 17. Northern- (35) and southern (36) adenosine analogues.
By activating either the A1 or A3 adenosine receptors in cardiac myocytes in several species
a cardioprotective effect mimicking the one induced by ischemic preconditioning could be
achieved.74 In contrast to a single activation of either A1 or A3, the coactivation of A1 and A3
adenosine receptors seems to be cardioprotective to a higher degree than activation of either
subtype alone. In agreement with their earlier work, Jacobson and co-workers deduced a
preference of A1 and A3 receptors for a northern sugar pucker72, 75 by synthesizing and
testing conformationally restricted (N)-methanocarba 2N-disubstituted adenine nucleoside
analogues as dual acting A1 and A3 adenosine receptor agonists (Fig. 18).

Introduction
21
Figure 18. Dual acting A1 and A3-receptor agonists
The derivatives 37-41 are all dually selective agonists for A1 and A3 receptors with nearly
balanced affinities, (Ki values in the range of 4 nM – 0.1 µM).
For the A2B receptor,76 there possibly exists a preference for one particular sugar pucker as
well, but this preference has not been elucidated.
1.4.3.2 P2Y Receptors
P2Y receptors are G-protein coupled receptors of which eight mammalian subtypes
(P2Y1,2,4,6,11-14) are currently known. For the activation of the P2Y1, P2Y11-13 subtypes adenine
nucleotides are required; while uracil nucleotides activate P2Y2, P2Y4, and P2Y6 subtypes.
P2Y receptor ligands are exploited for therapeutic applications in the cardiovascular and
endocrine systems and in relation to Alzheimer’s disease, and new ligands are also needed
as pharmacological tools for more basic research regarding these receptors, since detailed
structure-activity relationship (SAR) studies have only been established for P2Y1 and P2Y12
receptors.77
For the P2Y1 receptor, a preference of the receptor for the northern conformation of the
interacting ribose ring could be identified by testing northern bicyclo[3.1.0]hexane based
analogues78 43 and 44 of the known antagonist N-methyl-2’-deoxyadenosine 3’,5’-
bisphosphate (Fig. 19, relevant functional groups shown in green).
Figure 19. P2Y1 agonist 42 (relevant functional groups shown in red) and antagonists 43, 44 (green).

Introduction
22
The presence of the bicyclo[3.1.0]hexane moiety in these nucleotide analogues both
enhanced receptor affinity and improved stability towards nucleotidases. The same
conformational constraint of the ribose moiety that enhances antagonist action also improves
the potency and selectivity of P21 nucleotide agonists. The northern bicyclo[3.1.0]hexane
based compound 42 proved to be the much more potent and selective agonist of the P2Y1
receptor (Fig. 19, functional groups shown in red) than the parent nucleotide 2-methylthio
adenosine diphosphate.79 A comparison of adenosine triphosphate analogues with a
constrained northern or southern conformation had hinted at this preference of the P2Y1
receptor for a northern sugar pucker earlier: the southern analogue was less active than the
northern one, and the same was found for the P2Y2 receptor.80
A similar preference can be assumed for the P2Y14 receptor; here a bicyclo[3.1.0]hexane-
based analogue with a constrained southern conformation of uridine diphosphate glucose, a
nucleoside agonist of P2Y14, was tested and found to have no activity at all, which leads to
the conclusion that this receptor subtype also prefers a northern conformation of the sugar
moiety.81 Using conformationally locked nucleotides, preferences for the northern
hemisphere of the pseudorotational cycle could also be deduced for the P2Y4, and P2Y11
receptors.82 The P2Y6 receptor, however, prefers a southern conformation which was
predicted by molecular modelling and confirmed by synthesis of a rigid southern
bicyclo[3.1.0]hexane-based analog of uridine diphosphate (UDP, Fig. 20).
Figure 20. Selective P2Y6 agonist with constrained southern conformation.
Southern UDP analogue 45 proved to be more potent than UDP itself, whereas a uridine 5’-
diphosphate analogue locked in the northern conformation was devoid of any activity,
supporting the unique preference of the P2Y6 receptor for a southern conformation.83
1.5 Synthesis of Bicyclo[3.1.0]hexane Systems
1.5.1 Natural Products
Liu & Nan have recently described the synthesis of the bicyclo[3.1.0]hexane–containing core
framework of lindenane-type sesquiterpenoids, such as, e.g., chlorajaponilide and
Introduction
23
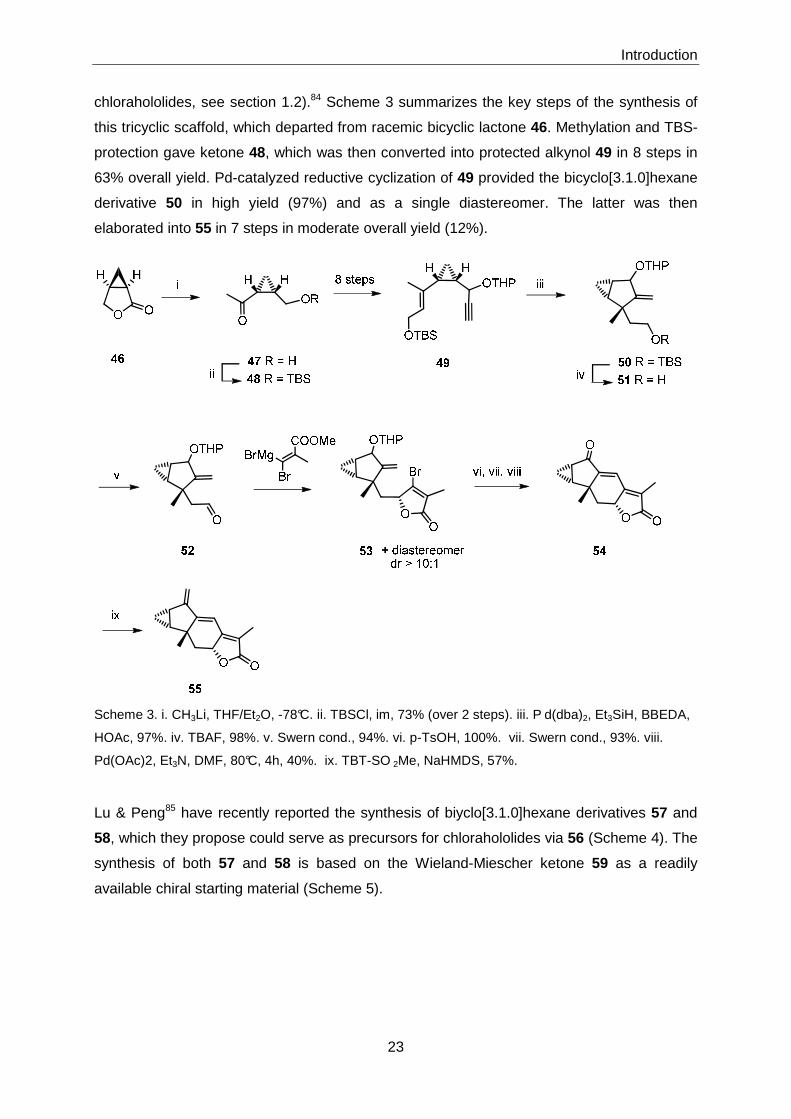
chlorahololides, see section 1.2).84 Scheme 3 summarizes the key steps of the synthesis of
this tricyclic scaffold, which departed from racemic bicyclic lactone 46. Methylation and TBS-
protection gave ketone 48, which was then converted into protected alkynol 49 in 8 steps in
63% overall yield. Pd-catalyzed reductive cyclization of 49 provided the bicyclo[3.1.0]hexane
derivative 50 in high yield (97%) and as a single diastereomer. The latter was then
elaborated into 55 in 7 steps in moderate overall yield (12%).
Scheme 3. i. CH3Li, THF/Et2O, -78°C. ii. TBSCl, im, 73% (over 2 steps). iii. P d(dba)2, Et3SiH, BBEDA,
HOAc, 97%. iv. TBAF, 98%. v. Swern cond., 94%. vi. p-TsOH, 100%. vii. Swern cond., 93%. viii.
Pd(OAc)2, Et3N, DMF, 80°C, 4h, 40%. ix. TBT-SO 2Me, NaHMDS, 57%.
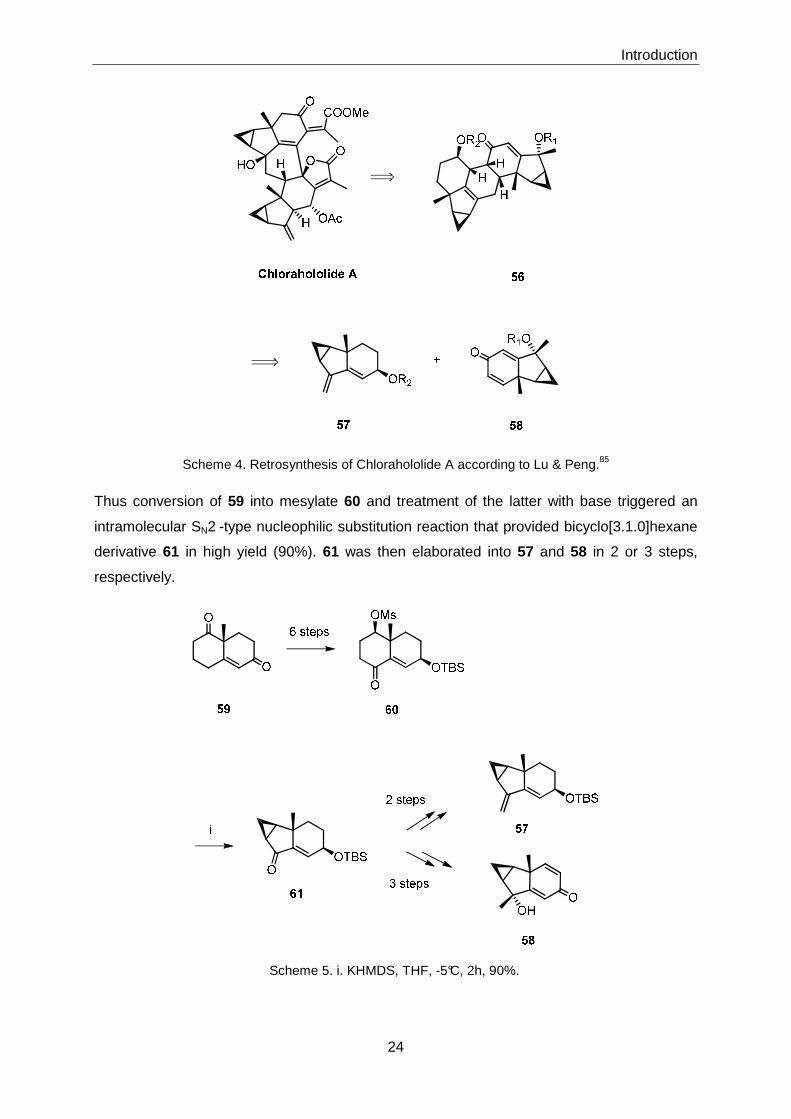
Lu & Peng85 have recently reported the synthesis of biyclo[3.1.0]hexane derivatives 57 and
58, which they propose could serve as precursors for chlorahololides via 56 (Scheme 4). The
synthesis of both 57 and 58 is based on the Wieland-Miescher ketone 59 as a readily
available chiral starting material (Scheme 5).
Introduction
24
Scheme 4. Retrosynthesis of Chlorahololide A according to Lu & Peng.85
Thus conversion of 59 into mesylate 60 and treatment of the latter with base triggered an
intramolecular SN2 -type nucleophilic substitution reaction that provided bicyclo[3.1.0]hexane
derivative 61 in high yield (90%). 61 was then elaborated into 57 and 58 in 2 or 3 steps,
respectively.
Scheme 5. i. KHMDS, THF, -5°C, 2h, 90%.

Introduction
25
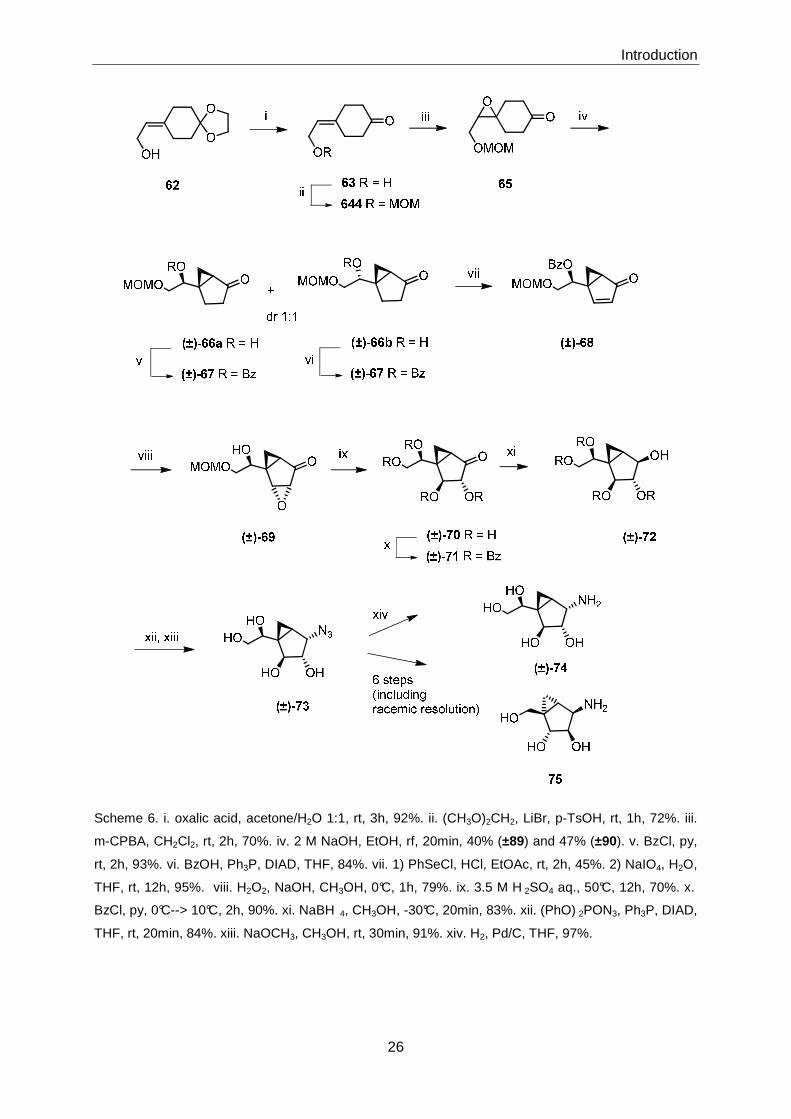
1.5.2 Sugar Analogues
In 2008, Li and co-workers86 synthesized (±)-74, a conformationally restricted amino
analogue of an α-(D)-galactofuranose ring. Using (±)-74 as a precursor, β-(D)-
arabinofuranose ring analogue 75 could also be prepared (Scheme 6). Both the analogues
were intended to be used as mimetics for UDP-α-(D)-galactofuranose, and decaprenyl β-(D)-
arabinofuranosylphosphate, respectively, the latter both playing important roles in the
synthesis of the cell wall of Mycobacterium tuberculosis and other mycobacterial
species.87
The synthesis departed from allylic alcohol 62 which can be accessed from cyclohexane-1,4-
dione in 3 steps.88 After acetal hydrolysis and MOM-protection epoxidation of 64 led to 65 as
the substrate for a ring contraction reaction that would install the bicyclo[3.1.0]hexane moiety
that was induced by treatment with base. This transformation gave a ~1:1 mixture of (±)-66a
and (±)-66b , which were separated by chromatography; both of these isomers could be
converted by different methods to the corresponding benzoates (±)-67. (±)-66b was
converted to (±)-67 by a Mitsunobu reaction. Subsequent α-selenation under acidic
conditions, oxidation of the selenide to the selenoxide and in situ elimination gave enone (±)-
68, which was then elaborated into azide (±)-73 by standard transformations in 6 steps. From
azide (±)-73, target structure 75 was received in 6 steps, including a racemic resolution, in a
high overall yield of 62%. (±)-74 was obtained from (±)-73 by reduction of the azide moiety.
Introduction
26
Scheme 6. i. oxalic acid, acetone/H2O 1:1, rt, 3h, 92%. ii. (CH3O)2CH2, LiBr, p-TsOH, rt, 1h, 72%. iii.
m-CPBA, CH2Cl2, rt, 2h, 70%. iv. 2 M NaOH, EtOH, rf, 20min, 40% (±89) and 47% (±90). v. BzCl, py,
rt, 2h, 93%. vi. BzOH, Ph3P, DIAD, THF, 84%. vii. 1) PhSeCl, HCl, EtOAc, rt, 2h, 45%. 2) NaIO4, H2O,
THF, rt, 12h, 95%. viii. H2O2, NaOH, CH3OH, 0°C, 1h, 79%. ix. 3.5 M H 2SO4 aq., 50°C, 12h, 70%. x.
BzCl, py, 0°C--> 10°C, 2h, 90%. xi. NaBH 4, CH3OH, -30°C, 20min, 83%. xii. (PhO) 2PON3, Ph3P, DIAD,
THF, rt, 20min, 84%. xiii. NaOCH3, CH3OH, rt, 30min, 91%. xiv. H2, Pd/C, THF, 97%.

Introduction
27

Introduction
28
1.5.3 Bicyclo[3.1.0]hexane-based Nucleoside Analogu es
In general, there are two main approaches to the synthesis of carbocyclic nucleoside
analogues, the first one employing carbohydrates as starting materials to afford optically pure
compounds, the second one would be a total synthesis approach starting either from racemic
or achiral compounds or from readily available chiral starting materials other than
carbohydrates. The first possibility bears the advantage of enantiomerically pure starting
materials which also possess the necessary polyoxygenated framework, but the
interconversion of one configuration to another is often not accomplishable without the
excessive use of protecting groups.
In the synthesis of carbocyclic nucleoside analogues, there is the additional question of how
the nucleobase is introduced best. The task can be accomplished by a convergent or a linear
strategy, as exemplified in Scheme 7 for the synthesis of N-type bicyclo[3.1.0]hexane-based
nucleoside analogs from a cyclopentene precursor. In the first approach, the complete
nucleobase is coupled directly to an activated carbocycle by a Mitsunobu reaction or by
displacement of a leaving group. A variety of carbocyclic nucleoside analogues can be
prepared in this way relatively fast, as the bases are introduced as ready moieties. The
disadvantage of this approach is the undesired formation of regioisomers, as the
nucleobases possess several nucleophilic sites, and the separation of the resulting mixtures
of isomers can be a tedious task.89
NH2
RO
RO
R1O
OH
RO
RO
NHR'
Convergent approach
(Mitsunobu or SN2 rct)
Linear approach
HO
RHO RHO
OH
HO
RHO OH or H
BASE
Scheme 7. Synthesis of northern bicyclo[3.1.0]hexane nucleoside analogues via a convergent vs.
linear strategy.
In the linear approach, a prefunctionalized carbocyclic amine has to be synthesized first, and
the heterocyclic base is then built up in a step-wise fashion by modification of the carbocyclic
amine. Employing this strategy, there are no regioisomers formed, however, each nucleoside
analogue has to be prepared by a separate multistep synthesis.
A convergent approach is only applicable for northern nucleoside analogues.

Introduction
29
1.5.3.1 Northern (N)-Type Analogues
Bicyclo[3.1.0]hexane-based nucleoside analogues with a fixed northern conformation are
more readily accessible and usually require shorter routes than the southern conformer, as
will be seen in the subsequent examples.
The first synthesis of a bicyclo[3.1.0]hexane based nucleoside analogue with northern
conformation was reported in 1994 by Altmann and co-workers.44a They synthesized
bicyclo[3.1.0]hexane-based thymidine analogue 82 that was incorporated into
oligonucleotides as a conformationally constrained sugar analogue (Scheme 7). Their
synthesis started with optically active cyclopentenone 76 which was accessible from D-ribose
in 7 steps.90
82
HO
N
HO
NH
O
O
OH
BnOO
O O
BnO
O O
OHBnO
O O
76 77
i ii iii, iv, v1'2'3'
4'5'
81
O
N
O
NBOM
O
O
Si
SiO
HO
HO OH
NNH
O
O
NH2BnO
O O
vi, vii, viii
ix, x, xi, xii xiii, xiv
80
78 79
Scheme 7. i. NaBH4, CeCl3, MeOH, 0°C, 10min, 97%. ii. Et 2Zn, CH2I2, CH2Cl2, 0°C to rt, 15h, 90%.
iii. TsCl, Et3N, CH2Cl2, DMAP, rt, 48h, 77%. iv. NaN3, DMF, 70°C, 18h, 88%. v. H 2, Lindlar’s cat., 4h,
100%. vi. MeOCH=C(Me)CONCO, CH2Cl2, -78°C-->rt, 30min, 95%. vii. 0.2 N HCl, EtOH/H 2O 9:1, rf,
20h, 80%. viii. H2, Pd/C, EtOAc/MeOH 1:1 (84% ee). ix. TIPSCl2, im, DMF, 67%. x. BOMCl, DBU,
CH3CN, rt, 1h, 85%. xi. CH3C6H4OC(S)Cl, DMAP, Et3N, CH2Cl2, rt, 3h, 40°C, 18h, 90%. xii. 1) Bu 3SnH,
AIBN, DME, 80°C, 3h. 2) prep. HPLC on Chiralgel OD, 250 x 4.8 mm, hexane/isopropanol 95:5, 85%
(100% ee). xiii. TBAF, THF, rt, 4h, 99%. xiv.1) H2, Pd/C, rt, 2h. 2) NaOMe, rt, 20h, 88%.
Subsequently, an additional stereocentre at C1 was installed via selective reduction to allylic
alcohol 77 by means of sodium borohydride and cerium(III)chloride, which afforded 77 in an
optical purity of approximately 50%.
Simmons-Smith cyclopropanation of 77 using Et2Zn and CH2I2 gave bicyclo[3.1.0]hexane
alcohol 78 as a single diastereosiomer, due to the directing effect of the allylic hydroxyl

Introduction
30
group.91 78 was converted to amine 79 via tosylation, azide formation and subsequent
reduction of the azido group. The base moiety was built up in a linear fashion by reacting 79
with (E)-3-methoxy-2-methylacryloyl isocyanate, followed by acid-catalyzed cyclization of the
resulting acryloyl urea with concomitant cleavage of the acetonide, and removal of the benzyl
group to afford 80. Simultaneous re-protection of the C5- and C3- hydroxyl groups in a cyclic
fashion with a TIPS-protecting group and BOM-protection of the thymine imido group allowed
for the removal of the C2-hydroxy group via radical deoxygenation to yield 81. Upon reaction
with tolyl chlorothioformate, the C2-hydroxy group underwent conversion to the
thiocarbonate, and radical reduction of the thiocarbonate with Bu3SnH in the presence of
AIBN furnished 81 as a partially racemic compound (84% ee). Complete racemic resolution
was achieved by preparative chiral HPLC, followed by removal of the TIPS- as well as the
BOM-protecting group with TBAF and catalytic hydrogenation over Pd/C, respectively, to
yield thymidine analogue 82.
Marquez and co-workers developed a synthesis of northern adenosine analogue 88 in 1995
(Scheme 8).92 They installed the bicyclo[3.1.0]hexane moiety in the same way as Altmann
and co-workers, starting from 76 with a stereoselective reduction and a Simmons-Smith
cyclopropanation to access 78 which was converted to the mesylate 83 quantitatively.
Following a convergent approach, condensation of 83 with adenine in the presence of K2CO3
and 18-crown-6 ether gave the desired N-9 product 84 as the major component (68%)
together with a small amount of the N-7 regioisomer (8%). Deprotection of the 5’-O-benzyl
ether using palladium black and formic acid, followed by the removal of the isopropylidene
group with 80% acetic acid, gave target compound 86. The primary alcohol function in 85
was oxidized to the aldehyde 87 using tetrapropylammonium perruthenate (TPAP), and
deprotection of the acetonide function afforded a second target compound 88.
Introduction
31
Scheme 8. i. MsCl, py, 0°C, 4h, 100%. ii. Adenine, K2CO3, 18-crown-6, DMF, 100°C, 18h, 68%. iii. Pd
black, HCO2H, MeOH, rt, 24h, 75%. iv. 80% AcOH, 80°C, 5h, 80%. v. TPAP, CH3CN, CH2Cl2, rt, 30h,
60%. vi. 88% HCO2H, rt, 5h ---> aq. 29% NH4OH, rt, 10min, 90%.
Starting with the same chiral cyclopentenone 76, the 2’-deoxy analogue of 86 was
synthesized by Siddiqui et al. in 1996 in a 7-step sequence with an overall yield of 25%.93 As
outlined in Scheme 9, acetonide 76 was converted into tert-butyl ether 89 by treatment with
AlMe3. The extra hydroxyl group was removed by a Barton deoxygenation in a 2-step
protocol to give 92, followed by deprotection of the TBS-group and subsequent modified
Simmons-Smith cyclopropanation to give 94.
Introduction
32
Scheme 9. i. AlMe3, CH2Cl2, -78°C -->rt, 18h. ii. TBSCl, im, DMF. iii. CS 2, NaH, MeI, THF, 1h, 82%. iv.
n-Bu3SnH, AIBN, tol, 120°C, 1.5h, 77%. v. TBAF, THF, rt, 12h, 92%. vi. Sm, HgCl2, ClCH2I, THF, -
78°C --> rt, 12h, 96%.
The construction of the base was also elaborated in a convergent approach, by a Mitsunobu
reaction of compound 94 with 6-chloropurine. Nucleophilic aromatic substitution of the
chloride atom and global deprotection with BCl3 concluded the synthesis of 2’-deoxy-
adenosine analogue 97 (Scheme 10).
Scheme 10. i. Ph3P, DEAD, 6-chloropurine, THF, rt, 3d, 58%. ii. NH4OH conc., dioxane (sealed tube),
65°C, 14h, 76%. iii. BCl 3, CH2Cl2, -78°C, 4h, 70%.

Introduction
33
In 1997, Marquez and co-workers developed a method that circumvented the 2-step
deoxygenation protocol to remove the extra hydroxyl group and that should allow the
synthesis of both conformers, southern and northern, proceeding through the same central
intermediate, azido-selenoxide NS-I (retrosynthesis depicted in Scheme 11).94
Scheme 11. Retrosynthesis of northern and southern nucleoside analogues according to Marquez et
al.
Theoretically, syn-elimination of NS-I could proceed in two directions, giving either the allylic
azide or the vinyl azide (abstraction of Ha or Hb, respectively). Experimentally, however, the
allylic azide predominated overwhelmingly over the vinyl azide, despite the inductive
polarization associated with the azido group, which might have been expected to facilitate
removal of Hb. Therefore, this strategy turned out to give access to northern conformers only,
via nucleoside analogue amine precursor 105 (Scheme 12).
The synthesis started with cyclopentadiene, which was stereoselectively converted to 98 via
deprotonation with NaH, subsequent quenching with benzyl chloromethyl ether and then
reaction with diisopinocampheylborane as the asymmetric hydroborating reagent. Oxidative
work-up gave cyclopentenol 98 in an enantiomeric excess of 94%-97%, however with
relatively low yield.95
Introduction
34
Scheme 12. i. NaH, BOMCl, THF, -60°C, 3h. ii. (-)-( ipc)2BH, -60°C --> rt, 15 h, 45%. iii. Me 3CPh2SiCl,
im, DMF, rt, 14h, 76%. iv. PhSeCl, NaN3, DMSO, rt, 12h, 87%. v. NaIO4, MeOH/H2O 9:1, rt, 36h, 76%.
vi. Ph3P, THF (wet), rf, 4h, 90%. vii. 1) C6H6(CO)2O, py, 90°C, 2h. 2) Ac 2O, 90°C, 2h, 77%. viii.
Et3N·HF, CH3CN, rf, 12h, 90%. ix. Et2Zn/CH2I2, CH2Cl2, -20°C, 10h, 96%. x. H 2NNH2, MeOH, 50°C,
3.5h, 100%.
TBDPS-protection of 98 was followed by azido-phenylselenylation, which proceeded with
complete stereochemical control. This stereochemical outcome was possibly due to the
preferred pseudoequatorial orientation of the two substituents in the cyclopentene ring, which
led to formation of the episelenonium intermediate from the bottom face of the ring to give a
stable chair-like transition state intermediate. In situ oxidation of the phenylselenide group
triggered the anticipated syn-elimination, giving almost exclusively the allylic azide 101. An
explanation for this finding is lacking, but there is some literature precedence for the
predominance of the allylic azide under similar oxidative conditions.96 Azide 101 was reduced
under Staudinger conditions and the resulting amine was protected with a phthalimido group.
Full amine protection proved necessary since an acyl-NH group, e.g., took precedence over
the hydroxyl group in directing delivery of the methylene group in the Simmons-Smith
reaction. After deprotection of the hydroxyl group the succeeding Simmons-Smith
cyclopropanation gave exclusively 105 in 96% yield. Hydrazinolysis of the phthalimido group
afforded bicyclo[3.1.0]hexane amine 106.

Introduction
35
A similar strategy also shortened access to northern conformer building block 112 allowing
for convergent approaches by starting from the same cyclopentenol synthon 98 that
Marquez’ group had used earlier for the synthesis of southern nucleoside analogues.
Scheme 13. i. NaH, BnBr, TBAI, THF, rt, 5h, 95%. ii. PhSeCl, AgCF3CO2, DMSO, rt, 1h. iii. 5%
KOH/EtOH, 0°C, 30min, 74%. iv. NaIO 4, MeOH/H2O 9:1, rt, 1h, 73%. v. Ph3P, DEAD, PhCOOH, rt,
30min, 85%. vi. K2CO3, MeOH, rt, 3h, 80%. vii. Et2Zn/CH2I2, CH2Cl2, -20°C--> rt, 6h, 80%.
In the same manner as with the PhSeCl/NaN3 system, the double bond in 107 reacted regio-
and stereoselectively with PhSeCl/AgCF3CO2 to give the trans-2-(phenylseleno)-
cyclopentanol 108 after hydrolysis of the trifluoroacetate ester. Oxidation and in situ
selenoxide-pyrolysis gave also exclusively allylic alcohol 109. This allylic alcohol was
inverted via a Mitsunobu esterification, providing the new alcohol 111 upon hydrolysis.
In 2002, Choi and co-workers presented alternative approaches to northern nucleoside
analogues, for both the 2’-, 3’-hydroxy derivatives as well as the 2’-deoxy compounds.97 Key
steps/methods included intramolecular cyclopropanation via a carbenoid intermediate to
generate the bicyclo[3.1.0]hexane scaffold and lipase-catalyzed racemic resolution of the
corresponding 2’-deoxy scaffold. The enantiospecific synthesis of 120, as outlined in Scheme
14, started with chiral precursor D-isoascorbic acid, from which 113 was obtained in 4 steps
and 60% overall yield.98 After a stepwise oxidation of the hydroxyl group, a Claisen
condensation of the activated acid with 2-lithio ethyl acetate afforded 114 with 14% of
undesired epimerization. Diazo transfer was accomplished with tosyl azide and triethylamine
to give compound 115, which underwent metal-catalyzed thermolysis to 116 in 44% yield (+
unwanted diastereomer in 19% yield) via a carbenoid intermediate. DIBALH-reduction of the
carbonyl moieties gave the corresponding diol, which was benzylated. Cleavage of the
acetonide afforded 117 and treatment with thionyl chloride then resulted in the formation of

Introduction
36
118. Subsequent reaction with sodium azide led to 82% of 119 and 10% of the alternative
regioisomer. Palladium-catalyzed hydrogenation of the azide gave amine 120.
Scheme 14. i. 1) COCl2, Et3N, DMSO, 30min. 2) NaClO4, 67%. ii. 1) Im2CO 2) LiCH2CO2Et, 67%. iii.
TsN3, TEA, 83%. iv. CuI, tol, rf, 63%. v. DIBALH, 55%. vi. BnBr, NaH, 99%. vii. H+, 74%. viii. SOCl2,
99%. ix. NaN3, 82%. x. H2, Pd/C, 81%.
An alternative approach to an aldehyde corresponding to ester 114 has been reported by
Jacobson and co-workers.99 The latter group has also employed L-ribose as a starting
material for northern-type bicyclo[3.1.0]hexane-based nucleoside analogs.
In this approach (Scheme 15) L-ribose is converted to iodides 121 (diastereomeric mixture
5:1); subsequent reductive cleavage of the mixture with Zn in refluxing methanol100
generated aldehyde 122 which was C-2 elongated in situ to 123. Next, the same key steps
as with Choi et al. were employed to install the bicyclo[3.1.0]hexane moiety (diazotization
and intramolecular cyclopropanation over a carbenoid intermediate) to give 125, along with
the diastereomer in a ratio of 4:1 in favor of the desired diastereoisomer. 125 was isolated
chromatographically, reduced stereoselectively with NaBH4 and equilibrated with TFA to the
isomeric acetonide 127, to which different bases can be attached via a convergent approach.
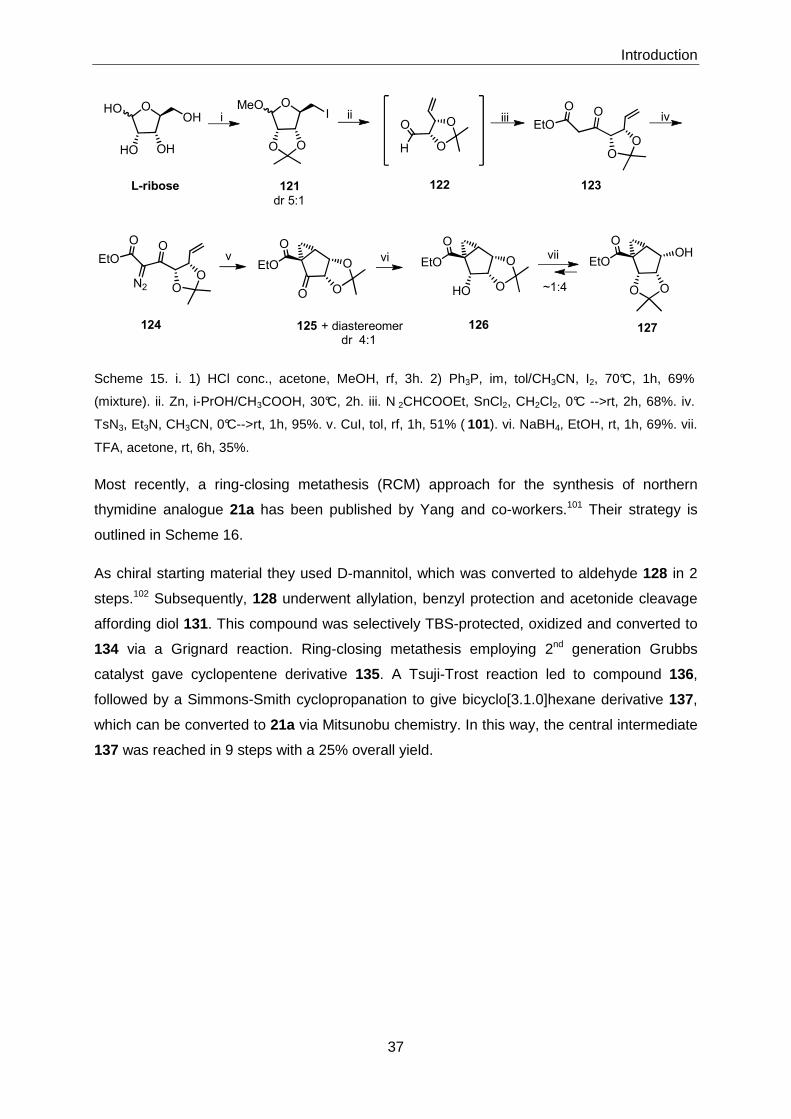
Introduction
37
O
HO OH
L-ribose
iOMeO
O O
121
iiOHHO I iii
OO
OEtO
O
OO
OEtO
O
123
124
N2
OEtO
O O
O
+ diastereomerdr 4:1
iv
v OEtO
HO O
OOH
EtO
O O
O
vi vii
125 126 127
122
dr 5:1
~1:4
O
O
O
H
Scheme 15. i. 1) HCl conc., acetone, MeOH, rf, 3h. 2) Ph3P, im, tol/CH3CN, I2, 70°C, 1h, 69%
(mixture). ii. Zn, i-PrOH/CH3COOH, 30°C, 2h. iii. N 2CHCOOEt, SnCl2, CH2Cl2, 0°C -->rt, 2h, 68%. iv.
TsN3, Et3N, CH3CN, 0°C-->rt, 1h, 95%. v. CuI, tol, rf, 1h, 51% ( 101). vi. NaBH4, EtOH, rt, 1h, 69%. vii.
TFA, acetone, rt, 6h, 35%.
Most recently, a ring-closing metathesis (RCM) approach for the synthesis of northern
thymidine analogue 21a has been published by Yang and co-workers.101 Their strategy is
outlined in Scheme 16.
As chiral starting material they used D-mannitol, which was converted to aldehyde 128 in 2
steps.102 Subsequently, 128 underwent allylation, benzyl protection and acetonide cleavage
affording diol 131. This compound was selectively TBS-protected, oxidized and converted to
134 via a Grignard reaction. Ring-closing metathesis employing 2nd generation Grubbs
catalyst gave cyclopentene derivative 135. A Tsuji-Trost reaction led to compound 136,
followed by a Simmons-Smith cyclopropanation to give bicyclo[3.1.0]hexane derivative 137,
which can be converted to 21a via Mitsunobu chemistry. In this way, the central intermediate
137 was reached in 9 steps with a 25% overall yield.
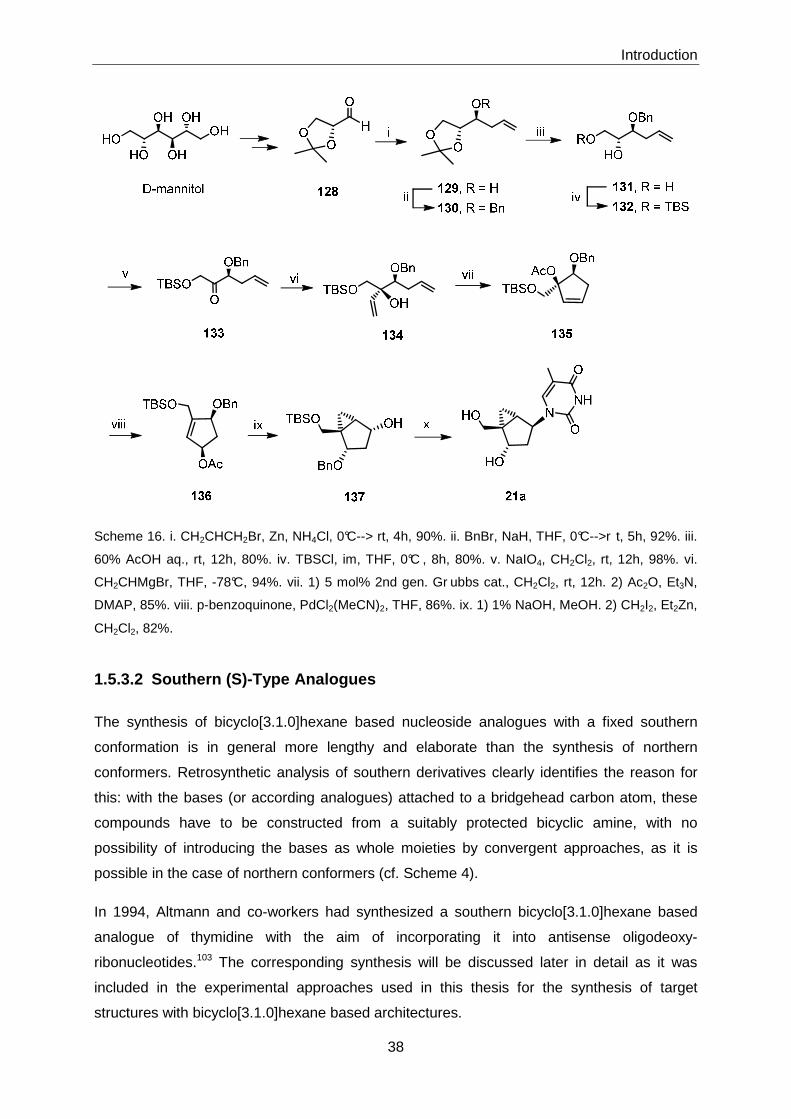
Introduction
38
Scheme 16. i. CH2CHCH2Br, Zn, NH4Cl, 0°C--> rt, 4h, 90%. ii. BnBr, NaH, THF, 0°C-->r t, 5h, 92%. iii.
60% AcOH aq., rt, 12h, 80%. iv. TBSCl, im, THF, 0°C , 8h, 80%. v. NaIO4, CH2Cl2, rt, 12h, 98%. vi.
CH2CHMgBr, THF, -78°C, 94%. vii. 1) 5 mol% 2nd gen. Gr ubbs cat., CH2Cl2, rt, 12h. 2) Ac2O, Et3N,
DMAP, 85%. viii. p-benzoquinone, PdCl2(MeCN)2, THF, 86%. ix. 1) 1% NaOH, MeOH. 2) CH2I2, Et2Zn,
CH2Cl2, 82%.
1.5.3.2 Southern (S)-Type Analogues
The synthesis of bicyclo[3.1.0]hexane based nucleoside analogues with a fixed southern
conformation is in general more lengthy and elaborate than the synthesis of northern
conformers. Retrosynthetic analysis of southern derivatives clearly identifies the reason for
this: with the bases (or according analogues) attached to a bridgehead carbon atom, these
compounds have to be constructed from a suitably protected bicyclic amine, with no
possibility of introducing the bases as whole moieties by convergent approaches, as it is
possible in the case of northern conformers (cf. Scheme 4).
In 1994, Altmann and co-workers had synthesized a southern bicyclo[3.1.0]hexane based
analogue of thymidine with the aim of incorporating it into antisense oligodeoxy-
ribonucleotides.103 The corresponding synthesis will be discussed later in detail as it was
included in the experimental approaches used in this thesis for the synthesis of target
structures with bicyclo[3.1.0]hexane based architectures.

Introduction
39
In 1996, Marquez and co-workers presented an approach to the synthesis of 2’-deoxy-
bicyclo[3.1.0]hexane based southern conformers bearing all common bases.104 The
synthesis was centered around the stable carbamate derivative 145, from which all the
heterocyclic bases could be constructed (Scheme 17). Starting point for the synthesis was
cyclopentene 98, which had been used before as chiral starting material in the synthesis of
northern-type conformers.94 Hydroxyl-directed stereoselective epoxidation and protection of
the free alcohol group95a gave compound 139.
Scheme 17. i. t-BuOOH, Va(acac)2, 1,2 dichloroethane, -30°C-->rt, 2h. ii. NaH, BnBr , TBAI, THF, rt,
12h, 81%. iii. KCN, LiClO4, CH3CN, rt, 3d, 77%. iv. Im2CS, DMAP, DMF, rt, 3h-->∆, 70°C, 30min, 98%.
v. CH2N2, Et2O, 0°C, 3d, 96%. vi. h ν, COPh2, 2h, 79%. vii. 25% aq. NaOH, MeOH, rf, 24h, 99%. viii.
Et3N, DPPA, tol, rt, 2h --> 80°C, 2h --> Me 3Si(CH2)2OH, 80°C, 2h --> rt, 12h, 64%. ix. TBAF,
CH3CN/THF 4:1, 70°C, 4h, 99%.
Nucleophilic opening of the epoxide ring occurred with excellent regioselectivity to give the
cyano intermediate 140, from which the desired α,β-unsaturated nitrile 141 was obtained
following the syn-β-elimination of the intermediate thiocarbonylimidazolide. The 1,3-dipolar
cycloaddition of diazomethane with 141 to give the cis-fused pyrazoline intermediate 142
occurred with the expected regioselectivity that is typical of diazomethane additions to
electron deficient alkenes in which the carbon atom of diazomethane functions as the
negative end of the dipole. The stereofacial selectivity is controlled by the the approach of
the diazomethane from the less hindered side of the double bond. Nitrile 143 was obtained
after nitrogen extrusion from 142 by photolysis. After hydrolysis of 143 to the acid, a modified
Curtius rearrangement of the corresponding acyl azide to the isocyanate, which was trapped
with 2-trimethylsilylethanol, gave stable carbamate 145.

Introduction
40
From the central intermediate 145 the elaboration of pyrimidine or purine bases could be
carried out by reacting amine 146 with 3-methoxy-2-methylacryloyl isocyanate, 3-
ethoxyacrylyol isocyanate, 5-amino-4,6-dichloroformylpyrimidine or 2,5-diamino-4,6-
dichlorodiformylpyrimidine, respectively, followed by final benzyl deprotection procedures to
give compounds 151-155. (Scheme 18).
Removal of the benzyl ethers was accomplished either by catalytic transfer hydrogenation, or
by treatment with BCl3. In the case of the target purine derivatives 154 and 155, the
nucleophilic substitution of the chloride attached to pyrimidine would only occur if 5-
formylamino-4,6-dichloropyrimidine was used instead of the free 5-amino-4,6-
dichloropyrimidine, the same was the case for 2,5-diamino-4,6-dichlorodiformylpyrimidine as
reagent.
Scheme 18. Elaboration of the base part of S-type bicylco[3.1.0]hexane-based nucleoside analogues.
More recently, Marquez and co-workers described the synthesis of a 4’-deoxy-
bicyclo[3.1.0]hexane template (160) by an approach making use of an intramolecular variant
of the Kulinkovich reaction (Scheme 19).105 Thus, (R)-epichlorohydrin was subjected to a
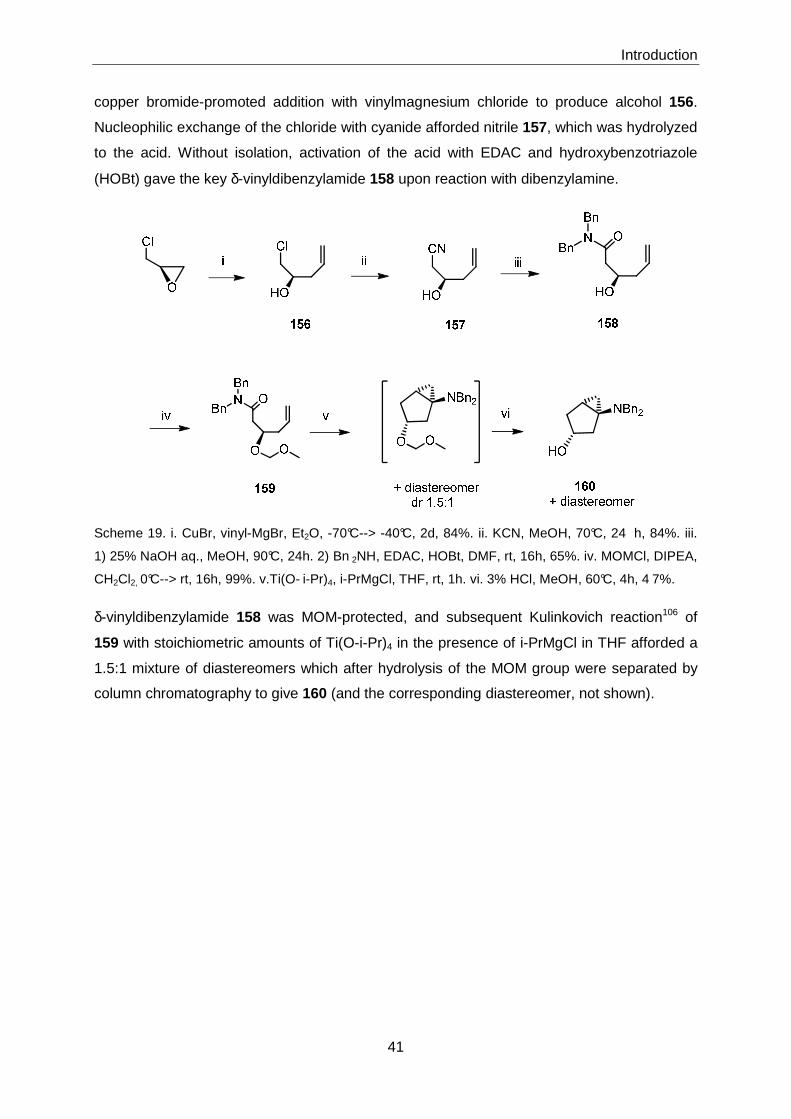
Introduction
41
copper bromide-promoted addition with vinylmagnesium chloride to produce alcohol 156.
Nucleophilic exchange of the chloride with cyanide afforded nitrile 157, which was hydrolyzed
to the acid. Without isolation, activation of the acid with EDAC and hydroxybenzotriazole
(HOBt) gave the key δ-vinyldibenzylamide 158 upon reaction with dibenzylamine.
Scheme 19. i. CuBr, vinyl-MgBr, Et2O, -70°C--> -40°C, 2d, 84%. ii. KCN, MeOH, 70°C, 24 h, 84%. iii.
1) 25% NaOH aq., MeOH, 90°C, 24h. 2) Bn 2NH, EDAC, HOBt, DMF, rt, 16h, 65%. iv. MOMCl, DIPEA,
CH2Cl2, 0°C--> rt, 16h, 99%. v.Ti(O- i-Pr)4, i-PrMgCl, THF, rt, 1h. vi. 3% HCl, MeOH, 60°C, 4h, 4 7%.
δ-vinyldibenzylamide 158 was MOM-protected, and subsequent Kulinkovich reaction106 of
159 with stoichiometric amounts of Ti(O-i-Pr)4 in the presence of i-PrMgCl in THF afforded a
1.5:1 mixture of diastereomers which after hydrolysis of the MOM group were separated by
column chromatography to give 160 (and the corresponding diastereomer, not shown).

Introduction
42
1.6 Conclusions
Exploiting the unique conformational characteristics of the bicyclo[3.1.0]hexane scaffold, the
conformational preferences of a number of nucleoside- and nucleotide-interacting proteins
could be elucidated. This includes viral RT enzymes, viral and host kinases, and also
GPCR’s such as the adenosine or P2Yx receptors. The bicyclo[3.1.0]hexane moiety’s special
feature, the preference for a boat-like conformation of the six-membered ring, leads to a
valuable degree of structural preorganization of substituents attached to this scaffold. This
feature might also be exploited for the presentation of pharmacophoric groups in a well-
defined arrangement.

Aims and Scope
43
2 Aims and Scope
In light of its special conformational properties, we wanted to make use of the
bicyclo[3.1.0]hexane scaffold in this Ph. D. project in two ways: First by the synthesis of a
small screening library of bicyclo[3.1.0]hexane based small molecules that would contain
various pharmacophoric groups in pre-defined orientations. The compounds should then be
submitted to biological screening in a variety of assays. As pointed out above, the
bicyclo[3.1.0]hexane scaffold has not been widely explored in drug discovery outside the
area of modified nucleosides and any hits from these screens would represent new types of
entry points into inhibitor (antagonist, agonist) development. Secondly, the effects of the
replacement of the ribose or 2’-deoxyribose moiety in S-adenosylhomocysteine and
pentostatin, respectively, on biological activity were to be investigated.
2.1 Chemical Library Synthesis
For the first objective of this research project we envisaged the construction of a chemical
library of 150-200 compounds based on a bicyclo[3.1.0]hexane scaffold with general
structures 1 and 2 (Fig. 21).
Figure 21. Target structures for the chemical library.
As discussed in the introduction, the bicyclo[3.1.0]hexane system exhibits a strong
preference for a boat-like conformation of the six-membered ring, thus leading to a significant
degree of structural preorganization of substituents attached to this scaffold. This feature
should be exploited by the attachment of a range of chemical entities as possible
pharmacophores with defined exit vectors from the core structure. These compounds should
then be made available for biological screening. The various substituent groups present in 1
and 2 were chosen according to their prevalence of occurrence in known drugs and such as
to ensure adequate physico-chemical properties.107 The heterocycles directly attached to the
bridgehead carbon atom in 2 were also selected based on their frequent occurrence in
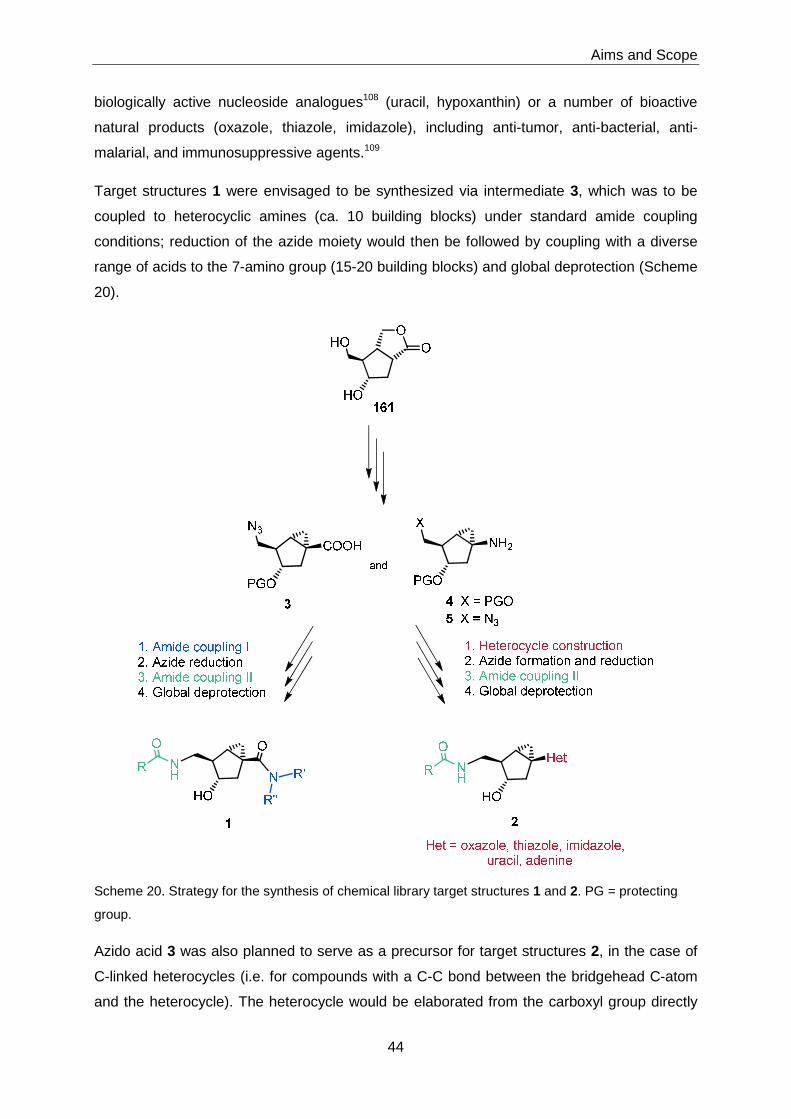
Aims and Scope
44
biologically active nucleoside analogues108 (uracil, hypoxanthin) or a number of bioactive
natural products (oxazole, thiazole, imidazole), including anti-tumor, anti-bacterial, anti-
malarial, and immunosuppressive agents.109
Target structures 1 were envisaged to be synthesized via intermediate 3, which was to be
coupled to heterocyclic amines (ca. 10 building blocks) under standard amide coupling
conditions; reduction of the azide moiety would then be followed by coupling with a diverse
range of acids to the 7-amino group (15-20 building blocks) and global deprotection (Scheme
20).
Scheme 20. Strategy for the synthesis of chemical library target structures 1 and 2. PG = protecting
group.
Azido acid 3 was also planned to serve as a precursor for target structures 2, in the case of
C-linked heterocycles (i.e. for compounds with a C-C bond between the bridgehead C-atom
and the heterocycle). The heterocycle would be elaborated from the carboxyl group directly
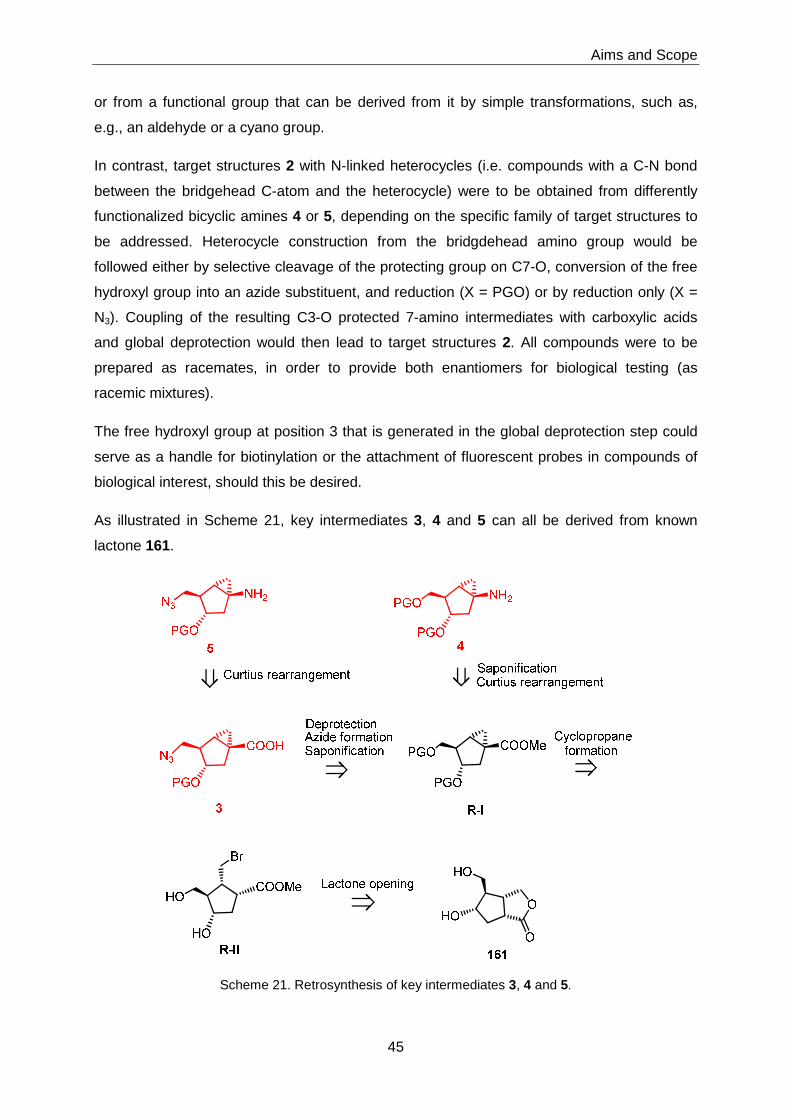
Aims and Scope
45
or from a functional group that can be derived from it by simple transformations, such as,
e.g., an aldehyde or a cyano group.
In contrast, target structures 2 with N-linked heterocycles (i.e. compounds with a C-N bond
between the bridgehead C-atom and the heterocycle) were to be obtained from differently
functionalized bicyclic amines 4 or 5, depending on the specific family of target structures to
be addressed. Heterocycle construction from the bridgdehead amino group would be
followed either by selective cleavage of the protecting group on C7-O, conversion of the free
hydroxyl group into an azide substituent, and reduction (X = PGO) or by reduction only (X =
N3). Coupling of the resulting C3-O protected 7-amino intermediates with carboxylic acids
and global deprotection would then lead to target structures 2. All compounds were to be
prepared as racemates, in order to provide both enantiomers for biological testing (as
racemic mixtures).
The free hydroxyl group at position 3 that is generated in the global deprotection step could
serve as a handle for biotinylation or the attachment of fluorescent probes in compounds of
biological interest, should this be desired.
As illustrated in Scheme 21, key intermediates 3, 4 and 5 can all be derived from known
lactone 161.
Scheme 21. Retrosynthesis of key intermediates 3, 4 and 5.

Aims and Scope
46
The synthesis diverges at the stage of ester R-I, which can be elaborated into 3 by selective
deprotection of the primary 7-hydroxyl group, conversion of the free hydroxyl group into an
azide moiety, and ester saponification. Intermediate 4 is obtained by ester saponification and
subsequent Curtius rearrangement; depending on the exact conditions for the Curtius
rearrangement, the reaction may lead to a protected amino group and would thus have to be
followed by a deprotection step.
5 can be obtained from 3 by way of a Curtius rearrangement.
Intermediate R-I with PG = TBS has been described previously in the context of the
synthesis of a southern bicyclo[3.1.0]hexane-based thymidine analogue.103 The synthesis of
R-I proceeds through bromo ester R-II; treatment of the latter with base leads to
cyclopropane ring formation. R-II in turn is obtained from lactone 161, which is ultimately
derived from cis-tetrahydrophthahlic anhydride.110
2.2 Synthesis of Bicyclo[3.1.0]hexane-based Analogu es of
Pentostatin and S-Adenosylhomocysteine
A second objective of this thesis project was the synthesis of bicyclo[3.1.0]hexane-based
analogues of pentostatin (6) and of S-adenosylhomocysteine (7) (Fig. 22).
Fig. 22. Target structures pentostatin analogue 6 and (SAH) analogue 7.
As outlined in the Introduction, pentostatin is a picomolar inhibitor of adenosine deaminase
(ADA), the enzyme catalyzing the deamination of adenosine and 2’-deoxyadenosine,
respectively, to the corresponding inosines. Plasmodial ADA additionally accepts 5’-
methylthioadenosine as a substrate and the structural basis for this discrimination remained
unclear until the crystal structures of plasmodial ADA in complex with its natural substrate
adenosine, but also with guanosine, and the picomolar inhibitor pentostatin were reported by
Merritt and co-workers in 2008.57 Their data showed that in all complexes, including the one
with pentostatin, the (deoxy-)ribose ring clearly adopts a C2’-endo conformation, while
mammalian adenosine deaminase preferentially interacts with C3’-endo puckered
nucleosides.57 Thus, a southern bicyclo[3.1.0]hexane-based pentostatin analogue, in
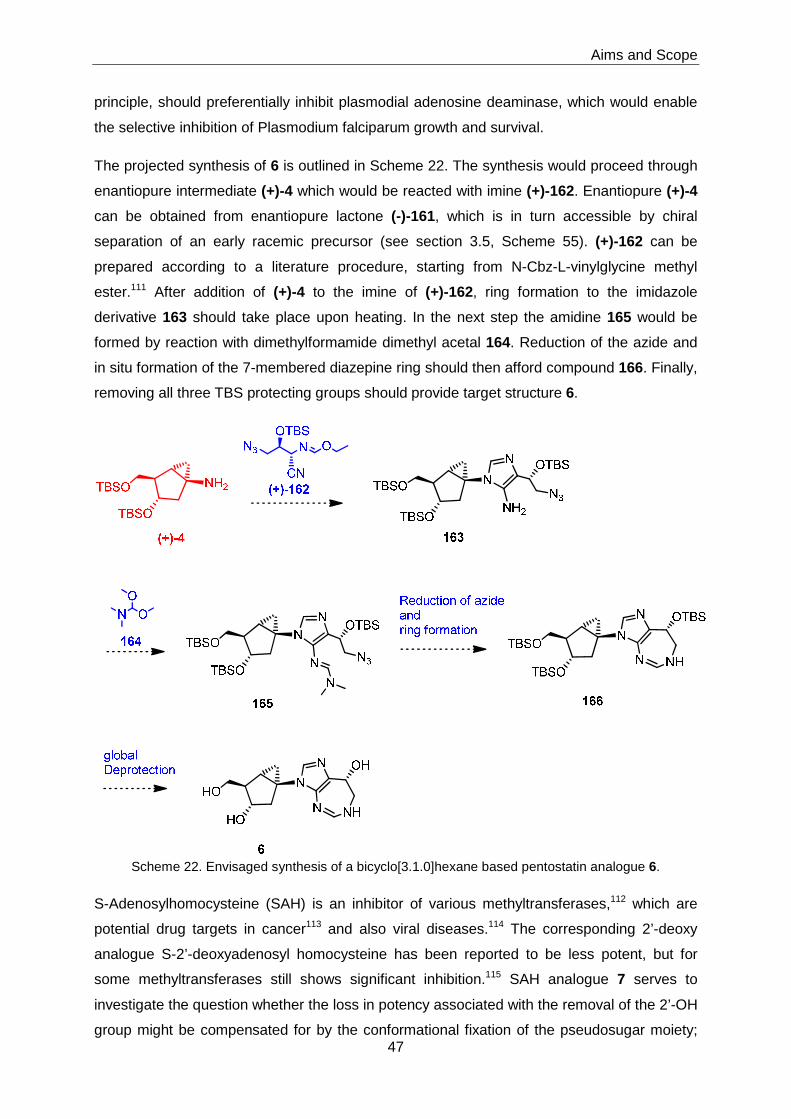
Aims and Scope
47
principle, should preferentially inhibit plasmodial adenosine deaminase, which would enable
the selective inhibition of Plasmodium falciparum growth and survival.
The projected synthesis of 6 is outlined in Scheme 22. The synthesis would proceed through
enantiopure intermediate (+)-4 which would be reacted with imine (+)-162. Enantiopure (+)-4
can be obtained from enantiopure lactone (-)-161, which is in turn accessible by chiral
separation of an early racemic precursor (see section 3.5, Scheme 55). (+)-162 can be
prepared according to a literature procedure, starting from N-Cbz-L-vinylglycine methyl
ester.111 After addition of (+)-4 to the imine of (+)-162, ring formation to the imidazole
derivative 163 should take place upon heating. In the next step the amidine 165 would be
formed by reaction with dimethylformamide dimethyl acetal 164. Reduction of the azide and
in situ formation of the 7-membered diazepine ring should then afford compound 166. Finally,
removing all three TBS protecting groups should provide target structure 6.
Scheme 22. Envisaged synthesis of a bicyclo[3.1.0]hexane based pentostatin analogue 6.
S-Adenosylhomocysteine (SAH) is an inhibitor of various methyltransferases,112 which are
potential drug targets in cancer113 and also viral diseases.114 The corresponding 2’-deoxy
analogue S-2’-deoxyadenosyl homocysteine has been reported to be less potent, but for
some methyltransferases still shows significant inhibition.115 SAH analogue 7 serves to
investigate the question whether the loss in potency associated with the removal of the 2’-OH
group might be compensated for by the conformational fixation of the pseudosugar moiety;
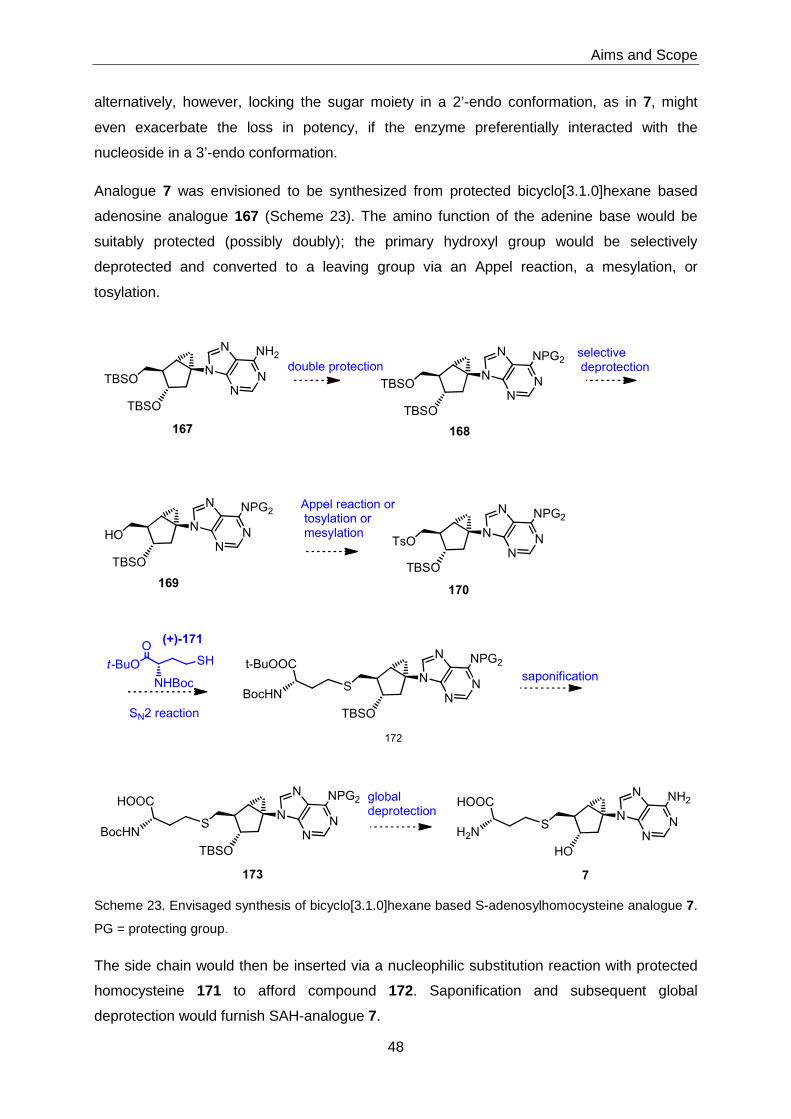
Aims and Scope
48
alternatively, however, locking the sugar moiety in a 2’-endo conformation, as in 7, might
even exacerbate the loss in potency, if the enzyme preferentially interacted with the
nucleoside in a 3’-endo conformation.
Analogue 7 was envisioned to be synthesized from protected bicyclo[3.1.0]hexane based
adenosine analogue 167 (Scheme 23). The amino function of the adenine base would be
suitably protected (possibly doubly); the primary hydroxyl group would be selectively
deprotected and converted to a leaving group via an Appel reaction, a mesylation, or
tosylation.
TBSO
TBSON
N
NN
NH2
TBSO
TBSON
N
NN
NPG2
TBSO
HON
N
NN
NPG2
TBSO
TsON
N
NN
NPG2
TBSO
SN
N
NN
NPG2
BocHN
t-BuOOC
167 168
169170
172
double protectionselectivedeprotection
Appel reaction ortosylation ormesylation
SN2 reaction
HO
SN
N
NN
NH2
H2N
HOOC
7
globaldeprotection
SH
NHBoc
O
t-BuO
(+)-171
TBSO
SN
N
NN
NPG2
BocHN
HOOC
173
saponification
Scheme 23. Envisaged synthesis of bicyclo[3.1.0]hexane based S-adenosylhomocysteine analogue 7.
PG = protecting group.
The side chain would then be inserted via a nucleophilic substitution reaction with protected
homocysteine 171 to afford compound 172. Saponification and subsequent global
deprotection would furnish SAH-analogue 7.

Aims and Scope
49
2.3 Alternative Synthesis of (+)-4
A possible alternative strategy to (+)-4 is outlined in Scheme 24 and is based on
cyclopentadiene as a possible starting material (cf. section 1.6). The stereoselective
conversion of cyclopentadiene into 98 via deprotonation with NaH, quenching of the
cyclopentadienyl anion with benzyl chloromethyl ether and in situ asymmetric hydroboration
with diisopinocamphenylborane had been described.116 Directed epoxidation of 98 then leads
to intermediate R-VI.95a, 95b An epoxide opening reaction with cyanide anion would result in
compound R-V, which would then be dehydrated to R-IV. A Simmons-Smith
cyclopropanation or other types of cyclopropanation reactions should then provide R-III
which would be converted into (+)-4 by nitrile hydrolysis and Curtius rearrangement (vide
supra).
Scheme 24. Retrosynthesis of (+)-4 starting from cyclopentadiene.

Aims and Scope
50
2.4 Biological Screening of the Bicyclo[3.1.0]hexan e-based
Chemical Library
The library of compounds 1 and 2 was intended to be screened for activity against discrete
kinases and proteases; in addition, whole cell screens were foreseen, including simple
cytotoxicity testing against cancer cells, screening against Mycobacterium tuberculosis, and
the assessment of activity in a series of pathway assays with specifically manipulated cancer
cell lines. This work would be carried out in collaboration. Additionally, the library would be
made available for testing by other interested research groups.

Results and Discussions
51
3 Results and Discussions
3.1 Chemical Library Synthesis
3.1.1 Synthesis of Advanced Intermediates 3, 4 and 5
As outlined in the section 2.1, the synthesis of advanced intermediates 3, 4 and 5 was to
proceed through lactone 161, for which a synthesis had been described in the literature
starting from cis-tetrahydrophthalic anhydride (174).117 The synthesis of 161 is summarized
in Scheme 25. Since 3, 4 and 5 were needed in gram quantities, the synthesis of 161 was
performed on the largest scale possible with conventional laboratory equipment; this involved
starting with approximately 100 g of 174 and the synthesis was carried out multiple times.
The opening of anhydride 174 with MeOH in the presence of catalytic amounts of p-toluene
sulphonic acid worked well to afford a racemic half ester 175 in quantitative yield. Selective
reduction of the acid moiety was possible through conversion to the acid chloride with oxalyl
chloride followed by treatment with NaBH4 to produce alcohol 176. As has been shown by
others, this reaction, when carefully controlled, proceeds without epimerization.117a Alcohol
176 was not isolated, but directly treated with acid to induce cyclization to lactone 177. The
latter was obtained in an excellent 93% yield from 175. Oxidative cleavage of the double
bond resulted in diacid 178, which could be isolated in 73% yield. Treatment of 178 with
MeOH/sulfuric acid not only produced the expected diester 179a, but also its rearranged
isomer 179b. Fortunately, both isomers 179a and 179b upon treatment with tert-BuOK
produced only a single Dieckmann cyclization product, namely enol 180. However, the
outcome of the Dieckmann cyclization depended to a great extent on a very careful work-up.
The formation of a brown residue at the bottom of the flask during quenching remained
undetected despite temperature monitoring in the first trials, as quenching resulted (upon
formation of KCl) in a thick suspension. The formation of this brown residue, probably the
result of local overheating of the mixture, could be avoided by intense mechanical stirring
and a very slow addition of the quenching acid. Thus, the desired product could be isolated
after work-up by recrystallization as colorless crystals in a yield of 72%. Assuming first the
water content of the used sulphuric acid to be problematic, we had switched to the use of dry
hydrochloric acid, but this had not given better results. The enolate double bond was next
reduced with sodium borohydride, which gave hydroxy ester 181 selectively (73%), with a
trans arrangement of the ester and the hydroxyl group. The selectivity of the reaction is the
result of the attack of the reducing agent from the exo side of the bicylic ring system in 180,
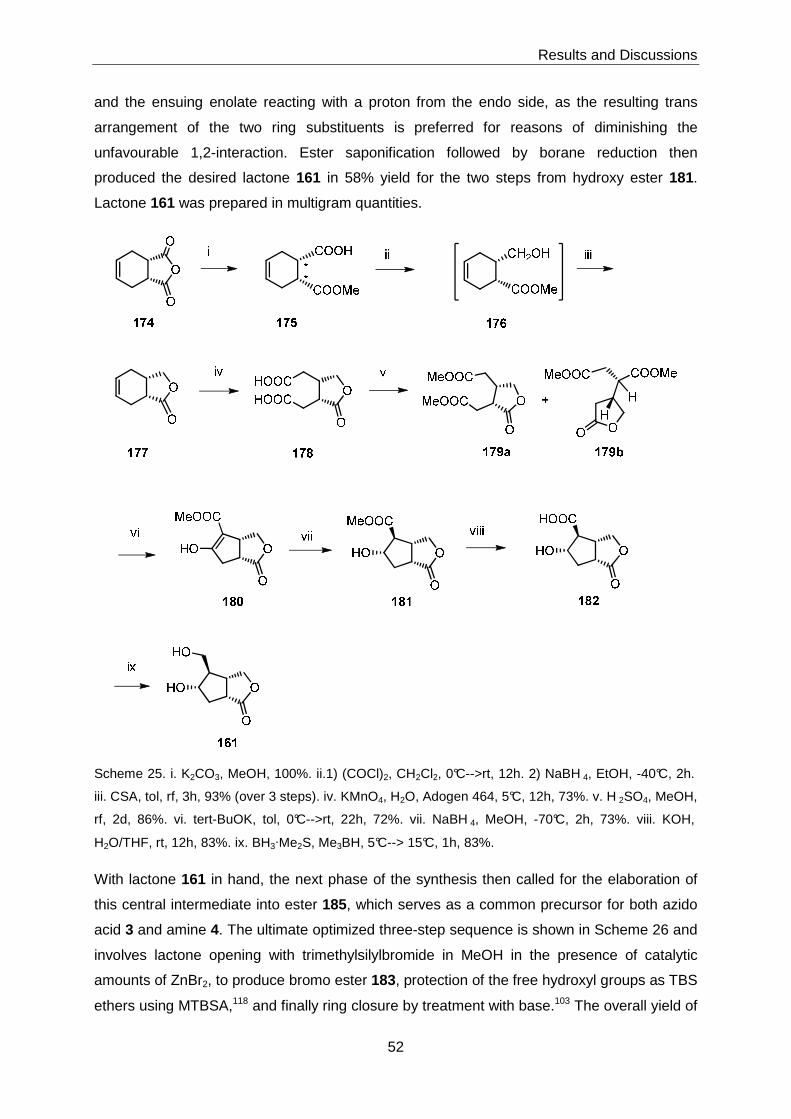
Results and Discussions
52
and the ensuing enolate reacting with a proton from the endo side, as the resulting trans
arrangement of the two ring substituents is preferred for reasons of diminishing the
unfavourable 1,2-interaction. Ester saponification followed by borane reduction then
produced the desired lactone 161 in 58% yield for the two steps from hydroxy ester 181.
Lactone 161 was prepared in multigram quantities.
Scheme 25. i. K2CO3, MeOH, 100%. ii.1) (COCl)2, CH2Cl2, 0°C-->rt, 12h. 2) NaBH 4, EtOH, -40°C, 2h.
iii. CSA, tol, rf, 3h, 93% (over 3 steps). iv. KMnO4, H2O, Adogen 464, 5°C, 12h, 73%. v. H 2SO4, MeOH,
rf, 2d, 86%. vi. tert-BuOK, tol, 0°C-->rt, 22h, 72%. vii. NaBH 4, MeOH, -70°C, 2h, 73%. viii. KOH,
H2O/THF, rt, 12h, 83%. ix. BH3·Me2S, Me3BH, 5°C--> 15°C, 1h, 83%.
With lactone 161 in hand, the next phase of the synthesis then called for the elaboration of
this central intermediate into ester 185, which serves as a common precursor for both azido
acid 3 and amine 4. The ultimate optimized three-step sequence is shown in Scheme 26 and
involves lactone opening with trimethylsilylbromide in MeOH in the presence of catalytic
amounts of ZnBr2, to produce bromo ester 183, protection of the free hydroxyl groups as TBS
ethers using MTBSA,118 and finally ring closure by treatment with base.103 The overall yield of

Results and Discussions
53
this sequence varied between 13% and 29%, with the most reliable step being the formation
of the cyclopropane ring. Quite remarkably, this step could be achieved in yields of up to
94%.
Bromide 184 proved to be unstable above 30°C and had the tende ncy to re-form the lactone
ring. For this reason, the best yields achieved for the lactone opening were around 50%.
Likewise, the simultaneous protection of the two hydroxyl functions gave moderate yields
only (between 33% and 58%).
Scheme 26. i. TMSBr, MeOH, ZnBr2 (cat.), rt, 25h, 53%. ii. MTBSA, CH2Cl2, rt, 4h, 58%. iii. tert-BuOK,
tert-BuOH, rt, 1h, 94%.
Different reagents were tested for the introduction of the tert-butyl dimethylsilyl protecting
groups and the best proved to be MTBSA (Table 1); MTBSA can be readily prepared from N-
methylacetamide and TBSCl.118
Table 1. Reagents and conditions of the TBS protection of 183 to 184.
Reagents/conditions yield
TBDMSCl, imidazole, DMF, 0°C, 12h 0%
TBDMSOTf, 2,6-lutidine, CH2Cl2, -78°C, 2h 33%
MTBSA, CH2Cl2, rt, 4h 33-58%
Other reagents tested included TBSOTf and TIPDSCl2, the latter leading to a TIPDS-
protection of the two hydroxyl groups in a cyclic fashion.119 TIPDSCl2 was reacted with diol
161 before the lactone opening step. Due to the low yield obtained 186 was not subjected to
the subsequent lactone opening with trimethylsilylbromide (Scheme 27).

Results and Discussions
54
Scheme 27. i. 1) AgNO3, py, THF, 1h. 2) TIPDSCl2, rt, 8h, 35%.
However, the TIPDS-protecting group would have required an exchange to a different set of
protecting groups at a later stage of the synthesis, as it does not allow selective deprotection
of the primary hydroxyl group, which was required in order to proceed with the synthesis in
the projected way, and also the yield of the TIPDS protection step was rather low (35%).
The tert-butyl dimethylsilyl group was thus our first choice for the protection of both hydroxyl
groups in 183, as it should survive the conditions required for subsequent transformations; in
addition, a primary TBS ether can be selectively cleaved over a secondary one under
relatively mild acidic conditions.120 Nevertheless, we also contemplated the use of the benzyl
ether protecting group, which can be cleaved reductively under neutral conditions121 and
would have been stable to the conditions needed for the remaining functionalizations. Most
importantly, we assumed that benzyl ethers would enhance the stability of the bromo ester
and (in contrast to the TBS ethers) would be stable under the acid conditions employed for
lactone opening. This led us to attempt protecting the two hydroxyl functionalities of 161 as
benzyl ethers before lactone opening with MeOH/TMSBr. Unfortunately, the reaction of 161
with sodium hydride, benzyl bromide and tetrabutylammonium iodide as a catalyst gave low
yields of the monobenzylated product at best. This is surprising as this transformation has
been reported in literature to proceed in 54% yield122, a result that could simply not be
reproduced here.
Scheme 28. i. TMSCl, MeOH, ZnCl2, rt, 24h, 62%. ii. MTBSA, CH2Cl2, rt, 16h, 62%. iii. tert-BuOK, tert-
BuOH, rt, 2h, 64%.
In an attempt to further improve the moderate yields in the steps from 161 to 185, we also
prepared the chloro derivative 187 by opening lactone 161 with TMSCl (Scheme 28).123 Due
to the higher stability of compound 187, it could be isolated in a somewhat higher yield of

Results and Discussions
55
62%, in comparison with 53% for bromide 183. Likewise, chloride 187 could also be TBS-
protected in higher yields (61-100%) than 183 (Scheme 35). However, the yield in the
subsequent cyclopropanation reaction was only 64%, compared to 94% in the case of the
bromide 184. These findings added up to approximately the same yields over the three
steps; as the reaction sequence from 161 to 189 via bromide 184, in spite of the lower
stability of the bromide was more reproducible, this approach was routinely used for the
preparation of 185.
The conversion of ester 185 into the key intermediate for the preparation of target structures
1, i.e. 3, proceeded uneventful. As illustrated in Scheme 29, this involved cleavage of the
primary TBS ether followed by conversion of the ensuing free hydroxyl group into a mesylate,
displacement of the mesyloxy group with azide anion, and finally ester cleavage with TMSOK
in THF at 80°C. 3 was thus obtained in 45% overall yield for the 4 steps from ester 185.
Scheme 29. i. CSA, CH2Cl2/MeOH 1:1, 0°C, 2.5h, 67%. ii. MsCl, Et 3N, Et2O, 0°C, 3h, 96%. iii. NaN 3,
DMF, 70°C, 20h, 76%. iv. TMSOK, THF, 80°C,))), 2h, 92%. [))) = microwave irradiation]
In principle, azido acid 3 could serve as the precursor for the preparation of intermediate 5
(with PG = TBS). However, for reasons that will be explained in section 3.1.3.1.1 subsequent
transformations mandated a C3-O-protecting group that would be stable under acidic
conditions, a condition which is not fulfilled for a TBS protecting group. 5 was thus prepared
with a benzyl protecting group on C3-O; to this end, ester 192 was converted into C3-O
benzyl-protected azido acid 194 by TBS-removal with TBAF, followed by treatment with
BnBr/NaH and cleavage of the methyl ester moiety with TMSOK in 75% overall yield for the
3-step sequence from ester 192 (Scheme 30). Acid 194 was then transformed into the
desired amine 5 by acyl azide formation and in situ Curtius rearrangement.124 The ensuing
isocyanate was quenched with aqueous sodium hydroxide, thus affording directly the primary
amine. This approach avoids the removal of an amine protecting group, which could have

Results and Discussions
56
been problematic in the presence of a benzyl group, but it has the disadvantage that the
amine 5 had to be used to the next step immediately after its preparation, due to its limited
stability.
Scheme 30. i.TBAF, THF, rt, 1h, 90%. ii.NaH, BnBr, DMF, -15°C-->rt, 1.5h, 92%. iii. TMSOK,
THF/DMF 4:1, ))), 90°C, 4h, 91%. iv. 1) DPPA, Et 3N, tol, 0°C-->rt-->80°C, 12h. 2) 2 N NaOH, THF,
30 min, 82%.
For the elaboration of 185 into our second key intermediate 4 the methyl ester moiety was
cleaved with KOH/EtOH to produce acid 195 in 90% yield. Acid 195 was then converted into
the corresponding acyl azide with DPPA; the azide was submitted to an in situ Curtius
rearrangement124a and the resulting isocyanate was directly reacted with benzyl alcohol to
give the Cbz-protected amine 196 in excellent yield (88% based on 195).
Scheme 31. i. KOH, EtOH, 100°C, ))), 2.5h, 90%. ii. 1) DPPA, Et3N, tol, 0°C--> 80°C, 2h. 2) BnOH, di-
n-butyltindilaurate (cat.), 80°C, 2.5h, 88%. iii. H 2 (1 bar), 10% Pd/C, tol/MeOH 1:1, rt, 4h, 88%.
The benzyloxycarbonyl (Cbz-) protecting group could be smoothly removed from 196 by
catalytic hydrogenation over Pd/C to furnish 4 in 88% yield. An attempt to convert the
isocyanate directly to primary amine 4 by quenching the reaction with water resulted in a
40% yield only. The route involving conversion of the isocyanate to the Cbz-protected amine
196 was thus preferable. It also allowed the amine to be stored in protected form; removal of
the Cbz protecting group in general was only performed immediately before use of the free
amine 4.
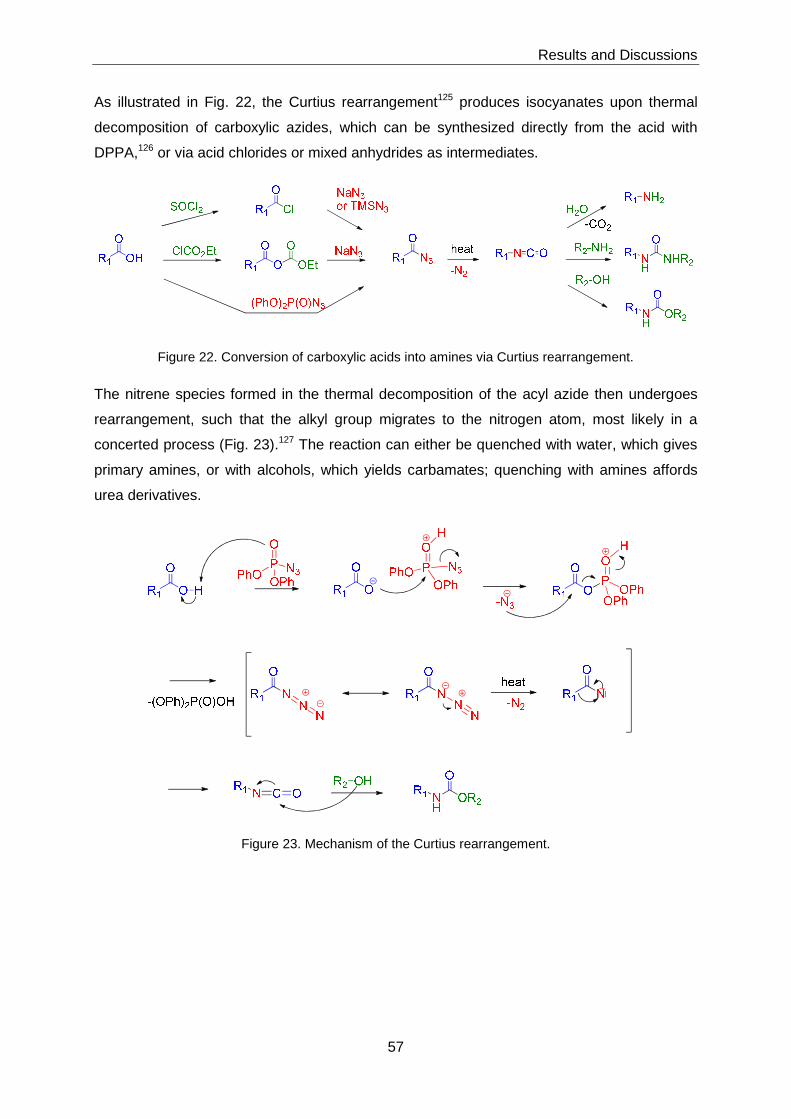
Results and Discussions
57
As illustrated in Fig. 22, the Curtius rearrangement125 produces isocyanates upon thermal
decomposition of carboxylic azides, which can be synthesized directly from the acid with
DPPA,126 or via acid chlorides or mixed anhydrides as intermediates.
Figure 22. Conversion of carboxylic acids into amines via Curtius rearrangement.
The nitrene species formed in the thermal decomposition of the acyl azide then undergoes
rearrangement, such that the alkyl group migrates to the nitrogen atom, most likely in a
concerted process (Fig. 23).127 The reaction can either be quenched with water, which gives
primary amines, or with alcohols, which yields carbamates; quenching with amines affords
urea derivatives.
Figure 23. Mechanism of the Curtius rearrangement.

Results and Discussions
58
3.1.2 Synthesis of Library Members with General Str ucture 1
3.1.2.1 Target Structures Incorporating Heterocycli c Amines
As illustrated in Scheme 32 the general strategy of the preparation of library members of
general structure 1 involved (i) coupling of acid 3 with the desired amine; (ii) reduction of the
azide moiety to a primary amino group; (iii) coupling of each individual amine with a set of
carboxylic acids; and (iv) cleavage of the TBS ether at position 3 of the bicyclo[3.1.0[hexane
skeleton.
Scheme 32. General strategy for the preparation of library members with general structure 1.
Scheme 33 summarizes the coupling of 3 with 5 different amines 197-201. Activation of 3
was generally carried out with HATU128 and couplings proceeded in yields of 80-100% to
provide amides 202a-e. No other coupling methods were investigated in light of the excellent
product yields obtained with HATU. Catalytic hydrogenation of 202a-e over 10% palladium
on carbon resulted in clean conversion of the azido moiety into a primary amino group to
furnish amines 203a-e in 83 to 100% yield. The reduction products were generally obtained
in purities >85% (based on NMR) and employed in the next coupling step without
chromatographic purification (in view of the good purity of the crude products and to avoid
loss of material upon flash chromatography of these very polar compounds).
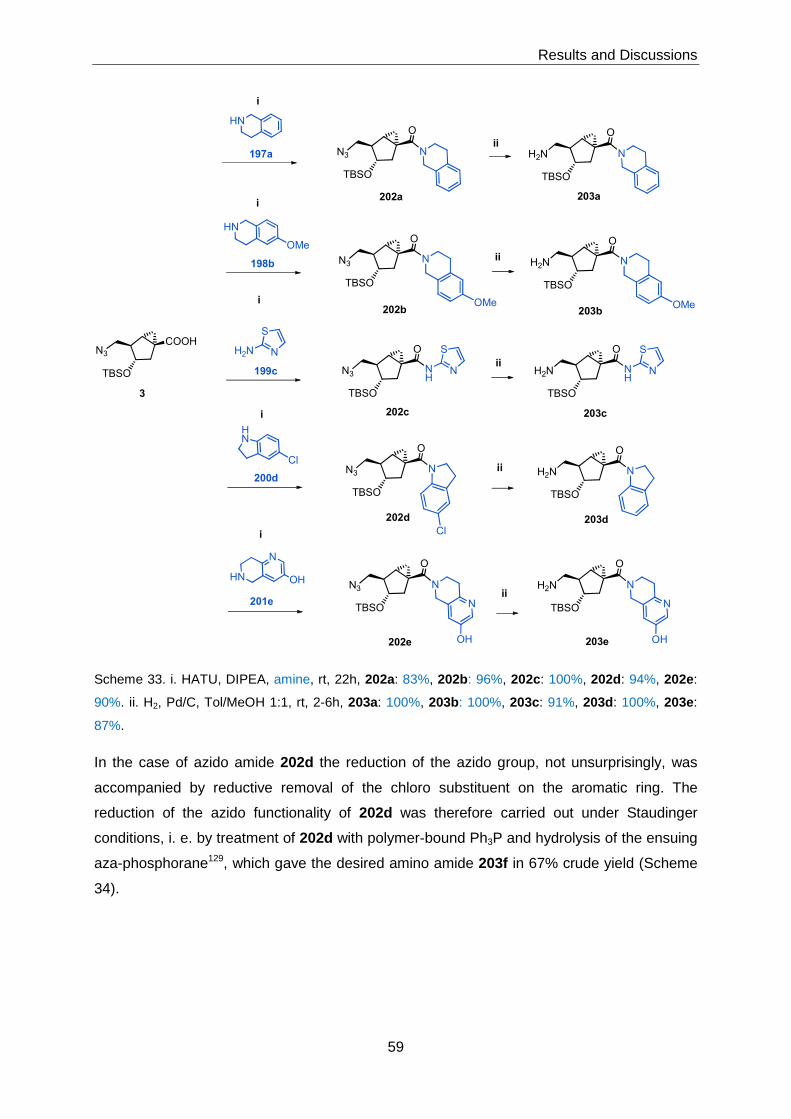
Results and Discussions
59
3
COOH
TBSO
N3
TBSO
N3
O
N
202d
TBSO
N3
O
N
N
OH
HN
N
OH
N
S
NH
TBSO
N3
O
TBSO
N3
O
N
N
OMe
TBSO
N3
O
Cl
HN
OMe
HN
Cl
HN
N
S
H2N
202a
202b
202c
202e
i
197a
198b
199c
200d
201e
TBSO
H2N
O
N
203d
TBSO
H2N
O
N
N
OH
N
S
NH
TBSO
H2N
O
TBSO
H2N
O
N
N
OMe
TBSO
H2N
O
203a
203b
203c
203e
ii
i
i
i
i
ii
ii
ii
ii
Scheme 33. i. HATU, DIPEA, amine, rt, 22h, 202a: 83%, 202b: 96%, 202c: 100%, 202d: 94%, 202e:
90%. ii. H2, Pd/C, Tol/MeOH 1:1, rt, 2-6h, 203a: 100%, 203b: 100%, 203c: 91%, 203d: 100%, 203e:
87%.
In the case of azido amide 202d the reduction of the azido group, not unsurprisingly, was
accompanied by reductive removal of the chloro substituent on the aromatic ring. The
reduction of the azido functionality of 202d was therefore carried out under Staudinger
conditions, i. e. by treatment of 202d with polymer-bound Ph3P and hydrolysis of the ensuing
aza-phosphorane129, which gave the desired amino amide 203f in 67% crude yield (Scheme
34).

Results and Discussions
60
Scheme 34. i. 1) PPh3 polymer-bound, dioxane, rt, 2h. 2) NH3 (32% in H2O), rt, 12h, 67%.
As shown in Scheme 33, the different amines (including the dehalogenated reduction product
203d), where then submitted to coupling with a set of up to 20 diverse carboxylic acids,
including 5 desamino-(natural) amino acids (Scheme 35).
Scheme 35. Structures of acids used in the coupling steps.
As for the coupling of acid 3 with amines 197-201, the different acids were generally
preactivated with HATU for 30 min; the coupling products were obtained in yields varying
between 45 and 100% after extractive work-up and in most cases the crude diamides were
directly submitted to deprotection. It was aimed at coupling all acids to all prepared amines;
however, coupling with acids 204h, 204l, 204m and 204s tended to give low yields by trend,
independent of the kind of amine they were coupled to. Coupling of these acids was then
avoided in the last coupling series, the one to amine 203e, as these derivatives already
contain an amino function that could possibly act as a hydrogen bond acceptor in the
heterocycle. Acid 204c also gave low yields in all the couplings, and acid 204i gave a low
yield in the coupling with 203b.

Results and Discussions
61
The derivatization of the 7-amino group and the subsequent deprotection step are
exemplified in Scheme 36 for amine 203d and 5 different carboxylic acids, spanning a range
of coupling yields.
Scheme 36. i. R-COOH, HATU, DIPEA, CH2Cl2, rt, 12h.ii. Dowex 50W X8, MeOH, 12h, 1db : 32%
(over 2 steps), 1dg : 55% (over 2 steps), 1dc : 26% (over 2 steps), 1de: 40% (over 2 steps), 1do : 35%
(over 2 steps).
Different conditions were investigated for the final TBS deprotection steps, including HF·py,
TBAF, and Dowex-50W-8X cation exchange resin. Deprotection was found to be most
efficient with Dowex 50W-X8,130 which gave the best yields of products 1d; in addition the
use of an insoluble reagent also proved to be advantageous technically, as it could be
removed by filtration and no other work-up was required.
All final products 1 were purified by flash column chromatography and characterized by high
resolution mass spectrometry; in addition, approximately one third of the library was fully
characterized by 1H and 13C NMR spectroscopy. The purity of all compounds was assessed
by RP-HPLC on a C18 column and for the majority was found to be ≥85%, based on
integration of peaks in the HPLC chromatogram obtained at 220 nm. Figure 24 shows the
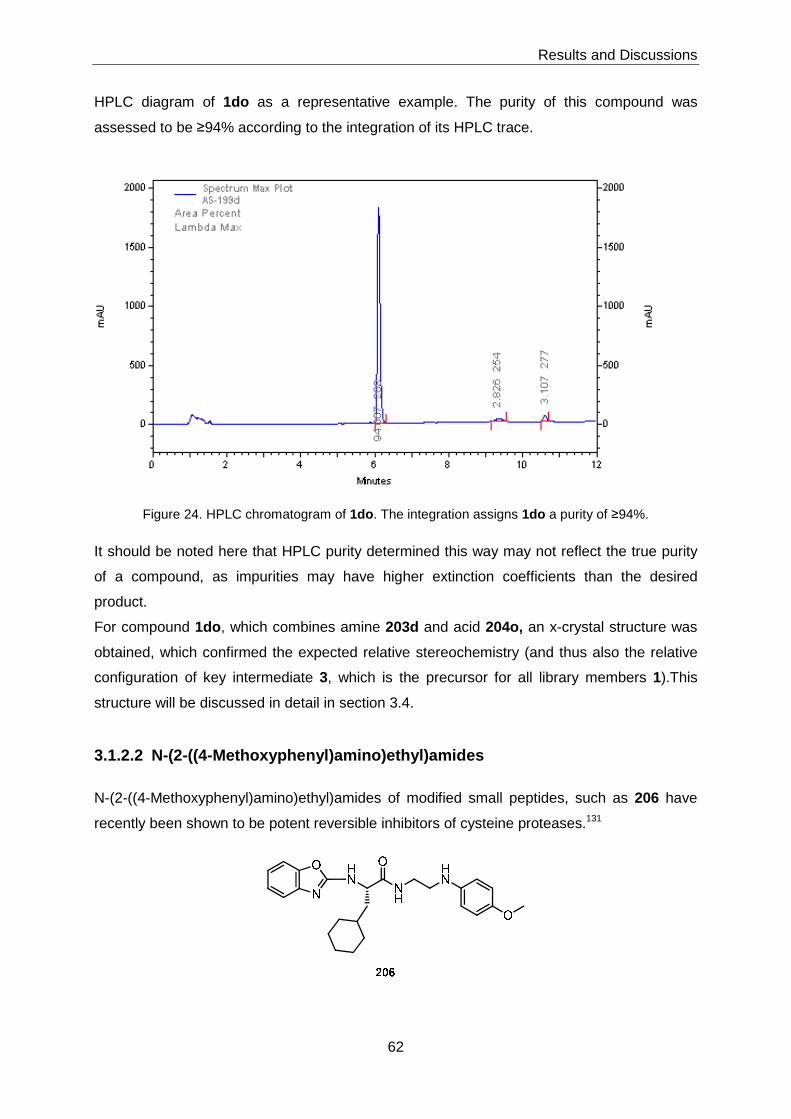
Results and Discussions
62
HPLC diagram of 1do as a representative example. The purity of this compound was
assessed to be ≥94% according to the integration of its HPLC trace.
Figure 24. HPLC chromatogram of 1do . The integration assigns 1do a purity of ≥94%.
It should be noted here that HPLC purity determined this way may not reflect the true purity
of a compound, as impurities may have higher extinction coefficients than the desired
product.
For compound 1do , which combines amine 203d and acid 204o, an x-crystal structure was
obtained, which confirmed the expected relative stereochemistry (and thus also the relative
configuration of key intermediate 3, which is the precursor for all library members 1).This
structure will be discussed in detail in section 3.4.
3.1.2.2 N-(2-((4-Methoxyphenyl)amino)ethyl)amides
N-(2-((4-Methoxyphenyl)amino)ethyl)amides of modified small peptides, such as 206 have
recently been shown to be potent reversible inhibitors of cysteine proteases.131
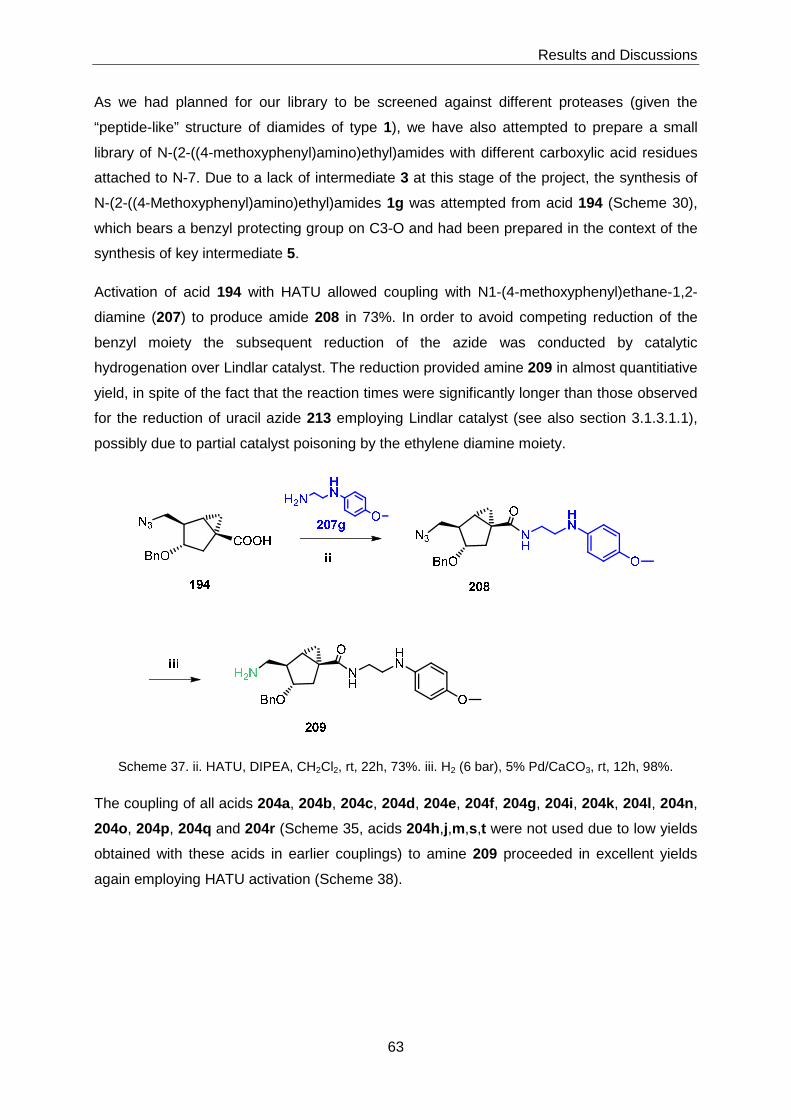
Results and Discussions
63
As we had planned for our library to be screened against different proteases (given the
“peptide-like” structure of diamides of type 1), we have also attempted to prepare a small
library of N-(2-((4-methoxyphenyl)amino)ethyl)amides with different carboxylic acid residues
attached to N-7. Due to a lack of intermediate 3 at this stage of the project, the synthesis of
N-(2-((4-Methoxyphenyl)amino)ethyl)amides 1g was attempted from acid 194 (Scheme 30),
which bears a benzyl protecting group on C3-O and had been prepared in the context of the
synthesis of key intermediate 5.
Activation of acid 194 with HATU allowed coupling with N1-(4-methoxyphenyl)ethane-1,2-
diamine (207) to produce amide 208 in 73%. In order to avoid competing reduction of the
benzyl moiety the subsequent reduction of the azide was conducted by catalytic
hydrogenation over Lindlar catalyst. The reduction provided amine 209 in almost quantitiative
yield, in spite of the fact that the reaction times were significantly longer than those observed
for the reduction of uracil azide 213 employing Lindlar catalyst (see also section 3.1.3.1.1),
possibly due to partial catalyst poisoning by the ethylene diamine moiety.
Scheme 37. ii. HATU, DIPEA, CH2Cl2, rt, 22h, 73%. iii. H2 (6 bar), 5% Pd/CaCO3, rt, 12h, 98%.
The coupling of all acids 204a, 204b, 204c, 204d, 204e, 204f, 204g, 204i, 204k, 204l, 204n,
204o, 204p, 204q and 204r (Scheme 35, acids 204h,j,m,s,t were not used due to low yields
obtained with these acids in earlier couplings) to amine 209 proceeded in excellent yields
again employing HATU activation (Scheme 38).

Results and Discussions
64
Scheme 38. i. R-COOH, HATU, DIPEA, CH2Cl2, rt, 12h, 92-100%.
The critical step in the synthesis of N-(2-((4-methoxyphenyl)amino)ethyl)amides 1g proved to
be the cleavage of the benzyl ether moiety at position 3 of the bicyclic core structure from
intermediates 209. Different conditions were investigated for this transformation; as
compounds 210a-210t were only available in milligram quantities, the trial reactions were
performed only on a very small scale, therefore some of the reactions were solely monitored
by mass spectrometry. Otherwise thin layer chromatography (TLC) was employed.
Treatment of compound 210c with Pd-C and formic acid as the hydrogen source in MeOH
gave some product formation according to TLC and mass spectrometry, but substantial
amounts of starting material were still left after 5 h and decomposition was visible upon TLC
analysis by the formation of several spots (Scheme 39 and Table 2). If the reaction was run
longer, the product spot could not be detected anymore.

Results and Discussions
65
Scheme 39. Debenzylation trials with selected benzyl ethers 210g.
Increasing the catalyst load (with 210a as substrate) led to decomposition only. Attempted
debenzylation of 210b with molecular hydrogen over Pd-C gave recovered starting material
only or, at high catalyst loads, a product that appeared to be overreduced (desired mass plus
6 mass units). In an attempt to avoid overreduction, 210p was treated with H2 at 8 bar over
Lindlar catalyst, but no reaction was observed. The use of Raney-Nickel instead of Pd-C led
to complete decomposition of the starting material 210b (Table 2).
Treatment of 210b with Lewis acids, such as iron trichloride or tin tetrachloride either resulted
in decomposition (FeCl3) or returned unchanged starting material. In contrast, boron
trichloride provided the desired deprotected compound 1gp (Scheme 39). In light of these
results no further efforts were made to debenzylate intermediates 210; instead these
compounds were directly used in different screening campaigns.
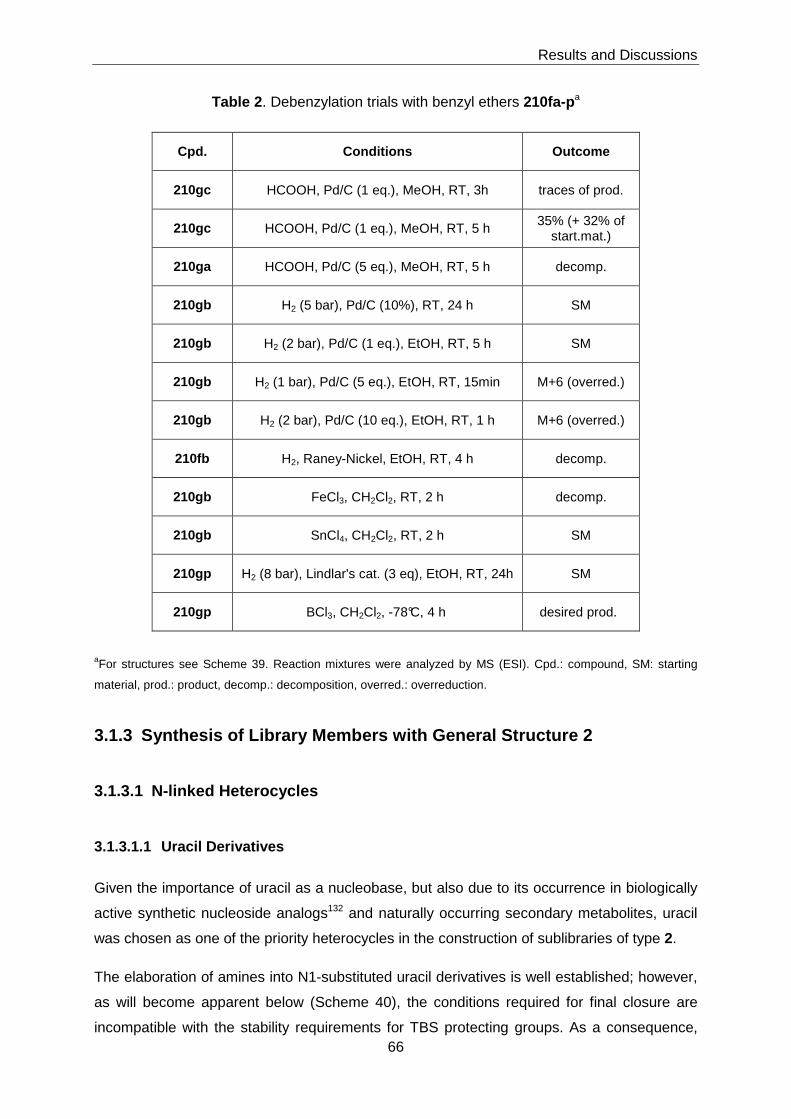
Results and Discussions
66
Table 2 . Debenzylation trials with benzyl ethers 210fa-p a
Cpd. Conditions Outcome
210gc HCOOH, Pd/C (1 eq.), MeOH, RT, 3h traces of prod.
210gc HCOOH, Pd/C (1 eq.), MeOH, RT, 5 h 35% (+ 32% of
start.mat.)
210ga HCOOH, Pd/C (5 eq.), MeOH, RT, 5 h decomp.
210gb H2 (5 bar), Pd/C (10%), RT, 24 h SM
210gb H2 (2 bar), Pd/C (1 eq.), EtOH, RT, 5 h SM
210gb H2 (1 bar), Pd/C (5 eq.), EtOH, RT, 15min M+6 (overred.)
210gb H2 (2 bar), Pd/C (10 eq.), EtOH, RT, 1 h M+6 (overred.)
210fb H2, Raney-Nickel, EtOH, RT, 4 h decomp.
210gb FeCl3, CH2Cl2, RT, 2 h decomp.
210gb SnCl4, CH2Cl2, RT, 2 h SM
210gp H2 (8 bar), Lindlar's cat. (3 eq), EtOH, RT, 24h SM
210gp BCl3, CH2Cl2, -78°C, 4 h desired prod.
aFor structures see Scheme 39. Reaction mixtures were analyzed by MS (ESI). Cpd.: compound, SM: starting
material, prod.: product, decomp.: decomposition, overred.: overreduction.
3.1.3 Synthesis of Library Members with General Str ucture 2
3.1.3.1 N-linked Heterocycles
3.1.3.1.1 Uracil Derivatives
Given the importance of uracil as a nucleobase, but also due to its occurrence in biologically
active synthetic nucleoside analogs132 and naturally occurring secondary metabolites, uracil
was chosen as one of the priority heterocycles in the construction of sublibraries of type 2.
The elaboration of amines into N1-substituted uracil derivatives is well established; however,
as will become apparent below (Scheme 40), the conditions required for final closure are
incompatible with the stability requirements for TBS protecting groups. As a consequence,

Results and Discussions
67
the use of key intermediate 5 was deemed most appropriate for the construction of this sub-
library, as loss of the protecting group on C3-O might have led to side reactions in the crucial
coupling step by competing acylation of the free hydroxyl group. Build-up of the uracil base
from amine 5 was accomplished in a two- step sequence that involved first reaction with acyl
isocyanate 211 and subsequent ring closure of the resulting N-acyl urea 212 under acidic
conditions in a maximum 65% overall yield. Azide 213a was then converted into amine 214a
in quantitative yield by catalytic hydrogenation; in order to avoid competing reduction of the
double bond of the uracil moiety, the reduction was carried out with Lindlar catalyst under a
hydrogen pressure of 3 bar. Amine 214a was sufficiently pure as judged by TLC analysis to
be used in the following coupling reactions without further purification.
Scheme 40. i. CH2Cl2, 0°C-->rt, 12h, 77%. ii. H 2SO4 1 M aq., dioxane, rf, 2h, 84%. iii. H2 (3 bar),
Lindlar cat. (5% Pd/CaCO3), EtOH, 4h, 100%.
Acyl-isocyanate 211 was obtained from commercially available methyl 3-methoxyacryloate
215 in three steps according to Scheme 41.133
Scheme 41. i. KOH, H2O, 50°C, 12h, 67%. ii. SOCl 2, CH2Cl2, rf, 3h, 74%. iii. AgOCN, tol, rf, 30min.

Results and Discussions
68
HATU-mediated coupling of amine 214 with carboxylic acids 204a-204r (for structures see
Scheme 35) in dichloromethane proceeded smoothly and afforded the desired amides
218a-r in yields between 47-100% (Scheme 42).
Scheme 42. i. R-COOH, HATU, DIPEA, CH2Cl2, rt, 12h, 47-100%.
Unfortunately, the clean removal of the benzyl protecting group on C3-O of compounds 218a
by catalytic hydrogenation proved to be impossible under a variety of conditions, due to
competing reduction of the double bond in the uracil base. As illustrated in Scheme 43 for
amide 218c, cleavage of the benzyl ether was feasible with boron trichloride according to
mass spectrometry and TLC analysis.
Scheme 43. i. BCl3, CH2Cl2, -78°C, 5h, yield not determined.
In light of the difficulties associated with the cleavage of the benzy ether moiety in amides
218, no further efforts were made to prepare the corresponding C3-hydroxyl derivatives.
Rather, the benzyl ethers as such were used for screening purposes.
3.1.3.1.2 Hypoxanthine Derivatives
In contrast to the uracil derivatives described above, intermediate 4 was employed as the
amine precursor for the construction of hypoxanthine derivatives of type 2. The elaboration of
the hypoxanthine base from a substituted amine does not involve any acid-promoted
transformations134 and, therefore, is fully compatible with the protection of the 3- and 7-
hydroxyl groups as TBS ethers.

Results and Discussions
69
The assembly of the hypoxanthine heterocycle started with the reaction of amine 4 with N-
(4,6-dichloropyrimidin-5-yl)formamide,135 which was followed by the in situ cyclization of the
resulting diamino-pyrimidine 220 with diethoxymethyl acetate under heating, to provide
chloropurine derivative 221 (Scheme 44). Unfortunately, the yield for this two-step
transformation did not exceed 45%; as a consequence only small quantities of 221 could be
prepared. Treatment of 221 with of 2-mercaptoethanol and sodium methoxide in MeOH gave
protected hypoxanthine derivative 222.
TBSO
TBSON
N
N N
Cl
NH2
TBSO
TBSO
HN
TBSO
TBSO
N N
NH
Cl
O
H
N
NHN
Cl
Cl
O
H
TBSO
TBSON
N
NN
OH
i
ii iii
219
220
221 222
4
Scheme 44. i. 219, Et3N, 1,4-dioxane, rf, 20h. ii. diethoxymethyl acetate (neat), 110°C, 20h, 45% (over
2 steps). iii. NaOMe, 2-mercaptoethanol, MeOH, ))), 80°C, 2h, 61%.
The mechanism for the conversion of 221 into 222 (Scheme 45) presumably involves
displacement of the 6-chloro substituent by the thiolate anion derived from 2-
mercaptoethanol to produce 223.134 The resulting 6-(2-hydroxyethyl)-purine anion 224 then
rearranges to produce 227. The latter eliminates episulfide, thus, finally leading to
hypoxanthine 222.
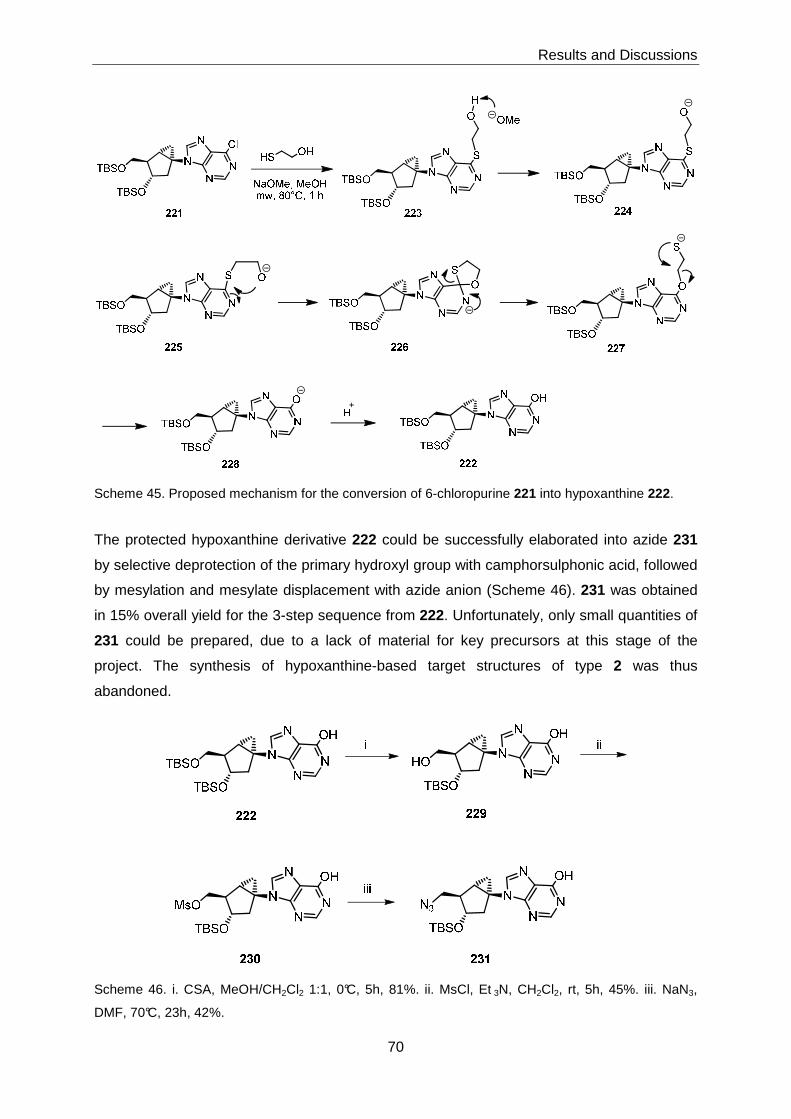
Results and Discussions
70
Scheme 45. Proposed mechanism for the conversion of 6-chloropurine 221 into hypoxanthine 222.
The protected hypoxanthine derivative 222 could be successfully elaborated into azide 231
by selective deprotection of the primary hydroxyl group with camphorsulphonic acid, followed
by mesylation and mesylate displacement with azide anion (Scheme 46). 231 was obtained
in 15% overall yield for the 3-step sequence from 222. Unfortunately, only small quantities of
231 could be prepared, due to a lack of material for key precursors at this stage of the
project. The synthesis of hypoxanthine-based target structures of type 2 was thus
abandoned.
Scheme 46. i. CSA, MeOH/CH2Cl2 1:1, 0°C, 5h, 81%. ii. MsCl, Et 3N, CH2Cl2, rt, 5h, 45%. iii. NaN3,
DMF, 70°C, 23h, 42%.

Results and Discussions
71
3.1.3.2 C-linked Heterocycles
Due to their known occurrence in biologically active synthetic molecules and natural
products,109 oxazole, (N-substituted) imidazole, thiazole, and tetrazole (or their partially
reduced congeners) were also of interest as heterocycle components of library structures of
type 2. While N-alkylation of tetrazole and (unsubstituted) imidazole is possible without the
formation of quaternary salts, oxazole (oxazoline) and thiazole (thiazoline) cannot be
connected to an alkyl residue by a C-N bond, except in quaternary salts (which were not of
interest here). As a consequence, our attempts to produce bicyclo[3.1.0]hexane derivatives
of oxazole (oxazoline) and thiazole (thiazoline), but also of imidazole (imidazoline) and
tetrazole all departed from acid 3 or ester 185, whose carbonyl carbon atom would become
part of the desired heterocycle. In the following, these attempts are briefly summarized.
Our first attempt at the synthesis of an oxazole derivative involved the reaction of methyl
ester 185 with methyl isocyanide in the presence of n-butyl lithium136, as the shortest
conceivable route (Scheme 47). The synthesis of oxazoles by the reaction of esters with in
situ prepared lithiomethyl isocyanide has been used by Jacobi and co-workers136 in their
synthesis of (+)-gnididione; unfortunately, we did not obtain any of the desired product 232
using this methodology. As an alternative, we have also investigated the reaction of aldehyde
234 (obtained from ester 185 by DIBAL-H reduction and subsequent PCC oxidation of the
resulting alcohol 233) with tosylmethyl isocyanide.137 While this approach did indeed deliver
the desired product 235, the yield of the reaction did not exceed 16%. As such this process
was not sufficiently effective to serve as the basis of the construction of an oxazole-based
sublibrary of type 2.
With aldehyde 234 at our disposal, we also investigated its conversion into imidazole
derivative 236 with glyoxal in aqueous ammonia solution.138 The reaction failed to produce
any of the desired product and was not investigated further.
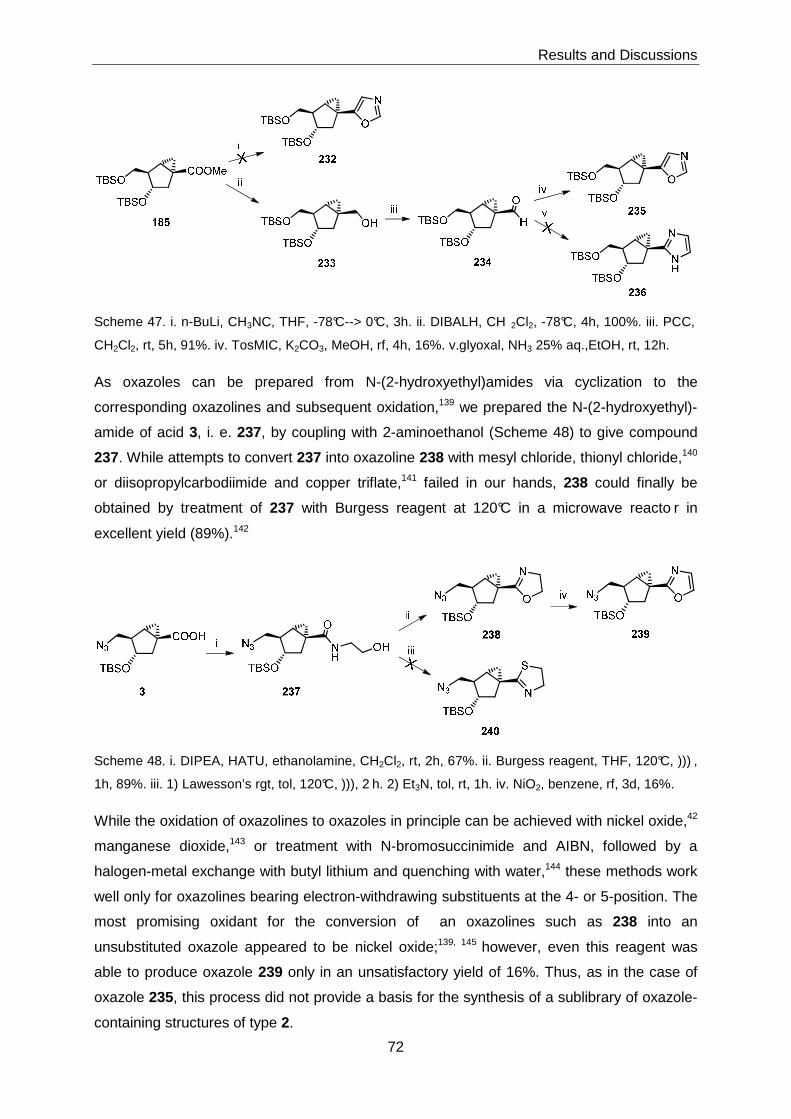
Results and Discussions
72
Scheme 47. i. n-BuLi, CH3NC, THF, -78°C--> 0°C, 3h. ii. DIBALH, CH 2Cl2, -78°C, 4h, 100%. iii. PCC,
CH2Cl2, rt, 5h, 91%. iv. TosMIC, K2CO3, MeOH, rf, 4h, 16%. v.glyoxal, NH3 25% aq.,EtOH, rt, 12h.
As oxazoles can be prepared from N-(2-hydroxyethyl)amides via cyclization to the
corresponding oxazolines and subsequent oxidation,139 we prepared the N-(2-hydroxyethyl)-
amide of acid 3, i. e. 237, by coupling with 2-aminoethanol (Scheme 48) to give compound
237. While attempts to convert 237 into oxazoline 238 with mesyl chloride, thionyl chloride,140
or diisopropylcarbodiimide and copper triflate,141 failed in our hands, 238 could finally be
obtained by treatment of 237 with Burgess reagent at 120°C in a microwave reacto r in
excellent yield (89%).142
Scheme 48. i. DIPEA, HATU, ethanolamine, CH2Cl2, rt, 2h, 67%. ii. Burgess reagent, THF, 120°C, ))) ,
1h, 89%. iii. 1) Lawesson’s rgt, tol, 120°C, ))), 2 h. 2) Et3N, tol, rt, 1h. iv. NiO2, benzene, rf, 3d, 16%.
While the oxidation of oxazolines to oxazoles in principle can be achieved with nickel oxide,42
manganese dioxide,143 or treatment with N-bromosuccinimide and AIBN, followed by a
halogen-metal exchange with butyl lithium and quenching with water,144 these methods work
well only for oxazolines bearing electron-withdrawing substituents at the 4- or 5-position. The
most promising oxidant for the conversion of an oxazolines such as 238 into an
unsubstituted oxazole appeared to be nickel oxide;139, 145 however, even this reagent was
able to produce oxazole 239 only in an unsatisfactory yield of 16%. Thus, as in the case of
oxazole 235, this process did not provide a basis for the synthesis of a sublibrary of oxazole-
containing structures of type 2.

Results and Discussions
73
N-(2-Hydroxyethyl)amide 237 was also investigated as a possible starting material for the
synthesis of 2-substituted thiazolines and, ultimately, thiazoles. According to Nishio et al., the
conversion of 2-hydroxyethylamides to 2-mercaptoethylthioamides should be feasible with
Lawesson reagent; upon treatment with triethylamine in toluene the latter would then yield
the corresponding thiazolines.146 In our hands, this reaction did not work for N-(2-
hydroxyethyl)amide 237 and thiazoline 240 was not obtained.
While oxazoles 235 and 239 were not obtained in sufficient quantities to consider their
elaboration into the desired amides of type 2, oxazoline N-(2-hydroxyethyl)amide 238 was
reduced to amine 241, which was subsequently coupled with a limited number of carboxylic
acids (Scheme 49). The reduction of the azide moiety with hydrogen over Pd-C proceeded in
excellent yield; the subsequent coupling step gave amides 242f-o in yields between 38% and
85%.
Scheme 49. i. H2 (1 bar), 10% Pd/C, Tol/MeOH 1:1, rt, 2h, 98%. ii. R-COOH, DIPEA, HATU, CH2Cl2,
rt, 12h, 38-82%.
Unfortunately, the deprotection of 242f,g,m,n,o under our previously developed standard
conditions (Dowex 50W-X8, MeOH) gave only low to moderate yields of the desired target
structures 2 (Scheme 50), thus providing only 0.5-1 mg amounts of material. Most likely,
these yields reflect the limited stability of the oxazoline ring under acidic conditions.147
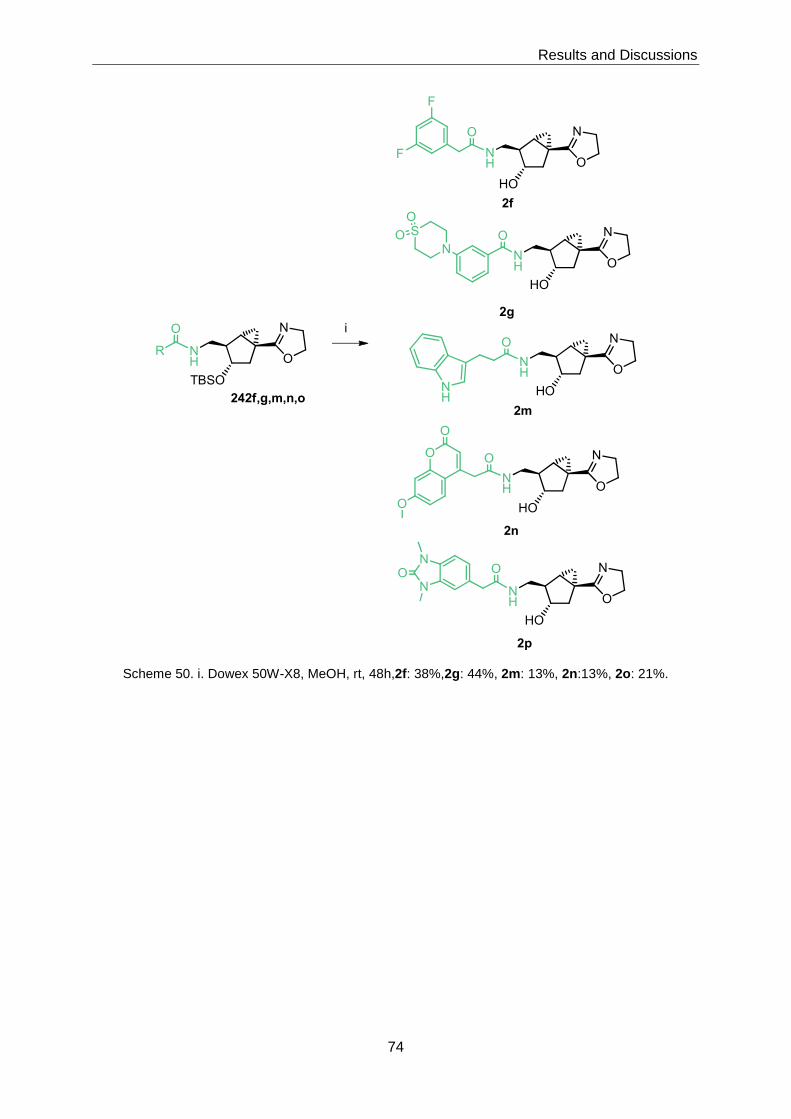
Results and Discussions
74
HO
NH O
N
TBSO
NH O
N
R
O iO
NH
F
F
O
HO
NH O
N
N
SO
O O
HO
NH O
N
242f,g,m,n,o2m
2g
2f
OO
O
O HO
NH O
N
ON
NO
HO
NH O
N
2n
2p
Scheme 50. i. Dowex 50W-X8, MeOH, rt, 48h,2f: 38%,2g: 44%, 2m: 13%, 2n:13%, 2o: 21%.

Results and Discussions
75
3.1.4 Final Library
Figure 25 shows one representative example for each of the bicyclo[3.1.0]hexane-based
sublibraries that have been prepared in the course of this project together with the number of
compounds in each sublibrary. In total, 110 compounds could be prepared in quantities that
were useful for screening purposes.
Figure 25. Structure and size of individual bicyclo[3.1.0]hexane-based sublibraries of types 1 and 2.
Single representatives are shown for each sublibrary.

Results and Discussions
76
3.2 Synthesis of Bicyclo[3.1.0]hexane-based 5-Alkyn yl-
deoxypyrimidine Nucleoside Analogue 243
In 2005 Rai and co-workers showed that 5-(1-alkynyl)-2’-deoxy uridine derivatives can have
significant in vitro antimycobacterial activity,148 with MIC90 values against Mycobacterium
bovis in the 10-100 µg/mL range. The activity was found to depend on the size of the 1-
alkynyl substituent and was postulated to be based on the interference with mycobacterial
DNA and/or RNA synthesis.
As 2’-deoxyribonucleosides preferentially adopt a 2’-endo (southern) sugar conformation,
which essentially corresponds to the conformation of the bicyclo[3.1.0]hexane moiety in
bicyclo[3.1.0]hexane-based 2’-deoxyribonucleoside analogues with the base attached to
position 1 of the bicyclic core, we have prepared analog 243 as a potential antimycobacterial
agent. The MIC90 of the corresponding 2’-deoxyribonucleoside 244 against M. bovis is
50 µg/mL.
Figure 26. 5-alkynynl-deoxypyrimidine nucleoside 244 and bicyclo[3.1.0]hexane based analogue 243.
The synthesis of 243 proceeded via 2’-deoxy uridine analog 246, which was obtained from
amine 4 and acyl isocyanate 204 in 2 steps and 71% overall yield (Scheme 51; see also
section 3.1.3.1.1). Compound 245 was cyclized to uracil derivative 246 by heating with
sulfuric acid with concomitant cleavage of both TBS protecting groups.
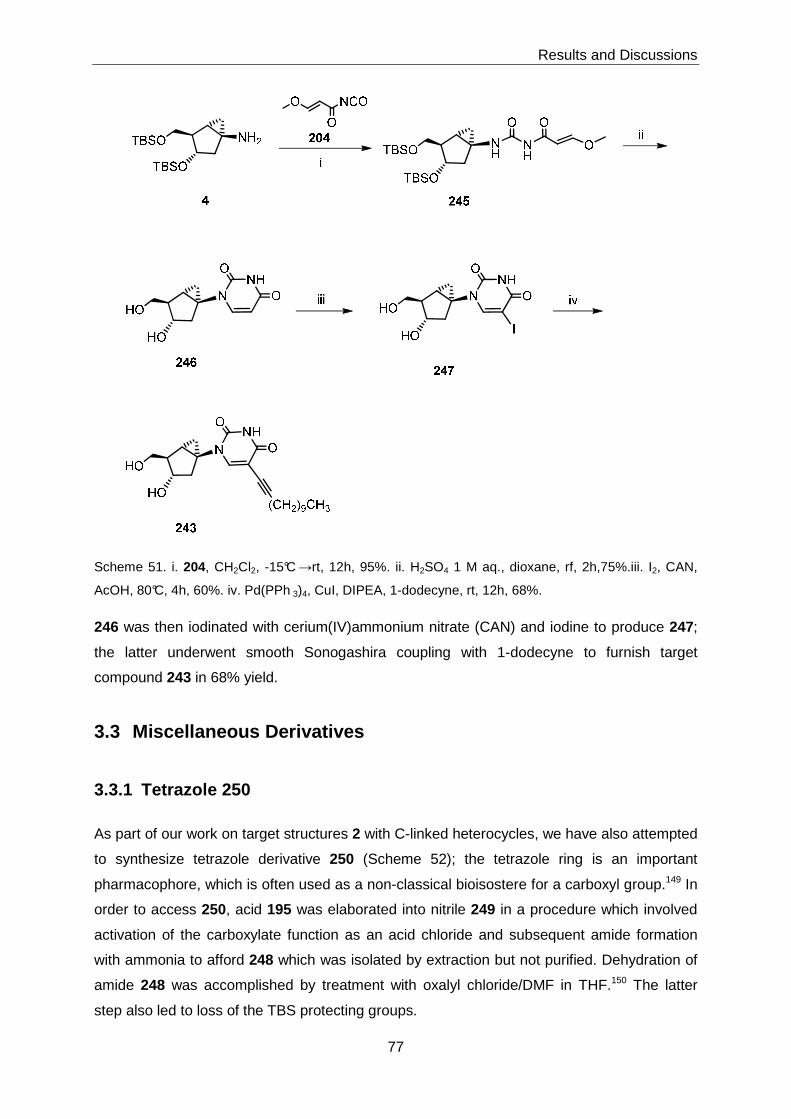
Results and Discussions
77
Scheme 51. i. 204, CH2Cl2, -15°C →rt, 12h, 95%. ii. H2SO4 1 M aq., dioxane, rf, 2h,75%.iii. I2, CAN,
AcOH, 80°C, 4h, 60%. iv. Pd(PPh 3)4, CuI, DIPEA, 1-dodecyne, rt, 12h, 68%.
246 was then iodinated with cerium(IV)ammonium nitrate (CAN) and iodine to produce 247;
the latter underwent smooth Sonogashira coupling with 1-dodecyne to furnish target
compound 243 in 68% yield.
3.3 Miscellaneous Derivatives
3.3.1 Tetrazole 250
As part of our work on target structures 2 with C-linked heterocycles, we have also attempted
to synthesize tetrazole derivative 250 (Scheme 52); the tetrazole ring is an important
pharmacophore, which is often used as a non-classical bioisostere for a carboxyl group.149 In
order to access 250, acid 195 was elaborated into nitrile 249 in a procedure which involved
activation of the carboxylate function as an acid chloride and subsequent amide formation
with ammonia to afford 248 which was isolated by extraction but not purified. Dehydration of
amide 248 was accomplished by treatment with oxalyl chloride/DMF in THF.150 The latter
step also led to loss of the TBS protecting groups.

Results and Discussions
78
Scheme 52. i. 1) SOCl2, THF, rf, 2h. 2) NH3 aq. ii. (COCl)2, DMF, THF, 0°C, 10min, >100%.
Nitrile 249 was difficult to purify, mainly due to its volatility. In light of these experimental
difficulties work on target structure 250 was abandoned.
3.3.2 Iodides 238 and 239
Bridgehead iodides 251 and 252 were prepared as possible precursors for transition metal-
catalyzed cross-coupling reactions. The syntheses were based on the Hundsdiecker-type
oxidative iododecarboxylation of acids 195 and 3 employing iodosobenzene diacetate and
iodine (Scheme 53).151 The reaction proceeds through a radical intermediate.
Scheme 53. i. Iodosobenzene diacetate, I2, C6H12, rf, hν, 3.5h, 39%. ii. Iodosobenzene diacetate, I2,
C6H12, rf, hν, 2.5h, 52%.
Iodides 251 and 252 were obtained in moderate yields only and no attempts were made at
cross-coupling reactions based on these intermediates.

Results and Discussions
79
3.3.3 C3-O-Alkyl Derivatives
As indicated in section 2.1, the free hydroxyl group at position 3 of our library structures was
meant to serve as a possible handle for biotinylation or the attachement of fluorescent tags in
compounds of biological interest. For this reason we conducted a series of preliminary
experiments on the alkylation of azido ester 253 as a model compound at an early stage of
the project (Scheme 54).
Scheme 54. i. TBAF, THF, rt, 1h, 90%. ii. See Table 3.
Alkylating reagents 255-262 (Table 3) were prepared in one or two steps from commercially
available starting materials and various conditions for the alkylation reaction were
investigated. As the only electrophile, idodide 261 produced any of the desired alkylation
product, albeit in low yield only (18%). In general, the starting material was recovered
unchanged. These findings indicate that the reactivity of primary alkyl halides of the types
investigated here is not sufficient for the alkylation of the C3-OH group and that other types
of linkages would have to be investigated for the preparation of conjugates of structures 1 or
2.

Results and Discussions
80
Table 3. Screening of alkylation reagents and conditions.
alkylating reagent conditions R Yield (%)
255
Et3N, Et2O, rt, 5h or NaH, DMF, 70°C, 12h
NHAloc 0 0
256
NaH, CH2Cl2, rt, 12h or DTBMP, CH2Cl2 72°C, 12h
NHAloc 0 0
257
NaH, DMF,-15°C-->rt, 2d or NaH, DMF, TBAI, rt, 2d
NHAloc 0 0
258
NaH, THF, rt, 5h NHBn 0
259
NaH, THF, rt, 5h NHBn 0
260
NaH, DMF, 70°C, 24h Br 0
261
NaH, DMF, rt, 12h or DTBMP, DMF, rt, 20h
NHCbz 18 0
262
NaH, THF, rt, 12h NH2 0
In contrast to 255-262, alkylation of 253 with methyl iodide in the presence of NaH gave the
expected C3 methyl ether in 63% yield; the benzylation of 253 gave even higher yields (see
section 3.1.1).

Results and Discussions
81
3.4 Structural Studies
To confirm the overall relative stereochemistry and the postulated boat conformation of the
bicyclo[3.1.0]hexane core of the synthesized bicyclo[3.1.0]hexane derivatives, structural
studies with three representative specific library members were performed, namely 1aa, 1ag
and 1do (Fig. 27).
Figure 27. Structure drawings of 1aa, 1do and 1ag.
3.4.1 Conformational Analysis of 1aa and 1ag by Sol ution NMR and
Simulated Annealing (SA)
The NMR experiments and computational analysis were carried out by Dr. Bernhard Pfeiffer.
Simulated Annealing as a specific computer-based simulation method refers to a randomized
algorithm executing a theoretical stepwise cooling of a structure, in order to find its global
internal energy minimum.152 Each point s of the search space is analogous to a state of the
structure, and the functions E(s) that are to be minimized correspond to the internal energy of
the structure in that state. If the cooling rate is slow enough, the probability distribution of the
current state is near thermodynamic equilibrium at all times and the system indeed relaxes
into its local energy minimum in each iteration step.153 This method was combined with a
Metropolis Monte Carlo/Stochastic Dynamics Simulation.154 The latter was designed as a
means for the examination of statistic-mechanic systems within certain parameter
boundaries.

Results and Discussions
82
In our case, the dynamic parameters represented torsion angles and distances between the
hydrogens attached to the bicyclo[3.1.0]hexane core. The boundaries of these parameters
were determined experimentally from solution NMR experiments. In the scope of this study,
the structures of compounds 1aa and 1ag (Fig. 27) were determined.
The boundaries of hydrogen-hydrogen distances of the bicyclo[3.1.0]hexane core could be
directly deduced from Rotating frame Overhauser effect spectroscopy (ROESY)
experiments. As the strength of the ROE signals is proportional to the inverse sixth power of
the distance between the hydrogens (I~1/r6), the experimentally obtained ROE peaks were
classified according to their intensity to correspond to the denoted distance ranges, (Fig. 28;
values given for 1ag, the same procedure was followed for 1aa).
Figure 28.
H’s
intensity Ia
derived distance r b ((((Å))))
3-2b s 2.15 ± 0.35
3-7a s 2.15 ± 0.35
3-7b s 2.15 ± 0.35
7-5 m 2.65 ± 0.85
4-5 m 2.65 ± 0.85
4-6b m 2.65 ± 0.85
2b-6b m 2.65 ± 0.85
3-5 w 3.40 ± 1.60
2a-3 s 2.15 ± 0.35
6b-3 m 2.65 ± 0.85 Spectra recorded in MeOH-d4.
a: s: strong,
m: medium, w: weak; b: derived from I ~ 1/r6.
The range of the torsion angles was determined by sampling the coupling constants of vicinal
hydrogen atoms, and processing them via the Karplus curve which outputs the range for
possible torsion angles (Fig. 29).
The torsion angles were calculated according to Altona and co-workers.155
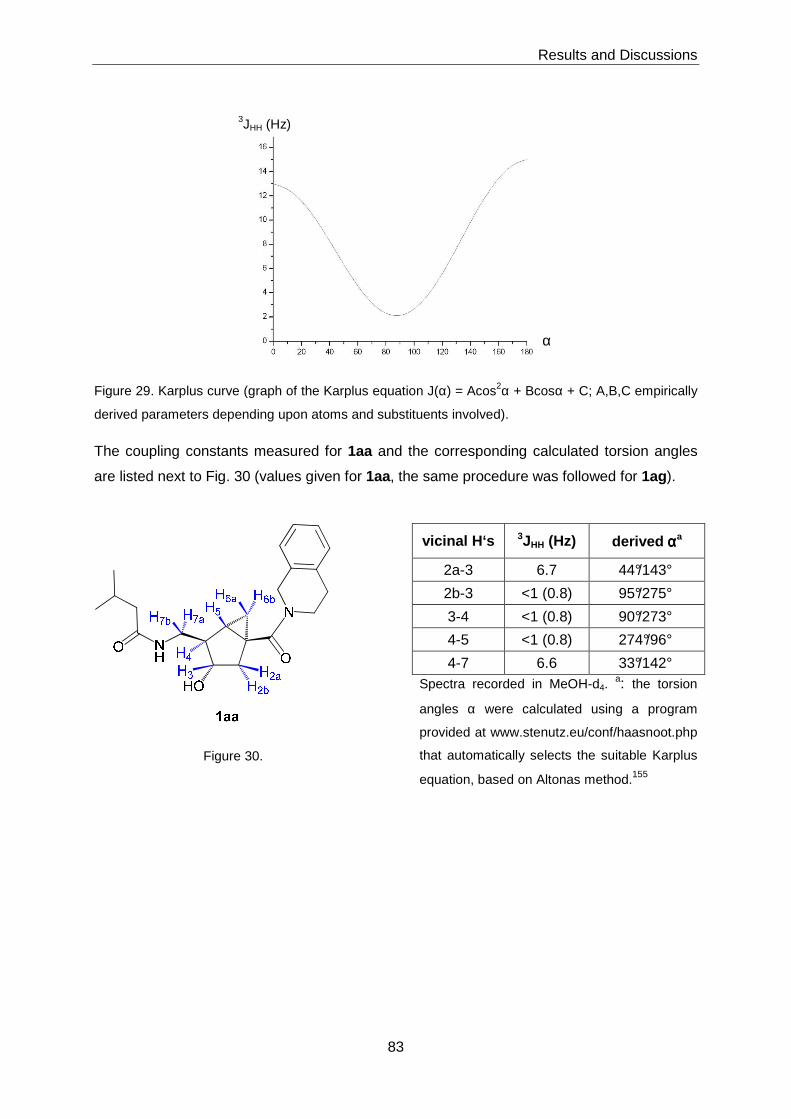
Results and Discussions
83
Figure 29. Karplus curve (graph of the Karplus equation J(α) = Acos2α + Bcosα + C; A,B,C empirically
derived parameters depending upon atoms and substituents involved).
The coupling constants measured for 1aa and the corresponding calculated torsion angles
are listed next to Fig. 30 (values given for 1aa, the same procedure was followed for 1ag).
Figure 30.
vicinal H‘s 3JHH (Hz) derived ααααa
2a-3 6.7 44°/143°
2b-3 <1 (0.8) 95°/275°
3-4 <1 (0.8) 90°/273°
4-5 <1 (0.8) 274°/96°
4-7 6.6 33°/142° Spectra recorded in MeOH-d4.
a: the torsion
angles α were calculated using a program
provided at www.stenutz.eu/conf/haasnoot.php
that automatically selects the suitable Karplus
equation, based on Altonas method.155
3JHH (Hz)
α
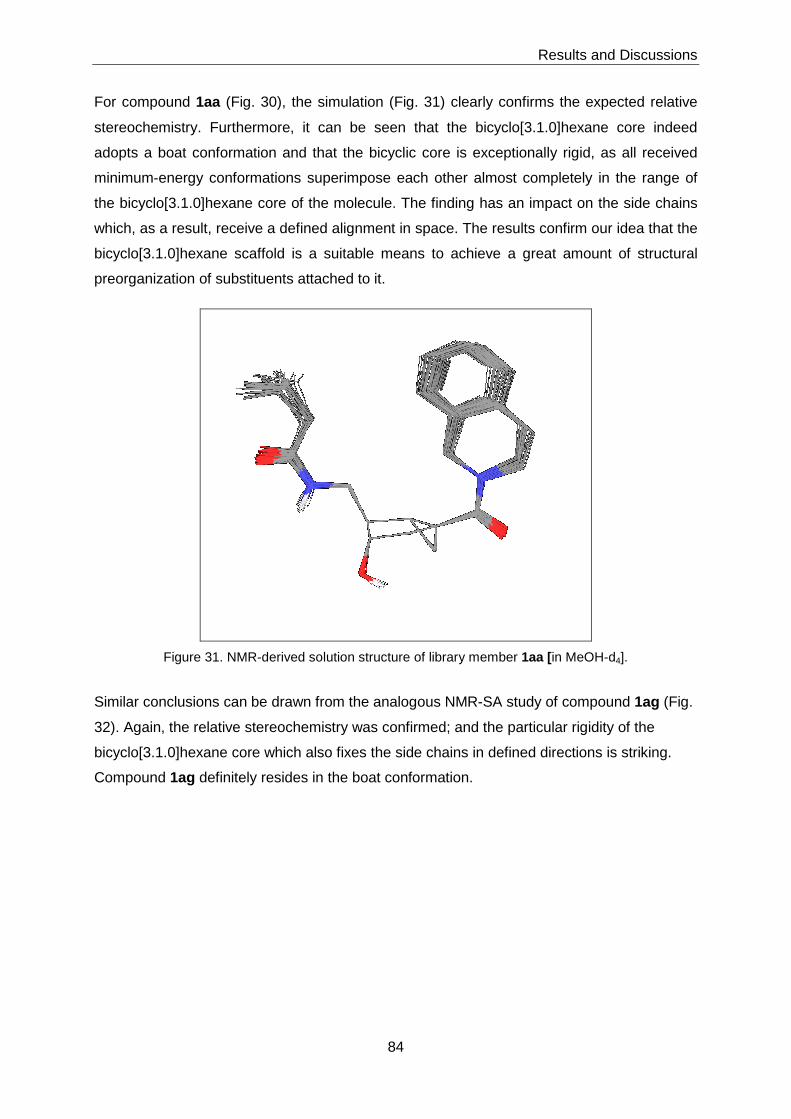
Results and Discussions
84
For compound 1aa (Fig. 30), the simulation (Fig. 31) clearly confirms the expected relative
stereochemistry. Furthermore, it can be seen that the bicyclo[3.1.0]hexane core indeed
adopts a boat conformation and that the bicyclic core is exceptionally rigid, as all received
minimum-energy conformations superimpose each other almost completely in the range of
the bicyclo[3.1.0]hexane core of the molecule. The finding has an impact on the side chains
which, as a result, receive a defined alignment in space. The results confirm our idea that the
bicyclo[3.1.0]hexane scaffold is a suitable means to achieve a great amount of structural
preorganization of substituents attached to it.
Figure 31. NMR-derived solution structure of library member 1aa [in MeOH-d4].
Similar conclusions can be drawn from the analogous NMR-SA study of compound 1ag (Fig.
32). Again, the relative stereochemistry was confirmed; and the particular rigidity of the
bicyclo[3.1.0]hexane core which also fixes the side chains in defined directions is striking.
Compound 1ag definitely resides in the boat conformation.

Results and Discussions
85
Figure 32. NMR-derived solution structure of library member 1ag [ in MeOH-d4].
3.4.2 Conformational Analysis by X-ray Crystallogr aphy
For compound 1do a monocrystal suitable for X-ray structural analysis was obtained by slow
evaporation of the solvent (MeOH). The analysis was performed by Dr. Bernd Schweizer and
Michael Solar from LOC, ETH Zurich.
The crystallographic data indicated the existence of a twinned crystal, suggesting two
conformational minima for compound 1do in the solid state; however, only very slight
differences were observed between the two structures. These differences mostly relate to the
torsion angles around the bonds that connect the indoline ring and the phthalazinone moiety,
respectively, to the bicyclo[3.1.0]hexane core (Figure 33). The differences in the
corresponding torsion angles were small, ranging from 3-8° (Table 4, entries 5-12).

Results and Discussions
86
Figure 33. Atom numbering for twinned crystal 1do . Torsion angle deviations for the 2 structures are
listed in table 4.
The bicyclo[3.1.0]hexane core adopts the expected boat conformation with the following
torsion angles: C5-C1-C2-C3 15.2°, C3-C4-C5-C1 -12. 3°, C2-C1-C6-C5 97.3°, C4-C5-C6-C1
-95.3° (Table 4, entries 1-4).
Table 4. Torsion angles of twin structures of 1do .
bond
torsion angle structure 1
torsion angle structure 2
C5-C1-C2-C3 15.2° 15.2°
C3-C4-C5-C1 -12.3° -12.3°
C2-C1-C6-C5 97.3° 97.3°
C4-C5-C6-C1 -95.3° -95.3°
C10-C12-C13-N14 109.6° 113.3°
C10-C12-C13-C21 -67.3° -64.4°
C34-N26-C24-C5 174.1° 170.7°
C4-C5-C24-N26 -59.3° -67.1°
The approximate angle between the planes spanned by C2-C1-C5 and C1-C6-C5 is 94°, the
angle between the planes spanned by C4-C5-C1 and C1-C6-C is 91°, for the planes
between C1-C2-C5 and C2-C3-C4, the angle is 151°, a nd the one between C4-C5-C1 and
C2-C3-C4 planes is 154°. These values are in good a greement with the ones that were
received in quantum-mechanical calculations with unsubstituted bicyclo[3.1.0]hexane,26 with
the slight difference that in our case the cyclopropane moiety is almost perpendicular in
relation to the connection of carbons C2, C1, C5, and C4 (they can be viewed approximately
as spanning a plane), whereas a slightly wider angle of about 110° was usually received in
the calculations (c.f. section 1.3).
The crystal structure also confirms the relative stereochemistry of 1do (Fig. 34), and it
reveals the absence of any intramolecular H-bonds.

Results and Discussions
87
Figure 34. Ball and stick depiction of the X-ray structure of 1do .
The crystal structure of 1do is in good agreement with the solution structures obtained for
compounds 1aa and 1ag, as both analyses confirm the expected boat conformation of the
bicyclo[3.1.0]hexane core as well as the overall relative stereochemistry. The existence of
two conformational minima in the solid state is in good agreement with the solution structures
obtained, which also show the existence of several conformational minima, differing only
slightly in the torsion angles for those bonds connecting the side chains to the
bicyclo[3.1.0]hexane core.

Results and Discussions
88
3.5 Synthesis of Bicyclo[3.1.0]hexane-based Analogu es of
Pentostatin and S-Adenosylhomocysteine
3.5.1 Bicyclo[3.1.0]hexane-based Pentostatin 6
As outlined in section 2.1, bicyclo[3.1.0]hexane-based pentostatin analog 6 was to be
obtained from the (1S, 3S, 4S, 5S)-enantiomer of racemic intermediate 4. Amine (+)-4 was
obtained via bicyclic lactone (-)-161; as for the racemic case, the latter can be accessed from
tetrahydrophthalic acid anhydride (174). Entry into the desired enantiomeric series is based
on the racemic resolution of rac-175 (Scheme 55) by conversion into a pair of
diastereoisomeric salts with (-)-ephedrine, which can be separated by fractional
crystallization according to Lindner et al.117b
Scheme 55. Racemic resolution of 175 via fractional crystallization.
After 2-3 recrystallizations and liberation of the acid from the salt by an acidic extraction with
diethylether, (-)-175 was obtained in 49% yield (based on rac-175) and with an ee of 85%,
based on HPLC analysis using a DAICEL chiral column (Fig 35).
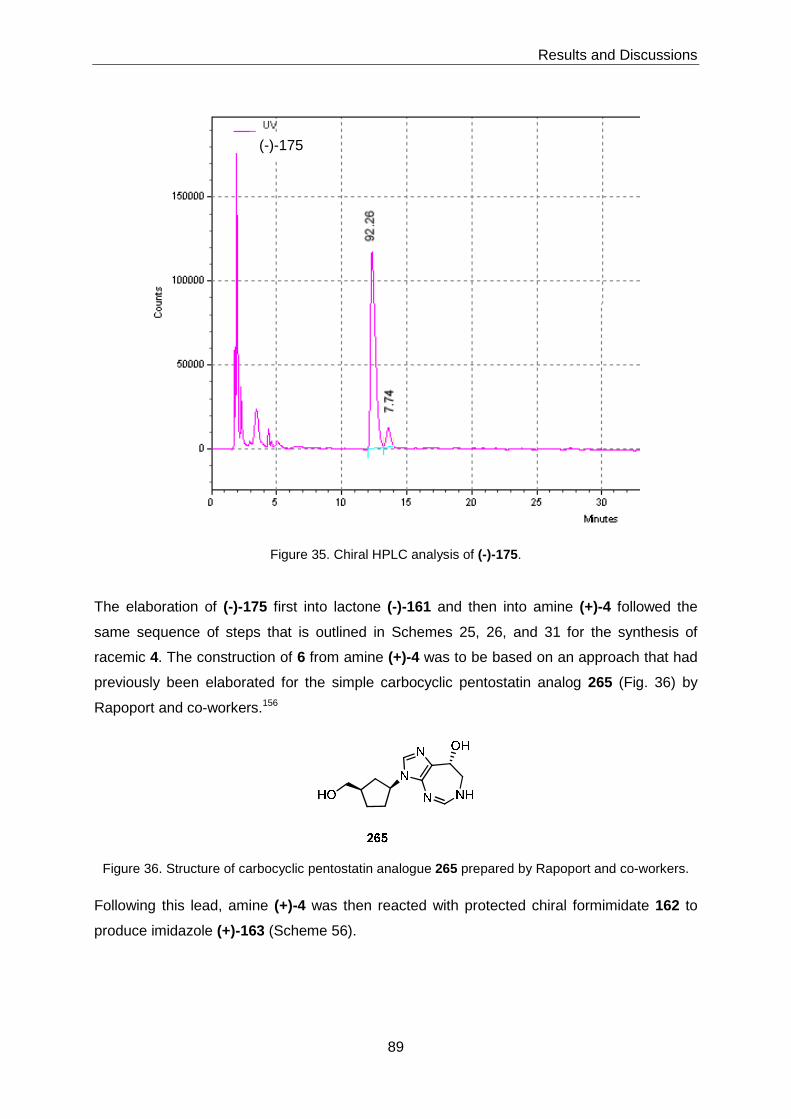
Results and Discussions
89
Figure 35. Chiral HPLC analysis of (-)-175.
The elaboration of (-)-175 first into lactone (-)-161 and then into amine (+)-4 followed the
same sequence of steps that is outlined in Schemes 25, 26, and 31 for the synthesis of
racemic 4. The construction of 6 from amine (+)-4 was to be based on an approach that had
previously been elaborated for the simple carbocyclic pentostatin analog 265 (Fig. 36) by
Rapoport and co-workers.156
Figure 36. Structure of carbocyclic pentostatin analogue 265 prepared by Rapoport and co-workers.
Following this lead, amine (+)-4 was then reacted with protected chiral formimidate 162 to
produce imidazole (+)-163 (Scheme 56).
(-)-175
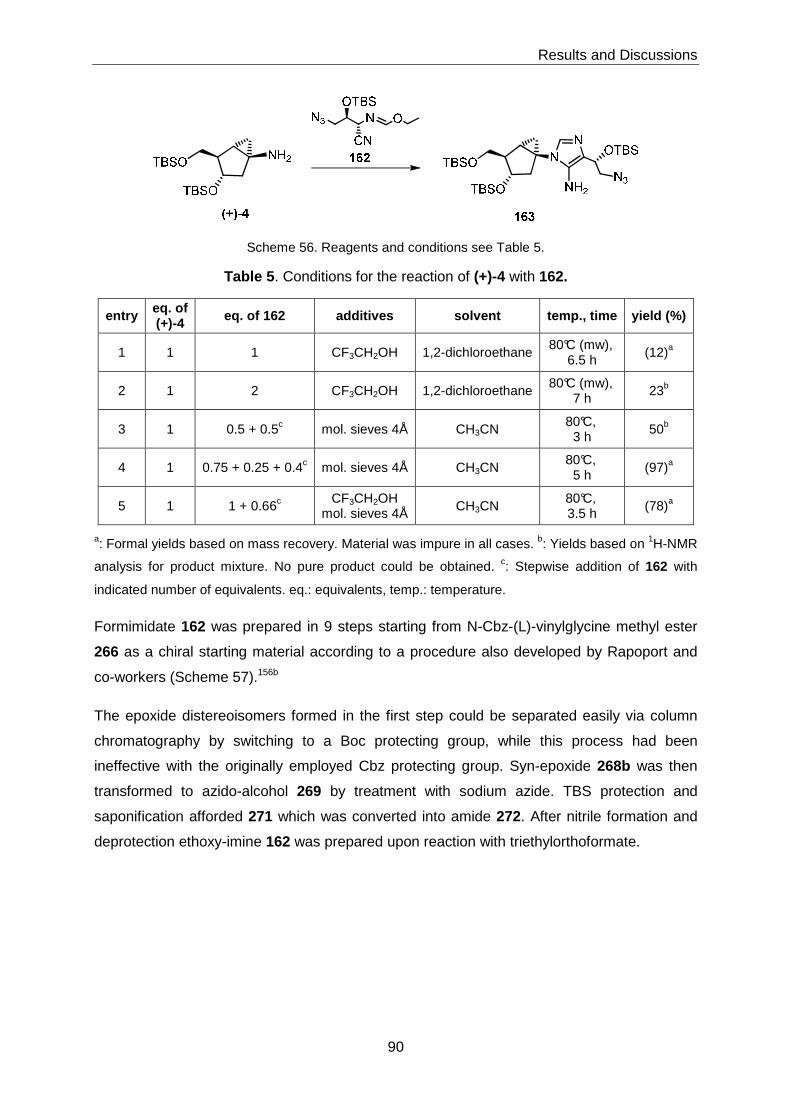
Results and Discussions
90
Scheme 56. Reagents and conditions see Table 5.
Table 5 . Conditions for the reaction of (+)-4 with 162.
entry eq. of (+)-4 eq. of 162 additives solvent temp., time yield (%)
1 1 1 CF3CH2OH 1,2-dichloroethane 80°C (mw),
6.5 h (12)a
2 1 2 CF3CH2OH 1,2-dichloroethane 80°C (mw),
7 h 23b
3 1 0.5 + 0.5c mol. sieves 4Å CH3CN 80°C,
3 h 50b
4 1 0.75 + 0.25 + 0.4c mol. sieves 4Å CH3CN 80°C,
5 h (97)a
5 1 1 + 0.66c CF3CH2OH mol. sieves 4Å CH3CN
80°C, 3.5 h (78)a
a: Formal yields based on mass recovery. Material was impure in all cases. b: Yields based on 1H-NMR
analysis for product mixture. No pure product could be obtained. c: Stepwise addition of 162 with
indicated number of equivalents. eq.: equivalents, temp.: temperature.
Formimidate 162 was prepared in 9 steps starting from N-Cbz-(L)-vinylglycine methyl ester
266 as a chiral starting material according to a procedure also developed by Rapoport and
co-workers (Scheme 57).156b
The epoxide distereoisomers formed in the first step could be separated easily via column
chromatography by switching to a Boc protecting group, while this process had been
ineffective with the originally employed Cbz protecting group. Syn-epoxide 268b was then
transformed to azido-alcohol 269 by treatment with sodium azide. TBS protection and
saponification afforded 271 which was converted into amide 272. After nitrile formation and
deprotection ethoxy-imine 162 was prepared upon reaction with triethylorthoformate.

Results and Discussions
91
Scheme 57. i. m-CPBA, CH2Cl2, rt, 96h, 81%. ii. H2, Pd/C, Boc2O, MeOH, rt, 45min, 50%. iii. NaN3,
NH4Cl, MeOH, rt-->rf, 6h, 84%. iv. TBSCl, im, DMF, rt, 72h, 90%. v. LiOH, dioxane/H2O, 0°C, 3h, 85%.
vi. N-methylmorpholin, i-BuCO2Cl , NH3, THF, -20°C, 3h, 80%. vii. TsCl, py, CH 2Cl2, rt, 72h, 75%. viii.
TFA, CH2Cl2, 0°C, 45min, 53%. ix. HC(OEt 3)3 (neat), 160°C, 2h, 54%.
The yields obtained in the reaction of (+)-4 with 162 were highly variable and strongly
depended upon the exact reaction conditions (Table 5); in addition, the reaction showed poor
reproducibility. The yields reported in entries 2 and 3 of Table 5 are based on 1H-NMR
analysis of material that had been subjected to purification by flash chromatography; even in
these case, however, the purity of the product obtained did not exceed 50%. For entries 1, 4,
and 5, yields are reported for crude products that were used immediately in the subsequent
amidination step. The 1H-NMR spectra of 163 that were recorded after purification by column
chromatography showed a partially decomposed material already upon recording. The poor
reproducibility of the reaction was initially ascribed to the potentially limited thermal stability
of 162 (the reaction was carried out at reflux temperature) and attempts were made to
overcome this problem by the addition of an excess of 162 to the reaction mixture in several
portions, until amine (+)-4 was no longer detectable. This stepwise addition was employed in
entries 3 (addition of 1 eq. in 2 portions), 4 (addition of 1.4 eq. in 3 portions) and 5 (addition
of 1.67 eq. in 2 portions). While this procedure showed a trend toward improvements in the
yield of 163, the latter could never be obtained in purities >50%, presumably due to the
instability of the compound. It was thus used in the next step immediately, either after
purification by column chromatography or as a crude material (Scheme 58).
As illustrated in Scheme 62, 163 was to be reacted with dimethylformamide dimethylacetal to
form 165, which would then be reduced and cyclized to give the bis-TBS-protected target
structure 6.

Results and Discussions
92
Scheme 58. Attempted synthesis of protected pentostatin analog 6.
Unfortunately, the amidination step turned out to be highly problematic. Different reagents
and conditions were sampled for this transformation (163 was treated with 164 and
trifluoroethanol in dichloroethane, microwave irradiation was employed, the solvent was
changed to CH3CN, molecular sieves were added, reaction times were varied), but the
desired 165 was never obtained in pure form. This may be related to a pronounced instability
of the compound, although this has not been investigated in any detail. Attempts to convert
crude preparations into the target structure under a variety of conditions (by the addition of
propanedithiol as reducing agent with Et3N in MeOH at 50°C, various reaction times) proved
to be unsuccessful.
3.5.2 Bicyclo[3.1.0]hexane-based S-Adenosylhomocyst eine 7
The projected synthesis of bicyclo[3.1.0]hexane-based S-adenosylhomocysteine 7, as for
pentostatin analog 6, was to proceed through amine (+)-4 as a key intermediate.
As summarized in Scheme 60, (+)-4 was elaborated into (-)-221 in analogy to the synthesis
of rac-221 (section 3.1.3.1.2, Scheme 44) in 45% overall yield.
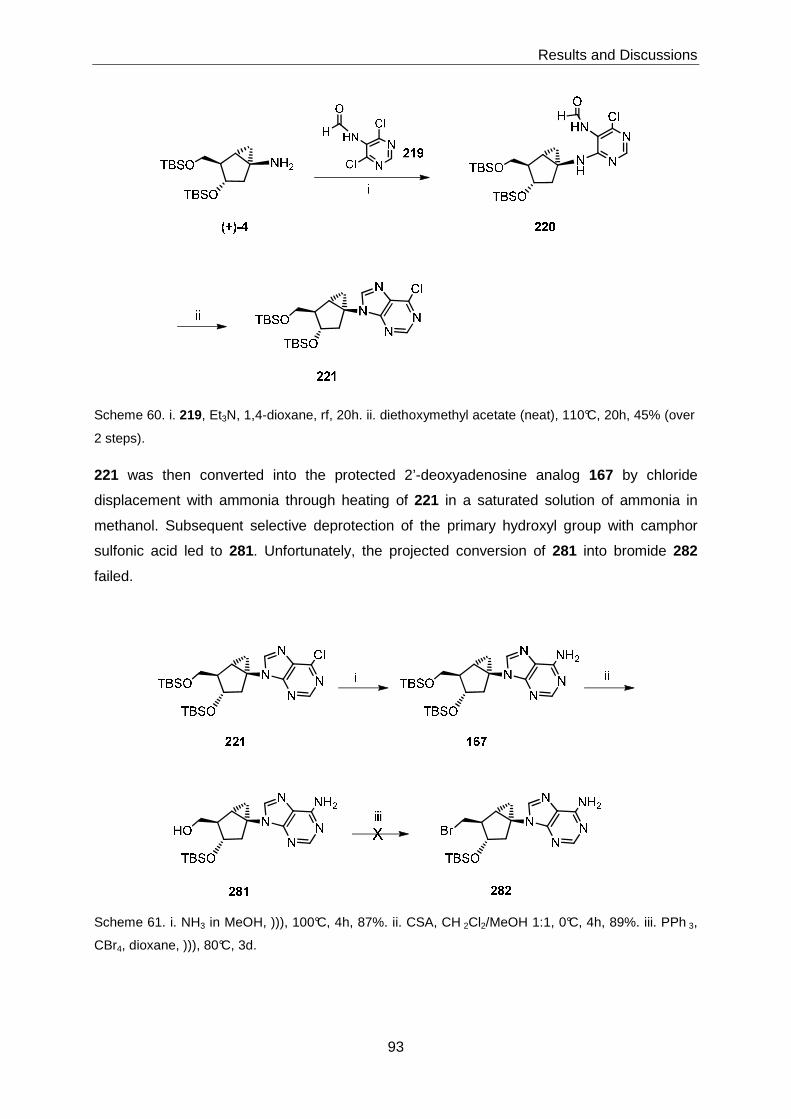
Results and Discussions
93
Scheme 60. i. 219, Et3N, 1,4-dioxane, rf, 20h. ii. diethoxymethyl acetate (neat), 110°C, 20h, 45% (over
2 steps).
221 was then converted into the protected 2’-deoxyadenosine analog 167 by chloride
displacement with ammonia through heating of 221 in a saturated solution of ammonia in
methanol. Subsequent selective deprotection of the primary hydroxyl group with camphor
sulfonic acid led to 281. Unfortunately, the projected conversion of 281 into bromide 282
failed.
Scheme 61. i. NH3 in MeOH, ))), 100°C, 4h, 87%. ii. CSA, CH 2Cl2/MeOH 1:1, 0°C, 4h, 89%. iii. PPh 3,
CBr4, dioxane, ))), 80°C, 3d.
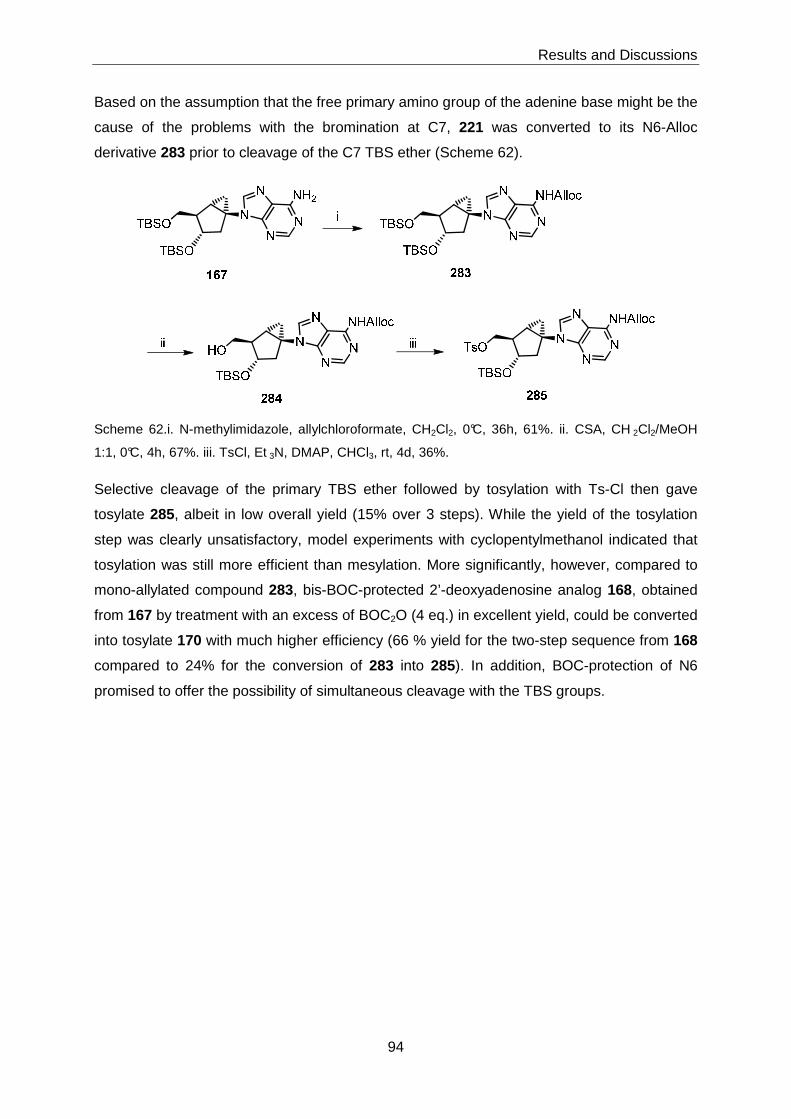
Results and Discussions
94
Based on the assumption that the free primary amino group of the adenine base might be the
cause of the problems with the bromination at C7, 221 was converted to its N6-Alloc
derivative 283 prior to cleavage of the C7 TBS ether (Scheme 62).
Scheme 62.i. N-methylimidazole, allylchloroformate, CH2Cl2, 0°C, 36h, 61%. ii. CSA, CH 2Cl2/MeOH
1:1, 0°C, 4h, 67%. iii. TsCl, Et 3N, DMAP, CHCl3, rt, 4d, 36%.
Selective cleavage of the primary TBS ether followed by tosylation with Ts-Cl then gave
tosylate 285, albeit in low overall yield (15% over 3 steps). While the yield of the tosylation
step was clearly unsatisfactory, model experiments with cyclopentylmethanol indicated that
tosylation was still more efficient than mesylation. More significantly, however, compared to
mono-allylated compound 283, bis-BOC-protected 2’-deoxyadenosine analog 168, obtained
from 167 by treatment with an excess of BOC2O (4 eq.) in excellent yield, could be converted
into tosylate 170 with much higher efficiency (66 % yield for the two-step sequence from 168
compared to 24% for the conversion of 283 into 285). In addition, BOC-protection of N6
promised to offer the possibility of simultaneous cleavage with the TBS groups.
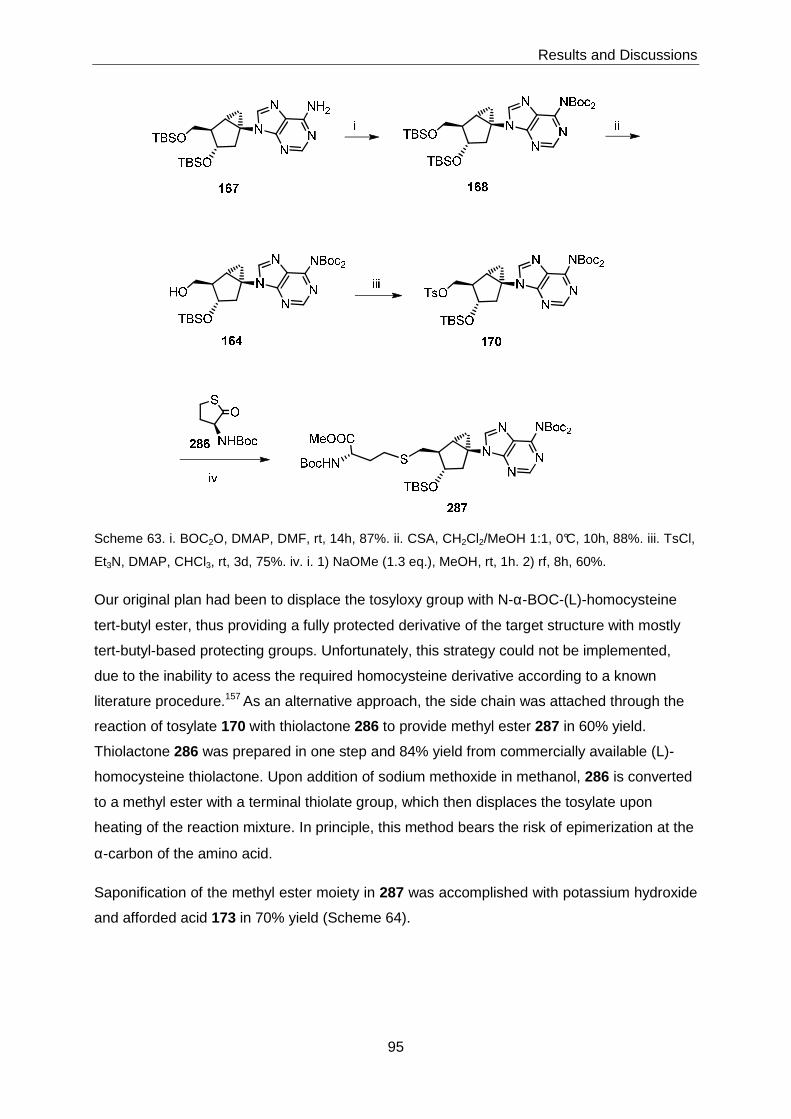
Results and Discussions
95
Scheme 63. i. BOC2O, DMAP, DMF, rt, 14h, 87%. ii. CSA, CH2Cl2/MeOH 1:1, 0°C, 10h, 88%. iii. TsCl,
Et3N, DMAP, CHCl3, rt, 3d, 75%. iv. i. 1) NaOMe (1.3 eq.), MeOH, rt, 1h. 2) rf, 8h, 60%.
Our original plan had been to displace the tosyloxy group with N-α-BOC-(L)-homocysteine
tert-butyl ester, thus providing a fully protected derivative of the target structure with mostly
tert-butyl-based protecting groups. Unfortunately, this strategy could not be implemented,
due to the inability to acess the required homocysteine derivative according to a known
literature procedure.157 As an alternative approach, the side chain was attached through the
reaction of tosylate 170 with thiolactone 286 to provide methyl ester 287 in 60% yield.
Thiolactone 286 was prepared in one step and 84% yield from commercially available (L)-
homocysteine thiolactone. Upon addition of sodium methoxide in methanol, 286 is converted
to a methyl ester with a terminal thiolate group, which then displaces the tosylate upon
heating of the reaction mixture. In principle, this method bears the risk of epimerization at the
α-carbon of the amino acid.
Saponification of the methyl ester moiety in 287 was accomplished with potassium hydroxide
and afforded acid 173 in 70% yield (Scheme 64).

Results and Discussions
96
Scheme 64. i. KOH aq. (2 eq.), THF/H2O 1:1, rt, 24h, 70%.
During two trials for the conversion of tosylate 170 to 287, a product corresponding to 287
but lacking two BOC-groups was obtained. This product (287a) was also subjected to ester
hydrolysis, yielding 173a, and the complete deprotection of this material was attempted with
HCl in ethyl acetate, it was hoped that the final product would precipitate from the solution as
its hydrochloride, which would have obviated the need for chromatographic purification of the
final product. (Given the high polarity of 7 we were skeptical about the prospects for the
purification of this material, which turned out to be justified). As the deprotection with HCl did
not go to completion in 3 days and the resulting hydrochloride salt was impure and poorly
soluble even in water, the final deprotection with trifluoroacetic acid was investigated as an
alternative. Upon stirring of 173 with a large excess of trifluoroacetic acid in dichloromethane
the TBS and the three Boc groups could be cleaved within 32 hours (Scheme 65).
Scheme 65. i. TFA, CH2Cl2, rt, 32h, yield not determined.
The resulting TFA-salt of 7 was purified by semi-preparative HPLC on a Zorbax-SB-Phenyl
reversed-phase column. As the analytical HPLC trace of crude 7 showed 2 peaks with the
same mass under certain conditions, we had to reconsider the possibility that epimerization
of the chiral centre at C-8 had taken place earlier during the nucleophilic substitution reaction
of 170 with 286 and that we had obtained a mixture of diastereoisomers (Fig. 36). The two
peaks showed an integration ratio of 2:1. They were separated in several batches using a
precisely determined injection volume and ratio of the two eluents water and acetonitrile; the
slightest change in these two parameters or in the HPLC method led to the loss of
separation.
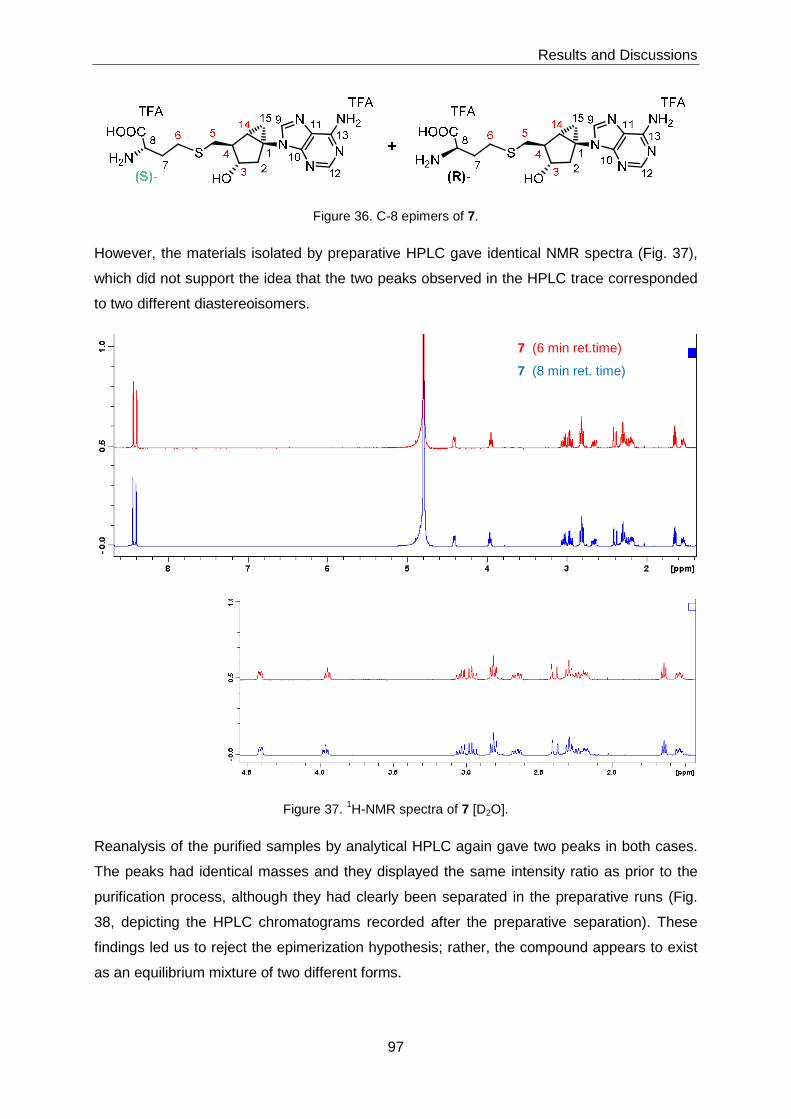
Results and Discussions
97
Figure 36. C-8 epimers of 7.
However, the materials isolated by preparative HPLC gave identical NMR spectra (Fig. 37),
which did not support the idea that the two peaks observed in the HPLC trace corresponded
to two different diastereoisomers.
Figure 37. 1H-NMR spectra of 7 [D2O].
Reanalysis of the purified samples by analytical HPLC again gave two peaks in both cases.
The peaks had identical masses and they displayed the same intensity ratio as prior to the
purification process, although they had clearly been separated in the preparative runs (Fig.
38, depicting the HPLC chromatograms recorded after the preparative separation). These
findings led us to reject the epimerization hypothesis; rather, the compound appears to exist
as an equilibrium mixture of two different forms.
7 (6 min ret.time)
7 (8 min ret. time)
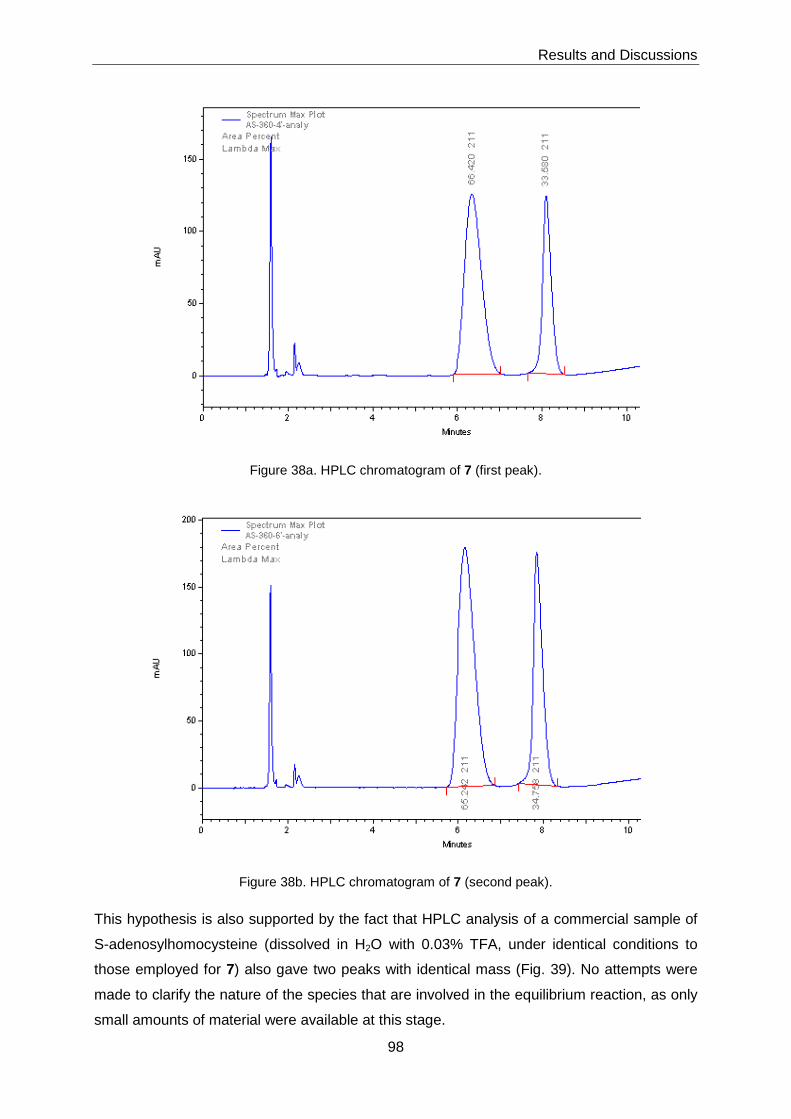
Results and Discussions
98
Figure 38a. HPLC chromatogram of 7 (first peak).
Figure 38b. HPLC chromatogram of 7 (second peak).
This hypothesis is also supported by the fact that HPLC analysis of a commercial sample of
S-adenosylhomocysteine (dissolved in H2O with 0.03% TFA, under identical conditions to
those employed for 7) also gave two peaks with identical mass (Fig. 39). No attempts were
made to clarify the nature of the species that are involved in the equilibrium reaction, as only
small amounts of material were available at this stage.
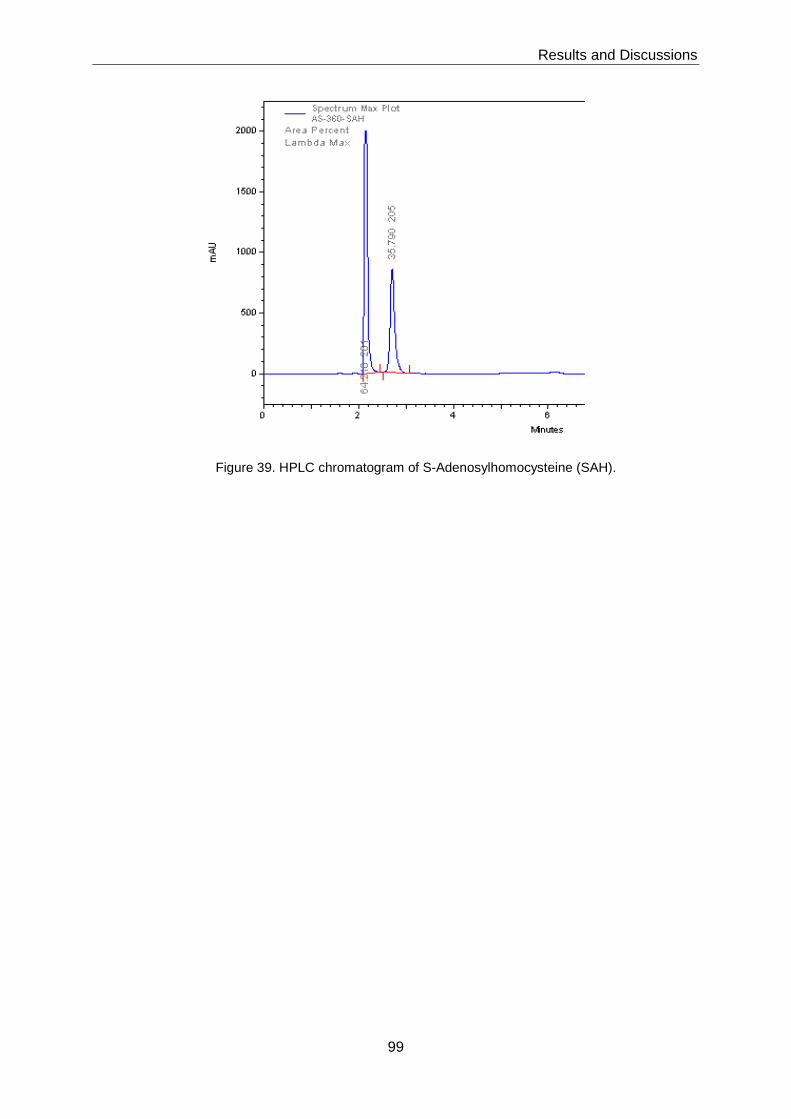
Results and Discussions
99
Figure 39. HPLC chromatogram of S-Adenosylhomocysteine (SAH).

Results and Discussions
100
3.6 Biological Evaluation of the Bicyclo[3.1.0]hexa ne-based
Chemical Library
3.6.1 Reporter Gene Assay Screening for Inhibitory Effects on Selected
Cellular Signaling Pathways
40 library members were tested against a panel of 9 reporter gene assays158 reflecting the
transcriptional output of different cellular signaling networks. (These experiments were
performed at the Novartis Institiute for Biomedical Research in Basel in collaboration with Dr.
Doriano Fabbro and Dr. Daniel D’Orazio at the Novartis Institute for Biomedical Research, in
Basel). The 40 compounds of the library were selected such as to achieve maximal structural
diversity and thus included representatives of each ‘sub family’ of target structures 1 and 2.
Reporter gene assays (RGAs) in the most general sense are cellular detection systems that
rely on the transfection of cells with genes, whose expression results in a readily recordable
physical signal, such as fluorescence. By providing integrated readouts for the activity of
signalling pathways or networks, RGAs, among other things, allow to study the interference
of small molecules with such signaling cascades or networks in live cells.159 The assays
employed in this study involved transcriptional readouts requiring the activation of TFAP1
(via EGF or TNFα stimulation), NFκB (via TNFα stimulation), the tumor suppressor protein
p53 (via TGFβ-stimulation), the cAMP pathway response element CRE, the TGFb signaling
pathway response element SBE, the ARE response element, reflecting oxidative stress, and
the control CMV promoter element to monitor for non signaling pathway-related interactions
within the assay system. In all cases, the different transcriptional response elements control
the expression of a β-lactamase reporter gene, whose expression results in the cleavage of
an exogenously added substrate. Due to fluorescence resonance energy transfer (FRET)
this substrate emits green fluorescence upon excitation at 409 nm; cleavage by the β-
lactamase enzyme eliminates the FRET and the cleavage product exhibits blue
fluorescence. The ratio of blue vs. green fluorescence is a direct measure for reporter gene
activity and thus for the activity of the pathway or network under investigation. The substrate
is added to the cells as a lipophilic pro-form, which penetrates the cell membrane by passive
diffusion and is subsequently converted to the charged substrate by cellular esterases. Due
to its charged nature the substrate remains trapped within the cells. These principles are
summarized in Scheme 66.
Results and Discussions
101
Scheme 66. Schematic representation of the reporter gene assay principle. (depiction adopted from
Invitrogen)
Across all compounds and pathways/networks investigated in this thesis, inihibitory activity
was observed only in the EGF- and/or TNFα-triggered AP1 assay (13 compounds) and in the
TGFβ-sensitive SBE assay (3 compounds). The structures of the active compounds together
with their IC50 values are depicted in Table 6. In particular, compounds 1dc , 1de and 1dm
showed sub-µM inhibition of AP1 activation after stimulation with TNFα. Quite intriguingly,
none of these compounds (or any of the other actives identified) showed any measurable
inhibition of TNFα-triggered NFκB activation at concentrations < 10 µM.
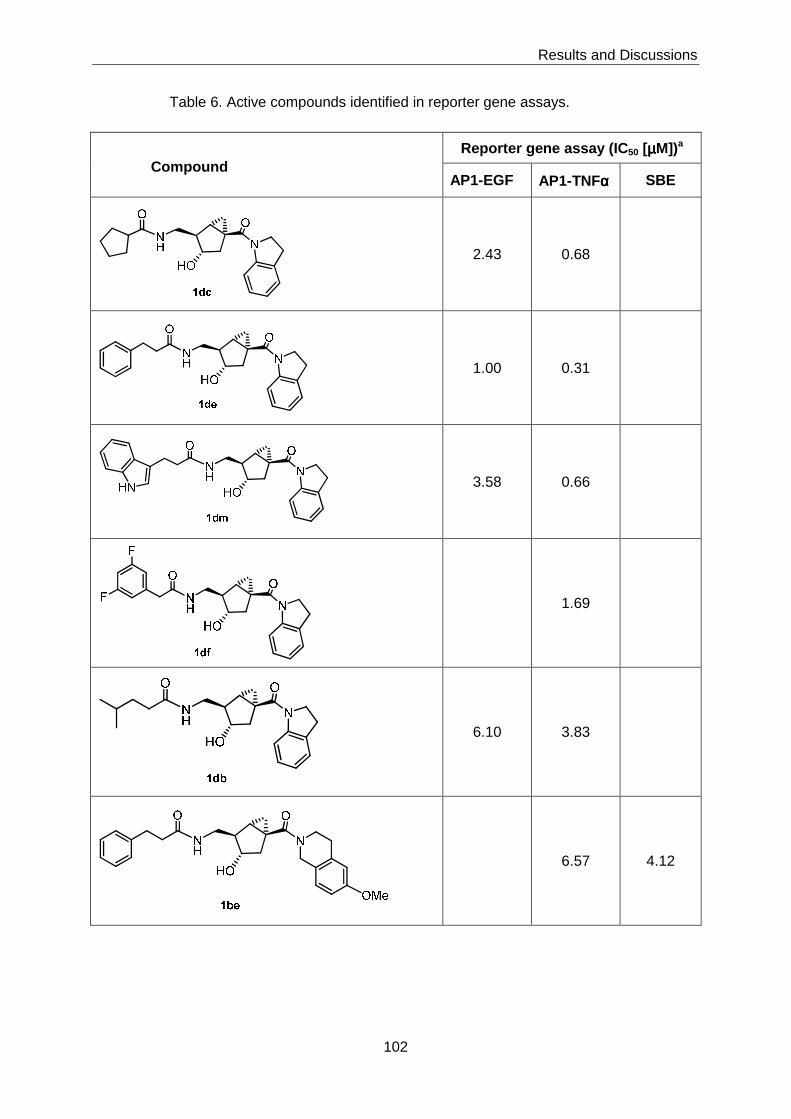
Results and Discussions
102
Table 6. Active compounds identified in reporter gene assays.
Compound Reporter gene assay (IC 50 [µµµµM])a
AP1-EGF AP1-TNFαααα SBE
2.43 0.68
1.00 0.31
3.58 0.66
1.69
6.10 3.83
6.57 4.12
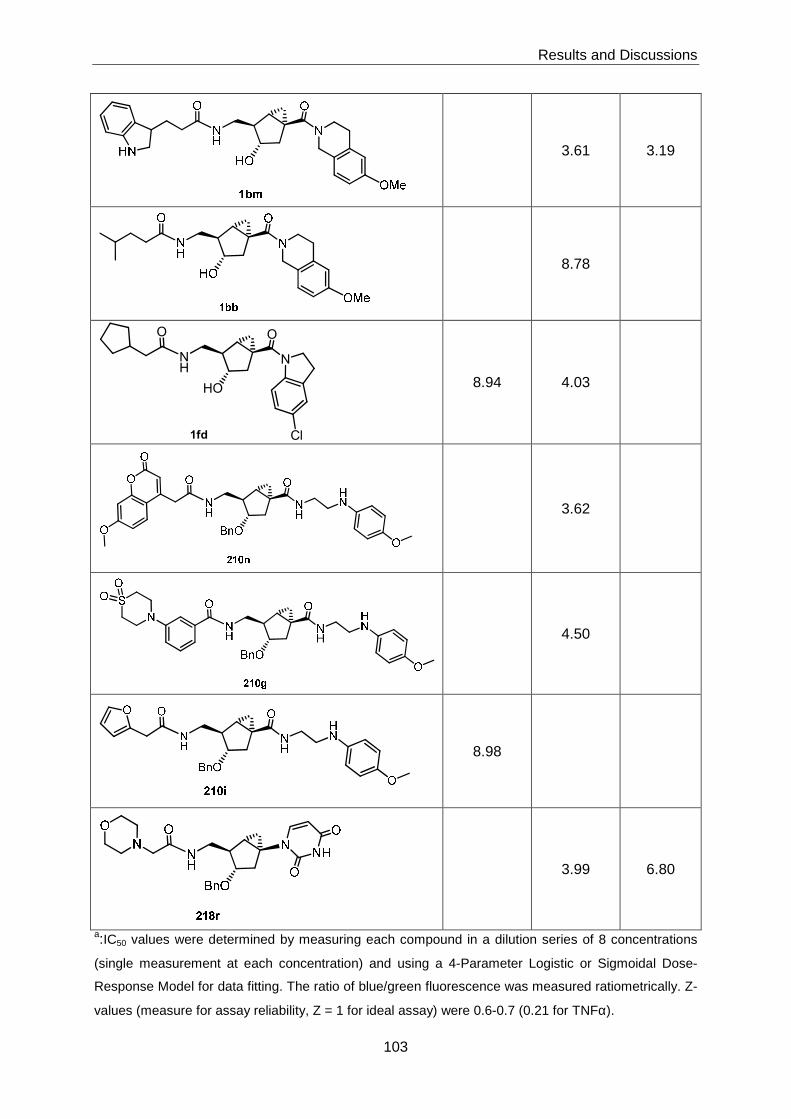
Results and Discussions
103
3.61 3.19
8.78
HO
NH
O
N
1fd Cl
O
8.94 4.03
3.62
4.50
8.98
3.99 6.80
a:IC50 values were determined by measuring each compound in a dilution series of 8 concentrations
(single measurement at each concentration) and using a 4-Parameter Logistic or Sigmoidal Dose-
Response Model for data fitting. The ratio of blue/green fluorescence was measured ratiometrically. Z-
values (measure for assay reliability, Z = 1 for ideal assay) were 0.6-0.7 (0.21 for TNFα).

Results and Discussions
104
The epidermal growth factor (EGF) receptor is a tyrosine kinase and together with the three
additional members ERBB2-ERBB4 constitutes the EGF receptor family. Activating mutants
and over-expression of these family members contribute to oncogenesis by inducing cells to
proliferate and to resist apoptosis. EGFR signals through the RAS/ERK-, the PI3-kinase and
the JAK/STAT pathways (Fig. 40).160
Figure 40. EGF-mediated signaling pathways.
TNFα acts through three main pathways upon binding to the TNFα-receptor type 1 and
subsequent TRADD binding; the pathways involving NFκB, the MAPK pathways, both
associated with cell proliferation and inflammation, and the pathways signaled through
FADD, leading to apoptosis.
Activity against EGF- or TNFα-mediated AP-1 activation seems to benefit from an indoline
moiety at the carboxy terminus, as all compounds active in the sub-micromolar range share
this structural feature; only two compounds with a structurally similar (methoxylated)
tetrahydroisoquinoline moiety were found to exhibit effects below a concentration of 10 µM.
Of these two compounds, 1be bears the same N-terminal capping group as 1de, but is ca.
20-fold less potent in the AP1-TNFα assay than the latter.

Results and Discussions
105
No specific conclusions can be drawn with regard to the nature of the N-terminal moiety,
except that polar acyl residues like in compounds 1dg and 1do are not tolerated.
3.6.2 In Vitro Protease Inhibition
By cleaving proteins, proteases are involved in a large number of key physiological
processes such as cell-cycle progression, cell proliferation and cell death, DNA replication,
tissue re-modelling, haemostasis (coagulation) and the immune response. We chose to
investigate the potential inhibitory activity of the bicyclo[3.1.0]hexane based chemical library
on the proteases thrombin, cathepsin B and urokinase (uPA). As our small chemical library
consists of compounds representing general peptidic structures, some of them also including
natural desamino acids, an interaction with proteases was well conceivable.
Thrombin is a serine protease and a potential target for antithrombotic agents.161 Known
inhibitors include benzamidines, benzoxazinones and macrocyclic peptides. Urokinase (uPA)
is a serine protease whose expression is upregulated in a number of diseases, e.g. in
cancer.162 Cathepsin B is a cysteine protease whose overexpression has been associated
with certain tumors163 and Alzheimer’s disease.164 The potential inhibitory activities against
these proteases were determined in kinetic assays using fluorometric substrates: if the
protease is inhibited by a certain compound, it cannot cleave the substrate to the same
extent anymore and therefore the fluorescence, which is released upon cleavage, drops.165
3.6.2.1 Thrombin Inhibition Assay
Thrombin (100 nM) in PBS (20 mM NaH2PO4, 30 mM Na2HPO4, 100 mMNaCl, pH 7.4) was
incubated with 84 library members as potential inhibitors at a concentration of 10 µM in 384-
well microtiter plates for 30 min at room temperature. The reaction was started by the
addition of the fluorogenic substrate Z-GGR-AMC (Bachem, Bubendorf, Switzerland, Fig. 43)
dissolved in PBS and 1% DMSO to a final concentration of 0.25 mM in a total volume of
50 µl.

Results and Discussions
106
O
HN O
NH
O
O NH
O
NH
O ONH
H2N
NH
Figure 43. Structure of thrombin and uPA substrate Z-GGR-AMC.
The change in fluorescence signal (ex: 360 nm, em: 460 nm) was recorded over 30 min
using a SpectraMaxmicroplate reader (Molecular Devices). As a positive control a 0.5 µM
solution of the known inhibitor hirudin fragment 54-65 was employed.
No thrombin inhibitory activity was found for any of the 84 tested library members at a
concentrations of 10 µM (Fig. 44). Figure 33 shows the dispersion of the fluorescence
intensities between 10’000 and 20’000 RFU for individual compounds, but this finding had to
be attributed partly to the assay conditions. The measurements were repeated at least three
times under identical conditions and gave a similar dispersion each time, but no correlating
with individual compounds was observed. The library compounds corresponding to lower
fluorescence values in at least two of three or four repeat experiments were additionally
investigated in dilution series, in which the concentrations of the library compounds were
increased up to 1 mM, but no inhibitor was found even at this concentration.
The large dispersion is also caused by the fact that certain library members such as 1bb ,
1bo , 1cg , 1cn , and 218a, 218ab, 218ac boosted fluorescence. The mechanism underlying
this effect remains unclear. The enhanced fluorescence could not be attributed to auto-
fluorescence of the corresponding compounds, as measurements showed.
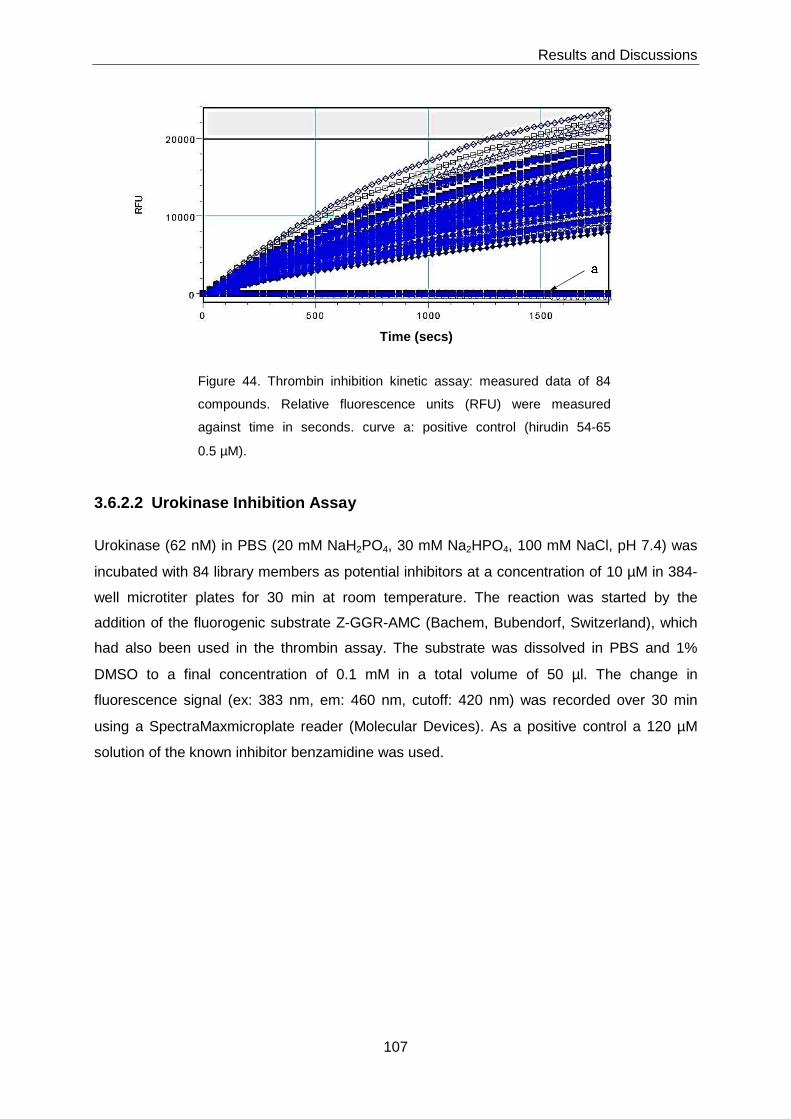
Results and Discussions
107
Figure 44. Thrombin inhibition kinetic assay: measured data of 84
compounds. Relative fluorescence units (RFU) were measured
against time in seconds. curve a: positive control (hirudin 54-65
0.5 µM).
3.6.2.2 Urokinase Inhibition Assay
Urokinase (62 nM) in PBS (20 mM NaH2PO4, 30 mM Na2HPO4, 100 mM NaCl, pH 7.4) was
incubated with 84 library members as potential inhibitors at a concentration of 10 µM in 384-
well microtiter plates for 30 min at room temperature. The reaction was started by the
addition of the fluorogenic substrate Z-GGR-AMC (Bachem, Bubendorf, Switzerland), which
had also been used in the thrombin assay. The substrate was dissolved in PBS and 1%
DMSO to a final concentration of 0.1 mM in a total volume of 50 µl. The change in
fluorescence signal (ex: 383 nm, em: 460 nm, cutoff: 420 nm) was recorded over 30 min
using a SpectraMaxmicroplate reader (Molecular Devices). As a positive control a 120 µM
solution of the known inhibitor benzamidine was used.
Time (secs)
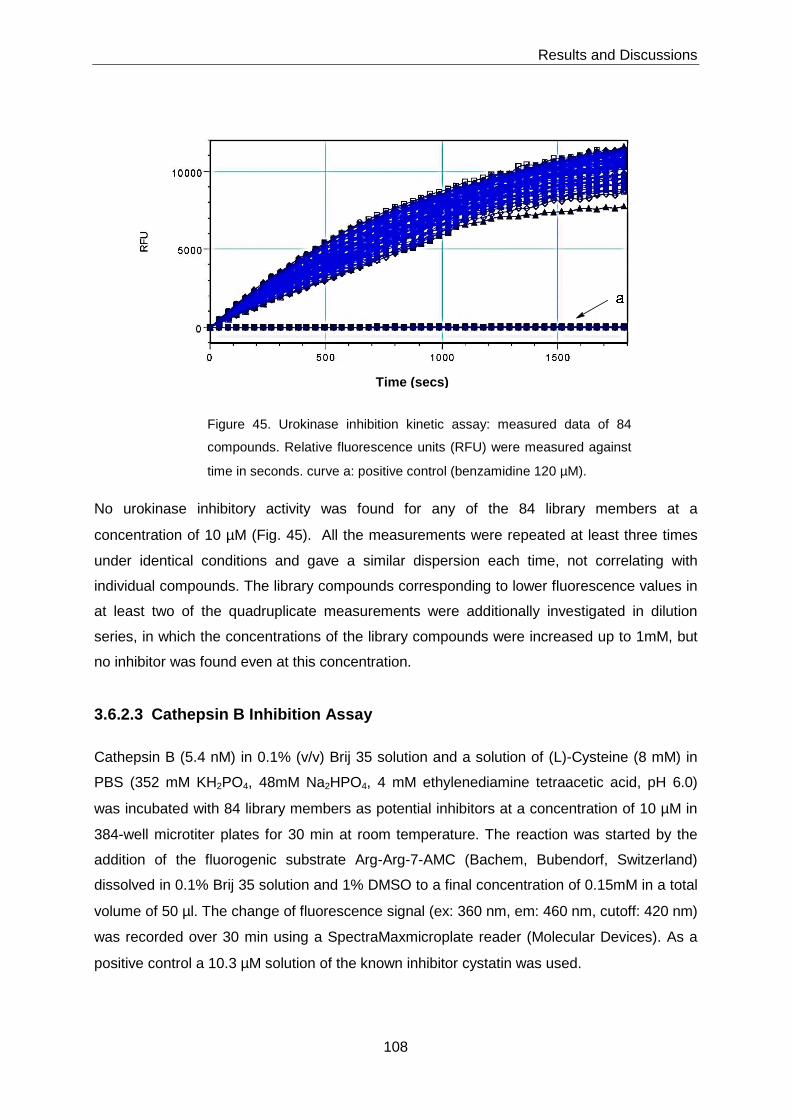
Results and Discussions
108
Figure 45. Urokinase inhibition kinetic assay: measured data of 84
compounds. Relative fluorescence units (RFU) were measured against
time in seconds. curve a: positive control (benzamidine 120 µM).
No urokinase inhibitory activity was found for any of the 84 library members at a
concentration of 10 µM (Fig. 45). All the measurements were repeated at least three times
under identical conditions and gave a similar dispersion each time, not correlating with
individual compounds. The library compounds corresponding to lower fluorescence values in
at least two of the quadruplicate measurements were additionally investigated in dilution
series, in which the concentrations of the library compounds were increased up to 1mM, but
no inhibitor was found even at this concentration.
3.6.2.3 Cathepsin B Inhibition Assay
Cathepsin B (5.4 nM) in 0.1% (v/v) Brij 35 solution and a solution of (L)-Cysteine (8 mM) in
PBS (352 mM KH2PO4, 48mM Na2HPO4, 4 mM ethylenediamine tetraacetic acid, pH 6.0)
was incubated with 84 library members as potential inhibitors at a concentration of 10 µM in
384-well microtiter plates for 30 min at room temperature. The reaction was started by the
addition of the fluorogenic substrate Arg-Arg-7-AMC (Bachem, Bubendorf, Switzerland)
dissolved in 0.1% Brij 35 solution and 1% DMSO to a final concentration of 0.15mM in a total
volume of 50 µl. The change of fluorescence signal (ex: 360 nm, em: 460 nm, cutoff: 420 nm)
was recorded over 30 min using a SpectraMaxmicroplate reader (Molecular Devices). As a
positive control a 10.3 µM solution of the known inhibitor cystatin was used.
Time (secs)
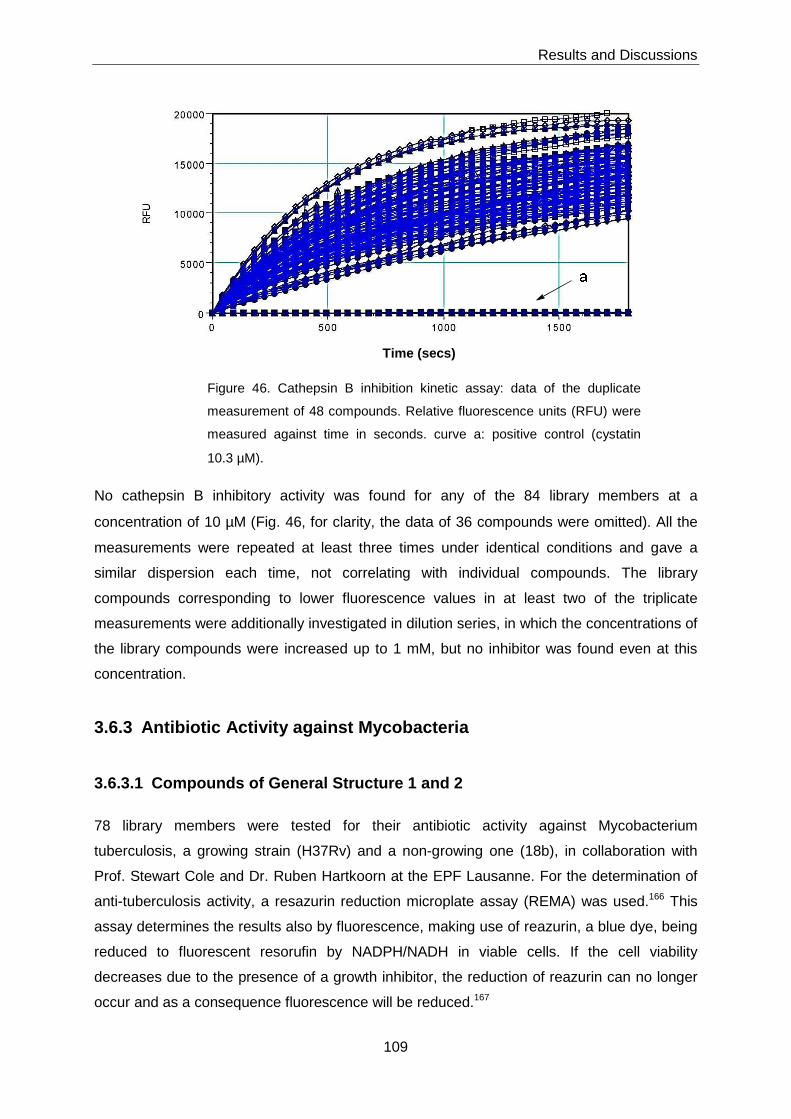
Results and Discussions
109
Figure 46. Cathepsin B inhibition kinetic assay: data of the duplicate
measurement of 48 compounds. Relative fluorescence units (RFU) were
measured against time in seconds. curve a: positive control (cystatin
10.3 µM).
No cathepsin B inhibitory activity was found for any of the 84 library members at a
concentration of 10 µM (Fig. 46, for clarity, the data of 36 compounds were omitted). All the
measurements were repeated at least three times under identical conditions and gave a
similar dispersion each time, not correlating with individual compounds. The library
compounds corresponding to lower fluorescence values in at least two of the triplicate
measurements were additionally investigated in dilution series, in which the concentrations of
the library compounds were increased up to 1 mM, but no inhibitor was found even at this
concentration.
3.6.3 Antibiotic Activity against Mycobacteria
3.6.3.1 Compounds of General Structure 1 and 2
78 library members were tested for their antibiotic activity against Mycobacterium
tuberculosis, a growing strain (H37Rv) and a non-growing one (18b), in collaboration with
Prof. Stewart Cole and Dr. Ruben Hartkoorn at the EPF Lausanne. For the determination of
anti-tuberculosis activity, a resazurin reduction microplate assay (REMA) was used.166 This
assay determines the results also by fluorescence, making use of reazurin, a blue dye, being
reduced to fluorescent resorufin by NADPH/NADH in viable cells. If the cell viability
decreases due to the presence of a growth inhibitor, the reduction of reazurin can no longer
occur and as a consequence fluorescence will be reduced.167
Time (secs)

Results and Discussions
110
Compound 1ea showed some activity against the growing H37Rv strain (table 7), but its
activity is substantially lower than that of rifampicin, which was used as the reference
compound in the assay and is a standard drug in tuberculosis treatment. None of the tested
compounds showed any activity against the non-growing M. tuberculosis strain 18b.
Table 7. Results of the screening of 78 library compounds for anti-tuberculosis activity. 1ea
showed a moderate inhibiting effect on growing M.tuberculosis (H37Rv).
Conc. compound
growing
M.tuberculosis
(H37Rv)
non-growing
M.tuberculosis
(18b)
20 µM
3742 RFUa 20'000 RFUb
0.5 µM rifampicin 2754 RFU 6487 RFU
a: Average value of a duplicate measurement. b: Against non-growing M.tuberculosis, single measurements were carried out. RFU = relative fluorescence units.
3.6.3.2 Antibiotic Activity against Mycobacterium Tuberculosis of 5-Alkynyl-
deoxypyrimidine Nucleoside Analogue 243
Bicyclo[3.1.0]hexane based 5-alkynyl-deoxypyrimidine nucleoside analogue 243 was tested
for antimycobacterial activity (collaboration with Dr. Thomas Keller at the Novartis Institute for
Tropical Diseases, in Singapore).
Figure 47. Structures of 2’-deoxy-nucleoside 244 and its bicyclo[3.1.0]hexane-based analogue 243.
The corresponding 5-alkynyl-2’-deoxypyrimidine nucleoside 244 was reported in 2005 (see
section 3.2) to inhibit M.bovis at a minimum inhibiting concentration (MIC90) of 50 µg per ml,
with some activity against other mycobacteria, e.g. M.avium (Table 8).
Table 8. Antimycobacterial activity of 5-alkynyl-dideoxypyrimidine168 nucleoside 244.

Results and Discussions
111
compound
M.bovis
% inhibition
(concentration, µg/ml)a
MIC90 (µg/ml)
M.avium
% inhibition
(concentration, µg/ml)a
244 90 (100, 50), 70 (10), 25(1) 50 75 (100), 0 (50)
MIC90: Concentration of compound exhibiting 90% inhibition on mycobacterial growth a: Antimicobacterial activity was determined at concentrations 100, 50, 10, and 1 µg/ml.
However, the synthesized bicyclo[3.1.0]hexane-based analogue 243, corresponding to a
southern conformation of the nucleoside, exhibited no antimycobacterial activity at all.
This finding can mean that the molecular target(s) of 244 in the bacteria interact(s)
preferentially or even exclusively with the northern conformer of the nucleoside. Rai and co-
workers found in 2007 that the corresponding 5-alkynyl-dideoxy-pyrimidine nucleoside was
even more active than 244 by a factor of 25-50 regarding the inhibition of M.bovis.169
3.6.4 Cytotoxicity
A subset of 78 library compounds were tested for their ability to inhibit human cancer cell
proliferation (collaboration with Prof. Jürg Gertsch and Dr. Andrea Chicca at the University of
Bern). The cytotoxicity of the library compounds against MCF-7 (breast cancer) and U937
(lymphoma) cells was determined after 72 hours incubation time in a WST-1 (4-[3-(4-
iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzen disulphate) based cell viability
assay.170 The assay is based on the enzymatic cleavage of the tetrazolium salt WST-1 to
formazan by cellular mitochondrial dehydrogenases present in viable cells, and the extent of
the cleavage is then measured colorimetrically. All compounds were tested in a
concentration of 5 µM. Every test was performed in triplicate.
There was no significant effect detectable on the proliferation of U937 cells for any of the
library compounds (Fig. 48). However, proliferation of MCF-7 cells was inhibited by library
members 218r, 1ai, 1fd , 1bc , 1cg , 1be, 1bm and 210i in the range of 35-42%. Noticeably,
compound 1fd increased cell proliferation by 42%. In MCF-7 cells, cell-growth is known to be
regulated by estrogen receptors.171 The experimental finding regarding compound 1fd may
suggest that this compound is exhibiting its activity through these receptors. The compounds
diminishing cell viability may also act on these receptors.
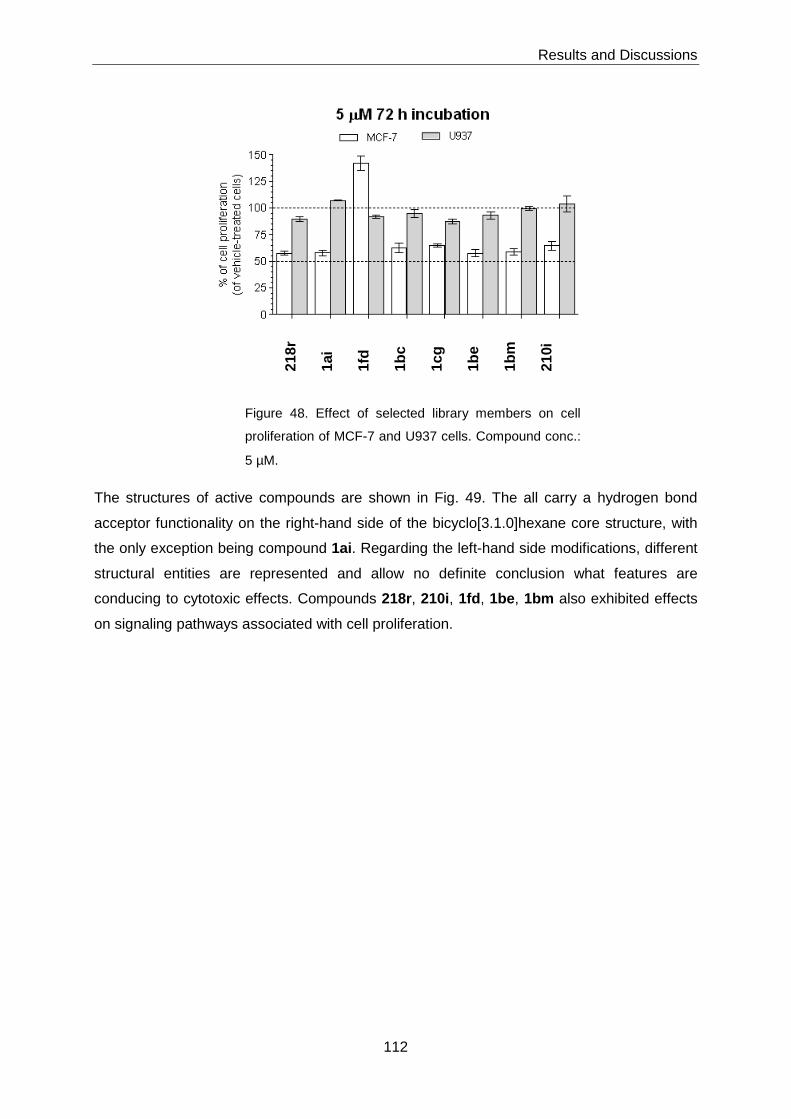
Results and Discussions
112
Figure 48. Effect of selected library members on cell
proliferation of MCF-7 and U937 cells. Compound conc.:
5 µM.
The structures of active compounds are shown in Fig. 49. The all carry a hydrogen bond
acceptor functionality on the right-hand side of the bicyclo[3.1.0]hexane core structure, with
the only exception being compound 1ai. Regarding the left-hand side modifications, different
structural entities are represented and allow no definite conclusion what features are
conducing to cytotoxic effects. Compounds 218r, 210i, 1fd , 1be, 1bm also exhibited effects
on signaling pathways associated with cell proliferation.
218
r
1ai
1fd
1bc
1cg
1be
1bm
210
i
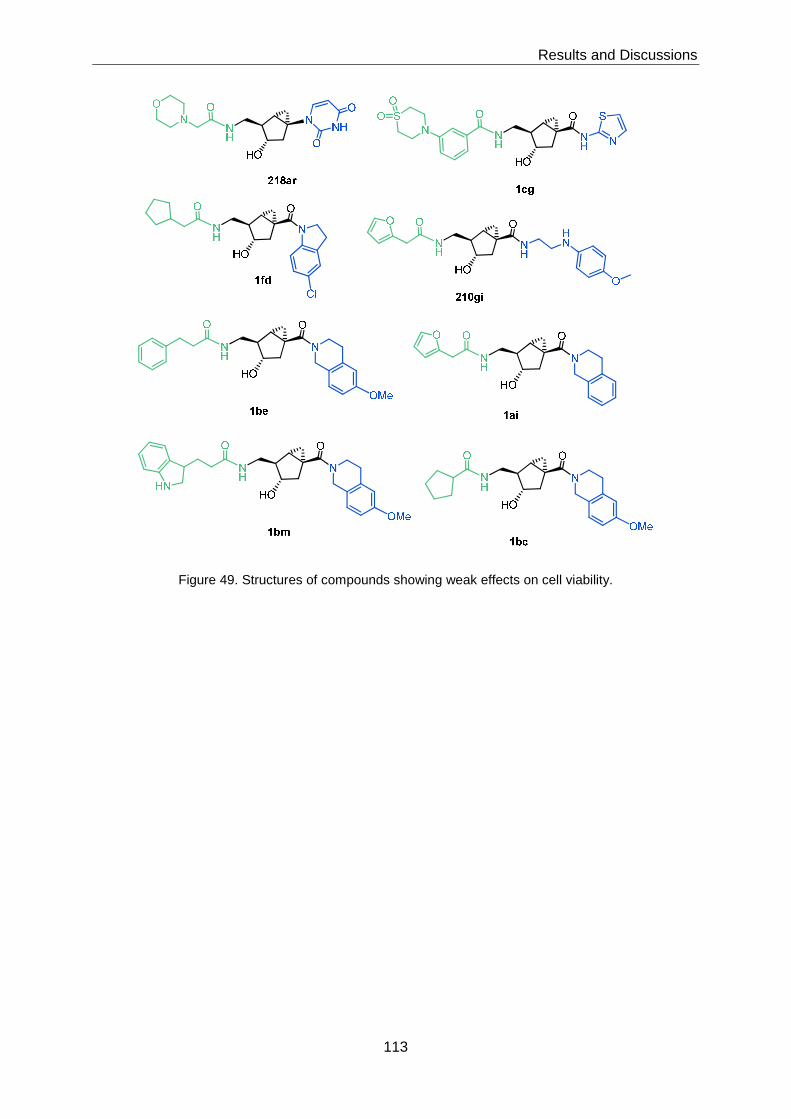
Results and Discussions
113
Figure 49. Structures of compounds showing weak effects on cell viability.

Results and Discussions
114
3.7 Biological Evaluation of the Bicyclo[3.1.0]hexa ne-based S-
Adenosylhomocysteine Analogue
3.7.1 Inhibition of AdoMet-dependant Methyltransfer ases by S-
Adenosylhomocysteine analogue 7
The group of AdoMet-dependant methyltransferases includes 7 DNA methyltransferases, 5
RNA methyltransferases, 4 protein methyltransferases and 4 small molecule methyl-
transferases acting on the carbon, oxygen or nitrogen atoms of their substrates. For the
biological evaluation of bicyclo[3.1.0]hexane based S-adenosylhomocysteine analogue 7, the
conformationally restricted analogue of (S)-Adenosylhomocysteine, we chose to investigate
the inhibitory effect on dengue N-7 and O-2’ methyltransferases, as this enzyme represents a
valid target for antiviral agents, due to a virus-conserved cavity located next to its co-
substrate, S-adenosylmethionine (AdoMet). The cavity has been shown to be occupied by
product inhibitor S-adenosylhomocysteine and related compounds, whereas these small
molecules had no inhibitory effect on related human enzymes.172
Dengue methyltransferase sequentially methylates viral RNA at the N7 and O-2’ positions,
using adenosylmethionine as a co-substrate. Using different RNA substrates and buffer
conditions, it is possible to examine the N7 and the O-2’ methylation by Dengue
methyltransferase separately. After incubation of the enzyme with various concentrations of
7, radioactively labeled AdoMet [S-adenosyl-L-(methyl-3H)-methionine] was added and the
levels of methylation were monitored (Fig. 50) in a scintillation proximity assay.173 The assay
employs biotinylated RNA substrates bound to streptavidin beads which emit light upon
methylation (binding of the radioactive methyl group). This event enables photometric
detection of methylation.
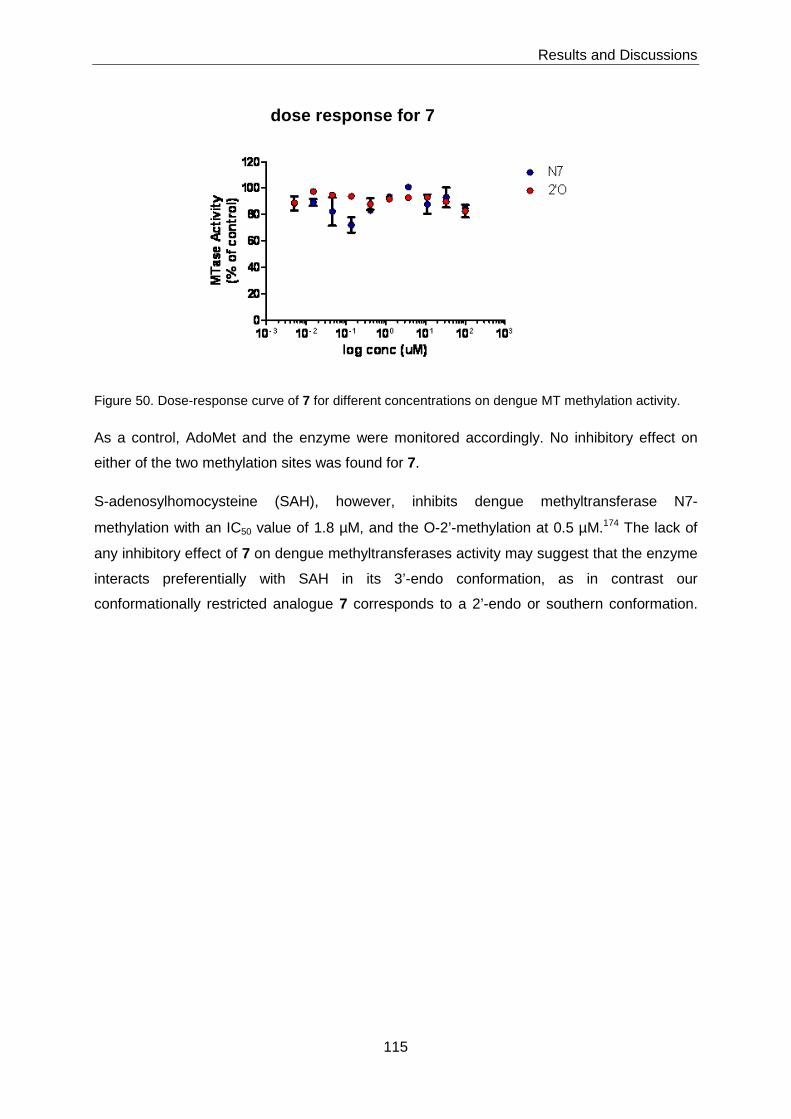
Results and Discussions
115
Figure 50. Dose-response curve of 7 for different concentrations on dengue MT methylation activity.
As a control, AdoMet and the enzyme were monitored accordingly. No inhibitory effect on
either of the two methylation sites was found for 7.
S-adenosylhomocysteine (SAH), however, inhibits dengue methyltransferase N7-
methylation with an IC50 value of 1.8 µM, and the O-2’-methylation at 0.5 µM.174 The lack of
any inhibitory effect of 7 on dengue methyltransferases activity may suggest that the enzyme
interacts preferentially with SAH in its 3’-endo conformation, as in contrast our
conformationally restricted analogue 7 corresponds to a 2’-endo or southern conformation.
dose response for 7

Conclusions
116
4 Conclusions and Outlook
The synthesis of a small, structurally unique chemical library of 110 new bicyclo[3.1.0]hexane
derivatives has been accomplished. Due to the unique conformational properties of the
bicyclo[3.1.0]hexane system, these derivatives were considered to provide an attractive
basis for the identification of new lead structures for drug discovery. The synthesis has
exploited the efficient conversion of the bicyclic lactone 161 into acid 3 and amine 4, which
were then elaborated into individual library members. Structural studies on 3 representative
library members confirmed the expected boat conformation of the bicyclo[3.1.0]hexane core
in these derivatives, and it is very resonable to assume that this conformational preference is
also prevalent in the majority, if not all other library members.
The chemisty developed in the course of the library work was also exploited for the synthesis
of S-adenosyl homocysteine (SAH) analog 7, based on chiral amine (+)-4. SAH analog 7
appears to exist as an equlibrium mixture of two species, at least under certain conditions. In
contrast to 7, the projected synthesis of pentostatin analog 6 from amine (+)-4 could not be
implemented. Based on the experiments performed in this thesis, it would appear that the
envisioned route from (+)-4, which parallels an appraoch that had been successfully applied
to the synthesis of a cyclopentane-based pentostatin analog, is not viable.
Screening of the library against 3 proteases (thrombin, urokinase, and cathepsin B) did not
reveal any inhibitory activity. However, not definitive conclusions are possible on the basis of
the limited selection of enzymes with respect to the general potential of compounds of type 1
to act as protease inhibitors.
Investigation of the antiproliferative activity of a selection of compounds 1 against MCF-7
human breast carcinoma cells provided 8 library members with weak inhibitory activity
(inihibition of proliferation by 35-42% at 5 µM compound concentration). Interestingly, one
compound increased cell proliferation by 42% (at 5 µM); it remains to be investigated
whether these findings my reflect interaction of (at least some) of the compounds with
estrogen receptors.
The most promising biological effects were observed in cellular reporter gene assays. Thus,
4 compounds, 1dc , 1dm , 1de, and 1df , were found to selectively inhibit the EGF- or
TNFα−mediated activation of the transcription factor AP-1. The exact molecular
mechanism(s) underlying these effects are unknown at this point.

Conclusions
117
Future work should include the screening of our unique library against a variety of additional
biological targets, the elucidation of the mechanism(s) underlying the inhibition of AP-1
activation, and the establishment of a more comprehensive SAR for the inhibition of these
pathways. The chemistry established in this thesis provides a sound basis for the efficient
synthesis of additional derivatives of type 1, as would be required as part of the SAR work.

Experimental Section
118
5 Experimental Section
5.1 General Methods
All non-aqueous reactions were carried out using oven-dried or flame-dried glassware under
a positive pressure of dry argon or nitrogen unless otherwise stated. Tetrahydrofuran,
acetonitrile, toluene, diethyl ether, dichloroethane, N, N-dimethylform-amide, dimethyl
sulfoxide and methylene chloride were purchased as anhydrous from Fluka. All chemicals
were purchased from Acros, Aldrich, Fluka, Merck, Lancaster, ABCR or TCI and used as
such unless otherwise stated. Deuterated solvents were obtained from Aldrich and
Cambridge Isotope Labs.
Reactions were magnetically stirred if not indicated otherwise and monitored by thin layer
chromatography using Merck silica gel 60 F254 TLC aluminium backed plates and visualized
by fluorescence quenching under UV light. In addition, TLC plates were stained with cerium
molybdate or potassium permanganate stain. Chromatographic purification was performed
as flash chromatography on Fluka Silica Gel 60 (230-400 mesh) using a forced flow of eluant
at 0.3 bar. Technical grade solvents were employed, which were distilled prior to use.
Concentration under reduced pressure was performed by rotary evaporation at 40 °C at the
appropriate pressure. Purified compounds were further dried for 12 – 48 h under high
vacuum (0.01 – 0.05 Torr). Yields refer to chromatographically purified and spectroscopically
pure compounds, unless stated otherwise.
Melting points: Melting points were measured on a Büchi B-540 melting point apparatus
using open glass capillaries and are uncorrected.
Optical rotations: Optical rotations were measured on a JASCO P-1020 digital polarimeter at
the sodium D line with a 100 mm or 10 mm path length cell, and are reported as follows:
[α]DT, concentration (g/100 ml), and solvent.
NMR spectra: 1H- and 13C-NMR spectra were recorded on a Bruker AV-400 400 MHz and a
Bruker DRX-500 500MHz spectrometer at room temperature. Chemical shifts (δ) are
reported in ppm with the solvent resonance as the internal standard relative to chloroform (δ
7.26 ppm for 1H and 77.0 ppm for 13C). All 13C spectra were measured with complete proton
decoupling. Data are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m =
multiplet, br = broad, coupling constants in Hz, integration.
IR spectra: IR spectra were recorded on a Jasco FT/IR-6200 instrument as thin film.
Absorptions are given in wavenumbers (cm–1).

Experimental Section
119
Mass spectra: Mass spectra were recorded by the MS service at ETH Zürich. EI-MS (m/z):
EI-HIRES Micromass Autospel-ULTIMA spectrometer at 70 eV. ESI-MS (m/z): IONSPEC
Ultima ESI-FT-ICR spectrometer at 4.7 T. MALDI-MS (m/z): Ion Spec Ultima HR. Low
resolution mass spectra were acquired as ESI-MS (m/z) with a Waters ZQ mass
spectrometer coupled to a Waters LC unit.
HPLC analyses: HPLC analyses were carried out on a Merck-Hitachi device using a Waters
Symmetry C18 column (3.5 µm, 4.6 x 100 mm) or an Agilent Zorbax SB-Phenyl column (3.5
µm, 4.6 x 150 mm). Semi-preparative HPLC was carried out using an Agilent Zorbax SB-
Phenyl column (5 µm, 9.4 x 150 mm). Preparative HPLC was carried out using Waters
Symmetry C18 columns (5 µm, 7.8 x 100 mm and C18, 5 µm, 19 x 100 mm).

Experimental Section
120
5.2 Experimental Procedures and Analytical Data
5.2.1 Synthesis of Bicyclo[3.1.0]hexane-based Chemi cal Library
5.2.1.1 Synthesis of Lactone 161
Lactone 161 in racemic form was prepared as described in section 6.2.4.1, except for the
fractionate crystallization step that was not carried out. Instead, 175 was directly converted to
176 and 177, respectively. In general, for the preparation of 161, procedures were followed
as described in 117b and in 117a. The analytical data were the same except for chiroptical data
that were not measured.
5.2.1.2 Synthesis of Advanced Intermediates 3, 4, a nd 5
183, 184, and 185 were prepared from 161 in the same way as described for the
enantiomeric series in section 6.2.4.2. Their analytical data were identical except for
chiroptical data that were not measured.
187
(1R,2S,3R,4S)-methyl 2-(chloromethyl)-4-hydroxy-3-
(hydroxymethyl)cyclopentanecarboxylate
To 161 dissolved in MeOH (3 ml), TMSCl (0.63 g, 5.810 mmol) was added under stirring,
then ZnCl2 (8 mg, cat.) was added and the reaction mixture was stirred at RT for 24 h. MeOH
was evaporated and the crude product, a green oil, was purified by flash column
chromatography (EtOAc/MeOH 11:1) to afford 80 mg (0.359 mmol, 62%) of the product as a
colorless oil.
Rf = 0.4 (EtOAc/MeOH 11:1).
1H-NMR (400 MHz, CDCl3): δ = 4.10-4.04 (m, 1H), 3.79 (dd, J = 10.7, 5.0 Hz, 1H), 3.70 (s,
3H), 3.71-3.66 (m, 1H), 3.66-3.61 (m, 1H), 3.58 (dd, J = 11.0, 8.0 Hz, 1H), 3.06-3.01 (m, 1H),
2.38-2.29 (m, 1H), 2.20-2.12 (m , 1H), 2.05-1.98 (m, 1H), 1.97-1.89 (m, 1H).
13C-NMR (100 MHz, CDCl3): δ = 175.9, 75.3, 63.3, 53.2, 52.2, 45.5, 45.0, 44.2, 37.5.

Experimental Section
121
HR-MS (ESI): calcd for C9H16ClO4 [M+H]+: 223.0737, found: 223.0732.
188
(1R,2S,3R,4S)-methyl 4-((tert-butyldimethylsilyl)ox y)-3-(((tert-
butyldimethylsilyl)oxy)methyl)-2-(chloromethyl)cycl opentanecarboxylate
To a solution of 187 (0.49 g, 2.201 mmol) in 6 ml of CH2Cl2 MTBSA (1.72 g, 9.903 mmol) was
added drop by drop (over 10 min) under cooling to 0°C. The reaction mixture was stirred at
RT for 16 h and diluted with 20 ml of CH2Cl2. The organic phase was washed with water (3 x
40 ml), dried over MgSO4 and evaporated. The residue was purified by flash column
chromatography (CH2Cl2) to afford 0.61 g (1.338 mmol, 62%) of the product as a colorless
oil.
Rf = 0.5 (CH2Cl2).
1H-NMR (400 MHz, CDCl3): δ = 4.03-3.96 (m, 1H), 3.72-3.67 (m, 2H), 3.69 (s, 3H), 3.66-
3.63 (m, 2H), 2.95-2.86 (m, 1H), 2.54-2.41 (m, 1H), 2.16-2.08 (m, 1H), 1.96-1.87 (m, 2H),
0.89 (s, 9H), 0.87 (s, 9H) 0.05 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H), 0.04 (s, 3H).
13C-NMR (100 MHz, CDCl3): 174.1, 72.6, 61.7, 53.2, 51.8, 45.7, 43.2, 42.5, 37.6, 25.9, 25.8,
18.2, 17.9, -4.5, -4.9, -5.5, -5.6.
MS (ESI): calcd for C21H44ClO4Si2 [M+H]+: 451.25, found: 450.79.
185
(1S,3S,4R,5S)-methyl 3-((tert-butyldimethylsilyl)ox y)-4-(((tert-
butyldimethylsilyl)oxy)methyl) bicyclo[3.1.0]hexane -1-carboxylate
188 (8.6 g, 0.019 mol) was dissolved in 60 ml of tert-BuOH, then tert-BuOK (2.87 g, 0.025
mol) was added slowly under stirring. The resulting suspension was stirred for 20h at RT,
then additional 660 mg of tert-BuOK were added. After additional 2h of stirring the reaction
mixture was poured on 600 ml of Et2O and extracted with ice water (3x 200 ml). The organic
phase was dried over MgSO4 and evaporated. Purification by flash column chromatography
(CH2Cl2/Et3N 0.5%) yielded 5.06 g (0.012 mmol, 64%) of the product as a white solid.

Experimental Section
122
The analytical data were identical to the ones stated for 185 in enantiomeric form (section
6.2.4.2), except for chiroptical data that were not measured.
190
(1S,3S,4R,5S)-methyl 3-((tert-butyldimethylsilyl)ox y)-4-
(hydroxymethyl)bicyclo[3.1.0]hexane-1-carboxylate
To a solution of 185 (4.5 g, 0.011 mol) in CH2Cl2/MeOH 1:1 (100 ml) CSA (3.1 g, 0.013 mol)
was added at 0°C. The reaction mixture was stirred for 2.5 h at 0°C. The reaction was
quenched with saturated aqueous NaHCO3 –solution and extracted with CH2Cl2 (4 x 120 ml).
The combined organic phases were dried over MgSO4 and evaporated. The residue was
purified by flash column chromatography (EtOAc/hex 1:2) to give 2.21 g (0.007 mol, 67%) of
the product as a colorless oil.
Rf = 0.34 (EtOAc/Hex 1:1)
1H-NMR (400 MHz, CDCl3): δ = 4.16 (d, J = 6.6 Hz, 1H), 3.63 (s, 3H), 3.53 (dd, J = 10.7, 6.5
Hz, 1H), 3.42 (dd, J = 10.7, 7.7 Hz, 1H), 2.51 (ddd, J = 14.2, 6.8, 1.1 Hz, 1H), 2.07 (t, J = 7.2
Hz, 1H), 1.80 (d, J = 14.2 Hz, 1H), 1.75 (dd, J = 8.9, 5.6 Hz, 1H), 1.58 (s, br, 1H), 1.47-1.41
(m, 2H), 1.24 (t, 7.3 Hz, 1H), 0.84 (s, 9H), 0.01 (s, 6H).
13C-NMR (100 MHz, CDCl3): δ = 175.1, 75.5, 64.6, 53.1, 51.6, 37.0, 32.2, 30.6, 25.8, 20.7,
17.6, -4.8.
MS (ESI): calcd for C15H29O4Si [M+H]+: 301.18, found: 301.13 (I = 100%).
191
(1S,3S,4R,5S)-methyl 3-((tert-butyldimethylsilyl)ox y)-4-(((methylsulfonyl)oxy)methyl)
bicyclo[3.1.0]hexane-1-carboxylate
Mesylchloride (0.5 g, 4.413 mmol) was added dropwise at 0°C to a stirred solution of 190
(0.884 g, 2.942 mmol) and Et3N (0.62 ml, 4.413 mmol) in Et2O (20 ml). The resulting
suspension was stirred for 3 h, then it was diluted with water and Et2O and the phases were

Experimental Section
123
separated. The organic phase was washed with water, dried over MgSO4 and evaporated to
afford 1.1 g (2.816 mmol, 96%) of the product as a colorless oil.
Rf = 0.31 (EtOAc/Hex 1:1)
1H-NMR (400 MHz, CDCl3): δ = 4.19 (d, J = 6.5 Hz, 1H), 4.12 (dd, J = 9.9, 5.5 Hz, 1H), 4.02
(dd, J = 10.2, 7.9 Hz, 1H), 3.66 (s, 3H), 3.02 (s, 3H), 2.57 (dd, J = 14.2, 6.5 Hz, 1H), 2.31
(7.6, 5.8 Hz, 1H), 1.86 (d, J = 14.4 Hz, 1H), 1.75 (t, J = 6.8 Hz, 1H), 1.50 (d, J = 6.8 Hz, 2H),
0.86 (s, 9H), 0.03 (s, 6H).
13C-NMR (100 MHz, CDCl3): δ = 174.3, 75.2, 69.6, 51.9, 49.9, 37.7, 36.7, 31.0, 30.6, 25.5,
20.6, 18.1, -4.8, -4.9.
MS (ESI): calcd for C16H31NaO6SSi [M+H]+: 401.14, found: 401.19 (I = 90%).
192
(1S,3S,4R,5S)-methyl 4-(azidomethyl)-3-((tert-
butyldimethylsilyl)oxy)bicyclo[3.1.0]hexane-1-carbo xylate
Sodium azide (1.195 g, 0.018 mol) was added to a solution of 191 (2.91 g, 0.007 mol) in dry
DMF (40 ml) and stirred for 20 h at 70°C. The react ion mixture was poured on water and
extracted with Et2O (4x 70 ml). The phases were separated, the combined organic phases
were dried over MgSO4 and evaporated. Purification of the residue by flash column
chromatography (Hex/EtOAc 10:1) yielded 1.82 g (0.006 mol, 76%) of the product as a
colorless oil.
Rf = 0.6 (EtOAc/Hex 1:1)
1H-NMR (400 MHz, CDCl3): δ = 4.09 (d, J = 6.5 Hz, 1H), 3.64 (s, 3H), 3.26 (dd, J = 12.1, 6.0
Hz, 1H), 3.09 (dd, J = 12.1, 8.3 Hz, 1H), 2.53 (ddd, J = 14.4, 6.9, 1.1 Hz, 1H), 2.11 (t, J = 7.3
Hz, 1H), 1.82 (d, J = 14.1 Hz, 1H), 1.72 (dd, J = 8.8, 5.6 Hz, 1H), 1.49-1.43 (m, 2H), 0.84 (s,
9H), 0.02 (s, 3H), 0.02 (s, 3H).
13C-NMR (400 MHz, CDCl3): δ = 174.6, 76.1, 53.9, 50.4, 36.9, 31.9, 30.9, 25.7, 20.4, 17.9, -
4.8, -4.9.
HR-MS (ESI): calcd for C15H28N3O3Si [M+H]+: 326.1894, found: 326.1903.

Experimental Section
124
3
(1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimethy lsilyl)oxy)bicyclo[3.1.0]hexane-1-
carboxylic acid
192 (240 mg) was dissolved in THF (5 ml) and 3 drops of DMF and potassium
trimethylsilanolate (350 mg) was added immediately. The reaction mixture was heated in the
microwave to 80°C for 2 h. The reaction mixture was quenched by the addition of ice and 2 N
HCl that was added dropwise. The water phase was extracted with EtOAc (3x 30 ml), the
combined organic phases were dried over MgSO4 and evaporated to give 212 mg (92%) of a
colorless oil that crystallized to a white solid upon storage in the fridge.
Rf = 0.18 (CH2Cl2)
1H-NMR (400 MHz, CDCl3): δ = 4.11 (d, J = 7.0 Hz, 1H), 3.28 (dd, J = 12.3, 6.0 Hz, 1H), 3.12
(dd, J = 12.3, 8.2 Hz, 1H), 2.53 (ddd, J = 14.5, 7.0, 1.2 Hz, 1H), 2.13 (dd, J = 8.4, 7.0 Hz,
1H), 1.87-1.79 (m, 2H), 1.59-1.53 (m, 2H), 0.85 (s, 9H), 0.03 (s, 3H), 0.02 (s, 3H).
13C-NMR (300 MHz, CDCl3): δ = 180.8, 75.9, 54.1, 50.5, 36.4, 33.4, 31.1, 25.8, 21.6, 18.2, -
4.8.
IR (film): ν 2950, 2929, 2858, 2099, 1683, 1449, 1253, 1165, 1084, 1047, 907, 837, 776,
730, 539.
MS (ESI): calcd for C14H24N3O3Si [M-H]-: 310.16, found: 310.16 (I = 100%).
253
(1S,3S,4R,5S)-methyl 4-(azidomethyl)-3-hydroxybicyc lo[3.1.0]hexane-1-carboxylate
192 (0.85 g) was dissolved in THF (20 ml), then TBAF (3.3 ml, 1 M sol. in THF) was added
slowly under stirring. The reaction mixture was stirred for 1 h at RT. 20 ml of a saturated
NH4Cl-sol. was added and the mixture was extracted with EtOAc (3x 50 ml). The organic
phases were combined, driedover MgSO4 and evaporated. Purification by flash column
chromatography (EtOAc/hex 1:1) yielded 0.5 g (90 %) of the title compound as a colorless
oil.

Experimental Section
125
Rf = 0.31 (EtOAc/hex 1:1)
1H-NMR (400 MHz, CDCl3): δ = 4.17 (dd, J = 6.9, 1.2 Hz, 1H), 3.63 (s, 3H), 3.27 (dd, J =
12.3, 6.6 Hz, 1H), 3.19 (dd, J = 12.3, 7.5 Hz, 1H), 2.61 (ddd, J = 14.6, 6.9, 1.7 Hz, 1H), 2.14
(t, J = 7.0 Hz, 1H), 2.04 (s, br, 1H), 1.84 (dd, J = 14.5, 1.2 Hz, 1H), 1.76 (ddd, J = 9.0, 5.2,
1.0 Hz, 1H), 1.52 (ddd, J = 9.0, 4.3, 1.7 Hz, 1H), 1.39 (t, J = 4.9 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 174.3, 76.0, 53.9, 52.0, 49.5, 36.6, 32.5, 31.0, 21.4.
HR-MS (ESI): calcd for C9H13N3NaO3 [M-Na]+: 234.0849, found: 234.0844.
193
(1S,3S,4R,5S)-methyl 4-(azidomethyl)-3-(benzyloxy)b icyclo[3.1.0]hexane-1-carboxylate
253 (1.68 g) was dissolved in anhydrous DMF (90 ml) and NaH (412 mg, 60% dispersion in
oil) was added under cooling to -15°C; after 5 min benzylbromide (1.79 g) was added, then
the mixture was warmed to RT and stirred for 1.5 h. The reaction was quenched with H2O
and extracted with EtOAc. The combined organic phases were dried over MgSO4 and
evaporated to afford the crude product which was purified by flash column chromatography
(EtOAc/Hex 1:12-->1:1) to yield 2.2 g (92%) of 193 as a slightly yellow oil.
Rf = 0.3 (EtOAc/Hex 1:10).
1H-NMR (400 MHz, CDCl3): δ = 7.34-7.21 (m, 5H), 4.37 (s, 2H), 3.82 (dd, J = 6.9, 0.8 Hz,
1H), 3.62 (s, 3H), 3.25 (dd, J = 12.2, 6.2 Hz, 1H), 3.12 (dd, J = 12.2, 8.0 Hz, 1H), 2.50 (ddd, J
= 14.6, 6.9, 1.7 Hz, 1H), 2.33 (t, J = 7.0 Hz, 1H), 2.05 (dd, J = 14.5, 0.6 Hz, 1H), 1.73 (ddd, J
= 9.0, 5.3, 1.0 Hz, 1H), 1.46 (ddd, J = 9.0, 4.3, 1.7 Hz, 1H), 1.38 (t, J = 4.9 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 174.6, 138.3, 128.4, 127.7, 127.5, 83.3, 71.2, 53.9, 52.0,
47.4, 33.4, 31.9, 30.9, 20.6.
IR (film): ν 3031, 2955, 2866, 2096, 1717, 1494, 1442, 1355, 1266, 1208, 1143, 1072, 1029,
904, 732, 693.
MS (ESI): calcd for C16H19N3NaO3 [M-Na]+: 324.13 , found: 323.95 (I = 60%).

Experimental Section
126
194
(1S,3S,4R,5S)-4-(azidomethyl)-3-(benzyloxy)bicyclo[ 3.1.0]hexane-1-carboxylic acid
193 (300 mg) was dissolved in THF (8 ml) and DMF (2 ml) and TMSOK (425 mg) was added
immediately. The reaction mixture was heated in the microwave at 90°C for 4 h. The reaction
mixture was quenched by adding ice, along with dropwise addition of 2 N aqueous HCl. The
mixture was extracted with EtOAc (3 x 30 ml) and the combined organic phases were dried
and evaporated. Purification by flash chromatography (CH2Cl2/MeOH 50:1) yielded 262 mg
(91%) of the product as a slightly yellow oil.
Rf = 0.11 (CH2Cl2/MeOH 50:1)
1H-NMR (400 MHz, CDCl3): δ = 7.37 – 7.26 (m, 5H), 4.42 (s, 2H), 3.88 (d, J = 6.6 Hz, 1H),
3.32 (dd, J = 12.3, 6.4 Hz, 1H), 3.18 (dd, J = 12.3, 8.0 Hz, 1H), 2.54 (ddd, J = 14.6, 7.0, 1.5
Hz, 1H), 2.44 – 2.37 (m, 1H), 2.09 (d, J = 14.7 Hz, 1H), 1.90 – 1.82 (m, 1H), 1.59 (ddd, J =
9.0, 4.4, 1.7 Hz, 1H), 1.54 – 1.48 (m, 1H).
13C-NMR (100 MHz, CDCl3): δ = 179.8, 138.4, 129.2, 128.5, 128.2, 127.8, 127.5, 125.2,
82.9, 71.3, 54.3, 47.2, 32.9, 30.8, 21.3.
IR (film): ν 2928, 2901, 2619, 2358, 2099, 1718, 1683, 1452, 1430, 1353, 1274, 1227, 1213,
1166, 1077, 1028, 912, 737, 697, 545.
HR-MS (ESI): calcd for C15H17N3NaO3 [M-Na]+: 310.1162, found: 310.1153.
5
(1S,3S,4R,5S)-4-(azidomethyl)-3-(benzyloxy)bicyclo[ 3.1.0]hexan-1-amine
194 (80 mg) was dissolved in toluene (3 ml) and cooled to 0°C, then DPPA (86 mg) and
Et3N (46 ul) were added. The reaction mixture was stirred for 1 h at 0°C, then 1.5 h at RT
and for 12 h at 80°C. During the reaction, the colo rless solution turned slightly yellow. The
crude isocyanate was diluted with THF, treated with 2 N NaOH and stirred for 30min. The
solvent was evaporated and the water phase was extracted with CH2Cl2 (3 x 50 ml). The
combined organic phases were dried over MgSO4 and evaporated. Purification by flash

Experimental Section
127
chromatography (CH2Cl2/MeOH 30:1-->CH2Cl2/MeOH 20:1) afforded 59 mg (82%) of the
product as a yellow oil.
Rf = 0.3 (EtOAc/Hex 1:1)
1H-NMR (400 MHz, MeOD): δ = 7.39 – 7.21 (m, 5H), 4.40 (s, 2H), 3.84 (d, J = 7.0 Hz, 1H),
3.54 (dd, J = 12.3, 6.4 Hz, 1H), 3.42 (ddd, J = 12.2, 8.3, 3.8 Hz, 1H), 2.31 – 2.18 (m, 2H),
2.15 (d, J = 13.8 Hz, 1H), 1.33 – 1.30 (m, 1H), 1.18 (td, J = 4.8, 1.5 Hz, 1H), 0.85 (ddd, J =
9.2, 4.7, 2.3 Hz, 1H).
13C-NMR (100 MHz, MeOD): δ = 139.1, 129.2, 128.8, 128.5, 83.6, 71.4, 55.1, 55.0, 41.7,
38.8, 28.6, 19.8.
IR (film): ν 3337, 2925, 2856, 2356, 2096, 1623, 1454, 1351, 1268, 1210, 1092, 1061, 963,
736, 697.
MS (ESI): calcd for C14H19N4O [M+H]+: 259.16, found: 259.17 (I = 100%).
4
(1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4-(( (tert-
butyldimethylsilyl)oxy)methyl)bicyclo[3.1.0] hexan- 1-amine
4 was prepared analogeously to the procedure described for (+)-4 (section 6.2.4.2) in 3 steps
from 185. The analytical data were the same, except for chiroptical data that were not
measured.

Experimental Section
128
5.2.1.3 Synthesis of library members of general str ucture 1
202a
((1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(3,4-dihydro isoquinolin-2(1H)-yl)methanone
3 (200 mg) was dissolved in CH2Cl2 (6 ml) and DIPA (272 ul) was added under stirring, after
5 min HATU (366 mg) was added. 1,2,3,4-tetrahydroisoquinoline (197a, 166 ul) was added
and the reaction mixture was stirred at RT for 24 h. The mixture was diluted with CH2Cl2, the
phases were separated and the organic phase was washed with NaHCO3 sol. and with brine,
then the organic phase was dried over MgSO4 and evaporated to dryness. The residue was
purified by flash column chromatography (Hex/EtOAc 2:1) to give 226 mg (83%) of the
product as a slightly yellow oil.
Rf = 0.33 (Hex/EtOAc 2:1)
1H-NMR (400 MHz, CDCl3): δ = 7.24-7.10 (m, 4H), 4.80-4.65 (m, 2H), 4.18 (dd, J = 6.7, 0.8
Hz, 1H), 3.94-3.76 (m, 2H), 3.44 (dd, J = 12, 6 Hz, 1H), 3.10 (dd, J = 12.5, 9.5 Hz, 1H), 2.87
(t, J = 5.8 Hz, 2H), 2.70 (ddd, J = 14.5, 6.5, 1.0 Hz, 1H), 2.12 (dd, J = 9.4, 6 Hz, 1H), 1.90
(dd, J = 13.7, 0.6 Hz, 1 H), 1.63 (ddd, J = 8.9, 4.8, 1.0 Hz, 1H), 1.39 (t, J = 4.5 Hz, 1H), 0.96
(ddd, J = 8.6, 4.2, 1.0 Hz, 1H), 0.87 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ = 171.7, 133.2, 128.8, 126.7, 126.5, 76.2, 53.9, 50.4, 39.4,
32.4, 27.2, 25.8, 18.8, 17.9, -4.7, -4.8.
IR (film): ν 2927, 2855, 2096, 1637, 1432, 1254, 1167, 1086, 1036, 931, 832, 776, 747, 674.
HR-MS (ESI): calcd for C23H35N4O2Si [M-H]+: 427.2524, found: 427.2522.

Experimental Section
129
203a
((1S,3S,4R,5S)-4-(aminomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(1,2,3,4-tetrahydronaphthalen-2-yl)methanone
202a (57 mg) was dissolved in Tol/MeOH 1:1 (1.5 ml), Pd/C (28 mg) was added and the
reaction mixture was stirred under H2 for 2 h. The reaction mixture was filtered through celite,
the residue was washed with EtOAc and the solvents were evaporated to afford 54 mg
(100%) of the crude product as a yellow oil that was immediately used to prepare
compounds 1ax.
Rf = 0.13 (EtOAc/MeOH 60:1)
1H-NMR (400 MHz, CDCl3): δ = 7.21-7.09 (m, 4H), 4.84-4.58 (m, 2H), 4.18 (d, J = 6.5 Hz,
1H), 3.88 (s, br, 2H), 3.45-3.34 (m, 1H), 2.92-2.85 (m, 2H), 2.78 (dd, J =11.5, 5.5 Hz, 1H),
2.31 (dd, J = 13.5, 6.5 Hz, 1H), 2.20-1.97 (m, 1H), 1.92-1.86 (m, 1H), 1.65 (dd, J = 8.9, 4.7
Hz, 1H), 1.39-1.34 (m, 1H), 0.99-0.94 (m, 1H), 0.87 (s, 9H), 0.03 (s, 6H).
13C-NMR (100 MHz, CDCl3): δ = 133.5, 128.8, 126.3, 77.8, 69.5, 53.2, 45.4, 40.9, 33.2, 27.7,
25.7, 18.2, 17.7, -4.4, -4.7.
HR-MS (ESI): calcd for C23H37N2O2Si [M-H]+: 401.2619, found: 401.2625.
1ax
The acids (in the amounts stated below, 0.038 mmol each) were dissolved in separate
reaction tubes in CH2Cl2 (0.3 ml each). 20-30 µl of DMF were added to each reaction tube.
204a 204b 204d 204e 204f 204g 204i 204m 204n 204p 204t
2.5ul 2.8ul 2.8 ul 3.3 mg 2.5 mg 9.7 mg 5 mg 7 mg 9 mg 8.4 mg 3 mg
DIPEA (32 ul, 0.188 mmol each) and HATU (15 mg, 0.040 mmol each) were added to each
reaction tube. The reaction mixtures were stirred for 30 min, then 203a (70 mg) was added
by syringe in equimolar amounts (10 mg, 0.025 mmol) to each reaction tube. The reaction

Experimental Section
130
mixtures were stirred for 12 h. The solutions were evaporated to dryness. The compounds
were subjected to TBS deprotection by being shaken with Dowex-50W-X8 (40 mg each) in
MeOH (0.3 ml each) for 12 h. The resin-bound acid was filtered off, the filtrates were
evaporated to dryness and the crude products were purified by flash column chromatography
to yield library members 1ax in the amounts stated below.
Amount : 4 mg. Purity : ≥ 98%. TLC: Rf 0.35 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C22H31N2NaO3 [M-Na]+: 393.22, found: 392.97 (I = 100%). 1H-NMR (400 MHz, acetone): δ = 7.52 (s,
br, 1H), 7.23-7.16 (m, 4H), 4.76 (s, br, 2H), 4.15 (d, J = 6.6 Hz, 1H), 3.97-3.86 (m, 2H), 3.54 (ddd, J =
12.7, 7.6, 4.9 Hz, 1H), 3.10 (ddd, J = 13.4, 5.6, 3.8 Hz, 1H), 2.93-2.87 (m, 2H), 2.21 (ddd, J = 13.4,
6.7, 1.4 Hz, 1H), 2.19 (t, J = 5.2 Hz, 1H), 2.11-2.07 (m, 1H), 2.04-1.97 (m, 3H), 1.54 (ddd, J = 9.0, 4.8,
1.0 Hz, 1H), 1.40 (t, J = 4.6 Hz, 1H), 1.04 (ddd, J = 9.0, 4.3, 1.4 Hz, 1H), 0.91 (d, J = 6.6 H, 3H), 0.89
(d, J = 6.6 Hz, 3H). IR (film): ν 3355, 2926, 2867, 2359, 2341, 2100, 1635, 1518, 1455, 1236, 1066,
1034, 822, 738, 698.
Amount : 6 mg. Purity : ≥ 97%. TLC: Rf 0.22 (EtOAc/MeOH 20:1, KMnO4). MS (ESI): calcd for
C25H26F2N2NaO3 [M-H]-: 439.18, found: 438.96 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 7.20-7.13
(m, 4H), 6.92 (d, J = 2.0 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.76 (td, J = 9.3, 2.0 Hz, 1H), 4.89-4.60 (m,
2H), 4.06 (d, J = 6.3 Hz, 1H), 4.01-3.79 (s, br, 2H), 3.53 (s, 2H), 3.33 (dd, J = 13.7, 5.9 Hz, 1H), 3.21
(dd, J = 13.7, 6.6 Hz, 1H), 2.89 (t, J = 5.9 Hz, 2H), 2.15 (t, J = 6.2 Hz, 1H), 2.11 (dd, J = 13.8, 6.4 Hz,
1H), 1.96 (dd, J = 13.9, 1.0 Hz, 1H), 1.62 (dd, J = 8.8, 4.4 Hz, 1H), 1.34 (t, J = 4.7 Hz, 1H), 1.04-0.98
(m, 1H).
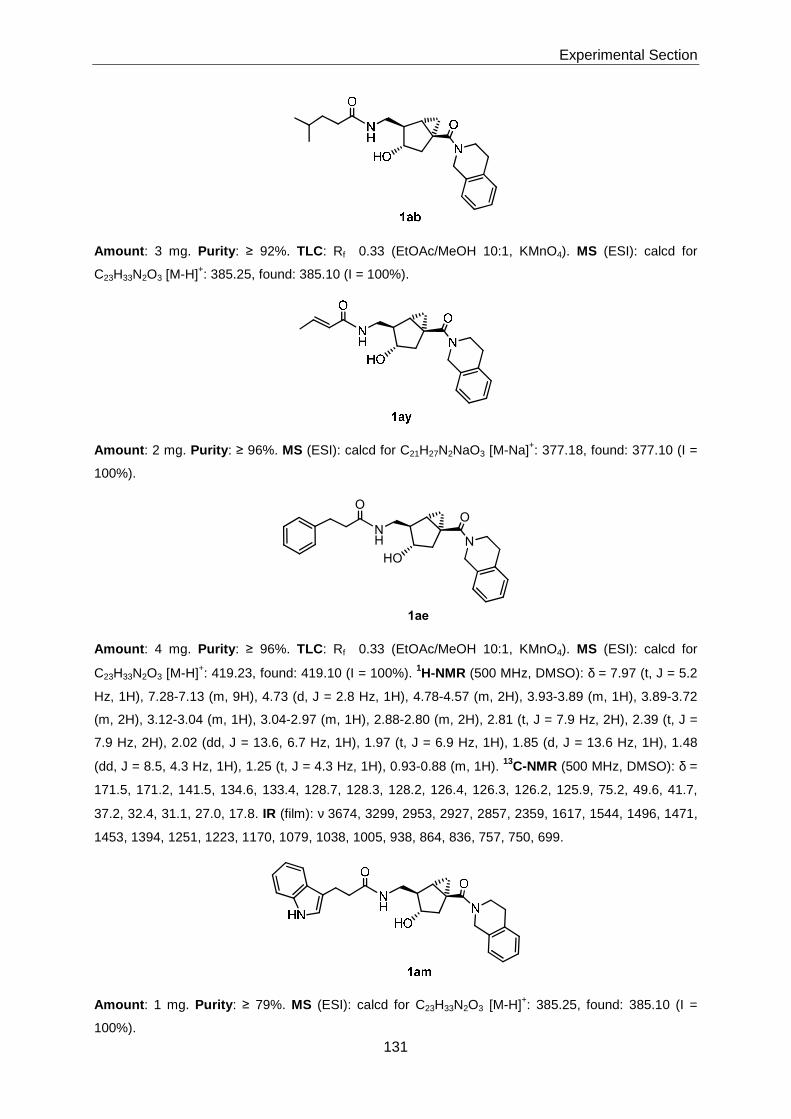
Experimental Section
131
Amount : 3 mg. Purity : ≥ 92%. TLC: Rf 0.33 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C23H33N2O3 [M-H]+: 385.25, found: 385.10 (I = 100%).
Amount : 2 mg. Purity : ≥ 96%. MS (ESI): calcd for C21H27N2NaO3 [M-Na]+: 377.18, found: 377.10 (I =
100%).
HO
NH
O
N
O
1ae
Amount : 4 mg. Purity : ≥ 96%. TLC: Rf 0.33 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C23H33N2O3 [M-H]+: 419.23, found: 419.10 (I = 100%). 1H-NMR (500 MHz, DMSO): δ = 7.97 (t, J = 5.2
Hz, 1H), 7.28-7.13 (m, 9H), 4.73 (d, J = 2.8 Hz, 1H), 4.78-4.57 (m, 2H), 3.93-3.89 (m, 1H), 3.89-3.72
(m, 2H), 3.12-3.04 (m, 1H), 3.04-2.97 (m, 1H), 2.88-2.80 (m, 2H), 2.81 (t, J = 7.9 Hz, 2H), 2.39 (t, J =
7.9 Hz, 2H), 2.02 (dd, J = 13.6, 6.7 Hz, 1H), 1.97 (t, J = 6.9 Hz, 1H), 1.85 (d, J = 13.6 Hz, 1H), 1.48
(dd, J = 8.5, 4.3 Hz, 1H), 1.25 (t, J = 4.3 Hz, 1H), 0.93-0.88 (m, 1H). 13C-NMR (500 MHz, DMSO): δ =
171.5, 171.2, 141.5, 134.6, 133.4, 128.7, 128.3, 128.2, 126.4, 126.3, 126.2, 125.9, 75.2, 49.6, 41.7,
37.2, 32.4, 31.1, 27.0, 17.8. IR (film): ν 3674, 3299, 2953, 2927, 2857, 2359, 1617, 1544, 1496, 1471,
1453, 1394, 1251, 1223, 1170, 1079, 1038, 1005, 938, 864, 836, 757, 750, 699.
Amount : 1 mg. Purity : ≥ 79%. MS (ESI): calcd for C23H33N2O3 [M-H]+: 385.25, found: 385.10 (I =
100%).
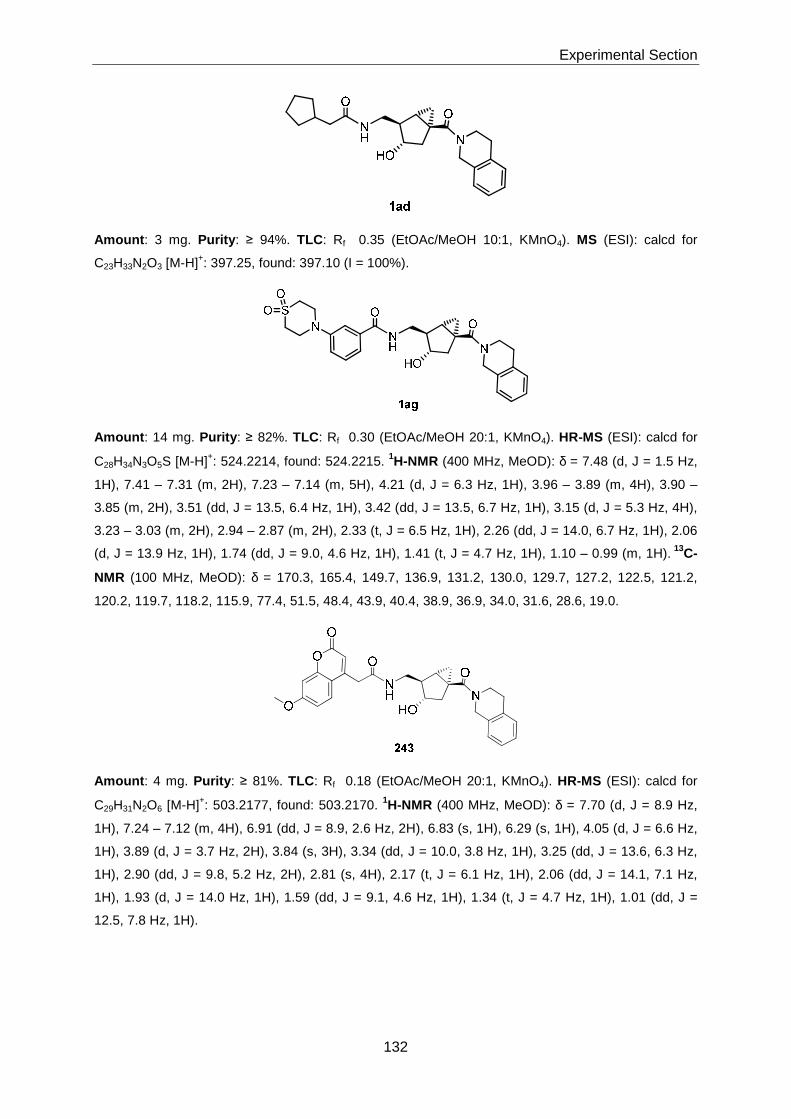
Experimental Section
132
Amount : 3 mg. Purity : ≥ 94%. TLC: Rf 0.35 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C23H33N2O3 [M-H]+: 397.25, found: 397.10 (I = 100%).
Amount : 14 mg. Purity : ≥ 82%. TLC: Rf 0.30 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C28H34N3O5S [M-H]+: 524.2214, found: 524.2215. 1H-NMR (400 MHz, MeOD): δ = 7.48 (d, J = 1.5 Hz,
1H), 7.41 – 7.31 (m, 2H), 7.23 – 7.14 (m, 5H), 4.21 (d, J = 6.3 Hz, 1H), 3.96 – 3.89 (m, 4H), 3.90 –
3.85 (m, 2H), 3.51 (dd, J = 13.5, 6.4 Hz, 1H), 3.42 (dd, J = 13.5, 6.7 Hz, 1H), 3.15 (d, J = 5.3 Hz, 4H),
3.23 – 3.03 (m, 2H), 2.94 – 2.87 (m, 2H), 2.33 (t, J = 6.5 Hz, 1H), 2.26 (dd, J = 14.0, 6.7 Hz, 1H), 2.06
(d, J = 13.9 Hz, 1H), 1.74 (dd, J = 9.0, 4.6 Hz, 1H), 1.41 (t, J = 4.7 Hz, 1H), 1.10 – 0.99 (m, 1H). 13C-
NMR (100 MHz, MeOD): δ = 170.3, 165.4, 149.7, 136.9, 131.2, 130.0, 129.7, 127.2, 122.5, 121.2,
120.2, 119.7, 118.2, 115.9, 77.4, 51.5, 48.4, 43.9, 40.4, 38.9, 36.9, 34.0, 31.6, 28.6, 19.0.
Amount : 4 mg. Purity : ≥ 81%. TLC: Rf 0.18 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C29H31N2O6 [M-H]+: 503.2177, found: 503.2170. 1H-NMR (400 MHz, MeOD): δ = 7.70 (d, J = 8.9 Hz,
1H), 7.24 – 7.12 (m, 4H), 6.91 (dd, J = 8.9, 2.6 Hz, 2H), 6.83 (s, 1H), 6.29 (s, 1H), 4.05 (d, J = 6.6 Hz,
1H), 3.89 (d, J = 3.7 Hz, 2H), 3.84 (s, 3H), 3.34 (dd, J = 10.0, 3.8 Hz, 1H), 3.25 (dd, J = 13.6, 6.3 Hz,
1H), 2.90 (dd, J = 9.8, 5.2 Hz, 2H), 2.81 (s, 4H), 2.17 (t, J = 6.1 Hz, 1H), 2.06 (dd, J = 14.1, 7.1 Hz,
1H), 1.93 (d, J = 14.0 Hz, 1H), 1.59 (dd, J = 9.1, 4.6 Hz, 1H), 1.34 (t, J = 4.7 Hz, 1H), 1.01 (dd, J =
12.5, 7.8 Hz, 1H).

Experimental Section
133
Amount : 8.5 mg. Purity : ≥ 96%. TLC: Rf 0.14 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C28H33N4O4 [M-H]+: 489.2496, found: 489.2496.
Amount : 11.5 mg. Purity : ≥ 95%. TLC: Rf 0.23 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C28H32N3O3 [M-H]+: 458.2438, found: 458.2422. 1H-NMR (400 MHz, MeOD): δ = 7.53 (d, J = 7.9 Hz,
1H), 7.27 (d, J = 8.2 Hz, 1H), 7.23-7.1 (m, 4H), 7.00 (s, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.86 (t, J = 7.1
Hz, 1H), 4.65 (s, br, 2H), 3.86 (d, J = 6.2 Hz, 1H), 3.82 (s, br, 2H), 3.36-3.32 (m, 1H), 3.12 (dd, J =
13.5, 6.0 Hz, 1H), 3.09-3.03 (m, 2H), 2.89-2.86 (m, 2H), 2.58 (o, J = 7.0 Hz, 2H), 2.08 (t, J = 5.8 Hz,
1H), 1.70 (t, J = 12.0 Hz, 1H), 1.65 (dd, J = 14.2, 6.2 Hz, 1H), 1.45 (dd, J = 8.9, 4.5 Hz, 1H), 1.26 (t, J
= 4.6 Hz, 1H), 0.98-0.90 (m, 1H). 13C-NMR (100 MHz, MeOD): δ = 176.1, 174.2, 138.4, 129.2, 128.5,
127.0, 123.1, 122.3, 119.5, 118.6, 110.6, 78.4, 51.3, 43.6, 39.9, 38.5, 34.3, 28.6, 22.6.
Amount : 13 mg. Purity : ≥ 94%. TLC: Rf 0.27 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C23H27N2O4 [M-H]+: 395.1965, found: 395.1977. 1H-NMR (400 MHz, MeOD): δ = 7.34 (d, J = 1.0 Hz,
1H), 7.23-7.15 (m, 4H), 6.28 (s, br, 1H), 6.20 (d, J = 2.9 Hz, 1H), 4.72 (s, br, 2H), 4.08 (dd, J = 6.8, 0.9
Hz, 1H), 3.90 (s, br, 2H), 3.57 (s, 2H), 3.35-3.31 (m, 1H), 3.23 (dd, J = 13.8, 6.8 Hz, 1H), 2.91 (t, J =
5.5 Hz, 2H), 2.21-2.14 (m, 2H), 1.99 (dd, J = 14.1, 1.0 Hz, 1H), 1.64 (dd, J = 9.1, 4.8 Hz, 1H), 1.37 (t, J
= 4.6 Hz, 1H), 1.02 (dd, J = 7.9, 3.7 Hz, 1H). 13C-NMR (100 MHz, MeOD): δ = 174.1, 171.7, 150.8,
143.2, 135.4, 130.1, 128.0, 127.4, 111.4, 108.4, 77.7, 51.1, 43.4, 40.3, 39.4, 36.6, 34.1, 28.4, 19.0.

Experimental Section
134
202d
((1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(5-chloro- indolin-1-yl)methanone
3 (70 mg) was dissolved in 2 ml of CH2Cl2 and DIPA (95 ul, 0.674 mmol) was added under
stirring and after 5 min HATU (128 mg, 0.338 mmol) was added. 5-Chloroindoline (200d, 44
mg) was added and the reaction mixture was stirred at RT for 24 h. It was diluted with CH2Cl2
and poured on 5% aqueous citric acid solution. The water phase was extracted with CH2Cl2
(3x 60 ml). The combined organic phases were washed with saturated aqueous NaHCO3-
solution and with brine, dried over MgSO4 and evaporated. Purification of the residue by flash
column chromatography (Hex/EtOAc 5:1) yielded 95 mg (94%) of the product as a slightly
green oil that solidified upon storage at -20°C.
Rf = 0.28 (Hex/EtOAc 5:1)
1H-NMR (400 MHz, CDCl3): δ = 7.94 (d, J = 8.2 Hz, 1H), 7.17-7.11 (m, 2H), 4.35-4.26 (m,
1H), 4.23-4.09 (m, 2H), 3.48 (dd, J = 12.3, 6.0 Hz, 1H), 3.19-3.11 (m, 3H), 2.33 (dd, J =13.3,
6.0 Hz, 1H), 2.15 (dd, J = 9.7, 6.0 Hz, 1H), 2.01 (d, J = 13.6 Hz, 1H), 1.79 (dd, J = 9.4, 5.2
Hz, 1H), 1.47 (t, J = 4.8 Hz, 1H), 1.09 (ddd, J = 9.0, 4.5, 1.4 Hz, 1H), 0.88 (s, 9H), 0.06 (s,
3H), 0.05 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 170.4, 141.7, 133.3, 128.7, 127.3, 125.0, 117.5, 76.2, 53.7,
50.0, 48.3, 38.2, 34.5, 28.2, 27.5, 25.9, 19.3, 17.8, -4.8, -4.9.
HR-MS (ESI): calcd for C22H32ClN4O2Si [M+H, M+Na]+: 447.1978, found: 447.1977.
203d
202d (113 mg, 0.253 mmol) was dissolved in Tol/MeOH 1:1 (2.5 ml), then 10% Pd/C (54 mg,
0.051 mmol) was added and the reaction mixture was stirred vigorously under a hydrogen
atmosphere (1 bar) for 2 h. The reaction mixture was filtered through celite, the residue was
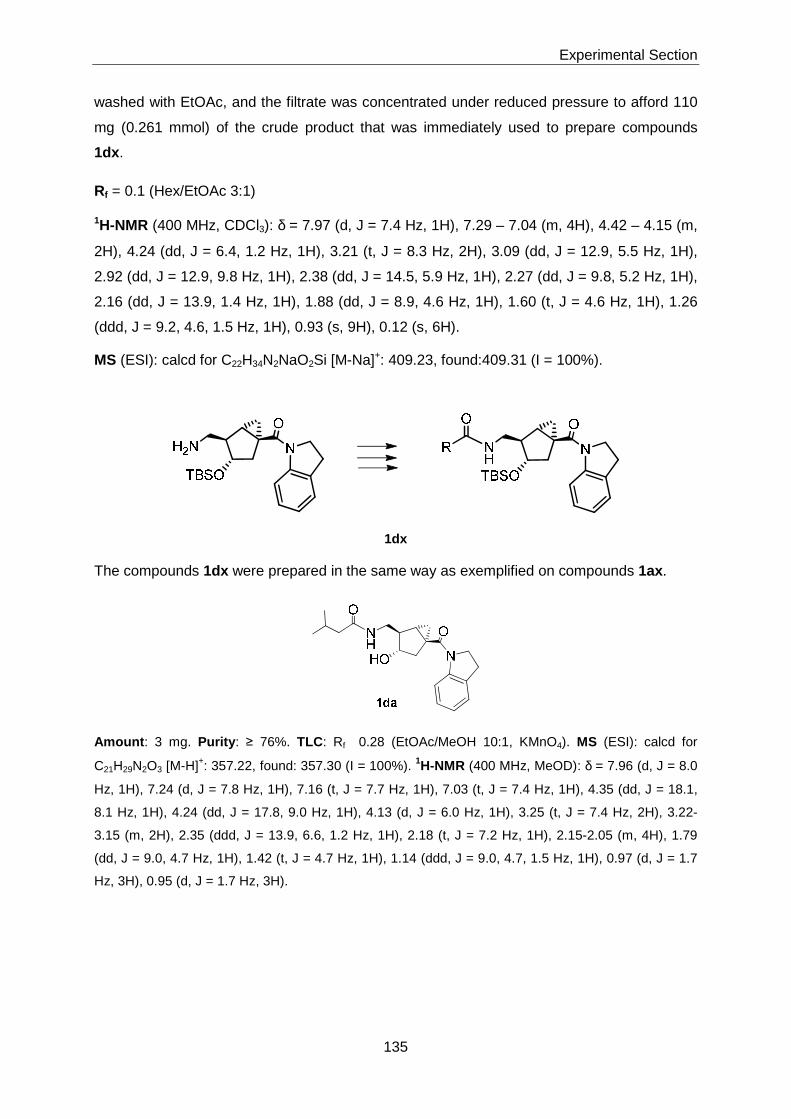
Experimental Section
135
washed with EtOAc, and the filtrate was concentrated under reduced pressure to afford 110
mg (0.261 mmol) of the crude product that was immediately used to prepare compounds
1dx .
Rf = 0.1 (Hex/EtOAc 3:1)
1H-NMR (400 MHz, CDCl3): δ = 7.97 (d, J = 7.4 Hz, 1H), 7.29 – 7.04 (m, 4H), 4.42 – 4.15 (m,
2H), 4.24 (dd, J = 6.4, 1.2 Hz, 1H), 3.21 (t, J = 8.3 Hz, 2H), 3.09 (dd, J = 12.9, 5.5 Hz, 1H),
2.92 (dd, J = 12.9, 9.8 Hz, 1H), 2.38 (dd, J = 14.5, 5.9 Hz, 1H), 2.27 (dd, J = 9.8, 5.2 Hz, 1H),
2.16 (dd, J = 13.9, 1.4 Hz, 1H), 1.88 (dd, J = 8.9, 4.6 Hz, 1H), 1.60 (t, J = 4.6 Hz, 1H), 1.26
(ddd, J = 9.2, 4.6, 1.5 Hz, 1H), 0.93 (s, 9H), 0.12 (s, 6H).
MS (ESI): calcd for C22H34N2NaO2Si [M-Na]+: 409.23, found:409.31 (I = 100%).
1dx
The compounds 1dx were prepared in the same way as exemplified on compounds 1ax.
Amount : 3 mg. Purity : ≥ 76%. TLC: Rf 0.28 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C21H29N2O3 [M-H]+: 357.22, found: 357.30 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 7.96 (d, J = 8.0
Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.16 (t, J = 7.7 Hz, 1H), 7.03 (t, J = 7.4 Hz, 1H), 4.35 (dd, J = 18.1,
8.1 Hz, 1H), 4.24 (dd, J = 17.8, 9.0 Hz, 1H), 4.13 (d, J = 6.0 Hz, 1H), 3.25 (t, J = 7.4 Hz, 2H), 3.22-
3.15 (m, 2H), 2.35 (ddd, J = 13.9, 6.6, 1.2 Hz, 1H), 2.18 (t, J = 7.2 Hz, 1H), 2.15-2.05 (m, 4H), 1.79
(dd, J = 9.0, 4.7 Hz, 1H), 1.42 (t, J = 4.7 Hz, 1H), 1.14 (ddd, J = 9.0, 4.7, 1.5 Hz, 1H), 0.97 (d, J = 1.7
Hz, 3H), 0.95 (d, J = 1.7 Hz, 3H).

Experimental Section
136
Amount : 3 mg. Purity : ≥ 92%. TLC: Rf 0.34 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C22H31N2O3 [M-H]+: 371.2335, found: 371.2348. 1H-NMR (400 MHz, MeOD): δ = 7.95 (d, J = 8.0 Hz,
1H), 7.23 (d, J = 7.8 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 4.38-4.29 (m, 1H), 4.27-
4.18 (m, 1H), 4.12 (d, J = 6.0 Hz, 1H), 3.24 (d, J = 6.7 Hz, 2H), 3.21-3.14 (m, 2H), 2.32 (ddd, J = 13.9,
6.7, 1.3 Hz, 1H), 2.22 (dd, J = 8.5, 7.2 Hz, 2H), 2.16 (t, J = 6.8 Hz, 1H), 2.11 (dd, J = 13.8, 0.9 Hz, 1H),
1.78 (dd, J = 9.0, 4.7 Hz, 1H), 1.59-1.47 (m, 3H), 1.40 (t, J = 4.7 Hz, 1H), 1.22 (t, J = 7.2 Hz, 1H), 1.14
(ddd, J = 9.0, 4.6, 1.4 Hz, 1H), 0.91 (s, 3H), 0.90 (s, 3H).
Amount : 3.5 mg. Purity : ≥ 77%. TLC: Rf 0.42 (EtOAc/MeOH 30:1, KMnO4). MS (ESI): calcd for
C22H29N2O3 [M-H]+: 369.22, found: 369.00 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 7.94 (d, J = 8.2
Hz, 1H), 7.24 (d, J = 7.3 Hz, 1H), 7.14 (td, J = 7.8, 1.0 Hz, 1H), 7.02 (td, J = 7.4, 1.0 Hz, 1H), 4.38-
4.30 (m, 1H), 4.27-4.18 (m, 1H), 4.12 (d, J = 6.2 Hz, 1H), 3.25 (dd, J = 7.9, 6.9 Hz, 2H), 3.21-3.14 (m,
2H), 2.32 (ddd, J = 13.9, 6.7, 1.3 Hz, 1H), 2.72-2.63 (m, 1H), 2.33 (ddd, J = 13.9, 6.6, 1.3 Hz, 1H),
2.17 (t, J = 6.9 Hz, 1H), 2.13 (dd, J = 13.9, 1.1 Hz, 1H), 1.93-1.82 (m, 2H), 1.78 (dd, J = 9.1, 4.8 Hz,
1H), 1.76-1.67 (m, 4H), 1.65-1.55 (m, 2H), 1.41 (t, J = 4.7 Hz, 1H), 1.14 (ddd, J = 9.1, 4.5, 1.5 Hz, 1H).
HO
NH
O
O
N
1de
Amount : 4 mg. Purity : ≥ 87%. TLC: Rf 0.33 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C25H29N2O3 [M-H]+: 405.22, found: 405.00 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 8.72 (dd, J =
4.5, 1.2 Hz, 1H), 8.42 (dd, J = 8.4, 1.3 Hz, 1H), 7.51 (dd, J = 8.6, 4.5 Hz, 1H), 7.23 (d, J = 7.3 Hz, 1H),
7.18-7.14 (m, 3H), 7.13-7.09 (m, 1H), 7.03 (td, J = 7.5, 1.0 Hz, 1H), 4.34-4.26 (m, 1H), 4.21-4.13 (m,
1H), 4.00 (dd, J = 6.4, 1.3 Hz, 1H), 3.27-3.14 (m, 4H), 2.93-2.88 (m, 2H), 2.54 (t, J = 7.6 Hz, 2H), 2.10
(t, J = 6.4 Hz, 1H), 2.03 (ddd, J = 14.0, 6.7, 1.3 Hz, 1H), 1.96 (dd, J = 14.1, 1.7 Hz, 1H), 1.63 (ddd, J =
9.2, 4.7, 1.0 Hz, 1H), 1.33 (t, J = 4.7 Hz, 1H), 1.09 (ddd, J = 9.2, 4.6, 1.5 Hz, 1H).
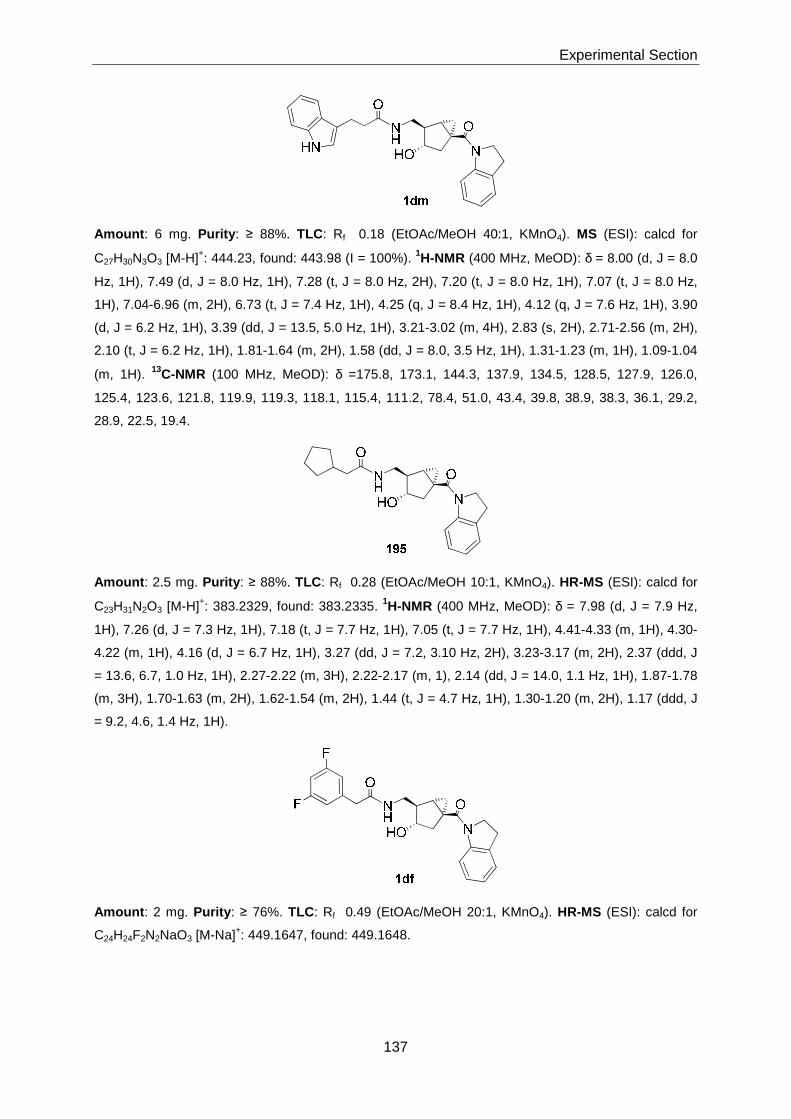
Experimental Section
137
Amount : 6 mg. Purity : ≥ 88%. TLC: Rf 0.18 (EtOAc/MeOH 40:1, KMnO4). MS (ESI): calcd for
C27H30N3O3 [M-H]+: 444.23, found: 443.98 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 8.00 (d, J = 8.0
Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.28 (t, J = 8.0 Hz, 2H), 7.20 (t, J = 8.0 Hz, 1H), 7.07 (t, J = 8.0 Hz,
1H), 7.04-6.96 (m, 2H), 6.73 (t, J = 7.4 Hz, 1H), 4.25 (q, J = 8.4 Hz, 1H), 4.12 (q, J = 7.6 Hz, 1H), 3.90
(d, J = 6.2 Hz, 1H), 3.39 (dd, J = 13.5, 5.0 Hz, 1H), 3.21-3.02 (m, 4H), 2.83 (s, 2H), 2.71-2.56 (m, 2H),
2.10 (t, J = 6.2 Hz, 1H), 1.81-1.64 (m, 2H), 1.58 (dd, J = 8.0, 3.5 Hz, 1H), 1.31-1.23 (m, 1H), 1.09-1.04
(m, 1H). 13C-NMR (100 MHz, MeOD): δ =175.8, 173.1, 144.3, 137.9, 134.5, 128.5, 127.9, 126.0,
125.4, 123.6, 121.8, 119.9, 119.3, 118.1, 115.4, 111.2, 78.4, 51.0, 43.4, 39.8, 38.9, 38.3, 36.1, 29.2,
28.9, 22.5, 19.4.
Amount : 2.5 mg. Purity : ≥ 88%. TLC: Rf 0.28 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C23H31N2O3 [M-H]+: 383.2329, found: 383.2335. 1H-NMR (400 MHz, MeOD): δ = 7.98 (d, J = 7.9 Hz,
1H), 7.26 (d, J = 7.3 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.05 (t, J = 7.7 Hz, 1H), 4.41-4.33 (m, 1H), 4.30-
4.22 (m, 1H), 4.16 (d, J = 6.7 Hz, 1H), 3.27 (dd, J = 7.2, 3.10 Hz, 2H), 3.23-3.17 (m, 2H), 2.37 (ddd, J
= 13.6, 6.7, 1.0 Hz, 1H), 2.27-2.22 (m, 3H), 2.22-2.17 (m, 1), 2.14 (dd, J = 14.0, 1.1 Hz, 1H), 1.87-1.78
(m, 3H), 1.70-1.63 (m, 2H), 1.62-1.54 (m, 2H), 1.44 (t, J = 4.7 Hz, 1H), 1.30-1.20 (m, 2H), 1.17 (ddd, J
= 9.2, 4.6, 1.4 Hz, 1H).
Amount : 2 mg. Purity : ≥ 76%. TLC: Rf 0.49 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C24H24F2N2NaO3 [M-Na]+: 449.1647, found: 449.1648.

Experimental Section
138
HO
NH
O
O
N
1dg
N
SO
O
Amount : 7 mg. Purity : ≥ 96%. TLC: Rf 0.28 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C27H31N3NaO5S [M-Na]+: 532.19, found: 531.91 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 7.96 (d, J
= 8.1 Hz, 1H), 7.52-7.49 (m, 1H), 7.39-7.36 (m, 2H), 7.28-7.16 (m, 3H), 7.06 (t, J = 7.4 Hz, 1H), 4.40-
4.32 (m, 1H), 4.29-4.23 (m, 1H), 3.95-3.89 (m, 5H), 3.51 (dd, J = 14.2, 6.7 Hz, 1H), 3.23-3.16 (m, 2H),
3.16-3.12 (m, 4H), 2.83 (s, 1H), 2.41-2.32 (m, 2H), 2.18 (d, J = 14.2 Hz, 1H), 1.89 (dd, J = 9.0, 4.7 Hz,
1H), 1.46 (t, J = 4.7 Hz, 1H), 1.18 (ddd, J = 9.1, 4.5, 1.3 Hz, 1H). 13C-NMR (100 MHz, MeOD): δ =
173.3, 169.7, 150.1, 136.9, 134.1, 130.8, 128.4, 125.7, 125.3, 120.6, 119.9, 118.1, 77.8, 61.2, 51.5,
51.2, 44.3, 39.5, 38.7, 36.8, 29.6, 28.9, 21.4, 19.9, 14.2.
Amount : 4 mg. Purity : ≥ 94%. TLC: Rf 0.16 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C23H31N2O3 [M-H]+: 459.20, found: 459.16 (I = 100%). 1H-NMR (400 MHz, MeOD): δ = 8.26 (d, J = 7.9
Hz, 1H), 7.90-7.79 (m, 4H), 7.76-7.72 (m, 1H), 7.13 (d, J = 7.4 Hz, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.92
(t, J = 7.3 Hz, 1H), 4.27-4.19 (m, 1H), 4.16-4.08 (m, 1H), 4.03 (d, J = 6.4 Hz, 1H), 3.89 (d, J = 1.7 Hz,
2H), 3.27-3.13 (m, 2H), 3.10-3.04 (m, 2H), 2.24 (ddd, J = 13.8, 6.8, 1.1 Hz, 1H); 2.09 (t, J = 6.6 Hz,
1H), 2.00 (dd, J = 13.8, 1.1 Hz, 1H), 1.66 (dd, J = 9.0, 4.6 Hz, 1H), 1.30 (t, J = 4.8 Hz, 1H), 1.03 (ddd,
J = 9.0, 4.8, 1.4 Hz, 1H). 13C-NMR (100 MHz, MeOD): δ = 173.5, 171.7, 1144.9, 135.5, 133.8, 132.7,
129.0, 128.1, 127.4, 126.6, 126.0, 125.2, 117.9, 77.7, 51.2, 43.5, 40.9, 39.5, 36.0, 29.2, 29.0, 19.8,
14.4.
203f
202d was dissolved in dioxane, polymer-bound PPh3 added and the suspension slightly
shaken for 2 h (11:30 -13:30) at RT. Then ammonia was added and RM shaken overnight.

Experimental Section
139
Resin filtered off and washed with dichloromethane (3x 1 ml) and solvents evaporated to
dryness to give 7 mg (67%) of the crude amine as a yellow oil that was used for the
preparation of compounds 1fx .
Rf = 0.1 (EtOAc, KMnO4).
MS (ESI): calcd for C22H34ClN2O2Si [M-H]+: 421.2078, found: 420.99 (I = 100%).
Amount : 2.5 mg. Purity : ≥ 87%. TLC: Rf 0.31 (EtOAc/MeOH 20:1, KMnO4). MS (ESI): calcd for
C23H30ClN2O3 [M-H]+: 417.19, found: 416.97 (I = 100%). 1H-NMR (400 MHz, DMSO): δ = 7.43 – 7.36
(m, 2H), 6.78 (s, 1H), 6.66 (d, J = 8.6 Hz, 1H), 3.76 (dd, J = 17.1, 9.5 Hz, 1H), 3.69 (dd, J = 18.3, 9.6
Hz, 1H), 3.45 (d, J = 6.2 Hz, 1H), 2.63 (t, J = 8.3 Hz, 2H), 1.99 (s, 2H), 1.70 (dd, J = 13.5, 6.5 Hz, 1H),
1.61 (dd, J = 14.6, 7.3 Hz, 1H), 1.56 (d, J = 7.3 Hz, 2H), 1.45 (dd, J = 13.7, 5.4 Hz, 2H), 1.21 – 1.12
(m, 2H), 1.07 – 1.02 (m, 2H), 0.99 – 0.91 (m, 2H), 0.79 – 0.75 (m, 2H), 0.63 – 0.55 (m, 2H), 0.51 (dd, J
= 8.7, 3.4 Hz, 1H), 0.39 – 0.32 (m, 1H). 13C-NMR (100 MHz, DMSO): δ =172.0, 170.3, 142.1, 134.4,
126.9, 126.3, 125.0, 117.4, 74.8, 49.4, 48.3, 41.8, 41.5, 37.9, 36.7, 33.9, 31.9, 31.8, 278, 27.5, 24.5,
18.4.
Amount : 0.5 mg. Purity : ≥ 66%. TLC: Rf 0.16 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C22H24ClN3NaO4 [M-Na]+: 452.1348, found: 452.1341.
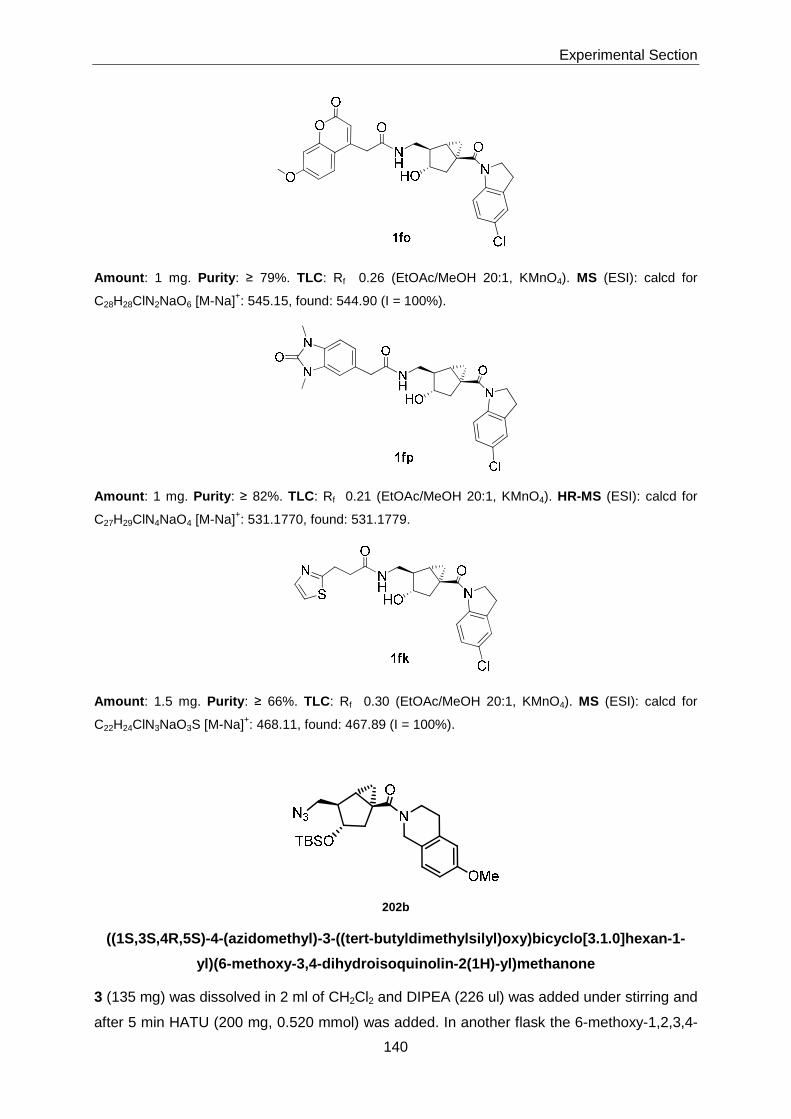
Experimental Section
140
Amount : 1 mg. Purity : ≥ 79%. TLC: Rf 0.26 (EtOAc/MeOH 20:1, KMnO4). MS (ESI): calcd for
C28H28ClN2NaO6 [M-Na]+: 545.15, found: 544.90 (I = 100%).
Amount : 1 mg. Purity : ≥ 82%. TLC: Rf 0.21 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C27H29ClN4NaO4 [M-Na]+: 531.1770, found: 531.1779.
Amount : 1.5 mg. Purity : ≥ 66%. TLC: Rf 0.30 (EtOAc/MeOH 20:1, KMnO4). MS (ESI): calcd for
C22H24ClN3NaO3S [M-Na]+: 468.11, found: 467.89 (I = 100%).
202b
((1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(6-methoxy-3,4-dihydroisoquinolin-2(1H)-yl)metha none
3 (135 mg) was dissolved in 2 ml of CH2Cl2 and DIPEA (226 ul) was added under stirring and
after 5 min HATU (200 mg, 0.520 mmol) was added. In another flask the 6-methoxy-1,2,3,4-

Experimental Section
141
tetrahydroisoquinoline hydrochloride (198a) was dissolved in CH2Cl2 (2 ml) and DMF (1 ml),
then DIPEA (166 ul, 0.953 mmol) was added, after 10 min of stirring this mixture was added
to the solution of AS-115. The reaction mixture was stirred at RT for 22 h. It was diluted with
CH2Cl2 and poured on saturated aqueous NaHCO3 –solution. The water phase was
extracted with CH2Cl2 (3x 60 ml), the combined organic phases were dried over MgSO4 and
evaporated. Purification of the residue by flash column chromatography (Hex/EtOAc 2:1)
yielded 190 mg (96%) of the product as a colorless oil.
Rf = 0.32 (Hex/EtOAc 2:1, CPS).
1H-NMR (400 MHz, CDCl3): δ = 7.02 (d, J = 8.1 Hz, 1H), 6.76 (dd, J = 8.5, 2.6 Hz, 1H), 6.67
(d, J = 2.8 Hz, 1H), 4.76-4.55 (m, 2H), 4.17 (dd, J = 6.5, 1.1 Hz, 1H), 3.91-3.79 (m, 2H), 3.78
(s, 3H), 3.45 (dd, J = 12.3, 5.9 Hz, 1H), 3.11 (dd, J = 12.7, 9.5 Hz, 1H), 2.85 (t, J = 6.3 Hz,
1H), 2.26 (ddd, J = 14.1, 6.9, 1.1 Hz, 1H), 2.12 (dd, J = 9.7, 6.2 Hz, 1H), 1.90 (dd, J = 13.6,
1.1 Hz, 1H), 1.63 (dd, J = 9.1, 4.5 Hz, 1H), 1.40 (t, J = 4.6 Hz, 1H), 0.97 (J = 9.03, 4.40 Hz,
1H), 0.88 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 171.9, 158.5, 127.2, 125.6, 113.7, 112.4, 76.4, 55.4, 54.4,
49.9, 39.9, 32.9, 26.9, 25.9, 21.2, 18.1, 17.7, -4.7, -4.9.
IR (film): υ 2951, 2929, 2856, 2096, 1635, 1505, 1462, 1431, 1389, 1257, 1225, 1168, 1084,
1037, 936, 862, 836, 808, 776, 670.
HR-MS (ESI): calcd for C24H37N4O3Si [M-H]+: 457.2629, found: 457.2638.
203b
((1S,3S,4R,5S)-4-(aminomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(6-methoxy-3,4-dihydroisoquinolin-2(1H)-yl)metha none
AS-142-1 (115 mg) was dissolved in Tol/MeOH 1:1 (0.5 ml), Pd/C (54 mg, 10% Pd on
charcoal) was added and the mixture was stirred under a hydrogen atmosphere (1 bar) for 3
h. The reaction mixture was filtered through celite and the residue was washed with EtOAc.
The filtrate was concentrated under reduced pressure to afford 130 mg (0.302 mmol, >100%)
of the crude amine as a yellow oil that was immediately used for the preparation of
compounds 1bx .
Rf = 0.24 (EtOAc/MeOH 10:1)
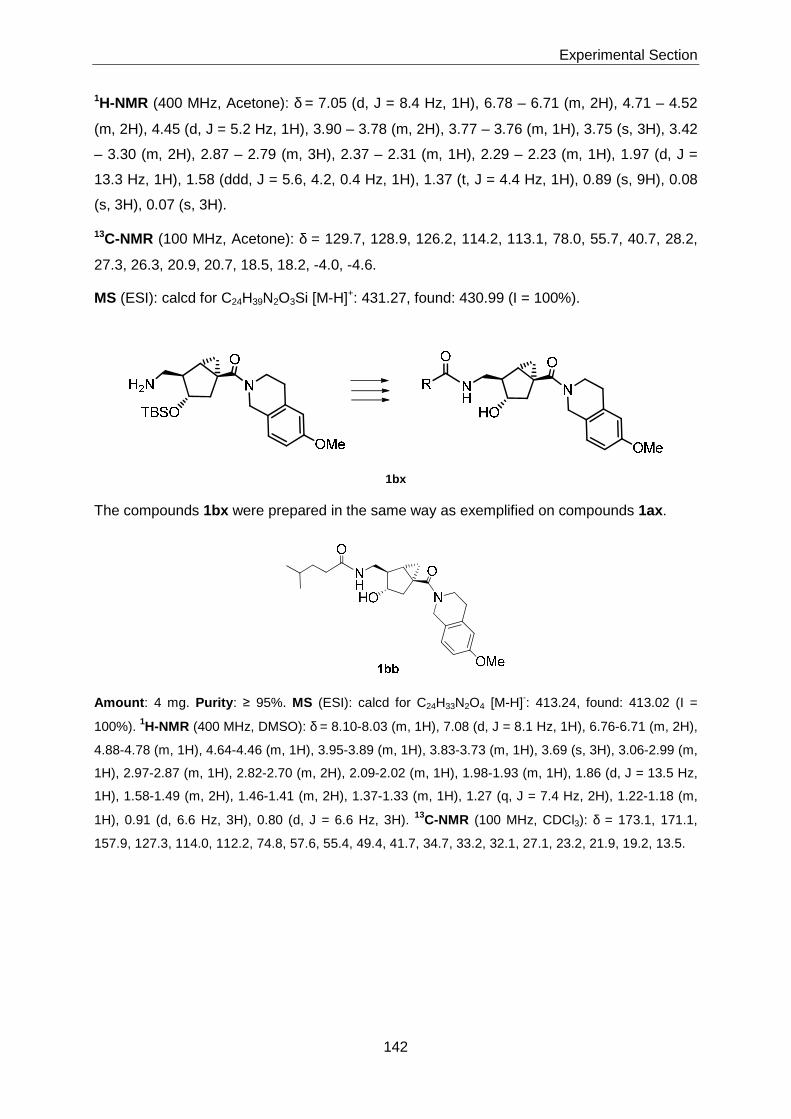
Experimental Section
142
1H-NMR (400 MHz, Acetone): δ = 7.05 (d, J = 8.4 Hz, 1H), 6.78 – 6.71 (m, 2H), 4.71 – 4.52
(m, 2H), 4.45 (d, J = 5.2 Hz, 1H), 3.90 – 3.78 (m, 2H), 3.77 – 3.76 (m, 1H), 3.75 (s, 3H), 3.42
– 3.30 (m, 2H), 2.87 – 2.79 (m, 3H), 2.37 – 2.31 (m, 1H), 2.29 – 2.23 (m, 1H), 1.97 (d, J =
13.3 Hz, 1H), 1.58 (ddd, J = 5.6, 4.2, 0.4 Hz, 1H), 1.37 (t, J = 4.4 Hz, 1H), 0.89 (s, 9H), 0.08
(s, 3H), 0.07 (s, 3H).
13C-NMR (100 MHz, Acetone): δ = 129.7, 128.9, 126.2, 114.2, 113.1, 78.0, 55.7, 40.7, 28.2,
27.3, 26.3, 20.9, 20.7, 18.5, 18.2, -4.0, -4.6.
MS (ESI): calcd for C24H39N2O3Si [M-H]+: 431.27, found: 430.99 (I = 100%).
1bx
The compounds 1bx were prepared in the same way as exemplified on compounds 1ax.
Amount : 4 mg. Purity : ≥ 95%. MS (ESI): calcd for C24H33N2O4 [M-H]-: 413.24, found: 413.02 (I =
100%). 1H-NMR (400 MHz, DMSO): δ = 8.10-8.03 (m, 1H), 7.08 (d, J = 8.1 Hz, 1H), 6.76-6.71 (m, 2H),
4.88-4.78 (m, 1H), 4.64-4.46 (m, 1H), 3.95-3.89 (m, 1H), 3.83-3.73 (m, 1H), 3.69 (s, 3H), 3.06-2.99 (m,
1H), 2.97-2.87 (m, 1H), 2.82-2.70 (m, 2H), 2.09-2.02 (m, 1H), 1.98-1.93 (m, 1H), 1.86 (d, J = 13.5 Hz,
1H), 1.58-1.49 (m, 2H), 1.46-1.41 (m, 2H), 1.37-1.33 (m, 1H), 1.27 (q, J = 7.4 Hz, 2H), 1.22-1.18 (m,
1H), 0.91 (d, 6.6 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ = 173.1, 171.1,
157.9, 127.3, 114.0, 112.2, 74.8, 57.6, 55.4, 49.4, 41.7, 34.7, 33.2, 32.1, 27.1, 23.2, 21.9, 19.2, 13.5.
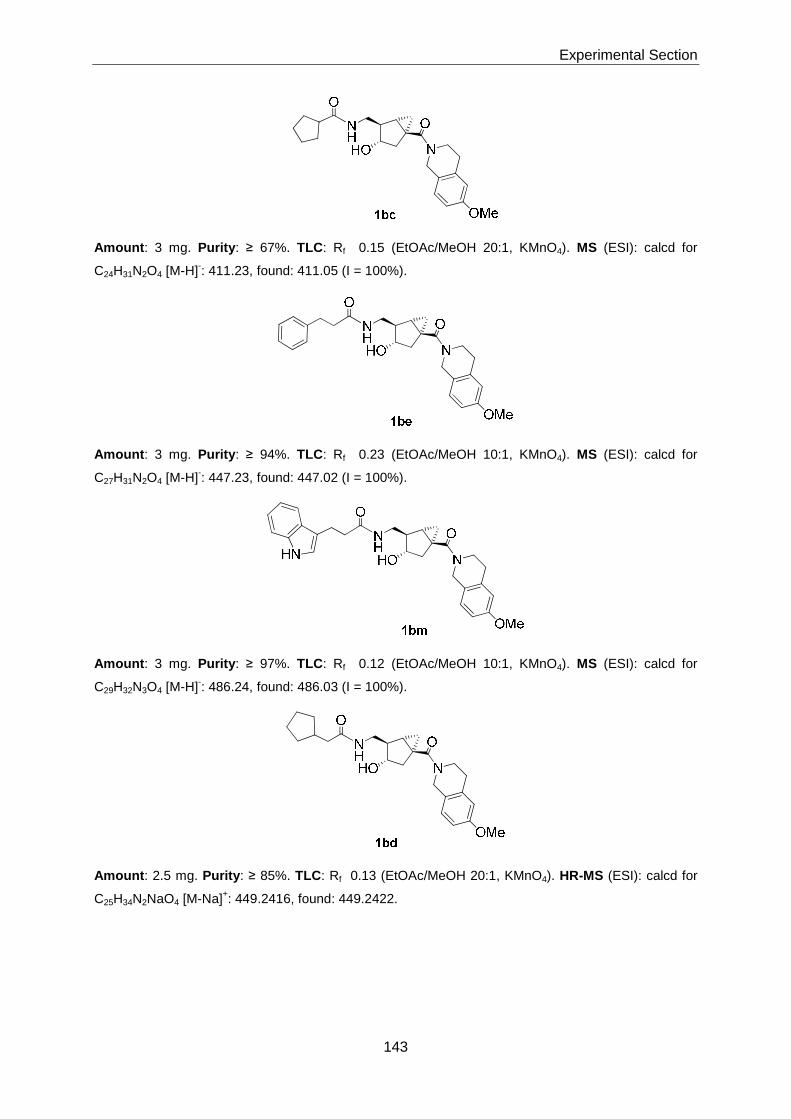
Experimental Section
143
Amount : 3 mg. Purity : ≥ 67%. TLC: Rf 0.15 (EtOAc/MeOH 20:1, KMnO4). MS (ESI): calcd for
C24H31N2O4 [M-H]-: 411.23, found: 411.05 (I = 100%).
Amount : 3 mg. Purity : ≥ 94%. TLC: Rf 0.23 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C27H31N2O4 [M-H]-: 447.23, found: 447.02 (I = 100%).
Amount : 3 mg. Purity : ≥ 97%. TLC: Rf 0.12 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C29H32N3O4 [M-H]-: 486.24, found: 486.03 (I = 100%).
Amount : 2.5 mg. Purity : ≥ 85%. TLC: Rf 0.13 (EtOAc/MeOH 20:1, KMnO4). HR-MS (ESI): calcd for
C25H34N2NaO4 [M-Na]+: 449.2416, found: 449.2422.
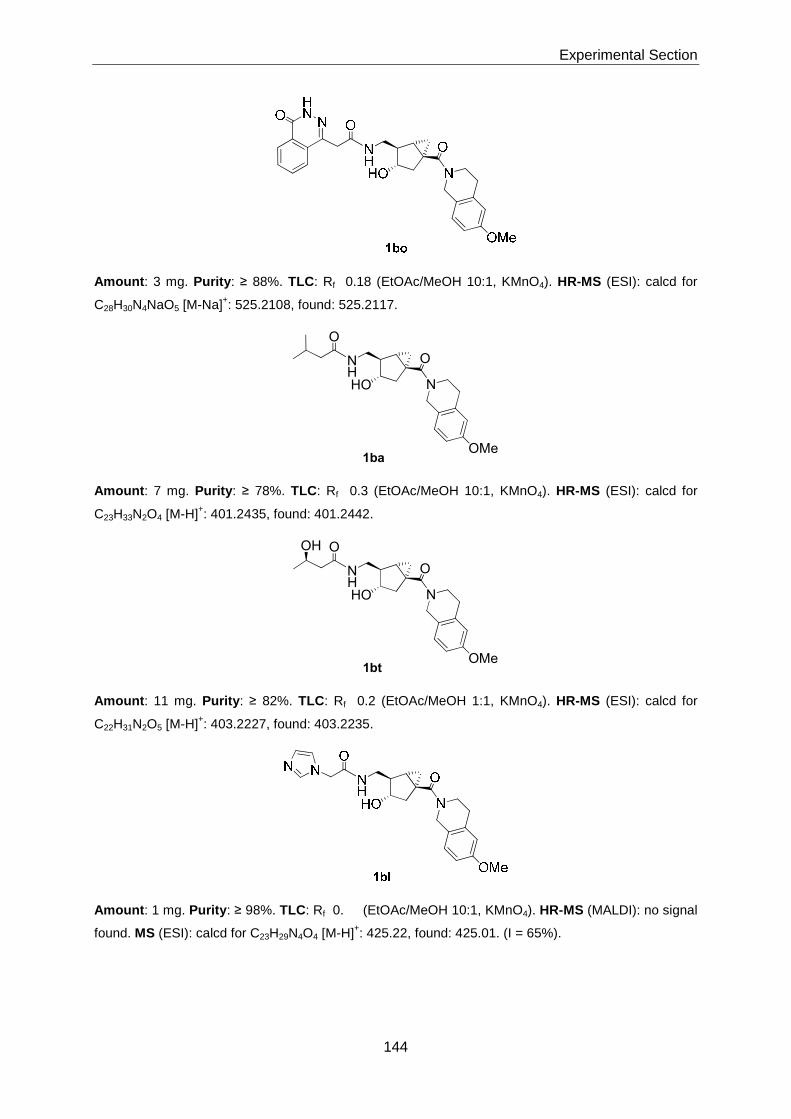
Experimental Section
144
Amount : 3 mg. Purity : ≥ 88%. TLC: Rf 0.18 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C28H30N4NaO5 [M-Na]+: 525.2108, found: 525.2117.
HO
NH
O
N
O
OMe1ba
Amount : 7 mg. Purity : ≥ 78%. TLC: Rf 0.3 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C23H33N2O4 [M-H]+: 401.2435, found: 401.2442.
HO
NH
O
N
O
OMe
OH
1bt
Amount : 11 mg. Purity : ≥ 82%. TLC: Rf 0.2 (EtOAc/MeOH 1:1, KMnO4). HR-MS (ESI): calcd for
C22H31N2O5 [M-H]+: 403.2227, found: 403.2235.
Amount : 1 mg. Purity : ≥ 98%. TLC: Rf 0. (EtOAc/MeOH 10:1, KMnO4). HR-MS (MALDI): no signal
found. MS (ESI): calcd for C23H29N4O4 [M-H]+: 425.22, found: 425.01. (I = 65%).

Experimental Section
145
Amount : 3.5 mg. Purity : ≥ 85%. TLC: Rf 0.37 (EtOAc/MeOH 40:1, KMnO4). HR-MS (ESI): calcd for
C26H29F2N2O4 [M-H]+: 471.2090, found: 471.2098. 1H-NMR (400 MHz, DMSO): δ = 8.29-8.24 (m, 1H),
7.13-7.03 (m, 2H), 6.99-6.93 (m, 2H), 6.78-6.72 (m, 2H), 4.78 (s, br, 1H), 4.67-4.46 (m, 2H), 3.92 (d, J
= 6.5 Hz, 1H), 3.74-3.68 (m, 1H), 3.71 (s, 3H), 3.68-3.62 (m, 1H), 3.06-3.02 (m, 2H), 2.81-2.76 (m,
2H), 2.68 (s, 2H), 2.07 (dd, J = 13.4, 7.0 Hz, 1H), 1.97 (t, J = 6.7 Hz, 1H), 1.86 (d, J = 13.4 Hz, 1H),
1.50 (dd, J = 8.5, 4.4 Hz, 1H), 1.17-1.13 (m, 1H), 0.91-0.87 (m, 1H).
Amount : 4 mg. Purity : ≥ 70%. TLC: Rf 0.12 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): no signal
found. MS (ESI): calcd for C24H30N3O5 [M-H]+: 440.22, found: 440.00.
Amount : 11 mg. Purity : ≥ 79%. TLC: Rf 0.34 (EtOAc/MeOH 40:1, KMnO4). HR-MS (ESI): calcd for
C29H36N3O6S [M-H]+: 554.2319, found: 554.2306.
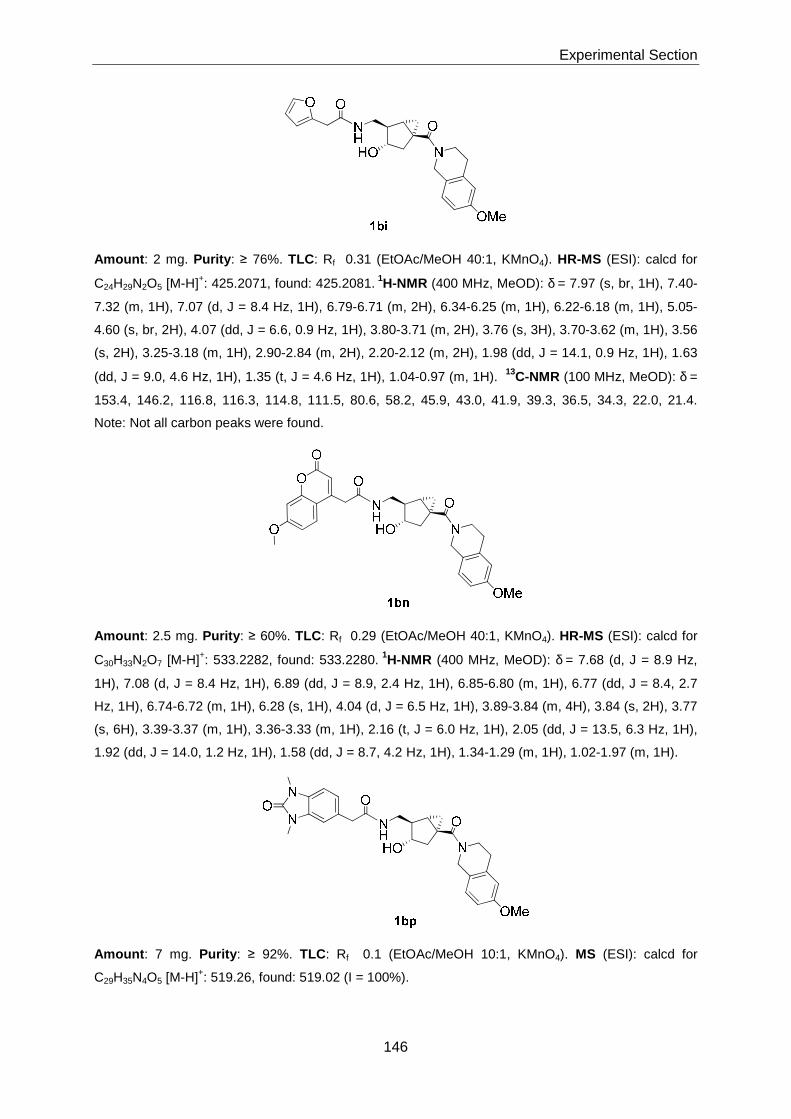
Experimental Section
146
Amount : 2 mg. Purity : ≥ 76%. TLC: Rf 0.31 (EtOAc/MeOH 40:1, KMnO4). HR-MS (ESI): calcd for
C24H29N2O5 [M-H]+: 425.2071, found: 425.2081. 1H-NMR (400 MHz, MeOD): δ = 7.97 (s, br, 1H), 7.40-
7.32 (m, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.79-6.71 (m, 2H), 6.34-6.25 (m, 1H), 6.22-6.18 (m, 1H), 5.05-
4.60 (s, br, 2H), 4.07 (dd, J = 6.6, 0.9 Hz, 1H), 3.80-3.71 (m, 2H), 3.76 (s, 3H), 3.70-3.62 (m, 1H), 3.56
(s, 2H), 3.25-3.18 (m, 1H), 2.90-2.84 (m, 2H), 2.20-2.12 (m, 2H), 1.98 (dd, J = 14.1, 0.9 Hz, 1H), 1.63
(dd, J = 9.0, 4.6 Hz, 1H), 1.35 (t, J = 4.6 Hz, 1H), 1.04-0.97 (m, 1H). 13C-NMR (100 MHz, MeOD): δ =
153.4, 146.2, 116.8, 116.3, 114.8, 111.5, 80.6, 58.2, 45.9, 43.0, 41.9, 39.3, 36.5, 34.3, 22.0, 21.4.
Note: Not all carbon peaks were found.
Amount : 2.5 mg. Purity : ≥ 60%. TLC: Rf 0.29 (EtOAc/MeOH 40:1, KMnO4). HR-MS (ESI): calcd for
C30H33N2O7 [M-H]+: 533.2282, found: 533.2280. 1H-NMR (400 MHz, MeOD): δ = 7.68 (d, J = 8.9 Hz,
1H), 7.08 (d, J = 8.4 Hz, 1H), 6.89 (dd, J = 8.9, 2.4 Hz, 1H), 6.85-6.80 (m, 1H), 6.77 (dd, J = 8.4, 2.7
Hz, 1H), 6.74-6.72 (m, 1H), 6.28 (s, 1H), 4.04 (d, J = 6.5 Hz, 1H), 3.89-3.84 (m, 4H), 3.84 (s, 2H), 3.77
(s, 6H), 3.39-3.37 (m, 1H), 3.36-3.33 (m, 1H), 2.16 (t, J = 6.0 Hz, 1H), 2.05 (dd, J = 13.5, 6.3 Hz, 1H),
1.92 (dd, J = 14.0, 1.2 Hz, 1H), 1.58 (dd, J = 8.7, 4.2 Hz, 1H), 1.34-1.29 (m, 1H), 1.02-1.97 (m, 1H).
Amount : 7 mg. Purity : ≥ 92%. TLC: Rf 0.1 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C29H35N4O5 [M-H]+: 519.26, found: 519.02 (I = 100%).

Experimental Section
147
Amount : 1 mg. Purity : ≥ 30%. MS (ESI): calcd for C24H30N3NaO4S [M-Na]+: 478.18, found: 477.96 (I =
100%).
202c
(1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimethyl silyl)oxy)-N-(thiazol-2-
yl)bicyclo[3.1.0]hexane-1-carboxamide
3 (70 mg, 0.225 mmol) was dissolved in CH2Cl2 (2 ml) and a few drops of DMF, then DIPEA
(192 ul, 1.124 mmol) and after 5 min HATU (96 mg, 0.248 mmol) were added. After 5 min 2-
amino-thiazole (199c, 30 mg) was added and the reaction mixture was stirred at RT
overnight. The solvents were evaporated and purification by flash chromatography
(CH2Cl2/MeOH 100:1) afforded 89 mg (0.226 mmol, >100%) of the product as a yellow oil.
Rf = 0.29 (CH2Cl2); 0.48 (CH2Cl2/MeOH 50:1)
1H-NMR (400 MHz, CDCl3): δ = 7.44 (d, J = 3.4 Hz, 1H), 6.96 (d, J = 3.1 Hz, 1H), 4.20 (d, J =
6.5 Hz, 1H), 3.36 (dd, J = 12.4, 5.7 Hz, 1H), 3.13 (dd, J = 12.4, 8.3 Hz, 1H), 2.48 (ddd, J =
13.1, 6.5, 1.6 Hz, 1H), 2.19 (dd, J = 8.3, 5.7 Hz, 1H), 1.98 (d, J = 13.2 Hz, 1H), 1.92 (ddd, J =
9.1, 5.2, 1.1 Hz, 1H), 1.69 – 1.62 (m, 1H), 1.54 (ddd, J = 9.1, 4.4, 1.7 Hz, 1H), 0.87 (s, 9H),
0.05 (s, 3H), 0.05 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 170.9, 162.0, 137.5, 113.9, 99.3, 75.8, 53.8, 49.3, 39.5,
36.3, 32.2, 25.7, 21.3, 17.9, -5.0.
IR (film): ν 3667, 2953, 2929, 2858, 2358, 2338, 2099, 1672, 1537, 1470, 1362, 1320, 1254,
1162, 1084, 936, 839, 778.
HR-MS (ESI): calcd for C17H28N5O2SSi [M-H]+: 394.1733, found: 394.1728.
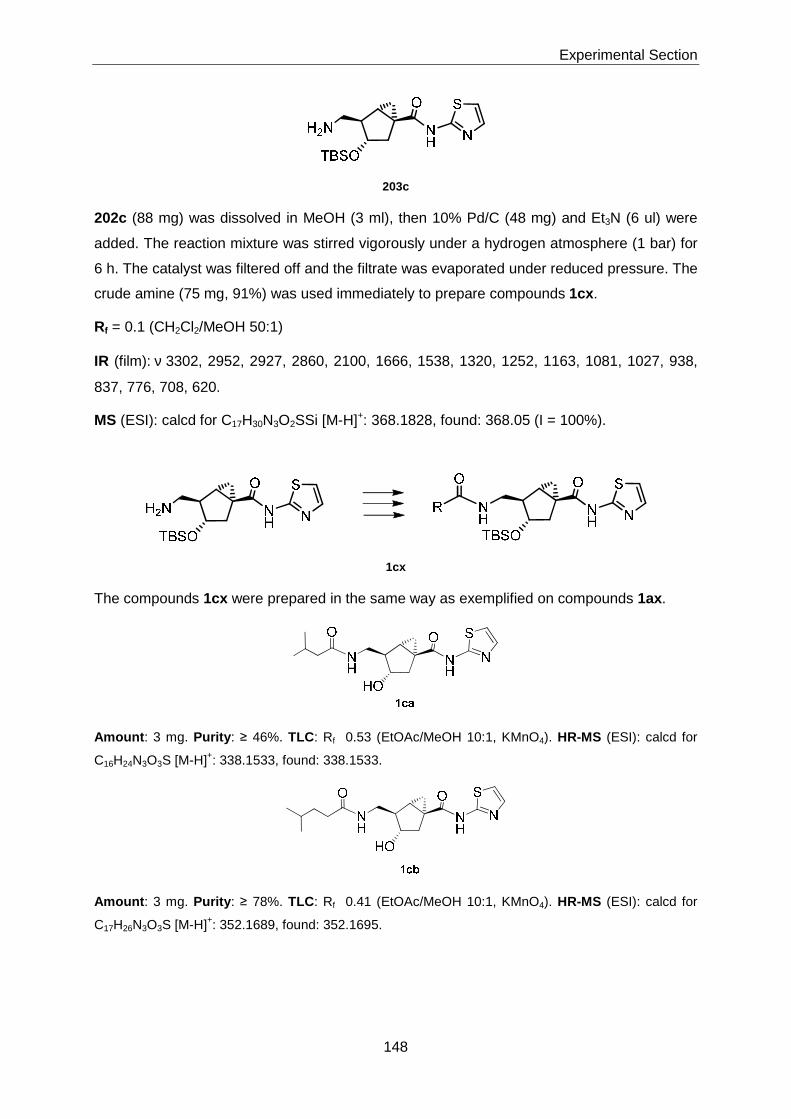
Experimental Section
148
203c
202c (88 mg) was dissolved in MeOH (3 ml), then 10% Pd/C (48 mg) and Et3N (6 ul) were
added. The reaction mixture was stirred vigorously under a hydrogen atmosphere (1 bar) for
6 h. The catalyst was filtered off and the filtrate was evaporated under reduced pressure. The
crude amine (75 mg, 91%) was used immediately to prepare compounds 1cx .
Rf = 0.1 (CH2Cl2/MeOH 50:1)
IR (film): ν 3302, 2952, 2927, 2860, 2100, 1666, 1538, 1320, 1252, 1163, 1081, 1027, 938,
837, 776, 708, 620.
MS (ESI): calcd for C17H30N3O2SSi [M-H]+: 368.1828, found: 368.05 (I = 100%).
1cx
The compounds 1cx were prepared in the same way as exemplified on compounds 1ax.
Amount : 3 mg. Purity : ≥ 46%. TLC: Rf 0.53 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C16H24N3O3S [M-H]+: 338.1533, found: 338.1533.
Amount : 3 mg. Purity : ≥ 78%. TLC: Rf 0.41 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C17H26N3O3S [M-H]+: 352.1689, found: 352.1695.
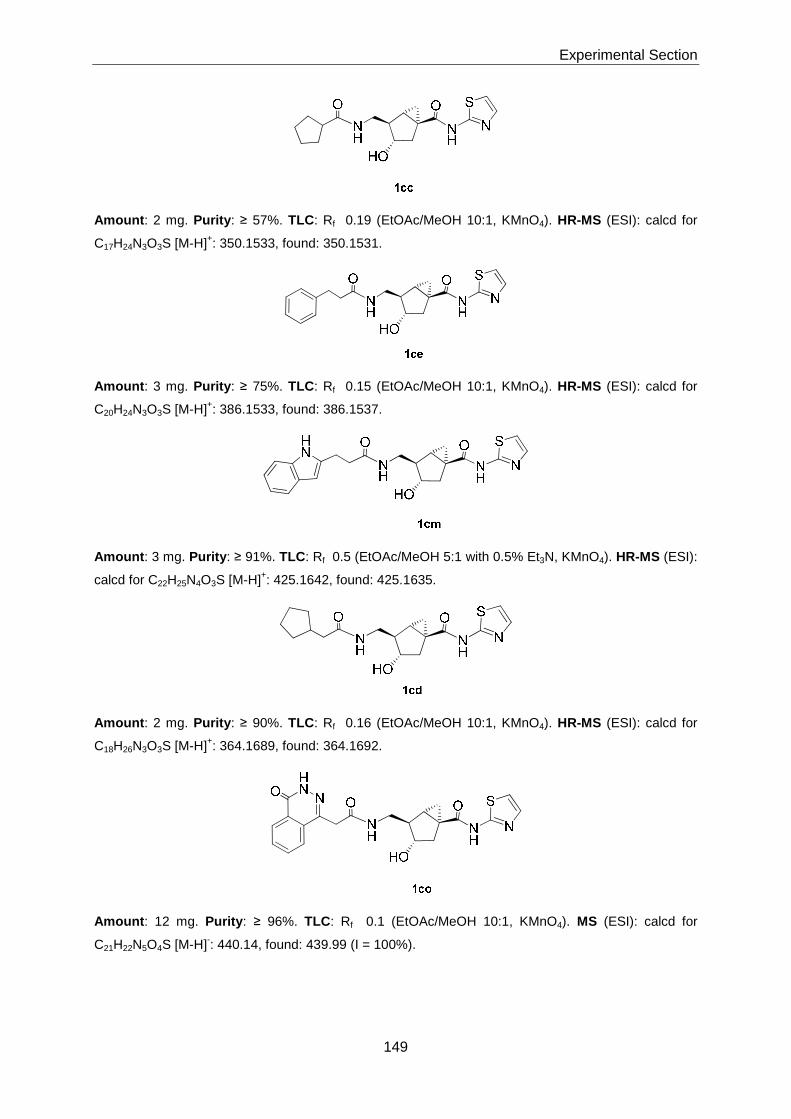
Experimental Section
149
Amount : 2 mg. Purity : ≥ 57%. TLC: Rf 0.19 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C17H24N3O3S [M-H]+: 350.1533, found: 350.1531.
Amount : 3 mg. Purity : ≥ 75%. TLC: Rf 0.15 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C20H24N3O3S [M-H]+: 386.1533, found: 386.1537.
Amount : 3 mg. Purity : ≥ 91%. TLC: Rf 0.5 (EtOAc/MeOH 5:1 with 0.5% Et3N, KMnO4). HR-MS (ESI):
calcd for C22H25N4O3S [M-H]+: 425.1642, found: 425.1635.
Amount : 2 mg. Purity : ≥ 90%. TLC: Rf 0.16 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C18H26N3O3S [M-H]+: 364.1689, found: 364.1692.
Amount : 12 mg. Purity : ≥ 96%. TLC: Rf 0.1 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C21H22N5O4S [M-H]-: 440.14, found: 439.99 (I = 100%).
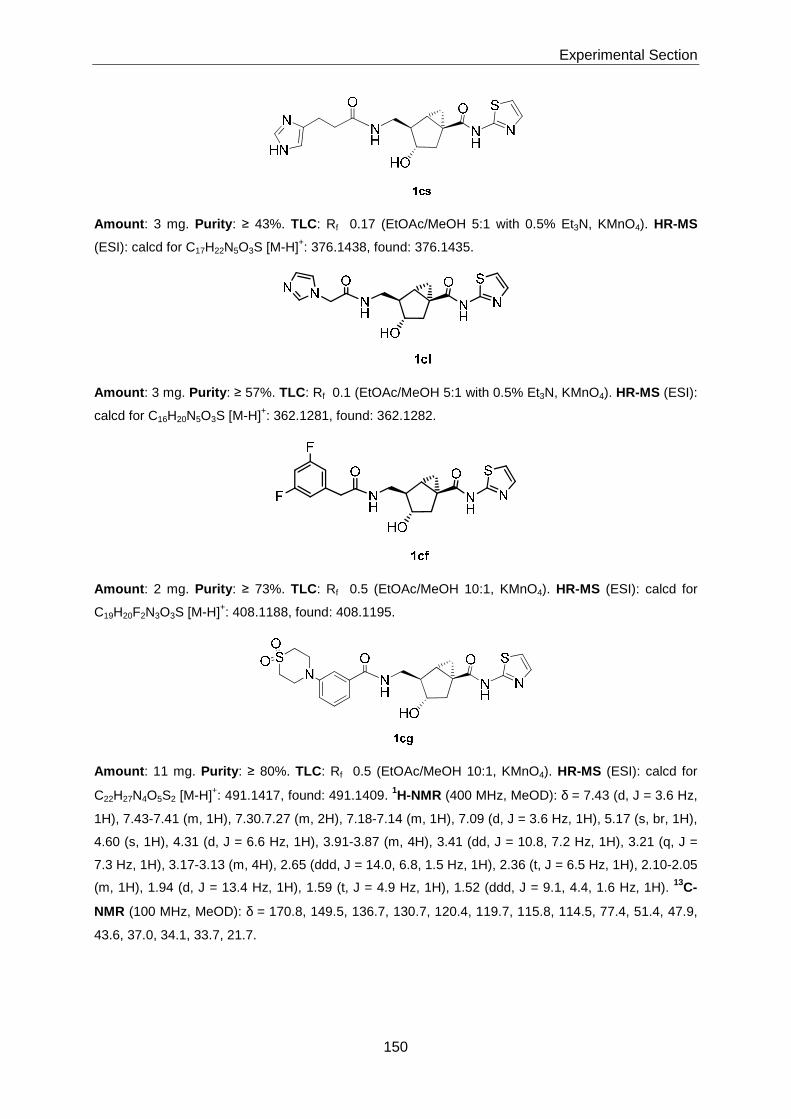
Experimental Section
150
Amount : 3 mg. Purity : ≥ 43%. TLC: Rf 0.17 (EtOAc/MeOH 5:1 with 0.5% Et3N, KMnO4). HR-MS
(ESI): calcd for C17H22N5O3S [M-H]+: 376.1438, found: 376.1435.
Amount : 3 mg. Purity : ≥ 57%. TLC: Rf 0.1 (EtOAc/MeOH 5:1 with 0.5% Et3N, KMnO4). HR-MS (ESI):
calcd for C16H20N5O3S [M-H]+: 362.1281, found: 362.1282.
Amount : 2 mg. Purity : ≥ 73%. TLC: Rf 0.5 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C19H20F2N3O3S [M-H]+: 408.1188, found: 408.1195.
Amount : 11 mg. Purity : ≥ 80%. TLC: Rf 0.5 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C22H27N4O5S2 [M-H]+: 491.1417, found: 491.1409. 1H-NMR (400 MHz, MeOD): δ = 7.43 (d, J = 3.6 Hz,
1H), 7.43-7.41 (m, 1H), 7.30.7.27 (m, 2H), 7.18-7.14 (m, 1H), 7.09 (d, J = 3.6 Hz, 1H), 5.17 (s, br, 1H),
4.60 (s, 1H), 4.31 (d, J = 6.6 Hz, 1H), 3.91-3.87 (m, 4H), 3.41 (dd, J = 10.8, 7.2 Hz, 1H), 3.21 (q, J =
7.3 Hz, 1H), 3.17-3.13 (m, 4H), 2.65 (ddd, J = 14.0, 6.8, 1.5 Hz, 1H), 2.36 (t, J = 6.5 Hz, 1H), 2.10-2.05
(m, 1H), 1.94 (d, J = 13.4 Hz, 1H), 1.59 (t, J = 4.9 Hz, 1H), 1.52 (ddd, J = 9.1, 4.4, 1.6 Hz, 1H). 13C-
NMR (100 MHz, MeOD): δ = 170.8, 149.5, 136.7, 130.7, 120.4, 119.7, 115.8, 114.5, 77.4, 51.4, 47.9,
43.6, 37.0, 34.1, 33.7, 21.7.
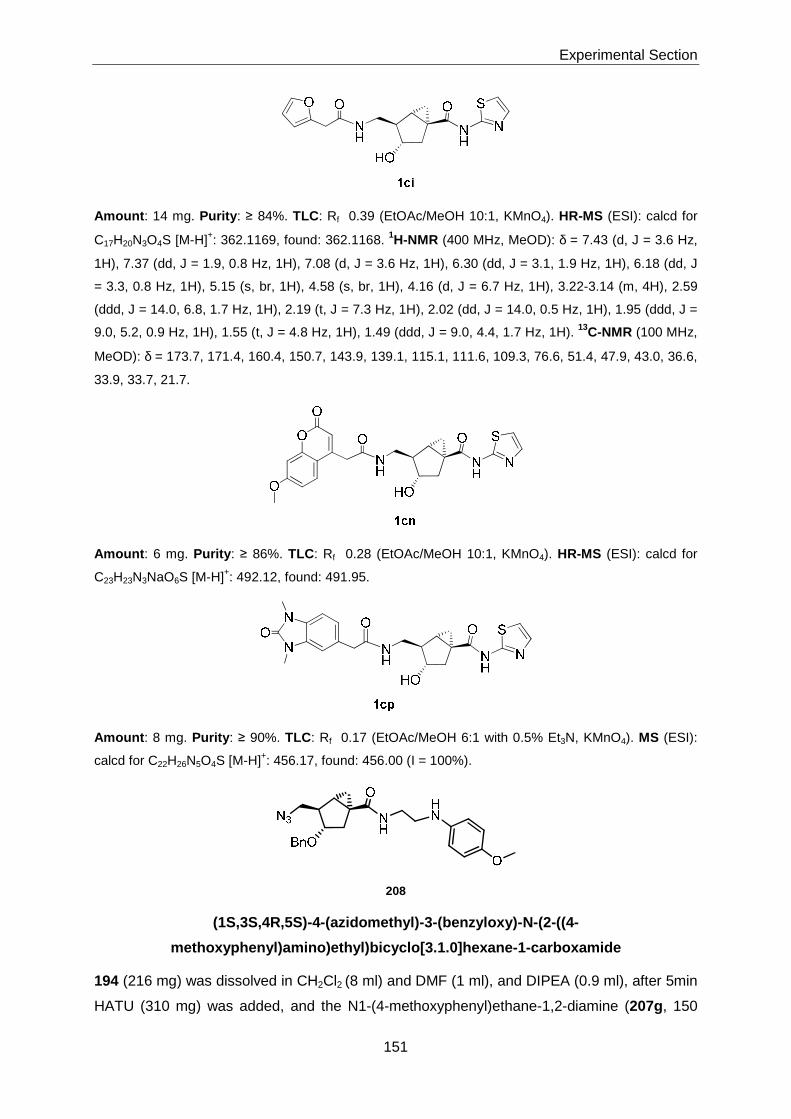
Experimental Section
151
Amount : 14 mg. Purity : ≥ 84%. TLC: Rf 0.39 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C17H20N3O4S [M-H]+: 362.1169, found: 362.1168. 1H-NMR (400 MHz, MeOD): δ = 7.43 (d, J = 3.6 Hz,
1H), 7.37 (dd, J = 1.9, 0.8 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 6.30 (dd, J = 3.1, 1.9 Hz, 1H), 6.18 (dd, J
= 3.3, 0.8 Hz, 1H), 5.15 (s, br, 1H), 4.58 (s, br, 1H), 4.16 (d, J = 6.7 Hz, 1H), 3.22-3.14 (m, 4H), 2.59
(ddd, J = 14.0, 6.8, 1.7 Hz, 1H), 2.19 (t, J = 7.3 Hz, 1H), 2.02 (dd, J = 14.0, 0.5 Hz, 1H), 1.95 (ddd, J =
9.0, 5.2, 0.9 Hz, 1H), 1.55 (t, J = 4.8 Hz, 1H), 1.49 (ddd, J = 9.0, 4.4, 1.7 Hz, 1H). 13C-NMR (100 MHz,
MeOD): δ = 173.7, 171.4, 160.4, 150.7, 143.9, 139.1, 115.1, 111.6, 109.3, 76.6, 51.4, 47.9, 43.0, 36.6,
33.9, 33.7, 21.7.
Amount : 6 mg. Purity : ≥ 86%. TLC: Rf 0.28 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C23H23N3NaO6S [M-H]+: 492.12, found: 491.95.
Amount : 8 mg. Purity : ≥ 90%. TLC: Rf 0.17 (EtOAc/MeOH 6:1 with 0.5% Et3N, KMnO4). MS (ESI):
calcd for C22H26N5O4S [M-H]+: 456.17, found: 456.00 (I = 100%).
208
(1S,3S,4R,5S)-4-(azidomethyl)-3-(benzyloxy)-N-(2-(( 4-
methoxyphenyl)amino)ethyl)bicyclo[3.1.0]hexane-1-ca rboxamide
194 (216 mg) was dissolved in CH2Cl2 (8 ml) and DMF (1 ml), and DIPEA (0.9 ml), after 5min
HATU (310 mg) was added, and the N1-(4-methoxyphenyl)ethane-1,2-diamine (207g, 150

Experimental Section
152
mg) was added and the reaction mixture was stirred for 22 h at RT. The solvents were
evaporated and the crude product was purified by flash chromatography (EE/Hex 4:1) to
afford 239 mg (73%) of the product as a very slightly brown oil.
Rf = 0.28 (CH2Cl2/MeOH 50:1)
1H-NMR (400 MHz, MeOD): δ = 7.36 – 7.24 (m, 5H), 6.78 – 6.70 (m, 2H), 6.69 – 6.61 (m,
2H), 4.42 (s, 2H), 3.89 (d, J = 7.0 Hz, 1H), 3.69 (s, 3H), 3.44 – 3.29 (m, 3H), 3.23 – 3.15 (m,
3H), 2.43 – 2.29 (m, 2H), 2.06 (d, J = 14.0 Hz, 1H), 1.72 (ddd, J = 8.8, 5.2, 1.0 Hz, 1H), 1.31
– 1.27 (m, 2H).
13C-NMR (400 MHz, MeOD): δ = 176.5, 153.8, 143.9, 139.9, 129.7, 128.8, 128.5, 115.9,
115.4, 84.7, 71.8, 56.2, 55.0, 47.9, 45.6, 40.7, 34.4, 33.7, 31.6, 19.7.
MS (ESI): calcd for C24H30N5O3 [M-H]+: 436.2349, found: 436.09 (I = 100%).
209
(1S,3S,4R,5S)-4-(aminomethyl)-3-(benzyloxy)-N-(2-(( 4-methoxyphenyl)amino)ethyl)
bicyclo[3.1.0]hexane-1-carboxamide
208 (90 mg) was dissolved in EtOH (2.5 ml), Lindlar’s catalyst (88 mg, ~5% Pd on CaCO3)
was added and the reaction mixture was stirred under an atmosphere of hydrogen (6 bar) for
12 h. The solution was filtrated over celite and the filter cake was washed with MeOH/EtOAc.
The solvents were evaporated to yield 83 mg of the crude product (98%) as a slightly brown
oil that was immediately used for the preparation of compounds 210x.
Rf = 0.10 (CH2Cl2/MeOH 50:1)
MS (ESI): calcd for C24H32N3O3 [M-H]+: 410.2444, found: 410.09 (I = 100%).
210x
The compounds 210x were prepared in the same way as described for compounds 1ax,
without the deprotection step employing DOWEX-50W.
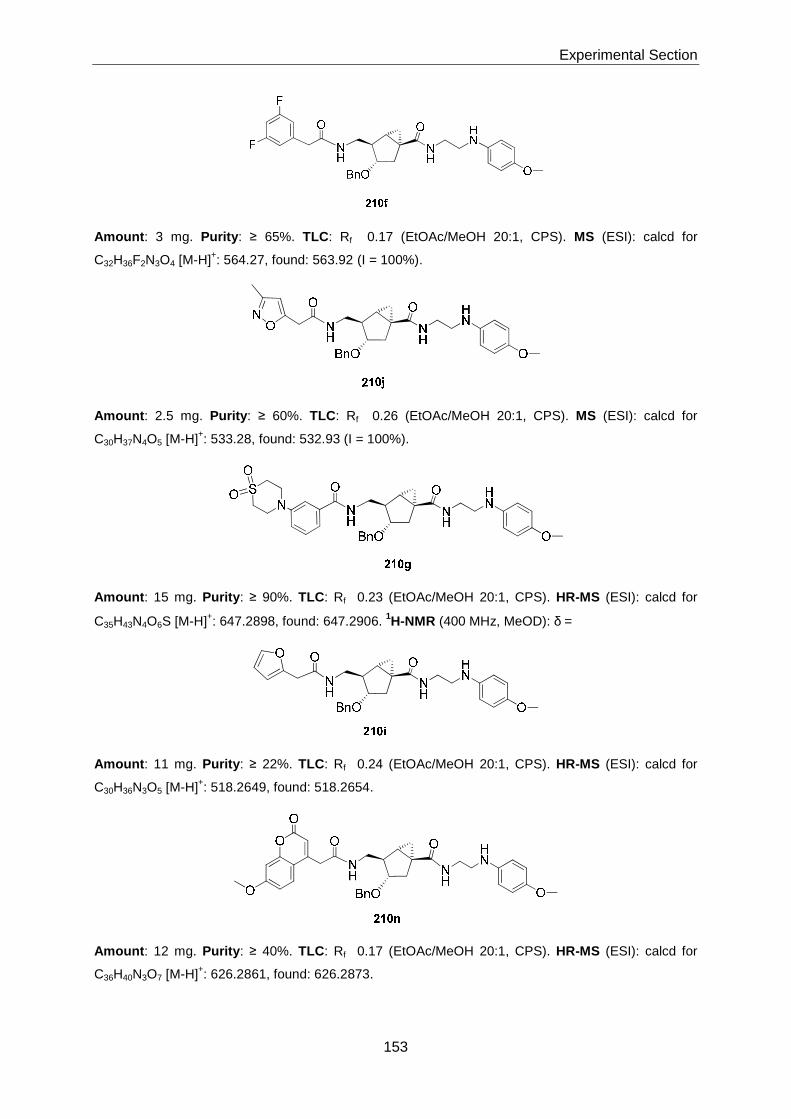
Experimental Section
153
Amount : 3 mg. Purity : ≥ 65%. TLC: Rf 0.17 (EtOAc/MeOH 20:1, CPS). MS (ESI): calcd for
C32H36F2N3O4 [M-H]+: 564.27, found: 563.92 (I = 100%).
Amount : 2.5 mg. Purity : ≥ 60%. TLC: Rf 0.26 (EtOAc/MeOH 20:1, CPS). MS (ESI): calcd for
C30H37N4O5 [M-H]+: 533.28, found: 532.93 (I = 100%).
Amount : 15 mg. Purity : ≥ 90%. TLC: Rf 0.23 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C35H43N4O6S [M-H]+: 647.2898, found: 647.2906. 1H-NMR (400 MHz, MeOD): δ =
Amount : 11 mg. Purity : ≥ 22%. TLC: Rf 0.24 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C30H36N3O5 [M-H]+: 518.2649, found: 518.2654.
Amount : 12 mg. Purity : ≥ 40%. TLC: Rf 0.17 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C36H40N3O7 [M-H]+: 626.2861, found: 626.2873.

Experimental Section
154
Amount : 9 mg. Purity : ≥ 90%. TLC: Rf 0.13 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C34H38N5O5 [M-H]+: 596.2867, found: 596.2883. 1H-NMR (500 MHz, DMSO): δ = 12.60 (s, 1H), 8.41 (t,
J = 5.6 Hz, 1H), 8.25 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 3.6 Hz, 2H), 7.85-7.78 (m, 1H), 7.60 (t, J = 5.6
Hz, 1H), 7.37-7.32 (m, 2H), 7.30-7.23 (m, 3H), 6.69 (d, J = 8.6 Hz, 2H), 6.52 (d, J = 8.6 Hz, 2H), 5.19
(t, J = 5.1 Hz, 1H), 4.28 (s, 2H), 3.82 (s, 2H), 3.76 (d, J = 6.4 Hz, 1H), 3.61 (s, 3H), 3.26-3.18 (m, 2H),
3.04-3.95 (m, 4H), 2.37 (dd, J = 14.0, 6.6 Hz, 1H), 2.23 (t, J = 7.6 Hz, 1H), 1.96 (d, J = 14.1 Hz, 1H),
1.61 (dd, J = 8.6, 4.9 Hz, 1H), 1.19 (dd, J = 9.0, 2.8 Hz, 1H), 1.11 (t, J = 4.2 Hz, 1H). 13C-NMR (500
MHz, DMSO): δ = 172.6, 168.5, 159.4, 150.6, 143.0, 141.7, 138.2, 133.1, 131.7, 129.5, 128.2, 127.6,
127.3, 127.2, 126.0, 125.6, 114.7, 112.9, 82.7, 69.7, 55.4, 46.6, 43.3, 41.6, 38.6, 33.0, 31.9, 29.6,
18.2.
Amount : 6 mg. Purity : ≥ 68%. TLC: Rf 0.16 (EtOAc/MeOH 10:1 with 0.5% Et3N, CPS). HR-MS (ESI):
calcd for C35H42N5O5 [M-H]+: 612.3180, found: 612.3166.
1gp
210gp (5 mg) was dissolved in CH2Cl2 and cooled to -78°C. BCl 3 (131 ul, 1M sol. in CH2Cl2) was
added dropwise and the reaction mixture was stirred for 5 h at -78°C. The reaction mixture was slowly
warmed to -25°C, quenched with MeOH (250 ul) and st irred at RT overnight. The solvents were
evaporated and the crude product was purified by flash chromatography (EtOAc/MeOH 5:1 0.5%
Et3N) to give 1 mg (24%) of 2gp .
TLC: Rf 0.10 (EtOAc/MeOH 6:1 with 0.5% Et3N, CPS). MS (ESI): calcd for C28H36N5O5 [M-H]+:
522.27, found: 521.86 (I = 100%).
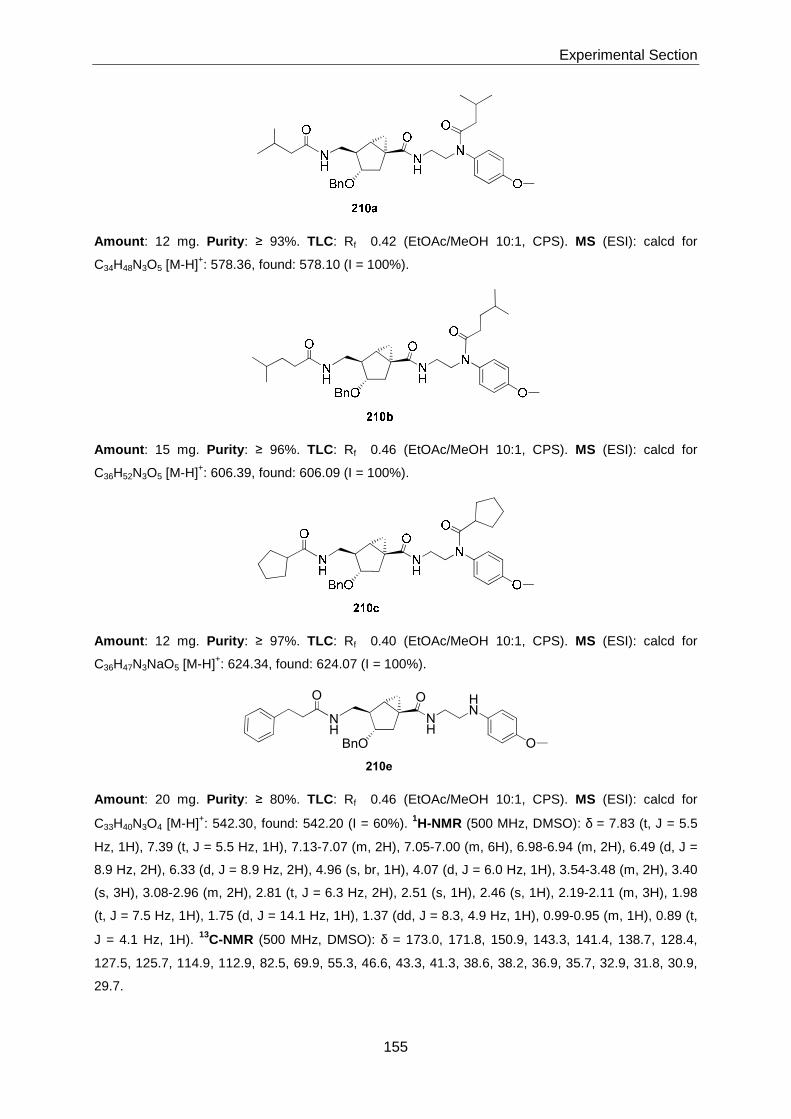
Experimental Section
155
Amount : 12 mg. Purity : ≥ 93%. TLC: Rf 0.42 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C34H48N3O5 [M-H]+: 578.36, found: 578.10 (I = 100%).
Amount : 15 mg. Purity : ≥ 96%. TLC: Rf 0.46 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C36H52N3O5 [M-H]+: 606.39, found: 606.09 (I = 100%).
Amount : 12 mg. Purity : ≥ 97%. TLC: Rf 0.40 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C36H47N3NaO5 [M-H]+: 624.34, found: 624.07 (I = 100%).
NH
O
210e
BnO
O
NH
HN
O
Amount : 20 mg. Purity : ≥ 80%. TLC: Rf 0.46 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C33H40N3O4 [M-H]+: 542.30, found: 542.20 (I = 60%). 1H-NMR (500 MHz, DMSO): δ = 7.83 (t, J = 5.5
Hz, 1H), 7.39 (t, J = 5.5 Hz, 1H), 7.13-7.07 (m, 2H), 7.05-7.00 (m, 6H), 6.98-6.94 (m, 2H), 6.49 (d, J =
8.9 Hz, 2H), 6.33 (d, J = 8.9 Hz, 2H), 4.96 (s, br, 1H), 4.07 (d, J = 6.0 Hz, 1H), 3.54-3.48 (m, 2H), 3.40
(s, 3H), 3.08-2.96 (m, 2H), 2.81 (t, J = 6.3 Hz, 2H), 2.51 (s, 1H), 2.46 (s, 1H), 2.19-2.11 (m, 3H), 1.98
(t, J = 7.5 Hz, 1H), 1.75 (d, J = 14.1 Hz, 1H), 1.37 (dd, J = 8.3, 4.9 Hz, 1H), 0.99-0.95 (m, 1H), 0.89 (t,
J = 4.1 Hz, 1H). 13C-NMR (500 MHz, DMSO): δ = 173.0, 171.8, 150.9, 143.3, 141.4, 138.7, 128.4,
127.5, 125.7, 114.9, 112.9, 82.5, 69.9, 55.3, 46.6, 43.3, 41.3, 38.6, 38.2, 36.9, 35.7, 32.9, 31.8, 30.9,
29.7.
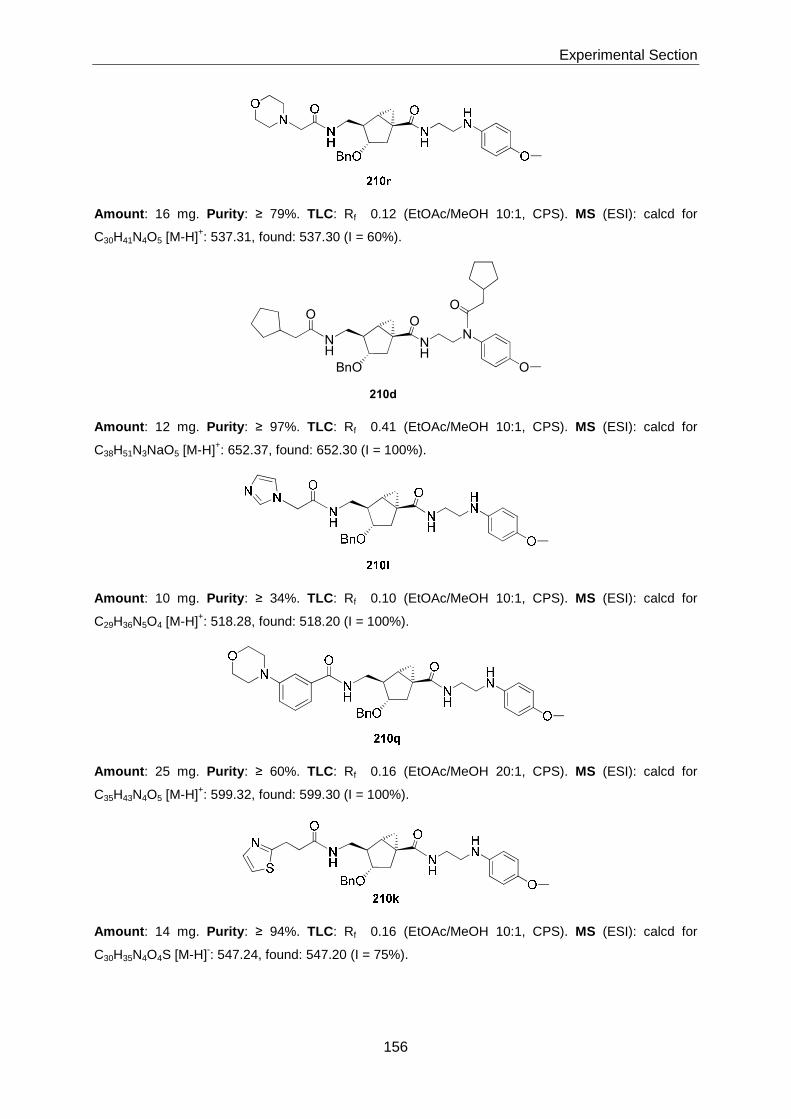
Experimental Section
156
Amount : 16 mg. Purity : ≥ 79%. TLC: Rf 0.12 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C30H41N4O5 [M-H]+: 537.31, found: 537.30 (I = 60%).
NH
O
BnO
O
NH
N
O
210d
O
Amount : 12 mg. Purity : ≥ 97%. TLC: Rf 0.41 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C38H51N3NaO5 [M-H]+: 652.37, found: 652.30 (I = 100%).
Amount : 10 mg. Purity : ≥ 34%. TLC: Rf 0.10 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C29H36N5O4 [M-H]+: 518.28, found: 518.20 (I = 100%).
Amount : 25 mg. Purity : ≥ 60%. TLC: Rf 0.16 (EtOAc/MeOH 20:1, CPS). MS (ESI): calcd for
C35H43N4O5 [M-H]+: 599.32, found: 599.30 (I = 100%).
Amount : 14 mg. Purity : ≥ 94%. TLC: Rf 0.16 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C30H35N4O4S [M-H]-: 547.24, found: 547.20 (I = 75%).

Experimental Section
157
202e
((1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimeth ylsilyl)oxy) icycle[3.1.0]hexan-1-
yl)(3-hydroxy-7,8-dihydro-1,6-naphthyridin-6(5H)-yl )methanone
3 (25 mg, 0.080 mmol) was dissolved in CH2Cl2 (1 ml) and DIPEA (42 ul, 0.241 mmol) was
added under stirring and after 5 min HATU (37 mg, 0.096 mmol) was added. In another flask
the 5,6,7,8-tetrahydro-1,6-naphthyridin-3-ol hydrochloride was dissolved in CH2Cl2 (0.5 ml)
and DMF (0.5 ml), then DIPEA (31 ul, 0.180 mmol) was added, after 10 min of stirring this
mixture was added to the solution of AS-115. The reaction mixture was stirred at RT for 22 h.
It was diluted with CH2Cl2 and poured on saturated aqueous NaHCO3 -solution. The water
phase was extracted with CH2Cl2 (3x 20 ml), the combined organic phases were dried over
MgSO4 and evaporated. Purification of the residue by flash column chromatography
(Hex/EtOAc 1:1--> EtOAc/MeOH 95:5) yielded 32 mg (0.072 mmol, 90%) of the product as a
colorless oil.
Rf = 0.29 (EtOAc/MeOH 20:1)
1H-NMR (400 MHz, CDCl3): δ = 8.13 (d, J = 2.6 Hz, 1H), 7.03 (d, J = 2.5 Hz, 1H), 4.69 (q, J =
17.3 Hz, 2H), 4.17 (d, J = 6.0 Hz, 1H), 4.01 (t, J = 5.9 Hz, 1H), 3.95 (s, 1H), 3.45 (dd, J =
12.5, 6.1 Hz, 1H), 3.12 (dd, J = 12.4, 9.2 Hz, 1H), 2.99 (t, J = 5.6 Hz, 2H), 2.29 (dd, J = 13.4,
6.4 Hz, 1H), 2.13 (dd, J = 9.0, 6.3 Hz, 1H), 1.92 (d, J = 13.4 Hz, 1H), 1.66 (dd, J = 8.9, 4.6
Hz, 1H), 1.44 (t, J = 4.6 Hz, 1H), 1.00 (dd, J = 8.6, 4.1 Hz, 1H), 0.88 (s, 9H), 0.06 (s, 3H),
0.05 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 172.4, 153.1, 136.3, 129.8, 121.7, 76.1, 54.0, 50.4, 40.0,
38.6, 32.5, 30.8, 27.1, 26.0, 18.2, 17.9, -4.5, -4.9.
IR (film): ν 2952, 2928, 2856, 2359, 2341, 2098, 1617, 1455, 1388, 1302, 1258, 1168, 1087,
1038, 928, 837, 766, 669.
MS (ESI): cacld for C22H34N5O3Si [M-H]+: 444.24, found: 444.12 (I = 100%).
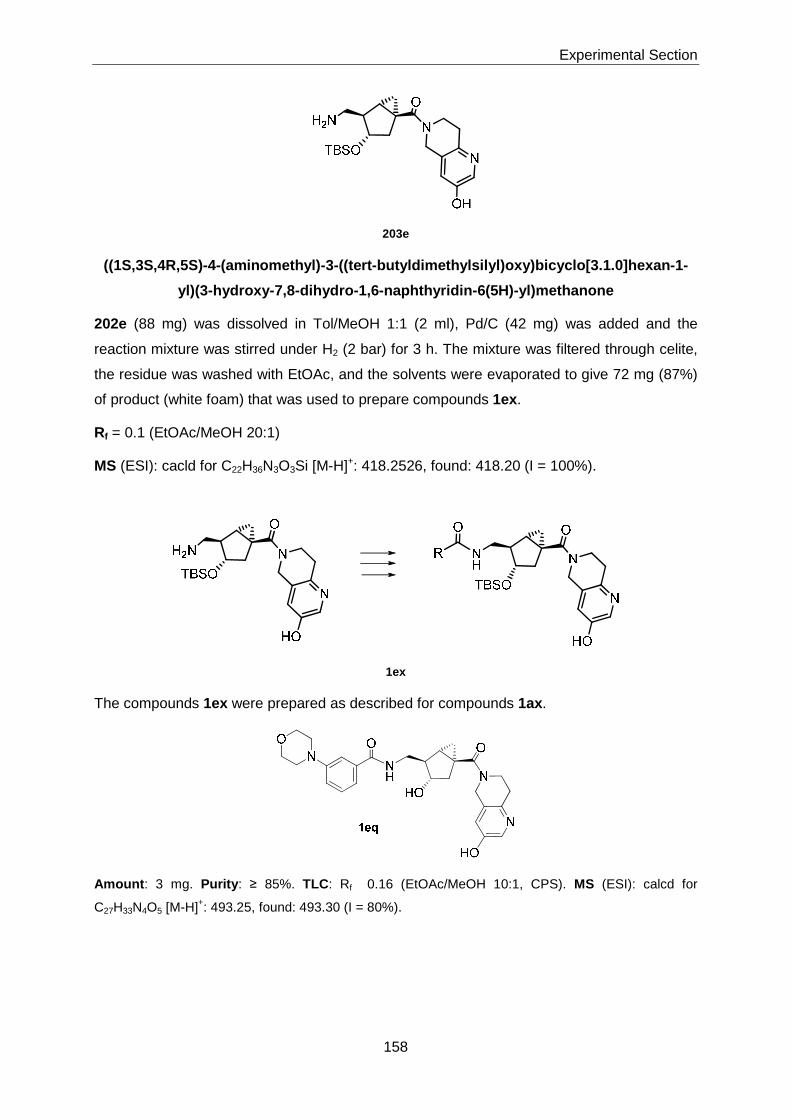
Experimental Section
158
203e
((1S,3S,4R,5S)-4-(aminomethyl)-3-((tert-butyldimeth ylsilyl)oxy)bicyclo[3.1.0]hexan-1-
yl)(3-hydroxy-7,8-dihydro-1,6-naphthyridin-6(5H)-yl )methanone
202e (88 mg) was dissolved in Tol/MeOH 1:1 (2 ml), Pd/C (42 mg) was added and the
reaction mixture was stirred under H2 (2 bar) for 3 h. The mixture was filtered through celite,
the residue was washed with EtOAc, and the solvents were evaporated to give 72 mg (87%)
of product (white foam) that was used to prepare compounds 1ex.
Rf = 0.1 (EtOAc/MeOH 20:1)
MS (ESI): cacld for C22H36N3O3Si [M-H]+: 418.2526, found: 418.20 (I = 100%).
1ex
The compounds 1ex were prepared as described for compounds 1ax.
Amount : 3 mg. Purity : ≥ 85%. TLC: Rf 0.16 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C27H33N4O5 [M-H]+: 493.25, found: 493.30 (I = 80%).
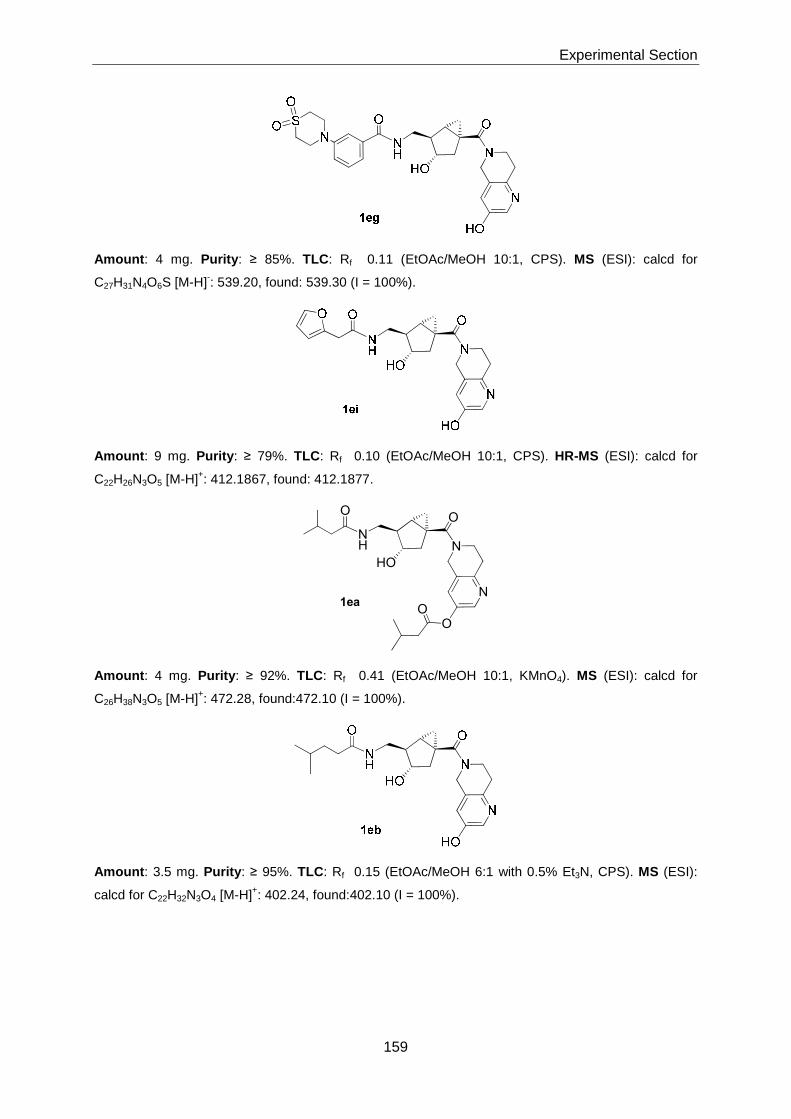
Experimental Section
159
Amount : 4 mg. Purity : ≥ 85%. TLC: Rf 0.11 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C27H31N4O6S [M-H]-: 539.20, found: 539.30 (I = 100%).
Amount : 9 mg. Purity : ≥ 79%. TLC: Rf 0.10 (EtOAc/MeOH 10:1, CPS). HR-MS (ESI): calcd for
C22H26N3O5 [M-H]+: 412.1867, found: 412.1877.
NH
O
1ea
N
N
O
HO
O
O
Amount : 4 mg. Purity : ≥ 92%. TLC: Rf 0.41 (EtOAc/MeOH 10:1, KMnO4). MS (ESI): calcd for
C26H38N3O5 [M-H]+: 472.28, found:472.10 (I = 100%).
Amount : 3.5 mg. Purity : ≥ 95%. TLC: Rf 0.15 (EtOAc/MeOH 6:1 with 0.5% Et3N, CPS). MS (ESI):
calcd for C22H32N3O4 [M-H]+: 402.24, found:402.10 (I = 100%).
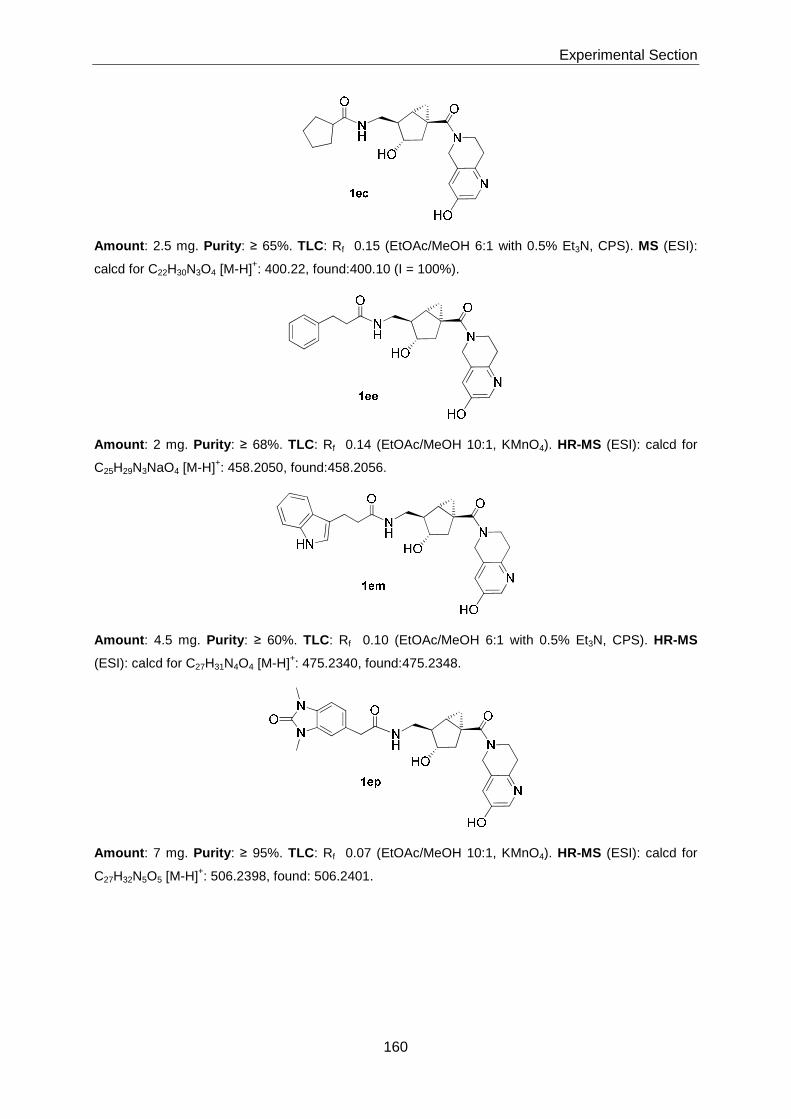
Experimental Section
160
Amount : 2.5 mg. Purity : ≥ 65%. TLC: Rf 0.15 (EtOAc/MeOH 6:1 with 0.5% Et3N, CPS). MS (ESI):
calcd for C22H30N3O4 [M-H]+: 400.22, found:400.10 (I = 100%).
Amount : 2 mg. Purity : ≥ 68%. TLC: Rf 0.14 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C25H29N3NaO4 [M-H]+: 458.2050, found:458.2056.
Amount : 4.5 mg. Purity : ≥ 60%. TLC: Rf 0.10 (EtOAc/MeOH 6:1 with 0.5% Et3N, CPS). HR-MS
(ESI): calcd for C27H31N4O4 [M-H]+: 475.2340, found:475.2348.
Amount : 7 mg. Purity : ≥ 95%. TLC: Rf 0.07 (EtOAc/MeOH 10:1, KMnO4). HR-MS (ESI): calcd for
C27H32N5O5 [M-H]+: 506.2398, found: 506.2401.

Experimental Section
161
Amount : 4 mg. Purity : ≥ 77%. TLC: Rf 0.43 (EtOAc/MeOH 10:1, CPS). MS (ESI): calcd for
C24H26F2N3NaO4 [M-Na]+: 480.17, found: 480.00 (I = 100%).
5.2.1.4 Synthesis of library members of general str ucture 2
233
((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4-( ((tert-butyldimethylsilyl)oxy)methyl)
bicyclo[3.1.0]hexan-1-yl)methanol
185 (60 mg, 0.145 mmol) was dissolved in CH2Cl2 (1 ml) and cooled to -78°C and DIBALH
(435 ul, 0.435 mmol) was added dropwise. The reaction mixture was stirred at -78°C for 4 h,
then it was quenched at this temperature with saturated aqueous NaHCO3 –solution and
extracted with CH2Cl2. The organic phases were dried and evaporated to afford 56 mg (0.145
mmol, 100%) of the product as a colorless oil.
Rf = 0.61 (Hex/EtOAc 1:1)
1H-NMR (400 MHz, CDCl3): δ = 4.14 (d, J = 6.5 Hz, 1H), 3.74-3.70 (m, 1H), 3.63 (d, J = 11.1
Hz, 1H), 3.54 (dd, J = 10.1, 5.1 Hz, 1H), 3.43-3.38 (m, 2H), 2.15 (ddd, J = 13.5, 6.6, 1.6 Hz,
1H), 2.00 (dd, J = 6.6, 5.4 Hz, 1H), 1.69 (d, J = 13.5 Hz, 1H), 1.07 (t, J = 4.3 Hz, 1H), 0.96
(ddd, J = 9.1, 3.9, 1.2 Hz, 1H), 0.88 (s, 9H), 0.83 (s, 9H), 0.53 (ddd, J = 8.7, 4.4, 1.7 Hz, 1H),
0.04 (s, 3H), 0.03 (s, 3H), -0.01 (s, 3H), -0.01 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 76.4, 68.1, 67.1, 65.7, 38.6, 31.8, 26.0, 25.8, 24.5, 18.6,
17.9, 15.3, -4.6, -4.8, -5.4, -5.7.
IR (film): ν 3358, 2928, 2856, 2359, 1471, 1462, 1255, 1082, 834, 773, 665.
MS (ESI): calcd for C20H42NaO3Si2 [M-Na]+: 409.26, found: 409.14 (I = 100%).

Experimental Section
162
234
(1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4-(( (tert-butyldimethylsilyl)oxy)methyl)
bicyclo[3.1.0]hexane-1-carbaldehyde
233 (56 mg, 0.145 mmol) was dissolved in CH2Cl2, pyridinium chlorochromate (94 mg, 0.435
mmol) was added and the reaction mixture was stirred at RT for 5 h, then it was diluted with
EtOAc/Hex 1:5 and filtered through a short column (EtOAc/Hex 1:5) to afford 51 mg of the
product (0.133 mmol, 91%) as a slightly yellow oil.
Rf = 0.40 (Hex/EtOAc 10:1)
1H-NMR (400 MHz, CDCl3): δ = 8.84 (s, 1H), 4.20 (d, J = 6.5 Hz, 1H), 3.74 (dd, J = 10.3, 6.3
Hz, 1H), 3.33 (dd, J = 10.3, 7.7 Hz, 1H), 2.52 (ddd, J = 14.2, 6.4, 1.5 Hz, 1H), 2.14 (t, J = 6.9
Hz, 1H), 1.85-1.75 (m, 2H), 1.69 (d, J = 14.3 Hz, 1H), 1.46 (ddd, J = 8.6, 4.4, 1.6 Hz, 1H),
0.88 (s, 9H), 0.85 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 199.9, 76.1, 63.8, 52.5, 42.4, 34.4, 30.4, 25.9, 25.7, 19.2,
18.4, 17.8, -4.8, -4.9, -4.4, -5.5.
HR-MS (ESI): no signal found. MS (ESI): calcd for C20H41O3Si2 [M-H]+: 385.26 found: 385.12
(I = 100%).
235
5-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4 -(((tert-butyldimethylsilyl)oxy)methyl)
bicyclo[3.1.0]hexan-1-yl)oxazole
234 (51 mg) was dissolved in MeOH (2 ml), K2CO3 (55 mg) and TosMIC (27 mg) were added
and the reaction mixture was refluxed for 4 h, then it was diluted with EtOAc/hex 1:5 and
purified by flash column chromatography (EtOAc/Hex 1:5) to give 9 mg of the product as a
colorless oil.
Rf = 0.31 (Hex/EtOAc 5:1)
1H-NMR (400 MHz, CDCl3): δ = 7.67 (s, 1H), 6.69 (s, 1H), 4.25 (d, J = 6.4 Hz, 1H), 3.55 (dd,
J = 10.1, 5.7 Hz, 1H), 3.36 (dd, J = 10.1, 8.1 Hz, 1H), 2.37 (ddd, J = 13.5, 6.4, 1.5 Hz, 1H),
2.16 (dd, J = 8.1, 5.7 Hz, 1H), 1.95 (d, J = 13.5 Hz, 1H), 1.54 (t, J = 4.5 Hz, 1H), 1.49 (ddd, J

Experimental Section
163
= 8.5, 4.6, 0.9 Hz, 1H), 1.14 (ddd, J = 8.6, 4.3, 1.7 Hz, 1H), 0.88 (s, 9H), 0.87 (s, 9H), 0.04 (s,
3H), 0.03 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 149.0, 120.1, 74.8, 64.7, 54.3, 39.7, 33.0, 25.9, 25.7, 24.5,
17.8, 17.6, -4.8, -4.9, -5.5.
MS (ESI): calcd for C22H42NO3Si2 [M-H]+: 424.27, found: 424.00 (I = 100%).
237
(1S,3S,4R,5S)-4-(azidomethyl)-3-((tert-butyldimethy lsilyl)oxy)-N-(2-
hydroxyethyl)bicyclo[3.1.0] hexane-1-carboxamide
3 (60 mg, 0.193 mmol) was dissolved in CH2Cl2 (2 ml) and a few drops of DMF. DIPEA (165
ul, 0.963 mmol) was added, then after 5 min HATU (82 mg, 0.212 mmol) and ethanolamine
(14 ul, 0.232 mmol) were added. The reaction mixture was stirred for 2 h at RT. The reaction
mixture was then diluted with CH2Cl2 and quenched with water. The water phase was
extracted with CH2Cl2, the combined organic phases were dried over MgSO4 and
evaporated. The residue was purified by flash column chromatography (EtOAc/MeOH 20:1)
to give 46 mg (0.139 mmol, 67%) of the product as a yellow oil.
Rf = 0.24 (EtOAc/MeOH 20:1)
1H-NMR (400 MHz, CDCl3): 6.09-6.02 (m, 1H), 4.18-4.14 (d, J = 6.5 Hz, 1H), 3.73-3.69 (t, 4.8
Hz, 1H), 3.47-3.37 (m, 2H), 3.35-3.30 (dd, J = 12.4, 5.4 Hz, 1H), 3.10-3.04 (dd, J = 12.4, 8.3
Hz, 1H), 2.37-2.30 (m, 1H), 2.16-2.11 (dd, J = 8.5, 5.5 Hz, 1H), 1.88-1.85 (d, J = 13.1 Hz,
1H), 1.72-1.67 (ddd, J = 9.0, 4.8, 1 Hz, 1H), 1.46-1.42 (t, J = 4.5 Hz, 1H), 1.36-1.32 (ddd, J =
9.2, 4.1, 1.5 Hz, 1H), 0.85 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H).
13C-NMR (100 MHz, : 175.0, 76.2, 62.2, 54.4, 50.2, 42.4, 37.5, 32.9, 30.8, 25.4, 20.5, 18.4, -
4.8, -4.9.
MS (ESI): calcd for C16H31N4O3Si [M-H]+: 355.22, found: 355.05 (I = 100%).
238
237 (100 mg, 0.282 mmol) was dissolved in THF (3 ml) and Burgess reagent (83 mg, 0.338
mmol, ((Methoxycarbonyl)sulfamoyl)triethylammonium hydroxide) was added. The reaction
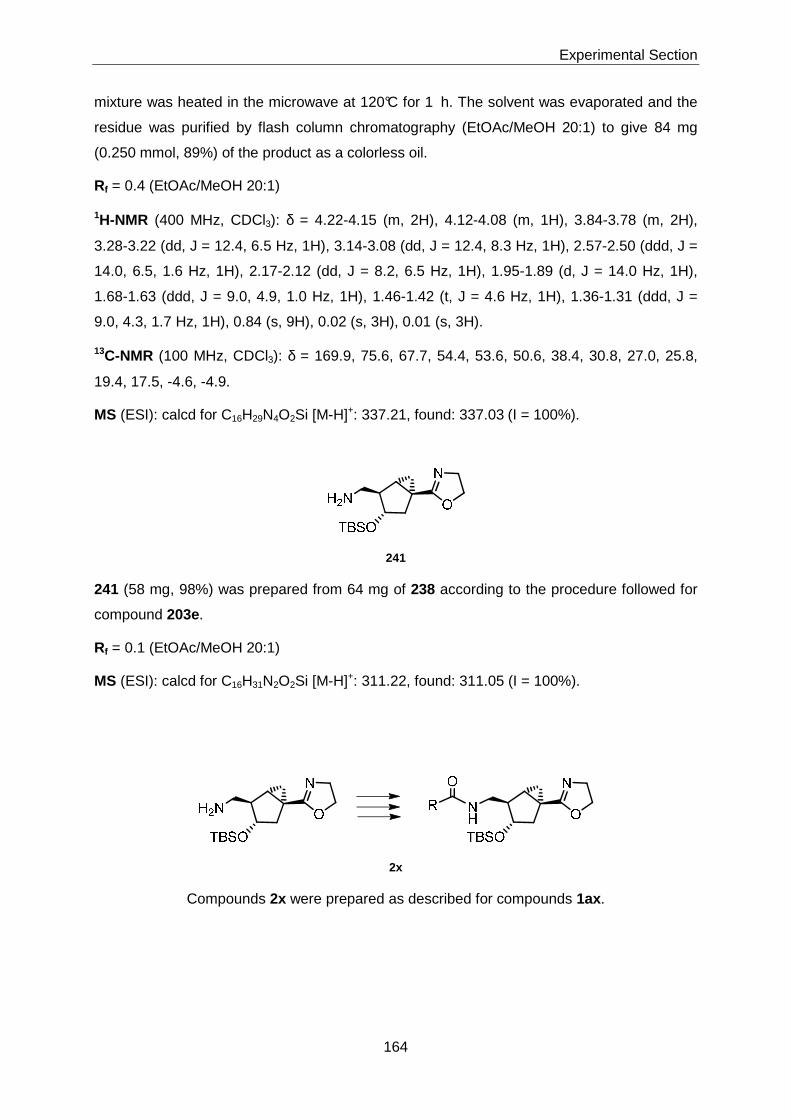
Experimental Section
164
mixture was heated in the microwave at 120°C for 1 h. The solvent was evaporated and the
residue was purified by flash column chromatography (EtOAc/MeOH 20:1) to give 84 mg
(0.250 mmol, 89%) of the product as a colorless oil.
Rf = 0.4 (EtOAc/MeOH 20:1)
1H-NMR (400 MHz, CDCl3): δ = 4.22-4.15 (m, 2H), 4.12-4.08 (m, 1H), 3.84-3.78 (m, 2H),
3.28-3.22 (dd, J = 12.4, 6.5 Hz, 1H), 3.14-3.08 (dd, J = 12.4, 8.3 Hz, 1H), 2.57-2.50 (ddd, J =
14.0, 6.5, 1.6 Hz, 1H), 2.17-2.12 (dd, J = 8.2, 6.5 Hz, 1H), 1.95-1.89 (d, J = 14.0 Hz, 1H),
1.68-1.63 (ddd, J = 9.0, 4.9, 1.0 Hz, 1H), 1.46-1.42 (t, J = 4.6 Hz, 1H), 1.36-1.31 (ddd, J =
9.0, 4.3, 1.7 Hz, 1H), 0.84 (s, 9H), 0.02 (s, 3H), 0.01 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 169.9, 75.6, 67.7, 54.4, 53.6, 50.6, 38.4, 30.8, 27.0, 25.8,
19.4, 17.5, -4.6, -4.9.
MS (ESI): calcd for C16H29N4O2Si [M-H]+: 337.21, found: 337.03 (I = 100%).
241
241 (58 mg, 98%) was prepared from 64 mg of 238 according to the procedure followed for
compound 203e.
Rf = 0.1 (EtOAc/MeOH 20:1)
MS (ESI): calcd for C16H31N2O2Si [M-H]+: 311.22, found: 311.05 (I = 100%).
2x
Compounds 2x were prepared as described for compounds 1ax.
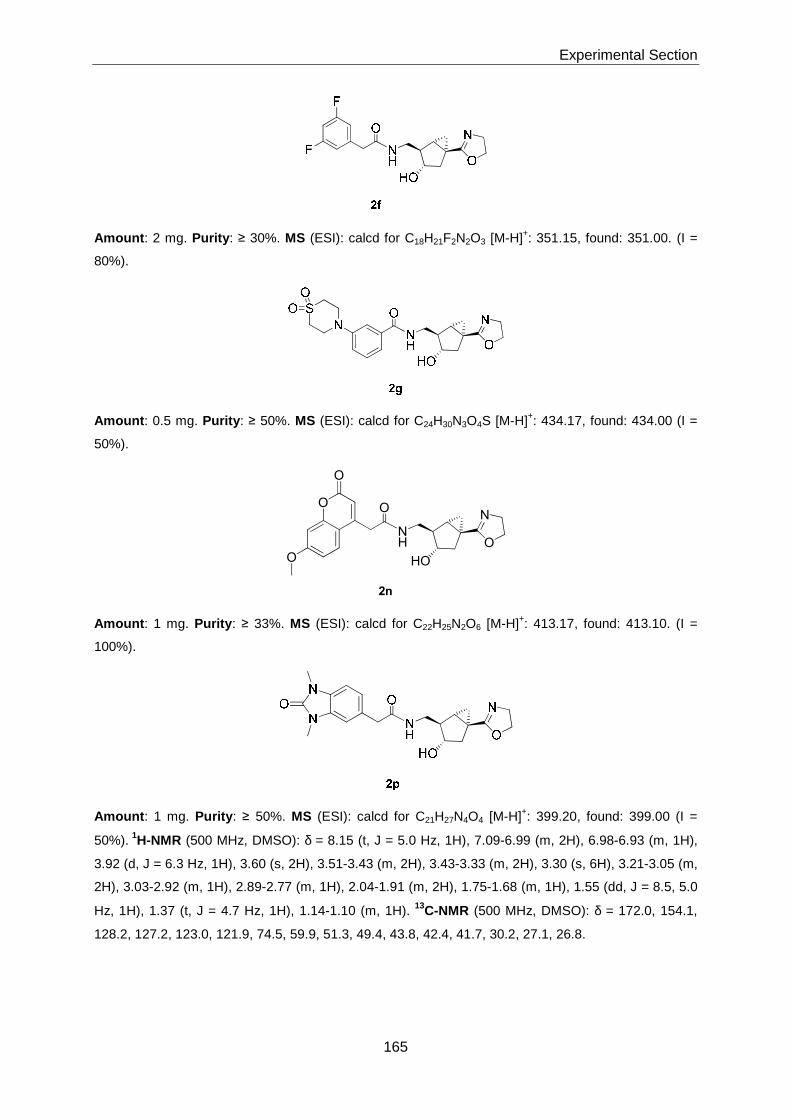
Experimental Section
165
Amount : 2 mg. Purity : ≥ 30%. MS (ESI): calcd for C18H21F2N2O3 [M-H]+: 351.15, found: 351.00. (I =
80%).
Amount : 0.5 mg. Purity : ≥ 50%. MS (ESI): calcd for C24H30N3O4S [M-H]+: 434.17, found: 434.00 (I =
50%).
HO
NH
O
2n
O
NO
O
O
Amount : 1 mg. Purity : ≥ 33%. MS (ESI): calcd for C22H25N2O6 [M-H]+: 413.17, found: 413.10. (I =
100%).
Amount : 1 mg. Purity : ≥ 50%. MS (ESI): calcd for C21H27N4O4 [M-H]+: 399.20, found: 399.00 (I =
50%). 1H-NMR (500 MHz, DMSO): δ = 8.15 (t, J = 5.0 Hz, 1H), 7.09-6.99 (m, 2H), 6.98-6.93 (m, 1H),
3.92 (d, J = 6.3 Hz, 1H), 3.60 (s, 2H), 3.51-3.43 (m, 2H), 3.43-3.33 (m, 2H), 3.30 (s, 6H), 3.21-3.05 (m,
2H), 3.03-2.92 (m, 1H), 2.89-2.77 (m, 1H), 2.04-1.91 (m, 2H), 1.75-1.68 (m, 1H), 1.55 (dd, J = 8.5, 5.0
Hz, 1H), 1.37 (t, J = 4.7 Hz, 1H), 1.14-1.10 (m, 1H). 13C-NMR (500 MHz, DMSO): δ = 172.0, 154.1,
128.2, 127.2, 123.0, 121.9, 74.5, 59.9, 51.3, 49.4, 43.8, 42.4, 41.7, 30.2, 27.1, 26.8.

Experimental Section
166
Amount : 1 mg. MS (ESI): calcd for C21H26N3O3 [M-H]+: 368.20, found: 368.05 (I = 50%).
219
N-(4,6-dichloropyrimidin-5-yl)formamide
Acetic anhydride (1.12 ml) was added dropwise over 5min to a solution of 4,6-dichloro-5-
aminopyrimidine (240 mg) in formic acid (3 ml) at 0°C, then it was stirred at RT for 4 h. The
solvent was removed and the residue was co-evaporated with toluene (3x) to yield 302 mg of
219 (1.573 mmol, >100%).
Rf = 0.13 (Hex/EtOAc 2:1)
1H NMR (400 MHz, DMSO): δ = 10.49 (s, br, 1H), 8.85 (s, 1H), 8.34 (s, 1H).
222
9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4 -(((tert-butyldimethylsilyl)oxy)
methyl)bicyclo [3.1.0]hexan-1-yl)-1H-purin-6(9H)-on e
221 (52 mg) was dissolved in MeOH (15 ml), 2-mercaptoethanol (145 ul) and NaOMe (110
mg) were added. The reaction mixture was heated to 80°C in the microwave for 1.5 h. Acetic
acid (117 ul) was added and the mixture was stirred for 10 min. The solvents were
evaporated and the residue was purified by flash column chromatography (EE/MeOH 20:1)
to yield 32 mg (61%) of the product as a white solid.
Rf = 0.16 (EtOAc/MeOH 20:1)
1H NMR (400 MHz, MeOD): δ = 8.03 (s, 1H), 7.99 (s, 1H), 4.39 (d, J = 6.4 Hz, 1H), 3.92 (dt, J
= 10.4, 7.5 Hz, 2H), 2.58 (ddd, J = 13.3, 6.5, 2.1 Hz, 1H), 2.25 (d, J = 13.4 Hz, 1H), 2.17 (dd,

Experimental Section
167
J = 8.5, 5.7 Hz, 1H), 1.84 – 1.76 (m, 1H), 1.71 (t, J = 5.1 Hz, 1H), 1.39 (ddd, J = 9.7, 5.3, 2.1
Hz, 1H), 0.95 (s, 6H), 0.91 (s, 6H), 0.13 (s, 3H), 0.13 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H).
13C-NMR (100 MHz, MeOD): δ = 159.2, 150.5, 144.3, 141.2, 125.2, 74.2, 60.5, 53.0, 43.4,
42.0, 41.4, 26.1, 25.8, 18.5, 17.9, 16.9, -4.7, -4.8, -5.3, -5.3.
IR (film): ν 2929, 2857, 1686, 1401, 1254, 1083, 1041, 907, 837, 778, 731, 649.
HR-MS (ESI): calcd for C24H42N4NaO3Si2 [M-Na]+: 513.2688, found: 513.2690.
229
9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4 -(hydroxymethyl)bicyclo[3.1.0]hexan-
1-yl)-1H-purin-6(9H)-one
To a solution of 222 (120 mg, 0.245 mmol) in CH2Cl2/MeOH 1:1 (6 ml) CSA (74 mg, 0.319
mmol) was added at 0°C. The reaction mixture was st irred for 3 h at 0°C, additional 25 mg of
CSA were added and the reaction mixture was stirred for additional 2 h at 0°C. The reaction
mixture was quenched with Et3N (55 ul, 0.392 mmol) and evaporated. The residue was
purified by flash chromatography (EE/MeOH 10:1) to afford 80 mg of the product as a white
solid. 1H-NMR revealed that the product contained CSA. It was therefore re-dissolved in
aqueous NaHCO3 solution and extracted with a mixture of EtOAc/MeOH 5% to give 75 mg
(0.199 mmol, 81%) of the pure product as a white solid.
Rf = 0.4 (EtOAc/MeOH 10:1)
1H-NMR (400 MHz, CDCl3): δ = 8.22 (s, 1H), 7.95 (s, 1H), 5.42 (d, J = 11.7 Hz, 1H), 4.43 (d,
J = 5.5 Hz, 1H), 4.02 (d, J = 11.4 Hz, 1H), 3.83 (t, J = 10.0 Hz, 1H), 2.60 (ddd, J = 8.7, 6.3,
1.7 Hz, 1H), 2.16 (d, J = 13.1 Hz, 2H), 1.77 (dd, J = 8.5, 4.9 Hz, 1H), 1.72 (t, J = 5.0 Hz, 1H),
1.30 (ddd, J = 10.8, 5.7, 2.8 Hz, 1H), 0.89 (s, 9H), 0.05 (s, 3H), 0.05 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 158.6, 148.9, 144.5, 142.4, 75.9, 64.9, 53.6, 44.5, 44.2,
26.9, 25.2, 17.9, 16.9, -4.5.
IR (film): ν 2952, 2927, 2856, 2365, 1714, 1591, 1546, 1467, 1404, 1344, 1253, 1210, 1171,
1084, 1048, 1011, 939, 963, 913, 863, 833, 775, 742, 647, 611.
HR-MS (ESI): calcd for C18H28N4NaO3Si [M-Na]+: 399.1823, found: 399.1829.

Experimental Section
168
216
(E)-3-methoxyacrylic acid
KOH (7.4 g, 0.132 mol) and Methyl 3-methoxyacrylate (215, 11 ml, 0.102 mol) were
dispensed in 30 ml of H2O, and the reaction mixture was stirred overnight at 50°C. The
reaction mixture was cooled to 0°C and acidified sl owly to pH ~1 with 1 N aqueous HCl. The
water phase was extracted with Et2O (3 x 200 ml), the combined organic phases were dried
and evaporated to yield the crude product as a light yellow solid. Purification by
recrystallization from hexane/CHCl3 gave 4.44 g (0.034 mol, 33%) of the pure product as
light yellow crystals.
Rf = 0.3 (EtOAc/Hex 1:1)
1H NMR (400 MHz, CDCl3): δ = 11.82 (s, br, 1H), 7.69 (d, J = 12.6 Hz, 1H), 5.17 (d, J = 12.6
Hz, 1H), 3.71 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 173.6, 165.3, 95.6, 57.5.
O
NCOO
211
(E)-3-methoxyacryloyl isocyanate
Thionyl chloride (3.6 ml) was added to a solution of 216 (2.93 g) in dry CH2Cl2 (300 ml). The
mixture was refluxed for 4 h. The solvent was evaporated to give a brown liquid that was
purified by kugelrohr distillation to afford 2.6 g (74%) of 3-methoxyacryloyl chloride (217) as a
colorless liquid. The liquid was dissolved in toluene (20 ml), silver cyanate (5.9 g, 0.039 mol))
was added and this mixture was refluxed at 130°C fo r 45 min in the dark. The suspension
was cooled with ice and the supernatant was transferred by syringe to the solution of 5 in
CH2Cl2 at -15°C.
212
(E)-N-(((1S,3S,4R,5S)-4-(azidomethyl)-3-(benzyloxy) bicyclo[3.1.0]hexan-1-
yl)carbamoyl)-3-methoxyacrylamide

Experimental Section
169
3-methoxyacryloyl chloride (0.6 g, 5 mmol) was dissolved in toluene and 1.4 g of silver
cyanate (dried over P2O5 at 100°C for 2.5 h in the dark before use) was add ed and the
mixture was refluxed at 120°C in the dark for 30 mi n. The suspension was cooled with ice
and the supernatant was transferred by syringe to the sol. of 5 (130 mg, 0.503 mmol) in
CH2Cl2 at 0°C. The reaction mixture was slowly warmed to RT, then stirred overnight at RT.
The mixture had turned into a dispersion. The solvents were evaporated and the crude
product was purified by flash chromatography (Hex/EtOAc 2:1--> Hex/EtOAc 1:1) to yield
150 mg (0.389, 77%) of the product as a white solid.
Rf = 0.3 (EtOAc/Hex 1:1)
1H-NMR (400 MHz, CDCl3): δ = 8.84 (s, 1H), 8.63 (s, 1H), 7.64 (d, J = 12.1 Hz, 1H), 7.37 –
7.27 (m, 5H), 5.24 (d, J = 12.1 Hz, 1H), 4.42 (s, 2H), 3.84 (d, J = 7.2 Hz, 1H), 3.71 (s, 3H),
3.54 (dd, J = 12.3, 6.5 Hz, 1H), 3.39 (dd, J = 12.3, 9.3 Hz, 1H), 2.45 (ddd, J = 13.8, 7.1, 2.0
Hz, 1H), 2.31 (dd, J = 9.2, 6.6 Hz, 1H), 2.18 (d, J = 13.9 Hz, 1H), 1.40 (dd, J = 8.4, 4.8 Hz,
1H), 1.28 (dd, J = 11.2, 6.2 Hz, 2H), 0.97 (ddd, J = 9.2, 5.2, 2.1 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 164.1, 138.8, 128.4, 127.6, 127.4, 97.9, 82.6, 70.9, 58.6,
53.9, 47.9, 39.9, 36.3, 26.9.
MS (ESI): calcd for C19H22N5O4 [M-H]- : 384.1672, found: 384.10 (I = 100%).
213
1-((1S,3S,4R,5S)-4-(azidomethyl)-3-(benzyloxy)bicyc lo[3.1.0]hexan-1-yl)pyrimidine-
2,4(1H,3H)-dione
A 1 M aq. H2SO4 sol.(3.4 ml) and dioxane (3 ml) were added to 212 and the reaction mixture
was refluxed for 2 h. After cooling, the mixture was neutralized with 2 M aq. NaOH sol. (3.4
ml) to pH 7 and the solvents were evaporated to give the crude product, ~0.8 g, that was
purified by flash chromatography (EtOAc/Hex 2:1) to afford 100 mg (0.283 mmol, 84%) of the
product.
Rf = 0.25 (EtOAc/Hex 2:1)
1H-NMR (400 MHz, CDCl3): δ = 9.10 (d, J = 10.0 Hz, 1H), 7.35 – 7.28 (m, 5H), 5.67 (d, J =
10.0 Hz, 1H), 4.43 (q, J = 11.8 Hz, 2H), 3.90 (d, J = 7.4 Hz, 1H), 3.66 (dd, J = 12.4, 6.8 Hz,
1H), 3.56 (dd, J = 12.4, 8.9 Hz, 1H), 2.47 (ddd, J = 13.8, 7.1, 2.4 Hz, 1H), 2.38 (dd, J = 8.8,
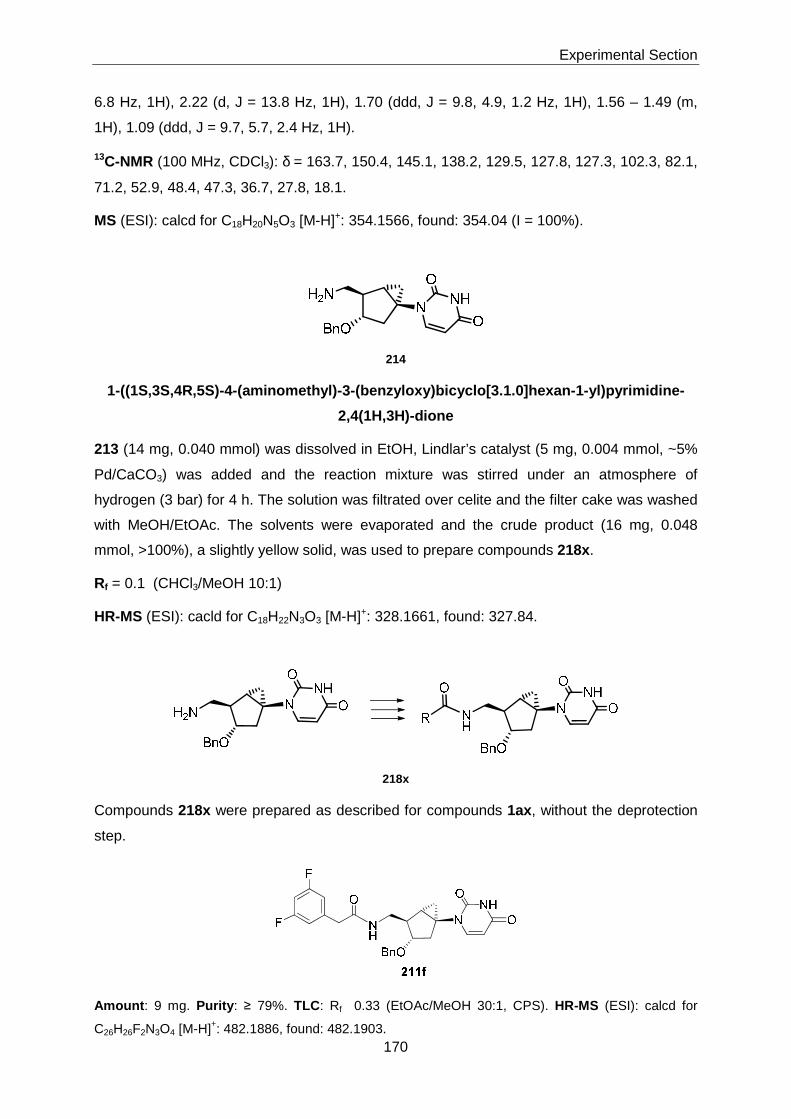
Experimental Section
170
6.8 Hz, 1H), 2.22 (d, J = 13.8 Hz, 1H), 1.70 (ddd, J = 9.8, 4.9, 1.2 Hz, 1H), 1.56 – 1.49 (m,
1H), 1.09 (ddd, J = 9.7, 5.7, 2.4 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 163.7, 150.4, 145.1, 138.2, 129.5, 127.8, 127.3, 102.3, 82.1,
71.2, 52.9, 48.4, 47.3, 36.7, 27.8, 18.1.
MS (ESI): calcd for C18H20N5O3 [M-H]+: 354.1566, found: 354.04 (I = 100%).
214
1-((1S,3S,4R,5S)-4-(aminomethyl)-3-(benzyloxy)bicyc lo[3.1.0]hexan-1-yl)pyrimidine-
2,4(1H,3H)-dione
213 (14 mg, 0.040 mmol) was dissolved in EtOH, Lindlar’s catalyst (5 mg, 0.004 mmol, ~5%
Pd/CaCO3) was added and the reaction mixture was stirred under an atmosphere of
hydrogen (3 bar) for 4 h. The solution was filtrated over celite and the filter cake was washed
with MeOH/EtOAc. The solvents were evaporated and the crude product (16 mg, 0.048
mmol, >100%), a slightly yellow solid, was used to prepare compounds 218x.
Rf = 0.1 (CHCl3/MeOH 10:1)
HR-MS (ESI): cacld for C18H22N3O3 [M-H]+: 328.1661, found: 327.84.
218x
Compounds 218x were prepared as described for compounds 1ax, without the deprotection
step.
Amount : 9 mg. Purity : ≥ 79%. TLC: Rf 0.33 (EtOAc/MeOH 30:1, CPS). HR-MS (ESI): calcd for
C26H26F2N3O4 [M-H]+: 482.1886, found: 482.1903.
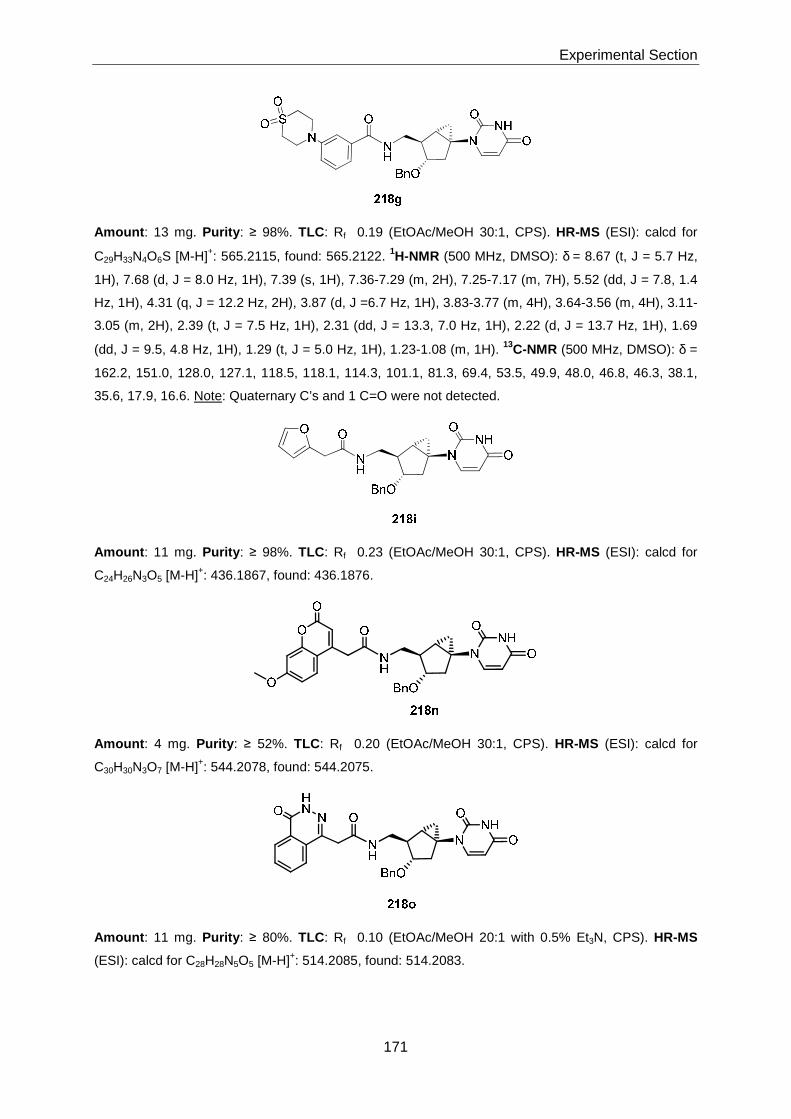
Experimental Section
171
Amount : 13 mg. Purity : ≥ 98%. TLC: Rf 0.19 (EtOAc/MeOH 30:1, CPS). HR-MS (ESI): calcd for
C29H33N4O6S [M-H]+: 565.2115, found: 565.2122. 1H-NMR (500 MHz, DMSO): δ = 8.67 (t, J = 5.7 Hz,
1H), 7.68 (d, J = 8.0 Hz, 1H), 7.39 (s, 1H), 7.36-7.29 (m, 2H), 7.25-7.17 (m, 7H), 5.52 (dd, J = 7.8, 1.4
Hz, 1H), 4.31 (q, J = 12.2 Hz, 2H), 3.87 (d, J =6.7 Hz, 1H), 3.83-3.77 (m, 4H), 3.64-3.56 (m, 4H), 3.11-
3.05 (m, 2H), 2.39 (t, J = 7.5 Hz, 1H), 2.31 (dd, J = 13.3, 7.0 Hz, 1H), 2.22 (d, J = 13.7 Hz, 1H), 1.69
(dd, J = 9.5, 4.8 Hz, 1H), 1.29 (t, J = 5.0 Hz, 1H), 1.23-1.08 (m, 1H). 13C-NMR (500 MHz, DMSO): δ =
162.2, 151.0, 128.0, 127.1, 118.5, 118.1, 114.3, 101.1, 81.3, 69.4, 53.5, 49.9, 48.0, 46.8, 46.3, 38.1,
35.6, 17.9, 16.6. Note: Quaternary C’s and 1 C=O were not detected.
Amount : 11 mg. Purity : ≥ 98%. TLC: Rf 0.23 (EtOAc/MeOH 30:1, CPS). HR-MS (ESI): calcd for
C24H26N3O5 [M-H]+: 436.1867, found: 436.1876.
Amount : 4 mg. Purity : ≥ 52%. TLC: Rf 0.20 (EtOAc/MeOH 30:1, CPS). HR-MS (ESI): calcd for
C30H30N3O7 [M-H]+: 544.2078, found: 544.2075.
Amount : 11 mg. Purity : ≥ 80%. TLC: Rf 0.10 (EtOAc/MeOH 20:1 with 0.5% Et3N, CPS). HR-MS
(ESI): calcd for C28H28N5O5 [M-H]+: 514.2085, found: 514.2083.
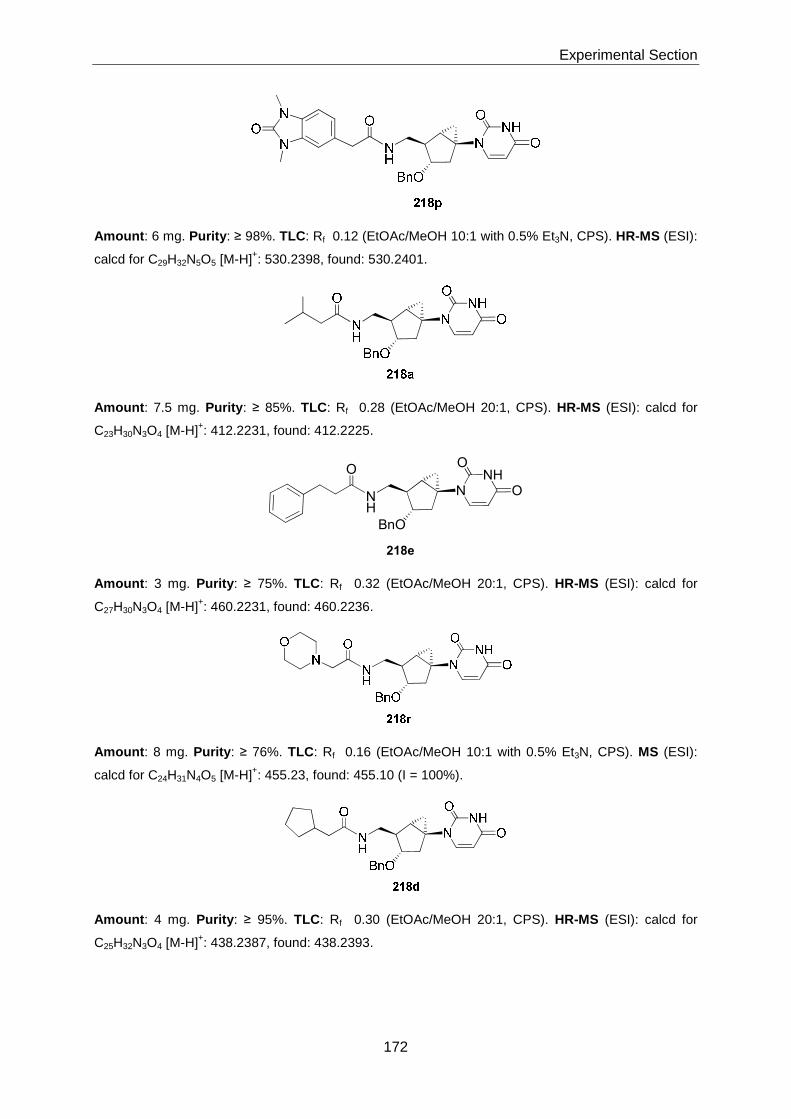
Experimental Section
172
Amount : 6 mg. Purity : ≥ 98%. TLC: Rf 0.12 (EtOAc/MeOH 10:1 with 0.5% Et3N, CPS). HR-MS (ESI):
calcd for C29H32N5O5 [M-H]+: 530.2398, found: 530.2401.
Amount : 7.5 mg. Purity : ≥ 85%. TLC: Rf 0.28 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C23H30N3O4 [M-H]+: 412.2231, found: 412.2225.
NH
O
218e
N
BnO
NHO
O
Amount : 3 mg. Purity : ≥ 75%. TLC: Rf 0.32 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C27H30N3O4 [M-H]+: 460.2231, found: 460.2236.
Amount : 8 mg. Purity : ≥ 76%. TLC: Rf 0.16 (EtOAc/MeOH 10:1 with 0.5% Et3N, CPS). MS (ESI):
calcd for C24H31N4O5 [M-H]+: 455.23, found: 455.10 (I = 100%).
Amount : 4 mg. Purity : ≥ 95%. TLC: Rf 0.30 (EtOAc/MeOH 20:1, CPS). HR-MS (ESI): calcd for
C25H32N3O4 [M-H]+: 438.2387, found: 438.2393.
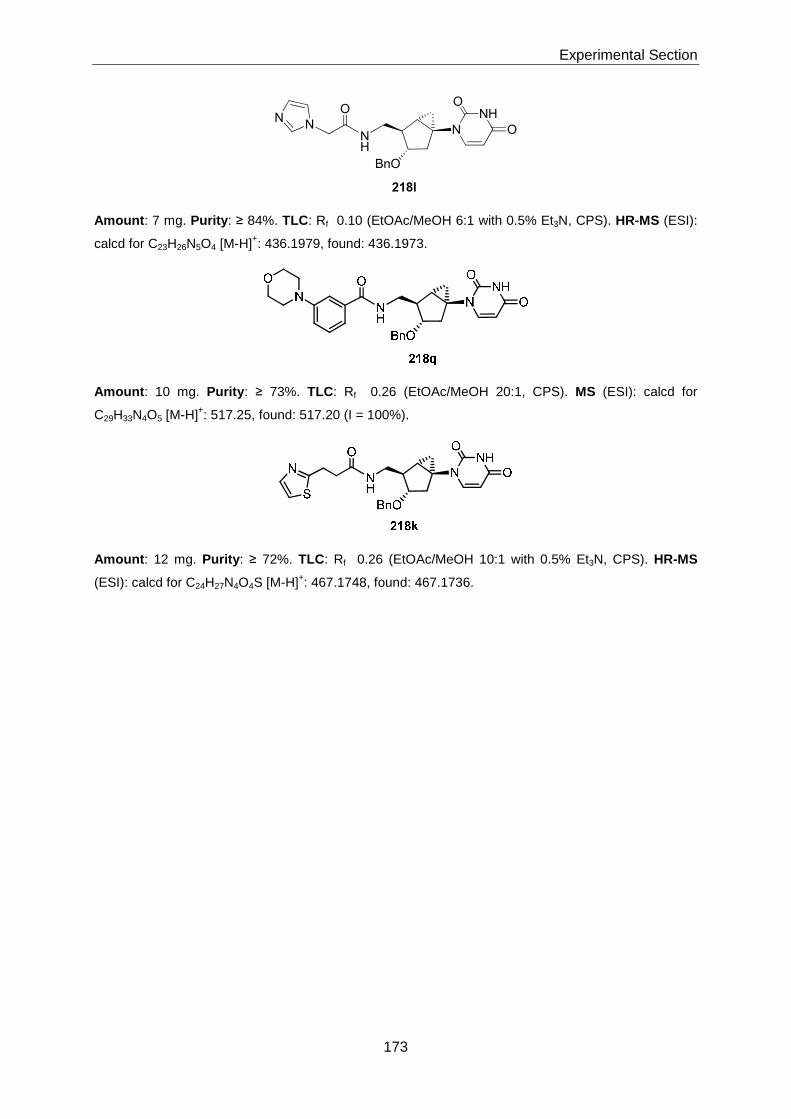
Experimental Section
173
NH
O
N
218l
NN
BnO
NHO
O
Amount : 7 mg. Purity : ≥ 84%. TLC: Rf 0.10 (EtOAc/MeOH 6:1 with 0.5% Et3N, CPS). HR-MS (ESI):
calcd for C23H26N5O4 [M-H]+: 436.1979, found: 436.1973.
Amount : 10 mg. Purity : ≥ 73%. TLC: Rf 0.26 (EtOAc/MeOH 20:1, CPS). MS (ESI): calcd for
C29H33N4O5 [M-H]+: 517.25, found: 517.20 (I = 100%).
Amount : 12 mg. Purity : ≥ 72%. TLC: Rf 0.26 (EtOAc/MeOH 10:1 with 0.5% Et3N, CPS). HR-MS
(ESI): calcd for C24H27N4O4S [M-H]+: 467.1748, found: 467.1736.
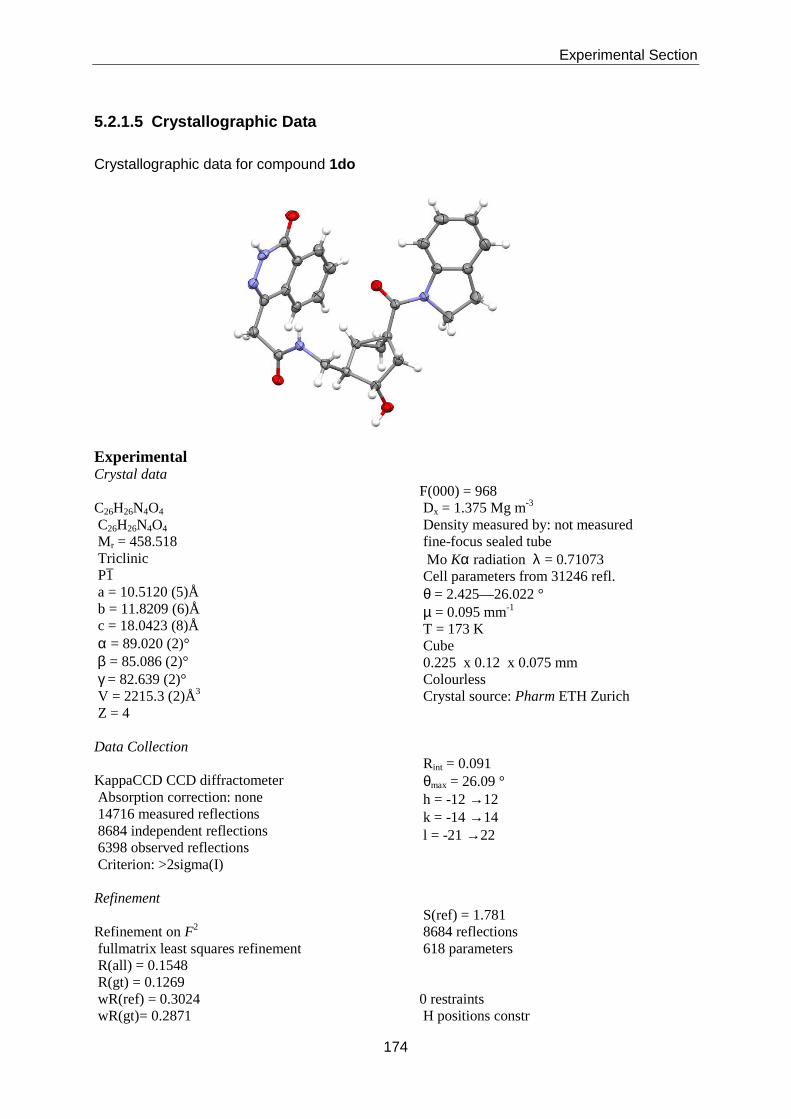
Experimental Section
174
5.2.1.5 Crystallographic Data
Crystallographic data for compound 1do
Experimental Crystal data C26H26N4O4 C26H26N4O4 Mr = 458.518 Triclinic P 1̄ a = 10.5120 (5)Å b = 11.8209 (6)Å c = 18.0423 (8)Å α = 89.020 (2)° β = 85.086 (2)° γ = 82.639 (2)° V = 2215.3 (2)Å3 Z = 4
F(000) = 968 Dx = 1.375 Mg m-3 Density measured by: not measured fine-focus sealed tube Mo Kα radiation λ = 0.71073 Cell parameters from 31246 refl. θ = 2.425—26.022 ° µ = 0.095 mm-1 T = 173 K Cube 0.225 x 0.12 x 0.075 mm Colourless Crystal source: Pharm ETH Zurich
Data Collection KappaCCD CCD diffractometer Absorption correction: none 14716 measured reflections 8684 independent reflections 6398 observed reflections Criterion: >2sigma(I)
Rint = 0.091 θmax = 26.09 ° h = -12 →12 k = -14 →14 l = -21 →22
Refinement Refinement on F2 fullmatrix least squares refinement R(all) = 0.1548 R(gt) = 0.1269 wR(ref) = 0.3024 wR(gt)= 0.2871
S(ref) = 1.781 8684 reflections 618 parameters 0 restraints H positions constr

Experimental Section
175
Calculated weights 1/[σ2(Io)+(Io+Ic)2/900]
∆/σmax = 0.001 ∆ρmax = 1.067eÅ3 ∆ρmin = -0.705eÅ3 Extinction correction: none Atomic scattering factors from International
Tables Vol C Tables 4.2.6.8 and 6.1.1.4
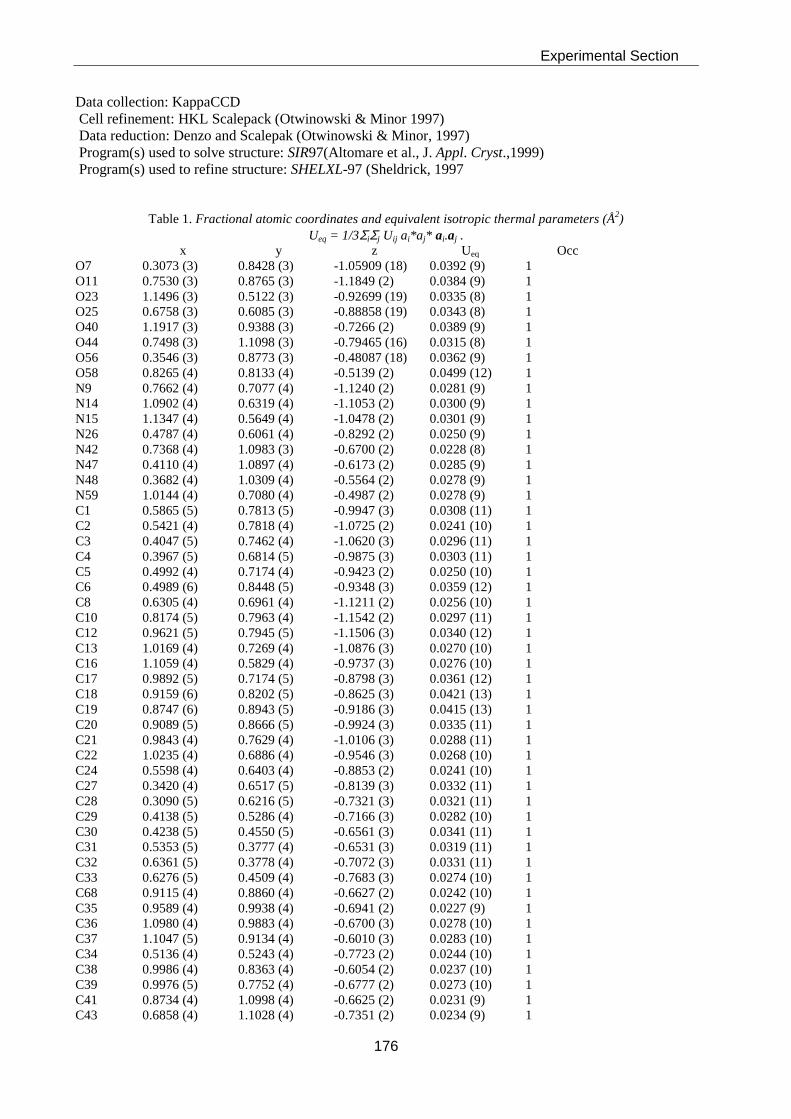
Experimental Section
176
Data collection: KappaCCD Cell refinement: HKL Scalepack (Otwinowski & Minor 1997) Data reduction: Denzo and Scalepak (Otwinowski & Minor, 1997) Program(s) used to solve structure: SIR97(Altomare et al., J. Appl. Cryst.,1999) Program(s) used to refine structure: SHELXL-97 (Sheldrick, 1997
Table 1. Fractional atomic coordinates and equivalent isotropic thermal parameters (Å2) Ueq = 1/3ΣiΣj Uij ai*a j* ai.aj .
x y z Ueq Occ O7 0.3073 (3) 0.8428 (3) -1.05909 (18) 0.0392 (9) 1 O11 0.7530 (3) 0.8765 (3) -1.1849 (2) 0.0384 (9) 1 O23 1.1496 (3) 0.5122 (3) -0.92699 (19) 0.0335 (8) 1 O25 0.6758 (3) 0.6085 (3) -0.88858 (19) 0.0343 (8) 1 O40 1.1917 (3) 0.9388 (3) -0.7266 (2) 0.0389 (9) 1 O44 0.7498 (3) 1.1098 (3) -0.79465 (16) 0.0315 (8) 1 O56 0.3546 (3) 0.8773 (3) -0.48087 (18) 0.0362 (9) 1 O58 0.8265 (4) 0.8133 (4) -0.5139 (2) 0.0499 (12) 1 N9 0.7662 (4) 0.7077 (4) -1.1240 (2) 0.0281 (9) 1 N14 1.0902 (4) 0.6319 (4) -1.1053 (2) 0.0300 (9) 1 N15 1.1347 (4) 0.5649 (4) -1.0478 (2) 0.0301 (9) 1 N26 0.4787 (4) 0.6061 (4) -0.8292 (2) 0.0250 (9) 1 N42 0.7368 (4) 1.0983 (3) -0.6700 (2) 0.0228 (8) 1 N47 0.4110 (4) 1.0897 (4) -0.6173 (2) 0.0285 (9) 1 N48 0.3682 (4) 1.0309 (4) -0.5564 (2) 0.0278 (9) 1 N59 1.0144 (4) 0.7080 (4) -0.4987 (2) 0.0278 (9) 1 C1 0.5865 (5) 0.7813 (5) -0.9947 (3) 0.0308 (11) 1 C2 0.5421 (4) 0.7818 (4) -1.0725 (2) 0.0241 (10) 1 C3 0.4047 (5) 0.7462 (4) -1.0620 (3) 0.0296 (11) 1 C4 0.3967 (5) 0.6814 (5) -0.9875 (3) 0.0303 (11) 1 C5 0.4992 (4) 0.7174 (4) -0.9423 (2) 0.0250 (10) 1 C6 0.4989 (6) 0.8448 (5) -0.9348 (3) 0.0359 (12) 1 C8 0.6305 (4) 0.6961 (4) -1.1211 (2) 0.0256 (10) 1 C10 0.8174 (5) 0.7963 (4) -1.1542 (2) 0.0297 (11) 1 C12 0.9621 (5) 0.7945 (5) -1.1506 (3) 0.0340 (12) 1 C13 1.0169 (4) 0.7269 (4) -1.0876 (3) 0.0270 (10) 1 C16 1.1059 (4) 0.5829 (4) -0.9737 (3) 0.0276 (10) 1 C17 0.9892 (5) 0.7174 (5) -0.8798 (3) 0.0361 (12) 1 C18 0.9159 (6) 0.8202 (5) -0.8625 (3) 0.0421 (13) 1 C19 0.8747 (6) 0.8943 (5) -0.9186 (3) 0.0415 (13) 1 C20 0.9089 (5) 0.8666 (5) -0.9924 (3) 0.0335 (11) 1 C21 0.9843 (4) 0.7629 (4) -1.0106 (3) 0.0288 (11) 1 C22 1.0235 (4) 0.6886 (4) -0.9546 (3) 0.0268 (10) 1 C24 0.5598 (4) 0.6403 (4) -0.8853 (2) 0.0241 (10) 1 C27 0.3420 (4) 0.6517 (5) -0.8139 (3) 0.0332 (11) 1 C28 0.3090 (5) 0.6216 (5) -0.7321 (3) 0.0321 (11) 1 C29 0.4138 (5) 0.5286 (4) -0.7166 (3) 0.0282 (10) 1 C30 0.4238 (5) 0.4550 (5) -0.6561 (3) 0.0341 (11) 1 C31 0.5353 (5) 0.3777 (4) -0.6531 (3) 0.0319 (11) 1 C32 0.6361 (5) 0.3778 (4) -0.7072 (3) 0.0331 (11) 1 C33 0.6276 (5) 0.4509 (4) -0.7683 (3) 0.0274 (10) 1 C68 0.9115 (4) 0.8860 (4) -0.6627 (2) 0.0242 (10) 1 C35 0.9589 (4) 0.9938 (4) -0.6941 (2) 0.0227 (9) 1 C36 1.0980 (4) 0.9883 (4) -0.6700 (3) 0.0278 (10) 1 C37 1.1047 (5) 0.9134 (4) -0.6010 (3) 0.0283 (10) 1 C34 0.5136 (4) 0.5243 (4) -0.7723 (2) 0.0244 (10) 1 C38 0.9986 (4) 0.8363 (4) -0.6054 (2) 0.0237 (10) 1 C39 0.9976 (5) 0.7752 (4) -0.6777 (2) 0.0273 (10) 1 C41 0.8734 (4) 1.0998 (4) -0.6625 (2) 0.0231 (9) 1 C43 0.6858 (4) 1.1028 (4) -0.7351 (2) 0.0234 (9) 1

Experimental Section
177
C45 0.5400 (5) 1.0987 (4) -0.7316 (3) 0.0293 (11) 1 C46 0.4876 (4) 1.0317 (4) -0.6670 (2) 0.0254 (10) 1 C49 0.3993 (4) 0.9187 (4) -0.5397 (2) 0.0264 (10) 1 C50 0.5254 (5) 0.7401 (5) -0.5854 (3) 0.0327 (11) 1 C51 0.6038 (5) 0.6779 (5) -0.6403 (3) 0.0335 (11) 1 C52 0.6428 (5) 0.7319 (5) -0.7063 (3) 0.0332 (11) 1 C53 0.6037 (4) 0.8471 (4) -0.7162 (3) 0.0258 (10) 1 C54 0.5258 (4) 0.9102 (4) -0.6604 (2) 0.0242 (10) 1 C55 0.4865 (4) 0.8534 (4) -0.5957 (2) 0.0262 (10) 1 C57 0.9391 (4) 0.7862 (4) -0.5361 (2) 0.0244 (10) 1 C60 1.1476 (6) 0.6592 (6) -0.5249 (3) 0.0552 (19) 1 C61 1.1865 (5) 0.5675 (5) -0.4691 (3) 0.0401 (13) 1 C62 1.0840 (5) 0.5812 (5) -0.4069 (3) 0.0308 (11) 1 C63 1.0766 (5) 0.5320 (5) -0.3370 (3) 0.0377 (12) 1 C64 0.9682 (6) 0.5610 (5) -0.2877 (3) 0.0400 (13) 1 C65 0.8667 (5) 0.6363 (5) -0.3095 (3) 0.0366 (12) 1 C66 0.8711 (5) 0.6888 (4) -0.3793 (3) 0.0313 (11) 1 C67 0.9818 (5) 0.6626 (4) -0.4271 (3) 0.0270 (10) 1 H7 0.2911 0.8633 -1.1031 0.06 (2) 1 H40 1.1933 0.9830 -0.7642 0.09 (3) 1 H9 0.8178 0.6527 -1.1045 0.034 1 H15 1.1879 0.5033 -1.0604 0.036 1 H42 0.6852 1.0943 -0.6292 0.027 1 H48 0.3138 1.0705 -0.5239 0.033 1 H1 0.6740 0.7471 -0.9926 0.037 1 H2 0.5391 0.8559 -1.0954 0.029 1 H3 0.3929 0.6975 -1.1022 0.036 1 H4A 0.3127 0.7008 -0.9623 0.036 1 H4B 0.4113 0.6009 -0.9968 0.036 1 H6A 0.5498 0.8802 -0.9024 0.043 1 H6B 0.4416 0.9024 -0.9575 0.043 1 H8A 0.6022 0.7058 -1.1703 0.031 1 H8B 0.6162 0.6219 -1.1024 0.031 1 H12A 1.0066 0.7621 -1.1955 0.041 1 H12B 0.9813 0.8711 -1.1454 0.041 1 H17 1.0151 0.6655 -0.8408 0.043 1 H18 0.8932 0.8409 -0.8114 0.051 1 H19 0.8230 0.9654 -0.9062 0.050 1 H20 0.8796 0.9178 -1.0310 0.040 1 H27A 0.3309 0.7325 -0.8227 0.040 1 H27B 0.2918 0.6156 -0.8463 0.040 1 H28A 0.3095 0.6879 -0.7022 0.039 1 H28B 0.2255 0.5964 -0.7260 0.039 1 H30 0.3548 0.4575 -0.6174 0.041 1 H31 0.5434 0.3222 -0.6137 0.038 1 H32 0.7140 0.3271 -0.7025 0.040 1 H33 0.6982 0.4495 -0.8059 0.033 1 H68 0.8244 0.8979 -0.6407 0.029 1 H35 0.9610 0.9956 -0.7474 0.027 1 H36 1.1146 1.0641 -0.6595 0.033 1 H37A 1.1884 0.8697 -0.6024 0.034 1 H37B 1.0909 0.9598 -0.5572 0.034 1 H39A 0.9462 0.7159 -0.6862 0.033 1 H39B 1.0550 0.7844 -0.7211 0.033 1 H41A 0.9019 1.1646 -0.6888 0.028 1 H41B 0.8882 1.1044 -0.6109 0.028 1 H45A 0.4957 1.1748 -0.7281 0.035 1 H45B 0.5199 1.0636 -0.7760 0.035 1 H50 0.4981 0.7045 -0.5395 0.039 1
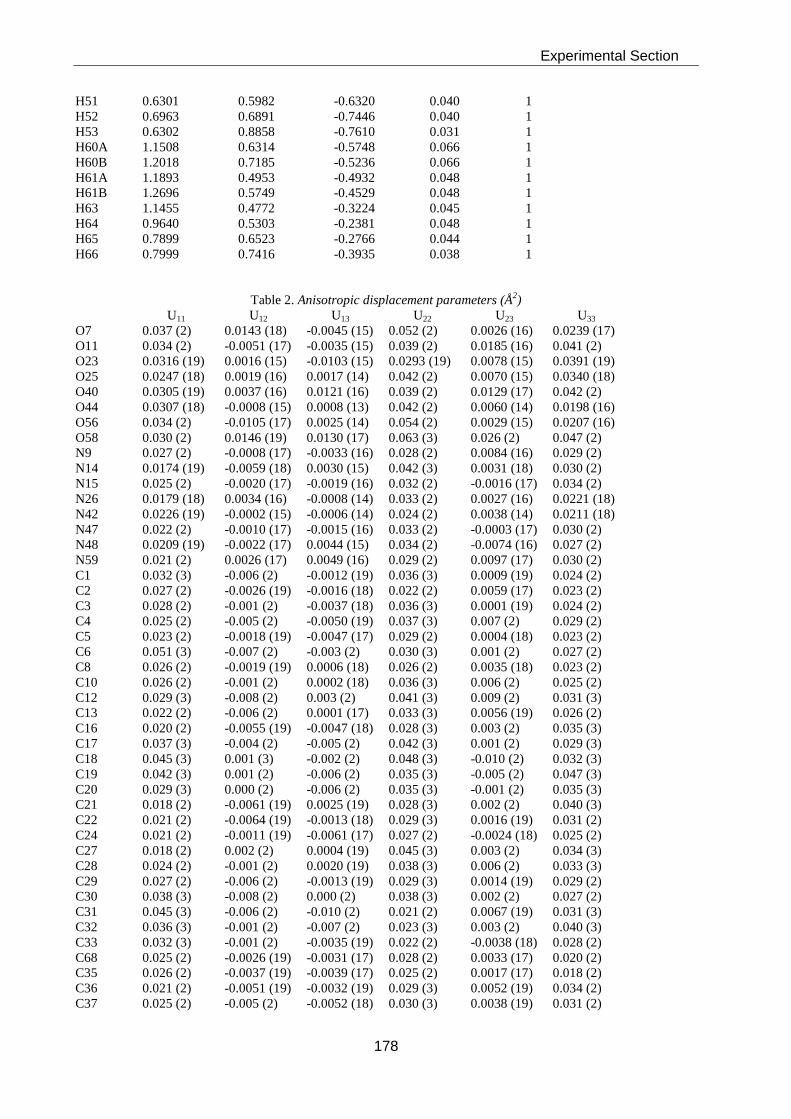
Experimental Section
178
H51 0.6301 0.5982 -0.6320 0.040 1 H52 0.6963 0.6891 -0.7446 0.040 1 H53 0.6302 0.8858 -0.7610 0.031 1 H60A 1.1508 0.6314 -0.5748 0.066 1 H60B 1.2018 0.7185 -0.5236 0.066 1 H61A 1.1893 0.4953 -0.4932 0.048 1 H61B 1.2696 0.5749 -0.4529 0.048 1 H63 1.1455 0.4772 -0.3224 0.045 1 H64 0.9640 0.5303 -0.2381 0.048 1 H65 0.7899 0.6523 -0.2766 0.044 1 H66 0.7999 0.7416 -0.3935 0.038 1
Table 2. Anisotropic displacement parameters (Å2) U11 U12 U13 U22 U23 U33
O7 0.037 (2) 0.0143 (18) -0.0045 (15) 0.052 (2) 0.0026 (16) 0.0239 (17) O11 0.034 (2) -0.0051 (17) -0.0035 (15) 0.039 (2) 0.0185 (16) 0.041 (2) O23 0.0316 (19) 0.0016 (15) -0.0103 (15) 0.0293 (19) 0.0078 (15) 0.0391 (19) O25 0.0247 (18) 0.0019 (16) 0.0017 (14) 0.042 (2) 0.0070 (15) 0.0340 (18) O40 0.0305 (19) 0.0037 (16) 0.0121 (16) 0.039 (2) 0.0129 (17) 0.042 (2) O44 0.0307 (18) -0.0008 (15) 0.0008 (13) 0.042 (2) 0.0060 (14) 0.0198 (16) O56 0.034 (2) -0.0105 (17) 0.0025 (14) 0.054 (2) 0.0029 (15) 0.0207 (16) O58 0.030 (2) 0.0146 (19) 0.0130 (17) 0.063 (3) 0.026 (2) 0.047 (2) N9 0.027 (2) -0.0008 (17) -0.0033 (16) 0.028 (2) 0.0084 (16) 0.029 (2) N14 0.0174 (19) -0.0059 (18) 0.0030 (15) 0.042 (3) 0.0031 (18) 0.030 (2) N15 0.025 (2) -0.0020 (17) -0.0019 (16) 0.032 (2) -0.0016 (17) 0.034 (2) N26 0.0179 (18) 0.0034 (16) -0.0008 (14) 0.033 (2) 0.0027 (16) 0.0221 (18) N42 0.0226 (19) -0.0002 (15) -0.0006 (14) 0.024 (2) 0.0038 (14) 0.0211 (18) N47 0.022 (2) -0.0010 (17) -0.0015 (16) 0.033 (2) -0.0003 (17) 0.030 (2) N48 0.0209 (19) -0.0022 (17) 0.0044 (15) 0.034 (2) -0.0074 (16) 0.027 (2) N59 0.021 (2) 0.0026 (17) 0.0049 (16) 0.029 (2) 0.0097 (17) 0.030 (2) C1 0.032 (3) -0.006 (2) -0.0012 (19) 0.036 (3) 0.0009 (19) 0.024 (2) C2 0.027 (2) -0.0026 (19) -0.0016 (18) 0.022 (2) 0.0059 (17) 0.023 (2) C3 0.028 (2) -0.001 (2) -0.0037 (18) 0.036 (3) 0.0001 (19) 0.024 (2) C4 0.025 (2) -0.005 (2) -0.0050 (19) 0.037 (3) 0.007 (2) 0.029 (2) C5 0.023 (2) -0.0018 (19) -0.0047 (17) 0.029 (2) 0.0004 (18) 0.023 (2) C6 0.051 (3) -0.007 (2) -0.003 (2) 0.030 (3) 0.001 (2) 0.027 (2) C8 0.026 (2) -0.0019 (19) 0.0006 (18) 0.026 (2) 0.0035 (18) 0.023 (2) C10 0.026 (2) -0.001 (2) 0.0002 (18) 0.036 (3) 0.006 (2) 0.025 (2) C12 0.029 (3) -0.008 (2) 0.003 (2) 0.041 (3) 0.009 (2) 0.031 (3) C13 0.022 (2) -0.006 (2) 0.0001 (17) 0.033 (3) 0.0056 (19) 0.026 (2) C16 0.020 (2) -0.0055 (19) -0.0047 (18) 0.028 (3) 0.003 (2) 0.035 (3) C17 0.037 (3) -0.004 (2) -0.005 (2) 0.042 (3) 0.001 (2) 0.029 (3) C18 0.045 (3) 0.001 (3) -0.002 (2) 0.048 (3) -0.010 (2) 0.032 (3) C19 0.042 (3) 0.001 (2) -0.006 (2) 0.035 (3) -0.005 (2) 0.047 (3) C20 0.029 (3) 0.000 (2) -0.006 (2) 0.035 (3) -0.001 (2) 0.035 (3) C21 0.018 (2) -0.0061 (19) 0.0025 (19) 0.028 (3) 0.002 (2) 0.040 (3) C22 0.021 (2) -0.0064 (19) -0.0013 (18) 0.029 (3) 0.0016 (19) 0.031 (2) C24 0.021 (2) -0.0011 (19) -0.0061 (17) 0.027 (2) -0.0024 (18) 0.025 (2) C27 0.018 (2) 0.002 (2) 0.0004 (19) 0.045 (3) 0.003 (2) 0.034 (3) C28 0.024 (2) -0.001 (2) 0.0020 (19) 0.038 (3) 0.006 (2) 0.033 (3) C29 0.027 (2) -0.006 (2) -0.0013 (19) 0.029 (3) 0.0014 (19) 0.029 (2) C30 0.038 (3) -0.008 (2) 0.000 (2) 0.038 (3) 0.002 (2) 0.027 (2) C31 0.045 (3) -0.006 (2) -0.010 (2) 0.021 (2) 0.0067 (19) 0.031 (3) C32 0.036 (3) -0.001 (2) -0.007 (2) 0.023 (3) 0.003 (2) 0.040 (3) C33 0.032 (3) -0.001 (2) -0.0035 (19) 0.022 (2) -0.0038 (18) 0.028 (2) C68 0.025 (2) -0.0026 (19) -0.0031 (17) 0.028 (2) 0.0033 (17) 0.020 (2) C35 0.026 (2) -0.0037 (19) -0.0039 (17) 0.025 (2) 0.0017 (17) 0.018 (2) C36 0.021 (2) -0.0051 (19) -0.0032 (19) 0.029 (3) 0.0052 (19) 0.034 (2) C37 0.025 (2) -0.005 (2) -0.0052 (18) 0.030 (3) 0.0038 (19) 0.031 (2)
Experimental Section
179
C34 0.025 (2) -0.0039 (19) -0.0053 (18) 0.024 (2) 0.0000 (18) 0.025 (2) C38 0.021 (2) -0.0017 (19) -0.0016 (17) 0.026 (2) 0.0028 (18) 0.023 (2) C39 0.031 (2) -0.002 (2) -0.0042 (19) 0.023 (2) -0.0003 (18) 0.028 (2) C41 0.024 (2) 0.0015 (19) -0.0032 (16) 0.027 (2) -0.0005 (17) 0.018 (2) C43 0.025 (2) 0.0011 (18) -0.0016 (17) 0.021 (2) -0.0004 (17) 0.023 (2) C45 0.027 (2) 0.002 (2) -0.0045 (19) 0.033 (3) 0.0048 (19) 0.027 (2) C46 0.021 (2) -0.0009 (19) -0.0060 (17) 0.030 (3) -0.0033 (18) 0.025 (2) C49 0.022 (2) -0.009 (2) -0.0041 (17) 0.036 (3) -0.0031 (19) 0.023 (2) C50 0.031 (3) -0.008 (2) -0.008 (2) 0.040 (3) 0.002 (2) 0.028 (2) C51 0.037 (3) -0.006 (2) -0.004 (2) 0.028 (3) -0.001 (2) 0.036 (3) C52 0.031 (3) -0.002 (2) 0.000 (2) 0.035 (3) -0.008 (2) 0.033 (3) C53 0.020 (2) -0.0064 (19) -0.0050 (18) 0.028 (3) -0.0018 (19) 0.031 (2) C54 0.021 (2) 0.0002 (19) -0.0047 (17) 0.030 (3) -0.0080 (18) 0.021 (2) C55 0.022 (2) -0.005 (2) -0.0028 (18) 0.033 (3) 0.0002 (19) 0.023 (2) C57 0.024 (2) 0.0013 (19) -0.0004 (17) 0.025 (2) 0.0028 (17) 0.022 (2) C60 0.032 (3) 0.021 (3) 0.014 (3) 0.070 (4) 0.030 (3) 0.052 (3) C61 0.035 (3) 0.010 (3) 0.001 (2) 0.043 (3) 0.007 (2) 0.037 (3) C62 0.027 (2) -0.008 (2) -0.0053 (19) 0.043 (3) 0.004 (2) 0.024 (2) C63 0.039 (3) -0.005 (2) -0.010 (2) 0.034 (3) 0.010 (2) 0.042 (3) C64 0.047 (3) -0.016 (3) -0.007 (2) 0.052 (4) 0.009 (2) 0.023 (2) C65 0.042 (3) -0.015 (3) 0.002 (2) 0.045 (3) -0.001 (2) 0.024 (2) C66 0.035 (3) -0.006 (2) 0.000 (2) 0.029 (3) 0.0013 (19) 0.031 (2) C67 0.029 (2) -0.003 (2) 0.0007 (19) 0.026 (2) 0.0017 (18) 0.025 (2)
Table 3 . Geometric parameters (Å, °) O7—C3 1.432 (6) O11—C10 1.244 (6) O23—C16 1.251 (6) O25—C24 1.226 (5) O40—C36 1.433 (6) O44—C43 1.226 (5) O56—C49 1.242 (6) O58—C57 1.221 (6) N9—C10 1.326 (6) N9—C8 1.447 (6) N14—C13 1.305 (6) N14—N15 1.371 (6) N15—C16 1.359 (6) N26—C24 1.358 (6) N26—C34 1.436 (6) N26—C27 1.474 (6) N42—C43 1.329 (6) N42—C41 1.456 (6) N47—C46 1.295 (6) N47—N48 1.365 (6) N48—C49 1.359 (7) N59—C57 1.349 (6) N59—C67 1.423 (6) N59—C60 1.484 (6) C1—C6 1.504 (7) C1—C2 1.515 (6) C1—C5 1.521 (7) C2—C8 1.519 (6) C2—C3 1.551 (7) C3—C4 1.536 (6) C4—C5 1.513 (7) C5—C24 1.491 (6) C5—C6 1.514 (7) C10—C12 1.526 (7)
C12—C13 1.491 (6) C13—C21 1.459 (7) C16—C22 1.455 (7) C17—C18 1.378 (8) C17—C22 1.403 (7) C18—C19 1.388 (8) C19—C20 1.383 (7) C20—C21 1.400 (7) C21—C22 1.382 (7) C27—C28 1.533 (7) C28—C29 1.494 (7) C29—C34 1.385 (6) C29—C30 1.388 (7) C30—C31 1.395 (7) C31—C32 1.378 (7) C32—C33 1.391 (7) C33—C34 1.393 (7) C68—C38 1.501 (6) C68—C39 1.508 (6) C68—C35 1.511 (6) C35—C41 1.533 (6) C35—C36 1.554 (6) C36—C37 1.517 (6) C37—C38 1.536 (7) C38—C57 1.497 (6) C38—C39 1.504 (7) C43—C45 1.536 (6) C45—C46 1.502 (7) C46—C54 1.447 (7) C49—C55 1.464 (6) C50—C55 1.364 (7) C50—C51 1.389 (7) C51—C52 1.397 (7) C52—C53 1.385 (7)
Experimental Section
180
C53—C54 1.403 (6) C54—C55 1.394 (7) C60—C61 1.507 (7) C61—C62 1.484 (7) C62—C63 1.380 (7) C62—C67 1.415 (7) C63—C64 1.394 (8) C64—C65 1.379 (8) C65—C66 1.394 (7) C66—C67 1.392 (7) O7—H7 0.8500 O40—H40 0.8500 N9—H9 0.8800 N15—H15 0.8800 N42—H42 0.8800 N48—H48 0.8800 C1—H1 0.9600 C2—H2 0.9601 C3—H3 0.9600 C4—H4A 0.9599 C4—H4B 0.9600 C6—H6A 0.9600 C6—H6B 0.9601 C8—H8A 0.9600 C8—H8B 0.9600 C12—H12A 0.9600 C12—H12B 0.9601 C17—H17 0.9601 C18—H18 0.9599 C19—H19 0.9599 C20—H20 0.9600
C27—H27A 0.9601 C27—H27B 0.9599 C28—H28A 0.9600 C28—H28B 0.9600 C30—H30 0.9599 C31—H31 0.9601 C32—H32 0.9600 C33—H33 0.9599 C68—H68 0.9599 C35—H35 0.9599 C36—H36 0.9599 C37—H37A 0.9600 C37—H37B 0.9599 C39—H39A 0.9600 C39—H39B 0.9600 C41—H41A 0.9601 C41—H41B 0.9599 C45—H45A 0.9600 C45—H45B 0.9601 C50—H50 0.9599 C51—H51 0.9600 C52—H52 0.9600 C53—H53 0.9600 C60—H60A 0.9600 C60—H60B 0.9600 C61—H61A 0.9600 C61—H61B 0.9600 C63—H63 0.9600 C64—H64 0.9600 C65—H65 0.9600 C66—H66 0.9600
C10—N9—C8 123.9 (4) C13—N14—N15 117.1 (4) C16—N15—N14 127.4 (4) C24—N26—C34 125.7 (4) C24—N26—C27 125.6 (4) C34—N26—C27 108.6 (4) C43—N42—C41 123.5 (4) C46—N47—N48 116.7 (4) C49—N48—N47 128.1 (4) C57—N59—C67 125.8 (4) C57—N59—C60 125.0 (4) C67—N59—C60 109.1 (4) C6—C1—C2 118.3 (4) C6—C1—C5 60.1 (3) C2—C1—C5 109.5 (4) C1—C2—C8 110.4 (4) C1—C2—C3 104.9 (4) C8—C2—C3 110.3 (4) O7—C3—C4 109.2 (4) O7—C3—C2 112.0 (4) C4—C3—C2 106.2 (4) C5—C4—C3 107.2 (4) C24—C5—C4 121.5 (4) C24—C5—C6 118.7 (4) C4—C5—C6 115.6 (4) C24—C5—C1 118.1 (4) C4—C5—C1 107.0 (4) C6—C5—C1 59.4 (3)
C1—C6—C5 60.5 (3) N9—C8—C2 115.6 (4) O11—C10—N9 122.8 (5) O11—C10—C12 120.9 (5) N9—C10—C12 116.3 (4) C13—C12—C10 115.0 (4) N14—C13—C21 122.4 (4) N14—C13—C12 116.2 (4) C21—C13—C12 121.4 (4) O23—C16—N15 120.8 (4) O23—C16—C22 124.1 (4) N15—C16—C22 115.1 (4) C18—C17—C22 119.6 (5) C17—C18—C19 120.4 (5) C20—C19—C18 120.2 (5) C19—C20—C21 119.9 (5) C22—C21—C20 119.7 (5) C22—C21—C13 118.3 (4) C20—C21—C13 121.9 (5) C21—C22—C17 120.2 (5) C21—C22—C16 119.4 (4) C17—C22—C16 120.2 (4) O25—C24—N26 121.3 (4) O25—C24—C5 122.6 (4) N26—C24—C5 116.1 (4) N26—C27—C28 104.8 (4) C29—C28—C27 103.2 (4) C34—C29—C30 119.9 (5)
Experimental Section
181
C34—C29—C28 110.5 (4) C30—C29—C28 129.6 (5) C29—C30—C31 118.6 (5) C32—C31—C30 120.9 (5) C31—C32—C33 121.2 (5) C32—C33—C34 117.3 (5) C38—C68—C39 60.0 (3) C38—C68—C35 109.0 (4) C39—C68—C35 117.4 (4) C68—C35—C41 111.0 (4) C68—C35—C36 104.6 (4) C41—C35—C36 111.1 (4) O40—C36—C37 108.7 (4) O40—C36—C35 111.7 (4) C37—C36—C35 106.9 (4) C36—C37—C38 105.2 (4) C29—C34—C33 122.0 (4) C29—C34—N26 109.2 (4) C33—C34—N26 128.8 (4) C57—C38—C68 118.3 (4) C57—C38—C39 118.4 (4) C68—C38—C39 60.2 (3) C57—C38—C37 120.3 (4) C68—C38—C37 108.0 (4) C39—C38—C37 116.3 (4) C38—C39—C68 59.8 (3) N42—C41—C35 113.8 (4) O44—C43—N42 122.9 (4) O44—C43—C45 121.3 (4) N42—C43—C45 115.8 (4) C46—C45—C43 114.5 (4) N47—C46—C54 122.8 (4) N47—C46—C45 116.1 (4) C54—C46—C45 121.0 (4) O56—C49—N48 120.8 (4) O56—C49—C55 124.0 (5) N48—C49—C55 115.1 (4) C55—C50—C51 120.3 (5) C50—C51—C52 119.7 (5) C53—C52—C51 119.6 (4) C52—C53—C54 120.6 (5) C55—C54—C53 118.4 (4) C55—C54—C46 119.3 (4) C53—C54—C46 122.4 (4) C50—C55—C54 121.3 (4) C50—C55—C49 120.8 (4) C54—C55—C49 117.9 (4) O58—C57—N59 120.5 (4) O58—C57—C38 122.0 (4) N59—C57—C38 117.5 (4) N59—C60—C61 105.6 (4) C62—C61—C60 105.6 (4) C63—C62—C67 119.2 (5) C63—C62—C61 131.4 (5) C67—C62—C61 109.4 (4) C62—C63—C64 120.0 (5) C65—C64—C63 120.0 (5) C64—C65—C66 121.7 (5) C67—C66—C65 117.8 (5) C66—C67—C62 121.1 (4)
C66—C67—N59 129.5 (4) C62—C67—N59 109.5 (4) C3—O7—H7 109.5 C36—O40—H40 109.5 C10—N9—H9 118.1 C8—N9—H9 118.1 C16—N15—H15 116.3 N14—N15—H15 116.3 C43—N42—H42 118.2 C41—N42—H42 118.2 C49—N48—H48 116.0 N47—N48—H48 116.0 C6—C1—H1 128.4 C2—C1—H1 112.8 C5—C1—H1 109.1 C1—C2—H2 112.0 C8—C2—H2 109.3 C3—C2—H2 109.8 O7—C3—H3 109.6 C4—C3—H3 110.6 C2—C3—H3 109.1 C5—C4—H4A 110.7 C3—C4—H4A 109.3 C5—C4—H4B 110.9 C3—C4—H4B 109.1 H4A—C4—H4B 109.5 C1—C6—H6A 109.2 C5—C6—H6A 124.9 C1—C6—H6B 108.9 C5—C6—H6B 125.3 H6A—C6—H6B 109.5 N9—C8—H8A 109.2 C2—C8—H8A 106.1 N9—C8—H8B 109.8 C2—C8—H8B 106.5 H8A—C8—H8B 109.5 C13—C12—H12A 106.7 C10—C12—H12A 109.4 C13—C12—H12B 106.8 C10—C12—H12B 109.3 H12A—C12—H12B 109.5 C18—C17—H17 119.9 C22—C17—H17 120.4 C17—C18—H18 120.0 C19—C18—H18 119.6 C20—C19—H19 119.8 C18—C19—H19 119.9 C19—C20—H20 120.0 C21—C20—H20 120.1 N26—C27—H27A 109.6 C28—C27—H27A 112.2 N26—C27—H27B 109.1 C28—C27—H27B 111.5 H27A—C27—H27B 109.5 C29—C28—H28A 112.6 C27—C28—H28A 109.2 C29—C28—H28B 112.1 C27—C28—H28B 110.1 H28A—C28—H28B 109.5 C29—C30—H30 120.2
Experimental Section
182
C31—C30—H30 121.2 C32—C31—H31 118.2 C30—C31—H31 120.9 C31—C32—H32 119.6 C33—C32—H32 119.3 C32—C33—H33 120.3 C34—C33—H33 122.3 C38—C68—H68 109.4 C39—C68—H68 128.9 C35—C68—H68 113.2 C68—C35—H35 111.3 C41—C35—H35 109.2 C36—C35—H35 109.6 O40—C36—H36 109.7 C37—C36—H36 111.0 C35—C36—H36 108.9 C36—C37—H37A 108.7 C38—C37—H37A 111.5 C36—C37—H37B 110.0 C38—C37—H37B 111.8 H37A—C37—H37B 109.5 C38—C39—H39A 125.6 C68—C39—H39A 109.7 C38—C39—H39B 124.6 C68—C39—H39B 109.0 H39A—C39—H39B 109.5 N42—C41—H41A 109.5 C35—C41—H41A 106.9 N42—C41—H41B 109.9 C35—C41—H41B 107.2 H41A—C41—H41B 109.5
C46—C45—H45A 107.4 C43—C45—H45A 109.5 C46—C45—H45B 106.8 C43—C45—H45B 109.1 H45A—C45—H45B 109.5 C55—C50—H50 118.8 C51—C50—H50 120.9 C50—C51—H51 119.1 C52—C51—H51 121.1 C53—C52—H52 120.3 C51—C52—H52 120.1 C52—C53—H53 120.8 C54—C53—H53 118.6 N59—C60—H60A 110.6 C61—C60—H60A 113.4 N59—C60—H60B 108.0 C61—C60—H60B 109.6 H60A—C60—H60B 109.5 C62—C61—H61A 111.1 C60—C61—H61A 107.4 C62—C61—H61B 112.0 C60—C61—H61B 111.1 H61A—C61—H61B 109.5 C62—C63—H63 120.2 C64—C63—H63 119.8 C65—C64—H64 119.3 C63—C64—H64 120.7 C64—C65—H65 119.9 C66—C65—H65 118.5 C67—C66—H66 121.5 C65—C66—H66 120.6

Experimental Section
183
C13—N14—N15—C16 3.8 (7) C46—N47—N48—C49 2.3 (7) C6—C1—C2—C8 -169.4 (4) C5—C1—C2—C8 -103.6 (5) C6—C1—C2—C3 -50.5 (6) C5—C1—C2—C3 15.2 (5) C1—C2—C3—O7 96.7 (4) C8—C2—C3—O7 -144.5 (4) C1—C2—C3—C4 -22.5 (5) C8—C2—C3—C4 96.4 (4) O7—C3—C4—C5 -99.2 (5) C2—C3—C4—C5 21.7 (5) C3—C4—C5—C24 -152.1 (4) C3—C4—C5—C6 51.3 (5) C3—C4—C5—C1 -12.3 (5) C6—C1—C5—C24 -108.4 (5) C2—C1—C5—C24 139.4 (4) C6—C1—C5—C4 110.1 (4) C2—C1—C5—C4 -2.0 (5) C2—C1—C5—C6 -112.1 (5) C2—C1—C6—C5 97.3 (5) C24—C5—C6—C1 107.4 (5) C4—C5—C6—C1 -95.3 (4) C10—N9—C8—C2 -66.5 (6) C1—C2—C8—N9 -53.5 (5) C3—C2—C8—N9 -169.0 (4) C8—N9—C10—O11 -2.1 (7) C8—N9—C10—C12 178.8 (4) O11—C10—C12—C13 154.6 (5) N9—C10—C12—C13 -26.3 (7) N15—N14—C13—C21 1.0 (7) N15—N14—C13—C12 -175.9 (4) C10—C12—C13—N14 109.6 (5) C10—C12—C13—C21 -67.3 (6) N14—N15—C16—O23 177.1 (4) N14—N15—C16—C22 -3.3 (7) C22—C17—C18—C19 0.8 (9) C17—C18—C19—C20 -1.2 (9) C18—C19—C20—C21 0.6 (9) C19—C20—C21—C22 0.3 (8) C19—C20—C21—C13 176.5 (5) N14—C13—C21—C22 -5.8 (7) C12—C13—C21—C22 171.0 (5) N14—C13—C21—C20 178.0 (5) C12—C13—C21—C20 -5.2 (7) C20—C21—C22—C17 -0.7 (7) C13—C21—C22—C17 -177.0 (5) C20—C21—C22—C16 -177.7 (5) C13—C21—C22—C16 6.0 (7) C18—C17—C22—C21 0.2 (8) C18—C17—C22—C16 177.1 (5) O23—C16—C22—C21 177.6 (5) N15—C16—C22—C21 -1.9 (7) O23—C16—C22—C17 0.7 (8) N15—C16—C22—C17 -178.8 (4) C34—N26—C24—O25 -5.6 (7) C27—N26—C24—O25 170.6 (5) C34—N26—C24—C5 174.1 (4) C27—N26—C24—C5 -9.8 (7) C4—C5—C24—O25 120.4 (5) C6—C5—C24—O25 -83.8 (6)

Experimental Section
184
C1—C5—C24—O25 -15.2 (7) C4—C5—C24—N26 -59.3 (6) C6—C5—C24—N26 96.5 (5) C1—C5—C24—N26 165.1 (4) C24—N26—C27—C28 -159.0 (5) C34—N26—C27—C28 17.7 (5) N26—C27—C28—C29 -19.0 (5) C27—C28—C29—C34 14.6 (6) C27—C28—C29—C30 -168.2 (5) C34—C29—C30—C31 -0.2 (8) C28—C29—C30—C31 -177.2 (5) C29—C30—C31—C32 3.1 (8) C30—C31—C32—C33 -3.2 (8) C31—C32—C33—C34 0.5 (8) C38—C68—C35—C41 -104.4 (4) C39—C68—C35—C41 -169.7 (4) C38—C68—C35—C36 15.5 (5) C39—C68—C35—C36 -49.8 (5) C68—C35—C36—O40 94.0 (4) C41—C35—C36—O40 -146.2 (4) C68—C35—C36—C37 -24.8 (5) C41—C35—C36—C37 95.0 (4) O40—C36—C37—C38 -96.4 (4) C35—C36—C37—C38 24.3 (5) C30—C29—C34—C33 -2.5 (8) C28—C29—C34—C33 175.0 (5) C30—C29—C34—N26 178.5 (4) C28—C29—C34—N26 -4.0 (6) C32—C33—C34—C29 2.4 (7) C32—C33—C34—N26 -178.8 (5) C24—N26—C34—C29 167.7 (4) C27—N26—C34—C29 -9.0 (5) C24—N26—C34—C33 -11.2 (8) C27—N26—C34—C33 172.1 (5) C39—C68—C38—C57 -108.4 (5) C35—C68—C38—C57 140.3 (4) C35—C68—C38—C39 -111.3 (4) C39—C68—C38—C37 110.5 (4) C35—C68—C38—C37 -0.7 (5) C36—C37—C38—C57 -154.9 (4) C36—C37—C38—C68 -14.7 (5) C36—C37—C38—C39 50.3 (5) C57—C38—C39—C68 108.1 (4) C37—C38—C39—C68 -96.6 (4) C35—C68—C39—C38 97.1 (4) C43—N42—C41—C35 -66.1 (6) C68—C35—C41—N42 -52.2 (5) C36—C35—C41—N42 -168.1 (3) C41—N42—C43—O44 -0.8 (7) C41—N42—C43—C45 179.2 (4) O44—C43—C45—C46 149.3 (4) N42—C43—C45—C46 -30.7 (6) N48—N47—C46—C54 1.1 (7) N48—N47—C46—C45 -176.5 (4) C43—C45—C46—N47 113.3 (5) C43—C45—C46—C54 -64.4 (6) N47—N48—C49—O56 178.4 (4) N47—N48—C49—C55 -2.0 (7) C55—C50—C51—C52 0.0 (8) C50—C51—C52—C53 0.5 (8) C51—C52—C53—C54 0.3 (7)
Experimental Section
185
C52—C53—C54—C55 -1.5 (7) C52—C53—C54—C46 177.2 (4) N47—C46—C54—C55 -4.6 (7) C45—C46—C54—C55 172.9 (4) N47—C46—C54—C53 176.7 (4) C45—C46—C54—C53 -5.7 (7) C51—C50—C55—C54 -1.3 (7) C51—C50—C55—C49 177.3 (5) C53—C54—C55—C50 2.0 (7) C46—C54—C55—C50 -176.7 (4) C53—C54—C55—C49 -176.6 (4) C46—C54—C55—C49 4.7 (6) O56—C49—C55—C50 -0.7 (7) N48—C49—C55—C50 179.7 (4) O56—C49—C55—C54 177.9 (4) N48—C49—C55—C54 -1.7 (6) C67—N59—C57—O58 -10.0 (8) C60—N59—C57—O58 172.8 (6) C67—N59—C57—C38 170.7 (4) C60—N59—C57—C38 -6.5 (8) C68—C38—C57—O58 -22.5 (7) C39—C38—C57—O58 -92.0 (6) C37—C38—C57—O58 113.7 (6) C68—C38—C57—N59 156.7 (4) C39—C38—C57—N59 87.2 (5) C37—C38—C57—N59 -67.1 (6) C57—N59—C60—C61 -174.1 (5) C67—N59—C60—C61 8.3 (7) N59—C60—C61—C62 -9.6 (7) C60—C61—C62—C63 -170.2 (6) C60—C61—C62—C67 7.9 (7) C67—C62—C63—C64 1.2 (8) C61—C62—C63—C64 179.1 (6) C62—C63—C64—C65 1.9 (9) C63—C64—C65—C66 -2.6 (9) C64—C65—C66—C67 0.2 (8) C65—C66—C67—C62 3.0 (8) C65—C66—C67—N59 -176.1 (5) C63—C62—C67—C66 -3.7 (8) C61—C62—C67—C66 177.9 (5) C63—C62—C67—N59 175.6 (5) C61—C62—C67—N59 -2.8 (6) C57—N59—C67—C66 -1.9 (9) C60—N59—C67—C66 175.6 (6) C57—N59—C67—C62 178.9 (5) C60—N59—C67—C62 -3.6 (6)

Experimental Section
186
5.2.2 Biological Evaluation of Bicyclo[3.1.0]hexane -based Chemical
Library
Reporter Gene Assay Screening for Inhibitory Effect s on Cellular Signaling Pathways:
The assays were performed by Dr. Dorian Fabbro and Dr. Daniel D’Orazio. For the EGF and
TNFα-involving pathways a reporter gene assay with AP-1-bla ME180 cells containing the β-
lactamase reporter gene under the control of the AP-1 promoter was used. The cell line
(catalogue nr. K1185) was purchased by Invitrogen, and the assay was modified and
validated for the use to profile kinase inhibitors that interact with cellular pathways leading to
the activation of AP-1 driven transcription based on the Invitrogen manual
(https://www.invitrogen.com/content/sfs/manuals/cellsensor_ap1blame180_man.pdf).
Protease Inhibition Assays : Either thrombin (100nM) in PBS (20 mM NaH2PO4, 30 mM
Na2HPO4, 100 mMNaCl, pH 7.4) or urokinase (62 nM) in PBS or cathepsin B (5.4 nM) in
0.1% (v/v) Brij 35 solution and a solution of (L)-Cysteine (8 mM) in PBS (352 mM KH2PO4,
48mM Na2HPO4, 4 mM ethylenediamine tetraacetic acid, pH 6.0) was incubated with 84
library members at a concentration of 10 µM in 384-well microtiter plates for 30 min at room
temperature. For the thrombin and urokinase assays, the reaction was started by the
addition of the fluorogenic substrate Z-GGR-AMC (Bachem, Bubendorf, Switzerland)
dissolved in PBS and 1% DMSO to a final concentration of 0.25 mM in a total volume of 50
µl. As positive control, for thrombin a 0.5 µM solution of the known inhibitor hirudin fragment
54-65 was employed. As a positive control for urokinase, a 120 µM solution of known
inhibitor benzamidine was used. For the cathepsin B assay, the reaction was started by the
addition of the fluorogenic substrate Arg-Arg-7-AMC (Bachem, Bubendorf, Switzerland)
dissolved in 0.1% Brij 35 solution and 1% DMSO to a final concentration of 0.15 mM in a
total volume of 50 µl. As a positive control a 10.3 µM solution of known inhibitor cystatin was
used.
The change of fluorescence signal (ex: 360 nm, em: 460 nm, cutoff: 420 nm) was recorded
over 30 min using a SpectraMaxmicroplate reader (Molecular Devices).
Every assay was performed as a triplicate.
Inhibition of Mycobacterium Tuberculosis: The assay was carried out by Dr. Ruben
Haartkoorn. Two bacterial strains were used, H37Rv (growing) and 18b (non-growing), in a
standard resazurine assay as described in 166.

Experimental Section
187
Cytoxicity Screening : The measurements were carried out by Dr. Andrea Chicca. The
cytotoxicity of the library compounds against MCF-7 (breast cancer) and U937 (lymphoma)
cells was determined after 72 hours incubation time in a WST-1 based cell viability assay as
described in 170. All compounds were tested in a concentration of 5 µM. Every test was
performed as a triplicate.
5.2.3 Synthesis of Miscellaneous Derivatives and Bi cyclo[3.1.0]hexane-
based 5-Alkynyl-deoxypyrimidine Nucleoside Analogue 243
COOMe
HO
N3
253
(1S,3S,4R,5S)-methyl 4-(azidomethyl)-3-hydroxybicyc lo[3.1.0]hexane-1-carboxylate
192 (850 mg, 2.612 mmol) was dissolved in THF (20 ml), then TBAF (3.3 ml, 3.134 mmol)
was added slowly under stirring. The reaction mixture was stirred for 1 h at RT. The reaction
was quenched by the addition of saturated aqueous NH4Cl-solution and the water phase was
extracted with EtOAc (3x 50 ml). The combined organic phases were dried over MgSO4 and
evaporated. The residue was purified by flash column chromatography (EtOAc/hex 1:1) to
give 495 mg (2.344 mmol, 90%) of the product as a colorless oil.
Rf = 0.31 (Hex/EtOAc 1:1).
1H-NMR (400 MHz, CDCl3): δ = 4.19 (dd, J = 7.0, 1.2 Hz, 1H), 3.64 (s, 3H), 3.28 (dd, J =
12.5, 6.6 Hz, 1H), 3.20 (dd, J = 12.5, 7.6 Hz, 1H), 2.62 (ddd, J = 14.6, 7.0, 1.8 Hz, 1H), 2.16
(t, J = 7.2 Hz, 1H), 1.95 (s, br, 1H), 1.86 (dd, J = 14.7, 1.2 Hz, 1H), 1.76 (ddd, J = 9.2, 5.2,
1.2 Hz, 1H), 1.53, (ddd, J = 9.0, 4.6, 1.8 Hz, 1H), 1.40, (t, J = 5.0 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 174.4, 76.3, 53.9, 52.1, 49.9, 36.8, 32.5, 31.4, 21.2.
IR (film): ν 3440, 2949, 2101, 1699, 1439, 1272, 1150, 1071.
HR-MS (ESI): calcd for C9H13N3NaO3 [M-H]+: 234.0849, found: 234.0844.

Experimental Section
188
(1S,3S,4R,5S)-methyl 4-(azidomethyl)-3-methoxybicy clo[3.1.0]hexane-1-carboxylate
NaH (38 mg, 60% dispersion in oil) was added to a solution of 240 (40 mg, 0.189 mmol) in 3
ml of DMF and the reaction mixture was stirred for 30 min at RT. The reaction mixture turned
slightly orange. CH3I (134 mg, 0.945 mmol) was added and the reaction mixture was stirred
at RT for 20 h. The mixture was poured on water and extracted with EtOAc (3x30 ml), the
combined organic phases were washed with brine, dried over MgSO4 and evaporated. The
residue, an orange oil, was purified by flash column chromatography (Hex/EtOAc 5:1) to give
27 mg (0.120 mmol, 63%) of the product as a colorless oil.
Rf = 0.37 (Hex/EtOAc 3:1)
1H-NMR (400 MHz, CDCl3): δ = 3.65 (s, 3H), 3.62 (d, J = 7.4 Hz, 1H), 3.28 (dd, J = 12.5, 6.6
Hz, 1H), 3.19 (s, 3H), 3.17 (dd, J = 12.5, 7.9 Hz, 1H), 2.48 (ddd, J = 14.7, 7.0, 1.8 Hz, 1H),
2.28 (t, J = 7.2 Hz, 1H), 2.00 (d, J = 14.6 Hz, 1H), 1.74 (ddd, 8.9, 5.2, 1.0 Hz, 1H), 1.46 (ddd,
J = 9.0, 4.4, 1.8 Hz, 1H), 1.26 (t, J = 4.8 Hz, 1H).
13C-NMR (100 MHz, CDCl3): δ = 174.4, 85.4, 56.5, 53.9, 51.9, 46.2, 32.8, 31.9, 30.9, 20.3.
MS (ESI): calcd for C10H16N3O3 [M-H]+: 226.12, found: 226.05 (I = 100%).
249
To a stirred solution of 195 (30 mg) in THF(0.5 ml) thionyl chloride (90 ul) was added
dropwise at rt and the resulting mixture was refluxed for 2 h. After cooling the mixture was
added dropwise to NH3 (aq., 0.5 ml) at 0°C. The solution was dried, filtr ated, the filter cake
was washed and the filtrate was added dropwise to a stirred solution of oxalyl chloride (13 ul)
with DMF (12 ul) in 0.5 ml of THF. The solvents were evaporated to afford 16 mg (>100%) of
249.
1H-NMR (400 MHz, MeOD): δ = 4.18 (d, J = 5.6 Hz, 1H), 3.48-3.38 (m , 2H), 2.69 (s, br, 1H),
2.43 (dd, J = 13.7, 6.6 Hz, 1H), 2.09 (t, J = 6.4 Hz, 1H), 1.92-1.81 (m, 2H), 1.47-1.35 (m, 2H).
13C-NMR (100 MHz, MeOD): δ = 118.7, 75.9, 64.7, 53.5, 32.9, 31.8, 21.4.

Experimental Section
189
252
(((1S,3S,4R,5S)-4-(azidomethyl)-1-iodobicyclo[3.1.0 ]hexan-3-yl)oxy)(tert-
butyl)dimethylsilane
A solution of 3 (50 mg, 0.160 mmol), iodosobenzene diacetate (56 mg, 0.176 mmol) and
iodine (44 mg, 0.176 mmol) was stirred placed above a 500 W tungsten lamp. The mixture
was refluxed and illuminated for 1 h, then additional iodosobenzene diacetate (56 mg) and
iodine (44 mg) were added and the reaction mixture was refluxed for additional 1.5 h. The
reaction mixture was cooled to RT and diluted with 10% aqueous sodium thiosulfate solution
(5 ml) and Et2O (10 ml), the phases were separated and the organic phase was dried over
MgSO4 and evaporated. The residue was purified by flash column chromatography (H/EE
100:1) to give 33 mg (0.084 mmol, 52%) of the product as colorless crystals.
1H-NMR (400 MHz, CDCl3): δ = 3.91 (d, J = 6.5 Hz, 1H), 3.40 (dd, J = 12.5, 5.8 Hz, 1H), 3.18
(dd, J = 12.2, 8.4 Hz, 1H), 2.51 (ddd, J = 14.1, 6.7, 2.0 Hz, 1H), 2.26 (d, J = 14.1 Hz, 1H),
2.11 (dd, J= 8.6, 6.0 Hz, 1H), 1.54-1.48 (m , 2H), 1.09 (ddd, J = 10.6, 7.0, 2.0 Hz, 1H), 0.85
(s, 9H), 0.03 (s, 6H).
13C-NMR (100 MHz, CDCl3): δ = 76.3, 54.2, 50.9, 49.5, 31.5, 25.7, 22.8, 17.8, 1.1, -4.8, -4.9.
IR (film): ν 2953, 2927, 2856, 2363, 2099, 1462, 1385, 1257, 1135, 1089, 1026, 930, 852,
837, 777, 744, 682.
MS (ESI): calcd for C13H25IN3OSi [M-H]+: 394.08, found: 394.05 (I = 40%).
251
tert-butyl(((1S,2R,3S,5S)-3-((tert-butyldimethylsil yl)oxy)-5-iodobicyclo[3.1.0]hexan-2-
yl)methoxy) dimethylsilane
A solution of 195 (60 mg, 0.150 mmol), iodosobenzene diacetate (53 mg, 0.165 mmol) and
iodine (42 mg, 0.165 mmol) in cyclohexane (2 ml) was stirred placed above a 500 W
tungsten lamp. The mixture was refluxed and illuminated for 2 h, then additional
iodosobenzene diacetate (53 mg) and iodine (42 mg) were added and the reaction mixture
was refluxed for additional 1.5 h. The reaction mixture was cooled to RT and diluted with 5%

Experimental Section
190
aqueous sodium thiosulfate solution (5 ml) and Et2O (20 ml). The phases were separated,
the organic phase was dried over MgSO4 and evaporated. The residue was purified by flash
column chromatography (Hex/EtOAc 200:1) to afford 28 mg (0.058 mmol, 39%) of the
product.
1H-NMR (400 MHz, CDCl3): 3.97 (d, J = 6.5 Hz, 1H), 3.61 (dd, J = 10.2, 5.7 Hz, 1H), 3.43
(dd, J = 10.2, 7.9 Hz, 1H), 2.47 (ddd, J = 13.8, 6.7, 2.0 Hz, 1H), 2.23 (d, J = 13.9 Hz, 1H),
2.06 (dd, J= 8.1, 5.7 Hz, 1H), 1.51-1.44 (m , 2H), 1.06 (ddd, J = 10.6, 7.0, 1.5 Hz, 1H), 0.93
(s, 9H), 0.85 (s, )H), 0.09 (s, 3H), 0.09 (s, 3H), 0.01 (s, 6H).
13C-NMR (100 MHz, CDCl3): 75.8, 64.7, 53.4, 49.6, 30.8, 26.0, 25.7, 22.4, 18.9, 17.5, 2.4, -
4.8, -4.9. -5.3, -5.4.
IR (film): ν 2953, 2927, 2886, 2856, 1462, 1385, 1253, 1136, 1115, 1076, 1005, 933, 835,
775, 689.
MS (ESI): calcd for C19H40IO2Si2 [M-H]+: 483.16, found: 483.05 (I = 50%).
245
(E)-N-(((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)o xy)-4-(((tert-
butyldimethylsilyl)oxy)methyl)bicyclo[3.1.0]hexan-1 -yl)carbamoyl)-3-
methoxyacrylamide
245 (271 mg, 95%) was prepared from 213 mg of 4 and freshly prepared 211 (from 1.2 g of
217) analogeously to the procedure described for 212.
Rf = 0.38 (Hex/EtOAc 2:1).
1H-NMR (400 MHz, CDCl3): δ = 9.30 (s, 1H), 8.80 (s, 1H), 7.64 (d, J = 12.4 Hz, 1H), 5.28 (d,
J = 12.4 Hz, 1H), 4.14 (d, J = 6.6 Hz, 1H), 3.70 (s, 3H), 3.63 (dd, J = 10.2, 5.6 Hz, 1H), 3.53
(dd, J = 10.2, 9.4 Hz, 1H), 2.33 (ddd, J = 13.4, 6.6, 1.7 Hz, 1H), 2.00-1.94 (m, 2H), 1.32 (t, J
= 4.5 Hz, 1H), 1.26-1.21 (m, 2H), 0.88 (s, 9H), 0.83 (s, 9H), 0.04 (s, 6H), -0.01 (s, 3H), -0.02
(s, 3H).
13C-NMR (100 MHz, CDCl3 ): δ = 167.6, 163.7, 155.4, 97.9, 74.4, 65.1, 57.4, 53.8, 40.8, 39.7,
26.8, 26.0, 25.9, 18.6, 18.3, 17.8, -4.7, -4.8, -5.3, -5.4.
MS (ESI): calcd for C24H46N2NaO5Si2 [M-H]+: 521.28, found: 521.30 (I = 100%).

Experimental Section
191
153
1-((1S,3S,4R,5S)-3-hydroxy-4-(hydroxymethyl)bicyclo [3.1.0]hexan-1-yl)pyrimidine-
2,4(1H,3H)-dione
153 (48 mg, 75%) was prepared from 135 mg of 245 with 2.7 ml of H2SO4 (aq., 1 M) in 1 ml
of dioxane analogeously to the procedure described for 213a.
Rf = 0.40 (EtOAc/MeOH 6:1).
1H-NMR (400 MHz, MeOD): δ = 7.66 (d, J = 10.1 Hz, 1H), 5.61 (d, J = 10.1 Hz, 1H), 4.21 (d,
J = 6.6 Hz, 1H), 3.75 (dd, J = 10.1, 5.6 Hz, 1H), 3.67 (dd, J = 10.2, 5.6 Hz, 1H), 2.38 (ddd, J
= 13.4, 6.6, 1.7 Hz, 1H), 2.12-2.05 (m, 2H), 1.73 (ddd, J = 10.2, 6.6, 1.0 Hz, 1H), 1.48 (t, J =
4.5 Hz, 1H), 1.15 (ddd, J = 13.4, 6.6, 1.7 Hz, 1H).
13C-NMR (100 MHz, MeOD): δ = 165.5, 152.9, 147.8, 101.8, 74.9, 69.7, 64.1, 53.6, 49.5,
47.4, 40.6, 28.3, 19.2.
MS (ESI): calcd for C11H15N2O2 [M-H]+: 239.10, found: 239.12 (I = 100%).
247
1-((1S,3S,4R,5S)-3-hydroxy-4-(hydroxymethyl)bicyclo [3.1.0]hexan-1-yl)-5-
iodopyrimidine-2,4(1H,3H)-dione
Iodine (14 mg, 0.053 mmol) and CAN (29 mg, 0.053 mmol) were added to a solution of 153
(25 mg, 0.105 mmol) in AcOH (3 ml). The mixture was stirred at 80°C for 4 h. The solvent
was evaporated and the residue was purified by flash column chromatography
(EtOAc/MeOH 20:1) to yield 23 mg (0.063 mmol, 60%) of the product as a yellow solid.
Rf = 0.38 (Hex/EtOAc 20:1)
1H-NMR (400 MHz, MeOD): δ = 8.20 (s, 1H), 4.27-4.24 (d, J = 7.5 Hz, 1H), 3.83-3.78 (dd, J =
10.5, 5.0 Hz, 1H), 3.77-3.72 (dd, J = 10.5, 5.0 Hz, 1H), 2.44-2.37 (ddd, J = 13.3, 7.0, 2.3 Hz,

Experimental Section
192
1H), 2.16-2.07 (m, 2H), 1.81-1.76 (ddd, J = 9.7, 4.8, 1.2 Hz, 1H), 1.53-1.48 (t, J = 5.2 Hz,
1H), 1.24-1.19 (m, 1H).
13C-NMR (300 MHz, MeOD): δ = 163.2, 153.1, 151.7, 75.6, 67.9, 64.9, 54.3, 50.2, 41.2, 28.9,
19.1.
IR (film): ν 2985, 2905, 2364, 2335, 1699, 1409, 1295, 1246, 1053, 439.
MS (ESI): calcd for C11H14IN2O2 [M-H]+: 365.00, found: 364.78 (I = 100%).
243
5-(dodec-1-yn-1-yl)-1-((1S,3S,4R,5S)-3-hydroxy-4-(h ydroxymethyl)bicyclo[3.1.0]hexan-
1-yl) pyrimidine-2,4(1H,3H)-dione
247 (20 mg, 0.055 mmol) was dissolved in DMF (2 ml), Pd(PPh3)4 (7 mg, 0.006 mmol), CuI
(2 mg, 0.011 mmol), DIPEA (19 ul, 0.165 mmol) and 1-Dodecyne (35 ul, 0.165 mmol) were
added and the reaction mixture was stirred overnight at RT. Two drops of 5% disodium salt
of an aqueous EDTA solution was added (colors turns from orange to brown) and the solvent
was evaporated. The residue was purified by flash column chromatography (EtOAc/MeOH
20:1) to afford 17 mg of product that was still containing some minor impurities. Subsequent
purification by preparative HPLC yielded 3 mg (0.008 mmol, 14%) of the product as a white
solid.
Rf = 0.22 (EtOAc/MeOH 20:1)
1H-NMR (400 MHz, MeOD): δ = 7.53 (s, 1H), 4.45-4.41 (d, J = 6.5 Hz, 1H), 3.91-3.87 (dd, J =
11.3, 2.7 Hz, 1H), 3.78-3.74 (dd, J = 11.3, 3.8 Hz, 1H), 2.54-2.47 (ddd, J = 13.4, 6.6, 2.3 Hz,
1H), 2.40-2.35 (t, J = 7.2 Hz, 2H), 2.15-2.13 (t, J = 3.3 Hz, 1H), 2.08-2.02 (d, J = 13.4 Hz,
1H), 1.84-1.79 (dd, J = 9.6, 4.7 Hz, 1H), 1.60-1.51 (m, 3H), 1.42-1.34 (m, 2H), 1.25 (br s,
12H), 1.12-1.07 (ddd, J = 9.8, 5.5, 2.4 Hz, 1H), 0.89-0.84 (t, J = 6.8 Hz, 3H).
13C-NMR (100 MHz, MeOD): δ = 167.0, 154.9, 152.0, 104.3, 97.7, 78.5, 75.0, 68.4, 56.6,
52.9, 44.0, 35.9, 33.3, 33.1, 33.0, 32.8, 32.5, 32.2, 31.5, 26.4, 22.7, 21.3, 16.9.
MS (ESI): calcd for C23H33N2O4 [M-H]-: 401.24, found: 401.04 (I = 75%).

Experimental Section
193
Biological Evaluation of 243 :
The test for inhibiting activity on mycobacterium tuberculosis was carried out by Dr. Thomas
Keller. The compound was found to be inactive.

Experimental Section
194
5.2.4 Synthesis of Bicyclo[3.1.0]hexane based Pento statin- and (S)-
Adenosylhomocysteine Analogues
5.2.4.1 Preparation of lactone (-)-44 (enantiomeric series)
In general, for the preparation of 161, procedures were followed as described in 117b and in
117a.
(1R,6S)-6-(methoxycarbonyl)cyclohex-3-enecarboxyli c acid
1,2,3,6-tetrahydrophthalic anhydride (83 g) was dispersed in MeOH, then K2CO3 (15 g) was
slowly added and the reaction mixture was stirred for 15 h at RT. The solvent was
evaporated and the residue was extracted with EtOAc. The organic phases were combined,
dried over MgSO4 and evaporated to yield 100.5 g (100%) of 175 (rac) as a white solid.
1H-NMR (400 MHz, CDCl3): δ = 5.62 (m, 2H), 3.65 (s, 3H), 3.08-3.02 (m, 2H), 2.59-2.50 (m,
2H), 2.39-2.30 (m, 2H).
13C-NMR (100 MHz, CDCl3): δ = 179.7, 173.8, 125.3, 125.1, 52.0, 39.6, 39.5, 25.8, 25.6.
COOMe
COO HNH2
HOH
Bn
264
175 (rac) (100 g) and (-)-ephedrine (90 g) each dissolved in 270 ml of EtOH at 70°C were
slowly combined. After 12 h at RT the crystals that had formed were titrated with cold
EtOH/EtOAc and filtrated and then recrystallized from 380 ml of EtOH , then a second and a
third time recrystallized from 250 ml of EtOH to afford 45 g (47 %, corresponding to the
racemate) of 264 as white crystals.
Mp. 147°C.

Experimental Section
195
[αααα]D20 = -35.7° (c = 2, MeOH).
1H-NMR (400 MHz, CDCl3): δ = 8.03 (s, br, 3H), 7.36-7.19 (m, 5H), 5.68-5.57 (m, 2H), 5.23
(d, J = 2 Hz, 1H), 3.59 (s, 3H), 3.10 (dd, J = 6.8, 2.2 Hz, 1H), 2.97-2.90 (m, 2H), 2.65 (s, 3H),
2.60-2.47 (m, 2H), 2.33-2.23 (m, 2H), 1.03 (d, J = 6.6 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 180.5, 175.3, 140.5, 128.2, 127.3, 126.3, 125.8, 125.1, 70.9,
61.6, 51.5, 41.8, 40.3, 31.3, 27.1, 26.1, 9.2.
264 was dissolved in cold 2 N H2SO4 (~200 ml) and extracted with Et2O (2x 400 ml), the
organic phases were washed with 2 N H2SO4 (~50 ml), then with water (~50 ml), dried over
MgSO4 and evaporated to give 24.3 g (47%, corresponding to the racemate) of (-)-175 as a
colorless oil.
[αααα]D20 = -10.6° (c = 2.18, acetone).
1H-NMR (400 MHz, CDCl3): δ = 5.61 (m, 2H), 3.67 (s, 3H), 3.08-3.00 (m, 2H), 2.60-2.51 (m,
2H), 2.39-2.30 (m, 2H).
13C-NMR (100 MHz, CDCl3): δ = 179.7, 173.7, 125.2, 125.0, 52.0, 39.6, 39.5, 25.8, 25.6.
177
(3aS,7aR)-3a,4,7,7a-tetrahydroisobenzofuran-1(3H)-o ne
To a solution of 175 (75.1 g) in dichloromethane, oxalylchloride (52 ml) was added slowly at
0°C under stirring. After gas development had cease d (1-2 h) the reaction mixture was stirred
at RT under Ar overnight. The mixture was concentrated under educed pressure to afford
80.6 g (98%) of the acid chloride derivative as a slightly yellow liquid.
1H-NMR (400 MHz, CDCl3): δ = 5.71 (m, 2H), 3.69 (s, 3H), 3.50-3.14 (m, 2H), 2.68-2.42 (m,
4H).
13C-NMR (100 MHz, CDCl3): δ = 174.3, 172.4, 125.5, 123.7, 52.3, 40.5, 26.0.

Experimental Section
196
NaBH4 (28.6 g) was dissolved in 1600 ml of EtOH and cooled to -40°C , then the acid
chloride derivative (80.6 g) in 160 ml of dry THF was added slowly under stirring. After gas
evaporation had ceased the reaction mixture was stirred for 2 h at -40°C, then it was
quenched at this temperature with 4N H2SO4 until a pH of 2 was reached. The solvents were
evaporated and the residue was mixed with 600 ml of water and extracted with Et2O. The
organic phases were washed with aq. NaHCO3 and brine, dried over MgSO4 and
concentrated under reduced pressure to yield approx. 80 g (the product was not completely
dried) of a mixture of 177 and the hydroxyl-methylester derivative. The mixture was dissolved
in 480 ml of toluene, 10-campher sulfonic acid (160 mg) was added and the reaction mixture
was refluxed in the water separator for approx. 3 h. The mixture was concentrated under
reduced pressure and purified by a Kugelrohrdistillation to give 51.9 g (94%, over 2 steps) of
the product as a colorless oil.
[αααα]D20 = +59.50° (c = 2, EtOAc)
ee ≥ 85%. The enantiomeric purity was determined by HPLC analysis using a DAICEL chiral
column.
1H-NMR (400 MHz, CDCl3): δ = 5.61 (m, 2H), 3.67 (s, 3H), 3.08-3.00 (m, 2H), 2.60-2.51 (m,
2H), 2.39-2.30 (m, 2H).
13C-NMR (100 MHz, CDCl3): δ = 179.3, 130.0, 125.1, 72.5, 37.2, 32.1, 24.9, 22.2.
From 177, lactone 161 was prepared according to procedures described in 117b and 117a, in 6
steps with an overall yield of 23%.
161
(3aS,4R,5S,6aR)-5-hydroxy-4-(hydroxymethyl)hexahydr o-1H-cyclopenta[c]furan-1-one
To a solution of 182 (5.2 g) and trimethylborate (9.4 ml, 0.084 mol, distilled prior to use) in 75
ml of THF a solution of boran-dimethylsulfid complex (5.3 ml, 10-10.2 M solution of SMe2 in
BH3) in 25 ml of THF was added drop by drop at 5°C. Af ter gas evaporation ceased the
reaction mixture was slowly, during 45 min, warmed to 15°C. The reaction was quenched
with 35 ml of H2O that was added slowly and the mixture was evaporated to dryness. The
residue was dissolved in 100 ml of MeOH and evaporated to remove boric acid (3x). The

Experimental Section
197
crude product was purified by flash column chromatography (EtOAc/MeOH 6:1) to afford 4 g
(83 %) of the product as a colorless oil.
Rf = 0.16 (EtOAc/MeOH 9:1)
[αααα]D20 = -54.60° ± 0.96° (c = 1.07, MeOH)
1H-NMR (400 MHz, D6-acetone): δ = 4.45 (dd, J = 9.1, 8.0 Hz, 1H), 4.22 (dd, J = 9.1, 2.8 Hz,
1H), 4.15-4.11 (m, 1H), 3.60 (dd, J = 10.7, 6.3 Hz, 1H), 3.51 (dd, J = 10.7, 6.8 Hz), 2.97 (tdd,
J = 11.5, 9.9, 3.3 Hz, 1H), 2.81 (dddd, J = 10.5, 8.1, 5.3, 2.7 Hz, 1H), 2.24 (ddd, J = 13.4,
9.8, 5.6 Hz, 1H), 2.00 (q, J = 5.6 Hz, 1H), 1.92 (dt, J = 13.6, 3.5 Hz, 1H)
13C-NMR (100 MHz, D6-acetone): δ = 181.3, 75.6, 75.3, 63.5, 57.7, 42.6, 41.7, 38.1.
HR-MS (ESI): calcd for C8H12O4 [M+H]+: 172.0808, found: 173.0800.
5.2.4.2 Preparation of Advanced Intermediate (+)-4
183
(1R,2S,3R,4S)-methyl 2-(bromomethyl)-4-hydroxy-3-
(hydroxymethyl)cyclopentanecarboxylate
To a solution of 161 (4 g, 0.023 mol) in MeOH (120 ml) bromotrimethylsilane (17.8 g, 0.116
mol) was added slowly during 10 min at 0°C. Then Zn Br2 (0.52 g, 0.002 mol) was added and
the reaction mixture was stirred at RT for 25 h. The solvent was evaporated at a water bath
temperature below 30°C and the crude product, an or ange oil, was purified by flash column
chromatography (EtOAc/MeOH 12:1) to yield 3.26 g (0.012 mol, 53%) of the product as a
slightly yellow oil.
Rf = 0.45 (EtOAc/MeOH 12:1)
1H-NMR (400 MHz, CDCl3): δ = 5.00 (s, br, 2H), 4.29-4.23 (m, 1H), 3.96-3.89 (m, 1H), 3.75
(s, 3H), 3.75-3.69 (m, 1H), 3.60-3.55 (m, 1H), 3.47-3.41 (m, 1H), 3.16-3.09 (m, 1H), 2.43-
2.34 (m, 1H), 2.26-2.19 (m, 1H), 2.15-2.10 (m, 1H), 2.20-1.96 (m, 1H).
13C-NMR (300 MHz, CDCl3): δ = 176.3, 76.1, 63.4, 54.3, 52.5, 45.9, 45.3, 37.1, 33.4.

Experimental Section
198
HR-MS (ESI): calcd for C9H16BrO4 [M+H]+ : 267.0232, found: 267.0231.
184
(1R,2S,3R,4S)-methyl 2-(bromomethyl)-4-((tert-butyl dimethylsilyl)oxy)-3-(((tert-
butyldimethylsilyl)oxy)methyl)cyclopentanecarboxyla te
To a solution of 183 (430 mg, 1.610 mmol) in 30 ml of DMF MTBSA (3 g, 0.016 mol) was
added drop by drop (over 10min) under cooling to 0°C. The reaction mixture was stirred for
24 h at room temperature. The CH2Cl2 was evaporated and the resulting oil was dried under
high vacuum conditions for 12 h to remove excessive MTBSA. The crude product was
purified by flash column chromatography (CH2Cl2) to afford 463 mg (0.934 mmol, 58%) of the
product as a very slightly yellow oil.
Rf = 0.65 (CH2Cl2).
[αααα]D20 = -11.83° ± 0.06° (c = 1, EtOAc).
1H-NMR (400 MHz, CDCl3): δ = 4.04-3.97 (m, 1H), 3.68 (s, 3H), 3.66-3.61 (m, 2H), 3.58-
3.54 (m, 2H), 2.94-2.86 (m, 1H), 2.16-2.07 (m, 1H),1.95-1.85 (m, 2H), 0.88 (s, 9H), 0.85 (s,
9H), 0.04 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H), 0.01 (s, 3H).
13C-NMR (300 MHz, CDCl3): δ = 1714.3, 72.5, 61.5, 54.2,51.6, 43.6, 43.2, 37.9, 34.7, 26.1,
25.7, 18.1, 17.6, -4.4, -4.8, -5.5, -5.9.
IR (film): ν 2929, 2857, 1735, 1471, 1435, 1361, 1252, 1109, 883, 833, 774, 669.
HR-MS (ESI): calcd for C21H43BrO4Si2 [M+H]+:495.1956, found: 495.1940.
185
(1S,3S,4R,5S)-methyl 3-((tert-butyldimethylsilyl)ox y)-4-(((tert-
butyldimethylsilyl)oxy)methyl) bicyclo[3.1.0]hexane -1-carboxylate
184 (90 mg, 0.181 mmol) was dissolved in 1 ml of tert-BuOH, then tert-BuOK (23 mg, 0.200
mmol) dissolved in 1 ml of tert-BuOH was added slowly under stirring. The resulting

Experimental Section
199
suspension was stirred for 1.5 h at room temperature. The reaction mixture was poured on
40 ml of Et2O and washed with ice water (3x 7 ml). The phases were separated and the
organic phase was dried over MgSO4 and evaporated. Purification by flash column
chromatography (CH2Cl2) yielded 71 mg (0.170 mmol, 94%) of the product as a colorless oil.
Rf = 0.58 (CH2Cl2).
[αααα]D24 = -20.21° (c = 1.35, CHCl 3).
1H-NMR (400 MHz, CDCl3): δ = 4.16 (d, J = 6.6 Hz, 1H), 3.62 (s, 3H), 3.51 (dd, J = 9.9, 5.7
Hz, 1H), 3.33 (dd, J= 10.1, 7.8 Hz, 1H), 2.49 (ddd, J = 14.2, 6.7, 1.6 Hz, 1H), 2.06 (dd, J =
7.8, 5.7 Hz, 1H), 1.79 (d, J = 14 Hz, 1H), 1.69 (ddd, 8.8, 5.0, 1.3 Hz, 1H), 1.46 (dd, J = 5.4,
4.2 Hz, 1H), 1.42-1.38 (m, 1H), 0.88 (s, 9H), 0.84 (s, 9H), 0.03 (s, 3H), 0.02 (s, 3H), 0.01 (s,
6H).
13C-NMR (100 MHz, CDCl3): δ = 175.1, 75.8, 64.8, 53.6, 50.7, 36.3, 31.9, 30.9, 25.9, 25.8,
20.2, 18.4, 17.3, -4.7, -4.8, -5.5.
IR (film): ν 2930, 2857, 2357, 1726, 1467, 1439, 1364, 1253, 1147, 1079, 1004, 939, 835,
776, 748, 662.
HR-MS (ESI): calcd for C21H43O4Si2 [M+H]+: 415.2694, found: 415.2693.
195
(1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4-(( (tert-
butyldimethylsilyl)oxy)methyl)bicyclo [3.1.0] hexan e-1-carboxylic acid
185 (0.97 g) was dissolved in EtOH (15 ml) and KOH (0.39 g) was added. The reaction
mixture was heated in the microwave at 100°C for 2. 5 h, then it was stirred at RT overnight.
The solvent was evaporated and the residue was mixed with 50 ml of EtOAc and 15 ml of
iced water, then 2 N aqueous HCl was added under vivid stirring until the pH reached 2. The
phases were separated and the water phase was extracted with EtOAc (2x 50 ml). The
combined organic phases were dried and evaporated. The residue was purified by flash
column chromatography (CH2Cl2/Et2O 4:1--> CH2Cl2) to yield 0.84 g (90%) of the product as
a slightly yellow solid.
Rf = 0.13 (CH2Cl2), 0.41 (Et2O)
[a] 24D = -24.65° (c = 1, CHCl 3)

Experimental Section
200
1H-NMR (400 MHz, CDCl3): δ = 4.17 (d, J = 6.5 Hz, 1H), 3.52 (dd, J = 10.2, 5.6 Hz, 1H),
3.33 (dd, J = 10.0, 7.8 Hz, 1H), 2.48 (ddd, J = 14.0, 6.7, 1.0 Hz, 1H), 2.08 (dd, J = 7.9, 5.6
Hz, 1H), 1.81-1.75 (m, 2H), 1.55 (dd, J = 5.3, 4.0 Hz, 1H), 1.49 (ddd, J = 9.2, 4.2, 1.4 Hz,
1H), 0.88 (s, 9H), 0.84 (s, 9H), 0.03 (s, 3H), 0.02 (s, 3H), 0.01 (s, 3H), 0.01 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 180.5, 75.0, 64.1, 53.2, 36.9, 33.1, 30.1, 25.9, 25.8, 21.3,
18.3, 17.8, -4.7, -4.9, -5.5.
IR (film): ν 2931, 2857, 2361, 1686, 1467, 1254, 1082, 1044, 917, 837, 776, 660.
HR-MS (ESI): calcd for C20H41Na2O4Si2 [M+H]+: 445.2177, found: 445.2178.
196
benzyl ((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)o xy)-4-(((tert-
butyldimethylsilyl)oxy)methyl) bicyclo[3.1.0]hexan- 1-yl)carbamate
195 (0.69 g) was dissolved in toluene (17 ml) and cooled to 0°C, then DPPA (398 ul) and
Et3N (333 ul) were added and the reaction mixture was stirred for 1 h at 0°C, then 1.5 h at RT
and for 2 h at 80°C. The colorless solution turned slightly yellow upon formation of the
isocyanate. The reaction mixture was cooled to RT, then BnOH (455 ul) and Di-n-
butyltindilaurate (114 mg) were added and the mixture was stirred at 80°C for 2 h, then at
100°C for 15 min. The mixture was cooled to RT, dil uted with Et2O (100 ml) and washed with
saturated aqueous NaHCO3 solution (2x 15 ml) and water (2x 30 ml). The combined organic
phases were dried and evaporated to yield the crude product as a yellow oil that was purified
by flash column chromatography (CH2Cl2 --> CH2Cl2/MeOH 15:1) to afford 0.77 g (88%) of
the product as a colorless oil.
Rf = 0.26 (CH2Cl2)
[a] 25D = -5.66° (c = 1.3 in CHCl 3)
1H-NMR (400 MHz, CDCl3): δ = 7.38-7.29 (m, 5H), 5.07 (s, br, 2H), 4.16 (d, J = 5.5 Hz, 1H),
3.72-3.52 (m, 2H), 2.39-2.29 (m, 1H), 2.01-1.95 (m, 1H), 1.92 (d, J = 13.3 Hz, 1H), 1.59-1.50
(m, 1H), 1.34-1.30 (m, 1H), 0.92 (s, 9H), 0.86 (s, 9H), 0.84-0.81 (m , 1H), 0.08 (m, 6H), 0.02
(s, 3H), 0.02 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 136.6, 128.6, 128.0, 74.3, 64.7, 54.0, 29.4, 26.0, 25.6, 18.3,
18.0, -4.6, -4.8, -5.3, -5.4.

Experimental Section
201
IR (film): ν 2928, 2856, 2359, 1714, 1498, 1462, 1255, 1219, 1083, 835, 774, 666.
MS (ESI): calcd for C27H48NO4Si2 [M+H]+: 506.31, found: 506.32 (I = 100%).
TBSO
TBSONH2
4
(1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4-(( (tert-
butyldimethylsilyl)oxy)methyl)bicyclo[3.1.0] hexan- 1-amine
To a solution of 196 (25 mg) in toluene/MeOH 1:1 (0.5 ml) 10% Pd/C (11 mg) was added and
the reaction mixture was stirred vigorously under H2 (1 bar) for 4 h at RT. The reaction
mixture was directly put on the column and purified by flash chromatography (EtOAc/MeOH
20:1) to afford 16 mg (88%) of the product as a yellow oil.
Rf = 0.24 (EtOAc/MeOH 20:1)
[a] 24D = +11.42° (c = 0.8 in MeOH)
1H-NMR (400 MHz, MeOD): δ = 4.16 (d, J = 6.5 Hz, 1H), 3.60 (dd, J = 10.1, 5.6 Hz, 1H),
3.40 (dd, J = 10.3, 8.9 Hz, 1H), 2.08 (ddd, J = 13.5. 6.6, 2.0 Hz, 1H), 1.90 (dd, J = 8.9, 5.6
Hz, 1H), 1.86 (d, J = 13.5 Hz, 1H), 1.06 (t, J = 4.3 Hz, 1H), 0.97 (ddd, J = 9.1, 3.9, 1.2 Hz,
1H), 0.92 (s, 9H), 0.86 (s, 9H), 0.69 (ddd, J = 9.1, 4.4, 2.2 Hz, 1H), 0.08 (m, 6H), 0.02 (s,
3H), 0.02 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 75.0, 65.5, 54.1, 45.4, 42.9, 27.7, 26.1, 25.6, 18.8, 18.3,
17.7, -4.7, -4.9, -5.4.
HR-MS (ESI): calcd for C19H42NO2Si2 [M-H]+: 372.2749, found: 372.2743.

Experimental Section
202
5.2.4.3 Attempted Preparation of Pentostatin Analog ue 6
162
Compound 162 was prepared by Kurt Hauenstein according to a literature procedure156b
starting from N-Cbz-(L)-vinylglycine methyl ester in an amount of 170 mg.
163
4-((R)-2-azido-1-((tert-butyldimethylsilyl)oxy)ethy l)-1-((1S,3S,4R,5S)-3-((tert-
butyldimethyl silyl)oxy)-4-(((tert-butyldimethylsil yl)oxy)methyl)bicyclo[3.1.0]hexan-1-
yl)-1H-imidazol-5-amine
After co-evaporation with toluene, 4 (22 mg) was dissolved in acetonitrile (0.6 ml) and added
to a sol. of 162 (9 mg) with mol. sieves (4 Å) in acetonitrile. The reaction mixture was refluxed
at 90°C for 2 h. Additional 9 mg of 162 were added and the reaction mixture was stirred for 1
h. The solvent was evaporated and the residue was purified by flash column chromatography
(EtOAc/Hex 2:3, 0.2% Et3N) to afford 27 mg (71%, based on mass recovery) of 163 as a light
brown oil.
Note: Additional peaks in the 1H-NMR indicated a partial decomposition. Purity was
estimated to be approx. 50%.
Rf = 0.51 (Hex/EtOAc 3:2, 0.2% Et3N )
1H-NMR (500 MHz, MeOD): δ = 7.80 (s, 1H), 7.18 (s, 1H), 4.58 (s, 2H), 4.23 – 4.13 (m, 1H),
3.88-3.81 (m, 1H), 3.68 – 3.65 (m, 1H), 3.58-3.54 (m, 1H), 2.25-2.20 (m, 1H), 2.15-2.11 (m,
1H), 1.92-1.88 (m, 1H), 1.76-1.73 (m, 1H), 1.57-1.53 (m, 1H), 1.15-1.12 (m, 1H), 0.96-0.86
(m, >27H), 0.20-0.02 (m , 18H).
MS (ESI): calcd for C30H61N6O3Si3 [M-H]+: 637.41, found: 637.70 (I = 100%).

Experimental Section
203
165
(E)-N'-(4-((R)-2-azido-1-((tert-butyldimethylsilyl) oxy)ethyl)-1-((1S,3S,4R,5S)-3-((tert-
butyldimethyl silyl)oxy)-4-(((tert-butyldimethylsil yl)oxy)methyl)bicyclo[3.1.0]hexan-1-
yl)-1H-imidazol-5-yl)-N,N-dimethylformimidamide
After co-evaporation with toluene (2x 3 ml), 4 (16 mg, 0.043 mmol) was dissolved in
acetonitrile (0.5 ml) and added to a sol. of 162 (20 mg, 0.065 mmol) and 2,2,2-trifluoro-
ethanol (43 ul, 0.004 mmol) with mol. sieves (4 Å) in acetonitrile. The reaction mixture was
refluxed at 90°C for 6 h. Dimethylformamide dimethy l acetal (23 ul, freshly distilled) was
added and the reaction mixture was refluxed for 13 h to give 28 mg (0.040 mmol, 94% based
on mass recovery) of the product as a brown oil.
Note: The 1H-NMR spectrum indicated the presence of both 163 and 165 in an approx. 1:1
mixture. Additional peaks indicated a partial decomposition.
Rf = 0.33 (Hex/EtOAc 10:1, 0.2% Et3N)
[a] 24D = -13.88° (c = 1 in CHCl 3)
1H-NMR (500 MHz, C6D6): δ = 7.33 (s, 1H), 6.99 (s, 1H), 5.66 – 5.47 (m, 1H), 4.21 (d, J = 5.8
Hz, 1H), 4.00 – 3.96 (m, 1H), 3.96 – 3.91 (m, 1H), 3.62 – 3.54 (m, 1H), 3.42 – 3.37 (m, 1H),
3.18 (d, J = 4.7 Hz, 3H), 2.52 (s, 3H), 2.25 (dd, J = 15.3, 7.8 Hz, 1H), 2.21 – 2.10 (m, 2H),
1.83 – 1.71 (m, 1H), 1.66-1.50 (m, 1H), 1.15-1.12 (m, 1H), 1.00 (s, 9H), 0.99 (s, 9H), 0.98 (s,
9H), 0.22 (s, 3H), 0.17 (s, 3H), 0.15 (s, 3H), 0.11 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H).
IR (film): ν 2929, 2858, 2363, 2336, 1769, 1651, 1458, 1386, 1249, 839, 791, 779, 658.
MS (ESI): calcd for C33H66N7O3Si3 [M-H]+: 692.45, found: 692.60 (I = 100%).

Experimental Section
204
6
(R)-8-(tert-butyldimethylsilyloxy)-3-((1S,3S,4R,5S) -3-(tert-butyldimethylsilyloxy)-4-((tert-
butyldimethylsilyloxy)methyl)bicyclo[3.1.0]hexan-1- yl)-3,6,7,8-tetrahydroimidazo[4,5-
d][1,3]diazepine
After co-evaporation with toluene, 4 (10 mg, 0.027 mmol) was dissolved in 1,2-
dichloroethane (0.3 ml), 162 (12 mg) and trifluoroethanol (27 ul) were added and the reaction
mixture was stirred at 80°C for 4 h. TLC indicated still starting material, so an additional 6 mg
of 162 were added and the mixture was stirred for additional 3 h at 80°C. TLC indicated that
all starting material had been consumed. The solvent was evaporated and the imidazole
intermediate was purified by flash chromatography (Hex/EtOAc 3:2,0.2% Et3N) to afford 4 mg
(0.006 mmol, 23%) of 163 that were immediately reacted with dimethylformamide dimethyl
acetal (15 ul) in dichloroethane (0.3 ml) at 80°C f or 14 h to afford crude 165. The
dichloroethane was evaporated and MeOH (0.3 ml), propanedithiol (19 ul) and Et3N (38 ul)
were added. The reaction mixture was stirred at 50°C for 12 h. Evaporation of the solvent
and purification by flash chromatography (Hex/EtOAc 10:1, 0.2% Et3N) yielded 1.3 mg of the
product (0.002 mmol, 7% over 3 steps).
Rf = 0.4 (EtOAc/MeOH 20:1)
MS (ESI): calcd for C33H66N7O3Si3 [M-H]+: 621.41, found: 621.50 (I = 100%).
1H-NMR indicated the presence of the title compound with 2 of the TBS groups removed, in
small quantity, along with large amounts of grease and additional peaks in the region of δ =
2.6 and 4.0.

Experimental Section
205
5.2.4.4 Preparation of S-Adenosylhomocysteine Analo gue 7
280
9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4 -(((tert-butyldimethylsilyl)oxy)methyl)
bicyclo[3.1.0]hexan-1-yl)-6-chloro-9H-purine
A mixture of 4 (118 mg), 4,6-dichloro-5-formamidopyrimidine (219, 122 mg), Et3N (176 ul) in
1,4-dioxane (3 ml) with mol. sieves was gently refluxed for 24 h at 110°C. The solvent was
evaporated to give approximately 250 mg of crude 279 (brown oil) that was purified by flash
column chromatography (H/EE 2:1) to give 112 mg of 279 as a yellow oil that was
immediately treated with diethoxymethyl acetate (3 ml) and heated to 110°C for 20 h. The
solvents were evaporated and the residue was purified by flash column chromatography
(H/EE 7:1) to yield 72 mg (45% over 2 steps) of the product as a white solid.
Rf = 0.2 (Hex/EtOAc 7:1).
[a] 24D = -30.13° ± 1.86° (c = 1 in CHCl 3).
1H-NMR (400 MHz, CDCl3): δ = 8.71 (s, 1H), 8.19 (s, 1H), 4.30 (d, J = 6.5 Hz), 3.92 (dd, J =
10.2, 7.8 Hz, 1H), 3.85 (dd, J = 10.2, 5.8 Hz, 1H), 2.55 (ddd, J = 13.3, 6.5, 2.0 Hz, 1H), 2.23-
2.16 (m, 2H), 1.81-1.71 (m, 2H), 1.36-1.31 (m, 1H), 0.91 (s, 9H), 0.87 (s, 9H), 0.10 (s,3H),
0.09 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 151.5, 150.9, 146.4, 131.5, 74.4, 64.6, 53.0, 43.4, 41.6,
26.5, 26.0, 25.7, 18.5, 17.8, 16.9, -4.7, -4.8, -5.2, -5.3.
IR (film): ν 2953, 2928, 2885, 2857, 2363, 1699, 1589, 1557, 1490, 1472, 1427, 1407, 1389,
1361, 1337, 1254, 1220, 1091, 1070, 1023, 934, 849, 835, 776, 636, 567.
HR-MS (ESI): calcd for C24H42ClN4O2Si2 [M+H]+: 509.2529, found: 509.2535.

Experimental Section
206
167
9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl)oxy)-4 -(((tert-
butyldimethylsilyl)oxy)methyl)bicyclo[3.1.0]hexan-1 -yl)-9H-purin-6-amine
A saturated solution of ammonia in methanol was prepared and 280 (25 mg) was dissolved
in 5 ml of it. The reaction mixture was heated to 100°C in the microwave for 4 h. The solvent
was evaporated and the crude product was purified by flash chromatography (CH2Cl2/MeOH
20:1 & 0.5% Et3N) to yield 21 mg (87%) of the product as a colorless oil that turned into a
white solid at -20°C.
Rf = 0.1 (CH2Cl2/MeOH 20:1 0.5% Et3N)
[a] 20D = -23.94° ± 0.38° (c = 1.13 in CHCl 3)
1H-NMR (400 MHz, MeOD): δ = 8.17 (s, 1H), 8.08 (s, 1H), 4.40 (d, J = 7.1 Hz, 1H), 3.99 (dd,
J = 10.3, 8.3 Hz, 1H), 3.91 (dd, J = 10.3, 5.5 Hz, 1H), 2.57 (ddd, J = 13.3, 6.5, 2.1 Hz, 1H),
2.26 (d, J = 13.4 Hz, 1H), 2.18 (dd, J = 8.2, 5.6 Hz, 1H), 1.79 (ddd, J = 9.5, 4.8, 1.1 Hz, 1H),
1.73 (t, J = 5.1 Hz, 1H), 1.41 – 1.35 (m, 1H), 0.95 (s, 9H), 0.91 (s, 9H), 0.15 (s, 3H), 0.14 (s,
3H), 0.09 (s, 3H), 0.08 (s, 3H).
13C-NMR (100 MHz, MeOD): δ = 153.7, 142.8, 76.4, 66.1, 54.9, 44.8, 42.6, 27.4, 26.6, 26.4,
20.2, 18.8, 17.1, -4.3, -4.6.
IR (film): ν 3325, 2952, 2928, 2856, 1614, 1573, 1471, 1390, 1294, 1253, 1190, 1085, 1040,
958, 835, 776, 650.
HR-MS (ESI): calcd for C24H44N5O2Si2 [M+H]+: 490.3028, found: 490.3025.
TBSO
TBSON
N
NN
NHAloc
283
Allyl-(9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl )oxy)-4-(((tert-
butyldimethylsilyl)oxy)methyl) bicyclo[3.1.0]hexan- 1-yl)-9H-purin-6-yl)carbamate
To a solution of 167 (10 mg, 0.020mmol) in CH2Cl2 (0.3 ml), N-methylimidazole (5 ul, 0.060
mmol) and allychloroformate (7 ul, 0.060 mmol) were added. The reaction mixture was stirred

Experimental Section
207
for 41 h at RT. The solvent was evaporated to afford the crude product that was purified by
flash chromatography (EtOAc/Hex 1:1) to yield 7 mg (0.012 mmol, 61%) of the product as a
white solid.
Rf = 0.4 (Hex/EtOAc 1:1)
1H-NMR (400 MHz, MeOD): δ = 8.58 (s, 1H), 8.30 (s, 1H), 6.05 (ddt, J = 17.2, 10.6, 5.6 Hz,
1H), 5.43 (dq, J = 17.2, 1.6 Hz, 1H), 5.28 (ddd, J = 10.5, 2.7, 1.3 Hz, 1H), 4.75 (dt, J = 5.6,
1.4 Hz, 2H), 4.42 (d, J = 7.0 Hz, 1H), 4.04 (dd, J = 10.3, 8.9 Hz, 1H), 3.94 (dd, J = 10.3, 5.8
Hz, 1H), 2.65 (ddd, J = 13.3, 6.6, 2.0 Hz, 1H), 2.26 (d, J = 13.4 Hz, 1H), 2.20 (dd, J = 8.8, 5.7
Hz, 1H), 1.82 (ddd, J = 9.4, 4.9, 1.0 Hz, 1H), 1.77 (t, J = 5.1 Hz, 1H), 1.44 (ddd, J = 9.4, 5.1,
2.1 Hz, 1H), 0.97 (s, 9H), 0.92 (s, 9H), 0.16 (s, 3H), 0.15 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H).
13C-NMR (100 MHz, MeOD): δ = 153.4, 146.6, 120.9, 119.1, 75.7, 67.7, 65.9, 54.6, 44.3,
42.7, 27.9, 26.6, 26.4, 19.4, 18.8, 17.9, -4.3, -4.7.
MS (ESI): calcd for C28H48N5O4Si2 [M+H]+: 574.32, found: 574.30 (I = 100%).
284
Allyl-(9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl )oxy)-4-
(hydroxymethyl)bicyclo[3.1.0]hexan-1-yl)-9H-purin-6 -yl)carbamate
283 (15 mg, 0.026 mmol) was dissolved in CH2Cl2/MeOH 1:1 (0.3 ml) and cooled to 0°C.
CSA (15 mg, 0.065 mmol) was added and the reaction mixture was stirred at 0°C for 4 h.
Additional 8 mg of CSA were added and the reaction mixture was stirred for additional 1h.
The reaction was quenched with sat. aq. NaHCO3-sol. until pH reached 8 and evaporated to
give the crude product (78 mg) as a white solid. Purification by flash chromatography
(EtOAc/MeOH 30:1) gave 8 mg (0.017 mmol, 67%) of the product as a yellow oil.
Rf = 0.4 (EtOAc/MeOH 20:1)
MS (ESI): calcd for C22H34N5O4Si [M-H]+: 460.24, found: 460.20 (I = 100%).

Experimental Section
208
285
Allyl-(9-((1S,3S,4R,5S)-3-((tert-butyldimethylsilyl )oxy)-4-((methylbenzenesulfonate)
methyl) bicyclo[3.1.0]hexan-1-yl)-9H-purin-6-yl)ca rbamate
Tosyl chloride (3.2 mg) was added to a stirred solution of 284 (4 mg) and triethylamine (5 ul)
in CHCl3 (0.3 ml) under Argon at 0°C, then DMAP (0.1 mg) was added. The reaction mixture
was warmed to room temperature and stirred for 4 d. Evaporation of the solvent and
purification by flash column chromatography (EtOAc/Hex 3:2) gave 2 mg of the desired
product (36%).
Rf = 0.15 (EtOAc/Hex 3:2).
1H-NMR (400 MHz, CDCl3): δ = 8.65 (s, 1H), 8.03 (s, 1H), 7.86 (d, J = 8.3 Hz, 2H), 7.37 (d, J
= 8.0 Hz, 2H), 6.00 (ddd, J = 16.2, 10.4, 5.7 Hz, 1H), 5.42 (dd, J = 17.2, 1.4 Hz, 1H), 5.30
(dd, J = 10.4, 1.2 Hz, 1H), 4.78 (d, J = 5.8 Hz, 2H), 4.52 – 4.42 (m, 1H), 4.32 (dd, J = 10.2,
5.9 Hz, 1H), 4.21 (d, J = 7.0 Hz, 1H), 2.54 (ddd, J = 13.9, 6.7, 2.3 Hz, 1H), 2.46 (s, 3H), 2.38
(dd, J = 8.4, 5.9 Hz, 1H), 2.15 (d, J = 13.5 Hz, 1H), 1.83 – 1.77 (m, 1H), 1.68 (dd, J = 9.2, 4.8
Hz, 1H), 1.44 – 1.38 (m, 1H), 0.87 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H).
HR-MS (ESI): calcd for C29H39N5NaO6SSi2 [M-Na]+: 636.2288, found: 636.2285.
((1S,2R,3S,5S)-5-(6-amino-9H-purin-9-yl)-3-((tert-
butyldimethylsilyl)oxy)bicyclo[3.1.0]hexan-2-yl)met hanol
To a solution of 167 (6 mg, 0.012 mmol) in CH2Cl2/MeOH 1:1 (0.2 ml) CSA (10 mg, 0.042
mmol) was added at 0°C and the reaction mixture was stirred for 4 h at 0°C. The reaction
was quenched with sat. aq. NaHCO3-sol. until pH reached 8 and the solvents were
evaporated to afford ~29 mg of the crude product as a slightly yellow solid. Purification by
flash chromatography (EtOAc/MeOH 20:1 & 0.5% Et3N) afforded 4 mg (0.011 mmol, 89%) of
the product as a white solid.
Rf = 0.2 (Hex/EtOAc 20:1 0.5% Et3N)

Experimental Section
209
1H-NMR (400 MHz, MeOD): δ = 8.18 (s, 1H), 8.08 (s, 1H), 4.56 (s, 1H), 4.42 (dd, J = 6.6, 1.3
Hz, 1H), 3.96 (dd, J = 11.5, 3.9 Hz, 1H), 3.82 (dd, J = 11.5, 4.5 Hz, 1H), 3.72 – 3.62 (m, 1H),
3.59 – 3.54 (m, 1H), 2.57 (ddd, J = 13.2, 6.6, 2.3 Hz, 1H), 2.23 (d, J = 13.3 Hz, 1H), 2.17 (t, J
= 4.1 Hz, 1H), 1.84 (ddd, J = 9.6, 4.8, 1.3 Hz, 1H), 1.71 (t, J = 5.1 Hz, 1H), 1.40 (ddd, J = 9.6,
5.2, 2.3 Hz, 1H), 0.92 (s, 9H), 0.09 (s, 3H), 0.09 (s, 3H).
HR-MS (ESI): cacld for C18H30N52Si [M+H]+: 376.2169, found: 376.2158.
MS (ESI): cacld for C18H30N52Si [M+H]+: 376.2169, found: 375.89 (I = 100%).
168
Di-tert-butyl 9-((1S,3S,4R,5S)-3-(tert-butyldimethy lsilyloxy)-4-((tert-
butyldimethylsilyloxy)methyl) bicyclo[3.1.0]hexan-1 -yl)-9H-purin-6-ylcarbamate
167 (40 mg, 0.082 mmol) and DMAP (40 mg) were dissolved in DMF and evaporated under
reduced pressure to remove atmospheric water, Boc2O (71 mg) was added and the reaction
mixture was stirred for 14 h at RT. The reaction mixture as such was purified by flash column
chromatography (Hex/EtOAc 5:1, 0.5% Et3N) to afford the diprotected product 161 (49 mg,
87%).
Rf = 0.3 (Hex/EtOAc 10:1 0.5% Et3N)
[a] 24D = -19.62° (c = 1 in CHCl 3)
1H-NMR (400 MHz, CDCl3): δ = 8.83 (s, 1H), 8.11 (s, 1H), 4.33 (d, J = 6.8 Hz, 1H), 3.96 (dd,
J = 10.2, 8.1 Hz, 1H), 3.88 (dd, J = 10.2, 5.9 Hz, 1H), 2.59 (ddd, J = 13.3, 6.6, 2.1 Hz, 1H),
2.26 – 2.15 (m, 2H), 1.78 (t, J = 5.0 Hz, 1H), 1.73 (ddd, J = 9.3, 4.8, 1.0 Hz, 1H), 1.46 (s,
18H), 1.36 (ddd, J = 9.1, 5.0, 1.9 Hz, 1H), 0.94 (s, 9H), 0.89 (s, 9H), 0.12 (s, 3H), 0.11 (s,
3H), 0.06 (s, 3H), 0.05 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 154.1, 151.9, 150.7, 150.2, 145.7, 129.4, 83.7, 74.3, 64.7,
53.1, 42.9, 41.5, 27.8, 26.4, 26.1, 25.8, 18.5, 17.7, 16.8, -4.7, -4.8, -5.3, -5.4.
IR (film): ν 2928, 2856, 2359, 2338, 1734, 1460, 1368, 1253, 1101, 837, 778.
HR-MS (ESI): calcd for C34H59N5NaO6Si2 [M+H]+: 712.3896, found: 712.3917.

Experimental Section
210
169
Di-tert-butyl (9-((1S,3S,4R,5S)-3-(( tert-butyldimethylsilyl)oxy)-4-(hydroxymethyl)-
bicyclo[3.1.0]hexan-1-yl)-9H-purin-6-yl)carbamate
To a solution of 168 (41 mg, 0.059 mmol) in CH2Cl2/MeOH1:1 (0.8 ml) CSA (14 mg, 0.059
mmol) was added at 0°C and the reaction mixture was stirred for 4 h at 0°C and for additional
2 h at 10°C. Additional 2 mg of CSA were added and the reaction mixture was stirred for
additional 3 h at 0°C. The reaction was quenched wi th sat. aq. NaHCO3-sol. until the pH
reached a value of 8 and the solvents were evaporated to give the crude product as white
solid (approx.70 mg). Purification by flash column chromatograph (Hex/EtOAc 2:1) afforded
30 mg (0.052 mmol, 88%) of the product as a colorless oil.
Rf = 0.23 (Hex/EtOAc 2:1)
[a] 24D = -13.88° (c = 1 in CHCl 3)
1H-NMR (400 MHz, CDCl3): δ = 8.83 (s, 1H), 8.17 (s, 1H), 5.65 (d, J = 10.6 Hz, 1H), 4.47 (dd,
J = 6.5, 1.3 Hz, 1H), 4.08 (d, J = 12.3 Hz, 1H), 3.86 (td, J = 11.9, 3.3 Hz, 1H), 2.65 (ddd, J =
13.0, 6.6, 2.3 Hz, 1H), 2.20 (dd, J = 2.3, 0.7 Hz, 1H), 2.15 (d, J = 13.1 Hz, 1H), 1.81 (ddd, J =
9.5, 4.8, 1.2 Hz, 1H), 1.75 (t, J = 5.0 Hz, 1H), 1.48 (s, 18H), 1.31 (ddd, J = 9.5, 5.0, 2.4 Hz,
1H), 0.90 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
13C-NMR (400 MHz, CDCl3): δ = 151.6, 146.2, 84.3, 76.3, 65.5, 53.1, 44.3, 43.8, 27.8, 26.8,
25.9, 17.1, -4.8, 4.9.
IR (film): ν 2963, 2928, 2856, 2363, 2321, 1760, 1599, 1368, 1259, 1090, 1024, 799, 672,
461.
HR-MS (ESI): calcd forC28H46N5O6Si [M-H]+: 576.3212, found: 576.3213.

Experimental Section
211
170
((1S,2R,3S,5S)-5-(6-((di- tert-butoxycarbonyl)amino)-9H-purin-9-yl)-3-(( tert-
butyldimethylsilyl)- oxy)bicyclo[3.1.0]hexan-2-yl)m ethyl 4-methylbenzenesulfonate
169 (30 mg) was dissolved in CHCl3 (0.4 ml). Triethylamine (10 ul), DMAP (0.5 mg) and TsCl
(13 mg, 0.066 mmol) were added and the reaction mixture was stirred for 3 d at RT (after 1 d
again 13 mg of TsCl and 18 ul of Et3N were added). The reaction mixture was directly put on
the column and purified by flash column chromatography (Hex/EtOAc 5:2) to afford 18 mg
(0.025 mmol, 75%) of the product as a colorless oil.
Rf = 0.37 (Hex/EtOAc 2:1)
[αααα]24D = -12.22° (c = 0.87 in CHCl 3)
1H-NMR (400 MHz, CDCl3): δ = 8.74 (s, 1H), 8.07 (s, 1H), 7.88 – 7.82 (m, 2H), 7.36 (dd, J =
8.5, 0.6 Hz, 2H), 4.47 (dd, J = 10.1, 8.7 Hz, 1H), 4.33 (dd, J = 10.1, 6.1 Hz, 1H), 4.20 (dd, J =
6.4, 1.0 Hz, 1H), 2.53 (ddd, J = 13.6, 6.6, 2.2 Hz, 1H), 2.45 (s, 3H), 2.37 (dd, J = 8.6, 6.1 Hz,
1H), 2.13 (d, J = 13.6 Hz, 1H), 1.79 (t, J = 5.3 Hz, 1H), 1.66 (ddd, J = 9.8, 4.9, 1.2 Hz, 1H),
1.48 (s, 18H), 1.40 (ddd, J = 9.7, 5.6, 2.2 Hz, 1H), 0.87 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H).
13C-NMR (100 MHz, CDCl3): δ = 153.9, 151.9, 150.7, 150.2, 145.3, 145.0, 132.9, 129.9,
129.3, 128.0, 83.9, 73.8, 70.7, 49.9, 42.8, 42.0, 40.9, 27.9, 25.7, 21.7, 17.9, 16.6, 14.2, -4.8,
-4.9.
IR (film): ν 2927, 2856, 2362, 2337, 1790, 1730, 1598, 1459, 1367, 1255, 1175, 1139, 1101,
1025, 957, 939, 815, 785, 696, 659, 572, 553, 530.
HR-MS (ESI): calcd for C35H52N5O8SSi [M+H]+: 730.3300, found: 730.3286.
286
(S)-tert-butyl (2-oxotetrahydrothiophen-3-yl)carbam ate
(L)-homocysteine thiolactone hydrochloride (70 mg) was slowly added to a mixture of
NaHCO3 (191 mg) in 1 ml of dioxane/H2O (1:1). The mixture was stirred for 30 min and then

Experimental Section
212
Boc2O (152 mg) was added and the reaction mixture was stirred overnight. The reaction
mixture was diluted with EtOAc, the phases were separated and the water phase was
extracted with EtOAc, the combined organic phases were washed with brine, dried and
evaporated to afford the crude product (approx. 150 mg). The crude product was purified by
flash column chromatography (EtOAc/Hex 1:4) to give 83 mg of product (white solid, 84%).
Rf = 0.3 (CHCl3/MeOH 10:1)
1H-NMR (400 MHz, CDCl3): δ = 4.96 (s, 1H), 4.36 – 4.17 (m, 1H), 3.31 (td, J = 11.7, 5.1 Hz,
1H), 3.23 (ddd, J = 11.3, 7.0, 1.2 Hz, 1H), 2.96 – 2.75 (m, 1H), 1.97 (qd, J = 12.5, 7.2 Hz,
1H), 1.45 (s, 9H).
MS (ESI): calcd for C9H15NNaO3S [M-Na]+: 240.0670, found: 240.10 (I = 100%).
287
(S)-methyl 4-((((1S,2S,3S,5S)-5-(6-(bis(tert-butoxy carbonyl)amino)-9H-purin-9-yl)-3-
((tert-butyldimethylsilyl)oxy)bicyclo[3.1.0]hexan-2 -yl)methyl)thio)-2-((tert-
butoxycarbonyl)amino)butanoate
170 (6 mg) was co-evaporated with DMF to remove atmospheric water. 286 (3 mg) was co-
evaporated with toluene. NaOMe (1.5 mg) was added to a solution of 286 in MeOH (0.2 ml)
and stirred for 1h, then 170 was added and the reaction mixture was refluxed (75°C) for 8 h.
The reaction mixture was directly put on the column and purified by flash chromatography
(Hex/EtOAc 2:1) to yield 4 mg (60%) of the product.
Note: An impurity probably derived from the reagent could not be removed, leading to
additional peaks at 5.02, 4.32, 3.50, 2.66, 2.16, 1.92, and 1.38.
Rf = 0.3 (Hex/EtOAc 2:1)
1H-NMR (400 MHz, CDCl3): δ = 8.77 (s, 1H), 8.05 (s,1H), 4.23 (d, J = 6.0 Hz, 1H), 3.74 (s,
3H), 3.53 – 3.49 (m, 1H), 3.40-3.36 (m, 1H), 2.93 – 2.88 (m, 1H), 2.62 – 2.59 (m, 1H), 2.58-
2.55 (m, 1H), 2.21 – 2.13 (m, 2H), 2.12 – 2.07 (m, 1H), 1.96-1.89 (m, 2H), 1.86-1.82 (m, 1H),
1.78 – 1.73 (m, 1H), 1.47 (s, 9H), 1.41 (s, 9H), 1.38 (s, >9H), 1.34-1.26 (m, 1H), 0.84 (s, 9H),
0.02 (s, 3H), 0.01 (S, 3H).
13C-NMR: (100 MHz, MeOD): δ = 151.9, 147.1, 84.1, 76.3, 51.4, 50.2, 49.9, 43.2, 40.3, 34.9,
31.3, 28.4, 28.1, 27.3, 26.9, 26.5, 24.9, 17.1, 16.6, -5.6, -6.0.

Experimental Section
213
MS (ESI): calcd for C38H63N6NaO9SSi [M-Na]+: 829.40, found: 829.40 (I = 100%).
173
(S)-4-((((1S,2S,3S,5S)-5-(6-(bis(tert-butoxycarbony l)amino)-9H-purin-9-yl)-3-((tert-
butyldi methylsilyl)oxy)bicyclo[3.1.0]hexan-2-yl)me thyl)thio)-2-((tert-
butoxycarbonyl)amino)butanoic acid
Methyl ester 287 (8 mg) was dissolved in THF (0.5 ml) and cooled to 0°C, then KOH (20 ul, 1
M aq. sol.) and water (50 ul) were added. The reaction mixture was stirred for 14 h. As TLC
and MS indicated the end of the reaction, the reaction mixture was cooled to 0°C and
quenched by adding HCl (20 ul, 1M aq.sol.), concentrated under reduced pressure and
purified by FC (CHCl3/MeOH/H2O 8:1:0.2%) to yield the product as a colorless solid (5.5 mg,
70%).
Note: The 1H-NMR spectrum displayed a splitting of TBS and aromatic peaks in the ratio of
2:3; and still contained some minor impurity derived from the reagent of the previous step.
The impurity was removed in the next step.
Rf = 0.3 (CHCl3/MeOH 10:1)
1H-NMR (500 MHz, MeOD): δ = 8.88 (s, 0.6H), 8.62 (s, 0.4H), 8.57 (s, 0.6H), 8.33 (s, 0.4H),
4.62 (s, 1H), 4.39 (dd, J = 20.3, 6.2 Hz, 1H), 4.17 (s, 1H), 3.72 – 3.66 (m, 1H), 3.06 (dd, J =
13.4, 7.0 Hz, 1H), 3.01 – 2.91 (m, 1H), 2.76 – 2.62 (m, 3H), 2.32 – 2.11 (m, 3H), 2.08 – 1.93
(m, 2H), 1.83 (dt, J = 21.5, 4.9 Hz, 1H), 1.59 (s, 5H), 1.50 (dd, J = 13.5, 7.1 Hz, 1H), 1.47 –
1.42 (m, 11H), 1.40 (s, 11H), 0.93 (s, 6H), 0.93 (s, 3H), 0.13 (s, 1H), 0.12 (s, 2H), 0.11 (s,
1H), 0.10 (s, 2H).
13C-NMR (400 MHz, MeOD): δ = 180.5, 151.6, 150.0, 131.8, 128.8, 83.2, 72.4, 60.7, 37.8,
36.3, 30.8, 28.9, 28.2, 27.8, 27.3, 25.6, 23.1, 22.4, 17.5, 13.8, 10.7, -4.6, -5.0.
HR-MS (ESI): calc. for C37H61N6O9SSi [M+H]+ : 793.3985, found: 793.3985.

Experimental Section
214
287a
(S)-methyl 4-((((1S,2S,3S,5S)-5-(6-amino-9H-purin-9- yl)-3-((tert-
butyldimethylsilyl)oxy)bicyclo[3.1.0]hexan-2-yl)met hyl)thio)-2-((tert-
butoxycarbonyl)amino)butanoate
NaOMe (1.5 mg) was added to a solution of 286 (3 mg) in MeOH and stirred for 1 h, then
170 was added and the reaction mixture was refluxed (75°C) for 18 h. 0.2 ml of aq. NaHCO 3-
sol. was added and the mixture was concentrated under reduced pressure. Purification by
flash column chromatography (EtOAc/Hex 5:1 � EtOAc/MeOH 20:1) gave 3 mg (60%) of the
title compound.
Rf = 0.48 (EtOAc/MeOH 20:1)
1H-NMR (400 MHz, CDCl3): δ = 8.77 (s, 1H), 8.05 (s,1H), 4.23 (d, J = 6.0 Hz, 1H), 3.74 (s,
3H), 3.53 – 3.49 (m, 1H), 3.40-3.36 (m, 1H), 2.93 – 2.88 (m, 1H), 2.62 – 2.59 (m, 1H), 2.58-
2.55 (m, 1H), 2.21 – 2.13 (m, 2H), 2.12 – 2.07 (m, 1H), 1.96-1.89 (m, 2H), 1.86-1.82 (m, 1H),
1.78 – 1.73 (m, 1H), 1.41 (s, 9H), 1.34-1.26 (m, 1H), 0.84 (s, 9H), 0.02 (s, 3H), 0.01 (S, 3H).
MS (ESI): calcd for C28H47N6O5SSi [M-H]+: 607.31, found: 607.40 (I = 100%).
173a
(S)-4-((((1S,2S,3S,5S)-5-(6-amino-9H-purin-9-yl)-3 -((tert-
butyldimethylsilyl)oxy)bicyclo[3.1.0]hexan-2-yl)met hyl)thio)-2-((tert-
butoxycarbonyl)amino)butanoic acid
287a (7 mg) was dissolved in THF (0.4 ml) and cooled to 0°C, then KOH-sol. (23 ul, 1M aq.
sol.) and water (50 ul) were added and the reaction mixture was stirred for 14 h. The reaction
mixture was cooled to 0°C and quenched by adding HC l (23 ul, 1M aq. sol.). The solvents
were evaporated and the crude product was purified by flash column chromatography

Experimental Section
215
(CHCl3/MeOH/H2O/acetic acid 8:1:0.1%:0.1%) to afford 3.5 mg (51%) of the title compound
as a white solid.
Rf = 0.2 (CHCl3/MeOH 10:1)
1H-NMR (500 MHz, MeOD): δ = 8.21 (s, 1H), 8.14 (s, 1H), 4.62 (s, 1H), 4.63-4.51 (m, 2H),
4.35 (d, J = 6.4 Hz, 1H), 4.09 (s, 1H), 3.03-2.96 (m, 1H), 2.93-2.86 (m, 1H), 2.68-2.59 (m,
3H), 2.26 – 2.21 (m, 1H), 2.19-2.12 (m, 2H), 1.78 (t, J = 5.1 Hz, 1H), 1.63-1.51 (m, 2H), 1.43
(s, 9H), 1.38-1.35 (m, 1H), 1.28 (s, 9H), 0.92 (s, 12H), 0.11 (s, 3H), 0.09 (s, 3H).
MS (ESI): calc. for C27H45N6O5SSi [M-H]+ : 593.29, found: 593.20 (I = 100%).
173b
(S)-2-amino-4-((((1S,2S,3S,5S)-5-(6-amino-9H-purin- 9-yl)-3-((tert-
butyldimethylsilyl)oxy)bicyclo[3.1.0]hexan-2-yl)met hyl)thio)butanoic acid
TBS-protected acid 173a (3.5 mg) was dissolved in EtOAc (0.2 ml), HCl (360 ul, 1M- sol. in
EtOAc) was added and the reaction mixture was stirred for 3 d at 40°C. A white precipitate
formed. The solvent was evaporated and the crude product was dried under high vacuum
conditions to give 4 mg (>100%) of a white solid.
Rf = 0.45 (acetic acid/BuOH/H2O 1:3:1, ninhydrin)
MS (ESI): calc. for C22H37N6O3SSi [M+H]+ : 493.24, found: 493.10 (I = 100%).
Note: Compound 7 also formed but the deprotection reaction did not go to completion with
the reagent used. TLC analysis indicated a 1:1 mixture of 173b and 7.
7
(S)-2-amino-4-((((1S,2S,3S,5S)-5-(6-amino-9H-purin- 9-yl)-3-hydroxybicyclo[3.1.0]hexan-
2-yl)methyl)thio)butanoic acid
Fully protected acid 173 (5 mg) was dissolved in CH2Cl2 (0.5 ml), TFA (200 ul) was added
and RM stirred for 24 h at RT. After this time, TLC and MS indicated that all Boc-groups had

Experimental Section
216
been removed but that the TBS group was still on. H2O (20 ul) was added and the reaction
mixture was stirred at RT for additional 18 h. TLC and MS indicated end of reaction. The
solvents and the TFA were evaporated under high vacuum conditions. The residue was
dissolved in water and extracted with EtOAc. The water phase was lyophylized to yield 7 mg
(>100%) of the crude product as a TFA salt in the form of a colorless solid.
HPLC analysis indicated the presence of two compounds with retention times 6.3 min and 8
min; they were separated by semi-preparative HPLC using an Agilent Zorbax SB-Phenyl
column (5 µm, 9.4 x 150 mm) using CH3CN with 0.025 % TFA and water with 0.03% TFA as
eluents. Subsequent analytical HPLC (Agilent Zorbax SB-Phenyl column, 3.5 µm, 4.6 x 150
mm) displayed the same two peaks (after 6.3 min and 8 min). Since the NMR data were
identical, we believe that compound 7 exists in a pH-dependent equilibrium of two forms.
Alternatively, 7 was prepared from partially deprotected acid 173b. 173b (4 mg) was
dissolved in CH2CH2 (0.5 ml), TFA (50 ul) was added and the reaction mixture was stirred for
12 h at RT. Additional 50 ul of TFA and 10 ul of H2O were added and the reaction mixture
was stirred for additional 8 h. The solvents were evaporated and the crude product was dried
under high vacuum conditions to yield 5 mg of a slightly green solid that was dissolved in
water (0.5 ml) and extracted with EtOAc (0.3 ml). The organic phase was removed and the
water phase was lyophilizised to afford 4.2 mg (>100%) of the crude product as a TFA salt in
the form of a colorless solid. This batch was also subjected to purification by semi-
preparative HPLC using the same conditions as above. Both batches combined yielded 2.8
mg of 7 after purification.
Rf = 0.3 (acetic acid/BuOH/H2O 1:3:1, ninhydrin)
1H NMR (500 MHz, D2O): δ = 8.44 (s, 1H), 8.40 (s, 1H), 4.41 (dd, J = 6.9, 1.0 Hz, 1H), 3.96
(dt, J = 6.7, 3.4 Hz, 1H), 3.04 (dd, J = 13.1, 7.3 Hz, 1H), 2.95 (dd, J = 13.1, 7.8 Hz, 1H), 2.81
(t, J = 7.6 Hz, 2H), 2.65 (ddd, J = 14.4, 7.0, 2.1 Hz, 1H), 2.39 (d, J = 14.2 Hz, 1H), 2.29 (t, J =
7.4 Hz, 2H), 2.25 – 2.14 (m, 2H), 1.67 – 1.61 (m, 1H), 1.54 (ddd, J = 9.7, 5.9, 2.1 Hz, 1H).
13C-NMR (500 MHz, D2O): δ = 173.8, 150.4, 149.5, 145.3, 144.6, 118.6, 75.9, 53.8, 48.8,
42.9, 40.2, 34.3, 30.5, 28.1, 26.9, 17.9.
HR-MS (ESI): calcd for C16H23N6O3S [M-H]+: 379.1547, found: 379.1554.
Biological Evaluation of S-Adenosylhomocysteine Ana logue 7:
SAH Analogue 7 has been tested for activity on dengue N-7 and O-2’ methyltransferases by
Dr. Sebastian Sonntag. Protocols as described in 173 and 172 were used, with the following
modifications in RNA substrate and buffer conditions:

Experimental Section
217
Buffer conditions: the O-2’ assay buffer was 50 mM Tris (pH 7.5), 10 mM KCl, 2mM MgCl2,
0.05% CHAPS; the N-7 assay buffer was 50 mM Tris (pH 7.5), 75 mM NaCl, 0.01% TritonX-
100.
RNA substrates: For the N-7 assay, the biotinylated substrate was 110 nucleotides in length,
and used at a final concentration of 240 nM. For the O-2’ assay, the biotinylated RNA was an
8mer, and was used at a final concentration of 40 nM.

Curiculum Vitae
218
6 Curriculum Vitae
PERSONAL DETAILS
Name Anna Schlegel
Date of Birth 9.11.1977
Place of Birth Arbon, Switzerland
Nationality Swiss
EXPERIENCE
05.2005 – present PhD student in the group of Prof. Dr. Karl-Heinz Al tmann,
Institut of Pharmaceutical Sciences, Departement of Chemistry and Applied Biosiences (D-CHAB), ETH Zurich
• „Synthesis of Chemical Libraries based on a Bicyclo[3.1.0]hexane Scaffold and Studies on the Synthesis of Bicyclo[3.1.0]hexane- based S-Adenosylhomocysteine and Pentostatin Analogues“
02.2010 – 11.2010 Teacher of Mathematics and Physics
at “Katholisches Gymnasium Zürich“
• teaching of physics and mathematics
01.2005 - 05.2005 Associate Researcher, Flisom AG, Technopark Zurich
• development of thin film solar cells
06.2004 - 10.2004 Trainee, section research and development, medicinal chemistry,
F. Hoffmann - La Roche AG
• up-scaling and optimisation of organic syntheses
07.2002 - 09.2002 Trainee at University of Manchester
• design and construction of a detector of molecular negative ions
10.2001 – 06.2003 Junior Assistant at the Department of Mathematics, ETH Zurich
• teaching of Analysis I und II

Curiculum Vitae
219
EDUCATION
01.2004 – 04.2004 Diploma thesis in the group of Prof. Dr. Francois D iederich,
Institute of Organic Chemistry, Departement of Chem istry and Applied Biosiences (D-CHAB), ETH Zurich
„Long-arm Cavitands via Sonogashira Couplings: Synthesis and NMR Studies”
1998 - 2004 Master of Science ETH
in Interdisciplinary Sciences, ETH Zurich
with majors
organic chemistry, physical chemistry
quantum electronics, particle physics
1992 - 1998 Grammar School in Romanshorn
PUBLICATIONS
Vladimir A.Azov, Anna Schlegel, Francois Diederich
Functionalized calix[4]resorcinarene cavitands. Ver satile platforms for the modular
construction of extended molecular switches
Bulletin of the Chemical Society of Japan 2006, 79(12), 1926-1940
Vladimir A.Azov, Anna Schlegel, Francois Diederich
Geometrically precisely defined multinanometer expa nsion/contraction motions in a
resorcin[4]arene cavitand based molecular switch
Angewandte Chemie Int. Ed. 2005, 44(29), 4635-4638

Bibliography
220
7 Bibliography
[1] K. H. Bleicher, H.-J. Bohm, K. Muller, A. I. Alanine, Nat Rev Drug Discov 2003, 2, 369-378.
[2] G. M. Keseru, G. M. Makara, Drug Discovery Today 2006, 11, 741-748.
[3] D. C. Rees, M. Congreve, C. W. Murray, R. Carr, Nat Rev Drug Discov 2004, 3, 660-672.
[4] S. Ghosh, A. Nie, J. An, Z. Huang, Current Opinion in Chemical Biology 2006, 10, 194-202.
[5] M. R. Reddy, L. Parrill Abby, in Rational Drug Design, Vol. 719, American Chemical Society,
1999, pp. 1-11.
[6] R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R.
P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer, G. S. Sittampalam, Nat Rev Drug Discov
2011, 10, 188-195.
[7] O. Roche, P. Schneider, J. Zuegge, W. Guba, M. Kansy, A. Alanine, K. Bleicher, F. Danel, E.-M.
Gutknecht, M. Rogers-Evans, W. Neidhart, H. Stalder, M. Dillon, E. Sjögren, N. Fotouhi, P.
Gillespie, R. Goodnow, W. Harris, P. Jones, M. Taniguchi, S. Tsujii, W. von der Saal, G.
Zimmermann, G. Schneider, Journal of Medicinal Chemistry 2001, 45, 137-142.
[8] T. W. J. Cooper, I. B. Campbell, S. J. F. Macdonald, Angewandte Chemie International Edition
2010, 49, 8082-8091.
[9] aK. M. Hoeld, N. S. Sirisoma, J. E. Casida, Chem. Res. Toxicol. 2001, 14, 589-595; bT. Deiml,
2003.
[10] P.-L. Fang, Y.-L. Cao, H. Yan, L.-L. Pan, S.-C. Liu, N.-B. Gong, Y. Lü, C.-X. Chen, H.-M. Zhong, Y.
Guo, H.-Y. Liu, Journal of Natural Products 2011, 74, 1408-1413.
[11] S.-P. Yang, Z.-B. Gao, Y. Wu, G.-Y. Hu, J.-M. Yue, Tetrahedron 2008, 64, 2027-2034.
[12] J. Kawabata, Y. Fukushi, S. Tahara, J. Mizutani, Phytochemistry 1990, 29, 2332-2334.
[13] X. S. Peng, Y. S. Lu, Org Lett 2011, 13, 2940-2943.
[14] A. F. Cameron, K. K. Cheung, G. Ferguson, Robertso.Jm, J Chem Soc B 1969, 559-&.
[15] W. Haas, H. Sterk, M. Mittelbach, J. Nat. Prod. 2002, 65, 1434-1440.
[16] W. Haas, H. Sterk, M. Mittelbach, Journal of Natural Products 2002, 65, 1434-1440.
[17] aD. A. D. Eric V. Anslyn, p. 101; bK. S. Pitzer, W. E. Donath, Journal of the American Chemical
Society 1959, 81, 3213-3218.
[18] R. L. Cook, T. Malloy, Journal of the American Chemical Society 1974, 96, 1703-1707.
[19] V. S. Mastryukov, E. L. Osina, L. V. Vilkov, R. L. Hilderbrandt, Journal of the American Chemical
Society 1977, 99, 6855-6861.
[20] M. F. Grostic, D. J. Duchamp, C. G. Chidester, The Journal of Organic Chemistry 1971, 36,
2929-2932.
[21] P. K. Freeman, M. F. Grostic, F. A. Raymond, The Journal of Organic Chemistry 1965, 30, 771-
777.
[22] S. Winstein, E. C. Friedrich, R. Baker, Y.-I. Lin, Tetrahedron 1966, 22, 621-645.
[23] R. J. Abraham, C. M. Holden, P. Loftus, D. Whittaker, Organic Magnetic Resonance 1974, 6,
184-189.
[24] K. Siam, J. D. Ewbank, L. Schäfer, C. V. Alsenoy, Journal of Molecular Structure: THEOCHEM
1987, 150, 121-128.
[25] P. Aped, N. L. Allinger, Journal of the American Chemical Society 1992, 114, 1-16.
[26] R. Okazaki, J. Niwa, S. Kato, B Chem Soc Jpn 1988, 61, 1619-1624.
[27] aL. L. Koh, H. H. Huang, K. Y. Sim, Acta Crystallographica Section C 1992, 48, 1641-1645; bR.
W. W. Hooft, J. Kroon, Acta Crystallographica Section C 1995, 51, 721-723.
[28] aV. G. Puranik, Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1988, C44, 163-164; bD. G.
Morris, P. Murray-Rust, J. Murray-Rust, J. Chem. Soc., Perkin Trans. 2 1977, 1577-1580.
[29] C. Altona, Sundaral.M, Journal of the American Chemical Society 1972, 94, 8205-&.

Bibliography
221
[30] V. E. Marquez, H. R. Moon, M. A. Siddiqui, G. Sun, I. V. Filippov, N. A. Landsman, Y. C. Lee, K.
M. Adams, J. J. Barchi, J. R. Deschamps, M. C. Nicklaus, J. A. Kelley, Tetrahedron 2010, 66,
6707-6717.
[31] J. Plavec, W. Tong, J. Chattopadhyaya, Journal of the American Chemical Society 1993, 115,
9734-9746.
[32] V. E. Marquez, M. A. Siddiqui, A. Ezzitouni, P. Russ, J. Y. Wang, R. W. Wagner, M. D.
Matteucci, Journal of Medicinal Chemistry 1996, 39, 3739-3747.
[33] aV. E. Marquez, A. Ezzitouni, P. Russ, M. A. Siddiqui, H. Ford, R. J. Feldman, H. Mitsuya, C.
George, J. J. Barchi, Journal of the American Chemical Society 1998, 120, 2780-2789; bV. E.
Marquez, P. Russ, R. Alonso, M. A. Siddiqui, K. J. Shin, C. George, M. C. Nicklaus, F. Dai, H.
Ford, Nucleosides Nucleotides & Nucleic Acids 1999, 18, 521-530.
[34] V. E. Marquez, A. Ezzitouni, M. A. Siddiqui, P. Russ, H. Ikeda, C. George, Nucleosides
Nucleotides 1997, 16, 1431-1434.
[35] V. E. Marquez, Abstr Pap Am Chem S 2008, 236.
[36] E. De Clercq, Current Opinion in Pharmacology 2010, 10, 507-515.
[37] P. A. Furman, J. A. Fyfe, M. H. St Clair, K. Weinhold, J. L. Rideout, G. A. Freeman, S. N.
Lehrman, D. P. Bolognesi, S. Broder, H. Mitsuya, Proceedings of the National Academy of
Sciences 1986, 83, 8333-8337.
[38] G. R. Painter, A. E. Aulabaugh, C. W. Andrews, Biochemical and Biophysical Research
Communications 1993, 191, 1166-1171.
[39] Y. Choi, C. George, M. J. Comin, J. J. Barchi, H. S. Kim, K. A. Jacobson, J. Balzarini, H. Mitsuya,
P. L. Boyer, S. H. Hughes, V. E. Marquez, Journal of Medicinal Chemistry 2003, 46, 3292-3299.
[40] aJ. Balzarini, P. Herdewijn, E. Declercq, J Biol Chem 1989, 264, 6127-6133; bW. Y. Gao, R.
Agbaria, J. S. Driscoll, H. Mitsuya, J Biol Chem 1994, 269, 12633-12638.
[41] V. E. Marquez, S. H. Hughes, S. Sei, R. Agbaria, Antiviral Research 2006, 71, 268-275.
[42] M. J. Comin, B. C. Vu, P. L. Boyer, C. Liao, S. H. Hughes, V. E. Marquez, Chemmedchem 2008,
3, 1129-1134.
[43] V. E. Marquez, Y. Choi, M. J. Comin, P. Russ, C. George, M. Huleihel, T. Ben-Kasus, R. Agbaria,
Journal of the American Chemical Society 2005, 127, 15145-15150.
[44] aK. H. Altmann, R. Kesselring, E. Francotte, G. Rihs, Tetrahedron Lett 1994, 35, 2331-2334;
bV. E. Marquez, T. Ben-Kasus, J. J. Barchi, Jr., K. M. Green, M. C. Nicklaus, R. Agbaria, Journal
of the American Chemical Society 2004, 126, 543-549.
[45] W. Saenger, Springer, 1984, pp. 51-104.
[46] V. E. Marquez, Y. Choi, M. J. Comin, P. Russ, C. George, M. Huleihel, T. Ben-Kasus, R. Agbaria,
Journal of the American Chemical Society 2005, 127, 15145-15150.
[47] M. J. Comin, R. Agbaria, T. Ben-Kasus, M. Huleihel, C. Liao, G. Sun, M. C. Nicklaus, J. R.
Deschamps, D. A. Parrish, V. E. Marquez, Journal of the American Chemical Society 2007, 129,
6216-6222.
[48] aK. M. Reilly, D. F. Kisor, Onco Targets Ther 2009, 2, 219-228; bM. S. Czuczman, P. Porcu, J.
Johnson, D. Niedzwiecki, M. Kelly, E. D. Hsi, J. R. Cook, G. Canellos, B. D. Cheson, Leuk
Lymphoma 2007, 48, 97-103; cM. S. Hershfield, Eur J Immunol 2005, 35, 25-30.
[49] G. Hasko, B. N. Cronstein, Trends Immunol 2004, 25, 33-39.
[50] R. Franco, R. Pacheco, J. M. Gatell, T. Gallart, C. Lluis, Crit Rev Immunol 2007, 27, 495-509.
[51] M. Asakura, H. Asanuma, J. Kim, Y. Liao, K. Nakamaru, M. Fujita, K. Komamura, T. Isomura, H.
Furukawa, H. Tomoike, M. Kitakaze, Hypertens Res 2007, 30, 781-787.
[52] R. H. Adamson, D. W. Zaharevitz, D. G. Johns, Pharmacology 1977, 15, 84-89.
[53] R. P. Agarwal, T. Spector, R. E. Parks, Jr., Biochem Pharmacol 1977, 26, 359-367.
[54] aT. Robak, E. Lech-Maranda, A. Korycka, E. Robak, Curr Med Chem 2006, 13, 3165-3189; bP.
P. Major, R. P. Agarwal, D. W. Kufe, Blood 1981, 58, 91-96.
[55] R. S. Hosmane, Research Signpost, 2002, pp. 133-151.
[56] Z. Wang, F. A. Quiocho, Biochemistry 1998, 37, 8314-8324.

Bibliography
222
[57] E. T. Larson, W. Deng, B. E. Krumm, A. Napuli, N. Mueller, W. C. Van Voorhis, F. S. Buckner, E.
Fan, A. Lauricella, G. DeTitta, J. Luft, F. Zucker, W. G. J. Hol, C. L. M. J. Verlinde, E. A. Merritt,
Journal of Molecular Biology 2008, 381, 975-988.
[58] L.-M. Ting, W. Shi, A. Lewandowicz, V. Singh, A. Mwakingwe, M. R. Birck, E. A. T. Ringia, G.
Bench, D. C. Madrid, P. C. Tyler, G. B. Evans, R. H. Furneaux, V. L. Schramm, K. Kim, J Biol
Chem 2005, 280, 9547-9554.
[59] K. Kim, L. M. Ting, W. X. Shi, A. Lewandowicz, V. Singh, A. Mwakingwe, M. R. Birck, E. A. T.
Ringia, G. Bench, D. C. Madrid, P. C. Tyler, G. B. Evans, R. H. Furneaux, V. L. Schramm, J Biol
Chem 2005, 280, 9547-9554.
[60] P. C. Tyler, E. A. Taylor, R. F. G. Fröhlich, V. L. Schramm, Journal of the American Chemical
Society 2007, 129, 6872-6879.
[61] T. Cacciamani, A. Vita, G. Cristalli, S. Vincenzetti, P. Natalini, S. Ruggieri, A. Amici, G. Magni,
Arch Biochem Biophys 1991, 290, 285-292.
[62] M. Lemaire, L. F. Momparler, M. L. Bernstein, V. E. Marquez, R. L. Momparler, Anticancer
Drugs 2005, 16, 301-308.
[63] O. R. Ludek, G. K. Schroeder, C. Liao, P. L. Russ, R. Wolfenden, V. E. Marquez, Journal of
Organic Chemistry 2009, 74, 6212-6223.
[64] aS. Xiang, S. A. Short, R. Wolfenden, C. W. Carter, Biochemistry 1995, 34, 4516-4523; bA.-H.
Teh, M. Kimura, M. Yamamoto, N. Tanaka, I. Yamaguchi, T. Kumasaka, Biochemistry 2006, 45,
7825-7833; cS. J. Chung, J. C. Fromme, G. L. Verdine, Journal of Medicinal Chemistry 2005, 48,
658-660.
[65] F. Lyko, R. Brown, Journal of the National Cancer Institute 2005, 97, 1498-1506.
[66] P. A. Jones, S. B. Baylin, Nat Rev Genet 2002, 3, 415-428.
[67] S. P. Lim, L. S. Sonntag, C. Noble, S. H. Nilar, R. H. Ng, G. Zou, P. Monaghan, K. Y. Chung, H.
Dong, B. Liu, C. Bodenreider, G. Lee, M. Ding, W. L. Chan, G. Wang, Y. L. Jian, A. T. Chao, J.
Lescar, Z. Yin, T. R. Vedananda, T. H. Keller, P.-Y. Shi, J. Biol. Chem. 2011, 286, 6233-6240.
[68] R. J. Roberts, X. Cheng, Annu Rev Biochem 1998, 67, 181-198.
[69] P. Wang, A. S. Brank, N. K. Banavali, M. C. Nicklaus, V. E. Marquez, J. K. Christman, A. D.
MacKerell, Journal of the American Chemical Society 2000, 122, 12422-12434.
[70] S. A. Poulsen, R. J. Quinn, Bioorg Med Chem 1998, 6, 619-641.
[71] C. E. Muller, Curr Med Chem 2000, 7, 1269-1288.
[72] S. Tchilibon, B. V. Joshi, S. K. Kim, H. T. Duong, Z. G. Gao, K. A. Jacobson, Journal of Medicinal
Chemistry 2005, 48, 1745-1758.
[73] K. A. Jacobson, X. D. Ji, A. H. Li, N. Melman, M. A. Siddiqui, K. J. Shin, V. E. Marquez, R. G. Ravi,
Journal of Medicinal Chemistry 2000, 43, 2196-2203.
[74] C. S. Carr, R. J. Hill, H. Masamune, S. P. Kennedy, D. R. Knight, W. R. Tracey, D. M. Yellon,
Cardiovascular Research 1997, 36, 52-59.
[75] K. A. Jacobson, Z. G. Gao, S. Tchilibon, H. T. Duong, B. V. Joshi, D. Sonin, B. T. Liang, Journal of
Medicinal Chemistry 2005, 48, 8103-8107.
[76] M. V. Sitkovsky, A. Ohta, Trends Immunol 2005, 26, 299-304.
[77] K. A. Jacobson, J.-M. Boeynaems, Drug Discovery Today 2010, 15, 570-578.
[78] H. S. Kim, M. Ohno, B. Xu, H. O. Kim, Y. Choi, X. D. Ji, S. Maddileti, V. E. Marquez, T. K. Harden,
K. A. Jacobson, Journal of Medicinal Chemistry 2003, 46, 4974-4987.
[79] D. M. Bourdon, S. K. Mahanty, K. A. Jacobson, J. L. Boyer, T. K. Harden, J Thromb Haemost
2006, 4, 861-868.
[80] K. A. Jacobson, S. Costanzi, A. A. Ivanov, S. Tchilibon, P. Besada, Z. G. Gao, S. Maddileti, T. K.
Harden, Biochem Pharmacol 2006, 71, 540-549.
[81] H. Ko, I. Fricks, A. A. Ivanov, T. K. Harden, K. A. Jacobson, Journal of Medicinal Chemistry
2007, 50, 2030-2039.
[82] K. A. Jacobson, S. Costanzi, B. V. Joshi, P. Besada, D. H. Shin, H. Ko, A. A. Ivanov, L.
Mamedova, Agonists and Antagonists for P2 Receptors, John Wiley & Sons, Ltd, 2008.

Bibliography
223
[83] S. Costanzi, B. V. Joshi, S. Maddileti, L. Mamedova, M. J. Gonzalez-Moa, V. E. Marquez, T. K.
Harden, K. A. Jacobson, Journal of Medicinal Chemistry 2005, 48, 8108-8111.
[84] Y. Liu, F.-J. Nan, Tetrahedron Lett 2010, 51, 1374-1376.
[85] Y.-S. Lu, X.-S. Peng, Org Lett 2011, 13, 2940-2943.
[86] J. Li, T. L. Lowary, Org Lett 2008, 10, 881-884.
[87] aS. Berg, D. Kaur, M. Jackson, P. J. Brennan, Glycobiology 2007, 17, 35-56R; bM. Belanova, P.
Dianiskova, P. J. Brennan, G. C. Completo, N. L. Rose, T. L. Lowary, K. Mikusova, J Bacteriol
2008, 190, 1141-1145.
[88] A. Srikrishna, P. Praveen Kumar, Tetrahedron 2000, 56, 8189-8195.
[89] O. R. Ludek, V. E. Marquez, Synthesis 2007, 2007, 3451,3460.
[90] V. E. Marquez, M. I. Lim, C. K. H. Tseng, A. Markovac, M. A. Priest, M. S. Khan, B. Kaskar, The
Journal of Organic Chemistry 1988, 53, 5709-5714.
[91] J. A. Staroscik, B. Rickborn, J. Org. Chem. 1972, 37, 738-740.
[92] L. S. Jeong, V. E. Marquez, C. S. Yuan, R. T. Borchardt, Heterocycles 1995, 41, 2651-2656.
[93] M. A. Siddiqui, H. Ford, C. George, V. E. Marquez, Nucleosides & Nucleotides 1996, 15, 235-
250.
[94] A. Ezzitouni, P. Russ, V. E. Marquez, The Journal of Organic Chemistry 1997, 62, 4870-4873.
[95] aK. Biggadike, A. D. Borthwick, D. Evans, A. M. Exall, B. E. Kirk, S. M. Roberts, L. Stephenson,
P. Youds, Journal of the Chemical Society, Perkin Transactions 1 1988, 549-554; bO. R. Ludek,
C. Meier, Nucleosides Nucleotides Nucleic Acids 2003, 22, 683-685.
[96] N. Kondo, H. Fueno, H. Fujimoto, M. Makino, H. Nakaoka, I. Aoki, S. Uemura, Journal of
Organic Chemistry 1994, 59, 5254-5263.
[97] Y. Choi, H. R. Moon, Y. Yoshimura, V. E. Marquez, Nucleosides, Nucleotides and Nucleic Acids
2003, 22, 547 - 557.
[98] N. Cohen, Organic Synthesis 1985, 63.
[99] B. V. Joshi, A. Melman, R. L. Mackman, K. A. Jacobson, Nucleosides Nucleotides Nucleic Acids
2008, 27, 279-291.
[100] L. A. Paquette, S. Bailey, The Journal of Organic Chemistry 1995, 60, 7849-7856.
[101] Y.-X. Yang, Z. Li, H.-J. Feng, G.-R. Chen, Y.-C. Li, Tetrahedron Lett 2010, 51, 3848-3851.
[102] S. Takano, A. Kurotaki, M. Takahashi, K. Ogasawara, Synthesis 1986, 1986, 403,406.
[103] K.-H. Altmann, R. Imwinkelried, R. Kesselring, G. Rihs, Tetrahedron Lett 1994, 35, 7625-7628.
[104] A. Ezzitouni, V. E. Marquez, Journal of the Chemical Society, Perkin Transactions 1 1997,
1073-1078.
[105] H. Saneyoshi, J. R. Deschamps, V. E. Marquez, J. Org. Chem. 2010, 75, 7659-7669.
[106] S.-H. Kim, M. J. Sung, K. C. Jin, Org. Synth. 2003, 80, No pp. given.
[107] T. H. Keller, A. Pichota, Z. Yin, Current Opinion in Chemical Biology 2006, 10, 357-361.
[108] P. Das, C. P. Spears, A. H. Shahinian, S. K. Dasgupta, N. G. Kundu, Bioorganic & Medicinal
Chemistry Letters 1996, 6, 2477-2480.
[109] aZ. Jin, Natural Product Reports 2009, 26, 382-445; bM. V. N. de Souza, Journal of Sulfur
Chemistry 2005, 26, 429-449.
[110] H.-J. Gais, H. J. Lindner, T. Lied, K. L. Lukas, W. A. Ball, B. Rosenstock, H. Sliwa, Liebigs Annalen
der Chemie 1986, 1986, 1179-1212.
[111] T. Van Truong, H. Rapoport, The Journal of Organic Chemistry 1993, 58, 6090-6096.
[112] R. T. Borchardt, J. A. Huber, Y. S. Wu, Journal of Medicinal Chemistry 1976, 19, 1094-1099.
[113] R. T. Borchardt, Journal of Medicinal Chemistry 1980, 23, 347-357.
[114] M. Milani, E. Mastrangelo, M. Bollati, B. Selisko, E. Decroly, M. Bouvet, B. Canard, M.
Bolognesi, Antiviral Research 2009, 83, 28-34.
[115] R. T. Borchardt, Y. S. Wu, Journal of Medicinal Chemistry 1975, 18, 300-304.
[116] A. Ezzitouni, V. E. Marquez, Journal of the Chemical Society-Perkin Transactions 1 1997, 1073-
1078.

Bibliography
224
[117] aH. J. Gais, H. J. Lindner, T. Lied, K. L. Lukas, W. A. Ball, B. Rosenstock, H. Sliwa, Liebigs
Annalen Der Chemie 1986, 1179-1212; bH. J. Gais, K. L. Lukas, W. A. Ball, S. Braun, H. J.
Lindner, Liebigs Annalen Der Chemie 1986, 687-716.
[118] T. P. Mawhinney, M. A. Madson, The Journal of Organic Chemistry 1982, 47, 3336-3339.
[119] S. Dong, L. A. Paquette, The Journal of Organic Chemistry 2006, 71, 1647-1652.
[120] aY. Mori, K. Yaegashi, H. Furukawa, The Journal of Organic Chemistry 1998, 63, 6200-6209;
bA. Fürstner, C. Mathes, C. W. Lehmann, Chemistry – A European Journal 2001, 7, 5299-5317.
[121] T. M. Willson, P. Kocienski, K. Jarowicki, K. Isaac, P. M. Hitchcock, A. Faller, S. F. Campbell,
Tetrahedron 1990, 46, 1767-1782.
[122] K.-H. Altmann, R. Kesselring, U. Pieles, Tetrahedron 1996, 52, 12699-12722.
[123] Y. Li, Synlett 2010, 2010, 837,838.
[124] aM. Takebayashi, Kagakushi Kenkyu (Narashino, Jpn.) 1989, 16, 149-153; bA. Bertho, Journal
für Praktische Chemie 1928, 120, 89-118; cT. Curtius, Journal für Praktische Chemie 1916, 94,
273-299.
[125] J. H. Saunders, R. J. Slocombe, Chemical Reviews 1948, 43, 203-218.
[126] T. Shioiri, K. Ninomiya, S. Yamada, Journal of the American Chemical Society 1972, 94, 6203-
6205.
[127] S. Linke, G. T. Tisue, W. Lwowski, Journal of the American Chemical Society 1967, 89, 6308-
6310.
[128] A. Speicher, T. Klaus, T. Eicher, Journal für Praktische Chemie/Chemiker-Zeitung 1998, 340,
581-583.
[129] T. Holletz, D. Cech, Synthesis 1994, 1994, 789,791.
[130] E. J. Thomas, J. W. F. Whitehead, Journal of the Chemical Society, Perkin Transactions 1 1989,
507-518.
[131] aP. B. Alper, H. Liu, A. K. Chatterjee, K. T. Nguyen, D. C. Tully, C. Tumanut, J. Li, J. L. Harris, T.
Tuntland, J. Chang, P. Gordon, T. Hollenbeck, D. S. Karanewsky, Bioorganic & Medicinal
Chemistry Letters 2006, 16, 1486-1490; bD. C. Tully, H. Liu, A. K. Chatterjee, P. B. Alper, R.
Epple, J. A. Williams, M. J. Roberts, D. H. Woodmansee, B. T. Masick, C. Tumanut, J. Li, G.
Spraggon, M. Hornsby, J. Chang, T. Tuntland, T. Hollenbeck, P. Gordon, J. L. Harris, D. S.
Karanewsky, Bioorganic & Medicinal Chemistry Letters 2006, 16, 5112-5117.
[132] A. Melman, M. Zhong, V. E. Marquez, K. A. Jacobson, The Journal of Organic Chemistry 2008,
73, 8085-8088.
[133] G. Shaw, R. N. Warrener, Journal of the Chemical Society (Resumed) 1958, 157-161.
[134] B. N. Yoo, H. O. Kim, H. R. Moon, S. K. Seol, S. K. Jang, K. M. Lee, L. S. Jeong, Bioorganic &
Medicinal Chemistry Letters 2006, 16, 4190-4194.
[135] D. J. P. Pinto, M. J. Orwat, S. Koch, K. A. Rossi, R. S. Alexander, A. Smallwood, P. C. Wong, A.
R. Rendina, J. M. Luettgen, R. M. Knabb, K. He, B. Xin, R. R. Wexler, P. Y. S. Lam, Journal of
Medicinal Chemistry 2007, 50, 5339-5356.
[136] P. A. Jacobi, H. G. Selnick, Journal of Organic Chemistry 1990, 55, 202-209.
[137] P. R. Krishna, V. V. R. Reddy, R. Srinivas, Tetrahedron 2007, 63, 9871-9880.
[138] G. Stupka, L. Gremaud, A. F. Williams, Helvetica Chimica Acta 2005, 88, 487-495.
[139] D. L. Evans, D. K. Minster, U. Jordis, S. M. Hecht, A. L. Mazzu, A. I. Meyers, The Journal of
Organic Chemistry 1979, 44, 497-501.
[140] S. Kreimerman, I. Ryu, S. Minakata, M. Komatsu, Comptes Rendus de l'Académie des Sciences
- Series IIC - Chemistry 2001, 4, 497-503.
[141] S. Crosignani, A. C. Young, B. Linclau, Tetrahedron Lett 2004, 45, 9611-9615.
[142] N. A. Braun, M. Ousmer, J. D. Bray, D. Bouchu, K. Peters, E.-M. Peters, M. A. Ciufolini, The
Journal of Organic Chemistry 2000, 65, 4397-4408.
[143] T. Aoyama, N. Sonoda, M. Yamauchi, K. Toriyama, M. Anzai, A. Ando, T. Shioiri, Synlett 1998,
1998, 35,36.
[144] C. Kashima, H. Arao, Synthesis 1989, 1989, 873,874.

Bibliography
225
[145] C. Wu, E. R. Decker, N. Blok, H. Bui, T. J. You, J. Wang, A. R. Bourgoyne, V. Knowles, K. L.
Berens, G. W. Holland, T. A. Brock, R. A. F. Dixon, Journal of Medicinal Chemistry 2004, 47,
1969-1986.
[146] Y. Kodama, M. Ori, T. Nishio, Helvetica Chimica Acta 2005, 88, 187-193.
[147] A. I. Meyers, D. L. Temple, Jr., J. Amer. Chem. Soc. 1970, 92, 6644-6646.
[148] D. Rai, M. Johar, T. Manning, B. Agrawal, D. Y. Kunimoto, R. Kumar, Journal of Medicinal
Chemistry 2005, 48, 7012-7017.
[149] C. F. Matta, A. A. Arabi, D. F. Weaver, European Journal of Medicinal Chemistry 2010, 45,
1868-1872.
[150] H. Mizufune, M. Nakamura, H. Mitsudera, Tetrahedron 2006, 62, 8539-8549.
[151] J. I. Concepcion, C. G. Francisco, R. Freire, R. Hernandez, J. A. Salazar, E. Suarez, J. Org. Chem.
1986, 51, 402-404.
[152] S. Kirkpatrick, C. D. Gelatt, M. P. Vecchi, Science 1983, 220, 671-680.
[153] V. Granville, M. Krivanek, J. P. Rasson, Pattern Analysis and Machine Intelligence, IEEE
Transactions on 1994, 16, 652-656.
[154] N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, E. Teller, Equation of State
Calculations by Fast Computing Machines, Vol. 21, AIP, 1953.
[155] C. A. G. Haasnoot, L. F. A. A. M. De, C. Altona, Tetrahedron 1980, 36, 2783-2792.
[156] aH. Rapoport, Y. W. Chen, R. M. Mohareb, J. H. Ahn, T. B. Sim, J. Z. Ho, Chem Pharm Bull
2003, 51, 1153-1156; bT. Vantruong, H. Rapoport, Journal of Organic Chemistry 1993, 58,
6090-6096.
[157] P. Serafinowski, E. Dorland, K. R. Harrap, J. Balzarini, C. E. De, J. Med. Chem. 1992, 35, 4576-
4583.
[158] K. A. Houck, W. P. Bocchinfuso, M. S. Dowless, K. M. Borchert, Drug Discovery Ser. 2006, 5,
357-369,, 351 plate.
[159] M. Whitney, E. Rockenstein, G. Cantin, T. Knapp, G. Zlokarnik, P. Sanders, K. Durick, F. F.
Craig, P. A. Negulescu, Nat Biotech 1998, 16, 1329-1333.
[160] A. Citri, Y. Yarden, Nat Rev Mol Cell Biol 2006, 7, 505-516.
[161] A. B. Tsoupras, M. Chini, N. Tsogas, A. Lioni, G. Tsekes, C. A. Demopoulos, M. C. Lazanas, J.
Inflammation (London, U. K.) 2011, 8, 17.
[162] S. Ulisse, E. Baldini, S. Sorrenti, M. D'Armiento, Curr. Cancer Drug Targets 2009, 9, 32-71.
[163] Q. Xiao, S. Tang, X. Fan, Xiandai Zhongliu Yixue 2008, 16, 674-676.
[164] V. Hook, G. Hook, M. Kindy, Biol. Chem. 2010, 391, 861-872.
[165] S. Ludewig, M. Kossner, M. Schiller, K. Baumann, T. Schirmeister, Curr. Top. Med. Chem.
(Sharjah, United Arab Emirates) 2010, 10, 368-382.
[166] C. Sala, N. Dhar, R. C. Hartkoorn, M. Zhang, Y. H. Ha, P. Schneider, S. T. Cole, Antimicrob.
Agents Chemother. 2010, 54, 4150-4158.
[167] A. Martin, M. Camacho, F. Portaels, J. C. Palomino, Antimicrob. Agents Chemother. 2003, 47,
3616-3619.
[168] D. Rai, M. Johar, T. Manning, B. Agrawal, D. Y. Kunimoto, R. Kumar, J. Med. Chem. 2005, 48,
7012-7017.
[169] D. Rai, M. Johar, N. C. Srivastav, T. Manning, B. Agrawal, D. Y. Kunimoto, R. Kumar, J Med
Chem 2007, 50, 4766-4774.
[170] A. Chicca, F. Pellati, B. Adinolfi, A. Matthias, I. Massarelli, S. Benvenuti, E. Martinotti, A. M.
Bianucci, K. Bone, R. Lehmann, P. Nieri, Br J Pharmacol 2008, 153, 879-885.
[171] N. A. Englert, B. C. Spink, D. C. Spink, J. Steroid Biochem. Mol. Biol. 2011, 123, 140-150.
[172] S. P. Lim, L. S. Sonntag, C. Noble, S. H. Nilar, R. H. Ng, G. Zou, P. Monaghan, K. Y. Chung, H. P.
Dong, B. P. Liu, C. Bodenreider, G. Lee, M. Ding, W. L. Chan, G. Wang, Y. L. Jian, A. T. Chao, J.
Lescar, Z. Yin, T. R. Vedananda, T. H. Keller, P. Y. Shi, J Biol Chem 2011, 286, 6233-6240.
[173] S. P. Lim, D. Wen, T. L. Yap, C. K. Yan, J. Lescar, S. G. Vasudevan, Antiviral Res. 2008, 80, 360-
369.
[174] S. P. Lim, K. Y. Chung, H. P. Dong, A. T. Chao, P. Y. Shi, J. Lescar, Virology 2010, 402, 52-60.

Bibliography
226


















