
Thermodynamics spring 2013
372
CY 11001 CHEMISTRY Professor: Dr. Amita (Pathak) Mahanty Office: Room C-301 (2nd Floor, Chemistry Department) e-mail: [email protected] phone: 83312 (office) Ground Rules for the Class • Mobile Phones in switched off mode • Attendance is a must and will be checked regularly • Vast syllabus and fast paced, advised to take class notes
-
Upload
abhishek-tripathi -
Category
Documents
-
view
2.147 -
download
13
Transcript of Thermodynamics spring 2013

CY 11001 CHEMISTRY
Professor: Dr. Amita (Pathak) MahantyOffice: Room C-301 (2nd Floor, Chemistry Department)e-mail: [email protected]: 83312 (office)
Ground Rules for the Class• Mobile Phones in switched off mode• Attendance is a must and will be checked regularly• Vast syllabus and fast paced, advised to take class notes

Physical: 3rd Jan, 2013 – 1st Feb, 2013 (11 lectures)
Inorganic: 7th February, 2013- 15th March, 2013 (12 lectures)
MID TERM SEMESTER EXAM … 18.02.2013 - 26.02.2013(Monday -Tuesday)
Organic: 18th March, 2013 - 12th April, 2013 (12 classes)
SCHEDULE CHEMISTRY THEORY COURSESECTIONS 1, 2 & 3
Room No. Day TimeMonday 4:30-5:25Thursday 4:30-5:25F-116
Friday 4:30-5:25Lecture Timings:

Sec & Group
Tutorial Room No
Tutorial Day
TutorialTimings
(AM)
Roll No
Sec: 11A S-126 Thursday 10:30-11.25 AE, AG, BT, CE, CH, CS, CY
Sec: 11B S-127 Thursday 10:30-11.25 EC, EE, EX, GG, HS, IE, IM, MA, ME10008
Sec: 11C F-131 Thursday 10:30-11.25 ME10022, MF, MI, MT, NA, PH, QD
Sec: 12A S-126 Monday 8.30 - 9.25 AE, AG, BT, CE, CH, CS, CY20010
Sec: 12B S-127 Monday 8.30 - 9.25 CY20024, EC, EE, EX, GG, HS, IE, IM, MA20032
Sec: 12C F-131 Monday 8.30 - 9.25 MA20046, ME, MF, MI, MT, NA, PH, QD
Tutorial Schedule for Spring 2012-13

Distribution of Marks:Mid Semester Exam (Only Physical): 30%End Semester Exam (Inorganic & Organic): 50%
Teachers Assessment (TA): 20%Break up: [Physical (6%), Inorganic (7%), Organic (7%)]
TA marks to be based on a CLASS-TEST: 30th January 2013
Text Book:Atkin’s Physical Chemistry 8th Ed
Reference Books:Physical Chemistry by Silbey, Alberty & Bawendy; Wiley.Physical Chemistry by I. N. Levine; McGrew.Physical Chemistry David W. Ball; Thompson.
CY 11001 CHEMISTRY

Thermodynamics of Chemical Processes:• Review of Essential Concepts and Definitions• Revision: Heat, Work & Energy; First Law of Thermodynamics• The Second Law of Thermodynamics • Concept of Entropy• Gibbs Free Energy • Equilibrium conditions for Closed Systems• Maxwell Relations • The Chemical Potential• Definition and Concept of Open Systems• Phase and Reaction Equilibria
Electrochemical Systems: • Electrochemical Cells and EMF• Applications of EMF Measurements: Thermodynamic data,
Activity Coefficients, Solubility product and pH
Kinetics of Chemical Reactions: • Reversible, Consecutive and Parallel Reactions, • Steady State Approximation, • Chain Reactions, Photochemical Kinetics
CY 11001 CHEMISTRY (L-T-P : 3-1-0; Credit : 4)

Thermodynamics provides a framework of relating the macroscopicproperties of a system to one another.
Thermodynamic: ↔ Classical, Statistical and Irreversible
Classical Thermodynamics, to the study of macroscopic properties of system/matter at equilibrium using the laws governing transformation of energy from one form to another.
It is concerned only with macroscopic quantities and ignores themicroscopic variables that characterize individual molecules.
THE BASIC CONCEPTS:System, the part of the world in which we have a special interest.Surroundings, the region outside the system where we make our
measurements.Boundary, a hypothetical/real barrier between system and
surroundings.
Thermodynamic

Thermodynamic Systems
Open systems can exchange both matter and energy with the environment. Closed systems exchange energy but not matter with the environment.Isolated systems can exchange neither energy nor matter with the environment.

System : Open, Closed, Isolated Surroundings Boundary / Wall• Rigid and Non rigid (movable)• Permeable and Impermeable (no matter is allowed to pass
through it) • Adiabatic and Diathermic boundary ( - permits the passage of
energy as heat )
A system surrounded by a rigid, impermeable, adiabatic wall –Isolated system Example: Universe as a whole, a thermo flask
Thermodynamic Systems

Thermodynamic Equilibrium
• Macroscopic properties do not change with time
• Thermodynamic State only defined by the present values of the state variables, not by the history
• A system in thermodynamic equilibrium is an equilibrium system
For Isolated system:
For Non isolated system:a) Macroscopic properties do not change with timeb) Removal of the system from contact with its surroundings causes
no change in the properties of the system
What are conditions of Thermodynamic Equilibrium?

The condition of equality of pressure on either side of a movable wall / boundary.
No unbalanced forces act on or within the system – does not undergo acceleration or no turbulence inside the system
Mechanical equilibrium

Thermal equilibrium
Thermal equilibrium between system and the surroundings, is a condition in which there is no change in the properties of the system or surroundings when they are separated by a thermally conducting wall.
In other words, Thermal Equilibrium is a condition in which no change of state occurs when two objects A to B are in contact through a diathermic boundary.

a) No net chemical reactions are occurring in the system
Concentrations of the chemical species in the various parts of the system are constant with time
Material Equilibrium
b) There is no net transfer of matter from one part (phase) of the system to another or between the system and its surroundings
For thermodynamic equilibrium, all three kinds of equilibrium must be present
Phase: a homogeneous part of a system is called a phase
No matter what is the initial state of an isolated system, eventually it will reach the state of thermodynamic equilibrium

How do we define a system? State of a System
Macroscopic state of a system can be specified by the values of a small number of macroscopic Properties/Parameters/Physical characteristics or attributes of a system - Thermodynamic variables
On the other hand, Microscopic state of a system needs description of each molecule – a very complicated picture
Thermodynamic variables which are experimentally measurable• Composition – mass of each chemical species that is present in each
phases, • pressure (P), volume (V), Temperature (T), density, etc.• field strength, if magnetic/electrical field act on the system• gravitational field
(N.B.: only the average values are considered, all fluctuation and perturbations are ignored).

For instance: The thermodynamic state of a single-phase system of fixed amounts of non reacting substances can be described by any two of the three state functions (P, V and T)
Thermodynamic State
If the value of every thermodynamic property in system A is same as in system B – then they are in the same thermodynamic state
The state of a thermodynamic system is defined by specifying the values of its thermodynamic properties – However, it is not necessary to specify all the properties to define the state of a system.
In fact, the macroscopic state of a system can be specified by the values of a small number of macroscopic variables – these called the Properties / Parameters / Thermodynamic variables of a system.
Equation of state, an equation that interrelates pressure, volume, temperature, and amount of substance: P = f (T, V, n).

The Equation of State of Ideal Gases
P – pressure [Newtons/m2]V – volume [m3]n – number of moles of gasT – the temperature in Kelvins [K]
R – a universal constantKmol
JR⋅
≈ 31.8
nRTPV =The ideal gas equation of state:
An equation of state - an equation that relates macroscopic variables (e.g., P, V, and T) for a given substance in thermodynamic equilibrium.In equilibrium (≡ No macroscopic motion), just a few macroscopic parameters are required to describe the state of a system.
Geometrical representation of the equation of state: f (P,V,T) = 0
P
V
T
an equilibriumstate
the equation-of-state surface

RecapThe gas laws
Boyle’s law: At constant temperature, the pressure of a sample of gas is inversely proportional to its volume, p ∝1/V at constant temperature.
Charles’ law: At constant pressure, the volume of a gas is proportional to the absolute tem perature, V ∝ T at constant pressure.
Avogadro’s principle: Equal volumes of gases at the same pressure and temperature contain the same numbers of molecules.
Dalton’s law: The pressure exerted by a mixture of gases isthe sum of the partial pressures of the gases.

Thermodynamic Systems
Homogeneous system, a system having its properties uniform throughout – a single phase solution / pure.
Heterogeneous system, a system with more than one phase.
Intensive properties Does not depend upon size (the quantity/amount of matter present) of
the system [e.g. Pressure, Temp, Viscosity, Density etc.]Total Property = property of part of the system
Extensive properties Depends upon size of the system [e.g. n, V, surface area etc ]
Extensive properties depend on the amount of substance in the system.]Total Property = Σ property (in each part of the system)
Other variables:Compositions, Surface area, Electrical/magnetic strengthGravitational field can/are generally ignored

A very important macroscopic parameter: Temperature
Temperature is a property associated with random motion ofmany particles. Introduction of the concept of temperature in thermodynamics is based on the the 0’th law of thermodynamics:
A well-defined quantity called Temperature exists such that two systems will be in thermal equilibrium if and only if both have the same temperature.
B acts as Thermometer.A, B, C all are at the same “temperature”.

Thermodynamic State Variables / Properties

Thermodynamic State Variables / Properties
• A few experimentally measureable macroscopic properties: p, T, V, n, m, ...• Knowledge if System is Homogeneous or Heterogeneous • Knowledge if System is in Equilibrium State • Knowledge of the number of components
Therefore, For describing the State of a System we require:

Change of State: Transformation
e.g., 3 H2 (g, 5 bar, 100 °C) → 3 H2 (g, 1 bar, 50 °C) (initial state) (final state)
Path: Sequence of Intermediate states through which a system passes to attain the final state
Process: Operation through which a system undergoes a change in state.Process describes the path.
Path can be:Reversible (always in Equilibrium)Irreversible (describes direction of time)Cyclic(initial and final state have the same value)
Processes can be:• Adiabatic (No heat transfer between system and surrounding)• Isobaric (Constant pressure process)• Isothermal (Constant temp process)• Constant Volume process • Endothermic & Exothermic process (q<0 & q>0)
Thermodynamic Process: Operation for Change in State
Path

Essential concepts / DefinitionsProcesses : When one or more of the parameters of a system changes, the state of the system also changes and the system is said to have undergone a process.
Various types of processes:• Isothermal (T), • Isobaric (P), • Isochoric (V)• Adiabatic (q),• Isenthalpic (H)• Exothermic & Endothermic• Cyclic
Each process can be either carried out Reversibly or Irreversibly

Thermodynamic Processes can be :
Irreversible Process: Real / Spontaneous• Occurs suddenly or spontaneously without the restriction of occurring in successive stages of infinitetesimal quantities. Irreversible processes cannot be undone by exactly reversing the change to the system.
• Do not remain in the virtual equilibrium during the transition.• The work (w) in the forward and backward processes would be unequal.
Reversible process: Ideal process• Change must occur in successive stages of infinitesimal quantities • Infinite duration
• Changes of the thermodynamic quantities in the different stages will be the same as in the forward direction but opposite in sign w.r.t. forward direction. The system and surroundings can be put back in their original states by exactly reversing the process.
• Virtual thermodynamic equilibrium, at each of the small stages.
Cyclic process: • Is one in which the initial and final states are the same. In such process:
• There is no change in the state variables (e.g., ∫dU = Uf-Ui = 0)• In contrast, path functions generally have non-zero values for cyclic
processes, dependent on the path (e.g., ∫δw ≠ (wf – wi) ≠ 0]
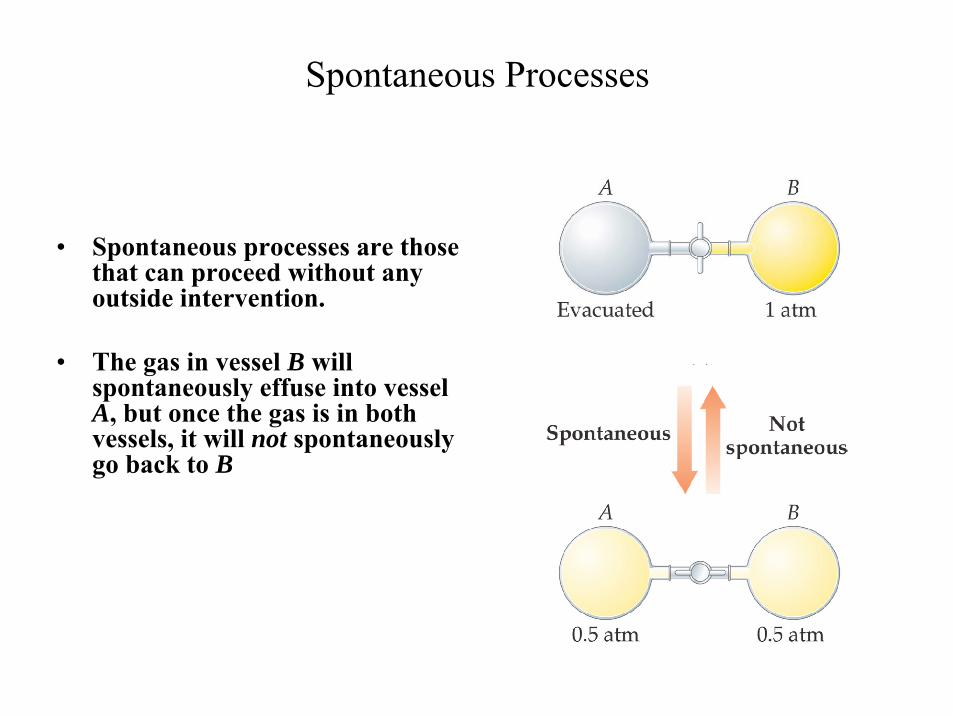
Spontaneous Processes
• Spontaneous processes are those that can proceed without any outside intervention.
• The gas in vessel B will spontaneously effuse into vessel A, but once the gas is in both vessels, it will not spontaneously go back to B

Reversible Processes
In a reversible process the system changes in such a way that the system and surroundings can be put back in their original states by exactly reversing the process.
Changes are infinitesimally small in a reversible process.

Irreversible Processes
• Irreversible processes cannot be undone by exactly reversing the change to the system.
• All Spontaneous processes are irreversible.• All Real processes are irreversible.

Energy is the capacity to do work.
Energy classification into: kinetic potential(by motion) (by position)
Is purely arbitrary! e.g.thermal chemical, electrical
• Heat and work are NOT “types” of ENERGY, but are processes involving TRANSFER of energy.
• They appear and disappear at the system boundary. They are path variables.• Heat is the transfer of energy from one body to another based on temperature
difference. They are path variables.• Heat stimulates random motion.• Convention: if heat flows into the system, q > 0.
• Work is the transfer of energy by some mechanism other than temperature difference.
• They appear and disappear at the system boundary. They are path variables.• Convention: if work is done on the system, w > 0.• Work stimulates organized motion.• Work “degrades” into heat.
Qualitative observations by Count Rumford (Ben Thompson)Quantitative measurements by James Joule
Energy, Work and Heat

Concept of Internal Energy
• E = K + V + U where, K and V are the macroscopic (not molecular) kinetic and potential energies of the system
• U is the internal energy of the body (due to molecular motions and intermolecular interactions)
• Internal Energy,U, is the total kinetic energy due to motion of molecules (translational, rotational, vibrational) the total potential energy associated with the vibrational and electric energy of atoms within molecules or crystals.
• Um (molar internal energy) = U/n, where n= no. of moles of the substance
• Um = Utr, m + Urot, m + Uvib, m + Uinternal, m + Urest, m (where,Urest,m = rest mass energy of electrons & nuclei = mrelC2= constant)
Energy

Properties of Internal Energy
• U is a State function, a property that: Depends only on the current state of the system and is independent of how or through path that state has been reached.
• It is an exact differential and its fundamental variables are V and TU = f (V, T);
• For cyclic process,
• U is an Extensive property
0=∫ dU
dTTUdV
VUdU
VT⎟⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=
∂∂
∂∂
Energy

The First Law of Thermodynamics: Work, Heat & Energy
T1 → T2
hT1 → T2
Joule ExperimentJoule Equivalent of Heat: 1 cal = 4.184 J

• Expansion work, work of expansion (or compression)• Non-expansion / additional work: any other work
• pex = external pressure• A = piston area• dz = displacement• dV = Adz = volume change for the gas
dVpwf
i
v
vex∫−=Total work done,
Work
• Work done by the system - ve• Work done on the system + ve
i.e., Expansion
i.e., Compression
where

1st Law of Thermodynamics
Consider an example system of a piston and cylinder with an enclosed dilute gas characterized by P,V,T & n.
What happens to the gas if the piston is moved inwards? Compression
•If the container is insulated the temperature will rise, the atoms move faster and the pressure rises.
•Is there more internal energy in the gas? Yes
External agent did work in pushing the piston inward.w = - F d = -(PA) (-Δx)w = P Δ V
Work done on the gas equals the change in the internal energy of the gas w = Δ U
∆x

• Change the situation:• Keep the piston fixed at its
original location.• Place the cylinder on a hot plate.
What happens to gas?• Heat flows into the gas.• Atoms move faster, internal energy
increases.• Q = heat (in Joules) flows in∆U = change in internal energy in Joules.Q = ∆U
1st Law of Thermodynamics

What if we added heat and pushed the pistonin at the same time?
F
• Work is done on the gas, • Heat is added to the gas and• the internal energy of the gas
increases:
ΔU = w + Q
1st Law of Thermodynamics
U INCREASES with INCREASE in TEMP. of the system, when:
1. work is done on the system by the surrounding or, 2. heat flows into the system from the surrounding or,3. both, heat and work flows from the surr. into the system

The First Law: Sign convention
• Work done by the system by surr - ve
• Work done on the system by surr + ve
i.e., Expansion
i.e., Compression

Consider a function of two variables f (x,y)
Definition: A partial derivative of one of the variables: the rate of change of the function w.r.t. that variable with all other variables kept fixed.
x 0y
df f ( x x,y ) f ( x,y )Limdx xΔ
ΔΔ→
+ −⎛ ⎞ =⎜ ⎟⎝ ⎠
y 0x
df f ( x,y y ) f ( x,y )Limdy yΔ
ΔΔ→
⎛ ⎞ + −=⎜ ⎟
⎝ ⎠
Suppose: f = f (x,y),
• For example: f (x, y) = x2y 2
y x
f f2xy, xx y
⎛ ⎞∂ ∂⎛ ⎞ = =⎜ ⎟⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠
Partial Differentiation: A primer

Properties of partial derivatives
Consider a function of two variables f (x, y)
∴ Its differential form will be:
y x
df dfdf dx dydx dy
⎛ ⎞⎛ ⎞= + ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠
•The Chain rule:
Let s be an independent parameter and if change of s results in changes in the values of x and y.
i.e., x = x (s); y = y (s); ∴ we can write, f (x, y) ≡ f (s)
y x
df df dx df dyds dx ds dy ds
⎛ ⎞⎛ ⎞= + ⎜ ⎟⎜ ⎟⎝ ⎠ ⎝ ⎠

Properties of partial derivatives
• The Cyclic Rule:
If z is a function two variable, then we can write: z = z (x, y),
V p T
dp dT dV 1dT dV dp
⎛ ⎞⎛ ⎞ ⎛ ⎞= −⎜ ⎟⎜ ⎟ ⎜ ⎟
⎝ ⎠ ⎝ ⎠ ⎝ ⎠
Example:
If V = f (p, T); p = f (V, T); T= f (p, V), then we can write
x y z
dy dz dxthen, 1dz dx dy
⎛ ⎞⎛ ⎞ ⎛ ⎞ = −⎜ ⎟⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠ ⎝ ⎠
Example 1:
Use the Equation of state for Ideal gas to verify the cyclic rulep = nRT/V; V= nRT/p; T= pV/nR
2V p T
2V p T
dp nR dT p dV nRT; ;dT V dV nR dp p
dp dT dV nR p nRTdT dV dp V nR p
nRT 1pV
⎛ ⎞⎛ ⎞ ⎛ ⎞= = = −⎜ ⎟⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠ ⎝ ⎠
⎛ ⎞ ⎛ ⎞⎛ ⎞ ⎛ ⎞ ⎛ ⎞ ⎛ ⎞∴ = −⎜ ⎟⎜ ⎟ ⎜ ⎟⎜ ⎟⎜ ⎟ ⎜ ⎟ ⎝ ⎠⎝ ⎠⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠
⎛ ⎞= − = −⎜ ⎟
⎝ ⎠

∴2f ( x ,y ) x y ,=
2f 2 x ,y x∂
=∂ ∂
2f 2 x ,y x∂
=∂ ∂
Verification: Let the function be:
The exact differential
2
x
df fx dy x y⎛ ⎞∂ ∂
=⎜ ⎟∂ ∂ ∂⎝ ⎠
2 2f fx y y x∂ ∂
=∂ ∂ ∂ ∂
•Two partial derivatives operating on a function:
Let f be function of two variables, i.e., f = f (x, y); Double partial derivative of the function f can be written as:
•In general, the total differential form of f(x,y) may be written as:df M( x ,y )dx N ( x ,y )dy= +
x
fNy
⎛ ⎞∂= ⎜ ⎟∂⎝ ⎠
where , andy
fMx∂⎛ ⎞= ⎜ ⎟∂⎝ ⎠
Properties of partial derivatives

Then, df = [M(x,y)dx + N(x,y)dy] represents an exact differential.
• Does any differential form of type [M(x,y)dx + N(x,y)dy]always represent the total differential form of a function?
Only ifyx
M Ny x
⎛ ⎞∂ ∂⎛ ⎞=⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠⎝ ⎠
Properties of partial derivatives
2 2 33 x y dx 2 x ydy+2 2
3
M( x ,y ) 3 x y ,N ( x ,y ) 2 x y
=
=
Example:Let us consider: where,
It can be easily verified that the above expression corresponds to an exact differential function of the type:
2 2 3 3 23 x y dx 2 x ydy d ( x y )+ =

Properties of partial derivatives
• The VALUE OF THE INTEGRAL OF AN EXACT DIFFERENTIAL isINDEPENDENT OF THE PATH taken in going from initial to final point and DEPENDS only on the values of f(x, y) at the initial and the final points.
Consequence:Let us move from a point (xi,yi) to another point (xf,yf)The corresponding change in f(x,y) can be written as:
f f
i if fi i
f f ( x ,y ) f ( x ,y ) df ( Mdx Ndy )Δ = − = = +∫ ∫
NOTE:Integral of an exact differential function,df, for a closed path (i.e., cyclic) where, i (initial state) = f (final state): ∫ =0df

Test for Exactness (State Variable)
dz will be a perfect differential when it is found
• dz is a single-valued function depending entirely on the instantaneous values of x and y.
• dz between any two specified points or states is independent of the path of transition.
• dz for a complete cyclic process is equal to zero
• δq and δw are NOT EXACT differential quantities therefore they are NOT STATE Variables.
y x
y x
yx
y xx
If z f( x,y),z zthen , dz dx dyx y
z zif, M ; and Nx y
then, dz M(x,y)dx N(x,y)dy
Then for exact differential,M Ny x
z zi.e.,y x x y
∂ ∂∂ ∂
∂ ∂∂ ∂
∂ ∂ ∂ ∂∂ ∂ ∂ ∂
=
⎛ ⎞⎛ ⎞= +⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎝ ⎠
⎛ ⎞∂ ∂⎛ ⎞ = =⎜ ⎟ ⎜ ⎟∂ ∂⎝ ⎠ ⎝ ⎠= +
⎛ ⎞ ⎛ ⎞⇒ =⎜ ⎟ ⎜ ⎟⎝ ⎠⎝ ⎠
⎡ ⎤⎡ ⎤ ⎛ ⎞⎛ ⎞ = ⎢⎢ ⎥⎜ ⎟ ⎜ ⎟⎝ ⎠ ⎢ ⎝ ⎠⎣ ⎦ ⎣ ⎦y⎥⎥

Properties of partial derivatives

The gas lawsisotherm, curves in a graph corresponding to a single temperature.
isobar, curves in a graph corresponding to a single pressure.
isochore, curves in a graph corresponding to a single volume.
gas constant, R (with R = NAk, where NA is Avogadro’s constant and k is Boltzmann’s constant).

The gas law
perfect/ideal gas equation, pV = nRT.
perfect/ideal gas, a gas that obeys pV = nRT exactly under all conditions.real gas, an actual gas. e.g.,
standard ambient temperature and pressure (SATP), 298.15 K (25 °C) and 1 bar (or, 0.986 atm or 100 kPa).
standard temperature and pressure (STP)273.15 K (0 °C) and 1 bar (100 kPa, 0.986 atm.)
1 atm =(101.325 kN/m2, 101.325 kPa, 14.7 psi, 29.92 in Hg, 760 torr).
Normal temperature and pressure (NTP)273.15 K (0 °C) and 1 atm (101.325 kPa, 760 Torr)

Vapour pressure, the pressure of a vapour in equilibrium with its condensed phase
Mole fraction, the amount of i species, expressed as a fraction of moles of the moles of ith species (ni) to the total amount of moles, n, in the sample, xi = ni/n
Partial pressure , of a gas is the pressure the gas would exert if it occupied the container alone; in general, pi = xi P

partial pressure: of an ideal gas, the pressure a gas would exert if it occupied the container alone; in general, pi = xi p.
Dalton’s law: The pressure exerted by a mixture of gases is the sum of the partial pressures of the gases.

Thermodynamics
Thermodynamics:→ Describes macroscopic properties of equilibrium
systems
→ Entirely Empirical
→ Built on 4 Laws and “simple” mathematics
0th Law Defines Temperature (T) 1st Law Defines Energy (U) 2nd Law Defines Entropy (S) 3rd Law Gives Numerical Value to Entropy
These laws are UNIVERSALLY VALID, they cannot be circumvented

• Expansion work, work of expansion (or compression)• Non-expansion / additional work: any other work
• pex = external pressure• A = piston area• dz = displacement• dV = Adz = volume change for the gas
dVpwf
i
v
vex∫−=Total work done,
Work
• Work done by the system - ve• Work done on the system + ve
i.e., Expansion
i.e., Compression
where

What if we added heat and pushed the pistonin at the same time?
F
• Work is done on the gas, • Heat is added to the gas and• the internal energy of the gas increases:
ΔU = w + Q
1st Law of Thermodynamics
U INCREASES with INCREASE in TEMP. of the system, when:
1. work is done on the system by the surrounding or, 2. heat flows into the system from the surrounding or,3. both, heat and work flows from the surr. into the system
Consider an example system of a piston and cylinder with an enclosed dilute gas characterized by P,V,T & n.
Statement: Change of internal energy in a closed system of is equal to the energy flow across its boundary as heat or work

The First Law: Sign convention
• Work done by the system by surr - ve
• Work done on the system by surr + ve
i.e., Expansion
i.e., Compression
Alternate Statement: If a system is subjected to a CYCLIC Transformation, the work produced in the surroundings is equal to the heat withdrawn from the surroundings.
Q = -w (change in U is zero as it’s a State function)
If w is +ve (compression, work done on the system) then q will be –ve(heat will be released by the system) and vice versa

w = - mg.A.dzw = - mg.dVw = - pex.dV =-pex (Vf-Vi)
piston held down by pins
Pi , Vi
m
Pf , Vf
dz
F= mgpex= mg/AF= pex.A
Area =A
• Expansive work, work of expansion (or compression)• Non-expansive / additional work : any other work
Work
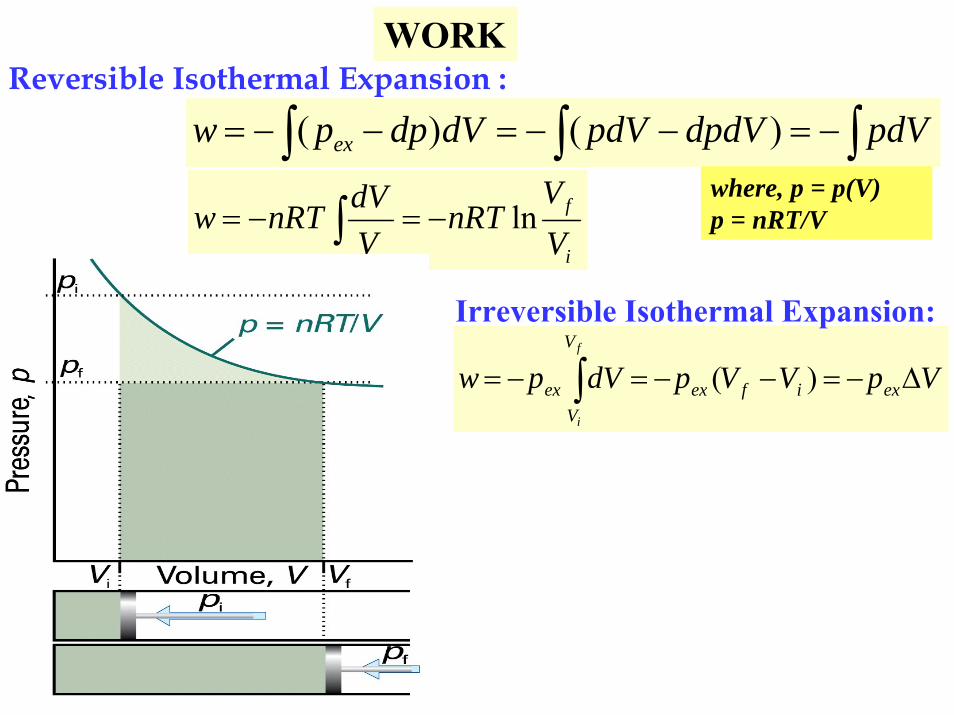
Reversible Isothermal Expansion :
∫ ∫ ∫−=−−=−−= pdVdpdVpdVdVdppw ex )()(
i
f
VV
nRTVdVnRTw ln−=−= ∫
VpVVpdVpw exifex
V
Vex
f
i
Δ−=−−=−= ∫ )(
Irreversible Isothermal Expansion:
WORK
where, p = p(V)p = nRT/V

Irreversible
Irreversible



• For a reversible process Work on expansion = work of compression (same path)• For an irreversible process Work on expansion ≠ work of compression (same path)
Expansion
Irreversible
Irreversible Reversible
Reversible
Compression

• For a reversible process Work on expansion = work of compression (same path)• For an irreversible process Work on expansion ≠ work of compression (same path)
• Work done on expansion by the system on surroundings (sign negative) operating between specified initial and final states and passing through and specified path is maximum work available from a system when the changes take place reversibly
• Work is a path dependent parameter, path function
Work
(so as Heat)
wrev > wirr

State and Path Functions
• Some state functions, e.g., U and H etc.– Value of a state function is independent of
path– Depends only on initial and final state,
e.g. ΔU = Ufinal - Uinitial– Overall Change
final
initialU dU dU is an exact differentialΔ = ⇒∫
final
initial,pathq q q is an inexact differential= ∂ ⇒ ∂∫
Path 1 -Adiabatic w≠0;q=0
Path 2 - Non-adiabatic w≠0;q≠0
ΔUpath1 = ΔUpath2 = Uf -Ui
• Path from initial to final state has a functional dependence– For ∆U, work may be done, or, heat may
flow or, both– Equations that describe how you get to final
state are path functions– Path functions are path specific

Work is a STATE FUNCTION?
Compression through two paths
1st Path:Ar(g, p1, V1) → Ar(g, p1, V2) → Ar(g, p2, V2) First V1 → V2 when p held constant at p1; then p1 → p2 when V held constant at V2
2nd Path:Ar(g, p1, V1) → Ar(g, p2, V1) → Ar(g, p2, V2) First p1 → p2 when V held constant at V1; then V1 → V2 when p held constant at p2
Note for the closed cycle, total work = w[path (1)] - [path (2)]
Assume a Reversible process so that pext = p where, Ar (gas) under goes change of state through compression, from Ar (g, p1, V1) (initial) → Ar (g, P2, V2) (final); such that V1 > V2 and p1< p2

2 2
1 2
2
1
1 2
1 1
2
1
V V
ext extV V
V
1 1 2 1 1 1 2V
V V
ext extV V
V
2 2 2 1 2 1 2V
w(1) p dV p dV
w(1) pdV p (V V ) p (V V )
w(2) p dV p dV
w(2) p dV p (V V ) p (V V )
= − −
= − = − − = −
= − −
= − = − − = −
∫ ∫
∫
∫ ∫
∫NOTE:
• Work done on the system to compress it is NOT EQUAL through the two paths. i.e., w(1) ≠ w (2)
• ∴ Net work done on the system in the complete cycle ≠ 0i.e., [w(1) - w(2)] ≠ 0;
• This means w is NOT a state function ∴ we CAN NOT write w = f(V, p)
)(0 cycleclosedw∫ ≠∂
Work is a STATE FUNCTION?

Ideal gas, closed system, reversible process
10 Pa, 1 m3, T 10 Pa, 10 m3, T3
1 Pa, 10 m3, T
10 Pa, 1 m3, T 1 Pa, 10 m3, Tisothermal
10 Pa, 1 m3, T 1 Pa, 1 m3, T2
1 Pa, 10 m3, T
isobaric
isochoric
isochoric
isobaric
Calculate wf and wb

25 L of gas is enclosed in a cylinder/piston apparatus at 2 atm of pressure and 300 K. A 100 kg of mass is placed on the piston causing the gas to compress to 20 L at constant pressure. This is done by allowing heat to flow out of the gas. What is the work done on the gas? What is the change in internal energy of the gas? How much heat flowed out of the gas?• Po = 202,600 Pa, Vo = 0.025 m3, • To = 300 K, Pf = 202,600 Pa, • Vf=0.020 m3, Tf= ?
• n = PV/RT W = -P∆V ∆ U = 3/2 nR ∆T Q = W + ∆ U
W = - P ∆ V = -202,600 Pa (0.020 – 0.025)m3
= 1013 J energy added to the gas
∆ U =3/2 nR ∆T=1.5(2.03)(8.31)(-60)= -1518 J
Q = W + ∆ U = 1013 – 1518 = -505 J heat out

addexp dwdwdqdU ++=
- For dV = 0; dwexp = 0 and if dwadd = 0, thendU = dq (constant volume, no additional work) = dqv
For a measurable change, ΔU = qv
- molar heat capacity, the heat capacity divided by the amount of substance, Cm = C /n. - specific heat capacity, the heat capacity divided by the mass, Cs = C /m
In general change in the internal energy of a closed system is given by
Heat Transactions
adiabatic bomb calorimeter
-heat capacity, C, the ratio of heat supplied to the temperature rise it causes. C = dq/dT;
C = q/ ΔT
- heat capacity at constant volume, CV = (∂U/∂T)V

Heat Capacity for Constant Volume Processes (Cv)
• Heat is added to a substance of mass m in a fixed volumeenclosure, which causes a change in internal energy, U. Thus,
Q = U2 - U1 = ∆U = m Cv ∆T
The v subscript implies constant volume
Heat, Qaddedm m
ΔTinsulation

Heat Capacity for Constant Pressure Processes (Cp)
• Heat is added to a substance of mass m held at a fixed pressure, which causes a change in internal energy, U, AND some PV work.
Heat, Qadded
ΔT
m m
Δx

For dV ≠ 0; dwexp≠ 0 even if dwadd = 0, dU ≠ dqthen
Heat Transactions and Enthalpy
At constant p, dH = dqp (constant pressure, no additional work)
•Differential scanning calorimeter•Isobaric calorimeter•Adiabatic flame calorimeter
Empirical expression, Cp,m = a + bT + c/T2
Define H = U + pV
dH = dU + pdV + Vdp= dU - w (at constant p)= dqp
ΔU = qp - pΔV qp = ΔU +pΔV = U2 – U1 + p(V2 – V1) = (U2 + pV2) – (U1 + pV1)
- Heat capacity at constant pressure, Cp = (∂H/∂T)p- Enthalpy change and temperature change, dH = nCp,m dT;
ΔH = nCp,m ΔT (if Cp independent of T)

Heat Transactions
- for ideal/perfect gas; relation between ∆U and ∆H, ∆H = ∆U + ∆ngRT.
- relation between heat capacities of a perfect gas, Cp – CV = nR

For ideal gas
dU = CV dT (for ideal gas)
dTTUdV
VU
VT⎟⎠⎞
⎜⎝⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
=dU
TVU⎟⎠⎞
⎜⎝⎛∂∂
= Tπ Internal pressure
dTCdV VT += π
0 T =⎟⎠⎞
⎜⎝⎛∂∂
=TV
Uπ
For any process
dTTHdP
PHdH
PT⎟⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=
∂∂
∂∂
),( TVfU = ),( TPfH =
dTCdPPH
PT
+⎟⎠⎞
⎜⎝⎛=∂∂
For ideal gas
dH = Cp dT (for ideal gas)
0 =⎟⎠⎞
⎜⎝⎛∂∂
TPH
Joule Experiment
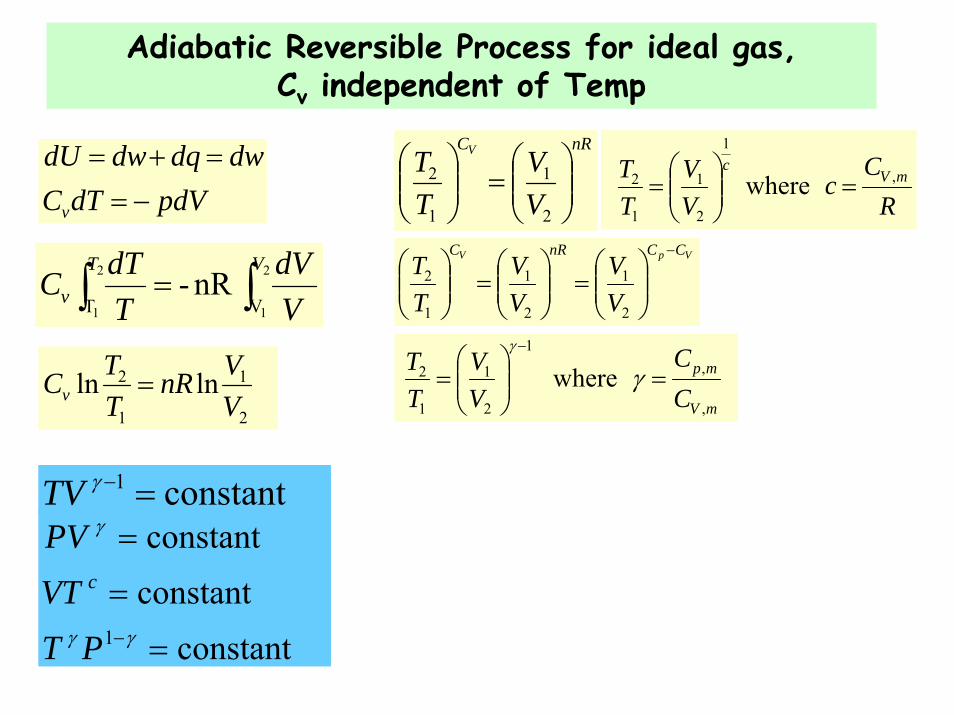
Adiabatic Reversible Process for ideal gas, Cv independent of Temp
pdVdTCdwdqdwdU
v −==+=
where ,
,1
2
1
1
2
mV
mp
CC
VV
TT
=⎟⎟⎠
⎞⎜⎜⎝
⎛=
−
γγ
∫∫ = 2
1
2
1 VT nR-
VT
v VdV
TdTC
lnln2
1
1
2
VVnR
TTCv =
RC
cVV
TT mV
c,
1
2
1
1
2 where =⎟⎟⎠
⎞⎜⎜⎝
⎛=
nRC
VV
TT V
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛
2
1
1
2
VpV CCnRC
VV
VV
TT
−
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛
2
1
2
1
1
2
constant constantconstant
1 =
=
=
−γγ
γ
PTVTPV
c
constant 1 =−γTV

Conclusion of Ist Law of Thermodynamics
• The energy of the universe is constant.
• When a system undergoes a process that leads to a change of state then the sum of the heat q transferred and the work wperformed, is equal to the change in the internal energy dU of the system.
• A system contains ONLY internal energy. A system does not contain heat or work.
For isolated system : dU = 0; (as q = 0, w = 0» No perpetual motion machines! )
• The internal energy of an isolated system is a constant
(Closed system, through a number of steps)dU q w= ∂ + ∂
∆Usystem = −∆Usurroundings

Conclusion of Ist Law of Thermodynamics
• For an Adiabatic Process δ wad = dU ; (as δq = 0)
• Cyclic Process (Exact differential)0=∫ dU
• In a cyclic process the work done by a system on its surroundings is equal to the heat withdrawn from the surroundings .
- δ w = δq
• For a Non-Adiabatic Processδq = dU − δw ≡ δq = (δwad – δw); (as, δwad = dU)

ΔU
ΔH
reversible phase change at const. T, p
const p heating, no phase change
const V heating, no phase change
w
q latent heat
VpdVp Δ−=− ∫
q + w
q
VpdVp Δ−=− ∫
∫= dTTCp )(
Hqp Δ=
q + w
0
UqV Δ=
∫= dTTCV )(
ΔU + VΔp
Calculation of thermodynamic functions for various processes

Calculation of thermodynamic function for various processes for Ideal Gas
ΔU
ΔH
Reversible, no phase change rev, isothermal rev, adiabatic
w
q ΔU-w
∫− PdV 1
2lnVVnRT−
∫= dTTCV )(
Vp f Δ−
∫= dTTCV )(
∫= dTTCp )(
1
2lnVVnRT
0
0
0
∫= dTTCV )(
∫= dTTCp )(
VpexΔ−
rev irrev, const final p
rev, const ext. p





• Reversible Adiabatic Expansion (or compression) of an Ideal Gas
1 mole gas (V1,T1) = 1 mole gas (V2,T2)
adiabatic ⇒ đq = 0 Reversible ⇒ đw = -pdV Ideal gas ⇒ dU = CvdT
∴ From 1st Law dU = -pdV ⇒ CvdT = -pdV along path
V = −pdV ⇒ V dT dVC dT C = −R
RT T Vp= V C
T V ⎛ ⎞ ⎛ ⎞R CV ⎛ ⎞ ⎛ ⎞CV
p −12 dT 2 dV T2 V1 Cp −CV =R for i.g. T2 V1C = −R ⇒ = ⎯⎯⎯⎯⎯⎯→ = V ∫ ∫ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟T T1 T1
T1 V1 V ⎝ ⎠ ⎝ ⎠V2 ⎝ ⎠ ⎝ ⎠V2
Cp ⎛ ⎞ ⎛ ⎞T2 V1 γ −1
Define γ ≡ ⇒ =⎜ ⎟ ⎜ ⎟C T1 V2V ⎝ ⎠ ⎝ ⎠
C = 3R ⎫
For monatomic ideal gas: V 2 ⎪⎪
⎬ γ = 5 ( > 1 generally)
Cp = 5R ⎪ 32 ⎪⎭
In an adiabatic expansion (V2 > V1), the gas cools (T2 > T1). And in an adiabatic compression (V2 < V1), the gas heats up.
⎛ ⎞ ⎛ ⎞For an ideal gas (one mole) T = pV ⇒
p2 = V1
γ
⇒ 1 1 γ = pV γ⎜ ⎟ ⎜ ⎟ pV 2 2R ⎝ ⎠ ⎝ ⎠V2p1
pV γ is constant along a reversible adiabat
For an isothermal process T = constant ⇒ pV = constant
Dr.Amita Pathak
Text Box
SUMMRAY OF ADIABATIC WORK (with solved example)

p V2
adiabat < V2isotherm p1
because the gas cools during reversible adiabatic expansion p2
V1 V2ad V2
iso
• Irreversible Adiabatic Expansion of an ideal gas against a constant external pressure
1 mol gas (p1,T1) = 1 mol gas (p2,T2) (pext=p2)
adiabatic ⇒ đq = 0 Constant pext = p2 ⇒ đw = -p2dVIdeal gas ⇒ dU = CvdT1st Law ⇒ dU = -p2dV
∴ CvdT = - p2dV
Integrating: Cv (T2 - T1) = - p2 (V2 - V1)
Using pV = RT T2 (CV + R ) =T1 ⎜⎛CV +
p2 R ⎟⎞
⎝ p1 ⎠
Note p2 < p1 ⇒ T2 < T1 Again, expansion cools
Note also (-wrev) > (-wirrev) Less work is recovered through an irreversible process
Prof.Amita Pathak
Highlight

Some Thermodynamic Cycles • Reversible Ideal Gas processes: Find ∆U, ∆H , q, w, ∫
đq T
( )I II( )p p
( )T1 ( )T1
A
D (const. p) ( )T3p1 p1
E(const. V) A (isotherm)
B(rev.
( )T1p2 (isotherm) ( )T1C (const. V) p2
p3 adiabat) ( )T2
V1 V2 V1 V2
[A] 1 mol gas (p1,V1,T1) const. T
1 mol gas (p2,V2,T1)=
Ideal gas isotherm: ∆UA = 0 ∆HA = 0
wA = −RT 1 lnVV
2
1
qA = RT 1 lnVV
2
1 ∫đT
= R ln VV
2
1
[B] 1 mol gas (p1,V1,T1) rev.adiabat
1 mol gas (p3,V 2,T2)=
Adiabat: qB = 0
∆U = C T( −T )Ideal gas: B V 2 1
∆H = C T( −T )B p 2 1
1st Law: wB = CV (T2 −T1 )
∫đ TqB = 0
qA
Prof.Amita Pathak
Text Box
as, ΔH = Cp ΔT = 0

reversible [C] 1 mol gas (p3,V2,T2) = 1 mol gas (p2,V2,T1)
const. V
∆U = C T −TConstant V: wC = 0 Ideal gas: C
C
V
p
((
1
1
2
2
))∆H = C T −T
1st Law: q = C T −TC V ( 1 2 )
đqC ⎛ ⎞T1∫ = CV ln ⎜ ⎟T T2⎝ ⎠
[A] vs. [B] + [C]
∆UA = 0 ∆UB + ∆UC = = 0 ∆UA
∆HA = 0 ∆HB + ∆HC = = 0 ∆HA
RT ln V2 q + = C ( T )q = q T − ≠ qA 1 B C V 1 2 AV1
w RT ln V2 w +w = C ( − ≠ )= − T T wA 1 B C V 2 1 AV1
VA 2 B C 2 Ađq = R ln V đq
+ đq
= R ln ⎛ ⎞
= đq
∫ ∫ ∫ ⎜ ⎟ ∫T V T T V1 T1 ⎝ ⎠
revThis result suggests that ⎜⎛
⎝∫đ Tq
⎟⎞
⎠ is a state function!
∆U = C T( −T )[D] D V 3 1 qD = C T T1∆H = C T( −T ) p ( 3 − )
D p 3 1
đq ⎛ ⎞= −R T T
TD = Cp ln T
T3
1
wD ( 3 − 1 ) ∫ ⎜ ⎟ ⎝ ⎠
Prof.Amita Pathak
Note
Accepted set by Prof.Amita Pathak
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Stamp

∆U = C T −TE V ( 1 3 )[E] E p ( 1 3 )
wE = 0∆H = C T −T
qE = CV (T1 −T3 )
đqE ⎛ ⎞T1∫ = CV ln ⎜ ⎟T T3⎝ ⎠
[A] vs. [D] + [E]
∆UA = 0 ∆UD + ∆UE = ∆UA
∆HA = 0 ∆HD + ∆HE = ∆HA
V2 q 3 − ≠ qAqA = RT 1 ln qD + = E R T ( T1 )V1
RT ln V2 +w = − ( ) wwA = − 1 wD E R T 3 − ≠ T1 AV1
đqA = R ln V2 đqD + đqE = R ln
⎛ ⎞V2 = đqA∫ ∫ ∫ ⎜ ⎟ ∫T V T T V1 T1 ⎝ ⎠
Here again ⎜⎝
⎛∫đ Tqrev
⎟⎠
⎞ looks like a state function!
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line
Prof.Amita Pathak
Line

Conclusion of Ist Law of Thermodynamics
• The energy of the universe is constant.
• When a system undergoes a process that leads to a change of state then the sum of the heat q transferred and the work wperformed, is equal to the change in the internal energy dU of the system.
• A system contains ONLY internal energy. A system does not contain heat or work.
For isolated system : dU = 0; (as q = 0, w = 0» No perpetual motion machines! )
• The internal energy of an isolated system is a constant
(Closed system, through a number of steps)dU q w= ∂ + ∂
∆Usystem = −∆Usurroundings

Conclusion of Ist Law of Thermodynamics
• For an Adiabatic Process δ wad = dU ; (as δq = 0)
• Cyclic Process (Exact differential)0=∫ dU
• In a cyclic process the work done by a system on its surroundings is equal to the heat withdrawn from the surroundings .
- δ w = δq
• For a Non-Adiabatic Processδq = dU − δw ≡ δq = (δwad – δw); (as, δwad = dU)


ΔU
ΔH
reversible phase change at const. T, p
const p heating, no phase change
const V heating, no phase change
w
q latent heat
VpdVp Δ−=− ∫
q + w
q
VpdVp Δ−=− ∫
∫= dTTCp )(
Hqp Δ=
q + w
0
UqV Δ=
∫= dTTCV )(
ΔU + VΔp
Calculation of thermodynamic functions for various processes

Calculation of thermodynamic function for various processes for Ideal Gas
ΔU
ΔH
Reversible, no phase change rev, isothermal rev, adiabatic
w
q ΔU-w
∫− PdV 1
2lnVVnRT−
∫= dTTCV )(
Vp f Δ−
∫= dTTCV )(
∫= dTTCp )(
1
2lnVVnRT
0
0
0
∫= dTTCV )(
∫= dTTCp )(
VpexΔ−
rev irrev, const final p
rev, const ext. p

dTTUdV
VU
VT⎟⎠⎞
⎜⎝⎛
∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=dU ),( TVfU =
Variation of U with system variables:
1. Change in U w.r.t. T at constant volume
C UT
q C T UvV
v v= ⎛⎝⎜
⎞⎠⎟
= =∂∂
; Δ Δ
As For ideal gasdU = CV dT (for ideal gas) 0 T =⎟⎠⎞
⎜⎝⎛
∂∂
=TV
Uπ
2. Change in U w.r.t. V at constant temp: dTCdV VT += π For real gas
∂∂UV T
⎛⎝⎜
⎞⎠⎟
3. Change in U w.r.t. T at constant pressure
dU C dT UV
dV
UT
C UV
VT
vT
Pv
T P
= + ⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
= + ⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
∂∂
∂∂
∂∂
∂∂
UT
C V UVP
vT
⎛⎝⎜
⎞⎠⎟
= + ⎛⎝⎜
⎞⎠⎟
∂∂
α ∂∂
where

Change in Internal Energy at Constant P
• Suppose we want to know how the internal energy, U, changes with temperature at constant pressure
– But the change in volume with temperature at constant pressure is related to the Isobaric thermal expansion / expansion coefficient of the gas, α
• Large α means sample responds strongly to changes in T• For an ideal gas, πT = 0 so
dU = π TdV + CVdT
∂U∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
=∂ πTdV + CVdT( )
∂T⎛
⎝ ⎜
⎞
⎠ ⎟
p
∂U∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
= πT
∂V∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
+ CV
α = 1V
∂V∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
or ∂V∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
= αV
so ∂U∂T
⎛ ⎝ ⎜
⎞ ⎠ ⎟
p
= π TαV + CV
[ ]definitionCTU
Vp
=⎟⎠⎞
⎜⎝⎛
∂∂

Variation of V with system variables: Experimentally known that the Volume of an isotropic material is a function of Temperature and Pressure.
isothermal compressibility
coefficient of Isobaric thermal expansion / expansion coefficient
For solid and liquid α and κT are approx. independent of T & p
For an ideal gas : PV = nRT
κT = 1/psimilarly
relative/fractionalchange of Volume

Another quantity of interest which can be obtained from α and κT is:
This derivative tells us how fast the pressure rises when we tryto keep the volume constant while increasing the temp.
Exercise:How much pressure would be generated in a mercury thermometer if we tried to heat the thermometer higher than the temperature where the mercury has reached the top of the thermometer. Given: For Hg, α = 1.82 Ø 10×4 K×1 and κT= 3.87 Ø 10×5 atm×1
We see that each 1 C increases the pressure by 4.7 atm, about 69 lb/sq in. This is a lot of pressure for a glass tube to withstand. It wouldn't take very many degrees of temperature increase to break the glass thermometer.!!!
Using a variation of Euler's chain rule we can write

dTTHdP
PHdH
PT⎟⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=
∂∂
∂∂ ),( TPfH =Variation of H with system variables:
1. Change in H w.r.t. T at constant pressure
C HT
q C T HpP
p p= ⎛⎝⎜
⎞⎠⎟
= =∂∂
; Δ Δ
2. Change in H w.r.t. P at constant temperature: ∂∂HP T
⎛⎝⎜
⎞⎠⎟
H f P THP
PT
TH
HP
TP
HT
C
T H P
T H PJT p
=
⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
= −
∴⎛⎝⎜
⎞⎠⎟
= −⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
= −
( , )∂∂
∂∂
∂∂
∂∂
∂∂
∂∂
μ
1dTCdP
PH
PT
+⎟⎠⎞
⎜⎝⎛=
∂∂
For ideal gas
dH = Cp dT (for ideal gas)
0 =⎟⎠⎞
⎜⎝⎛
∂∂
TPH
Alternately,
3. Change in H w.r.t. T at constant volume: ∂∂HT V
⎛⎝⎜
⎞⎠⎟
dH C dT HP
dP
HT
C HP
PT
PT
VP
T V
= + ⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
= + ⎛⎝⎜
⎞⎠⎟
⎛⎝⎜
⎞⎠⎟
∂∂
∂∂
∂∂
∂∂
HP
C PT
HT
C
TJT p
V
VP
JT
⎛⎝⎜
⎞⎠⎟
= − ⎛⎝⎜
⎞⎠⎟
=
∴⎛⎝⎜
⎞⎠⎟
= −⎡⎣⎢
⎤⎦⎥
∂∂
μ ∂∂
ακ
∂∂
αμκ
;
1
From earlier results

The Joule-Thomson Expansion Expt
we need to decide what is being held constant when we take the partial derivative. In this case, since q =0, the change in internal energy of the gas is equal to w.
The Joule-Thomson experiment consists of forcing a gas at constant backing pressure passing through a porous plug under adiabatic condition and measuring the change in temperature.
The Joule-Thomson coefficient (μJT ) =HP
T⎟⎠⎞
⎜⎝⎛
∂∂
pT
ΔΔ
≈
The most straightforward way to measure the coefficient is to measure the temperature of the gas entering and leaving the porous plug and divide the change in temperature by thechange in pressure:

Joule-Thompson Experiment: Inversion Temperature
dTTHdP
PHdHTPfH
PT⎟⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛==
∂∂
∂∂ );,(
In this experiment
w = - pf (Vf – 0) - pi (0 - Vi)
q = 0
ΔU = Uf – Ui = q + w = - pfVf + piVi
Uf + pfVf = Ui + piVi
Hf = Hi , ΔH = 0 Isenthalpic process
μ∂∂
∂∂
∂∂
×−=⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛−=⎟⎟
⎠
⎞⎜⎜⎝
⎛p
HpT
CpT
TH
pH
dTCdPCdH pp +−= μ
Joule-Thompson Coefficient , μ (or μJT ) = HP
T⎟⎠⎞
⎜⎝⎛
∂∂
pT
ΔΔ
≈ in this J-T expt.
Applying Euler’s Chain Rule, we get:
dTTHdP
PHdHTPfH
PT⎟⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛==
∂∂
∂∂ );,(

THE JOULE-THOMPSON EXPERIMENT
HpT
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=μ
If μ > 0 then this indicates that the gas cools (-ve δT) when gas expanded (-ve δp).
If μ < 0 then this indicates that the gas heats (+ve δT) when expanded (-ve δp).
The "inversion temperature" indicates where on a (T, p) isenthalpic plot the (dT/dP)H flips from +ve to –ve. Or,point where µchanges sign from + to –ve and the point is the maximum inversion temperature
For a real gas µ is non-zero (except at the maximum inversion temperature)

The Linde process
HpT
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=μ
Prof.Amita Pathak
Text Box
2
Prof.Amita Pathak
Text Box
2

• µ can be either (+) or (-)– Positive µ means dT is negative when
dp is negative•Gas cools on expansion
– Negative µ means that means dT is positive when dp is negative•Gas warms on expansion
– Transition between positive and negative µ is called the Joule-Thompson inversion temperature•Gases have both high and low inversion temperatures
– Ideal gas µ = 0, T unchanged by change in p on expansion
• Practical applications:– Gas liquefaction– Isotope separation
GasTinversion
(K)
µ(K atm-1)
CO2 1500 1.11N2 621 0.25He 621 0.25
Joule-Thompson Coefficient , μ (or μJT ) =
HPT
⎟⎠⎞
⎜⎝⎛
∂∂
pT
ΔΔ
≈
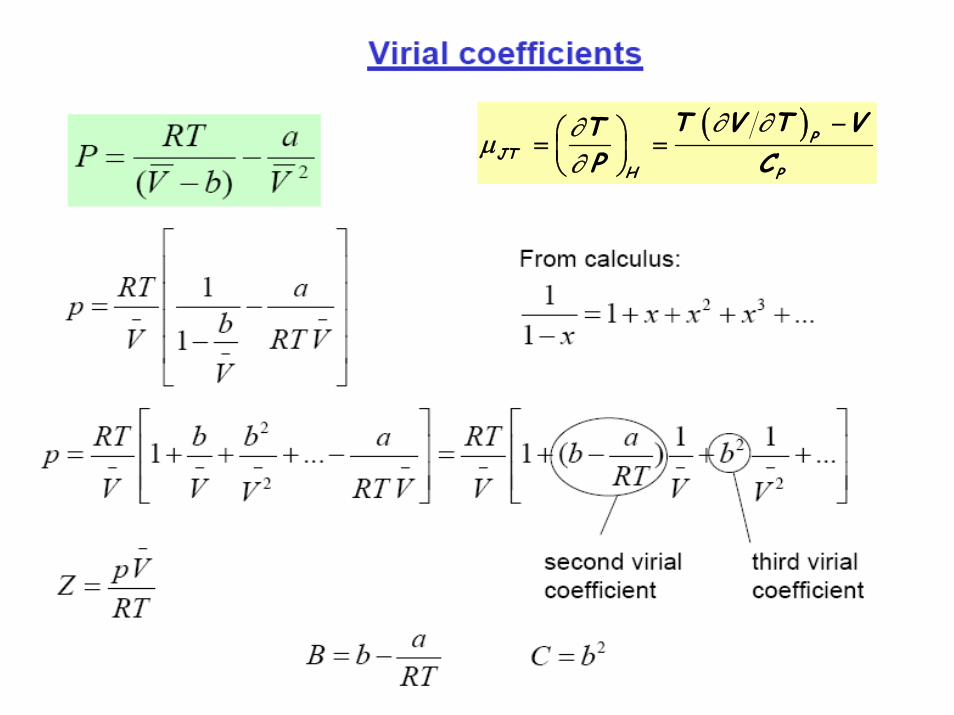
( )PJT
H P
T V T VTP C
∂ ∂∂μ∂
−⎛ ⎞= =⎜ ⎟⎝ ⎠

( )PJT
H P
T V T VTP C
∂ ∂∂μ∂
−⎛ ⎞= =⎜ ⎟⎝ ⎠

( )PJT
H P
T V T VTP C
∂ ∂∂μ∂
−⎛ ⎞= =⎜ ⎟⎝ ⎠
JTP
1 2a bC RT
μ ⎛ ⎞= −⎜ ⎟⎝ ⎠
For a van der Waal gas:Therefore, μJT depends critically on the constants a and b, which are the indices of non zero interaction and finite volume of the gas molecules
)(02, observedcoolingthenbRT
aIf >> μ
)(02, observedheatingthenbRT
aIf << μ
TTwhen inversionJT 0 ==μ
bRT
aorbRT
aC inversionp
2,210 =⎟⎟⎠
⎞⎜⎜⎝
⎛⎥⎦⎤
⎢⎣⎡ −=∴
RbaTinversion
2=∴
Inversion temperature
• If initial Temp (Ti) is belowTinversion only then cooling is observed.
• And the final temp T2 is lower than T1

N2 and O2 will cool upon expansion at room temperature, but He and Ne will warm upon expansion at room temperature.

Derive the Following expressions
κα
∂∂ =⎟
⎠⎞
⎜⎝⎛
VTP
κα TVCV
2
=−pC
Monoatomic ideal gas CV,m = 3R/2, Cp,m = 5R/2
For ideal gas
Isothermal Joule-Thompson Coefficient, μT = TP
H⎟⎠⎞
⎜⎝⎛
∂∂
μT = - Cpμ
TpJT P
HC
⎟⎠⎞
⎜⎝⎛−=∴
∂∂μ 1
( )PJT
H P
JT
JTP
T V T VTP C
for ideal gases : 0
1 2a bC RT
∂ ∂∂μ∂
μ
μ
−⎛ ⎞= =⎜ ⎟⎝ ⎠
=
⎛ ⎞= −⎜ ⎟⎝ ⎠

The Perfect gas and πT, α, κT, μ
Property PD Value for Perfect Gas Info:
TT V
U⎟⎠⎞
⎜⎝⎛
∂∂
=πInternal Pressure πT=0 Strength/nature of
interactions between molecules
pTV
V⎟⎠⎞
⎜⎝⎛
∂∂
=1α
Expansion Coefficient α =1 / T The higher T, the less
responsive is its volume to a change in temperature
TT p
VV ⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
−=1κIsothermal
Compressibility κT=1 / p The higher the p, the lower its compressibility
HpT
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=μJoule-Thomson Coefficient μ=0 Another indication of
molecular interactions.

Ist Law of Thermodynamics:• The different forms of energy are inter convertible.• When one form of energy disappears an equivalent of another kind
must appear. Conversion of energy.• Energy can be transferred / transformed, keeping the total energy
fixed.qsys = - qsurrwsys = - wsurr
ΔU = q + w (= – pexdV)∆U = q – pexdV
Incompleteness of Ist Law: It Does not say anything about:
• Whether any transfer / transformation of energy would occur at all?
• If it does, then in which direction would the change occur?• If any transfer / transformation of energy occurs in a particular
direction, then how long will it be sustained?• If any transfer / transformation of energy occurs in a particular
direction, then how fast will it proceed?
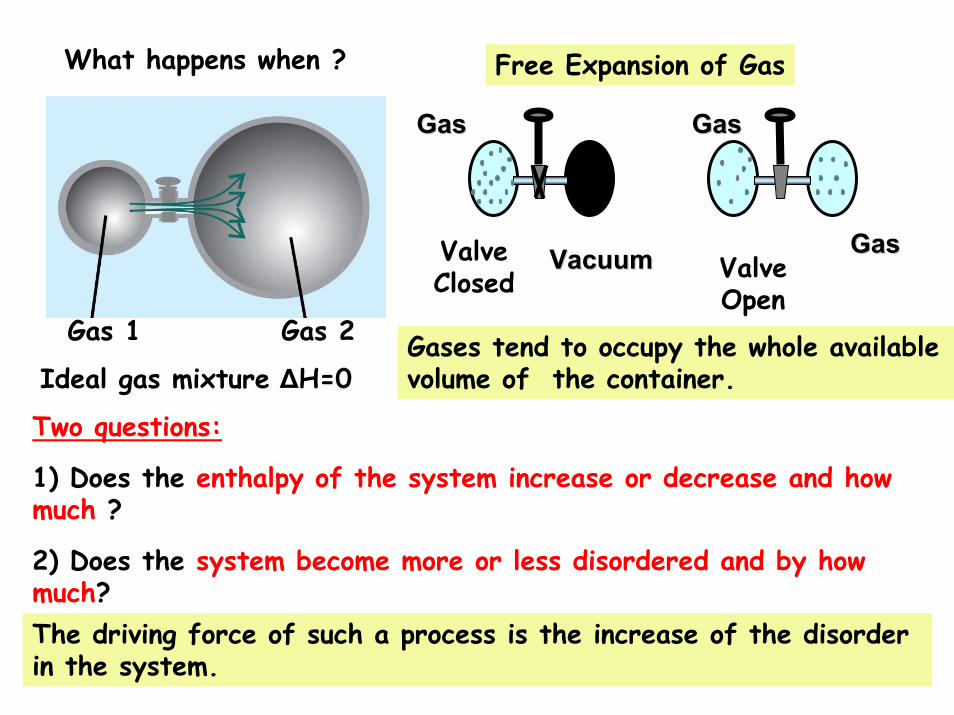
What happens when ?
Gas 1 Gas 2
Ideal gas mixture ΔH=0
ValveClosed
VacuumVacuum
GasGas
ValveOpen
GasGas
GasGas
Free Expansion of Gas
Gases tend to occupy the whole available volume of the container.
Two questions:
1) Does the enthalpy of the system increase or decrease and how much ?
2) Does the system become more or less disordered and by how much?The driving force of such a process is the increase of the disorder in the system.

War
mH
otC
old
dQ
Ther
mal C
ondu
ction
The two liquids (with very similar molecules) form an ideal solution (ΔH ≈ 0), in which the molecules are randomly and uniformly mixed.
Suppose two bodies are brought in contact. Then from the Ist law, we ONLY know if q heat was lost by one, exactly q heat would be gained by the other. The law does not specify if the Ist one will lose heat or, the other one. To know the direction of flow of heat, we need another information, namely the temp of the two bodies.

First Law of Thermodynamics
• Conservation of Energy• Says Nothing About Direction of Energy Transfer
Second Law of Thermodynamics
• Preferred (or Natural) Direction of Energy Transfer• Determines Whether a Process Can Occur
• Thermodynamic Processes can be classified into Three Types :– Natural (or Irreversible)– Impossible– Reversible

Disorder increase for the following processes:• Solids melt to liquids
• Solids or liquids vaporize to form gases
• Solids or liquids dissolve in a solvent to form non-electrolyte solution
• A substance is heated (increased temperature increases the molecular motions and the disorder)
• A chemical reaction produces an increase in the number of molecules of gases
• The 2nd Law puts forward the criteria for the direction of the spontaneous chemical/ physical change of a thermodynamic system.
• The historical roots of the second law stems from the invention of heat engine (namely, the steam engine). It is known that work can be fully converted to heat (joule’s expt), the question is can heatbe converted to work???

• The entropy is an extensive property, i.e. increases with the amount of matter in the system.
Entropy• The thermodynamic property of a system that is related to its
degree of randomness or disorder is called entropy (S).
• The entropy S and the entropy change ΔS=S2-S1 are state functions
• The entropy S has a unique value, once the pressure P, the temperature T and the composition n of the system are specified, S = S (p,T,n)

Carnot Engine: Introduction to Heat Engines
• One of the primary applications of thermodynamics is to Turn heat into work
• The standard heat engine works on a reversible cyclic process:
1. extract heat from a hot reservoir, 2. perform work, 3. dump excess heat into a cold
reservoir (often the environment).
A “reservoir” is a large body whose temperature doesn’t change when it absorbs or gives up heat
Carnot and then by Clausius: Statement of the Second law :It states that it is impossible to construct a device that operates in a cycle and produces no effect other than the transfer of heat from a lower-temperature body to a higher-temperature body.

QL
WOUT
QH
TH
TL
HE
QL
WOUT
QH
TH
TL
R
Thermal efficiency = HQW−
=ε
For a complete cycle: dU = 0
Qcy = - Wcy
Qcy = QH - Qc
∴
∴H
c
H
cH
Hcyc Q
QQQ
W−=
−=
−= 1ε
The engine will have 100% efficiency only when Qc = 0

Heat Engines
• By conservation of energy:|QH| = |W| + |QL|.
• The high and low temperatures TH and TL are called the operating temperatures of the engine.
• We will considering only engines that run in a repeating cycle, that is, the system returns repeatedly to its starting point, and thus can run continuously.
• Absolute value signs are used because we are interested only in the magnitudes.

Second Law of Thermodynamics.The Carnot Cycle.
• The Carnot cycle is an example of a reversible process, which is a process that can be done in reverse.
• A reversible process requires that any changes are made infinitely slowly.
• Real processes are not reversible due to friction , turbulence in the gas, etc.

Second Law of Thermodynamics: The Carnot Cycle

• Four steps• Isothermal expansion: a to b• Adiabatic expansion: b to c• Isothermal compression: c to d• Adiabatic compression: d to a
1 C
H H
QWefficiencyQ Q
ε= = = −
Define the efficiency ε
cost""work""
=≡εreservoirfromextractedheat
systembydonework
Valid for all heat engines.
(∆U = 0 for closed cycle)
where, W = QH - Qc; following conservation of energy

The Carnot Cycle
• Step 1: a to b.
– The gas is in contact with a heat bath at temperature THand weight is removed from the piston.
– The gas expands, while maintaining a constant temperature, isothermal
– the change in the internal energy is thus equal to zero
11
1
0
ln
qwdU
VVnRTw
a
bH
−==
−=

• Step 2: b to c.
– The gas is isolated from the environment and some more weight is removed from the piston.
– The gas expands and during the adiabatic expansion, the temperature of the gas will decrease.
– For adiabatic expansion pVγ
is constant, and we can thus related state b to state c: 0
)(
2
2
11
=−=
= −−
qTTnCw
VTVT
HLV
cLbHγγ
The Carnot Cycle

• Step 3: c to d.
– The gas is in contact with a heat bath at temperature TC and weight is added to the piston.
– The gas is compressed, while maintaining a constant temperature, isothermal
– the change in the internal energy is thus equal to zero
33
3
0
ln
qwdU
VVnRTw
c
dL
−==
−=
The Carnot Cycle

The Carnot Cycle
• Step 4: d to a.
– The gas is isolated from the environment and some more weight is added to the piston.
– The gas is compressed and during the adiabatic compression, the temperature of the gas will increase.
0)(
4
4
11
=−=
= −−
qTTnCw
VTVT
CHV
dLaHγγ

11
1
0
ln
qwdU
VVnRTw
a
bH
−==
−=
0)(
2
2
11
=−=
= −−
qTTnCw
VTVT
HLV
cLbHγγ
33
3
0
ln
qwdU
VVnRTw
c
dL
−==
−=
0)(
4
4
11
=−=
= −−
qTTnCw
VTVT
CHV
dLaHγγ
• Four steps• Isothermal expansion: a to b
• Adiabatic expansion: b to c
• Isothermal compression: c to d
• Adiabatic compression: d to a

4321 wwwwwcycle +++=
c
dH
a
bHcycle V
VnRTVVnRTq
dU
lnln
0
+=
=∫c
dL
a
bHcycle
LHvc
dLHLv
a
bHcycle
VVnRT
VVnRTw
TTnCVVnRTTTnC
VVnRTw
lnln
)(ln)(ln
−−=
−+−−+−=
a
d
b
c
a
d
b
c
L
H
aHdL
cLbH
VV
VV
VV
VV
TT
VTVT
VTVT
=∴==
=
=
−
−
−
−
−−
−−
1
1
1
1
11
11
γ
γ
γ
γ
γγ
γγ
b
aLHcycle
c
dL
b
aHcycle
VVTTnRw
VVnRT
VVnRTw
ln)(
lnln
−=
−=

1 C
H H
QWefficiencyQ Q
ε= = = −
11
1
0
ln
qwdU
VVnRTw
a
bH
−==
−=
But, QH = q1 = -w1
33
3
0
ln
qwdU
VVnRTw
c
dL
−==
−=
And, Qc = q3 = -w3
a
d
b
c
a
d
b
c
L
H
aHdL
cLbH
VV
VV
VV
VV
TT
VTVT
VTVT
=∴==
=
=
−
−
−
−
−−
−−
1
1
1
1
11
11
γ
γ
γ
γ
γγ
γγ
ε = 1 – Qc/ QH = 1 – TLln (Vd/Vc)/ THln (Vb/Va)
1 – Qc/ QH = 1 – TL/ TH
Qc/ QH = TL/ TH Qc/ TL = QH / TH
Qc/ TL – QH / TH = 0 (here, Qc ≡ QL)
00 =∂
⇒= ∫∑ Tq
Tq
i
i Thus, the term δq/T is a state function

tconsTq
Tq
Tq
Tq
Tq
TqbyMultiplyTT
TT
TTT
qqq
qqq
TT
TTT
L
L
H
H
H
H
L
L
LH
H
L
H
L
H
L
H
L
H
LH
H
LH
H
LH
H
L
H
LH
tan;0
011
1
=⇒==+−
=+−⇒−=−
−=
−∴
−=
−=−
=
η
η
Carnot’s theorem tells us that no real engine can have an efficiency more than that of the Carnot engine.

When the Carnot cycle is operating as a heat engine (going around clockwise), w < 0, so that -w > 0 and q > 0.
H
L
H
LH
a
bH
b
aLH
cyclecycle
b
aLHcycle
TT
TTT
VVnRT
VVTTnR
qw
qw
orEfficiency
VVTTnRw
−=−
=−−
=
=−
=
−=
1ln
ln)(
,
ln)(
11
η
ηε
• Efficiency of Carnot engine < 1 • No cycle can have an efficiency greater than a Carnot cycle• All reversible engines operates between same 2 temperatures have same
efficiency

const.; 1 =−γTV 11 −− = γγCCBh VTVT 11 −− = γγ
DCAh VTVT11 −−
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛γγ
D
C
A
B
VV
VV
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛
D
C
A
B
VV
VV
)/( ln)/( ln ACD VVVV B−=
A (pA, VA, Th)
B (pB, VB, Th)
C (pC, VC, TC)
(pD, VD, TC) D
A (pA, VA, Th)
B (pB, VB, Th)
C (pC, VC, TC)
(pD, VD, TC) D
Th > Tc
Carnot Cycle (Ideal gas, All the steps are reversible)
1 (A to B) 2 (B to C) 3 (C to D) 4 (D to A) Total (A to A)
W - nRThln(VB/VA) CV(TC-Th) - nRTC ln(VD/VC) CV(Th-TC) + nR(TC– Th) ln(VB/VA)
q + nRTh ln(VB/VA) Zero + nRTC ln(VD/VC) Zero + nR(Th– TC) ln(VB/VA)
DU Zero CV(TC-Th) Zero CV(Th-TC) Zero
(qrev / T) + nR ln(VB/VA) Zero + nR ln(VD/VC) Zero Zero

Entropy
• Our study of the Carnot cycle showed that over the cycle q/T = 0.
• Since any reversible cycle can be approximated by a series of Carnot cycles, we expect that the integral of dQ/T along the closed path of the cycle = 0.
• One consequence is that the integral of dQ/Tbetween a and b is independent of the path.
• The quantity dQ/T is called the entropy dS.
• The entropy difference between a and b is thus path independent, and entropy is a state variable.

Entropy
• Entropy is a state variable, but Q and W are not state variables since they depend on the path used to get from a to b.
• For a reversible process, the change in the entropy of the engine is opposite to the change in entropy of the environment that provides the heat required (or absorbs the heat generated). The net change in the total entropy = 0.
• For an irreversible process, the net change in the total entropy will be larger than 0.

Entropy :– Is a measure of the disorderness of a system.– In a reversible thermodynamic process, entropy of a system
is related the change in the extent to which energy is dispersed in a disorderly manner, which in turn depends on the amount of energy transferred as heat (as heat stimulates motion) to the surroundings.
– Is a measure of the energy in a system or process that is unavailable to do work.
• For a process occurring at constant temperature (an isothermal process):
qrev = the heat that is transferred when the process is carried out reversibly at a constant temperature.T = temperature at which the heat is transferred (in Kelvin).

Second Law of Thermodynamics
Entropy of the universe (i.e., Total Entropy):
• Does not change for reversible processes• Increases for spontaneous processes. • But, entropy can decrease for individual systems
Reversible (ideal):
Irreversible (real, spontaneous):

dSUniv = dSsys + dSsurr > 0dSUniv = dSsys + dSsurr = 0dSUniv = dSsys + dSsurr < 0
Spontaneous process
Equilibrium processImpossible process
2nd law of thermodynamics
all spontaneous processes are irreversible
dSsys + dSsurr ≥ 0
For any process“=“ for reversible, equilibrium“>” spontaneous (irreversible) (real)

Example• Show the Carnot cycle on a T-S diagram and identify the heat
transfer at both the high and low temperatures, and the work output from the cycle.
S
TTH
TL
S1=S4 S2=S3
1 2
34
A B
• 1-2, reversible isothermal heat transferQH = ∫TdS = TH(S2-S1) area 1-2-B-A
• 2-3, reversible, adiabatic expansionisentropic process, S=constant (S2=S3)
• 3-4, reversible isothermal heat transferQL = ∫TdS = TL(S4-S3), area 3-4-A-B
• 4-1, reversible, adiabatic compressionisentropic process, S1=S4
• Net work Wnet = QH - QL, the area enclosed by 1-2-3-4, the shaded area

Efficiency of an heat engine
WbyQC
Qh
TH
TC
2. What is efficiency of reverse engine (refrigerator)? Coefficient of Performance
a. QC / W b. Tc/ (TH-TC) c. 1-TH/TC
3. When can the heat engine convert the absorbed heat into work completely?.
a. At TH = -273 ˚C b. At Tc = O ˚C c. when TC =-273 ˚C
Consider a Carnot heat engine.1.What fraction of the total heat transferred Qh
to engine is converted into work in the cycle?
a. ∆T/TH b. ∆T/TC c. TC/TH

a.η th rev
L
H
TT
KK
or
,
( )( )
. .
= −
= −++
=
1
1 30 273652 273
0 672 67 2%
TT
KK
Q kJkJ
L
H
L
H
L
=
=++
=
==
( )( )
.
( . )
30 273652 273
0 328
500 0 328164
b.QL
WOUT
QH
TH = 652oC
TL = 30oC
HE
A Carnot heat engine receives 500 kJ of heat per cycle from a high-temperature heat reservoir at 652oC and rejects heat to a low-temperature heat reservoir at 30oC. Determine(a) The thermal efficiency of this Carnot engine.(b) The amount of heat rejected to the low-temperature heat reservoir.

Suppose a mole of a diatomic gas, such as O2, is compressed adiabatically so the final volume is half the initial volume. The starting state is V = 1 liter, T = 300 K. What are the final temperature and pressure?
Equation relating p,V for adiabatic process in α-ideal gasγ is the ratio of Cp/CV for diatomic gas in this case
Solve for pf from first equation
Substitute for pi
Use ideal gas law to calculate final temperature
OR use the equation relating T,V for an adiabatic process to get the final temperature (α = 5/2 for diatomic gas)
Solve for Tf
K
VVTT
VTVTK
nRVp
T
Pax
VV
VnRT
VVpp
VpVp
f
iif
ffii
fff
f
i
i
i
f
iif
ffii
395
395
1057.6
4.12/52/7
1
6
=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
=
=
=
=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
==
=
α
αα
γ
γ
γγ
γ

Entropy change in heat engine
Consider a Carnot heat engine.1. What is the sign of the entropy change of
the hot reservoir during one cycle?a. ΔSh < 0 b. ΔSh = 0 c. ΔSh > 0
WbyQC
Qh
Th
TcBecause energy is flowing out of the hot reservoir, its entropy (the number of microstates) is decreasing.2. What is the sign of the entropy change of the cold reservoir?a. ΔSc < 0 b. ΔSc = 0 c. ΔSc > 0
Because energy is flowing into the cold reservoir, it’s entropy (number of microstates) is increasing.
3. Compare the magnitudes of the two changes.a. |ΔSc| < |ΔSh| b. |ΔSc| = |ΔSh| c. |ΔSc| > |ΔSh|

Example Problem (1)
Solution:
How much heat is absorbed by 3 moles of helium when it expands from V = 10 liters to V = 20 liters and the temperature is kept at a constant 350 K? What are the initial and final pressures?
Q = -Won
Won = -nRT ln(Vf/Vi)= -6048 J
pi = nRT/Vi = 8.72×105 Papf = pi/2 = 4.36 ×105 Pa
From first law. For an ideal gas, isothermal means ∆U = 0. Positive Q means heat flows into the gas.
This was derived in lecture.R = 8.31 J/mole·K
pV = nRT
Where is the heat coming from? In order to keep the gas at a constant temperature, it must be put in contact with a large object having that temperature. That object is called a “heat reservoir”, and it supplies heat to the gas (or absorbs heat, if necessary) in order to keep the gas temperature constant.

Carnot Engine: Introduction to Heat Engines
• One of the primary applications of thermodynamics is to Turn heat into work
• The standard heat engine works on a reversible cyclic process:
1. extract heat from a hot reservoir, 2. perform work, 3. dump excess heat into a cold
reservoir (often the environment).
A “reservoir” is a large body whose temperature doesn’t change when it absorbs or gives up heat
Carnot and then by Clausius: Statement of the Second law :It states that it is impossible to construct a device that operates in a cycle and produces no effect other than the transfer of heat from a lower-temperature body to a higher-temperature body.

Heat Engines
• By conservation of energy:|QH| = |W| + |QL|.
• The high and low temperatures TH and TL are called the operating temperatures of the engine.
• We will considering only engines that run in a repeating cycle, that is, the system returns repeatedly to its starting point, and thus can run continuously.
• Absolute value signs are used because we are interested only in the magnitudes.

Second Law of Thermodynamics.The Carnot Cycle.
• The Carnot cycle is an example of a reversible process, which is a process that can be done in reverse.
• A reversible process requires that any changes are made infinitely slowly.
• Real processes are not reversible due to friction , turbulence in the gas, etc.

Second Law of Thermodynamics: The Carnot Cycle

• Four steps• Isothermal expansion: a to b• Adiabatic expansion: b to c• Isothermal compression: c to d• Adiabatic compression: d to a
1 C
H H
QWefficiencyQ Q
ε= = = −
Define the efficiency ε
cost""work""
=≡εreservoirfromextractedheat
systembydonework
Valid for all heat engines.
(∆U = 0 for closed cycle)
where, W = QH - Qc; following conservation of energy

The Carnot Cycle
• Step 1: a to b.
– The gas is in contact with a heat bath at temperature THand weight is removed from the piston.
– The gas expands, while maintaining a constant temperature, isothermal
– the change in the internal energy is thus equal to zero
1
1 1
ln
0
ln ln
bH
a
b aH H H
a b
Vw nRT
VdU
V Vw q q nRT nRT
V V
= −
=
= − ≡ − = ≡ −

• Step 2: b to c.
– The gas is isolated from the environment and some more weight is removed from the piston.
– The gas expands and during the adiabatic expansion, the temperature of the gas will decrease.
– For adiabatic expansion pVγ
is constant, and we can thus related state b to state c: 0
)(
2
2
11
=−=
= −−
qTTnCw
VTVT
HLV
cLbHγγ
The Carnot Cycle

• Step 3: c to d.
– The gas is in contact with a heat bath at temperature TC and weight is added to the piston.
– The gas is compressed, while maintaining a constant temperature, isothermal
– the change in the internal energy is thus equal to zero
3
3 3
ln
0
ln
dL
c
dL L
c
Vw nRT
VdU
Vw q q nRT
V
= −
=
= − ≡ − =
The Carnot Cycle

The Carnot Cycle
• Step 4: d to a.
– The gas is isolated from the environment and some more weight is added to the piston.
– The gas is compressed and during the adiabatic compression, the temperature of the gas will increase.
0)(
4
4
11
=−=
= −−
qTTnCw
VTVT
CHV
dLaHγγ

11
1
0
ln
qwdU
VVnRTw
a
bH
−==
−=
0)(
2
2
11
=−=
= −−
qTTnCw
VTVT
HLV
cLbHγγ
33
3
0
ln
qwdU
VVnRTw
c
dL
−==
−=
0)(
4
4
11
=−=
= −−
qTTnCw
VTVT
CHV
dLaHγγ
• Four steps• Isothermal expansion: a to b
• Adiabatic expansion: b to c
• Isothermal compression: c to d
• Adiabatic compression: d to a

4321 wwwwwcycle +++=
c
dH
a
bHcycle V
VnRTVVnRTq
dU
lnln
0
+=
=∫c
dL
a
bHcycle
LHvc
dLHLv
a
bHcycle
VVnRT
VVnRTw
TTnCVVnRTTTnC
VVnRTw
lnln
)(ln)(ln
−−=
−+−−+−=
a
d
b
c
a
d
b
c
L
H
aHdL
cLbH
VV
VV
VV
VV
TT
VTVT
VTVT
=∴==
=
=
−
−
−
−
−−
−−
1
1
1
1
11
11
γ
γ
γ
γ
γγ
γγ
b
aLHcycle
c
dL
b
aHcycle
VVTTnRw
VVnRT
VVnRTw
ln)(
lnln
−=
−=

H
WefficiencyQ
ε= =
11
1
0
ln
qwdU
VVnRTw
a
bH
−==
−=
But, QH = q1 = -w1
33
3
0
ln
qwdU
VVnRTw
c
dL
−==
−=
And, Qc = q3 = -w3
a
d
b
c
a
d
b
c
L
H
aHdL
cLbH
VV
VV
VV
VV
TT
VTVT
VTVT
=∴==
=
=
−
−
−
−
−−
−−
1
1
1
1
11
11
γ
γ
γ
γ
γγ
γγ
00 =∂
⇒= ∫∑ Tq
Tq
i
i
Thus, the term δq/T is a state function
cost""work""
=≡εreservoirfromextractedheat
systembydonework
⎟⎟⎠
⎞⎜⎜⎝
⎛
⎟⎟⎠
⎞⎜⎜⎝
⎛
−=−=−
==
a
bh
c
dc
h
c
h
ch
h
VVT
VVT
QQQ
Qw
ln
ln11)(ε
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛⇒=⇒
−=−=
h
h
c
c
h
c
h
c
h
c
h
c
TQ
TQ
TT
TT
QQ )1()1(ε
0=⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟
⎠
⎞⎜⎜⎝
⎛⇒
h
h
c
c
TQ
TQ

Kelvin: It is impossible for any system to operate in a cycle that takes heat from a hot reservoir and converts it to work in the surroundingswithout at the same time transferring some heat to a colder reservoir.
QL
WOUT
QH
TH
TL
HE
Thermal efficiency = HQW−
=ε
For a complete cycle: dU = 0Qcy = - Wcy
Qcy = QH - Qc
∴
∴H
c
H
cH
Hcyc Q
QQQ
W−=
−=
−= 1ε
The engine will have 100% efficiency only when Qc = 0
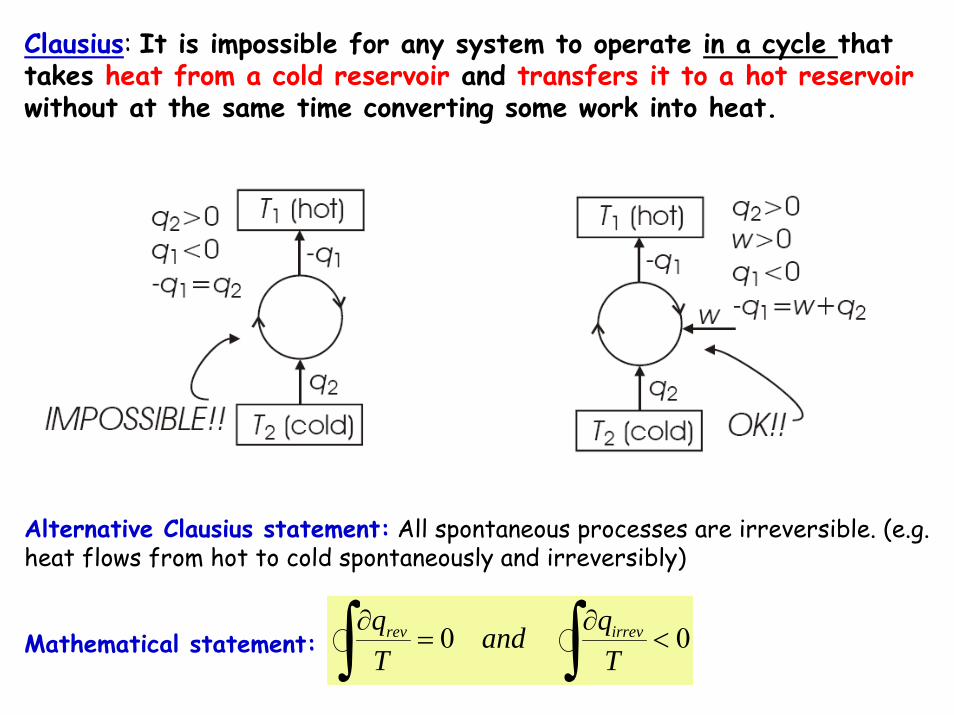
Alternative Clausius statement: All spontaneous processes are irreversible. (e.g. heat flows from hot to cold spontaneously and irreversibly)
Mathematical statement:
Clausius: It is impossible for any system to operate in a cycle that takes heat from a cold reservoir and transfers it to a hot reservoirwithout at the same time converting some work into heat.
00 <∂
=∂ ∫∫ T
qandTq irrevrev

WIN
WOUT
QH
HE TLTHQL QL QH
HPTL TH
A “Heat pump” is a machine or device that moves heat from one location (the 'source') at a lower temperature to another location (the 'sink' or 'heat sink') at a higher temperatureusing mechanical work or a high-temperature heat source.
“Refrigerator" if the objective is to cool the heat source (as in the normal operation of a freezer).
Thus a heat pump may be thought of a "heater" if the objective is to warm the heat sink or a "refrigerator" if the objective is to cool the heat source (as in the normal operation of a freezer)
The efficiency of a refrigerator or heat pump is given by a parameter called the coefficient of performance (COP).
In HP and R, in both cases, the operating principles are identical. Heat is moved from a cold place to a warm place.

tconsTq
Tq
Tq
Tq
Tq
TqbyMultiplyTT
TT
TTT
qqq
qqq
TT
TTT
L
L
H
H
H
H
L
L
LH
H
L
H
L
H
L
H
L
H
LH
H
LH
H
LH
H
L
H
LH
tan;0
011
1
=⇒==+−
=+−⇒−=−
−=
−∴
−=
−=−
=
η
η
Carnot’s theorem tells us that no real engine can have an efficiency more than that of the Carnot engine.

When the Carnot cycle is operating as a heat engine (going around clockwise), w < 0, so that -w > 0 and q > 0.
H
L
H
LH
a
bH
b
aLH
cyclecycle
b
aLHcycle
TT
TTT
VVnRT
VVTTnR
qw
qw
orEfficiency
VVTTnRw
−=−
=−−
=
=−
=
−=
1ln
ln)(
,
ln)(
11
η
ηε
• Efficiency of Carnot engine < 1 • No cycle can have an efficiency greater than a Carnot cycle• All reversible engines operates between same 2 temperatures have same
efficiency

const.; 1 =−γTV 11 −− = γγCCBh VTVT 11 −− = γγ
DCAh VTVT11 −−
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛γγ
D
C
A
B
VV
VV
⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟⎟
⎠
⎞⎜⎜⎝
⎛
D
C
A
B
VV
VV
)/( ln)/( ln ACD VVVV B−=
A (pA, VA, Th)
B (pB, VB, Th)
C (pC, VC, TC)
(pD, VD, TC) D
A (pA, VA, Th)
B (pB, VB, Th)
C (pC, VC, TC)
(pD, VD, TC) D
Th > Tc
Carnot Cycle (Ideal gas, All the steps are reversible)
1 (A to B) 2 (B to C) 3 (C to D) 4 (D to A) Total (A to A)
W - nRThln(VB/VA) CV(TC-Th) - nRTC ln(VD/VC) CV(Th-TC) + nR(TC– Th) ln(VB/VA)
q + nRTh ln(VB/VA) Zero + nRTC ln(VD/VC) Zero + nR(Th– TC) ln(VB/VA)
DU Zero CV(TC-Th) Zero CV(Th-TC) Zero
(qrev / T) + nR ln(VB/VA) Zero + nR ln(VD/VC) Zero Zero

Entropy
• Our study of the Carnot cycle showed that over the cycle q/T = 0.
• Since any reversible cycle can be approximated by a series of Carnot cycles, we expect that the integral of dQ/T along the closed path of the cycle = 0.
• One consequence is that the integral of dQ/Tbetween a and b is independent of the path.
• The quantity dQ/T is called the entropy dS.
• The entropy difference between a and b is thus path independent, and entropy is a state variable.

Entropy
• Entropy is a state variable, but Q and W are not state variables since they depend on the path used to get from a to b.
• For a reversible process, the change in the entropy of the engine is opposite to the change in entropy of the environment that provides the heat required (or absorbs the heat generated). The net change in the total entropy = 0.
• For an irreversible process, the net change in the total entropy will be larger than 0.

Entropy :– Is a measure of the disorderness of a system.– In a reversible thermodynamic process, entropy of a system
is related the change in the extent to which energy is dispersed in a disorderly manner, which in turn depends on the amount of energy transferred as heat (as heat stimulates motion) to the surroundings.
– Is a measure of the energy in a system or process that is unavailable to do work.
• For a process occurring at constant temperature (an isothermal process):
qrev = the heat that is transferred when the process is carried out reversibly at a constant temperature.T = temperature at which the heat is transferred (in Kelvin).

• The entropy is an extensive property, i.e. increases with the amount of matter in the system.
Entropy• The thermodynamic property of a system that is related to its
degree of randomness or disorder is called entropy (S).
• The entropy S and the entropy change ΔS=S2-S1 are state functions
• The entropy S has a unique value, once the pressure P, the temperature T and the composition n of the system are specified, S = S (p,T,n)

RefrigeratorsHeat Engines

Mathematical definition of entropy
dS = dqrev /T
dSsurr = -dq /Tsurr
∫=Δ2
1 TdqS rev
surrsurr T
qS −=Δ
dqrev ≥ dq
(dqrev/T) – (dq/T) ≥ 0
dS ≥ (dq/T)
dSsys + dSsurr > 0 Clausius inequality
Thermal equilibrium between system and surroundings

dSUniv = dSsys + dSsurr > 0dSUniv = dSsys + dSsurr = 0dSUniv = dSsys + dSsurr < 0
Spontaneous process
Equilibrium processImpossible process
2nd law of thermodynamics
all spontaneous processes are irreversible
dSsys + dSsurr ≥ 0
For any process“=“ for reversible, equilibrium“>” spontaneous (irreversible) (real)

More energy flows as work under reversible condition:
Now, ∆U from the two processes can be represented as:
Consider ∆U for the cyclic process 1→2→1. Internal energy being astate function, ∆U = 0
0)( ,
≥∂−∂∴
∂−≥∂− ∂∂
rev
irrevrev
wwww wrepresentswwhere
)12()21(
→∂+∂=Δ
→∂+∂=Δ
pathrevforqwUpathrevforqwU
revrev
irrevirrev
0,
0
≥∂
−∂
≥∂−∂=∂−∂∴
∂+∂=∂+∂∴
Tq
Tqor
qqwwqwqw
irrevrev
irrevrevrevirrev
irrevirrevrevrev
0≥∂−
TqdS irrev ∂
≥TqdS irrev 0<∂
∫ Tqirrev
Various forms of the Clausius Inequality
δq represents δqirrev
dSsys + dSsurr ≥ 0

dS + dSsurr ≥ 0 “=“ for reversible, equilibrium“>” spontaneous (irreversible) (real)
(closed system)
“=“ for reversible, equilibrium“>” for irreversible, spontaneous“<” for non-spontaneous
dS - dq/T ≥ 0
dU - TdS - dw ≤ 0
TdS - dq ≥ 0
dU - TdS – dwp-V – dwnon p-V ≤ 0
dq = dU - dw
dS = difference in entropy between two equilibrium states.Thermal equilibrium between system and surroundings
2nd LAW : Clausius inequality

Second Law of Thermodynamics
Entropy of the universe (i.e., Total Entropy):
• Does not change for reversible processes• Increases for spontaneous processes. • But, entropy can decrease for individual systems
Reversible (ideal):
Irreversible (real, spontaneous):

How do you calculate entropy change?
Calculate the actual q for the process from state 1 to state 2 and apply the above formula
For surrounding: A reservoir of large constant Volume
dSsurr = - dq /Tsurrsurr
surr TqS −
=Δ
When heat of q quantity is transferred to the Surrounding in a process,
• Then the U changes (∆Usurr) and it is independent whether thechange is Rev or Irrev.)
• Volume is constant, dV = 0 or, w = 0 ; So, ∆Usurr = dqrev
So, regardless of how the change was carried out, the change of entropy of the surrounding will also be given by amount of heat transferred reversibly divided by the temp at which heat is transferred
dSsurr = 0 and dqsurr = 0(ISENTROPIC)In an adiabatic process entropy change

In a process, where a given quantity of heat q is transferred to the system from a cold body and hot body, the Entropy change when it is TRANSFERRED TO a colder body will be always greater .
dq
A TH
B TC
For dq amount of heat leaving A, entropy change: ⎟⎟⎠
⎞⎜⎜⎝
⎛−
HTdq
For dq amount of heat enters B, entropy change:
⎟⎟⎠
⎞⎜⎜⎝
⎛
CTdqOverall change in Entropy = ⎟⎟
⎠
⎞⎜⎜⎝
⎛−
HTdq + ⎥
⎦
⎤⎢⎣
⎡−=
Hc TTdqdS 11
dS > 0 when TH > Tc; which means transfer of heat from a hot body to cold body is a spontaneous process.
When temp of the two system are equal, dStot =0, the two system are in thermal equilibrium
⎟⎟⎠
⎞⎜⎜⎝
⎛
CTdq
pliedisheatwhichattempTdS
sup(1
∝

Entropy Change in an Isolated System
ISOLATED SYSTEM : for irreversible change stateWe know: dS - dq/T ≥ 0 (Clausius inequality) where, dS = dqrev/T
In Isolate system: 1. Surrounding is NOT affected, dSsurr = 0 2. dq = 0 (for both Rev. and Irrev. change)So,dS > 0 (from Clausius inequality)
Therefore, •For any process occurring in an ISOLATED system, the ENTROPY INCREASES ( dS > 0)
• ENTROPY of the system will continue to INCREASE as long as changes occur. When no more changes takes place, the system is said to attain equilibrium and the entropy has reached the maximum value.
•Thus, the condition of equilibrium in an ISOLATED SYSTEM is that the ENTROPY has the MAXIMUM VALUE.

Entropy Change in an Isothermal Process: Ideal gas
dStotal > 0
From our experience:
For a spontaneous process, V2 > V1
dq = 0
dqrev = 1
2lnVVnRT
dSsys = dqrev / Tsys = 1
2lnVVnR
dStotal = 1
2lnVVnR
dSsurr = - dq / Tsurr= 0

Entropy Change
T
pdVdTCdVVU
VT
++⎟⎠⎞
⎜⎝⎛∂∂
=
TwdU
TdqdS revrev −
==
dVTp
TdTCdV
VU
T VT
++⎟⎠⎞
⎜⎝⎛∂∂
=1
Entropy Change in a Process for ideal gas
V dVnR
TdTC
TwdU
TdqdS V
revrev +=−
==
1
2
1
2
VVln ln nR
TTCS V +=Δ
1
2
2
1 lnln TTC
ppnRS p+=Δ

surrsurr T
qS −=Δ
TSTH
THSSuniv
)( Δ−Δ−=
Δ−+Δ=Δ
TGSuniv
Δ−=Δ
TqSsurr
−=Δ
THSsurr
Δ−=Δ
Tsurr = T
P const, only p-V work
P const, only p-V work,T const,
Tsurr = T
When Tsurr ≠ T0=Δ surrS
adiabatic

Thermodynamic Equations of State
U = f (V, T) ∫∫ −−=Δ dVPTdTCU V )(κα
∫∫ −−=Δ dPTVVdTCH P )( αH = f (P, T)
TC
TS V
V
=⎟⎠⎞
⎜⎝⎛∂∂
TC
TS P
P
=⎟⎠⎞
⎜⎝⎛∂∂
S = f (P, T) ∫∫ −=Δ VdPdTTCS P α
V α∂∂
−=⎟⎠⎞
⎜⎝⎛
TPS
∫=Δ dTTCS P
p1
2lnTTCP=
Heating at constant volume
∫=Δ dTTCS V
V1
2lnTTCV=
Heating at constant pressure

Calculation of Entropy change for Phase Transition
As degree of disorder changes at phase transition, an entropy change is associated with phase transition
qp = ΔH(reversible)
A wide range of liquid give approx. the same standard entropy of vaporization ~ 85 J K-1 mol-1
Trouton’s Rule
(unless there are strong molecular interactions in the liquid)
Normal Transition point is Ttrs at 1 atm
trs
trs
trs
psurrtrs T
HTq
S Δ−=−=Δ
trs
trs
trs
psurrtrs T
HTq
S Δ==Δ qtrs = ΔHtrs



Second law of thermodynamics
• It is impossible for heat to move spontaneously from a cold bodyto a hot body with no other result.
• It is impossible to convert heat quantitatively into work with no other resu"It is impossible to make a perpetual motion machine of the second kind." (A perpetual motion machine of the second kind is a machine thatconverts heat into work without doing anything else.)
• Mathematical statement of the second law of thermodynamics.
dS ≥ dq/T
dSsys + dSsurr ≥ 0
dS + (- dq / T) ≥ 0 .
dSiso ≥ 0 = for reversible process> for spontaneous (irreversible) process, all
real process dSuniv ≥ 0
Clausius inequality

Q. 1 mole of liquid water and ice at -5 °C and 1atm. Which one is more stable?
ΔS = ΔSl + ΔStrs + ΔSs ΔSsurr = Homework
Calculate Entropy Change in a Process

Calculate Entropy Change in a Adiabatic Irreversible Process
A (10 bar, 5 L)A: (10 bar, 5 L, ? K, )
B (1 bar)
TA = PA VA / n R = (106 x 5.10-3) / (2 x 8.3145) = 300.68 K
Q. 2 mole of monoatomic ideal gas undergoing irreversible adiabatic expansion against constant final pressure of 1 bar, calculate ΔS
dTCV=→BAdU
BABABAdU →→→ += wq
)(0)(C V ABBAB VVpTT −−=−
)2()(R232 A
B
BBAB V
pRTpTT −−=−×
TB = 192.44 KVB = 32 L
>
T
V
C (10 bar, 32 L)(300.6 K)
(32 L, 192.4 K)
ΔSA C = nCP,m ln (TC/TA)= 77.1 J K-1
ΔSC B = nCV,mln(TB/TC) = -57.43 J K-1
TC = PCVC / nR.= 1924.3 K
dVw exBA p −=→
REV
REV
-1BCCABA K J 19.73 =Δ+Δ=Δ →→→ SSS
D>E

dS – δq/Tsurr ≥ 0
TsurrdS – δq ≥ 0
TsurrdS – (dU- δw) ≥ 0
TsurrdS – (dU + pextdV) ≥ 0






Example• Show the Carnot cycle on a T-S diagram and identify the heat
transfer at both the high and low temperatures, and the work output from the cycle.
S
TTH
TL
S1=S4 S2=S3
1 2
34
A B
• 1-2, reversible isothermal heat transferQH = ∫TdS = TH(S2-S1) area 1-2-B-A
• 2-3, reversible, adiabatic expansionisentropic process, S=constant (S2=S3)
• 3-4, reversible isothermal heat transferQL = ∫TdS = TL(S4-S3), area 3-4-A-B
• 4-1, reversible, adiabatic compressionisentropic process, S1=S4
• Net work Wnet = QH - QL, the area enclosed by 1-2-3-4, the shaded area

Efficiency of an heat engine
WbyQC
Qh
TH
TC
2. What is efficiency of reverse engine (refrigerator)? Coefficient of Performance
a. QC / W b. Tc/ (TH-TC) c. 1-TH/TC
3. When can the heat engine convert the absorbed heat into work completely?.
a. At TH = -273 ˚C b. At Tc = O ˚C c. when TC =-273 ˚C
Consider a Carnot heat engine.1.What fraction of the total heat transferred Qh
to engine is converted into work in the cycle?
a. ∆T/TH b. ∆T/TC c. TC/TH

a.η th rev
L
H
TT
KK
or
,
( )( )
. .
= −
= −++
=
1
1 30 273652 273
0 672 67 2%
TT
KK
Q kJkJ
L
H
L
H
L
=
=++
=
==
( )( )
.
( . )
30 273652 273
0 328
500 0 328164
b.QL
WOUT
QH
TH = 652oC
TL = 30oC
HE
A Carnot heat engine receives 500 kJ of heat per cycle from a high-temperature heat reservoir at 652oC and rejects heat to a low-temperature heat reservoir at 30oC. Determine(a) The thermal efficiency of this Carnot engine.(b) The amount of heat rejected to the low-temperature heat reservoir.

Suppose a mole of a diatomic gas, such as O2, is compressed adiabatically so the final volume is half the initial volume. The starting state is V = 1 liter, T = 300 K. What are the final temperature and pressure?
Equation relating p,V for adiabatic process in α-ideal gasγ is the ratio of Cp/CV for diatomic gas in this case
Solve for pf from first equation
Substitute for pi
Use ideal gas law to calculate final temperature
OR use the equation relating T,V for an adiabatic process to get the final temperature (α = 5/2 for diatomic gas)
Solve for Tf
K
VVTT
VTVTK
nRVp
T
Pax
VV
VnRT
VVpp
VpVp
f
iif
ffii
fff
f
i
i
i
f
iif
ffii
395
395
1057.6
4.12/52/7
1
6
=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
=
=
=
=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
==
=
α
αα
γ
γ
γγ
γ

Entropy change in heat engine
Consider a Carnot heat engine.1. What is the sign of the entropy change of
the hot reservoir during one cycle?a. ΔSh < 0 b. ΔSh = 0 c. ΔSh > 0
WbyQC
Qh
Th
TcBecause energy is flowing out of the hot reservoir, its entropy (the number of microstates) is decreasing.2. What is the sign of the entropy change of the cold reservoir?a. ΔSc < 0 b. ΔSc = 0 c. ΔSc > 0
Because energy is flowing into the cold reservoir, it’s entropy (number of microstates) is increasing.
3. Compare the magnitudes of the two changes.a. |ΔSc| < |ΔSh| b. |ΔSc| = |ΔSh| c. |ΔSc| > |ΔSh|

Example Problem (1)
Solution:
How much heat is absorbed by 3 moles of helium when it expands from V = 10 liters to V = 20 liters and the temperature is kept at a constant 350 K? What are the initial and final pressures?
Q = -Won
Won = -nRT ln(Vf/Vi)= -6048 J
pi = nRT/Vi = 8.72×105 Papf = pi/2 = 4.36 ×105 Pa
From first law. For an ideal gas, isothermal means ∆U = 0. Positive Q means heat flows into the gas.
This was derived in lecture.R = 8.31 J/mole·K
pV = nRT
Where is the heat coming from? In order to keep the gas at a constant temperature, it must be put in contact with a large object having that temperature. That object is called a “heat reservoir”, and it supplies heat to the gas (or absorbs heat, if necessary) in order to keep the gas temperature constant.



⇒
substituting ⇒




It is not possible to calculate the entropy change ∆S = SB - SAfor an irreversible process between A and B, by integrating δq / T, the ratio of the heat increment over the temperature, along the actual irreversible path A-B characterizing the process. Indeed, the Second Law states that: dS > δq / T for an irreversible process.
However, since the entropy is a state function, the entropy change ∆S does not depend on the path chosen. Since we know how to calculate entropy changes only along reversible paths, wemust imagine another path between states A and B, an “imagined”or hypothetical path that is reversible. We will now consider some examples to illustrate this concept.

Example 1:Q. Two blocks of metal A, and B, of masses mA and mB, initially at two different temperatures TA and TB are placed in contact with one another in a thermally insulated container.Ans. Heat is exchanged only between the two blocks until they exhibit the same final temperature, TF . Considering the two blocks to form the System, and no heat is exchanged between system and surroundings.
Therefore,
If MA and MB are the molar mass of A and B, and by CP,m(A) and CP,m(B) be the molar heat capacities at constant pressure for A and B, respectively, then heat is exchanged at constant pressure will be:
and the same final temperature, TF will be:
Note: if A and B are made of the same material and have the same mass, then the final temp, TF, is half-way between the initial temp, TA and TB.

Now to calculate the changes in entropy, lets us imagine a reversible path from the initial state to the final state. A reversible path could be aconstant-pressure very slow heating of A from TA to TF and a constant-pressure very slow cooling of B from TB to TF.
For such processes, the changes in entropy will be given as:
As the process is adiabatic so, ∆Ssurr= 0
Therefore, ∆Ssur = ∆Ssurr + ∆Ssys = ∆SA + ∆SB
Example 1:Two blocks A and B made of the same material of molar mass 100 g/mol and of mass (1000 g) and molar heat capacity to be 24.44 J.mol-1.K-1 are brought in contact in an insulated jacket. If the initial temperatures of the blocks A and B are respectively, 0°C and 100°C, then the final temperature is TF = 50°C , what will be the entropy change for the two blocks ?

∆Ssur = 0, then ∆ ST = ∆ S. Therefore, ∆ ST > 0, in accord with the requirement that ∆ ST > 0 for a spontaneous process.
Therefore, heat transfer and temperature equilibration processes are spontaneous.
Example #2:Consider what happens when you mix under a pressure of 1 bar, 500 g of iceat -10°C with 100 g of H2O vapor at +110°C. Assume the mixing takes place in a thermally insulated container, so that heat transfer occurs exclusively between the ice and the vapor. What is the final temperature and what is the change in entropy for the process (system + surroundings) ?Assume the latent heat of vaporization (standard enthalpy change for vaporization) is 44 kJ/mol and the latent heat of fusion is 6 kJ/mol. Theseprocesses occur reversibly at 1 bar at 373 K and 273 K, respectively. Assume the molar heat capacity at constant pressure of water vapor, liquid and solids are 33.58, 75.29 and 36.8 J.K-1.mol-1, respectively.We can now calculate the entropy change for the system by following the reversible path consisting of:Heating slowly the ice from -10°C to 0°C→transforming the ice reversibly to water at 0°C and 1 bar→ heating up slowly the molten ice (water liquid) from 0°C to 44°C → Cooling slowly the vapor from 110°C to 100°C → transforming reversibly the water vapor to water liquid at 100°C and 1 bar → cooling slowly the water liquid from 100°C to 44°C.

N.B. In the above problem, we assumed that the final state was water liquid at some temperature TF, (i.e. that all the vapor had condensed while all the ice melted). Other final states were possible !! If the mass of ice was very small compared to that of vapor and warm up the resulting liquid to 100°C, at which temperature some or all of that molten ice could now vaporize. The final state could therefore have been 600 g of vapor at some temperature between 100 and 110°C or a mass m (m less than 500 g) of ice, the final state could have been 600 g of ice at a temperature between 0 and -10°C or a mass m of ice in equilibrium at 0°C with a mass 600 – m of liquid. Whatever assumption is used, it leads to the existence of one unknown, (either the mass when two phases are expected in the final state or the final temperature if only one phase is present). Using qtotal = 0, enables one to solve for the unknown and to check whether our assumption is valid.Once the assumption is verified and the final state is “unambiguousl characterized”, then the entropy calculation along the reversible path can be carried out.

Example #3Consider the adiabatic irreversible expansion of two moles of an ideal monoatomic gas under an external pressure, Pext = 1 bar. In the initial state, the gas occupies a volume VA = 5 L under a pressure of PA = 10 bars. In thefinalstate the pressure is PB = 1 bar.
We know initial pressure, volume and number of moles, therefore we can calculate the initial temperature TATA = PA VA / n R = (106 x 5.10-3) / (2 x 8.3145) = 300.68 K
Final state is given by writing: dU = δw + δq = δw (adiabatic process).
dU = CV dT (CV = 2 x 3R/2 which is constant for an ideal monoatomic gas).
Since the expansion occurs under constant external pressure therefore,w = -Pext ∆V
Therefore from : dU = w, we get3R (TB - TA) = -Pext (VB - VA) ; Since VB = n R TB / PB and PB = Pext we get:TB = (TA + Pext VA / 3R) / (1 + n / 3) TB = 192.44 K and VB = 32 L.
Now that we know the final and initial states, we can design any number ofreversible processes between A and B.

1) We can envision, an isobaric slow heating from state A to a state C, characterized by: PC = PA and VC = VB The temperature at state C is defined by TC = PC VC / nR.Therefore, TC = PA VB / nR = 1924.3 K.
Then we carry out an isochoric slow cooling from state C to state B.The change in entropy between A and B, is given by: ∆SA-B = ∆ SA-C + ∆ SC-B∆ SA-C = n CPm ln(TC / TA) = 2 (5R/2) ln (1924.3/300.68) = 77.16 J/K and∆ SC-B = n Cvm ln(TB / TC) = 2 (3R/2) ln (192.44/1924.3) = -57.43 J/K.The change in entropy for the A-B process is therefore = 19.73 J/K
2) We could have also envisioned the path A → D → B, which consists in a slow isochoric cooling from A to D followed by a slow isobaric heating from D to B (see figure).State D is characterized by PD = PB and VD = VA. In this case TD = PD VD / nR which is to say TD = PB VA./ n R = 30.07 K
The entropy change between A and B is therefore the sum of the entropy changes between A and D and between D and B. Therefore:∆ SA-D = n CVm ln(TD / TA) = 2 (3R/2) ln (30.07/300.68) = -57.43 J/K and∆ SD-B = n CPm ln(TB / TD) = 2 (5R/2) ln (192.44/30.07) = 77.16 J/K.The change in entropy for the A-B process is therefore = 19.7 J/K

Thus, you could envision any other reversible path involving combinations of adiabatic reversible, isothermal reversible, etc… and get the same answer.
You may want to practice this type of calculation on your own following the path A → E → B where the transformation A → E is isothermal (TA = TE)and reversible to a volume VE = VB and the process E → B is a slow isochoric cooling from TE to TB. (as shown in the figure). You should again find that ∆SA→B is identical to that found by the other paths.

Entropy :– Is a measure of the disorderness of a system.– In a reversible thermodynamic process, entropy of a system
is related the change in the extent to which energy is dispersed in a disorderly manner, which in turn depends on the amount of energy transferred as heat (as heat stimulates motion) to the surroundings.
– Is a measure of the energy in a system or process that is unavailable to do work.
• For a process occurring at constant temperature (an isothermal process):
qrev = the heat that is transferred when the process is carried out reversibly at a constant temperature.T = temperature at which the heat is transferred (in Kelvin).

Second Law of Thermodynamics
Entropy of the universe (i.e., Total Entropy):
• Does not change for reversible processes• Increases for spontaneous processes. • But, entropy can decrease for individual systems
Reversible (ideal):
Irreversible (real, spontaneous):

dSUniv = dSsys + dSsurr > 0dSUniv = dSsys + dSsurr = 0dSUniv = dSsys + dSsurr < 0
Spontaneous process
Resversible processImpossible process
2nd law of thermodynamics
all spontaneous processes are irreversible
dSsys + dSsurr ≥ 0
For any process“=“ for reversible, equilibrium“>” spontaneous (irreversible) (real)

Entropy Change in a Process for ideal gas
V dVnR
TdTC
TwdU
TdqdS V
revrev +=−
==
1
2
1
2
VVln ln nR
TTCS V +=Δ
2
1
1
2 ln lnppnR
TTCS p +=Δ
For Reversible Process
∫=Δ dTTCS P
p1
2lnTTCP=
Heating at constant pressure Heating at constant volume
∫=Δ dTTCS V
V1
2lnTTCV=

Entropy Change for any Reversible Process
T
pdVdTCdVVU
VT
++⎟⎠⎞
⎜⎝⎛∂∂
=T
wdUT
dqdS revrev −==
dVTp
TdTCdV
VU
T VT
++⎟⎠⎞
⎜⎝⎛∂∂
=1
Thermodynamic Equations of State
U = f (V, T) ∫∫ −−=Δ dVPTdTCU V )(κα
∫∫ −−=Δ dPTVVdTCH P )( αH = f (P, T)
TC
TS V
V
=⎟⎠⎞
⎜⎝⎛∂∂
TC
TS P
P
=⎟⎠⎞
⎜⎝⎛∂∂
S = f (P, T) ∫∫ −=Δ VdPdTTCS P α V α
∂∂
−=⎟⎠⎞
⎜⎝⎛
TPS

Example: Entropy Change in an Isothermal Process: Ideal gas
dStotal > 0
From our experience:
For a spontaneous process, V2 > V1
dq = 0
dqrev = 1
2lnVVnRT
dSsys = dqrev / Tsys = 1
2lnVVnR
dStotal = 1
2lnVVnR
dSsurr = - dq / Tsurr= 0

Calculation of Entropy change for Phase Transition
As degree of disorder changes at phase transition, an entropy change is associated with phase transition
qp = ΔH(reversible)
A wide range of liquid give approx. the same standard entropy of vaporization ~ 85 J K-1 mol-1
Trouton’s Rule
(unless there are strong molecular interactions in the liquid)
Normal Transition point is Ttrs at 1 atm
trs
trs
trs
psurrtrs T
HTq
S Δ−=−=Δ
trs
trs
trs
psystrs T
HTq
S Δ==Δ qtrs = ΔHtrs
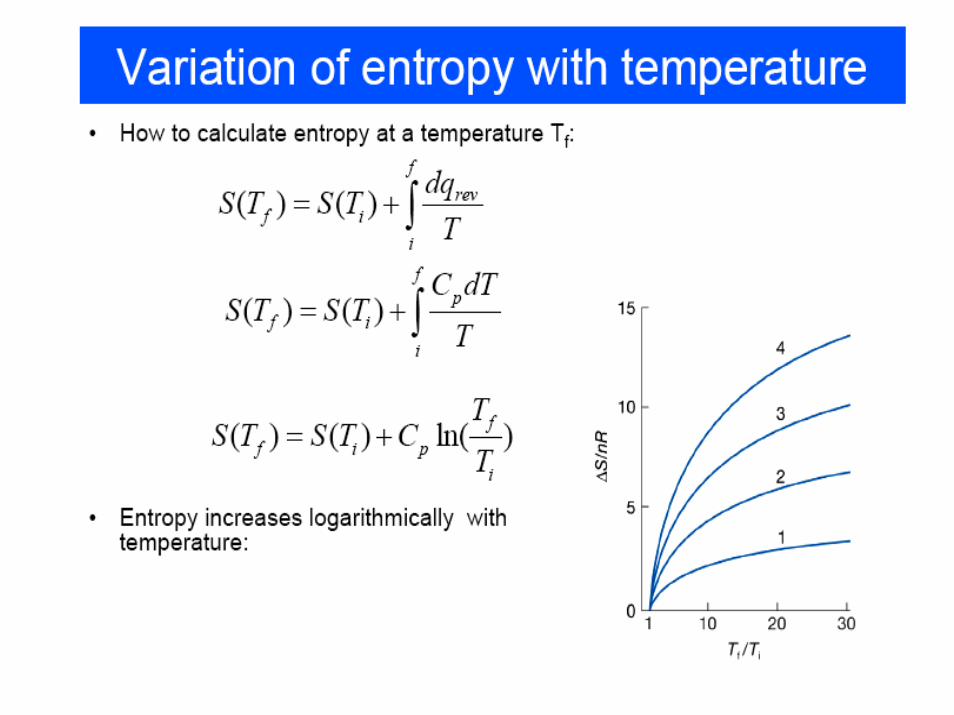


Calculation of Entropy Change in a Process
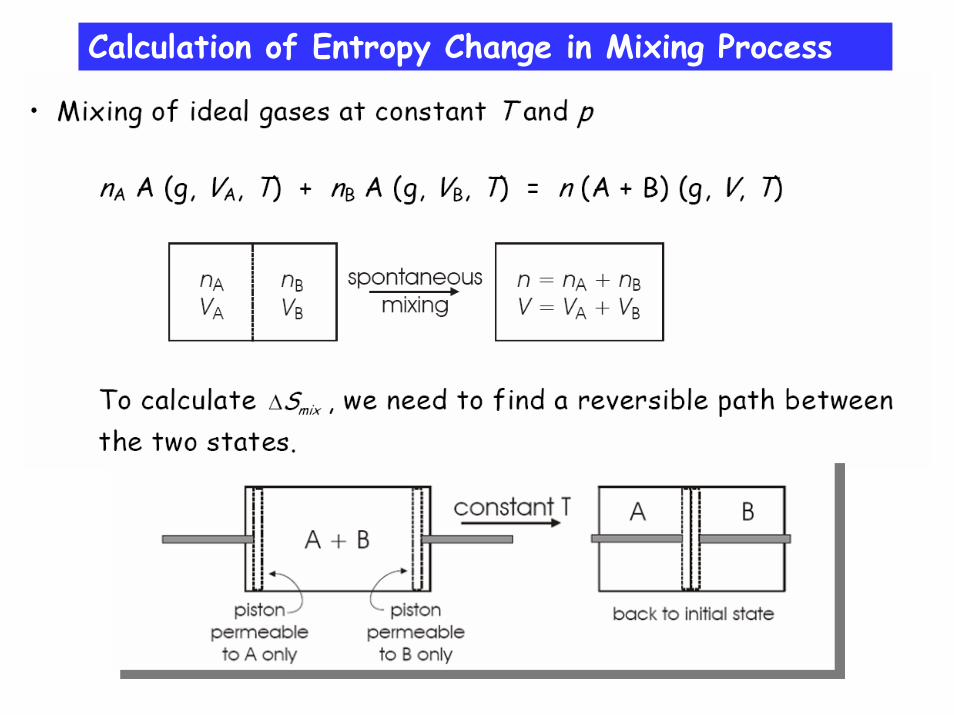
Calculation of Entropy Change in Mixing Process

Calculation of Entropy Change in Mixing Process

Entropy is a state function, but to calculate ∆S we require a reversible path
.
** An irreversible isothermal expansion requires less heat than a reversible one
As ∆U (state function) = 0
(q <0)
•An irreversible Carnot (or any other) cycle is less efficient than a reversible one
Irrev. Isothermal Expansionq1
q2

The Entropy of an Isolated System NEVER Decreases
(A): The system is isolated and irreversibly(spontaneously) changes from [1] to [2] Path (A): qirrev =0 (isolated)
(B): The system is brought into contact with a heat reservoir and reversibly brought back from [2] to [1]
From Clausius Inequality we know,
This gives the direction of spontaneous change!

Here, ∆Ssys (Entropy change of aSYSTEM) is INDEPENDENT of path of process
But! ∆Ssurr DEPENDS on whether the process is reversible or irreversible
Consider the UNIVERSE as an isolated system containing our initial system and its surroundings, for any process which is
• Irreversible:. (b) Reversible:.
Entropy Change for System and Surrounding

Calculation of Entropy Change in a Spontaneous ProcessTwo metal blocks (of mass mA and mB) thermally connected are placed in an Insulated Chamber under constant pressure
Considering the two blocks to form the System, and no heat is exchanged between system and surroundings.Isolated system ↔(∆U = 0)Therefore: δq = q1→2 + q2→1 = 0 or, δq1 =- δq2
The change in entropy for the surroundings is zero because the process was adiabatic (total heat is zero). Therefore, the total change in entropy for system + surroundings is equal to the sum of the entropy change for 1 and the entropy change for 2.

Joule Expansion with an Ideal Gas: Expansion into Vacuum
Adiabatic irreversible expansion of Ideal gas into vacuum
Note that to calculate ∆S for the irreversible process, we needed to find a reversible path so we could determine
Free expansion

Summary of Second law of thermodynamics
• It is impossible for heat to move spontaneously from a cold body to a hot body with no other result.
• It is impossible to convert heat quantitatively into work with no other result."It is impossible to make a perpetual motion machine of the second kind." (A perpetual motion machine of the second kind is a machine that converts heat into work without doing anything else.)
• Mathematical statement of the second law of thermodynamics.
dS ≥ dq/T
dSsys + dSsurr ≥ 0
dS + (- dq / T) ≥ 0 .
dSiso ≥ 0 = for reversible process> for spontaneous (irreversible) process, all
real process dSuniv ≥ 0
Clausius inequality

dS – δq/Tsurr ≥ 0
TsurrdS – δq ≥ 0
TsurrdS – (dU- δw) ≥ 0
TsurrdS – (dU + pextdV) ≥ 0







Criteria for spontaneity
For Closed system (constant composition), NO non p-V work
dSU,V > 0
dUS,V < 0 dHS,p < 0 dAV,T < 0
dGp,T < 0dSH,p > 0
dSsys + dSsurr > 0
At normal lab condition of const. T & p; G should decrease for spontaneous processes
Reversible processes will have an equality sign

, qor dST∂
≥



dU - TdS – dwp-V – dwnon p-V ≤ 0
For a closed system (constant composition)
Const. V dwp-V = 0
dU - TdS - dwnon p-V ≤ 0
Const. T A = U - TS
dAV, T ≤ dwnon p-V
for dwnon p-V = 0
dAV, T, no non p-V work ≤ 0
Const. T
dA- dw tot ≤ 0
A = U - TS
dA ≤ dwtot
dA = (- dwtot) max
dA ≥ (- dwtot)
for dwnon p-V = 0
dU - TdS ≤ 0
dUS, V, no non p-V work ≤ 0
dSU, V, no non p-V work ≥ 0
Helmoltz’s energy / Helmoltz’s free energy / Helmoltz’s function
Summary of Spontaneity Conditions (General form) at Constant V and T

dU - TdS – dwp-V – dwnon p-V ≤ 0
dH - TdS - dwnon p-V ≤ 0
Const. T G = H - TS
dGp, T ≤ dwnon p-V
for dwnon p-V = 0
dGp, T, no non p-V work ≤ 0
dGp, T = (- dwnon p-V) max
dGp,T ≥ (- dwnon p-V)
dHS, p, no non p-V work ≤ 0
dSH, p, no non p-V work ≥ 0
Const. p H = U + pV Const. p H = U + pV
dH - TdS - dwnon p-V ≤ 0
Gibbs’s energy / Gibbs’s free energy / Gibbs’s function
for dwnon p-V = 0
dH - TdS ≤ 0
For a closed system (constant composition)
Summary of Spontaneity Conditions (General form) at Constant P and T

Fundamental Equations from H, A, G
Combination of 1st and 2nd Law(true for any path)
For a closed system (const. composition), no non p-V (additional) work
PdVTdSdUdwdqdU−=+=
VdPTdSdHVdPPdVPdVTdSdH
VdPPdVdUdHPVUH
+=
++−=
++=
+=
SdTPdVdASdTTdSPdVTdSdA
SdTTdSdUdATSUA
−−=−−−=
−−=−=
SdTVdPdGSdTTdSVdPTdSdG
SdTTdSdHdGTSHG
−=
−−+=
−−=
−=

TdSVdpdTnCp +−=
TdTnCdp
TVdS p+=
TdTnC
pdpnRdS p+−=
TdSpdVdTnCV +−=
TdTnCdV
TpdS V+=
TdTnC
VdVnRdS V+=
1
2
1
2 lnVVln
TTnCnRS V+=Δ
1
2
2
1 lnln TTnC
ppnRS p+=Δ
Entropy Change Derived from the Fundamental Equations of U & H

The Maxwell Relations
If Z=f(x,y),and Z has continuous second partial derivatives, then
yxxy yZ
xxZ
y ⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∂∂
=⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛∂∂
∂∂
That is:
xyZ
yxZ
∂∂∂
=∂∂
∂ 22
dyyxNdxyxMdf ),(),( +=
yx xN
yM
⎟⎠⎞
⎜⎝⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂For exact differential

The Maxwell RelationsdU = f(V,S)
dSSUdV
VUdU
VS⎟⎠⎞
⎜⎝⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
=
TSU
V
=⎟⎠⎞
⎜⎝⎛∂∂p
VU
S
−=⎟⎠⎞
⎜⎝⎛∂∂
As dU is an exact differential
VS Sp
VT
⎟⎠⎞
⎜⎝⎛∂∂
−=⎟⎠⎞
⎜⎝⎛∂∂ Therefore,
dSSVNdVSVMdU ),(),( +=
V T
S p
- ve
- ve
Comparing with

V T- ve
S p- ve

These are the Maxwell Relations
VT TP
VS
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
PT TV
PS
⎟⎠⎞
⎜⎝⎛−=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
PS SV
PT
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
SV VT
SP
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛−
∂∂
∂∂
The first two are little used.
The last two are extremely valuable.
The equations relate the isothermal pressure and Isobaricvolume variations of entropy to measurable properties
that can be obtained from an equation of state.


Now suppose (G/T) is differentiated w.r.t. T
we will get: Substitute (4) in (5)
we get the Gibbs-Helmholtz Equation
Alter
nate
for
m By applying the Gibbs-Helmholtz equation to both reactants and products of a chemical reaction,


Criteria for spontaneity
For Closed system (constant composition), NO non p-V work
dSU,V > 0
dUS,V < 0 dHS,p < 0 dAV,T < 0
dGp,T < 0dSH,p > 0
dSsys + dSsurr > 0
At normal lab condition of const. T & p; G should decrease for spontaneous processes
Reversible processes will have an equality sign

, qor dST∂
≥

)( InequalityClausiusTqdS ∂
≥

Similarly, dG is gives the Maximum non-PV Work

Fundamental Equations from H, A, G
Combination of 1st and 2nd Law(true for any path)
For a closed system (const. composition), no non p-V (additional) work
PdVTdSdUdwdqdU−=+=
VdPTdSdHVdPPdVPdVTdSdH
VdPPdVdUdHPVUH
+=
++−=
++=
+=
SdTPdVdASdTTdSPdVTdSdA
SdTTdSdUdATSUA
−−=−−−=
−−=−=
SdTVdPdGSdTTdSVdPTdSdG
SdTTdSdHdGTSHG
−=
−−+=
−−=
−=

TdSVdpdTnCp +−=
TdTnCdp
TVdS p+=
TdTnC
pdpnRdS p+−=
TdSpdVdTnCV +−=
TdTnCdV
TpdS V+=
TdTnC
VdVnRdS V+=
1
2
1
2 lnVVln
TTnCnRS V+=Δ
1
2
2
1 lnln TTnC
ppnRS p+=Δ
Entropy Change Derived from the Fundamental Equations of U & H

The Maxwell Relations
If Z=f(x,y),and Z has continuous second partial derivatives, then
yxxy yZ
xxZ
y ⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
∂∂
=⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛∂∂
∂∂
That is:
xyZ
yxZ
∂∂∂
=∂∂
∂ 22
dyyxNdxyxMdf ),(),( +=
yx xN
yM
⎟⎠⎞
⎜⎝⎛∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂For exact differential

The Maxwell RelationsdU = f(V,S)
dSSUdV
VUdU
VS⎟⎠⎞
⎜⎝⎛∂∂
+⎟⎠⎞
⎜⎝⎛∂∂
=
TSU
V
=⎟⎠⎞
⎜⎝⎛∂∂p
VU
S
−=⎟⎠⎞
⎜⎝⎛∂∂
As dU is an exact differential
VS Sp
VT
⎟⎠⎞
⎜⎝⎛∂∂
−=⎟⎠⎞
⎜⎝⎛∂∂ Therefore,
dSSVNdVSVMdU ),(),( +=
V T
S p
- ve
- ve
Comparing with


V T- ve
S p- ve

These are the Maxwell Relations
VT TP
VS
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
PT TV
PS
⎟⎠⎞
⎜⎝⎛−=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
PS SV
PT
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
∂∂
∂∂
SV VT
SP
⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛−
∂∂
∂∂
The first two are little used.
The last two are extremely valuable.
The equations relate the isothermal pressure and Isobaricvolume variations of entropy to measurable properties
that can be obtained from an equation of state.

where




Now suppose (G/T) is differentiated w.r.t. T
we will get: Substitute (4) in (5)
we get the Gibbs-Helmholtz Equation
Alter
nate
for
m By applying the Gibbs-Helmholtz equation to both reactants and products of a chemical reaction,

For solids and liquids V does not change much with P, so
where,

Standard Gibbs energy, G˚, varies with Temp.
It is defined as the free energy change accompanying the formation of a compound from the element, when all the reactant and product are in their standard state.

Similarly,



OPEN SYSTEMS

• At equilibrium, the chemical potential of a species is the same everywhere in the system
• µi is the Gibbs free energy per mole of component “i ”, i.e. the partial molar Gibbs free energy
• G is extensive but µi (or, Gm) is Intensive property, i.e.,

OPEN SYSTEMS
U f S V n n
dU US
dS UV
dV Un
dn
dU TdS PdV dn where Un
V n S n i S V ni
i
n
i ii
n
ii S V n
i i J
J
=
= ⎛⎝⎜
⎞⎠⎟
+ ⎛⎝⎜
⎞⎠⎟
+⎛⎝⎜
⎞⎠⎟
= − + =⎛⎝⎜
⎞⎠⎟
=
=
∑
∑
( , , , ,...)
,
, , , ,
, ,
1 2
1
1
∂∂
∂∂
∂∂
μ μ ∂∂
The chemical potential:dU TdS PdV dn where U
n
dH TdS VdP dn where Hn
dA SdT PdV dn where An
dG SdT VdP dn where Gn
i ii
n
ii S V n
i ii
n
ii S P n
i ii
n
ii T V n
i ii
n
ii T P n
J
J
J
J
= − + =⎛⎝⎜
⎞⎠⎟
= + + =⎛⎝⎜
⎞⎠⎟
= − − + =⎛⎝⎜
⎞⎠⎟
= − + + =⎛⎝⎜
⎞⎠⎟
=
=
=
=
∑
∑
∑
∑
μ μ ∂∂
μ μ ∂∂
μ μ ∂∂
μ μ ∂∂
1
1
1
1
,
,
,
,
, ,
, ,
, ,
, ,
Special case for n moles of a pure substance:G nG G molar Gibbs free energy
Gm m
m
= =∴ =
,μ

Properties of Chemical Potential:
1. Chemical potential in a pure (1-component or pure subs): n moles ideal gas
G G nRT PP
RT PP
oo
oo
= +
= +
ln
lnμ μ
µi,A µi,B
A BdG dn dG dnAi A i
Bi B i= − =μ μ, ,( ) &
2. Chemical potential of a species during transfer of matter
net change in Gibbs free energydG dG dG dnA B
i B i A i= + = −( ), ,μ μ
μ μi A i B dG, ,> < 0μ μi A i B dG, ,< > 0
μ μi A i B dG, ,= = 0
Case I: Case II:
Case III:
•Spontaneous transfer of matter from region of high µ to region of low µ•At equilibrium µ is the same everywhere



3. Chemical potential in a mixture of ideal gases
of the underlying
Chemical potential of “A” in the mixture is less than if it were pure, under the same (T,p) conditions, because of the underlying entropy change!


Gibbs energy for ideal gas at constant Temp was given as:
bar1Pwhere;PlnRTμμand;PPlnnRTGG oo
oo =+=+=
For Real gases: Lewis recognized that it would be convenient to keep same formal relation by defining a new function – the fugacity function, such that:
( ) t)coefficien(fugacityPfwhere,
;PflnRTμμand;
PflnnRTGG o
oo
o
=
⎟⎠⎞
⎜⎝⎛+=⎟
⎠⎞
⎜⎝⎛+=
φ
1Limor,1PfLim 0P0P ==⎟⎠⎞
⎜⎝⎛
→→ )(φ
In solution : we use Activity
)tcoefficienactivity(where,caGenerally,
Pf
PPa);(alnRTμμ
iii
oi
oi
iio
ii
==
==+=
γγ
Non-ideality:

Fugacity:
• Has units of pressure and if function of T and P
• As p → 0, the gas approaches ideality and f (fugacity) → P
• It is the measure of the molar Gibbs free energy of a Real Gas
• Real gas behaves like ideal gas in the limits of LOW Pressure and HIGH Temp and they deviate at High P and Low Temp.
• Compressibility factor , Z, is a measure of the deviation from ideality and it is defines as,
Vol)molarV(where,RTVP
VVzid
===
1Limor,1PfLim 0P0P ==⎟⎠⎞
⎜⎝⎛
→→ )(φ
Fugacity of a Real gas can be calculated at constant T and a particular P from the equation of state
Non-ideality:

=
==∴
=
+−=+−=
+=+=
idid
oo
dPVdμ and,
0)dT(whendPVdμT)constant(atVdPdG
dPVdTSdμor,VdPSdTdGlnfRTμμor,lnfnRTGG
⎥⎦⎤
⎢⎣⎡ −
=⎟⎠⎞
⎜⎝⎛∴
==
⎟⎠⎞
⎜⎝⎛=−
=⎟⎠⎞
⎜⎝⎛−=−
∫
∫
→
P0
id
P0
id
0P
idid
dPP1zexp
Pf
PRTV;
PRTzVwhere,
PflnRT)dPVV(
1Pfputting,)dPVV()μd(μ lim
Vol)molarV(where,RTVP
VVzid
===
To Calculate Fugacity or, fugacity coeff. at constant T for a given P

P →
f →
Standard state
P = 1 bar
1
Fugacity coefficient
Fugacity
Compressibilityfactor

How to describe physical properties of mixtures knowing the properties of their pure ingredients
Simple Mixtures
Simple mixtures:example: the volume of a mixture
a)perfect (ideal) mixtures: no interactions
b)real mixtures: interactions
Imagine a huge volume of water, and add 1 mol H2O. The volume will increase by 18 cm3, because18 cm3 mol-1 is the molar volume of water.
•

• Now, add 1 mol H2O to a huge volume of pure ethanol. The volume will increase by 14 cm3 mol-1, which is the partial molar volume of water in pure ethanol. The reason for the different increase is that the volume occupied by a given number of water molecules depends on the identity of the molecules that surround them.
• In general, the partial molar volume of a substance A in a mixture is the change in volume per mole of A added to a large volume of the mixture.
• The partial molar volumes of the components of the mixture vary with composition because the environment of each type of molecule changes as the composition changes from pure A to pure B.
The partial molar volumes of water and ethanol at 25°C.Note the different scales (water on the left, ethanol on the right).

The partial molar volume of a substance is the slope of the variation of the total volume of with the composition, as shown by the different slopes at thecompositions a and b. Note that the partial molar volume at b is negative: the overall volume of the sample decreases as a is added.

)( InequalityClausiusTqdS ∂
≥

Similarly, dG is gives the Maximum non-PV Work

where, PA = χA P; PB = χB PχA = nA/n; χA < 1



Perfect gases mix spontaneously in all proportions
nT
mixmix
np
mixmix
pGV
TGS
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ=Δ
⎟⎠⎞
⎜⎝⎛ Δ
−=Δ
,
,
)(
)(
∂∂
∂∂
mixmixmix STGH Δ+Δ=Δ


where,

OPEN SYSTEMS

• At equilibrium, the chemical potential of a species is the same everywhere in the system
• µi is the Gibbs free energy per mole of component “i ”, i.e. the partial molar Gibbs free energy
• G is extensive but µi (or, Gm) is Intensive property, i.e.,

OPEN SYSTEMS
ji
Jii
nVSii
n
iii
n
ii
nVSinSnV
nUwherednPdVTdSdU
dnnUdV
VUdS
SUdU
nnVSfU
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛=+−=
⎟⎟⎠
⎞⎜⎜⎝
⎛+⎟
⎠⎞
⎜⎝⎛+⎟
⎠⎞
⎜⎝⎛=
=
∑
∑
=
=
,,1
1 ,,,,
21
,;
,...),,,(
∂∂μμ
∂∂
∂∂
∂∂
The chemical potential:
ij
ji
ji
ji
nPTii
n
iii
nVTii
n
iii
nPSii
n
iii
nVSii
n
iii
nGwherednVdPSdTdG
nAwherednPdVSdTdA
nHwherednVdPTdSdH
nUwherednPdVTdSdU
≠
≠
≠
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛=++−=
⎟⎟⎠
⎞⎜⎜⎝
⎛=+−−=
⎟⎟⎠
⎞⎜⎜⎝
⎛=++=
⎟⎟⎠
⎞⎜⎜⎝
⎛=+−=
∑
∑
∑
∑
=
=
=
=
,,1
,,1
,,1
,,1
,;
,;
,;
,;
∂∂μμ
∂∂μμ
∂∂μμ
∂∂μμ
Special case for n moles of a pure substance (i=1):
G nG G molar Gibbs free energyGm m
m
= =∴ =
,μ
( )m
m Gn
nG=⎥⎦
⎤⎢⎣⎡
∂∂
=μ

Properties of Chemical Potential:
1. Chemical potential in a pure (1-component or pure subs): n moles ideal gas,Gm and μindep P for liquid and solids
G G nRT PP
RT PP
oo
oo
= +
= +
ln
lnμ μ
µi,A µi,B
A BdG dn dG dnAi A i
Bi B i= − =μ μ, ,( ) &
2. Chemical potential of a species during transfer of matter (T,p)
net change in Gibbs free energydG dG dG dnA B
i B i A i= + = −( ), ,μ μ
μ μi A i B dG, ,> < 0μ μi A i B dG, ,< > 0
μ μi A i B dG, ,= = 0
Case I: Case II:
Case III:
•Spontaneous transfer of matter from region of high µ to region of low µ•At equilibrium µ is the same everywhere

Change of μ when water vapourize at 1 atm and 25°C
dμ= Gm° (H2O,g) –Gm°(H2O, l)
dμ =8.56 kJ mol-1
Change of μ when water vapourize at 1 atm and 100°C:
dμ = 0


3. Chemical potential in a mixture of ideal gases
Chemical potential of “A” in the mixture is less than if it were pure, under the same (T,p) conditions, because of the underlying entropy change!

Relation of Chemical Potential with Composition

where, PA = χA P; PB = χB PχA = nA/n; χA < 1

∆Gmix is independent of pressure, so the derivative is zero ; hence, ∆Vmix = 0
Ideal mixtures are formed without any volume change.

Perfect gases mix spontaneously in all proportions

Perfect gases mix spontaneously in all proportions
nT
mixmix
np
mixmix
pGV
TGS
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ=Δ
⎟⎠⎞
⎜⎝⎛ Δ
−=Δ
,
,
)(
)(
∂∂
∂∂
mixmixmix STGH Δ+Δ=Δ

How to describe physical properties of mixtures knowing the properties of their pure ingredients
Simple Mixtures
Simple mixtures:example: the volume of a mixture
a) perfect (ideal) mixtures: no interactions
b) real mixtures: interactions
Imagine a huge volume of water, and add 1 mol H2O. The volume will increase by 18 cm3, because18 cm3 mol-1 is the molar volume of water.
•

• Now, add 1 mol H2O to a huge volume of pure ethanol. The volume will increase by 14 cm3 mol-1, which is the partial molar volume of water in pure ethanol. The reason for the different increase is that the volume occupied by a given number of water molecules depends on the identity of the molecules that surround them.
• In general, the partial molar volume of a substance A in a mixture is the change in volume per mole of A added to a large volume of the mixture.
• The partial molar volumes of the components of the mixture vary with composition because the environment of each type of molecule changes as the composition changes from pure A to pure B.
The partial molar volumes of water and ethanol at 25°C.Note the different scales (water on the left, ethanol on the right).

The partial molar volume of a substance is the slope of the variation of the total volume of with the composition, as shown by the different slopes at thecompositions a and b. Note that the partial molar volume at b is negative: the overall volume of the sample decreases as a is added.

Gibbs energy for ideal gas at constant Temp was given as:
bar1Pwhere;PlnRTμμand;PPlnnRTGG oo
oo =+=+=
For Real gases: Lewis recognized that it would be convenient to keep same formal relation by defining a new function – the fugacity function, such that:
( ) t)coefficien(fugacityPfwhere,
;PflnRTμμand;
PflnnRTGG o
oo
o
=
⎟⎠⎞
⎜⎝⎛+=⎟
⎠⎞
⎜⎝⎛+=
φ
1Limor,1PfLim 0P0P ==⎟⎠⎞
⎜⎝⎛
→→ )(φ
In solution : we use Activity
)tcoefficienactivity(where,caGenerally,
Pf
PPa);(alnRTμμ
iii
oi
oi
iio
ii
==
==+=
γγ
Non-ideality:

Fugacity:
• Has units of pressure and if function of T and P
• As p → 0, the gas approaches ideality and f (fugacity) → P
• It is the measure of the molar Gibbs free energy of a Real Gas
• Real gas behaves like ideal gas in the limits of LOW Pressure and HIGH Temp and they deviate at High P and Low Temp.
• Compressibility factor , Z, is a measure of the deviation from ideality and it is defines as,
Vol)molarV(where,RTVP
VVzid
===
1Limor,1PfLim 0P0P ==⎟⎠⎞
⎜⎝⎛
→→ )(φ
Fugacity of a Real gas can be calculated at constant T and a particular P from the equation of state
Non-ideality:

=
==∴
=
+−=+−=
+=+=
idid
oo
dPVdμ and,
0)dT(whendPVdμT)constant(atVdPdG
dPVdTSdμor,VdPSdTdGlnfRTμμor,lnfnRTGG
⎥⎦⎤
⎢⎣⎡ −
=⎟⎠⎞
⎜⎝⎛∴
==
⎟⎠⎞
⎜⎝⎛=−
=⎟⎠⎞
⎜⎝⎛−=−
∫
∫
→
P0
id
P0
id
0P
idid
dPP1zexp
Pf
PRTV;
PRTzVwhere,
PflnRT)dPVV(
1Pfputting,)dPVV()μd(μ lim
Vol)molarV(where,RTVP
VVzid
===
To Calculate Fugacity and fugacity coeff. at constant T for a given P
dpp
zdpp
z pp
∫∫−
=⇔⎥⎦
⎤⎢⎣
⎡ −=⇔
00
)1(ln)1(exp φφ
⎟⎟⎠
⎞⎜⎜⎝
⎛=
pfwhere φ,
Fugacity is a measure of the chemical potential (μ ) of a real gas: μ may vary from -∞ to ∞ and f from 0 to ∞
( for pure substances in gaseous state)
Note: Φ is measure of non ideality of a gas in connection with the phase and chemical equilibrium properties

P →
f →
Standard state
P = 1 bar
1
Fugacity coefficient
Fugacity
Compressibility factor

To Calculate Fugacity and fugacity coeff.
Example 1: If the equation of state is as follows: (Virial Equation) dpp
zp
∫−
=0
)1(lnφK+++==
RTPC
RTBP
RTVPz
2'1 K++=−
RTPC
RTB
PzThen '1,
dpRTpC
RTBpp
∫ ⎥⎦
⎤⎢⎣
⎡++=∴
0
2
2'ln Kφ K++=⇒
RTpC
RTBp
2'ln
2
φ pfand ×= φ,
⎥⎦
⎤⎢⎣
⎡++×=⎥
⎦
⎤⎢⎣
⎡++=⇒ KK
RTpC
RTBppfand
RTpC
RTBp
2'exp
2'exp
22
φ
Example I1: If the equation of state is as follows: (van der Waals Equation)
a)RTp
RTabz ⎥⎦⎤
⎢⎣⎡ −+= 1
RTRTabdp
RTRTab
p 1ln1ln0
⎥⎦⎤
⎢⎣⎡ −=⇒⎥⎦
⎤⎢⎣⎡ −=∴ ∫ φφ
General Form
b) In the limiting case where, the attractive interactions may be neglected: P (Vm- b) = RT
⇒ PVm- bp = RT ⇒ PVm = RT + bp ⇒ PVm/RT = 1 + (bp/RT) z = 1 + (bp/RT)

To Calculate Fugacity and fugacity coeff.
⎟⎠⎞
⎜⎝⎛+=
RTbp 1 z dp
pzinzngSubstituti
p
∫−
=0
)1(ln: φ
⎟⎠⎞
⎜⎝⎛×=⇒⎟
⎠⎞
⎜⎝⎛=∴=⇒
RTbppf
RTbp
RTbp expexpln φφ
c) In the limiting case where, attractive interactions predominate: p + (a/Vm2 ) = RT/ Vm
⇒ pVm= RT – (a/Vm) z = 1 – (a/RTVm)
0,;ln,2
0=+−−= ∫ aVRTVpnowdp
VRTaThen
p
φ
⎥⎦
⎤⎢⎣
⎡−+=⇒
−+=∴ 2
2
)(412;
24)(
RTapRTRTVp
papRTRT
V
If p is sufficiently small, then : RTVpRTVpwritemayweandRTap
=⇔=⟨⟨ 22:;1)(
42
⎟⎟⎠
⎞⎜⎜⎝
⎛−×=−= 22 )(
exp)(
ln,RTappfand
RTapThus φ
dpRTbgetwe
p
∫ ⎟⎠⎞
⎜⎝⎛=
0ln: φ

Gibbs Paradox: The mixing of NON-IDENTICAL gases
Before Mixing After Mixing
Shows obvious increase in entropy (disorder)
Now consider the mixing of IDENTICAL gasesBefore Mixing After Mixing
Shows zero increase in entropy as action is reversible

• When 2 non-identical gases mix and entropy increase, we imply that the gases can be separated and returned to their original state
• When 2 identical gases mix, it is impossible to separate the two gases into their original state as there is no recognizable difference between the gases
Gibbs Paradox: Entropy of mixing
• Thus, these two cases stand on different footing and should not be compared with each other
• The mixing of gases of different kinds that resulted in the entropy change was independent of the nature of the gases
• Hence independent of the degree of similarity between them

Suppose we have a set of 4 cells each of which can contain 1 ball.The set of 4 cells is then divided in half; each half has 2 cells
We place 2 balls in the cells ; the arrangements that are possible are:( ⃝ indicates an empty cell,⊗ indicates an occupied cell)
Of these 6 arrangements, 4 correspond to uniform filling ; that is, 1 ball in each half of the box. The probability of uniform filling is therefore,
while the probability of finding both balls on 1 side of the box isThe probability of any particular arrangement is 1/6 But 4 particular arrangements lead to uniform filling ; only 2 particular arrangements lead to no-nuniform filling.
32
64=
31
62=
Gibbs Paradox: Entropy of mixing

Suppose that there are 8 cells and 2 balls ; then the total number of arrangements is 28. Of the 28 arrangements, 16 of them correspond to one ball in each half of the box. The probability of the uniform distribution is therefore 16/28 = 4/7
It can be shown that, as the number of cells increases without limit, the probability of finding 1 ball in one half of the box and the other in the other half of the box approaches the value 1/2
The entropy of a system in a specified state can be defined in termsof the number of possible arrangements of the particles composing the system that are consonant with the state of the system.
Each such possible arrangement is called a complexion of the system. Following Boltzmann, we define the entropy by the equation:
where k is the Boltzmann constant, k = R/NA, and Ω is the number of complexions of the system that are consonant with the specified state of the system. Since the probability of a specified state of a system is proportional to the number of complexions which make up that state, it is clear from Equation that the entropy depends on the logarithm of the probability of the state.

Situation 1: The 2 balls are confined to the left half of the box. There is only 1 arrangement (complexion) that produces this situation ; hence, Ω = 1, and from
The entropy of this state is zero.
Situation 2: The 2 balls may be anywhere in the box. As we have seen, there are 6 complexions corresponding to this situation ; hence, Ω = 6, and from
The entropy increase associated with the expansion of the system from 2 cells to 4 cells is then

This result is readily generalized to apply to a box having N cells. How many arrangements are possible for two balls in N cells ? There are N choices for the placement of the first ball ; for each choice of cell for the first ball there are (N – 1) choices for the second ball.The total number of arrangements of 2 balls in N cells is apparently N(N - 1).
However, since we cannot distinguish between ball 1 in position x, ball 2 in position y , and the arrangement ball 1 in y, ball 2 in x, this number must be divided by 2 to obtain the number of distinct arrangements"; hence
The entropy of this system is,
If we increase the number of cells available to N',
then Ω2 = N'(N' - 1), and21
The increase in entropy associated with increasing the number of cells from N to N' is


The minimum in G occurs at the point where ∆Gmixdecreases as rapidly as Gpureincreases

at constant T and P
dxdnxnn
dxdnxnn
BBBB
AAAA
B
A
νν
νν
=⇒+=
−=⇒−=°
°

where,

Chemical equilibrium in a mixture of Ideal gases at constant T & P
PTrxn
GG,
⎟⎠
⎞⎜⎝
⎛=Δ
∂ξ∂ ∑ +==Δ∴
iiiiiirxn plnRTwhereG 0,, μμμν
00000
0 ,ln
BADCrxn
BA
DCrxnrxn
G
ppppRTGG
βμαμδμγμ
βα
δγ
−−+=Δ
+Δ=Δ
eqBA
DCrxnrxn pp
ppRTGG ⎟⎟⎠
⎞⎜⎜⎝
⎛−=Δ⇒=Δ βα
δγ
ln,0 0
αA + βB γC + δD
At equilibrium,
The reaction Gibbs free energy can be written as:,

Some books still refer to the pressure equilibrium constant
b
eqPZp
a
eqPAp
z
eqPZp
y
eqPYp
i
iν
eqPiPK
⎟⎟⎠
⎞⎜⎜⎝
⎛°⎟
⎟⎠
⎞⎜⎜⎝
⎛°
⎟⎟⎠
⎞⎜⎜⎝
⎛°⎟
⎟⎠
⎞⎜⎜⎝
⎛°
=∏ ⎟⎟⎠
⎞⎜⎜⎝
⎛
°=°
( )
( )
( )
( )beqBp
zeqZp
aeqAp
yeqYp
PK = In general pressure units

Kc is a quotient of equilibrium concentrations ; Kc is a function of temperature only.





• Chemical Potential μ controls phase transitions and phase equilibrium.
• Equilibrium condition – at equilibrium μ must be identical throughout the system
• If there are several phases present, the chemical potential of each species must have the same value in every phase in which it appears.
Phase Equilibrium and Phase Transitions conditions for a Single Component System

• At T’ (below Tm), Liquid (µa ) is greater than solid (µb), so liquid freezes spontaneously, since this will decrease the Gibbs energy of the system..• At a temperature above Tm the situation is reversed : the µb of solid is greater than that of the µa of liquid and the solid melts spontaneously to decrease the Gibbs energy. Consequence : heat is absorbed in the transformation from solid to liquid, and from liquid to gas.
Melting (freezing) temp. – the temp at which the liquid and solid phases coexist in equilibrium (at given P). Melting temp at 1 atm – called normal m.p., Melting temp at 1 bar – called standard m.p
Triple point (T3) the point at which the three phases coexist.
At the critical point the gasliquid line stops. Beyond the Tc the liquid and gas phase become indistinguishable, they merge into asingle phase.
ive as from 3rd Law, the entropy of a substanceis always +ive .
Consequently, the plot of µ versus T at constant pressure is a curve (should be slightly concave downwards) with a negative slope
At any temperature,
gasS
solidS
liguidS
µa
µb
T’Sequence of phase changes observed if a solid is heated under constant pressure
nG=μ
solidliquidgas SSS >≥

If the pressure is decreased, dp is negative, V is positive ; hence dµ is negative, and the µ decreasesin proportion to the volume of the phase.
solidliq VV >
• Figure shows that the equilibrium temperatures ( intersection points) have shifted with change in Pressure. • Since , and are very small, the value of µ is decreased only slightly. The volume of the gas is roughly 1000 times larger than that of the solid or liquid, so the µ of the gas decreases greatly.
• At the lower pressure the range of stability of a phase is noticeably decreased.• If the pressure is reduced to a sufficiently low value, the boiling point of a liquid may even fall belowthe melting point of the solid. Then there is no temperature at which the liquid is stable ; the solid sublimes.
• At the sublimation temperature, Ts, the solid and vapor coexist in equilibrium.. Ts is very much P dependent.
ligV solidV

•Whether or not a particular material will sublime under reduced pressure rather than melt dependsentirely on the individual properties of the substance.
• Water, for example, sublimes at pressures below 611 Pa.• The higher the melting point, and the smaller the difference between the melting point and boiling pointat 1 atm pressure, the higher will be the pressure below which sublimation is observed.
• The pressure (in atm) below which sublimation is observed can be estimated for substances obeying Trouton's rule by the formula
⎟⎟⎠
⎞⎜⎜⎝
⎛ −−=
m
mb
TTTp 8.10ln
Bold lines are higher P µ vs T plot for subtance that sublimes
Tm


Clapeyron Equation
Using this then we obtain the two forms of the Clapeyron Equation

Now use the Clapeyron equation to understand the phase diagram




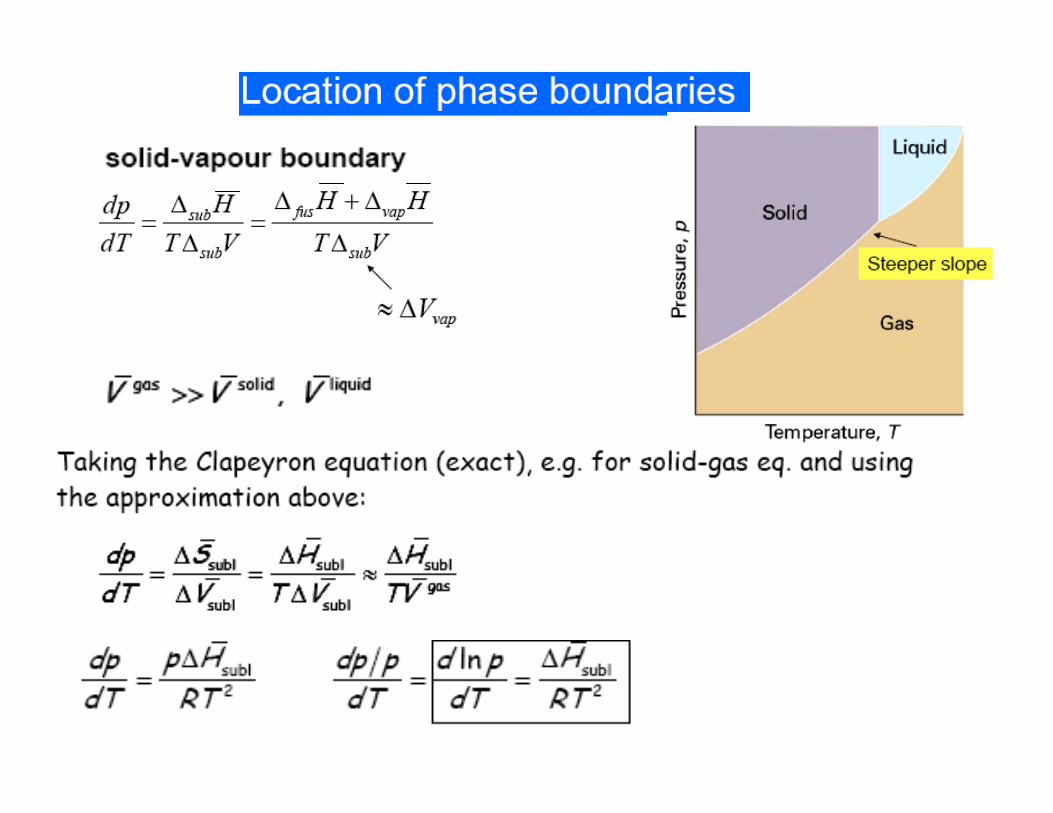


F= 1 (represented by a line), 2phases in equilibrium
F= 2 (represented by a region), 1phases in equilibrium
s-g
s-l g-l
F= 0(represented by a point), 3 phases in equilibrium
Four phases cannot mutually coexist in equilibrium.
There are other solid phases at much higher pressures.

F= 1 (represented by a line), 2 phases in equilibrium
F= 2 (represented by a region), 1 phases in equilibrium
s-g
s-l
g-l
F= 0 (represented by a point), 2 phases in equilibrium

Critical Point and Supercritical Fluids

At atmospheric pressure “dryice”, forms directlyform gas
Note that, as the triple point lies at pressures well above atmospheric, liquid carbon dioxide does not exist under normal conditions (a pressure of at least 5.11 atm must be applied).






• Chemical Potential μ controls phase transitions and phase equilibrium.
• Equilibrium condition – at equilibrium μ must be identical throughout the system
• If several phases are in equilibrium, then the chemical potential of each species must have the same value in every phase in which it appears.
Phase Equilibrium and Phase Transitions Conditions for a Single Component System

• At T’ (below Tm), Liquid (µa ) is greater than solid (µb), so liquid freezes spontaneously, since this will minimize the Gibbs energy (or, µ) of the system.• At a temperature above Tm the situation is reversed : the µb of solid is greater than that of the µa of liquid and the solid melts spontaneously to decrease the Gibbs energy. Consequence : heat is absorbed in the transformation from solid to liquid, and from liquid to gas.
Melting (freezing) temp. – the temp at which the liquid and solid phases coexist in equilibrium (at given P). Melting temp at 1 atm – called normal m.p., Melting temp at 1 bar – called standard m.p
Triple point (T3) the point at which the three phases coexist.
At the critical point the gasliquid line stops. Beyond the Tc the liquid and gas phase become indistinguishable, they merge into asingle phase.
ive as from 3rd Law, the entropy of a substanceis always +ive .
Consequently, the plot of µ versus T at constant pressure is a curve (should be slightly concave downwards) with a negative slopeAt any temperature,
gasS
solidS
liguidS
µa
µb
T’Sequence of phase changes observed if a solid is heated under constant pressure
solidliquidgas SSS >≥
nGμ =

The following curves are obtained if a SOLID is HEATED under constant pressure to form LIQUID and finally to GAS
gasS
solidS
liguidS
µa
µb
T´
µc
At any temperature: solidliquidgas SSS >≥
T´ < Tm, (below )SOLID is the stable phase
T´ > Tm, (below )LIQUID is the stable phase
Tm < T < Tb LIQUID is the stable phase
T > Tb GAS is the stable phase

If the pressure is decreased, dp is negative, as Vcannot be negative ; hence dµ is negative, and the µdecreases in proportion to the volume of the phase.
solidliq VV >
• Figure shows that the equilibrium temperatures ( intersection points) have shifted with change in Pressure. • Since , and are very small, the value of µ is decreased only slightly. The volume of the gas is roughly 1000 times larger than that of the solid or liquid, so the µ of the gas decreases greatly.
• At the lower pressure, the range of stability of a phase is noticeably decreased.• If the pressure is reduced to a sufficiently low value, the boiling point of a liquid may even fall below the melting point of the solid. Then there is no temperature at which the liquid is stable ; the solid sublimes.
• At the sublimation temperature, Ts, the solid and vapor coexist in equilibrium. Ts is very much P dependent.
ligV solidV

•Whether or not a particular material will sublime under reduced pressure rather than melt dependsentirely on the individual properties of the substance.
• Water, for example, sublimes at pressures below 611 Pa.• The higher the melting point, and the smaller the difference between the melting point and boiling pointat 1 atm pressure, the higher will be the pressure below which sublimation is observed.
• The pressure (in atm) below which sublimation is observed can be estimated for substances obeying Trouton's rule by the formula
⎟⎟⎠
⎞⎜⎜⎝
⎛ −−=
m
mb
TTTp 8.10ln
Bold lines are higher P µ vs T plot for substance that sublimes.
Tm
If the pressure is reduced to a sufficiently low value, the boiling point of a liquid may even fall below the melting point of the solid.


nGμ =
Clapeyron Equation

Alternately, we can derive another expression of Clapeyron Equation
βαβα →→ Δ= STHΔ as,( )
VTH
VS
dTdp
ΔΔ
=ΔΔ
=⎟⎠⎞
⎜⎝⎛ →
→
→ βα
βα
βα
coexists
Using this then we obtain the two forms of the Clapeyron Equation
Therefore we can rewrite: as

Now use the Clapeyron equation to understand the phase diagram







Phase Diagram describe the phase properties as a function of state variables, for example in terms of (T, p).
Phase Diagram
For the melting line, for example, solid and liquid coexist, and μs(T, p) = μℓ(T, p) One equation, two variables (T, p). This means that coexistence of two phases is described by T(p) or p(T). e.g. a line in the (T, p) phase diagram.
At the triple point, the chemical potentials of all three phases are the same…solid, liquid and gas coexist. μs(T, p) = μℓ(T, p) = μg(T, p)Two equations, two variables. This defines a unique point (Tt, pt) in the (T, p) phase diagram. In the single phase (planar) regions of the diagram, one of the chemical potentials is lower than the other two. T and p can be changed independently without changing phases.

F= 1 (represented by a line), 2phases in equilibrium
F= 2 (represented by a region), 1phases in equilibrium
s-g
s-l g-l
F= 0(represented by a point), 3 phases in equilibrium
Four phases cannot mutually coexist in equilibrium.
There are other solid phases at much higher pressures.
All these cases can be summarized by the Phase Rule.

F= 1 (represented by a line), 2 phases in equilibrium
F= 2 (represented by a region), 1 phases in equilibrium
s-g
s-l
g-l
F= 0 (represented by a point), 2 phases in equilibrium

Critical Point and Supercritical Fluids

At atmospheric pressure “dryice”, forms directlyform gas
Note that, as the triple point lies at pressures well above atmospheric, liquid carbon dioxide does not exist under normal conditions (a pressure of at least 5.11 atm must be applied).






















