
Thermo Fisher Scientific Visit - Microsoft · Detailed Manual Analysis (uses .PUF files) Drill ......
56
PROSIGHT INTRODUCTION Neil Kelleher
Transcript of Thermo Fisher Scientific Visit - Microsoft · Detailed Manual Analysis (uses .PUF files) Drill ......

PROSIGHT INTRODUCTION Neil Kelleher

OVERVIEW
History and terms
How ProSight Handles Variability
Creating DBs
Search Logic (Modes)
Scoring
Targeted Workflow
HT Workflow
New Stuff !!

ProSightPC v1.0 (released Fall, 2006)
ProSightPC 1.0, 2.0, 3.0 (Developed Since 2004)

WHAT DOES PROSIGHT DO?
Requires:
Input database
MS Data
Searches data against input database
Provides Results
Input Data(Sequence+ MS Data)
Searching Results
400 600 800 1000 1200 1400m/z
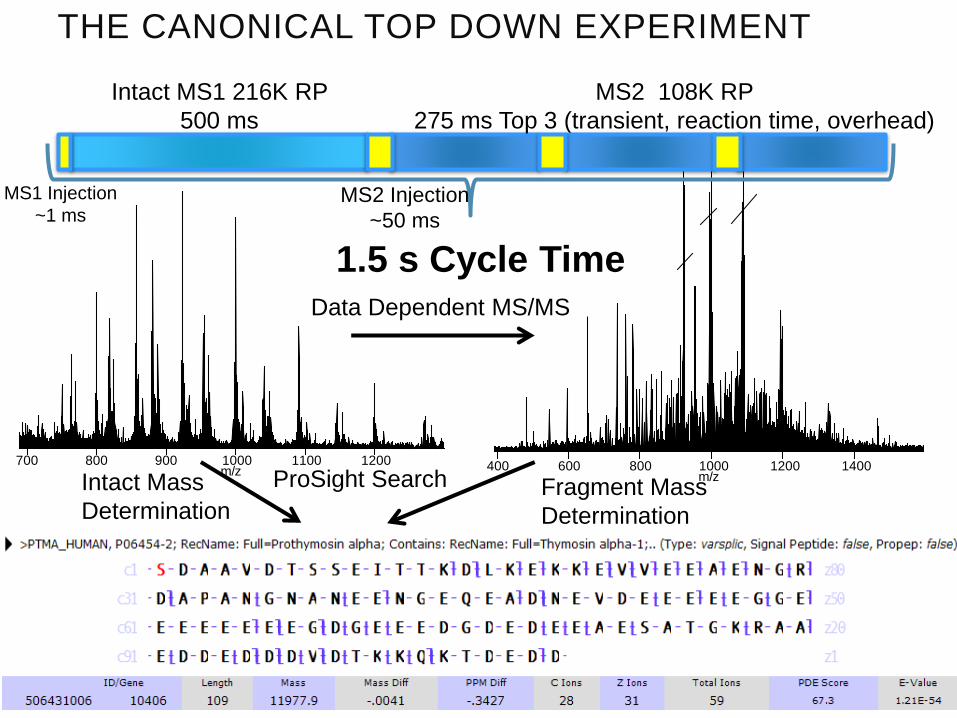
THE CANONICAL TOP DOWN EXPERIMENT
Intact MS1 216K RP
500 ms
MS2 108K RP
275 ms Top 3 (transient, reaction time, overhead)
1.5 s Cycle Time
700 800 900 1000 1100 1200m/z
Data Dependent MS/MS
ProSight SearchIntact Mass
DeterminationFragment Mass
Determination
MS1 Injection
~1 msMS2 Injection
~50 ms



How Most Search Engines Handle
Variation
AATASAFGSK (Pot. Phospho on T3, S5, & S9)
AATASAFGSK
AATASAFGSK
AATASAFGSK AATASAFGSK
AATASAFGSK
AATASAFGSK AATASAFGSK
AATASAFGSK
AATASAFGSK
AATASAFGSK AATASAFGSK
AATASAFGSK
AATASAFGSK AATASAFGSK
T3
S5
S9
Unmod S9 S5 S5,S9 T3 T3,S9 T3,S5 T3,S5,S9

How ProSightPC/PD Handles Protein
Variation
• 4 Proteoforms
– PDCD5
– PDCD5 + K63Ac
– PDCD5 + S119Ph
– PDCD5 + K63Ac +
S119Ph
• 262,144 Proteoforms
in Typical Search
Engine using
variable
modifications

Sources of
variability• Alternative Splices
• Endogenous
Cleavages
• Other large sequence
changes
Isoform: Large changes to amino acid sequence
Protein Isoform

Proteoform: The complete set of modifications on an amino acid sequence
Proteoform (Observed)

What is a Proteoform, Exactly?

The Proteoform:
A distinct molecular form of a protein product arising from a single gene
-- The Consortium for Top DownProteomics
CannonicalSequence(UniProtKB)
Endogenous proteolysis mRNA splicingMutationsSNPs
Site specific features-govern activity, localization, interactions, half-life
Smith, L.S., Kelleher, N.L., & The Consortium for Top Down Proteomics, Nat. Methods, 2013, 10, 186–187.
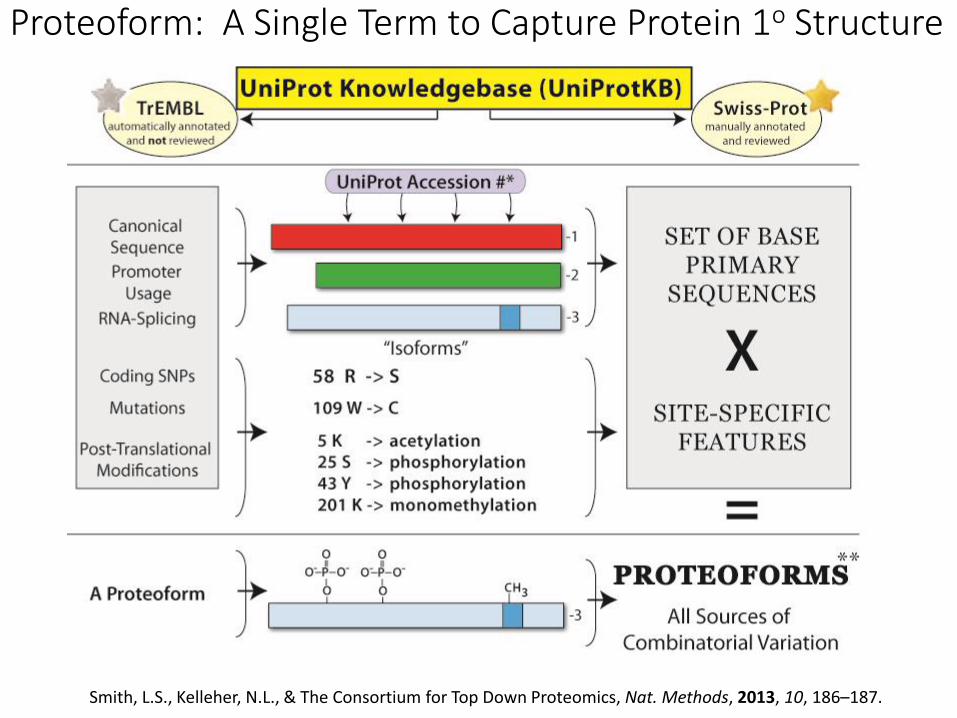
Proteoform: A Single Term to Capture Protein 1o Structure
Smith, L.S., Kelleher, N.L., & The Consortium for Top Down Proteomics, Nat. Methods, 2013, 10, 186–187.

Proteoform: A Single Term to Capture Protein 1o Structure
Smith, L.S., Kelleher, N.L., & The Consortium for Top Down Proteomics, Nat. Methods, 2013, 10, 186–187.

Swiss-Prot Entry for HMGA1
ID HMGA1_HUMAN Reviewed; 107 AA.
AC P17096; P10910; Q5T6U9; Q9UKB0;
DT 01-AUG-1990, integrated into UniProtKB/Swiss-Prot.
DT 23-JAN-2007, sequence version 3.
DT 05-APR-2011, entry version 140.
DE RecName: Full=High mobility group protein HMG-I/HMG-Y;
DE Short=HMG-I(Y);
DE AltName: Full=High mobility group AT-hook protein 1;
DE Short=High mobility group protein A1;
DE AltName: Full=High mobility group protein R;
GN Name=HMGA1; Synonyms=HMGIY;
OS Homo sapiens (Human).
OX NCBI_TaxID=9606;
FT INIT_MET 1 1 Removed.
FT CHAIN 2 107 High mobility group protein HMG-I/HMG-Y.
FT MOD_RES 2 2 N-acetylserine (By similarity).
FT MOD_RES 15 15 N6-acetyllysine.
FT MOD_RES 26 26 Asymmetric dimethylarginine; in isoform HMG-I alternate.
FT MOD_RES 26 26 Omega-N-methylarginine; in isoform HMG-I alternate.
FT MOD_RES 26 26 Symmetric dimethylarginine; in isoform HMG-I; alternate.
FT MOD_RES 36 36 Phosphoserine.
FT MOD_RES 39 39 Phosphothreonine.
FT MOD_RES 44 44 Phosphoserine.
FT MOD_RES 49 49 Phosphoserine.
FT MOD_RES 53 53 Phosphothreonine.
FT MOD_RES 58 58 Asymmetric dimethylarginine; by PRMT6; alternate.
FT MOD_RES 58 58 Omega-N-methylarginine; by PRMT6; alternate.
FT MOD_RES 60 60 Asymmetric dimethylarginine; by PRMT6;
FT MOD_RES 60 60 Omega-N-methylarginine; by PRMT6;
FT MOD_RES 99 99 Phosphoserine; in isoform HMG-I and isoform HMG-Y.
FT MOD_RES 102 102 Phosphoserine; by CK; in isoform HMG-I and isoform HMG-Y.
FT MOD_RES 103 103 Phosphoserine; by CK; in isoform HMG-I and isoform HMG-Y.
FT VAR_SEQ 35 45 Missing (in isoform HMG-Y).
FT VAR_SEQ 66 107 NKGAAKTRKTTTTPGRKPRGRPKKLEKEEEEGISQESSEEE
FT Q -> KNWRRRKRRASRRSPRRRSSDPCVPPAPHWRSSFLL
FT GLDSFAPLPPPPPLPQAHHHHRLWPPPPSSTCALTTTLHST
FT PAAAGLPWAEWGAVFPWPQFPAPPAHPRIHTCPPGQG (in
FT isoform HMG-R).
SQ SEQUENCE 107 AA; 11676 MW; E9C4E3F2200914B8 CRC64;
MSESSSKSSQ PLASKQEKDG TEKRGRGRPR KQPPVSPGTA LVGSQKEPSE VPTPKRPRGR
VAR_SEQ indicates splice variants
MOD_RES indicates post-translational
modifications
This entry will
produce 3 ProSight
Base Sequences and
32,256 ProSight
Protein Forms
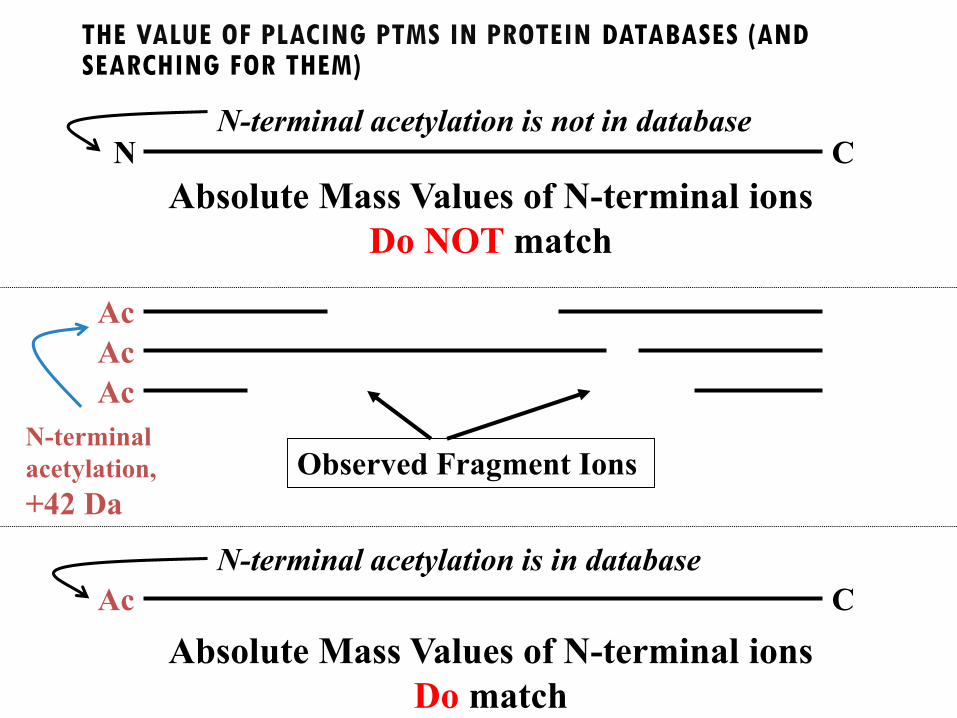
THE VALUE OF PLACING PTMS IN PROTEIN DATABASES (AND SEARCHING FOR THEM)
N CN-terminal acetylation is not in database
Observed Fragment Ions
Ac
N-terminal
acetylation,
+42 Da
Absolute Mass Values of N-terminal ions
Do NOT match
C
N-terminal acetylation is in database
Absolute Mass Values of N-terminal ions
Do match
Ac
Ac
Ac
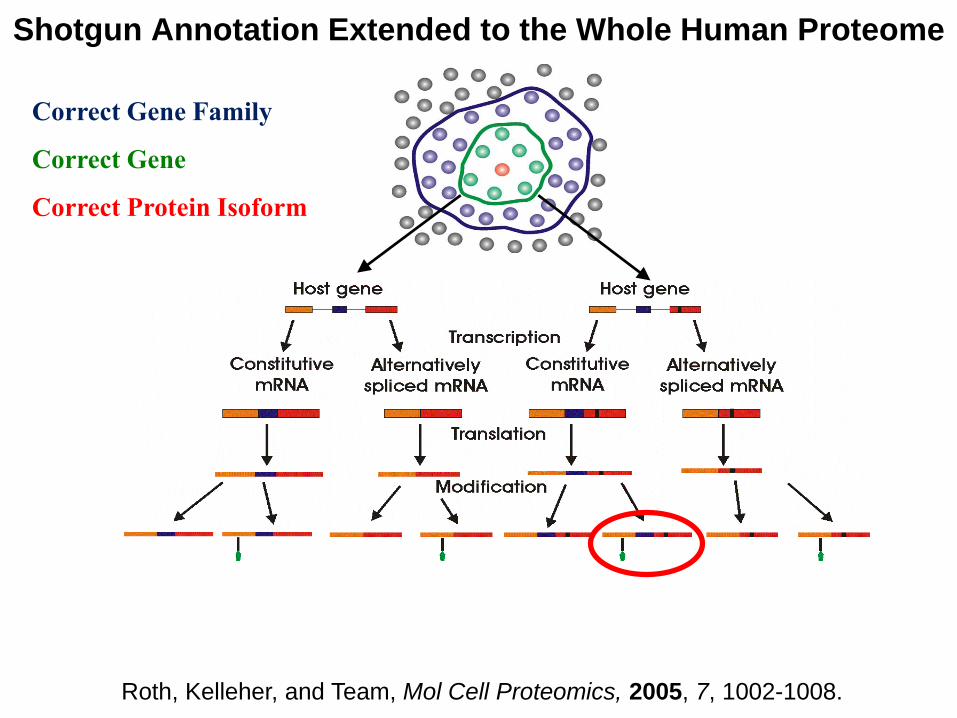
Correct Gene Family
Correct Gene
Correct Protein Isoform
Shotgun Annotation Extended to the Whole Human Proteome
Roth, Kelleher, and Team, Mol Cell Proteomics, 2005, 7, 1002-1008.
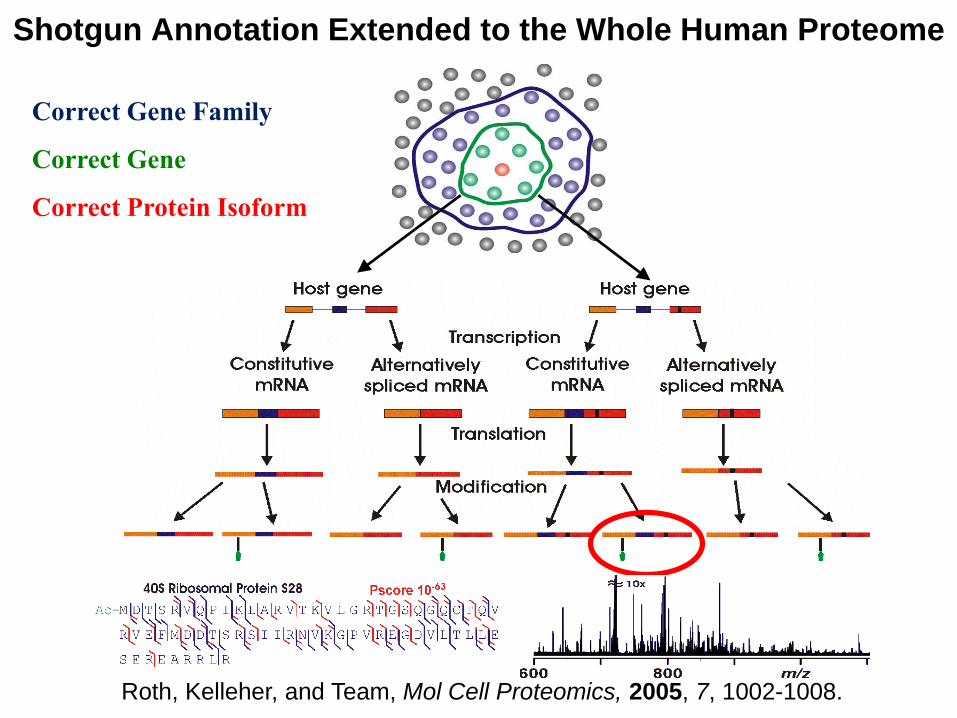
Correct Gene Family
Correct Gene
Correct Protein Isoform
Shotgun Annotation Extended to the Whole Human Proteome
Roth, Kelleher, and Team, Mol Cell Proteomics, 2005, 7, 1002-1008.
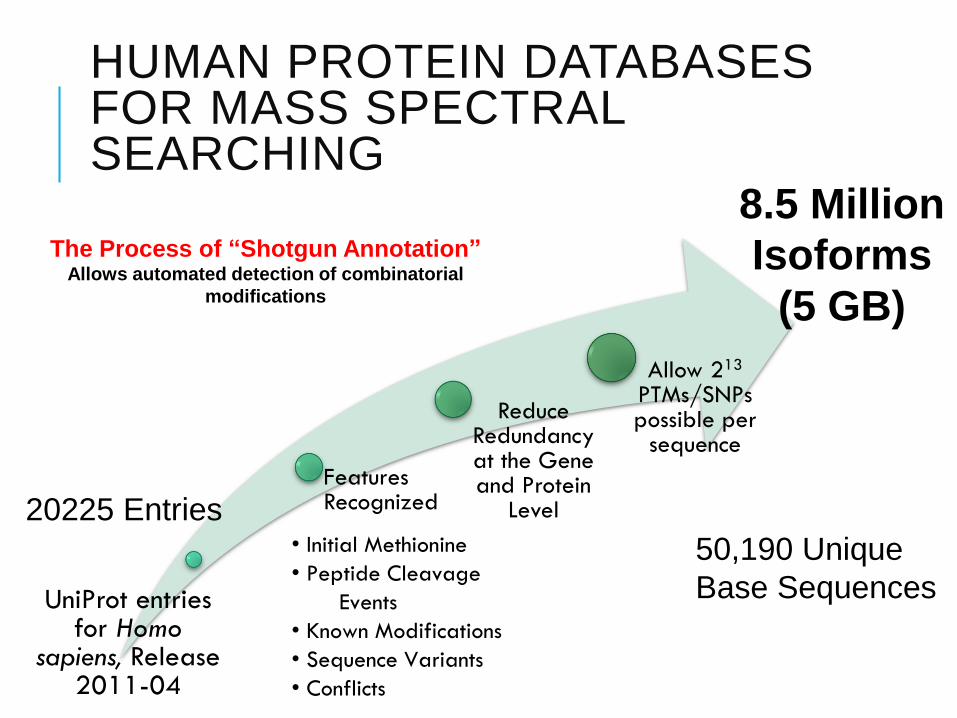
HUMAN PROTEIN DATABASES FOR MASS SPECTRAL SEARCHING
UniProt entries for Homo
sapiens, Release 2011-04
Features Recognized
Reduce Redundancy at the Gene and Protein
Level
Allow 213
PTMs/SNPs possible per
sequence
50,190 Unique
Base Sequences
20225 Entries
8.5 Million
Isoforms
(5 GB)
• Initial Methionine
• Peptide Cleavage
Events
• Known Modifications
• Sequence Variants
• Conflicts
The Process of “Shotgun Annotation”Allows automated detection of combinatorial
modifications
DATABASE MANAGER

DATABASE CREATION
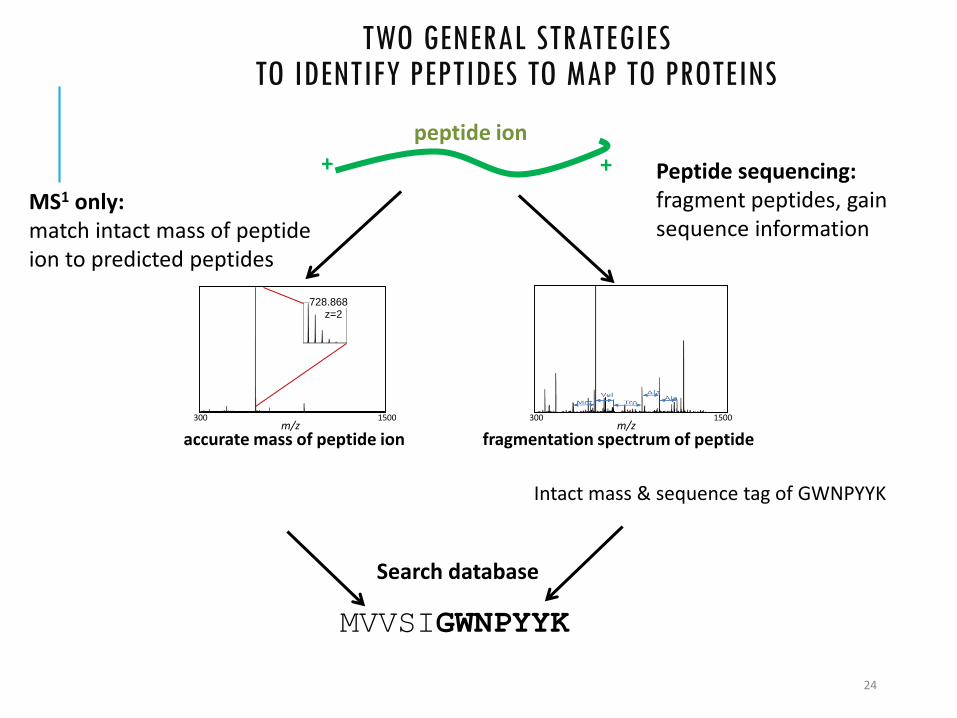
TWO GENERAL STRATEGIES TO IDENTIFY PEPTIDES TO MAP TO PROTEINS
24
MS1 only: match intact mass of peptide ion to predicted peptides
peptide ion
accurate mass of peptide ion
Peptide sequencing: fragment peptides, gain sequence information
fragmentation spectrum of peptide
Intact mass & sequence tag of GWNPYYK
MVVSIGWNPYYK
+ +
Search database
300 1500m/z
300 1500m/z
728.868z=2

SEARCH MODES (THERE ARE 5 IN PROSIGHTPC)
Absolute Mass:
Interrogates all protein forms within a window of
the observed intact mass.
Biomarker:
Searches all possible sub-sequences to find
those with the observed intact mass.

DELTA M MODEError tolerant search: A delta mass is placed at both termini and fragment matching is performed.
∆M = observed precursor mass – candidate mass
N C
N C
N C
∆M
∆M
Match both directions
Match towards N terminus
Match towards C terminus
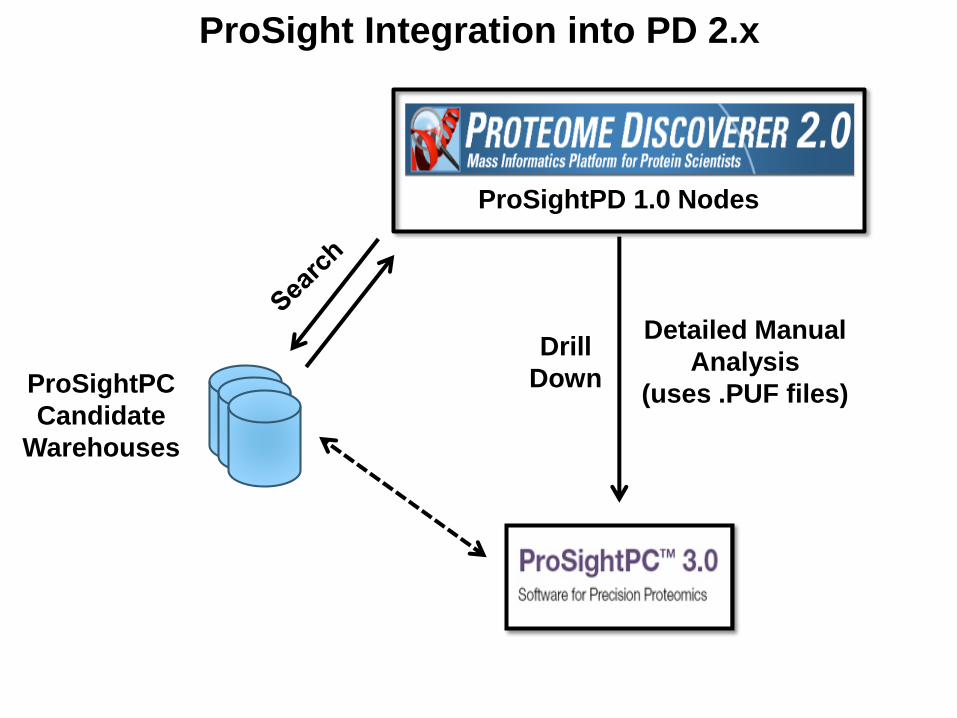
ProSightPC
Candidate
Warehouses
Detailed Manual
Analysis
(uses .PUF files)
Drill
Down
ProSightPD 1.0 Nodes
ProSight Integration into PD 2.x

BETTER GRAPHICAL SUPPORT IN PROSIGHTPD(PROTEOME DISCOVERER NEEDS TO SUPPORT TOP DOWN BETTER!)
AX – green, angled flag
BY – blue, angled flag
CZ* – red, square flag
If both AB or XY, displays as
BY

TOP DOWN DATA TYPES
Lo/Hi – Respect in ProSightPD
Med/Hi – Respect in ProSightPD
Hi/Hi – cRAWler in ProSightPD and
ProSightPC 3.0
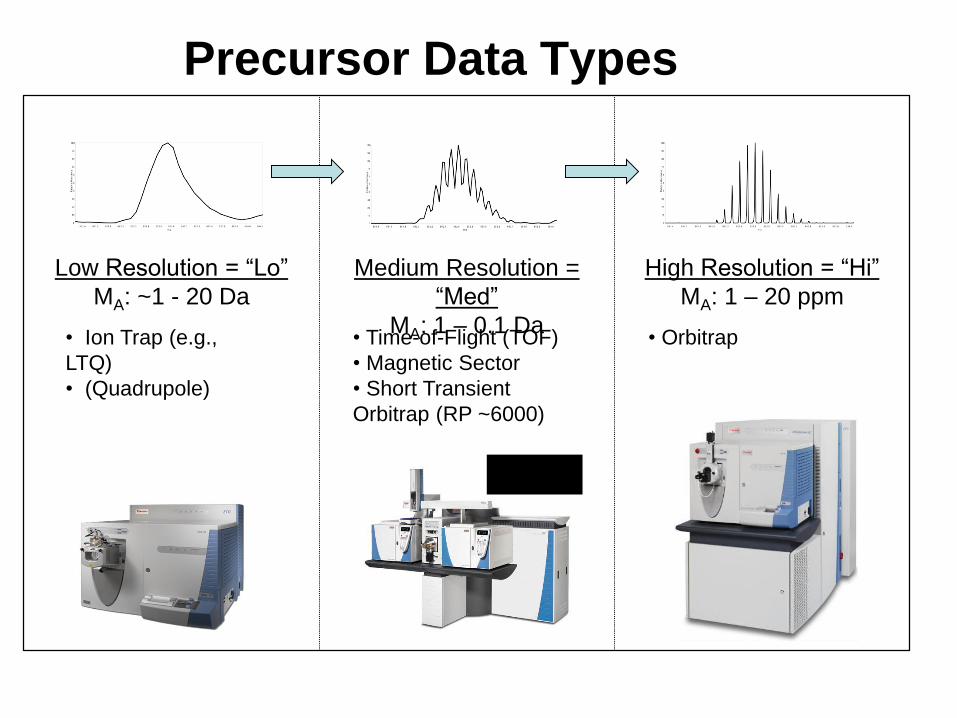
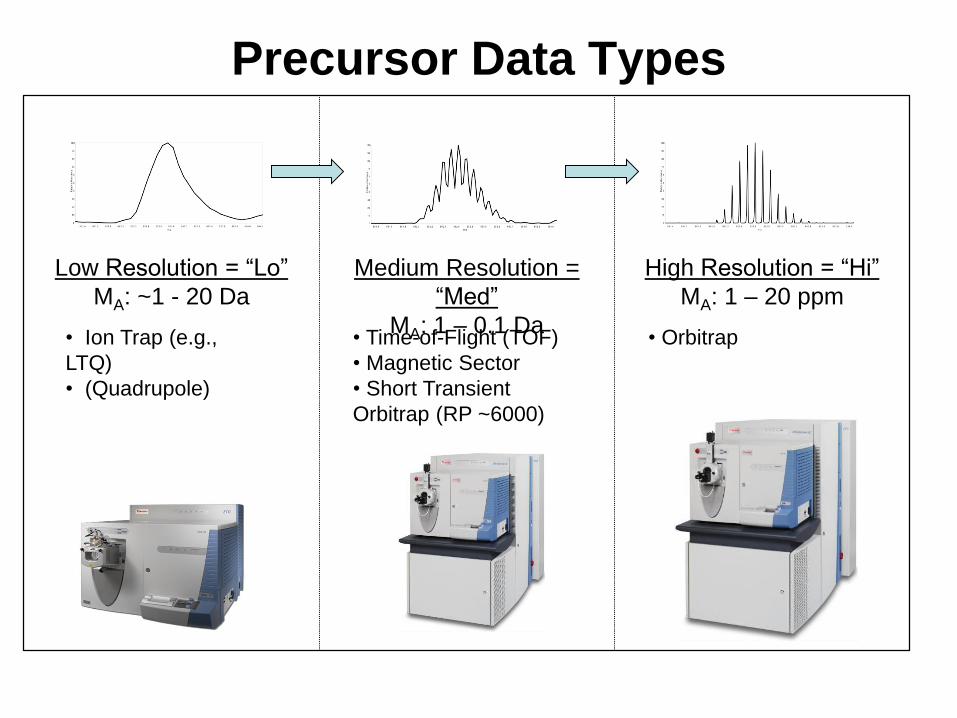
Precursor Data Types
Low Resolution = “Lo”
MA: ~1 - 20 Da
Medium Resolution =
“Med”
MA: 1 – 0.1 Da
High Resolution = “Hi”
MA: 1 – 20 ppm
• Ion Trap (e.g.,
LTQ)
• (Quadrupole)
• Time-of-Flight (TOF)
• Magnetic Sector
• Short Transient
Orbitrap (RP ~6000)
• Orbitrap

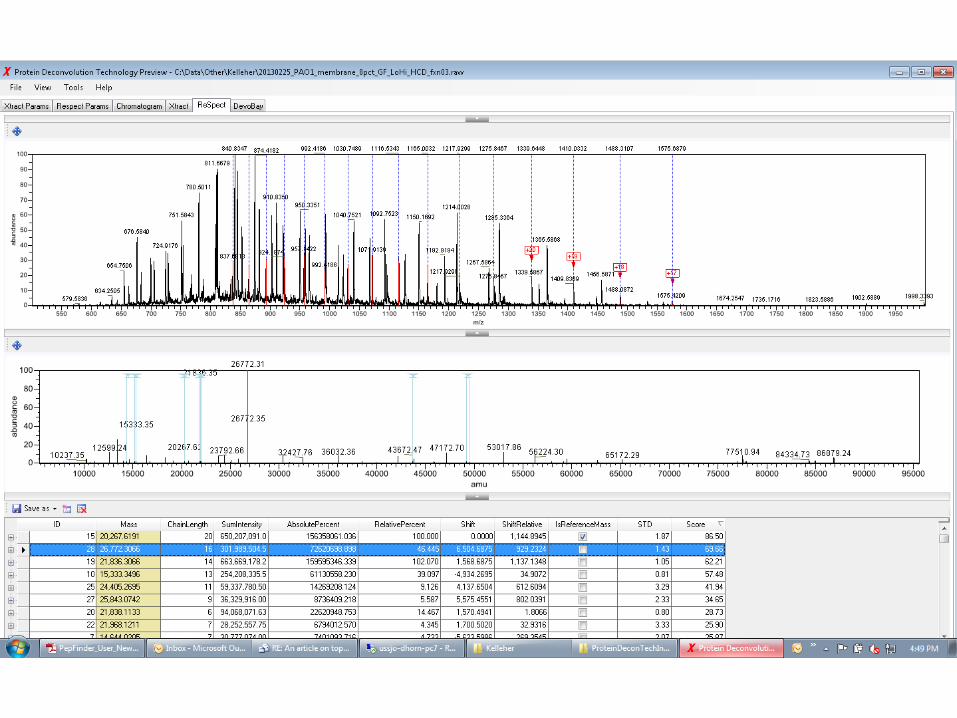
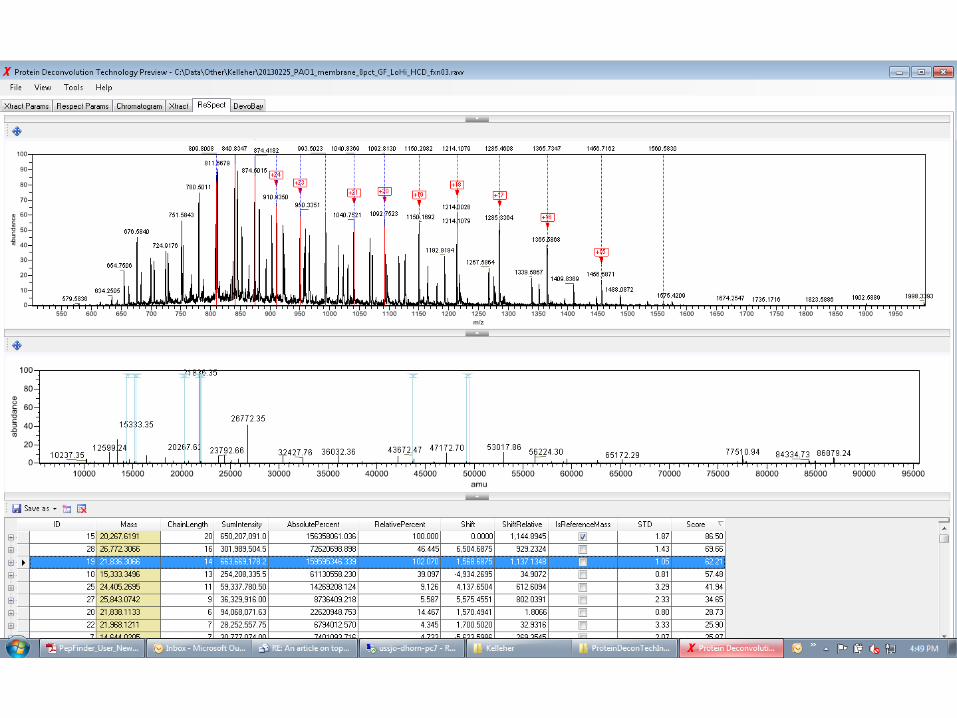
APPLICATION OF THE RESPECT ALGORITHM TO IT-FT (LO-HI) DATA FROM PSEUDOMONAS AERUGINOSA
(PROVIDED BY IOANNA NTAI, PCE , NORTHWESTERN U.)
JANUARY, 24TH 2014




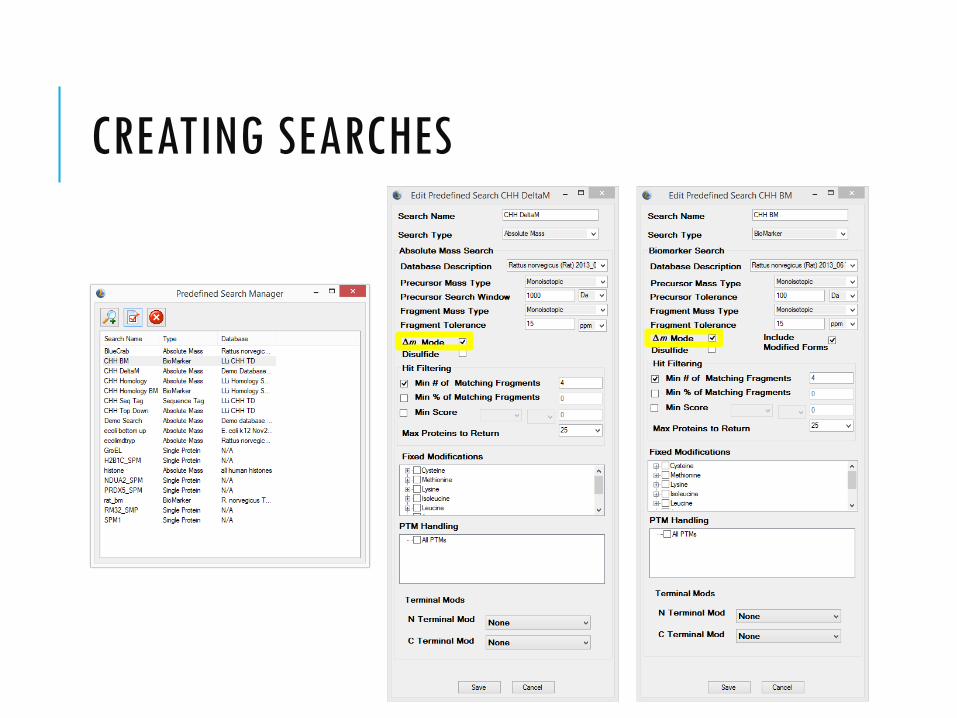
CREATING SEARCHES

SCORING MODEL
Statistical Model based on the Poisson Distribution• If an event is expected to occur times, then the probability of it occurring k times is
given by the Poisson distribution:
• If f is the total number of observed fragments, and n is the number of matching fragments, we may estimate the probability of a random protein match given f and n:
where x is the mean probability that a mass of an observed fragment ion will randomly match one from a generic protein.
!
,k
ekP
k
221.111
1
!),(
a
xfn
Mx
n
exffnP
[Eq.1]
[Eq.2]

SCORING MODEL
Given the probability of a random protein match with a particular n, f, and Ma, we can now estimate the probability of getting a match at least as good as we have by random chance:
where p is the p-value assuming our Poisson model – what we call the P-Score.
The Expectation Value (e-value) reported in ProSightPC is simply the P-Score scaled to the size of the search space.
1
0 !1
n
i
xfn
n
exfp [Eq.3]

CRAWLER ALGORITHM
Creates MS/MS Experiments during a high throughput run
Implementation
Groups fragment scans based on their isolation m/z and retention times
For each fragment scan group
Averages fragment scans
Runs Xtract/THRASH to create neutral fragment masses
Averages precursor scans based on proximity
Runs Xtract/THRASH in isolation window to create neutral precursor masses
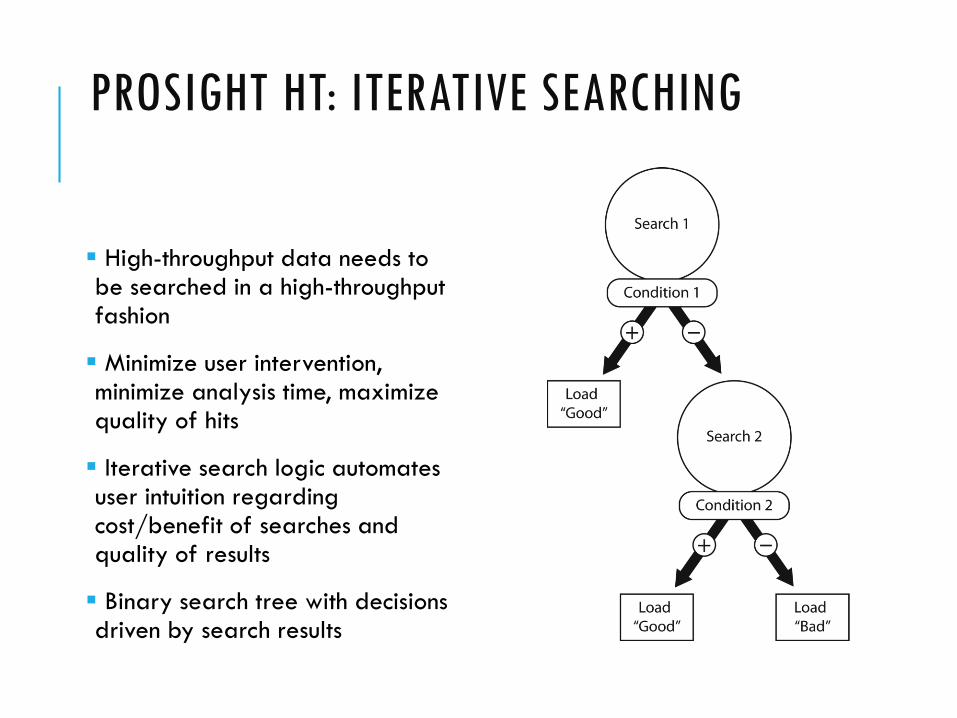
PROSIGHT HT: ITERATIVE SEARCHING
High-throughput data needs to be searched in a high-throughput fashion
Minimize user intervention, minimize analysis time, maximize quality of hits
Iterative search logic automates user intuition regarding cost/benefit of searches and quality of results
Binary search tree with decisions driven by search results

PROSIGHT HT: ITERATIVE SEARCHING
• Fast, Narrow, Specific Searches on Small Databases
• Contaminant Filtering (Bottom-Up/Middle-Down)
• Targeted Protein Identification/Characterization
• Narrow Absolute Mass Searches on Large Databases
• Proteomic Investigations
• Wide Absolute Mass Searches on Small Databases
• Characterization of Unknown cSNPs and PTMs on
Targeted Proteins
• Wide Absolute Mass Searches on Large Databases
• Proteomic Characterization of Unknown cSNPs and PTMs
• Narrow Biomarker Searches
• Identification of Unknown Proteolytic Products
• Wide Biomarker Searches
• Identification of Unknown Proteolytic Products with Unknown
cSNPs and PTMs
1
2
3
4
Searc
h C
om
ple
xity
Se
arc
h S
pe
ed
Load
Load
Load
Load Load

HIGH THROUGHPUT WIZARD

HIGH THROUGHPUT LOGIC

HIGH THROUGHPUT RESULTS
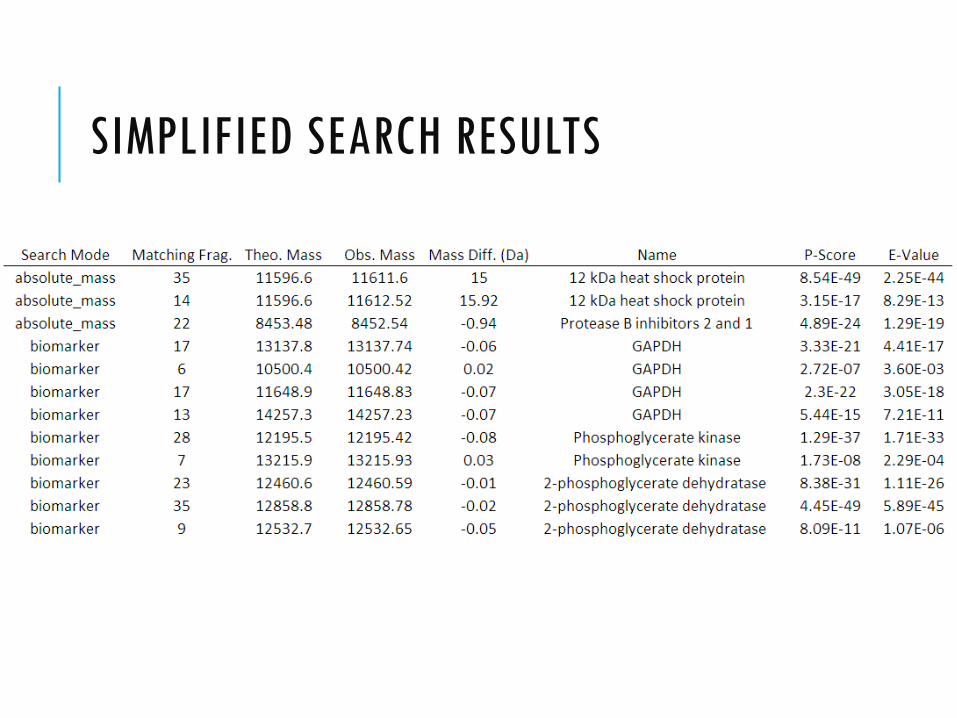
SIMPLIFIED SEARCH RESULTS

Top Down Data Types
Lo/Hi – Respect in ProSightPD
Mid/Hi – Respect in ProSightPD
(Express Scans for MS1)
High/High – cRAWler in
ProSightPD and ProSightPC 3.0
Precursor Data Types
Low Resolution = “Lo”
MA: ~1 - 20 Da
Medium Resolution =
“Med”
MA: 1 – 0.1 Da
High Resolution = “Hi”
MA: 1 – 20 ppm
• Ion Trap (e.g.,
LTQ)
• (Quadrupole)
• Time-of-Flight (TOF)
• Magnetic Sector
• Short Transient
Orbitrap (RP ~6000)
• Orbitrap

The “C-Score”: Improving Proteoform Characterization
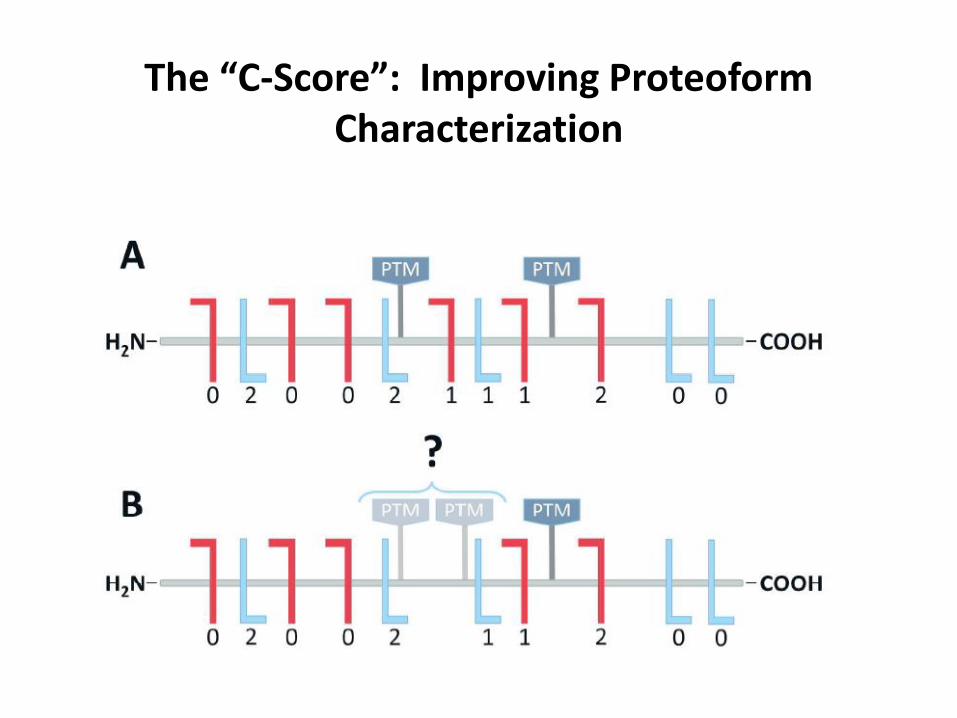
The “C-Score”: Improving Proteoform Characterization

The “C-Score”: Improving Proteoform Characterization

UVPD Now fully
Supported in
ProSightPC 4.0
and
ProSightPD 1.x
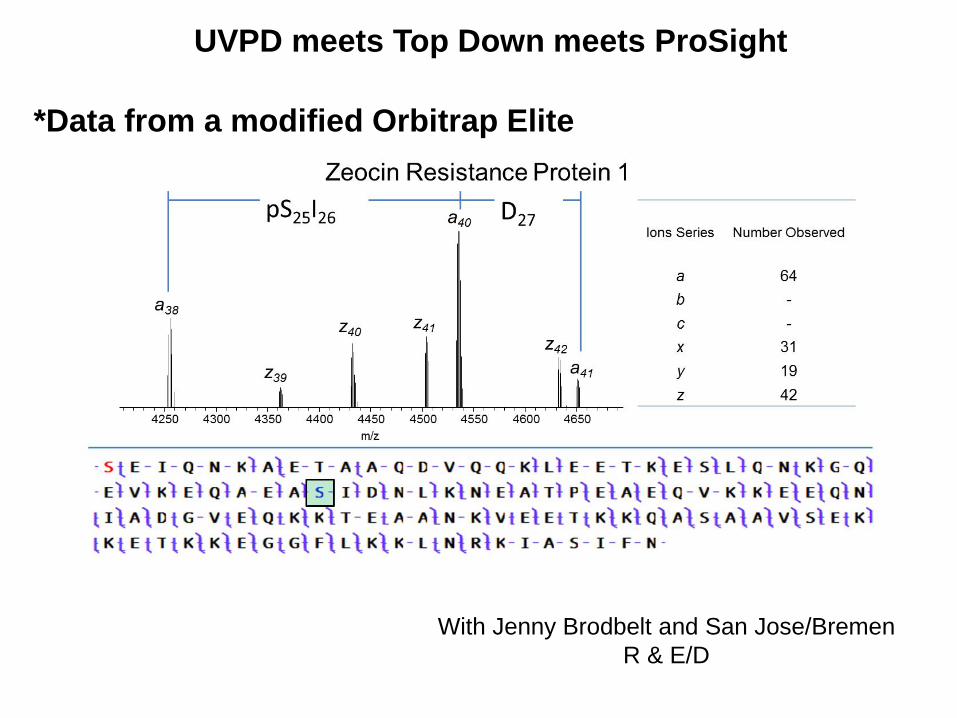
UVPD meets Top Down meets ProSight
With Jenny Brodbelt and San Jose/Bremen
R & E/D
*Data from a modified Orbitrap Elite

Fragment Map Comparison
UVPD of RL29_ECOBW, 50S
ribosomal protein L29
Ions Searched
A, A+, X, X+
B, Y, Y-
C, Z*

Better Graphical Support in ProSightPD
(Proteome Discoverer needs to support
top down better!)
AX – green, angled flag
BY – blue, angled flag
CZ* – red, square flag
If both AB or XY, displays
as BY


















