
Theoretical Studies of Anti-cancer Drug Tamoxifen and ...571758/FULLTEXT01.pdf · Theoretical...
68
Theoretical Studies of Anti-cancer Drug Tamoxifen and Estrogen Receptor Alpha (ERα) Li Gao (郜莉) Doctoral Thesis in Theoretical Chemistry and Biology School of Biotechnology Royal Institute of Technology Stockholm, Sweden 2012
-
Upload
truongphuc -
Category
Documents
-
view
214 -
download
0
Transcript of Theoretical Studies of Anti-cancer Drug Tamoxifen and ...571758/FULLTEXT01.pdf · Theoretical...

Theoretical Studies of Anti-cancer Drug
Tamoxifen and Estrogen Receptor Alpha (ERα)
Li Gao
(郜莉)
Doctoral Thesis in Theoretical Chemistry and Biology
School of Biotechnology
Royal Institute of Technology
Stockholm, Sweden 2012

© Li Gao, 2012
ISBN 978-91-7501-586-6
ISSN 1654-2312
TRITA-BIO Report 2012:23
Printed by Universitetsservice US-AB
Stockholm, Sweden 2012

To my family


I
Abstract
For decades tamoxifen (TAM) has been widely used for treatment of breast cancer by
mediating mainly the estrogen receptor α (ERα) signaling pathways, whereby it suppresses
estrogen stimulated cancer cell growth. The clinical response of TAM has been linked to
cytochrome P450 2D6 (CYP2D6), which is the main isoform responsible for the conversion
of TAM to the active metabolites 4-hydroxyTAM (OHT) and endoxifen. Numerous clinical
studies have thus attempted to assess the effects of CYP2D6 genetic variants on patients
treated by TAM. However, the studies have resulted in contradictive conclusions. This thesis
focuses on computational investigations of TAM and its main target ERα. The results
obtained describe how the ligands contact with the ERα ligand binding domain (LBD), and
provide possible mechanisms responsible for the CYP2D6 activating in TAM treatment. In
addition, the CYP-mediated biotransformation of TAM-like compounds is investigated. All
studies in this thesis aim to a step towards developing improved therapeutic agents for breast
cancer treatment. In paper I, molecular dynamics simulations of ligand-LBD complexes have
been performed. The results indicate that although OHT is a high affinity metabolite, it may
have more undesired estrogen-like properties than the parent drug TAM, as a consequence of
the additional 4-hydroxy group. In papers II and V, quantum mechanics calculations have
been performed to study how the ligands are bound to ERα LBD. It is found that different
conformational isomers of TAM-like ligands are discriminated by the LBD. The interactions
between ligands and His524-Leu525 in the LBD are correlated with the transcriptional
activity of estrogen agonist compounds. In papers III and IV, different CYP-mediated
biotransformations of TAM and derivatives are studied. Based on the results from the
computations, we suggest two modified compounds which are highly possible to be activated
by other CYP isoforms besides CYP2D6, thereby avoiding CYP2D6 genetic polymorphism.
Overall, the results generally agree with the hitherto available experimental results. Further
experimental studies are needed to verify the proposed principles of ligands signaling through
ERα, and to test the suggested CYP-mediated reactions and the bioactivity of the modified
compounds.

II
List of papers
Following papers are included in this thesis:
Paper I Gao, L.; Tu, Y. and Eriksson, L. A. More Stable, more Estrogenic: the
SERM-ERα LBD complex. J. Biophys. Chem. 2011, 2(3):233-243.
Reprinted with permission from the Scientific Research Publishing Inc.
Doi:10.4236/jbpc.2011.23029
Paper II Gao, L.; Tu, Y.; Wegman P.; Wingren, S. and Eriksson, L. A.
Conformational enantiomerization and estrogen receptor alpha binding of
anti-cancer drug tamoxifen and its derivatives. J. Chem. Inf. Model. 2011,
51(2):306-314.
Reprinted with permission from the American Chemical Society.
Doi:10.1021/ci100401t
Paper III Gao, L.; Tu, Y.; Wegman P.; Wingren, S. and Eriksson, L. A. A mechanistic
hypothesis for the cytochrome P450-catalyzed cis-trans isomerization of 4-
hydroxytamoxifen: an unusual redox reaction. J. Chem. Inf. Model. 2011,
51(9):2293-2301.
Reprinted with permission from the American Chemical Society.
Doi:10.1021/ci2001082
Paper IV Gao, L.; Tu, Y.; Agren, H. and Eriksson, L. A. Modification of the anti-
cancer drug tamoxifen to avoid CYP2D6 polymorphism. In manuscript.
Paper V Gao, L.; Tu, Y.; Agren, H. and Eriksson, L. A. Characterization of agonist
binding to His524 in the estrogen receptor α ligand binding domain. J.
Phys. Chem. B 2012, 116(16):4823-4830.
Reprinted with permission from the American Chemical Society.
Doi:10.1021/jp300895g
All the papers are the results of teamwork. I am responsible for the computations and a large
part of the writing in all papers.

III
Acknowledgement
The present work has been carried out at the Division of Theoretical Chemistry and Biology,
School of Biotechnology at the Royal Institute of Technology, and at Örebro Life Science
Center, School of Science and Technology at Örebro University. I would like to thank
everyone in both departments for making my studies in Sweden such an unforgettable time.
I am extremely grateful to my supervisor Prof. Hans Ågren, for taking me as your student,
and for your excellent guide during the time.
I am also deeply indebted to my supervisor Prof. Leif A. Eriksson, for your help,
encouragement, and patience.
I would like to thank my supervisor Dr. Yaoquan Tu, for your scientific knowledge and
generous support.
I would also like to thank Prof. Yi Luo, Dr. Ying Fu, Dr. Zilvinas Rinkevicius and Prof. Boris
Minaev, for your help and the nice lectures.
I was fortunate to collaborate with Dr. Pia Palmebäck Wegman and Prof. Sten Wingren.
Thanks for initiating us doing the research of anti-cancer drug. I have enjoyed doing the
interesting project.
I have had the pleasure to work in great groups, and would like to thank the former and
present members: Boxue, Emma, Edvin, Min, Viraja, Chunxia, Klefah, Magnus, Oles, Salama,
Dragan, Xin, Qiong, Lu, Xianqiang, Yan, Peng, Ying, Liqin, Xiuneng, Yong, Ce, Qiang,
Weijie, Wei, Xinrui, Sai, Lijun, Xing, Chunze, Yuejie, Guangjun, Yongfei, Xiao, Hongbao,
LiLi, Li Li, Junfeng, Hongbao, Quan, Jing, Guanglin, Xifeng, Bogdan, Johannes, Rocio,
Jaime, Kestutis, Irina, Murugan, Mårten, Staffan, Faris, Xiangjun, Zhihui, Zhijun, Anuar,
Ignat, Kayathri, Xu, Xiaofei, Vinicius, Ananda, Robert, Nina, Olav, Kersti, and Keyan. All
my colleagues and friends, thank you for just being who you are.
The School of Biotechnology at Royal Institute of Technology, the School of Science and
Technology at Örebro University, and the Swedish Science Council (VR) are gratefully
acknowledged for financial support.
Finally, I would like to express my deepest gratitude to my family, for all your love, and for
always being there for me.

IV
Abbreviations
AI Aromatase inhibitor
Cpd I Compound I
CYP Cytochrome P450
DES Diethylstilbestrol
DFT Density functional theory
E2 Estradiol
ER Estrogen receptor
GAFF The general amber force field
GEN Genistein
HSD Hydroxysteroid dehydrogenase
LBD Ligand binding domain
MTA 4-methyl-tamoxifen
MTO 4-methyl-toremifene
MD Molecular dynamics
NDT N-desmethyl-tamoxifen
OHT 4-hydroxy-tamoxifen
PDB Protein Data Bank
PME Particle mesh Ewald
RBA Relative binding affinity
RMSD Root-mean-square deviation
SERM Selective estrogen receptor modulators
TAM Tamoxifen, (Z)-2-[4-(1,2-diphenyl-1-butenyl)phenoxy]-N,N-
dimethylethanamine
TOR Toremifene
ZPE Zero-point vibrational energy
W Way-169916

V
Contents
CHAPTER 1. INTRODUCTION ................................................................................................... 1
1.1 Breast Cancer .................................................................................................................... 1
1.2 TAM: Chemical Structures ............................................................................................... 1
1.3 Estrogen ............................................................................................................................. 3
1.4 Estrogen Receptor (ER) .................................................................................................... 3
1.5 ERα Ligands ...................................................................................................................... 6
1.6 Endocrine therapy ............................................................................................................. 8
1.6.1 TAM for breast cancer treatment ................................................................................... 9
1.6.2 Aromatase inhibitor ...................................................................................................... 10
1.7 TAM: in vivo Metabolism ............................................................................................... 10
1.7.1 The major TAM metabolic pathways ............................................................................ 10
1.7.2 CYP-mediated α-hydroxylation and DNA adducts ....................................................... 11
1.7.3 CYP2D6-mediated activation of TAM: 4-hydroxylation .............................................. 12
1.7.4 CYP2D6: the debate over TAM therapy ....................................................................... 14
1.7.5 CYP-mediated cis-trans isomerization of OHT ............................................................ 14
1.7.6 CYP reactions ............................................................................................................... 15
1.8 Comparisons of Selected SERMs .................................................................................... 17
1.8.1 Geometries of SERMs in complex with ERα LBD ........................................................ 18
1.8.2 SERMs with indication of breast cancer ...................................................................... 20
1.9 Aims of the Study ............................................................................................................ 21
CHAPTER 2. COMPUTATIONAL METHODS ......................................................................... 22
2.1 Quantum Mechanics Approaches .................................................................................... 22
2.2 Molecular Mechanics Approaches .................................................................................. 25
CHAPTER 3. ANTAGONISTS AND THE ANTAGONIST-ERα LBD COMPLEXES ............ 28
3.1 More Stable, More Estrogenic―the SERM Profiles ...................................................... 28

VI
3.2 The SERM Profiles of TAM vs OHT .............................................................................. 32
3.3 Flip-Flop: Rotation about the C(Ar)―C(sp2) Bond ........................................................ 33
3.4 CYP-Catalyzed Rotation about the C(sp2)═C(sp2) Bond ............................................... 37
3.5 Modified SERMs ............................................................................................................. 40
CHAPTER 4. AGONISTS AND THE AGONIST-ERα LBD COMPLEXES ............................. 45
CONCLUSIONS ........................................................................................................................... 49
REFERENCES .............................................................................................................................. 50

1
CHAPTER 1. INTRODUCTION
The main focus of this work is the anti-cancer drug Tamoxifen (TAM), which is widely used
to treat breast cancer, and for which the primary target is the estrogen receptor α (ERα). This
chapter gives a brief introduction of TAM therapy for breast cancer and the related
mechanistic pictures of small compounds signaling through ERα. TAM is a prodrug, and its
biotransformation paths are introduced with particular focus on the in vivo activation of the
parent compound.
1.1 Breast Cancer
Breast cancer is the most prevalent cancer in women, especially in the western countries. As
an example, in the United States, it is expected that 39 920 breast cancer patients will die
during 2012, and at the same time the number of new cases among women is expected to be
290 170. Due to the earlier diagnosis and progress in treatment, deaths from breast cancer
have decreased in the last decades, with the current 5-year survival rate of 90% compared to
that of 63% in the early 1960s.1
The discovery of the link between breast cancer and estrogen has made a remarkable
contribution to improve the cancer treatment and reduce the mortality rate. Approximately
70% of breast cancers are estrogen dependent, expressing estrogen receptors (ERs). The ER
positive breast cancer cells exert an estrogen promoted proliferation through ER-regulated
gene transcription. If the growth stimulated by estrogen can be blocked, then the breast cancer
may be controlled. For example, TAM is a commonly used drug to treat breast cancer by
directly blocking the actions of endogenous estrogen.
1.2 TAM: Chemical Structures
TAM (ICI 46,474) was first synthesized in the 1960s by the pharmaceutical company ICI
(now known as AstraZeneca). It is an old drug with the brand name of Nolvadex, which has
been used as first-line therapy for breast cancer since the 1970s, and is until now still widely
used. It was initially marketed to treat breast cancer in 1973, and approved for breast cancer
treatment and prevention by the Food and Drug Administration in 1977 and 2007,
respectively.

Chapter 1. Introduction
2
Scheme 1.1 The atropisomers of trans- and cis-TAM: trans- and cis-TAM in ‘counterclockwise’
propellers; trans'- and cis'-TAM in ‘clockwise’ propellers.
ON
ON
ON
ON
C(Ar) C(sp2)
trans'-TAM
cis'-TAMcis-TAM
trans-TAM
C(Ar) C(sp2)
The chemical structures of TAM are seen in Scheme 1.1. The trans-isomer ((Z)-2-[4-(1,2-
diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine) but not the cis form is used for breast
cancer treatment. The core structure of TAM is a triarylethylene, which has been found to
exist in a molecular propeller.2 It is a highly conjugated system as the ethylene substituted by
three aromatic rings. To stay in a low energy conformation, conjugation effects require the
three rings to be coplanar in the ethylene plane, but this leads to increased steric repulsion of
the rings. The aryl rings are thus forced to twist away from the ethylene plane to compromise
between conjugation and steric repulsion, thereby the system stays in the low energy
conformations, i.e., the ‘clockwise’ and ‘counterclockwise’ propellers (Figure 1.1).3,4 The two
propellers differ in helicity, as the rings twist in opposite directions forming atropisomers.
The interconversion between the atropisomers has been studied experimentally for more
crowded triarylvinyl propellers, in which the rotational barriers were found to be in the range
of 15-20 kcal/mol for the correlated rotations about the C(Ar)―C(sp2) bonds.3,5-7 The thermal
equilibrium between the ‘clockwise’ and ‘counterclockwise’ TAM propellers was too fast to
be measured experimentally even at -75 ˚C.8 Therefore, the atropisomeric propellers of TAM
are expected to have equivalent populations under physiological conditions. In such
circumstance, the isolation of either TAM isomer is not possible, and the bioactivity of each

Chapter 1. Introduction
3
isomer is not clear. The pharmacological profiles of TAM atropisomers are thus
compensatively studied by computational approaches in Paper II.
1.3 Estrogen
The endogenous estrogens in women are steroid hormones, which are biosynthesized from
cholesterol via multiple enzymatic steps (Scheme 1.2). Aromatase is one of the most
important enzymes catalyzing the biotransformation to eventually produce estrogens, of
which 17β-estradiol (E2) is the most potent female hormone. In premenopausal women,
estrogens are produced primarily in the ovaries. Oppositely, the ovaries almost cease to
secrete estrogen in postmenopausal women and the serum concentration of estrogen thus
decreases dramatically. Possible consequences of the lack of estrogen in postmenopausal
women are frequently reported, including postmenopause symptoms, increased risks of
osteoporosis, coronary heart disease and Alzheimer's disease.9-13 On the other hand, a
cumulative exposure to estrogen does encourage development of female reproductive cancers.
Such examples include breast cancer and uterus cancer, which are found associated with
hormone replacement therapy, early menarche and late menopause.14 The contribution of
estrogens in various physiological and pathological pathways highly depends on their binding
to estrogen receptors and activating transcription of estrogen responsive genes.15-18
Scheme 1.2 The major biosynthetic paths of endogenous estrogens.
HO
Estradiol
OH
HOCholesterol
O
HO
Estrone
O
O
Androstenedione
OH
OTestosterone
OH
HOAndrostenediol
O
HODehydroepi-androsterone
Aromatase
Aromatase
3β-HSD
3β-HSD
17β-HSD 17β-HSD 17β-HSD
1.4 Estrogen Receptor (ER)
ERs belong to the large superfamily of nuclear receptors and reside in the nuclei of estrogen
target cells. They are expressed in two subtypes: ERα19 and ERβ.20 The focus of the current
work is the ERα isoform, which is primarily expressed in breast and uterine tissues. ERα is
found intimately associated with breast cancer initiation and progression, and is hence very

Chapter 1. Introduction
4
important for breast cancer prevention and treatment. Breast cancer cells possess much higher
levels of ERα than normal breast epithelial cells.21,22 The increase of ERα expression in
benign breast epithelium has been linked to an increased risk of breast cancer.23,24
ERs are composed of five function domains (Figure 1.1), referred to as the N-terminal A/B
domain, the DNA binding domain (C), the hinge domain (D), the ligand binding domain
(LBD, E), and the C-terminal F domain. As ERα and ERβ are encoded by homologous genes,
their sequences are significantly similar at the amino acid level, in particular the DNA binding
domain and the LBD, with identity of 97% and 56%, respectively. The DNA binding domain
binds to specific estrogen response elements, and the resulting gene transcription is initiated
through a ligand-independent or -dependent manner mediated by transcriptional activation
domain 1 or 2 (AF-1 or AF-2), respectively.25
N'
N'
hERβ C'
C'
AF-1 AF-2
A/B C D E F
A/B C D E F
hERα
Figure 1.1 The human estrogen receptor α and β (hERα and hERβ).
The ligand-dependent manner of the gene transcription through AF-2 makes it possible to
interfere with the expression of estrogen responsive genes by small molecule ligands, which
have the potential of activating or repressing ER signaling in estrogen-related diseases. Great
efforts have been made in related studies, and a branch thereof is structural biology studies of
ER ligands and the LBD. Since the crystallographic structures of the ligand-ERα LBD
complexes were initially determined in the late 1990s, about 100 LBD structures of ERs have
been deposited in the RCSB Protein Data Bank (PDB). However, the other domains, except
the DNA binding domain, have not yet been solved, and the complete structure of the five ER
domains as a whole is still lacking.
Of particular interest is that the LBD shows binding properties of trapping a variety of
exogenous compounds besides the endogenous steroid hormones. As a result, a wide range of
compounds have been cocrystalized with the wild type or mutated LBD, and have shown to
induce different conformational changes of the receptor. The crystallographic structures of the
ligand-ERα LBD complexes are generally classified as agonist or antagonist conformations
based on the positions of the C-terminal Helix 12 (H12, Figure 1.2), which displays a high
degree of dynamic flexibility. H12 closes the binding site in the agonist conformation (Figure
1.2A), which enables co-activator binding and the dimerization process of the ligand-ERα

Chapter 1. Introduction
5
complex to form a transcription unit that triggers gene expression or cell proliferation. This
closed/agonist conformation is thus asserted to be an active conformation, and is frequently
found in the cases of LBD in complex with E2 and agonist ligands. Conversely, antagonists
have bulky tails and bind to the same site of the LBD as agonists do, but force H12 to project
towards the open/antagonist conformation (Figure 1.2B), whereby they occupy part of the co-
activator binding groove. The co-activator binding is blocked with the open/antagonist-LBD,
which slows or stops ER-mediated transcription.26,27 The open/antagonist-LBD is a relatively
inactive conformation compared to the closed/agonist-LBD, but it is not completely inactive.
The contradictory bioactivities have been the key issues in the studies of estrogen antagonists,
and more detailed information is given later in this summary.
Figure 1.2 The closed/agonist E2-ERα LBD complex (A, 1GWR28) and the open/antagonist TAM-
ERα LBD complex (B, 3ERT26), H12 in cyan and coactivator in blue.
It is worthwhile to note that the ligand activated transcription is completely silent for the
unbound LBD. However, the crystallization of an unbound ERα LBD (without any ligand) is
hampered by the conformational flexibility of the LBD. A large part of the LBD is formed by
rigid helixes as seen in Figure 1.2, while extreme flexibility is mainly due to the variation of
H12. The apo conformation (Figure 1.3) of a crystallized ERα LBD (1a5229) is suggested to
be a representative of the unbound LBD, in which the Helix 12 is solvent exposed and
extended away from the domain, and is thus very flexible. The reorientation of H12 occurs in
the capture of a ligand, resulting in the corresponding closed/agonist- or open/antagonist-LBD
depending on the ligand molecules.
H12
H12 A B

Chapter 1. Introduction
6
Figure 1.3 The apo-ERα LBD (1a5229).
1.5 ERα Ligands
Beside steroid hormones, numerous exogenous compounds are able to bind to ERα due to the
versatile binding properties of the LBD, albeit these ligands are structurally very different,
and result in anti-estrogen and/or estrogen like properties.30 According to the differences in
functional activities, ERα ligands are generally classified into four orders: full agonist, partial
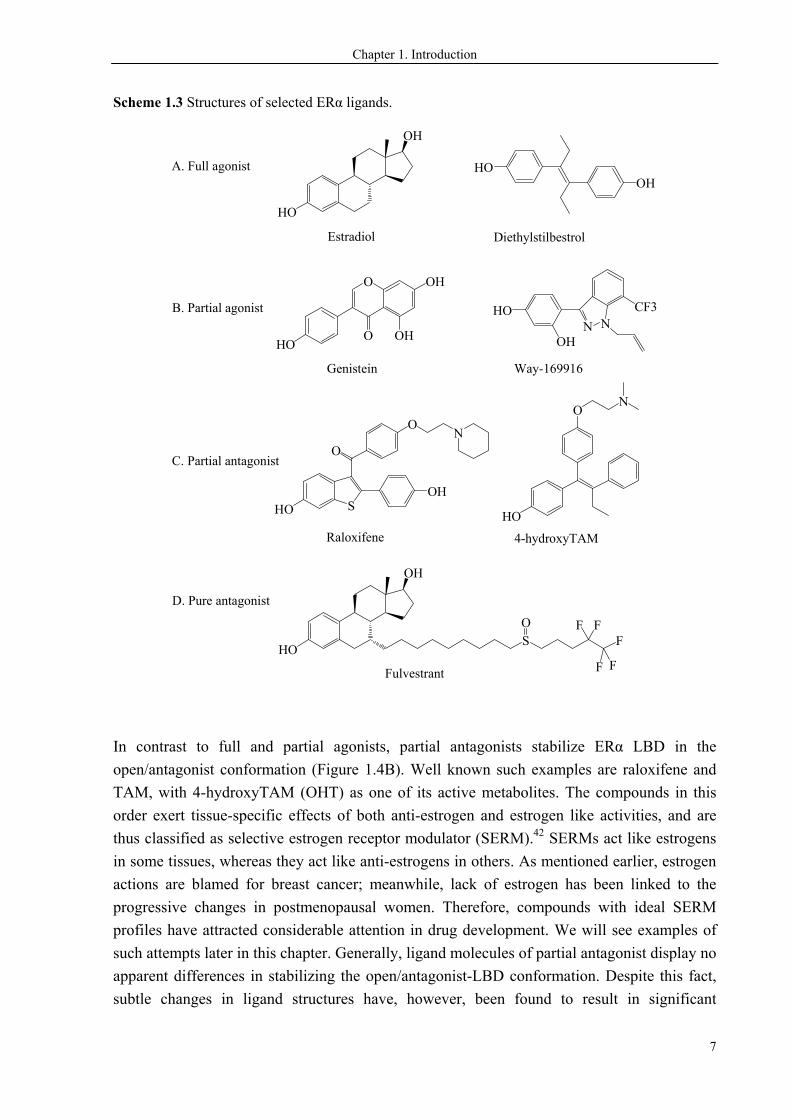
agonist, partial antagonist and pure antagonist, with examples in Scheme 1.3.
Full and partial agonists stabilize ERα LBD in the closed/agonist conformation (Figure 1.4A).
E2 is known as the most potent female hormone in women, and is frequently used as a
standard full agonist in experimental studies. Diethylstilbestrol (DES) is a synthetic full
agonist, which has been used in pregnant women and blamed for many adverse effects on the
offspring.31,32 The bioactivity profiles of partial agonists are much more complicated due to
the mixed agonist/antagonist properties. The soy phytoestrogen genistein (GEN) is a partial
agonist, and the consumption of soy food has been suggested to reduce the risk of developing
breast cancer.33,34 However, GEN has also been found to stimulate breast cancer cell growth
in some assays.35,36 Way-169916 (W) is an even weaker partial agonist, and has broad anti-
inflammatory activity instead of the conventional agonistic effects of transcriptional
activation.37-39 Besides the reduced capacity in transcriptional activation, partial agonists were
experimentally found more difficult to be cocrystallized with the LBD than full agonists. With
the aid of mutational techniques for success in the cocrystallization, the crystal structures of
partial agonists in complex with LBD have the same closed/agonist conformation as that of
full agonists.40,41 Little is known about how the ligands affect the allosteric conformational
changes in H12, and subsequently affect the transcriptional or regular bioactivities. We thus
discuss what may elicit such differences between full and partial agonists in Paper V.
H12
Chapter 1. Introduction
7
Scheme 1.3 Structures of selected ERα ligands.
OH
HO
O
OHO
OH
OH
OHHO
N NHO
OH
CF3
Estradiol Diethylstilbestrol
Genistein Way-169916
A. Full agonist
B. Partial agonist
C. Partial antagonist
D. Pure antagonist
ON
HO S
O
OH
ON
4-hydroxyTAMRaloxifene
OH
HOS F
Fulvestrant
HO
O F F
F F
In contrast to full and partial agonists, partial antagonists stabilize ERα LBD in the
open/antagonist conformation (Figure 1.4B). Well known such examples are raloxifene and
TAM, with 4-hydroxyTAM (OHT) as one of its active metabolites. The compounds in this
order exert tissue-specific effects of both anti-estrogen and estrogen like activities, and are
thus classified as selective estrogen receptor modulator (SERM).42 SERMs act like estrogens
in some tissues, whereas they act like anti-estrogens in others. As mentioned earlier, estrogen
actions are blamed for breast cancer; meanwhile, lack of estrogen has been linked to the
progressive changes in postmenopausal women. Therefore, compounds with ideal SERM
profiles have attracted considerable attention in drug development. We will see examples of
such attempts later in this chapter. Generally, ligand molecules of partial antagonist display no
apparent differences in stabilizing the open/antagonist-LBD conformation. Despite this fact,
subtle changes in ligand structures have, however, been found to result in significant

Chapter 1. Introduction
8
differences in bioactivities (agonism and/or antagonism).43 The means by which the
compounds share similar crystal structures of the ligand-LBD complexes, but differ in their
ER signaling are poorly understood. We here discuss in Paper I the possible mechanism
which may govern the agonist and antagonist properties of SERMs.
Pure antagonists competitively bind to ERα as partial antagonists do, but unlike SERMs, they
exert anti-estrogen actions in all tissues. The mechanism of pure antagonists has been
described as downregulation of ER levels and inhibition of dimerization, and hence the
ligand-ERα complex does not work as a transcription initiation unit. The pure antagonist
fulvestrant is used in breast cancer patients who have failed in TAM therapy. The ligands in
this order are not a focus of the current study.
Figure 1.4 Superposed crystallographic structures of ligands in complex with ERα LBD: A. full and
partial agonists, 1G50 in red (E2),44 3ERD in gray (DES),26 1X7R in blue (GEN),41 and 2QZO in green
(W).40 B. partial antagonists, OHT-LBD (3ERT in red,26 2JF9.A in gray,45 and 2BJ4.A in yellow46)
and raloxifene-LBD (1ERR.A in blue,27 and 2JFA.A in green45). Reproduced with permission of the
copyright owner.47
1.6 Endocrine therapy
Endocrine therapy is also called hormone therapy. In this treatment hormones are usually
added, blocked or removed. In the case of breast cancer, the ER-positive cancer cell
proliferation is stimulated by estrogens, and hence hormone therapy for breast cancer is in fact
anti-hormone treatment. This is an approach through which the tumor growth is suppressed by
preventing ERα from binding the endogenous estrogens, which the tumor cells require to
grow.
Different strategies are used in endocrine therapy. Surgical removal of ovaries
(oophorectomy), which decreases the levels of circulating estrogens in premenopausal women,
A B

Chapter 1. Introduction
9
was applied to breast cancer patients even before the discovery of ERα and corresponding
estrogen responsiveness in the cancer cells. As simpler and safer alternatives to surgical
approaches, such as oophorectomy, adrenalectomy, and hypophysectomy, drugs are
commonly used to block the estrogen stimulated cancer cell proliferation. Based on different
mechanisms, these drugs can either work as estrogen antagonism to block estrogen binding to
ERα, or directly block the estrogen biosynthetic process. TAM treatment represents the most
commonly used approach by competitively binding to ERα, thereby TAM functions as
estrogen antagonist in breast tissue. For the other approach, aromatase inhibitors (AIs) are
usually applied to inhibit the aromatase enzyme which catalyzes the essential conversion to
produce estrogens.
1.6.1 TAM for breast cancer treatment
TAM is the first clinically used SERM for breast cancer treatment. The SERM profiles, the
mixed estrogen agonist and antagonist properties depending on the specific tissue, and related
effects of TAM48 are briefly summarized below:
Anti-estrogenic properties in breast tissue: protect against tumor relapse in breast cancer
treatment, and reduce the incidence of breast cancer as a preventive drug.
Estrogenic properties in the cardiovascular system: decrease heart disease and reduce
serum concentration of cholesterol.
Estrogenic properties in bone: increase the bone density in postmenopausal women and
thus reduce the fracture rate.
Estrogenic properties in uterine tissue: increase the risk of endometrial cancer and increase
endometrial thickness.
Menopausal symptoms are most frequently reported adverse effects of TAM, such as hot
flashes and atrophic vaginitis, which may be caused by deprivation of estrogen due to the
antagonist effects of TAM. Other adverse effects include slightly increased risks of
cataract and thromboembolic.
Overall, TAM significantly reduces the risk of cancer recurrence and death49 with beneficial
effects on bone and the cardiovascular system, and has no serious side effects except the
slightly increased incidence of endometrial cancer. Compared to other cancer therapies, such
as radiation or chemotherapy, TAM therapy specifically targets the ERα signaling pathways,
which does not damage normal cells and tissues, and thus causes relatively fewer and milder
side effects. The drug is well tolerated by breast cancer patients with benefits that are much
larger than its adverse effects.

Chapter 1. Introduction
10
1.6.2 Aromatase inhibitor
Anastrozole, exemestane and letrozole are commonly used AIs for breast cancer treatment,
which block the biosynthesis of endogenous estrogens by inhibiting the aromatase enzyme
and thus significantly decrease the levels of circulating estrogen. The mechanism of this
approach is based on estrogen deprivation all over the body, in some sense close to that of
pure antagonists, and is not discussed further in this thesis.
1.7 TAM: in vivo Metabolism
TAM undergoes in vivo Phase I and Phase II reactions and the processes can vary
significantly among individuals due to patients carrying different genotypes of the metabolic
enzymes, resulting in dramatic differences in plasma levels of TAM and its metabolites
among patients. Phase I reactions of TAM mainly consist of oxidations catalyzed by
cytochrome P450 (CYP) enzymes, which produce both active and toxic metabolites.50-52
Phase II reactions are also known as conjugation reactions, which usually combine polar
functional groups with the Phase I products, and thus facilitate drug elimination. The
conjugation of TAM and its metabolites is most commonly catalyzed by sulfotransferase
(SULT) and UDP-glucuronosyltransferase (UGT). Genetic variation in patients occurs with
the enzymes participating in Phase I or Phase II reactions, and affects the plasma levels of
TAM and its metabolites. Hence, the influences of enzyme genotypes in clinical response of
TAM therapy have been the focus of studies on TAM treatment for decades. We will see a
more detailed introduction in the following section.
1.7.1 The major TAM metabolic pathways
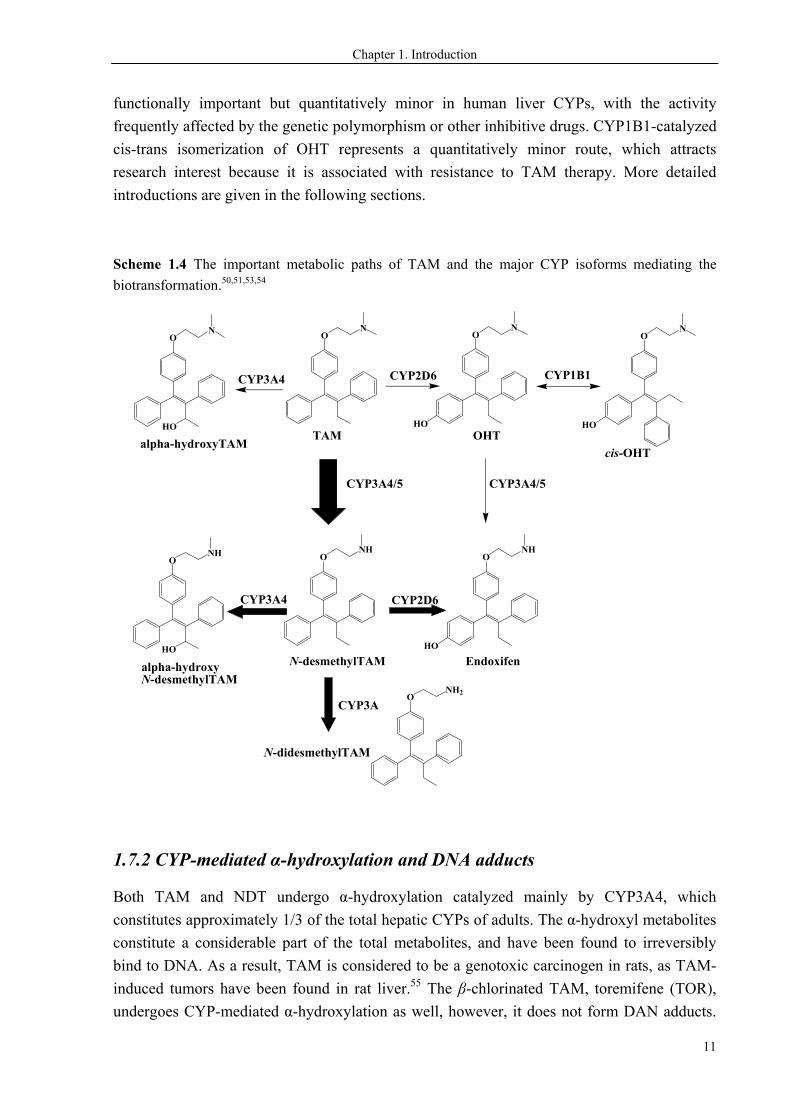
TAM undergoes extensive hepatic oxidations, i.e. the CYP-mediated Phase I reactions
(Scheme 1.4). The sequential biotransformation mainly constitutes CYP-mediated N-
demethylation, 4-hydroxylation, and α-hydroxylation, yielding the active or toxic
metabolites.50-52 The primary metabolites, including N-didesmethyl-TAM (NDT), α-hydroxy-
TAM, and OHT, are generated via direct oxidation of the parent drug TAM. The metabolites
may undergo further oxidations into secondary metabolites, including N-didesmethyl-TAM,
endoxifen, and α-hydroxy-NDT. A variety of CYP isoforms are involved in the TAM
biotransformation, including CYP1A2, CYP1B1, CYP2B6, CYP2C9, CYP2C19, CYP2D6,
CYP3A4, CYP3A5 and CYP3A4, of which CYP3A4/5 and CYP2D6 play prominent roles.
CYP3A4 is the most abundant isoform in adult humans. It does not have a clinically
significant polymorphism, and mainly catalyzes N-demethylation and α-hydroxylation. With
CYP3A5 also contributing to the N-demethylation, N-desmethyl-TAM (NDT) is the most
prevalent in vivo metabolite. CYP2D6 is responsible for 4-hydroxylation, hence it is of
particular importance in the metabolic activation of the prodrug. The CYP2D6 isoform is
Chapter 1. Introduction
11
functionally important but quantitatively minor in human liver CYPs, with the activity
frequently affected by the genetic polymorphism or other inhibitive drugs. CYP1B1-catalyzed
cis-trans isomerization of OHT represents a quantitatively minor route, which attracts
research interest because it is associated with resistance to TAM therapy. More detailed
introductions are given in the following sections.
Scheme 1.4 The important metabolic paths of TAM and the major CYP isoforms mediating the
biotransformation.50,51,53,54
ON
HO
ON
ONH
HO
ONH
TAM OHT
N-desmethylTAM Endoxifen
CYP3A4/5
CYP2D6
CYP2D6
CYP3A4/5
ON
HO
ONH2
ONH
HO
HO
ON
cis-OHT
CYP1B1
CYP3A4
CYP3A
N-didesmethylTAM
CYP3A4
alpha-hydroxyTAM
alpha-hydroxyN-desmethylTAM
1.7.2 CYP-mediated α-hydroxylation and DNA adducts
Both TAM and NDT undergo α-hydroxylation catalyzed mainly by CYP3A4, which
constitutes approximately 1/3 of the total hepatic CYPs of adults. The α-hydroxyl metabolites
constitute a considerable part of the total metabolites, and have been found to irreversibly
bind to DNA. As a result, TAM is considered to be a genotoxic carcinogen in rats, as TAM-
induced tumors have been found in rat liver.55 The β-chlorinated TAM, toremifene (TOR),
undergoes CYP-mediated α-hydroxylation as well, however, it does not form DAN adducts.

Chapter 1. Introduction
12
The postulated mechanism of forming DNA adducts is related to the Phase II conjugation of
the α-hydroxyl group. The allylic carbocation electrophile subsequently formed is capable of
binding DNA, illustrated in Scheme 1.5.53,54 TOR was developed with the purpose to improve
the safety of TAM with regard to forming DNA adducts.56,57 Experimentally, DNA adducts
were not clearly found in patients treated by TAM or TOR.58,59 TOR was proven no better
than TAM, as it showed equivalent potency to TAM in suppressing breast cancer and in
encouraging the development of endometrial cancer.56 The incidence of endometrial cancer
was thus proven to be a consequence of TAM or TOR actions via the ER signaling pathway
rather than genotoxicity of DNA adducts.
Scheme 1.5 α-hydroxy and DNA adducts.53,54
ON
TAM
ON
HO
CYP3A4
alpha-hydroxyTAM
ON
+
DNA adducts
ON
DNA
Carbocation intermediate
DNA
ON
TOR
ON
HO
CYP3A4
alpha-hydroxyTOR
Cl
ON
+
Carbocation intermediate
ClCl
1.7.3 CYP2D6-mediated activation of TAM: 4-hydroxylation
Both TAM and NDT undergo 4-hydroxylation predominantly catalyzed by CYP2D6.50,51
Being the opposite of the CYP3A4 distribution, CYP2D6 is present in less than 5% of total
CYPs in the liver. However, the importance of CYP2D6 in metabolizing TAM does not
parallel its hepatic abundance. CYP2D6 catalyzes the 4-hydroxylation of TAM and NDT to
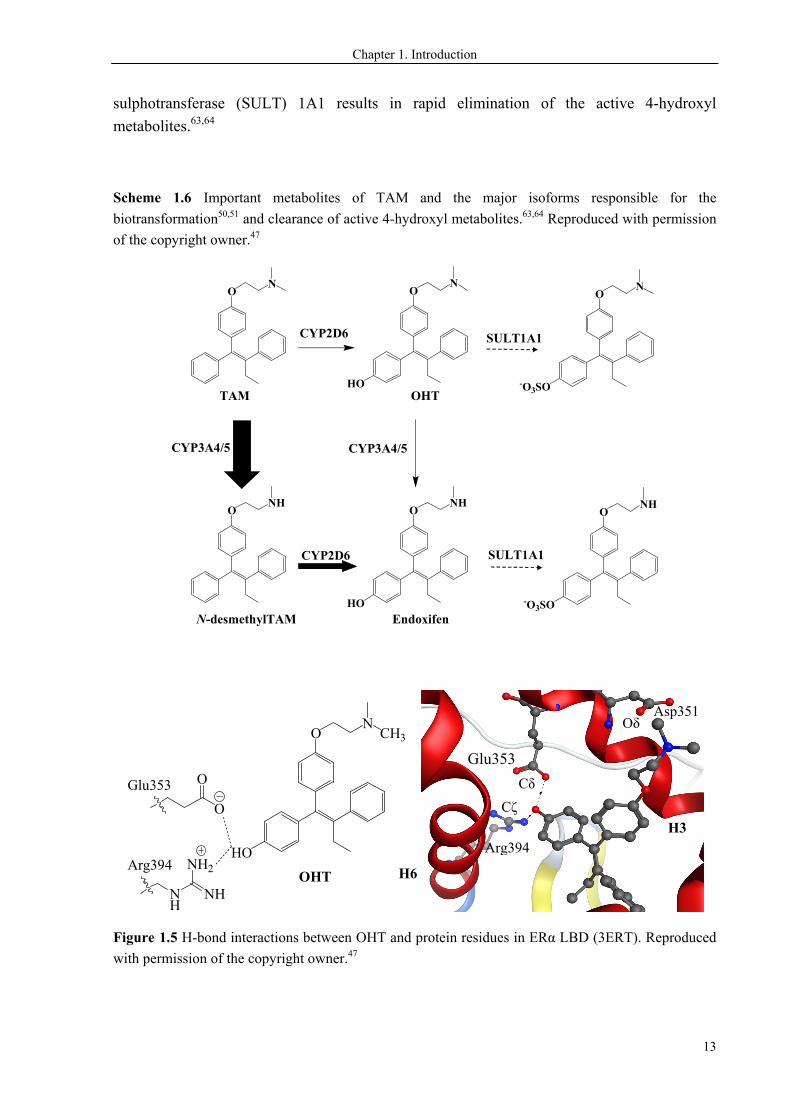
produce the active metabolites OHT and endoxifen, respectively (Scheme 1.6). The additional
4-hydroxyl group forms hydrogen bonds with the side chains of Glu353 and Arg394 in ERα
(Figure 1.5). OHT binds to ERα with affinity equivalent to that of E2, and so does endoxifen.
OHT and endoxifen are thus much more potent than the TAM parent drug, which binds to
ERα with fairly weak affinity.60-62 The conversion of TAM to 4-hydroxy metabolites results in
the metabolic activation of the prodrug, thereby it is intuitively seen to be positive for breast
cancer treatment. In contrast, the subsequent sulphate conjugation mediated by
Chapter 1. Introduction
13
sulphotransferase (SULT) 1A1 results in rapid elimination of the active 4-hydroxyl
metabolites.63,64
Scheme 1.6 Important metabolites of TAM and the major isoforms responsible for the
biotransformation50,51 and clearance of active 4-hydroxyl metabolites.63,64 Reproduced with permission
of the copyright owner.47
ON
HO
ON
ONH
HO
ONH
TAM OHT
N-desmethylTAM Endoxifen
CYP3A4/5
CYP2D6
CYP2D6
SULT1A1
CYP3A4/5
-O3SO
ONH
SULT1A1
-O3SO
ON
HO
ON
CH3
OHT
O
O
NH
NH
NH2
Glu353
Arg394
Figure 1.5 H-bond interactions between OHT and protein residues in ERα LBD (3ERT). Reproduced
with permission of the copyright owner.47
Arg394
Asp351
Glu353
H6
H3
Cδ
Cζ
Oδ

Chapter 1. Introduction
14
1.7.4 CYP2D6: the debate over TAM therapy
The key step, 4-hydroxylation, in the bioactivation of TAM is predominantly, though not
exclusively, catalyzed by CYP2D6. CYP2D6 is not an inducible isoform, with individual
activity in patients under the control of genetic variants. A variety of genotypes of CYP2D6
have been found in humans. The genetic polymorphism of CYP2D6 is particularly
problematic, because most allelic variants of CYP2D6 show diminished or absent catalytic
activity compared to the wild type CYP2D6*1. CYP2D6*4 is the most common
polymorphism in Caucasian women, encoding inactive CYP2D6. CYP2D6*10 is frequently
carried by Asian women with decreased CYP2D6 activity. The polymorphic variants affect
the plasma concentrations of the active metabolites through the genetically determined
CYP2D6 activities, and hence intimately affect the clinical response to TAM treatment. The
conjugation of the active 4-hydroxyl metabolites mediated by SULT1A1 may also affect the
outcome. Therefore, an intuitive implication in individualized therapy for breast cancer is that
patients carrying null CYP2D6 alleles and high-activity SULT1A1*1 allele might response
poorly to TAM treatment, whereupon they would be best treated by other approaches.
To establish a more personalized treatment, the link between CYP2D6 genotypes and the
clinical outcome of TAM treatment has been extensively investigated in breast cancer patients.
Patients carrying null CYP2D6 alleles were frequently reported to have an increased risk of
cancer recurrence than women with two-wide type CYP2D6*1 alleles.65-70 These findings
were intuitively right, but conflicting with other studies, which reported that patients carrying
one or two null CYP2D6*4 alleles benefited more than those with two functional CYP2D6*1
alleles.71,72 It was also reported that CYP2D6 activity had no effect on breast cancer
recurrence in other studies,73,74 and even in two recent large studies.75,76 Another
counterintuitive clinical result was that patients with low SULT1A activity had higher risk of
death than those with high SULT1A1 activity.77 The effects of higher levels of active
metabolites remained elusive, meanwhile markedly lower intracellular levels of the prodrug
TAM were found in resistant tumors.78
1.7.5 CYP-mediated cis-trans isomerization of OHT
Many patients suffer from the recurrences in TAM therapy due to acquired TAM resistance.
In TAM-resistant tumors, the relatively high ratio of cis:trans-OHT was clinically observed
besides the observation of a lower cellular concentration of TAM.78,79 Cis-OHT was found to
be isomerized directly from the active metabolite trans-OHT, with much weaker anti-estrogen
activity compared to the trans form.80 In contrast to the facile cis-trans isomerization of OHT,
the isomeric interconversion was not observed for the parent compound TAM (Scheme 1.7).
The isomerization of OHT was proven to be catalyzed by human liver microsomes in the
presence of NADPH, which served as a typical cofactor for CYP-mediated reactions.81 A

Chapter 1. Introduction
15
more detailed study by recombinant individual CYP isoforms found that CYP1B1 was the
major isoform catalyzing this isomerization, with CYP2B6 and CYP2C19 also involved.82
The cis-trans isomerization of OHT elicits no changes in the redox (reduction-oxidation) state
between the cis and trans isomers, however, a CYP-mediated redox process is argued by the
requirement for NADPH. Less clear is the mechanistic picture of this CYP-mediated cis-trans
isomerization, which is a rarely reported CYP reaction. A postulated isomerization process is
thus discussed in Paper III.
Scheme 1.7 The cis-trans isomerization.
ON
trans-TAM
ON
cis-TAM
HO
ON
trans-OHT
HO
ON
cis-OHT
CYP1B1
NADPH
1.7.6 CYP reactions
As seen in the TAM metabolism, the CYP superfamily consists of a great variety of CYP
enzyme forms (isoforms). These enzymes are particularly versatile and perform diverse types
of transformations, such as N-dealkylation, hydroxylation, epoxidation, and desaturation.83,84
The CYP-mediated reactions are of vital importance in drug metabolism as well as in other
biotranformations. The activation and detoxification of a drug may depend on the activity of
the related CYP isoforms, which controls the rate and the amount of a compound that is
metabolized. In contrast, the activity of CYP isoforms can be affected by many drugs, either
decreased by directly inhibiting the activity of a specific isoform or increased by inducing the
biosynthesis of an inducible isoform.

Chapter 1. Introduction
16
Figure 1.6 The heme group (iron protoprophyrin, heme b).
Most CYP isoforms have a heme group (Figure 1.6) tethering to a cysteine side chain in the
active site. The prosthetic heme group most commonly contains an iron atom in the center of
a large porphyrin ring, and has been the essential focus of theoretical studies of CYP reactions
for decades.85-90 A generally accepted catalytic pathway of CYP reactions is illustrated in
Scheme 1.8. The heme group is activated by dioxygen in the presence of substrate molecules.
The resulting ferrous-dioxygen complex may evolve through the ferric-hydroperoxide, and
finally be activated to the high-valent iron-oxo intermediate, Compound I (Cpd I), which is
well asserted as the driving force in the efficient CYP oxidations. The chemical nature of the
highly active Cpd I is elctrophilic, and hence the reactions are initiated from either
electrophilic attack of the π system or abstraction of an H atom from the substrate. Some
generally consensus pathways are illustrated in Scheme 1.9.83,85,86,91-97
Scheme 1.8 An overall equation and a representative pathway of CYP-mediated hydroxylation of
hydrocarbon C—H bond.
OO
H
Fe H+
OO
H
Fe
O
Fe
H
Cpd 0 Cpd I
RHO
Fe
R
-H2O H
abstr.
OH-
rebound
Fe
Prod.
H ORH
Fe
O2+NADPH
NADP+Heme
RHRHRH
RH + O2 + NADPH + H+ ROH + NADP+ + H2OCYP

Chapter 1. Introduction
17
Scheme 1.9 The CYP catalyzed reaction paths of hydroxylation and dealkylation.
HR RO
RH
H2C
XCH3
CH3
XCH2
CH3
XH
CH3
O
Alkane hydroxylation
Dealkylation
Fe
S
O
Fe
S
Fe
S
O
Fe
S
OH
Fe
S
OH
Fe
S
Arene hydroxylation Fe
S
O
Fe
S
Fe
S
O OHNIH-shift
While the explicit heme-containing oxidation steps are intriguing, they are, however, not a
focus of the current studies. The CYP-mediated Phase I metabolism of TAM is investigated
based on the most well justified initial step, the H atom abstraction from the individual
reactive sites leading to different metabolites. Different CYP reactions are thus compared at
the substrate levels with low computational cost.
1.8 Comparisons of Selected SERMs
An ideal SERM should fulfill certain criteria, such as (1) having antagonist actions in breast
tissues with a potency high enough to suppress the estrogen-stimulated cancer cell growth, (2)
having estrogenic effects on bone to protect against bone loss and fracture, on the
cardiovascular system to protect against coronary heart disease, on the nervous system to
improve cognitive function and protect against Alzheimer’s, (3) having less undesired agonist
properties in the uterus and being safe in the long run. Great effort has been made for such
attempts with many SERMs under development or abandoned, and some of which are briefly
compared herein (Figure 1.7 and Scheme 1.10). New structures are proposed in Paper IV.

Chapter 1. Introduction
18
1.8.1 Geometries of SERMs in complex with ERα LBD
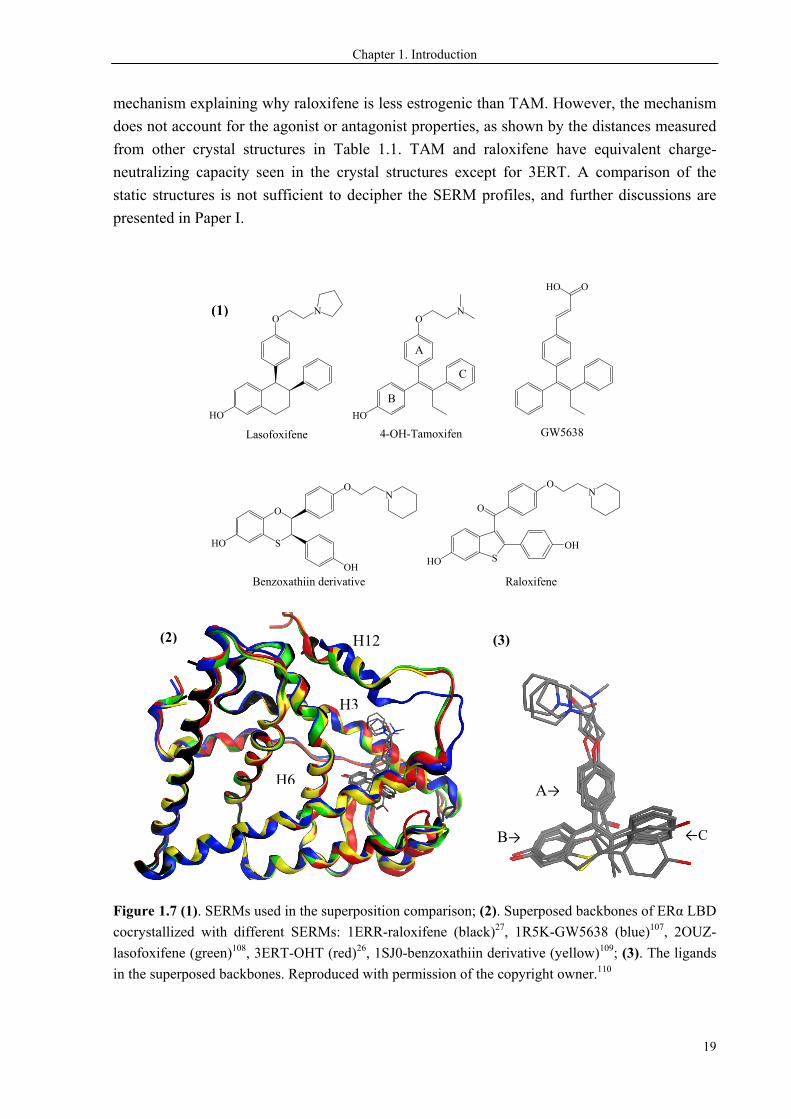
We performed geometric comparison of five structurally different SERMs in complex with
ERα LBD (Figure 1.7). Lasofoxifene can be viewed as a fixed ring analogue to OHT, and for
which the isomerization to the cis-isomer is prohibited. GW5638 is a triphenylethylene
derivative, and is thus structurally analogous to TAM. The lack of the high affinity 4-hydroxy
group does not affect the binding mode of GW5638 to LBD. It binds to the receptor in the
same site and orientation as OHT does. For this reason, OHT-LBD can represent the anti-
cancer drug TAM and its major metabolites bound to the receptor. The binding of raloxifene
and the benzoxathiin derivative is also in a similar manner, albeit they are structurally very
different from TAM-like compounds. The positioning of SERMs in the binding site is
compared in Figure 1.13(3), showing identical binding behavior of the B-rings, slight
deviations of the A-rings, and most flexible orientations of the C-rings. The comparison
indicates that ERα ligangs require the proper orientations of A- and B-rings, which have
proven by experiments as affecting the relative binding affinity (RBA) to ERα.8,98
The ligand-induced allosteric changes lead to identical structures of these SERM-LBD
complexes except for GW5638, as the superimposed backbones are shown in Figure 1.13(2).
The GW5638-LBD complex (1R5K, Figure 1.13(2), in blue) shows a distinct H12
conformation, of which H12 gets closer to the binding pocket than it does in others. This may
be due to the shorter acrylate side chain of GW5638 compared to the bulky tails of other
SERMs, whereby the allosteric changes of the LBD are again proven to depend on the ligand
molecules.
Of particular interest is the comparison between raloxifene and TAM. The piperidinoethoxy
side chain of raloxifene stabilizes H12 exactly in the same geometry as the
dimethylaminoethoxy tail of TAM does. The positioning of H12 affects the recruitment of co-
regulators and subsequently up/down regulates the target gene transcription. Therefore,
similar physiological functions of raloxifene and TAM have been found experimentally. They
both have estrogenic properties in bone showing similar effects on maintaining bone density,
and have anti-estrogenic properties in breast cells showing equivalent potency in preventing
breast cancer. In addition, raloxifene also displays cross-resistance in TAM-resistant breast
cancer.99-101 However, raloxifene is less estrogenic than TAM in uterine tissues,102 in which
TAM works as an agonist and encourages development of endometrial cancer.103,104
Raloxifene is thus a safer preventive drug than TAM in health women, and is approved for the
prevention and treatment of osteoporosis with beneficial side effect of preventing breast
cancer. The SERM profiles of raloxifene have been rigorously studied and analyzed, and
finally ascribed to the charge neutralization between the basic amine of raloxifene and the
negatively charged residue Asp351 in the LBD 105,106 The distance between the basic amine
(Nlig.) and Asp351 (Oδ351) has been found to be ~1 Å shorter of raloxifene-LBD (1ERR)
compared to that of OHT-LBD (3ERT, Figure 1.5), which is thus assumed to be the
Chapter 1. Introduction
19
mechanism explaining why raloxifene is less estrogenic than TAM. However, the mechanism
does not account for the agonist or antagonist properties, as shown by the distances measured
from other crystal structures in Table 1.1. TAM and raloxifene have equivalent charge-
neutralizing capacity seen in the crystal structures except for 3ERT. A comparison of the
static structures is not sufficient to decipher the SERM profiles, and further discussions are
presented in Paper I.
HO
ON
Lasofoxifene
HO
ON
4-OH-Tamoxifen
OHO
GW5638
HO S
O
OH
ON
Raloxifene
HO S
O
OH
ON
Benzoxathiin derivative
A
B
C
Figure 1.7 (1). SERMs used in the superposition comparison; (2). Superposed backbones of ERα LBD
cocrystallized with different SERMs: 1ERR-raloxifene (black)27, 1R5K-GW5638 (blue)107, 2OUZ-
lasofoxifene (green)108, 3ERT-OHT (red)26, 1SJ0-benzoxathiin derivative (yellow)109; (3). The ligands
in the superposed backbones. Reproduced with permission of the copyright owner.110
A→
B→ ←C
H3
H6
H12(2) (3)
(1)

Chapter 1. Introduction
20
Table 1.1 Distances (Å) between Asp351 and the N atom of the tertiary amine of OHT and Raloxifene
in complex with ERα LBD.
OHT Raloxifene
3ERT26 2JF9(A. B. C.)45 2BJ4(A. B.)46 2JFA(A. B.)45 1ERR(A. B.)27
3.82 2.74 2.59 2.69 2.68 2.58 2.83 2.79 2.66 2.77
1.8.2 SERMs with indication of breast cancer
The area of SERMs for the treatment of breast cancer has been the focus of research due to
problems found with TAM, such as the genetic polymorphism of CYP2D6, aquired resistance,
and the undesired uterine stimulation. In order to avoid CYP-mediated activation of prodrug,
many SERMs (Figure 1.7 and Scheme 1.10) have a hydroxyl group on the B-rings to achieve
high affinity to ERα. Hence, the Phase II conjugation of these compounds is much faster than
that of TAM, resulting in more rapid clearance and much shorter in vivo lifetime.111,112 As a
result, droloxifene,113-115 miproxifene116, raloxifene117 and arozoxifene 118 were all proven
inferior to TAM in breast cancer treatment. It suggests that the therapeutic agents in breast
cancer are required to be long-acting. In contrast, idoxifene and nafoxidine are metabolically
more stable, and they were stopped from further development due to the side effect of uterine
prolapse119 and liver toxicity,120 respectively. Unfortunately, none of the new developed
SERMs has proven to be superior to TAM in treating breast cancer. The studies of the
SERMs launched or rejected provide a wealth of information for the pursuit of ideal SERMs.
Scheme 1.10 Structures of selected SERMs.
ON
HO
ON
HO
I
O N
O
O N
Droloxifene Miproxifene
Idoxifene Nafoxidine
HO S
O
O
ON
Arzoxifene

Chapter 1. Introduction
21
1.9 Aims of the Study
The overall aim of this project was to elucidate the possible explanations for the CYP2D6
debate in the TAM-treated breast cancer patients, in order to provide new information that
might be useful to develop novel and improved therapeutic agents.
Paper I — To investigate what governs the estrogenic or anti-estrogenic SERM profiles of
compounds which stabilize ERα LBD in the canonical open/antagonist conformation.
Paper II — To explore how the TAM-like ligands contact ERα LBD. We aimed to gain a
better knowledge on the three-dimensional geometry of the ERα ligand binding pocket.
Paper III — To study the mechanism of the CYP-catalyzed cis-trans isomerization of OHT,
which is a rarely reported CYP-reaction. Understanding the reaction mechanism might be a
step forward to avoiding the cis-trans isomerization problem, which is related to increased
risk of cancer recurrence.
Paper IV — To provide more insight into the comprehensive CYP-mediated metabolism of
TAM. We aimed to create new SERMs to avoid being activated by CYP2D6.
Paper V — To study the agonists binding to ERα LBD, and to understand the signaling
differences of small ligands which stabilize the LBD in the canonical closed/agonist
conformation.

22
CHAPTER 2. COMPUTATIONAL METHODS
All studies presented in this thesis were carried out by computational approaches to
investigate the issues introduced in Chapter 1. Quantum Mechanics (QM) calculations were
performed in Papers II-V to investigate the properties of small systems, which contain one to
several molecules in the studies. Of these computations, energies are generated from the
distribution of electrons, which are explicitly modeled. In contrast, electrons are treated
implicitly in Molecular Mechanics (MM) calculations, which are widely used to model larger
systems closer to real objects, such as proteins. As the systems consist of many nuclei and
several times more electrons, atoms are treated as a whole using “classical” hard spheres. MM
calculations are thus based on “classical” mechanics mainly of Newton’s second law, and
have been performed in Papers I, II, IV, and V.
The computational methods and programs used in the current studies are briefly introduced in
the following section.
2.1 Quantum Mechanics Approaches
In QM, Schrödinger equation121 is used to describe the quantum states of physical systems.
Equation 2.1 is the time-independent Schrödinger equation,
Ψ = EΨ (2.1)
where the Hamiltonian operator acts on the wavefunction Ψ, yielding the total energy E of
the system multiplied by Ψ.
= + (2.2)
= N + e + NN + Ne + ee (2.3)
The Hamiltonian is the total energy operator as a combination of kinetic and potential
energy operators (Equation 2.2), consisting of the kinetic energies of nuclei N, the kinetic
energies of electrons e, and three terms of potential energies, NN, Ne, and ee, describing
the interactions of nuclei and electrons in the system (Equation 2.3).
By solving the Schrödinger equation, the eigenfunction Ψ and eigenvalue E are found, from
which various properties of the system are derived. However, the Schrödinger equation
cannot be exactly solved for many-electron systems, which are usually the systems of interest.
Hartree-Fock (HF) method is a primary approach to solve the Schrödinger equation (2.1) in
large systems. A series of approximations has been applied in the HF method, such as (1) the
Born-Oppenheimer approximation: splitting of the movements of nuclei and electrons based

Chapter 2. Computational methods
23
on the fact that nuclei move much slower than electrons; (2) the relativistic effects are not
considered; (3) electrons are assumed to be uncorrelated and move in the mean field
independently of each other, and the eigenfunction of a many-electron system is thus
described by a single Slater determinant; (4) the LCAO-MO approximation: the wavefunction
of each molecular orbital (MO) can be constructed by a linear combination of atomic orbitals
(LCAO), expressed in terms of a finite number of basis functions.
HF methods are referred to as wavefunction-based ab initio approaches. Based on certain
approximations, the Schrödinger equation is solved iteratively using the self-consistent field
(SCF) approach. The iteration process starts from an initial guess of the wavefunction, which
is refined in each energy minimization step until convergence is reached. As guaranteed by
the variational principle, the obtained HF energy of a given system is an upper bound to the
energy of the exact wavefunction. The HF energies of many-electron systems are always
overestimated since electrons are assumed to be uncorrelated, which is apparently an over
approximation accounting for a major portion of energy errors in HF calculations. Of the
system being described, the energy gap between the HF energy EHF and the exact
nonrelativistic energy Eexact gives rise to the definition of correlation energy Ecorr (Equation
2.4), which is always negative.
Ecorr = Eexact – EHF (2.4)
The correlation energy is relatively small and usually less than 1% of the total energy of the
system. However, the chemical properties of interest, such as reaction barriers, are practically
described by the relative energies, i.e., the energy changes between different states. Not as in
the absolute total energy, the effects of correlation energies are not negligible in the relative
energy.
In addition to the primitive HF methods, many computational methods have been developed,
among which Density Functional Theory (DFT) methods have evolved as widely used
approaches. Different from HF theory, DFT is based on the electron density ρ instead of the
explicit wavefunction Ψ. The energy of a given system E[ρ] is thus described by a functional
of the electron density ρ. Therefore, in an N-electron system, only three spatial variables are
required in DFT, whereas in wavefunction-based methods (3+1)N variables are required to
describe the spatial and spin wavefuntions. DFT computations are hence much faster than
wavefunction-based methods, and are even more efficient in systems consisting of more
electrons.
DFT in principal is an accurate method, and in practice, the computations are most commonly
performed using the Kohn-Sham approach, in which the electron density is generated by non-
interacting electrons to model the interacting electrons of a real system. The Kohn-Sham
equation is solved iteratively in a similar manner to HF calculations. The DFT energy E[ρ] is
expressed in terms of the kinetic energy T[ρ] of the non-interacting electrons, the attraction

Chapter 2. Computational methods
24
energy ENe[ρ] between nuclei and electrons, the Coulomb energy among electrons J[ρ], and
the exchange-correlation energy EXC[ρ] representing the interactions not included in the other
terms (Equation 2.5).
E[ρ] = T[ρ] + ENe[ρ] + J[ρ] + EXC[ρ] (2.5)
The main focus in DFT methods is the challenge of describing the exchange-correlation
energy, in which approximation has to be made. The accuracy of DFT functionals highly
depends on the exchange-correlation functional, which is usually decomposed into an
exchange functional and a correlation functional (Equation 2.6).
EXC[ρ] = EX[ρ] + EC[ρ] (2.6)
Hence, efforts have been made to improve the exchange-correlation functional, and various
types of exchange-correlation functionals have been developed. The approximations used
include the local density approximation (LDA), generalized gradient approximation (GGA),
and meta-GGA. LDA depends only on the local density at a given coordinate, and in general
does not yield sufficient accuracy. As an improvement, GGA takes the gradient of the local
density into account. One such example is B88122 which is a frequently used exchange
functional, and usually in conjunction with the GGA correlation functional LYP developed by
Lee, Yang and Parr.123 A further development is meta-GGA, which includes the Laplacian
(the second derivative) of the electron density as the additional ingredients. Besides these
DFT methods, exchange energy could be extracted by the HF method, in which exchange
energy is fully accounted for and considered as exact exchange. Hybrid exchange-correlation
functionals are thus developed by incorporating a portion (usually not 100%) of HF exchange
with DFT exchange and correlation. Such examples are B3LYP,123-125 a hybrid GGA; and
M06-2X,126,127 a hybrid meta-GGA. The hybrid functionals represent a great improvement in
DFT functionals and provide satisfactory accuracy in many situations.
All QM calculations in this thesis were performed using the Gaussian09128 program packages.
In Paper II, the geometry of TAM was optimized using a range of DFT functionals with the 6-
31+G(d,p) basis set. The pure GGA functional BLYP122,123 and the hybrid GGA functional
B3LYP123-125 gave rise to the optimized structures in best agreement with the X-ray geometry,
and were thus used to investigate the flip-flop process of the TAM propeller and derivatives.
The more extended 6-311++G(2d,2p) basis set was applied in the computations of the
rotational barriers about the C(Ar)―C(sp2) single bond. Frequency calculations were
performed at the same level of theory to analyse the obtained structures to be local minima or
transition states. The solvent effects were considered using the SMD continuum solvation
model129 with single-point calculations. Water (ε = 78.36) was used as a solvent to model the
physiological conditions.
In Paper III, IV, and V, the hybrid meta-GGA functional M06-2X126,127 was used, as it is
highly parameterized for main group atoms and recommended for studies of weak interactions,

Chapter 2. Computational methods
25
such as hydrogen binding and dispersive interactions. The 6-31+G(d,p) basis set was used for
most calculations. In Paper III and IV, the systems of single molecules were optimized both in
vacuo and in nonpolar medium (ε = 4.24) mimicking the protein environment of CYP
enzymes. Corresponding harmonic vibrational frequency calculations were performed to
extract zero-point vibrational energies (ZPE) and thermal corrections to the Gibbs free
energies. In paper V, the systems containing one or more molecules were optimized in vacuo,
and corresponding harmonic vibrational frequencies were calculated in vacuo and in solvents
(ε = 78.36 and ε = 4.24). The ZPE-corrected binding energies between ligands and part of the
ligand binding region were calculated by Equation 2.7:
∆∆Ebinding = ∆Etotal - ∆Eligand - ∆Eresidues (2.7)
2.2 Molecular Mechanics Approaches
The requirement of computational studies of systems with thousands to millions of atoms has
evoked the development of MM methods. In MM, the system is constructed with classical
spheres representing the atoms, whereby only the motion of nuclei is modeled and the
electrons are not explicitly considered as in QM methods. The MM calculations are thus
simplified to handle large systems, of which, Newton’s second law (Equation 2.8) is the main
principle of modeling a system evolving with time. It gives rise to the concept of Molecular
Dynamics (MD) simulation of motions of atoms and molecules.
F = ma (2.8)
where F is the force acting on a body with mass m, yielding an acceleration a. The
propagation of the system is calculated from Equation 2.9.
2
2ixi
i
Fd x
dt m (2.9)
The computation accuracy thus highly depends on the force F generated from the force field
which is used to describe the potential energy of the system. The potential energy defined by a
force field usually contains contributions of covalent-bonded interactions and noncovalent-
bonded interactions (Equation 2.10),
V = Vbonded + Vnon-bonded (2.10)
in terms of (Equation 2.11 and 2.12):
Vbonded = Vbond + Vangle + Vtorsion (2.11)
Vnon-bonded = VvdW + Velec (2.12)

Chapter 2. Computational methods
26
The covalent-bonded interactions are generally described by Equations 2.13~2.15:
2
,0
1
2bond b i iV k l l (2.13)
2
,0
1
2angle i iV k (2.14)
1 cos2
ntorsion
VV n (2.15)
Another term of improper torsion is described in similar ways.
The noncovalent-bonded interactions are decomposed into van de Waals interactions and
electrostatic interactions, modeled by the Lenard-Jones potential (Equations 2.16) and
Coulomb’s law (Equations 2.17), respectively:
12 6
4 ij ijLJ ij
ij ij
Vr r
(2.16)
04i j
elecij
q qV
r (2.17)
MM methods used in the current studies:
Molecular Operating Environment (MOE) program130 was used to visualize and analyze the
crystal structures. Comparisons were made of the cocrystallized ligand-LBD structures
through structural alignments with the root-mean-square deviation (RMSD) calculated from
Cα atoms.
YASARA structure program131 was used to perform MD simulations of the ligand-LBD
complexes. All starting geometries were constructed from crystal structures, and in some
cases the ligands in the crystal structures were manually modified. Unrestrained all-atom MD
simulations were conducted using a predefined macro (md_run) within the YASARA package.
In all simulations, the ligand-LBD complexes were solvated with TIP3P132 water molecules in
a periodic box, which extended 10 Å outside the protein. The AM1-BCC model133 was used
to calculate partial atomic charges of each ligand. The systems were then neutralized by
adding counter ions,134 Na+ or Cl-, and additional ions were added to give the NaCl
concentration of 0.9 % to mimic the physiological solution. Long-range Coulomb interactions
were included using particle-mesh Ewald (PME) summation with a cut-off of 7.86 Å. All runs
were carried out at 298 K using the AMBER03135 force field for proteins and the general
amber force field136 (GAFF) for ligands. The systems evolved with multiple time steps, 1.25
fs for intramolecular forces and 2.5 fs for intermolecular forces.

Chapter 2. Computational methods
27
The dynamic behaviours of the ligands in contacting with the LBD were analyzed from the
MD trajactories. Geometry changes were directly monitored from the atomic coordinates in
each trajactory. The conformational drift of the structures was compared according to RMSD
values of Cα atoms throughout the simulation time. The RMSDs were calculated with regard
to the initial conformation by Equations 2.18 (R is the vector linking n corresponding Cα-
atom pairs in space):
2
1
n
iiR
RMSDn
(2.18)

28
CHAPTER 3. ANTAGONISTS AND THE ANTAGONIST-ERα LBD
COMPLEXES
Paper I
3.1 More Stable, More Estrogenic―the SERM Profiles
As it is introduced in Chapter 1, the estrogen-related diseases have provoked a wide use of
SERMs in both patients and healthy women, such as therapeutic and preventive agents for
breast cancer. SERMs generally act as partial antagonists. The rationale behind the SERM
actions is the fact that they stabilize ERα LBD in the open/antagonist conformation which is
significantly less active compared to the closed/agonist conformation. The SERM profiles, i.e.
how potent they work as agonists or antagonists, vary significantly by individual compounds.
However, they are not distinguishable from the crystal structures of the SERM-LBD
complexes.
The ERα-mediated estrogenic and antiestrogenic properties are of vital importance for the
clinical applications of SERMs, and have been tested and compared for many
compounds.43,137-143 The results indicated the crucial effects of the basic side chain on the
biological activity in uterine tissue, as seen in dihydrobenzoxathiin analogous (Scheme 3.1)
compounds 15, 16, 18 and 19 (Table 3.1, numbering from ref [43]). Significant differences in
the SERM profiles have been found for compounds 15 vs 16 and 18 vs 19, which have subtle
changes in the ligand structures as they differ only in the orientation of one or two methyl
substituents.
Scheme 3.1 Dihydrobenzoxathiin.
HO S
O
OH
OR
Fortunately, the crystal structures were solved for these ligands bound to ERα LBD, which
made it possible to study the SERM profiles by modeling techniques. The overall structures
are compared by structure alignments (Table 3.1 and Figure 3.1). A highly conserved
open/antagonist conformation is found for the compounds in complex with the LBD, with

Chapter 3. Antagonists and the antagonist-ERα LBD complexes
29
fairly slight deviations computed from Cα atoms of the LBD. The charge-neutralizing
capacity, the once believed mechanism for the SERM profiles, are again compared by the
distances measured between Oδ351 and Nlig. of the crystal structures. The distances of 2.6~2.8
Å are found in all SERM-LBD complexes except 3ERT, showing no apparent differences of
these ligands in shielding the charge of Asp351. Hence, it cannot be the rationale behind the
different SERM profiles of compounds 15 vs 16, 18 vs 19, or raloxifen vs TAM.
Table 3.1 Biodata43,137,140 of SERMs and geometry comparisons of the cocrystallized structures.
Reproduced with permission of the copyright owner.47
SERM IC50 (nM) Uterine acivityc
PDB Oδ351-Nlig.
(Å)
RMSD
(Cα, Å) hERαa MCF-7b %Ant. %Ag.
E2 1.3 — — 100 — — —
OHT — — — —
3ERT26 3.8 —
2JF945 2.6~2.7 —
2BJ446 2.6~2.7 —
Lasofoxifene 1.4 0.1 74 26 2OUZ108 2.7 —
Raloxifene 1.8 0.8 81 24 1ERR27 2.7~2.8 —
2JFA45 2.8 —
N
Comp. 15
0.5 0.4 103 -10 1XP143 2.7
0.16
N
Comp. 16
0.4 — 52 39 1XP643 2.6
N
Comp. 18
1.3 0.3 101 0 1XP943 2.6
0.25
N
Comp. 19
1.7 0.5 18 71 1XPC43 2.8
a In the ERα ligand binding assays used tritiated estradiol. b The in vitro MCF-7 breast cancer cell
proliferation assays were performed in the presence of low levels of estradiol. c The %Ant.
(%antagonism) and %Ag. (%agonism of estradiol control as 100% agonism) were compared in the
uterine weight assay with compounds dosed orally.43
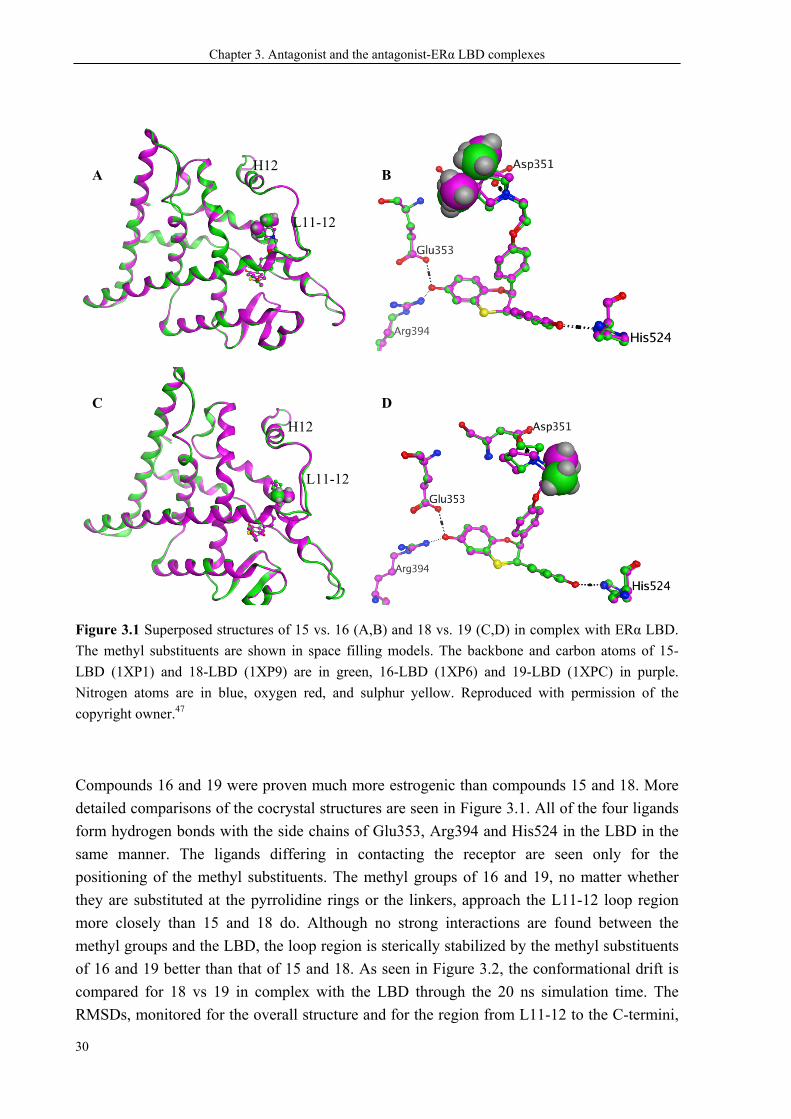
Chapter 3. Antagonist and the antagonist-ERα LBD complexes
30
Figure 3.1 Superposed structures of 15 vs. 16 (A,B) and 18 vs. 19 (C,D) in complex with ERα LBD.
The methyl substituents are shown in space filling models. The backbone and carbon atoms of 15-
LBD (1XP1) and 18-LBD (1XP9) are in green, 16-LBD (1XP6) and 19-LBD (1XPC) in purple.
Nitrogen atoms are in blue, oxygen red, and sulphur yellow. Reproduced with permission of the
copyright owner.47
Compounds 16 and 19 were proven much more estrogenic than compounds 15 and 18. More
detailed comparisons of the cocrystal structures are seen in Figure 3.1. All of the four ligands
form hydrogen bonds with the side chains of Glu353, Arg394 and His524 in the LBD in the
same manner. The ligands differing in contacting the receptor are seen only for the
positioning of the methyl substituents. The methyl groups of 16 and 19, no matter whether
they are substituted at the pyrrolidine rings or the linkers, approach the L11-12 loop region
more closely than 15 and 18 do. Although no strong interactions are found between the
methyl groups and the LBD, the loop region is sterically stabilized by the methyl substituents
of 16 and 19 better than that of 15 and 18. As seen in Figure 3.2, the conformational drift is
compared for 18 vs 19 in complex with the LBD through the 20 ns simulation time. The
RMSDs, monitored for the overall structure and for the region from L11-12 to the C-termini,
A
C
B
D
H12
L11-12
H12
L11-12
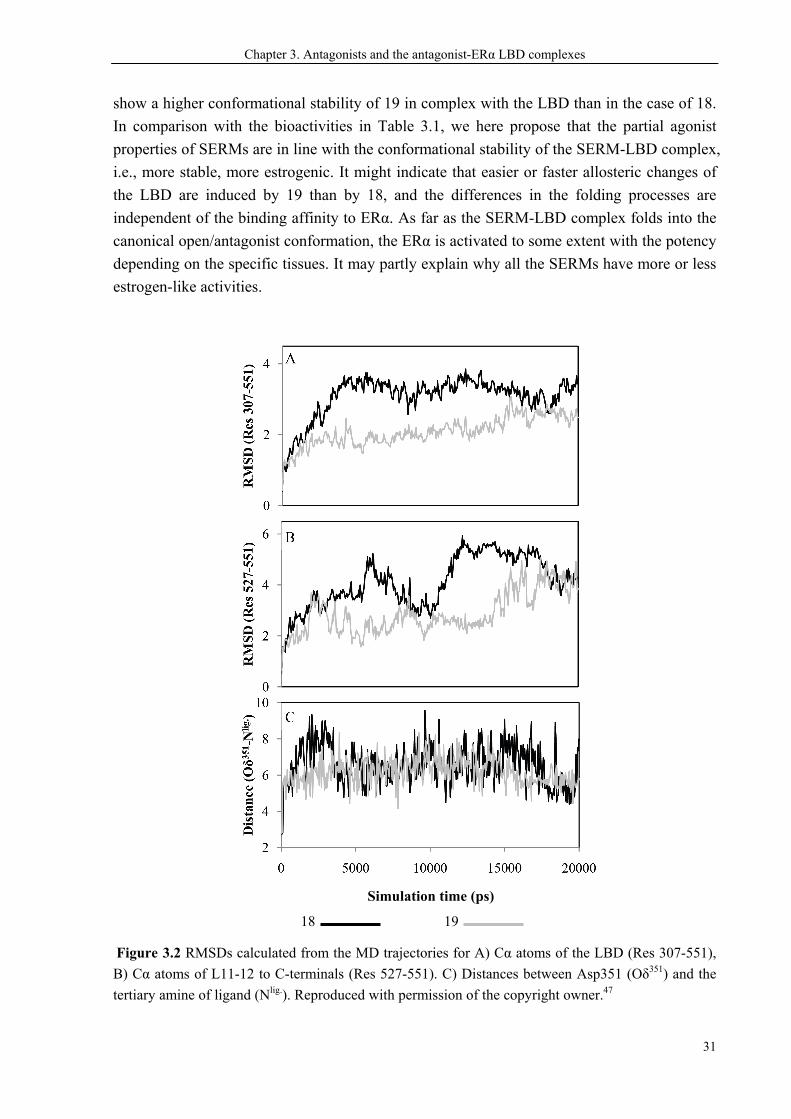
Chapter 3. Antagonists and the antagonist-ERα LBD complexes
31
show a higher conformational stability of 19 in complex with the LBD than in the case of 18.
In comparison with the bioactivities in Table 3.1, we here propose that the partial agonist
properties of SERMs are in line with the conformational stability of the SERM-LBD complex,
i.e., more stable, more estrogenic. It might indicate that easier or faster allosteric changes of
the LBD are induced by 19 than by 18, and the differences in the folding processes are
independent of the binding affinity to ERα. As far as the SERM-LBD complex folds into the
canonical open/antagonist conformation, the ERα is activated to some extent with the potency
depending on the specific tissues. It may partly explain why all the SERMs have more or less
estrogen-like activities.
Figure 3.2 RMSDs calculated from the MD trajectories for A) Cα atoms of the LBD (Res 307-551),
B) Cα atoms of L11-12 to C-terminals (Res 527-551). C) Distances between Asp351 (Oδ351) and the
tertiary amine of ligand (Nlig.). Reproduced with permission of the copyright owner.47
Simulation time (ps)
18 19

Chapter 3. Antagonist and the antagonist-ERα LBD complexes
32
3.2 The SERM Profiles of TAM vs OHT
TAM is highly successful in the treatment of ER-positive breast cancer, with antiestrogenic
properties in breast tissue. The 4-hydroxy metabolites, OHT and endoxifen, have ~100 fold
higher affinity to ERα than TAM itself, and are thus simply assumed to be more active
antagonists in breast cancer cells. As CYP2D6 is the primary isoform responsible for
producing the 4-hydroxy metabolites, patients who have null alleles for inactive CYP2D6
resulting in limited amounts of OHT and endoxifen, have been believed to benefit less from
TAM treatment than patients who have wide type CYP2D6 alleles. However, this intuitively
right concept has been greatly challenged through many clinical studies, in particular the very
recent large studies which suggest that CYP2D6 activity does not predict clinical response to
TAM therapy.75,76 The effects of higher-affinity metabolites on TAM treatment have
remained elusive. However, the decreased concentration of the parent drug TAM has been
related to the failure in TAM treatment despite the fact that it binds to ERα with fairly low
affinity.78
OHT and endoxifen have been assumed to work as potent anti-estrogens in breast cancer cells,
but the truth lies in very complex actions, because they have both estrogen agonist and
antagonist properties, as seen in compounds 15, 16, 18 and 19 (Table 3.1). However, there is
as yet no report of experimental comparisons on the SERM profiles between TAM and OHT,
since the experiments are not easy to carry out due to TAM is in vivo metabolized to OHT, etc.
We thus performed in silico comparisons of the SERM profiles based on the principle of more
stable, more estrogenic. As seen in Figure 3.3A, a higher conformational drift is found for
TAM than OHT in complex with the LBD. The difference in dynamic stability between
TAM- and OHT-LBD complexes is a result of the additional 4-hydroxyl group, which
stabilizes the inter-helix interactions (Figure 1.5) through hydrogen bond interactions as seen
in Figure 3.3B. Constant distances between Cδ353 (on Helix 3) and Cζ394 (on Helix 6) are
found for OHT but not TAM in the modeled systems. The simulation results show that TAM-
LBD complex is less dynamically stable than OHT-LBD. TAM may thus have more
antagonist properties than OHT does. Hence, the conversion of TAM into OHT mediated by
CYP2D6 has complex effects on TAM treatment. The 4-hydroxyl group increases the affinity
to ERα, and also results in more estrogen-like properties. The mixed agonist-antagonist
properties of OHT are necessary to be testified experimentally. Conclusive experimental
studies are still lacking to clarify the clinical effects of CYP2D6 activity on TAM therapy.
In addition, the salt bridges between Nlig. and the negatively charged Asp351 are found not
retained in solvent in all the simulations (Figures 3.2 C and 3.3 C). The side chain of Asp351
is more likely to be solvent exposed, facilitating the interaction with other domains of ERα or
coregulators, and thus contributes to the overall folding of the ERα transcription unit. It has
proven by experiments that mutations of Asp351 into noncharged residues (Ala, Val and Gly)
result in reduced agonism of SERMs.144-146
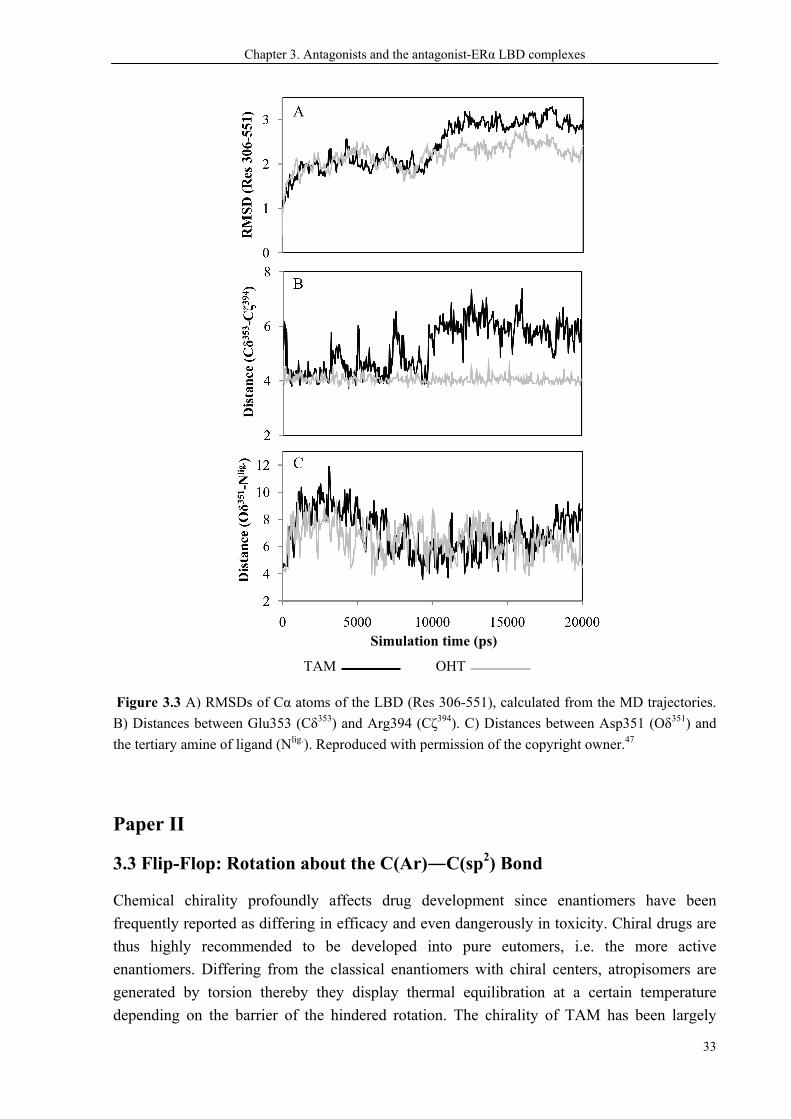
Chapter 3. Antagonists and the antagonist-ERα LBD complexes
33
Figure 3.3 A) RMSDs of Cα atoms of the LBD (Res 306-551), calculated from the MD trajectories.
B) Distances between Glu353 (Cδ353) and Arg394 (Cζ394). C) Distances between Asp351 (Oδ351) and
the tertiary amine of ligand (Nlig.). Reproduced with permission of the copyright owner.47
Paper II
3.3 Flip-Flop: Rotation about the C(Ar)―C(sp2) Bond
Chemical chirality profoundly affects drug development since enantiomers have been
frequently reported as differing in efficacy and even dangerously in toxicity. Chiral drugs are
thus highly recommended to be developed into pure eutomers, i.e. the more active
enantiomers. Differing from the classical enantiomers with chiral centers, atropisomers are
generated by torsion thereby they display thermal equilibration at a certain temperature
depending on the barrier of the hindered rotation. The chirality of TAM has been largely
Simulation time (ps)
TAM OHT

Chapter 3. Antagonist and the antagonist-ERα LBD complexes
34
overlooked due to the fast interconversion between the atropisomeric properllers (clockwise
and counterclockwise triarylvinyl propellers) through the rotation about the C(Ar)―C(sp2)
bonds (Scheme 3.2).
The spontaneous racemization of the TAM propellers is too fast to be investigated by
dynamic NMR methods,8 whereas computational methods are applied in the current studies.
The possible pathways of the helicity reversal, through the simultaneous rotations of the three
rings, are illustrated in Scheme 3.2, with corresponding barriers summarized in Table 3.2. The
rotation of the rings is referred to as ‘flip’ or ‘nonflip’ according to the pass through the plane
perpendicular or coplanar to the vinyl plane, respectively. The lowest activation energies of
the helical inversion of TAM-like compounds are in the range of 2–4 kcal/mol through the
three-ring flip pathway. Besides this threshold pathway, the middle ring noflip pathway has
the barrier of ~5 kcal/mol, which could be an alternative route for the helicity reversal.
Scheme 3.2 Idealized depictions of the competing flip-flop mechanisms. The transition states are
illustrated in the middle. The ‘flip’ rings via the perpendicular plane are denoted as . Reproduced
with permission of the copyright owner.110
R
R
R
R
R
Clockwise
R
A
A
B
C
A
B
C
C
C
C
B
B
A
A
B
B-ring nonflip
C-ring nonflip
A-ring nonflip
3-ring flip
2-ring flip
1
2
34
5
671'
2'
3'4'
5'
6'
1''
2''
3'' 4''
5''
6''8
9 10
Counterclockwise
R = OCH2CH2 NMe2
α = 2-1-7-8
β = 2′-1′-7-8
γ = 2″-1″-8-7
δ = 7-8-9-10
Chapter 3. Antagonists and the antagonist-ERα LBD complexes
35
Table 3.2 Enantiomerization free energies of TAM and OHT in gas phase and in aqueous solution at
298 K, calculated at B3LYP and BLYP levels in conjunction with the 6-311++G(2d,2p) basis set.a
Reproduced with permission of the copyright owner.110
∆Ggas298 ∆∆Ggas
298 ∆Gaq298 ∆∆Gaq
298
TAM -1138.451268 0 -1138.546663 0
TSA-ring nonflip -1138.443265 5.02 -1138.536915 6.12
B3LYP TSB-ring nonflip -1138.437699 8.51 -1138.531152 9.73
TSC-ring nonflip -1138.436200 9.46 -1138.532617 8.81
TSThree-ring flip -1138.447269 2.51 -1138.541852 3.02
TAM -1137.933528 0 -1138.026927 0
TSA-ring nonflip -1137.925389 5.11 -1138.017351 6.01
BLYP TSB-ring nonflip -1137.919486 8.81 -1138.011767 9.51
TSC-ring nonflip -1137.918868 9.20 -1138.014482 7.81
TSThree-ring flip -1137.929204 2.71 -1138.02288 2.54
OHT -1213.698635 0 -1213.801621 0
TSA-ring nonflip -1213.691089 4.74 -1213.793122 5.33
B3LYP TSB-ring nonflip -1213.684152 9.09 -1213.786731 9.34
TSC-ring nonflip -1213.683738 9.35 -1213.785962 9.83
TSThree-ring flip -1213.693212 3.40 -1213.795477 3.86
OHT -1213.176659 0 -1213.277634 0
TSA-ring nonflip -1213.169456 4.52 -1213.268342 5.83
BLYP TSB-ring nonflip -1213.162487 8.89 -1213.262283 9.63
TSC-ring nonflip -1213.162523 8.87 -1213.26128 10.26
TSThree-ring flip -1213.171214 3.42 -1213.273524 2.58
a Absolute energies in a.u., and relative energies in kcal/mol.
As seen in Table 3.2, the threshold barriers are too low to allow for the resolution of TAM
atropisomers, resulting in that TAM appears as an achiral compound. Atropisomers were
defined by Oki in 1983, as the isomers interconverted slowly enough to be resolved, with a
half life not less than 1000 seconds.147 However, recognizing the existence of atropisomers of
TAM and its metabolites is of great importance, because these atropisomers are discriminated
by the drug target, ERα. As seen in Figure 3.4 and Table 3.3, only the clockwise propellers of

Chapter 3. Antagonist and the antagonist-ERα LBD complexes
36
the ligands are found in the cocrystallized structures, while the counterclockwise
conformation is found in crystalline TAM. The counterclockwise propeller is not tolerable by
the ligand binding pocket of ERα, as tested by MD simulations with the detailed results seen
in Paper II. Therefore, the counterclockwise isomers of TAM and its metabolites do not bind
to ERα directly, but rapidly flip forward to the clockwise isomers to cater for the ligand
binding pocket of ERα. Although the fast interconversion between the atropisomers of TAM-
like compounds makes no distinguishable atropisomeric conformations in the solvent, the
clockwise atropisomers are the pharmacologically functional isomers, which work as agonists
or antagonists in TAM treatment through ERα signaling pathways.
Figure 3.4 Superposed crystal structures of OHT in complex with ERα LBD solved by three
independent research groups , 3ERT (red)26, 2JF9.A (blue)45, and 2BJ4.A (green)46. Reproduced with
permission of the copyright owner.110
Table 3.3 Torsional angles measured from the crystal structures of crystalline TAM and
OHT/GW5638 cocrystallized with ERα LBD. Reproduced with permission of the copyright owner.110
Conformer
Torsional angle (º)
α β γ δ
TAM2 Counterlockwise -48 -64 -55 114
Ligand OHT (3ERT) Clockwise 47 47 51 -115
Ligand OHT (2JF9.A) Clockwise 61 57 64 -133
Ligand OHT (2BJ4.A) Clockwise 56 55 58 -127
Ligand GW5638 (1R5K) Clockwise 31 53 47 -127
α
βδ
γ

Chapter 3. Antagonists and the antagonist-ERα LBD complexes
37
Paper III
3.4 CYP-Catalyzed Rotation about the C(sp2)═C(sp2) Bond
Trans-TAM is prescribed to treat ER-positive breast cancer as an estrogen antagonist in breast
tissue, whereas cis-TAM is an estrogen agonist. Cis-4-hydroxyTAM (cis-OHT) was, besides
trans-OHT, found in patients who took TAM drug in the trans-form. Cis-OHT was a direct
product from trans-OHT mainly due to the CYP-mediated cis-trans isomerization of OHT.
The analogous isomerization process was, however, not observed with TAM itself. Of these
isomers, the relative binding affinities to ER (which was 100% of estradiol) were reported as
0.3 vs 2.5% for cis- vs trans-TAM, and 2.8 vs 310% for cis- vs trans-OHT.80 The trans-to-cis
isomerization of OHT results relatively high cis:trans-OHT ratio in vivo, which has been
associated with an increased risk of tumor relapse possibly owning to the less potent cis-OHT
in suppressing tumor growth.78,79
The CYP-catalyzed interconversion between cis- and trans-OHT implies a redox process by
the attendance of NADPH as cofactor, thereby the oxidation steps are most likely to be
initiated from the additional 4-OH group, since the isomerization is forbidden between cis-
and trans-TAM. Hence, the 4-OH substituent is assumed to undergo an initial H-atom
abstraction, which has been regarded as a typical initiation step of many CYP reactions,
resulting in a radical intermediate. This putative radical intermediate is also possible to be
further oxidized into a cationic intermediate by the heme-oxygen complex. In both cases, the
C(sp2)═C(sp2) bond is disturbed due to the oxidation of the 4-hydroxy group in OHT or
endoxifen, and the subsequent rotation process is allowed to occur via either the radical or the
cationic intermediate (Scheme 3.3A).
Hence, as the key step of the isomerization process, rotations about the disturbed alkene bond
were studied via the radical (Scheme 3.3B) or cationic intermediates (Scheme 3.3C). The
rotations via the radical intermediates have similar barriers (15–18 kcal/mol) for cis→trans
and trans→cis isomerizations. Alternatively, the barriers are much lower via the cationic
intermediates, estimated at 8~11 vs 2–5 kcal/mol for cis→trans vs trans→cis isomerization.
The rotational barriers indicate a faster conversion of trans→cis than cis→trans via the
cationic pathway. The experimental studies were performed by Williams et al. using human
CYP’s. Pure cis- or trans-OHT were incubated with human liver microsomes in the presence
of NADPH cofactor, and the conversion rates were 51–64% and 22–27% for trans→cis and
cis→trans reactions, respectively.81 The rotational energy differences between the trans→cis
and cis→trans processes are seen only in the cationic pathways, mainly due to the cis-d+
isomers are energetically favored than the trans-d+ isomers (Figure 3.5), and are in agreement
with the order of the experimental conversion rates. The optimized geometries of cis/trans-
cation intermediates and the detailed isomerization potentials are depicted in Figure 3.5.

Chapter 3. Antagonist and the antagonist-ERα LBD complexes
38
Scheme 3.3 Possible radical and cationic intermediates for the trans-to-cis isomerization of
OHT/endoxifen catalyzed by CYP. The cis-to-trans reaction may proceed by analogous mechanisms,
via the cis-c• or cis-d+ intermediates. Reproduced with permission of the copyright owner.148
HO
R
-e-
HO
R
+
-H+
O
R
a b+.. .
c .
-e-
d+
OHT R=OCH2CH2NMe2
EdoxifenR=OCH2CH2NHMe
O O O
+ ++
c
d+
.
. . .
R R R
O O O
R R R
O
.
R
+
O
R
B. Radical pathway
C. Cationic pathway
A.
Hitherto, the cationic pathways haven’t verified experimentally. Comparisons are made between CYP-mediated isomerization and desaturation, which appear as distinctly different reactions, but derive from the same phenolic hydroxyl group (Scheme 3.4). Instead of isomerization, 4-hydroxyacetanilide and raloxifen undergo CYP-catalyzed dehydrogenation, in which process CYP enzymes abstract two hydrogen atoms from the substrate. However, the second abstractable proton is absent in cis- or trans-OHT, thereby the stable dehydrogenation product is not yielded by the CYP oxidation steps, and a radical or cationic intermediate is instead formed, resulting in cis-trans isomerization. We propose that
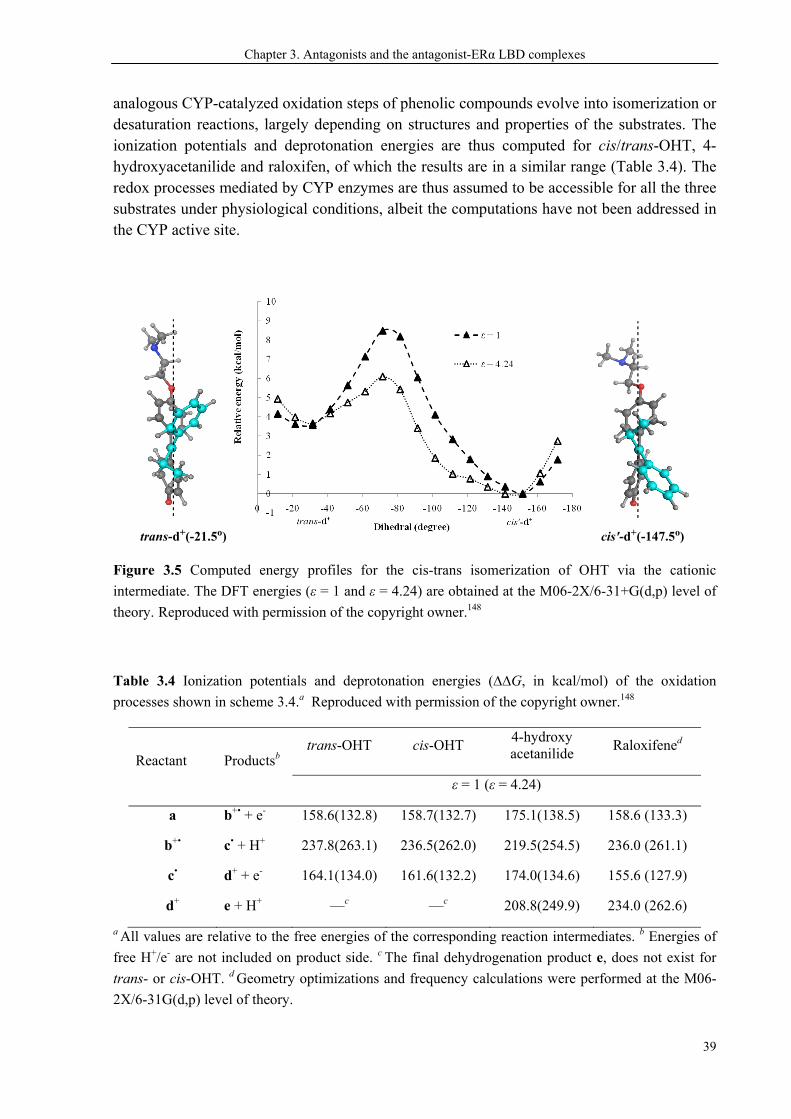
Chapter 3. Antagonists and the antagonist-ERα LBD complexes
39
analogous CYP-catalyzed oxidation steps of phenolic compounds evolve into isomerization or desaturation reactions, largely depending on structures and properties of the substrates. The ionization potentials and deprotonation energies are thus computed for cis/trans-OHT, 4-hydroxyacetanilide and raloxifen, of which the results are in a similar range (Table 3.4). The redox processes mediated by CYP enzymes are thus assumed to be accessible for all the three substrates under physiological conditions, albeit the computations have not been addressed in the CYP active site.
Figure 3.5 Computed energy profiles for the cis-trans isomerization of OHT via the cationic
intermediate. The DFT energies (ε = 1 and ε = 4.24) are obtained at the M06-2X/6-31+G(d,p) level of
theory. Reproduced with permission of the copyright owner.148
Table 3.4 Ionization potentials and deprotonation energies (∆∆G, in kcal/mol) of the oxidation
processes shown in scheme 3.4.a Reproduced with permission of the copyright owner.148
Reactant Productsb trans-OHT cis-OHT
4-hydroxy acetanilide
Raloxifened
ε = 1 (ε = 4.24)
a b+• + e- 158.6(132.8) 158.7(132.7) 175.1(138.5) 158.6 (133.3)
b+• c• + H+ 237.8(263.1) 236.5(262.0) 219.5(254.5) 236.0 (261.1)
c• d+ + e- 164.1(134.0) 161.6(132.2) 174.0(134.6) 155.6 (127.9)
d+ e + H+ —c —c 208.8(249.9) 234.0 (262.6)
a All values are relative to the free energies of the corresponding reaction intermediates. b Energies of
free H+/e- are not included on product side. c The final dehydrogenation product e, does not exist for
trans- or cis-OHT. d Geometry optimizations and frequency calculations were performed at the M06-
2X/6-31G(d,p) level of theory.
trans-d+(-21.5o) cis'-d+(-147.5o)
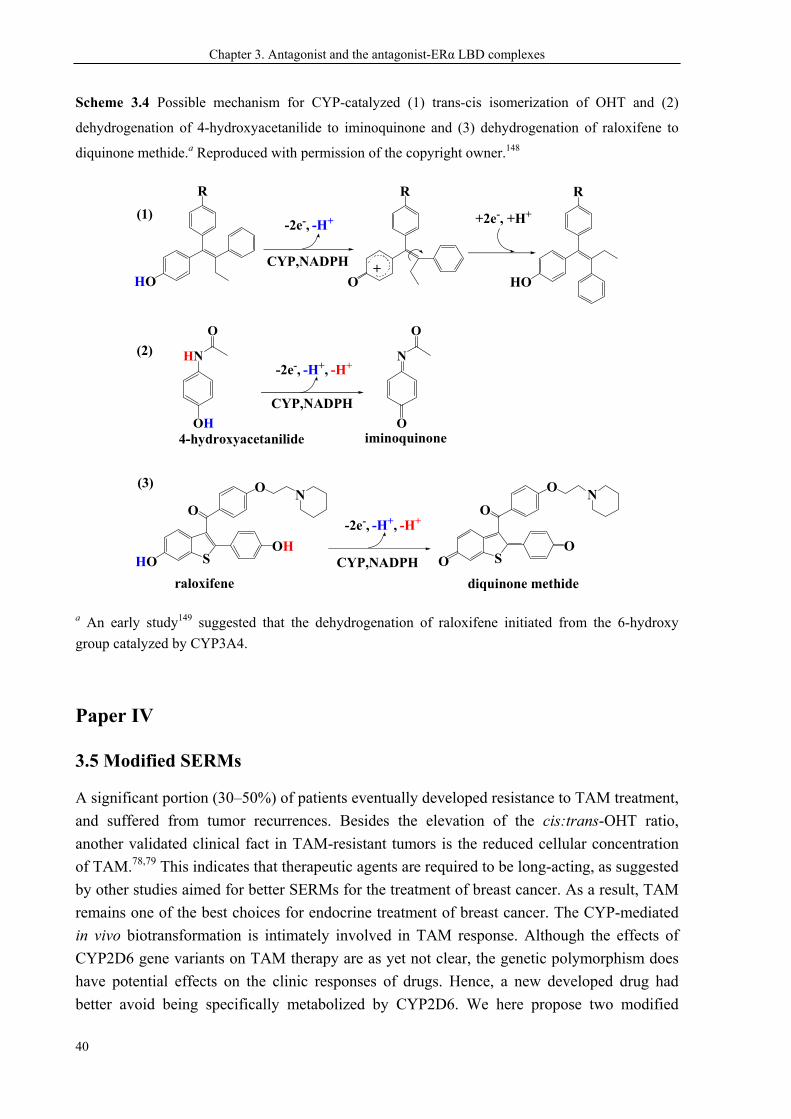
Chapter 3. Antagonist and the antagonist-ERα LBD complexes
40
Scheme 3.4 Possible mechanism for CYP-catalyzed (1) trans-cis isomerization of OHT and (2)
dehydrogenation of 4-hydroxyacetanilide to iminoquinone and (3) dehydrogenation of raloxifene to
diquinone methide.a Reproduced with permission of the copyright owner.148
HO
R
HO
R
-2e-, -H+ +2e-, +H+
O
R
+CYP,NADPH
HO S
O
OH
ON
O S
O
O
ON
4-hydroxyacetanilide
raloxifene
OH
HN
O
O
N
O
-2e-, -H+, -H+
-2e-, -H+, -H+
CYP,NADPH
CYP,NADPH
iminoquinone
diquinone methide
(1)
(2)
(3)
a An early study149 suggested that the dehydrogenation of raloxifene initiated from the 6-hydroxy
group catalyzed by CYP3A4.
Paper IV
3.5 Modified SERMs
A significant portion (30–50%) of patients eventually developed resistance to TAM treatment,
and suffered from tumor recurrences. Besides the elevation of the cis:trans-OHT ratio,
another validated clinical fact in TAM-resistant tumors is the reduced cellular concentration
of TAM.78,79 This indicates that therapeutic agents are required to be long-acting, as suggested
by other studies aimed for better SERMs for the treatment of breast cancer. As a result, TAM
remains one of the best choices for endocrine treatment of breast cancer. The CYP-mediated
in vivo biotransformation is intimately involved in TAM response. Although the effects of
CYP2D6 gene variants on TAM therapy are as yet not clear, the genetic polymorphism does
have potential effects on the clinic responses of drugs. Hence, a new developed drug had
better avoid being specifically metabolized by CYP2D6. We here propose two modified

Chapter 3. Antagonists and the antagonist-ERα LBD complexes
41
structures, i.e., MTA and MTO (Scheme 3.5), of which both the cis-trans isomerization and
the CYP2D6 polymorphism are most likely avoided according to the computational
comparisons of the CYP-mediated biotransformation.
Scheme 3.5 Structures and reaction sites labeling of TAM and derivatives.
HO
ON
CH3
HCHC
HC
CH
CHCH
H2CCH3
ON
CH3
HC H2C
O
HN
CH3
OHTTAM NDT
H2C
ON
TOR
Cl
2
3
4 2'
3'
4'
4
CH
H2C
ON
CH3
MTA
H3C
4' CH
H2C
ON
CH3
MTO
H3C
4'
Cl
β
4α
α α
α α α
α′ α′ α′
α′ α′
4α
4α
TAM-like compounds undergo several types of CYP-mediated oxidations, involving N-
demethylation, 4-hydroxylation, α-hydroxylation, and cis-trans isomerization. The CYP
reactions are initiated by electrophilic-type attack due to the electron deficiency of the high-
valent iron-oxo complex (Por+•Fe(IV)O), Cpd I, which is an important intermediate in the
CYP catalytic cycle. Different reactivity patterns have been found, among which the initial
abstraction of a H atom has been frequently suggested.83,85,92 Although computational studies
of CYP reactions have been performed extensively, the mechanistic studies of CYP-mediated
TAM metabolism are seldom reported, partly due to that the compound is relatively large. In
order to gain a better understanding of the biotransformation of TAM-like compounds,
whereby they evolve into different products by CYP reactions, the initial H-abstraction is
assumed to be the mechanistic choice for all the reactions studied herein. The reaction
energies of the H-abstractions are computed for the distinctively different CYP reactions
(Scheme 3.5), and results are shown in Tables 3.5.
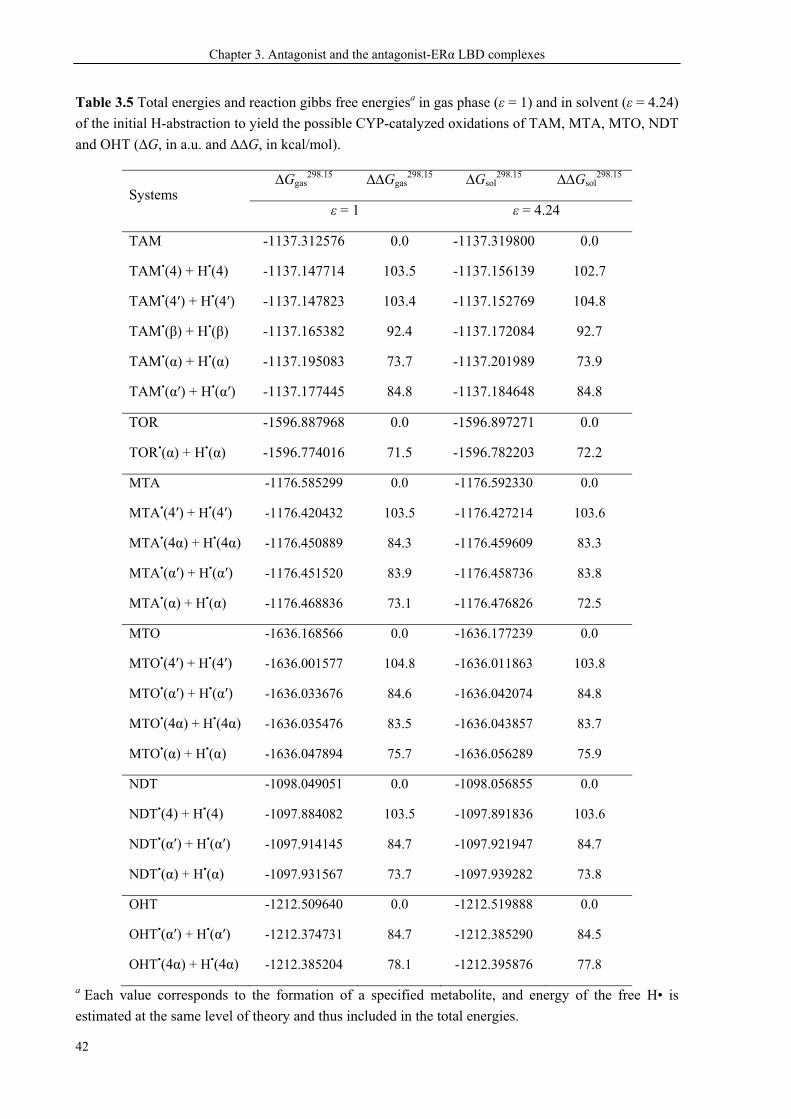
Chapter 3. Antagonist and the antagonist-ERα LBD complexes
42
Table 3.5 Total energies and reaction gibbs free energiesa in gas phase (ε = 1) and in solvent (ε = 4.24)
of the initial H-abstraction to yield the possible CYP-catalyzed oxidations of TAM, MTA, MTO, NDT
and OHT (∆G, in a.u. and ∆∆G, in kcal/mol).
Systems ∆Ggas
298.15 ∆∆Ggas298.15 ∆Gsol
298.15 ∆∆Gsol298.15
ε = 1 ε = 4.24
TAM -1137.312576 0.0 -1137.319800 0.0
TAM•(4) + H•(4) -1137.147714 103.5 -1137.156139 102.7
TAM•(4′) + H•(4′) -1137.147823 103.4 -1137.152769 104.8
TAM•(β) + H•(β) -1137.165382 92.4 -1137.172084 92.7
TAM•(α) + H•(α) -1137.195083 73.7 -1137.201989 73.9
TAM•(α′) + H•(α′) -1137.177445 84.8 -1137.184648 84.8
TOR -1596.887968 0.0 -1596.897271 0.0
TOR•(α) + H•(α) -1596.774016 71.5 -1596.782203 72.2
MTA -1176.585299 0.0 -1176.592330 0.0
MTA•(4′) + H•(4′) -1176.420432 103.5 -1176.427214 103.6
MTA•(4α) + H•(4α) -1176.450889 84.3 -1176.459609 83.3
MTA•(α′) + H•(α′) -1176.451520 83.9 -1176.458736 83.8
MTA•(α) + H•(α) -1176.468836 73.1 -1176.476826 72.5
MTO -1636.168566 0.0 -1636.177239 0.0
MTO•(4′) + H•(4′) -1636.001577 104.8 -1636.011863 103.8
MTO•(α′) + H•(α′) -1636.033676 84.6 -1636.042074 84.8
MTO•(4α) + H•(4α) -1636.035476 83.5 -1636.043857 83.7
MTO•(α) + H•(α) -1636.047894 75.7 -1636.056289 75.9
NDT -1098.049051 0.0 -1098.056855 0.0
NDT•(4) + H•(4) -1097.884082 103.5 -1097.891836 103.6
NDT•(α′) + H•(α′) -1097.914145 84.7 -1097.921947 84.7
NDT•(α) + H•(α) -1097.931567 73.7 -1097.939282 73.8
OHT -1212.509640 0.0 -1212.519888 0.0
OHT•(α′) + H•(α′) -1212.374731 84.7 -1212.385290 84.5
OHT•(4α) + H•(4α) -1212.385204 78.1 -1212.395876 77.8
a Each value corresponds to the formation of a specified metabolite, and energy of the free H• is
estimated at the same level of theory and thus included in the total energies.

Chapter 3. Antagonists and the antagonist-ERα LBD complexes
43
Although the reaction energies do not account for activation energies or reaction rates,
comparisons can be made between different reactions regarding to how endothermic the H-
abstraction processes are. The H-abstraction energies decrease in the order of 4(4′)-
hydroxylation (~103 kcal/mol) > β-hydroxylation (~93 kcal/mol) > N-demethylation ~ 4α-
hydroxylation (~84 kcal/mol) > cis-trans isomerization (~78 kcal/mol) > α-hydroxylation
(~74 kcal/mol). The most endothermic process is the aromatic 4(4′)-H-abstraction yielding the
CYP-mediated aromatic hydroxylation. This hydroxylation has been proposed through a more
efficient reactivity pattern, which initiates from a direct attack of the π electrons forming the
Fe-O-Ph intermediate.93 The β-H-abstraction energy is 10 kcal/mol lower than that of the
4(4′)-H-abstraction, however, the β-hydroxyl product of TAM hasn’t been found
experimentally. Therefore, the β-H-abstraction is not a rational reactivity step, and neither is
the 4(4′)-H-abstraction. For N-demethylation, 4α-hydroxylation, cis-trans isomerization, and
α-hydroxylation, the energies of the initial H-abstraction are in the range of 74~84 kcal/mol,
which are 20~30 kcal/mol lower than that of aromatic H-abstraction. These H-abstractions
with relatively low reaction energies are thus assumed to be accessible, as analogous H-
abstractions have been frequently proposed in other studies.83,85
In the experimental studies of TAM biotransformation, CYP2D6, CYP3A4/5, and CYP3A4
were found to be the main isoforms responsible for 4-hydroxylation, N-demethylation, and α-
hydroxylation, respectively. The members of CYP3A are able to catalyze N-demethylation
and α-hydroxylation, but not 4-hydroxylation. CYP2D6 is hence the main isoform for 4-
hydroxylation, which eventually produces the high affinity metabolites OHT and endoxifen.51
The alkane 4α-hydroxylation of the modified structures, MTA and MTO, are highly possible to
be catalyzed by CYP3A through the initial H-abstraction step, which has relatively low
reaction energy. In this process, the CYP2D6 polymorphism is circumvented by the CYP3A
mediated activation of the modified compounds. The subsequent 4-hydroxymethyl products
are different from the 4-hydroxy products of TAM, without the phenolic para-hydroxy group,
and are not likely to undergo the cis-trans isomeriztion. More experimental studies are
required to testify the proposed metabolic paths of MTA and MTO, and to examine the anti-
cancer efficacy and other beneficial or adverse side effects, because of which most SERMs
have failed in the clinical trials.
The SERM profiles and related ER-dependent side effects are difficult to estimate through
computational approaches. An ER-independent side effect of genotoxicity is, however,
possible to be investigated by quantum mechanics calculations. TAM genotoxicity is found in
rats, whereby it is caused by the DNA adducts forming from the α-hydroxy metabolites of
TAM (Scheme 1.5). The formation of DNA adducts has been proposed to occur through the
allylic carbocation intermediate, of which a high formation enthalpy results in low stability
and thus leads to low possibility to form DNA adducts.4 This physicochemical property has
been calculated, and the results are shown in Table 3.6. Regarding to formation of DNA
adducts, the computed potentials decrease in the order of OHT > MTA > TAM > TOR ~
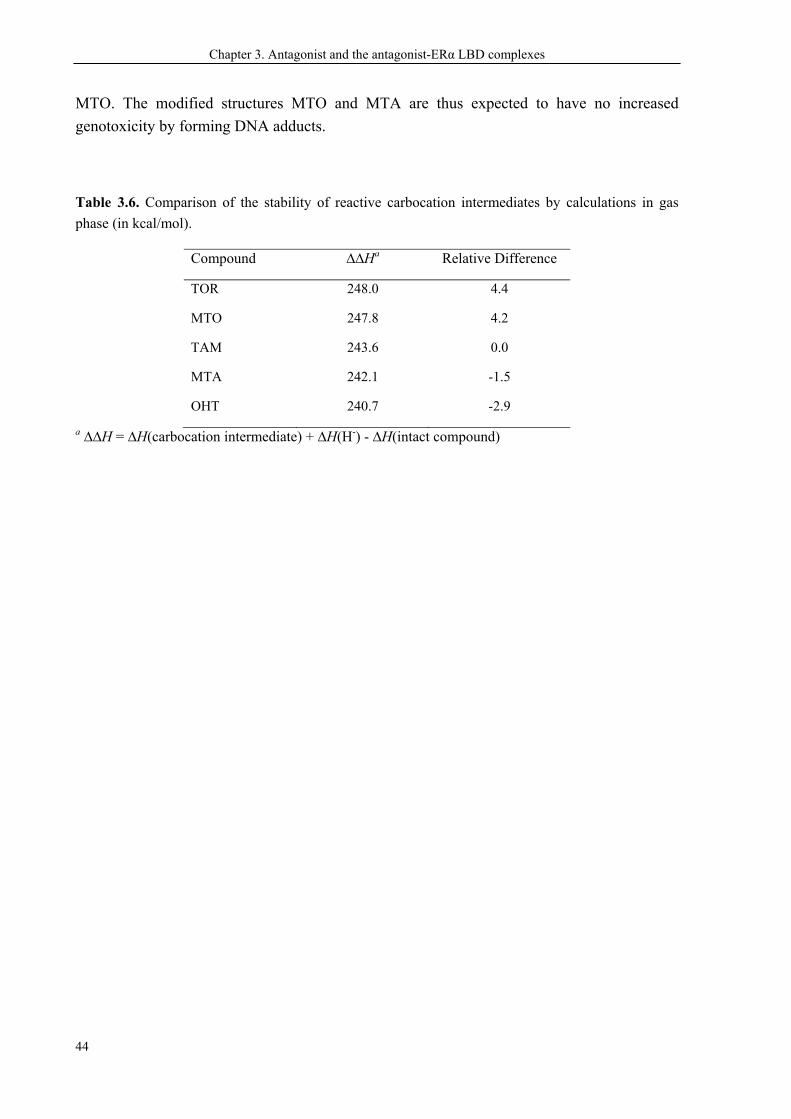
Chapter 3. Antagonist and the antagonist-ERα LBD complexes
44
MTO. The modified structures MTO and MTA are thus expected to have no increased
genotoxicity by forming DNA adducts.
Table 3.6. Comparison of the stability of reactive carbocation intermediates by calculations in gas
phase (in kcal/mol).
Compound ∆∆Ha Relative Difference
TOR 248.0 4.4
MTO 247.8 4.2
TAM 243.6 0.0
MTA 242.1 -1.5
OHT 240.7 -2.9
a ∆∆H = ∆H(carbocation intermediate) + ∆H(H-) - ∆H(intact compound)

45
CHAPTER 4. AGONISTS AND THE AGONIST-ERα LBD
COMPLEXES
Paper V
As shown in Figure 1.4A, full and partial agonists in complex with ERα LBD resemble each
other in the canonic closed/agonist conformation. Comparisons are made between full and
partial agonists, including the well known natural and synthetic full estrogens E2 and DES, the
partial agonists of phytoestrogen GEN and another synthetic compound W, with structures
seen in Schemes 4.1, and 4.2. One of the experimentally found differences is that partial
agonists are more difficult to cocrystallize with the LBD, since they may stabilize the LBD in
mixed closed/agonist and open/antagonist conformations. Therefore, mutation techniques
were applied to aid the cocrystallization of partial agonists with the LBD, by facilitating the
folding process of H12 into the closed/agonist conformation.40,41
Full and partial agonists differ mainly in the ligand-dependent transcription activity, i.e., the
conventional agonist action of proliferative effects, which decreases in the order of E2 ~
DES > GEN > W (not detectable) for the ligands studied herein.37-39 Molecular modeling
techniques were used in this study to investigate how the full and partial agonists
communicate ERα LBD. The cocrystal structures are compared among E2–LBD (1G50),
DES–LBD (3ERD), GEN–LBD (1X7R), and W–LBD (2QZO). Figure 4.1 represents the
main interactions of agonists binding to the receptor displayed by the E2–LBD cocrystal
structure.
Figure 4.1 E2 in complex with the agonist-ERα LBD, which represents in general how agonists form
contacts with the receptor; PDB code 1G50.44 Reproduced with permission of the copyright owner.150
Glu353
Arg394
Leu525
His524
Glu419
Nδ
Nε
Olig

Chapter 4. Agonists and the agonist-ERα LBD complexes
46
The interactions of highly consensus are found for the phenolic hydroxyl group of each ligand
bound to the side chains of Glu353 and Arg394. In the other end of the ligands, they
communicate with His524-Leu525 in different manners, which are expected to be responsible
for different bioactivities. As it was found experimentally, His524 and Leu525 were
intimately involved in ligand-induced transcriptional response of ERα agonists, as mutations
in either of them to alanine resulted in large reductions in binding affinity and subsequent
ligand-induced transcriptional activation.151,152 The interactions have thus been studied in
terms of ligands bound to His524 only (Scheme 4.1) and together with Leu525 (Scheme 4.2).
Scheme 4.1 Side chain of histidine and the structures of systems modeled for the
ligand…His524…Glu419 H-bonding network. Asterisks indicate the centers which are frozen in the
geometry optimization steps. Reproduced with permission of the copyright owner.150
O
HO
N
N
His524H O
HO
H
N
N
H
*
* *
*
O
OHO
OH
OH
N
N
O
OHO
OH
O
H
N
N
H
*
* *
*
O
HN
HGlu419
*
*
H H
H
His524
O
HN
Glu419
*
*
His524
O
HN
Glu419
*
*
His524
O
HN
Glu419
*
*
*HON
N
His524
*
O
HN
HGlu419
*
*
E2
GEN
DES
N
N
H
*
H
His524
O
HN
Glu419
*
*
N NHO
OH
F
F
FN
N
H
*
H
His524
O
HN
Glu419
*
*
*
OH
*HOO
H
His
C. Protonated B. δ-tautomer A. ε-tautomer
HN
N
HN
NH ε ε
δ δ
α α β γ
N
NH ε
δ
α β γ
β
γ
W
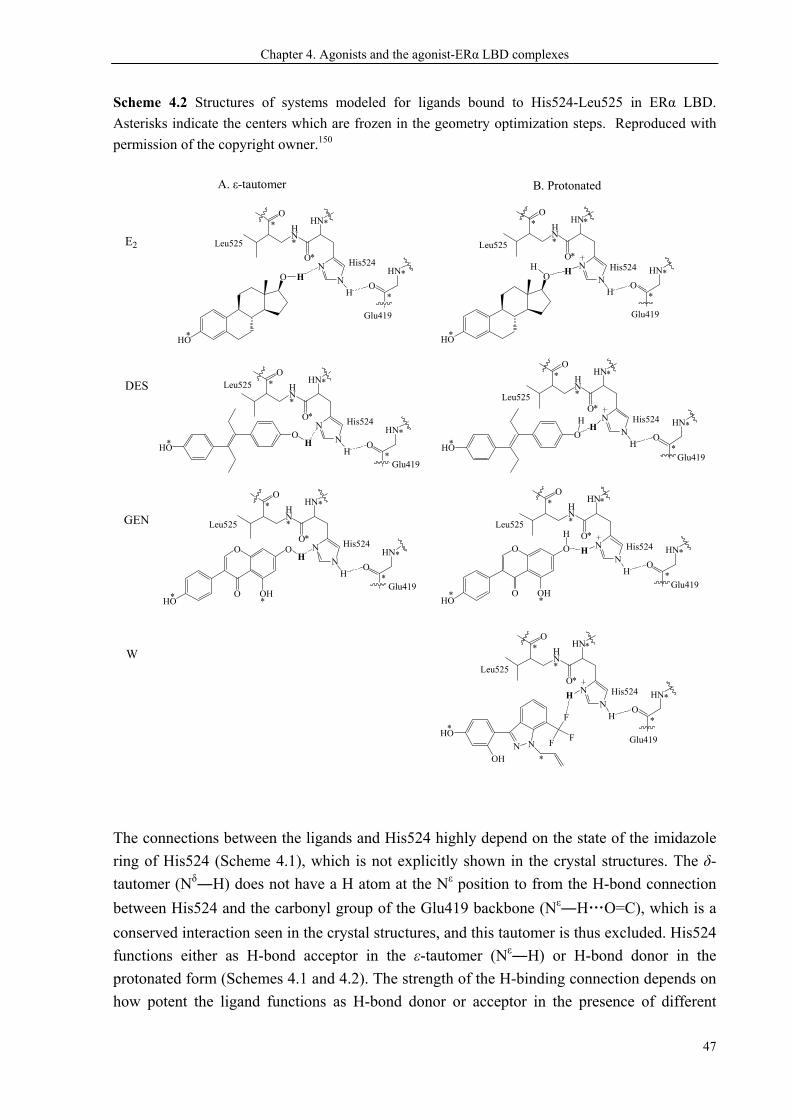
Chapter 4. Agonists and the agonist-ERα LBD complexes
47
Scheme 4.2 Structures of systems modeled for ligands bound to His524-Leu525 in ERα LBD.
Asterisks indicate the centers which are frozen in the geometry optimization steps. Reproduced with
permission of the copyright owner.150
A. ε-tautomer B. Protonated
E2
GEN
DES
O
HO
N
N
His524
H
*
O
HN
H
*
*
HNHN
O
O
*
*
* *
Leu525
Glu419
O
HO
H N
NH
*
H
His524
O
HN*
*
HNHN
O
O
*
*
* *
Leu525
Glu419
N
NH
H
His524
O
HN*
*
HNHN
O
O
*
*
* *
Leu525
N NHO
OH
F
F
F
*
Glu419*
N
N
His524
O
HN
HGlu419
*
*
HNHN
O
O
*
*
* *Leu525
*HOO
H
N
NH
H
His524
O
HN
Glu419
*
*
HNHN
O
O
*
*
* *
Leu525
*HOO
H
N
N
His524
O
HN
H
Glu419
*
*
HNHN
O
O
*
*
* *
Leu525
O
OHO
OH
OH
* *
N
NH
H
His524
O
HN
Glu419
*
*
HNHN
O
O
*
*
* *
Leu525
O
OHO
OH
O
H
**
W
The connections between the ligands and His524 highly depend on the state of the imidazole
ring of His524 (Scheme 4.1), which is not explicitly shown in the crystal structures. The δ-
tautomer (Nδ―H) does not have a H atom at the Nε position to from the H-bond connection
between His524 and the carbonyl group of the Glu419 backbone (Nε―H…O=C), which is a
conserved interaction seen in the crystal structures, and this tautomer is thus excluded. His524
functions either as H-bond acceptor in the ε-tautomer (Nε―H) or H-bond donor in the
protonated form (Schemes 4.1 and 4.2). The strength of the H-binding connection depends on
how potent the ligand functions as H-bond donor or acceptor in the presence of different
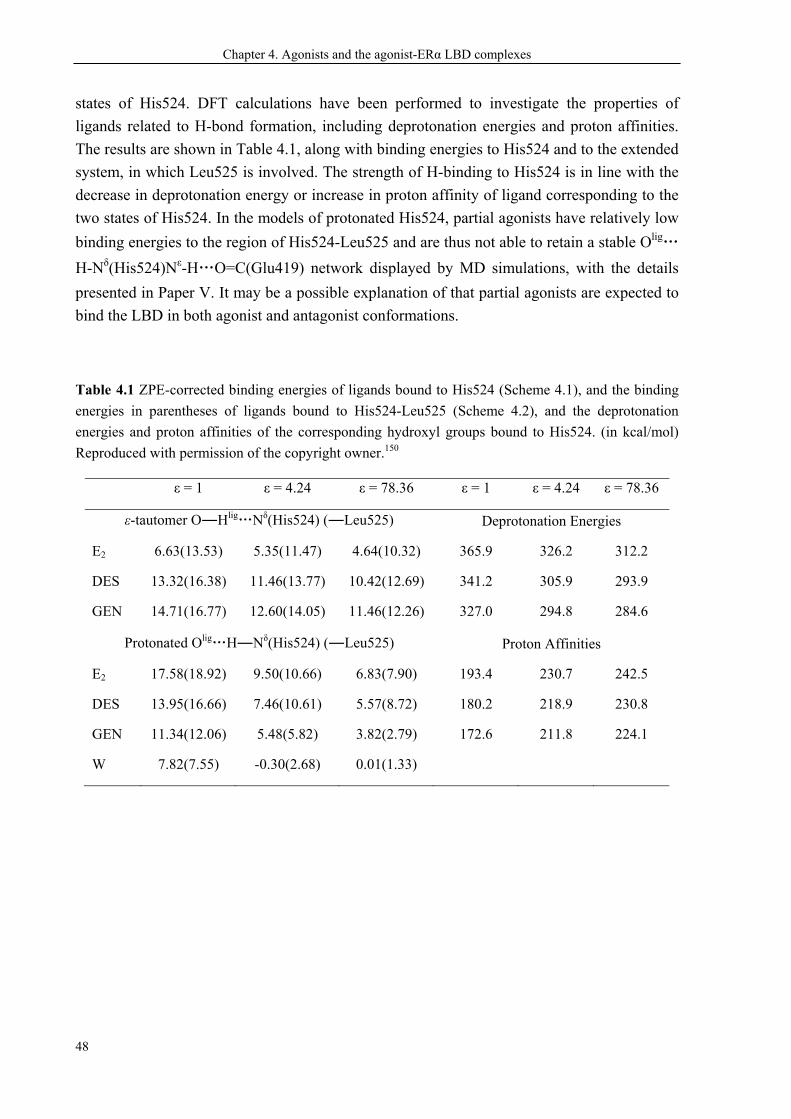
Chapter 4. Agonists and the agonist-ERα LBD complexes
48
states of His524. DFT calculations have been performed to investigate the properties of
ligands related to H-bond formation, including deprotonation energies and proton affinities.
The results are shown in Table 4.1, along with binding energies to His524 and to the extended
system, in which Leu525 is involved. The strength of H-binding to His524 is in line with the
decrease in deprotonation energy or increase in proton affinity of ligand corresponding to the
two states of His524. In the models of protonated His524, partial agonists have relatively low
binding energies to the region of His524-Leu525 and are thus not able to retain a stable Olig…
H-Nδ(His524)Nε-H…O=C(Glu419) network displayed by MD simulations, with the details
presented in Paper V. It may be a possible explanation of that partial agonists are expected to
bind the LBD in both agonist and antagonist conformations.
Table 4.1 ZPE-corrected binding energies of ligands bound to His524 (Scheme 4.1), and the binding
energies in parentheses of ligands bound to His524-Leu525 (Scheme 4.2), and the deprotonation
energies and proton affinities of the corresponding hydroxyl groups bound to His524. (in kcal/mol)
Reproduced with permission of the copyright owner.150
ε = 1 ε = 4.24 ε = 78.36 ε = 1 ε = 4.24 ε = 78.36
ε-tautomer O―Hlig…Nδ(His524) (―Leu525) Deprotonation Energies
E2 6.63(13.53) 5.35(11.47) 4.64(10.32) 365.9 326.2 312.2
DES 13.32(16.38) 11.46(13.77) 10.42(12.69) 341.2 305.9 293.9
GEN 14.71(16.77) 12.60(14.05) 11.46(12.26) 327.0 294.8 284.6
Protonated Olig…H―Nδ(His524) (―Leu525) Proton Affinities
E2 17.58(18.92) 9.50(10.66) 6.83(7.90) 193.4 230.7 242.5
DES 13.95(16.66) 7.46(10.61) 5.57(8.72) 180.2 218.9 230.8
GEN 11.34(12.06) 5.48(5.82) 3.82(2.79) 172.6 211.8 224.1
W 7.82(7.55) -0.30(2.68) 0.01(1.33)

49
CONCLUSIONS
SERMs have been more and more widely used as multifunctional drugs acting as ERα
signaling modulators. A very successful example is TAM, which has been used to treat breast
cancer for decades. Computational studies in this research have led to new information on
how ligands act on ERα, and have provided better understanding of the CYP-mediated
biotransformation of TAM. The studies aimed for new SERMs with optimized in vivo
metabolism. The main results thus obtained are:
— The SERM profiles of partial antagonists are related to the conformational flexibility of the
SERM-ERα LBD complex. The principle of more stable, more estrogenic is used to interpret
between MD simulation and experimental results.
— Base on this principle, a plausible explanation thus obtained for the CYP2D6 debate is that
the CYP2D6-mediated activation of TAM has contradictive effects on TAM treatment: the
positive side is that OHT has much higher binding affinity to ERα than the prodrug TAM
does, while the negative side is that OHT may provide more estrogenic properties.
— Rotation about the C(Ar)―C(sp2) single bond of TAM is a lower energy cost process,
resulting in spontaneous helicity reversion of the molecular propeller. Only the clockwise
propellers fit the ERα binding pocket.
— Rotation about the C(sp2)═C(sp2) double bond of OHT (cis-trans isomerization), a rarely
reported CYP reaction, evolves from a conventional CYP-mediated redox process analogous
to the CYP-catalyzed dehydrogenation. The evolution towards distinctly different reactions is
mainly a result of the chemical properties of the substrate compounds.
— TAM and TOR are modified by adding a methyl substituent to the para-position.
Activation of the resulting structures is highly possible to be a CYP3A-mediated aliphatic
hydroxylation. The modified structures are likely to be used in a wider range than TAM, as
both the CYP2D6 genetic polymorhpism and the undesired cis-trans isomerization are
possibly avoided.
— The full agonists E2 and DES display sufficiently strong interactions with His524-Leu525
so as to stabilize the agonist conformation of the LBD, whereas the partial agonist GEN does
not, which explains the different transcriptional activities of full and partial agonists.
The complete mechanism of SERM actions is still quite a mystery. Much more research is
required for deciphering small ligands signaling via ERα pathways in order to develop idea
SERMs. Computational methods are powerful tools to analyze chemical properties of
compounds, reaction mechanisms, et al., and can assist development of new drugs in addition
to experimental methods.

50
REFERENCES
1 American cancer society. cancer facts and figures 2012 2 Precigoux G, Courseille C, Geoffre S, et al. [p-(Dimethylamino-2 -ethoxy)phenyl]-1-trans-diphenyl-
1,2-butene-1(Tamoxifen)(ICI-46474). Acta Crystallographica Section B-Structural Science 1979; 35:3070-3072
3 Rappoport Z, Biali SE. Threshold rotational mechanisms and enantiomerization barriers of polyarylvinyl propellers. Accounts of Chemical Research 1997; 30:307-314
4 Kuramochi H. Conformational studies and electronic structures of tamoxifen and toremifene and their allylic carbocations proposed as reactive intermediates leading to DNA adduct formation. Journal of Medicinal Chemistry 1996; 39:2877-2886
5 Biali SE, Rappoport Z. Stable Simple Enols .3. Static and Dynamic Nmr Behavior of Crowded Triarylethenols and Related-Compounds - 3-Ring Flip as the Threshold Mechanism for Enantiomerization of Crowded Triarylvinyl Propellers. Journal of the American Chemical Society 1984; 106:477-496
6 Rochlin E, Rappoport Z. Mapping the Enantiomerization Routes of Triarylvinyl Propellers - Barriers for the 3-Ring Flip and the 3 Different 2-Ring Flips of M-Methoxy-Substituted Trimesitylvinyl Isopropyl Ethers. Journal of Organic Chemistry 1994; 59:3857-3870
7 Rochlin E, Rappoport Z, Kastner F, et al. Four enantiomerization routes of 1,2,2-trimesitylvinyl acetate. Enantioselective liquid chromatography of (E)- and (Z)-2-m-methoxymesityl-1,2-dimesitylvinyl acetates. Journal of Organic Chemistry 1999; 64:8840-8845
8 McCague R, Kuroda R, Leclercq G, et al. Synthesis and estrogen receptor binding of 6,7-dihydro-8-phenyl-9-[4-[2-(dimethylamino)ethoxy] phenyl]-5H-benzocycloheptene, a nonisomerizable analogue of tamoxifen. X-ray crystallographic studies. Journal of Medicinal Chemistry 1986; 29:2053-2059
9 Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science 2005; 308:1583-1587
10 Cauley JA, Seeley DG, Ensrud K, et al. Estrogen Replacement Therapy and Fractures in Older Women. Annals of Internal Medicine 1995; 122:9-16
11 Mora S, Kershner DW, Vigilance CP, et al. Coronary Artery Disease in Postmenopausal Women. Current treatment options in cardiovascular medicine 2001; 3:67-79
12 Riggs BL, Khosla S, Melton LJ. Sex steroids and the construction and conservation of the adult skeleton. Endocrine Reviews 2002; 23:279-302
13 Sherwin BB. Estrogen and cognitive aging in women. Trends in Pharmacological Sciences 2002; 23:527-534
14 Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis 1996; 17:2279-2284 15 Korach KS, Hewitt SC, Couse JF. Estrogen receptor transcription and transactivation Estrogen
receptor knockout mice: what their phenotypes reveal about mechanisms of estrogen action. Breast Cancer Research 2000; 2:345-352
16 Ettinger B. Overview of estrogen replacement therapy: A historical perspective. Proceedings of the Society for Experimental Biology and Medicine 1998; 217:2-5
17 Korach KS, Deroo BJ. Estrogen receptors and human disease. Journal of Clinical Investigation 2006; 116:561-570
18 Gustafsson JA, Heldring N, Pike A, et al. Estrogen receptors: How do they signal and what are their targets. Physiological Reviews 2007; 87:905-931

References
51
19 Green S, Walter P, Greene G, et al. Cloning of the human oestrogen receptor cDNA. Journal of Steroid Biochemistry 1986; 24:77-83
20 Kuiper GG, Enmark E, Pelto-Huikko M, et al. Cloning of a novel receptor expressed in rat prostate and ovary. Proceedings of the National Academy of Sciences of the United States of America 1996; 93:5925-5930
21 Ricketts D, Turnbull L, Ryall G, et al. Estrogen and progesterone receptors in the normal female breast. Cancer Research 1991; 51:1817-1822
22 Leygue E, Dotzlaw H, Watson PH, et al. Altered estrogen receptor alpha and beta messenger RNA expression during human breast tumorigenesis. Cancer Research 1998; 58:3197-3201
23 Ali S, Coombes RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. Journal of Mammary Gland Biology and Neoplasia 2000; 5:271-281
24 Khan SA, Rogers MA, Khurana KK, et al. Estrogen receptor expression in benign breast epithelium and breast cancer risk. Journal of the National Cancer Institute 1998; 90:37-42
25 Dahlman-Wright K, Cavailles V, Fuqua SA, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacological Reviews 2006; 58:773-781
26 Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998; 95:927-937
27 Brzozowski AM, Pike AC, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997; 389:753-758
28 Warnmark A, Treuter E, Gustafsson JA, et al. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. Journal of Biological Chemistry 2002; 277:21862-21868
29 Sigler PB, Tanenbaum DM, Wang Y, et al. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proceedings of the National Academy of Sciences of the United States of America 1998; 95:5998-6003
30 Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, et al. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocrine Reviews 2009; 30:293-342
31 O'Reilly EJ, Mirzaei F, Forman MR, et al. Diethylstilbestrol Exposure in Utero and Depression in Women. American Journal of Epidemiology 2010; 171:876-882
32 Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol Revisited - a Review of the Long-Term Health-Effects. Annals of Internal Medicine 1995; 122:778-788
33 Warri A, Saarinen N, Makela S, et al. The role of early life genistein exposures in modifying breast cancer risk. British Journal of Cancer 2008; 98:1485-1493
34 Messina MJ, Persky V, Setchell KDR, et al. Soy Intake and Cancer Risk - a Review of the in-Vitro and in-Vivo Data. Nutrition and Cancer-an International Journal 1994; 21:113-131
35 Matsumura A, Ghosh A, Pope GS, et al. Comparative study of oestrogenic properties of eight phytoestrogens in MCF7 human breast cancer cells. Journal of Steroid Biochemistry and Molecular Biology 2005; 94:431-443
36 Ju YH, Allred KF, Allred CD, et al. Genistein stimulates growth of human breast cancer cells in a novel, postmenopausal animal model, with low plasma estradiol concentrations. Carcinogenesis 2006; 27:1292-1299
37 Booth EA, Marchesi M, Knittel AK, et al. The pathway-selective estrogen receptor ligand WAY-169916 reduces infarct size after myocardial ischemia and reperfusion by an estrogen receptor dependent mechanism. Journal of Cardiovascular Pharmacology 2007; 49:401-407
38 Keith JC, Albert LM, Leathurby Y, et al. The utility of pathway selective estrogen receptor ligands that inhibit nuclear factor-kappa B transcriptional activity in models of rheumatoid arthritis. Arthritis Research & Therapy 2005; 7:R427-R438

References
52
39 Chadwick CC, Chippari S, Matelan E, et al. Identification of pathway-selective estrogen receptor ligands that inhibit NF-kappaB transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America 2005; 102:2543-2548
40 Bruning JB, Parent AA, Gil G, et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nature Chemical Biology 2010; 6:837-843
41 Manas ES, Xu ZB, Unwalla RJ, et al. Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure 2004; 12:2197-2207
42 Riggs BL, Hartmann LC. Drug therapy: Selective estrogen-receptor modulators - Mechanisms of action and application to clinical practice. New England Journal of Medicine 2003; 348:618-629
43 Blizzard TA, DiNinno F, Morgan JD, et al. Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side chains and linkers. Bioorganic & Medicinal Chemistry Letters 2005; 15:107-113
44 Eiler S, Gangloff M, Duclaud S, et al. Overexpression, purification, and crystal structure of native ER alpha LBD. Protein Expression and Purification 2001; 22:165-173
45 Heldring N, Pawson T, McDonnell D, et al. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. Journal of Biological Chemistry 2007; 282:10449-10455
46 Kong EH, Heldring N, Gustafsson JA, et al. Delineation of a unique protein-protein interaction site on the surface of the estrogen receptor. Proceedings of the National Academy of Sciences of the United States of America 2005; 102:3593-3598
47 Gao LT, Y. and Eriksson, L. A. More Stable, more Estrogenic: the SERM-ERα LBD complex. Journal of Biophysical Chemistry 2011; 2:233-243
48 Osborne CK. Tamoxifen in the treatment of breast cancer. New England Journal of Medicine 1998; 339:1609-1618
49 Clarke M, Collins R, Davies C, et al. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet 1998; 351:1451-1467
50 Jin Y, Desta Z, Stearns V, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. Journal of the National Cancer Institute 2005; 97:30-39
51 Desta Z, Ward BA, Soukhova NV, et al. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: Prominent roles for CYP3A and CYP2D6. Journal of Pharmacology and Experimental Therapeutics 2004; 310:1062-1075
52 Potter GA, Mccague R, Jarman M. A Mechanistic Hypothesis for DNA Adduct Formation by Tamoxifen Following Hepatic Oxidative-Metabolism. Carcinogenesis 1994; 15:439-442
53 Potter GA, McCague R, Jarman M. A mechanistic hypothesis for DNA adduct formation by tamoxifen following hepatic oxidative metabolism. Carcinogenesis 1994; 15:439-442
54 Boocock DJ, Brown K, Gibbs AH, et al. Identification of human CYP forms involved in the activation of tamoxifen and irreversible binding to DNA. Carcinogenesis 2002; 23:1897-1901
55 Williams GM, Iatropoulos MJ, Djordjevic MV, et al. The Triphenylethylene Drug Tamoxifen Is a Strong Liver Carcinogen in the Rat. Carcinogenesis 1993; 14:315-317
56 Pagani O, Gelber S, Price K, et al. Toremifene and tamoxifen are equally effective for early-stage breast cancer: first results of International Breast Cancer Study Group Trials 12-93 and 14-93. Annals of Oncology 2004; 15:1749-1759
57 da Costa GG, Pereira PC, Churchwell MI, et al. DNA adduct formation in the livers of female Sprague-Dawley rats treated with toremifene or alpha-hydroxytoremifene. Chemical Research in Toxicology 2007; 20:300-310

References
53
58 Bartsch H, Phillips DH, Nair J, et al. Lack of evidence for tamoxifen- and toremifene-DNA adducts in lymphocytes of treated patients. Carcinogenesis 2000; 21:845-847
59 Martin EA, Rich KJ, White INH, et al. P-32 Postlabeled DNA-Adducts in Liver Obtained from Women Treated with Tamoxifen. Carcinogenesis 1995; 16:1651-1654
60 Fabian C, Tilzer L, Sternson L. Comparative Binding Affinities of Tamoxifen, 4-Hydroxytamoxifen, and Desmethyltamoxifen for Estrogen-Receptors Isolated from Human-Breast Carcinoma - Correlation with Blood-Levels in Patients with Metastatic Breast-Cancer. Biopharmaceutics & Drug Disposition 1981; 2:381-390
61 Johnson MD, Zuo H, Lee KH, et al. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Research and Treatment 2004; 85:151-159
62 Coezy E, Borgna JL, Rochefort H. Tamoxifen and Metabolites in Mcf7 Cells - Correlation between Binding to Estrogen-Receptor and Inhibition of Cell-Growth. Cancer Research 1982; 42:317-323
63 Seth P, Lunetta KL, Bell DW, et al. Phenol sulfotransferases: hormonal regulation, polymorphism, and age of onset of breast cancer. Cancer Res 2000; 60:6859-6863
64 Falany CN, Wheeler J, Oh TS, et al. Steroid Sulfation by Expressed Human Cytosolic Sulfotransferases. Journal of Steroid Biochemistry and Molecular Biology 1994; 48:369-375
65 Goetz MP, Rae JM, Suman VJ, et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. Journal of Clinical Oncology 2005; 23:9312-9318
66 Schroth W, Antoniadou L, Fritz P, et al. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. Journal of Clinical Oncology 2007; 25:5187-5193
67 Goetz MP, Knox SK, Suman VJ, et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Research and Treatment 2007; 101:113-121
68 Kiyotani K, Mushiroda T, Sasa M, et al. Impact of CYP2D6*10 on recurrence-free survival in breast cancer patients receiving adjuvant tamoxifen therapy. Cancer Science 2008; 99:995-999
69 Xu Y, Sun Y, Yao L, et al. Association between CYP2D6*10 genotype and survival of breast cancer patients receiving tamoxifen treatment. Annals of Oncology 2008; 19:1423-1429
70 Newman WG, Hadfield KD, Latif A, et al. Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clinical Cancer Research 2008; 14:5913-5918
71 Wegman P, Vainikka L, Stal O, et al. Genotype of metabolic enzymes and the benefit of tamoxifen in postmenopausal breast cancer patients. Breast Cancer Research 2005; 7:R284-R290
72 Wegman P, Elingarami S, Carstensen J, et al. Genetic variants of CYP3A5, CYP2D6, SULT1A1, UGT2B15 and tamoxifen response in postmenopausal patients with breast cancer. Breast Cancer Research 2007; 9:R7
73 Nowell SA, Ahn JY, Rae JM, et al. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Research and Treatment 2005; 91:249-258
74 Okishiro M, Taguchi T, Kim SJ, et al. Genetic Polymorphisms of CYP2D6*10 and CYP2C19*2,*3 Are not Associated With Prognosis, Endometrial Thickness, or Bone Mineral Density in Japanese Breast Cancer Patients Treated With Adjuvant Tamoxifen. Cancer 2009; 115:952-961
75 Regan MM, Leyland-Jones B, Bouzyk M, et al. CYP2D6 Genotype and Tamoxifen Response in Postmenopausal Women with Endocrine-Responsive Breast Cancer: The Breast International Group 1-98 Trial. Journal of the National Cancer Institute 2012; 104:441-451

References
54
76 Rae JM, Drury S, Hayes DF, et al. CYP2D6 and UGT2B7 Genotype and Risk of Recurrence in Tamoxifen-Treated Breast Cancer Patients. Journal of the National Cancer Institute 2012; 104:452-460
77 Nowell S, Sweeney C, Winters M, et al. Association between sulfotransferase 1A1 genotype and survival of breast cancer patients receiving tamoxifen therapy. Journal of the National Cancer Institute 2002; 94:1635-1640
78 Osborne CK, Coronado E, Allred DC, et al. Acquired Tamoxifen Resistance - Correlation with Reduced Breast-Tumor Levels of Tamoxifen and Isomerization of Trans-4-Hydroxytamoxifen. Journal of the National Cancer Institute 1991; 83:1477-1482
79 Osborne CK, Wiebe VJ, Mcguire WL, et al. Tamoxifen and the Isomers of 4-Hydroxytamoxifen in Tamoxifen-Resistant Tumors from Breast-Cancer Patients. Journal of Clinical Oncology 1992; 10:304-310
80 Katzenellenbogen BS, Norman MJ, Eckert RL, et al. Bioactivities, Estrogen-Receptor Interactions, and Plasminogen Activator-Inducing Activities of Tamoxifen and Hydroxytamoxifen Isomers in Mcf-7 Human-Breast Cancer-Cells. Cancer Research 1984; 44:112-119
81 Williams ML, Lennard MS, Martin IJ, et al. Interindividual Variation in the Isomerization of 4-Hydroxytamoxifen by Human Liver-Microsomes - Involvement of Cytochromes P450. Carcinogenesis 1994; 15:2733-2738
82 Crewe HK, Notley LM, Wunsch RM, et al. Metabolism of tamoxifen by recombinant human cytochrome P450 enzymes: Formation of the 4-hydroxy, 4 '-hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifen. Drug Metabolism and Disposition 2002; 30:869-874
83 Sono M, Roach MP, Coulter ED, et al. Heme-containing oxygenases. Chemical Reviews 1996; 96:2841-2887
84 Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chemical Research in Toxicology 2001; 14:611-650
85 Shaik S, Kumar D, de Visser SP, et al. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chemical Reviews 2005; 105:2279-2328
86 Shaik S, Cohen S, Wang Y, et al. P450 Enzymes: Their Structure, Reactivity, and Selectivity-Modeled by QM/MM Calculations. Chemical Reviews 2010; 110:949-1017
87 Nussinov R, Fishelovitch D, Shaik S, et al. How Does the Reductase Help To Regulate the Catalytic Cycle of Cytochrome P450 3A4 Using the Conserved Water Channel? Journal of Physical Chemistry B 2010; 114:5964-5970
88 Schyman P, Lai W, Chen H, et al. The directive of the protein: how does cytochrome p450 select the mechanism of dopamine formation? Journal of the American Chemical Society 2011; 133:7977-7984
89 Friesner RA, Bochevarov AD, Li JN, et al. Insights into the Different Dioxygen Activation Pathways of Methane and Toluene Monooxygenase Hydroxylases. Journal of the American Chemical Society 2011; 133:7384-7397
90 Han KL, Li DM, Huang XQ, et al. Catalytic Mechanism of Cytochrome P450 for 5 '-Hydroxylation of Nicotine: Fundamental Reaction Pathways and Stereoselectivity. Journal of the American Chemical Society 2011; 133:7416-7427
91 Guengerich FP, Macdonald TL. Chemical Mechanisms of Catalysis by Cytochromes-P-450 - a Unified View. Accounts of Chemical Research 1984; 17:9-16
92 Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chemical Reviews 2004; 104:3947-3980

References
55
93 de Visser SP, Shaik S. A proton-shuttle mechanism mediated by the porphyrin in benzene hydroxylation by cytochrome P450 enzymes. Journal of the American Chemical Society 2003; 125:7413-7424
94 Groves JT, Mcclusky GA. Aliphatic Hydroxylation Via Oxygen Rebound - Oxygen-Transfer Catalyzed by Iron. Journal of the American Chemical Society 1976; 98:859-861
95 Groves JT. Key Elements of the Chemistry of Cytochrome-P-450 - the Oxygen Rebound Mechanism. Journal of Chemical Education 1985; 62:928-931
96 Li DM, Huang XQ, Han KL, et al. Catalytic Mechanism of Cytochrome P450 for 5 '-Hydroxylation of Nicotine: Fundamental Reaction Pathways and Stereoselectivity. Journal of the American Chemical Society 2011; 133:7416-7427
97 Schyman P, Lai WZ, Chen H, et al. The Directive of the Protein: How Does Cytochrome P450 Select the Mechanism of Dopamine Formation? Journal of the American Chemical Society 2011; 133:7977-7984
98 Muthyala RS, Sheng S, Carlson KE, et al. Bridged bicyclic cores containing a 1,1-diarylethylene motif are high-affinity subtype-selective ligands for the estrogen receptor. Journal of Medicinal Chemistry 2003; 46:1589-1602
99 Vogel VG, Costantino JP, Wickerham DL, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. Journal of the American Medical Association 2006; 295:2727-2741
100 Gao ZO, Gao ZP, Fields JZ, et al. Development of cross-resistance to tamoxifen in raloxifene-treated breast carcinoma cells. Anticancer Research 2002; 22:1379-1383
101 O'Regan RM, Gajdos C, Dardes RC, et al. Effects of raloxifene after tamoxifen on breast and endometrial tumor growth in athymic mice. Journal of the National Cancer Institute 2002; 94:274-283
102 Sato M, Rippy MK, Bryant HU. Raloxifene, tamoxifen, nafoxidine, or estrogen effects on reproductive and nonreproductive tissues in ovariectomized rats. Faseb Journal 1996; 10:905-912
103 DeMichele A, Troxel AB, Berlin JA, et al. Impact of raloxifene or tamoxifen use on endometrial cancer risk: A population-based case-control study. Journal of Clinical Oncology 2008; 26:4151-4159
104 Assikis VJ, Neven P, Jordan VC, et al. A realistic clinical perspective of tamoxifen and endometrial carcinogenesis. European Journal of Cancer 1996; 32A:1464-1476
105 Grese TA, Sluka JP, Bryant HU, et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proceedings of the National Academy of Sciences of the United States of America 1997; 94:14105-14110
106 Liu H, Park WC, Bentrem DJ, et al. Structure-function relationships of the raloxifene-estrogen receptor-alpha complex for regulating transforming growth factor-alpha expression in breast cancer cells. Journal of Biological Chemistry 2002; 277:9189-9198
107 Wu YL, Yang X, Ren Z, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Molecular Cell 2005; 18:413-424
108 Vajdos FF, Hoth LR, Geoghegan KF, et al. The 2.0 A crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Science 2007; 16:897-905
109 Kim S, Wu JY, Birzin ET, et al. Estrogen receptor ligands. II. Discovery of benzoxathiins as potent, selective estrogen receptor alpha modulators. Journal of Medicinal Chemistry 2004; 47:2171-2175
110 Gao L, Tu Y, Wegman P, et al. Conformational enantiomerization and estrogen receptor alpha binding of anti-cancer drug tamoxifen and its derivatives. J Chem Inf Model 2011; 51:306-314

References
56
111 Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. American Journal of Health-System Pharmacy 2000; 57:1669-1675
112 Devos D, Slee PHTJ, Stevenson D, et al. Serum Elimination Half-Life of Tamoxifen and Its Metabolites in Patients with Advanced Breast-Cancer. Cancer Chemotherapy and Pharmacology 1992; 31:76-78
113 Hasmann M, Rattel B, Loser R. Preclinical Data for Droloxifene. Cancer Letters 1994; 84:101-116
114 Buzdar A, Hayes D, El-Khoudary A, et al. Phase III randomized trial of droloxifene and tamoxifen as first-line endocrine treatment of ER/PgR-positive advanced breast cancer. Breast Cancer Research and Treatment 2002; 73:161-175
115 Grill HJ, Pollow K. Pharmacokinetics of Droloxifene and Its Metabolites in Breast-Cancer Patients. American Journal of Clinical Oncology-Cancer Clinical Trials 1991; 14:S21-S29
116 Nomura Y, Nakajima M, Tominaga T, et al. [Late phase II study of TAT-59 (miproxifene phospate) in advanced or recurrent breast cancer patients (a double-blind comparative study with tamoxifen citrate)]. Gan To Kagaku Ryoho 1998; 25:1045-1063
117 Gradishar W, Glusman J, Lu Y, et al. Effects of high dose raloxifene in selected patients with advanced breast carcinoma. Cancer 2000; 88:2047-2053
118 Deshmane V, Krishnamurthy S, Melemed AS, et al. Phase III double-blind trial of Arzoxifene compared with tamoxifen for locally advanced or metastatic breast cancer. Journal of Clinical Oncology 2007; 25:4967-4973
119 Arpino G, Krishnan MN, Dinesh CD, et al. Idoxifene versus tamoxifen: a randomized comparison in postmenopausal. patients with metastatic breast cancer. Annals of Oncology 2003; 14:233-241
120 Heuson JC, Engelsman E, Blonkvanderwijst J, et al. Comparative Trial of Nafoxidine and Ethinylestradiol in Advanced Breast-Cancer - Eortc Study. British Medical Journal 1975; 2:711-713
121 Schrödinger E. An undulatory theory of the mechanics of atoms and molecules. Physical review 1926; 28:1049
122 Becke AD. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Physical Review A 1988; 38:3098-3100
123 Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Physical Review B 1988; 37:785-789
124 Becke AD. Density-Functional Thermochemistry .3. The Role of Exact Exchange. Journal of Chemical Physics 1993; 98:5648-5652
125 Stephens PJ, Devlin FJ, Chabalowski CF, et al. Ab-Initio Calculation of Vibrational Absorption and Circular-Dichroism Spectra Using Density-Functional Force-Fields. Journal of Physical Chemistry 1994; 98:11623-11627
126 Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts 2008; 120:215-241
127 Zhao Y, Truhlar DG. Density functionals with broad applicability in chemistry. Accounts of Chemical Research 2008; 41:157-167
128 Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.02. Gaussian, Inc., Wallingford CT 2009
129 Marenich AV, Cramer CJ, Truhlar DG. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. Journal of Physical Chemistry B 2009; 113:6378-6396

References
57
130 Molecular Operating Enviroment (MOE). Chemcal Computing Group, Montreal, Canada 131 Krieger E, Darden T, Nabuurs SB, et al. Making optimal use of empirical energy functions: Force-
field parameterization in crystal space. Proteins-Structure Function and Bioinformatics 2004; 57:678-683
132 Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics 1983; 79:926-935
133 Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of Computational Chemistry 2002; 23:1623-1641
134 Krieger E, Nielsen JE, Spronk CA, et al. Fast empirical pKa prediction by Ewald summation. Journal of Molecular Graphics and Modelling 2006; 25:481-486
135 Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of Computational Chemistry 2003; 24:1999-2012
136 Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. Journal of Computational Chemistry 2004; 25:1157-1174
137 Blizzard TA, Morgan JD, Chan WD, et al. Estrogen receptor ligands. Part 14: Application of novel antagonist side chains to existing platforms. Bioorganic & Medicinal Chemistry Letters 2005; 15:5124-5128
138 Blizzard TA, DiNinno F, Chen HY, et al. Estrogen receptor ligands. Part 13: Dihydrobenzoxathiin SERAMs with an optimized antagonist side chain. Bioorganic & Medicinal Chemistry Letters 2005; 15:3912-3916
139 Tan Q, Blizzard TA, Morgan JD, et al. Estrogen receptor ligands. Part 10: Chromanes: old scaffolds for new SERAMs. Bioorganic & Medicinal Chemistry Letters 2005; 15:1675-1681
140 Liu J, Birzin ET, Chan WD, et al. Estrogen receptor ligands. Part 11: Synthesis and activity of isochromans and isothiochromans. Bioorganic & Medicinal Chemistry Letters 2005; 15:715-718
141 Blizzard TA, DiNinno F, Morgan JD, et al. Estrogen receptor ligands. Part 7: Dihydrobenzoxathiin SERAMs with bicyclic amine side chains. Bioorganic & Medicinal Chemistry Letters 2004; 14:3861-3864
142 Blizzard TA, DiNinno F, Morgan JD, et al. Estrogen receptor ligands. Part 8: Dihydrobenzoxathiin SERAMs with heteroatom-substituted side chains. Bioorganic & Medicinal Chemistry Letters 2004; 14:3865-3868
143 Tan Q, Birzin ET, Chan W, et al. Estrogen receptor ligands. Part 5: The SAR of dihydrobenzoxathiins containing modified basic side chains. Bioorganic & Medicinal Chemistry Letters 2004; 14:3747-3751
144 MacGregor Schafer J, Liu H, Bentrem DJ, et al. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Research 2000; 60:5097-5105
145 Anghel SI, Perly V, Melancon G, et al. Aspartate 351 of estrogen receptor alpha is not crucial for the antagonist activity of antiestrogens. J Biological Chemistry 2000; 275:20867-20872
146 Dayan G, Lupien M, Auger A, et al. Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor alpha. Molecular Pharmacology 2006; 70:579-588
147 Oki M. Recent Advances in Atropisomerism. Topics in Stereochemistry 1983; 14:1-81 148 Gao L, Tu Y, Wegman P, et al. A mechanistic hypothesis for the cytochrome P450-catalyzed cis-
trans isomerization of 4-hydroxytamoxifen: an unusual redox reaction. Journal of Chemical Information and Modeling 2011; 51:2293-2301

References
58
149 Yost GS, Moore CD, Shahrokh K, et al. Improved Cytochrome P450 3A4 Molecular Models Accurately Predict the Phe215 Requirement for Raloxifene Dehydrogenation Selectivity. Biochemistry 2010; 49:9011-9019
150 Gao L, Tu Y, Agren H, et al. Characterization of Agonist Binding to His524 in the Estrogen Receptor alpha Ligand Binding Domain. Journal of Physical Chemistry B 2012; 116:4823-4830
151 Ekena K, Weis KE, Katzenellenbogen JA, et al. Different residues of the human estrogen receptor are involved in the recognition of structurally diverse estrogens and antiestrogens. Journal of Biological Chemistry 1997; 272:5069-5075
152 Ekena K, Weis KE, Katzenellenbogen JA, et al. Identification of amino acids in the hormone binding domain of the human estrogen receptor important in estrogen binding. Journal of Biological Chemistry 1996; 271:20053-20059


















