
The Therapeutic Role of Turmeric in Treatment and ...
62
Southeastern University FireScholars Selected Honors eses 4-2015 e erapeutic Role of Turmeric in Treatment and Prevention of Alzheimer’s Disease Rylan M. McQuade Southeastern University - Lakeland Follow this and additional works at: hp://firescholars.seu.edu/honors Part of the Alternative and Complementary Medicine Commons , Nervous System Diseases Commons , Neurology Commons , and the Neurosciences Commons is esis is brought to you for free and open access by FireScholars. It has been accepted for inclusion in Selected Honors eses by an authorized administrator of FireScholars. For more information, please contact fi[email protected]. Recommended Citation McQuade, Rylan M., "e erapeutic Role of Turmeric in Treatment and Prevention of Alzheimer’s Disease" (2015). Selected Honors eses. Paper 23.
Transcript of The Therapeutic Role of Turmeric in Treatment and ...

Southeastern UniversityFireScholars
Selected Honors Theses
4-2015
The Therapeutic Role of Turmeric in Treatmentand Prevention of Alzheimer’s DiseaseRylan M. McQuadeSoutheastern University - Lakeland
Follow this and additional works at: http://firescholars.seu.edu/honors
Part of the Alternative and Complementary Medicine Commons, Nervous System DiseasesCommons, Neurology Commons, and the Neurosciences Commons
This Thesis is brought to you for free and open access by FireScholars. It has been accepted for inclusion in Selected Honors Theses by an authorizedadministrator of FireScholars. For more information, please contact [email protected].
Recommended CitationMcQuade, Rylan M., "The Therapeutic Role of Turmeric in Treatment and Prevention of Alzheimer’s Disease" (2015). Selected HonorsTheses. Paper 23.

The Therapeutic Role of Turmeric in Treatment
and Prevention of Alzheimer’s Disease Rylan M. McQuade
Department of Natural Sciences. Southeastern University, Lakeland, Florida, USA

McQuade 2
Abstract: As a devastating neurological condition that expends millions of lives each
year, Alzheimer’s disease (AD) is a subject of intense investigation.1 Although AD has
been known for over a century, the precise mechanisms that underlie AD pathogenesis
and development are still poorly understood. The Alzheimer phenotype is typified by
extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs),
causing researchers to notice several key enzymes implicated in this process.1 Most
notable are β and γ secretases (which drive Aβ plaque production) and phospholipase
A2 (which stimulates major cascade activation through the specific cleavage of fatty acyl
esters).2 Both acidic and neutral sphingomyelinases of the ceramide pathway are also
strongly linked to AD development and progression.3,4
As a specific type of esterase,
PLA2 specializes in phospholipid catabolism by specifically cleaving fatty acyl bonds at
the sn-2 position, producing a free fatty acid and lysophospholipids as products.3,5
This
stimulates the arachidonic acid (AA) cascade, while lipid cleavage by sphingomyelinase
upregulates the ceramide pathway.3 Cyclooxygenase (COX) and lipooxygenase (LOX)
are suspected to be key players in oxidative and neurological damage and are involved in
both these metabolic pathways.4,5
A further investigation of these molecular messengers
and their cellular environments is absolutely essential for therapeutic intervention and
effective medical treatments. Recently, the role of herbal medicines has been investigated
to produce possible leads for Alzheimer’s treatment. Turmeric contains over 20
medicinally useful compounds, of which curcumin is a key component.6 In this review,
we hypothesize that turmeric will prove especially useful in preventing amyloid-β plaque
deposition in early stages of AD and improvement of learning and coordination in middle
stages of the disease. Further evidence suggests curcumin’s efficacy in resisting ROS,
preventing tau aggregation, and limiting the spread of pro-inflammatory molecules
throughout the body. Because of its highly beneficial effects as an Alzheimer therapeutic,
we commend that turmeric be studied in greater detail and incorporated into Western
medical treatments. Additionally, a prospective study will be presented to determine the
efficacy of turmeric therapies, both in improving physical activities and overall
neurological health. Although turmeric’s precise mechanisms are not yet understood,
multiple studies have confirmed its beneficial effects in preventing amyloidogenesis by
modulating esterase and sphingomyelinase activities and also resisting misdirected

McQuade 3
cleavage of APP.2 Turmeric has also been found to downregulate abnormal GSK-3β
function and prevent formation of SPs and NFTs. Interestingly, murine models subjected
to a turmeric regimen not only displayed a reversal of AD symptoms, but also
demonstrated a lower susceptibility to neurodegenerative disease.3 Collectively, these
findings strongly support turmeric’s therapeutic use in Western medicine.
Chapter 1
Introduction
As a widespread ailment that affects millions worldwide, AD wreaks a deadly
impact on the elderly population.4 Recently, the Western world has experienced a
dramatic peak in Alzheimer’s incidence.5 In this year alone (2015), over five million
Americans are currently living with AD. Certainly, Alzheimer’s is nothing new. Cases of
pre-senile dementia have been reported from early Mesopotamia and ancient Egypt when
external manifestations of the disease were noticed without much (if any) understanding
of its inner workings.6 Recently, however, AD has struck with a dogged intensity to
become one of the leading causes of death among Western cultures. What are the reasons
for AD’s sudden appearance? How does this disease begin and what are its hallmark
symptoms? Genomic comparisons and intensive genetic research reveal that humans have
always possessed the potential for AD development.6 Indeed, AD pathology seems to be
permanently etched on the Homo sapiens genome, consistently conserved from one
generation to the next. Because of the body’s intrinsic ability to produce β and γ
secretases, anyone can develop insoluble plaque deposition when secretases are
overstimulated. Similarly, every individual has the ability to develop NFTs from
excessive tau protein phosphorylation.7 With improved living conditions and medical
treatment, the geriatric population has increased substantially in recent years and
correlates closely with increasing AD incidence. AD largely affects the elderly, usually
65 years or older.5
Alzheimer’s disease progression is split into three overlapping categories: First, the
subject find is difficult to remember recent events. This period usually lasts around two to

McQuade 4
four years and is typified by mild bouts of confusion concerning daily routine and
activities.8 Next, the patient enters a stage of persistent confusion, often forgetting the
identities of close friends and family members. This part of disease progression is
especially devastating since it often robs the patient of his or her self-identity as neuronal
necrosis occurs more rapidly.9 Finally, the individual enters a sudden onset of physical
and cognitive decline, ultimately leading to death. Disease progression is a prolonged and
bitter defeat, stripping away memories and leaving feelings of social isolation and a lost
personal identity in their place. Countless therapies have been developed for AD, ranging
from COX-2 inhibitors to more progressive drugs like memantine and NSAIDs.10
Despite
displaying some promise, these drugs are largely inefficient in addressing the root cause
of AD pathology. The failure of modern AD therapies has driven researchers to delve
further, searching for the most effective treatment. Recent studies suggest that turmeric
may possess the clinical and pharmacological effects that reverse AD symptoms while
also preventing disease pathology. This chapter will review AD’s enormous impact on
the Western world, the rising cost of research, and the lipase enzymes involved in driving
amyloidogenesis. It will be shown precisely how enzymes like secretases and PLA2
directly impact AD development by producing Aβ peptides and pro-inflammatory
molecules respectively.
Alzheimer’s and the Western World
Due to the rising cost of neuropathological research and increasing incidence of
neurodegenerative diseases, much of current research is turning to homeopathic remedies,
largely derived from plant-based materials.3 The use of turmeric corresponds with many
aspects of Adurvedic (Indian) medicine and is frequently used in tandem with additional
Adurvedic compounds, such as Convolvulus pluricaulis (CP), liquiritin (obtained from
liquorice), and shankhpushpi (gathered from the alowe weed). Despite its frequent
medicinal use in much of Eastern medicine and its promising neurological benefits from
multiple studies, turmeric (to this date) is still not used therapeutically in Western
medicine. As researchers are desperately screening novel compounds and developing

McQuade 5
molecular analogues for the treatment of AD, turmeric (specifically curcumin) must be
examined more thoroughly as a potential candidate for AD medicines. Before turmeric
can be investigated in further detail, however, the basic mechanisms underlying AD must
be presented and understood.
As a fatal neurological disease that expends millions of lives each year, AD is a
major health concern among developed countries. It is especially prevalent across North
America and Europe, causing significant health concerns worldwide.11
In Western
cultures, individuals of 65 years exhibit an AD occurrence of 8%; this figure escalates
dramatically with age, affecting over 45% of 85-year-olds.4 With increasing medical
costs and rising expenditure of drug screening, many researchers are searching for
simpler, more efficient alternatives. Rather than halting disease conditions, current
treatments are merely palliative, hoping to ease symptoms in spite of internal tissue
necrosis. In this regard, even the most developed therapeutics are incompetent to prevent
AD pathogenesis and development.10
Researchers are desperately searching for a cure but
so far have been left without any conclusive results. It seems that the search for designing
an effective AD drug regimen has all but exhausted its efforts and research is turning to
components of Eastern medicine that exhibit curative potential. As a key medicinal
compound and major dietary component in Eastern cultures, turmeric may be useful in
preventing the pathogenesis of AD, as well as easing symptoms. After presenting relevant
studies and outlining current understandings of the disease, the efficacy of turmeric
(specifically curcumin) will be discussed and evaluated. First, a comprehensive overview
of AD—its hallmark symptoms, its molecular and genetic origins within the body, and
molecular inhibitors to stop AD progression—will all be presented and discussed in
greater detail. Finally, turmeric’s novel properties and its exceptional use as a dose-
dependent therapeutic will be presented and examined.
Basics of AD
A devastating and dehabilitating illness, AD is the subject of intense
investigation. This disease is becoming increasingly common with millions of cases each

McQuade 6
year and exorbitant costs expended in treating the disease.10
A chronically progressive
category of dementia, AD drastically impacts memory and spatial learning capabilities.12
Patients diagnosed with AD typically survive only three to nine years following AD
onset, manifesting both physical and social deterioration.1 Although the subject of
exhaustive research, the exact cause of AD still remains uncertain. Approximately 5% of
cases are suspected to be genomically linked and are subject to the influence of many
polygenomic sequences.13
Other cases are presumed to be environmental, either linked to
diet or lifestyle choices that are detrimental to overall homeostasis. Understandably, head
trauma can lead to neural disruption, but more obscure factors (such as high blood
pressure) can easily form a complex web of AD’s causative factors. Alzheimer’s complex
ontology coupled with its multiple phenotypic effects and interlinked signaling cascades
provides a difficult scenario for researchers.14
Each new finding underscores the
conviction that AD is a multifactorial illness, stimulated by a host of complex molecular
interactions. Discovered over a century ago by Alois Alzheimer— a Bavarian psychiatrist
and neuropathologist— the mysteries behind AD remain enigmatic.15
Originally
categorizing the disease as “pre-senile dementia,” it was later termed “Alzheimer’s
disease” by Dr. Emil Kraepelin. AD typically afflicts elderly patients (65 years or older),
although pre-senile dementia in younger patients is also a well-documented
phenomenon.9 As mentioned earlier these pre-senile cases are often linked to genetic
abnormalities or to head trauma early in life. Alzheimer’s disease progression is split into
seven distinct stages, ranging from pre-onset of disease symptoms to severe cognitive,
social, and cellular dysfunction.15
As the most common form of dementia, AD patients typically display signs of
forgetfulness and short-term memory (STM) impairment.16
Over the years, symptoms
progressively worsen until the patient displays dramatic behavioral changes,
motor/kinetic imbalance, and gradual loss of coordinated movement. Left untreated, the
patient deteriorates and death inevitably follows within three to nine years.17
In nearly all
cases, AD onset is associated with internal neurofibrillary tangles (NFTs) and external
senile plaques (SPs).11
NFTs are mainly composed of excessively phosphorylated tau
protein, which is overexpressed and has become insoluble in the intracellular

McQuade 7
environment.18
This is most likely due to erroneous expression of the tau gene, causing
protein overexpression and misfolding. Alteration of protein configuration mainly occurs
when defects occurring in the protein’s 3-dimensional structure are not corrected by
molecular chaperones. Because of this buildup of misfolded proteins, synapse
degradation is a common phenomenon of most tauopathies.19
As neural junctions become
clogged and the axon and cell body become crowded with proteinaceous debris, neural
tissues deteriorate and the overall health of the patient decreases. Under normal
conditions, tau proteins help support microtubules and facilitate molecular transport
across the axon. In AD pathology, tau aggregates to form large secondary structures
called paired helical filaments that become tangled and interwoven, causing impairment
of neuronal functions.20
Contrastingly, SPs are mainly present extracellularly and consist almost entirely
of amyloid-β (Aβ) peptide. Ultimately, Aβ peptides originate from the APP gene, which
encodes the amyloid protein precursor (APP).21
Although APP is the substrate in a
myriad of enzymatic reactions, it is largely cleaved through the actions of three main
secretase enzymes-- α, β, and γ. In multiple studies, α-secretase has been noted for its
beneficial effects involving neural growth and protection.7 A disintegrin and
metalloprotease domain protein (ADAM), α-secretase is normally found near the cell
glycocalyx or imbedded as a transmembrane protein within the phospholipid bilayer.22
α-
secretase cuts APP (using ADAM10 and ADAM17) which creates the C-terminus (C83)
portion and the secretion of larger α APP, which collectively regulate brain function. The
C83 part is retained within the lipid bilayer where it is enzymatically cleaved by γ-
secretase, producing a P3 peptide.23
Although found mainly in the plasma membrane,
ADAM10 is also found in significant amounts in the Golgi apparatus. When a
transmembrane protein is cleaved so that its extracellular domain is freed, this is called
“ectodomain shedding.” In contrast to α-secretase, β- and γ-secretases are known to
stimulate the production of Aβ1-40 and Aβ1-42 oligopeptides, which are equally implicated
in AD development.24
In AD pathogenesis, β-secretase (BASE1) acts upon APP first,
cleaving APP at its N-terminus by a catalytic aspartate residue in its active site. This
produces sAPPβ and a C-terminal fragment (CTFβ) called C99 is produced. sAPPβ is

McQuade 8
then cleaved by γ -secretase at the C-terminus to make multiple Aβ oligopeptides.25
Studies overwhelmingly demonstrate that Aβ1-40 and Aβ1-42 are most detrimental to neural
health and are key players in AD pathology. Although originally soluble, Aβ plaques
eventually aggregate to assume their insoluble form.26
This is due to steric considerations
involving surface area, as well as intramolecular interactions that make the aggregates
insoluble. Although soluble polymers have been the main subject of concern, recent
research suggests that soluble plaques may be equally (if not more) deadly than the
insoluble form.27
Although the precise details behind their neurotoxicity still remain a
mystery, researchers have found that soluble (s) Aβ peptides exert a lethal impact on
miRNA, exerting their efforts through N-methyl-D-aspartate (NMDA) receptors and
through reactive oxygen species (ROS).28
This finding highlights the need for total
prevention of Aβ peptide production, both soluble and insoluble forms.
A portion of γ-secretase (presenilin-1) is a central component for APP’s activity.
Researchers have found that even slight structural changes in presenilin-1’s structure are
linked with familial AD (which appears early in life).29
Since Aβ plaque detection is
often obscured by confounding factors, AD diagnosis is often difficult and SPs are
usually observed in deceased specimens. Several methods, however, exist for AD
detection in vivo. Positron emission tomography (PET) allows recording of abnormalities
in glucose metabolism, while MRIs can relay shrinkage of neural tissue that is noticeably
smaller than healthy brain tissue.30
Behavioral studies reveal that AD primarily affects
hippocampal functions involving memory storage and the entorhinal cortex (a processing
portion of the temporal lobe). Further investigations reveal that TNF-α may indirectly
encourage AD development.31
TNF-α converting enzyme (TACE) is a type of ADAM
whose activity liberates TNF-α into the extracellular environment. These proteins usually
contain large amounts of cysteine residues, which are implicated in joining of cells and
protein aggregation caused by disulfide bonding. In recent years, tumor necrosis factor
(TNF) α is a subject of interest for researchers. As a master-regulator of the immune
system, it is involved in critical reactions involving bodily defense systems,
carcinogenesis, inflammation, and regulated cell death.32
It is mainly secreted by
macrophages that have been alerted to exogenous threats to immunity. The immune

McQuade 9
system is well known for its rapid response to exogenous threats and external insults to
the body. An invading bacterium, a virus, or even an epidermal abrasion are common
stimulatory factors of the immune response.33
The immune system’s control of
endogenous systems, however, is only beginning to be understood. Indeed, science has
only touched the tip of the vast iceberg in its understanding of internal immune
modulation.34
An immune response mounted against internal bodily components was
first noted by Dr. William Coley in 1968 when he found that macrophages could mount a
response to carcinogenesis.35
This proved a notable discovery, opening a range of
unknown possibilities for the body’s defense system. For centuries, physicians had
understood the concept of bodily defense against external insults (albeit poorly). Finding
of modulatory defense mechanisms against internal threats radically altered medicine’s
approach to treating illness. In fact, Dr. Coley’s finding brought about a substantial
paradigm shift, turning investigative research to consider internal mechanisms that
stimulated pathogenesis. As Susan Sontag describes in her treatise “Illness as Metaphor,”
pinpointing an internal origin of disease drastically changed patient self-perception.
Rather than being viewed as a foreign entity, illness suddenly assumes an esoteric and
mystical indwelling as part of the host organism, so that fighting illness is a struggle
waged against one-self. Coley’s discovery was roughly concurrent with Nixon’s war on
cancer—a federally-legislated struggle against an internal pathology—and the innovative
campaigns of Mary Lasker in her own political struggle against cancer.36
These
generously funded campaigns, however, did not live up to their potential because their
magnified intensity was severely limited in scope. Since then, research has broadened its
viewpoint in most aspects, approaching cancer as a multifaceted disease or even many
diseases confined within a shared category. Today’s cancer research even works in
tandem with the human genome project to “map” out a cancer cell’s genome, and
research is currently investigating the precise layout of the basal cell carcinoma genome.6
These systematic, top-down approaches will offer a more comprehensive understanding
of disease while simultaneously noting unique “fingerprint” aspects of cancers that are
individually unique. Rather than a monogenic disease, cancer is a body in cellular
disarray where cells are nonresponsive to their normal signaling components or are
overly stimulated to excessive growth.37
In carcinogenesis, normal protective functions

McQuade 10
involving cell division have been damaged or downregulated through dynamic molecular
factors. Based on this approach, similar tactics are being adopted for understanding
neurodegenerative diseases. The seminal observation that internal disarray of molecular
messengers and signaling factors is responsible for cancer is a prime corollary for AD.
Although all AD patients demonstrate characteristic features of the disease state, many
unique factors also emerge. This key aspect underlies the failure behind current drug
regimens, which typically treat external symptoms rather than addressing the root
molecular cause.38
Phospholipase A2 & Lipid Metabolism
Of all biological molecules, lipids are adept as energy-rich storage molecules,
forming lipid bilayers and micelles of cells, and are involved in many biochemical
cascades within the body.39
In fact, lipids are the body’s main energy source and (mole
for mole) pack more energy than their polysaccharide counterparts.40
However, the role
of lipids as mere storage molecules is growing rapidly antiquated due to recent research.
Lipids are critically important to overall health and wellbeing and that lipid composition
alone (both in cell membranes and plasma free fatty acids) is vital for health. In fact,
researchers have found that free fatty acids (FFAs) and their metabolites can even act as
secondary messengers to impact countless molecular interactions.41
Pathways involving
the normal metabolism of fats include the AA cascade and the ceramide pathway
(sphingomyelinase cascade).27
Taken together, these biochemical pathways are essential
for normal homeostatic conditions and are critically important in producing a wide
variety of signaling molecules. Imbalances in these cascades, however, can be
detrimental and even fatal in some cases. These two major cascades (the AA cascade and
the ceramide pathway) display frequent abnormalities in Alzheimer’s disease and other
neurological conditions.42
The arachidonic acid (AA) cascade is a highly complex
molecular signaling pathway that mediates a wide variety of bodily functions, using
lipids as its primary stimulation. The ceramide (or sphingosine) pathway involves the use
of COX and LOX enzymes for lipid metabolism.10
Thus, a more thorough investigation
of these pathways and their involvement in neuropathogenesis is crucially important for

McQuade 11
medicinal and therapeutic value. In particular, PLA2 and its role in the AA cascade will
be described in greater detail.
Phospholipase A2 (PLA2) is a critically important enzyme for the regulation of
lipid types within the body.43
Occurring in virtually all organisms, PLA2 is a ubiquitous
enzyme that exists in six major types: Secreted (sPLA2), cytosolic (Ca2+
-dependent),
calcium-independent, platelet activating factor acetylhydrolase (Groups VII and VIII),
lysosomal PLA2, and adipose-specific PLA2 (AdPLA2).44
Members of the PLA2 family
are organized according to disulfide bonding trends and their order of discovery.43
With
greater refinement of crystallography techniques in the 1970s, researchers made further
inroads into the precise molecular structure of sPLA2. Utilizing X-ray crystallography,
researchers uncovered an uncommonly high amount of cysteine residues in PLA2 (over
10% of its total amino acids), all of which contribute in disulfide bonding.45
Collectively,
PLA2 enzymes play vitally important roles throughout the body. Through modulating
lipid composition of phospholipid bilayers, controlling protein expression, and making
critically vital molecules for the body, PLA2 is intimately involved in a variety of bodily
functions.46
Recent research investigating lipid-signaling cascades has highlighted that
tight regulation of PLA2 is essential for proper bodily functions.21
This is accomplished
through chemical modification (phosphorylation) by MAPK (mitogen-activated protein
kinase) at Serine-505.45,47,48
Likewise, the presence of high levels of calcium ions (acting
simultaneously with phosphorylation of chemical groups) is thought to activate PLA2’s
enzymatic activity.46,21
The overall three-dimensional structure of sPLA2 is shown in
Figure 1. PLA2’s modulatory activity in the AA cascade is especially important for
preventing neurodegeneration and the creation of pro-inflammatory eicosanoid
molecules. The overall molecular structure of sPLA2 is shown in Figure 1. Observe the
N-terminal (positioned near the lower right corner) and the HI, H2, and H3 α-helices
(shown as long red coils).43
The β-wing (strangely absent in some plant species) is
depicted by antiparallel yellow arrows. The calcium-binding loop (shown in green) is
essential in binding and catalyzing substrate.22
Notice that sPLA2 consists of only one
polypeptide. The C-terminus is located near the top left of the diagram. Seven disulfide
bonds are shown in tan, which lend stability to PLA2’s overall structure. The two helical

McQuade 12
short turns (SH4 and SH5) are depicted as tight red loops. Interestingly, most aspects of
PLA2’s secondary structure (α helices and beta sheets) are conserved from one species to
another. This molecular homology underscores the extreme structural and functional
importance of PLA2’s secondary and tertiary configurations.
Arachidonic acid (AA) is a polyunsaturated fatty acid (PUFA) exhibiting a long, 20-
carbon skeleton with four cis double bonds (shown below in Figure 2). It cannot be
synthesized de novo in humans so it must be obtained exogenously from meats, eggs, and
other animal products, or synthesized from its precursor, linoleic acid. Under normal
circumstances, the body produces α-linolenic acid. Although some conversion does occur
to DHA, it does not occur readily.49
Therefore DHA is categorized as an essential fatty
Figure 1: The three-dimensional structure of
sPLA2. Notice that the α helices (shown in red)
and the β sheets (shown in yellow) are vital to the
protein’s overall conformation. As shown, the
cysteine residues are vital to the protein’s function.
Each cysteine contains a thiol group, which is
adept at bonding with other molecules and/or
protonating in solution. Additionally, thiol groups
possess the ability to form disulfide bridges—a
special biological function.

acid that must be obtained through diet. Fish and certain plant materials like coconut and
olive oils supply n-3 PUFAs.
expression, inhibit inflammation and oxidation, a
dynamics.49
AA is a fatty acid acyl chain with four
common fatty acids in the brain, AA has been linked to spatial learning, image
recognition, memory, and critical thinking skills. A study conducted Birch
demonstrated that infants (18
brain development compared to controls.
especially apparent in this study, as infants taking AA supplementation exhibited higher
thinking abilities than other infants of the same age.
(DHA) was provided to supplement AA, learning capabilities were accelerated even more
dramatically.50
PLA2, Acetyl-
metabolism and processing of AA and DHA. AA can be produced by linoleic acid
(18:2n-6), but the chemical reaction proceeds slowly in the brain although somewhat
faster in the liver.51
These molecules are involved in a series of interrelated pathways all
linked to a wide variety of biochemical activity.
McQuade
acid that must be obtained through diet. Fish and certain plant materials like coconut and
3 PUFAs.50
Researchers believe that DHA can modulate gene
expression, inhibit inflammation and oxidation, and correctly modulate membrane
AA is a fatty acid acyl chain with four cis double bonds. As one of the most
common fatty acids in the brain, AA has been linked to spatial learning, image
recognition, memory, and critical thinking skills. A study conducted Birch et al.
demonstrated that infants (18-months) receiving doses of AA demonstrated more rapid
ain development compared to controls.50
AA’s role in cognitive development became
especially apparent in this study, as infants taking AA supplementation exhibited higher
thinking abilities than other infants of the same age.50
When docosahexaneoic acid
(DHA) was provided to supplement AA, learning capabilities were accelerated even more
-CoA, and COX-2 mediators are all involved in the
metabolism and processing of AA and DHA. AA can be produced by linoleic acid
6), but the chemical reaction proceeds slowly in the brain although somewhat
These molecules are involved in a series of interrelated pathways all
linked to a wide variety of biochemical activity.
McQuade 13
acid that must be obtained through diet. Fish and certain plant materials like coconut and
Researchers believe that DHA can modulate gene
nd correctly modulate membrane
the most
common fatty acids in the brain, AA has been linked to spatial learning, image
et al.
months) receiving doses of AA demonstrated more rapid
AA’s role in cognitive development became
especially apparent in this study, as infants taking AA supplementation exhibited higher
ic acid
(DHA) was provided to supplement AA, learning capabilities were accelerated even more
2 mediators are all involved in the
metabolism and processing of AA and DHA. AA can be produced by linoleic acid
6), but the chemical reaction proceeds slowly in the brain although somewhat
These molecules are involved in a series of interrelated pathways all

McQuade 14
Arachidonic acid cascade—Lipid Catabolism & Inflammatory
Eicosanoids
As described previously, AA can be obtained exogenously through diet or through
chemical modulation of linoleic acid (which is also an omega-6 fatty acid).51
This occurs
through desaturation (addition of double bonds) and subsequent elongation of linoleic
acid’s hydrocarbon chain. Once produced, esterification then occurs to incorporate AA
into a plasma membrane phospholipid. Along with other 20-carbon fats like EPA and
DGLA, AA can form the hydrophobic tail of the phospholipid molecule, which remains
buried in the lipophilic portion of the membrane.52
Recently, researchers have discovered
that AA cascade is closely involved in inflammatory activity. When pro-inflammatory
environmental factors (exogenous) or endogenous factors are present, phospholipase is
stimulated to cleave the bond joining AA with the phospholipid.49
This liberates AA,
leaving it free for oxygenation to occur. Frequently, oxygenation of FFA is carried out by
cyclooxygenase (COX) molecules, which cleave two of FA’s alkene groups to add
oxygen atoms and increase overall saturation of the molecule. Collectively, these changes
produce a wide class of pro-inflammatory molecules called eicosanoids, which are used
as secondary messengers that signal inflammation and swelling within the body.53
As
Figure 3 depicts, COX enzymes produce thromboxanes (TX), prostaglandins (PGs), and
prostacyclins (PGIs). Since two of AA’s four double bonds have been cleaved,
eicosanoids produced in this way contain two double bonds and are classified as “series-
2” molecules.53
Notice that promiscuous enzymes called synthases are used in the
creation of new compounds. Cytochrome P450 is a heme-containing protein contained
within the mitochondrial inner membrane. It’s activity is crucial for the creation of
Figure 2: Arachidonic acid’s molecular structure.
As shown, AA has the ability in vivo to fold in on
itself, naturally decreasing its overall steric
hindrance and surface area. As a 20-carbon fatty
acid, AA is produced through carboxylation of the
16-carbon linoleic acid.

stimulatory hormones, cholesterol
bilirubin in the liver.54
leukotrienes (LTs).25
LTs are intimately involved in the 5
which activates LTC4, LTD4, and LTE4 (all three are classes of LTs that use cysteine
residues’ thiol groups for their enzymatic activity).
LTCs’ involvement in weakeni
murine models, neurons were systematicall
concentration). This was found to stimulate
dependent manner (shown below in
Figure 3: Production of pro
molecules from lipid cleavage by PLA
epoxides are created through the enzymatic
activity of CytP450 on AA. COX enzymes produce
PGs and TXs, which are present in various forms.
McQuade
stimulatory hormones, cholesterol and vitamin D production, and modification of
Lipooxogenase (LOX),
on the other hand, does not
tamper with the alkene bonds
and merely oxygenates the FA
molecule, ultimately producing
LTs are intimately involved in the 5-lipooxygenase (5LO) pathway,
LTC4, LTD4, and LTE4 (all three are classes of LTs that use cysteine
for their enzymatic activity).55
Further studies have confirmed
LTCs’ involvement in weakening of memory and learning abilities.56
In one study using
models, neurons were systematically exposed to Abeta1-42 oligopeptides (10
concentration). This was found to stimulate cysteinyl LTR expression in a concentration
below in Figure 4).
Production of pro-inflammatory
molecules from lipid cleavage by PLA2. As shown,
epoxides are created through the enzymatic
on AA. COX enzymes produce
PGs and TXs, which are present in various forms.
McQuade 15
modification of
Lipooxogenase (LOX),
on the other hand, does not
tamper with the alkene bonds
merely oxygenates the FA
molecule, ultimately producing
lipooxygenase (5LO) pathway,
LTC4, LTD4, and LTE4 (all three are classes of LTs that use cysteine
Further studies have confirmed
In one study using
oligopeptides (10 µM
concentration-

normal conditions, AA can exert negative genomic control on its own production.
When control of molecular messengers runs amok, however, AA often demonstrates
positive control to drive production of LTs and upregulate LOX and COX enzymes.
Discussion
Collectively, these data suggest that methods of modulating the AA cascade
essential for AD therapies.52,57
important part of lipid metabolism that must occur for regular homeostasis.
lipase activity is not only involved in controlling membrane lipid composition, but also
modulating inflammatory factors involved in the immune response. TXs
isolated from thrombocytes) and LTs (initially found in white blood cells) are key
components of the immunity and cell
Figure 4: LTRs stimulate amyloid
deposition. LTR signaling is frequently associated
with histamine and PG expression, which induce
further swelling and inflammation. LTRs, for
example, can stimulate smooth muscle of the
respiratory system, but excessive
is associated with asthma and allergic reactions.
McQuade
Besides chemical modulation
through its metabolites,
itself also exhibits
transcriptional control by
passing the nuclear membrane
and directly influencing
transcription factors.
rmal conditions, AA can exert negative genomic control on its own production.
When control of molecular messengers runs amok, however, AA often demonstrates
positive control to drive production of LTs and upregulate LOX and COX enzymes.
Collectively, these data suggest that methods of modulating the AA cascade
57 PLA2-mediated cleavage of membrane phospholipids
important part of lipid metabolism that must occur for regular homeostasis. It appears that
lipase activity is not only involved in controlling membrane lipid composition, but also
modulating inflammatory factors involved in the immune response. TXs (originally
isolated from thrombocytes) and LTs (initially found in white blood cells) are key
components of the immunity and cell-cell signaling.55
This finding suggests that AD
LTRs stimulate amyloid-beta plaque
deposition. LTR signaling is frequently associated
with histamine and PG expression, which induce
further swelling and inflammation. LTRs, for
example, can stimulate smooth muscle of the
respiratory system, but excessive LTR production
is associated with asthma and allergic reactions.
McQuade 16
Besides chemical modulation
through its metabolites, AA
also exhibits
transcriptional control by
passing the nuclear membrane
and directly influencing
transcription factors. Under
rmal conditions, AA can exert negative genomic control on its own production.57
When control of molecular messengers runs amok, however, AA often demonstrates
positive control to drive production of LTs and upregulate LOX and COX enzymes.
Collectively, these data suggest that methods of modulating the AA cascade are
mediated cleavage of membrane phospholipids is an
It appears that
lipase activity is not only involved in controlling membrane lipid composition, but also
(originally
isolated from thrombocytes) and LTs (initially found in white blood cells) are key
This finding suggests that AD

McQuade 17
therapies must specifically modulate these signaling cascades by allowing a homeostatic
amount of lipase-mediated cleavage to occur.46
The precise amount of PLA2 needed is
difficult to determine since it varies between individuals and is largely contingent upon
diet.50
As evident from the data presented in this section, a total paradigm shift is needed
to bring about substantially persistent results for AD patients. Future therapies must delve
to the root cause of AD by addressing the origins and subsequent development of the
disease. Despite exhaustive research, a foolproof medication has not been developed.17 It
is clear that abnormal PLA2 cleavage is a pivotal event for AD development and that the
rate of cleavage is essential for determining the fine balance between normal health and
AD development. Despite all the collective knowledge about AD, researchers are still
uncertain about its precise cause. Its effects (SPs and NFTs) can be easily noticed, but the
precise factors driving this disease still remain a mystery. There is a temptation among
researchers to develop therapies that target specific portions of the disease phenotype.
This position is untenable, however, since AD is a multifactorial illness, causing a
plethora of unbalanced activities within the body. Thus, it becomes evident that a therapy
that strongly impacts pathogenesis at a very early stage of illness is paramount. Precisely
what this therapy entails will be discussed in later chapters.
Chapter 2
Introduction
In addition to the AA cascade, a number of other mechanisms contribute to AD
ontology. The major players in this disease include overregulated secondary messengers
(such as PGs, TXs, and LTs), a specific kinase called glycogen synthase kinase-3β (GSK-
3β), and iron atoms that assault neural tissue.58
All these molecular factors are implicated
in stimulating aberrant secretase activity, upregulating signaling cascades and
extracellular plaque deposition, and accommodating neural toxicity.59
Originally named
for its ability to regulate glycogen synthase (a key enzyme in gluconeogenesis), GSK-3β
is a kinase enzyme present as two different forms (α and β) that has been implicated in
countless bodily processes.60
Under normal circumstances, it controls energy distribution,

McQuade 18
glycogen formation, and establishing the overall body plan during fetal development.61
Interestingly, GSK-3β is also involved in neuronal differentiation and its dysfunction is
strongly linked to down syndrome (DS) and Parkinson’s disease (PD).2 Researchers
testing pre-neuronal cells in murine DS models and DS human embryos found that GSK-
3β function was increased due to lowered phosphorylation of the enzyme.2 By exhibiting
a third copy of the AICD portion of the APP gene, DS leads to significant problems with
enzyme expression.59
Administration of lithium, however, was found to increase
phosphorylation to normal levels. This finding may lend credence to the role of lithium
therapy in treating AD. This is discussed in greater detail in “Lithium: A Curative
Agent?” In this chapter, GSK-3β’s involvement in AD will be examined as well as the
varied roles of secondary messengers and the neurotoxic effects of iron in driving AD
progression. The role of lithium as a therapeutic will be evaluated and determined in
chapter 3.
Secondary Messengers, GSK-3β, and the Role of Iron
AA, PGs, and LTs are all secondary messengers that relay chemical signals
throughout the body. Dysfunctions involving AA have been linked to many pathologies
and chemical reactions that involve PUFAs. Recent research suggests that PUFAs like
AA and DHA are deeply involved in membrane dynamics as well as influencing the
enzyme conformation and activity, regulating protein expression, and produce vitally
important products for use in the cellular environment. These fats are called “essential”
because they cannot be produced de novo, but rather must be obtained from an exogenous
source.53
Of all body organs, the brain has an exceptionally large amount of long-chain
PUFAs (LCPUFAs). Collectively, AA and DHA occupy over 90% of fats in the brain,
occupying over half of its non-aqueous mass.62
In the initial stages of this cascade, PLA2
is activated to cut phospholipids in the sn-2 position, liberating AA. PLA2 is upregulated
when G-protein coupled receptors (GPCRs) are occupied or when NMDA receptors are
activated by glutamate to allow for entry of calcium.28
In a widely used, generic form of
signal transduction, G-proteins activate adenylyl cyclase (AC), which drives cAMP to
interact with protein kinase A (PKA). Once activated, PKA is able to phosphorylate

McQuade 19
nearby enzymes to upregulate their activity.63
Once released, AA can either be
reincorporated back into plasma membranes or can undergo further reactions to form a
wide class of eicosanoid molecules. These include prostaglandins and thromboxanes (via
COX-2); epoxides (via cytochrome P450); leukotrienes (via lipoxygenases); or
hydroperoxy acids (via autooxidation).57
Although the precise method of conversion is
poorly understood, researchers found that AA can also be converted to anandamide.64
Collectively, the production of LTs and COX lead to underexpression of DHA, which is
important for brain vitality. In fact, multiple studies suggest that DHA may prevent
oxidation.65
Although several trials have shown that supplemental DHA seemed to
encourage oxidation in vitro instead of impeding it, most other research shows DHA’s
positive effect in counteracting inflammation.62
DHA may encourage oxidation in some
organisms due to its high levels of unsaturation, but in humans most studies have shown
beneficial results.66
Since DHA is useful in treating asthma, researchers speculate that it
might favorably impact the cPLA2 cascade and act to inhibit inflammation. Several in
vitro studies revealed that AA is still produced even in the presence of DHA, suggesting
that it is not effective in halting the cPLA2 signaling pathway. DHA and eicosapentaenoic
acid (EPA; n-3 C20:5) both stimulate production of docosatrienes and resolvins, which
both aid in neural function.1 Most notably, DHA is converted to neuroprotectin D1
(whose production can also occur via sAPPα).46
In addition to PKA, another kinase called GSK-3β exhibits regulatory activity
through excessive phosphorylation of presenilin-1.67
Researchers found abnormally
increased levels of this kinase within AD brains. Through its enzymatic control of
presenilin-1, it is closely linked to secretase cleavage of APP and the production of Aβ
oligopeptides.27
This enzyme plays a key role in the insulin signal transduction pathway
and AD symptoms develop when this system runs awry.68
In fact, recent research
suggests an increasingly close connection between type II diabetes and AD.69
A down-
regulated response to insulin is associated with biochemical changes in the
phosphatidylinositol-3 (PI3) kinase pathway. Neural tissue carries receptors for insulin
and these are regulated through the PI3 kinase pathway and by mitogen-activated protein
(MAP) kinase pathway.68
These pathways are highly dynamic and are chemically
regulated on a microsecond basis. Researchers found, for instance, that Akt/PKB

McQuade 20
activates the glucose transporter (GLUT) 4 to change its location on the neurolemma
plasma membranes.70(p4)
Akt/PKB also negatively impacts GSK-3 through the addition of
phosphate groups at the serine 9 position. Interestingly, insulin receptors (IRs) in neural
tissue are not only involved in meeting the brain’s need for sugar, but are also closely
involved with memory and understanding new concepts.68
The rate of APP cleavage and
tau phosphorylation is closely modulated by IRs. Collectively, these data indicate a closer
correlation between type II diabetes and AD than was previously assumed. Glucose
uptake by neuronal cells is critically important: on average, the brain consumes an
estimated 80% of ingested glucose.66
Finding efficient methods of controlling the MAP
and PI3 kinase signaling pathways is critically important in finding a cure for AD, since it
may also impact regulation of secretase activity. Results are currently conflicted
regarding concentrations of PI3 kinase: some AD brains demonstrate excessive PI3,
while others show a deficit of PI3 kinase.71
Disruption of these pathways causes
increased lactate production and lower ATP production because of disrupted glycolytic
pathways. In one significant study, steptozotocin (STZ) was administered in sufficient
amounts to cause onset of diabetes mellitus in murine test subjects.72
Interestingly, mice
administered STZ developed disruptions in short-term memory (STM) and several
behavioral/social alterations as well. These findings highlight the critical roles of insulin
and IRs for neural health, deepening a possible connection between diabetes and AD.41
These data show that IRs’ collective response to glucose impacts the nervous system
tremendously. Further research into neural IRs and their role in STM is essential for
discovering the connection between diabetes and AD.
Iron is another factor suspected in the ontology of AD.4 When APP is transcribed
to form mRNA, it can then be translated into an amino acid sequence. When iron is
present in large amounts in the brain, it over stimulates translation of the mRNA through
iron-responsive factors that are especially sensitive to iron stimulation.4 Therapeutics are
desperately needed to address the potentially harmful role of excess iron in CSF and
surrounding neural tissue. Multiple studies demonstrate that iron accumulation occurs
rapidly in neural tissue and can encourage Aβ plaque production. Iron and calcium levels
within the cell were increased when APP was overexpressed. The precise molecular
details behind this process still remain unknown. It is suspected, however, that metals

McQuade 21
already present in the extracellular environment are ingested and interact with APP.
Researchers also discovered lowered superoxide dismutase (SOD) concentrations in cells
with disregulated APP. This is probably due to the overexpression of metal-transporting
proteins. Contrary to expectations, transferrin was found to play a minimal role in this
process. Iron controls APP functions within the cell. It shares many features for how iron
response element (IRE) regulates the translation of ferritin L- and H mRNAs using its 5’
UTRs.
Sphingomyelinases, the Ceramide Pathway, & Soluble Aβ--
Pathogenesis of Inflammation
Ceramides are a class of fatty compounds made of sphingosine and a fatty acid.
Present in abundant amounts within the phospholipid bilayer, recent evidence has found
that ceramides are not merely building components of cells but also participate in a wide
variety of cellular activities.73
This activity ranges from differentiation, division, and
apoptosis. Ceramides (literally “waxy amides”) are created through several different
pathways, one of which is through enzymatic (SMase) breakdown of sphingomyelin.
Interestingly, ceramide production upregulates SOCS3, a gene that suppresses kinase
signaling. This leads directly to leptin and insulin insensitivity, further establishing the
diabetes-AD correlation.74
Low oxygen, reactive oxygen species (ROS), and UV
radiation are all factors that increase SMase activity, leading to the creation of ceramides.
When neural tissue is lacking sufficient oxygen, excessive ceramide production was
noticed in astrocytes of the hippocampus.68
This is most likely a response to swelling
caused by ischemic conditions.74
nSMases are mainly located in the phospholipid bilayer, while aSMases are found
in endosomal-lysosomal portions of the cell.75
Researchers hypothesize that nSMases
(especially nSMase2) are involved in AD pathogenesis. nSMase consists of a phosphate
group covalently bonded to a protein that contains five serine residues (S173, S208,
S289, S292, S299) that are remarkably consistent between species. In the presence of
certain stress stimuli, these residues can be phosphorylated to activate the protein.74
Tumor necrosis factor (TNF)- α, IL-1beta, and IL-6 are all secondary messengers

McQuade 22
implicated in stimulated ceramide production. The AA cascade joins the ceramide
pathway (which is highly activated in AD patients) through the interaction of these ILs.73
Researchers noticed major changes in sphingolipid pathways of patients displaying
neurodegenerative disorders.52
Plasma levels of these enzymes were determined by
increased levels of SMase and acid ceramidase, resulting in higher ceramide and
sphingosine concentrations.76
Both of these are secondary messengers are intimately
involved in cell maturation and death. Both ceramide and sphinogosine, for instance, can
activate caspases 3 and 9 that trigger apoptosis. It is thought that Aβ upregulates neutral
and acid SMases via cPLA2 and AA.77
Both kinds of SMases provide structural support
for BAC-1, which cuts the β-site of APP. Lowered amounts of SPs in Alzheimer’s
patients have been shown to prevent Aβ activity, producing a beneficial neurological
effect.8
Under normal conditions, AA regulates sphingomyelinase (SMase) function.75
In
AD pathology, however, Aβ peptides activate SMases (both acidic and neutral forms) to
induce neural apoptosis. Likewise, antioxidants inhibit SMase by blocking the activity of
cPLA2 or by targeting specific genes using antisense oligonucleotides.73
Sphingosine-1-
phosphate protects the brain by preventing stimulation of acidic sphingomyelinase. Many
researchers propose that AD begins with small modifications to the hippocampus as
insoluble plaques build up in the extracellular environment. This causes problems with
cell-cell signaling through synapses, ultimately causing neuronal death. This theory falls
short, however, when one realizes that hardly any correlation exists between dementia
and plaque formation. Also, recent research suggests that neuronal apoptosis is linked to
soluble concentrations of Aβ peptide oligomers, rather than the fibrous, insoluble kind.27
Using sections of mouse cerebrum, researchers determined that soluble Aβ oligomers are
toxic to the CA1 region of the hippocampus.68
Several additional studies created
transgenic murine models lacking plaque accumulation. Even in the absence of neural
plaques, these mice still developed neuropathies and learning problems.78
Collectively,
these studies lend credence to the theory known as the “soluble Aβ” hypothesis.79
Sphingolipids (ceramides) and their metabolic products are extremely important aspect of
this hypothesis. Ceramide is made through two major methods of production. It can either
be made through the breakdown of sphingomyelin by neutral sphingomyelinase (N-

McQuade 23
SMase) and acidic sphingomyelinase (A-SMase), which degrade sphingomyelin or
manufactured through ceramide synthase.73
This signal cascade is present in nearly all
cells, but ceramide is present in abnormally high concentrations in AD patients and
sphingomyelin amounts were low. This suggests that metabolism of sphingolipids and
production of cereamide leads to AD progression. Soluble Aβ leads to cell death because
of the cPLA2 pathway that makes AA. Also, excessive oxidation of fats, nucleic acids,
and proteins has been linked to damage caused by soluble and fibrous Aβ peptides.24, 26
In one significant study, immunoblot analysis and SDS-PAGE were used to
determine the histological location of Aβ peptides in vitro. After this, MAFP (methyl
arachidonyl fluorophosphate) was used to inhibit cPLA2. By stopping the activity of
cPLA2, AA liberation was prohibited. Through this study, the researchers found that
excessive production of AA is harmful to neurons of the cortex by stimulating SMase
activity. By using an MTT assay, the study found an inverse relationship between
arachidonic acid concentration and neuronal viability.13
Also, neurons exposed to AA
longer suffered more toxic effects. Neurons exposed to 5 µM of AA, for instance,
demonstrated an upregulation of N- and A-SMase activities even after only one hour of
incubation.80
Maximum SMase production occurred after 6 hours of exposure.
Desipramine and 3-O-methyl-sphingomyelin (3OMeSM) inhibit SMases and these
stopped apoptosis caused by AA. Results were also tested using different fatty acids
others than AA (stearic acid, oleic acid, and linoleic acid). Interestingly, the introduction
of these fatty acids did not lead to apoptosis or upregulation of SMase activity. These
results are supported by a similar study (Florent et al.) showing that docosahexaenoic and
eicosopentaenoic acids did not induce apoptotic activity in neurons.49
In fact, the results
suggested that these additional fatty acids were helpful for neuronal activity, while Aβ
oligomers may induce A- and N-SMase production. Antisense oligomers, Aβ (40-1),
were used as a control and did not induce SMase production. 3OMeSM and N-acetyl
cysteine (NAC) inhibit SMase and helped preserve neuronal integrity. Desipramine and
imipramine both inhibit A-SMase and also lead to lower rates of cell death. Strangely,
however, 3OMeSM and desipramine both encouraged cell death.73
This finding puzzled
researchers and still cannot be properly explained. Fumosin B1 inhibits ceramide
synthesis and inhibition of serine-palmitoyl transferase (a gateway enzyme for creating

McQuade 24
sphingolipids) caused no changes in rates of cell death. From this data, researchers
concluded that prevention of SMase activity greatly benefitted neuronal function by
lowering caspase-3 and caspase-9 activity (caused by soluble Aβ peptides).75
Amazingly,
1 µM concentrations of soluble Aβ peptides caused a 51% lowering of cell livelihood
after 24 hours of incubation. 3OMeSM and desipramine lowered soluble Aβ1-42-induced
apoptosis to 75 and 81%. Also, a study found that ceramide utilizes cathepsin-D (which is
manufactured by lysosomal A-SMase).27
When cathepsin-D was inhibited by an
octapeptide, this proved highly beneficial to neurons. Thus, overexpression of the
ceramide pathway (and associated overproduction of N- and A-SMases) are detrimental
to neural health and can stimulate development and progression of AD.
Genetics & Environmental Risk Factors
The risk of AD and cerebrovascular disease (CVD) is highly increased with the
possession of Apolipoprotein ε4 (APOE4) gene. Strokes are often a common side affect
with ApoE4 because CVD usually exhibits a phenotypic impact (like a cerebral
infarction) that cannot be noticed clinically. Unfortunately, many cerebral infarctions
remain unnoticed and can lead to strongly detrimental effects like stroke.81
Interestingly,
researchers have correlated white matter hyperintensities (WMH) to stroke incidence. For
individuals older than 65-years-old, AD is the fourth leading cause of morbidity. In the
initial stages, AD first affects thinking and memory and later muscle control.14
Hypertension, diabetes, smoking, high blood homocysteine, and previous strokes are all
thought to increase chances of AD onset.82
“Late-onset AD” is more common and more
closely corrlated to APOE4. “Early-onset AD” is correlated to a set of three genes. The
first codes for β-amyloid precursor protein (APP) of chromosome 21.42
The second codes
for presenilin-1 (PS1) on chromosome 14.77
The third codes for presenlin-2 (PS2) on
chromosome 1, which aids in presenilin degradation. These genes drive upregulation of
astrocytes and microglia, ultimately stimulating AD onset. Interestingly, demographic
studies have noticed AD’s infrequence in India and other parts of Asia. It is often debated
whether this is a result of diet or is genetically linked and/or cased by some other

McQuade 25
unnoticed factor. Genomic testing reveals that the ApoE4 gene is extremely low in India.
Because of their genetic variability, most Indian AD patients demonstrated differences in
disease progression, showing lower levels of oligometric Aβ1-42 peptides than their
American counterparts, as well as less heavily phosphorylated tau. These studies still
remain unclear and further testing is needed to determine the importance of these studies
for current Alzheimer research.
Impact of Neuroinflammation
Although tau aggregation and SP deposition are important in disease pathology,
neural inflammation seems to be the driving factor. This is hypothesized to occur through
excessive stimulation of AA and the production of pro-inflammatory eicosanoids
mentioned earlier. In one significant study, researchers used spontaneously hypertensive
rats (SHRs), which are characterized by hypertensivity, diabetic symptoms, and other
metabolic disorders. SHRs were chose because of their propensity to create eicosanoid
molecules, while Wistar-Kyoto (WKY) controls exhibited normal blood pressure and
produced fewer inflammatory factors. SHRs were found to demonstrate a more rapid
progression of AD, most likely due to widespread microglial activation. Microglial are a
special type of phagocytic cell within the nervous system that is stimulated to consume
threats to homeostasis. In the presence of inflammatory stimuli, microglia are alerted and
produce a variety of cytokines. Unfortunately, in the SHR rats this only lead to further
stimulation of tissue swelling. Under inflammatory stimuli, mitogen-activated protein
kinase (MAPK) is hyper-activated, leading to increased NFTs. This overall process is
shown below in Figure 5.

studies show that NFTs stimulate inflammation, while others suggest that pro
inflammatory cytokines precede disease onset.
demonstrated these results using tau transgenic (Tg) mice.
questions was whether inflammation or neural necrosis is the driving force behind AD.
Researchers found that abnormal tau expression may be caused by cytokine
Figure 5: Tau aggregation in AD neural tissue.
Abnormal tau expression leads to microglial
activation, which contribute to neuronal death.
Notice that the microglia clump together in
response to chemokine signals from neighboring
cells.
McQuade
Results are currently
conflicted regarding
inflammation’s role in
neurodegeneration. Some
studies show that NFTs stimulate inflammation, while others suggest that pro
inflammatory cytokines precede disease onset.12
A similar study (Yoshiyama
demonstrated these results using tau transgenic (Tg) mice.20
One of the researchers’ key
questions was whether inflammation or neural necrosis is the driving force behind AD.
Researchers found that abnormal tau expression may be caused by cytokine
Tau aggregation in AD neural tissue.
Abnormal tau expression leads to microglial
activation, which contribute to neuronal death.
Notice that the microglia clump together in
response to chemokine signals from neighboring
McQuade 26
Results are currently
licted regarding
inflammation’s role in
neurodegeneration. Some
studies show that NFTs stimulate inflammation, while others suggest that pro-
A similar study (Yoshiyama et al.) also
One of the researchers’ key
questions was whether inflammation or neural necrosis is the driving force behind AD.

McQuade 27
overproduction, a phenomenon that Zilka et al. have creatively called “cytokine storm.”19
When brain cells are damaged through ROS, for example, they can stimulate over-
activation of microglia, which upregulate their phagocytic activity to form large clusters
in the nervous system. This triggers an immune system response, which crowds blood
plasma with macrophages.83
Strangely, researchers found that immune system activation
can actually accelerate tau aggregation—ironically stimulating the very process it was
striving to prevent. By manipulating the fractalalkine-receptor for microglia (CX3CR1)
and the ligand for microglia (CX3CL1), researchers hoped to find a method of opposing
immune-stimulated neuroinflammation.19
Although extracellular levels of SPs decreased
in CX3CR1 knockout mice, intracellular tau protein concentrations increased.19
Hence,
Zilka et al’s study demonstrated mixed results: inhibition of the receptor lowered
insoluble plaques, but lead to an increase in NFTs. Although largely unsure why this
occurred, Zilka et al. concluded that their data supports the hypothesis of inflammation-
induced AD.19,74
This is certainly a matter of debate that cannot be conclusively decided;
however, the most current data suggests that neurodegeneration and the inflammatory
cascade can be largely attributed to the actions of the esterase PLA2 and its enzymatic
functions in the AA.45
Discussion
More than any other single finding, GSK-3β’s effect on neural insulin receptors
underscores the relationship between diabetes and AD. The precise molecular factors that
link these two illnesses must be researched in greater detail. How caloric (and sugar)
intake affects the metabolism of membrane lipids will be especially pertinent to this area
of study. Also, cholinergic receptors (typified by a positive response to acetylcholine) are
prime targets for future therapies. Finding methods of artificially stimulating these
receptors (especially through Galantine) may prove promising and must certainly be
studied in greater detail. Any potentially harmful effects of Galantine overstimulation
must be researched in greater detail. As a majorly activated cascade in AD, the ceramide
pathway must be targeted by a therapy that effectively addresses the disease. In
particular, SMases must be brought to normal plasma concentrations and ceramide
recruitment of caspase-3 and -9 must be inhibited. Also, although SPs and NFTs are a

McQuade 28
major issue, neuroinflammation seems to be a deeper problem. Research seems to
indicate that inflammatory signals are the motive driving force behind plaque deposition
and NFT accumulation.84
Therapies that specifically impede internal inflammation by
inhibiting enzyme overactivity (and the related “cytokine storm”) are sorely needed.
Also, patients who have suffered a stroke must be subjected to preemptive treatments to
halt pathogenesis and monitor the development of WHM. The immune-mediated
response of microglia is not intrinsically detrimental, but overactivated glia leads to
apoptotic activity.85
Following this theme, chapter 3 will present various current therapies
used to address AD symptoms. Finally, the therapeutic efficiency of turmeric will be
noted in comparison to other treatments.
Chapter 3
Introduction
Drawing from copious amounts of data, researchers are hoping to form a
constructive framework for an increased understanding of AD pathogenesis and
development. By direct chemical modulation of the AA and ceramide cascades (and
interception of the secondary messengers involved), researchers hope to design effective
drug regimens for AD patients. Thus far, the search for a cure has proven only
moderately successful. Due to the body’s complex molecular interchanges and formation
of intermediary structures, development of effective therapies with minimal side effects is
an extremely difficult endeavor.84
COX-2 inhibitors are a subject of special interest for
researchers, since modulation of these oxygenase molecules will prove vitally useful in
treating AD.86
However, as this chapter shows, even the most carefully concocted
therapies fall short of their desired intent. This failure is due mainly to ineffective
metabolism, misdirected tissue targeting, or through complications caused by adverse
effects. As discussed in this chapter, rofecoxib and celecoxib both exhibit therapeutic
promise, but are no longer widely used because of their potential fatality.87
This chapter
will examine many common pharmaceutical regimens used for AD patients, their
mechanism of action (if known), and their comparative success. Also, the cholinergic

McQuade 29
receptor hypothesis will be discussed relative to AD and a brief evaluation of how
different drug classes influence brain activity will be presented. Finally, the need to
incorporate turmeric into Western medicine will be presented and curcumin’s influence
on body systems will be examined in greater detail. Although not exhaustively
researched, turmeric’s medicinal benefits can be strongly inferred from numerous studies.
These studies indicate that turmeric offers a therapeutic remedy to replace ineffective
COX inhibitors, NSAID drugs, and other compounds like memantine.10,88
Searching for a Cure—Inhibitors and Molecular Intervention
Current medical treatments recognize the extreme importance of decreasing
inflammation, since it causes (or at the very least aggravates) neurodegeneration. Also,
treatments that address neurotoxins (such as rising iron concentrations and
overexpression of Aβ) are essential for treatment of AD. Current treatments seek to
address these issues using a variety of methods. First, there are molecular inhibitors that
specifically antagonize the activity of particular enzymes. COX-2 inhibitors are perhaps
the most widely known of this category. Cox-2 inhibitors (rofecoxib, celecoxib, and
others) have been used therapeutically, but exhibit less than desirable results (in some
cases even causing death). Likewise, glucocorticoids like prednisone have demonstrated
no observable improvements for memory, learning, and social interactions.89
COX-1 has
been shown to cause some inflammatory activity but it is also highly involved in
digestion, so its activity cannot be inhibited without causing gastrointestinal problems.19
Although NSAIDs were found to reduce swelling, they were inefficient at reducing Aβ
plaques.10
The inefficiency of most pharmaceutical drugs is due to a great deal of
educated guesswork and trial-and-error. In many ways, the drug design process itself is
experimental, based upon hypothesis and tweaking of the final drug product based upon
experimental results.90
Up to this point, these therapies have yielded highly discouraging
results. The most efficient therapy would effectively address swelling, reduction of
neuritic plaques, downregulation of tau protein, and normal regulation of PLA2. Once
these issues are addressed, AD can be effectively controlled. Also, an effective drug

McQuade 30
regimen for AD would address genomic regulation, which is often the source of enzyme
dysfunction.
Lithium—A Curative Agent?
Since the 1950s, lithium has been advanced as a neurological therapeutic for
mood and general neural homeostasis. Lithium has a long history of therapeutic use for
neurodegenerative diseases. Originally used to treat gout in the 1840’s, lithium has also
been clinically administered to bipolar patients for over 50 years.91
It has also been used
as an adjunct for blood pressure medication. The precise molecular mechanism of lithium
within the brain is poorly understood and it was prohibited after demonstrating negative
side effects, including the deaths of four patients.92
Many researchers hope it may prove
effective in treating neuropathies like AD. Lithium—originating from the Greek “Lithos”
(stone) since it was originally found in rocks-- was first discovered by Arfwedson, a
Swedish chemist, in 1817.93
Lithium exists as a positively-charged monovalent atom that
is interchangeable with other cations like potassium, sodium, and even divalent atoms
like magnesium and calcium.61 In the laboratory environment, lithium acts catalytically
on uric acid in the kidneys, although significant amounts are needed to induce these
results in vivo.94
By the 1880s, it was clinically prescribed for chronic depression and
other psychological abnormalities. By 1940, physicians realized a correlation between
sodium-rich diets and hypertension, and lithium was used to substitute sodium chloride as
a treatment for blood pressure.30
But in 1949, Corcoron et al. showed that lithium could
cause widespread tissue damage and even death, and its use as a therapeutic was
prohibited across America.95
This large-scale discrimination against lithium began to fade, however, when a
study conducted by Cade noted the sedentary effect of lithium on guinea pigs.91
Cade
theorized that mental problems were caused by a hitherto unrecognized poisonous agent,
which was then excreted in urine. In one notable study, Cade injected lithium urate into
guinea pigs, causing them to become sedated. Lithium chloride is the only dissolvable
salt in uric acid, which lead to sedation. This study reinvigorated the therapeutic use of
lithium carbonate in the treatment of neurological diseases. Interestingly, lithium

McQuade 31
carbonate showed no effect in schizophrenia or depression, but it was effective in all ten
manic patients in one study.93
After these hopeful results, lithium chloride became the
first FDA-approved drug for bipolar disorder.96
The practice of prescribing lithium may
have historical precedent. In the 5th century A.D., Caelius Aurelianus recommended
alkaline water for mentally disturbed patients, which may have contained high
concentrations of dissolved lithium ions.91
At this point, research remains inconclusive
and further studies must be done to determine the precise mechanisms of lithium’s
clinical efficacy. However, recent studies have uncovered the chemical interactions
involved in lithium’s usefulness as a therapeutic. Recently, lithium has been shown to
impact adenylyl cyclase (AC), which is an important cellular modulator for countless
chemical reactions.61
Both cyclic adenosine monophosphate (cAMP) and AC modulate
the effects of cAMP in the body for countless biological reactions. AC transforms ATP to
cAMP by way of a multistep process. Then cyclic nucleotide phosphoidiesterase changes
cAMP to AMP.95
This is a common event in signal transduction pathways, followed by
subsequent phosphorylation of specific protein targets. Protein kinase A (PKA) is
involved in this process through the removal of phosphate molecules.91
Lithium
upregulates cAMP and PKA activities by increasing enzyme binding efficiencies. Thus,
lithium serves as a metal catalyst that improves reaction conditions and accelerates the
reaction. Mori et al. found that lithium prohibits the transfer of phosphate groups by PKA
in microtubule sections of rat brains. This is accomplished when lithium vies with
magnesium at the C subunit of PKA.
Recent research suggests that lithium may play crucial roles in protecting neurons
and may be therapeutically valuable in treating AD, Parkinson’s, Huntington’s disease,
Amyotrophic lateral sclerosis, and ischemic conditions. Also, it can block N-methyl-D-
aspartate (NMDA) receptors and helps neuroregulatory proteins. As one of the first
proteins discovered that regulates apoptotic activity, B-cell lymphoma/leukemia-2 (Bcl-
2) gene can antagonize potential hazards like absence of growth factors, radiation,
glucocorticoid hormones, and oxidative agents like hydrogen peroxide.97
Bcl-2
upregulates caspase and increases mitochondrial calcium influx. Lithium causes higher
Bcl-2 levels in the cortex, hippocampus, and striatum and also lowers concentrations of
protein 53, which normally induces apoptosis.97
It also stimulates Akt, a class of protein

McQuade 32
kinases that specifically utilizes serine and threonine and is controlled by the
phosphatidylinositol kinase molecular cascade.97
GSK-3β prevents overproduction of β-
catenin by degrading it over time. Long-term administration of lithium increases β-
catenin in murine models. AD is frequently associated with misreadings in presenilin
protein, which leads to lower levels of β-catenin. This is linked to apoptosis by Aβ plaque
accumulation. Presenilin-1 joins with β-catenin, leading to a lower energy state. Low
levels of GSK-3α are directly correlated to Aβ concentrations, whereas GSK-3β produces
the inverse effect. Prevents Aβ being made from APP. A study by Feyt et al., however,
offers conflicting data to these studies involving lithium as a therapeutic agent for AD.97
Thus, although lithium should be considered a viable candidate in treating AD, much
more research is needed to prevent detrimental effects. Lithium may be incorporated as a
therapeutic, but only after plentiful research has been conducted to verify its beneficial
effects while preventing neurotoxicity.
New Frontiers-- Memantine & NSAIDS for AD Therapy
Cholinergic neurons are characterized by their responsiveness to acetylcholine
and upregulating cholinergic neuron activity is a prime target for therapies. Molecules
that antagonize cholinesterases are the most efficient type of inhibitor used because they
prevent the enzymatic deconstruction of cholinergic receptors.98
Through conservation of
these receptors, acetylcholine is encouraged to remain within the synaptic cleft and
participate in vital neural functions. Galantamine, Rivastigmine, Donepezil, and Tacrine
are four of the most commonly used compounds to prevent cholinesterase activity. Drugs
that exhibit this type of activity are currently the only officiated drug for AD.98
As AD
progresses, however, these treatments grow less potent, gradually losing their efficiency
over time. It is hypothesized that cholinesterases gradually lose their responsiveness to
treatments or find methods of avoiding the intended therapy. Thus, although
cholinesterase inhibitors are headed in the right direction, they are not a viable, long-term
solution. NMDA inhibitors are also a topic of interest. Memantine, in particular, is
especially effective through its prohibition of excessive AA release.88
The process of
synthetic memantine production is shown below in Figure 6. Although memantine may

prove promising, researchers are concerned that it may prove too constricting on AA
production.88
commonly used NSAID, is now prescribed for a variety of ailments. Now it is often taken
to preemptively stop the occurrence of strokes and myocardial infarctions.
commonly utilized drug in Western medicine, aspirin is useful for a variety of activities,
including reducing fever, pain, and inflammation. Multiple studies suggest that aspirin
taken in combination with DHA may prevent the pro
When placed in the same chemical environment as omega
production of resolvins, protectins, and oxoderivatives that collectively protect
neurological tissues.99
AD is often associated with vascular dementia (VaD), which is typified by abnormal,
undetectable growth of the brain’s capillaries.
often remains undiscovered clinically. In fact, VaD phenotype is so internalized that
autopsies are the most frequent method
display inflammation as a normal sign of aging; unchecked, however, inflammation can
quickly turn pathological over time. As plasma cytokine concentrations rise, astrocytes
Figure 6: The synthetic production of memantine. As a
therapeutic used in AD treatment, memantine is an incredibly
important compound.39,70 Once obtaining the starting reactant,
its three-step laboratory synthesis is relatively straightforward.
The final production of HCl (shown beside the memantine
product) is also medicinally useful in low doses.
McQuade
prove promising, researchers are concerned that it may prove too constricting on AA
Nonsteroidal anti-inflammatory
drugs (NSAIDs) are compounds
meant to counteract AD
pathology. Aspirin, the most
commonly used NSAID, is now prescribed for a variety of ailments. Now it is often taken
to preemptively stop the occurrence of strokes and myocardial infarctions.19
commonly utilized drug in Western medicine, aspirin is useful for a variety of activities,
including reducing fever, pain, and inflammation. Multiple studies suggest that aspirin
nation with DHA may prevent the pro-inflammatory activity of AD.
en placed in the same chemical environment as omega-3 PUFAs, aspirin leads to the
production of resolvins, protectins, and oxoderivatives that collectively protect
AD is often associated with vascular dementia (VaD), which is typified by abnormal,
wth of the brain’s capillaries.81
This occurs early on in the disease and
overed clinically. In fact, VaD phenotype is so internalized that
utopsies are the most frequent method of VaD detection.100
Elderly patients often
ation as a normal sign of aging; unchecked, however, inflammation can
turn pathological over time. As plasma cytokine concentrations rise, astrocytes
The synthetic production of memantine. As a
therapeutic used in AD treatment, memantine is an incredibly
Once obtaining the starting reactant,
step laboratory synthesis is relatively straightforward.
The final production of HCl (shown beside the memantine
product) is also medicinally useful in low doses.70
McQuade 33
prove promising, researchers are concerned that it may prove too constricting on AA
inflammatory
SAIDs) are compounds
meant to counteract AD
Aspirin, the most
commonly used NSAID, is now prescribed for a variety of ailments. Now it is often taken
As the most
commonly utilized drug in Western medicine, aspirin is useful for a variety of activities,
including reducing fever, pain, and inflammation. Multiple studies suggest that aspirin
inflammatory activity of AD.99
3 PUFAs, aspirin leads to the
production of resolvins, protectins, and oxoderivatives that collectively protect
AD is often associated with vascular dementia (VaD), which is typified by abnormal,
This occurs early on in the disease and
overed clinically. In fact, VaD phenotype is so internalized that
Elderly patients often
ation as a normal sign of aging; unchecked, however, inflammation can
turn pathological over time. As plasma cytokine concentrations rise, astrocytes

McQuade 34
swell in a phenomenon known as astrogliosis.74
Found in the central nervous system
(CNS), astrocytes are star-shaped cells that participate in a wide variety of reactions, such
as providing structural support and nourishment to neurons. They also constitute the
lining of the blood-brain barrier (BBB), exchange substances between neural parenchyma
and blood plasma, communicate through surface contact with other neurons, increase
synapse connections, and facilitate signal transduction from one neuron to another.75
Astrogliosis stimulates a vicious cycle where inflamed tissues release more pro-
inflammatory molecules, leading to deadly results.
Receptors in the brain are divided into two major categories: nicotinic and
muscarinic. Nicotinic receptors are typified by ligand-gated ion channels embedded in the
neurolemma and at the neuromuscular junction. Both receptor types are cholinergic,
meaning that they respond to acetylcholine to propagate a signal along the axon to a
neighboring neuron, or to directly stimulate muscle contraction.98
Although both are
responsive to ACh, they are differentiated by their response to other compounds. For
instance, nicotinic acetylcholine receptors (nAChRs) are characterized by their response
to nicotine, while muscarinic acetylcholine receptors (mAChRs) are stimulated by
muscarine-- a substance originally found in the poisonous mushroom Amanita
muscaria.98
mAChRs are G-protein coupled receptors (GPCRs), which is the starting
point for the types of signal transduction described earlier. nAChRs are severely depleted
in AD. As discussed previously, cholinesterase inhibitors (ChEIs) are frequently used
during early stages of the disease, but they do not provide optimal results and show
limited efficiency. Researchers suspect that poor response may be due to gene variability.
Single nucleotide repeats (SNRs) were noted in PRKCE and NBEA, which are both gene
regions associated with cholinergic receptor functionality.101
α-nAChR is mainly
conserved between individuals and plays key roles throughout the CNS, originating from
CHRNA7 (on chromosome 15q14).101
Researchers noted that genetic differences in this
gene can impede AD development, slowing down disease progression in some patients.
Interestingly, gender differences can contribute genetic variations for how these receptors
are encoded and expressed. More than likely, this is due to the influence of sex hormones

upon these receptors.101
Control of nAChR by Galantamine (a ChEI) is depicted below in
Figure 7.
Many studies noted the connection that many nonsteroidal drug
inflammation demonstrated lower AD incidence.
noticed that Aβ and tau protein concentrations decreased upon administration of these
nonsteroidal drugs. Although the underlying mechanism f
unknown, it is suspected that
Figure 7: Modulatory Control of Neural Receptors by
Galantamine. As noted in the figure, binding of Galantamine
to receptors stimulates nAChR receptors, improves
memory, and promotes overall neural health.
McQuade
Control of nAChR by Galantamine (a ChEI) is depicted below in
Many studies noted the connection that many nonsteroidal drugs to prevent arthritic
demonstrated lower AD incidence.6 Likewise, multiple animal studies
and tau protein concentrations decreased upon administration of these
Although the underlying mechanism for these drugs’ efficiency is still
that γ-secretase is antagonized by these drugs. Inhibition of
Modulatory Control of Neural Receptors by
. As noted in the figure, binding of Galantamine
to receptors stimulates nAChR receptors, improves
memory, and promotes overall neural health.
McQuade 35
Control of nAChR by Galantamine (a ChEI) is depicted below in
arthritic
animal studies
and tau protein concentrations decreased upon administration of these
or these drugs’ efficiency is still
Inhibition of γ-

McQuade 36
secretase would prevent cleavage of APP (and subsequent production of Aβ
oligopeptides).22
Although some progress has been made in recent years, a definitive treatment that
is viable for AD has not been proposed. Current treatments (such as NSAIDS and COX-2
inhibitors) are merely palliative, dealing only with disease symptoms rather than
addressing the root cause. Also, patient variability is another factor to be considered:
individuals respond to drugs differently because of various environmental and genomic
factors. Because molecular conditions vary so drastically between individuals, there is no
cure-all treatment for AD, just as there is no “silver bullet” to defeat cancer.102
If
Alzheimer’s is to be cured, a drug (or drug regimen) must be found to deal with the
symptoms while also attacking the root cause of the disease. Current medicinal methods
of treatment (although possibly delaying AD onset) do nothing to prevent neural damage
and cannot ultimately save the patient.90
Against the background of failed medicinal
treatments, turmeric emerges as a novel therapeutic compound that may prove highly
efficient in preventing and permanently curing AD. The therapeutic values of turmeric as
well as common critiques against its efficiency will now be evaluated in greater detail
Turmeric Absorption & Bioavailability
Turmeric is often critiqued for its poor bioavailability levels, especially in elderly
test subjects.103
This is an expected phenomenon: phenolic ring systems and their
associated hydrocarbon chains in curcumin are not easily dissolvable in the aqueous
plasma solution.104
Although the hydroxyl and ether groups are water-soluble, these
represent a very small portion of the curcumin molecule. Also, research suggests that
metabolism of turmeric occurs too quickly to produce healthful effects.105
Phenolic
groups have traditionally been viewed as relatively inactive. Often found in flowers,
fruits, nuts, and roots, phenols have been seen as unimportant to overall wellbeing.
Recently, however, researchers have found that these groups may play essential roles in
stopping cancer, protecting against oxidative damage, and preventing inflammation.106
In
fact, they may prove especially important in brain and liver health. Phenols strongly lend

McQuade 37
electrons and form sturdy intermediates due to hyperconjugation of their aromatic
systems.107
This provides them with a high degree of chemical stability. Some studies
suggest that a turmeric regimen supplemented with probiotics may produce enhanced
curcumin absorption from the stomach.108
When turmeric is obtained and freshly ground,
it is often accompanied by lactic acid bacteria (LAB) that aid substantially in its
metabolism and eventual digestion. In one study, a ferric-reducing antioxidant power
(FRAP) assay was used to determine the degree to turmeric (in the presence of LAB)
counteracted ROS.3 Rats were given a turmeric-containing solution that was fermented
with LAB. These rats demonstrated much higher absorption levels than humans who
imbibed turmeric without LAB. This finding suggests that therapeutic turmeric should be
as fresh as possible (so it contains LAB), or should be fortified with LAB probiotics.109
Also, like most other compounds, turmeric is best absorbed when ingested with a meal.
This leads to greater production of bile salts within the small intestine that can help
emulsify water-insoluble portions of curcumin.110
Certainly, future studies are needed to
elucidate optimal methods of turmeric delivery. Also, to date no studies have been
conducted regarding optimal dosage of turmeric for mammalian systems. Although many
of turmeric’s molecular mechanisms still remain hazy, substantial evidence supports the
hypothesis that turmeric will prove an effective therapeutic for AD.
Curcumin—A Therapeutic Solution
Turmeric (Curcuma longa) is a plant grown in much of southeast Asia.
Especially common in India, it is related to the ginger family and its dried rhizomes
are usually consumed as a spice.111 Turmeric is a yellow-brown color and exhibits a
mildly spicy taste. As a diarylheptanoid molecule, curcumin (diferuloylmethane) exists as
a substituted heptene chain flanked by two substituted benzene molecules.106
Curcumin is
categorized as a beta-diketone molecule, which lends special chemical and medicinal
activities. These phenolic groups contribute to turmeric’s brown-yellow color. As a
racemic molecule, it exists in both keto and enol forms.108
The enol form exists in higher
concentrations since it is more energetically favorable. In Curcuma longa, curcumin’s
biochemical production uses phenylalanine as a starting reactant. Turmeric contains over

McQuade 38
20 bioactive molecules, of which curcumin occupies approximately 3% of all turmeric’s
biomolecules.112
Circuminoids exist as three major forms: diferuloylmethane,
demethoxycurcumin, and bisdemethoxycurcumin. The first of these, diferuloylmethane,
is the circuminoid found in turmeric. Turmeric has been evaluated in many different
studies. One study measured the effects of suberoylanilidehydroxamic acid (SAHA) and
curcumin used together on Aβ plaques.113
Used in tandem, they were found to increase
learning and memory in transgenic rat models.113
Also, both these compounds were
discovered to protect neurons from the toxic effects of soluble and insoluble plaques. In a
separate study, researchers demonstrated that BDMC33 (a modified form of curcumin)
prevented the production of nitric oxide among macrophage cells, thus partially inhibiting
inflammation among body systems.114
PGs are common cellular components that are
naturally manufactured by nearly every cell type. They are made from AA and serve as
important lipid-derived secondary messengers in the body. As a vitally important PG, the
E2 variety is chiefly modulated by COX, which is also known as prostaglandin
endoperoxide H2 (PGH2) synthase.115
Current research has uncovered three major COX
molecules. Under normal conditions, COX-1 is active without any form of enzymatic
stimulation. COX-2, on the other hand, is inducible and is upregulated by the presence of
cytokines, cancer-stimulating genes, and various growth factors. Although not thoroughly
understood, COX-3 was found to possess intron-1. Researchers found that cellular
overexpression of PGE2 and COX-2 lead to a number of diseases, such as asthma,
arthritis, and AD—all related to pro-inflammatory activity.116
In multiple studies,
turmeric was found to prevent biological synthesis of PGE2. NSAIDS are designed to
specifically block pain caused by inflammatory disorders. COX-1 plays vital roles in
protecting the GI tracts, so researchers must be careful that NSAIDS do not block this
activity as well. Turmeric therapy solves this problem by effectively inhibiting PGH2
synthase (COX-2) activity, while not interfering with the normal homeostatic role of
COX-1.117
Figure 8 shows the pharmaceutical production of BDMC33. Notice that the
basic cyclical structure of curcumin is conserved and that a number of oxygen atoms with
lone-pair electrons (in the methoxy and carbonyl groups) are present.

BDMC33 in particular was found to stop nitric oxide synthesis from IFN
macrophages.114
Also, it partially inhibited COX
acts against free radical species that can cause major damage within the body.
the intermediary activity of curcumin, ROS could damage neural proteins, fats,
genetic sequences.111
Left unchecked, free radical species ultimately lead to
carcinogenesis or widespread apoptosis.
curcumin is able to “catch” free
neural tissue.116
Entrapment of metal ions like copper and iron prevents activation
NF-B pathway, thereby inhibiting
In multiple studies, curcumin has also been shown to
through downregulation of eicosanoid molecules, both in
bloodstream.53
In fact, curcumin
in many Eastern cultures.118
Ingestion of turmeric causes greater vasodilation (
produces the same health effect caused by an aerobic workout). In fact, when
amounts of turmeric ingestion is paired with aerobic exercise, results are markedly
improved.103
Thus, the potential benefits of aspirin in preventing inflammation are
miniscule compared to curcumin’s potent effects. It is critical that future studi
turmeric’s effect on swelling and blood pressure in greater detail.
Figure 8: BDMC33’s basic molecular structure. Notice that
its chemical preparation comes from addition of ethanol and a
strong base to cyclohexanone and a methoxy
benzaldehyde. The resulting product is a symmetric ketone
molecule that exhibits many medicinal effects.
McQuade
Curcumin analogues are especially
adept at preventing tumor activation,
excessive blood vessel growth, and
eicosanoid-stimulated inflammation.
BDMC33 in particular was found to stop nitric oxide synthesis from IFN- γ/LPS
Also, it partially inhibited COX-2 and PGE2 activity. Additionally, it
acts against free radical species that can cause major damage within the body.
the intermediary activity of curcumin, ROS could damage neural proteins, fats,
Left unchecked, free radical species ultimately lead to
carcinogenesis or widespread apoptosis. Also because of its unique molecular structure,
“catch” free-floating metal ions, thus preventing them from damaging
Entrapment of metal ions like copper and iron prevents activation
inhibiting production of Aβ plaques.78
In multiple studies, curcumin has also been shown to decrease inflammation
through downregulation of eicosanoid molecules, both in neural tissues and in the
In fact, curcumin is also known as an effective blood pressure medication
Ingestion of turmeric causes greater vasodilation (
produces the same health effect caused by an aerobic workout). In fact, when
turmeric ingestion is paired with aerobic exercise, results are markedly
the potential benefits of aspirin in preventing inflammation are
d to curcumin’s potent effects. It is critical that future studi
turmeric’s effect on swelling and blood pressure in greater detail.
BDMC33’s basic molecular structure. Notice that
its chemical preparation comes from addition of ethanol and a
strong base to cyclohexanone and a methoxy-substituted
benzaldehyde. The resulting product is a symmetric ketone
inal effects.
McQuade 39
urcumin analogues are especially
adept at preventing tumor activation,
excessive blood vessel growth, and
stimulated inflammation.
/LPS
Additionally, it
acts against free radical species that can cause major damage within the body. If not for
the intermediary activity of curcumin, ROS could damage neural proteins, fats, and
ecause of its unique molecular structure,
floating metal ions, thus preventing them from damaging
Entrapment of metal ions like copper and iron prevents activation of the
decrease inflammation
neural tissues and in the
is also known as an effective blood pressure medication
Ingestion of turmeric causes greater vasodilation (which
produces the same health effect caused by an aerobic workout). In fact, when moderate
turmeric ingestion is paired with aerobic exercise, results are markedly
the potential benefits of aspirin in preventing inflammation are
d to curcumin’s potent effects. It is critical that future studies examine

McQuade 40
By modulating key molecular players early in the pathway of amyloidogenesis,
curcumin therapy cuts straight to the heart of the issue. It precisely targets vital molecular
messengers like PGH2 and COX-2, directly impacts excessive enzyme activity by
targeting PLA2 and ceramide, and prevents ROS damage.93
Unless taken in exorbitantly
high doses (> 1 kg) intravenously, curcumin poses no known threats to body systems.106
Excessive ingestion of turmeric merely leads to discoloration of feces and diarrhea
without any other known complications.112
Compared to the side effects of COX-2
inhibitors (which can cause severe indigestion, insomnia, and headaches), the secondary
effects of turmeric are virtually nonexistent when taken in normal amounts. Rather than
attempting to treat AD symptoms, curcumin delves to the source of the problem by
stopping APP’s production of Aβ peptides.109
It also plays a modulatory role through
precise mediation of lipid catabolism pathways. Multiple studies show that curcumin
lowers inflammation, disposes of ROS, and prevents accumulation of plaques. It is also
suspected to inhibit a wide variety of cancers. Demographics that consume high amounts
of turmeric tend to demonstrate lower rates of Alzheimer’s. Although demographics
cannot conclusively prove that a curcumin-rich diet is responsible for a healthful
phenotype, this correlation cannot be overlooked in light of previous data.106
Turmeric demonstrates considerable medicinal activity for treatment of oncogenic
activity.111
In one study, murine macrophage RAW264.7 cells and HT-29 carcinogenic
human colon cells were both tested to determine turmeric’s effect on lipid cascade
activity and its impact on concentrations of select proteins. Cystolic phospholipase A2
(cPLA2) levels were measured as wells as COX and 5-lipoxygenase (5-LOX).106
First,
cells were activated using lipopolysaccharides (LPS), which upregulated COX-2
production. Tetrahydrocurcumin (THC) and curcumin both exerted an inhibitory effect
on the AA cycle so that AA was not liberated to produce pro-inflammatory eicosanoid
molecules.3 It seems that this was largely due to prevention of cPLA2 phosphorylation.
The enzyme itself was not inhibited; rather, activation through phosphorylation was
prevented. This is an important distinction because it means that curcumin did not knock
out total enzyme function. Even in the presence of curcumin and THC, cPLA2 was still
able to conduct its normal activities, but without excessive phosphorylation. Every
curcuminoid tested in this study caused lower levels of prostaglandin E (PGE2). Thus,
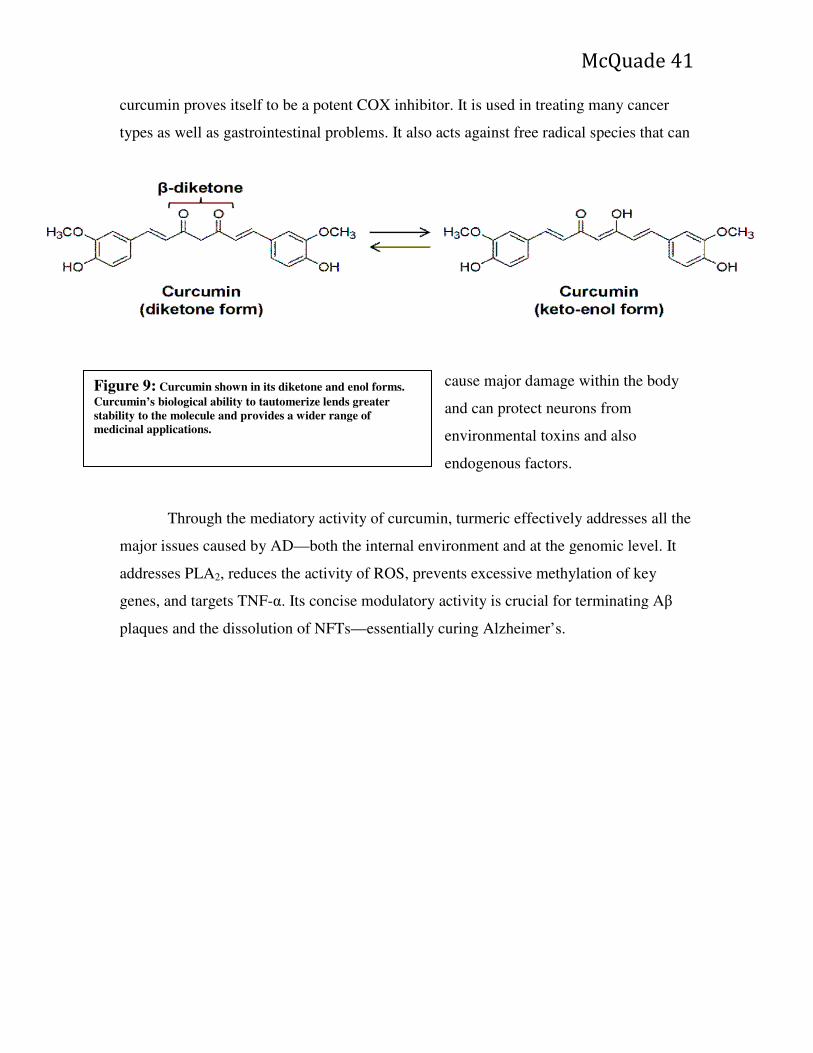
curcumin proves itself to be a potent COX inhibitor. It is used in treating many cancer
types as well as gastrointestinal problems. It also acts against free radical species that can
Through the mediatory activity of curcumin, turmeric effectively addresses all the
major issues caused by AD—
addresses PLA2, reduces the acti
genes, and targets TNF-α. Its concise
plaques and the dissolution of NFTs
Figure 9: Curcumin shown in its diketone and enol forms.
Curcumin’s biological ability to tautomerize lends greater
stability to the molecule and provides a wider range of
medicinal applications.
McQuade
itself to be a potent COX inhibitor. It is used in treating many cancer
types as well as gastrointestinal problems. It also acts against free radical species that can
cause major damage within the body
and can protect neurons from
environmental toxins and also
endogenous factors.
Through the mediatory activity of curcumin, turmeric effectively addresses all the
—both the internal environment and at the genomic level. It
, reduces the activity of ROS, prevents excessive methylation of key
Its concise modulatory activity is crucial for terminating A
plaques and the dissolution of NFTs—essentially curing Alzheimer’s.
Curcumin shown in its diketone and enol forms.
Curcumin’s biological ability to tautomerize lends greater
stability to the molecule and provides a wider range of
McQuade 41
itself to be a potent COX inhibitor. It is used in treating many cancer
types as well as gastrointestinal problems. It also acts against free radical species that can
e major damage within the body
and can protect neurons from
environmental toxins and also
Through the mediatory activity of curcumin, turmeric effectively addresses all the
both the internal environment and at the genomic level. It
vity of ROS, prevents excessive methylation of key
terminating Aβ

Figure 10 shows the multiple methods by which curcumin modulates molecular factors
Figure 10: Therapeutic roles of turmeric. As
section, curcumin directly impacts secretase, presenilin, GSK
3β activity and also provides increased neural protection
against exogenous metals, tau proteins, and extracellular
plaques.
McQuade
shows the multiple methods by which curcumin modulates molecular factors
Therapeutic roles of turmeric. As outlined in this
section, curcumin directly impacts secretase, presenilin, GSK-
activity and also provides increased neural protection
against exogenous metals, tau proteins, and extracellular
McQuade 42
shows the multiple methods by which curcumin modulates molecular factors

McQuade 43
involved in AD ontology and progression. Turmeric offers a safer and more effective
treatment than NSAIDS, cholinesterase inhibitors, or anti-inflammatory medications.108
Future studies must investigate the role of turmeric paired with other therapeutic
compounds, such as liquiritin, CP, and shankhpushpi. To date, several studies have
underscored the importance of CP and liquiritin, but none have ever investigated
potential synergistic effects when used in tandem.119
Prospects for Future Studies
Study Purpose: The main purpose of this study is to determine the efficacy of turmeric
for the treatment of AD. Hence, the aims of this prospective study are two-fold. First, this
study will determine turmeric’s impact on amyloid-β plaque deposition in transgenic AD
models in very early stages of the disease. Secondly, it will determine the effect of
turmeric on improvement of spatial learning, coordination, and balance in stage 2
Alzheimer mice.
Experimental Design: This study will be preformed across a span of six months using a
total of 100 test subjects (Mus musculus) divided into four groups. Results from each
group will be collected on a daily basis. This study will be performed during a period
of six months using a total of 100 test subjects (Mus musculus) divided into four
groups. Results from each group will be collected on a daily basis. Before beginning
the study, all mice (AD and healthy) will be pre-screened to measure blood pressure,
coordination and balance, etc. Conduct brain scans of normal mice and Stage 1
transgenic Alzheimer mice. Scans measuring brain activity between the two groups
will be compared and evaluated. Four groups will be created (each containing 25
subjects)
1). Group A1 will consist of healthy mice that consume standard lab-grade rodent
feed.

McQuade 44
2). Group A2 will consist of healthy mice that consume a mixture of standard lab-
grade rodent feed with 5 mg/kg of freshly ground turmeric added. Two grams of
turmeric will be added to every liter of water.
3). Group B1 will contain Alzheimer’s mice and will only consume lab-grade rodent
feed.
4). Group B2 will contain Alzheimer’s mice, but will consume lab-grade feed with 5
mg/kg of turmeric added to food and 2 grams of freshly ground turmeric added per
liter of drinking water. Each mouse will receive its own separate cage with
individual food and water. Amount of food/water consumed will be measured and
appropriated for each mouse. All other elements besides food will be kept as
constant as possible. SMase activity will be closely monitored (both acidic and basic
forms). Plasma levels of SMase and a fluorescent assays will be used to collect
enzyme activity. COX-2 activity will be measured as well as reactive oxygen species
(ROS). Enzyme activity will be determined using Michaelis-Menten kinetics and
evaluated appropriately. Enzymes in blood serum and CSF will also be collected and
measured. PLA2 and ceramide activities will be evaluated for each of the four
groups. Data will be statistically evaluated using appropriate parameters.
Mice will be tested periodically every 2 weeks and be subject to a variety of
balance/strength/coordination tests. Rotarod test Morris water navigation task, the
grip test meter test, maze tests, and a balance beam will all be used. All results will
be evaluated using appropriate statistical methods. Extensive collection of data will
be conducted throughout the course of the experiment. After a period of six months,
all mice will be sacrificed and overall brain appearance will be evaluated by a team
of researchers. Brains will be tested for plaques, lesions, demyelination, tissue
shrinkage, or any other pathological or abnormal signs.
Necropsy: After 60 days, all subjects will be terminated via CO2 asphyxiation and a
necropsy will be preformed to determine the degree to which amyloid-beta plaques have
accumulated in the brain.

McQuade 45
Hypothesis: Mice consuming a turmeric-containing diet will demonstrate lowered
susceptibility to development of AD. Also, AD mice on the turmeric diet will show fewer
motor/coordination abnormalities than the AD group that did not receive turmeric.
Prevention of Unnecessary Pain: If mice display intense pain or suffering, they will be
terminated humanely using CO2 asphyxiation.
Problems/Obstacles: The most probable foreseen obstacle in this study is whether
positive results can be pinpointed to turmeric’s medicinal activity or due to some other
unforeseen factor. Although a quantitative evaluation of results will be conducted, this
study cannot rule out some other confounding factor that influenced turmeric’s activity.
Also, it is unknown whether some component of the lab-grade rat feed exerts a
beneficial/negative effect upon turmeric’s activity, both through absorption in the
bloodstream and activity in the nervous system. Although environmental factors can be
carefully manipulated, there is no guarantee that all conditions for every group will be
equivalent.
Significance of Study: Many researchers suspect that turmeric possesses incredible
potential as a medicinal chemical compound. This must be determined experimentally
before turmeric is implemented in a hospital or clinical setting. The future of turmeric as
a healthful compound is dependent upon this study and other related investigations.
Future Directions: Avenues of future research are largely contingent upon experimental
results. If the turmeric-fed mice exhibit retardation of AD progression, future research
should determine turmeric’s optimal dosage for Alzheimer’s patients. Although
investigating this issue to some extent, this current study will not elucidate all details of
effective drug regimen. Also, future studies must determine if turmeric is best taken alone
or in combination with another compound. In particular, CP and liquiritin are two
compounds are special interest Methods will be suggested as to certain novel compounds
that can be screened for medicinal efficiency. Many current studies complain that
turmeric is poorly absorbed because of its relatively poor solubility in water.

McQuade 46
Discussion
As multiple studies demonstrate, NSAIDs can mediate immune response, but they
do not effectively reduce Aβ plaques.10
Although well-intentioned, current drug designs
suffer from a fundamental flaw in their production-- they are derived largely from
empirical methods and do not usually begin with a presuppositional framework in mind.
This directly leads to the treatment of specific disease symptoms (COX-2 reduction,
thromboxane inhibition, etc.) without targeting the basic cause of the disease. Since these
therapies are limited in scope, they also demonstrate numerous secondary effects, some
of which are deadly. Although offering great potential, memantine suffers from short-
term activity as neural receptors quickly accommodate. Likewise, lithium fails to offer
consistently reliable results (and also demonstrates mixed experimental results). Lithium
has also garnished concern over its potentially toxic effect to body organs.95
Therefore,
neither lithium nor memantine can be employed for patient care or for any kind of long-
term treatment. In contrast to pharmaceutical compounds, turmeric offers a wide range of
healthful effects. Despite some studies that demonstrate poor solubility, research has
determined that fortification with LAB significantly increases turmeric’s bioavailability.
This finding suggests that turmeric used for AD therapy should be intentionally grown in
the presence of LAB and should be consumed in the freshest form possible. Further
studies must verify and add further details regarding how LAB increases curcumin’s
effectiveness.
The proposed turmeric study will elucidate further details about how turmeric
impacts AD murine models. Adjusting for any confounding factors, these findings can be
directly applied to human patients. Based on current research, it is hypothesized that
turmeric ingestion will greatly increase cognition and memory and also boost
coordination and muscle control. Future studies will investigate dosage considerations
and whether turmeric is best used in combination with other compounds or taken by
itself. Other transgenic model organisms will be tested to determine if turmeric’s effects
are confined to one species or can impact many different species. It is presumed that the
latter scenario will be proven correct and that each organism will utilize similar cascades
and molecular mechanisms in metabolism of turmeric.

McQuade 47
Conclusion
Despite the valiant efforts of researchers and physicians, AD still remains an
incurable and deadly disease. Impacting millions around the world each year, it is
especially prevalent in America and Europe and targets the elderly demographic.81
AD is
a complex, multifactorial disease that is typified by external Aβ plaques and internal
NFTs (due to faulty secretase cleavage of APP and excessive tau phosphorylation
respectively).20
Historically, the Homo sapiens genome has always displayed a potential
for AD development; widespread Alzheimer’s, however, is a relatively recent
phenomenon due to longer lifespans and harmful environmental factors.13
It is
hypothesized that upregulation of inflammatory cascades (driven by AA and ceramide
production) are the major stimulant in driving disease pathology.74
A complex interplay
of diet, gene expression, and organism variability is implicated in this process. It is
believed that this internal inflammation provides the motive force behind SP and NFT
accumulation. By means of a vicious cycle, even the body’s own immune response
stimulates disease conditions through microglial activation.85
Likewise, the “cytokine
storm” produced by AA and ceramide pathways only increase inflammation, caspase
activity, and cell necrosis. Multiple drug regimens have been developed for treating AD,
but so far are only mildly effective (or even detrimental in many cases). Although
demonstrating some potential, COX-2 and cholinesterase inhibitors are not yet suitable
for patient use and their efficacy in treating AD is doubtful.87
Even NSAIDs (perhaps the
most effective of AD therapeutics) and memantine are extremely time-dependent, and
their usefulness rapidly declines with each use.10,88
In the absence of a useful AD
therapeutic, researchers are desperately searching for novel compounds that produce
clinical results. As multiple studies show, the burden of evidence weighs heavily on
turmeric, warranting the need for further clinical and experimental investigations.3,105,106
It is hoped that turmeric will turn the tide of disease progression and restore cognitive,
learning, and STM functions.16
Additionally, a turmeric-based drug regimen will inhibit
neuroinflammation by retroactively preventing exorbitant eicosanoid expression by
deactivation of PGH2 synthase (COX-2).114
Although most neural damage is irreversible,
curcumin will demolish existent Aβ plaques, unravel NFTs, and prevent apoptotic

McQuade 48
activity.118
It is tantamount that turmeric therapies be administered in the earliest stages
of the disease since this promises the greatest chance of a full recovery. These
conclusions will be investigated further in the “Proposed Study” section, which will test
how turmeric affects motor skills, coordination, and balance of transgenic Alzheimer
mice. Because of its widespread effects in targeting SPs and NFTs, impacting AA and
sphingomyelinase cascades, and halting ROS, turmeric warrants incorporation into
Western medicinal practice. It is hoped that turmeric may be used in tandem with another
medicinally useful compounds (natural or synthetic) to ebb the tide of AD.108
Of all
potential therapies, turmeric will halt the Alzheimer epidemic and its ravaging grip on
Western society.

McQuade 49
References
1. De Strooper B, Woodgett J. Alzheimer’s disease: Mental plaque removal. Nature. 2003;423(6938):392.
2. Trazzi S, Fuchs C, De Franceschi M, Mitrugno VM, Bartesaghi R, Ciani E. APP-dependent alteration of GSK3β activity impairs neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis. 2014;67:24-36. doi:10.1016/j.nbd.2014.03.003.
3. Srivastava KC, Bordia A, Verma SK. Curcumin, a major component of food spice turmeric (Curcuma longa) inhibits aggregation and alters eicosanoid metabolism in human blood platelets. Prostaglandins Leukot Essent Fatty Acids. 1995;52(4):223-227.
4. Bandyopadhyay S, Rogers JT. Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: Maintenance of brain iron homeostasis. Biochem Pharmacol. 2014;88(4):486-494. doi:10.1016/j.bcp.2014.01.032.
5. Davidson JE, Lockhart A, Amos L, et al. Plasma lipoprotein-associated phospholipase A2 activity in Alzheimer’s disease, amnestic mild cognitive impairment, and cognitively healthy elderly subjects: a cross-sectional study. Alzheimers Res Ther. 2012;4(6):1-7. doi:10.1186/alzrt154.
6. Beecham GW, Hamilton K, Naj AC, et al. Genome-Wide Association Meta-analysis of Neuropathologic Features of Alzheimer’s Disease and Related Dementias. PLoS Genet. 2014;10(9):1-15. doi:10.1371/journal.pgen.1004606.
7. Pietri M, Dakowski C, Hannaoui S, et al. PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat Med. 2013;19(9):1124-1131. doi:10.1038/nm.3302.
8. Henry A, Li Q-X, Galatis D, et al. Inhibition of platelet activation by the Alzheimer’s disease amyloid precursor protein. Br J Haematol. 1998;103(2):402-415.
9. Lambert MA, Bickel H, Prince M, et al. Estimating the burden of early onset dementia; systematic review of disease prevalence. Eur J Neurol. 2014;21(4):563-569. doi:10.1111/ene.12325.
10. Trepanier CH, Milgram NW. Neuroinflammation in Alzheimer’s Disease: Are NSAIDs and Selective COX-2 Inhibitors the Next Line of Therapy? J Alzheimers
Dis. 2010;21(4):1089-1099.

McQuade 50
11. Igarashi M, Kaizong Ma, Fei Gao, Hyung-Wook Kim, Rapoport SI, Rao JS. Disturbed Choline Plasmalogen and Phospholipid Fatty Acid Concentrations in Alzheimer’s Disease Prefrontal Cortex. J Alzheimers Dis. 2011;24(3):507-517. doi:10.3233/JAD-2011-101608.
12. Cheng JS, Craft R, Yu G-Q, et al. Tau Reduction Diminishes Spatial Learning and Memory Deficits after Mild Repetitive Traumatic Brain Injury in Mice. PLoS
ONE. 2014;9(12):1-17. doi:10.1371/journal.pone.0115765.
13. Keleshian VL, Modi HR, Rapoport SI, Rao JS. Aging is associated with altered inflammatory, arachidonic acid cascade, and synaptic markers, influenced by epigenetic modifications, in the human frontal cortex. J Neurochem. 2013;125(1):63-73. doi:10.1111/jnc.12153.
14. Ferreira LK, Tamashiro-Duran JH, Squarzoni P, et al. The link between cardiovascular risk, Alzheimer’s disease, and mild cognitive impairment: support from recent functional neuroimaging studies. Rev Bras Psiquiatr. 2014;36(4):344-357. doi:10.1590/1516-4446-2013-1275.
15. Engstrom EJ. Researching Dementia in Imperial Germany: Alois Alzheimer and the Economies of Psychiatric Practice. Cult Med Psychiatry. 2007;31(3):405-413. doi:10.1007/s11013-007-9060-4.
16. Caza N, Belleville S. Reduced short-term memory capacity in Alzheimer’s disease: The role of phonological, lexical, and semantic processing. Memory. 2008;16(4):341-350. doi:10.1080/09658210701881758.
17. Gnjidic D, Hilmer SN, Hartikainen S, et al. Impact of High Risk Drug Use on Hospitalization and Mortality in Older People with and without Alzheimer’s Disease: A National Population Cohort Study. PLoS ONE. 2014;9(1):1-8. doi:10.1371/journal.pone.0083224.
18. Panchal M, Loeper J, Cossec J-C, et al. Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J Lipid Res. 2010;51(3):598-605. doi:10.1194/jlr.M001859.
19. Zilka N, Kazmerova Z, Jadhav S, et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47. doi:10.1186/1742-2094-9-47.
20. Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337-351. doi:10.1016/j.neuron.2007.01.010.

McQuade 51
21. Mirza Z, Pillai VG, Kamal MA. Protein Interactions Between the C-terminus of Aβ-Peptide and Phospholipase A2 - A Structure Biology Based Approach to Identify Novel Alzheimer Therapeutics. CNS Neurol Disord Drug Targets. 2014.
22. Grimm MOW, Haupenthal VJ, Rothhaar TL, et al. Effect of Different Phospholipids on α-Secretase Activity in the Non-Amyloidogenic Pathway of Alzheimer’s Disease. Int J Mol Sci. 2013;14(3):5879-S13. doi:10.3390/ijms14035879.
23. Fragkouli A, Tzinia AK, Charalampopoulos I, Gravanis A, Tsilibary EC. Matrix Metalloproteinase-9 Participates in NGF-Induced α-Secretase Cleavage of Amyloid-β Protein Precursor in PC12 Cells. J Alzheimers Dis. 2011;24(4):705-719. doi:10.3233/JAD-2011-101893.
24. Karr JW, Szalai VA. Cu(II) Binding to Monomeric, Oligomeric, and Fibrillar Forms of the Alzheimer’s Disease Amyloid-β Peptide. Biochemistry (Mosc). 2008;47(17):5006-5016.
25. Joshi YB, Di Meco A, Praticó D. Modulation of Amyloid-β Production by Leukotriene B4 via the γ-Secretase Pathway. J Alzheimers Dis. 2014;38(3):503-506. doi:10.3233/JAD-131223.
26. Li JJ, Dolios G, Wang R, Liao F-F. Soluble Beta-Amyloid Peptides, but Not Insoluble Fibrils, Have Specific Effect on Neuronal MicroRNA Expression. PLoS
ONE. 2014;9(3):1-14. doi:10.1371/journal.pone.0090770.
27. Malaplate-Armand C, Florent-Béchard S, Youssef I, et al. Soluble oligomers of amyloid-β peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol Dis. 2006;23(1):178-189. doi:10.1016/j.nbd.2006.02.010.
28. Shelat PB, Chalimoniuk M, Wang J-H, et al. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem. 2008;106(1):45-55. doi:10.1111/j.1471-4159.2008.05347.x.
29. Yi Li, Bohm C, Dodd R, et al. Structural biology of presenilin 1 complexes. Mol
Neurodegener. 2014;9(1):1-21. doi:10.1186/1750-1326-9-59.
30. Brendel M, Jaworska A, Grießinger E, et al. Cross-Sectional Comparison of Small Animal [18F]-Florbetaben Amyloid-PET between Transgenic AD Mouse Models. PLoS ONE. 2015;10(2):1-21. doi:10.1371/journal.pone.0116678.
31. Chang Liu, Jianwei Tang. Expression levels of tumor necrosis factor-α and the corresponding receptors are correlated with trauma severity. Oncol Lett. 2014;8(6):2747-2751. doi:10.3892/ol.2014.2575.

McQuade 52
32. Fatima A, Hj. Abdul AB, Abdullah R, Karjiban RA, Lee VS. Binding Mode Analysis of Zerumbone to Key Signal Proteins in the Tumor Necrosis Factor Pathway. Int
J Mol Sci. 2015;16(2):2747-2766. doi:10.3390/ijms16022747.
33. Rosenthal JA, Huang C-J, Doody AM, et al. Mechanistic Insight into the TH1-Biased Immune Response to Recombinant Subunit Vaccines Delivered by Probiotic Bacteria-Derived Outer Membrane Vesicles. PLoS ONE. 2014;9(11):1-24. doi:10.1371/journal.pone.0112802.
34. Dolcino M, Patuzzo G, Barbieri A, et al. Gene Expression Profiling in Peripheral Blood Mononuclear Cells of Patients with Common Variable Immunodeficiency: Modulation of Adaptive Immune Response following Intravenous Immunoglobulin Therapy. PLoS ONE. 2014;9(5):1-9. doi:10.1371/journal.pone.0097571.
35. Lundin JI, Checkoway H. Endotoxin and Cancer. Environ Health Perspect. 2009;117(9):1344-1350. doi:10.1289/ehp.0800439.
36. Nguyen A, Louisa Ho, Yonghong Wan. Chemotherapy and oncolytic virotherapy: advanced tactics in the war against cancer. Front Oncol. 2014;4:1-10. doi:10.3389/fonc.2014.00145.
37. Hsien-San Hou, Hsieh-Fu Tsai, Hsien-Tai Chiu, Ji-Yen Cheng. Simultaneous chemical and electrical stimulation on lung cancer cells using a multichannel-dual-electric-field chip. Biomicrofluidics. 2014;8(5):1-15. doi:10.1063/1.4896296.
38. An Zhou, Hongfei Wu, Jian Pan, et al. Synthesis and Evaluation of Paeonol Derivatives as Potential Multifunctional Agents for the Treatment of Alzheimer’s Disease. Molecules. 2015;20(1):1304-1318. doi:10.3390/molecules20011304.
39. Grösgen S, Grimm MOW, Frieß P, Hartmann T. Role of amyloid beta in lipid homeostasis. BBA - Mol Cell Biol Lipids. 2010;1801(8):966-974. doi:10.1016/j.bbalip.2010.05.002.
40. Straub BK, Gyoengyoesi B, Koenig M, et al. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology. 2013;62(4):617-631. doi:10.1111/his.12038.
41. Atsuhiko Ichimura, Sae Hasegawa, Mayu Kasubuchi, Ikuo Kimura, Hudson B, Cho TY. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol. 2014;5:1-6. doi:10.3389/fphar.2014.00236.
42. Tajima Y, Ishikawa M, Maekawa K, et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor

McQuade 53
protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013;12(1):1-14. doi:10.1186/1476-511X-12-68.
43. Mirza Z, Pillai VG, Zhong W-Z. Structure of N-terminal sequence Asp-Ala-Glu-Phe-Arg-His-Asp-Ser of Aβ-peptide with phospholipase A2 from venom of Andaman Cobra sub-species Naja naja sagittifera at 2.0 Å resolution. Int J Mol
Sci. 2014;15(3):4221-4236. doi:10.3390/ijms15034221.
44. Quach ND, Arnold RD, Cummings BS. Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol. 2014;90(4):338-348. doi:10.1016/j.bcp.2014.05.022.
45. Dennis EA, Cao J, Hsu Y-H, Magrioti V, Kokotos G. Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chem Rev. 2011;111(10):6130-6185. doi:10.1021/cr200085w.
46. Okumura K, Ohno A, Nishida M, Hayashi K, Ikeda K, Inoue S. Mapping the Region of the α-Type Phospholipase A₂ Inhibitor Responsible for Its Inhibitory Activity. J Biol Chem. 2005;280(45):37651-37659. doi:10.1074/jbc.M507250200.
47. Haowei Song, Wohltmann M, Min Tan, Shunzhong Bao, Ladenson JH, Turk J. Group VIA PLA2 (iPLA2β) Is Activated Upstream of p38 Mitogen-activated Protein Kinase (MAPK) in Pancreatic Islet β-Cell Signaling. J Biol Chem. 2012;287(8):5528-5541. doi:10.1074/jbc.M111.285114.
48. Moscardó A, Vallés J, Piñón M, Aznar J, Martínez-Sales V, Santos M-T. Regulation of cytosolic PlA 2 activity by PP1/PP2A serine/threonine phosphatases in human platelets. Platelets. 2006;17(6):405-415. doi:10.1080/09537100600757869.
49. Ryan VH, Primiani CT, Rao JS, Ahn K, Rapoport SI, Blanchard H. Coordination of Gene Expression of Arachidonic and Docosahexaenoic Acid Cascade Enzymes during Human Brain Development and Aging. PLoS ONE. 2014;9(6):1-12. doi:10.1371/journal.pone.0100858.
50. Birch EE, Garfield S, Hoffman DR, Uauy R, Birch DG. A randomized controlled trial of early dietary supply of long-chain polyunsaturated fatty acids and mental development in term infants. Dev Med Child Neurol. 2000;42(3):174-181. doi:10.1111/j.1469-8749.2000.tb00066.x.
51. Stawarska A, Białek A, Stanimirova I, Stawarski T, Tokarz A. The Effect of Conjugated Linoleic Acids (CLA) Supplementation on the Activity of Enzymes Participating in the Formation of Arachidonic Acid in Liver Microsomes of Rats—Probable Mechanism of CLA Anticancer Activity. Nutr Cancer. 2015;67(1):145-155. doi:10.1080/01635581.2015.967875.

McQuade 54
52. Frisardi V, Panza F, Seripa D, Farooqui T, Farooqui AA. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer’s disease pathology. Prog Lipid Res. 2011;50(4):313-330. doi:10.1016/j.plipres.2011.06.001.
53. Yin H, Zhou Y, Zhu M, et al. Role of mitochondria in programmed cell death mediated by arachidonic acid-derived eicosanoids. Mitochondrion. 2013;13(3):209-224. doi:10.1016/j.mito.2012.10.003.
54. Sarkar P, Narayanan J, Harder D r. Differential effect of amyloid beta on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience. 2011;194:241-249. doi:10.1016/j.neuroscience.2011.07.058.
55. Tang S-S, Hong H, Chen L, et al. Involvement of cysteinyl leukotriene receptor 1 in Aβ1–42-induced neurotoxicity in vitro and in vivo. Neurobiol Aging. 2014;35(3):590-599. doi:10.1016/j.neurobiolaging.2013.09.036.
56. Yang G, Haczku A, Chen H. Transgenic smooth muscle expression of the human CysLT1, receptor induces enhanced responsiveness of murine airways to leukotriene D4. Am J Physiol. 2004;286(5):L992-L1001.
57. Samuelsson B. Arachidonic acid metabolism: role in inflammation. Z Für
Rheumatol. 1991;50 Suppl 1:3-6.
58. Hayashi L, Sheth M, Young A, Kruger M, Wayman GA, Coffin AB. The effect of the aquatic contaminants bisphenol-A and PCB-95 on the zebrafish lateral line. NeuroToxicology. 2015;46:125-136. doi:10.1016/j.neuro.2014.12.010.
59. Phiel CJ, Wilson CA, Lee VM-Y, Klein PS. GSK-3a regulates production of Alzheimer’s disease amyloid-b peptides. Nature. 2003;423(6938):435.
60. Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114(2-3):147-162.
61. Teixeira Mendes C, Borges Mury F, De Sá Moreira E, et al. Lithium reduces Gsk3b mRNA levels: implications for Alzheimer Disease. Eur Arch Psychiatry
Clin Neurosci. 2009;259(1):16-22. doi:10.1007/s00406-008-0828-5.
62. Janssen CIF, Kiliaan AJ. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: The influence of LCPUFA on neural development, aging, and neurodegeneration. Prog Lipid Res. 2014;53:1-17. doi:10.1016/j.plipres.2013.10.002.
63. Shimizu T, Wolfe LS. Arachidonic Acid Cascade and Signal Transduction. J Neurochem. 1990;55(1):1-15. doi:10.1111/j.1471-4159.1990.tb08813.x.

McQuade 55
64. Beaulieu E, Ioffe J, Watson SN, Hermann PM, Wildering WC. Oxidative-stress induced increase in circulating fatty acids does not contribute to phospholipase A2-dependent appetitive long-term memory failure in the pond snail Lymnaea stagnalis. BMC Neurosci. 2014;15(1):2-28. doi:10.1186/1471-2202-15-56.
65. Upadhyay P, Panjwani D, Yadav AK. Neuropathology staging and treatment strategies of Alzheimer’s disease: An update. Int J Nutr Pharmacol Neurol Dis. 2014;4(1):28-42. doi:10.4103/2231-0738.124612.
66. Cheng K, Delghingaro-Augusto V, Nolan CJ, et al. High Passage MIN6 Cells Have Impaired Insulin Secretion with Impaired Glucose and Lipid Oxidation. PLoS
ONE. 2012;7(7). doi:10.1371/journal.pone.0040868.
67. DaRocha-Souto B, Coma M, Pérez-Nievas B g., et al. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol Dis. 2012;45(1):425-437. doi:10.1016/j.nbd.2011.09.002.
68. Salkovic-Petrisic M, Tribl F, Schmidt M, Hoyer S, Riederer P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J Neurochem. 2006;96(4):1005-1015. doi:10.1111/j.1471-4159.2005.03637.x.
69. Jelenik T, Roden M. Mitochondrial Plasticity in Obesity and Diabetes Mellitus. Antioxid Redox Signal. 2013;19(3):258-268. doi:10.1089/ars.2012.4910.
70. Teng L, Meng Q, Lu J, et al. Liquiritin modulates ERK‑ and AKT/GSK‑3β‑dependent pathways to protect against glutamate‑induced cell damage in differentiated PC12 cells. Mol Med Rep. 2014;10(2):818-824. doi:10.3892/mmr.2014.2289.
71. Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694-2701. doi:10.1016/j.cellsig.2014.08.019.
72. Rahmati M, Gharakhanlou R, Movahedin M, et al. Treadmill Training Modifies KIF5B Moter Protein in the STZ-induced Diabetic Rat Spinal Cord and Sciatic Nerve. Arch Iran Med AIM. 2015;18(2):94-101.
73. Panchal M, Gaudin M, Lazar AN, et al. Ceramides and sphingomyelinases in senile plaques. Neurobiol Dis. 2014;65:193-201. doi:10.1016/j.nbd.2014.01.010.
74. LiZe Gu, BaoSheng Huang, Wei Shen, et al. Early activation of nSMase2/ceramide pathway in astrocytes is involved in ischemia-associated

McQuade 56
neuronal damage via inflammation in rat hippocampi. J Neuroinflammation. 2013;10(1):1-16. doi:10.1186/1742-2094-10-109.
75. Chatterjee S. Neutral sphingomyelinase action stimulates signal transduction of tumor necrosis factor-alpha in the synthesis of cholesteryl esters in human fibroblasts. J Biol Chem. 1994;269(2):879-882.
76. Haughey NJ, Bandaru VVR, Bae M, Mattson MP. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. BBA - Mol
Cell Biol Lipids. 2010;1801(8):878-886. doi:10.1016/j.bbalip.2010.05.003.
77. Grimm MOW., Grimm HS., Pätzold AJ., et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat Cell Biol. 2005;7(11):1118-1123. doi:10.1038/ncb1313.
78. Tabner BJ, Mayes J, Allsop D. Hypothesis: Soluble Aβ Oligomers in Association with Redox-Active Metal Ions Are the Optimal Generators of Reactive Oxygen Species in Alzheimer’s Disease. Int J Alzheimers Dis. 2011;2011:1-6. doi:10.4061/2011/546380.
79. Perez J l., Carrero I, Gonzalo P, et al. Soluble oligomeric forms of beta-amyloid (Aβ) peptide stimulate Aβ production via astrogliosis in the rat brain. Exp
Neurol. 2010;223(2):410-421. doi:10.1016/j.expneurol.2009.10.013.
80. Kornhuber J, Muehlbacher M, Trapp S, et al. Identification of Novel Functional Inhibitors of Acid Sphingomyelinase. PLoS ONE. 2011;6(8):1-13. doi:10.1371/journal.pone.0023852.
81. Yagami T. Cerebral Arachidonate Cascade in Dementia: Alzheimer’s Disease and Vascular Dementia. Curr Neuropharmacol. 2006;4(1):87-100. doi:10.2174/157015906775203011.
82. Frisardi V, Panza F, Farooqui AA. Late-life depression and Alzheimer’s disease: The glutamatergic system inside of this mirror relationship. Brain Res Rev. 2011;67(1/2):344-355. doi:10.1016/j.brainresrev.2011.04.003.
83. Choi S-H, Aid S, Kim H-W, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120(2):292-301. doi:10.1111/j.1471-4159.2011.07572.x.
84. Aïd S, Bosetti F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie. 2011;93(1):46-51. doi:10.1016/j.biochi.2010.09.009.

McQuade 57
85. Baron R, Babcock AA, Nemirovsky A, Finsen B, Monsonego A. Accelerated microglial pathology is associated with Aβ plaques in mouse models of Alzheimer’s disease. Aging Cell. 2014;13(4):584-595. doi:10.1111/acel.12210.
86. Lee YS, Lee SJ, Seo KW, Bae JU, Park SY, Kim CD. Homocysteine induces COX-2 expression in macrophages through ROS generated by NMDA receptor-calcium signaling pathways. Free Radic Res. 2013;47(5):422-431. doi:10.3109/10715762.2013.784965.
87. Patrignani P, Tacconelli S, Sciulli MG, Capone ML. New insights into COX-2 biology and inhibition. Brain Res Rev. 2005;48(2):352-359. doi:10.1016/j.brainresrev.2004.12.024.
88. Parsons C, Danysz W, Dekundy A, Pulte I. Memantine and Cholinesterase Inhibitors: Complementary Mechanisms in the Treatment of Alzheimer’s Disease. Neurotox Res. 2013;24(3):358-369. doi:10.1007/s12640-013-9398-z.
89. Lee SJ, Ritchie CS, Yaffe K, Stijacic Cenzer I, Barnes DE. A Clinical Index to Predict Progression from Mild Cognitive Impairment to Dementia Due to Alzheimer’s Disease. PLoS ONE. 2014;9(12):1-15. doi:10.1371/journal.pone.0113535.
90. Wang J, Tan L, Wang H-F, et al. Anti-Inflammatory Drugs and Risk of Alzheimer’s Disease: An Updated Systematic Review and Meta-Analysis. J Alzheimers Dis. 2015;44(2):385-396. doi:10.3233/JAD-141506.
91. Forlenza OV, de Paula VJ, Machado-Vieira R, Diniz BS, Gattaz WF. Does Lithium Prevent Alzheimer’s Disease? Drugs Aging. 2012;29(5):335-342. doi:10.2165/11599180-000000000-00000.
92. Lawrence D. Lithium as a potential Alzheimer’s treatment? Lancet. 2003;361(9371):1796.
93. Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F. Lithium Improves Hippocampal Neurogenesis, Neuropathology and Cognitive Functions in APP Mutant Mice. PLoS ONE. 2010;5(12):1-19. doi:10.1371/journal.pone.0014382.
94. Verzola D, Ratto E, Villaggio B, et al. Uric Acid Promotes Apoptosis in Human Proximal Tubule Cells by Oxidative Stress and the Activation of NADPH Oxidase NOX 4. PLoS ONE. 2014;9(12):1-19. doi:10.1371/journal.pone.0115210.
95. Nakashima H, Ishihara T, Suguimoto P, et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol (Berl). 2005;110(6):547-556. doi:10.1007/s00401-005-1087-4.

McQuade 58
96. Xie C, Zhou K, Wang X, Blomgren K, Zhu C. Therapeutic Benefits of Delayed Lithium Administration in the Neonatal Rat after Cerebral Hypoxia-Ischemia. PLoS ONE. 2014;9(9):1-6. doi:10.1371/journal.pone.0107192.
97. Marmol F. Lithium: Bipolar disorder and neurodegenerative diseases Possible cellular mechanisms of the therapeutic effects of lithium. Prog
Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1761-1771. doi:10.1016/j.pnpbp.2008.08.012.
98. Fisher A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: perspectives and challenges in treatment of Alzheimer’s disease. J Neurochem. 2012;120:22-33. doi:10.1111/j.1471-4159.2011.07507.x.
99. Pomponi MFL, Gambassi G, Pomponi M, Masullo C. Alzheimer’s Disease: Fatty Acids We Eat may be Linked to a Specific Protection via Low-dose Aspirin. Aging Dis. 2010;1(1):37-59.
100. Xu X, Li Z, Yang Z, Zhang T. Decrease of synaptic plasticity associated with alteration of information flow in a rat model of vascular dementia. Neuroscience. 2012;206:136-143. doi:10.1016/j.neuroscience.2011.12.050.
101. Weng P-H, Chen J-H, Chen T-F, et al. CHRNA7 Polymorphisms and Response to Cholinesterase Inhibitors in Alzheimer’s Disease. PLoS ONE. 2013;8(12):1-8. doi:10.1371/journal.pone.0084059.
102. Altena R, Fehrmann RSN, Boer H, de Vries EGE, Meijer C, Gietema JA. Growth Differentiation Factor 15 (GDF-15) Plasma Levels Increase during Bleomycin- and Cisplatin-Based Treatment of Testicular Cancer Patients and Relate to Endothelial Damage. PLoS ONE. 2015;10(1):1-15. doi:10.1371/journal.pone.0115372.
103. Jäger R, Lowery RP, Calvanese AV, Joy JM, Purpura M, Wilson JM. Comparative absorption of curcumin formulations. Nutr J. 2014;13(1):1-18. doi:10.1186/1475-2891-13-11.
104. Belkov M, Polozov G, Skornyakov I, Tolstorozhev G, Shadyro O. Infrared spectra and pharmacological activity of hindered phenols. J Appl Spectrosc. 2011;78(3):400-405. doi:10.1007/s10812-011-9472-3.
105. Tayyem RF, Heath DD, Al-Delaimy WK, Rock CL. Curcumin content of turmeric and curry powders. Nutr Cancer. 2006;55(2):126-131. doi:10.1207/s15327914nc5502_2.
106. Mishra S, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann Indian Acad Neurol. 2008;11(1):13-19. doi:10.4103/0972-2327.40220.

McQuade 59
107. Chandra AK, Parveen S, Das S, Zeegers-Huyskens T. Blue shifts of the C&bond;H stretching vibrations in hydrogen-bonded and protonated trimethylamine. Effect of hyperconjugation on bond properties. J Comput Chem. 2008;29(9):1490-1496. doi:10.1002/jcc.20910.
108. Pianpumepong P, Anal AK, Doungchawee G, Noomhorm A. Study on enhanced absorption of phenolic compounds of Lactobacillus-fermented turmeric ( Curcuma longa Linn.) beverages in rats. Int J Food Sci Technol. 2012;47(11):2380-2387. doi:10.1111/j.1365-2621.2012.03113.x.
109. Potter PE. Curcumin: a natural substance with potential efficacy in Alzheimer’s disease. J Exp Pharmacol. 2013;5:23-31. doi:10.2147/JEP.S26803.
110. Welak SR, Rentea RM, Teng R-J, et al. Intestinal NADPH Oxidase 2 Activity Increases in a Neonatal Rat Model of Necrotizing Enterocolitis. PLoS ONE. 2014;9(12):1-15. doi:10.1371/journal.pone.0115317.
111. Hong J, Bose M, Ju J, et al. Modulation of arachidonic acid metabolism by curcumin and related beta-diketone derivatives: effects on cytosolic phospholipase A(2), cyclooxygenases and 5-lipoxygenase. Carcinogenesis. 2004;25(9):1671-1679. doi:10.1093/carcin/bgh165.
112. Pari L, Tewas D, Eckel J. Role of curcumin in health and disease. Arch Physiol
Biochem. 2008;114(2):127-149. doi:10.1080/13813450802033958.
113. Meng J, Li Y, Camarillo C, et al. The Anti-Tumor Histone Deacetylase Inhibitor SAHA and the Natural Flavonoid Curcumin Exhibit Synergistic Neuroprotection against Amyloid-Beta Toxicity. PLoS ONE. 2014;9(1):1-11. doi:10.1371/journal.pone.0085570.
114. Ka-Heng Lee, Abas F, Mohamed Alitheen NB, Shaari K, Lajis NH, Ahmad S. A Curcumin Derivative, 2,6-Bis(2,5-dimethoxybenzylidene)-cyclohexanone (BDMC33) Attenuates Prostaglandin E2 Synthesis via Selective Suppression of Cyclooxygenase-2 in IFN-γ/LPS-Stimulated Macrophages. Molecules. 2011;16(11):9728-9738. doi:10.3390/molecules16119728.
115. Jäger R, Lowery RP, Calvanese AV, Joy JM, Purpura M, Wilson JM. Comparative absorption of curcumin formulations. Nutr J. 2014;13(1):1-18. doi:10.1186/1475-2891-13-11.
116. Ahmad W, Kumolosasi E, Jantan I, Bukhari SNA, Jasamai M. Effects of Novel Diarylpentanoid Analogues of Curcumin on Secretory Phospholipase A2, Cyclooxygenases, Lipo-oxygenase, and Microsomal Prostaglandin E Synthase-1. Chem Biol Drug Des. 2014;83(6):670-681. doi:10.1111/cbdd.12280.

McQuade 60
117. Pecchi E, Dallaporta M, Jean A, Thirion S, Troadec J-D. Prostaglandins and sickness behavior: Old story, new insights. Physiol Behav. 2009;97(3/4):279-292. doi:10.1016/j.physbeh.2009.02.040.
118. Abdel Shakor AB, Atia M, Ismail IA, et al. Curcumin induces apoptosis of multidrug-resistant human leukemia HL60 cells by complex pathways leading to ceramide accumulation. BBA - Mol Cell Biol Lipids. 2014;1841(12):1672-1682. doi:10.1016/j.bbalip.2014.09.006.
119. Sethiya NK, M. K. Mohan Maruga Raja, Mishra H. Antioxidant markers based TLC-DPPH differentiation on four commercialized botanical sources of Shankhpushpi (A Medhya Rasayana): A preliminary assessment. J Adv Pharm
Technol Res. 2013;4(1):25-30. doi:10.4103/2231-4040.107497.
Figures
1. Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling.
J. Lipid Res. 2008;50(Supplement):S237-S242. doi:10.1194/jlr.R800033-JLR200.
2. Shimizu T, Wolfe LS. Arachidonic Acid Cascade and Signal Transduction. J
Neurochem. 1990;55(1):1-15. doi:10.1111/j.1471-4159.1990.tb08813.x.
3. Shelat PB, Chalimoniuk M, Wang J-H, et al. Amyloid beta peptide and NMDA induce
ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in
cortical neurons. J Neurochem. 2008;106(1):45-55. doi:10.1111/j.1471-
4159.2008.05347.x.
4. Malaplate-Armand C, Florent-Béchard S, Youssef I, et al. Soluble oligomers of
amyloid-β peptide induce neuronal apoptosis by activating a cPLA2-dependent
sphingomyelinase-ceramide pathway. Neurobiol Dis. 2006;23(1):178-189.
doi:10.1016/j.nbd.2006.02.010.
5. Zilka N, Kazmerova Z, Jadhav S, et al. Who fans the flames of Alzheimer’s disease
brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory
pathways. J Neuroinflammation. 2012;9:47. doi:10.1186/1742-2094-9-47.6

McQuade 61
6. Parsons C, Danysz W, Dekundy A, Pulte I. Memantine and Cholinesterase Inhibitors:
Complementary Mechanisms in the Treatment of Alzheimer’s Disease. Neurotox
Res. 2013;24(3):358-369. doi:10.1007/s12640-013-9398-z.
7. Yang G, Haczku A, Chen H. Transgenic smooth muscle expression of the human
CysLT1, receptor induces enhanced responsiveness of murine airways to
leukotriene D4. Am J Physiol. 2004;286(5):L992-L1001.
8. Pianpumepong P, Anal AK, Doungchawee G, Noomhorm A. Study on enhanced
absorption of phenolic compounds of Lactobacillus-fermented turmeric (
Curcuma longa Linn.) beverages in rats. Int J Food Sci Technol.
2012;47(11):2380-2387. doi:10.1111/j.1365-2621.2012.03113.x.
9. Srivastava KC, Bordia A, Verma SK. Curcumin, a major component of food spice
turmeric (Curcuma longa) inhibits aggregation and alters eicosanoid metabolism in
human blood platelets. Prostaglandins Leukot Essent Fatty Acids. 1995;52(4):223-
227.
10. Potter PE. Curcumin: a natural substance with potential efficacy in Alzheimer’s
disease. J Exp Pharmacol. 2013;5:23-31. doi:10.2147/JEP.S26803.


















