
The Stimulation of Protein Degradation in Muscle by Ca2 ... · The Stimulation of Protein...
8
The Stimulation of Protein Degradation in Muscle by Ca2* Is Mediated by Prostaglandin E2 and Does Not Require the Calcium-activated Protease* (Received for publication, November 18, 1981) H. Peter RodemannS, Lloyd Waxman, and Alfred L. Goldberg From the Department of Physiology a n d Biophysics, Haruard Medical School, Boston, Massachusetts 02115 Treatment of isolated rat skeletal muscles with the Ca2+ ionophores, A23181 or ionomycin, increased over- all protein degradation 45-140%. Removal of extracel- lular Ca2+ reduced overall proteolysis and most of the stimulation by A23187. Treatment of the muscles with the sulfhydryl inhibitor, mersalyl, completely inacti- vated the Ca2’-activated protease without altering overall protein breakdown or the stimulation by A23187. This agent did not inhibit the lysosomal pro- tease, cathepsin B, in the muscle; however, leupeptin and Ep-475, which inhibit this enzyme in intact cells, decreased the stimulation of proteolysis by Ca2+. Thus, this effect does not require the Ca2+-activated enzyme, but seems to involve lysosomal proteases, Prostaglandin E, (PGE2) and its precursor, arachi- donic acid, were previously shown to stimulate protein degradation in rat muscle through an effect on lyso- somal function. We tested whether the enhancement of muscle proteolysis by CaZ+ ionophores may result from increased synthesis of PGE2. A23187 increased release of PGEz and PGF2, by the muscles 3-4-fold. High extra- cellular potassium also markedly promotes muscle pro- teolysis, apparently by increasing intracellular Ca2+, and this treatment also stimulates prostaglandin pro- duction. Indomethacin and aspirin, which inhibit the cyclooxygenase, and mepacrine, which inhibits the Ca“-activated phospholipase A,, markedly reduced the increase in prostaglandin production. These agents also reduced the enhancement of protein degradation by Ca2+ or high K+. Thus, Ca2+ appears to promote protein breakdown by stimulating synthesis of PGE2, which in turn activates the lysosomal apparatus. Although much has been learned about the effects of hor- mones and other physiological factors on protein breakdown in muscle, the intracellular mechanisms that regulate this process have not been elucidated. Further knowledge about these regulatory mechanisms is not only of scientific interest, but also may be of appreciable medical importance, since many pathological states involve muscle atrophy or hypertro- phy. Recently, Kameyama and Etlinger and co-workers (1,2) * This work has been supported by research grants from the National Institute of Neurological Disease and Stroke and the Mus- cular Dystrophy Association of America. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. Recipient of a fellowship from the Deutsche Forschungsgemein- schaft (Ro 527/1). Present address, University of Hohenheim, Insti- tute for Genetics, D7000 Stuttgart 70, Postfach 106, Emil-Wolff- Strasse 14, West Germany. reported that an increase in the intracellular level of Ca2+ induced with the ionophore A23187 stimulates protein syn- thesis and breakdown in isolated rat skeletal muscles. A similar enhancement of protein breakdown was observed when these muscles were exposed to high levels of K+ (Z), which can depolarize the muscle membraneand induce a release of Ca2+ from the sarcoplasmic reticulum intothe cytoplasm (2). The primary goal of the present studies was to learn more about the mechanisms of these effects of Ca2+ on protein turnover. It was originally proposed (1, 2) that the enhanced proteolysis resulted from binding of Ca” to the Ca2’-activated protease. This enzyme is present in skeletal muscle and other mammalian cells in two forms, one which requires 20 PM Ca” (4) for activity and one requiring about 250 p~. Treatment of rat muscles with leupeptin, an inhibitor of the Ca2’-activated protease (3), was found to reduce the effect of the ionophore on protein degradation (I), but this agent also inhibits lyso- somal thiol proteases(3). Several other reportshave also postulated a role for CaZ+ and the Ca2+-activated protease in the breakdown of myofibrillar proteins (5-10) underboth physiological and pathological conditions (11-14). This possi- bility is of particular interest because of the reports that dystrophic and denervated muscle, in which overall proteol- ysis is enhanced, show increased permeability to Ca2+ (15-17) or decreased ability of the sarcoplasmic reticulum to sequester Ca2+ (18, 19). These investigations were undertaken to clarify whether changes in the Ca’+ levels affect protein breakdown by acti- vation of the Ca’+-activated protease or by some alternative mechanism. CaS+ ionophores are known to affect a wide vari- ety of cell processes. For example, in several types of mam- malian ceIls the Ca’+ ionophore A23187 enhances prostaglan- din production (20-23), because phospholipase Az, the rate- limiting step in the biosynthesis of prostaglandins, prostacy- clin, and thromboxanes (21, 23), is Ca2+-dependent. This en- zyme releases arachidonic acid from membrane phospholipids and thus supplies the precursors for the synthesis of prosta- glandin endoperoxides by the cyclooxygenases (21, 23). The possible existence of a similar pathwayin muscle was of special interest, because we have recently demonstrated that arachidonic acid, PGHa,‘ and PGE2 increase rates of protein breakdown in muscle (24), while PGF2- enhances protein synthesis. Studies with leupeptin and other inhibitors of ly- sosomal proteases suggested thatthis increase in protein breakdown involves the lysosomal pathway (24). Therefore, ‘ The abbreviations used are: PG, prostaglandin; EGTA, ethylene glycol bis(P-aminoethyl ether)-N,N,N’,N”tetraacetic acid; CBZ-Ala- Arg-Arg-MNA, carbobenzoxy-alanyl-arginyl-arginyl-4-methoxy-,8- naphthylamine. 8716
Transcript of The Stimulation of Protein Degradation in Muscle by Ca2 ... · The Stimulation of Protein...

The Stimulation of Protein Degradation in Muscle by Ca2* Is Mediated by Prostaglandin E2 and Does Not Require the Calcium-activated Protease*
(Received for publication, November 18, 1981)
H. Peter RodemannS, Lloyd Waxman, and Alfred L. Goldberg From the Department of Physiology and Biophysics, Haruard Medical School, Boston, Massachusetts 02115
Treatment of isolated rat skeletal muscles with the Ca2+ ionophores, A23181 or ionomycin, increased over- all protein degradation 45-140%. Removal of extracel- lular Ca2+ reduced overall proteolysis and most of the stimulation by A23187. Treatment of the muscles with the sulfhydryl inhibitor, mersalyl, completely inacti- vated the Ca2’-activated protease without altering overall protein breakdown or the stimulation by A23187. This agent did not inhibit the lysosomal pro- tease, cathepsin B, in the muscle; however, leupeptin and Ep-475, which inhibit this enzyme in intact cells, decreased the stimulation of proteolysis by Ca2+. Thus, this effect does not require the Ca2+-activated enzyme, but seems to involve lysosomal proteases,
Prostaglandin E, (PGE2) and its precursor, arachi- donic acid, were previously shown to stimulate protein degradation in rat muscle through an effect on lyso- somal function. We tested whether the enhancement of muscle proteolysis by CaZ+ ionophores may result from increased synthesis of PGE2. A23187 increased release of PGEz and PGF2, by the muscles 3-4-fold. High extra- cellular potassium also markedly promotes muscle pro- teolysis, apparently by increasing intracellular Ca2+, and this treatment also stimulates prostaglandin pro- duction. Indomethacin and aspirin, which inhibit the cyclooxygenase, and mepacrine, which inhibits the Ca“-activated phospholipase A,, markedly reduced the increase in prostaglandin production. These agents also reduced the enhancement of protein degradation by Ca2+ or high K+. Thus, Ca2+ appears to promote protein breakdown by stimulating synthesis of PGE2, which in turn activates the lysosomal apparatus.
Although much has been learned about the effects of hor- mones and other physiological factors on protein breakdown in muscle, the intracellular mechanisms that regulate this process have not been elucidated. Further knowledge about these regulatory mechanisms is not only of scientific interest, but also may be of appreciable medical importance, since many pathological states involve muscle atrophy or hypertro- phy. Recently, Kameyama and Etlinger and co-workers ( 1 , 2 )
* This work has been supported by research grants from the National Institute of Neurological Disease and Stroke and the Mus- cular Dystrophy Association of America. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Recipient of a fellowship from the Deutsche Forschungsgemein- schaft (Ro 527/1). Present address, University of Hohenheim, Insti- tute for Genetics, D7000 Stuttgart 70, Postfach 106, Emil-Wolff- Strasse 14, West Germany.
reported that an increase in the intracellular level of Ca2+ induced with the ionophore A23187 stimulates protein syn- thesis and breakdown in isolated rat skeletal muscles. A similar enhancement of protein breakdown was observed when these muscles were exposed to high levels of K+ (Z), which can depolarize the muscle membrane and induce a release o f Ca2+ from the sarcoplasmic reticulum into the cytoplasm (2).
The primary goal of the present studies was to learn more about the mechanisms of these effects of Ca2+ on protein turnover. It was originally proposed (1, 2) that the enhanced proteolysis resulted from binding of Ca” to the Ca2’-activated protease. This enzyme is present in skeletal muscle and other mammalian cells in two forms, one which requires 20 PM Ca” (4) for activity and one requiring about 250 p ~ . Treatment of rat muscles with leupeptin, an inhibitor of the Ca2’-activated protease (3), was found to reduce the effect of the ionophore on protein degradation (I), bu t this agent also inhibits lyso- somal thiol proteases (3). Several other reports have also postulated a role for CaZ+ and the Ca2+-activated protease in the breakdown of myofibrillar proteins (5-10) under both physiological and pathological conditions (11-14). This possi- bility is of particular interest because of the reports that dystrophic and denervated muscle, in which overall proteol- ysis is enhanced, show increased permeability to Ca2+ (15-17) or decreased ability of the sarcoplasmic reticulum to sequester Ca2+ (18, 19).
These investigations were undertaken to clarify whether changes in the Ca’+ levels affect protein breakdown by acti- vation of the Ca’+-activated protease or by some alternative mechanism. CaS+ ionophores are known to affect a wide vari- ety of cell processes. For example, in several types of mam- malian ceIls the Ca’+ ionophore A23187 enhances prostaglan- din production (20-23), because phospholipase Az, the rate- limiting step in the biosynthesis of prostaglandins, prostacy- clin, and thromboxanes (21, 23), is Ca2+-dependent. This en- zyme releases arachidonic acid from membrane phospholipids and thus supplies the precursors for the synthesis of prosta- glandin endoperoxides by the cyclooxygenases (21, 23). The possible existence of a similar pathway in muscle was of special interest, because we have recently demonstrated that arachidonic acid, PGHa,‘ and PGE2 increase rates of protein breakdown in muscle (24), while PGF2- enhances protein synthesis. Studies with leupeptin and other inhibitors of ly- sosomal proteases suggested that this increase in protein breakdown involves the lysosomal pathway (24). Therefore,
‘ The abbreviations used are: PG, prostaglandin; EGTA, ethylene glycol bis(P-aminoethyl ether)-N,N,N’,N”tetraacetic acid; CBZ-Ala- Arg-Arg-MNA, carbobenzoxy-alanyl-arginyl-arginyl-4-methoxy-,8- naphthylamine.
8716

Stimulation of Protein Degradation in Muscle by Ca2+ Involues PGE, 8717
we examined to what extent the Ca2+-induced stimulation of protein degradation may be due to enhanced production of prostaglandin E, and thereby to an activation of lysosomal proteolysis.
EXPERIMENTAL PROCEDURES
Methods-Young CD strain rats (60-80 g) were obtained from Charles River Breeding Laboratories, Inc. and were maintained on Purina Lab Chow and water ad libitum. Quarter diaphragms or the hind limb muscles, soleus, and extensor digitorum longus were dis- sected as described previously (25, 26). The muscles were preincu- bated at 37 "C for 30 min, prior to the final 2-h incubation. Both incubations were carried out in Krebs-Ringer bicarbonate buffer, equilibrated with 5% C02 and 95% oxygen. Except where indicated, this buffer was supplemented with 5 mM glucose, 0.1 unit/ml of insulin, and five times the normal plasma concentration of branched chain amino acids. In different experiments, either cycloheximide (0.5 mM), an inhibitor of protein synthesis, or [i4C]phenylalanine (0.05 pCi/ml) plus unlabeled phenylalanine (0.5 mM) were added to the medium. Rates of protein synthesis and protein degradation were measured as described earlier (26, 27). In studies using inhibitors of the cyclooxygenase, aspirin or indomethacin (29, 30), these agents were present during the 30-min preincubation at the same concentra- tion as in the subsequent incubation. When mepacrine, an irreversible inhibitor of phospholipases (31), was used, this agent was only added to the preincubation medium, since it interfered with the fluorometric tyrosine assay (28). Muscles preincubated with mepacrine were washed carefully before transfer to the final incubation medium.
The calcium ionophore A23187 was dissolved in dimethyl sulfoxide and stored frozen at -30 "C. When A23187 was added to the incu- bation medium, the final concentration of dimethyl sulfoxide did not exceed 0.1%; the control muscles were incubated in medium contain- ing the same amount of dimethyl sulfoxide. However, the final con- centration of dimethyl sulfoxide did not affect basal rates of protein synthesis or degradation (data not shown). When rat skeletal muscles were exposed to medium with high levels of potassium, Krebs-Ringer bicarbonate buffer was prepared by substituting 100 mM KC1 for 100 mM NaCl in Krebs-Ringer bicarbonate buffer. When muscles were incubated in Ca'+-free medium, the buffer was prepared without CaC12 and 1.0 mM EGTA was added.
The amount of Ca'+-activated protease activity in individual soleus muscles was determined after the standard 2-h incubation. Each muscle (20-30 mg wet weight) was rinsed in ice-cold saline and then homogenized at 4 "C with 10 strokes in a ground glass homogenizer in 2 ml of buffer consisting of 5 m~ Tris-HC1 (pH 7.4), 25 mM NaCl, 10 mM 2-mercaptoethanol, and 0.1 n m EDTA. Particulate matter was removed by centrifugation at 30,000 X g for 30 min and 100-pl aliquots (50-100 pg of protein) of the supernatant were assayed for proteolytic activity by measuring the production of acid-soluble w- dioactivity from r3H]casein in the presence and absence of 2 mM CaC12. The reaction mixture contained 20 p1 of 0.1 M Tris-HC1 (pH 7.4), 20 pg of [methyl-"Hlcu-casein (50,ooO-100,000 cpm) (32), and 20 pl of 20 mM CaC12 or H20 in a final volume of 200 pl. Following incubation at 25 "C for 20 or 40 min, the reaction was stopped by the addition of 25 pl of 10% bovine serum albumin and 575 pl of 10% trichloroacetic acid. After standing 1 h on ice, the mixture was centrifuged to remove insoluble protein and 400 pl of the supernatant were counted in 4 ml of Liquiscint (National Diagnostics).
In these assays, no attempt was made to remove the endogenous inhibitor of the Ca'+-activated protease and in those experiments which involved the sulfhydryl reagent mersalyl, 2-mercaptoethanol was omitted from the extraction buffer. Mersalyl also has no effect on the activity of the endogenous inhibitor.'
For the measurement of cathepsin B activity, crude homogenates of rat soleus muscles were prepared by washing the freshly excised tissues or muscles after a 2-h incubation in uitro in saline solution (pH 7.4), placing them in buffer (0.15 M KCI, pH 5.0), and disrupting them with four 30-s bursts of a Tekrnar homogenizer. A 10% homog- enate (w/v) was prepared and frozen at -30 "C to disrupt lysosomes. Cathepsin B activity was measured by monitoring the hydrolysis of the substrate CBZ-ala-arg-arg-MNA fluorometrically in 75 mM K7HPO4, 2.25 mM citric acid buffer (pH 6.0) containing 1 mM EDTA and in the presence or absence of 0.5 m~ dithiothreitol. When mersalyl was added to the muscle homogenate, dithiothreitol was
' L. Waxman, unpublished observations.
omitted from the assay buffer. The net production of prostaglandins Ez and F2,\ was measured by
radioimmunoassay of the released PGEz and PGFp, recovered in the medium, during the 2-h incubation. For measurements of PGEz and PGF2,, rabbit anti-PGE2 or anti-PGFz, was used. Although these antibodies were produced by repeated injections of prostaglandin EZ or Fze, they also reacted strongly with PGEI and PGF,,, respectively. The cross-reactivity of the anti-PGEz antibody with PGEl is 100% but with PGF2, less than 5%. while the anti-PGFp,, antibody cross- reacts with PGF!,, 100W, but with PGE2 less than 0.01% (Product Information Service, Miles Biochemicals). For the radioimmunoassay, aliquots of the incubation media were used without solvent extraction (33). The sensitivity of this assay is not sufficient to detect less than 10 pg of antigen per sample.
In all experiments, differences between experimental groups were evaluated using the paired Student's t-test. Values given are for the means & S. E. of at least six determinations.
Materials-Calcium ionophore A23187 was a product of Calbi- ochem-Behring. Ionomycin was kindly provided by Dr. Robert Perl- man (University of Illinois Medical School, Chicago). Indomethacin was purchased from Sigma. Mepacrine was a product of ICN Phar- maceuticals, Inc., Plainview, NY. ['HIPGEz (165 mCi/mmol) and [JH] PGF2, (150 mCi/mmol) were obtained from New England Nuclear. ['4C]Phenylalanine (460 mCi/mmol) was a product of Schwarz/Mann. Antisera against PGEa and PGF, were purchased from Miles Bio- chemicals, Elkhart, IN.
The protease inhibitor Ep-475 (E-64-C) was kindly provided to us by Dr. K. Hanada of the Taisho Pharmaceutical Co. Ltd., Saitama, Japan, and leupeptin by Professors T. Aoyagi and H. Umezawa, Institute of Microbial Chemistry, Shinagawa-ku, Tokyo, Japan. Mer- salyl (sodium O-[3-(hydroxymercury)-2-methoxypropyl]carbamoyl phenoxyacetate) was purchased from Sigma. The fluorometric sub- strate, CBZ-Ala-Arg-Arg-MNA, was purchased from Enzyme Sys- tems Products, Livermore, CA.
RESULTS
Effects of Ca'+ Ionophores and High K' on Muscle Protein Turnover-Upon the addition of the Cat' ionophore A23187 to the incubation medium, rates of tyrosine release from the diaphragm, red soleus, and white extensor digitorum longus muscles increased between 63 and 116% (Table I) in accord with earlier reports (1). In a series of control experiments, A23187 did not alter the amount of intracellular tyrosine. Therefore, the increase in tyrosine release by the muscles reflects increased net protein breakdown and not enhanced leakage of amino acids from intracellular pools.
In contrast with previous reports of Kameyama and Etlin- ger ( I ) , in our experiments the rates of protein synthesis did not increase significantly after treatment with A23187 in any muscle (although in the diaphragm, mean rates appeared to be higher after A23187 treatment). Thus, the overall rates of protein degradation increased in all muscle types tested be- tween 42% and 63% ( p < 0.001) (Table I). A similar increase in tyrosine production has been obtained when absolute rates of protein degradation were measured in the presence of cycloheximide (data not shown). Because no significant change in protein synthesis was observed with A23187 (Table I), in subsequent experiments with this agent, only tyrosine release was used as a measure of protein breakdown.
These results with A23187 are supported by findings ob- tained with another specific Ca2+ ionophore, ionomycin. This ionophore enhanced absolute rates of protein degradation in rat diaphragm by about 160% (Table I). These data were obtained in the presence of cycloheximide; in related studies;' no significant change in protein synthesis was observed in accord with the findings with A23187 (Table I). Since A23187 is better characterized and is more readily available, all sub- sequent studies used this agent.
In order to test whether the effects of -4423187 on protein
'' L. Waxman, H. P. Rodemann, and A. L. Goldberg, manuscript in preparation.
8718 Stimulation of Protein Degradation in Muscle by Ca2' Involves PGE2
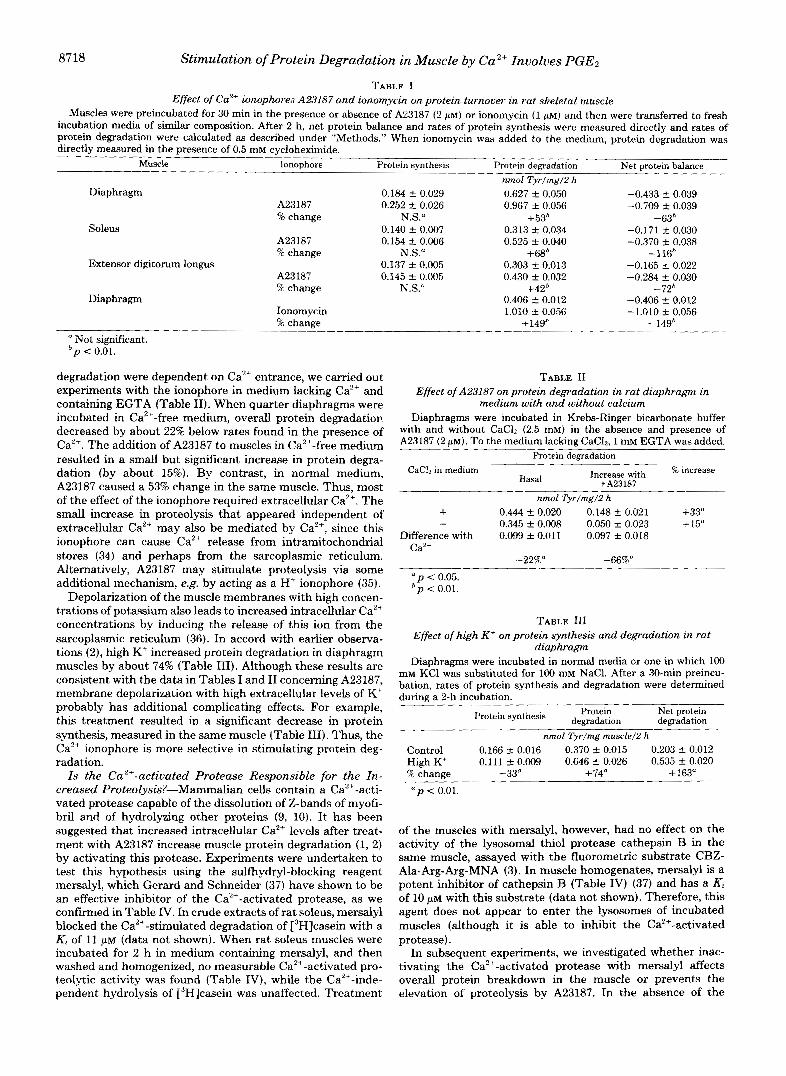
TABLE I Effect of eaZ+ ionophore; A23187 and ionomycin on protein turnover in rat skeletal muscle
Muscles were preincubated for 30 min in the presence or absence of A23187 (2 p ~ ) or ionomycin (1 p ~ ) and then were transferred to fresh incubation media of similar composition. After 2 h, net protein balance and rates of protein synthesis were measured directly and rates of protein degradation were calculated as described under "Methods." When ionomycin was added to the medium, protein degradation was directly measured in the presence of 0.5 mM cycloheximide.
Muscle Ionophore Protein synthesis Protein demadation Net protein balanc-
Diaphragm
Soleus
Extensor digitorum longus
Diaphragm
A23187 '% change
A23187 5% change
A23187 % change
Ionomycin '% change
0.184 C 0.029 0.252 f 0.026
N.S." 0.140 f 0.007 0.154 f 0.006
N.S." 0.137 f 0.005 0.145 f 0.005
N.S."
nmol Tyr/mg/2 h 0.627 f 0.050 0.967 f 0.056
+53' 0.313 f 0.034 0.525 f 0.040
+68' 0.303 f 0.013 0.430 f 0.032
+42' 0.406 C 0.012 1.010 f 0.056
+149'
-0.433 f 0.039 -0.709 k 0.039
-63h -0.171 f 0.030 -0.370 f 0.038
-116' -0.165 f 0.022 -0.284 f 0.030
-72' -0.406 5 0.012 -1.010 2 0.056
-14gh Not significant.
bp < 0.01.
degradation were dependent on Ca" entrance, we carried out experiments with the ionophore in medium lacking Ca2+ and containing EGTA (Table 11). When quarter diaphragms were incubated in Ca2+-free medium, overall protein degradatior decreased by about 22% below rates found in the presence of Ca". The addition of A23187 to muscles in Ca"-free medium resulted in a small but significant increase in protein degra- dation (by about 15%). By contrast, in normal medium, A23187 caused a 53% change in the same muscle. Thus, most of the effect of the ionophore required extracellular Ca2+. The small increase in proteolysis that appeared independent of extracellular Ca" may also be mediated by Ca", since this ionophore can cause Ca2+ release from intramitochondrial stores (34) and perhaps from the sarcoplasmic reticulum. Alternatively, A23187 may stimulate proteolysis via some additional mechanism, e.g. by acting as a H' ionophore (35).
Depolarization of the muscle membranes with high concen- trations of potassium also leads to increased intracellular Ca2+ concentrations by inducing the release of this ion from the sarcoplasmic reticulum (36). In accord with earlier observa- tions (2), high K' increased protein degradation in diaphragm muscles by about 74% (Table 111). Although these results are consistent with the data in Tables I and I1 concerning A23187, membrane depolarization with high extracellular levels of K+ probably has additional complicating effects. For example, this treatment resulted in a significant decrease in protein synthesis, measured in the same muscle (Table 111). Thus, the Caz+ ionophore is more selective in stimulating protein deg- radation. Is the Ca2+-activated Protease Responsible for the In-
creased Proteolysis?-Mammalian cells contain a Ca2+-acti- vated protease capable of the dissolution of Z-bands of myofi- bril and of hydrolyzing other proteins (9, 10). It has been suggested that increased intracellular Ca2+ levels after treat- ment with A23187 increase muscle protein degradation (1, 2) by activating this protease. Experiments were undertaken to test this hypothesis using the sulfhydryl-blocking reagent mersalyl, which Gerard and Schneider (37) have shown to be an effective inhibitor of the Ca'+-activated protease, as we confirmed in Table IV. In crude extracts of rat soleus, mersalyl blocked the Ca"-stimulated degradation of r3H]casein with a K, of 11 FM (data not shown). When rat soleus muscles were incubated for 2 h in medium containing mersalyl, and then washed and homogenized, no measurable Ca"-activated pro- teolytic activity was found (Table IV), while the Ca'+-inde- pendent hydrolysis of ["]casein was unaffected. Treatment
TABLE I1 Effect of A23187 on protein degradation in rat diaphragm in
medium with and without calcium Diaphragms were incubated in Krebs-Ringer bicarbonate buffer
with and without CaC12 (2.5 mM) in the absence and presence of A23187 (2 PM). To the medium lacking CaC12,l rn EGTA was added.
Protein degradation CaCL in medium
Basal Increase with ?& increase +A23187 .~"~.
nmol Tyrlmg/Z h + 0.444 f 0.020 0.148 f 0.021 +33" - 0.345 f 0.008 0.050 f 0.023 +15"
Difference with 0.099 f 0.011 0.097 f 0.018 Ca2+
-22%10" ""/oh
' p < 0.05. hp < 0.01.
TABLE I11 Effect of high K' on protein synthesis and degradation in rat
diaphragm Diaphragms were incubated in normal media or one in which 100
mM KC1 was substituted for 100 m NaC1. After a 30-min preincu- bation, rates of protein synthesis and degradation were determined during a 2-h incubation.
Protein synthesis dg:E:on Net protein degradation
nmol Tyr/mg muscle/2 h
Control 0.166 2 0.016 0.370 f 0.015 0.203 f 0.012 High K' 0.111 f 0.009 0.646 f 0.026 0.535 f 0.020 '% change -33" + 74" + 163"
" p < 0.01.
of the muscles with mersalyl, however, had no effect on the activity of the lysosomal thiol protease cathepsin B in the same muscle, assayed with the fluorometric substrate CBZ- Ala-Arg-Arg-MNA (3). In muscle homogenates, mersalyl is a potent inhibitor of cathepsin B (Table IV) (37) and has a K, of 10 PM with this substrate (data not shown). Therefore, this agent does not appear to enter the lysosomes of incubated muscles (although it is able to inhibit the Ca2+-activated protease).
In subsequent experiments, we investigated whether inac- tivating the Ca2'-activated protease with mersalyl affects overall protein breakdown in the muscle or prevents the elevation of proteolysis by A23187. In the absence of the
Stimulation of Protein Degradation in Muscle by Ca2+ Involves PGE2 8719
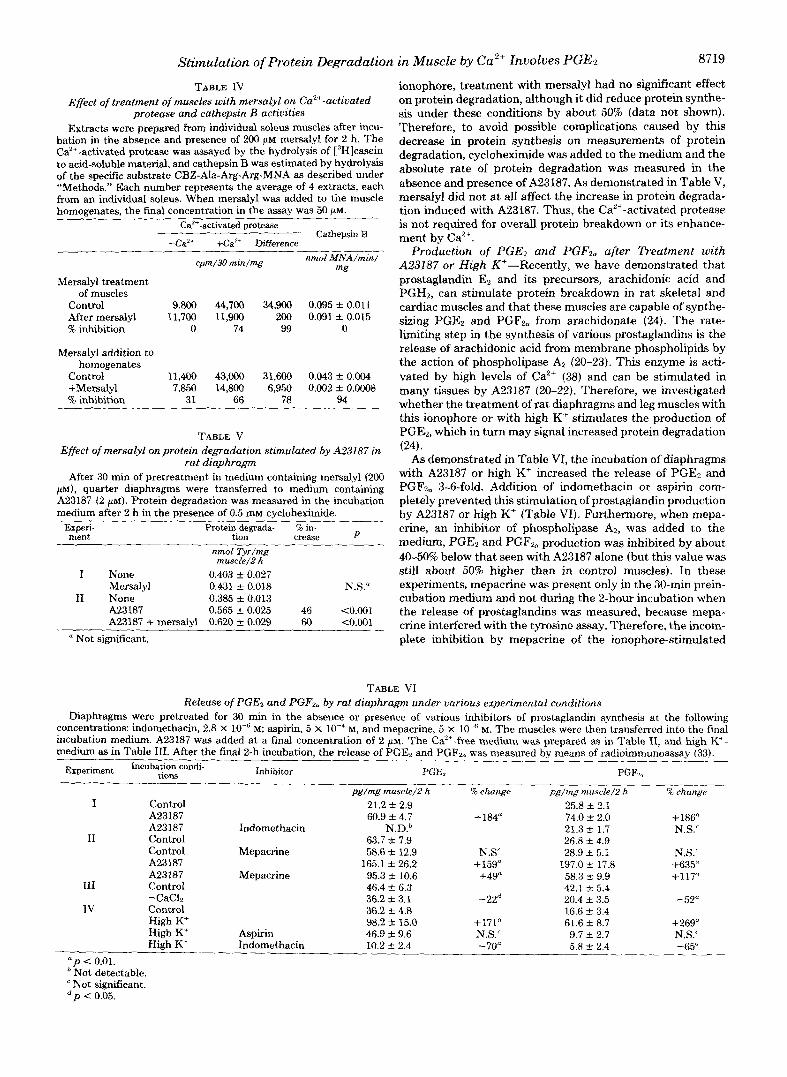
TABLE IV Effect of treatment of muscles with mersalyl on Ca"+-activated
protea.se and cathepsin B activities Extracts were prepared from individud soleus muscles after incu-
bation in the absence and presence of 200 p~ mersalyl for 2 h. The Ca2+-activated protease was assayed by the hydrolysis of ["]casein to acid-soluble material, and cathepsin B was estimated by hydrolysis of the specific substrate CBZ-Ala-Arg-Arg-MNA as described under "Methods." Each number represents the average of 4 extracts, each from an individual soleus. When mersalyl was added to tile muscle homoeenates. the final concentration in the assav was 50 uM.
~ ~~
ea'+-activated protease Cathepsin B
-Ca" +Ca'+ Difference
cpm/30 mm/mg nmol .WNA/min/ mg
Mersalyl treatment of muscles
Control 9,800 44,700 34,900 0.095 f 0.011 After mersalyl 11,700 11,900 200 0.091 f 0.015 % inhibition 0 74 99 0
Mersalyl addition to homogenates
Control 11,400 43,W 31,600 0.043 f 0.004 +Mersalyl 7,850 14,800 6,950 0.002 f 0.0008 '% inhibition 31 66 78 94
TABLE V Effect of mersalyl onprotein degradation stimulated by A23187 in
rat diaphragm After 30 min of pretreatment in medium containing mersalyl (200
PM), quarter diaphragms were transferred to medium containing A23187 (2 p l ) . Protein degradation was measured in the incubation medium after 2 h in the presence of 0.5 m cycloheximide.
_I_
Experi- ment
Protein degrada- % in- tion crease P
nmol Tyr/mg muscle/Z h
I None 0.403 & 0.027 Mersalyl 0.431 _t 0.018 N.S."
I1 None 0.385 t- 0.013 A23187 0.565 * 0.025 46 ~0.001 A23187 + mersalyl 0.620 & 0.029 60 tO.OO1
Not significant.
ionophore, treatment with mersalyl had no significant effect on protein degradation, although it did reduce protein synthe- sis under these conditions by about 50% (data not shown). Therefore, to avoid possible complications caused by this decrease in protein synthesis on measurements of protein degradation, cycloheximide was added to the medium and the absolute rate of protein degradation was measured in the absence and presence of A23187. As demonstrated in Table V, mersalyl did not a t all affect the increase in protein degrada- tion induced with A23187. Thus, the Ca"-activated protease is not required for overall protein breakdown or its enhance- ment by Ca2+.
Production of PGE2 and PGF,, after Treatment with A23187 or High K'-Recently, we have demonstrated that prostaglandin Ez and its precursors, arachidonic acid and PGH2, can stimulate protein breakdown in rat skeletal and cardiac muscles and that these muscles are capable of synthe- sizing PGE2 and PGR, from arachidonate (24). The rate- limiting step in the synthesis of various prostaglandins is the release of arachidonic acid from membrane phospholipids by the action of phospholipase A2 (20-23). This enzyme is acti- vated by high levels of Ca2' (38) and can be stimulated in many tissues by A23187 (20-22). Therefore, we investigated whether the treatment of rat diaphragms and leg muscles with this ionophore or with high K' stimulates the production of PGEz, which in turn may signal increased protein degradation
As demonstrated in Table VI, the incubation of diaphragms with A23187 or high K' increased the release of PGE, and PGFz, 3-6-fold. Addition of indomethacin or aspirin com- pletely prevented this stimulation of prostaglandin production by A23187 or high K' (Table VI). Furthermore, when mepa- crine, an inhibitor of phospholipase AP, was added to the medium, PG& and PGF,, production was inhibited by about 40-50% below that seen with A23187 alone (but this value was still about 50% higher than in control muscles). In these experiments, mepacrine was present only in the 30-min prein- cubation medium and not during the 2-hour incubation when the release of prostaglandins was measured, because mepa- crine interfered with the tyrosine assay. Therefore, the incom- plete inhibition by mepacrine of the ionophore-stimulated
(24).
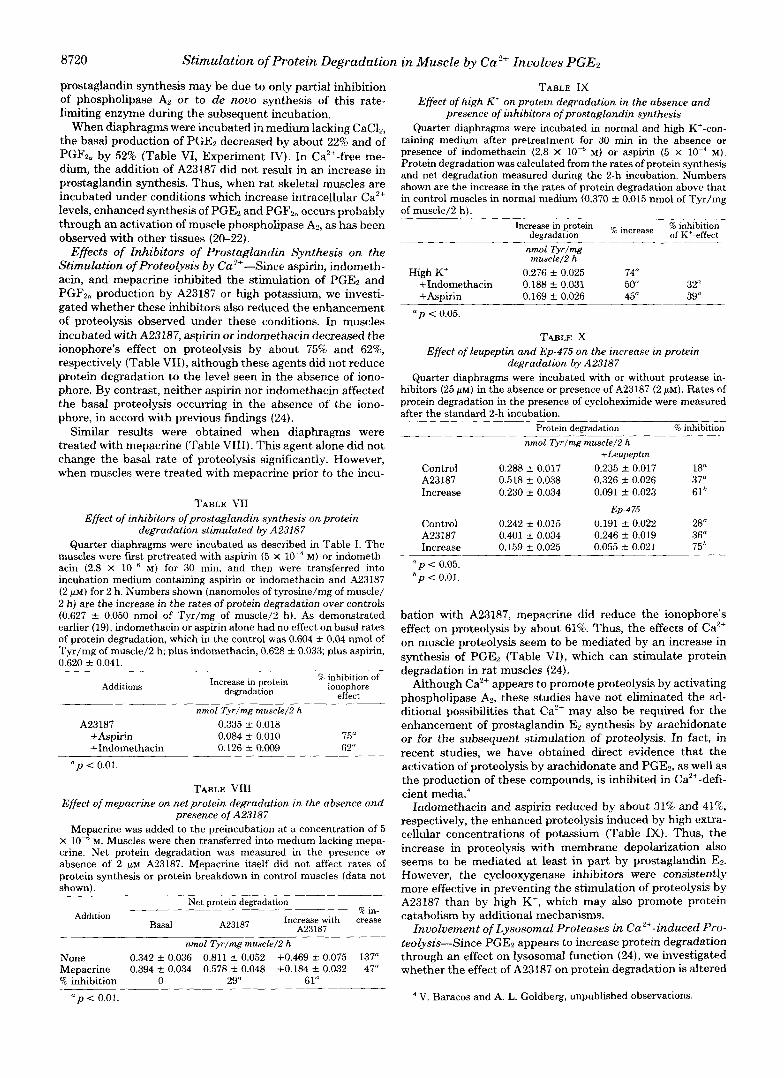
TABLE VI Release of PGEz and PGF,, by rat diaphragm under various experimental conditions
Diaphragms were pretreated for 30 min in the absence or presence of various inhibitors of prostaglandin synthesis at the following concentrations: indomethacin, 2.8 X M; aspirin, 5 X M, and mepacrine, 5 x M. The muscles were then transferred into the final incubation medium. A23187 was added at a final concentration of 2 PM. The Ca2'-free me.dium was prepared as in Table 11, and high K+- medium as in Table 111. After the final 2-h incubation, the release of PGE, and PGF2,, was measured by means of radioimmunoassay (33).
Experiment Incubation condi- tions Inhibitor PGE, PGFL,
I Control 21.2 f 2.9 25.8 k 2.1 60.9 f 4.7 + m u 74.0 2 2.0
A23187 Indomethacin N.D.b I1
21.3 +- 1.7 Control
N.S.' 63.7 & 7.9
Control Mepacrine 58.6 -c_ 12.9 N.S' 28.9 k 5.1 N.S.' A23187 A23187
165.1 & 26.2 + 159" 197.0 k 17.8 +635" Mepacrine 95.3 t- 10.6 +49" 58.3 9.9
111 Control +11Y
-CaCL 42.1 S 5.4
IV -22" 20.4 k 3.5
Control -52"
High K' 16.6 2 3.4
High K' High K'
Aspirin 46.9 k 9.6 N.S.' 9.7 k 2.7 Indomethacin
pg/mg muscle/Z h % change pg/mg muscle/2 h % change
~ 2 3 1 8 7 + 186
26.8 k 4.9
46.4 t- 6.3 36.2 k 3.1 36.2 k 4.8 98.2 s_ 15.0 +171" 61.6 k 8.7 +269"
N.S.' 10.2 +. 2.4 - 70" 5.8 2 2.4 -65"
" p 0.01. Not detectable. Not significant.
d p c 0.05.
Stimulation of Protein Degradation in Muscle by ea2+ Involves PGE2
prostaglandin synthesis may be due to only partial inhibition of phospholipase A2 or to de novo synthesis of this rate- limiting enzyme during the subsequent incubation.
When diaphragms were incubated in medium lacking CaC12, the basal production of PGE2 decreased by about 22% and of PGF2, by 52% (Table VI, Experiment IV). In Ca2+-free me- dium, the addition of A23187 did not result in an increase in prostaglandin synthesis. Thus, when rat skeletal muscles are incubated under conditions which increase intracellular Ca" levels, enhanced synthesis of PGEz and PGF2, occurs probably through an activation of muscle phospholipase Az, as has been observed with other tissues (20-22).
Effects of Inhibitors of Prostaglandin Synthesis on the Stimulation of Proteolysis by Ca2'-Since aspirin, indometh- acin, and mepacrine inhibited the stimulation of PGE2 and PGFz, production by A23187 or high potassium, we investi- gated whether these inhibitors also reduced the enhancement of prot.eolysis observed under these conditions. In muscles incubated with A23187, aspirin or indomethacin decreased the ionophore's effect on proteolysis by about 75% and 62%, respectively (Table VII), although these agents did not reduce protein degradation to the level seen in the absence of iono- phore. By contrast, neither aspirin nor indomethacin affected the basal proteolysis occurring in the absence of the iono- phore, in accord with previous findings (24).
Similar results were obtained when diaphragms were treated with mepacrine (Table VIII). This egent alone did not change the basal rate of proteolysis significantly. However, when muscles were treated with mepacrine prior to the incu-
TABLE VI1 Effect of inhibitors ofprostaglandin synthesis on protein
degradation stimulated by A23187 Quarter diaphragms were incubated as described in Table I. The
muscles were Fist pretreated with aspirin (5 X M) or indometh- acin (2.8 x M ) for 30 min, and then were transferred into incubation medium containing aspirin or indomethacin and A23187 (2 PM) for 2 h. Numbers shown (nanomoles of tyrosine/mg of muscle/ 2 h) are the increase in the rates of protein degradation over controls (0.627 & 0.050 nmol of 'ryr/mg of muscle/2 h). As demonstrated earlier (19), indomethacin or aspirin alone had no effect on basal rates of protein degradation, which in the control was 0.604 -t- 0.04 nmol of Tyr/mg of muscle/2 h plus indomethacin, 0.628 -+ 0.033; plus aspirin, 0.620 rt 0.041.
Additions Increase in protein B inhibition of
degradation ionophore effect
nmol Tyr/mg muscle/Z h A23187 0.335 -t- 0.018
+Aspirin 0.084 S 0.010 75" +Indomethacin 0.126 & 0.009 62"
rzp < 0.01.
TABLE VI11 Effect of mepacrine on net protein degradation in the absence and
presence of A23187 Mepacrine was added to the preincubation at a concentration of 5
X M. Muscles were then transferred into medium lacking mepa- crine. Net protein degradation was measured in the presence or absence of 2 WM A23187. Mepacrine itself did not affect rates of protein synthesis or protein breakdown in control muscles (data not shown).
Net protein degradation Addition R in-
A23187 Increase with crease Basal A23187
nmol Tyr/mg muscle/Z h
None 0.342 C 0.036 0.811 2 0.052 +0.469 -t- 0.075 137" Mepacrine 0.394 C 0.034 0.578 f 0.048 +0.184 C 0.032 47" % inhibition 0 29" 61"
" p < 0.01.
TABLE IX Effect of high K' on protein degradation in the absence and
presence of inhibitors ofprostaglandin synthesis Quarter diaphragms were incubated in normal and high K'-con-
taining medium after pretreatment for 30 min in the absence or presence of indomethacin (2.8 X M) or aspirin (5 x IO" M ) . Protein degradation was calculated from the rates of protein synthesis and net degradation measured during the 2-h incubation. Numbers shown are the increase in the rates of protein degradation above that
of muscle/2 h) . in control muscles in normal medium (0.370 s 0.015 nmol of Tyr/mg
Increase in protein increase W inhibition degradation of K'-effect
nmol Tyr/mg muscle/2 h
High K' 0.276 -t 0.025 74" +Indomethacin 0.188 rt 0.031 5 0 32" +Aspirin 0.169 C 0.026 45" 39"
a p < 0.05.
TABLE X Effect of leupeptin and Ep-475 on the increase in protein
degradation by A23187
hibitors (25 PM) in the absence or presence of A23187 (2 PM). Rates of Quarter diaphragms were incubated with or without protease in-
protein degradation in the presence of cycloheximide were measured after the standard 2-h incubation.
Protein degradation % inhibition
nmol Tyrlmg muscle/Z h +Leupeptm
Control 0.288 -t- 0.017 0.235 f 0.017 18 A23187 0.518 f 0.038 0.326 f 0.026 37" Increase 0.230 f 0.034 0.091 f 0.023 61h
Ep-475
Control 0.242 2 0.015 0.191 f 0.022 28" A23187 0.401 -t- 0.034 0.246 f 0.019 36" Increase 0.159 f 0.025 0.055 j , 0.021 75h
" p < 0.05. h p < 0.01.
bation with A23187, mepacrine did reduce the ionophore's effect on proteolysis by about 61%. Thus, the effects of Ca" on muscle proteolysis seem to be mediated by an increase in synthesis of PGE2 (Table VI), which can stimulate protein degradation in rat muscles (24).
Although Ca2+ appears to promote proteolysis by activating phospholipase A*, these studies have not eliminated the ad- ditional possibilities that Ca" may also be required for the enhancement of prostaglandin E, synthesis by arachidonate or for the subsequent stimulation of proteolysis. In fact, in recent studies, we have obtained direct evidence that the activation of proteolysis by arachidonate and PGE2, as well as the production of these compounds, is inhibited in Ca2+-defi- cient media.4
Indomethacin and aspirin reduced by about 31% and 41% respectively, the enhanced proteolysis induced by high extra- cellular concentrations of potassium (Table IX). Thus, the increase in proteolysis with membrane depolarization also seems to be mediated at least in part by prostaglandin E*. However, the cyclooxygenase inhibitors were consistently more effective in preventing the stimulation of proteolysis by A23187 than by high K', which may also promote protein catabolism by additional mechanisms.
Involvement of Lysosomal Proteases in Ca2'-induced Pro- teolysis-Since PGE2 appears to increase protein degradation through an effect on lysosomal function (24), we investigated whether the effect of A23187 on protein degradation is altered
V. Baracos and A. L. Goldberg, unpublished observations.

by inhibitors of lysosomal thiol proteases. Leupeptin and Ep- 475 decrease protein degradation in rat skeletal muscle appar- ently by an effect on intralysosomal proteolysis, without af- fecting protein synthesis" (3). Table X shows that the addition of leupeptin or Ep-475 to medium containing A23187 reduced sipificantly the enhancement of proteolysis due to the iono- phore. When muscles were incubated either with leupeptin (25 p ~ ) or Ep-475 (25 p ~ ) for 2 h, subsequent measurements of the activity of cathepsin B showed a complete inhibition of this enzyme. In related studies: we have found that at these concentrations Ep-475 does not affect the Ca"-activated pro- tease within the muscle. Furthermore, our findings with mer- salyl (Tables IV and V) indicate that complete inhibition of that enzyme does not reduce proteolysis. These results strongly suggest that A23187 stimulates protein breakdown by activation of the lysosomal apparatus, as was found with arachidonic acid and PGE2 (24).
DISCUSSION
The present studies c o n f i i and extend the earlier finding by Kameyama and Etlinger (1,2) that an increase in intracel- lular Ca2+ promotes protein breakdown in skeletal muscle. Enhanced proteolysis was evident upon t,reatment with the Ca2+ ionophores, A23187 and ionomycin (Table I), and fell significantly when the muscles were incubated in a Ca"-free medium (Table 11). Although the marked increase in prote- olysis with A23187 required a high extracellular concentration of calcium, the ionophore did have some stimulatory effect in the Ca'+-free medium. Under such conditions, A23187 may promote release of calcium from the sarcoplasmic reticulum or the mitochondria of the muscle. This agent has in fact been shown to release Ca"+ from the mitochondria of other tissues (34). We cannot, however, eliminate the additional possibility that the ionophore may also promote proteolysis in part by some mechanism not involving Ca".
In contrast to prior reports (1, 2), our studies failed to demonstrate a significant stimulation of protein synthesis in either red or white skeletal muscles with these ionophores (Table I). The reasons for these discrepant findings are not clear. The lack of a consistent stimulation of protein synthesis in our various experiments is also surprising, since the Ca'+ ionophore did cause a large increase in the production of prostaglandin Fz, (Table VI). This agent was found in our earlier studies (24) to stimulate protein synthesis in these muscles (24). Presumably, some other consequences of the ionophore treatment prevented this effect of the prostaglan- din.
The enhancement of muscle proteolysis by the Ca2+ iono- phore was initially explained as a stimulation of the Ca2+- activated protease (1,2). This enzyme is capable of catalyzing dissolution of the Z-band of the myofibril (9), and it has often been proposed to play an essential first step in the degradation of myofibrils (8, 9). Accordingly, treatment with Ca2+ iono- phores (2) and depolarization of the neuromuscular junction (39) can cause disappearance of the 2-bands in the muscle in a similar fashion as occurs with the isolated enzyme. One finding that seemed to support involvement of the Ca2+-acti- vated protease in the ionophore-induced stimulation of pro- teolysis was the inhibition of this effect by leupeptin (I) , which is a potent inhibitor of this enzyme in muscle homogenates (3). However, leupeptin also inhibits strongly the lysosomal thiol proteases, cathepsins B and L, and treatment of intact rat muscles with leupeptin causes almost a complete inhibition of intracellular cathepsin B (3). Therefore, use of leupeptin cannot distinguish between the enhancement of proteolysis by the lysosomal apparatus or by the Ca2+-activated protease.
Our experiments with mersalyl (Table IV) resolved this
issue in a definitive way. Treatment of muscle with this sulfhydryl blocking agent selectively inactivated the Ca2'- activated protease without reducing overall proteolysis or the stimulation by A23187. By contrast, mersalyl had no effect on the activity of cathepsin B in the incubated muscles even though it strongly inhibits this enzyme if added to tissue extracts (Table IV). Presumably, mersalyl cannot reach the thiol proteases within the lysosome, and the Ca2+-activated protease must be in a more accessible location, perhaps even on the cell surface as suggested by certain workers (40). The conclusion that the Ca2+-activated protease was not involved was further supported by the experiments using Ep-475 (Table X). In related studies," we found that treatment of the muscles with this highly specific inhibitor of thiol proteases (41) inactivated cathepsin B but did not affect the Ca2+-activated protease (at the concentrations used in Table IV). Ep-475 and leupeptin, unlike mersalyl, inhibited both basal protein break- down in the muscle and the stimulation by the ionophore. Together these various observations indicate that increased intracellular Ca2+ leads to enhanced protein breakdown by an effect on lysosomal function, mediated by prostaglandins.
Our studies indicate that in skeletal muscle the synthesis of PGE2 and PGF2,, is controlled in a similar fashion and is sensitive to the same inhibitors as in other tissues. In several types of cells (21, 22), as in rat muscle (Table VI), treatment with A23187 leads to enhanced synthesis of prostaglandins, apparently through a stimulation by Ca'+ of phospholipase A2 (21, 22, 38). For maximal activity (38), this enzyme requires approximately 10 p~ Ca2+, which is above the concentration normally found within muscles, but is similar to the levels reached during contraction or treatment with the ionophore (see below). In the rat muscles, A23187 stimulated the pro- duction of PGE2 and PGFzu 3-6-fold (Table VI) and this effect was sensitive to inhibitors of both the phospholipase A2 (Table VI) and the cyclooxygenase.
Since these same inhibitors also reduced the stimulation by Ca2+ of protein breakdown (without affecting basal rates of proteolysis), it appears very likely that this effect is mediated by increased production of prostaglandin E*. Accordingly, the use of Ca2'-free medium reduced the increases in both pros- taglandins and proteolysis (Tables I1 and VI). Furthermore, the stimulation of protein breakdown induced by prostaglan- din E2 appears to involve the lysosomal apparatus (24), like the stimulation by Ca" ionophores (Table X). One specific prediction of our conclusion is that the production of prosta- glandins and the acceleration of protein breakdown should show a similar Ca2+ dependence as phospholipase A,. Because this enzyme seems to involve calmodulin (42), it should be activated by the same concentrations of Ca'+ as are necessary for force generation (43). In fact, Etlinger et al. (44) have found that concentrations of the Ca'+ ionophore that stimu- late muscle proteolysis also caused tension development.
Although these studies indicate that PGE2 mediates most of the effects of Ca2+ on protein degradation, it is noteworthy that indomethacin and aspirin blocked completely the iono- phore-induced stimulation of prostaglandin release, but only partially reduced the concomitant increase in proteolysis. This discrepancy may mean that prostaglandin release into the medium is not an accurate reflection of intracellular levels of PGE,. I t is also possible that Ca2+ enhances protein break- down in part by an additional effect not involving prostaglan- dins. For example, high intracellular levels of Ca'+ may modify the conformation of muscle proteins or cause them to be cross- linked (45,46), and mammalian cells are known to selectively degrade polypeptides with highly abnormal conformations (47).
In the incubated muscles, high concentrations of K+ stim-

8 722 Stimulation of Protein Degradation in Muscle by ea2+ Involves PGE,
ulated the production of PGE2 and protein breakdown in a similar fashion to A23187, and these effects of K' were also reduced by inhibitors of phospholipase AZ and the cyclooxy- genase. Depolarization should increase intracellular Ca2+ lev- els, and Etlinger et al. (2) found that the stimulation of muscle proteolysis by K' was reduced by dantrolene, an inhibitor of ea2+ release from the sarcoplasmic reticulum. In cultured rat pituitary cells (48), membrane depolarization with K' has also been shown to activate phospholipase A2 and thereby cause an accumulation of PGEP (48). However, our experiments with indomethacin showed only a partial inhibition of the effect of high K' potassium on proteolysis (although a highly signifi- cant one). This observation and related ones' suggest that exposure to high potassium has additional effects, unrelated to PGE2, that lead to enhanced protein breakdown.
The stimulatory effect of Ca" on protein breakdown in muscle is of particular interest for understanding muscle dis- ease in animals or humans. In the inherited muscular dystro- phies (47, 49-53), upon denervation (54, 55) and in certain types of tissue injury (56), muscle protein breakdown appears to rise, and this increase seems responsible for the progressive muscle wasting (45, 54-56). An increase in intracellular Ca"' has been reported in dystrophic muscle by several workers (15, 16, 18, 19). It is attractive to suggest that the excessive protein breakdown in such muscles may be due to increased levels of PGE2 resulting from the enhanced intracellular ea2+. Unfortunately, data on the levels of PGEZ and PGFza in dystrophic or damaged muscle are not available (57).
In any case, inhibitors of prostaglandin synthesis (e.8. as- pirin, indomethacin, or mepacrine) are not toxic and clearly deserve evaluation as possibly useful therapeutic agents for the treatment of muscular dystrophy or other forms of muscle wasting. Elsewhere, we shall present evidence that the en- hanced muscle proteolysis during fever involves a Ca2'-de- pendent increase in prostaglandin prod~ction.~ Similarly, many physiologic factors enhance overall protein breakdown and stimulate lysosomal function (58) in muscle and other tissues; possibly they also act via prostaglandins and changes in intracellular ea2+ levels.
Acknowledgments-We are grateful to Robin Levine and Maureen Rush for their expert assistance in preparing this manuscript and to Maureen Rush and Timothy Meixsell for assistance in several of these experiments.
1. 2.
3. 4. 5.
6.
7.
8.
9.
10. 11.
12.
REFERENCES Kameyama, T., and Etlinger, J . D. (1979) Nature 279, 344-346 Etlinger, J. D., Kameyama, T., Toner, K., van der Westhuyzen,
D., and Matsumoto, K. (1980) in Plasticity of Muscle (Pette, D., ed) pp. 541-558, Walter de Gruyter, New York
Libby, P., and Goldberg, A. L. (1978) Science 199, 534-536 Waxman, L. (1982) Methods Enrymol. 80,664-680 Busch, W. A., Stromer, M. H., Goll, D. E., and Suzuki, A. (1972)
Okitani, A., Otsuka, Y., Sugitani, M., and Fujimaki, M. (1974)
Dayton, W. R., GoU, D. E., Zeece, M. G., Robson, R. M., and
Dayton, W. R., Reville, W . J . , Goll, D. E., and Stromer, M. H.
Reddy, M. K . , Etlinger, J . D., Rabinowitz, M., Fischman, D. A.,
Kar, N. C., and Pearson, C. M. (1978) Muscle Nerve 1, 308-313 Statham, H. E., Duncan, C. J., and Smith, J . L. (1976) Cell Tissue
Publicover, S. J., Duncan, C. J., and Smith, J . L. (1978) J.
J. Cell Biol. 52, 367-381
Agric. Biol. Chem. 38,573-579
Revdle, W. J . (1976) Biochemistry 15, 2150-2158
(1976) Biochemistry 15, 2159-2167
and Zak, R. (1975) J. Biol. Chem. 250,4278-4284
Res. 173, 193-209
Neuropathol. Exp. Neurol. 37, 544-557
V. Baracos, H. P. Rodemann, and A. L. Goldberg, submitted for publication.
13. Reed, P. W., and Lardy, H. A. (1972) J. Bioi. Chem. 247, 6970-
14. Sugden, P. H. (1980) Biochern. J . 190, 593-603 15. Engel, A. G., Mokri, B., Jerusalem, F., Sakakibara, H., and Paul-
son, 0. B. (1977) in Pathogenesis of Human Muscular Dystro- phies (Rowland, L. P., ed) pp. 310-324, Excerpta Medica, Am- sterdam
6977
16. Emery, A. E. H., and Burt, D. (1980) Br. Med. J. 280,355-357 17. Jaffe, M., Savage, N., and Isaacs, H. (1981) Biochem. J. 196,663-
18. Duncan, C. J. (1978) Experientia 34, 1531-1535 19. Samaha, F. J. (1977) in Pathogenesis of Human Muscular Dys-
trophies (Rowland, L. P., ed) pp. 633-639, Excerpta Medica, Amsterdam
20. Pickett, W. C., Jesse, R. L., and Cohen, P. (1977) Biochim. Biophys. Acta 486, 209-213
21. Zenser, T. V., Herman, C. A., and Davis, B. B. (1980) Am. J. Physiol. E371-E376
22. Billah, M. M., Lapetina, E. G., and Cuatrecasas, P. (1980) J. Biol. Chem. 255, 10227-10231
667
23.
24.
25.
26.
27.
28.
29. 30.
31.
32. 33.
34.
35. 36.
37.
38.
39.
40. 41.
42.
43.
44.
45. 46.
47.
48. 49.
50.
51. 52. 53.
Knapp, H. R:, Oelz, O., Roberts, L. J., Sweetman, B. J., Oates, J . A., and Reed, P. W. (1977) Proc. Natl. Acad. Sci. U. S. A. 74,
Rodemann, H. P., and Goldberg, A. L. (1982) J. Biol. Chem. 257,
Goldberg, A. L., Martel, S. M., and Kushmerick, M. J. (1975)
Fulks, R. M., Li, J. B., and Goldberg, A. L. (1975) J. Biol. Chem.
Tischler, M. E., Desautels, M., and Goldberg, A. L. (1982) J. Biol.
Waalkes, T. P., and Udenfriend, S. (1957) J. Lab. Cizn. Med. 50,
Vane, J. R. (1971) Nature 231,232-239 Whittle, B. J. R., Higgs, G. A,, Eakins, K. E., Moneda, S., and
Winocour, P. D., Kinlough-Rathbone, R. L., and Mustard, J. F.
Rice, R. H., and Means, G. E. (1971) J. Biol. Chem. 246,831-832 Jaffe, B. M., and Behrman, H. R. (1974) in Methods in Hormone
Radioimmunoassay (Jaffe, B. M., and Behrman, H. R., e&) pp. 19-34, Academic Press, New York
Blackmore, P. F., Brumley, F. T., Marks, J. L., and Exton, J. H. (1978) J. Biol. Chem. 253,4851-4858
M a , J. L. (1981) Science 213, 745-747 Ellis, K. O., and Bryant, S. H. (1972) Arch. Exp. Pathol. Phar-
Gerard, K. W., and Schneider, D. L. (1980) Biochem. Biophys.
Billah, M. M., Lapetina, E. G., and Cuatrecasas, P. J. (1981) J .
Leonard, J. P., and Salpeter, M. M. (1979) J. Cell Biol. 82, 811-
Barth, R., and Elce, J . S. (1981) Am. J. Physiol. 240, E493-E498 Hanada, K., Tami, M., Yamaguchi, M., Ohmura, S., Sawada, J.,
Wong, P. Y.-K., and Cheung, W . Y. (1979) Biochem. Biophys.
Mannherz, H. G., and Goody, R. S. (1976) Annu. Reu. Biochem.
Etlinger, J. D., Kameyama, T., van der Westhuysen, D., Erlij, D., and Matsumoto, K. (1982) in Mechanism ofMuscle Adaptation to Functional Requirements (Guba, F., Marechal, F., and Tak- acs, O., eds) Academia Kiado, Budapest, in press
4251-4255
1632-1638
Methods Enzymol. 39,82-93
250,290-298
Chem. 257, 1613-1621
733-736
Vane, J. R. (1980) Nature 284,271-273
(1979) Fed. Proc. 38,1271-1273
makol. 27,107-113
Res. Commun. 94, 1353-1361
Biol. Chem. 256,5399-5403
819
and Tanaka, N. (1978) Agric. Biol. Chem. 42,523-527
Res. Commun. 90,473-480
45,427-465
Folk, J. E., and Chung, S. I. (1973) Adu. Enzymol. 38, 109-191 Lorand, L., Weissman, L. B., Epel, D. L., and Bruner-Lorand, J.
Goldberg, A. L., and St. John, A. C. (1974) Annu. Rev. Biochem.
Betteridge, A. (1980) Biochem. J. 186,987-992 Simon, E. J., Gross, C. S., and Lessell, I. M. (1962) Arch. Biochern.
Weinstock, I. M., Soh, T. S., Freedman, H. A., and Cutler, M. E.
Strivastara, U. (1968) Cen. J. Biochem. 46, 35-41 Rourke, A. W. (1975) J. Cell Physiol. 86,343-352 Goldberg, A. L., Griffin, G. E., and Dice, J . F. (1977) in Pathogen-
esis of Human Muscular Dystrophies (Rowland, L. P., ed) pp.
(1976) Proc. Natl. Acad. Sci. U. S. A. 73, 4479-4481
45, 747-803
Biophys. 96,41-46
(1969) Biochem. Med. 2, 345-356

Stimulation of Protein Degradation in Muscle by Ca2+ Involves PGEz 8723
376-385, Excerpta Medica, Amsterdam M. S., Karmazyn, M., and Cunnane, S. C. (1979) in Muscular 54. Goldspink, D. F. (1978) Biochem. J. 174,595-602 Dystrophy and Other Inherited Diseases of Skeletal Muscles 55. Goldspink, D. F. (1976) Biochem. J. 156, 71-80 in Animals (Harris, J. B., ed) pp. 532-547, The New York 56. Seider, M. J., Kapp, R., Chen, C.-P., and Booth, F. W. (1980) Academy of Sciences, New York
Biochem. J. 188,247-254 58. Mortimore, G. E., and Schworer, Ch.M. (1980) Ciba Found. Symp. 57. Horrobin, D. F., Morgan, R. O., Karmali, R. A,, Ally, A. I., Manku, 75,281-305






![Combinations of Patch-Clamp and Confocal …indicator Oregon Green 488 BAPTA-6F (100 µM) via a patch pipet. Changes in postsynaptic [Ca2+]i induced by presynaptic stimulation at 20,](https://static.fdocuments.us/doc/165x107/5e4f21d21a023711ac01343d/combinations-of-patch-clamp-and-confocal-indicator-oregon-green-488-bapta-6f-100.jpg)











