The spectrum of human diseases Cystic fibrosis thalassemiaHuntington’s cancer
33
The spectrum of human diseases Cystic fibrosis thalassem ia Huntington ’s cancer <5%
-
Upload
alexia-rose -
Category
Documents
-
view
226 -
download
0
Transcript of The spectrum of human diseases Cystic fibrosis thalassemiaHuntington’s cancer
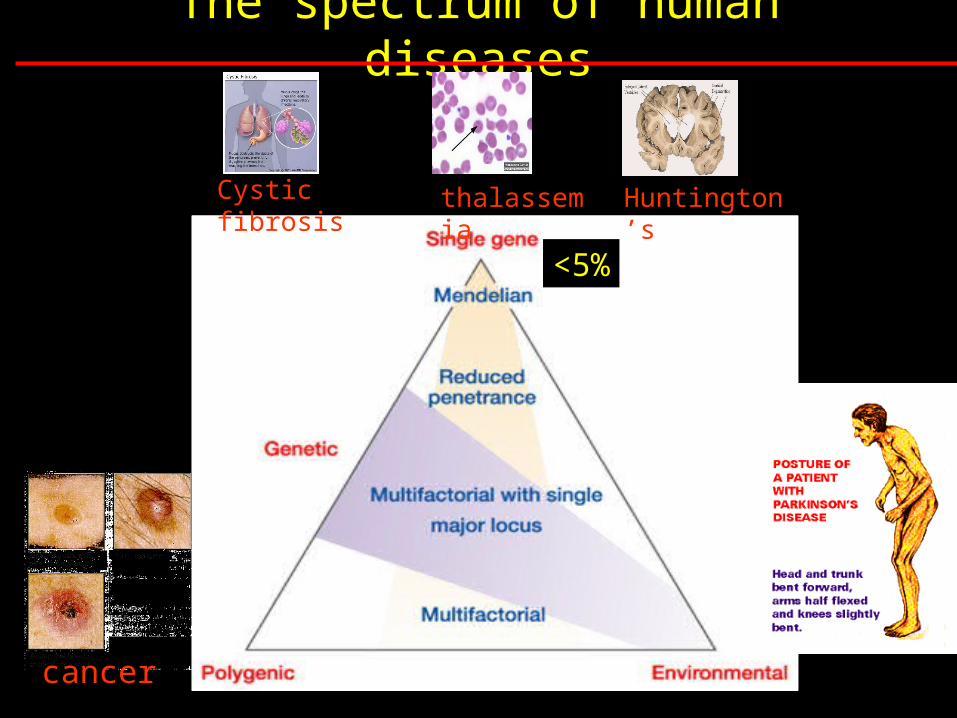
The spectrum of human diseases
Cystic fibrosis thalassemia
Huntington’s
cancer
<5%

Mapping complex loci
PAF – population attributable factor:
Fraction of the disease that would be eliminated if the
risk factor were removed
High PAF for single gene conditions (>50% for CF)
Low PAF for complex disease (< 5% for Alzheimer’s)

Identifying genes involved in complex diseases
Steps
Perform family, twin or adoption studies - check for genetic component
Segregation analysis- estimate type and frequency of susceptibility alleles
Linkage analysis- map susceptibility loci
Population association- identify candidate region
Identify DNA sequence variants conferring susceptibility

Linkage versus Assocciation
Association studies compare the allele frequency of a polymorphic marker, or a set of markers, in unrelated patients (cases) and healthy controls to identify markers that differ significantly between the two groups.
Used to identify common modest-risk disease variants
Higher density of markers needed
e.g. HapMap uses association data
Linkage analyses search for regions of the genome with a higher-than-expected number of shared alleles among affected individuals within a family.
Used to identify rare high-risk disease alleles
<500 markers needed for initial genome scan

Haplotype analysis• specific combination of 2 or more DNA marker
alleles situated close together on the same chromosome (cis markers)
• SNPs most commonly used markers in haplotypes.
• series of closely linked mutations accumulate over time in the surviving generation derived from a common ancestor.
• powerful genetic tool for identifying ancient genetic relationships.
• Alleles at separate loci that are associated with each other at a frequency that is significantly higher than that expected by chance, are said to be in linkage disequilibrium

Direct versus indirect association analysis.
a, In direct association analysis,all functional variants (red arrows) are catalogued and tested for association with disease. A GeneSNPs image of the CSF2 gene is shown. Genomic features are shown as boxes along the horizontal axis (for example, blue boxes indicate exons). Polymorphisms are shown as vertical bars below the axis, with the length of the line indicating allele frequency and colour indicating context (for example, red indicates coding SNPs that change amino acids). b, For indirect association analysis, all common SNPs are tested for function by assaying a subset of tagSNPs in each gene (yellow arrows), such that all unassayed SNPs (green arrows) are correlated with one or more tagSNPs. Effects at unassayed SNPs (green arrows) would be detected through linkage disequilibrium with tagSNPs. Images adapted from GeneSNPs (http://www.genome.utah.edu/genesnps).

Formation of haplotypes over time

Ancient disease loci are associated with haplotypes
• Start with population genetically isolated for a long time such as Icelanders or Amish
• Collect DNA samples from subgroup with disease• Also collect from equal number of people without
disease• Genotype each individual in subgroups for
haplotypes throughout entire genome• Look for association between haplotype and
disease phenotype• Association represents linkage disequilibrium• If successful, provides high resolution to narrow
parts of chromosomes

Haplotype analysis provides high resolution gene mapping

Genetic heterogeneityMutations at more than one locus cause
same phenotype
e.g. thalassemias – Caused by mutations in
either the or -globin genes.
– Linkage analysis studies therefore always combine data from multiple families
Why is it still so difficult?

Variable expressivity - Expression of a mutant trait differs from person to person

• Phenocopy– Disease phenotype is not caused by any
inherited predisposing mutation – e.g. BRCA1 mutations
• 33% of women who do not carry BRCA1 mutation develop breast cancer by age 55

Incomplete penetrance – when a mutant genotype does not always cause a mutant phenotype• No environmental factor associated with
likelihood of breast cancer• Positional cloning identified BRCA1 as
one gene causing breast cancer.– Only 66% of women who carry BRCA1
mutation develop breast cancer by age 55
• Incomplete penetrance hampers linkage mapping and positional cloning– Solution – exclude all nondisease individuals
form analysis– Requires many more families for study

• Polygenic inheritance– Two or more genes interact in the
expression of phenotype• QTLs, or quantitative trait loci
– Unlimited number of transmission patterns for QTLs» Discrete traits – penetrance may increase with
number of mutant loci» Expressivity may vary with number of loci
– Many other factors complicate analysis» Some mutant genes may have large effect» Mutations at some loci may be recessive while
others are dominant or codominant

Polygenic inheritance
E.g heart attacks or cholesterol levels
Sudden cardiac death (SCD)

Breast cancer
Although a genetic basis for familial BC identified, the causes of sporadic disease still unknown
Sudden cardiac death (SCD)
Mutations in 2 loci account for 20-25% of early onset (<45 years) breast cancer cases due to inherited factors– BRCA1: mutations found in 80-90% of families
with both breast and ovarian cancer– BRCA2: mutations mainly in male breast
cancer families
Common condition – familial or sporadic forms

Alzheimer’s disease
familial AD – mutations in APP, presenilin-1 and 2Sporadic AD – strong association with APO4, Apolipoprotein 4, which
affects age of onset rather than susceptibility
Sudden cardiac death (SCD)
Affects 5% of people >65 years and 20% of people over 80 has familial (early-onset) or sporadic (late-onset) forms, although
pathologically both are similarAetiology of sporadic forms unknown
3 major alleles (APO E2, E3, and E4)
Position
112 158
ApoE2 Cys Cys
ApoE3 Arg Cys
ApoE4 Arg Arg

Epigenetics – differential imprinting
Genetic conditions that are independent of the DNA
sequence

Epigenetics – differential imprinting
molecular defect involves a ~2 Mb imprinted domain at 15q11–q13 that contains both paternally and maternally expressed genes
Prader-Willi syndrome Angelman syndrome
defect lies within the imprinted domain at 15q11–q13
failure to thrive during infancy, hyperphagia and obesity during early childhood, mental retardation, and behavioural problems
characteristics include mental retardation, speech impairment and behavioural abnormalities

Genetic causes
70% have a deletion of the PWS/AS region on their paternal chromosome 15
25% have maternal uniparental disomy for chromosome 15 (the individual inherited both chromosomes from the mother, and none from the father)
5% have an imprintingdefect
<1% have a chromosome abnormality including the PWS/AS region
Prader-Willi syndrome Angelman syndrome70% have a deletion of the PWS/AS region on their maternal chromosome 15
7% have paternal uniparental disomy for chromosome 15 (the individual inherited both chromosomes from the father, and none from the mother)
3% have an imprinting defect
11% have a mutation in UBE3A
1% have a chromosome rearrangement
11% have a unknown genetic cause

Molecular pathology
NomenclatureEffect of mutant allele and not the sequenceLoss of functionGain of function
Gene to disease
Disease to geneChromosomal disorders

The Haemoglobinopathies
Thalassemias -Anaemias associated with impaired synthesis of Hb subunits
Thalassaemias can arise from different mutations causing a disease of varying severity.
a0/b0 thalassaemias – globin chain absenta+/b+ thalassaemias – normal globin chain in reduced amounts

Evolution of globin superfamily
Fig. 21.16

Organisation of globin genes
Fig. 21.16

Developmental variation in gene expression
Fig. 21.16
-like chains - -like chains - Adult human made of
– 97%; - ~2%;-~1% (fetal persistence)

Gene expression controlled by location
Fig. 21.16
– embryonic yolk sac– yolk sac & fetal liver
– adult bone marrow

thalassemias

thalassemias
GENOTYPE PHENOTYPEa+ a+ a+a+ Normala+a a+a+ Silent carrierasymptomatic condition.
a-thalassaemia – 2
a+ a a+a a-thalassaemia trait minor anaemic conditionsa+a+ a aa+a a a HbH mild – moderate anaemiaa a a a Hydrops foetalis foetus survives until around birth
deletion of one or both a globins in an a gene clusterSeverity depends on whether the individual has 1,2,3, or 4 missing a globin genes.

thalassemias

thalassemias
Non coding regulatory regions
Exons
Introns (InterVening Sequences)
3’ cleavage mutant
deletion
RNA splicing mutant
transcription mutant
nonsense mutation
frameshift insertion
frameshift deletion
3’ 5’
Mutations in globin cluster are of different types gene deletion transcriptional mutation RNA processing mutations RNA cleavage signal mutations Nonsense & frameshift mutations

thalassemias

Main genetic mechanisms that contribute to the phenotypic diversity of the -thalassaemias.

Reading
HMG3 by T Strachan & AP Read : Chapter 14
AND/OR
Genetics by Hartwell (2e) chapter 11 References on Cystic fibrosis: Science (1989) vol 245 pg 1059 by JM Rommens et al (CF mapping)J. Biol Chem (2000) vol 275 No 6 pp 3729 by MH Akabas (CFTR)
Optional Reading on Molecular medicine Nature (May2004) Vol 429 Insight series• human genomics and medicine pp439 (editorial)• Mapping complex disease loci in whole genome
studies by CS Carlson et al pp446-452