
The reactions of silylene in the infrared multiphoton-induced decomposition of silane—ethylene...
11
J. Photo&m. Photobiol. A: Chem., 58 (1991) 173-183 173 The reactions of silylene in the infrared multiphoton- induced decomposition of silane-ethylene mixtures J. R. Fisher and F. W. Lampe IS2 Davey Laboratory, Department of Chemistq The Pennsylvania Stare University, University Park, PA 16802 (U.S.A.) (Received July 23, 1990) Abstract A study of the competitive reactions of silylene, formed via the IR laser multiphoton decomposition of silane, with ethylene and silane shows that the relative rate constant for addition to ethylene is 0.19 times that for insertion into a silicon-hydrogen bond of silane. The sole product of the addition to ethylene was vinylsilane as identified by comparison of the product mass spectrum with that of a synthesized sample. A study of the temperature dependence of the rate constant ratio indicates that the activation energy for addition to the double bond of ethylene is 0.8 kcal mol-’ greater than that of insertion into a silicon-hydrogen bond of silane. 1. Introduction The reactions of SiHz with a variety of substrate molecules have become of considerable interest to gas-phase kineticists in recent years, most likely because of the technological importance of SiH4 decomposition processes in the formation of amorphous silicon films. The reactions appear to be of two major types, namely insertion into Si-H bonds and addition across multiple bonds in organic compounds. Our interest in the reaction of SiHz with C2H, arose because it is the simplest system representing SiH2 addition to an olefin and has not been examined under the conditions of reaction initiation by infrared laser multiphoton decomposition. Eley et nl. [I] generated SiHz by the 206 nm photodecomposition of C6HsSiH3 and measured its relative rate of reaction with a variety of molecules, including C2H+ They found that reaction of SiHr with C2H4 occurred with a rate constant 0.88 times that of its reaction with C6H5SiH3 but did not detect any reaction products. Rogers et al. [2] reported studies of the reactions of SiH2, generated by the pyrolysis of Si2H6, with CIH4 using several different experimental techniques in the overall temperature range of 600-987 K and for conversions in the range of 24-70%; depending on conditions, CZH3SiH3 and C2H,SiH3 were the major products found. Inoue and Suzuki [3] reported that SiH2, formed via the 193 nm photodecomposition of C6H5SiH3 [4, 51, reacted rapidly with C2H4 at room temperature; on the basis of a study of the laser-induced fluorescence of SiHr, they were able to report the absolute rate constant for the reaction of SiHz with C,H, to be 9.7X lo-” cm3 particle-’ s-l. Shortly thereafter, Jasinski and co-workers [6,7], using laser-resonance absorption flash-kinetic spectroscopy, in which SiH3 was generated by both the 193 nm photodecomposition of CaHsSiH3 and the 248 nm photodecomposition of SiH31, showed the rate constant to be pressure lOlO-6030/91/%3.50 0 Elsevier Sequoia/Printed in The Netherlands
Transcript of The reactions of silylene in the infrared multiphoton-induced decomposition of silane—ethylene...

J. Photo&m. Photobiol. A: Chem., 58 (1991) 173-183 173
The reactions of silylene in the infrared multiphoton- induced decomposition of silane-ethylene mixtures
J. R. Fisher and F. W. Lampe
IS2 Davey Laboratory, Department of Chemistq The Pennsylvania Stare University, University Park, PA 16802 (U.S.A.)
(Received July 23, 1990)
Abstract
A study of the competitive reactions of silylene, formed via the IR laser multiphoton decomposition of silane, with ethylene and silane shows that the relative rate constant for addition to ethylene is 0.19 times that for insertion into a silicon-hydrogen bond of silane. The sole product of the addition to ethylene was vinylsilane as identified by comparison of the product mass spectrum with that of a synthesized sample. A study of the temperature dependence of the rate constant ratio indicates that the activation energy for addition to the double bond of ethylene is 0.8 kcal mol-’ greater than that of insertion into a silicon-hydrogen bond of silane.
1. Introduction
The reactions of SiHz with a variety of substrate molecules have become of considerable interest to gas-phase kineticists in recent years, most likely because of the technological importance of SiH4 decomposition processes in the formation of amorphous silicon films. The reactions appear to be of two major types, namely insertion into Si-H bonds and addition across multiple bonds in organic compounds. Our interest in the reaction of SiHz with C2H, arose because it is the simplest system representing SiH2 addition to an olefin and has not been examined under the conditions of reaction initiation by infrared laser multiphoton decomposition.
Eley et nl. [I] generated SiHz by the 206 nm photodecomposition of C6HsSiH3 and measured its relative rate of reaction with a variety of molecules, including C2H+ They found that reaction of SiHr with C2H4 occurred with a rate constant 0.88 times that of its reaction with C6H5SiH3 but did not detect any reaction products. Rogers et al. [2] reported studies of the reactions of SiH2, generated by the pyrolysis of Si2H6, with CIH4 using several different experimental techniques in the overall temperature range of 600-987 K and for conversions in the range of 24-70%; depending on conditions, CZH3SiH3 and C2H,SiH3 were the major products found. Inoue and Suzuki [3] reported that SiH2, formed via the 193 nm photodecomposition of C6H5SiH3 [4, 51, reacted rapidly with C2H4 at room temperature; on the basis of a study of the laser-induced fluorescence of SiHr, they were able to report the absolute rate constant for the reaction of SiHz with C,H, to be 9.7X lo-” cm3 particle-’ s-l. Shortly thereafter, Jasinski and co-workers [6,7], using laser-resonance absorption flash-kinetic spectroscopy, in which SiH3 was generated by both the 193 nm photodecomposition of CaHsSiH3 and the 248 nm photodecomposition of SiH31, showed the rate constant to be pressure
lOlO-6030/91/%3.50 0 Elsevier Sequoia/Printed in The Netherlands

174
dependent in the range 1 to 10 Torr and to have the value of 5.3 +0.5X 10-r’ cm3 particle-’ s-i at 5 Torr at 298 K.
In the present study SiH2 was generated by the IR multiphoton decomposition of SiH4 in the presence of C2H4, with reactant and product concentrations being monitored continuously by mass spectrometry.
2. Experimental details
Reaction mixtures ranging from OS:1 to 3:l C2H4:SiH., were prepared on a Saunders-Taylor [8] apparatus with helium added so that an 80 Torr aliquot would contain a constant 20 Torr of SiH,. All experiments were conducted with samples of 80 Torr which resulted in an SiH4 decomposition of approximately 1% (0.5:1 mixture) to 0.1% (3:l mixture) per laser pulse.
The reactions were carried out in a stainless steel cell, 15.7 cm in length and 3.5 cm in diameter, fitted with NaCl windows held by “0’‘-ring compression fittings. Both a 20 pm gold pinhole and a glass pinhole of similar diameter were used as a leak into a Bendix time-of-flight mass spectrometer,
The radiation source was a Lumonics Research Ltd. Model 103 COa TEA laser which was operated at 0.5 Hz. The laser was tuned to the P(20) line of the 10.6 pm transition which produces 944.19 cm-’ photons. Experiments showed that neither QH4 nor C2D, absorbed laser energy at this frequency. The beam was unfocused and stopped down to a 2.54 cm diameter by means of an adjustable iris mounted in the optical path. Measurements of the absorption of laser energy were made on a Gen Tee ED- 500 Joulemeter positioned directly behind the exit window of the reaction cell. The incident energy of the laser beam is 3.2 J pulse-’ corresponding to a fluence of 0.65 J cme2.
The reaction products were identified mass spectrometrically by comparison of the mass spectra produced by the multiphoton decomposition of the mixtures to the mass spectra of authentic samples of these same molecules. Rates of SiH, depletion and product formation were determined from the initial slopes of continuously monitored ion currents <associated with SiH4 (m/q = 31, SiH,+), C2H3SiH3 (m/q =53, SiCaH’) and S&H6 (m/q = 61, Si2H5+).
SiH., was purchased from Matheson and subjected to several freeze-pump-thaw cycles in liquid N2 to remove non-condensible impurities prior to use. S&He, obtained from Merck and Company, was used as received. Helium and argon were supplied by Linde. C2Dd was purchased from Merck, Sharp, and Dohme.
C2H3SiH3 was synthesized by the conventional reduction of GH$iCla, obtained from Petrarch Systems, with LiAlH4 (1 M in THF) obtained from Aldrich Chemical Company. The synthetic procedure, which utilized the simple apparatus shown in Fig. 1, was as follows. To the reaction flask was added 0.027 mol of GHJSiC13 and the system evacuated by a mechanical pump. The reaction flask and collection bulk were isolated and 200 Torr of argon was admitted. Fifty milliliters of 1 M LiAl& in THF were injected into the addition funnel and this solution was slowly added to the C2H3SiC13. The gaseous product was allowed to expand freely through the system. Upon completion of the reaction, all condensable species were frozen with liquid nitrogen into the cold finger of the 1 L collection bulb. The collected gases, which included a large amount of THF, were vacuum distilled at -98 “C from a liquid N2-CH30H slush bath. The mass spectrum of the pure C7HJSiH3 so obtained was identical, within experimental error, with the mass spectrum of the reaction product,

175
f
Dry-Ice-acetone
In
Condenser o-eootorr
Wallace and Tiernonpaupe
Mechonicol Pump “Fl$
Fig. 1. Apparatus used in the synthesis of qH3SiH3.
I-..-_+ 30 35 40
J, 55
L--- 1 45 JO
Lt- 60
m/q ratio
Fig. 2. Mass spectrum of C2H3SiH3 at 70 eV.
corrected for the presence of S&H6 and Si3Hs. Since this mass spectrum does not appear to have been published, we present it in Fig. 2.
3. Results and discussion
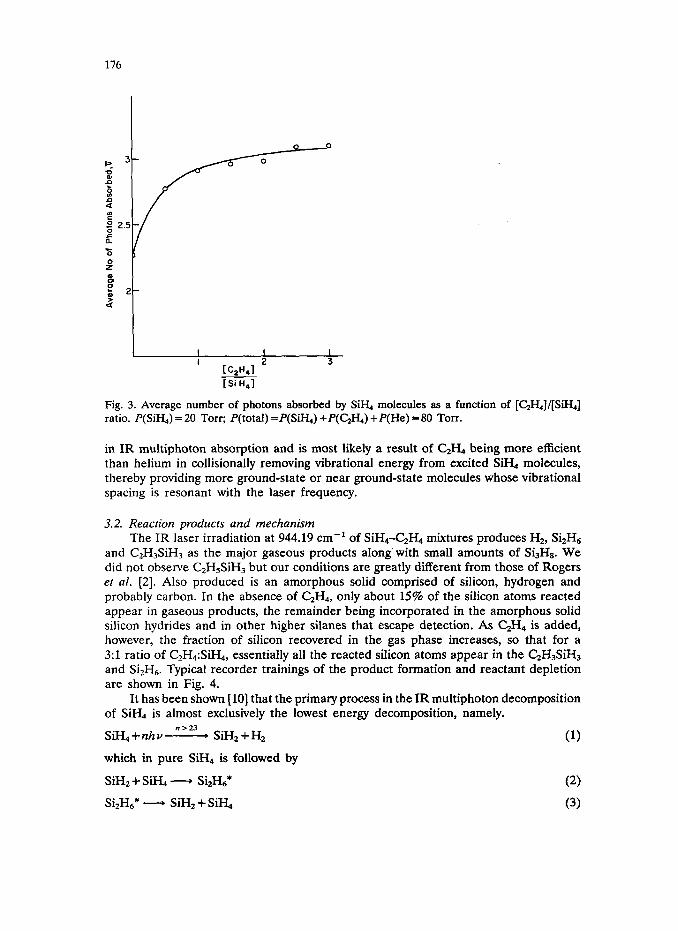
3.1. Energy absorption Although C2H4 does not absorb the laser radiation, the replacement of helium
by C& in a He-SiH, mixture increases the energy that is absorbed per pulse of the laser. As may be seen in Fig. 3, the average number of photons absorbed per Si& molecule per pulse, which is a useful value for calculation of the fraction of molecules in a Boltzmann distribution [9] that contains a given energy, increases from 2.3 for a 1:0:3 (Si%:C&:He) mixture to 3.1 for a 1:3:0 (SiH&H,:He) mixture, all at a constant total pressure of 80 Torr. This type of enhancement is commonly observed
176
I I I I
[CzH,l 2 3
[Si HaI
Fig. 3. Average number of photons absorbed by SiH, molecules as a function ratio. P(SiH,) = 20 Torr; P(tota1) =P(SiHJ +P(GHJ +P(He) = 80 Torr.
in IR multiphoton absorption and is most likely a result of CzH4 being more efficient than helium in collisionally removing vibrational energy from excited SiH, molecules, thereby providing more ground-state or near ground-state molecules whose vibrational spacing is resonant with the laser frequency.
3.2. Reaction products and mechanism The IR laser irradiation at 944.19 cm-’ of SiH&$H, mixtures produces Hz, S&H,
and CIHJSiH3 as the major gaseous products along with small amounts of S&Ha. We did not observe CrH5SiH3 but our conditions are greatly different from those of Rogers et al. [Z]. Also produced is an amorphous solid comprised of silicon, hydrogen and probably carbon. In the absence of GH,, only about 15% of the silicon atoms reacted appear in gaseous products, the remainder being incorporated in the amorphous solid silicon hydrides and in other higher silanes that escape detection. As C,H, is added, however, the fraction of silicon recovered in the gas phase increases, so that for a 3:l ratio of C2H4:SiH,, essentially all the reacted silicon atoms appear in the C,H,SiH, and SizH6. Typical recorder trainings of the product formation and reactant depletion are shown in Fig. 4.
It has been shown [lo] that the primary process in the IRmultiphoton decomposition of SiH4 is almost exclusively the lowest energy decomposition, namely.
Six+-nhv- n’23 SiH2 +H2 (I)
which in pure SiH4 is followed by
SiH2 + SiI& - Si,H,* (2)
Si2H6* - SiH, + SiI-& (3)

177
m/q=61 m/q=53 I ” I ’ 0 I XI x2 0 30 60s
Fig. 4. Recorder tracings of the formation of Si,H6 (m/q=61) and GH3SiH3 (m&=53) and depletion of SIH, (m/q=31).
S&He* - SiH$iH + HZ (4)
S&He* + M - S&H, + M (5)
SiH$iH + SiH, - Si3Hp* (6)
etc. The “etc.” indicates that reaction sequences analogous to that of reactions (4) and (6), coupled with collisional deactivation, lead to higher silanes and amorphous powder formation which are not detected in the gas phase.
In the presence of C2H,, some of the SiHz molecules are intercepted by C2H, before they can react with SiH4 and it is reasonably well established [ll, 121 that this reaction will proceed via the following sequence
SiH, + CzH4 + ,SiH,*,
CH2 CH, - CH2=CHSiH,* (7)
CH2=CHSiH3* - SiHz + CJ-L, (8)
CH2=CHSiH3* + M - CHz=CHSiHs + M (9)
The chemically activated silacyclopropane in reaction (7) is thought [ll-131 to be too labile to be detected at 298 K and 80 Torr total pressure because of its rapid isomerization to the more stable CH2=CHSiH3.
We have neglected decomposition of CH2=CHSiH3* to SiH4 and CzH,, although it is energetically feasible (i.e. A?zI’= -8 kcal mol-I), because we did not detect GH, among the products of the SiH2/C& reaction or deuterated silane among the products

178
SiH, CH=CH,
[CoH,l/[SiH, 1
Fig. 5. Depletion rate of SiH4 and formation rates of SiJ-& and C$H,SiH, as a [GHJ/[SiHJ at 300 K.
function of
of the SiH2/C2D4 reaction. Our failure to observe this decomposition is consistent with all reported studies of the reactions of silylene with olefins [ll, 121 and is probably because of the large activation barrier expected [14] in a reaction such as a unimolecular decomposition of CHZ=CHSiH3 to Si& and C&H,.
As expected, and as shown in Fig. 5, both the depletion rate of SiH, and the formation rate of S&Ha decreases as the ratio [C$I.,]/[SiH.,] increases, at constant total pressure and partial pressure of Sib; also, as expected, the formation rate of C,H$iH, increases with increasing [C&,]/[SiH.,]. This decrease in the depletion rate of SiH4 is not a result of collisional deactivation of Si&*(v) formed as an intermediate in reaction (1). Rather, as indicated by the increase in recovery of reacted silicon atoms as [C&]/[SiHJ increases, the depletion rate is reduced because the reactions of SiH2 and higher silylenes, such as SiH$iH, SiHsSiH*SiH, etc. to consume Si&, thereby forming higher silanes and amorphous powder are becoming less and less probable.
3.3. Kinetic treahnent A standard kinetic treatment of the mechanism embodied in reactions (2)-(g),
assuming that [Si&*J and [GH,SiH,*] are always at steady state, leads to the following expressions for the initial rates of formation of Si& and qH3SiH3

=R”( C2H3SiH3) = W~[SWOG~41[Ml
0 ks + k9Wl
179
(10)
(11)
where [SiH2],, may be viewed as an instantaneous initial concentration produced by the laser pulse, i.e. reaction (1).
It is more convenient to consider the ratio of initial rates of major product formation, which from eqns. (10) and (11) may be written as
where the ratio cfs/fg) represents the probability of collisional stabilization of Si2Ha* relative to that of C2H3SiH3*. That is
(13)
In our experiments the total pressure is fixed at 80 Torr but the gas composition is varied and, as mentioned in the experimental reaction, the average number of photons absorbed per pulse increases as [CJ&]/[SiH4] increases. Therefore, since the uni- molecular rate constants in eqn. (13) depend on the amount of absorbed energy, (fslf) will depend on the ratio [C21-&]/[Si&]. This will be true even for a fixed total pressure and with the simplifying approximation that the collisional stabilization terms, which strictly speaking should be written
ks[M] =ks[Sis] +ks’[C2H4] +k=,“[He] (14)
k,[M] = k,[SiI-IJ + k9’[GH4] + kg”[He] (15)
can be taken as average values independent of composition. We may estimate fs/f9 by using RRKM theory to calculate the unimolecular rate
constants k3, k4 and kg and use the simplifying approximation that ks =k9. As a reasonable value [14] for this average stabilization rate constant in our system, we take it to be l/l0 of the rate constant for collision, using an average collision diameter of 2.8 8, and an average reduced mass of 14 a.m.u. The value so estimated is: ks = k9 = 1.7 X 10-l’ cm3 particle-’ s-l. Although quite fortuitous, we may mention, as confirmation of the reasonableness of our estimate, that it agrees with the value found experimentally [15] for the collisional stabilization of vibrationally excited UF6.
3.4. RRKM Calculation of k3, k4 and k8 The internally excited silanes, Si2Hs* and CaH,SiHa*, that decompose unimolec-
ularly, contain distributions of internal energy that are determined by the energetics of reactions (3), (4) and (8) and by the internal energy distributions in the SiH2 and SiH4 molecules that are involved in these reactions. To approximate these distributions we make the following assumptions:
(i) the vibrational energy distribution in SiH, molecules that have absorbed IR photons is a Boltzmann distribution [9, lo], i.e.

where 0 is the average number of photons absorbed by SiH, and f(v) is the fraction of SIH, molecules that contain v photons;
(ii) all SiH4 molecules that contain 23 or more photons will decompose by reaction (1) before the next pulse occurs;
(iii) the excess vibrational energy in the decomposing SiH4 molecules is equi- partitioned among the internal degrees of freedom of the SiHz and H2 molecules formed;
(iv) any SiHz molecules formed that contain more than 39.6 kcal mol-’ [15] of internal energy will decompose to silicon + Hz and hence be inactive in further reaction of SiH, or C2H4: this means that those activated SiH, molecules for which 23 (v < 45 will decompose to SiH, molecules that will attack either C& or SiI!&;
(v) the unimolecular rate constant for decomposition of a molecule with an internal energy E* is given by [14]:
k(E*) = L*q??+)
hiV*(E *) (17)
where E* is the non-fixed excess energy of the activated molecule, N(E*) is the density of states at energy E *, E + is the excess energy of the transition state, P(E ‘) is the sum of vibrational states of the transition state with energy equal to or less than E+, and L+ is the number of equivalent decomposition paths.
With the above assumptions, the average rate constant for decomposition of C$H$rH,* becomes
(18)
where LI is the number of photons absorbed by SiH4 molecules that decompose, ii is the average number of photons in SiH, molecules that decompose, and E * = 2.7~ kcal mol-‘.
‘In the case of formation and decomposition of Si2H6*, both precursors, namely SiHz and SiHI, are internally excited, and hence the average rate constant becomes
where v is the number of photons absorbed by molecules of Si& that do not decompose and P is the average number of photons absorbed by non-decomposing SiH., molecules. As before E * = 2.7(u + v). An analogous expression results for k4(u + v).
Vibrational frequencies for the unimolecular rate constant calculations of Si2&* and C2H3SiH3* were taken from the literature [17-191 and the Whitten-Rabinovitch approximation [20] was used to compute the sums and densities of vibrational states in reaction (16). The reaction coordinates for reactions (3) and (4) were taken to be the Si-Si stretching mode and an Si-H stretching mode respectively, while that for reaction (8) was the GSi stretching mode; the corresponding vibrational frequencies were simply removed for the respective transition states.

181
The average unimolecular rate constants, k3, k4 and k5 so calculated are shown in Fig. 6(a) as a function of the reactant ratio [CzH4]/[SiH4]. By combining these unimolecular rate coefficients with the deactivation rate constants k5 = kg = 1.7 X 10-l’ cm3 particle-’ s-l (as discussed earlier) and [M] =2.58x 10” molecules cmd3, we obtain by eqn. (13) the ratio of the probability of deactivation of Si2H6* to the probability of deactivation of C2H3SiH3*, namely f51fg. The dependence of fslf9 on reactant ratio is shown in Fig. 6(b).
3.5. Evaluation of silylene rate constant ratio According to eqn. (ll), a plot of (fslfs) R”(C2H3SiH3)Ro(Si2H6) will yield a straight
line of slope k7/k2. Such a plot of our data for reaction temperatures of 300, 400 and 500 K is shown in Fig. 7. The observed linear plots add credence to our treatment and yield the rate coefficient ratio as a function of ambient temperature.
Our value of (k,/k,)=0.189 at 300 K from the slope in Fig. 7 is about half of the value obtained using the absolute rate coefficients reported by Jasinski and co- workers [6, 71. However, given the total pressure differences of the respective mea- surements (i.e. 80 Torr and 5 Torr), the fact that in our work SiHz and SiH, are vibrationally excited, and the uncertainty in our estimation of fslf9, the agreement must be considered satisfactory.
A plot of ln(k,/k,) vs. l/T is shown in Fig. 8. From this figure we may deduce the Arrhenius form of the rate constant ratio as
k, - =0.76+0.14 exp( - F) kz
s-l*iI x lo* (hi(E ))
2-
I I 2 3 rczu41 [Si t-L1
0.10 (L)
(20)
0.06t , , ,
I [W-b] 2 3
CSi &I
Fig. 6. (a) Calculated values of k3(E*), k&T*) and k&Y*) as a function of [C&l/[SiH,I. (b) Ratio of deactivation probabilities of Si&* and GH3SiH3* as a function of W-WSiH~l.

182
Fig. 7. Ratio of initial rates of attack of
Fig. 8. Arrhenlus plot of k7k2.
SiH, on SiH, and GH, as a function of [CJ5,,]/[SiH.,].
-1.6
or in other words the addition of SiH2 to C& has an activation energy that is 0.8 kcal mol-’ greater than that for insertion into an Si-H bond of SiH,.
3.6. Decomposition of SiH,-C2D4 mixtures A few experiments were carried out on equimolar mixtures of SM.,-&D4 for
the purpose of checking for the formation of Si&D4_,, in a possible decomposition channel of chemically activated GD3SiH2D*. As stated earlier no evidence for such a channel was found.
It was observed that the equimolar SZ&-GD, mixtures exhibited a marked reduction (approximately twofold) in the depletion rate of SiH., and, consequently, a corresponding decrease in the rate of product formation. It was also observed that substitution of GD, for C&L, in an equimolar mixture resulted in an approximately 20% decrease in the average number of photons absorbed per SiH., molecule. This magnitude of decrease in ?J has been observed previously [lo] to cause a reduction of at least a factor of two in !XL, decomposition rate. While we do not understand the phenomenon completely, it appears that GD4 is less effective than C&I., in collisionally removing vibrational energy from SiH,*(v) to provide a continuous supply of SiH4 molecules in the ground state or in low v states that consequently can absorb more laser energy during the pulse.
Acknowledgment
This work was supported by the U.S. Department of Energy through Grant No. DE-FGO2-88ER13835.
References
1 E. D. Eley, M. C. A. Rowe and R. Walsh, Chem. P&s. Lett., 126 (1986) 153. 2 D. S. Rogers, K. L. Walker, M. A. Ring and H. E. O’Neal, Oqpnometullics, 6 (1987) 2313.

183
3 G. Inoue and M. Suzuki, Chem. Phys. Letts., 122 (4) (1985) 361. 4 G. Inoue and M. Suzuki, Chem. Phys. Let&., 105 (1984) 641. 5 J. E. Baggort, H. M. Frey, P. D. Lightfoot and R. Walsh, Chem. Phys. Letts., 125 (1) (1986)
22. 6 J. 0. Chu, D. B. Beach and J. M. Jasinski, J. Phys. Chem., 91 (20) (1987) 5340. 7 J. M. Jasinski and J. 0. Chu, in E. R. Corey, J. Y. Corey and P. P. Gaspar (eds.), Silicon
Chemistry, Ellis Horwood, Chichester, England, 1988, Chap. 40. 8 K. W. Saunders and H. A. Taylor, J. Chem. Phys., 9 (1941) 616. 9 E. Grundwald, D. F. Denver and P. M. Keehn, Megawatt Infrared Laser Chemistry, Wiley,
New York, 1979. 10 P. A. Longeway and F. W. Lampe, J. Am. Chem. Sot., 103 (1981) 6813. 11 P. S. Skell and E. J. Goldstein, J. Am. Chem. Sot., 86 (1964) 1442. 12 P. P. Gaspar, in M. Jones, Jr. and R. A. Moss (eds.), Reactive Zntermediutes, Wiley, New
York, Vol. 1, 1978, p. 229; Vol. 2, 1981, p. 335; Vol. 3, 1985, p. 333. 13 R. Walsh, in E. R. Corey, J. Y. Corey and P. P. Gaspar (eds.), Silicon Chemistry, Ellis
Horwood, Chichester, England, 1988, Chap. 41. 14 P. J. Robinson and K. A. Holbrook, Unimolecular Reactions, Wiley-Interscience, New York,
1972. 15 H. E. Bass, F. D. Shields, W. D. Breshears and L. B. Asprey, J. Chem. Phys., 67 (1977)
1136. 16 P. Ho, M. E. Coltrin, J. S. Binkley and C. F. Melius, J. Am. Chem. Sot., 108 (1986) 2191. 17 A. J. Vandersielen, M. A. Ring and H. E. O’NeaI, J. Am. Chem. Sot., 97 (1975) 993. 18 T. L. Pollock, H. S. Andhu, A. Jodhau and 0. P. Strausz, .Z. Am. Chem. Sot., 95 (1973)
1017. 19 S. G. Frankiss, Spectrochim. Actu, 22 (1966) 295. 20 G. Z. Whitten and B. S. Rabinovitch, Z. Chem. Phys., 38 (1963) 2466.


















