
The pathology of hereditary polyposis syndromes - … · The pathology of hereditary polyposis...
10
REVIEW The pathology of hereditary polyposis syndromes Marco Novelli Department of Histopathology, UCLH NHS Foundation Trust, London, UK Novelli M (2015) Histopathology 66, 78–87. DOI: 10.1111/his.12590 The pathology of hereditary polyposis syndromes Our understanding of the genetics and pathology of familial colorectal cancer continues to evolve with both the discovery of underlying genetic defects and the description of entirely new entities. Genetic analy- sis has demonstrated phenotypic overlap between some of these syndromes, such that their nosology is rapidly becoming based on genetics with clinicopath- ological features playing a secondary, but important, role. Further clinical characterization of these syn- dromes has also demonstrated widely differing risks for the development of colorectal cancer and a range of other malignancies with implications for both the affected patient and members of their families. This review aims to outline the clinical, pathological and genetic features of this increasingly complex group of diseases. Keywords: colorectal cancer, genetics, hereditary, polyposis Introduction Before the advent of molecular biology the classifica- tion of hereditary colorectal cancer syndromes was relatively straightforward, being based largely on clinical criteria. There was colorectal cancer with polyposis [familial adenomatous polyposis (FAP)], the non-polyposis colorectal cancer syndromes, (Lynch syndromes 1 and 2) and the ‘benign’ hamartomatous polyp syndromes. This situation was slightly con- founded by some rarer clinical overlap syndromes, including polyposis with a variety of extra-colonic manifestations (Gardner’s syndrome) and polyposis with brain tumours (Turcot’s syndrome). In 1991 research on the genetics of FAP lad to the discovery of the adenomatous polyposis coli (APC) gene and to the outlining of the genetics of the adenoma–carci- noma sequence in sporadic colorectal cancer. 1 Since this time our understanding of the genetics of familial colorectal cancer has changed markedly, such that many of the older terms have effectively now become obsolete (e.g. Gardner’s syndrome is now generally accepted to be FAP, with some of its well-described extra colonic manifestations), and a number of novel colorectal cancer syndromes have been described. As our understanding of the genetics of familial colorec- tal cancer syndromes has improved it has become apparent that there is phenotypic overlap between many of the syndromes. The aim of this paper is not to describe the histopathology of intestinal polyps, which is covered extensively elsewhere, 2 but to out- line our current understanding of the inheritance, genetics and clinical features of colorectal cancer syn- dromes (see Table 1). Familial adenomatous polyposis (FAP) This is perhaps the best known and archetypal polyp- osis syndrome, and was the first to be characterized fully. FAP is due to a germline mutation in the APC gene located on the long arm of chromosome 5 (5q21-22). FAP is inherited in an autosomal domi- nant fashion with a high penetrance, although de-novo germline mutations may account for perhaps up to 25% of cases. 3 Mutation of the APC gene leads typically to the development of a full-blown polyposis with more than 100 (and typically thousands of) Address for correspondence: Professor M Novelli, Department of Histopathology, University College London, Rockefeller Building, University Street, London WC1E 6JJ, UK. e-mail: m.novelli@ucl. ac.uk © 2015 John Wiley & Sons Ltd. Histopathology 2015, 66, 78–87. DOI: 10.1111/his.12590
-
Upload
trinhquynh -
Category
Documents
-
view
227 -
download
1
Transcript of The pathology of hereditary polyposis syndromes - … · The pathology of hereditary polyposis...

REVIEW
The pathology of hereditary polyposis syndromes
Marco NovelliDepartment of Histopathology, UCLH NHS Foundation Trust, London, UK
Novelli M
(2015) Histopathology 66, 78–87. DOI: 10.1111/his.12590
The pathology of hereditary polyposis syndromes
Our understanding of the genetics and pathology offamilial colorectal cancer continues to evolve withboth the discovery of underlying genetic defects andthe description of entirely new entities. Genetic analy-sis has demonstrated phenotypic overlap betweensome of these syndromes, such that their nosology israpidly becoming based on genetics with clinicopath-ological features playing a secondary, but important,
role. Further clinical characterization of these syn-dromes has also demonstrated widely differing risksfor the development of colorectal cancer and a rangeof other malignancies with implications for both theaffected patient and members of their families. Thisreview aims to outline the clinical, pathological andgenetic features of this increasingly complex group ofdiseases.
Keywords: colorectal cancer, genetics, hereditary, polyposis
Introduction
Before the advent of molecular biology the classifica-tion of hereditary colorectal cancer syndromes wasrelatively straightforward, being based largely onclinical criteria. There was colorectal cancer withpolyposis [familial adenomatous polyposis (FAP)], thenon-polyposis colorectal cancer syndromes, (Lynchsyndromes 1 and 2) and the ‘benign’ hamartomatouspolyp syndromes. This situation was slightly con-founded by some rarer clinical overlap syndromes,including polyposis with a variety of extra-colonicmanifestations (Gardner’s syndrome) and polyposiswith brain tumours (Turcot’s syndrome). In 1991research on the genetics of FAP lad to the discoveryof the adenomatous polyposis coli (APC) gene and tothe outlining of the genetics of the adenoma–carci-noma sequence in sporadic colorectal cancer.1 Sincethis time our understanding of the genetics of familialcolorectal cancer has changed markedly, such thatmany of the older terms have effectively now become
obsolete (e.g. Gardner’s syndrome is now generallyaccepted to be FAP, with some of its well-describedextra colonic manifestations), and a number of novelcolorectal cancer syndromes have been described. Asour understanding of the genetics of familial colorec-tal cancer syndromes has improved it has becomeapparent that there is phenotypic overlap betweenmany of the syndromes. The aim of this paper is notto describe the histopathology of intestinal polyps,which is covered extensively elsewhere,2 but to out-line our current understanding of the inheritance,genetics and clinical features of colorectal cancer syn-dromes (see Table 1).
Familial adenomatous polyposis (FAP)
This is perhaps the best known and archetypal polyp-osis syndrome, and was the first to be characterizedfully. FAP is due to a germline mutation in the APCgene located on the long arm of chromosome 5(5q21-22). FAP is inherited in an autosomal domi-nant fashion with a high penetrance, althoughde-novo germline mutations may account for perhapsup to 25% of cases.3 Mutation of the APC gene leadstypically to the development of a full-blown polyposiswith more than 100 (and typically thousands of)
Address for correspondence: Professor M Novelli, Department of
Histopathology, University College London, Rockefeller Building,
University Street, London WC1E 6JJ, UK. e-mail: m.novelli@ucl.
ac.uk
© 2015 John Wiley & Sons Ltd.
Histopathology 2015, 66, 78–87. DOI: 10.1111/his.12590
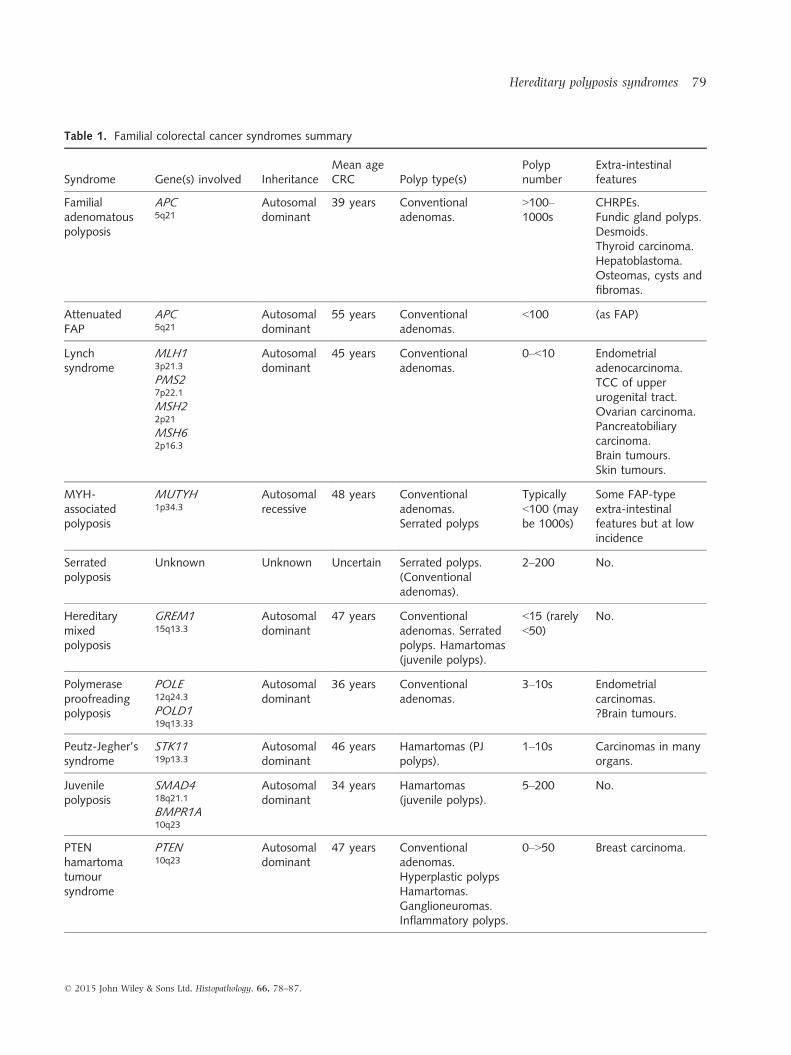
Table 1. Familial colorectal cancer syndromes summary
Syndrome Gene(s) involved InheritanceMean ageCRC Polyp type(s)
Polypnumber
Extra-intestinalfeatures
Familialadenomatouspolyposis
APC5q21
Autosomaldominant
39 years Conventionaladenomas.
>100–1000s
CHRPEs.Fundic gland polyps.Desmoids.Thyroid carcinoma.Hepatoblastoma.Osteomas, cysts andfibromas.
AttenuatedFAP
APC5q21
Autosomaldominant
55 years Conventionaladenomas.
<100 (as FAP)
Lynchsyndrome
MLH13p21.3
PMS27p22.1
MSH22p21
MSH62p16.3
Autosomaldominant
45 years Conventionaladenomas.
0–<10 Endometrialadenocarcinoma.TCC of upperurogenital tract.Ovarian carcinoma.Pancreatobiliarycarcinoma.Brain tumours.Skin tumours.
MYH-associatedpolyposis
MUTYH1p34.3
Autosomalrecessive
48 years Conventionaladenomas.Serrated polyps
Typically<100 (maybe 1000s)
Some FAP-typeextra-intestinalfeatures but at lowincidence
Serratedpolyposis
Unknown Unknown Uncertain Serrated polyps.(Conventionaladenomas).
2–200 No.
Hereditarymixedpolyposis
GREM115q13.3
Autosomaldominant
47 years Conventionaladenomas. Serratedpolyps. Hamartomas(juvenile polyps).
<15 (rarely<50)
No.
Polymeraseproofreadingpolyposis
POLE12q24.3
POLD119q13.33
Autosomaldominant
36 years Conventionaladenomas.
3–10s Endometrialcarcinomas.?Brain tumours.
Peutz-Jegher’ssyndrome
STK1119p13.3
Autosomaldominant
46 years Hamartomas (PJpolyps).
1–10s Carcinomas in manyorgans.
Juvenilepolyposis
SMAD418q21.1
BMPR1A10q23
Autosomaldominant
34 years Hamartomas(juvenile polyps).
5–200 No.
PTENhamartomatumoursyndrome
PTEN10q23
Autosomaldominant
47 years Conventionaladenomas.Hyperplastic polypsHamartomas.Ganglioneuromas.Inflammatory polyps.
0–>50 Breast carcinoma.
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
Hereditary polyposis syndromes 79

adenomatous polyps scattered throughout the colo-rectum. However, some germline mutations (particu-larly those at either the 50 or 30 ends of the gene)lead to a milder phenotype known as attenuatedFAP, with fewer adenomas (typically 20–30), whichare usually concentrated on the right side of thecolon (see Figure 1A). In both FAP and attenuatedFAP the adenomas usually develop during adoles-cence, and patients typically go on to develop colorec-tal cancer in their late 20s to 40s with classical FAP,or later with attenuated FAP (mean age 55 years).Both FAP and attenuated FAP patients may alsodevelop small intestinal adenomas/carcinomas (par-
ticularly around the ampulla of Vater), fundic glandpolyps and a variety of extra-intestinal manifesta-tions, including desmoid tumours, thyroid carcinomas(papillary, cribriform-morula variant), brain tumours,hepatoblastomas, epidermoid cysts and osteomas.The histopathology of adenomas and adenocarcino-
mas in FAP is identical to that of their sporadic colo-rectal counterparts. However, examination of thebackground colorectal mucosa of FAP patients typi-cally shows many microscopic adenomas (so-calledmicroadenomas), including single dysplastic crypts(unicryptal adenomas). The finding of a unicryptaladenoma is almost pathognomonic of FAP.
Table 1. (Continued)
Syndrome Gene(s) involved InheritanceMean ageCRC Polyp type(s)
Polypnumber
Extra-intestinalfeatures
Constitutionalmismatchrepairdeficiency
MLH13p21.3
PMS27p22.1
MSH22p21
MSH62p16.3
Autosomalrecessive
Childhood– youngadults
Conventionaladenomas.
Few–10s Caf�e-au-lait spots.High-grade braintumours.Leukaemias/lymphomas.
CHRPE: Congenital hypertrophy of the retinal pigment epithelium; CRC: colorectal cancer; FAP: familial adenomatouspolyposis; TCC: transitional cell carcinoma; APC: adenomatous polyposis coli; MLH1: MutL homologue 1; MSH2: MutShomologue 2; MSH6: MutS homologue 6; PMS2: postmeiotic segregation increased 2; PTEN: phosphatase and tensinhomologue; SMAD4: SMAD family member 4; BMPR1A: bone morphogenetic protein receptor, type IA; POLE: polymeraseepsilon; POLED1: polymerase (DNA directed), delta 1; STK11: serine/threonine kinase 11; GREM1: gremlin 1; MUTYH:
mutY homologue.
A B
Figure 1. Typical macroscopic appearances of: a, attenuated FAP; and b, Juvenile polyposis (please note atypical juvenile polyps, arrowed).
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
80 M Novelli
Lynch syndrome
As its previous name (‘hereditary non-polyposis colo-rectal cancer’) suggests, Lynch syndrome does notpresent typically with a polyposis. However, Lynchsyndrome patients have a higher incidence of adeno-matous polyps than age-matched controls,4 and nodescription of hereditary colorectal cancer syndromeswould be complete without the inclusion of Lynchsyndrome. Lynch syndrome is by far the most com-mon of the inherited colorectal cancer syndromes,accounting for up to 3% of all colorectal cancers.5
Patients typically develop colorectal and endometrialcancers between their 40s and 60s. They are also atincreased risk of developing adenocarcinomas of thestomach, small intestine, upper urinary tract, ovary,pancreatobiliary tract, brain and skin. Their tumoursare often synchronous or metachronous.Lynch syndrome is inherited in an autosomal
dominant fashion, although perhaps up to 5% ofcases3 may be due to new mutations. The syndromeis caused by a germline mutation in one of fourmismatch repair genes—MLH1 (MutL homologue 1),MSH2 (MutS homologue 2), MSH6 (MutS homolo-gue 6) and PMS2 (postmeiotic segregation increased2). A 5th gene, MSH3, is very rarely, if ever,involved. These mismatch repair proteins are respon-sible for recognizing and repairing mistakes in DNAtranscription, which occur particularly in areas ofrepeat DNA sequences (microsatellites) where DNApolymerases have a tendency to ‘slip’, either insert-ing extra or removing repeats. In the case of mono-nucleotide and dinucleotide repeat sequences thisoften leads to a frameshift mutation, resulting in ashortened non-functional protein.Lynch syndrome colorectal cancers are seen predom-
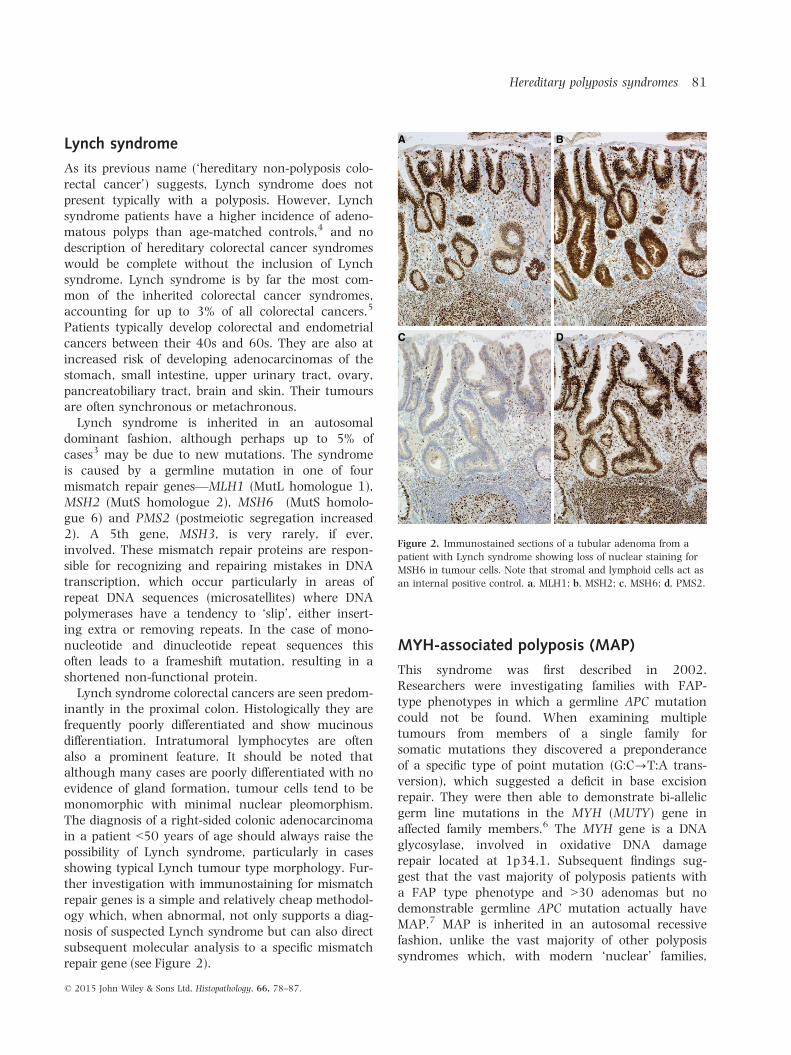
inantly in the proximal colon. Histologically they arefrequently poorly differentiated and show mucinousdifferentiation. Intratumoral lymphocytes are oftenalso a prominent feature. It should be noted thatalthough many cases are poorly differentiated with noevidence of gland formation, tumour cells tend to bemonomorphic with minimal nuclear pleomorphism.The diagnosis of a right-sided colonic adenocarcinomain a patient <50 years of age should always raise thepossibility of Lynch syndrome, particularly in casesshowing typical Lynch tumour type morphology. Fur-ther investigation with immunostaining for mismatchrepair genes is a simple and relatively cheap methodol-ogy which, when abnormal, not only supports a diag-nosis of suspected Lynch syndrome but can also directsubsequent molecular analysis to a specific mismatchrepair gene (see Figure 2).
MYH-associated polyposis (MAP)
This syndrome was first described in 2002.Researchers were investigating families with FAP-type phenotypes in which a germline APC mutationcould not be found. When examining multipletumours from members of a single family forsomatic mutations they discovered a preponderanceof a specific type of point mutation (G:C?T:A trans-version), which suggested a deficit in base excisionrepair. They were then able to demonstrate bi-allelicgerm line mutations in the MYH (MUTY) gene inaffected family members.6 The MYH gene is a DNAglycosylase, involved in oxidative DNA damagerepair located at 1p34.1. Subsequent findings sug-gest that the vast majority of polyposis patients witha FAP type phenotype and >30 adenomas but nodemonstrable germline APC mutation actually haveMAP.7 MAP is inherited in an autosomal recessivefashion, unlike the vast majority of other polyposissyndromes which, with modern ‘nuclear’ families,
A B
C D
Figure 2. Immunostained sections of a tubular adenoma from a
patient with Lynch syndrome showing loss of nuclear staining for
MSH6 in tumour cells. Note that stromal and lymphoid cells act as
an internal positive control. a, MLH1; b, MSH2; c, MSH6; d, PMS2.
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
Hereditary polyposis syndromes 81
means there is rarely any family history of polyposis.Many germline mutations in the MYH gene in MAPhave been described, but there are hotspots in differ-ent populations which allow for relatively simplegenetic screening (e.g. Caucasian populations havemutation hotspots at p.Y179C and/or p.G396D in90%8).Most MAP patients present with between 10 and
500 polyps, but some patients may have more than1000 polyps,9 and there are also reports of MAPpatients with colorectal cancer but no polyposis. Therisk of developing colorectal cancer increases withage, with a penetrance of 43% by age 60 and a meanage of presentation with colorectal cancer of 48 years(range 21–70).Although MAP was considered initially to closely
mimic attenuated FAP, it has become apparent thatMAP patients frequently develop serrated type polyps(hyperplastic polyps and sessile serrated lesions) inaddition to classical adenomas. Serrated-type polypsare said to be present in approximately half of MAPpatients (47%), with up to a fifth of MAP patients(18%) fulfilling the World Health Organization(WHO) diagnostic criteria for serrated polyposis.10 Ithas been hypothesized that the order of developingsomatic mutations in target genes [e.g. APC, KRAS]may determine which type of polyp develops subse-quently. FAP-type extra-intestinal manifestationshave also been reported, but at a relatively low level.9
There is also evidence that there may be an increased
risk of a variety of extra-intestinal cancers, includingthose of ovary, bladder and skin.11
Serrated polyposis (hyperplastic polyposis)
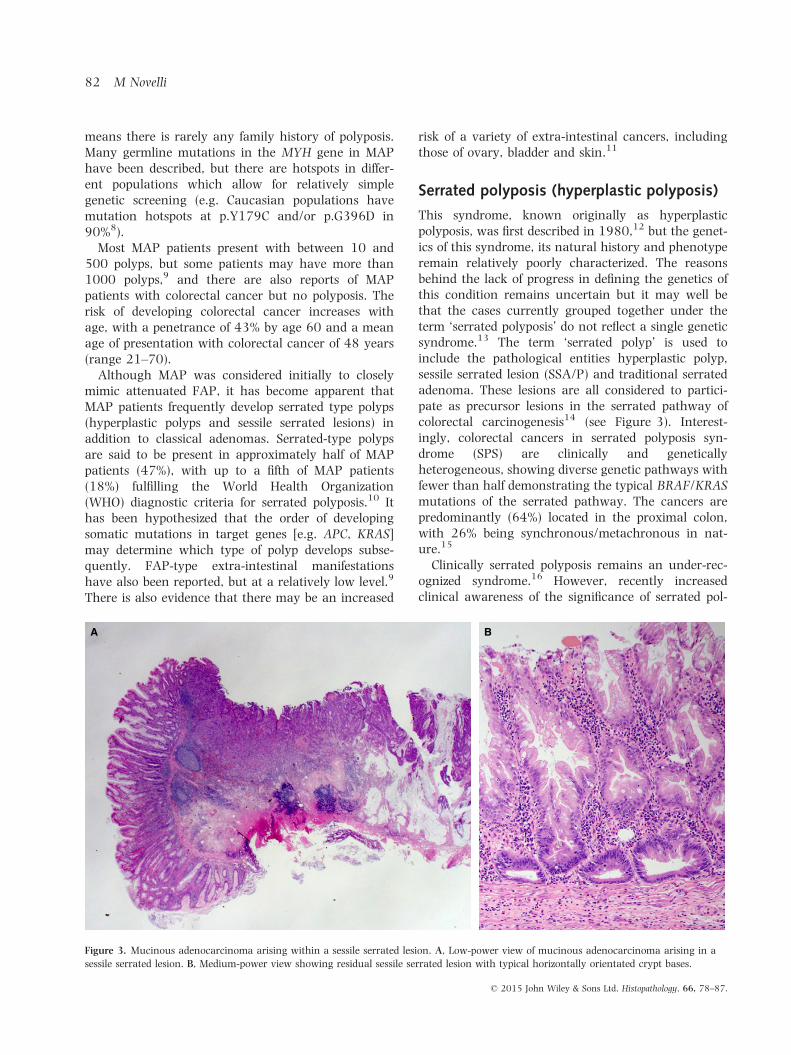
This syndrome, known originally as hyperplasticpolyposis, was first described in 1980,12 but the genet-ics of this syndrome, its natural history and phenotyperemain relatively poorly characterized. The reasonsbehind the lack of progress in defining the genetics ofthis condition remains uncertain but it may well bethat the cases currently grouped together under theterm ‘serrated polyposis’ do not reflect a single geneticsyndrome.13 The term ‘serrated polyp’ is used toinclude the pathological entities hyperplastic polyp,sessile serrated lesion (SSA/P) and traditional serratedadenoma. These lesions are all considered to partici-pate as precursor lesions in the serrated pathway ofcolorectal carcinogenesis14 (see Figure 3). Interest-ingly, colorectal cancers in serrated polyposis syn-drome (SPS) are clinically and geneticallyheterogeneous, showing diverse genetic pathways withfewer than half demonstrating the typical BRAF/KRASmutations of the serrated pathway. The cancers arepredominantly (64%) located in the proximal colon,with 26% being synchronous/metachronous in nat-ure.15
Clinically serrated polyposis remains an under-rec-ognized syndrome.16 However, recently increasedclinical awareness of the significance of serrated pol-
A B
Figure 3. Mucinous adenocarcinoma arising within a sessile serrated lesion. A, Low-power view of mucinous adenocarcinoma arising in a
sessile serrated lesion. B, Medium-power view showing residual sessile serrated lesion with typical horizontally orientated crypt bases.
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
82 M Novelli

yps (in particular of the sessile serrated lesion) andadvances in colonoscopic technique have led to anincreasing rate of discovery of serrated polyps andhence cases of SPS. There are widely utilized WHOdiagnostic criteria for serrated polyposis (see Table 2),but it should be noted that there is still relatively littleknown about this condition. The fact that patientsappear to develop new lesions with increasing agesuggests that, despite failing to make the strict WHOcriteria for serrated polyposis, young patients (e.g.<50 years old) with multiple right-sided hyperplasticpolyps or sessile serrated lesions should perhaps beconsidered for screening.Although considered a hereditary polyposis, the
exact mode of inheritance of SPS remains uncertain;40–60% of affected individuals report a family historyof colorectal cancer (CRC) in first- and/or second-degree relatives.17 In a recent study the mean age ofdiagnosis of serrated polyposis was 49 (range 18–77 years), with a median of 35 serrated polyps(range 8–180).18 It should be noted that patients ful-filling the diagnostic criteria for serrated polyposisalso often have conventional-type adenomas.13,19
There is undoubtedly an increased risk of the develop-ment of colorectal carcinoma in patients with SPS,but published prevalence rates range markedly(0–77%).18 Results from a multicentre cohort studyshowed a 7% risk of developing colorectal cancer dur-ing 5 years of colonoscopic follow-up.19 Althoughsuggested, there is currently little firm evidence forextra-colonic manifestations with this syndrome.18
Hereditary mixed polyposis
Hereditary mixed polyposis is a very rare colorectalcancer syndrome. This is predominantly a disease ofAshkenazi Jews, probably originating from one largeextended family. The syndrome is inherited in an auto-somal dominant fashion and has been shown to be dueto a duplication spanning the 30 end of the secretogra-nin V5 (SCG5) gene and a region upstream of thegremlin 1 (GREM1) locus. This mutation is thought toresult in increased GREM1 expression which thenreduces BMP pathway activity, causing tumorigenesis.
The BMP pathway is also thought to be involved injuvenile polyposis.20
Patients develop a variety of polyp types, includingconventional adenomas, juvenile-type polyps and ser-rated-type polyps. Polyps are distributed throughoutthe colorectum, typically numbering <15. Patients goon to develop colonic adenocarcinoma at a mean ageof 47 (range 32–74 years).21
Polymerase proofreading associatedpolyposis (PPAP)
Polymerase proofreading-associated polyposis (PPAP)is the most recently described familial polyposis syn-drome.22 Currently, there is a relative paucity of infor-mation about this syndrome, but initial studies suggestthat it is a colorectal cancer syndrome with autosomaldominant inheritance and a high penetrance. PPAPhas been shown to be due to germline mutations inthe DNA polymerases polymerase epsilon (POLE) andpolymerase delta (POLD1)]. Mutations in POLE havealso been demonstrated in sporadic colorectal cancers.Patients are said to develop multiple colorectal ade-
nomas (few large adenomas to 10s of adenomas).Colorectal adenocarcinomas, which may be multiple,present typically in the mid-30s. In female patientswith germline POLD1 mutations there is also anincreased risk of developing endometrial adenocarci-noma.23 There is also a suggestion that the germlinePOLD1 mutation may predispose to some braintumours. The adenomas and adenocarcinomas ofPPAP are said to be of conventional type with nocharacteristic morphological features (unlike the car-cinomas of Lynch syndrome). They are typicallymicrosatellite stable. Thus, polyposis, an early age ofdevelopment of colorectal cancer and a family historyof colorectal and endometrial adenocarcinoma arethe main clues to diagnosing this syndrome.
Hamartomatous polyp syndromes
There are a number of hamartomatous polyp syn-dromes including, among others, juvenile polyposis,
Table 2. World Health Organization diagnostic criteria for the diagnosis of serrated polyposis syndrome (SPS)
Any one of the following
(1) At least five serrated polyps proximal to the sigmoid colon, two of which are >10 mm in diameter
(2) Any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with SPS
(3) Greater than 20 serrated polyps of any size distributed throughout the colon
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
Hereditary polyposis syndromes 83

Peutz–Jeghers syndrome and PTEN-hamartomatumour syndrome (Cowden’s syndrome). These wereconsidered originally to be benign ‘hamartomatoussyndromes’, but over the years it has become appar-ent that these are full-blown hereditary cancer syn-dromes.
P E U T Z – J E G H E R S Y N D R O M E ( P J S )
Although perhaps best known for its characteristicarborizing polyps, PJS is a multisystem cancer syn-drome, with affected individuals being at risk of devel-oping carcinomas of the colorectum, pancreas,stomach, small intestine, oesophagus, breast, ovary,endometrium and lung. In a meta-analysis, 93%of Peutz–Jegher patients developed cancer by the ageof 65, with a mean age of cancer presentation of42 years.24 Thirty-nine per cent of these patientsdeveloped colorectal adenocarcinoma. PJS is inheritedin an autosomal dominant fashion and in 94% ofcases has been shown to be associated with a germ-line mutation in the serine/threonine kinase 11(STK11) gene on chromosome 19p.25
Diagnosis is based on family history, the finding oftypical Peutz–Jegher-type polyps and the presence ofmucocutaneous pigmentation, such pigmentationbeing present in 95% of patients (see Table 3).26
Polyps can occur anywhere in the gastrointestinaltract, but small intestine and colorectum are the mostcommon sites. Polyp numbers vary widely, rangingfrom single polyps to tens of polyps. The finding of asingle polyp with typical Peutz–Jeghers features shouldraise the possibility of this syndrome, with some
authors questioning the very existence of ‘sporadic’Peutz–Jeghers polyps.27 The presence of areas of floridepithelial misplacement and the development of con-ventional-type dysplasia are both well described inPeutz–Jegher polyps.28 The development of dysplasiain these polyps may well explain the increased risk ofcolorectal adenocarcinoma seen in PJS patients, but itshould also be noted that the combination of epithelialdysplasia and epithelial misplacement may both mimicand lead to an erroneous diagnosis of adenocarcinoma.
J U V E N I L E P O L Y P O S I S ( J P S )
Juvenile polyposis is considered a pure intestinalpolyposis syndrome lacking the extra-intestinal mani-festations seen in many of the other polyposis syn-dromes. JPS patients develop characteristic polypsthroughout the gastrointestinal (GI) tract, typicallyby the age of 20 years, with a reported cumulativelifetime risk for developing colorectal adenocarcinomaof 40–70%.29 The syndrome is inherited in an auto-somal dominant fashion, with a germline mutation inthe SMAD4 or BMPR1A genes being demonstrable inapproximately 50–60% of JPS patients.30 Diagnosis isbased on family history and the finding of juvenile-type polyps (see Table 4).Juvenile polyposis patients typically develop five to
200 polyps throughout the gastrointestinal tract withcolonic involvement in the vast majority of patients29
(see Figure 1B). The polyps are usually similar to, ifnot often indistinguishable from, sporadic inflamma-tory juvenile type polyps. However, they may beatypical, with multilobation and a relative lack of the
Table 4. World Health Organization diagnostic criteria for the diagnosis of juvenile polyposis syndrome
Any one of the following
(1) More than five juvenile polyps in the colon or rectum
(2) Juvenile polyps throughout the gastrointestinal tract
(3) Any number of juvenile polyps in a person with a family history of juvenile polyposis
Table 3. World Health Organization diagnostic criteria for the diagnosis of Peutz–Jeghers syndrome (PJS)
Any one of the following
(1) Three or more histologically confirmed Peutz–Jeghers polyps
(2) Any number of Peutz–Jeghers polyps with a family history of PJS
(3) Characteristic, prominent, mucocutaneous pigmentation with a family history of PJS
(4) Any number of Peutz–Jeghers polyps and characteristic, prominent, mucocutaneous pigmentation
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
84 M Novelli

expanded lamina propria seen usually in sporadicjuvenile polyps.29 Dysplasia can sometimes be seen inthe polyps.
P T E N - H A M A R T O M A T U M O U R S Y N D R O M E
The PTEN-hamartoma tumour syndrome is the termfor clinical syndromes caused by germline mutationsin the tumour suppressor phosphatase and tensinhomologue, situated on chromosome 10q23 (PTEN).This term includes Cowden syndrome (CS) and therarer clinical entities of Bannayan–Riley–Ruvalcaba(BRRS), PTEN-related Proteus syndrome (PS) and Pro-teus-like syndrome. CS and BRRS are now consideredby many to represent a single entity. CS is inherited inan autosomal dominant fashion with probably approx-imately 30% of patients having de-novo mutations.31
PTEN hamartoma tumour syndrome patients are saidto develop hamartomas in multiple organ systems andhave an increased risk of malignancy, including carci-nomas of the breast, thyroid, endometrium and kidneyand possibly melanomas.32 Approaching 95% ofpatients develop a polyposis, which is typically ofmixed type and may include hyperplastic-, adenoma-tous-, hamartomatous-, ganglioneuromatous- andinflammatory-type polyps. At least some hyperplasticpolyps are present in approximately half thesepatients.33 Colonic involvement is seen typically, andthere is an increased risk of colorectal cancer with aCRC prevalence of approximately 14% and a meanage of colorectal cancer diagnosis of 47.34
Constitutional mismatch repair deficiency(CMMRD)
This extremely rare syndrome effectively representsrecessively inherited Lynch syndrome. This disease isseen predominantly in the offspring of consanguine-ous marriages, where rare germline DNA mutationsare brought together and inherited in an autosomalrecessive fashion. The syndrome presents typically inchildhood, but may also present in young adults.Counterintuitively, there is rarely a family history ofLynch type tumours despite the fact that both of theaffected patient’s parents harbour germline mutationsin a mismatch repair gene. Patients are at risk ofdeveloping a spectrum of tumour types which, inmany cases, can be synchronous or metachronous.Commonly seen tumour types include colorectal ade-nocarcinomas, high-grade brain tumours and leukae-mias/lymphomas. CMMRD patients with acombination of colorectal and brain tumours most
probably account for a large proportion of cases clas-sified previously as Turcot’s syndrome. All patientsare said to have cafe0-au-lait spots, making dermato-logical examination a useful diagnostic adjunct.35
Although relatively poorly characterized, unlike thecolorectal adenocarcinomas of Lynch syndrome thoseof CMMRD do not appear to have a predilection for theproximal colon. They do, however, often show similarmicroscopic morphology to Lynch syndrome tumours(poorly differentiated, mucinous differentiation, etc.)(see Figure 4). Immunostaining for mismatch repairproteins is helpful in both confirming a suspected diag-nosis and directing subsequent genetic analysis. Itshould be noted that there is total loss of immunoposi-tivity for the affected protein(s), both in tumour andnormal cells (see Figure 4E). Ideally, the histopathol-ogy laboratory should be prewarned in suspected casesto avoid repeated attempts at optimizing apparentlyfailed immunostaining. Microsatellite instability studiesare reported to be unreliable in CMMRD patienttumours.35
Discussion
Research on familial colorectal cancer patients con-tinues to provide insights into the genetics of bothfamilial and sporadic colorectal cancer. While itremains pertinent for many CRCs, it has becomeapparent that the seminal work of the Vogelsteingroup in the 1990s outlining the adenoma carci-noma sequence cannot be applied to all colorectalcancers. A number of novel colorectal cancer path-ways have since been described, and the situation iscomplicated further by the fact that some tumoursseem to show features of more than one CRC path-way.14 This increasing complexity in the genetics ofCRC is reflected in the complexity of the colorectalcancer syndromes. It has also become apparent thatthe DNA repair machinery is crucial for prevention oftumours in normal cells with three separate colorec-tal cancer syndromes (Lynch, MAP and PPAP), allresulting from germline mutations in DNA ‘repair’proteins. What has yet to be explained is why muta-tions in these genes leads to a susceptibility todevelop colorectal cancers and (for Lynch syndromeand PPAP) endometrial carcinomas.In cases of a suspected colorectal cancer syndrome,
a family history of colorectal cancer or other associ-ated tumour types may be extremely informative.However, while most of the colorectal cancer syn-dromes are inherited in a Mendelian autosomal domi-nant fashion both MAP and CMMRD showautosomal recessive inheritance, so affected patients
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
Hereditary polyposis syndromes 85

often lack a family history of polyps or CRC. It shouldalso be noted that de-novo germline mutations mayaccount for up to 30% of cases of some familial CRCsyndromes. Thus, a family history is useful whenpresent, but otherwise the clinician/pathologist mayhave to depend upon other clues to warn them thatthey may be dealing with a familial cancer. Age ofonset of colorectal cancer is perhaps one of the mosthelpful of such clues. Sporadic colorectal cancers veryrarely occur before the age of 50, and most familialcolorectal cancers (with the exception of attenuatedFAP tumours) occur around or before this age (seeTable 1). Other clues include a right-sided location,histopathological features (in Lynch syndrome) andthe presence of a polyposis. The number and distribu-tion of the polyps taken together with the composi-tion of the polyposis may help to both heighten thesuspicion of a familial tumour and to narrow thespectrum of the most likely syndromes involved.However, it should be noted that there is phenotypi-cal overlap between the varying syndromes (e.g.some patients with MAP fulfil the WHO diagnosticcriteria for serrated polyposis). Ideally, a combinationof clinical, genetic, endoscopic and pathological fea-tures is required to allow for accurate disease classifi-cation (see Table 5), but ultimately the diagnosisneeds to be confirmed by genetic testing.
Learning points
• The clinical phenotypes of hereditary polyposissyndromes may overlap.
• De-novo mutations may account for up to 30% ofcases of some CRC syndromes.
• Serrated polyposis patients may develop conven-tional adenomas.
• MAP typically mimics attenuated FAP, butpatients may present with a serrated polyposis-typepicture.
Table 5. Does this patient have a familial cancer syn-drome?
Questions to address:
Age of diagnosis of colorectal cancer or polyposis?
Is there any family history of polyposis or colorectalcancer?
Is there any family history of other cancers (especiallyendometrial)?
What type of polyps are present?
What is the distribution of polyps?
Are there any extra-intestinal manifestations?
A B C
D E
Figure 4. Colonic pathology from 9-year-old male with constitutional mismatch repair deficiency (CMMRD). A, Subtotal colectomy specimen
showing two left-sided colonic adenocarcinomas and multiple adenomas. B, H&E-stained section showing moderately differentiated sigmoid ade-
nocarcinoma with mucinous differentiation. C, MLH1 immunostained section showing a normal staining pattern. D, MSH2 immunostained sec-
tion showing a normal staining pattern. E, MSH6 immunostained section showing complete loss of staining in both tumour and stromal cells.
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
86 M Novelli

Conflict of interests
The author has no competing interests to declare.
References
1. Kinzler KW, Nilbert MC, Vogelstein B et al. Identification of a
gene located at chromosome 5q21 that is mutated in colorectal
cancers. Science 1991; 251; 1366–1370.2. Clouston AD, Walker NI. Polyps and tumour-like lesions of the
large intestine. In Shepherd NA, Warren BF, Williams GT,
Greenson JK, Lauwers GY, Novelli MR eds. Morson and Daw-
son’s gastrointestinal pathology. 5th ed. Chichester: Wiley-Black-
well, 2013; 647–684.3. Win AK, Jenkins MA, Buchanan DD et al. Determining the fre-
quency of de novo germline mutations in DNA mismatch repair
genes. J. Med. Genet. 2011; 48; 530–534.4. Jass JR, Stewart SM. Evolution of hereditary non-polyposis colo-
rectal cancer. Gut 1992; 33; 783–786.5. de la Chapelle A. The incidence of Lynch syndrome. Fam. Can-
cer 2005; 4; 233–237.6. Al-Tassan N, Chmiel NH, Maynard J et al. Inherited variants of
MYH associated with somatic G:C–>T: a mutations in colorec-
tal tumors. Nat. Genet. 2002; 30; 227–232.7. Filipe B, Baltazar C, Albuquerque C et al. APC or MUTYH
mutations account for the majority of clinically well-
characterized families with FAP and AFAP phenotype and
patients with more than 30 adenomas. Clin. Genet. 2009; 76;
242–255.8. Nielsen M, Joerink-van de Beld MC, Jones N et al. Analysis of
MUTYH genotypes and colorectal phenotypes in patients with
MUTYH-associated polyposis. Gastroenterology 2009; 136; 471–476.
9. Nielsen M, Morreau H, Vasen HF, Hes FJ. MUTYH-associated
polyposis (MAP). Crit. Rev. Oncol. Hematol. 2011; 79; 1–16.10. Boparai KS, Dekker E, Van Eeden S et al. Hyperplastic polyps
and sessile serrated adenomas as a phenotypic expression of
MYH-associated polyposis. Gastroenterology 2008; 135; 2014–2018.
11. Vogt S, Jones N, Christian D et al. Expanded extracolonic
tumor spectrum in MUTYH-associated polyposis. Gastroenterol-
ogy 2009; 137; 1976–1985, e1–10.12. Williams GT, Arthur JF, Bussey HJ, Morson BC. Metaplastic
polyps and polyposis of the colorectum. Histopathology 1980;
4; 155–170.13. Carvajal-Carmona LG, Howarth KM, Lockett M et al. Molecular
classification and genetic pathways in hyperplastic polyposis
syndrome. J. Pathol. 2007; 212; 378–385.14. Noffsinger AE. Serrated polyps and colorectal cancer: new
pathway to malignancy. Annu. Rev. Pathol. 2009; 4;
343–364.15. Rosty C, Walsh MD, Walters RJ et al. Multiplicity and molecu-
lar heterogeneity of colorectal carcinomas in individuals with
serrated polyposis. Am. J. Surg. Pathol. 2013; 37; 434–442.16. Crowder CD, Sweet K, Lehman A, Frankel WL. Serrated polyp-
osis is an underdiagnosed and unclear syndrome: the surgical
pathologist has a role in improving detection. Am. J. Surg.
Pathol. 2012; 36; 1178–1185.17. Rosty C, Parry S, Young JP. Serrated polyposis: an enigmatic
model of colorectal cancer predisposition. Patholog. Res. Int.
2011; 2011; 157073.
18. Jasperson KW, Kanth P, Kirchhoff AC et al. Serrated polyposis:
colonic phenotype, extracolonic features, and familial risk in a
large cohort. Dis. Colon Rectum 2013; 56; 1211–1216.19. Boparai KS, Mathus-Vliegen EM, Koornstra JJ et al. Increased
colorectal cancer risk during follow-up in patients with hyper-
plastic polyposis syndrome: a multicentre cohort study. Gut
2010; 59; 1094–10100.20. Jaeger E, Leedham S, Lewis A et al. Hereditary mixed polyposis
syndrome is caused by a 40-kb upstream duplication that leads
to increased and ectopic expression of the BMP antagonist
GREM1. Nat. Genet. 2012; 44; 699–703.21. Whitelaw SC, Murday VA, Tomlinson IP et al. Clinical and
molecular features of the hereditary mixed polyposis syndrome.
Gastroenterology 1997; 112; 327–334.22. Palles C, Cazier JB, Howarth KM et al. Germline mutations
affecting the proofreading domains of POLE and POLD1 predis-
pose to colorectal adenomas and carcinomas. Nat. Genet.
2013; 45; 136–144.23. Briggs S, Tomlinson I. Germline and somatic polymerase epsi-
lon and delta mutations define a new class of hypermutated
colorectal and endometrial cancers. J. Pathol. 2013; 230;
148–153.24. van Lier MG, Mathus-Vliegen EM, Wagner A, van Leerdam ME,
Kuipers EJ. High cumulative risk of intussusception in patients
with Peutz-Jeghers syndrome: time to update surveillance guide-
lines? Am. J. Gastroenterol. 2011; 106; 940–945.25. Aretz S, Stienen D, Uhlhaas S et al. High proportion of large
genomic STK11 deletions in Peutz-Jeghers syndrome. Hum.
Mutat. 2005; 26; 513–519.26. Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syn-
drome: diagnostic and therapeutic approach. World J. Gastroen-
terol. 2009; 15; 5397–5408.27. Burkart AL, Sheridan T, Lewin M, Fenton H, Ali NJ, Montgomery
E. Do sporadic Peutz-Jeghers polyps exist? Experience of a large
teaching hospital. Am. J. Surg. Pathol. 2007; 31; 1209–1214.28. Petersen VC, Sheehan AL, Bryan RL, Armstrong CP, Shepherd
NA. Misplacement of dysplastic epithelium in Peutz-Jeghers
polyps: the ultimate diagnostic pitfall? Am. J. Surg. Pathol.
2000; 24; 34–39.29. Jass JR. Colorectal polyposes: from phenotype to diagnosis.
Pathol. Res. Pract. 2008; 204; 431–447.30. Brosens LA, Langeveld D, van Hattem WA, Giardiello FM, Of-
ferhaus GJ. Juvenile polyposis syndrome. World J. Gastroenterol.
2011; 17; 4839–4844.31. Mester J, Eng C. Estimate of de novo mutation frequency in pro-
bands with PTEN hamartoma tumor syndrome. Genet. Med.
2012; 14; 819–822.32. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C.
Lifetime cancer risks in individuals with germline PTEN muta-
tions. Clin. Cancer Res. 2012; 18; 400–407.33. Heald B, Mester J, Rybicki L, Orloff MS, Burke CA, Eng C. Fre-
quent gastrointestinal polyps and colorectal adenocarcinomas
in a prospective series of PTEN mutation carriers. Gastroenterol-
ogy 2010; 139; 1927–1933.34. Stanich PP, Pilarski R, Rock J, Frankel WL, El-Dika S, Meyer
MM. Colonic manifestations of PTEN hamartoma tumor syn-
drome: case series and systematic review. World J. Gastroenter-
ol. 2014; 20; 1833–1838.35. Bakry D, Aronson M, Durno C et al. Genetic and clinical deter-
minants of constitutional mismatch repair deficiency syn-
drome: report from the constitutional mismatch repair
deficiency consortium. Eur. J. Cancer 2014; 50; 987–996.
© 2015 John Wiley & Sons Ltd, Histopathology, 66, 78–87.
Hereditary polyposis syndromes 87


















