
The Official Journal of Saudi Society of Hematology · The Official Journal of Saudi Society of...
62
VOLUME 2 - ISSUE 1 - MARCH 2011 ISSN: 1658-5127 JOURNAL OF APPLIED HEMATOLOGY - VOLUME 2 - ISSUE 1 - MARCH 2011 The Official Journal of Saudi Society of Hematology Image of the Issue: page 44
Transcript of The Official Journal of Saudi Society of Hematology · The Official Journal of Saudi Society of...

VOLUME 2 - ISSUE 1 - MARCH 2011
ISSN: 1658-5127
JOU
RN
AL O
F APPLIED
HEM
ATOLO
GY
- VO
LUM
E 2 - ISSUE 1 - M
AR
CH
2011
The Official Journal of Saudi Society of Hematology
Image of the Issue: page 44

1

1
EDITORIAL BOARD:
Editor-in-Chief:Abdul Kareem M. Al-Momen
Associate Editors:Abdel Galil M. Abdel Gader
Mohamed H.Qari Tarek M. Owaidah
Advisory Board:
Anisa Abboud (Yemen)Mohamed Abdulaal (KSA)Saud Abu-Harbesh (KSA)Mirghani Ali M. Ahmed (KSA)Abdul Aziz Al-Abdulaaly (KSA)Ahmed Al-Askar (KSA)Baker Al-Awamy (KSA)Abdulmajeed Albanyan (KSA)Fatima Al-Batniji (KSA)Basem Al-Beiruti (KSA)Hind Al-Humaidan (KSA)Abdullah Al-Jefri (KSA) Salam Al-Kindi (Oman)Fahad Al-Mohareb (KSA)Naima Al-Mulla (Qatar)Fat-Haia Al Qurashi (Bahrain)Mohamad Al-Shahrani (KSA)Salem Al-Shemmari (Kuwait)Ahmed Al-Sulaiman (KSA)Hazzaa Al-Zahrani (KSA)Alan Burnett (UK)Naeem Chaudhri (KSA)Michael Copeman (Australia)Ghazi Damanhori (KSA)Mohsen El-Alfy (Egypt)
Magdy EL- Ekiaby (Egypt)Omar Fahmy (Egypt)Assad Haffar (Canada)Ulla Hedner ( Sweden)Ahmed Ibrahim (Lebanon) Soad Jaoni (KSA)Armand Keating (Canada)Salem Khalil (KSA)Abdullah Kutlar (USA)Pier Mannucci (Itay)Ghulam Mufti (UK)Shaker Musa (USA)Thomas Ortel (USA)Abdulla Owaidi (Jordan)George Rivard (Canada)Ahmed Rustomani (UAE)Giuseppe Saglio (Italy)Michele Samama (France)Faten Sayes (KSA)David Spence (UAE)Alison Street (Australia)Ali Taher (Lebanon)Ahmed Tarawah (KSA)Ayalew Tefferi (USA)
Scientist Publishing Group P.O. Box 91409 Riyadh 11633Saudi Arabia . Tel . : 4780312a a l e m f o r p r e s s @ y a h o o . c o m
Scientist Publishing Group

Instruction to Authors:Journal of Applied Hematology is an international journal that publishes articles covering both clinical and experimental research in hematology. The journal welcomes original as well as review articles, manuscripts, case reports, short communications, images and clinical pictures, letters and correspondence to the Editor; on non-malignant and malignant hematological diseases, hemostasis and thrombosis, immunology, stem cells biology, and transfusion medicine. Clinical studies describing novel therapeutic approaches to the diagnosis and treatment of hematological diseases are welcomed and encouraged as well. The journal follows the style of Uniform Requirements for Manuscripts Submitted to Biomedical Journals.Review Articles: Authors are invited by Editor-in-Chief for submission of review articles which should focus on recent scientific or clinical advances in an area of interest to those in the field of Hematology.Original Articles: Maximum length of 5000 words not counting the abstract, table and figures. The Abstract should not exceed 250 words and should be constructed a single paragraph with no subheadings. The Article should be in order of Abstract, Introduction, Methods, Results, Discussion, Acknowledgements, Authorship Contributions and Disclosure of Conflicts of Interest, References, Tables, Figure Legends, and Figures. Image in Hematology: The Journal of Applied of Hematology welcomes submission of photo image and brief case descriptions to serve as a regular teaching feature and comprehensive reference accessible to physicians and hematology students around the world.Letter to the Editor: Constructive comments on published articles or current topics in Hematology not more than 1000 words. No abstract is required, but please include a brief title. Pioneer in Hematology: Papers about one of the great contributors in the history of Hematology who had made great advancement and change in the understanding of Hematology. Case Report: Short manuscripts reporting one or more cases of informative clinical observation should not exceed 2000 words.Test Validation: Author can report the results of new test that had been scientifically validated and had clinical application. Submission of Manuscripts: Manuscripts are received with the understanding that they are not under simultaneous consideration by another publication. If an abstract of the work has previously been published or if manuscripts using the same database or relating to the same topic have been published or submitted by any of the authors, this should be disclosed. Authors who violate this requirement will be subject to an extended publication ban. An abstract published prior to a full report is not regarded as a duplicate publication. Accepted manuscripts may not be published elsewhere without the journal’s permission. Manuscripts should be submitted online at http://mc.mannuscriptcentral.com/jahem. to ensure that peer review is double-blinded, there should be no title page in the file of the manuscript that is submitted online. The file name should not include the author’s name or other identifying text. Portions of the submission (photographs, artwork, etc.) may be sent by mail if necessary, but we prefer that these be prepared in digital form and submitted online. If you have difficulty, contact us by e-mail at [email protected].
The Official Journal of Saudi Society of Hematology


Table of Contents
Review Articles
1 How I treat Paroxysmal Nocturnal Hemoglobinuria (PNH)
Alexander Röth
7 Blood Supply in Saudi Arabia–Self Sufficiency and Safety Considerations
Abel Galil M Abdel Gader, Farga H. Alqahtani, Abdulmajeed A. Albanayan
Original Articles
15 Impact of inherited thrombophilic factors on deep vein thrombosis in individuals in south Iran
Majid Yavarian, Mani Ramzi, Marym Zakernia, Mansor Hagshenas, Mehran Karimi
20 Endocrinopathies in Children and Adolescents with b-Thalassemia Major
Abdulmoein Al-Agha, Shadi A. Shabakah, Ali Ocheltree, Daniah El-fateh M. Abdullatif, Soad K. Al Jaouni
25 Evaluation of the Laboratory tests used in the Identification of Lupus Anticoagulants
Logman A. Gasmelsid, Abdel Galil M. Abel Gader, Anwar Y. Kordofani
34 Low bone mineral density in patients with sickle cell disease: Association with blunted parathyroid hormone response and accelerated bone turnover
Jalaluldin A. Jalal, Mohamed F. Elshal, Mohamed H. Qari, Maryam A. Al-Ghamdy, Amna E. Bernawi
Test Validation
41 Validation of a monospecific enzyme-linked immunosorbent assay as a screening test for heparin-induced thrombocytopenia and its comparison with immunological and functional assays
Rasheed Nasr, Randa Al Nounou, Mansoor Ahmed, Tarek Owaidah
Image of the Issue
44 T-cell acute lymphoblastic leukemia in a patient with Fanconi anemia
Najeeb Kamli, Tarek M. Owaidah
Case Report
46 Postpartum hemolytic uremic syndrome: A case report and review of the literature
Ghuzayel Al Dawsari, Abdul Rahman Jazieh
Letter to the Editor
51 Comprehensive Care of Hemophilia Nursing Prospective
Mahmoud I. Abu-Riash
Pioneers in Hematology
52 The Father of Blood Banking

review
Journal of Applied Hematology 2011 1
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, debilitating, and life-threatening ac-quired hematologic disorder. Clinically, PNH
is characterized by the classical triad of acquired Coombs-negative intravascular hemolytic anemia, thrombophilia, and bone marrow failure to various degrees.1-4
This clinical entity was first described in 1882 by the German physician Paul Strübing from Greifswald.5 In 1911, this form of hemolytic anemia was reported in conjunction with the characteristic hemoglobin-uria (Figure 1) by Marchiafava and Micheli,6,7 which finally gave rise to the eponymous name Strübing-Marchiafava-Micheli syndrome.
In PNH, the absence of the glycosylphosphati-dylinositol (GPI)-anchored complement inhibitory protein CD55 and most importantly CD59 from the
How I treat Paroxysmal Nocturnal Hemoglobinuria (PNH)
Alexander Röth
From the University Hospital Essen, Department of Hematology, West German Cancer Center, Essen, Germany
Correspondence: Alexander Rš th, M.D., Department of Hematology, West German Cancer Center, University Hospital Essen, Hufelandstrasse 55, D-45122 Essen, Germany. T: +49 (0)201-723-84219. F: +49 (0)201-723-1716. [email protected]
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare life-threatening and de-
bilitating clonal blood disorder and is caused by an acquired mutation of the
phosphatidylinositol glycan (PIG)-A gene of the pluripotent hematopoietic stem
cell, leading to a deficiency of glycosylphosphatidylinositol (GPI)-anchors and
GPI-anchored proteins on the surface of affected blood cells, including the com-
plement regulators CD55 and CD59. PNH red blood cells are highly vulnerable
to activated complement with the formation of the membrane attack complex
(MAC). This results in chronic intravascular hemolysis as the underlying cause
of morbidities and mortality in PNH. Until recently, the treatment of PNH has
been largely empirical and symptomatic with blood transfusions, anticoagulation,
and supplementation of folic acid or iron. Potentially, the only curative treatment
is allogeneic stem cell transplantation, in case of severe complication with a high
rate of mortality and morbidity. A new targeted and disease-modifying treatment
strategy is the inhibition of the terminal complement cascade with a humanized
monoclonal anti-C5 antibody (eculizumab). Therefore, the MAC formation along
with the intravascular hemolysis is effectively inhibited. Eculizumab has shown
significant efficacy in controlled studies leading to a marked decrease of anemia,
fatigue, transfusion requirements, renal impairment, pulmonary hypertension,
and the risk of severe thromboembolic events, thereby leading to an improve-
ment in the quality of life and survival.
Keywords: paroxysmal nocturnal hemoglobinuria, PNH, eculizumab, ter-
minal complement inhibitor, therapy
surface of PNH red blood cells (RBCs) renders them susceptible to terminal complement-mediated lysis.8-10 This is a consequence of mutations in the phosphati-dylinositol glycan-A gene resulting in a decrease in, or total deficiency of, GPI-anchored proteins.11,12
The gold standard for the diagnosis of PNH is flow cytometry with the measurement of GPI-anchored proteins as well as the GPI-anchor itself (FLAER) and has replaced the Ham test and the sucrose lysis test.3,13-
16 Flow cytometry allows sensitive and specific detec-tion and quantification of GPI-deficient populations in various cell lineages, and it can be used for diagnosis and monitoring during follow up. Ideally, at least the lack of 2 different GPI-anchored proteins on at least 2 different cell lineages should be used to diagnose PNH.1,17,18 Together with the assessment of hemolytic parameters and bone marrow investigation, including

review treatment of pnh
Journal of Applied Hematology 20112
an aspirate, cytogenetics, and a biopsy, the minimal essential criteria for the diagnosis and categorization of PNH are met.1
The chronic intravascular hemolysis in PNH is the cause for weakness, pallor, fatigue, anemia, dys-pnea on exertion, reduced quality of life, the need for transfusions, renal impairment, pulmonary hyperten-sion, and the risk of life-threatening thromboembolic complications.19,20 Free hemoglobin leads to deple-tion of nitric oxide in plasma, causing complications associated with smooth muscle dystonias, including abdominal pain, dysphagia, pulmonary hypertension, and erectile dysfunction.1,19
Eculizumab (Soliris™; Alexion Pharmaceuticals, Incorporated, Cheshire, CT, USA) is a humanized monoclonal antibody that binds to the complement protein C5. Eculizumab inhibits the terminal comple-ment cascade, thereby preventing complement-me-diated destruction of PNH RBCs.21 Before the ap-proval of eculizumab in March 2007 by the Food and Drug Administration in the USA and in June 2007 by the European Commission for the treatment of patients with PNH, therapeutic options were mainly directed towards palliation of symptoms rather than the underlying hemolytic process. With the availability of a targeted treatment in PNH, eculizumab has be-come the standard treatment for symptomatic PNH.
How to treat patients with PNH
Historical treatment of PNHHistorically, 2 treatment approaches were available in PNH, namely, symptomatic treatment and prophy-
laxis of complications or stem cell transplantation (SCT). However, with symptomatic treatment and prophylaxis, long-term control of the disease was rather unsatisfactory as well as SCT with the potential for cure but also a high treatment related morbidity and mortality due to infections, graft-versus-host dis-ease GVHD as well as graft failure.1,3,22 Possible treat-ment options today are listed in Table 1 in detail.
Based on the pathophysiology of PNH, inhibition of the complement system was a rational and targeted approach.
Inhibition of the terminal complementEculizumab (Soliris®, Alexion Pharmaceuticals) is a first-in-class, humanized, monoclonal antibody di-rected against the terminal complement protein C5. The germline human framework acceptor sequences were used to minimize immunogenicity and the hu-man IgG2/4 heavy chain constant regions to elimi-nate the ability of the antibody to bind Fc receptors and activate the complement.21,23 It has a very high binding affinity for human C5, and each molecule binds 2 C5 proteins. Hereby, terminal complement cascade with the formation of the membrane attack complex C5b-C9 is blocked by preventing its cleav-age to C5a and C5b (Figure 2). Importantly, the gen-eration of components in the early steps of comple-ment activation remains intact, which are critical for immunoregulation and protection against infec-tions.21,23
Results from the phase III, multicenter, double-blind, placebo-controlled TRIUMPH study demon-strated that eculizumab reduces hemolysis, transfu-
Figure 1. Hemoglobinuria in PNH. Hemoglobinuria is the hallmark of PNH. It is typically more obvious in the concentrated morning urine. However, 75% of PNH patients present without hemoglobinuria at the time point of diagnosis.1 With eculizumab treatment, hemoglobinuria nowadays has become a very rare phenomenon.
Table 1. Treatment options in paroxysmal nocturnal hemoglobinuria (PNH).
Symptomatic Treatment
- Red blood cell transfusions
- Supplementation of folic acid and vitamin B12 (if deficient)
- Iron replacement (based on iron stores)
- Prevention/early treatment of bacterial infections
- Prophylactic or post-thrombotic anticoagulation
- Corticosteroids
- Immunosuppressive treatment (only in aplastic anemia-PNH syndrome)
- Complement inhibition by eculizumab
Potentially Curative Treatment
- Allogeneic stem cell transplantation

reviewtreatment of pnh
Journal of Applied Hematology 2011 3
sion requirements, and improves fatigue in patients with PNH.24 The SHEPHERD study, an open-label, phase III, safety and efficacy trial that enrolled a more heterogeneous population of patients with PNH (including those with significant thrombocyto-penia and minimal transfusion requirements) showed similar benefits of eculizumab.25,26 Based on its ef-ficacy in those 2 phase III clinical trials, eculizum-ab was approved in the US and Europe for use in PNH in 2007.24,25 Eculizumab is highly effective in reducing intravascular hemolysis in PNH. However, it does not improve the associated bone marrow fail-ure and mild hemolysis via extravascular sequestra-tion of PNH RBCs loaded with C3 cleavage product becomes relevant.27-29 Therefore, eculizumab is the most effective in classical PNH patients as compared to PNH in the setting of another bone marrow fail-ure syndrome, e.g., aplastic anemia (AA), myelodys-plastic syndromes, or osteomyelofibrosis.1
Eculizumab is administered intravenously over 35 minutes as an infusion independent of the body weight at a dose of 600 mg weekly for the first 4 weeks, then 900 mg biweekly starting on week 5 (Figure 3). Inhibition of the complement at C5 in-creases the risk for infections with encapsulated microorganisms, especially Neisseria meningitidis and N. gonorrhoeae. Thus, all patients should be vaccinated at least 2 weeks before the start of the eculizumab treatment, preferentially with a tetrava-lent, conjugated meningococcal vaccine. Patients should also be revaccinated every 3 to 5 years and be instructed about early clinical signs or symptoms of meningococcal infections and then seek immedi-
ate medical attention. The most common side effect reported during eculizumab treatment was headache, which occurred in approximately half of the patients and most of the time within the first 24 hours after the first dose or two. Otherwise, eculizumab is safe and well tolerated.24,25
Treatment of PNH in the era of eculizumabEculizumab was approved for any patient diagnosed with PNH. However, eculizumab should preferen-tially be used for symptomatic PNH patients with severe fatigue, recurrent hemolytic crisis with ab-dominal pain, renal insufficiency, thromboembolic events, or other end-organ complications. PNH pa-tients with no or only mild symptoms could be fol-lowed by watchful waiting. In some patients, the ecu-lizumab dose must be adjusted due to breakthrough hemolysis. Typically, this occurs 1 or 2 days before the next scheduled dose together with a spike of the LDH level. This can be treated by either shortening of the interval from 14 to 12 days or increasing the eculizumab dose from 900 mg to 1200 mg biweekly. Bacterial infections can still trigger hemolytic crisis, even during treatment with eculizumab due to in-creased complement activation. Thus, an immediate and consequent antibiotic treatment in case of bacte-rial infections is recommended. In case of a hemolyt-ic crisis, patients should be administered intravenous fluids to ensure hydration. Increased complement ac-tivation can also be triggered by viral infections, sur-gery, trauma, or pregnancy. Despite the effective ecu-lizumab treatment, some PNH patients still require RBC transfusion based on symptoms of anemia.24
Figure 2. Eculizumab blocks the terminal complement cascade. Eculizumab and its mechanism of action, i.e., inhibition of the terminal complement cascade. The activation of the complement system by either classic, alternative, or lectin pathway leads ultimately to the formation of the membrane attack complex (MAC) and consequent cell lysis. Eculizumab is a monoclonal antibody that specifically binds to C5 preventing its cleavage to C5a and C5b, thereby preventing the generation of C5b-C9 or MAC.
Figure 3. Eculizumab dosing schedule. At least 2 weeks before the start of eculizumab treatment, all patients must be vaccinated against N. meningitidis. Eculizumab is administered over 35 minutes as an intravenous infusion according to the following dosing schedule: an induction dose of 600 mg every 7 (±2) days for the first 4 weeks, followed one week later by 900 mg, and then 900 mg every 14 (±2) days.

review treatment of pnh
Journal of Applied Hematology 20114
MonitoringA continuous monitoring remains essential in treated and untreated patients with PNH. I recommend moni-toring of complete blood count, including reticulo-cyte count, LDH, and more thorough parameters of hemolysis (haptoglobin, hemopexin, and bilirubin). Determination of the reticulocyte production index is also helpful to accurately reflect the marrow produc-tion of erythrocytes. To follow renal function, creati-nine levels should be measured.30 BNP levels can be a noninvasive parameter for pulmonary hypertension.31 Elevated D-dimers are associated with activated coagu-lation and thrombosis.32
Serum iron studies are recommended to rule out rel-evant iron deficiency or iron overload (1). PNH clone size measurement in PNH patients with an otherwise stable disease is recommended yearly. In the situation of changes in clinical parameters, however, reevaluation should be done.1 In case of inadequate response on ec-ulizumab treatment or continuous increase of transfu-sion requirements, a bone marrow investigation should be performed to rule out AA or clonal transformation.
After the start of the eculizumab treatment, intra-vascular hemolysis is reduced dramatically character-ized by the LDH levels returning to normal or near normal within days to weeks.24,25 Reticulocytes often remain elevated as extravascular hemolysis becomes relevant as mentioned before. Screening for extravas-cular hemolysis by using the monospecific direct agglu-tination test is useful during eculizumab treatment. If clinically relevant, low dose corticosteroids are reported to be a treatment option.27-29
Supplementation and depletionAs elevated reticulocytes indicate an increased regen-eration along with an erythroid hyperplasia in the bone morrow, a sufficient supply with folic acid and vitamin B12 should be warranted. I generally recommend the daily intake of 1–5 mg folic acid and vitamin B12 if deficient. A significant urinary iron loss from hemoglo-binuria and hemosiderinuria is common in hemolytic PNH patients with the need for oral iron supplemen-tation.33,34 However, as eculizumab blocks intravascular hemolysis effectively, there is no further urinary iron loss. Even more, there is an increase in iron storage es-pecially in PNH patients requiring blood transfusions. Routine supplementation of iron becomes obsolete, and in case of a relevant iron overload, iron depletion should be initiated.
Prevention and treatment of thrombosisThe most feared complications and the leading cause
of death in PNH are thromboembolic events.2,3 Eculizumab, however, has clearly demonstrated in the clinical trials that long-term treatment significantly reduces the risk for thrombosis in PNH from 7.37 events/100 patient-years to 1.07 events/100 patient-years (85%, P<0.01).32 Interestingly, in the subgroup of patients already on antithrombotic treatment, the thromboembolic events were reduced by 94% from 10.6 events/100 patient-years before eculizumab treat-ment to 0.62 events/100 patient-years during eculizum-ab treatment (P<0.01).32
In acute thrombotic events, anticoagulation with heparin along with eculizumab should be started im-mediately and sometimes even local or systemic throm-bolytic therapy.35,36
After a thromboembolic event, patients should be anticoagulated indefinitely1 and treatment with eculi-zumab initiated. In a retrospective study, the risk for thromboembolic events was related to the PNH clone size.37 Patients with a PNH clone size >50% (as mea-sured by GPI-deficient granulocytes) should prefer-entially be treated with eculizumab; however, those patients not indicated for eculizumab and normal platelets should be prophylactically anticoagulated.1,37 Furthermore, prophylactic anticoagulation preferential-ly with low molecular heparin is also recommended in high-risk situations for thrombosis like surgery or preg-nancy, despite eculizumab treatment. So far, discontin-uation of primary or even more secondary prophylaxis remains controversial, and more long-term results and further studies are needed. Individual decisions on pri-mary prophylaxis should be based on symptoms, clone size, other thrombophilic risk factors, platelet count, medication including eculizumab, age, activity level, compliance, and patient preferences.
Stem cell transplantationPNH in the setting of AA treatment should be directed to the underlying bone marrow failure. If the criteria for severe AA are met, patients should go for alloge-neic SCT or immunosuppressive therapy depending on the age of patient and availability of suitable HLA-matched donor.38,39 Furthermore, and according to the IPIG criteria, SCT should also be considered in the sit-uation of major complications of PNH (e.g., refractory, transfusion-dependent hemolytic anemia or recurrent life-threatening thromboembolic disease).1
ConclusionsThe advances in the understanding of the pathophysi-ology of PNH over the last decades have led to a highly effective and targeted therapy with eculizumab. With

reviewtreatment of pnh
Journal of Applied Hematology 2011 5
its approval, a targeted and disease-modifying treat-ment option is available that is well tolerated and reduces hemolysis, fatigue, anemia, transfusion re-quirements, renal impairment, pulmonary hyperten-sion, and the risk for thromboembolic events and improves anemia and the quality of life. Eculizumab has therefore become the gold standard treatment for hemolytic PNH patients, even leading to a major improvement in survival, as demonstrated previously by Kelly et al.40
Moreover, eculizumab has initiated an expanding new era of complement modification as a therapeu-tic strategy and will offer new options for the treat-
ment of other complement-mediated diseases such as atypical hemolytic uremic syndrome,41,42 antibody-mediated transplant rejection,43 and hemolytic cold agglutinin disease.44
AcknowledgementsThis review is dedicated to Prof. Dr. Günter Brittinger on the occasion of his 80th birthday.
Conflicts of interestA.R. received lecture fees from Alexion Pharmaceuticals and served on advisory boards for Alexion Pharmaceuticals.
1. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socie G. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699-709.2. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of parox-ysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333:1253-8.3. de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, Roth S, de Guibert S, Maury S, Cahn JY, Socie G. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112:3099-106.4. Rš th A, DŸ hrsen U, Schrezenmeier H, Schubert J. Paroxysmal nocturnal he-moglobinuria (PNH). Dtsch Med Wochenschr. 2009;134:404-9.5. StrŸ bing P. Paroxysmale Haemoglobinurie. Dtsch Med Wochenschr. 1882;8:1-17. [Article in German]6. Marchiafava E, Nazari A. Nuovo contributo allo studio degli itteri cronici emo-litici. Il Policlinico. 1911;18:241-54. [Article in Italian]7. Micheli F. Un caso di anemia emolitica con emosiderinuria perpetua. Accad Med (Torino). 1928;7:148. [Article in Italian]8. Parker CJ. Molecular basis of paroxysmal nocturnal hemoglobinuria. Stem Cells.1996;14:396-411.9. Yamashina M, Ueda E, Kinoshita T, Takami T, Ojima A, Ono H, Tanaka H, Kondo N, Orii T, Okada N. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1990;323:1184-9.10. Motoyama N, Okada N, Yamashina M, Okada H. Paroxysmal nocturnal he-moglobinuria due to hereditary nucleotide deletion in the HRF20 (CD59) gene. Eur J Immunol. 1992;22:2669-73.11. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703-11.12. Bessler M, Mason PJ, Hillmen P, Miyata T, Yamada N, Takeda J, Luzzatto L, Kinoshita T. Paroxysmal nocturnal hemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13:110-7.13. Bessler M, Fehr J. Fc III receptors (FcRIII) on granulocytes: a specific and sensitive diagnostic test for paroxysmal nocturnal hemoglobinuria (PNH). Eur J Haematol. 1991;47:179-84.14. Hall SE, Rosse WF. The use of monoclonal antibodies and flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria. Blood. 1996;87:5332-40.15. Brodsky RA, Mukhina GL, Li S, Nelson KL, Chiurazzi PL, Buckley JT, Borowitz MJ. Improved detection and characterization of paroxysmal nocturnal hemoglo-binuria using fluorescent aerolysin. Am J Clin Pathol. 2000;114:459-66.16. Richards SJ, Rawstron AC, Hillmen P. Application of flow cytometry to the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry. 2000;42:223-33.17. Richards SJ, Barnett D. The role of flow cytometry in the diagnosis of par-oxysmal nocturnal hemoglobinuria in the clinical laboratory. Clin Lab Med. 2007;27:577-90, vii.18. Borowitz MJ, Craig FE, Digiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, Wittwer CT, Richards SJ. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010;78:211-30.19. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653-62.20. Rosse WF, Nishimura J. Clinical manifestations of paroxysmal nocturnal he-moglobinuria: present state and future problems. Int J Hematol. 2003;77:113-20.
21. Thomas TC, Rollins SA, Rother RP, Giannoni MA, Hartman SL, Elliott EA, Nye SH, Matis LA, Squinto SP, Evans MJ. Inhibition of complement activity by human-ized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33:1389-1401.22. Saso R, Marsh J, Cevreska L, Szer J, Gale RP, Rowlings PA, Passweg JR, Nugent ML, Luzzatto L, Horowitz MM, Gordon-Smith EC. Bone marrow transplants for paroxysmal nocturnal hemoglobinuria. Br J Haematol. 1999;104:392-6.23. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and develop-ment of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256-64.24. Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, Rš th A, Szer J, Elebute MO, Nakamura R, Browne P, Risitano AM, Hill A, Schrezenmeier H, Fu CL, Maciejewski J, Rollins SA, Mojcik CF, Rother RP, Luzzatto L. The comple-ment inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. Engl J Med. 2006;355:1233-43.25. Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, Gaya A, Coyle L, de Castro C, Fu CL, Maciejewski JP, Bessler M, Kroon HA, Rother RP, Hillmen P. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglo-binuria. Blood. 2008;111:1840-7.26. Schubert J, Hillmen P, Rš th A, Young NS, Elebute MO, Szer J, Gianfaldoni G, Socie G, Browne P, Geller R, Rother RP, Muus P. Eculizumab, a terminal comple-ment inhibitor, improves anemia in patients with paroxysmal nocturnal haemo-globinuria. Br J Haematol. 2008;142:263-72.27. Risitano AM, Marando L, Seneca E, Rotoli B. Hemoglobin normalization after splenectomy in a paroxysmal nocturnal hemoglobinuria patient treated by ecu-lizumab. Blood. 2008;112:449-51.28. Risitano AM, Notaro R, Marando L, Serio B, Ranaldi D, Seneca E, Ricci P, Alfinito F, Camera A, Gianfaldoni G, Amendola A, Boschetti C, Di Bona E, Fratel-lanza G, Barbano F, Rodeghiero F, Zanella A, Iori AP, Selleri C, Luzzatto L, Rotoli B. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113:4094-100.29. Rš th A, Peine S, DŸ hrsen U. Paroxysmal nocturnal hemoglobinuria turning Coombs-positive. Int J Hematol. 2010;91:159-60.30. Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Hill A, Rother RP, Khursigara G, Fu CL, Omine M, Browne P, Rosse W. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85:553-9.31. Hill A, Rother RP, Wang X, Morris SM Jr, Quinn-Senger K, Kelly R, Richards SJ, Bessler M, Bell L, Hillmen P, Gladwin MT. Effect of eculizumab on haemolysis-associated nitric oxide depletion, dyspnea, and measures of pulmonary hyper-tension in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2010;149:414-25.32. Hillmen P, Muus P, DŸ hrsen U, Risitano AM, Schubert J, Luzzatto L, Schrezen-meier H, Szer J, Brodsky RA, Hill A, Socie G, Bessler M, Rollins SA, Bell L, Rother RP, Young NS. Effect of the complement inhibitor eculizumab on thromboembo-lism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;33. Dacie JV, Lewis SM. Paroxysmal nocturnal haemoglobinuria: clinical manifes-tations, hematology, and nature of the disease. Ser Haematol. 1972;5:3-23.34. Rosse WF. Treatment of paroxysmal nocturnal hemoglobinuria. Blood. 1982;60:20-3.35. Kuo GP, Brodsky RA, Kim HS. Catheter-directed thrombolysis and throm-bectomy for the Budd-Chiari syndrome in paroxysmal nocturnal hemoglobin-
References

review treatment of pnh
Journal of Applied Hematology 20116
uria in three patients. J Vasc Interv Radiol. 2006;17:383-7.36. McMullin MF, Hillmen P, Jackson J, Ganly P, Luzzatto L. Tissue plasminogen ac-tivator for hepatic vein thrombosis in paroxysmal nocturnal hemoglobinuria. J Intern Med. 1994;235:85-9.37. Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin pre-vents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003;102:3587-91.38. Marsh JC, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan J, Laurie A, Martin A, Mercieca J, Killick SB, Stewart R, Yin JA. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147:43-70.39. Kelly RJ, Hill A, Mitchell LD, Richards SJ, Arnold LM, Valters GL, Cullen M, Cohen DR, Gregory WM, Hillmen P. Long term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria (PNH): sustained efficacy and improved survival. Blood. 2010;116:639.
40. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transplant. 2010;16:S119-S125.41. NŸ rnberger J, Philipp T, Witzke O, Opazo SA, Vester U, Baba HA, Kribben A, Zimmerhackl LB, Janecke AR, Nagel M, Kirschfink M. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:542-4.42. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:544-6.43. Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, King KE, Kraus E, Lees LM, Melancon JK, Stewart ZA, Warren DS, Zachary AA, Montgomery RA. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9:231-5.44. Rš th A, HŸ ttmann A, Rother RP, DŸ hrsen U, Philipp T. Long-term effi-cacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:3885-6.

review
Journal of Applied Hematology 2011 7
Since the discovery of the ABO blood group system by Landsteiner and his co-workers in the early years of the 20th century, blood trans-
fusion has developed into a multifaceted medical dis-cipline based on scientific knowledge and highly de-veloped technology. This has been the result of the extensive interaction between blood transfusion and many basic sciences, including immunology, chem-istry, biochemistry, physiology, genetics as well as engineering and computer science and technology. Throughout and up to this date, blood transfusion kept its place as a vital supporting service to clini-cal medicine. As more and more advances are gained by clinical medicine, blood transfusion has kept pace
Blood supply in the Kingdom of Saudi Arabia–self-sufficiency and safety considerations
Abel Galil M. Abdel Gader, Farga H. Alqahtani, Abdulmajeed A. Albanayan
From the The Blood Bank, King Khalid University HospitalThe Blood Transfusion Research GroupKing Saud University Riyadh, Saudi Arabia
Correspondence:Professor A.M.A. Gader MB, BS, PhD, FRCP (London & Edinburgh)The Blood BankKing Khalid University HospitalKing Saud UniversityP.O. Box 2925Riyadh 11461Saudi ArabiaT: +9661 4671042F: +9661 4672549M: [email protected]@ksu.edu.sa
The transfusion of blood and its derivatives is a vital supporting service to clinical
medicine. However, over the years, 2 considerations have been of major concern
to both health planners as well as professionals in charge of blood banks, namely,
self-sufficiency and safety.
In the Kingdom of Saudi Arabia, the blood transfusion service is predominantly
a hospital-based blood banking system. Despite the shortcomings of this sys-
tem, self-sufficiency has been attained with respect to fresh cellular components
(packed red blood cells and platelet concentrates) and plasma derivatives (fresh
frozen plasma and cryoprecipitate). However, since the requirement for hemo-
therapy is phasic in nature and variable in quantity, hospital blood banks are
exposed to frequent shortages in the supply of single components when heavy
demands of that component arise.
As to the second issue of safety, specifically reducing the risk of infection with
transfusion-transmitted pathogens, it is addressed satisfactorily by undertaking
newly emerging screening assays, including nucleic acid testing[A4] for hepati-
tis B and C and human immunodeficiency viruses. The continuous expansion in
the number and sophistication of assay techniques designed to detect an ever-
increasing number of pathogens leaves a lot to be desired. Malaria, for which
there is no specific and sensitive screening test, remains a daunting challenge.
Additionally, viral inactivation of the frequently consumed fresh frozen plasma
as well as universal leucodepletion is yet to be implemented in all blood banks.
Current efforts led by the Ministry of Health towards establishing a unified na-
tional blood transfusion service, based on non-remunerated voluntary donors,
is a dream that should not take long to come true and will no doubt be the
ultimate answer for self-sufficiency and safety.
and has also scored uncountable developmental steps. For example, the constant advances in cardiac and transplant surgery, bone marrow transplanta-tion, and chemotherapy of malignancies, especially hematological malignancies, would not have been possible without the support of blood transfusion. Recent developments have led to better exploitation of the limited available blood resources, increased safety, new therapeutic options, and even alternatives to blood transfusion, especially oxygen carriers (so-called blood substitutes).
However, it is the danger of transmitting infec-tious diseases, particularly human immunodeficiency virus (HIV), that has provoked intense medical as

review blood supply in saudi arabia
Journal of Applied Hematology 20118
well as public concern on the safety of transfusion. Although it is understandable that patients and their treating physicians should worry about blood trans-fusion therapy, such worries have reached blood do-nors who request reassurance on the safety of even blood donation. These perspectives have prompted major changes in the field of blood transfusion in re-cent years. This is particularly evident in developing countries where blood transfusion service (BTS) is nowadays being managed on market basis, governed by good manufacturing practices, and the balance has shifted from blood transfusion being a service freely available to needy patients to a form of drug therapy with its consequent accountability and liability. This whole issue has been compounded further by finan-cial constraints1-3 that ended in the management of blood transfusion falling gradually into the hands of administrators from outside the blood transfusion specialty.4
While these developments are taking place in the industrial countries, with well-developed and very sophisticated national (or Red Cross) BTSs, the de-veloping countries have lagged behind and are strug-gling to sustain the minimum standards of BTS. They have also been isolated to a great extent from sharing the many advantages and developments prevailing in the industrial countries.
Where does BTS in the Kingdom of Saudi Arabia (KSA) stand? This review attempts to explore the current status of BTS in the KSA, addressing 2 ma-jor transfusion issues, namely, self-sufficiency and safety.
Where Does BTS in the KSA Stand?An important prerequisite to the answer to this ques-tion is the definition of the aims of the BTS that can be summarized as follows:
i. Provision of adequate, safe, and effective blood products
ii. Care of the donor, donation, and recipientsiii. The optimal use of the available donor blood.
These aims remained unchanged for decades. However, the extent to which they are fulfilled has been dominated by growing concerns about self-sufficiency and safety in the face of dwindling finan-cial support, a constantly changing BTS management structure (whether it being independent blood bank-ing system or national service or part of the hospital laboratory service) as well as more and more intru-sion by the legal system.5,6
Blood Transfusion in Saudi Arabia:The main provider of health services in the KSA is the Ministry of Health. There are also University, Military, National Guard, Security Forces Hospitals, as well as Private Hospitals. These different providers are total-ly independent of each other. Accordingly, the BTS, which is mainly a hospital-based blood banking sys-tem, is spread in the form of hospital blood banks all over the KSA, and every hospital (small or large) has its own blood bank to cover its needs of blood and its derivatives. Therefore, blood banks are everywhere because hospitals are everywhere. In the large provin-cial capital cities (Riyadh, Jeddah, and Dammam), BTS assumes regional character since the Central Banks in the major cities, which are part of the major Ministry of Health Central Laboratories, supply blood products not only to the main hospitals within the city boundar-ies but also to smaller provincial hospitals.
As expected the responsibilities of these wide-spread Hospital Blood Banks include:
i. The collection of blood from donorsii. Testing the blood for infective agentsiii. Processing of donated blood units and the
preparation of packed red blood cells (RBCs), fresh frozen plasma (FFP), and cryoprecipitate (in large blood banks)
iv. Storage and issue of blood products.
The main sources of blood donations are:
i. Relatives of patients admitted to hospitals and whose care requires hemotherapy, particularly elective surgery. Hospital Blood Banks are cur-rently following the rule: “No Blood - No Operation.” The major drawback of this system of forced donations is that the concern and re-sponse is short-lived.
ii. Voluntary donor recruitment: This takes the form of either the blood banks sending their collection teams to the various government departments, educational institutes particularly universities, higher colleges, security and mili-tary forces, factories and large commercial busi-nesses, etc. or the increasing number of volun-tary donors who are taking the habit of regular donations
iii. Autologous donation is also practiced but on a very limited scale.
As mentioned earlier, in most blood banks, the blood products that are prepared from whole blood
reviewblood supply in saudi arabia
Journal of Applied Hematology 2011 9
donations are both cellular components (packed RBC and platelet concentrate) and plasma derivatives (FFP and cryoprecipitate).
Sellf-sufficiency?
Definition:Self-sufficiency refers to the ability of the BTS to cover the patient’s requirement for blood derivatives, includ-ing plasma fractions. Accordingly, the KSA has not yet attained self-sufficiency with respect to certain blood derivatives, especially the following plasma fractions, which are all imported from commercial sources: clot-ting factor concentrates (especially FVIII), albumin, PPF, immunoglobulins, and vaccines.
The important question then arises: Is it possible for the KSA to attain self-sufficiency in these plasma derivatives? To answer this question let us take the ex-ample of clotting factor VIII concentrate, the main-stream of hemophilia replacement therapy, as an ex-ample.
Although the exact number of hemophiliacs in the KSA has not been ascertained, if we take the interna-tional estimate of 50 hemophiliacs per million, then the estimated number of hemophiliacs in KSA is ex-pected to be 900 patients. If we assume the annual requirement for each patient to be 200,000 IU of fac-tor VIII concentrate, the total requirement will be 18 million IU per year. The estimated need for donated plasma to prepare this quantity of factor VIII con-centrate will be 450,000 L/year. This means that the Ministry of Health blood banks (alone) should double its current number of collected blood donations to meet the plasma requirements for clotting factor VIII preparation by some manufacturing facility whether within or outside the KSA.
Such a large increase in the number of donors rais-es a more relevant question: Have we tested the extent of the donor potential in Saudi Arabia?7
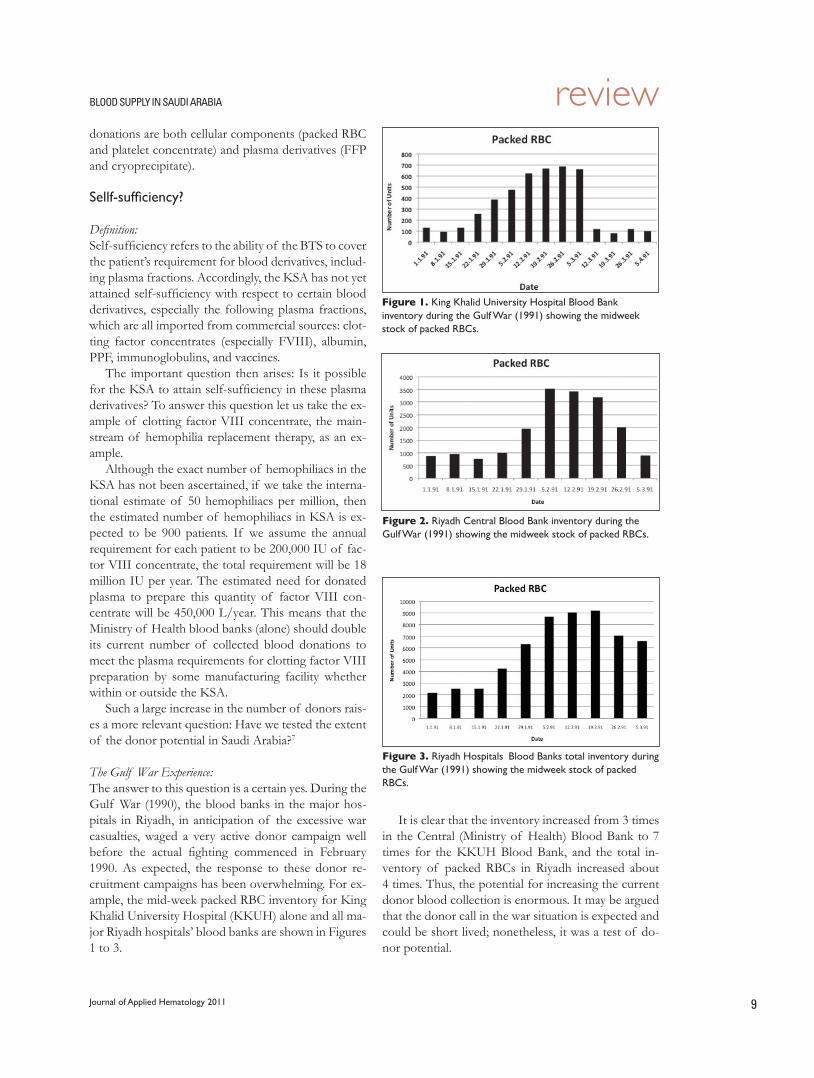
The Gulf War Experience:The answer to this question is a certain yes. During the Gulf War (1990), the blood banks in the major hos-pitals in Riyadh, in anticipation of the excessive war casualties, waged a very active donor campaign well before the actual fighting commenced in February 1990. As expected, the response to these donor re-cruitment campaigns has been overwhelming. For ex-ample, the mid-week packed RBC inventory for King Khalid University Hospital (KKUH) alone and all ma-jor Riyadh hospitals’ blood banks are shown in Figures 1 to 3.
It is clear that the inventory increased from 3 times in the Central (Ministry of Health) Blood Bank to 7 times for the KKUH Blood Bank, and the total in-ventory of packed RBCs in Riyadh increased about 4 times. Thus, the potential for increasing the current donor blood collection is enormous. It may be argued that the donor call in the war situation is expected and could be short lived; nonetheless, it was a test of do-nor potential.
Figure 1. King Khalid University Hospital Blood Bank inventory during the Gulf War (1991) showing the midweek stock of packed RBCs.
Figure 2. Riyadh Central Blood Bank inventory during the Gulf War (1991) showing the midweek stock of packed RBCs.
Figure 3. Riyadh HospitalsÕ Blood Banks total inventory during the Gulf War (1991) showing the midweek stock of packed RBCs.

review blood supply in saudi arabia
Journal of Applied Hematology 201110
King Saud University (KSU) Students Donor Drive–The Peace Experience:This donor drive, which is organized by the Deanship of Student Affairs, KSU, started in 1394 (1973) with 13 donors in its first year to reach 4500 donors in the academic session of 1407–1408 (1986–1987) (Figure 4). The donors are mainly university students. The incentives given to donors, include wristwatch, brief-case, and headdress (ghutra). A “University Blood do-nation Trophy” is awarded annually to the college that donates the maximum number of units. Each year the university rector is the first to donate, and his inaugu-ration of the campaign is given wide publicity within and outside the university.
The KSU donor drive is a model, which is gradually being followed by other educational, civil, and military institutes as well as government departments. It forms an excellent basis for future national voluntary blood donation. Two remarks need to be added on the enor-mous potential of the KSU donor drive: Firstly, most donors donate only once/year, i.e., the total number could easily be doubled if the current donors give their donations at least twice/year. Secondly, no tar-get figure for the total number of donations was set. Indeed, the current number of donors could easily be increased 4 to 5 times, if use could be made of the collected blood and its derivatives. As seen in Figure 4, the total number of donors dropped markedly in recent years, and this is due to the fact that donor collection teams started to get calls from numerous educational institutes, government departments, and private companies to come over to collect blood from their employees. A closer look at Figure 4 indicates that the potential at KSU alone could cover the an-nual needs of KKUH for blood and its derivatives without resorting to the forced recruitment of donors from relatives of patients, i.e., the “No Blood - No Operation” rule. The number of enrolled students at KSU which about 30,000 10 years ago, now stands at
around 120,000 students]. If we add the number of staff, both academic and nonacademic, and their fami-lies the total potential donor population at KSU could be well above 200,000.
What is needed now is the exploitation of this potential for expanding the donor pool, and the de-pendence in totally voluntary non-remunerated do-nor system. This entails proper planning backed by appropriate legislation to integrate the current donor recruitment activity, targeting a specific total donor input to cover the current needs for blood and its de-rivatives and also to help plan for the future plasma fractionation facility. This should eventually transpire into reliance on voluntary blood donations. Voluntary donor recruitment is currently very successful and is expanding as more and more donors are building the tradition of regular blood donation. Ultimately, when service is well integrated and organized, full scale vol-untary service could be established.
This discussion will not be complete without re-ferring to a widely referenced document relating the establishment of BTS in developing countries.
The World Health Assembly in 1975 passed the following resolution (Resolution 25.72):
“…member states to promote the development of National Blood Services based on voluntary non-remunerated donations of blood…”
However, various countries are facing some of these problems, and it is taking them too long to es-tablish a national BTS. Nonetheless, this resolution has set the stage for developing countries to take the necessary steps to reach this aim. Success has already been scored in Hong Kong, Zimbabwe, South Africa, Iran, where the following main features of a National Blood Transfusion Service are already established:8,9
* Blood is given free* BTS is organized without profit on a national
scale* Blood and its derivatives are made available to
patients at any time in the quantities needed.
Blood SafetyThe general approaches to secure safety of transfu-sion of blood and its derivatives9 can be summarized in the following:
* Education, questioning, and selection of donors* Routine serological screening of all donations for
a range of microbial infections
Figure 4. King Saud University Donor drive showing the total number of whole blood units collected from students in the period from 1394 to 1423.
reviewblood supply in saudi arabia
Journal of Applied Hematology 2011 11
* Enhancements in automation of testing* Computerization * Collation and monitoring of test performance,
on a national basis* Test kit evaluations for suitability* Viral inactivation of fractionated plasma prod-
ucts* Viral inactivation of full range of blood compo-
nents* Leucodepletion (impact on bacteria, CMV, EBV,
HHV, HTLV, and vCJD)* Detection of microbial nucleic acid (in mini-pools
or in individual samples).
Despite marked advances in medical sciences and the marked improvement in specificity and sensitivity of the pathogen detection techniques, blood transfu-sion is still fraught with risks, and zero-risk blood trans-fusion does not exist.10 The potential risks to blood transfusion include:
* Risks of infection: This remains the most signifi-cant risk of transfusion of blood derivatives. The infective microbial agents could be bacteria caus-ing septic toxic reactions, protozoa (syphilis and malaria), and viruses (HIV, hepatitis A, B, C, D, and E, retrovirus HTLV, and CMV). However, for practical purposes, screening is performed on a limited number of these infective agents (Table 1).11-13
* Risks related to technique and physics of trans-fusion (cooling, air embolism, microaggregation, and circulatory overload)
* Biochemical-metabolic risks (citrate intoxication, coagulation deficiencies).
General approaches to secure safety have been identified, and different blood banks follow these ap-proaches depending on the available financial resourc-es, alignment, and/or membership of international blood banking accreditation bodies, e.g., American Association of Blood Banks (AABB), and above all conforming to the guidelines of the Ministry of Health. These guidelines oblige all blood banks in the KSA to perform all the tests in Table 1. Nucleic acid testing (NAT) was recently introduced, and all blood banks are obliged to perform NAT for hepatitis viruses B and C as well HIV for all the collected donor blood and its derivatives. We may add here that NAT, which allowed the direct detection of HIV and HCV and HBV, has also helped in elimination of the window period before seroconversion for viral antibody.13,14
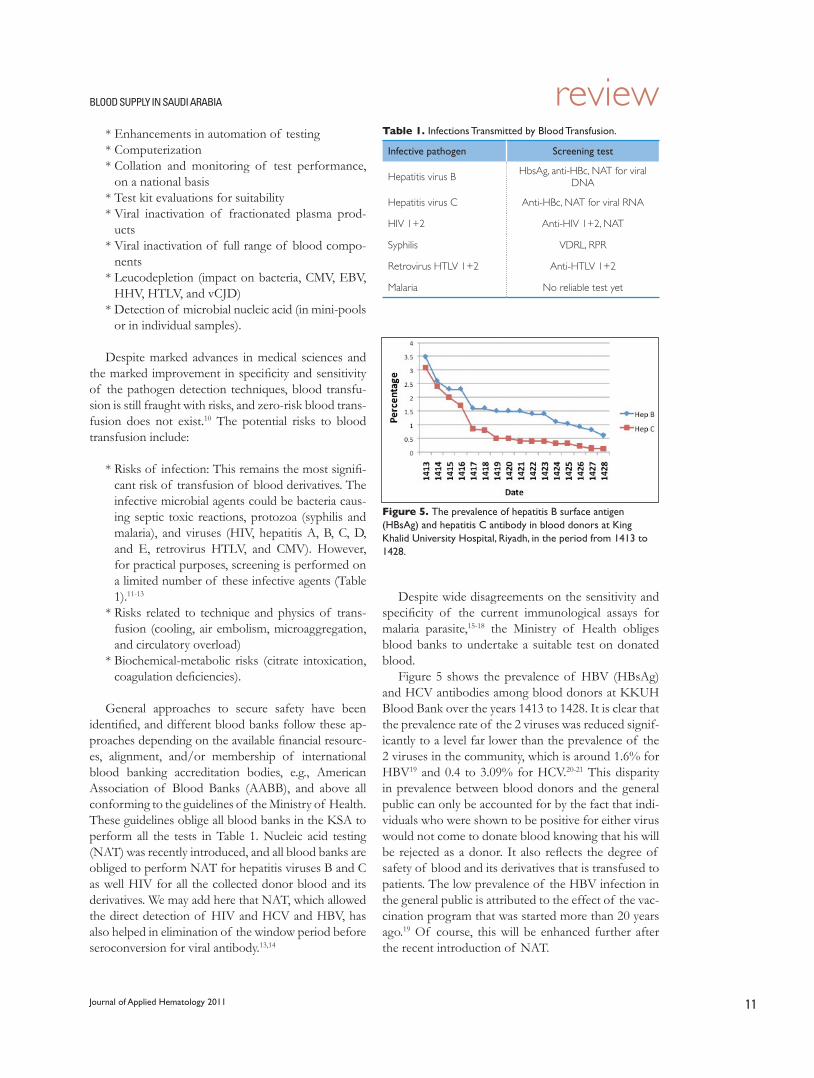
Despite wide disagreements on the sensitivity and specificity of the current immunological assays for malaria parasite,15-18 the Ministry of Health obliges blood banks to undertake a suitable test on donated blood.
Figure 5 shows the prevalence of HBV (HBsAg) and HCV antibodies among blood donors at KKUH Blood Bank over the years 1413 to 1428. It is clear that the prevalence rate of the 2 viruses was reduced signif-icantly to a level far lower than the prevalence of the 2 viruses in the community, which is around 1.6% for HBV19 and 0.4 to 3.09% for HCV.20-21 This disparity in prevalence between blood donors and the general public can only be accounted for by the fact that indi-viduals who were shown to be positive for either virus would not come to donate blood knowing that his will be rejected as a donor. It also reflects the degree of safety of blood and its derivatives that is transfused to patients. The low prevalence of the HBV infection in the general public is attributed to the effect of the vac-cination program that was started more than 20 years ago.19 Of course, this will be enhanced further after the recent introduction of NAT.
Table 1. Infections Transmitted by Blood Transfusion.
Infective pathogen Screening test
Hepatitis virus B HbsAg, anti-HBc, NAT for viral DNA
Hepatitis virus C Anti-HBc, NAT for viral RNA
HIV 1+2 Anti-HIV 1+2, NAT
Syphilis VDRL, RPR
Retrovirus HTLV 1+2 Anti-HTLV 1+2
Malaria No reliable test yet
Figure 5. The prevalence of hepatitis B surface antigen (HBsAg) and hepatitis C antibody in blood donors at King Khalid University Hospital, Riyadh, in the period from 1413 to 1428.

review blood supply in saudi arabia
Journal of Applied Hematology 201112
Other than the battery of screening tests (including the ascertainment of quality assurance of the profi-ciency of these test), viral inactivation of plasma prod-ucts, including FFP, and leucodepletion and its effect in removing the risk of transmitting CMV and HTLV viruses22-27 are waiting to be introduced in all blood banks in the KSA. Also, as the sensitivity of the screen-ing tests for various pathogens is getting to their limits, further refinements in these tests will yield little further reduction in the risk of viral infectivity, leaving more emphasis to be focused on health education and selec-tion of donors particularly the donor questionnaire.22-27 This approach should also focus on the identification and keeping of the so-called “safe” donors.
Ò SafeÓ Donors:By definition, safe blood donors are those whose regu-lar donations were negative on screening tests and who reported no behavior risk factors on post-donation survey.28 Further specific characteristics of a “safe” donor have been outlined recently;28-29 these character-istics include repeat donors, women, donors aged >45 years, and donors with more education.
Lastly, we have to bear in mind that since blood is a biologic product, it is unlikely that the risk for transfu-sion-transmitted infection will ever be reduced to zero. A recent review has identified 2 elements involved in the provision of safe (infection-free) blood products:29
i. Production Process:This concentrates on the donor area and deals with donor education, selection, testing, and exclusion. The concept of “safe” donor already dealt with above sum-marizes the production process satisfactorily.
ii. Clinical Supply Process:This involves the actual transfusion process from the moment a decision is made that a blood product should be transfused to a patient and then getting the right blood, to the right person, at the right place, at the right time. Examples of failures in the clinical process include patient receiving the wrong blood products, transfusing inappropriate doses of blood products, blood arriving late in an emergency, or errors and ad-verse reactions concealed.
Recently, the seriousness of administrative errors was reported; earlier by McClelland & Phillips30 and more recently by others21-33 who found the frequency of deaths due to patients receiving wrong blood to be 30-fold higher than current estimates of transfusion-transmitted HIV infection. Also, in a 5-year period, 22 young women with major obstetric bleeding died due
(at least in part) to delays in administrating RBC trans-fusion (Department of Health, UK, 1994).34 Lastly, it must be noted that most decisions of emergency trans-fusion are taken by busy clinicians who are young, in-experienced, fatigued, and poorly supported by senior colleagues.
The experience of the last 3 decades confirmed further the following steps to be most effective in reducing the risk of transmitting blood-borne infec-tions:12,13,23,26-29,33
* Elimination of paid donors* Refinements of methods of recruiting donors
(specificity of health history)* Implementation of highly sensitive continuously
refined blood screening tests, including molecular testing for viral agents that eliminate the window period for viral infections
* Viral inactivation of plasma for transfusion. It will not be too long before viral inactivation of cellular blood components will be devised.
Where Do We Go From Here?The experience of well-established BTS, particularly, in industrialized countries can allow us to find general answers to this question and that may activate direct efforts and generate practical steps for the future de-velopment of BTS in Saudi Arabia. First and foremost, BTS must be given a separate and independent identity (including administration) and not to be taken as part of a “laboratory service.” Given this identity, then BTS can move forward and fulfill the following goals:
1. Development and implementation of (evidence-based) standards for:
i. Blood transfusion ii. Clinical guidelines2. Quality Assurance3. Objective indicators of achieved safety and ef-
ficacy should be developed, validated, and used4. Clinicians, patients, and donors should be assured
and made aware of these steps5. Financial Support: All the above steps cannot
succeed without generous financial support. There is evidence that money is available should health planners take the right decision for the future development of BTS in Saudi Arabia and guarantee the fulfillment of self-sufficiency and safety of blood and its derivatives.
Dr. A F Britten, Former Head, Blood Program,
International Federation of the Red Cross and Red

reviewblood supply in saudi arabia
Journal of Applied Hematology 2011 13
Crescent Societies (International Blood Transfusion, Size of the problem).35 Countries have summarized the following problems of blood transfusion in the developing world:
* Cultural/Religious attitudes discourage blood do-nation
* Public not educated to the need for blood dona-tion
* Excess plasma but no facilities for plasma frac-tionation
* Money not available for major equipment* Transportation system inadequate for blood de-
liveries* Power failures cause equipment breakdown* Equipment maintenance not available* Person responsible for blood transfusion[A13]
is not given the authority/power to carry out re-sponsibility
* Blood donations inadequate to meet need
* Government does not recognize the importance of blood transfusion
* No building facility suitable for a blood center* No technical expertise* Competition for voluntary donors* No quality control exist* Military, private, social security, Red Cross, and
university blood banks do not cooperate.
It is clear that none of these problems exist in the KSA. Indeed, if one is to scale the international stand-ing of current services offered by the BTS in KSA, it lies comfortably ahead of most, if not all, developing countries. Most blood banks either are accredited by the AABB or have started the accrediting procedure. Steps towards unifying the BTS have already com-menced under the auspices of the Ministry of Health. So, the establishment of a Saudi National Blood Transfusion Service is a dream that should not take long to be true.

review blood supply in saudi arabia
Journal of Applied Hematology 201114
1. Beat R. Organization of blood services. The constancy of change. Trans Med. 1988;8(Suppl 1):2.2. Shander A, Hofmann A, Ozawa S, Theusinger OM, Gombotz H, Spahn DR. Activity-based costs of blood transfusions in surgical patients at four hospitals. Transfusion. 2010 Apr;50(4):753-65.3. .GlenngŒ rd AH, Persson U, Sš derman C. Costs associated with blood transfu-sions in Sweden--the societal cost of autologous, allogeneic and perioperative RBC transfusion. Transfus Med. 2005 Aug;15(4):295-306.4. Gunson HH. The national blood authority in England. Trans Med. 1998;8:165-7.5. Newdick C. Product liability for defective blood. Trans Med. 1991;1(Suppl 2):9-14.6. Angelotta C, McKoy JM, Fisher MJ, Buffie CG, Barfi K, Ramsey G, Frohlich L, Bennett CL. Legal, financial, and public health consequences of transfusion-transmitted hepatitis C virus in persons with haemophilia. Vox Sang. 2007 Aug;93(2):159-6.7. Gader AMA, Al Momen AK, Osman A, Al-Hori I. Blood donor potential in Saudi Arabia. The Ò WarÓ and Ò PeaceÓ experience. Transfusion Today. 2003;54:4-6.8. Cumming RA, Cash JD. The voluntary blood donor. Clin Haematol. 1976;5:3-12.9. Towards better, safer blood transfusion. A Report for the Australian Council for Safety and Quality in Health Care. 2005. ISBN: 0 642 82708 7. © Common-wealth of Australia 2005. Available from: http://www.dcita.gov.au/cca10. Farrugia A, Penrod J, Bult JM. Payment, compensation and replacement - the ethics and motivation of blood and plasma donation. Vox Sang. 2010;99:202-211. 11. Chamberland M, Khabbaz RF. Emerging issues in blood safety. Infect Dis Clin North Am. 1998;12:217-29.12. Epstein JS. Alternative strategies in assuring blood safety: an overview. Biologi-cals. 2010 Jan;38(1):31-5. Epub 2010 Jan 27.13. Vamvakas EC, Blajchman MA. Blood still kills: six strategies to further re-duce allogeneic blood transfusion-related mortality. Transfus Med Rev. 2010 Apr;24(2):77-124.14. Velati C, Roman L, Fomiatti L, Baruffi L, Zanetti AR; SIMTI Research Group. Impact of nucleic acid testing for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus on the safety of blood supply in Italy: a 6-year survey. Transfusion. 2008 Oct;48(10):2205-13. Epub 2008 Jul 8.15. Barbara JAJ. Microbiological safety of blood transfusion. Vox Sang. 1998;74 (Suppl 2):11-3.16. Saeed AA, Al Rasheed AM, Al Nasser I, Al Onaizi M, Al Kahtani S, Dubois L. Malaria screening of blood donors in Saudi Arabia. Ann Saudi Med. 2002;22(5-6):329-32.17. Mertens G, Tony Vervoort T, Sandra Heylen S, Muylle L. Malaria antibody ELISA insufficiently sensitive for blood donor screening. AuthorÕ s Reply. Vox Sang. 1999;77:237-8.
18. Seed CR, Kitchen A, Davis TM. The current status and potential role of labo-ratory testing to prevent transfusion-transmitted malaria. Transfus Med Rev. 2005 Jul;19(3):229-40.19. Alrowaily MA, Mostafa A, Abolfotouh MA, Ferwanah MS. Hepatitis B virus sero-prevalence among pregnant females in Saudi Arabia. Saudi J Gastroenterol. 2008 Apr;14(2):70-2. 20. El-Hazmi MM. Prevalence of HBV, HCV, HIV-1, 2 and HTLV-I/II infections among blood donors in a teaching hospital in the Central region of Saudi Arabia. Saudi Med J. 2004 Jan;25(1):26-33.21. Al-Faleh FZ, Ayoola A, Al-Jeffry M, Al-Rashed R, Al-Mofarreh M, Arif M, Ramia, S, Al-Karawi M, Al-Shabrawi M. Prevalence of antibody to hepatitis C virus among Saudi Arabian children: a community-based study. Hepatology. 1991;14:105-218. 22. Cheng A, Seed CR, Ismay AG. Malaria antibody testing of Australian blood donors. Vox Sang. 2011;100:252-253. 23. Blajchman MA. Protecting the blood supply from emerging pathogens: the role of pathogen inactivation. Transfus Clin Biol. 2009 May;16(2):70-4. Epub 2009 May 7.24. Luban NL. Transfusion safety: where are we today? Ann N Y Acad Sci. 2005;1054:325-41.25. Busch MP, Kleinman SH, Nemo GJ. Current and emerging infectious risks of blood transfusions. JAMA. 2003 Feb 26;289(8):959-62.26. Mushahwar IK. Verses, viruses, and the vulnerability of the blood supply in industrialized countries. J Med Virol. 2007 Aug;79(8):1229-37.27. Stramer SL. Current risks of transfusion-transmitted agents: a review. Arch Pathol Lab Med. 2007 May;131(5):702-7.28. Davey RJ. The Ò safeÓ blood donor and the national blood supply: is there a new interface? Transfusion. 1998;38:323-6.29. McClelland DBL, McMenamin JJ, Moores HM, Barbara JAJ. Reducing risks in blood transfusion: process and outcome. Transf Med. 1996;6:1-10.30. McClelland DBL, Phillips P. Errors in blood transfusion in Britain: survey of hospital haematology departments. Br Med J. 1994;308:1205-6.31. Krombach J, Kampe S, Gathof BS, Diefenbach C, Kasper S. Human error: the persisting risk of blood transfusion: a report of five cases. Anesth Analg. 2002;94:154-6.32. Elhence P, Veena S, Sharma RK, Chaudhary RK. Root cause analysis of transfusion error: identifying causes to implement changes. Transfusion. 2010 Dec;50(12 Pt 2):2772-7. doi: 10.1111/j.1537-2995.2010.02943.x.33. LaRocco M, Brient K. Interdisciplinary process improvement for enhancing blood transfusion safety. J Healthc Qual. 2010 Mar-Apr;32(2):29-34.34. Department of Health. Report on confidential enquiries into maternal deaths in the United Kingdom 1988-1990. HMSO, London 1994.35. Britten AFH. International blood transfusion, size of the problem. In: Mann J, Tarantola D, editors. AIDS in the world. Cambridge, Mass.USA. Harvard Univer-sity Press; 1992. p. 423-33.
References

original article
Journal of Applied Hematology 2011 15
Thromboembolic complications occur due to major surgery or several clinical conditions such as hypercoagulable state. The risk of
venous thrombosis due to acquired hypercoagulabil-ity is directly related to age and elder individuals are more susceptible to this condition. The probability of thrombosis increases by 100 fold from the age of 40 to 75.1 Multiple acquired factors such as obesity,2 hospitalization,3 trauma,4 surgery,4 and immobility5 can modify the onset age and/or the clinical presentation of thrombosis directly or in concert with inherited ele-ments.
Inherited thrombophilia due to natural anticoagu-lant deficiencies or gain of functions can modify the
Impact of inherited thrombophilic factors on deep vein thrombosis in individuals in south Iran
Majid Yavarian, Mani Ramzi, Marym Zakernia, Mansor Hagshenas, Mehran Karimi
From the Hematology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
Correspondence:Mehran Karimi MDProfessor of Pediatric Hematology-OncologyHematology Research Center, Nemazee HospitalShiraz University of Medical Sciences T: +98 711 6473239F: +98 711 [email protected]
Inherited thrombophilic factors are risk factors for venous thrombosis. This study
aimed to estimate the frequencies of factor V Leiden (FVL) and prothrombin
G20210A mutations and protein C, protein S, and antithrombin III deficiencies
in individuals from south Iran and the impact of these factors on the incidence
of venous thrombosis in this area. The study population included 135 patients
with venous thrombosis and 1200 healthy blood donors. The protein C, protein
S, and antithrombin III activities were measured, and the prothrombin 20210A
and FVL mutations were analyzed using polymerase chain reaction-based high-
resolution melting analysis and restriction fragment length polymorphism with
genomic DNA. The frequencies of protein C, protein S, and antithrombin defi-
ciencies in the control group were 0.33% (95% confidence interval [CI]=0.30Ð
0.36), 0.25% (95% CI=0.23Ð0. 27), and 0.08% (95% CI=0.06Ð0. 10), respectively,
and for the patient group were 5.3% (95% CI=1.5Ð9. 1), 3.8% (95% CI=0.5Ð7. 0),
and 4.5% (95% CI=0.9Ð8. 0), respectively. The allele frequencies of FV Leiden and
prothrombin 20210A in the control group were (0.0175 [CI=0.0122Ð0. 0227]
and 0.0200 [CI=0.0144Ð0. 0256], their frequencies in the patients were signifi-
cantly high 0.196 and 0.181, respectively. Thus, in the area studied, the genetic
background clearly affected the onset age and the clinical course of thrombosis.
Approximately 41.5% of the patients who were from the study area and had
DVT had at least 1 of these predisposing genetic factors.
Keywords: Activated protein C resistance, factor V Leiden, Prothrombin
21210A, Deep vein thrombosis, Thrombotic risk factor.
onset age and/or the clinical complications of throm-bosis.
Deficiency of natural coagulation inhibitors such as protein C (PC), protein S (PS), and antithrombin III (AT III) is reported to occur in less than 1%6-8 of the general population. However, the second group of genetic factors (i.e., gain of function), including factor V Leiden (FVL) and its HR2 haplotype,9 the prothrombin G20210A mutation (PT 20210A),10 and elevation of procoagulant factors such as factor VIII, von Willebrand factor, and factors V, VII, IX and XI11 are more prevalent than natural anticoagulants.
Depending on the nature of inherited thrombo-philic defect(s), the spectrum of clinical complica-

original article inherited thromboembolism in iran
Journal of Applied Hematology 201116
tions varies from mild to severe venous thrombosis.The frequency of each inherited risk factor varies
among different ethnic groups, and a high frequency of consanguinity increases the homozygosity of each defect separately or in combination with the others.
Only a few studies have been performed on the frequency of genetic defects in the Iranian popula-tion, and the impacts of genetic defects on the devel-opment of deep vein thrombosis (DVT) in individu-als in south Iran is not well defined. The aim of this study is to determine the frequency and influence of the following factors on DVT in individuals in south Iran: inherited PC and PS mutations, inherited AT III deficiency, and the presence of PT 20210A and FVL mutations.
Patients and MethodsDuring a period spanning 1 year, 135 patients with venous thrombosis and 1200 randomly selected healthy blood donors were enrolled in the study. The local ethics committee approved the study, and in-formed consent was obtained from all participants.
Laboratory testsBlood samples were collected before the patients started taking any medications and at 10 days after holding anticoagulant therapy with Na-citrate (109 mmol/L) and platelet-poor plasma was initiated; the samples were frozen and stored at -70ºC until testing. The activities of PC, PS, and AT III were measured in accordance with the manufacturer’s instructions (Albion, France). The resistance to proteolytic degra-dation by activated PC (APC-R) was measured using a kit (Instrumentation Laboratory Company), and the results were expressed in terms of normalized ratios.
Molecular analysisFor PT 20210A and FVL mutation analysis, genomic DNA was extracted from white blood cells12 and
amplified using polymerase chain reaction (PCR). Mutation analysis for both genes was performed using restriction fragment length polymorphism (RFLP)13,14 and high-resolution melting (HRM) anal-ysis (Rotor-gene 6000), as described in a previous study.15
Statistical AnalysisThe c2 test was used for statistical analysis. The prevalence of odds ratios (ORs) was considered as the prevalence of existing disease, and 95% confi-dence intervals (CIs) were calculated using normal ap-proximation; P values less than 0.05 were considered significant. Data were statistically analyzed using the student t test and Mann-Whitney U test, and a P value of <0.05 was considered significant.
ResultsAll the target genes were autosomal, and no gender priority was observed. From the 1200 healthy indi-viduals, 4 individuals with PC deficiency, 3 with PS deficiency, and 1 with AT III deficiency were identi-fied. The mean estimated frequencies of PC, PS, and AT III deficiencies with 95% CI were 0.33% (range, 0.30–0.36%), 0.25% (range, 0.23–0.27%), and 0.08% (range, 0.06–0.10%), respectively. Mutation analysis of the genomic DNA samples revealed 4 heterozygotes and 1 homozygote of PT 20210A; 46 individuals were FVL carriers, and 1 case of homozygosity was identi-fied (Table 1).
The sensitivity and specificity of HRM were better than those of RFLP but were extremely dependent on the DNA extraction method and DNA quality (Figure 1)
The patient group included 64 males and 71 fe-males; their ages ranged from 24 to 65 years. All the patients with lower limb DVT underwent Doppler color sonography or venography to confirm the diag-nosis of DVT.
The normal range was set at 70–130% for PC activ-
Table 1. Frequency of inherited thrombophilic factor deficiencies among 1200 healthy individuals and 135 patients with DVT.
Protein Cn (%)
95% CI
Protein Sn (%)
95% CI
Antithrombinn (%)
95% CI
PT 20210A Allele*n (%)
95% CI
1691 A (FVL) Allele*n (%)
95% CI
DVT 7 (5.2)1.5Ð9. 1
5 (3.7)0.5Ð7. 0
6 (4.4)0.9Ð8. 0
49 (18. 1)13.5Ð22. 6
53 (19.6)15.1Ð2 4.2
Control 4 (0.33)(0.30Ð0. 36)
3 (0.25)(0.23Ð0. 27)
1 (0.08)(0.06Ð0. 10)
42 (1.75)(1.22Ð2. 27)
48 (2.00)1.44Ð2 .56
*Alleles per chromatid

original articleinherited thromboembolism in iran
Journal of Applied Hematology 2011 17
ity, 65–140% for PS activity, and 80–120% for AT ac-tivity; values below 40% of the mean were considered indicative of inherited deficiency. The overall results revealed that 56 individuals (41.5%) with DVT had at least 1 of the predisposing genetic factors (Table 2).
The APC-R values for the FVL carriers (2.42±0.42) were lower than those for the control subjects (2.97±0.49). The difference between patients and con-trols was extremely significant (P< 0.001)”.
The expected genotype frequency in the control population was calculated using the Hardy-Weinberg equation. The observed genotype frequency for the patients and the expected value for the population are tabulated in Table 2.
DiscussionDVT is a multifactorial disease and is influenced by acquired factors, genetic factors, or both. The genetic and environment act synergistically in the develop-ment of DVT. The proportion of the genetic risk factors in certain populations, and consequently, the priority of each risk factor and the possibility of co-inheritance of these factors are variable. Although the incidence of thrombosis before the age of 40 years is approximately 1 in 10,000 individuals per year,1 throm-bosis can occur at a young age because of certain in-herent parameters.
The gene encoding PC is located on chromosome 2q13-q14 and has 262 reported mutations.16 The gene consists of 9 exons and encompasses 11 kb of DNA.7 Type I and II deficiencies can be diagnosed using func-tional analysis, but an antigen assay or DNA analysis is required for differential diagnosis.
The gene encoding PS has 15 exons, spans 80 kb, and is located on chromosome 3q11; it acts as a cofac-tor for activated PC.17 In 1994, Reitsma first reported mutations in the PROS1 gene;18 to date, approximately 200 mutations have been reported for this gene.16 All the types of PS gene mutations result in low protein activity; only type I and III deficiencies are character-ized by low antigen-free PS levels. As a rule, 60% of inactive PS is bound to C4b; its level decreases in type I deficiency but is normal in type II and III deficien-cies. We are now performing a DNA-based study on PC and PS deficiencies.
The interpretation of the results of the functional assay for the diagnosis of AT III deficiency is slightly complex in the case of type II deficiency and depends on whether the defect at the active site or heparin binding site have different patterns, but in type I defi-ciency, the results of both assays (antigenic and func-tional) are always low.
Table 2. Frequencies of factor V Leiden mutation and prothrombin (G20210A) genotypes in DVT patients from Shiraz University hospitals and in healthy controls (asymptomatic).
Observed Genotype in Patients (%)
Expected Genotype in the Population (%)
FVL 1691 A/A+ PT20210 A/G* 5.2 0.1376
FVL 1691 A/A+ PT20210A A/A** 2.2 0.0012
FVL1691 A/A*** 6.6 0.0400
FVL 1691 A/G 10.6 3.9200
PT20210 A/A 9.6 0.0300
PT20210 A/G 6.8 3.4400
*Compound homozygote FVL + Heterozygote PT, **Compound homozygote FVL + Homozygote PT,
***Homozygote FVL
Figure 1. High-resolution melting analysis (HRM) of Factor V Leiden by using the Rotor-gene 6000 analyzer.
APC-R is characterized by prolonged clotting after adding activated PC. Diagnosis based on conventional coagulation-based methods for APC resistance is not highly specific with respect to genetic defects. In con-trast, DNA-based assays are highly specific and are not affected by pregnancy, contraceptive use, antico-agulant therapy, or inhibitors.19 Approximately 7% of individuals who have abnormal APC-R do not have defects in any of the abovementioned factors.
The carrier frequency of FVL (R506Q), a missense mutation, is 1 in 25, and approximately 1 in 1000 ho-mozygous individuals in the south Iranian population are at risk of developing thrombosis in the. In our study, the frequency of the FVL allele in the patient group was significantly higher than that in the controls subjects (0.196 vs 0.021, P<.0001).
The prevalence of the FVL allele among Europeans

original article inherited thromboembolism in iran
Journal of Applied Hematology 201118
is also high: Greece, 0.070; United Kingdom, 0.044;20 Germany, 0.033; and France, 0.017.21 In our study, the frequency of this mutation was 0.021 and was significant; the thrombotic risk in the carriers was high (OR=11.9; 95% CI, 5.6–25.5). These results were in contrast to those of several studies that showed a low frequency of this mutation in people from Asia, Africa, and the Far East,20,22 and were in agreement with those of another study where a fre-quency of 0.014 was reported for individuals from west Iran.23, 24
The prothrombin G20210A mutation is also common among Europeans, but the data on its fre-quency in Asians and Africans are controversial.22,25,26 Currently available data suggest that the frequency of the PT 20210A mutation increases from the Northern Hemisphere to the Southern Hemisphere. The fre-quency of this allele is even higher in Saudi Arabia than in South of Iran. In our study, the PT 20210A mutation was detected in 23.7% of patients with DVT and in 3.1% of the control group. These values are higher than those reported for Sweden (7.1%, 1.8%) and the United Kingdom (5.5%, 1.2%), and the differ-ence between the groups was significant (P>.05).27,28 This might be related to the relatively high frequency of this allele in the population studied.
The overall risk of recurrent DVT in patients with compound heterozygous FVL and the PT 20210A mutation is 2.7 and 2.6 times higher, respectively, than that in patients who do not have these muta-tions or have FVL alone.29 In the current study, 10 of
56 patients had coinheritance of FVL and PT 20210A (Table 1).
An HR2 haplotype of factor V30 has been report-ed that can contribute by itself to a mild APC resis-tance phenotype and can synergistically interact with the FVL mutation or PT 20210A to produce a severe APC resistance phenotype. This haplotype is associ-ated with more than 12 polymorphisms of the factor V gene, which are collectively known as HR2.30,31 This haplotype is associated with an increased risk of de-veloping venous thrombosis as well as arterial throm-bosis.9,32 In our study, the frequency of this haplotype in the patients with DVT was significantly higher than that in the control group (data not included).
Considering that the incidence of PC, PS, and AT deficiencies in the general population is low, the num-ber of controls used in this study is not sufficient for estimating the frequency of these deficiencies in the south Iranian population.
A few studies have compared the combined heredi-tary risk factors according to the clinical manifesta-tions of venous thromboembolism.
In conclusion, we found that the frequencies of the FVL and PT 20210A mutations in the patient group were greater than those in the control group. Coexistence of hereditary thrombophilic risk factors lowers the onset age, increases clinical severity, and causes recurrent DVT. Family-based genetic counsel-ing and prophylaxis therapy can reduce the adverse effect of thrombosis. Identifying genetically predis-posed individuals would aid in achieving this goal.

original articleinherited thromboembolism in iran
Journal of Applied Hematology 2011 19
1 Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost. 1999;82(2):610-9.2 Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Car-diovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med. 2002;162:1182-9.3 Baglin TP, White K, Charles A. Fatal pulmonary embolism in hospitalised medi-cal patients. J Clin Pathol. 1997;50:609-10.4 White RH, Zhou H, Gage BF. Effect of age on the incidence of venous throm-boembolism after major surgery. J Thromb Haemost. 2004;2:1327-33. 5 Eekhoff EM, Rosendaal FR, Vandenbroucke JP. Minor events and the risk of deep venous thrombosis. Thromb Haemost. 2000;83:408-11.6 De Stefano V, Finazzi G, Mannucci PM. Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood. 1996;87:3531-44.7 Whilatch NL, Orfel TL. Thrombophilias: when should we test and how does it help? Seminars in Respiratory and Critical Care Medicine. 2008;29(1):27-36.8 Seligsohn U, Lubetsky A. Genetic susceptibility to venous thrombosis. N Engl J Med. 2001;344:1222-31.9 Castaman G, Faioni EM, Tosetto A, Bernardi F. The factor V HR2 haplo-type and the risk of venous thrombosis: a meta-analysis. Haematologica. 2003;88(10):1182-9.10 Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previ-ously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc NaH Acad Sci USA. 1993;90:1004-8.11 Reiner AP, Lange LA, Smith NL, Zakai NA, Cushman M, Folsom AR. Common hemostasis and inflammation gene variants and venous thrombosis in older adults from the Cardiovascular Health Study. J Thromb Haemost. 2009 Jun 22.12 Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988 Feb 11;16(3):1215.13 Koeleman BP, Reitsma PH, Allaart CF, Bertina RM. Activated protein C resis-tance as an additional risk factor for thrombosis in protein C-deficient families. Blood. 1994 Aug 15;84(4):1031-5.14 Denninger MH, Cha• t Y, Casadevall N, Hillaire S, Guillin MC, Bezeaud A, et al. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2000 Mar;31(3):587-91.15 Karimi M, Shahraki GRP, Yavarian M, Afrasiabi AR, Dehbozorgian J, Bordbar MR, Mannucci PM. Frequency of factor V Leiden and prothrombin polymor-phism in south of Iran. Iran J Med Sci 2009;34(2):137-40.16 http://www.hgmd.cf.ac.uk17 Edenbrandt CM, Lundwall A. Biochemistry. 1990;29:7861.18 Reitsma PH, Ploos van Amstel HK, Bertina RM: Three novel mutations in five unrelated subjects with hereditary protein S deficiency type I. J Clin Invest
1994; 93:486.19 Lutz CT, Foster PA, Noll WW, Voelkerding KV, Press RD, McGlennen RC, Kirschbaum NE. Multicenter evaluation of PCR methods for the detection of factor V Leiden(R506Q) genotypes. Clin Chem. 1998; 44:1356-8.20 Dzimiri N, Meyer B. World distribution of factor V Leiden. Lancet. 1996;347:481-2.21 Herrmann FH, Koesling M, Schr?der W, Altman R, JimŽ nez Bonilla R, Lopaciuk S, et al. Prevalence of factor V Leiden mutation in various populations. Genet Epidemiol. 1997;14(4):403-11.22 Rees DC. The population genetics of factor V Leiden (Arg506Gln). Br J Hae-matol. 1996;95:579-86.23 Rahimi Z, Vaisi-Raygani A, Mozafari H, Kharrazi H, Rezaei M, Nagel RL. Preva-lence of factor V Leiden (G1691A) and prothrombin (G20210A) among Kurd-ish population from Western Iran. J Thromb Thrombolysis. 2008 Jun;25(3):280-3.24 Rahimi Z, Mozafari H, Shahriari-Ahmadi A, Alimogaddam K, Ghavamzadeh A, Aznab M, et al. Deep venous thrombosis and thrombophilic mutations in western Iran: association with factor V Leiden. Blood Coagul Fibrinolysis. 2010;21(5):385-8.25 Zeinali S, Duca F, Zarbakhsh B, et al. Thrombophilic mutations in Iran. Thromb Haemost. 2000;83:351-2.26 Lin JS, Shen MC, Tsay W. The mutation at position 20210 in the 3Õ -untrans-lated region of the prothrombin gene is extremely rare in Taiwanese Chinese patients with venous thrombophilia. Thromb Haemost. 1998;80:343.27 Hillarp A, Zoller B, Svensson PJ, et al. The 20210 A allele of the prothrombin gene is a common risk factor among Swedish outpatients with verified deep venous thrombosis. Thromb Haemost. 1997;78:990-2.28 Cumming AM, Keeney S, Salden A, et al. The prothrombin gene G20210A variant: prevalence in a U.K. anticoagulant clinic population. Br J Haematol. 1997;98:353-5.29 De Stefano V, Martinelli I, Mannucci PM, Paciaroni K, Chiusolo P, Casorelli I, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med. 1999 Sep 9;341(11):801-6.30 Bernardi F, Faioni EM, Castoldi E, Lunghi B, Castaman G, Sacchi E, et al. A factor V gene component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood. 1997;90:1552-7.31 Castoldi E, Rosing J, Girelli D, Hoekema L, Lunghi B, Mingozzi F, et al. Muta-tions in the R2 FV gene affect the ratio between the two FV isoforms in plasma. Thromb Haemost 2000;83:362-5.32 Hoekema L, Castoldi E, Tans G, Manzato F, Bernardi F, Rosing J. Characteriza-tion of blood coagulation factor V (a) encoded by the R2-gene. Thromb Hae-most. 1999;82 Suppl:S68.
References

original article
Journal of Appplied Hematology 201120
Children who have b-thalassemia major (b-TM) and undergo multiple transfusions may develop severe endocrine complications be-
cause of iron overload.1 The anterior pituitary gland is particularly sensitive to iron overload, which disrupts hormonal secretion resulting in delayed puberty; short stature; adrenal insufficiency; and acquired, clinical, or subclinical hypothyroidism.2,3 Such endocrinopathies hinder growth, ultimately decreasing adult height.4 Delayed or absent puberty are common complica-tions affecting many adolescents with b-TM.4 Glucose
Endocrinopathies in Children and Adolescents with b-Thalassemia Major
Objectives: b-Thalassemia major (b-TM), a prevalent medical condition, is
associated with multiple endocrinopathies. We aimed to evaluate the prevalence
of endocrinopathies between 2006-2010 in children and adolescents with b-TM
and were 2-18 years old.
Patients and MethOds: This retrospective study involved children
and adolescents with b-TM (n=143, 62 females, 57.81% were pubertal, and 81
males, 56.96% were pubertal) presenting at the pediatric endocrine clinic at King
Abdul-Aziz University Hospital. The mean and standard deviation (SD) for age
were 10.96 and 4.4. A comprehensive review of patient serum analysis, and medi-
cal records were done.
Results: Vitamin D (Vit. D) deficiency was the commonest (56%) endocri-
nopathy in both children and adolescents with b-TM, followed by pubertal delay
(29.37%) and hypothyroidism (21%); 7.6% of the patients had no endocrinopa-
thies, and 45.5% had 3 or more endocrinopathies. Growth hormone deficiency
was observed in 12.58% of the patients. The overall mean and SD serum ferritin
levels were 3400.86 and 3067.43 ng/mL, respectively. Iron overload worsened as
the children grew older; the mean and SD serum ferritin levels were 2893 and
1919 ng/mL, respectively, for pre-adolescents and 4299 and 4276 ng/mL, respec-
tively, for adolescents (P=0.0368 [S]).
cOnclusiOn: Children and adolescents with b-TM are at risk of multiple
endocrinopathies. Vit. D deficiency, delayed puberty, short stature, and hypothy-
roidism are the prevalent complications of iron overload. We recommend the
promotion of early screening programs for iron overload to prevent endocri-
nopathies among children and adolescents with b-TM and prophylactic Vit. D
supplements.
KeywORds: b-Thalassemia major, Endocrinopathy, Vitamin D deficiency, Iron
overload, Children.
Abdulmoein Al-Agha,a Shadi A. Shabakah,a Ali Ocheltree,a Daniah El-fateh M. Abdullatif,a Soad K. Al Jaounib
From the aPediatric Department, bHematology Department, King Abdul-Aziz University Hospital, Faculty of Medicine, Jeddah, Kingdom of Saudi Arabia
Correspondence:Dr. Abdulmoien E. Al-AghaAssistant Professor and Consultant Pediatric Endocrinologist, Department of PediatricsKing Abdul-Aziz University Hospital PO Box 80215Jeddah 21589Saudi ArabiaT: +96626408327F: [email protected]
intolerance (GI) in adolescence and diabetes mellitus (DM) later in life are also frequent complications and mainly develop because of iron overload, chronic liver disease, and genetic predisposition.5
Early recognition of endocrinopathies and their prevention by performing early and regular chelation therapy is imperative for improving the quality of life and psychological outcomes of these patients.5 The purpose of this study was to determine the prevalence of endocrinopathies in children and adolescents who had b-TM and visited the pediatric endocrine clinic

original articleendocrinopathies in thalassemia
Journal of Applied Hematology 2011 21
at King Abdul-Aziz University (KAAU) Hospital be-tween 2006-2010.
Patients and Methods
Study design and site This is a retrospective cross-sectional study involv-ing children who had b-TM and visited the pediatrics endocrine clinic at KAAU Hospital between 2006-2010. We reviewed the data on endocrinopathies in the clinical files of all children and adolescents who had b-TM. All the laboratory test data were obtained from KAAU Hospital laboratory database.
Study SubjectsThe study population comprised 143 pediatric pa-tients who were between 2-18 years of age (female patients, 62; male patients, 81). Local patients of multiple nationalities were enrolled in this study (Saudi patients, 24.75%; non Saudi patients 75.25%). The mean, SD and median for the patient’s age were 10.96, ±4.4 and 11 years, respectively. Mean and standard deviation (SD) for female’s age were 11.35 and ±4.75 years, respectively, 37 (57.81%) of females were pubertal. Mean and SD for male’s age were 11.44 and ±4.06 years, respectively, 45 (56.96%) of males were pubertal.
The inclusion criteria were as follows: age be-tween 2-18 years; pretransfusion hemoglobin level >9 g/dL; high ferritin levels >1000 ng/mL. In addi-tion, all the patients enrolled in this study were diag-nosed with ?-TM both clinically and via laboratory methods (hemoglobin electrophoresis) and required regular monthly blood transfusions before 2 years of age.
Diagnosis of Endocrinopathies:The following parameters were reviewed: serum levels of calcium, phosphorous, alkaline phosphatase enzyme, parathyroid hormone, 25-hydroxyl Vit. D, fasting glucose, thyroid-stimulating hormone (TSH); free thyroxin, (fT4), adrenocorticotrophic hormone (ACTH), morning cortisol, serum level of insulin like growth factor -1 (IGF-1), growth hormone (GH). Stimulation test in those with short stature, luteinizing hormone (LH), follicular-stimulating hormone (FSH), and estradiol in girls above the age of 10 years and testosterone in boys above the age of 12 years were also reviewed. The normal reference ranges for the laboratory variables reviewed in this study are given in Table 2. We compared the serum ferritin levels of the patients with respect to age and the endocrinopa-
thies (normal vs. affected) that they had developed. We defined the onset of normal puberty as the devel-opment of thelarche by the age of 8 years or older in girls, and testicular enlargement, measured using the Prader orchidometer, by the age of 9 years or older in boys. Delayed puberty in girls was defined as the absence of breast development by the age of 13 years and/or absence of pubic hair by the age of 14 years, and primary amenorrhea was defined as the absence of menarche by the age of 16 years or a time gap of greater than 5 years between thelarche and menarche. In boys, puberty was defined as delayed if testicular enlargement, as measured using a Prader orchidom-eter, was less than 4 mL by the age of 14 years and/or pubic hair did not develop by the age of 15 years, or complete genital enlargement was delayed for more than 5 years.6 We defined short stature as an SD score of height less than - 2 below the mean for age, gender, and ethnicity. GH deficiency was defined by a maxi-mum peak of GH values less than 10 ng/mL by 2 pharmacological provocative agents (clonidine and glucagon agents were used as stimulants). The exis-tence of primary hypothyroidism was established via evidence such as greater than normal levels of thyroid stimulating hormone and low or normal free thyroxin levels.7 Hypocalcemia in our study population was de-fined as corrected total serum calcium values of less than 2.1 mmoL/L with respect to phosphate levels that were adjusted according to age. Vit. D deficiency was defined as levels less than 75 nmoL/L. We defined adolescents as children in the 13-18-year age group and preadolescents as children younger than 13 years.
Statistical analysis The clinical and laboratory information was collected on a datasheet from KAAU Hospital Phoenix data-base and was digitized on an IBM CPU. The mean, SD, and median values for age and serum ferritin lev-els, and the frequencies of endocrinopathies were cal-culated. Microsoft Excel 2010 software was used for the data analysis and table formulation and presenta-tion of the presented study findings. Student t-test was performed for qualitative data analysis. The level of significance was expressed in terms of P-values: P> 0.05=non significant (NS), P<0.05=significant (S), and P<0.001 = highly significant (HS).
ResultsIron overload worsened as the patients grew older. The mean, SD, and median values for serum ferri-tin for preadolescents were 2893, 1919, and 2387 ng/ml, respectively, and the mean, SD, and median
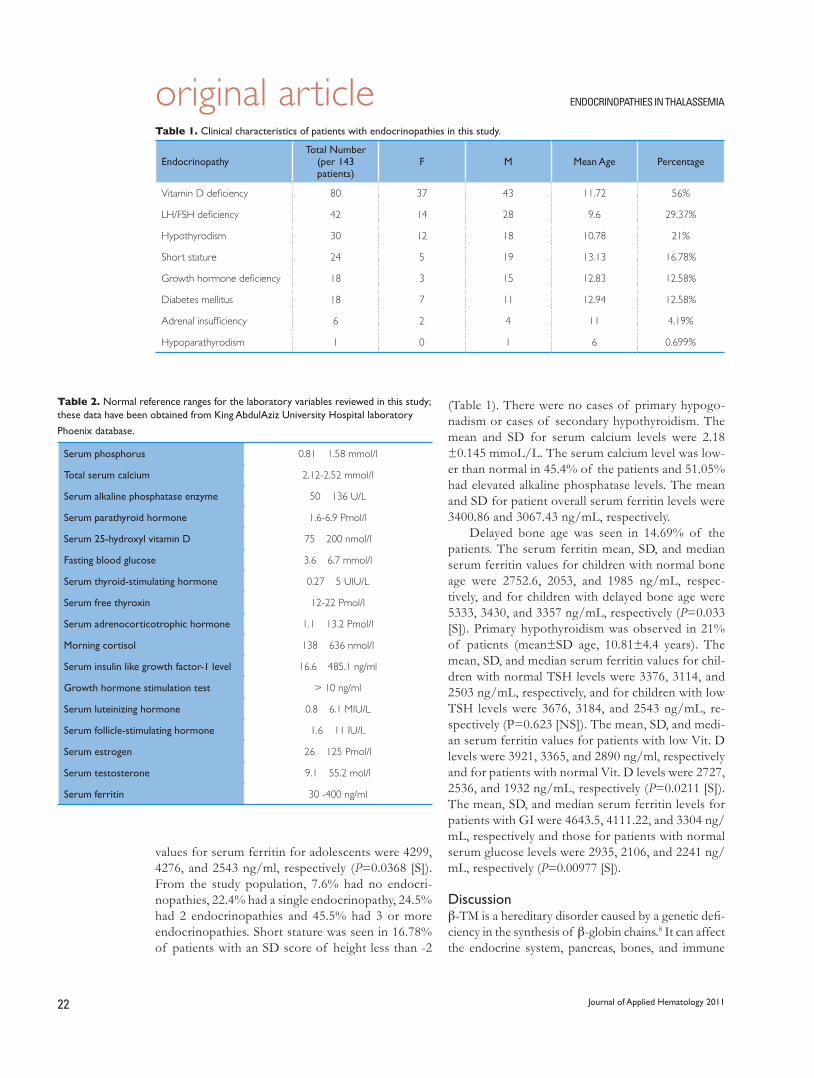
original article endocrinopathies in thalassemia
Journal of Applied Hematology 201122
values for serum ferritin for adolescents were 4299, 4276, and 2543 ng/ml, respectively (P=0.0368 [S]). From the study population, 7.6% had no endocri-nopathies, 22.4% had a single endocrinopathy, 24.5% had 2 endocrinopathies and 45.5% had 3 or more endocrinopathies. Short stature was seen in 16.78% of patients with an SD score of height less than -2
table 1. Clinical characteristics of patients with endocrinopathies in this study.
EndocrinopathyTotal Number
(per 143 patients)
F M Mean Age Percentage
Vitamin D deficiency 80 37 43 11.72 56%
LH/FSH deficiency 42 14 28 9.6 29.37%
Hypothyrodism 30 12 18 10.78 21%
Short stature 24 5 19 13.13 16.78%
Growth hormone deficiency 18 3 15 12.83 12.58%
Diabetes mellitus 18 7 11 12.94 12.58%
Adrenal insufficiency 6 2 4 11 4.19%
Hypoparathyrodism 1 0 1 6 0.699%
table 2. Normal reference ranges for the laboratory variables reviewed in this study; these data have been obtained from King AbdulAziz University Hospital laboratory
Phoenix database.
Serum phosphorus 0.81 Ð 1.58 mmol/l
Total serum calcium 2.12-2.52 mmol/l
Serum alkaline phosphatase enzyme 50 Ð 136 U/L
Serum parathyroid hormone 1.6-6.9 Pmol/l
Serum 25-hydroxyl vitamin D 75 Ð 200 nmol/l
Fasting blood glucose 3.6 Ð 6.7 mmol/l
Serum thyroid-stimulating hormone 0.27 Ð 5 UIU/L
Serum free thyroxin 12-22 Pmol/l
Serum adrenocorticotrophic hormone 1.1 Ð 13.2 Pmol/l
Morning cortisol 138 Ð 636 nmol/l
Serum insulin like growth factor-1 level 16.6 Ð 485.1 ng/ml
Growth hormone stimulation test > 10 ng/ml
Serum luteinizing hormone 0.8 Ð 6.1 MIU/L
Serum follicle-stimulating hormone 1.6 Ð 11 IU/L
Serum estrogen 26 Ð 125 Pmol/l
Serum testosterone 9.1 Ð 55.2 mol/l
Serum ferritin 30 -400 ng/ml
(Table 1). There were no cases of primary hypogo-nadism or cases of secondary hypothyroidism. The mean and SD for serum calcium levels were 2.18 ±0.145 mmoL/L. The serum calcium level was low-er than normal in 45.4% of the patients and 51.05% had elevated alkaline phosphatase levels. The mean and SD for patient overall serum ferritin levels were 3400.86 and 3067.43 ng/mL, respectively.
Delayed bone age was seen in 14.69% of the patients. The serum ferritin mean, SD, and median serum ferritin values for children with normal bone age were 2752.6, 2053, and 1985 ng/mL, respec-tively, and for children with delayed bone age were 5333, 3430, and 3357 ng/mL, respectively (P=0.033 [S]). Primary hypothyroidism was observed in 21% of patients (mean±SD age, 10.81±4.4 years). The mean, SD, and median serum ferritin values for chil-dren with normal TSH levels were 3376, 3114, and 2503 ng/mL, respectively, and for children with low TSH levels were 3676, 3184, and 2543 ng/mL, re-spectively (P=0.623 [NS]). The mean, SD, and medi-an serum ferritin values for patients with low Vit. D levels were 3921, 3365, and 2890 ng/ml, respectively and for patients with normal Vit. D levels were 2727, 2536, and 1932 ng/mL, respectively (P=0.0211 [S]). The mean, SD, and median serum ferritin levels for patients with GI were 4643.5, 4111.22, and 3304 ng/mL, respectively and those for patients with normal serum glucose levels were 2935, 2106, and 2241 ng/mL, respectively (P=0.00977 [S]).
Discussionb-TM is a hereditary disorder caused by a genetic defi-ciency in the synthesis of b-globin chains.8 It can affect the endocrine system, pancreas, bones, and immune

original articleendocrinopathies in thalassemia
Journal of Applied Hematology 2011 23
system. Iron overload in organs because of numer-ous blood transfusions is one of the leading causes of morbidity in all patients with severe forms of b-TM.9 On comparing our prevalence of endocrinopathies in children and adolescents with b-TM to other studies, we found that, in recent years, several studies have re-ported a high incidence of endocrinopathies in chil-dren, adolescents and young adults suffering from b-TM (10). A study conducted at KAAU Hospital be-tween 1990 and 2004 involved 360 patients with b-TM (male patients, 203; female patients, 157) the median age of the patients was 12.5 years. Endocrinopathies were found to be the second leading cause of morbid-ity among children with b-TM. Twenty-nine patients (10.4%) developed endocrinopathies. DM was the most common endocrinopathies (13 patients), fol-lowed by hypoparathyroidism (11 patients) and hypo-thyroidism (5 patients). Furthermore, serum ferritin levels were found to increase with age. In their cohort, the mean and SD serum ferritin levels were 2100, and 512.6 ng/mL, respectively for children who were 0-10 years old and 4920, and 2024.1 ng/mL, respectively among patient ages between 11_20 years old.11
An Iranian study involving 56 children who were more than 10 years of age, stated that the prevalence of endocrinopathies was as follows: DM in (8.9%), short stature (70%) in boys and (73%) in girls, hypo-calcemia in (41%), primary overt hypothyroidism in (16%). Only 8 patients (14.3%) had no endocrine ab-normalities. Delayed puberty was observed in approxi-mately 71% of the patients and it was the commonest endocrinopathy, followed by short stature (51.8%). Hypoparathyroidism is thought to be a rare complica-tion that is usually, but not always, accompanied by hypocalcemia.12 A North America study involving 361 patients who had b-TM and were above 6 years of age showed that such patients had a higher rate of mul-tiple endocrinopathies. Hypogonadism was the most frequent endocrinopathy and affected individuals of both genders; it was observed in 14.3% of the female patients and 25.5% of the male patients. GH deficien-cy was present in 9.6%, DM was in 14%, and 12% of subjects were Vit. D deficient.13
A Taiwanese study was performed on 82 patients whose age was 2 years or older and were receiving frequent transfusions, (15 ml packed erythrocytes per kg body wt given every 2–3 weeks to keep their hemoglobin level at a minimum of 10 g/dl before each transfusion), found that, the prevalence of im-paired glucose tolerance was (8.5%), 7 of 82, and that of DM was (19.5%), 16 of 82. Presentation with diabetic ketoacidosis was (31.1%), 5 out of 16, and
(72.0%) had normal glucose tolerance.14 In the pre-viously mentioned study, the incidence of diabetes was higher than in the current study (12.58%). In an Australian study all the subjects had at least 1 endo-crinopathy, with 16 patients (55%) having 3 or more endocrinopathies. Hypogonadism was the most prev-alent followed by osteoporosis and growth failure (less than 3rd centile) with a frequency of 16/29 (55%), 14/29 (48%) and 10/29 (35%) patients, respectively.15 Thirty seven patients, ages ≥14 years, with b-TM were studied in Turkey for endocrinopathy they concluded that (40%) of the patients had growth retardation. Gonadal dysfunction was detected in (47%) of pa-tients. Hypothyroidism was observed in (16%) of pa-tients. While, impaired glucose tolerance was observed in (10.8%) of patients.16
In a French study involving 267 patients with ?-TM (median age, 20 years), 6% of the patients devel-oped DM, 10% developed hypothyroidism, and 48% developed hypogonadism. The median height was also negatively affected in the youngest patients, and hypogonadism was the most frequent complication.17 The study conducted in France had a higher incidence of hypogonadism when compared to the presented study (21%), the incidence of both hypothyroidism and DM was less. A study conducted at the University of Ioannina in Greece involved 27 patients with b-TM who were segregated into 2 groups: group A with 15 patients aged 5-10 years, and group B with 22 patients with aged 11-23 years. The serum of 24,25-dihy-droxyvitamin D concentrations of the patients with b-TM in both age groups were found to be low.18
In the current study, we compared the serum fer-ritin levels of unaffected and affected children and adolescents with respect to multiple endocrinopathies. We found that the serum ferritin levels of children and adolescents with delayed bone age were significantly higher than those of children and adolescents with normal bone age. Furthermore, a similar finding was observed in connection with Vit. D deficiency; the mean and median serum ferritin of children and ado-lescents with low Vit. D levels were higher than those of children and adolescents who had normal Vit. D levels. Iron overload and GI were found to be corre-lated; the serum ferritin levels of children and adoles-cents with GI were higher than those of children and adolescents who did not have GI.
Vit. D deficiency was the most frequent endocrine complication of iron overload in patients with b-TM, followed by delayed puberty, hypothyroidism, short stature, DM, and GH deficiency. Adrenal insufficiency and hypoparathyroidism accounted for less than 5%

original article endocrinopathies in thalassemia
Journal of Applied Hematology 201124
of the endocrinopathies observed in patients with b-TM in the current study. In our cohort, the serum ferritin levels increased with age, which was similar to the findings obtained in other published studies.11 Furthermore, we observed similar finding with re-spect to the above mentioned studies in most aspects save one, a very high incidence of Vit. D deficiency due to poor appetite, decreased consumption of dairy products and lack of sun exposure. Given the high prevalence of Vit. D deficiency in the general popu-lation and the added burden of increased metabolic demands, chronic medical care, and ironoverload, it is not surprising that Vit. D deficiency is quite com-mon among patients with b-TM.18,19 We emphasize on the importance of establishing an endocrine evi-dence based practice guidelines for b-TM patients, that would perform routine physical and laboratory examinations on ?-TM patients. Such guidelines will improve both the morbidity and mortality rates of b-TM patients who have endocrinopathies and pro-mote compliance to medication and life style im-provements.
ConclusionThe current study provides evidence that endocri-nopathies frequently occur in patients with b-TM and
start early in childhood. We have identified children and adolescents with b-TM as a particularly vulnerable group for multiple endocrinopathies. Vit. D deficien-cy, delayed puberty, hypothyroidism, and short stature were the prevalent endocrineopathies in patients with b-TM. We emphasize on the importance of screening for endocrineopathies in children with b-TM, to pre-vent lifelong complications. Furthermore, we recom-mend that prophylactic Vit. D supplements be given to children with b-TM and that a healthier life style be promoted by encouraging sun exposure and a high intake of fortified dairy products to improve their overall Vit. D status.
AcknowledgmentsThe authors are grateful for the essential cooperation of both the Pediatric Department and the Hematology Department at KAAU. We would like to express our gratitude to both Dr. Mohamad Qari, head of the Hematology Department at KAAU and Dr. Fatin Al Sayes, hematology consultant at KAAU for their valued revision of the current study. In addition, we sincerely thank Dr. Areej Al Hasmi and Dr. Mohammad Al Muhayawi for their participation in data col-lection and assessment. We highly appreciate the assistance of the nurses at KAAU who helped us in data collection and without whom this study would not have been possible.
1. Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxaamine. Haematologica. 2004 Oct;89(10):1187-93. 2. Rehman M, Qureshi AA, Ain Q, Mehmood S. MRI findings of pituitary gland in beta thalassemia major patients. Pak Paed J. 2007 Sep;31(3):137-41.3. Multicenter study on prevalence of endocrine complications in thalassemia major. Italian working Group on Endocrine Complications in Non-endocrine Disease. Clin Endocrinal. 1995;42(6):581-60. 4. Raiola G, Galati MC, De Sanctis V, Caruso NM, Pintor C, De Simone M, et al. Growth and puberty in thalassemia major. J Pediatr Endocrinol Metab. 2003;16 Suppl 2:259-66. 5. Pediatr Endocrinol Rev. 2007 Dec;5(2):642-8. Endocrine complications in patients with Thalassaemia Major. Toumba M, Sergis A, Kanaris C, Skordis N.6. Chern JP, Lin KH, Tsai WY, Wang SC, Lu MY, Lin DT, et al. Hypogonado-tropic hypogonadism and hematologic phenotype in patients with transfusion dependent beta-thalassemia. J Pediatr Hemmatol Oncol. 2003;25(11):880-4.7. Filosa A, Di Maio S, Aloj G, Acampora C. Longitudinal study on thyroid function in patients with thalassemia major. J Pediatr Endocrinol Metab. 2006 Dec;19(12):1397-404. 8. Bens EI, Schwartz E. Thalassemia syndromes in blood disease of infancy and childhood. Mosby: Philadelphia Press; 1990;428-40. 9. Lukens JN. The thalassemia and related disorders. In: Jonathan W, Pine Jr, Editor. Wintrobe’s clinical hematology. 10th ed. USA: Williams and Wilkins 1999:1405-49. 10. Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalasse-mia major treated with transfusion and deferoxaamine. Haematologica. 2004 Oct;89(10):1187-93.
11. Al Jaouni SK. Survival and disease complication of thalassemia major: ex-perience of 14 Years at King Abdulaziz University Hospital, Jeddah, KSA. JKAU: Med. Scie. Vol. 17, No. 1, pp: 19-28 ;2010 A.D.1431 A.H. DOI:10,4197/Med. 17-1.312. Farzad N, Akbar A, Naser A, Amir B, Majid M, Mitra N, et al. A cross-sectional study of metabolic and endocrine complications in beta-thalassemia major. Annals of Saudi Medicine. 28. 13. Vogiatzi M, Macklin E, Trachtenberg F, Fung E, Cheung A, Vichinsky E, et al. Differences in the prevalence of growth, endocrine and vitamin D abnor-malities among the various thalassaemia syndromes in North America. British Journal of Haematology. 2009; 146(5):546-56.14. Chern JP, Lin KH, Lu MY, Lin DT, Lin KS, Chen JD, et al. Abnormal glucose tolerance in transfusion-dependent beta-thalassemic patients. Diabetes Care. 2001; 24(5):850-54.15. Perera NJ, Lau NS, Mathews S, Waite C, Ho PJ, Caterson ID. Overview of endocrinopathies associated with ?-thalassaemia major. Internal Medicine Journal. 40(10):689-696.16. Aydinok Y, Darcan S, Polat A, Kavakli K, Nisli G, Coker M, et al. Endocrine complications in patients with {beta}-thalassemia major. Journal of Tropical Pe-diatrics. 2002; 48(1):50.17. Thuret I, Pondarre C, Loundou A, Steschenko D, Girot R, Bachir D, et al. Complications and treatment of patients with {beta}-thalassemia in France: results of the National Registry. Haematologica. 95(5):724.18. Moulas A, Challa A, Chaliasos N, Lapatsanis PD. Vitamin D metabolites (25-hydroxyvitamin D, 24,25-dihydroxyvitamin D and 1,25-dihydroxyvitamin D) and osteocalcin in beta-thalassaemia. Acta Paediatrica.1997;86:594-99.19. Napoli N, Carmina E, Bucchieri S, Sferrazza C, Rini GB, Di Fede G. Low serum levels of 25-hydroxy vitamin D in adults affected by thalassemia major or intermedia. Bone. 2006;38:888-92.
References

original article
Journal of Applied Hematology 2011 25
Evaluation of the Laboratory tests used in the Identification of Lupus Anticoagulants
Logman A. Gasmelsid,a Abdel Galil M. Abel Gader,a Anwar Y. Kordofanib
From the aCoagulation Research Laboratory, Department of Physiology College of Medicine and King Khalid University Hospital Riyadh, Saudi Arabia King Saud University and bDepartment of Pathology, Faculty of Medicine, University of Khartoum, Sudan
Correspondence:Professor A M Abdel Gader, MD, PhD, FRCP (London & Edinburgh)The Coagulation Research Laboratory Department of PhysiologyCollege of Medicine and King Khalid University HospitalKing Saud UniversityRiyadh 11461, Saudi ArabiaT: 966 1 4671042F: 966 1 467 [email protected]; [email protected]
Background: Lupus anticoagulant (LA) refers to a group of autoanti-
bodies that inhibit certain phospholipid-dependent coagulation reactions, and
typically cause prolongation of activated partial thromboplastin time (APTT).
These are diverse laboratory tests for antiphospholipid antibodies (APAs). This
situation is compounded further by the lack of Ò a golden standardÓ for their
detection and this has resulted in wide variation in LA testing between labora-
tories. The aim of this study is to evaluate the sensitivity of a wide range of assay
procedures and reagents in common use for the detection of LA in patients
with recurrent fetal loss.
Patients: Citrated blood samples were collected from 110 women with
recurrent fetal loss (RFL) attending a special outpatient RFL Clinic, King Khalid
University Hospital, Riyadh. They had history of 3 or more consecutive spon-
taneous unexplained abortions before the 10th week of gestation. Their ages
ranged from 20 to 43 years (mean=30±7.5). Control group: 110 normal healthy
Blood Donors.
Blood samPle Processing: Citrated blood samples were subject-
ed to double 15 min centrifugation at 3000 rpm. The resulting platelet poor
plasma (PPP) was either tested immediately or stored in aliquots in the frozen
state at -40¡C for testing later.
results of laBoratory tests used for detection
la: the detection rates for LA among RFL patients versus healthy controls
are as follows:
* The activated partial thromboplastin time (APTT, Manchester Reagent)
(17.3% vs. 3.6%)
• Mixing studies either with normal plasma (NP) or the platelet neutralization
procedure (PNP):
* Prolonged APTT + NP: (11% vs. 2.7%);
* Prolonged APTT + PNP: (6.3% vs. 1.8%)
* The Staclot-LA test kit (Diagnostica Stago, France) (25.5% vs. 6.3%);
* PTT-LA (Diagnostica Stago, France): (4.5% vs 4.5%);
* The Kaolin Clotting Time (KCT) (28.1% vs. 1.8%);
* The Dilute RussellÕ s Viper Venom Test (dRVVT), (35.5% vs. 6.3%).
conclusions: The dRVVT followed by the KCT identified more patients
with LAC among those with RFL, than the other tests, particularly the low-
phosopholipid APTT (Manchester Reagent). Staclot LA is a complete system
of confirmatory and screening tests. Staclot LA is easy to perform and com-
mercially available as a complete test system, containing both testing and con-
firming for LA. PTT-LA is least sensitive for the detection of LAC. The other
more feasible confirmatory test is PNP combined with a sensitive APTT re-
agent. Depending on the available financial resources, laboratories may follow
different practices in diagnosing LAC ranging from one test to combinations
test procedures.

original article laboratory tests for lupus anticoagulants
Journal of Applied Hematology 201126
Antiphospholipid antibodies (APAs) are a heterogeneous group of antibodies directed against complexes of phospholipids and pro-
teins. Over the years, 2 laboratory procedures have been carried out for the detection of APAs, anticadio-lipin antibodies (ACAs), and the lupus anticoagulant (LAC). Both these procedures have assumed increas-ing importance and popularity in view of the estab-lished association between APAs and many disease states, particularly fetal loss, thrombocytopenia, and venous and arterial thrombosis.1,2
The diversity of these antibodies and their associa-tion with a wide variety of diseases stimulated mul-tidisciplinary interest in linking them with potential pathophysiologic mechanisms3 and generated wide interest in their utility as diagnostic tools. This has re-sulted in an increasing burden on hospital laboratories as tests for APAs, and particularly LAC, are more fre-quently ordered than before by clinicians of diverse specialties.
Lupus anticoagulant belongs to a heterogeneous group of immunoglobulins, of the IgG class, IgM class, or a combination of both the classes, directed against negatively charged phospholipids.3,4 It is an autoantibody that interferes with one or more phos-pholipids-dependent steps of blood coagulation, re-sulting in the prolongation of the prothrombin time (PT), activated partial thromboplastin time (APTT), kaolin clotting time (KCT), and diluted Russell’s Viper Venom Time (dRVVT). This prolongation is attrib-uted to the agglutination of phospholipids present in plasma by LAC, thereby preventing their participation as cofactors in coagulation activation pathways, partic-ularly the inhibition of the conversion of prothrom-bin to thrombin.5
The anticoagulant activity of these antibodies and the prolongation of in vitro phospholipid-dependent clotting tests are currently tested for by lowering the phospholipid concentration in these phospholipid-dependant tests.
The assay procedures and reagents used for the detection of APAs are on the increase and are very diverse, and given the heterogeneity of APAs, no single test is exclusively sensitive or specific for LAC. This situation is compounded further by the lack of a “golden standard” for their detection.
Therefore, there is need to throw more light on the currently available diagnostic tests for LAC, and this leaves the door open for comparative studies on the various testing procedures currently used for the de-tection of LAC.
Therefore, the aim of this study included the fol-
lowing: (i) to evaluate the sensitivity of a wide range of assay procedures and reagents in common use for the detection of LAC in patients with recurrent fetal loss (RFL) and in normal healthy controls; (ii) to find out whether one or more tests need to be undertaken to avoid missing cases positive for LAC.
Materials and Methods
Selection of subjects:Hundred-and-ten women with recurrent fetal loss (RFL) were recruited consecutively from the at-tendants to a special outpatient RFL clinic, King Khalid University Hospital, Riyadh, from January to December 2006. They had a history of 3 or more con-secutive spontaneous abortions before the 10th week of gestation. No obvious causes of abortion (ana-tomical, genetic infective, etc.) were identified i.e., they were considered to have unexplained RFL. Their ages ranged from 20 to 43 years (mean=30±7.5).
Patients were tested at least 3 months after the last abortion or fetal loss. They underwent general medical examination and extensive relevant laboratory and ra-diological tests before enrollment as unexplained fetal loss patients.
Control group: A total of 110 normal healthy blood donors (male n=100, female n=10) from the Blood Bank Donation Center at King Khalid University Hospital, Riyadh, were randomly selected. They were taking no medication. Their ages ranged from 18 to 52 years (±SD: 31±8.2).
Blood sampling and processing: Venous blood samples were collected in sodium citrate (0.11 M) to give a blood citrate ratio of 9:1. Samples were trans-ported without delay to the Coagulation Research Laboratory, College of Medicine. A 15-min double centrifugation at 3000 rpm was done before separat-ing the plasma by double centrifugation as follows: (1) citrated blood was centrifuged at 3000 rpm for 15 min. (2) A plastic transfer pipette was then used to remove the supernatant platelet rich plasma, which was placed in a second plastic tube and recentrifuged under iden-tical conditions. (3) The resulting platelet poor plasma (PPP) was either tested immediately or stored in ali-quots in the frozen state at -40°C for testing later.
Assay techniques:Assay techniques for detecting lupus anticoagulant closely followed the guidelines of the Scientific and Standardization Committee of the International Society for Thrombosis and Haemostasis (ISTH), with respect to both pre-analytical and analytical variables.6

original articlelaboratory tests for lupus anticoagulants
Journal of Applied Hematology 2011 27
The activated partial thromboplastin time (APTT)7 test was performed using a sensitive low phospho-lipid APTT reagent (APTT Manchester Comparative Reagents–UK, Activated PTT Kaolin; Catalogue Code: R9) according to the manufacturer’s instruc-tions. The clotting time was recorded in duplicates us-ing a coagulometer (Start Coagulometer, Diagnostica Stago; France). After the test is taken, LA is suspected if the patient’s APTT is more than 2 SDs (3.5 s) above the mean (48±7 s) of the normal range for healthy population, which is 55 (48+7=55 s). Mixing with normal pool plasma (NP) was undertaken in samples showing >5 s prolongation of the APTT, and those samples, which did not show correction of prolon-gation, were tested further with the platelet neutral-ization procedure (PNP) to confirm the presence of LAC.
The Staclot LA: The Staclot-LA test kit (Diagnostica Stago; France) is a reagent system de-signed for the qualitative detection of lupus anticoagu-lant (LAC) in the plasma by the use of hexagonal (II) phase molecules.8 The Staclot LA is an APTT-based assay that is based on the principle that LAC can be neutralized by hexagonal phase phospholipids (HPP). The Haemostasis Thrombosis Laboratory uses this test to confirm the presence of a LAC following the finding of a prolonged PTT-LA that does not correct fully with normal plasma. The test plasma is incubated with and without addition of HPP. An APTT is then performed on both samples using an LA-sensitive re-agent. If LAC is present, it is neutralized by HPP re-sulting in a shorter clotting time than the plasma with-out HPP. The Staclot LA is considered to be positive for the presence LAC if the difference in the clotting time between the 2 tubes is more than 8.0 s.
In 1993, Douglas Triplett and his colleagues evalu-ated the Staclot LA®, which utilized hexagonal (II) phase (e.g., phosphatidyethanolamine) as a confirma-tory test for LAC.9 The Staclot LA test was performed according to the instructions provided by the manu-facturer.
PTT-LA (Diagnostica Stago; France): The PTT-LA kit is intended for the determination of the APTT.10,11 The PTT-LA reagent is sensitized to aid the detection of LAC.
Test principle: LA exerts an inhibitory effect on phospholipids, which are normally required in clotting tests, such as the APTT. The principle of the PTT-LA test is based on the measurement of plasma recalcifi-cation time in the presence of cephalin and activator silica. The presence in the test plasma of LAC pro-longs the clotting time. Sensitization of the reagent,
which depends on the concentration and the source of the phospholipids and the activator, specifically en-hance the prolongation of the clotting time due to the LA in the test plasma.
The Kaolin Clotting Time (KCT) test was per-formed according to the method proposed by Extner et al.12 The KCT is an assay similar to APTT; however, exogenous phospholipid is not added to the reaction mixture, and the assay depends on the APAs to block the availability of trace quantities of phospholipid present in the centrifuged plasma. Some authors be-lieve that KCT depends on prothrombin as a cofac-tor, than the dRVVT test, which appears to be more dependent on bGP1.13
The results were expressed by a calculated index according to the formula:
An index of 10% or more is considered positive for the presence of LAC.
Confirmatory tests
APTT mixing study:The first step investigating prolonged APTT is to per-form 1:1 mixing study of normal pool plasma (NP) with the patient test plasma. The APTT is repeated, and if the result shows APTT within normal range, a factor deficiency is suggested; if “correction” is in-complete, an inhibitor is suspected.14
The Platelet Neutralization Procedure (PNP)7 was performed in conjunction with APTT. Shortening of the APTT by <5 s in the reaction mixture as com-pared to the saline control is considered confirmatory of positive for LAC. The results of all the tests were recorded in duplicates.
The Dilute Russel’s Viper Venom Test (dRVVT) was performed according to the method of Thiagarajan et al.15 The venom (Manchester Comparative Reagents; UK) was reconstituted using tris buffered saline, pH 7.5. Thiagarajan et al. (1986) were the first to describe the use of a modified dRVVT for the diagnosis of LAC. Their studies found the dRVVT to be more sen-sitive than APTT and the tissue thromboplastin inhi-bition test.
The principle of the assay is based on the use of snake venom to activate factor X and to initiate the co-

original article laboratory tests for lupus anticoagulants
Journal of Applied Hematology 201128
agulation cascade in the common pathway in the pres-ence of a low concentration of phospholipids. This eliminates any problems, including inhibitor or factor deficiency that occurs prior to factor X activation. The dRVVT may be prolonged as a result of hepa-rin therapy, coumardin, or decreased levels of V and X. The confirmatory test uses a high concentration of phospholipids, which should shorten the clotting time. The dRVVT has become a very popular screening test in both USA and Europe. It was performed by add-ing snake venom (Russell’s viper venom) to citrated plasma and recording the clotting time. LA will pro-long the dRVVT by interfering with the assembly of prothrombinase complex.16
The result is taken as positive for lupus anticoagu-lant if the index of patient clotting time divided by control plasma clotting time is >1.1.
Statistical analysis: The Chi-square test was em-ployed to determine the significance of differences in the prevalence of LA, based on the results of different laboratory tests.
Results
The Laboratory detection of LACTo facilitate the comparison between the sensitivities of various assay procedures, the results for both RFL patients as well as healthy controls (blood donors) will be presented as a frequency (number and percent) of positive results of each LAC test procedure.
The screening tests:
APTTIn the first group of tests, which are based on the APTT, we used 3 different reagents APTT, (Manchester Comparative Reagent; UK), PTT-LA (Diagnostica Stago; France), and Staclot LA (Diagnostica Stago; France), whose manufacturers claim their high sensi-tivity for the detection of LAC.
Normal Healthy ControlsThe results obtained for both healthy controls (Blood Donors n=110) and in women with history of RFL (n = 110) are shown in Table-1. In the control subjects, Staclot LA gave the highest frequency of positive re-sults (7 out of 110; 6.3%), which is significantly high-er than the results given by the APTT (Manchester) (3.6%), and the PTT-LA (4.5%.)
RFL Patients The Staclot LA gave the highest frequency of positive
results (25.5%), while the APTT Manchester gave a frequency of 17.3%. The PTT LA proved to be the least sensitive as it gave a frequency of 4.5%, which was similar to that of the controls (Table 1).
KCTThe KCT gave positive results in 28.1% of the RFL patients and in 1.8% of healthy controls (Table-1).
dRVVTThe dRVVT gave the highest frequency of posi-tive results (35.5%) in RFL patients and a frequency of 6.3% in controls, which is similar to that given by Staclot LA (6.3%). The results of the screening tests of RFL patients indicate that the sensitive test for the detection of LAC in a decreasing order of sensitiv-ity is as follows: dRVVT, KCT, Staclot LA, the APTT Manchester, and lastly PTT-LA, while in controls the most sensitive tests were Staclot LA and dRVVT (6.3%), PTT-LA, APTT Manchester, and lastly KCT (Table 1).
The combination of the positive tests: Despite the fact that different screening tests possibly detect different specificities of APAs, we analyzed the results to find out which combination of screening tests agree on positive results:
Staclot LA with dRVVT gave a frequency of posi-tive results of the 14.5% in RFL patients and a fre-quency of 0.9% in controls. Staclot LA + KCT gave a frequency of 10% in RFL patients and 0.9% in con-trols; Staclot LA in combination with dRVVT + KCT gave a frequency of positive results in 7.2% of the RFL patients and in 0.9% of the controls (Table-1).
APTT Manchester + dRVVT gave the highest fre-quency of positive results in 15.4% of the RFL pa-tients, which is similar to that of Staclot LA (14.5%), while the respective frequency in healthy controls is significantly lower (4.5%). On the other hand, the combination of APTT Manchester + KCT gave a frequency of positive results in 10.9% RFL patients, which is similar to that given by Staclot LA, (10%) and 1.8% in healthy controls. APTT Manchester + dRV-VT + KCT gave a frequency of positive KCT results 9% of in RFL patients, which is close to that given by Staclot LA (7.2%) and 0.9% in healthy controls, which is identical to Staclot LA (0.9%) (Table 1).
APTT Manchester + NP: Only 11% (out of the 17.3% who were positive with the APTT, Table 2) were confirmed positive in RFL patients and in 2.7% (out of 3.6%, Table 2) of the healthy controls.
APTT Manchester + PNP: Only 6.3% of RFL

original articlelaboratory tests for lupus anticoagulants
Journal of Applied Hematology 2011 29
table 1. The frequency of positive results of the screening tests for lupus anticoagulant (LAC), using different screening assay procedures, in patients with history of recurrent fetal loss (RFL) and healthy controls.
Assay Procedure(s)
Healthy Controls (n=110)
RFL Patients (n=110)
P valuesNo. of positive
ControlsPositive Controls
%No. of Positive
PatientsPositive Patients
%
APTT Manchester 4 3.6% 19 17.3% 0.0020*
Staclot LA 7 6.3% 28 25.5% 0.0020*
PTT-LA 5 4.5% 5 4.5% -
KCT 2 1.8% 31 28.1% <0.001*
dRVVT 7 6.3% 39 35.5% <0.001*
Staclot LA + dRVVT 1 0.9% 16 14.5% 0.0004*
Staclot -LA+ KCT 1 0.9% 11 10% 0.0075*
Staclot-LA + KCT + dRVVT 1 0.9% 8 7.2% 0.0353*
APTT(Manchester) + dRVVT 5 4.5% 17 15.4% 0.0134*
APTT(Manchester)+ KCT 2 1.8% 12 10.9% 0.0129*
APTT(Manchester) +KCT + dRVVT 1 0.9% 10 9% 0.0133*
* The percentage positive results between patients and controls differed significantly at 5% level of significance. (n=number of observations)
table 2. The frequency of positive results identified by KCT+ dRVVT, APTT-Manchester +PNP, APTT+NP, and the three screening
test (dRVVT+KCT+APTT).
Healthy Controls (n = 110)
RFL Patients (n= 110)
P valueCombinations ofassay procedures
No. of positive Control
Positive Control %
No. of Positive Patients
Positive Patients %
KCT+dRVVT 2 1.8 % 9 8.2 % 0.0634
APTT-Manchester +PNP 2 1.8 % 7 6.3 % 0.042*
APTT-Manchester +NP 3 2.7 % 12 11 % 0.001*
3 tests positive 2 1.8 % 14 12.6 % 0.001*
*The percentage positive results between patients and controls differed significantly at 5% level of significant.
(n= number of observations)
patients (out of the 17.3% which tested positive with APTT alone, Table 1) were confirmed as positive, and 1.8% of healthy controls were confirmed as positive (out of 3.6%, Table 1).
The three screening tests (dRVVT + KCT + APTT) gave positive results in 12.6% RFL patients and in 1.8% healthy controls (Table 2).
DiscussionPhospholipids play multiple roles in the coagulation
system as they participate at several critical points of the coagulation pathways, both extrinsic and intrinsic. In the intrinsic pathway, phospholipids are required for the activation of factor IX in the presence of FVIII (Antihaemophilic factor), platelets, and calcium ions, which in turn activates FX to active FXa. In the ex-trinsic pathway, tissue factor and FVII complex, in the presence of phospholipids, platelets and calcium ions, activate factor X to active FXa. In the final common pathway of coagulation, prothrombin is converted to

original article laboratory tests for lupus anticoagulants
Journal of Applied Hematology 201130
thrombin when bound to phospholipids. Therefore, APAs, whether LACs or ACAs, are expected to act at these numerous sites in the coagulation activation pathways, targeting membrane phospholipids as well as plasma protein co-factors.
LACs were given that name because they were orig-inally detected in patients with SLE. Their anticoagu-lant properties emerge from the fact that they cause prolongation of activated partial thromboplastin time (APTT). Prothrombin time (PT) reagents usually con-tain high concentrations of phospholipids that will mask the antigen/antibodies reaction and are rarely prolonged by LAC, whereas APTT reagent, particu-larly those with low concentration of phospholipids, do not bind all the circulating antibodies resulting in the prolongation of clotting times. This has led to the assumption that patients with LAC are prone to bleed-ing; on the contrary, LAC patients are predisposed to thrombosis.4,17
Requests for the detection of APAs are on the in-crease, so are the diverse laboratory assays used for their detection. This situation is compounded further by the wide diversity of these APAs and the lack of “a golden standard” for their detection, especially for the LACs. This leaves the door open for comparative studies on the available reagents and assay procedures used for the detection of LAC in the hope of identi-fying the most sensitive assay procedure(s), and also triggers numerous efforts to standardize the test pro-cedures employed in the detection of LAC.
Efforts to standardize the laboratory tests for LAC date back to 1992, when an intra-laboratory survey highlighted the variation in APA testing between labo-ratories.18 Although the basic laboratory techniques used were the phospholipids-dependent coagulation tests, APTT, KCT, and dRVVT, most centers in this survey used only one of these assays to test for LA. In later years, many studies that dealt with the evalu-ation of the laboratory tests for LAC19-25 highlighted the major difficulties testing LAC, which are still with us today. Basically, the wide variation in the test pro-cedures used results in a wide variation in the detec-tion rate of LAC. This dilemma triggered the ISTH to take the initiative and assembled world experts in co-agulation testing to work out a standard procedure for testing for LAC, by detailing all the steps from blood collection to the final step and deciding positive from negative results,6 and these guidelines were updated recently.26
Despite these guidelines and voluminous literature on LAC testing that has accumulated in the last 2 de-cades, we are still in a dilemma. This is best highlighted
in 2 very recent North American studies. The first was published in 2010, in which a review was conducted on the performance and practices by North American clinical laboratories.27 The study found that the APPT and the dRVVT constituted major testing methods, and that LAC-sensitive APTT methods were more sensitive to weak LA than dRVVT-based methods but were less specific. In confirmatory testing, dRVVT methods performed better, but the performance was LAC dependent. Noncompliance with recommenda-tions for LAC testing according to the International Society on Thrombosis and Haemostasis Guidelines was high (8%–38%), with the majority of noncom-pliant laboratories failing to report results of mixing studies. It was concluded that the survey provided new insights into LAC testing in North America and iden-tified opportunities for standardization.
In the results of the second study,28 2 question-naires were distributed to the clinical laboratory mem-bers of the North American Specialized Coagulation Laboratory Association (NASCOLA) and the ECAT Foundation (ECAT) to determine their LAC testing practices, and checked if they conformed with the published recommendations. The first and second questionnaires were completed by 113 and 96 labo-ratories, respectively. Commonly performed LAC tests included the dRVVT, LAC-sensitive APPT, and hexagonal phospholipid test. Although some labora-tories did single LAC tests, the majority complied with published recommendations: (1) to use platelet poor plasma for LAC tests; (2) to use two or more screening tests representing different assay principles, and one assay having a low phospholipid concentration to ex-clude LAC; (3) to confirm LAC phospholipid depen-dency by the method giving an abnormal LAC screen; (4) to document the inhibitor activity on pooled nor-mal plasma; (5) and not to use phospholipid antibod-ies to confirm LAC. A minority (<35%) followed the recommendations to exclude factor deficiencies and factor inhibitors as the cause of an abnormal LAC test. After participating, 32% of the laboratories had changed practices and 20% indicated that they would be changing practices. While most laboratories gener-ally follow published guidelines for LAC testing, few follow recommendations to evaluate for other coagu-lation abnormalities.
The results obtained in the present study, which is a single institute study employing multiple procedures for testing for LAC, showed that out of the many APTT-based tests, Staclot LA is the most sensitive as it gave the highest frequency of positive results in 28 (25.5%) out of 110 RFL patients, while LAC-sensitive

original articlelaboratory tests for lupus anticoagulants
Journal of Applied Hematology 2011 31
APTT (Manchester) gave a frequency 17.3%, PTT-LA gave only 5 (4.5%) positive results of 110 RFL pa-tients and failed to detect positive results, which were identified by others.
The reliability of the APPT got further sup-port from the findings of the Working Group on Haemostasis of the Sciété Française de Biologie Clinque,29 employing the APTT reagent (Organon-Teknika), tissue thromboplastin inhibitor assay (TTI) (Thromborel.Behring-werke) and Staclot LA. The study concluded that Staclot LA is the most sensitive and specific test. Similarly, Triplett et al9 in their study of 20 plasma samples previously identified as positive for LAC, found that all 20 samples were positive for the Staclot LA test; i.e., a sensitivity of 100% and con-cluded that the Staclot LA procedure has a very high sensitivity to LAC-positive plasmas.
Disagreements with the above findings have been reported. Thus, Schjetlein and Wisløff (1995)30 in their study of 30 known LAC positive plasma samples, found that the PTT-LA and Staclot dRVVT gave a sensitivity of 67% (20 out of 30). This disagreement can be attributed to the fact that they used known LA positive plasma samples. The determination of posi-tive PTT-LA test was defined by the manufacturer (i.e., >47 s). We established our own reference positive value for PTT-LA, which is 55.1 s (which is the mean of normal (48.1) + 2SD). On the other hand, the APTT-Manchester reagent gave positive results with a frequency of 17.3% in RFL patients. This is in line with the results of the LAC Working Party in United Kingdom,31,32 which defined Manchester APTT as a sensitive reagent for LAC.
PTT-LA (Diagnostica Stago; France) proved to be the least sensitive reagent as it failed to detect most of positive results identified by the other reagents (Staclot LA and APTT Manchester). This agrees with the findings of Denis-Magdelaine et al33 and Arnout et al34 that the responsiveness of PTT-LA appeared to be significantly lowered when performed on the KC10 Coagulometer (Amelung Lemgo; Germany). Instrument dependency of the LAC sensitivity has not been documented in other studies.
The dRVVT produced a remarkably higher rate of positive results than other tests; 35.5% positively in 110 RFL patients as compared to 6.3% in healthy controls. This agrees with the findings of Moor et al,35 who un-dertook the test in 2843 patients with a thrombemo-bolic disease, and found a high frequency of 40.7% (417 out of 1024) of positive results. Similarly, Ferro et al36 studied 53 consecutive patients, with either SLE, or a history of miscarriage or both; the dRVVT gave
a sensitivity of 36%. Monica et al37 employed 2 co-agulation assays, KCT and dRVVT, on the plasma of 72 patients who were previously diagnosed positive for LAC (using Staclot LA to confirm the presence of LAC); dRVVT gave a frequency of 41% (30 out of 72 patients), whereas KCT gave a frequency of 36% (26 out of 72 patients). Pengo et al37 also agree that the dRVVT is the most sensitive and specific test for LAC. Similarly, Anne et al38 prospectively studied 584 consecutive patients who were referred to Mayo’s Special Coagulation Laboratory for either suspected APAs or unexplained prolonged clotting times at the Mayo Clinic in Rochester, Minn Esota (USA). All the patients were screened for LAC using different assay procedures, APTT reagent (BioMèrieux; Durham, NC), dRVVT, and KCT. Among these, 61 patients (10.4%) were positive for LAC out of 584 patients. The dRVVT was the mostly positive LA assay (74% of the 61 patients with positive results for LA). On the other hand Rune et al30 studied 30 plasma samples known to be positive for LAC that belonged to pa-tients with thromboembolic disease, pregnancy com-plications, or autoimmune disease; the sample was considered positive for LAC if it gave a PTT-LA clot-ting time <47.7 s and dRVVT <32.9 s. The dRVVT gave a high frequency of the positive results reaching 80% (24 out of 30); the limit of the clotting time for the dRVVT was 32.9 s (mean+2 SD; 27.7±2.6). In our study, we considered a positive result a ratio < 1.2.
The results of the current study have shown that KCT is less sensitive than dRVVT, but more sensi-tive than APTT assays (APTT Manchester, Staclot LA, and PTT-LA). It gave a positivity of 29.1% among RFL patients, compared to 1.8% normal healthy con-trols. This finding agrees with Ferro et al35 who studied 53 consecutive patients with SLE, who had a history of miscarriage or both. They found a detection rate of KCT positivity of 27% compared to a much higher rate of 36% for the dRVVT.
In RFL patients, the combination of APTT Manchester, dRVVT, and KCT gave a LAC detection rate of 9.1%, which is not markedly different from that detected by the combination of Staclot LA, KCT, and dRVVT (8.2%).
The results of the present study further confirmed the limitations inherent in the laboratory detection of LAC, which in simple term; no 2 tests seem to agree on. Given the diversity of these autoantibodies along with similar diversity in the currently available reagents and test procedures, it is left to individual laborato-ries to use multiple tests to improve the detection of LAC. In a recent study,39 it was shown clearly that the

original article laboratory tests for lupus anticoagulants
Journal of Applied Hematology 201132
more tests are performed; the better is the chance of identifying LAC in patient samples, since no single test has sufficient specificity and sensitivity for the detec-tion of LAC.26 This could make a difference in the management of such enigmatic disease states, such as unexplained recurrent fetal loss. Finally, the new and more rigorous guidelines issued by the ISTH,26 if ap-plied strictly, could be instrumental in improving the laboratory diagnosis of LAC.
In conclusion, the results obtained in the current survey on the performance of commonly available re-agents and test procedures employed for the detection of LAC confirm what many other studies have already found that the dRVVT and KCT gave the highest de-tection rates and performed better than the low-phos-
pholipid APTT. As expected, mixing studies whether by the normal pooled plasma or PNP procedure when used to confirm the specificity of the coagulation in-hibitor reduced the detection rate. Guidelines have been proved difficult to follow, in part due to its cost and labor intensiveness. This could prove to be signifi-cance in a cost-conscious health service, particularly in private medical practice. Therefore, the conflict be-tween the ever-improving guidelines and what it en-tails of excessive laboratory effort and cost will remain a critical factor in drawing the line between what is positive or negative LAC test, and accordingly whether a patient with RFL or other conditions known to be associated with LAC, particularly arterial and venous thrombosis, will or will not receive directed therapy.

original articlelaboratory tests for lupus anticoagulants
Journal of Applied Hematology 2011 33
1. Asherson RA, Cervera R, Merrill JT, Erkan D. Antiphospholipid antibodies and the antiphospholipid syndrome: clinical significance and treatment. Semin Thromb Hemost. 2008; 34(3):256-66.2. Sefer Gezer MD. Antiphospholipid syndrome. Disease of Month. 2003; 49:691-742.3. Triplett DA Antiphospholipid antibodies. Clin Adv Hematol Oncol. 2003 Dec; 1(12):726-30.4. Vivaldi P, Rossetti G, Galli M, Finazzi G. Severe bleeding due to acquired hy-poprothrombinemia-lupus anticoagulant syndrome. Case report and review of literature. Haematologica. 1997; 82:345-347.5. Robert A.S. Roubey. Review Article: Autoantibodies to Phospholipid-binding Plasma Protein: A New View of Lupus Anticoagulant and “Other” Autoantibod-ies. Blood. 1994; 84: 2854-2867.6. Brandt JT, Triplett, DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: An update. On behalf of the Subcommittee on Lupus Anticoagu-lant/Antiphospholipid Antibody of the Scientific and Standardization Committee of the ISTH. Thromb Haemost. 1995;74:1185-1190.7. Triplett DA. The laboratory diagnosis of Lupus Anticoagulants. Fifth Interna-tional Symposium on Antiphospholipid Antibodies. September 9-12, 1992. San Antonio, Texas. 1-13. 8. Rauch J, Janoff AS. Phospholipids in the hexagonal II phase is immunogenic: evidence for immune-recognition of non-bilayer lipid phase in vivo. Proc. Nati. Acad. Sci. USA, 1990; 87:4112-4114. 9. Triplett DA, Barna LK, Unger GA. A Hexagonal (II) phase phospholipids neu-tralization assay for lupus anticoagulant identification. Thromb Haemostas 1993; 70:787-793.10. Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests. J Lab Clin Med. 1953; 41:637-647.11. Larrieu MJ, Weilland C.b Utilisation de la cephaline dans les tests de coagula-tion. Nouv Rev Fr Hematol 1957; 12:199-210.12. Exner T, Rickard KA, Kronenberg H. A sensitive test demonstrating lupus anticoagulant and its behavioral patterns. Br J Haematol. 1978; 40:143-151.13. Galli M, Finazzi G, Bevers EM, Barubui T. Kaolin clotting time and dilute Rus-sell’s viper venom time distinguish between prothrombin-dependent and beta 2-glycoprotein I-dependent antiphospholipid antibodies. Blood 1995; 86:617-623.14. Lie JT. Pathology of antiphospholipid syndrome. In: Asherson RA, Cervera R, Piette JC Shoenfeld Y, editors. The antiphoshpolipid syndrome. Boca Raton (Fla): CRC press; 1996; pp 89-104.15. Thiagarajan P, Pengo V, Sapiro SS. The use of the dilute Russel Vipor Venom Time for diagnosis of lupus anticoagulants. Blood. 1986; 68:869-874.16. Forastiero RR, Cerrato GS, Carreras LO. Evaluation of recently described tests for detection of the lupus anticoagulant. Thromb Haemostas. 1994; 72:728-733.17. Donna D. Castellone, MS, The Lupus Anticoagulant: Truth or consequence. Advance Newsmagazines issue Date 1/8/2001. 18. Peaceman AM, Silver RK, MacGregor SN, Socol ML. Interlaboratory variation in antiphospholipid antibody testing. Am J Obestet Gynecol. 1992; 166:1780-7.19. Chi-Shun Feng, Sophia S. F. Tsang and Edmind K.Li. Evaluation of Laboratory Tests for Lupus Anticoagulant in a group of Chinese Lupus Patients. Pathology. 1994; 26:40-42.20. Jacobsen EM, Barna-Cler L, Taylor JM, Triplett DA, Wisloff F. The evaluation of clotting times in the laboratory detection of lupus anticoagulants. Thromb Res. 2001; 104:275–28221. Jenning J, Greaves M, Mackie IJ, Kitchen S, Timothy A. Lupus anticoagulant test-ing: improvements in performance in a UK NEQAS proficiency testing exercise after dissemination of national guidelines on laboratory methods. British Journal of Haematology. 2002; 119:364-369.22. Favaloro EJ, Wong RC. Current clinical and laboratory practice for the inves-
tigation of the antiphospholipid syndrome: findings from the 2008 Australasian antiphospholipid antibody survey. Pathology. 2009; 41(7):666-75.23. Feng CS, Tsang SS, Li EK. Evaluation of laboratory tests for lupus anticoagu-lant in a group of Chinese lupus patients. Pathology. 1994; 26(1):40-2.24. Ricardo R. Forastiero, Graciela S. Cerrato, Luis O. Carreras. Evaluation of Recently Described Tests for Detection of the Lupus Anticoagulant. Thrombos Haemostas. 1994; 72(5):728-33.25. Devreese K, Hoylaerts MF. Laboratory diagnosis of the antiphospholipid syn-drome: a plethora of obstacles to overcome. Eur J Haematol. 2009; 83(1):1-16. 26. Pengo V, Tripodi A, Reber G, Rand JH, Ortel TL, Galli M, De Groot PG. Sub-committee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis.Update of the guidelines for lupus anticoagulant detection. Sub-committee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009; 7(10):1737-40.27. Dembitzer FR, Ledford Kraemer MR, Meijer P, Peerschke EI. Lupus antico-agulant testing: performance and practices by North American Clinical Labora-tories. Am J Clin Pathol. 2010;134(5):764-73.).28. Moffat KA, Ledford-Kraemer MR, Plumhoff EA, McKay H, Nichols WL, Meijer P, Hayward CP. Are laboratories following published recommendations for lupus anticoagulant testing? An international evaluation of practices. Thromb Haemost. 2009; 101(1):178-84.),29. Working Group on Hemostasis of the Sociètè Fran?aise de Biologie Clinique Comparison of a standardized Procedure with Current Laboratory Practices for the Detection of Lupus Anticoagulant in France. Thrombosis and Haemostasis. 1993; (5):781-786.30. Schjetlein R, Wisløff F. Detection of lupus anticoagulant: An evaluation of routines for preparation and storage of plasma Thromb Res.1995; 79:135-140. 31. Working Party in United Kingdom (1990 and 1992) The Lupus Anticoag-ulant Working Party. Detection of lupus like anticoagulant: current laboratory practice in the United Kingdom. J Clin Pathol. 1990; 43(1):73-5.32. Denis-Magdelaine et al (1995), Denis-Magdelaine A, Flahault A, Verdy E. Sensitivity of sixteen APTT reagents for the presence of lupus anticoagulants. Haemostasis. 1995; 25(3):98-105.33. Arnout J, Vanrusselt M, Huybrechts E, Vermylen J. Optimization of the dilute prothrombin time for the detection of lupus anticoagulant by use of a recombinant human tissue thormboplastin. Br J Heamatol. 1994; 87:94-99.34. Moore GW. Savidge GF, Smith MP. Improved detection of lupus antico-agulant by the dilute Rusell’s Viper venom time. Blood Coagul Fibrinolysis. 2000; 11(8):767-7435. Ferro D, Saliola M, Quintarelli C, Valesini G, Basili S, Grandilli AM, Bonavita MS, Violi F. Methods for detecting lupus anticoagulants and their relation to thrombosis and miscarriage in patients with systemic lupus erythematosus. J Clin Pathol. 1992; 45(4):332-836. Monica Galli, Tiziano Barbui. Antiphospholipid antibodies and pregnancy. Best Practice & Research Clinical Heamatology. 2003; 16:2; 211-225.37. Pengo V, Biasiolo A, Rampazzo P, Brocco T. dRVVT is more sensitive than KCT or TTI for detecting lupus anticoagulant activity of anti-beta2-glycopro-tein I autoantibodies. Thromb Haemost. 1999 Feb; 81(2):256-8.38. Proven A, Bartlett RP, Moder KG, Chang-Miller A, Cardel LK, Heit JA, Homburger HA, Petterson TM, Christianson TJ, Nichols WL. Clinical impor-tance of positive test results for lupus anticoagulant and anticardiolipin anti-bodies. Mayo Clin Proc. 2004; 79(4):467-75.39. Al-Mishari AA, Gader AG, Al-Jabbari AW, Al-Momen AK, El Rab MO, Babay ZH, Mahmoud N. The prevalence of lupus anticoagulant in normal pregnancy and in women with recurrent fetal loss-- recommendations for laboratory testing for lupus anticoagulant. Ann Saudi Med. 2004; 24(6):429-33.
References

original article
Journal of Appplied Hematology 201134
Bone involvement is a frequent cause of acute morbidity in sickle cell anemia and some of its variant hemoglobinopathies.1,2 Published stud-
ies suggest that children with sickle cell disease (SCD) often have undiagnosed osteopenia or osteoporosis.3-5 Osteoporosis is defined as a state of increased frac-ture risk because of low bone mineral density (BMD) and deterioration in the bone microarchitecture.6 In patients with b-thalassemia, a red blood cell disorder characterized by anemia and low BMD, the patho-physiology of low BMD is related in part to increased
Low bone mineral density in patients with sickle cell disease: Association with blunted parathyroid hormone response and accelerated bone turnover
Jalaluldin A. Jalal,a Mohamed F. Elshal,a Mohamed H. Qari,b Maryam A. Al-Ghamdy,a Amna E. Bernawia
From aBiochemistry Department, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia, bCenter of excellence for osteoporosis research andHematology department, Faculty of Medicine, King Abdulaziz University, Jeddah, KSA
Corresponding Author:Mohamed F. Elshal, Ph.D.Biochemistry DepartmentFaculty of Science and Center of excellence for os-teoporosis research King Abdulaziz UniversityJeddah, [email protected]
Conflict of interest:
The authors have no conflicts of interest to declare.
Background/aim: Bone complications in sickle cell disease (SCD)
have been well documented, but the mechanisms underlying SCD are poorly
characterized. Therefore, we conducted this study to elucidate the factors af-
fecting bone health in patients with SCD by measuring bone turnover markers
and correlating them with bone mineral density (BMD).
Patients and methods: Serum from 30 patients with confirmed
SCD and 20 age-matched healthy controls were included in the study. Dual
X-ray absorptiometry was used to determine the BMD of the lumbar spine
(L2ÐL 4) and of the whole body.
results: According to the WHO definitions for T-scores, 30% of HbSS pa-
tients were osteopenic (30%) or osteoporotic (50%) in at least 1 of the 3 stud-
ied locations. BMD, serum calcium, and parathyroid hormone (PTH) showed
a significant decrease, while PO4, osteocalcin (OC), and bone-specific alkaline
phosphatase (b-ALP) showed a significant increase in the patient group com-
pared to the control group (P<0.05, for all). Multivariate analysis identified only
serum PTH as an independent determinant of low whole-body BMD in HbSS
(P=0.0399), whereas it identified osteocalcin, magnesium, and CTX as indepen-
dent predictors for PTH.
conclusions: These findings suggest that low BMD is prevalent in SCD
and that blunted PTH response to reduced total calcium levels and accelerated
bone turnover may be the underlying mechanisms.
keyWords: Sickle cell anemia, osteoporosis, BMD, parathyroid hormone,
bone turnover markers.
bone resorption;7 however, the pathophysiology of similar bone manifestations in SCD is less well char-acterized.4,5
A substantial body of evidence suggests that mi-crovascular occlusion by sickled erythrocytes may cause ischemic modifications of osseous tissue and thereby lead to osteonecrosis.8,9 Moreover, impaired bone blood flow was found to increase the apopto-sis of osteoblasts and osteocytes, which may lead to osteoporosis.10,11 Although osteonecrosis accounts for most chronic severe pain and further deterioration of

original articleBlunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 2011 35
the quality of life in patients with SCD,12 other skeletal deformities including vertebral end-plate depression, mostly due to osteoporosis, have also been reported, especially in young patients.4,13-15
Recent studies have acknowledged BMD as an in-termediate osteoporosis marker that can be used to recognize the patient’s risk of fracture, but changes in the microarchitecture of the bones are not captured by BMD measurements.16 In this regard, the measure-ment of bone turnover markers may complement BMD testing. These markers have proven useful in screening for fracture risk in elderly patients, assess-ing therapeutic response to antiresorptive agents, and identifying patients with high bone turnover to predict rapid bone loss.17,18 Hormones play an essential role in skeletal health. Evidence suggests that PTH exerts anabolic action on bone partly through local growth factors and anti-apoptotic action on osteoblasts.19,20 Moreover, intermittent administration of PTH was found to be useful in the treatment of osteoporo-sis, since it improves bone architecture and strength without causing the appearance of abnormal bone elements.21 Nevertheless, scarce data exist on the re-lationship between PTH, bone turnover, and BMD, the gold standard measure for identifying patients at risk of osteoporosis, in particular, adults with SCD. Therefore, we aimed to investigate the possible rela-tionships between BMD, PTH, and several bone turn-over markers, including the bone formation markers osteocalcin (OC) and bone-specific alkaline phos-phatase (b-ALP), and the bone resorption markers C-terminal telopeptide of type I collagen (CTX) and N-terminal telopeptide of type I collagen (NTX).
Materials and MethodsSCD patients who regularly visited the outpatient clinic of King Abdulaziz University Hospital were enrolled in the study after ethical approval was ob-tained from the Research and Scientific Committee of College of Medicine, King Abdulaziz University, Jeddah, KSA. Informed consent was obtained from each participant prior to enrollment. The patients had their weight and height measured to calculate the body mass index (BMI). Their history was noted, and a clin-ical examination was conducted, which was followed by appropriate investigations to rule out secondary os-teoporosis. The following patients were excluded from the study: (1) those who were diagnosed and treated for osteopenia and osteoporosis; (2) those who were on steroids; (3) those who had anorexia nervosa, hy-perthyroidism, chronic obstructive pulmonary disease, liver disease, or inflammatory bowel disease; and (4)
those who had undergone organ transplantation.Whole-body BMD and T-score were determined
twice by dual X-ray absorptiometry (Hologic QDR 2000; Bedford, MA)22 at the Osteoporosis Center of Excellence, King Fahad Medical Research Center (KFMRC), King Abdulaziz University, Jeddah. All blood samples were collected at 10 AM. Serum sam-ples were separated by centrifugation at 3000 rpm for 10 min and were then were stored in the freezer at –20°C until analysis. The bone formation marker b-ALP was analyzed using the IDS BAP immunoas-say (Immunodiagnostic Systems Inc., Fountain Hills, AZ, USA). The analytical sensitivity for the BAP as-say was <1 U/L (reference range, 50–136 U/L) with intra- and interassay variability lower than 10.1% and 10%, respectively. The other bone-specific marker, osteocalcin (OC), was measured using BioSource, a human Osteocalcin Enzyme Amplified Sensitivity Immunoassay (hOST)-EASIA kit (BioSource Europe S.A., Belgium). The analytical sensitivity of OC was <0.4 ng/mL (reference range, 6.8-32.2 ng/mL), with intra- and interassay coefficient of varia-tion (CV) values of 5.2% and 6.7% respectively. Bone resorption markers included CTX, which was measured using Immunodiagnostic System Limited (Immunodiagnostic Systems Inc., Fountain Hills, AZ, USA), and NTX, which was analyzed using the Wampole Laboratories (USA). The reference range for CTX is between 0.13 and 4.1 ng/mL, and it is 10–60 nM BCE for NTX, with a CV of 4.6%.
Intact serum parathyroid hormone (iPTH) was analyzed by enzyme-linked immunosorbent assay (Immunodiagnostic Systems Inc., Fountain Hills, AZ, USA). The reference range for iPTH was 15–65 pg/mL (1.6–6.9 pmol/L), with an analytical sensitivity of <0.1 pmol/L and intra- and interassay CV values of less than 5% and 7%, respectively (Aloia et al. 2006). Serum calcium (Ca), magnesium (Mg), and inorganic phosphorus (iP) were measured using an endpoint assay in the Dade Behring Dimension-RxL Clinical Chemistry System (Dade Behring; International Incorporated, USA).23 The total calcium concentra-tion was adjusted for serum albumin. The laboratory reference ranges were 2.2–2.6 mmol/L for total Ca, 0.8–1.2 mmol/L for Mg, and 1.0–1.4 mmol/L for iP.
Statistical analysisResults are expressed as the mean±standard deviation (X ± SD). The statistical significance of the differ-ences between groups was determined using one-way analysis of variance (ANOVA) combined with the post-ANOVA Tukey-Kramer test for multiple com-

original article Blunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 201136
parisons. Uni- and multivariate regression analyses were performed to evaluate the strength of the rela-tionship between BMD and other studied parameters. P<0.05 was defined as statistically significant. A sam-ple size analysis demonstrated that 19 subjects would be required in each group to detect a statistically signif-icant difference of P<0.05 in BMD between groups (one SD of 0.13; power, 88%). Data were analyzed using SPSS software version 13.0 for Windows (SPSS Inc., Chicago, IL, USA).
ResultsThirty patients with SCD (19 female and 11 male) were included in the study; 20 age-matched healthy volunteers (11 female and 9 male) were included in the control group. There were no significant age or sex differences between the patients and the individu-als in the control group. However, the BMD results demonstrated that SCD patients showed a significant reduction of 17.6%, 15.45%, and 12.3% at L2, L4, and for the whole body in comparison with those of the healthy control group, respectively (P<0.001; Table 1). There was also a highly statistically significant differ-ence between the groups for the T-scores at L2 and for the whole body, and a more modest one for the T-score at L4. For all the T-scores, the values were lower in the SCD group than in the control group (Table 1). Cases studied were classified into nor-mal BMD (T-score greater than –1 SD), osteopenia (T-score greater than –2.5 SD and less than –1 SD), and osteoporosis (T-score less than –2.5 SD) accord-ing to the guidelines proposed by the World Health Organization (WHO) for the diagnosis of osteopo-rosis based on measurement of BMD.24 In general, the T-scores showed that 30% of the patients were osteopenic and 50% were osteoporotic in at least 1 of the 3 studied locations. In contrast, the T-scores of the control subjects were within normal range (T-score > –1).
As shown in Table 2, SCD patients had low, border-line serum Ca levels (2.2±0.39 mmol/L) and showed a significant increase in serum phosphorus levels com-pared to the control group. There were no significant differences in serum magnesium levels between the 2 groups. A highly significant decrease (P<0.0001) in se-rum PTH was found in the HbSS patients relative to the controls (Table 1).
The biochemical bone markers results showed a significant increase in the levels of the bone forma-tion markers b-ALP and OC in HbSS patients when compared to controls; however, the CTX and NTX levels were not significantly different between the
groups (Table 1). In terms of the correlation between PTH and the other studied parameters, BMD and os-teocalcin showed a significantly positive correlation (r=0.387, P<0.008; r=0.585, P<0.0001, respectively), whereas PTH showed a significantly negative corre-lation with CTX (r=0.343, P<0.05). Both PTH and BMD also correlated negatively with serum phospho-rus levels (r=0.447, P<0.0001; r=0.373, P<0.01 re-spectively) (Table 2).
The results of the multiple regression analyses with BMD as the dependent variable are presented in Table 3. Stepwise multiple regression analyses revealed that PTH was an independent determinant of BMD (b= 0.045, P=0.0399). Multiple linear regression anal-yses indicated a significant correlation of PTH with osteocalcin, magnesium, and CTX.
DiscussionBeyond the hematological severity and heterogene-ity of SCD,25 bone and joint complications are the most common manifestations.15 These complications include bone infarction, osteomyelitis, osteonecrosis, osteopenia, and osteoporosis.3,4 Accelerated hema-topoiesis and bone infarction probably contributed to the low BMD in these patients.26 Low BMD is a hallmark of osteoporosis and increased fracture risk.27 In the present study, patients with SCD had a signif-icantly lower BMD concentration at L2 (P< 0.001) and L4 (P< 0.05), and a lower total body BMD (P< 0.001) than the control group. These results are in ac-cordance with other studies.3,4,28
Lower T-scores have been reported to have a strong association with a higher fracture risk.29 In our study, the patients had a significantly lower T-score at L2 (P<0.001) and L4 (P<0.05), and a lower total body T-score (P<0.001) in comparison with the control in-dividuals. These results may indicate the prevalence of osteopenia or osteoporosis in the patients and a higher fracture risk. Furthermore, 30% of the patients were found to be osteopenic and 50% to be osteoporotic according to the WHO guidelines for T-scores. These results were accompanied by a significant reduction in PTH (P<0.001) in the SCD group relative to the con-trol group (P<0.05), as well as low borderline serum Ca levels (2.2±0.39 mmol/L). Low serum calcium levels in SCD patients have been reported in earlier studies30,31 and have been suggested to be a result of impairment of the intestinal absorption of calcium in SCD patients.30 In the present study, serum calcium showed positive correlation with serum phospho-rus and magnesium (r=0.390, P<0.032; r=0.736, P< 0.0001, respectively).
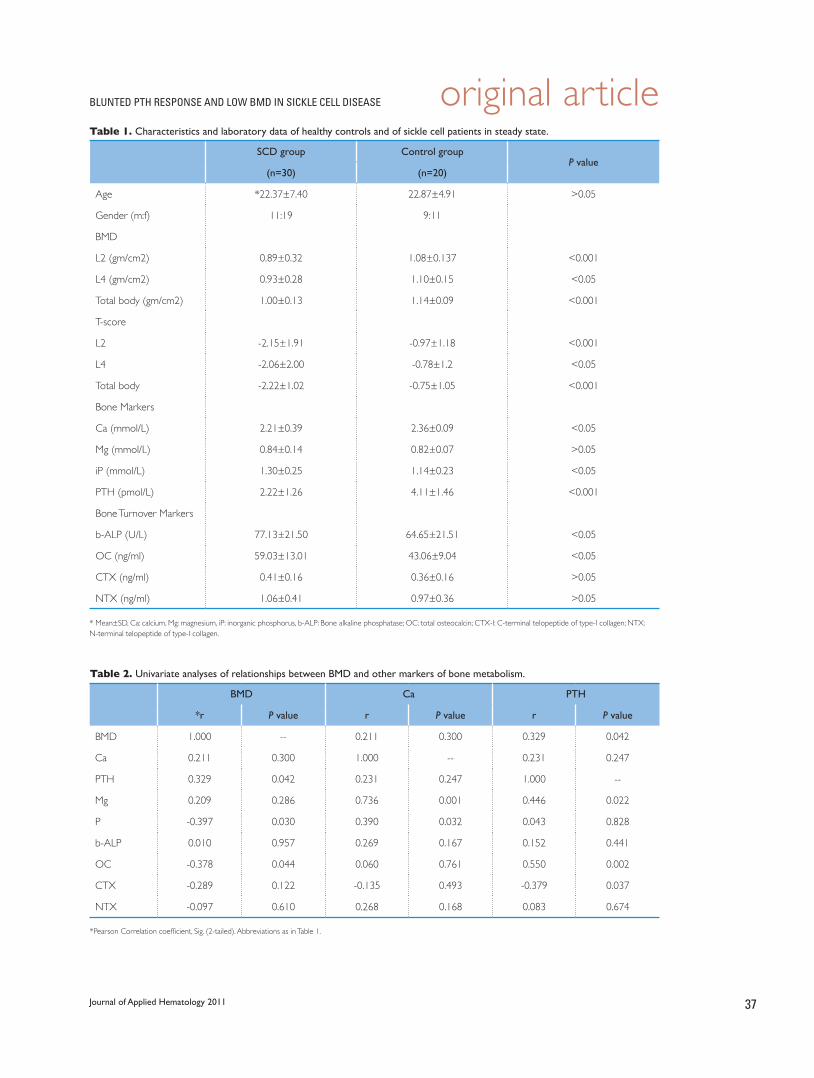
original articleBlunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 2011 37
table 1. Characteristics and laboratory data of healthy controls and of sickle cell patients in steady state.
SCD group Control groupP value
(n=30) (n=20)
Age *22.37±7.40 22.87±4.91 >0.05
Gender (m:f) 11:19 9:11
BMD
L2 (gm/cm2) 0.89±0.32 1.08±0.137 <0.001
L4 (gm/cm2) 0.93±0.28 1.10±0.15 <0.05
Total body (gm/cm2) 1.00±0.13 1.14±0.09 <0.001
T-score
L2 -2.15±1.91 -0.97±1.18 <0.001
L4 -2.06±2.00 -0.78±1.2 <0.05
Total body -2.22±1.02 -0.75±1.05 <0.001
Bone Markers
Ca (mmol/L) 2.21±0.39 2.36±0.09 <0.05
Mg (mmol/L) 0.84±0.14 0.82±0.07 >0.05
iP (mmol/L) 1.30±0.25 1.14±0.23 <0.05
PTH (pmol/L) 2.22±1.26 4.11±1.46 <0.001
Bone Turnover Markers
b-ALP (U/L) 77.13±21.50 64.65±21.51 <0.05
OC (ng/ml) 59.03±13.01 43.06±9.04 <0.05
CTX (ng/ml) 0.41±0.16 0.36±0.16 >0.05
NTX (ng/ml) 1.06±0.41 0.97±0.36 >0.05
* Mean±SD, Ca: calcium, Mg: magnesium, iP: inorganic phosphorus, b-ALP: Bone alkaline phosphatase; OC: total osteocalcin; CTX-I: C-terminal telopeptide of type-I collagen; NTX: N-terminal telopeptide of type-I collagen.
table 2. Univariate analyses of relationships between BMD and other markers of bone metabolism.
BMD Ca PTH
*r P value r P value r P value
BMD 1.000 -- 0.211 0.300 0.329 0.042
Ca 0.211 0.300 1.000 -- 0.231 0.247
PTH 0.329 0.042 0.231 0.247 1.000 --
Mg 0.209 0.286 0.736 0.001 0.446 0.022
P -0.397 0.030 0.390 0.032 0.043 0.828
b-ALP 0.010 0.957 0.269 0.167 0.152 0.441
OC -0.378 0.044 0.060 0.761 0.550 0.002
CTX -0.289 0.122 -0.135 0.493 -0.379 0.037
NTX -0.097 0.610 0.268 0.168 0.083 0.674
*Pearson Correlation coefficient, Sig. (2-tailed). Abbreviations as in Table 1.

original article Blunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 201138
table 3. Multivariate analysis of relationships between either BMD or PTH with other studied parameters.
aDependent: BMD bDependent: PTH
Beta coefficient P value Beta coefficient P value
Bone Mineral Density (BMD) 0.145 0.373
Parathyroid hormone (PTH) 0.413 0.04
Calcium (Ca) -0.055 0.792 -0.062 0.784
Magnesium (Mg) 0.161 0.457 0.397 0.012
Phosphorous (P) -0.288 0.133 -0.123 0.407
Osteocalcin (OC) -0.123 0.601 0.403 0.013
Bone specific alkaline phosphatase (b-ALP) -0.026 0.895 0.013 0.94
C-terminal telopeptides of type 1 collagen (CTX) -0.131 0.553 -0.399 0.013
N-terminal telopeptide of type 1 collagen (NTX) -0.13 0.508 0.039 0.806
Age 0.079 0.662 -0.016 0.918
Beta-coefficient: standardized regression coefficient representing the independent correlation between the respective variable and the dependent variable (after controlling for all other independent variables studied).
aDependent Variable: BMD, Predictor in the Model: PTH. bDependent Variable: PTH, Predictors in the Model: osteocalcin, magnesium, CTX.
On the other hand, our finding of decreased PTH in SCD contradicts the results of Mohammed et al.30 These conflicting data may be a result of subject se-lection. In his study, Mohammed et al noticed that PTH was significantly higher in the patients, with 31% having values above the normal range. In our study, PTH levels were within the low average to bor-derline ranges (2.2±1.26 pmol/L), with 30% of pa-tients showing PTH concentrations below the nor-mal range (1.6–6.9 pmol/L). Previous studies have attributed the reduced PTH levels in patients to vari-ous factors, including different types of hemoglobin-opathies and iron overload, as secondary to repeated blood transfusions. Iron overload was found to exert oxidative stress and to result in signs of toxicity and dysfunction for the vast majority of endocrine glands, including the parathyroid gland.32,33 Although most of our patients had received frequent blood transfusions as indicated in their medical records, we did not evalu-ate this possibility, as the assessment of iron overload constitutes a diagnostic challenge owing to the unreli-ability of serum ferritin levels and the risks associated with liver biopsy in patients with SCD. In addition, studies involving SCD have not shown any agreement on the relationship between ferritin and BMD.4,34
PTH deficiency may occur when parathyroid gland
tissue has been traumatized or its blood supply has been interrupted. Other causes include immune-me-diated destruction, congenital hypoplasia or aplasia, and end-organ resistance.35 Blunted PTH response to the correction in reduced calcium levels, acting secondary to magnesium deficiency, may be another possible explanation. In osteoporotic patients, it was found that hypomagnesemia is linked to vitamin D deficiency and can blunt the PTH response.36 In our study, although there was no significant difference in serum magnesium concentrations between SCD pa-tients and healthy controls, 30% of the SCD patients showed magnesium concentrations lower than the normal range (0.7–1.0 mmol/L). Moreover, magne-sium showed a significantly positive correlation with PTH (r=0.436, P<0.02) and Ca (r=0.329, P<0.05). This result is in agreement with that of Olukoga and coworkers, who reported low plasma magnesium lev-els in SCD.37 In addition, low PTH levels have been reported by other investigators studying impaired parathyroid gland function in magnesium deficien-cy.38,39
PTH has anabolic actions on bones as it acts on the proliferative capacity of osteoblastic cells.19,20 Osteoblasts were found to not only play an important role in bone formation, but also to stimulate osteo-

original articleBlunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 2011 39
clastic bone resorption under the action of PTH.40 Bone remodeling in healthy adults is based on the bal-anced actions between osteoclasts resorbing old bones and osteoblasts forming new ones.41 An imbalance in this process, which occurs whenever bone resorption exceeds bone formation, causes osteoporosis.42 To validate these data in our study, we measured bone-specific alkaline phosphatase (b-ALP) and OC, pheno-typic markers for osteoblasts in the early stage of dif-ferentiation and terminally differentiated osteoblasts, respectively.43 It was also found that both b-ALB and OC were significantly increased in the SCD group compared with the control (P<0.05). These findings are in agreement with previous studies.5,44 In addition, Wong et al45 found that OC was negatively correlated with cortical BMD.45 Our result is in line with this study, since OC, but not b-ALP, showed a significant but inverse correlation with BMD (r=–0.348, P<0.05). Furthermore, PTH correlated positively with OC (r=0.550, P= 0.002), but not with b-ALP.
The bone resorption markers CTX and NTX, which are released during the bone resorption pro-cess, were found to be increased in the patient group compared to the control group; however, none of these markers disclosed a statistically significant dif-ference between the 2 groups. These elevations in the bone turnover markers in our patients lead us to suggest that the overall bone turnover is increased in SCD as a compensatory reaction to low BMD in pa-tients with SCD.
In conclusion, the current findings demonstrate that low BMD is prevalent in SCD and that blunted PTH response to reduced total calcium levels and ac-celerated bone turnover may be the underlying mech-anisms. Moreover, our study points out a characteris-tic profile of SCD and stresses the great importance of monitoring serum magnesium concentrations and investigating the causes of its deficiency as a possible pathophysiological etiology of blunted PTH response and low BMD in SCD.

original article Blunted PtH resPonse and low BMd in sickle cell disease
Journal of Applied Hematology 201140
1. Huo, M. H., et al. (1990). Ò Orthopaedic manifestations of sickle-cell disease.Ó Yale J Biol Med 63(3): 195-207.2. Cordner, S. and K. De Ceulaer (2003). Ò Musculoskeletal manifestations of hemoglobinopathies.Ó Curr Opin Rheumatol 15(1): 44-47.3. Miller, R. G., et al. (2006). “High prevalence and correlates of low bone mineral density in young adults with sickle cell disease.” Am J Hematol 81(4): 236-241.4. Sarrai, M., et al. (2007). “Bone mass density in adults with sickle cell disease.” Br J Haematol 136(4): 666-672.5. Adewoye, A. H., et al. (2008). “Sickle cell bone disease: response to vitamin D and calcium.Ó Am J Hematol 83(4): 271-274.6. Legrand, E., et al. (2000). Ò Trabecular bone microarchitecture, bone mineral density, and vertebral fractures in male osteoporosis.Ó J Bone Miner Res 15(1): 13-19.7. Vichinsky, E. P. (1998). Ò The morbidity of bone disease in thalassemia.Ó Ann N Y Acad Sci 850: 344-348.8. Wingate, J., et al. (1996). Ò Osteonecrosis of the humeral head in sickle cell disease.Ó J South Orthop Assoc 5(2): 101-107.9. Adekile, A. D., et al. (2001). “Avascular necrosis of the hip in children with sickle cell disease and high Hb F: magnetic resonance imaging findings and influ-ence of alpha-thalassemia trait.Ó Acta Haematol 105(1): 27-31.10. Weinstein, R. S., et al. (2000). Ò Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip.Ó J Clin Endocrinol Metab 85(8): 2907-2912.11. Gangji, V., et al. (2003). Ò Abnormalities in the replicative capacity of osteo-blastic cells in the proximal femur of patients with osteonecrosis of the femoral head.Ó J Rheumatol 30(2): 348-351.12. Hernigou, P., et al. (1993). Ò Avascular necrosis of the femoral head in sickle-cell disease. Treatment of collapse by the injection of acrylic cement.Ó J Bone Joint Surg Br 75(6): 875-880.13. Al-Awamy, B., et al. (1986). “Pathological fracture of vertebral column in association with sickle cell anemia in Saudi Arabia.” Trop Geogr Med 38(4): 421-424.14. Omojola, M. F., et al. (1993). Ò Bone changes in sickle cell anaemia.Ó East Afr Med J 70(3): 154-158.15. Almeida, A. and I. Roberts (2005). Ò Bone involvement in sickle cell disease.Ó Br J Haematol 129(4): 482-490.16. Friedman, A. W. (2006). Ò Important determinants of bone strength: beyond bone mineral density.Ó J Clin Rheumatol 12(2): 70-77.17. Bonnick, S. L. and L. Shulman (2006). Ò Monitoring osteoporosis therapy: bone mineral density, bone turnover markers, or both?Ó Am J Med 119(4 Suppl 1): S25-31.18. Garnero, P. (2008). Ò Biomarkers for osteoporosis management: utility in di-agnosis, fracture risk prediction and therapy monitoring.Ó Mol Diagn Ther 12(3): 157-170.19. Cornish, J., et al. (1999). Ò Stimulation of osteoblast proliferation by C-terminal fragments of parathyroid hormone-related protein.Ó J Bone Miner Res 14(6): 915-922.20. Sowa, H., et al. (2003). “Parathyroid hormone-Smad3 axis exerts anti-apop-totic action and augments anabolic action of transforming growth factor beta in osteoblasts.Ó J Biol Chem 278(52): 52240-52252.21. Miki, T., et al. (2004). “Effect and safety of intermittent weekly administration of human parathyroid hormone 1-34 in patients with primary osteoporosis evaluated by histomorphometry and microstructural analysis of iliac trabecular bone before and after 1 year of treatment.Ó J Bone Miner Metab 22(6): 569-576.22. Kirk, J. K., et al. (2002). Ò Use of a peripheral dexa measurement for osteopo-rosis screening.Ó Fam Med 34(3): 201-205.23. Cuka, S., et al. (2001). Ò Evaluation of the Dade Behring Dimension RxL clini-
cal chemistry analyzer.” Clin Lab 47(1-2): 35-40.24. Kanis, J. A. (1994). Ò Assessment of fracture risk and its application to screen-ing for postmenopausal osteoporosis: synopsis of a WHO report. WHO Study Group.Ó Osteoporos Int 4(6): 368-381.25. Platt, O. S. (1994). Ò Easing the suffering caused by sickle cell disease.Ó N Engl J Med 330(11): 783-784.26. Nelson, D. A., et al. (2003). Ò Trabecular and integral bone density in adults with sickle cell disease.” J Clin Densitom 6(2): 125-129.27. Kanis, J. A., et al. (2006). Ò The use of multiple sites for the diagnosis of osteo-porosis.Ó Osteoporos Int 17(4): 527-534.28. Lal, A., et al. (2006). “Bone mineral density in children with sickle cell ane-mia.Ó Pediatr Blood Cancer 47(7): 901-906.29. Faulkner, K. G. and E. Orwoll (2002). “Implications in the use of T-scores for the diagnosis of osteoporosis in men.Ó J Clin Densitom 5(1): 87-93.30. Mohammed, S., et al. (1993). Ò Serum calcium, parathyroid hormone, and vitamin D status in children and young adults with sickle cell disease.” Ann Clin Biochem 30(Pt 1): 45-51.31. van der Dijs, F. P., et al. (1997). Ò Serum calcium and vitamin D status of pa-tients with sickle cell disease in Curacao.” Ann Clin Biochem 34(Pt 2): 170-172.32. Phillips, G., Jr., et al. (1992). “Hypothyroidism in adults with sickle cell anemia.” Am J Med 92(5): 567-570.33. Walter, P. B., et al. (2006). Ò Oxidative stress and inflammation in iron-over-loaded patients with beta-thalassaemia or sickle cell disease.” Br J Haematol 135(2): 254-263.34. Soliman, A., et al. (2008). “An adolescent boy with thalassemia major presenting with bone pain, numbness, tetanic contractions and growth and pubertal delay: panhypopituitarism and combined vitamin D and parathyroid defects.Ó Pediatr Endocrinol Rev 6(Suppl 1): 155-157.35. Shoback, D. (2008). Ò Clinical practice. Hypoparathyroidism.Ó N Engl J Med 359(4): 391-403.36. Sahota, O., et al. (2006). Ò Vitamin D insufficiency and the blunted PTH re-sponse in established osteoporosis: the role of magnesium deficiency.Ó Osteo-poros Int 17(7):1013-1021.37. Olukoga, A. O., et al. (1990). Ò Erythrocyte and plasma magnesium in sickle-cell anaemia.Ó East Afr Med J 67(5): 348-354.38. Anast, C. S., et al. (1972). Ò Evidence for parathyroid failure in magnesium deficiency.Ó Science 177(4049): 606-608.39. Suh, S. M., et al. (1973). Ò Pathogenesis of hypocalcemia in primary hypomag-nesemia: normal end-organ responsiveness to parathyroid hormone, impaired parathyroid gland function.Ó J Clin Invest 52(1): 153-160.40. McSheehy, P. M. and T. J. Chambers (1986). Ò Osteoblastic cells mediate os-teoclastic responsiveness to parathyroid hormone.Ó Endocrinology 118(2): 824-828.41. Manolagas, S. C. (2000). Ò Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteopo-rosis.Ó Endocr Rev 21(2): 115-137.42. Arlot, M., et al. (1984). Ò Impaired osteoblast function in osteoporosis: com-parison between calcium balance and dynamic histomorphometry.” Br Med J (Clin Res Ed) 289(6444): 517-520.43. Nakashima, K. and B. de Crombrugghe (2003). Ò Transcriptional mechanisms in osteoblast differentiation and bone formation.Ó Trends Genet 19(8): 458-466.44. Chapelon, E., et al. (2009). Ò Osteopenia and vitamin D deficiency in children with sickle cell disease.” Eur J Haematol 83(6): 572-578.45. Wang, Q., et al. (2006). Ò Differential effects of sex hormones on peri- and endocortical bone surfaces in pubertal girls.Ó J Clin Endocrinol Metab 91(1): 277-282.
References

test validation
Journal of Applied Hematology 2011 41
Heparin is an important anticoagulant drug widely used to treat and prevent thrombo-embolism. Heparin-induced thrombocytpe-
nia (HIT) is a major complication of heparin therapy, with an incidence of 1–5% in patients treated using heparin.1 There are 2 types of HIT: type 1 and type 2. Type 1 presents within the first 2 days after heparin exposure, and the platelet count normalizes with con-tinued heparin therapy; type I HIT is a non-immune disorder caused by the direct effect of heparin on platelet activation. Type 2 HIT is an immune-mediated
Validation of a monospecific enzyme-linked immunosorbent assay as a screening test for heparin-induced thrombocytopenia and its comparison with immunological and functional assays
Rasheed Nasr, Randa Al Nounou, Mansoor Ahmed, Tarek Owaidah
From the Department of Pathology and Laboratory Medicine, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia
Correspondence:Tarek M. Owaidah, MDDepartment of Pathology and Laboratory Medicine (MBC 10)King Faisal Specialist Hospital and Research Centre and Center of Excellence for Thrombosis and Homeostasis, King Saud of University PO Box 3354, Riyadh 11211, Saudi ArabiaT: +966-1-442-4328; F: [email protected]
Heparin-induced thrombocytopenia (HIT) is an adverse drug reaction caused
by antibodies to heparin/platelet factor 4 (PF4) complexes and results in the
prothrombotic state. Early diagnosis is important for clinical decision making.
Various laboratory tests have been examined as a tool for confirming the suspi-
cion of HIT. The monospecific PF4 immunoglobulin G (IgG) assay is a qualitative
screening assay for detecting heparin-associated IgG antibodies in human serum.
Of 100 patients with clinical suspicion of HIT, the blood samples of 20 patients
who had positive clinical pretest probability scores were tested at our institute
for particle gel immunoassay (PaGIA; Diamed ID; Switzerland), enzyme-linked
immunosorbent assay (ELISA) (HPF4 ELISA; Stago, Asnières sur Seine, France),
and the new monospecific ELISA (IgG) from GTI, USA. The optical density (OD)
of the color that developed was measured using a spectrophotometer. These
samples were collected from the patients at the same time as were samples
that were sent to a reference laboratory for a gold standard test (serotonin-
release test). Of the 20 samples tested, 1 was clearly positive and another 1 had
borderline positivity in the serotonin-release test. The results of all 4 tests were
in agreement for all samples, except for the sample with borderline positivity,
which was missed by ELISA (STAGO). The frequency of samples positive for HIT
antibodies was 10%. Although this study included a low number of cases with
clinical suspicion of HIT, our results confirm the good sensitivity and specificity
of the monospecific ELISA that could be more sensitive than polyspecific ELISA.
Keywords: Heparin, Thrombocytopenia, ELISA
disorder that typically occurs 4–10 days after heparin exposure and has a more serious effect than does Type 1 HIT, with grave thrombotic complications.2 Because of the high morbidity and mortality associated with HIT II, an immediate change of the heparin deriva-tive is indicated. This change is sometimes difficult because of the absence of available alternatives or the occurrence of clinical conditions, such as renal failure, that may compromise the use of alternative antico-agulation drugs. Laboratory data supporting the clini-cal suspicion are very helpful for making this deci-
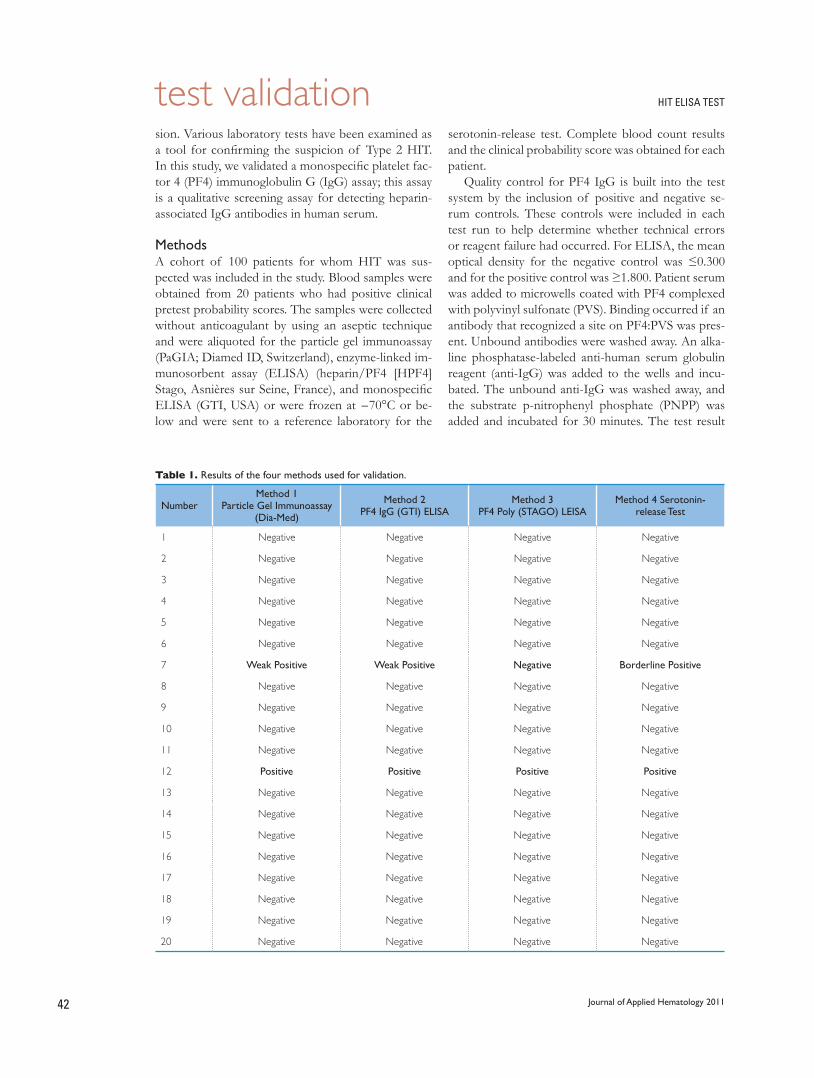
test validation HIT ELISA TEST
Journal of Applied Hematology 201142
sion. Various laboratory tests have been examined as a tool for confirming the suspicion of Type 2 HIT. In this study, we validated a monospecific platelet fac-tor 4 (PF4) immunoglobulin G (IgG) assay; this assay is a qualitative screening assay for detecting heparin-associated IgG antibodies in human serum.
MethodsA cohort of 100 patients for whom HIT was sus-pected was included in the study. Blood samples were obtained from 20 patients who had positive clinical pretest probability scores. The samples were collected without anticoagulant by using an aseptic technique and were aliquoted for the particle gel immunoassay (PaGIA; Diamed ID, Switzerland), enzyme-linked im-munosorbent assay (ELISA) (heparin/PF4 [HPF4]Stago, Asnières sur Seine, France), and monospecific ELISA (GTI, USA) or were frozen at −70°C or be-low and were sent to a reference laboratory for the
serotonin-release test. Complete blood count results and the clinical probability score was obtained for each patient.
Quality control for PF4 IgG is built into the test system by the inclusion of positive and negative se-rum controls. These controls were included in each test run to help determine whether technical errors or reagent failure had occurred. For ELISA, the mean optical density for the negative control was ≤0.300 and for the positive control was ≥1.800. Patient serum was added to microwells coated with PF4 complexed with polyvinyl sulfonate (PVS). Binding occurred if an antibody that recognized a site on PF4:PVS was pres-ent. Unbound antibodies were washed away. An alka-line phosphatase-labeled anti-human serum globulin reagent (anti-IgG) was added to the wells and incu-bated. The unbound anti-IgG was washed away, and the substrate p-nitrophenyl phosphate (PNPP) was added and incubated for 30 minutes. The test result
Table 1. Results of the four methods used for validation.
NumberMethod 1
Particle Gel Immunoassay (Dia-Med)
Method 2PF4 IgG (GTI) ELISA
Method 3 PF4 Poly (STAGO) LEISA
Method 4 Serotonin-release Test
1 Negative Negative Negative Negative
2 Negative Negative Negative Negative
3 Negative Negative Negative Negative
4 Negative Negative Negative Negative
5 Negative Negative Negative Negative
6 Negative Negative Negative Negative
7 Weak Positive Weak Positive Negative Borderline Positive
8 Negative Negative Negative Negative
9 Negative Negative Negative Negative
10 Negative Negative Negative Negative
11 Negative Negative Negative Negative
12 Positive Positive Positive Positive
13 Negative Negative Negative Negative
14 Negative Negative Negative Negative
15 Negative Negative Negative Negative
16 Negative Negative Negative Negative
17 Negative Negative Negative Negative
18 Negative Negative Negative Negative
19 Negative Negative Negative Negative
20 Negative Negative Negative Negative

test validationHIT ELISA TEST
Journal of Applied Hematology 2011 43
was reported as positive if the OD value was equal to or greater than 0.400.
The test has the following limitations: Erroneous results may be obtained in the case of bacterial con-tamination of test materials, inadequate incubation pe-riods, inadequate washing and decanting of test wells, exposure of substrate to stray light, omission of test reagents, exposure to temperatures higher or lower than those prescribed, or omission of steps. The pres-ence of immune complexes or other immunoglobulin aggregates in the patient sample may cause increased nonspecific binding and lead to false-positive results in
this assay. The results of this assay should not be used as the sole basis for a clinical decision but should be correlated with clinical conditions.
ResultsOf the 20 samples tested, 1 was clearly positive and another one had borderline positivity in the serotonin-release test. The results of all 4 tests were in agreement for all samples—except for the sample with borderline positivity, which was missed by ELISA (STAGO). The frequency of samples positive for HIT antibodies was 10%. The results have been provided in Table 1.
1. Visentin GP, Aster RH. Heparin-induced thrombocytopenia and thrombo-sis. Curr Opin Hematol. 1995;2:351-7.2. Chong BH. Heparin-induced thrombocytopenia. Blood Re. 1988;2:108-14.3. Warketin TE, Chong BH, Greinacher A. Heparin-induced thrombocyto-penia: towards consensus. Thromb Haemost. 1998;79:1-7.4. Chong BH, Burgess J, Ismail F. The clinical usefulness of the platelet aggre-gation test for the diagnosis of heparin-induced thrombocytopenia. Thromb Haemost. 1993;69:344-50.5. Sheridan D, Carter C, Kelton JG. A diagnostic test for heparin-induced thrombocytopenia. Blood. 1986;67:27-30.6. Amiral J, Bridey F, Dryfus M, Vissac AM, Fressinaud E, Wolf M, et al. Platelet factor 4 complexed to heparin is the target of antibodies generated in hep-arin-induced thrombocytopenia [letter]. Thromb Haemost. 1992;68:95-6.7. Visentin GP, Ford SE, Scott JP, Aster RH. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet fac-tor 4 complexed with heparin or bound to endothelial cells. J Clin Invest.
1994;93: 81-8.8. Greinacher A, Potzch B, Amiral J, Dummel V, Eichner A, Mueller-Eckhardt C. Heparin-induced thrombocytopenia: isolation of the antibody and char-acterization of a multimolecular PF-4 heparin complex as the major anti-gen. Thromb Haemost. 1994;71:247-51.9. Visentin GP, Moghaddam M, Collins JL, McFarland JG, Aster RH. Antibod-ies associated with heparin-induced thrombocytopenia (HIT) report con-formational changes in platelet factor 4 (PF4) induced by linear, polyanionic compounds. Blood. 1997; 90 Suppl 1:460a.10. Collins JL, Aster RH, Moghaddam M, Piotrwoski MA, Strauss TR, McFar-land JG. Diagnostic testing for heparin-induced thrombocytopenia (HIT): an enhanced platelet factor 4 complex enzyme linked immunosorbent assay (PFA ELISA). Blood. 1997;90 Suppl 1:461a.11. Visentin GP, et al. Heparin is not required for the detection of antibodies associated with heparin-induced thrombocytopenia/thrombosis. J Lab Clin Med. 2011 July;138:22-31.12. Aster RH. Unpublished Observations.
References

image of the issue
Journal of Appplied Hematology 201144
A 20-year-old man presented with a 2-month history of fever, epistaxis, weight loss, and progressive hearing loss. His initial workup
showed pancytopenia. Physical examination revealed no abnormalities ex-
cept for polydactyly of the left hand. His complete blood count (CBC) showed a white blood cell (WBC) count of 1.46×109/L (normal 3.90–11.00×109/L); hemoglobin, 86 g/L; platelet, 62 (150–350×109/L); polymorph, 1%; lymphocytes, 88%; blasts, 2%; mono-cytes, 4%; atypical lymphocytes, 5%; and basophil, 1%. Bone marrow analysis showed 50% of blasts ex-pressed CD45, CD5, and cytoplasmic CD3, CD2 with only partial expression of CD7, CD11b, which was consistent with pre-T-cell ALL. Chromosomal break-age study confirmed Fanconi anemia.
The patient was treated for acute lymphoblastic leukemia (ALL) with induction chemotherapy at 75% dose reduction. His disease remained active as expect-ed, but he developed severe liver impairment because of the extreme sensitivity and impaired tissue repair in Fanconi anemia patients. In spite of trials with repeat-ed reduced doses of chemotherapy and steroids, the condition of the patient deteriorated and he eventu-ally died because of active leukemia. This case demon-strates the rare form of transformation and progres-sion of Fanconi anemia. Typically, Fanconi anemia pa-tients progresses to myelodysplastic syndrome, which can then transform to acute myeloid leukemia.
T-cell acute lymphoblastic leukemia in a patient with Fanconi anemia
Hazzaa Al-Zahrani,a Tarek Owaidahb
From the aDepartment of Oncology and bDepart-ment of Pathology and Laboratory Medicine King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
Correspondence:Tarek M. Owaidah, MDDepartment of Pathology and Laboratory Medicine (MBC 10)King Faisal Specialist Hospital and Research Centre and Center of Excellence for Thrombosis and Homeostasis, King Saud of University PO Box 3354, Riyadh 11211, Saudi ArabiaT: +966-1-442-4328; F: [email protected]
Figure 1. Left hand showing polydectaly.
Figure 2. Bone marrow biopsy showing hypocellularity and collection of blasts.

image of the issue
Journal of Applied Hematology 2011 45
1. Daisuke Suzuki et al. Precursor-T Lymphoblastic Lymphoma after Unrelated Bone Marrow Transplantation in a Patient with Fanconi Anemia. Journal of Pediatric Hematology/Oncology Issue: Volume 33(1), January 2011, p 22Ð 24.
Reference
This case also demonstrates the need for early diagnosis of Fanconi anemia and offering stem cell transplantation in order to avoid progression of the
condition to such devastating outcome. These patients require chemotherapy dose adjustment in order to avoid organ toxicity.

case report
Journal of Appplied Hematology 201146
Postpartum non-diarrhea-associated HUS is an unusual complication of pregnancy. It oc-curs as a single episode, either immediately or
a few weeks after delivery. The disease is severe and is associated with microangiopathic hemolytic anemia (MAHA), thrombocytopenia, hypertension, and acute renal failure. It usually occurs in primigravida but has also been reported in multiparous women with a mean age of 27 years. Various triggering factors were im-plicated in causing postpartum HUS, especially pre-eclampsia, which was previously identified in 15% of patients with postpartum non-diarrhea-associated HUS.1
Early recognition is the key for successful treat-ment of this disease; however, the treatment of post-partum HUS remains controversial.
Case ReportA 22-year-old Saudi gravida 3, para 2 presented in labor at 39 weeks of gestation. She had no previous prenatal visits, and there was no history of bleeding, fever, or diarrhea. She had no medical history of hy-pertension. Physical examination was normal except for high blood pressure of 156/103 and petechiae of upper and lower limb.
The patient was diagnosed with mild preeclampsia and was found to have had intrauterine fetal death for
Postpartum hemolytic uremic syndrome: A case report and review of the literatureGhuzayel Al Dawsari, Abdul Rahman Jazieh
From the Department of Oncology, King Saud bin Abdulaziz University for Health Sciences/National Guard Health Affairs KSAU-HS/NGHA, Riyadh, KSA
Correspondence:Ghuzayel Al Dawsari, MDConsultant Adult Hematology, Assistant ProfessorDepartment of Oncology, KSAU-HS/NGHAP.O. Box 22490 Riyadh [email protected]
Postpartum hemolytic uremic syndrome (HUS) is a well-documented rare com-
plication after pregnancy with various triggering factors. We present the case
of a 22-year-old Saudi female admitted in labor at 39 weeks of gestation with
mild preeclampsia and a probable intrauterine fetal death. The patient underwent
spontaneous vaginal delivery and was found to have abruptio placenta. Within
48 hours, she developed microangiopathic hemolytic anemia, thrombocytopenia,
and acute renal failure. Plasma exchange using fresh frozen plasma was initi-
ated immediately, and she recovered completely after 11 sessions[Author4]. This
manuscript describes the details of this case in addition to a literature review of
the reported cases of postpartum HUS.
KEYWORDS: Postpartum hemolytic uremic syndrome (HUS), Abruptio pla-
centa, plasma exchange (PE).
which she underwent spontaneous normal vaginal de-livery that revealed an abruptio placenta. The delivery was complicated by immediate vaginal bleeding, and she received 4 units of packed red blood cell as her hemoglobin level dropped to 60 g/dL from a base line of 87 g/dL.
Forty-eight hours after delivery, laboratory inves-tigations revealed a leukocyte count of 14.9×109/L; hemoglobin, 76 g/dL; hematocrit, 0.225, mean cor-puscular volume (MCV), 70 fL, and random distribu-tion of red cell width (RDW) 20%; a drop in platelet count to 36×109/L from the base line of 161×109/L was observed. The renal function had deteriorated over this period with creatinine level increasing from base line 55 µmol/L to 350 µmol/L within 24 hours. Other serum electrolyte levels were normal. Lactate dehydrogenase (LDH) at presentation was 662 U/L and had increased to 2756 U/L within 48 hours.
Peripheral blood smear showed a significant num-ber of schistocytes with normal prothrombin time and activated partial thromboplastin time. Coombs test result was negative. Liver function test results showed a transient elevated alanine aminotransferase (ALT) level of 69 U/L while aspartate aminotransfer-ase (AST) and indirect bilirubin levels were normal. Other laboratory investigations, including antinuclear antibody (ANA), anti-DNA, and complement C3 and

case reportpostpartum hemolytic uremic syndrome
Journal of Applied Hematology 2011 47
C4 levels were within the normal limit. The results of antiphospholipid antibodies (APL) and lupus antico-agulants were negative. Hematology and nephrology teams were involved in the management and differ-ential diagnosis, which revealed anemia secondary to bleeding along with coexisting iron-deficiency anemia; dilutional thrombocytopenia; preeclampsia; hemolysis, elevated liver enzyme, and low platelet (HELLP) syn-drome; thrombotic thrombocytopenic purpura (TTP); and postpartum HUS.
The diagnosis of postpartum HUS was established with the exclusion of all the other differential diag-noses. Fresh frozen plasma (FFP), 15 mL/kg body weight of the patient, was given initially until plasma exchange (PE) started. PE was performed daily, and within 72 hours, the patient started responding as her hemoglobin count, platelet count as well as the LDH level improved.
After 11 sessions, her CBC and LDH normalized and her creatinine improved to a level of 99 umol/l and patient was discharged home on aspirin 81mg daily. After one month, she was seen in the outpatient clinic and she was in good health and her CBC, LDH, and creatinine levels were normal.
Postpartum Hemolytic Uremic Syndrome literature reviews:HUS and TTP are thrombotic microangiopathies (TMA) that are characterized by systemic and/or in-trarenal aggregation of platelets, thrombocytopenia, and mechanical injury to erythrocytes (schistocyte). There is usually some degree of overlap in the clinical presentation of both syndromes with a prominent re-nal involvement in HUS and a central nervous system involvement in TTP.2
HUS can be classified into diarrhea-associated HUS (D+HUS) and non-diarrhea-associated HUS (D−HUS).
The most common form of D+HUS is associated with infection by Escherichia coli and usually has an excellent prognosis in most cases, while D−HUS can be genetic, which is linked to mutation of gene factor H and/or gene membrane cofactor protein (MCP).3
Other D−HUS presentation could be secondary to connective tissue diseases such as systemic lupus erythematosus (SLE), scleroderma, malignant hyper-tension, radiation to the kidney, immunosuppressant therapy, pregnancy, and oral contraceptives. Usually, D−HUS has a worse prognosis compared to D+HUS, often resulting in uremia and/or death of the patient.4
The association of HUS with pregnancy is rare, but
a well documented event.Postpartum HUS is an even rarer complication of
pregnancy with unknown exact incidence but estimat-ed to be around 1/25,000 pregnancies.5
Robsen et al., first described postpartum HUS in 19686; and subsequently, different authors have re-ported few cases with similar presentation but differ-ent associated conditions, treatment approaches, and variable outcomes.
The reported cases and other relevant publications in the literature were reviewed and the salient findings are summarized in this manuscript. (Table 1).
The exact pathogenesis of postpartum HUS is unknown, but the disease can be triggered after an uncomplicated pregnancy as well as an abruptio pla-centa, spontaneous abortions, presence of circulating anticardiolipin antibody and lupus anticoagulant, and pregnancy-induced hypertension.7-9
The association of postpartum HUS with the aforementioned conditions indicates that increased concentration of procoagulant factor and decreased fibrinolytic activity, loss of endothelial cell thrombo-modulation, and decreased activity of von Willebrand factor-cleaving metalloprotease have major contribu-tions to the development of postpartum HUS.10
Postpartum HUS has also been associated with other deficiencies, including abnormalities of com-plement factor Hvi and antibodies produced in re-sponse to verocytotoxin-producing E. coli O157:H7 infection.12
In 1994, Sabai et al. postulated that the primary defect in postpartum HUS is an unidentified platelet-aggregating factor that causes deposition of micro-thrombin in the vessel wall causing occlusions in the microvasculature of the kidney, resulting in acute re-nal failure.13,14
Recently, several reports have pointed out the re-lationship of thrombotic microangiopathic hemolytic anemia (TMHA) with the presence of APL.15,16
SLE was the first autoimmune disease in which the association of TMHA with APL was recognized.17, 18
G. Espinosa reviewed 47 patients with TMHA associated with APL: HUS was the most common clinical presentation (26%), occurring postpartum pe-riod in 3 patients, followed by catastrophic antiphos-pholipid syndrome (APS) (23%), acute renal failure (15%), malignant hypertension (13%), TTP (13%), and HELLP syndrome (4%).19
The diagnosis of postpartum HUS can be diffi-cult, and it requires a very high index of suspicion. The presentation can be subtle and careful evaluation of CBC, peripheral blood smear, renal function in

case report postpartum hemolytic uremic syndrome
Journal of Applied Hematology 201148
Table 1. Summary of the reported cases of post-partum hemolytic uremic syndrome (HUS).
Author No. of Patients
Onset after delivery Clinical Presentation Treatment Response Reference
Segonds 3 10, 17, and 24 weeks
MAHA, thrombocytopenia, ARF, hypertension Heparin + HD
1 complete recovery1 developed Malignant
hypertension andSlight improvement in
renal function after HDESRD on H.D.
28
Kniaz 1Immediate after
spontaneous abortion
Severe preeclampsia, MAHA, ARF, thrombocytopenia PE Complete recovery 9
Pajor 1
Immediate after placental
abruption (24 weeks of
gestation)
ARF, MAHA, thrombocytopenia, fever and hypertension FFP + HD Complete recovery
after 7 weeks 29
Jeng-Jong 1
Proceeded by acute
enterocolitis after normal delivery,
Typical picture of HUS FFP + Heparin + HD
Partial recovery, ESRD on HD 8
Shemin 3 Postpartum Typical picture of HUS PE + Prednisone Complete recovery 30
Takahashi 2 Postpartum After successful cesarean delivery FFP1 complete recovery,1 complicated by sub
arachnoid hemorrhage 31
Wu et al 1 After abruptio placenta Typical picture of HUS PE Complete recovery 20
Francisco 1
After emergency cesarean section due to abruptio
placenta
Typical picture of HUS and hypertension
FFP + HD + pulse methylprednisone
Complete recovery and persistent renal
impairment21
Lampinen K. 4 After emergency cesarean section
Typical picture of HUS and hypertension
2 PE onlyPE + HD + IV IGgPE + LWH + IV IGg + steroid
2 complete recovery after 1Ð2 weeks
Complete recovery after 5 weeks
Complete recovery after 4 weeks
32
Yu-Chieh 1
Under the indication of
previous cesarean section
After the fourth episode of postpartum hemorrhage,
Full picture of HUS
PE + steroid + HD Died 22
Mariarosaria 1 After cesarean section for twins Full picture of HUS PE
Complete recovery and persistent renal
impairment4
Al Sina 1 10 days postpartum
MAHA, thrombocytopenia, ARF and nephrotic syndrome PE
Refractory hypertension and
infectious complications.33
Fakhari 15 Postpartum period ARF, MAHA, thrombocytopenia
All PE1 PE + FFP
1 PE + IV IGg
1 complete recovery2 chronic renal
impairment12 patients with ESRD
34
MAHAÐm icroangiopathic hemolytic anemia; ARFÐa cute renal failure; PEÐp lasma exchange; FFPÐf resh frozen plasma; ESRDÐen d stage renal disease; HDÐh emodialysis; LWH-low molecular weight heparin; IV IG-
intravenous immunoglobulin

case reportpostpartum hemolytic uremic syndrome
Journal of Applied Hematology 2011 49
1. Weiner CP. Thrombotic microangiopathy in pregnancy and the postpar-tum period. Semin Hematol 1987;24:119-1292. Moake JL. Thrombotic microangiopathies. N Eng J Med 2002;347:589-6003. Caprioli J, Bettinglio P, Zipfel PF et al. The molecular basis of familial hemo-lytic uremic syndrome: Mutation analysis of factor H gene reveals a hot spot in the short consensus repeat 20. J Amer Soc Nephrol 2001;12:297-3074. Mariarosaria I, Patriziz S, Antonio D, Maria C, Luigi T, Michela M. A post-partum hemolytic-uremic-like-syndrome in patient with pre-eclampsia: De-scription of a clinical case. Transfusion and Apheresis Science 2006;34:11-145. Dashe JS, Ramin SM, Cunnigham FG. The long-term consequences of thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome)in pregnancy. Obstet Gyneacol 1998;91:662-6686. Robson JS, Martin AM, Ruckley VA, MacDonald MK. Irreversible postpar-tum renal failure: A new syndrome. QJMed 1968;37:4237. Ribeiro FM, Rocha E, Maccariello E, Caldas ML, Gomes MV, Lugon JR. Early gestational hemolytic uremic syndrome: Case report and review of litera-ture. Ren Fail 1997;19:475-4798. Huang JJ, Chen MW, Sung JM, Lan RR, Wang MC, Chen F. Postpartum haemolytic uraemic syndrome associated with antiphospholipid antibody. Nephrology dialysis transplantation 1998;13:182-1869. Kniaz D, Eisenberg GM, Elrad H, Johnson CA, Valaitis J, Bregman H. Postpar-tum hemolytic uremic syndrome associated with antiphospholipid antibod-ies: A case report and review of the literature. Amer J Nephrol 1992;12:126-13310. Mannuci PM, Canciani MT, Rossi E et al. Changes in health and dis-ease of the metalloprotease that cleaves Von Willebrand factor. Blood 2001;98:2730-273511. Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tiele-mans CL, Goodship TH. Factor H mutations in hemolytic uremic syndrome cluster in exons 18-20, a domain important for host cell recognition. Am J Hum Genet 2001;68:485-49012. Chart H, Perry NT, Cheasty T, Wright PA. The kinetics of antibody pro-duction to antigen of Escherichia coli O157 in pregnant women with hae-molytic ureamic syndrome. J Med Microbiol 2002;51:522-52513. Sibai BM, Kustermam L, Velasco J. Current understanding of severe pre-eclampsia, pregnancy associated hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, hemolysis, elevated liver enzymes, low platelet syndrome, and postpartum acute renal failure: Different clinical syndromes or just different names? Curr Opin Nephrol Hypertens 1994;3:436-44514. Furlan M, Lammle B. Aetiology and pathogenesis of thrombotic throm-
bocytopenic purpura and haemolytic uraemic syndrome: The role of von Willebrand factor-cleaving protease. Best Pract Res Clin Haematol 2001;14:437-45415. Durand JM, Lefevre P, Kaplanski G, Souberyarnd J. Thrombotic mi-croangiopathy and antiphospholipid antibody syndrome. J Rheumatol 1991;18:1916-191816. Hess DC, Sethi K, Awad E. Thrombotic thrombocytopenic purpura in systemic lupus erythematosus and antiphospholipid antibodies: Effective treatment with plasma exchange and immunosuppression. J Rheumatol 1992;19:1474-147817. Glueck HI, Kant KS, Weiss MA, Pollak VE, Miller MA, Coots M. Thrombosis in systemic lupus erythrematosus: Relation to the presence of circulating anticoagulants. Arch Intern Med 1985;145:1389-139518. Love PE, Santoro SA. Antiphospholipid antibodies: Anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and non-SLE dis-orders. Prevalence and clinical significance. Ann Intern Med. 1990;112:682-9819. Esponosa G, Bucciarelli S, Cervera R, Lozano M, Reeverter J-C, de la Red G, Gil V, lngelmo M, Font J, Asherson RA. Thrombotic microangiopathic haemolytic anaemia and antiphospholipid antibodies. Ann Rheum Dis 2004;63;730-73620. Wu VC, Lin SL, Tsai CC, Tien HF. Postpartum hemolytic uremic syn-drome following abruptio placenta: Report of a case. J Formos Med Assoc. 2002;101:868-87021. Francisco E, Anacleto, Christina L, Cifra, Joel S, Elises. Postpartum he-molytic uremic syndrome in a 17- year-old Filipina primigravida. Pediatric Nephrol 2003;18:1283-128522. Chen YC, Chen HS, Shen CJ, Chang HM, Tsai EM. Delayed postpartum hemorrhageÐ A rare clinical presentation of thrombotic thrombocytopenic purpura- hemolytic uremic syndrome: A case report. Kaohsiung J Med Sci 2005;2123. Baha M, Sibai. Imitator of severe pre-eclampsia. Seminars in perinatology. 2009;33:196-20524. First MR, Pollak VE. Pregnancy and renal disease. In: Diseases of the kidney, 4th ed. (Schrier RW, Gottschalk CW, Eds.) Little Brown, Boston, 1988, p 255025. Churg J, Goldstein MH, Bernstein J. Thrombotic microangiopathy includ-ing hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, and postpartum renal failure. In: Renal pathology with clinical and functional correlations, (Tischer CC, Brener BM, Eds.) Lippincott Williams and Wilkins, Philadelphia, 1989, p 109126. Esplin MS, Branch DW. Diagnosis and management of thrombotic micro-angiopathies during pregnancy. Clin Obset Gynecol 1999;42:360-365
References
patients with abruptio placenta,20, 21 and delayed post-partum hemorrhage should be considered.22
Acute renal failure in late pregnancy in association with MAHA and thrombocytopenia were observed in 3 main disease entities, which include postpartum HUS, TTP, and severe preeclampsia with HELLP syndrome.23
Patient with or possible APS and a repeated history of fetal loss are another group of patients where the diagnosis of postpartum HUS should be suspected.
The prognosis of postpartum HUS is generally poor.24, 25
Most women died, or survived with severely im-paired renal function, or progressed to end stage-re-nal disease. One study showed that there was at least one serious long-term sequela in 78% of survivors.
The disease recurred in 50% of survivors. Fetal loss rate was as high as 80%; 10% of cases showed complete recovery of renal function.
However, early diagnosis and appropriate treat-ment, may reduce maternal mortality by 90%.26
At present, PE is the most important treatment option for TTP/HUS that results in an improved out-come of 80–90%.27
It is clear from the literature that most patients that recovered from postpartum HUS were treated with either plasma infusion along with hemodialysis or PE.
The role of steroid, heparin or other therapies re-main unclear and should be considered only if the patient shows no response to PE.
SummaryPostpartum HUS is a rare but serious complication of pregnancy. It should be considered in certain com-plications such as abruptio placenta, fetal death, and APS. Careful monitoring of CBC in addition to other expressed features of the syndrome in the postpartum period will lead to early detection of HUS.
Immediate treatment with FFP and PE will im-prove maternal mortality outcome as seen in our patient.

case report postpartum hemolytic uremic syndrome
Journal of Applied Hematology 201150
27. Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in throm-botic thrombocytopenic purpura-hemolytic uremic syndrome. N Engl J Med 1991;325-40328. Segonds A, Louradour N, Suc JM, Orfila C. Postpartum hemolytic uremic syndrome: A study of three cases with a review of literature. Clin Nephrol 1979;12(5):229-24229. Pajor A, Hintalan A, Bakos L, Linter F. Postpartum hemolytic uremic syn-drome following placental abruption. Eur J Obstet Gynecol Reprod Biol 1993;49(3):201-20430. Shamin D, Dworkin LD. Clinical outcome in three patients with postpar-tum hemolytic uremic syndrome treated with frequent plasma exchange. Ther Apher 1998;2(1):43-48
31. Takahashi Y, Imai A, Hayasaki Y, Kawabata I, Tamaya T. Postpartum micran-giopathic hemolytic anemia: cases of successful and dismal outcome assisted with plasma therapy. Eur J Obstet Gynecol Reprod Biol 2000;89(2):213-21532. Lampinen K, Peltonen S, Pettila V, Kaaja R. Treatment of postpartum thrombotic microangiopathy with plasma exchange using cryosupernatant as replacement. Acta Obstet Gynecol Scand 2004,83(2):175-17933. Al sina Segui E, Martin Conde ML, Craver Hospital L, Fernandez Giraldez E. Postpartum hemolytic uremic syndrome: A rare entity and a treatment challenge. Nefrologia 2008;28(1):120-12134. Fakhouri F, Roumenina L, Provot F, Sallee M, Caillad S, Couzi L, Essig M, Ribes D, Dragon-Durey MA, Bridoux F, Rondeau E, Fremeaux-Bacchi V. Preg-nancy-associated hemolytic uremic syndrome. J Am Soc Nephrol 2010;4

letter to the editor
Journal of Applied Hematology 2011 51
The goals of Comprehensive Care of Hemophilia (CCH) are to deliver the optimal care to patients with bleeding disorders and
educate the patients and their families about the dis-ease and how to minimize or prevent complications. Hemophilia is a rare bleeding disorder. Patients with hemophilia can develop many systemic complications. Although hematology service is the primary service for care of these patients, Hemophilia patients can-not be adequately treated in the settings of general hematology due to its complexity. The concept of multidisciplinary team care had changed the natural history of this disorder. This concept had been rec-ognized by World Health Organization (WHO) and World Federation of Hemophilia (WFH).
There are many benefits expected from such pro-gram including:
1. Decrease mortality rate by receiving treatment through multidisciplinary team in Hemophilia Treatment Centre (HTC) by 70% compared to patient receiving treatment in general Hematology department.
2. Decrease hospitalization rate up to 40%. 3. Provide safe management of complicated
cases such as seroconvert patients to HIV or Hepatitis C through contaminated blood prod-ucts.
4. Multidisciplinary team can coordinate this man-agement with better out come in (HTC) which minimize the need for excessive factor replace-ment during repeated bleeding episodes or pro-cedures
It is well known that more than 90% of the cost in hemophilia management is actual cost of factor so proper supervision and coordination in (HTC) can lead to optimal use of replacement therapy and de-
Comprehensive Care of Hemophilia Nursing Prospective
Mahmoud I. Abu-Riash
crease cost of treatment. Many studies had shown that organized and coordinated comprehensive care is less costly.
There are many approaches for establishment of Multidisciplinary team of CCH but most cen-ters would have a comprehensive team that con-sists of: Pediatric Hematology, Adult Hematology, Physical Therapist, Social worker, Hemophilia Nurse Coordinator, Dentist, Orthopedic Surgeon, Infectious disease Pediatric Surgeon, laboratory tech-nician, Psychologist and Genetic Counselor
The Role of Hemophilia Nurse CoordinatorHemophilia nurse consider being the main person in the team and he/she plays many roles. He/she is the Resource person for patient & families, responsible for arrange CHC visits. He Receive & assess patient according to his/her current situation (emergency or regular appt.), insure that necessary & regular F/U blood tests for (Factor assay, virology, Inhibitor sta-tus) and X-rays results are ready with the patient with each visits. He/she communicates with patients be-fore appointment and help patient with their refer-rals to other hospital services. The hemophilia nurse assists physicians in collecting data regarding various aspect of hemophilia and ensuring that the Registry is up-to-date, and continuo to be available for physi-cians, patient & families. Patient, families and com-munity education is an important participation of the nurse in hemophilia care program. In program were home therapy is available, the nurse has to con-duct regular home visits to e emphasis on the therapy and advice for changes in the home settings accord-ing to the needs of the patient. The role of the nurse in hemophilia care will always be evolving and will always be a challenge and be rewarding.
1. Sohail, Muhammad Tariq and Lily Heijnen, eds. 2001. Comprehensive Haemophilia Care in Developing Countries. Montreal: World Federation
Reference
From Nursing Affairs, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia

pioneers in hematology
Journal of Appplied Hematology 201152
Charles Drew was born on June 3, 1904, and lived all his life in Washington, DC, travelling a lot for his career and education. Charles
was one of those rare individuals who seemed to ex-cel at everything he did and on every level and would go on to become a pioneer in the field of medicine. Charles’ early interests were in education, particular-ly, in medicine, but he was also an outstanding ath-lete. As a youngster, he was an award-winning swim-mer and starred Dunbar High School in football, baseball, basketball, and track and field, winning the James E Walker Memorial medal as his school’s best all-around athlete.
In 1928, Charles decided to pursue his interest in medicine and enrolled at McGill University, Montreal, Canada. He was received as a member of the Medical Honorary Society and graduated in 1933 with Master of Surgery and Doctor of Medicine degrees, finish-ing second in his class of 127 students. He stayed in Montreal for a while as an intern at the Montreal General Hospital and at the Royal Victoria Hospital. In 1935, he returned to the United States, began working as an instructor of pathology at Howard University in Washington, DC, and was awarded the Rockefeller Foundation Research Fellowship.
Years back, while a student at McGill, he had saved a man by giving him a blood transfusion and had studied under Dr. John Beattie, an instructor of anatomy who was intensely interested in blood trans-fusions. Now, at Columbia, he wrote a dissertation on “Banked Blood” in which he described a tech-nique he developed for the long-term preservation of blood plasma. Prior to his discovery, blood could not be stored for more than 2 days because of the rapid breakdown of red blood cells. Drew had dis-covered that by separating the plasma (the liquid part of blood) from the whole blood (in which the red blood cells exist) and then refrigerating them sepa-rately, they could be combined up to a week later for a blood transfusion. He also discovered that everyone has a certain type of blood (A, B, AB, or O) and thus are prevented from receiving a full blood transfusion
The Father of Blood Bankingfrom someone with different blood; ev-eryone has the same type of plasma. Thus, in certain cases where a whole blood trans-fusion is not neces-sary, it was sufficient to give a plasma trans-fusion, which could be administered to anyone, regardless of their blood type. He convinced the Columbia University to establish a blood
bank and soon was asked to go to England to help set up that country’s first blood bank.
Drew created a central location for the blood col-lection process where donors could go to give blood. He made sure all blood plasma was tested before it was shipped out. He ensured that only skilled per-sonnel handled blood plasma to avoid the possibility of contamination. The Blood for Britain program operated successfully for 5 months, with total col-lections of almost 15,000 people donating blood and with over 5,500 vials of blood plasma. As a result, the Blood Transfusion Betterment Association ap-plauded Drew for his work. Out of his work came the American Red Cross Blood Bank.
Charles Drew died on April 1, 1950, when the automobile he was driving went out of control and turned over. Drew suffered extensive, massive in-juries, but contrary to popular legend, he was not denied a blood transfusion by an all-white hospital. He, indeed, received a transfusion but was beyond the help of the experienced physicians attending to him. His family later wrote letters to those physicians thanking them for the care they provided. Over the years, Drew has been considered one of the most honored and respected figures in the medical field and his development of the blood bank.
Charles Drew (1904-1950)
1. http://en.wikipedia.org/wiki/Charles_R._Drew, [Internet ] Wikipedia® is a registered trademark of the Wikimedia Foundation, Inc. , a non-profit organization. Updated 20 march 2011, Cited 31 March 2011 2. http://www.blackinventor.com/pages/charles-drew.html . [Internet] The Black Inventor Online Museum ™, is the first of numerous educational/information web sites presented by Adscape International(1998-2011), cited 31 March 2011 N.B. Due to scarcity of literature referring to Charles Drew’s biography, the websites are taken as references for this essay.
References

2
The Center of Excellence inThrombosis and Hemostasis organizes
the 2nd Symposium on Thromboembolism and Hemostasis
on 25-26 May 2011 (21-22 Jumada Al Thani 1432).
Many international speakers will beinvited.
Registration:[email protected]
+966 1 4671546





















