
The functions of long noncoding RNAs in development and...
13
REVIEW The functions of long noncoding RNAs in development and stem cells Rotem Ben-Tov Perry* and Igor Ulitsky* ABSTRACT Eukaryotic genomes are pervasively transcribed, with tens of thousands of RNAs emanating from uni- and bi-directional promoters and from active enhancers. In vertebrates, thousands of loci in each species produce a class of transcripts called long noncoding RNAs (lncRNAs) that are typically expressed at low levels and do not appear to give rise to functional proteins. Substantial numbers of lncRNAs are expressed at specific stages of embryonic development, in many cases from regions flanking key developmental regulators. Here, we review the known biological functions of such lncRNAs and the emerging paradigms of their modes of action. We also provide an overview of the growing arsenal of methods for lncRNA identification, perturbation and functional characterization. KEY WORDS: Long noncoding RNAs, Regulatory networks, Stem cell differentiation Introduction It is thought that virtually all protein-coding genes in vertebrate genomes have already been discovered, and it is established that the key drivers of differences between species, as well as the majority of genetic variants associated with human traits and diseases, map to regions that do not overlap with protein-coding exons. It is also established that much of the genomic sequence between protein- coding genes is transcribed into long noncoding RNAs (lncRNAs) (Clark et al., 2011). However, because the vast majority of the loci transcribed into lncRNAs [up to 50,000 in humans (Iyer et al., 2015)] are expressed at low levels and are poorly conserved in other species, there is uncertainty about how many human lncRNAs are functional. Nevertheless, there are ∼1000 human lncRNAs that are moderately to highly expressed and show signs of evolutionary constraint on their sequences and their transcription, and ∼300 of these are conserved outside mammals in other vertebrates (Hezroni et al., 2015). Other eukaryotes also express hundreds to thousands of lncRNAs, although none has so far been found to be orthologous to a vertebrate lncRNA. An increasing number of human and mouse lncRNAs have been implicated as key regulators in a variety of cellular processes, including proliferation, apoptosis and response to stress. Many lncRNAs are differentially expressed in human diseases (Huarte, 2015; Lorenzen and Thum, 2016; Zhao and Lin, 2015), an observation that boosts interest in their study as potential biomarkers and therapeutic targets. Several features of lncRNAs make them attractive candidates for having important roles in embryonic development: (1) stem and progenitor cells, which are typically associated with more open and active chromatin, produce numerous lncRNAs (Guttman et al., 2009); (2) lncRNAs are typically expressed in very specific patterns, both spatially and temporally (Cabili et al., 2011; Ulitsky et al., 2011); and (3) many lncRNAs are transcribed from large regions flanking transcription factor (TF) genes and other regulators that are important during embryonic development (Ulitsky et al., 2011). Indeed, the list of lncRNAs implicated in embryonic development and in the acquisition of cell identity during differentiation is rapidly growing. Lagging behind, but also progressing, is our understanding of how these functions are carried out. In this Review, we present an overview of the current approaches and methods for identifying and annotating lncRNAs. We then survey the diverse functions of lncRNAs in embryonic development, highlighting the insights into lncRNA functions and modes of action that have recently been obtained and the remaining challenges and open questions in the field. The identification and annotation of lncRNAs Approaches for reconstructing transcriptomes and sifting them for the purposes of identifying lncRNAs have been described in detail elsewhere (Housman and Ulitsky, 2015; Ulitsky and Bartel, 2013); here, we provide just an overview. The transcriptome of a tissue or a developmental state is typically reconstructed using RNA sequencing (RNA-seq) libraries prepared from either poly(A)- selected or rRNA-depleted (‘total’) RNA. The use of total RNA has the advantage of capturing non-polyadenylated lncRNAs, but these are generally non-abundant transcripts that are difficult to reconstruct accurately and, in our experience, tools for expression level estimation work better when applied to poly(A)-selected RNA-seq data. The RNA-seq reads are then assembled into transcripts, either with or without the use of a reference genome, and the resulting transcript models are annotated based on known protein-coding genes and small RNAs and the sequences and transcriptomes of related species. We recently developed PLAR – a pipeline for lncRNA annotation from RNA-seq data – (Hezroni et al., 2015), and other tools are also available (Chen et al., 2016). The accuracy of the exon-intron structures reconstructed from short-read RNA-seq data is limited by several factors, including algorithmic challenges, extensive alternative splicing and incomplete genome assembly (in practically all animals, except human and mouse). The use of orthogonal data, such as chromatin marks (Guttman et al., 2009; Ulitsky et al., 2011) or RNA fragments enriched for 5′ or 3′ termini of the transcripts (Brown et al., 2014; Hezroni et al., 2015; Ulitsky et al., 2011), can improve transcriptome quality, but these data are frequently difficult to obtain. Therefore, it is important to treat each reconstructed transcript of interest with caution and validate its structure both by manual inspection of all the available data and experimentally. Particular caution is warranted when dealing with single-exon Department of Biological Regulation, Weizmann Institute of Science, 234 Herzl St, Rehovot 76100, Israel. *Authors for correspondence ([email protected]; [email protected]) I.U., 0000-0003-0555-6561 3882 © 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 3882-3894 doi:10.1242/dev.140962 DEVELOPMENT
-
Upload
doannguyet -
Category
Documents
-
view
226 -
download
0
Transcript of The functions of long noncoding RNAs in development and...

REVIEW
The functions of long noncoding RNAs in development andstem cellsRotem Ben-Tov Perry* and Igor Ulitsky*
ABSTRACTEukaryotic genomes are pervasively transcribed, with tens ofthousands of RNAs emanating from uni- and bi-directional promotersand from active enhancers. In vertebrates, thousands of loci in eachspecies produce a class of transcripts called long noncoding RNAs(lncRNAs) that are typically expressed at low levels and do not appearto give rise to functional proteins. Substantial numbers of lncRNAs areexpressed at specific stages of embryonic development, in manycases from regions flanking key developmental regulators. Here, wereview the known biological functions of such lncRNAs and theemerging paradigms of their modes of action. We also provide anoverview of the growing arsenal of methods for lncRNA identification,perturbation and functional characterization.
KEY WORDS: Long noncoding RNAs, Regulatory networks,Stem cell differentiation
IntroductionIt is thought that virtually all protein-coding genes in vertebrategenomes have already been discovered, and it is established that thekey drivers of differences between species, as well as the majority ofgenetic variants associated with human traits and diseases, map toregions that do not overlap with protein-coding exons. It is alsoestablished that much of the genomic sequence between protein-coding genes is transcribed into long noncoding RNAs (lncRNAs)(Clark et al., 2011). However, because the vast majority of the locitranscribed into lncRNAs [up to 50,000 in humans (Iyer et al.,2015)] are expressed at low levels and are poorly conserved in otherspecies, there is uncertainty about how many human lncRNAs arefunctional. Nevertheless, there are ∼1000 human lncRNAs that aremoderately to highly expressed and show signs of evolutionaryconstraint on their sequences and their transcription, and ∼300 ofthese are conserved outside mammals in other vertebrates (Hezroniet al., 2015). Other eukaryotes also express hundreds to thousands oflncRNAs, although none has so far been found to be orthologous toa vertebrate lncRNA.An increasing number of human and mouse lncRNAs have been
implicated as key regulators in a variety of cellular processes,including proliferation, apoptosis and response to stress. ManylncRNAs are differentially expressed in human diseases (Huarte,2015; Lorenzen and Thum, 2016; Zhao and Lin, 2015), anobservation that boosts interest in their study as potentialbiomarkers and therapeutic targets. Several features of lncRNAsmake them attractive candidates for having important roles in
embryonic development: (1) stem and progenitor cells, which aretypically associated with more open and active chromatin, producenumerous lncRNAs (Guttman et al., 2009); (2) lncRNAs aretypically expressed in very specific patterns, both spatially andtemporally (Cabili et al., 2011; Ulitsky et al., 2011); and (3) manylncRNAs are transcribed from large regions flanking transcriptionfactor (TF) genes and other regulators that are importantduring embryonic development (Ulitsky et al., 2011). Indeed, thelist of lncRNAs implicated in embryonic development and in theacquisition of cell identity during differentiation is rapidly growing.Lagging behind, but also progressing, is our understanding of howthese functions are carried out.
In this Review, we present an overview of the current approachesand methods for identifying and annotating lncRNAs. We thensurvey the diverse functions of lncRNAs in embryonicdevelopment, highlighting the insights into lncRNA functions andmodes of action that have recently been obtained and the remainingchallenges and open questions in the field.
The identification and annotation of lncRNAsApproaches for reconstructing transcriptomes and sifting them forthe purposes of identifying lncRNAs have been described in detailelsewhere (Housman and Ulitsky, 2015; Ulitsky and Bartel, 2013);here, we provide just an overview. The transcriptome of a tissueor a developmental state is typically reconstructed using RNAsequencing (RNA-seq) libraries prepared from either poly(A)-selected or rRNA-depleted (‘total’) RNA. The use of total RNAhas the advantage of capturing non-polyadenylated lncRNAs, butthese are generally non-abundant transcripts that are difficult toreconstruct accurately and, in our experience, tools for expressionlevel estimation work better when applied to poly(A)-selectedRNA-seq data. The RNA-seq reads are then assembled intotranscripts, either with or without the use of a reference genome,and the resulting transcript models are annotated based on knownprotein-coding genes and small RNAs and the sequences andtranscriptomes of related species. We recently developed PLAR – apipeline for lncRNA annotation from RNA-seq data – (Hezroniet al., 2015), and other tools are also available (Chen et al., 2016).The accuracy of the exon-intron structures reconstructed fromshort-read RNA-seq data is limited by several factors, includingalgorithmic challenges, extensive alternative splicing andincomplete genome assembly (in practically all animals, excepthuman and mouse). The use of orthogonal data, such as chromatinmarks (Guttman et al., 2009; Ulitsky et al., 2011) or RNA fragmentsenriched for 5′ or 3′ termini of the transcripts (Brown et al., 2014;Hezroni et al., 2015; Ulitsky et al., 2011), can improvetranscriptome quality, but these data are frequently difficult toobtain. Therefore, it is important to treat each reconstructedtranscript of interest with caution and validate its structure bothby manual inspection of all the available data and experimentally.Particular caution is warranted when dealing with single-exon
Department of Biological Regulation, Weizmann Institute of Science, 234 Herzl St,Rehovot 76100, Israel.
*Authors for correspondence ([email protected];[email protected])
I.U., 0000-0003-0555-6561
3882
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

transcripts, antisense transcripts and those overlappingpseudogenes, as these are especially prone to errors in readmapping or transcript assembly.Once the transcriptome of a specific combination of tissues, cell
types or developmental stages is available, the next challenge is toidentify those transcripts that correspond to lncRNAs. BecauselncRNAs are rather loosely defined, different researchers adoptdifferent criteria for inclusion and exclusion of transcripts, inparticular those that overlap loci of protein-coding genes on eitherthe sense or antisense strand. A typical pipeline retains intergeniclncRNAs and those that overlap mRNAs on the other strand, andremoves those transcripts that: (1) have an ORF that is either long orhas a protein-like sequence composition; (2) have sequences similarto known proteins or domains; or (3) overlap regions wheresequence evolution suggests a selective pressure to preserve aparticular succession of amino acids [for a description of specifictools and algorithms, see Housman and Ulitsky (2015)]. Such acombination of filters is effective in removing candidates thatencode large or conserved peptides; in our experience, most of thesecorrespond to pseudogenes. Recent studies have identified a numberof conserved peptides of 34-58 amino acids in length encoded bytranscripts that were initially annotated by some pipelines aslncRNAs, and identified important physiological roles for thosepeptides (Anderson et al., 2015; Nelson et al., 2016; Pauli et al.,2014). Also, there are probably species-specific short proteins thatremain to be discovered, but we estimate that these are relativelyrare, in part because mRNAs and lncRNAs are distinguishable fromeach other in ribosome-footprinting data (Chew et al., 2013;Guttman et al., 2013). Perhaps counterintuitively, but consistentwith the cytoplasmic presence of many lncRNAs, there is evidencefor some degree of translation on most well expressed lncRNAs(Ingolia et al., 2014). This translation is similar to that occurring at5′ UTRs, and the vast majority of peptides produced by suchtranslation events in lncRNAs and 5′ UTRs are likely to beimmediately degraded and nonfunctional [as discussed at length byHousman and Ulitsky (2015)]. The presence of some ribosome-protected fragments overlapping an lncRNA, or its localization tothe polysome fraction (Carlevaro-Fita et al., 2016; van Heesch et al.,2014), do not, therefore, invalidate the noncoding nature of atranscript.
How many lncRNAs are evolutionarily conserved?Conservation in other species is currently one of the key indicatorsfor functionality of lncRNAs, and so the fraction of lncRNAs thatare conserved is an indicator of the fraction that is functional. Basedon the criteria listed above, the human genome encodes tens ofthousands of lncRNAs, as do the genomes of non-human primates,and at least thousands of lncRNAs are found in other vertebratespecies. Large numbers of lncRNAs have also been found in everymulticellular organism subjected to in-depth RNA-seq analysis,including vertebrates, insects, nematodes, sponges and plants(Kapusta and Feschotte, 2014; Ulitsky, 2016). Although there areno clear orthologous lncRNAs between these groups of species,there are some prominent similarities between them. For example,lncRNAs are shorter than mRNAs, span only a few exons (typically1-3), accumulate to levels at least an order of magnitude lower thanthose of mRNAs and in a more spatially and temporally specificmanner, and evolve much faster than mRNA (Hezroni et al., 2015).This rapid evolution manifests itself on multiple levels. Forexample, human and mouse share the vast majority of protein-coding genes and highly expressed microRNA (miRNA) genes. Bycontrast, most human lncRNAs do not have any recognizable
homologs in mice, and vice versa. In the homologous pairs that arepresent, many exons appear in one species but not in the other, andwithin those exons that do align the sequence similarity is typicallymuch lower than among homologous coding or UTR exons.Overall, the sequences of lncRNA exons are only marginally betterconserved than those in the introns of protein-coding genes or inrandom intergenic regions, and they are only marginally depleted ofsequences derived from transposable elements (Kapusta et al., 2013;Kelley and Rinn, 2012). There is some weak correlation betweenconservation levels and expression (Necsulea et al., 2014), andbetween conservation and expression breadth, and so lncRNAsdiscovered in larger and deeper sequencing efforts appear tobe less conserved than those discovered by earlier efforts, whichused relatively shallow sequencing. Overall, taking into accountboth genomic features and the conservation of lncRNAs in humanand in other species, it appears that many human lncRNAsarose recently through neutral sequence evolution andretrotransposition, and will likely be lost with time throughsimilar mechanisms. In this view, the lncRNAs that we witness inthe human genome today are likely to constitute only a smallsubset of those that appeared in our ancestors. They include asubset that is functional and has been selectively retained, but thissubset is likely to be relatively small. Unfortunately, even inrelatively well-conserved lncRNAs, the constrained regions arerather short and degenerate, so identifying conserved lncRNAsand the conserved regions within them remains a challenge in needof better computational tools, such as those described recently(Quinn et al., 2016).
Which lncRNAs aremore likely to be functionally important?Researchers interested in a particular developmental systemtypically need to employ various criteria for selecting lncRNAs,often from hundreds of dynamically expressed candidates, forexperimental follow-up. Possible parameters for selection includelevels and specificity of expression, the genomic context of thelncRNA, and the extent of its sequence conservation. Because ourexperience and that of others is still limited, it is difficult to specifythe right criteria for ‘successful’ selection, but several factors areimportant to keep in mind. First, tissue- or stage-specific expressiondoes not necessarily imply function; weak promoters randomlyplaced in the mouse genome have been shown to have very specificactivity patterns, depending on the context into which they areintroduced (Ruf et al., 2011) and the presence of active enhancers inthe vicinity of the insertion site. The expression levels of an lncRNAare, however, an important consideration and can limit the range ofpossibilities for its mode of action; for example, an RNA expressedat 1-2 copies per cell is unlikely to directly regulate many targets intrans. Because many lncRNAs are likely to regulate gene expressionin cis, genomic proximity to a gene with a known function in theprocess of interest can also indicate potential functionality andprovide a readily testable hypothesis for the mode of action. Closeproximity or overlap with promoters of known genes, or withregions bearing chromatin marks characteristic of enhancers, canhowever complicate the mechanistic dissection of the mode ofaction of an lncRNA, as genome editing or chromatin modulationcan have secondary effects on promoter or enhancer activity. Forthese reasons, conservation of an lncRNA in other species, which atleast in vertebrates is now relatively easy to test with the availabledata (Chen et al., 2016; Hezroni et al., 2015; Necsulea et al., 2014;Washietl et al., 2014), is arguably the strongest current indicator forfunctional relevance. The combination of some of these factorstypically allows the candidate list to be narrowed down from
3883
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

hundreds to a number that is feasible for some degree ofexperimental characterization.
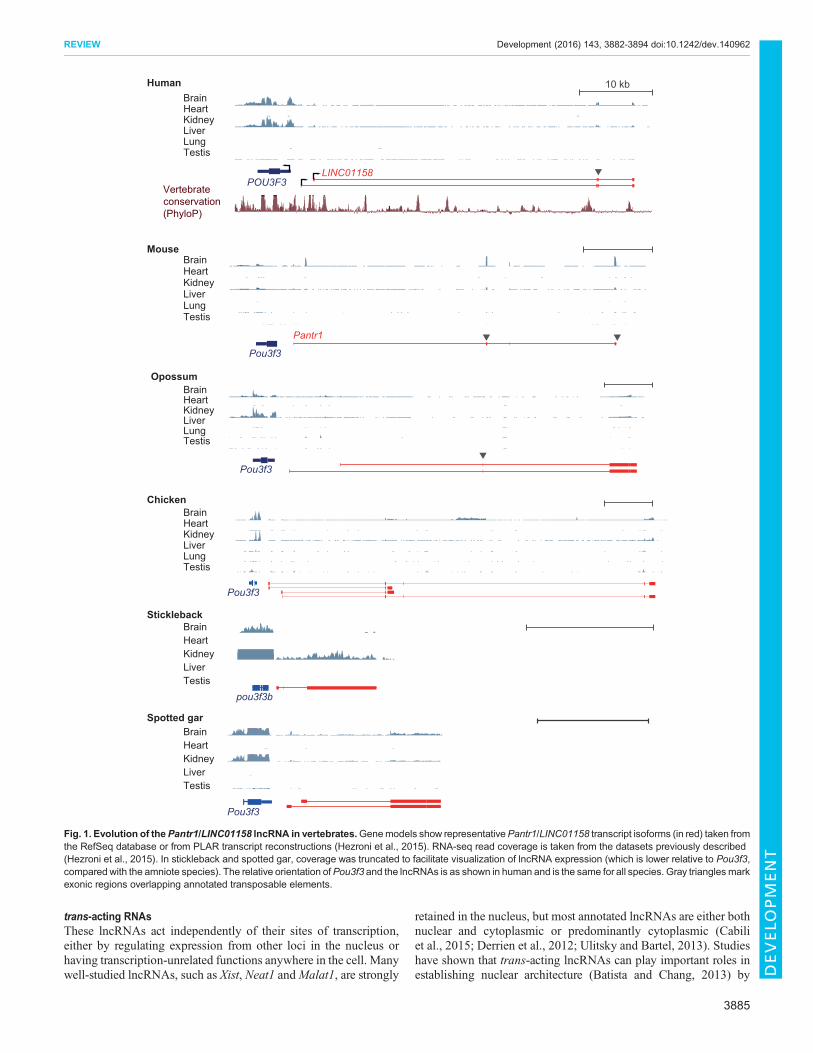
The identification of lncRNA homologsAs reviewed in detail elsewhere (Kapusta and Feschotte, 2014;Ulitsky, 2016), and as touched upon above, studies suggest thatlncRNAs evolve rapidly, with gains and losses occurring much morefrequently than in other gene classes. This complicates theidentification of homologs for study in model organisms. Theresources available for addressing this problem includewhole-genomealignments (such as those available in the UCSC genome browser orin Ensembl), databases of lncRNAs annotated in various species (Buet al., 2015; Chen et al., 2016; Hezroni et al., 2015; Necsulea et al.,2014), as well as groups of orthologous protein-coding genes (such asEnsembl Compara) for identifying positionally conserved (syntenic)lncRNAs. Several levels of lncRNA conservation are possible(Ulitsky, 2016). On the highest level, multiple regions within thelncRNA sequence are conserved together with splice sites and theoverall exon-intron architecture; the Miat lncRNA is an example thatexhibits this level of conservation (Ulitsky, 2016). However, suchcases are scarce between human and mouse, and are very rare whenconsidering more distantly related species (Chen et al., 2016; Hezroniet al., 2015). More common are cases in which just one or two shortsequence stretches are conserved, embedded in a rapidly evolvinglocus that underwent extensive rewiring of exon-intron architecture,typically with substantial contribution from transposable elements.When considering distantly related species (e.g. mammals and birds,or mammals and fish), the number of pairs of lncRNAs that exhibit‘positional conservation’ – two lncRNAs with the same relativeorientation to flanking conserved protein-coding genes – exceeds thenumber expected by chance and the number of lncRNAs withconserved sequences (Amaral et al., 2016 preprint; Hezroni et al.,2015; Ulitsky et al., 2011).The Pou3f3 adjacent noncoding transcript 1 (Pantr1; LINC01158
in human) lncRNA, which is transcribed divergently from thePou3f3 (Brn1) gene (Fig. 1) and plays a role in regulating geneexpression in the developing mouse brain (Goff et al., 2015),provides an illustrative example of the complexity of lncRNAevolution. Several parts of Pantr1, including its two mainpromoters, are well conserved among mammals. Similar lncRNAsdivergent with Pou3f3, which are expressed specifically in neuronaltissues and in the kidney, are also found throughout vertebrates, buttheir locus architectures, lengths and sequences are different.Furthermore, the human, mouse and opossum lncRNAs eachcontain exons derived from independent transposable elements(Fig. 1), and the 3′ end of Pantr1 in mouse is derived from atransposon. The sequence homology between the mammalianlncRNAs and the homologs in chicken and lizard is restricted to thefirst exon. In more distant species, such as teleost fish and thespotted gar, syntenic, kidney-expressed lncRNAs withoutdetectable sequence homology to amniote Pantr1 lncRNAs arefound in the same orientation near Pou3f3 orthologs (Fig. 1). Thisrapid turnover of sequence and expression status implies thatlncRNAs rarely depend on specific long sequences in their locifor function. It is possible that, in many of them, just the actof transcription across a certain region is important, or thatfunctionality is sequence dependent but relies on short sequencesthat are conserved but difficult to align across large evolutionarydistances. Another implication of rapid lncRNA evolution is thatsome human lncRNAs can only be studied in human cells, and thatfor those with homologs in model organisms the low sequence andgene structure homology may cast doubts on the conservation of
functionality. Few studies have looked into this question [reviewedin detail elsewhere (Ulitsky, 2016)] and, although it is still too earlyto draw definitive conclusions, it does appear that limited sequenceconservation can be sufficient to maintain conserved functionalityacross large evolutionary distances.
LncRNA mechanisms of actionThe potential modes by which lncRNAs function, and the relativeprevalence of each ‘mechanistic group’, remain unclear and are stillactively debated. However, lncRNAs can be broadly divided intothree groups based on their mechanisms of action (Fig. 2): (1) thosefor which only the act of transcription is important, and the RNAthat is produced carries no function (‘transcription only’); (2) thosefor which the RNA is important, but the activity is linked to the siteof transcription (‘cis-acting’); and (3) those for which the RNA actsindependently of the site of its transcription (‘trans-acting’). Below,we discuss each of these lncRNA groups, detailing their modes ofaction and highlighting how they can be distinguished from oneanother.
Transcription-only lncRNAsThe act of transcription itself can affect regulatory elementsoverlapping the lncRNA locus. Indeed, one potential functionaloutcome of transcription is changes in chromatin modifications inthe locus. For example, parts of transcribed regions (mostly from thesecond exon onwards) are typically demarcated with H3K36trimethylation (Wagner and Carpenter, 2012), which is associatedwith repression of transcription initiation (Carrozza et al., 2005;Fang et al., 2010). Therefore, if the lncRNA locus overlaps with thepromoter of another gene (such as in the case of Airn and Igf2r),transcription of the lncRNA can potentially lead to repression of thepromoter. Importantly, in these cases, the RNA product oftranscription, and hence its sequence or structure, do not have afunctional role.
cis-acting RNAsThe main mechanisms ascribed to the cis-acting group of lncRNAsinvolve the recruitment of factors, either to the site of lncRNAtranscription or to adjacent loci. The recruited factors can berepressive, as in the cases of ANRIL [CDKN2B-AS1; which isreported to recruit CBX7 from the PRC1 complex (Yap et al., 2010)and SUZ12 from the PRC2 complex (Kotake et al., 2011)], Airn[which is reported to recruit G9a (Ehmt2) (Nagano et al., 2008)] andH19 [which is reported to recruit MBD1 (Monnier et al., 2013)].Alternatively, such factors can be activating, as in the case ofHottip,which is reported to recruit WDR5 (Wang et al., 2011; Yang et al.,2014). There can be various consequences to the recruitment of suchfactors. For example, the lncRNA can increase the localconcentration of the factor in the locus, as has been suggested forYY1-binding RNAs (Sigova et al., 2015). By contrast, it can act as alocal decoy, diverting the factor from binding to DNA elsewhereacross the locus (Krawczyk and Emerson, 2014).
For many years, the prototypical example of an lncRNA actingthrough factor recruitment has been Xist, which has been proposed torecruit the PRC2 complex to the mammalian X chromosome duringX-inactivation (Zhao et al., 2008). However, recent studies havechallenged this model and shown that the picture is much morecomplicated, as PRC2 appears to bind RNA non-specifically(Brockdorff, 2013; Davidovich et al., 2013), Xist specifically bindsto other factors (Roth and Diederichs, 2015), and the temporaldynamics of silencing are inconsistent with the simple model of PRC2recruitment leading to gene silencing (Gendrel and Heard, 2014).
3884
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT
trans-acting RNAsThese lncRNAs act independently of their sites of transcription,either by regulating expression from other loci in the nucleus orhaving transcription-unrelated functions anywhere in the cell. Manywell-studied lncRNAs, such as Xist, Neat1 andMalat1, are strongly
retained in the nucleus, but most annotated lncRNAs are either bothnuclear and cytoplasmic or predominantly cytoplasmic (Cabiliet al., 2015; Derrien et al., 2012; Ulitsky and Bartel, 2013). Studieshave shown that trans-acting lncRNAs can play important roles inestablishing nuclear architecture (Batista and Chang, 2013) by
10 kb
LINC01158POU3F3
BrainHuman
Mouse
Opossum
Chicken
Stickleback
Spotted gar
HeartKidneyLiverLungTestis
BrainHeartKidneyLiverLungTestis
BrainHeartKidneyLiverLungTestis
Vertebrateconservation(PhyloP)
Pantr1
Pou3f3
Pou3f3
Pou3f3
Pou3f3
pou3f3b
Brain
Lung
Heart
Liver
Testis
Kidney
BrainHeartKidneyLiverTestis
BrainHeartKidneyLiverTestis
Fig. 1. Evolution of thePantr1/LINC01158 lncRNA in vertebrates.Genemodels show representativePantr1/LINC01158 transcript isoforms (in red) taken fromthe RefSeq database or from PLAR transcript reconstructions (Hezroni et al., 2015). RNA-seq read coverage is taken from the datasets previously described(Hezroni et al., 2015). In stickleback and spotted gar, coverage was truncated to facilitate visualization of lncRNA expression (which is lower relative to Pou3f3,compared with the amniote species). The relative orientation ofPou3f3 and the lncRNAs is as shown in human and is the same for all species. Gray trianglesmarkexonic regions overlapping annotated transposable elements.
3885
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

dictating the proximity of different loci, as in the case of Firre(Hacisuleyman et al., 2014), or by nucleating subnuclear bodies, asin the case of Neat1 (Clemson et al., 2009).Another trans-acting function that has been proposed to be
widespread is the recruitment of chromatin-altering complexes tospecific loci in trans (Koziol and Rinn, 2010). However, themechanisms by which lncRNAs can specifically recognizeelements in trans remain unknown, and it is unclear how manytarget loci can be efficiently reached by lncRNAs that accumulate atonly a few copies per cell. The same stoichiometric concerns existfor the cytoplasmic functions of lncRNAs and, in general, the rolesof cytoplasmic lncRNAs have been less explored. Somecytoplasmic lncRNAs, including the circular lncRNA CDR1as(Memczak et al., 2013) and NORAD (Lee et al., 2016; Tichon et al.,2016), are abundant enough to bind to and affect the function ofRNA-binding proteins (Argonaute and Pumilio, respectively) andthereby modulate their ability to regulate their other targets. Othercytoplasmic lncRNAs have been shown to bind and modulate thestability and translation of other RNAs by base-pairing with them(Carrieri et al., 2012; Gong and Maquat, 2011).
Distinguishing between lncRNA modes of actionIn many cases, it is relatively straightforward to experimentallydistinguish between the cis-acting and the trans-acting mechanisms.For example, if the phenotype caused by loss of a particular lncRNAcan be rescued by its expression from an exogenous locus [as in thecase of roX lncRNAs (Park et al., 2008; Quinn et al., 2014; Quinn
et al., 2016)], the lncRNA is most likely trans-acting. By contrast, itis difficult to distinguish between transcription-only and cis-actinglncRNAs, as this requires eliminating the possibility that the RNAproduct is important. For example, in the case of the lncRNA Airn, itwas shown that piecewise replacement of all parts of the Airntranscript does not compromise its allele-specific repression ofIgf2r, strongly implying that only the act of transcription and not theproduct of Airn transcription is important for Airn function.However, in most cases, such studies require extensive genomeengineering and precise phenotypic recoding, which often remainvery difficult despite the growing toolbox for lncRNAmanipulation(Box 1). It should also be noted that the same locus can havemultiple independent activities. For example, the Airn lncRNAregulates the expression of imprinted genes in the vicinity of itslocus via at least two mechanisms: the AirnRNA product acts on theSlc22a3 promoter by recruiting the G9a histone methyltransferase(Nagano et al., 2008) in a mechanism shared by other lncRNAsimplicated in imprinting (Pandey et al., 2008), but only transcriptionthrough the Airn locus is required for silencing Igf2r (Latos et al.,2012). Furthermore, even the transcription of mRNAs (which all‘act in trans’ to produce proteins) can have a cis-regulatory functionand affect the transcription of nearby genes (Ebisuya et al., 2008).
Roles for lncRNAs during mammalian developmentIn recent years, loss-of-function (LOF) and gain-of-function (GOF)studies have revealed that many lncRNAs are involved in a widevariety of biological processes during development (Ponting et al.,2009). The different techniques used in these studies are presentedin Box 1, although it should be noted that each of these has pros andcons [see Box 2 and the recent review by Bassett et al. (2014)]. Inthis section, we focus on the functional roles of lncRNAs inmammalian embryonic development and differentiation, as revealedmostly by in vivo LOF studies in mice.
A general survey of mammalian lncRNA functions in vivoThe largest systematic surveys of lncRNA knockout (KO) mousemodels have revealed important roles for lncRNAs in regulatingorganism viability as well as many specific developmentalprocesses. Three complementary publications characterized 20lncRNAKOmouse strains using expression pattern characterizationand LOF experiments (Goff et al., 2015; Lai et al., 2015; Sauvageauet al., 2013). The 20 candidates were selected based on acombination of stringent filters (e.g. the absence of protein-codingcapacity, a lack of overlap with protein-coding genes), expressionlevels and conservation; all 20 lncRNAs overlap sequencesconserved in the human genome, and at least 14 are homologousto annotated human lncRNAs (Sauvageau et al., 2013). Notably,however, these candidate lncRNAs are not necessarily arepresentative sample of mammalian lncRNAs. KO mouse strainsin which a lacZ reporter cassette replaced each lncRNA locus weregenerated using VelociGene technology (Valenzuela et al., 2003).These lacZ reporters revealed a wide spectrum of spatiotemporaland tissue-specific lncRNA transcription patterns, both in mouseembryos and in adult mice (Goff et al., 2015; Lai et al., 2015;Sauvageau et al., 2013). Importantly, since most of the locus isexcised in this approach, the phenotypes might result from theremoval of DNA elements in the locus rather than from the loss ofRNA expression (Bassett et al., 2014).
The initial characterization of 18 of these KO strains revealed thefunctional relevance of specific lncRNAs in mouse embryonicdevelopment, viability and growth. Indeed, three out of the 18lncRNA KO strains – Peril (Perl), Mdgt (Haglr) and Fendrr
Transcription process affects chromatin
RNA acts on genomically or spatially proximal regulatory elements and/or the factors bound by them
Regulation of a distal locus Binding other
RNAsNucleation of subcellular domains
B cis-acting
A Transcription only
C trans-acting
RNA and its sequence not important
Sequence may or may not be important
Parts of the sequence are important
Fig. 2. LncRNA modes of action. (A) For some lncRNA loci, the act oftranscription itself plays a role in mediating the function of the lncRNA, forexample by affecting the underlying chromatin structure of the locus. In thiscontext, the RNA product itself and its sequence are inconsequential. (B) Bycontrast, other lncRNAs act in the vicinity of their site of transcription, recruitingor diverting specific factors, which may recognize the RNA in sequence-specific or nonspecific ways. (C) Other lncRNAs leave their site of transcriptionand act elsewhere, typically in a sequence- or structure-dependent manner,and via interactions with protein and other RNA factors.
3886
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

(of which the latter two are conserved in humans) – exhibit viabilityphenotypes (Sauvageau et al., 2013). Two subsequent studies of the20 KO strains examined the expression patterns of these targetedlncRNA genes (Lai et al., 2015) and the effects of the KOs on braingene expression (Goff et al., 2015), suggesting roles for specificlncRNAs such as linc-Brn1b (Pantr2) and Peril in brain
development and function (as discussed in detail below). Theunique phenotypes and exquisitely specific expression patternsdescribed in these studies suggest that some lncRNAs performdistinct functions that are consequential on the organismal level.
LncRNAs implicated in dosage compensation: Xist and TsixOne of the first-discovered and best-characterized lncRNAs shownto have a specific developmental role and a robust LOF phenotypein vivo is X-inactive specific transcript (Xist). Xist is directlyinvolved in the process of X chromosome inactivation, which isinitiated by the induction of Xist expression. Xist is absolutelyrequired for X-inactivation to occur in cis, and an extended Xistlocus is sufficient for silencing when placed on an autosome (Leeand Bartolomei, 2013). Accordingly, the deletion of Xist in micecauses a loss of X-inactivation and female-specific lethality(Marahrens et al., 1997). The expression of Xist itself is controlledby other lncRNAs. For example, Tsix lncRNA, which is theantisense partner of Xist RNA, represses Xist expression (Lee andBartolomei, 2013). Tsix LOF in vivo results in ectopic Xistexpression, aberrant X-inactivation and early embryonic lethality(Sado et al., 2001). Xist also plays a role beyond early embryonicdevelopment. Because Xist activity is followed by epigeneticchanges, transient repression of Xist does not result in immediate Xreactivation (Wutz and Jaenisch, 2000). However, in Xist-deficientmouse hematopoietic stem cells, which undergo a large number ofcell divisions, Xist loss does cause X reactivation and subsequentgenome-wide changes that lead to cancer, thus potentially linkingthe X chromosome to cancer in mice (Yildirim et al., 2013).
Imprinting-associated lncRNAs: Kcnq1, Airn and H19A number of lncRNAs are associated with the process of genomicimprinting, which is crucial for normal development. During thisevent, genes are epigenetically silenced on the basis of their parentalorigin, resulting in monoallelic expression. Many imprinted clusterscontain protein-coding genes and lncRNAs that are expressed fromreciprocal alleles. The best-characterized examples of lncRNAs thatregulate imprinting are Kcnq1ot1 and Airn; both are paternallyexpressed and repress flanking protein-coding genes in cis. Thus,although the loss of these lncRNAs in the embryo is not lethal,
Box 2. Potential caveats to lncRNA perturbationapproachesWhen considering LOF approaches, the main distinction is betweenmethods that target the transcript and leave the DNA intact (e.g. RNAi,antisense oligonucleotides) and those that alter the DNA in the locususing genome engineering (e.g. TALEN, CRISPR). Both can be efficientat dramatically reducing transcript levels, but the former methods sufferfrom off-targeting while the latter can interfere with other activitiesencoded in the same locus, such as those exerted by enhancer elementsand/or elements regulating chromatin architecture. Methods based on acatalytically inactive CRISPR/Cas9 system (dCas9) have the potential ofharnessing the targeting specificity of CRISPR without altering thegenome (Dominguez et al., 2016), but they still may have lncRNA-unrelated effects on chromatin, in particular when the dCas9 is coupledwith a chromatin-modifying domain [as in dCas9-KRAB fusions (Gilbertet al., 2014)]. In the case of rescue experiments following GOF or LOFapproaches, the available techniques differ in their relevance forlncRNAs acting in cis, which requires the expression of the lncRNA inthe proximity of its endogenous site of transcription. Here too, the use ofdCas9 can assist in recruiting the lncRNA to the target site of interest,which can be sufficient for eliciting regulatory effects (Luo et al., 2016;Shechner et al., 2015).
Box 1. Tools for studying lncRNA functionTranscript characterization. Initial reconstruction of lncRNA transcriptsis typically performed usingRNA-seq data. RT-PCR, 3′ and 5′RACE andnorthern blots can be used to validate lncRNA transcript structure.Single-molecule RNA fluorescence in situ hybridization (smFISH) canthen be used to examine expression patterns, detect even low-abundance lncRNAs, and enable absolute quantification of thenumber and location of lncRNA molecules within cells (Dunagin et al.,2015).Binding partners. The dissection of lncRNA mechanisms typicallyrequires the identification of protein, DNA and RNA binding partners.Biotinylated RNA transcribed in vitro from a cDNA template can beincubated with cell lysates to identify protein binding partners (Hämmerleet al., 2013). Alternatively, endogenous RNAs can be enriched usingbiotinylated antisense oligos and the resulting material can be subjectedto mass spectrometry, DNA or RNA sequencing (Simon, 2016). RNAimmunoprecipitation can also be used to identify lncRNA species thatbind to a protein of interest (Cozzitorto et al., 2015).Transient LOF. Methods to transiently perturb lncRNA expressionwithout changing the underlying DNA sequence include RNAinterference (RNAi), antisense oligonucleotides (ASOs; which includesmorpholinos) and CRISPR interference (CRISPRi). RNAi has been usedto successfully knock down lncRNAs in many studies (Guttman et al.,2011). Recently, it was found that nuclear lncRNAs are more effectivelysuppressed using ASOs, cytoplasmic lncRNAs are more effectivelysuppressed using RNAi, and dual-localized lncRNAs are suppressedusing either method (Lennox and Behlke, 2016). CRISPRi is a muchnewer technology that has already been used successfully for targetinglncRNAs (Ghosh et al., 2016).Constitutive LOF via genome engineering. Genome engineering canbe performed using both traditional recombination-based techniquesand TALEN/CRISPR technologies to target lncRNA genes. Smallinsertions or deletions caused by Cas9-mediated double-strand breaksand inducing frameshifts in ORFs are the current method of choice forinactivating protein-coding genes. These are not well suited to knock outlncRNAs, and several other targeting strategies are used: deletion of thefull-length lncRNA locus [or its replacement by reporter genes orselection cassettes (Lai et al., 2015; Sauvageau et al., 2013)]; deletion ofthe promoter sequence; mutation of putative functional domains;engineered inversions (Li and Chang, 2014); or insertion oftranscriptional terminator sequences (i.e. STOP signals) (Bond et al.,2009; Grote et al., 2013). In all cases, it is important to minimize theremoval or reorganization of regulatory factor binding sites or otherregulatory elements within the DNA locus, and to control for the additionof novel DNA regulatory elements (Bassett et al., 2014). To prove that anlncRNA molecule has a direct functional role, rescue or GOF assaysinvolving transgene expression can be used (Grote et al., 2013).GOF assays. Exogenous overexpression of the lncRNA from a plasmidor a viral vector is the most common and simplest GOF approach, but ithas limited efficacy if the lncRNA acts in cis. Instead, CRISPR-on, whichis a variant of CRISPRi that combines a catalytically dead Cas9 (dCas9)with transcriptional activators such as the VP64 activator domain, can beused to increase lncRNA production from endogenous loci (Gilbert et al.,2014; Luo et al., 2016). Another possibility is to localize the exogenouslyexpressed lncRNA to a specific genomic locus by fusing it with theCRISPR gRNA targeted to the specific locus and to co-introduce thischimeric RNA into cells together with dCas9 (Luo et al., 2016; Shechneret al., 2015). Lastly, genome editing can be used to knock-in a strongpromoter upstream of the lncRNA to increase gene expression (Luoet al., 2016) or to knock-in the cDNA of an lncRNA for rescueexperiments (Yin et al., 2015).
3887
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

paternal inheritance of a LOF allele causes a loss of imprinting andhence gives rise to growth defects, whereas maternal inheritance ofthis allele has no effects on imprinting or growth (Fitzpatrick et al.,2002; Sleutels et al., 2002).The lncRNA H19 is encoded by a conserved imprinted gene that
is expressed exclusively from the allele of maternal origin. H19 isstrongly expressed in both mesoderm- and endoderm-derivedtissues during embryogenesis in mice, then becomes fullyrepressed after birth except in skeletal muscle and heart (Poirieret al., 1991). This expression pattern is similar to that of the majorfetal growth factor gene Igf2, which is paternally expressed(DeChiara et al., 1991). Two KO models have been established toexamine H19 function – one in which only the 3 kb transcriptionunit is deleted (H19Δ3 mice) and another in which 10 kb upstream ofthat region is also deleted (H19Δ13 mice). In both cases, maternalheterozygotes are viable and fertile but exhibit an overgrowthphenotype. In H19Δ13 mice, the maternal Igf2 allele is totallyreactivated in all expressing tissues (Leighton et al., 1995). In H19Δ3
mice, the maternal Igf2 allele is also reactivated, although itsexpression is only 25% of that of the paternal allele in wild-typemice and is only observed in mesoderm-derived tissues (skeletalmuscle, tongue, diaphragm and heart) (Ripoche et al., 1997). Theovergrowth phenotype can be rescued in H19Δ3 mice expressing anH19 transgene, with expression of Igf2 and other imprinted genesreturning to wild-type levels. This activity was recently associatedwith the ability of H19 to recruit MBD1 (Monnier et al., 2013),suggesting that H19 itself can act in trans to control imprinting(Gabory et al., 2009). Together, these findings highlight thatlncRNAs in imprinted gene clusters are not just imprinted in theirexpression patterns, but can also regulate the imprinting process.
Hox cluster lncRNAs: Hotair, Hottip and MdgtHox genes encode TFs that orchestrate the embryo body plan andcontribute to several adult cell fate specification processes (Barberand Rastegar, 2010). In addition to containing protein-coding andmiRNA genes, these clusters produce numerous lncRNAs thatexhibit spatiotemporal expression patterns resembling those of theirneighboring protein-coding genes. For example, Hotair is a 2.2 kblncRNA expressed from the HoxC cluster that can repress the HoxDlocus in trans in mammalian cells (Rinn et al., 2007). In linewith thekey function of HoxD genes, homeotic transformation of the fourthcaudal vertebra is observed inHotair−⁄−mice (Lai et al., 2015). TheHotair homeotic phenotype is also observed in mice carrying analternative Hotair KO allele (Li et al., 2013). In this model,disruption of Hotair also leads to the derepression of hundreds ofgenes, including those within the HoxD cluster. By contrast, nomajor skeletal transformations are observed in a mouse strain inwhich the entire HoxC gene cluster, including the Hotair gene, isdeleted (Schorderet and Duboule, 2011). However, it should benoted that the HoxC cluster produces a multitude of noncodingRNAs, and removal of the entire gene cluster may remove protein-coding genes and lncRNAs that could opposeHotair activity. Thus,although the phenotypes of Hotair mutants are not severe, theyprovide compelling evidence that lncRNAs in Hox clusters canregulate the expression patterns of Hox genes, and other genes,during development. Indeed, another lncRNA – Hottip – has beenshown to regulate the expression of posterior HoxA genes. It does soby interacting with the activating histone-modifying MLL1complex and via the formation of chromatin loops that connectdistally expressed Hottip transcripts with posterior HoxA genepromoters (Wang et al., 2011). Hottip−⁄− mice exhibit hind limbabnormalities, including muscle weakness and skeletal
malformations (Lai et al., 2015). Furthermore, the transfection ofshort hairpin RNAs (shRNAs) targeting a region with sequencesimilarity toHottip in chick embryos alters limbmorphology (Wanget al., 2011). The lncRNAMdgt [also calledHaglr (Lai et al., 2015;Yarmishyn et al., 2014)], which is transcribed from a bi-directionalpromoter that is shared with Hoxd1, has also been implicated in thecontrol of Hox gene expression. Homozygous Mdgt mutants diewithin 2 weeks after birth, with Mdgt−/− pups displaying a severegrowth retardation phenotype that may contribute to their lethality(Sauvageau et al., 2013).
LncRNAs required for neuronal developmentThe detailed analysis of lncRNAKOmouse models has revealed keyroles for three lncRNAs – Peril, Evf2 (Dlx6os1) and linc-Brn1b –during neural development. The Peril transcript is derived from an18.2 kb genomic locus that is located 110 kb downstream of Sox2,which encodes a key pluripotency factor. Peril is highly enriched inmouse embryonic stem cells (ESCs) but is also expressed at lowerlevels in the mouse adult brain and testes (Sauvageau et al., 2013).The deletion of Peril in mice leads to reduced viability; 50% ofPeril−/− pups die within 2-20 days of birth (Sauvageau et al., 2013).The expression of Sox2 and its overlapping lncRNA Sox2ot is notsignificantly affected in Peril KO brains, suggesting that the KOphenotype is not due to a defect in Sox2 function (Sauvageau et al.,2013). In a follow-up studyofPerilKOmice (Goff et al., 2015), usingRNA-seq analyses and β-gal staining, it was shown that Peril isexpressed in neural stem cells and may affect their biology.Furthermore, the neural stem cell-specific expression of Peril ismaintained in Peril+/− adult mice, with β-gal staining observed in theependymal lining of the ventricles and in the dentate gyrus of thehippocampus, both of which are regions associated with adultneurogenesis. Consistently, RNA-seq analyses ofPeril−/− embryonicbrains revealed a misregulation of cell cycle genes that are known tobe important for the correct maintenance and differentiation of neuralprogenitors. These results clearly demonstrate biological activity ofthe Peril transcript, although additional work needs to be done tounderstand the molecular mechanism by which Peril functions andthe extent to which it operates independently of Sox2.
The lncRNA Evf2 also appears to play a role in neuraldevelopment. Evf2 is transcribed antisense to Dlx6 and is locatedimmediately downstream of the Dlx5 genomic locus.Dlx5 and Dlx6,which are related to the Drosophila melanogaster Distal-less (Dll)gene, encode TFs that are expressed in the developing ventralforebrain and have been implicated in both forebrain and craniofacialdevelopment. Evf2 KO mice have been generated by inserting atranscriptional terminator consisting of three polyadenylation signalsinto the first exon. In these mice, the numbers of GABAergicinterneurons in the early postnatal hippocampus are reduced (Bondet al., 2009). Although GABAergic interneuron numbers and levelsof Gad1 mRNA (which encodes an enzyme involved in GABAsynthesis) return to normal in the adult hippocampus ofEvf2mutants,defects in synaptic inhibition are observed, indicating a crucial rolefor Evf2 in neuronal activity in vivo (Bond et al., 2009). To determinewhether Evf2 controlsDlx5/6CpGmethylation and hence expressionthrough trans or cis mechanisms, an Evf2 rescue transgenic modelwas developed. Using this model, it was shown that transcriptionthrough the Evf2 locus controls the levels of Dlx6 in cis; afterdisengaging the polymerase, Evf2 then acts in trans to modulatemethylation of the Dlx5/6 enhancer and transcription of Dlx5.Therefore, by regulating the cellular levels of the Dlx5 and Dlx6 TFs,Efv2 controls GABAergic interneuron activity (Berghoff et al., 2013;Bond et al., 2009).
3888
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

Finally, recent studies suggest a role for the lncRNA linc-Brn1b(also known asPantr2) in neural development. linc-Brn1b is locatednear the Pou3f3 (Brn1) gene, which is also adjacent to the lncRNAPantr1 (described above) (Sauvageau et al., 2013). linc-Brn1b−/−
mice exhibit distinct growth defects as well as defects in the cerebralcortex, especially in the development of upper layer II/III-IVneurons (Sauvageau et al., 2013).
LncRNAs required for the development of other organsSeveral lncRNAs have been shown to play roles in the developmentof specific organs and tissues during embryogenesis. Fendrr is a2397 nt transcript consisting of seven exons transcribed divergentlyfrom the TF gene Foxf1. Two approaches have been used to perturbFendrr in mice: KO mice (Lai et al., 2015; Sauvageau et al., 2013)have been obtained by genomic deletion (starting from the secondexon to the last annotated exon), and knock-in (KI) mice havebeen generated by replacing the first exon of Fendrr with apolyadenylation cassette without a reporter gene (Grote et al.,2013). KO pups exhibit multiple defects in the lung, heart andgastrointestinal tract (Sauvageau et al., 2013). Further investigationshighlighted that, at E13.5, the developing lungs of the KO are smallwith globular and disorganized lobes. Accordingly, these KO micesurvive to birth but die shortly thereafter due to breathing problems(Lai et al., 2015). By contrast, the KI mice display lethality atE13.75 due to heart and body wall (omphalocele) defects. Notably,resorbed embryos or omphalocele were not observed whenanalyzing E14.5 KO embryos. The differences between thestudies extended to the expression domain of Fendrr. Usingwhole-mount in situ hybridization and qPCR analysis, Grote et al.(2013) observed that endogenous Fendrr expression is restricted tonascent lateral plate mesoderm and cannot be detected in any othertissues or organs. However, expression profiling of the knocked-inlacZ reporter in E14.5 and E18.5 embryos showed expression ofFendrr in lung, colon, liver, spleen and brain, as well as in thetrachea and the gastrointestinal tract (Lai et al., 2015). Given thatboth studies used similar genetic background strains, the differenttargeting strategies used to remove the Fendrr gene provide themost probable explanation for the phenotypic discrepancies.Discrepancies in the reported expression patterns might be a resultof the different methods used to study the endogenous Fendrrexpression pattern. Regardless, both studies confirm that FendrrLOF is lethal in mice. Mechanistically, Fendrr was shown to act bybinding to the PRC2 and TrxG/MLL complexes, in turn modifyingthe chromatin signatures of genes involved in lateral mesodermlineage formation and differentiation (Grote et al., 2013).The lncRNA Neat1 has been implicated in mammary gland
development and pregnancy in mice.Neat1 is an essential constituentof paraspeckles – nuclear substructures that are found in all primarycells and cell lines examined to date, with the exception of ESCs(Clemson et al., 2009). It has been proposed that paraspeckles controlkey cellular processes, including differentiation, via their ability tointegrate transcriptional and post-transcriptional events (Hiroseand Nakagawa, 2012). Neat1 KO mice, which were generated byinsertion of lacZ and polyadenylation signals immediatelydownstream of the transcription start site, develop normally and areindistinguishable from their wild-type littermates with respect togrowth, viability and apparent behavior (Nakagawa et al., 2011).However, two follow-up studies on these mice revealed importantfunctions for Neat1 and paraspeckles in vivo. First, it wasdemonstrated that, during mammary gland development in mice,paraspeckles containingNeat1 are present inmammary gland luminalcells and that Neat1 is required for branching morphogenesis,
lobular-alveolar development and lactation (Standaert et al., 2014).Second, it was shown that, despite exhibiting normal ovulation,Neat1KO mice stochastically fail to become pregnant, potentially owing tocorpus luteum dysfunction and concomitant low progesterone levels(Nakagawa et al., 2014).
Finally, studies suggest that the lncRNA Pint (Lncpint) mightalso play a role in organ growth and development. Pint, which isubiquitously expressed in mice, is a direct transcriptional target ofp53 (Trp53) (Marín-Béjar et al., 2013) and positively regulates cellproliferation and survival by affecting the expression of hundreds ofgenes. Accordingly, Pint KO mice are smaller and exhibit lowerbody weights than their wild-type littermates (Sauvageau et al.,2013).
Roles for lncRNAs during zebrafish embryogenesisWhile KO studies in mice have provided key insights into thefunction of lncRNAs, it should be noted that zebrafish studies havealso been fruitful with regards to understanding the biology oflncRNAs in both developing embryos and adult tissues (Pauli et al.,2011). Indeed, several groups have shown that lncRNAs areexpressed dynamically in a spatiotemporal manner in zebrafishembryos and adult fish (Kaushik et al., 2013; Pauli et al., 2012;Ulitsky et al., 2011). Hundreds of zebrafish lncRNAs are found insyntenic positions to human lncRNAs, and a few dozen of themdisplay short sequences of high homology across vertebrateevolution (Ulitsky et al., 2011). Morpholino (MO)-mediatedknockdown of two lncRNAs that display such short stretches ofsequence conservation across vertebrates – cyrano (oip5-as1) andmegamind (birc6-as2) – results in defects in the central nervoussystem (Ulitsky et al., 2011). Importantly, the zebrafish, human andmouse homologs of these lncRNAs are able to rescue thesephenotypes, suggesting that the functionality of the lncRNAsequence has been conserved despite drastic changes in most ofthe sequence. However, a recent study reported that genetic deletionofmegamind does not give rise to the same phenotype, although themutant is still susceptible to megamind MO (Kok et al., 2015).Although this suggests that MO off-target effects might be involved,this appears to be unlikely given that the same phenotype isobserved with different MOs. One possible explanation is that theKO of megamind is compensated by the presence of two homologswith related sequences in the zebrafish genome (Ulitsky et al.,2011).
In a different study, three highly conserved lncRNAs –TERMINATOR, ALIEN [also known as lnc-FOXA2, LL35(Herriges et al., 2014) and DEANR1 (or LINC00261) (Jiang et al.,2015)] and PUNISHER – were identified and functionallycharacterized in zebrafish embryos as well as in mouse ESCs(Kurian et al., 2015). These lncRNAs are specifically expressed inpluripotent stem cells, cardiovascular progenitors and differentiatedendothelial cells. LOF analyses, using shRNAs in mouse ESCs andMOs against zebrafish orthologs in vivo, demonstrate that all threelncRNAs are involved in cardiovascular development (Kurian et al.,2015).
LncRNAs with cellular functions but no apparent in vivophenotypeThe lncRNA KO models discussed above indicate that some haveessential roles in development in vivo. However, this is not alwaysthe case. Some lncRNAs, despite exhibiting reasonably highexpression levels and conservation, fail to show a clear phenotypewhen deleted in mice (Oliver et al., 2015; Sauvageau et al., 2013).Interestingly, several lncRNA KO animals show subtle or no
3889
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

phenotypes during development, whereas in vitro studies of thesame lncRNA suggest an essential function in the cell. An exampleof such an lncRNA is Malat1, which is among the most abundant,widely expressed and conserved lncRNAs in vertebrate cells.Malat1 localizes to nuclear speckles and has been shown to regulatesynapse formation bymodulating a subset of genes that have roles innuclear and synapse function (Bernard et al., 2010), to regulatesplicing in a human cell line (Tripathi et al., 2010), and to haveseveral important roles in regulating metastasis potential in cancercells (Gutschner et al., 2013). To address the physiological functionof Malat1 in a living organism, three KO mouse models have beengenerated independently (Eissmann et al., 2012; Nakagawa et al.,2012; Zhang et al., 2012). Unexpectedly, these mice are viable andfertile, initially showing no apparent gross phenotypes.Interestingly, a study using one of these KO strains has shown aretinal vascularization phenotype (Michalik et al., 2014), andanother has showed that the Malat1 KO mice exhibit enhancedtumor differentiation and a reduced tendency to form mammarytumors in the background of transgenic expression of a strongoncogene (Arun et al., 2016). In one of the three models, minoreffects on the expression of several genes, including genesneighboring Malat1, are observed, indicating a potential cis-regulatory role for Malat1 in gene transcription (Zhang et al.,2012), although it is likely that these changes are a result ofremoving the strong Malat1 promoter rather than the Malat1 RNAitself. Therefore, despite its high expression and conservationbetween human and fish (Ulitsky et al., 2011), which, based on thecriteria described above, makes it an excellent candidate for havingan important function,Malat1 does not appear to be required for theproper development of whole-organism physiology. However, it ispossible thatMalat1 is important under suboptimal conditions or inspecific cell types, and there is strong evidence for its roles in cancerand metastasis (Schmitt and Chang, 2016).
Roles for lncRNAs during stem cell differentiationA large number of studies carried out in the past five years haveimplicated lncRNAs in regulating stem cell maintenance anddifferentiation. For example, specific lncRNAs have now beenimplicated in neuronal differentiation [e.g. RMST (Ng et al., 2013),Miat (Aprea et al., 2013), Tunar (also known as megamind orTUNA) (Lin et al., 2014)], epidermal differentiation [e.g. DANCR(Kretz et al., 2012)], cardiac differentiation [e.g. Braveheart(Klattenhoff et al., 2013)], endoderm differentiation [e.g. ALIEN(Jiang et al., 2015; Kurian et al., 2015)], endothelial differentiation[e.g. SENCR (Boulberdaa et al., 2016)], adipocyte differentiation[e.g. lnc-RAP1-10 (Sun et al., 2013)] and hematopoieticdifferentiation [e.g. hoxBlinc (Deng et al., 2016)]. These studieshave shown that lncRNAs play important roles in establishing andmaintaining cell identity throughout the mammalian body. In thesevarious studies, transcriptional profiling is typically first used tofind the lncRNAs that are differentially expressed during thedifferentiation process. Perturbation approaches, most commonlyusing RNAi, are then typically used to probe lncRNA functionsduring differentiation. More recent studies have employed acombination of post-transcriptional repression using RNAi andantisense oligonucleotides with CRISPR/Cas9 genome editing. Forinstance, this combination has recently been used to study the rolesof Haunt (Halr1 or linc-Hoxa1) in mouse ESCs and duringdifferentiation into embryoid bodies (Yin et al., 2015). Using anextensive allelic series of edited mouse ESC lines, it was shown thatHaunt lncRNA negatively modulates the activity of enhancersfound in the same locus, thus acting as a repressor during the early
activation of proximal genes in the HoxA cluster. This use of amixture of techniques and a combination of strategies in the contextof CRISPR-based editing is now expected to become standard in thefield.
Large-scale functional screens have also revealed roles forlncRNAs in the process of stem cell differentiation. For example,shRNA libraries have been used to target hundreds of shRNAs inmouse ESCs (Guttman et al., 2011; Lin et al., 2014), and haveimplicated numerous lncRNAs in the maintenance of pluripotencyand differentiation towards various lineages. In the future, CRISPR-based screens are expected to be of use in identifying functionallyimportant lncRNAs in various differentiation systems, as well as todelineate regions within lncRNAs that are functionally important.
Characteristic roles of lncRNAs in gene regulatory networksAs the number of lncRNAs that have been studied in the context ofdevelopment, stem cells and cell differentiation is still small,realistically it is too early to speak of their ‘characteristic’ roles.Furthermore, in contrast to TFs and miRNAs (Box 3), the fewexperimentally characterized lncRNAs appear to be very diversewith regards to their modes of action and regulation. Nonetheless,an emerging theme is that lncRNAs play predominantly local roles,acting on just a few targets that are mostly located in cis to the site oflncRNA transcription. In line with this, lncRNAs are enriched in thevicinity of genes encoding transcriptional regulators and, even moreso, TFs involved in embryonic development (Ulitsky et al., 2011).In addition, some lncRNAs, such as CDR1as, can affect the activityof specific miRNAs. Thus, various lncRNAs are likely to influencedevelopment and differentiation by affecting only a few specificmaster regulators. Several specific lncRNA activities can becurrently foreseen in this context (Fig. 3). In the progenitor state,prior to cell fate transition, lncRNAs can facilitate local repressionand establish a threshold for the activation of specific targetsrequired for differentiation. During the early stages of cell fatetransition, transcription through the lncRNA locus and/or thelncRNA product itself can change the local chromatin environmentto facilitate the binding of TFs and their co-factors to yield increasedgene activation or repression. At later stages, lncRNAs can also playroles in positive-feedback loops, which are known to be importantfor high and stable TF expression. In this case, the lncRNAwould be
Box 3. Transcription factors and miRNAs: general modesof actionTranscription factors (TFs) and microRNAs (miRNAs) are establishedkey players in developmental pathways, and they have both shared anddifferential characteristics (Hobert, 2004). TFs and miRNAs regulateexpression in trans via short cis-regulatory elements present in theirtargets. Each target integrates diverse signals coming from multipleregulators to yield precise protein expression levels. This combinatorialregulation facilitates a myriad of transcriptional programs essential forestablishing cell fate in multicellular organisms. In many cases, TFs actas master regulators that dictate the expression of genes supporting aparticular cell fate while repressing the expression of other genes. Bycontrast, miRNAs typically play a more refined role, modulating theexpression levels of genes that should be expressed in the same cells asthe miRNA, and repressing the expression of genes that should beexpressed in alternative lineages (Bartel, 2009). At least in modelorganisms, a substantially larger fraction of TFs than of miRNAs isessential for proper development in laboratory conditions (Hobert, 2008).TFs and miRNAs also typically act on hundreds to thousands of targetgenes and, accordingly, are expressed at hundreds to tens of thousandsof copies per cell.
3890
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

a transcriptional target of the TF and act to further increase TFexpression. Finally, and akin to some of their roles in imprinting,lncRNAs can play repressive roles, affecting the chromatinenvironment or recruiting repressive complexes to prevent thetranscription of genes driving alternative fates.
ConclusionsLncRNAs are emerging as an important class of regulators thatfunction during embryonic development and are endowed withcertain features that set them apart from other classes of regulators.The currently annotated lncRNAs are likely to constitute anagglomeration of multiple functional classes, each with their owncharacteristics and relative importance. To resolve these classes,researchers have to overcome several hurdles, including the need todistinguish between lncRNA functions and those ofDNA elements intheir loci, and the need to use multiple perturbation strategies toovercome inherent limitations and off-target effects of the variousavailable perturbation methods. Still, the field is progressing rapidly,and we expect that future studies will uncover a multitude ofimportant roles for lncRNAs in virtually any central differentiationprocess. As lncRNAs are emerging as crucial players in bothmaintaining progenitor identity and driving differentiation, it is also
expected that the manipulation of lncRNA genes will allow thedevelopment of more efficient cell differentiation protocols forregenerative medicine. Lastly, understanding the roles of lncRNAsin development and cell differentiation will undoubtedly beinstrumental for untangling their functions in those human diseasesfor which lncRNA dysregulation is reported.
AcknowledgementsWe thank Neta Degani and Binyamin Zuckerman for comments on the manuscriptand members of the I.U. lab for fruitful discussions.
Competing interestsThe authors declare no competing or financial interests.
FundingWork in the authors’ laboratories is funded by Israeli Centers for ResearchExcellence [1796/12]; Israel Science Foundation [1242/14 and 1984/14]; EuropeanResearch Council lincSAFARI; Minerva Foundation; Fritz Thyssen Stiftung; LaponRaymond; and the Abramson Family Center for Young Scientists. I.U. is incumbentof the Sygnet Career Development Chair for Bioinformatics and recipient of an AlonFellowship.
ReferencesAmaral, P. P., Leonardi, T., Han, N., Vire, E., Gascoigne, D. K., Arias-Carrasco,
R., Buscher, M., Zhang, A., Pluchino, S., Maracaja-Coutinho, V. et al. (2016).Genomic positional conservation identifies topological anchor point (tap)RNAslinked to developmental loci. bioRxiv doi: 10.1101/051052.
Anderson, D. M., Anderson, K. M., Chang, C.-L., Makarewich, C. A., Nelson,B. R., McAnally, J. R., Kasaragod, P., Shelton, J. M., Liou, J., Bassel-Duby, R.et al. (2015). A micropeptide encoded by a putative long noncoding RNAregulates muscle performance. Cell 160, 595-606.
Aprea, J., Prenninger, S., Dori, M., Ghosh, T., Monasor, L. S., Wessendorf, E.,Zocher, S., Massalini, S., Alexopoulou, D., Lesche, M. et al. (2013).Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J. 32,3145-3160.
Arun, G., Diermeier, S., Akerman, M., Chang, K.-C., Wilkinson, J. E., Hearn, S.,Kim, Y., MacLeod, A. R., Krainer, A. R., Norton, L. et al. (2016). Differentiation ofmammary tumors and reduction in metastasis upon Malat1 lncRNA loss. GenesDev. 30, 34-51.
Barber, B. A. and Rastegar, M. (2010). Epigenetic control of Hox genes duringneurogenesis, development, and disease. Ann. Anat. 192, 261-274.
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell136, 215-233.
Bassett, A. R., Akhtar, A., Barlow, D. P., Bird, A. P., Brockdorff, N., Duboule, D.,Ephrussi, A., Ferguson-Smith, A. C., Gingeras, T. R., Haerty, W. et al. (2014).Considerations when investigating lncRNA function in vivo. eLife 3, e03058.
Batista, P. J. and Chang, H. Y. (2013). Long noncoding RNAs: cellular addresscodes in development and disease. Cell 152, 1298-1307.
Berghoff, E. G., Clark, M. F., Chen, S., Cajigas, I., Leib, D. E. and Kohtz, J. D.(2013). Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancermethylation andthe differential transcriptional control of adjacent genes. Development 140,4407-4416.
Bernard, D., Prasanth, K. V., Tripathi, V., Colasse, S., Nakamura, T., Xuan, Z.,Zhang, M. Q., Sedel, F., Jourdren, L., Coulpier, F. et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating geneexpression. EMBO J. 29, 3082-3093.
Bond, A. M., VanGompel, M. J. W., Sametsky, E. A., Clark, M. F., Savage, J. C.,Disterhoft, J. F. and Kohtz, J. D. (2009). Balanced gene regulation by anembryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat.Neurosci. 12, 1020-1027.
Boulberdaa, M., Scott, E., Ballantyne, M., Garcia, R., Descamps, B., Angelini,G. D., Brittan, M., Hunter, A., McBride, M., McClure, J. et al. (2016). A role forthe long noncoding RNA SENCR in commitment and function of endothelial cells.Mol. Ther. 24, 978-990.
Brockdorff, N. (2013). Noncoding RNA and Polycomb recruitment. RNA 19,429-442.
Brown, J. B., Boley, N., Eisman, R., May, G. E., Stoiber, M. H., Duff, M. O., Booth,B.W., Wen, J., Park, S., Suzuki, A. M. et al. (2014). Diversity and dynamics of theDrosophila transcriptome. Nature 512, 393-399.
Bu, D. C., Luo, H. T., Jiao, F., Fang, S. S., Tan, C. F., Liu, Z. Y. and Zhao, Y. (2015).Evolutionary annotation of conserved long non-coding RNAs in major mammalianspecies. Sci. China Life Sci. 58, 787-798.
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A. andRinn, J. L. (2011). Integrative annotation of human large intergenic noncoding
Progenitor
Progenitor
TF
Fate B
Fate A
Progenitor
TF
Fate B
Fate A
Progenitor
Fate B
Fate A
Fate B
Fate A
TF
Prevents precocious differentiation by establishinga repressive environment
TF
Facilitates chromatin accessibility to enable TF binding
Positive-feedback loop reinforcing TF activation
Suppresses alternative cell fates
Fig. 3. Potential roles for lncRNAs in cell fate decisions duringdevelopment. LncRNAs can play various roles during the establishment ofcell identity. In the progenitor state, lncRNAs can repress differentiationprograms to prevent precocious differentiation (e.g. by establishing arepressive chromatin environment). At the onset of differentiation, thetranscription of lncRNAs (i.e. the act of transcription alone and/or the lncRNAproduct itself ) can alter the chromatin environment to facilitate the binding ofTFs. If the lncRNA target gene is also a TF, lncRNA activity can be reinforcedby expression of the target gene resulting in a positive-feedback loop. Inaddition, lncRNAs expressed during differentiation can repress regulatoryprograms required for the establishment of alternative cell fates.
3891
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

RNAs reveals global properties and specific subclasses. Genes Dev. 25,1915-1927.
Cabili, M. N., Dunagin, M. C., McClanahan, P. D., Biaesch, A., Padovan-Merhar,O., Regev, A., Rinn, J. L. and Raj, A. (2015). Localization and abundanceanalysis of human lncRNAs at single-cell and single-molecule resolution.Genome Biol. 16, 20.
Carlevaro-Fita, J., Rahim, A., Guigo, R., Vardy, L. A. and Johnson, R. (2016).Cytoplasmic long noncoding RNAs are frequently bound to and degraded atribosomes in human cells. RNA 22, 867-882.
Carrieri, C., Cimatti, L., Biagioli, M., Beugnet, A., Zucchelli, S., Fedele, S.,Pesce, E., Ferrer, I., Collavin, L., Santoro, C. et al. (2012). Long non-codingantisense RNA controls Uchl1 translation through an embedded SINEB2 repeat.Nature 491, 454-457.
Carrozza, M. J., Li, B., Florens, L., Suganuma, T., Swanson, S. K., Lee, K. K.,Shia, W.-J., Anderson, S., Yates, J., Washburn, M. P. et al. (2005). Histone H3methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppressspurious intragenic transcription. Cell 123, 581-592.
Chen, J., Shishkin, A. A., Zhu, X., Kadri, S., Maza, I., Guttman, M., Hanna, J. H.,Regev, A. andGarber, M. (2016). Evolutionary analysis acrossmammals revealsdistinct classes of long non-coding RNAs. Genome Biol. 17, 19.
Chew, G.-L., Pauli, A., Rinn, J. L., Regev, A., Schier, A. F. and Valen, E. (2013).Ribosome profiling reveals resemblance between long non-coding RNAs and 5′leaders of coding RNAs. Development 140, 2828-2834.
Clark, M. B., Amaral, P. P., Schlesinger, F. J., Dinger, M. E., Taft, R. J., Rinn, J. L.,Ponting, C. P., Stadler, P. F., Morris, K. V., Morillon, A. et al. (2011). The realityof pervasive transcription. PLoS Biol. 9, e1000625.
Clemson, C. M., Hutchinson, J. N., Sara, S. A., Ensminger, A. W., Fox, A. H.,Chess, A. and Lawrence, J. B. (2009). An architectural role for a nuclearnoncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol.Cell 33, 717-726.
Cozzitorto, J. A., Jimbo, M., Chand, S., Blanco, F., Lal, S., Gilbert, M., Winter,J. M., Gorospe, M. and Brody, J. R. (2015). Studying RNA-binding proteininteractions with target mRNAs in eukaryotic cells: native ribonucleoproteinimmunoprecipitation (RIP) assays. Methods Mol. Biol. 1262, 239-246.
Davidovich, C., Zheng, L., Goodrich, K. J. and Cech, T. R. (2013). PromiscuousRNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 20,1250-1257.
DeChiara, T. M., Robertson, E. J. and Efstratiadis, A. (1991). Parental imprintingof the mouse insulin-like growth factor II gene. Cell 64, 849-859.
Deng, C., Li, Y., Zhou, L., Cho, J., Patel, B., Terada, N., Li, Y., Bungert, J., Qiu, Y.and Huang, S. (2016). HoxBlinc RNA recruits Set1/MLL complexes to activateHox gene expression patterns and mesoderm lineage development.Cell Rep. 14,103-114.
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H.,Guernec, G., Martin, D., Merkel, A., Knowles, D. G. et al. (2012). TheGENCODE v7 catalog of human long noncoding RNAs: Analysis of their genestructure, evolution, and expression. Genome Res. 22, 1775-1789.
Dominguez, A. A., Lim, W. A. and Qi, L. S. (2016). Beyond editing: repurposingCRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol.Cell Biol. 17, 5-15.
Dunagin, M., Cabili, M. N., Rinn, J. and Raj, A. (2015). Visualization of lncRNA bysingle-molecule fluorescence in situ hybridization.Methods Mol. Biol. 1262, 3-19.
Ebisuya, M., Yamamoto, T., Nakajima, M. and Nishida, E. (2008). Ripples fromneighbouring transcription. Nat. Cell Biol. 10, 1106-1113.
Eissmann, M., Gutschner, T., Hammerle, M., Gunther, S., Caudron-Herger, M.,Gross, M., Schirmacher, P., Rippe, K., Braun, T., Zornig, M. et al. (2012). Lossof the abundant nuclear non-coding RNA MALAT1 is compatible with life anddevelopment. RNA Biol. 9, 1076-1087.
Fang, R., Barbera, A. J., Xu, Y., Rutenberg, M., Leonor, T., Bi, Q., Lan, F., Mei, P.,Yuan, G.-C., Lian, C. et al. (2010). Human LSD2/KDM1b/AOF1 regulates genetranscription by modulating intragenic H3K4me2 methylation. Mol. Cell 39,222-233.
Fitzpatrick, G. V., Soloway, P. D. and Higgins, M. J. (2002). Regional loss ofimprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat.Genet. 32, 426-431.
Gabory, A., Ripoche, M.-A., Le Digarcher, A., Watrin, F., Ziyyat, A., Forne, T.,Jammes, H., Ainscough, J. F. X., Surani, M. A., Journot, L. et al. (2009). H19acts as a trans regulator of the imprinted gene network controlling growth in mice.Development 136, 3413-3421.
Gendrel, A.-V. and Heard, E. (2014). Noncoding RNAs and epigeneticmechanisms during X-chromosome inactivation. Annu. Rev. Cell Dev. Biol. 30,561-580.
Ghosh, S., Tibbit, C. and Liu, J.-L. (2016). Effective knockdown of Drosophila longnon-coding RNAs by CRISPR interference. Nucleic Acids Res. 44, e84.
Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y., Whitehead,E. H., Guimaraes, C., Panning, B., Ploegh, H. L., Bassik, M. C. et al. (2014).Genome-scale CRISPR-mediated control of gene repression and activation. Cell159, 647-661.
Goff, L. A., Groff, A. F., Sauvageau, M., Trayes-Gibson, Z., Sanchez-Gomez,D. B., Morse, M., Martin, R. D., Elcavage, L. E., Liapis, S. C., Gonzalez-Celeiro,
M. et al. (2015). Spatiotemporal expression and transcriptional perturbations bylong noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. USA 112,6855-6862.
Gong, C. andMaquat, L. E. (2011). lncRNAs transactivate STAU1-mediatedmRNAdecay by duplexing with 3′ UTRs via Alu elements. Nature 470, 284-288.
Grote, P., Wittler, L., Hendrix, D., Koch, F., Wahrisch, S., Beisaw, A., Macura, K.,Blass, G., Kellis, M., Werber, M. et al. (2013). The tissue-specific lncRNA Fendrris an essential regulator of heart and body wall development in the mouse. Dev.Cell 24, 206-214.
Gutschner, T., Hammerle, M. and Diederichs, S. (2013). MALAT1 — a paradigmfor long noncoding RNA function in cancer. J. Mol. Med. 91, 791-801.
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., Huarte, M.,Zuk, O., Carey, B. W., Cassady, J. P. et al. (2009). Chromatin signature revealsover a thousand highly conserved large non-coding RNAs in mammals. Nature458, 223-227.
Guttman, M., Donaghey, J., Carey, B. W., Garber, M., Grenier, J. K., Munson, G.,Young, G., Lucas, A. B., Ach, R., Bruhn, L. et al. (2011). lincRNAs act in thecircuitry controlling pluripotency and differentiation. Nature 477, 295-300.
Guttman,M., Russell, P., Ingolia, N. T.,Weissman, J. S. and Lander, E. S. (2013).Ribosome profiling provides evidence that large noncoding RNAs do not encodeproteins. Cell 154, 240-251.
Hacisuleyman, E., Goff, L. A., Trapnell, C., Williams, A., Henao-Mejia, J., Sun,L., McClanahan, P., Hendrickson, D. G., Sauvageau, M., Kelley, D. R. et al.(2014). Topological organization of multichromosomal regions by the longintergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 21, 198-206.
Hammerle, M., Gutschner, T., Uckelmann, H., Ozgur, S., Fiskin, E., Gross, M.,Skawran, B., Geffers, R., Longerich, T., Breuhahn, K. et al. (2013).Posttranscriptional destabilization of the liver-specific long noncoding RNAHULC by the IGF2 mRNA-binding protein 1 (IGF2BP1). Hepatology 58,1703-1712.
Herriges, M. J., Swarr, D. T., Morley, M. P., Rathi, K. S., Peng, T., Stewart, K. M.and Morrisey, E. E. (2014). Long noncoding RNAs are spatially correlated withtranscription factors and regulate lung development. Genes Dev. 28, 1363-1379.
Hezroni, H., Koppstein, D., Schwartz, M. G., Avrutin, A., Bartel, D. P. andUlitsky, I. (2015). Principles of long noncoding RNA evolution derived from directcomparison of transcriptomes in 17 species. Cell Rep. 11, 1110-1122.
Hirose, T. and Nakagawa, S. (2012). Paraspeckles: possible nuclear hubs by theRNA for the RNA. Biomol. Concepts 3, 415-428.
Hobert, O. (2004). Common logic of transcription factor and microRNA action.Trends Biochem. Sci. 29, 462-468.
Hobert, O. (2008). Gene regulation by transcription factors and microRNAs.Science 319, 1785-1786.
Housman, G. and Ulitsky, I. (2015). Methods for distinguishing between protein-coding and long noncoding RNAs and the elusive biological purpose of translationof long noncoding RNAs. Biochim. Biophys. Acta 1859, 31-40.
Huarte, M. (2015). The emerging role of lncRNAs in cancer. Nat. Med. 21,1253-1261.
Ingolia, N. T., Brar, G. A., Stern-Ginossar, N., Harris, M. S., Talhouarne, G. J. S.,Jackson, S. E., Wills, M. R. and Weissman, J. S. (2014). Ribosome profilingreveals pervasive translation outside of annotated protein-coding genes.Cell Rep.8, 1365-1379.
Iyer, M. K., Niknafs, Y. S., Malik, R., Singhal, U., Sahu, A., Hosono, Y., Barrette,T. R., Prensner, J. R., Evans, J. R., Zhao, S. et al. (2015). The landscape of longnoncoding RNAs in the human transcriptome. Nat. Genet. 47, 199-208.
Jiang, W., Liu, Y., Liu, R., Zhang, K. and Zhang, Y. (2015). The lncRNA DEANR1facilitates human endoderm differentiation by activating FOXA2 expression. CellRep. 11, 137-148.
Kapusta, A. and Feschotte, C. (2014). Volatile evolution of long noncoding RNArepertoires: mechanisms and biological implications. Trends Genet. 30, 439-452.
Kapusta, A., Kronenberg, Z., Lynch, V. J., Zhuo, X., Ramsay, L., Bourque, G.,Yandell, M. and Feschotte, C. (2013). Transposable elements are majorcontributors to the origin, diversification, and regulation of vertebrate longnoncoding RNAs. PLoS Genet. 9, e1003470.
Kaushik, K., Leonard, V. E., Kv, S., Lalwani, M. K., Jalali, S., Patowary, A., Joshi,A., Scaria, V. and Sivasubbu, S. (2013). Dynamic expression of long non-codingRNAs (lncRNAs) in adult zebrafish. PLoS ONE 8, e83616.
Kelley, D. R. and Rinn, J. L. (2012). Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 13, R107.
Klattenhoff, C. A., Scheuermann, J. C., Surface, L. E., Bradley, R. K., Fields,P. A., Steinhauser, M. L., Ding, H., Butty, V. L., Torrey, L., Haas, S. et al. (2013).Braveheart, a long noncoding RNA required for cardiovascular lineagecommitment. Cell 152, 570-583.
Kok, F. O., Shin, M., Ni, C.-W., Gupta, A., Grosse, A. S., van Impel, A.,Kirchmaier, B. C., Peterson-Maduro, J., Kourkoulis, G., Male, I. et al. (2015).Reverse genetic screening reveals poor correlation between morpholino-inducedand mutant phenotypes in zebrafish. Dev. Cell 32, 97-108.
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M. andXiong, Y. (2011). Long non-coding RNA ANRIL is required for the PRC2recruitment to and silencing of p15(INK4B) tumor suppressor gene.Oncogene 30,1956-1962.
3892
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

Koziol, M. J. and Rinn, J. L. (2010). RNA traffic control of chromatin complexes.Curr. Opin. Genet. Dev. 20, 142-148.
Krawczyk, M. and Emerson, B. M. (2014). p50-associated COX-2 extragenic RNA(PACER) activates COX-2 gene expression by occluding repressive NF-kappaBcomplexes. eLife 3, e01776.
Kretz, M., Webster, D. E., Flockhart, R. J., Lee, C. S., Zehnder, A., Lopez-Pajares, V., Qu, K., Zheng, G. X., Chow, J., Kim, G. E. et al. (2012). Suppressionof progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev.26, 338-343.
Kurian, L., Aguirre, A., Sancho-Martinez, I., Benner, C., Hishida, T., Nguyen,T. B., Reddy, P., Nivet, E., Krause, M. N., Nelles, D. A. et al. (2015). Identificationof novel long noncoding RNAs underlying vertebrate cardiovascular development.Circulation 131, 1278-1290.
Lai, K. M., Gong, G., Atanasio, A., Rojas, J., Quispe, J., Posca, J., White, D.,Huang, M., Fedorova, D., Grant, C. et al. (2015). Diverse phenotypes andspecific transcription patterns in twenty mouse lines with ablated LincRNAs.PLoSONE 10, e0125522.
Latos, P. A., Pauler, F. M., Koerner, M. V., Senergin, H. B., Hudson, Q. J.,Stocsits, R. R., Allhoff, W., Stricker, S. H., Klement, R. M., Warczok, K. E. et al.(2012). Airn transcriptional overlap, but not its lncRNA products, inducesimprinted Igf2r silencing. Science 338, 1469-1472.
Lee, J. T. and Bartolomei, M. S. (2013). X-inactivation, imprinting, and longnoncoding RNAs in health and disease. Cell 152, 1308-1323.
Lee, S., Kopp, F., Chang, T.-C., Sataluri, A., Chen, B., Sivakumar, S., Yu, H., Xie,Y. andMendell, J. T. (2016). Noncoding RNANORAD regulates genomic stabilityby sequestering PUMILIO proteins. Cell 164, 69-80.
Leighton, P. A., Ingram, R. S., Eggenschwiler, J., Efstratiadis, A. and Tilghman,S. M. (1995). Disruption of imprinting caused by deletion of the H19 gene region inmice. Nature 375, 34-39.
Lennox, K. A. and Behlke, M. A. (2016). Cellular localization of long non-codingRNAs affects silencing by RNAi more than by antisense oligonucleotides.NucleicAcids Res. 44, 863-877.
Li, L. and Chang, H. Y. (2014). Physiological roles of long noncoding RNAs: insightfrom knockout mice. Trends Cell Biol. 24, 594-602.
Li, L., Liu, B., Wapinski, O. L., Tsai, M.-C., Qu, K., Zhang, J., Carlson, J. C., Lin,M., Fang, F., Gupta, R. A. et al. (2013). Targeted disruption of Hotair leads tohomeotic transformation and gene derepression. Cell Rep. 5, 3-12.
Lin, N., Chang, K.-Y., Li, Z., Gates, K., Rana, Z. A., Dang, J., Zhang, D., Han, T.,Yang, C.-S., Cunningham, T. J. et al. (2014). An evolutionarily conserved longnoncoding RNATUNA controls pluripotency and neural lineage commitment.Mol.Cell 53, 1005-1019.
Lorenzen, J. M. and Thum, T. (2016). Long noncoding RNAs in kidney andcardiovascular diseases. Nat. Rev. Nephrol. 12, 360-373.
Luo, S., Lu, J. Y., Liu, L., Yin, Y., Chen, C., Han, X., Wu, B., Xu, R., Liu,W., Yan, P.et al. (2016). Divergent lncRNAs regulate gene expression and lineagedifferentiation in pluripotent cells. Cell Stem Cell 18, 637-652.
Marahrens, Y., Panning, B., Dausman, J., Strauss, W. and Jaenisch, R. (1997).Xist-deficient mice are defective in dosage compensation but notspermatogenesis. Genes Dev. 11, 156-166.
Marın-Bejar, O., Marchese, F. P., Athie, A., Sanchez, Y., Gonzalez, J., Segura, V.,Huang, L., Moreno, I., Navarro, A., Monzo, M. et al. (2013). Pint lincRNAconnects the p53 pathway with epigenetic silencing by the Polycomb repressivecomplex 2. Genome Biol. 14, R104.
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., Maier, L.,Mackowiak, S. D., Gregersen, L. H., Munschauer, M. et al. (2013). CircularRNAs are a large class of animal RNAs with regulatory potency. Nature 495,333-338.
Michalik, K. M., You, X., Manavski, Y., Doddaballapur, A., Zornig, M., Braun, T.,John, D., Ponomareva, Y., Chen, W., Uchida, S. et al. (2014). Long noncodingRNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res.114, 1389-1397.
Monnier, P., Martinet, C., Pontis, J., Stancheva, I., Ait-Si-Ali, S. and Dandolo, L.(2013). H19 lncRNA controls gene expression of the imprinted gene network byrecruiting MBD1. Proc. Natl. Acad. Sci. USA 110, 20693-20698.
Nagano, T., Mitchell, J. A., Sanz, L. A., Pauler, F. M., Ferguson-Smith, A. C., Feil,R. and Fraser, P. (2008). The Air noncoding RNA epigenetically silencestranscription by targeting G9a to chromatin. Science 322, 1717-1720.
Nakagawa, S., Naganuma, T., Shioi, G. and Hirose, T. (2011). Paraspeckles aresubpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol.193, 31-39.
Nakagawa, S., Ip, J. Y., Shioi, G., Tripathi, V., Zong, X., Hirose, T. and Prasanth,K. V. (2012). Malat1 is not an essential component of nuclear speckles in mice.RNA 18, 1487-1499.
Nakagawa, S., Shimada, M., Yanaka, K., Mito, M., Arai, T., Takahashi, E., Fujita,Y., Fujimori, T., Standaert, L., Marine, J. C. et al. (2014). The lncRNA Neat1 isrequired for corpus luteum formation and the establishment of pregnancy in asubpopulation of mice. Development 141, 4618-4627.
Necsulea, A., Soumillon, M., Warnefors, M., Liechti, A., Daish, T., Zeller, U.,Baker, J. C., Grutzner, F. and Kaessmann, H. (2014). The evolution of lncRNArepertoires and expression patterns in tetrapods. Nature 505, 635-640.
Nelson, B. R., Makarewich, C. A., Anderson, D. M., Winders, B. R., Troupes,C. D., Wu, F., Reese, A. L., McAnally, J. R., Chen, X., Kavalali, E. T. et al.(2016). A peptide encoded by a transcript annotated as long noncoding RNAenhances SERCA activity in muscle. Science 351, 271-275.
Ng, S.-Y., Bogu, G. K., Soh, B. S. and Stanton, L. W. (2013). The long noncodingRNA RMST interacts with SOX2 to regulate neurogenesis.Mol. Cell 51, 349-359.
Oliver, P. L., Chodroff, R. A., Gosal, A., Edwards, B., Cheung, A. F. P., Gomez-Rodriguez, J., Elliot, G., Garrett, L. J., Lickiss, T., Szele, F. et al. (2015).Disruption of Visc-2, a brain-expressed conserved long noncoding RNA, does notelicit an overt anatomical or behavioral phenotype. Cereb. Cortex 25, 3572-3585.
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski,J., Nagano, T., Mancini-Dinardo, D. and Kanduri, C. (2008). Kcnq1ot1antisense noncoding RNA mediates lineage-specific transcriptional silencingthrough chromatin-level regulation. Mol. Cell 32, 232-246.
Park, S.-W., Kuroda, M. I. and Park, Y. (2008). Regulation of histone H4 Lys16acetylation by predicted alternative secondary structures in roX noncoding RNAs.Mol. Cell. Biol. 28, 4952-4962.
Pauli, A., Rinn, J. L. and Schier, A. F. (2011). Non-coding RNAs as regulators ofembryogenesis. Nat. Rev. Genet. 12, 136-149.
Pauli, A., Valen, E., Lin, M. F., Garber, M., Vastenhouw,N. L., Levin, J. Z., Fan, L.,Sandelin, A., Rinn, J. L., Regev, A. et al. (2012). Systematic identification of longnoncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22,577-591.
Pauli, A., Norris, M. L., Valen, E., Chew, G.-L., Gagnon, J. A., Zimmerman, S.,Mitchell, A., Ma, J., Dubrulle, J., Reyon, D. et al. (2014). Toddler: an embryonicsignal that promotes cell movement via Apelin receptors. Science 343, 1248636.
Poirier, F., Chan, C. T., Timmons, P. M., Robertson, E. J., Evans, M. J. andRigby, P.W. (1991). Themurine H19 gene is activated during embryonic stem celldifferentiation in vitro and at the time of implantation in the developing embryo.Development 113, 1105-1114.
Ponting, C. P., Oliver, P. L. and Reik, W. (2009). Evolution and functions of longnoncoding RNAs. Cell 136, 629-641.
Quinn, J. J., Ilik, I. A., Qu, K., Georgiev, P., Chu, C., Akhtar, A. and Chang, H. Y.(2014). Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat. Biotechnol. 32, 933-940.
Quinn, J. J., Zhang, Q. C., Georgiev, P., Ilik, I. A., Akhtar, A. and Chang, H. Y.(2016). Rapid evolutionary turnover underlies conserved lncRNA–genomeinteractions. Genes Dev. 30, 191-207.
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A.,Goodnough, L. H., Helms, J. A., Farnham, P. J., Segal, E. et al. (2007).Functional demarcation of active and silent chromatin domains in humanHOX lociby noncoding RNAs. Cell 129, 1311-1323.
Ripoche, M. A., Kress, C., Poirier, F. and Dandolo, L. (1997). Deletion of the H19transcription unit reveals the existence of a putative imprinting control element.Genes Dev. 11, 1596-1604.
Roth, A. and Diederichs, S. (2015). Molecular biology: Rap and chirp about Xinactivation. Nature 521, 170-171.
Ruf, S., Symmons, O., Uslu, V. V., Dolle, D., Hot, C., Ettwiller, L. and Spitz, F.(2011). Large-scale analysis of the regulatory architecture of the mouse genomewith a transposon-associated sensor. Nat. Genet. 43, 379-386.
Sado, T., Wang, Z., Sasaki, H. and Li, E. (2001). Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 128, 1275-1286.
Sauvageau, M., Goff, L. A., Lodato, S., Bonev, B., Groff, A. F., Gerhardinger, C.,Sanchez-Gomez, D. B., Hacisuleyman, E., Li, E., Spence, M. et al. (2013).Multiple knockout mouse models reveal lincRNAs are required for life and braindevelopment. eLife 2, e01749.
Schmitt, A. M. and Chang, H. Y. (2016). Long noncoding RNAs in cancerpathways. Cancer Cell 29, 452-463.
Schorderet, P. and Duboule, D. (2011). Structural and functional differences in thelong non-coding RNA hotair in mouse and human. PLoS Genet. 7, e1002071.
Shechner, D. M., Hacisuleyman, E., Younger, S. T. and Rinn, J. L. (2015).Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat.Methods 12, 664-670.
Sigova, A. A., Abraham, B. J., Ji, X., Molinie, B., Hannett, N. M., Guo, Y. E.,Jangi, M., Giallourakis, C. C., Sharp, P. A. and Young, R. A. (2015).Transcription factor trapping by RNA in gene regulatory elements. Science 350,978-981.
Simon, M. D. (2016). Insight into lncRNA biology using hybridization captureanalyses. Biochim. Biophys. Acta 1859, 121-127.
Sleutels, F., Zwart, R. and Barlow, D. P. (2002). The non-coding Air RNA isrequired for silencing autosomal imprinted genes. Nature 415, 810-813.
Standaert, L., Adriaens, C., Radaelli, E., Van Keymeulen, A., Blanpain, C.,Hirose, T., Nakagawa, S. and Marine, J.-C. (2014). The long noncoding RNANeat1 is required for mammary gland development and lactation. RNA 20,1844-1849.
Sun, L., Goff, L. A., Trapnell, C., Alexander, R., Lo, K. A., Hacisuleyman, E.,Sauvageau,M., Tazon-Vega, B., Kelley, D. R., Hendrickson, D. G. et al. (2013).Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 110,3387-3392.
3893
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT

Tichon, A., Gil, N., Lubelsky, Y., Solomon, T. H., Lemze, D., Itzkovitz, S., Stern-Ginossar, N. and Ulitsky, I. (2016). A conserved abundant cytoplasmic longnoncoding RNA modulates repression by Pumilio proteins in human cells. Nat.Commun. 7, 12209.
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M.,Bennett, C. F., Sharma, A., Bubulya, P. A. et al. (2010). The nuclear-retainednoncoding RNAMALAT1 regulates alternative splicing by modulating SR splicingfactor phosphorylation. Mol. Cell 39, 925-938.
Ulitsky, I. (2016). Evolution to the rescue: using comparative genomics tounderstand long non-coding RNAs. Nat. Rev. Genet. 17, 601-614.
Ulitsky, I. and Bartel, D. P. (2013). lincRNAs: genomics, evolution, andmechanisms. Cell 154, 26-46.
Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H. and Bartel, D. P. (2011).Conserved function of lincRNAs in vertebrate embryonic development despiterapid sequence evolution. Cell 147, 1537-1550.
Valenzuela, D. M., Murphy, A. J., Frendewey, D., Gale, N.W., Economides, A. N.,Auerbach,W., Poueymirou,W. T., Adams, N. C., Rojas, J., Yasenchak, J. et al.(2003). High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21, 652-659.
van Heesch, S., van Iterson, M., Jacobi, J., Boymans, S., Essers, P. B., deBruijn, E., Hao, W., MacInnes, A. W., Cuppen, E. and Simonis, M. (2014).Extensive localization of long noncoding RNAs to the cytosol and mono- andpolyribosomal complexes. Genome Biol. 15, R6.
Wagner, E. J. and Carpenter, P. B. (2012). Understanding the language of Lys36methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115-126.
Wang, K. C., Yang, Y. W., Liu, B., Sanyal, A., Corces-Zimmerman, R., Chen, Y.,Lajoie, B. R., Protacio, A., Flynn, R. A., Gupta, R. A. et al. (2011). A longnoncoding RNA maintains active chromatin to coordinate homeotic geneexpression. Nature 472, 120-124.
Washietl, S., Kellis, M. and Garber, M. (2014). Evolutionary dynamics and tissuespecificity of human long noncoding RNAs in six mammals. Genome Res. 24,616-628.
Wutz, A. and Jaenisch, R. (2000). A shift from reversible to irreversible Xinactivation is triggered during ES cell differentiation. Mol. Cell 5, 695-705.
Yang, Y. W., Flynn, R. A., Chen, Y., Qu, K., Wan, B., Wang, K. C., Lei, M. andChang, H. Y. (2014). Essential role of lncRNA binding for WDR5 maintenance ofactive chromatin and embryonic stem cell pluripotency. eLife 3, e02046.
Yap, K. L., Li, S., Mun oz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., Gil, J.,Walsh, M. J. and Zhou, M.-M. (2010). Molecular interplay of the noncoding RNAANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptionalsilencing of INK4a. Mol. Cell 38, 662-674.
Yarmishyn, A. A., Batagov, A. O., Tan, J. Z., Sundaram, G. M., Sampath, P.,Kuznetsov, V. A. and Kurochkin, I. V. (2014). HOXD-AS1 is a novel lncRNAencoded in HOXD cluster and a marker of neuroblastoma progression revealedvia integrative analysis of noncoding transcriptome. BMC Genomics 15, Suppl.9, S7.
Yildirim, E., Kirby, J. E., Brown, D. E., Mercier, F. E., Sadreyev, R. I., Scadden,D. T. and Lee, J. T. (2013). Xist RNA is a potent suppressor of hematologic cancerin mice. Cell 152, 727-742.
Yin, Y., Yan, P., Lu, J., Song, G., Zhu, Y., Li, Z., Zhao, Y., Shen, B., Huang, X.,Zhu, H. et al. (2015). Opposing roles for the lncRNA haunt and its genomic locusin regulating HOXA gene activation during embryonic stem cell differentiation.CellStem Cell 16, 504-516.
Zhang, B., Arun, G., Mao, Y. S., Lazar, Z., Hung, G., Bhattacharjee, G., Xiao, X.,Booth, C. J., Wu, J., Zhang, C. et al. (2012). The lncRNA Malat1 is dispensablefor mouse development but its transcription plays a cis-regulatory role in the adult.Cell Rep. 2, 111-123.
Zhao, X.-Y. and Lin, J. D. (2015). Long noncoding RNAs: a new regulatory code inmetabolic control. Trends Biochem. Sci. 40, 586-596.
Zhao, J., Sun, B. K., Erwin, J. A., Song, J.-J. and Lee, J. T. (2008). Polycombproteins targeted by a short repeat RNA to the mouse X chromosome. Science322, 750-756.
3894
REVIEW Development (2016) 143, 3882-3894 doi:10.1242/dev.140962
DEVELO
PM
ENT


















