
The effect of polymer composition and structure on the ...
91
Rochester Institute of Technology RIT Scholar Works eses esis/Dissertation Collections 4-1-2008 e effect of polymer composition and structure on the photo-Fries rearrangement Glen David Labenski Follow this and additional works at: hp://scholarworks.rit.edu/theses is esis is brought to you for free and open access by the esis/Dissertation Collections at RIT Scholar Works. It has been accepted for inclusion in eses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected]. Recommended Citation Labenski, Glen David, "e effect of polymer composition and structure on the photo-Fries rearrangement" (2008). esis. Rochester Institute of Technology. Accessed from
Transcript of The effect of polymer composition and structure on the ...

Rochester Institute of TechnologyRIT Scholar Works
Theses Thesis/Dissertation Collections
4-1-2008
The effect of polymer composition and structureon the photo-Fries rearrangementGlen David Labenski
Follow this and additional works at: http://scholarworks.rit.edu/theses
This Thesis is brought to you for free and open access by the Thesis/Dissertation Collections at RIT Scholar Works. It has been accepted for inclusionin Theses by an authorized administrator of RIT Scholar Works. For more information, please contact [email protected].
Recommended CitationLabenski, Glen David, "The effect of polymer composition and structure on the photo-Fries rearrangement" (2008). Thesis. RochesterInstitute of Technology. Accessed from

The Effect of Polymer Composition and Structure on
the Photo-Fries Rearrangement.
Glen David Labenski
April 2008
Thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Chemistry.
Approved: ___________________________________ Thomas W. Smith (Advisor)
___________________________________ Paul Rosenberg (Department Head)
Department of Chemistry
Rochester Institute of Technology
Rochester, New York 14623-5603

ii
Abstract: The objective of research presented in this thesis is to elucidate the effect of
polymer composition and structure on the photo-Fries rearrangement in polymers bearing
aryl ester moieties. Towards this end, a systematic study wherein constraint, proximity,
and dielectric constant were varied was carried out. This study was enabled by the
synthesis of poly(phenylacrylate), poly(p-acetoxystyrene) and copolymers of
phenylacrylate or p-acetoxystyrene with ethylacrylate, butylacrylate and 2-
hydroxyethylmethacrylate. In addition, siloxane polymers with pendant aryl ester groups
were also synthesized.
Varying constraint by tethering aryl esters to the polymer backbone at the ester
group, tethering aryl esters to the polymer backbone through the aryl group and varying
the Tg of polymers had no effect on the rate of the photo-Fries rearrangement. The
hydration state of hydrophilic copolymers had no effect on conversion but did increase
formation of cage escape products and changed the ratio of products formed. Decreasing
the proximity of aryl ester units relative to one another lead to increased ultimate
conversion in the rearrangement. Altering from a carbon to siloxane polymer backbone
may have had an effect on the initial rate of rearrangement but the effect could not be
isolated from other variables (proximity and Tg).
Refractive index changes (∆n) which occurred as the polymers underwent
rearrangement were also evaluated. Refractive index changes between 0.01 and 0.07
were observed, with the ∆n being controlled in a dose dependent manner. Findings
discussed here may be significant in the development of holographic imaging systems,
optical data storage devices, waveguides and intra-ocular lens implants where materials
capable of significant refractive index modulation are desirable.

iii
Table of Contents
Introduction........................................................................................................................ 1
Objective ..................................................................................................................................... 1
Photochemistry of Aryl Esters ................................................................................................... 1
The photo-Fries Rearrangement in Constrained Media .......................................................... 5
The photo-Fries Rearrangement in Polymers......................................................................... 12
Utility of the Current Research................................................................................................ 19
Experimental .................................................................................................................... 24
Materials................................................................................................................................... 24
Instrumentation........................................................................................................................ 25
Synthesis ................................................................................................................................... 26
Poly(p-acetoxy styrene)..................................................................................................................... 26
Poly(phenyl acrylate). ....................................................................................................................... 27
Poly(phenyl acrylate-co-ethyl acrylate)........................................................................................... 27
Poly(p-acetoxy styrene-co-butyl acrylate). ...................................................................................... 28
Poly(phenyl acrylate-co-butyl acrylate). ......................................................................................... 29
Poly(phenyl acrylate-co-hydroxyethyl methacrylate). ................................................................... 30
Poly(acetoxy styrene-co-hydroxyethyl methacrylate). ................................................................... 32
Acetoxystyrene functionalized polysiloxane. .................................................................................. 33
Phenyl 3-butenoate functionalized polysiloxane................................................................... 35
Phenyl 3-butenoate....................................................................................................................... 35 Hydrosilylation of phenyl 3-butenoate. ...................................................................................... 36
Kinetics Analysis ...................................................................................................................... 37
Variable angle spectroscopic ellipsometry (VASE) Analysis.................................................. 37
Sensitization of the photo-Fries rearrangement. .................................................................... 38
Synthesis of naphthalene-1-yl benzoate. ......................................................................................... 38
Photo-Fries rearrangement of naphthalene-1-yl benzoate. ........................................................... 39
Results and Discussion .................................................................................................... 39
Synthesis of Polymers .............................................................................................................. 40
Kinetics of the photo-Fries Rearrangement ............................................................................ 42
Constraint. ......................................................................................................................................... 44

iv
Proximity. .......................................................................................................................................... 51
Dielectric Constant............................................................................................................................ 54
Refractive Index Changes........................................................................................................ 56
Preliminary Sensitization Experiments ................................................................................... 62
Conclusions and Future Directions ................................................................................ 64
Appendix........................................................................................................................... 65
References ........................................................................................................................ 76

v
List of Figures
Figure 1: Acetoxybenzene undergoing the photo-Fries rearrangement........................................................ 2
Figure 2: Detailed mechanism of photo-Fries rearrangement...................................................................... 3
Figure 3: Comparison of thermal and photochemical catalyzed Fries rearrangements............................... 4
Figure 4: Representation of the structure of ZSM-5 zeolite. ......................................................................... 6
Figure 5: Structure of Nafion®. .................................................................................................................... 7
Figure 6: Morphology of water-swollen Nafion®......................................................................................... 7
Figure 7: Structure of β-cyclodextrin............................................................................................................ 8
Figure 8: Rearrangement of β-cyclodextrin naphthyl ester complexes......................................................... 9
Figure 9: Styrene-based amphiphilic homopolymer (polymer A) utilized by Ramamurthy.......................... 11
Figure 10: Photo-Fries rearrangement of polycarbonate........................................................................... 13
Figure 11: Photo-Fries rearrangement of poly(p-acetoxystyrene) and poly(p-formyloxystyrene) ............. 15
Figure 12: Novel photo-Fries active polymers synthesized by Kern. .......................................................... 16
Figure 13:FT-IR spectra of PAS and poly(bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, phenyl ester)
undergoing the photo-Fries rearrangement. .............................................................................. 17
Figure 14: Cauchy fit curve obtained from PAS. ........................................................................................ 18
Figure 15: Additional photo-Fries active polymers studied by Kern. ......................................................... 19
Figure 16: Refractive index change associated with photo-Fries rearrangement of acetoxybenzene. ....... 20
Figure 17: Structure of poly(naphthylene-1-yl 4-vinyl-benzoate). ............................................................... 21
Figure 18: Photo-acid catalysis of the photo-Fries rearrangement of PFS in the presence of
triarylsulfonium hexafluoroarsenate. ......................................................................................... 21
Figure 19:Energy transfer observed between poly(sodium styrenesolfonate-co-2-vinylfluorene) and
1-naphthyl acetate. ..................................................................................................................... 22
Figure 20:Antenna effect in the rearrangement of naphthalene-1-yl benzoate in the presence of fluorene. 23
Figure 21: Synthesis of poly(p-acetoxystyrene)........................................................................................... 26
Figure 22: Synthesis of poly(phenyl acrylate). ............................................................................................ 27
Figure 23: Synthesis of poly(phenyl acrylate-co-ethyl acrylate)................................................................. 27
Figure 24: Synthesis of poly(p-acetoxystyrene-co-butyl acrylate). ............................................................. 28
Figure 25: Synthesis of poly(phenyl acrylate-co-butyl acrylate)................................................................. 29
Figure 26: Synthesis of poly(phenyl acrylate-co-hydroxyethyl methacrylate). ........................................... 30
Figure 27: Synthesis of poly(p-acetoxystyrene-co-hydroxyethyl methacrylate). ......................................... 32
Figure 28: Synthesis of p-acetoxystyrene functionalized polysiloxane. ...................................................... 33
Figure 29: Synthesis of phenyl 3-butenoate. ............................................................................................... 35
Figure 30: Synthesis of phenyl 3-butenoate functionalized polysiloxane.................................................... 36
Figure 31: Synthesis of naphthalene-1-yl benzoate..................................................................................... 38
Figure 32: Initial polymerization conditions for HEMA copolymers.......................................................... 41
Figure 33: Overlaid FT-IR Spectra of PPA at 0, 30 and 60 minutes. ......................................................... 43

vi
Figure 34: Comparison of constraint of PAS and PPA............................................................................... 44
Figure 35: Conversion of PAS and PPA versus time. ................................................................................. 45
Figure 36: Conversion of PAS and AS-BA verses time. .............................................................................. 46
Figure 37: Conversion of PPA, PA-EA and PA-BA verses time. ................................................................ 47
Figure 38: Yield of ortho and para products in PPA and PA-EA rearrangements. .................................... 48
Figure 39: Conversion of dry and water swollen AS-HEMA over time. ..................................................... 49
Figure 40: Conversion of dry and water swollen PA-HEMA over time. ..................................................... 49
Figure 41: Yield of ortho product in AS-HEMA rearrangement. ................................................................ 50
Figure 42: Yield of ortho and para products in PA-HEMA rearrangement ............................................... 51
Figure 43: Difference in proximity demonstrated between PAS and PA-BA. ............................................ 54
Figure 44: Conversion of PAS and AS-SiO. ................................................................................................ 55
Figure 45: Conversion of PPA and PA-SiO. ............................................................................................... 55
Figure 46:Cauchy fit curves showing ∆n of PAS with increasing irradiation time with
Kern’s data shown in inset plot. ................................................................................................. 57
Figure 47: Cauchy fit curves showing ∆n in PPA with increasing irradiation time. .................................. 58
Figure 48: Cauchy fit curves showing ∆n in PA-EA with increasing irradiation time. .............................. 59
Figure 49: Cauchy fit curves showing ∆n in PA-HEMA with increasing irradiation time. ........................ 59
Figure 50: Refractive indices of isolated aryl ester groups, dyads and triads
calculated from group contributions. ........................................................................................ 61
Figure 51: Linear dose dependent modulation of ∆n in PA-EA. ................................................................. 62
Figure 52: Photo-Fries rearrangement of naphthalene-1-yl benzoate
in the presence of increasing amounts of fluorene. .................................................................... 63

vii
List of Tables
Table 1: Product Selectivity Exhibited in Unstretched and Stretched LDPE Films. ................................... 10
Table 2: Product Selectivity Exhibited in micelle-like structures of polymer A. ......................................... 12
Table 3: Quantum Yields Observed From Irradiation of Poly(phenyl acrylate) and Phenyl Acetate. ........ 14
Table 4: Properties of polymers under investigation. ................................................................................. 40
Table 5: Ultimate Conversion observed in Polymers Under Investigation. ................................................ 52
Table 6: Observed ∆n in Polymers Under Investigation. ............................................................................ 60

viii
List of Abbreviations
AS-BA : poly(p-acetoxystyrene-co-butyl acrylate)
AS-HEMA : poly(p-acetoxystyrene -co-2-hydroxyethyl methacrylate)
AS-SiO : p-acetoxystyrene functionalized polysiloxane
LDPE : low density polyethylene
PA-BA : poly(phenyl acrylate-co-butyl acrylate)
PA-EA : poly(phenyl acrylate-co-ethyl acrylate)
PA-HEMA : poly(phenyl acrylate-co-2-hydroxyethyl methacrylate)
PAS : poly(p-acetoxystyrene)
PA-SiO : phenyl-3-butenoate functionalized polysiloxane
PFS : poly(p-formyloxy styrene)
PPA : poly(phenyl acrylate)
THF : tetrahydrofuran

ix
Acknowledgements
I must thank many people for their assistance and support towards completion of this thesis. Most notably, I thank Dr. Thomas Smith, my adviser. Dr. Smith has given me a wonderful opportunity to learn and work in his research group at RIT. Dr. Smith has taught me things in the classroom and laboratory that will no doubt stick with me throughout my career. He provided me with answers when I asked questions but more importantly, drew the answers from me when he knew I had the knowledge hidden within. I thank the professors who taught me in the courses I have taken at RIT. Through their efforts, they challenged and exposed me to new knowledge in the field of chemistry. I especially thank Dr. Chris Collison and Dr. Christina Collison who have contributed to this thesis as members of my committee.
I thank Bausch & Lomb for their financial support which allowed me to immerse myself into this research deeper than I ever thought possible. Special thanks go to Dr. Jay Kunzler for taking time from his job with B&L to serve on my thesis committee. Dr. Jani Dharmendra and Dr. Jeff Linhardt of B&L also supplied a great deal of input in this work. I thank the stock room personnel: Glenn Robinson, Dave Lake, John Gallucci, Paul Allen, Erin Madden, and the staff of student workers. They have worked hard to address my countless requests for equipment and chemicals that were necessary for me to achieve what I have. Tom Alston also deserves special recognition for training and maintenance of the instrumentation used on a day-to-day basis at RIT. Without the important efforts of these people, progress would not have been possible. I thank all of my fellow graduate students whom I have worked beside during the past two years. Your friendship and encouragement has kept me motivated throughout. I wish you all nothing but the best in your personal endeavors. I thank the teachers from Lake Shore, Nazareth College and the University at Buffalo. The education and skills imparted upon me by these people served as a strong foundation for what I have achieved.
Finally, I thank my family, Ron, Carol and Jeremy. The unconditional love, support and encouragement from these people has shown me that I am capable of achieving great things in life. I also thank Emily, who has stood by me throughout some of the most personally demanding and stressful chapters in my life.

1
Introduction
The photo-Fries rearrangement is the photo-catalyzed rearrangement of an aryl
ester to hydroxyacetophenone and phenolic products. The mechanistic and kinetic
aspects of the photo-Fries rearrangement of small molecular aryl esters in constrained
media have been a subject of extensive study.1-9 The photo-Fries rearrangement occurring
in functional polymers bearing aryl ester moieties has also been studied.10-15 Building
upon this precedent, the goal of the present research is to study the effects of constraint,
dielectric constant and proximity on the photo-Fries rearrangement in polymers bearing
aryl ester functional groups.
An inherent change of refractive index (∆n) as aryl esters undergo the photo-Fries
rearrangement to hydroxyacetophenones has recently led to the application of this
rearrangement in developing homopolymers capable of adjustable refractive indices.16
Materials capable of refractive index modulation are desirable in holographic imaging
systems, optical data storage devices, waveguides and lenses with adjustable optical
properties. Towards these applications, the present research has assessed the change in
refractive index that can be realized in aryl ester-functionalized homopolymers and
copolymers of varying kind.
Photochemistry of Aryl Esters
The photo-Fries rearrangement was first reported by Kobsa,17 and Anderson and
Reese18 upon UV irradiation of substituted phenyl acetates in solution. In 1960,
Anderson and Reese reported the rearrangement of catechol monoacetate to 2,3- and 3,4-
dihydroxyacetophenone with the 2,3- substituted isomer being the dominant product.18 A
year later, Kobsa reported the rearrangement in twelve substituted phenyl esters of

2
varying structure.17 The simplest example of the photo-Fries rearrangement is shown in
Figure 1. Here acetoxybenzene rearranges to form ortho and para
hydroxyacetophenones and phenol.
Figure 1: Acetoxybenzene undergoing the photo-Fries rearrangement.
Immediately following its discovery there was much speculation regarding the
mechanism of the photo-Fries rearrangement. It was observed that presence of
substituents on the phenyl ring other than hydrogen (particularly electron withdrawing
groups) reduced the quantum yield of the rearrangement.19 Initial reports by Sandner et
al. showed the quantum yield of acetophenone products was not affected by solvent
polarity or viscosity.20, 21 This observation led to the suggestion that the rearrangement
occurs through a concerted transition state to generate the observed
hydroxyacetophenones. A concerted mechanism did not however explain the observation
of phenol as a product. Kobsa rationalized the formation of phenol by suggesting the
acyloxy bond is homolytically cleaved leading to the generation of an intermediate
“caged radical pair,” with phenols being generated only when the radical pair diffuses
away from one another.17
Recently, two-photon-two-color spectroscopy techniques were used to study the
mechanism of the photo-Fries rearrangement.22, 23 Figure 2 shows a detailed description
of the currently accepted mechanism of the photo-Fries rearrangement. The
rearrangement is triggered by UV light that excites the molecule to its first excited singlet

3
state (S0�S1) in which homolytic cleavage of the acyloxy bond occurs to generate a
geminal pair of radicals in a solvent cage. The radical pair within the solvent cage may
diffuse away from one another (“cage escape”) leading to the phenolic products (“cage
escape products”). The radical pair may also reorient relative to one another within the
solvent cage forming an enol which subsequently undergoes tautomerization to yield the
observed hydroxyacetophenone products (“cage products”). Jiménez et al. used two-
photon-two-color spectroscopic techniques to demonstrate the existence of short-lived
cyclohexadienone intermediates before they undergo tautomerization.22 Lochbrunner et
al. used similar techniques to demonstrate the excited state occurs through a π�π*
excitation with acyloxy bond cleavage occurring within 2 ps and radical recombination
occurring within 13 ps.23
OO
hν
OO
Solventcage
Cage escape
OH
OO
H
O
O
H
Tautomerization
OHOOH
O
Figure 2: Detailed mechanism of photo-Fries rearrangement.

4
As compared to the classic thermal, Lewis acid-catalyzed Fries rearrangement,
(often carried out at temperatures in excess of 100 °C)24, the photo-Fries rearrangement
occurs under mild conditions (room temperature, hν<260nm). For this reason, the photo-
Fries rearrangement has utility as a synthetic tool.25-27 Comparisons between the
thermally and photo-chemically catalyzed rearrangements have been reported which
highlight some of their major differences.28-31 While the mechanism of the Lewis acid-
catalyzed rearrangement has been debated, the accepted mechanism involves an
intermediate acylium ion capable of dealkylating positions on the aromatic ring as shown
in Figure 3 A.17 Since the analogous photo-Fries rearrangement proceeds through a
radical intermediate dealkylation is not observed. Additionally, dehalogenation shown in
Figure 3 B is not observed under acid-catalyzed conditions but is observed under
photochemical conditions due to the rearrangement proceeding through a radical
intermediate.17
Figure 3: Comparison of thermal and photochemical catalyzed Fries rearrangements.

5
The photo-Fries Rearrangement in Constrained Media
While the photo-Fries rearrangement has gained attention as a useful synthetic
reaction, the photo-Fries rearrangement, of acetoxybenzene leads to the formation of
multiple products of which the major product tends to be ortho-hydroxyacetophenone. In
order to improve the product selectivity of the photo-Fries rearrangement, the reaction
has been studied in the presence of a variety of constraining media. Constraining media
can be defined as any reaction medium that will restrict the motion of the molecules and
radicals involved in the photo-Fries rearrangement, thus, increasing the overall product
selectivity of the reaction. Studies have been reported in which zeolites,2,4 Nafion®,4,5
cyclodextrins,3 polyethylene,1,6 poly(vinyl acetate),7, 9 water soluble dendrimers,8 and
water soluble micelle-like polymers32 have acted as constraining media.
The use of zeolites as a constraining media for the photo-Fries rearrangement was
first reported by Tung, et al. Zeolites are a broad class of hydrated aluminosilicate
minerals that have an open micro-porous structure that can vary in shape and size,
depending on the composition of the mineral. This micro-porous structure can
accommodate a variety of small organic molecules, typified by phenyl acetate. In a 1997
publication, Tung reported studies of phenyl acetate substrates within the zeolites, ZSM-5
and NaY, undergoing the photo-Fries rearrangement.2, 4 A representation of the pore
structure of the ZSM-5 zeolite is depicted in Figure 4. Pore size and shape differs
substantially in ZSM-5 and NaY zeolites thus, leading to a difference in the distribution
of products formed upon rearrangement of phenyl acetate. Reactions in ZSM-5
preferentially yielded cage escape products (phenol) while reactions in the NaY
preferentially yielded the ortho-hydroxyacetophenone rearrangement product. This

6
indicates that the pores in ZSM-5 are too small to bind phenyl acetate. Accordingly, the
propensity for formation of cage escape products is comparable to that in solution. The
pore size of NaY, on the other hand, is large enough to bind phenyl acetate and the
formation of o-hydroxyacetophenone is enhanced.
Figure 4: Representation of the structure of ZSM-5 zeolite. http://en.wikipedia.org/wiki/Image:Zeolite-ZSM-5-3D-vdW.png
Tung also reported the effects of Nafion® as a reaction medium for the photo-
Fries rearrangement of phenyl acetates. Nafion® is the trade name of a family of
polymers consisting of a perfluorinated backbone with fluorinated pendant chains
terminated by sulfonic groups (Figure 5).

7
Figure 5: Structure of Nafion®.
Nafion® is thought to take on a micelle-like structure upon hydration in which the
negatively charged sulfonic groups accumulate at the interfaces exposed to water while
the fluorinated hydrocarbon tails are aggregated. Figure 6 is a common representation of
how the hydrocarbon tails might trap a pocket of water of about 40 Å in diameter that is
connected by narrower 10 Å channels to adjacent pockets. The proposed morphology is
called the cluster-network model.34 Water swollen Nafion® has been shown to
incorporate relatively large concentrations of hydrophobic molecules like phenyl
acetates.4, 5
Figure 6: Morphology of water-swollen Nafion®.35
Nafion® was shown to have a substantial effect on the product distribution of the
photo-Fries rearrangement of phenyl acetate. Non-hydrated protonated Nafion® films

8
doped with phenyl acetate yielded ortho rearranged product exclusively. The ortho
preference was less pronounced in hydrated, water-swollen protonated, Nafion®. The
observed product selectivity was reported to be a consequence of the restrictive effect
that Nafion® has on the motion of the geminal radical pair.4, 5
At the same time the work in zeolites and Nafion® was published, Banu, et al.
reported the effects of complexation of phenyl and naphthyl esters with cyclodextrins.
Cyclodextrins are cyclic oligosaccharide molecules capable of forming complexes with
apolar organic molecules that can fit within the central opening of their ring shaped
structure. Banu, et al. compared the photo-Fries rearrangement in hexane and methanol
solutions to that of 1:1 complexes of photo-Fries active molecules with β-cyclodextrin
(Figure 7) in the solid state.
Figure 7: Structure of β-cyclodextrin.
The results indicate a significantly lower yield in the solid-state complexes
compared to solution. Additionally, molecules in complexes only rearranged to form the

9
substitution product at the 2 position on the naphthalene ring.3 Figure 8 shows β-
cyclodextrin represented as a cup-like complex encircling the naphthalene ring system,
thus, preventing rearrangement to any position on the ring other than the 2 position. With
this work, Banu demonstrates that the fate of the geminal radical pair in the photo-Fries
rearrangement is highly dependent on how the motion of the radical pair is restricted by
the surrounding environment.
Figure 8: Rearrangement of β-cyclodextrin naphthyl ester complexes.3
A large body of work has been reported by Weiss, et al. at Georgetown University
regarding the photo-Fries rearrangement of various small molecules doped into
polyethylene1, 6 and poly(vinyl acetate).7, 9 A striking example of selectivity in the photo-
Fries rearrangement was reported by Weiss in low-density polyethylene (LDPE) films
doped with the naphthyl dodecanoate.
Table 1 shows the relative percentage of products 1-4 formed as naphthyl
dodecanoate underwent the photo-Fries rearrangement. The photo-conversion of ester to
products was kept low (<10%) in order to minimize the occurrence of undesirable side

10
reactions from the products. In hexane, 85% of the product formed is the cage escape
product, 2-naphthol (1). When dissolved in LDPE, the dominant product was the
rearrangement product in which recombination occurs at the 3 position (3) on the
naphthyl ring. When the LDPE film is stretched, the preference for the rearrangement
product at the 3 position increases. This is a unique example of a macroscopic applied
strain having an effect on the microscopic properties controlling the selectivity of a
reaction. The results are detailed in Table 1.
Table 1: Product Selectivity Exhibited in Unstretched and Stretched LDPE Films.1
O R
O
OH OH
COR
OH
COR
OH
ROC
R = -(CH2)12CH3
1 2 3 4
Photoproduct distribution (%)
Medium % conv. 1 2 3 4
Hexane 14 85 14 0 1
LDPE - Unstretched ≤7 0 0 75 25
LDPE - Stretched ≤7 0 0 92 8
In a 2005 publication, Ramamurthy et al. reports the use of an amphiphilic water-
soluble styrene-based polymer as a constraining medium for the photo-Fries
rearrangement in addition to α-clevage, and Norrish type I and II reactions.32 The
polymer, whose structure is shown in Figure 9 A, takes on a micelle-like structure with
hydrophilic groups towards the exterior and hydrophobic domains in the interior. These
water-soluble structures are formed when the polymer is in its sodium salt form at a pH
of 8.5. The hydrophobic domains are capable of hosting small hydrophobic molecules
like aryl esters (Figure 9 C).

11
Figure 9: Structure of styrene-based amphiphilic homopolymer (polymer A) utilized by Ramamurthy (A). Micelle-like structure taken on by Polymer A in aqueous solution (B). Reaction scheme of photo-reactions
occuring within polymer micelles (C). 32
When the aryl ester in Table 2 is irradiated with UV light in hexane solution it
undergoes rearrangement to form products 1-4. When this same substrate is absorbed
into the hydrophobic domains of the micelle-like structures formed by polymer A the
motions of the aryl ester is radically restricted. As a result, only product 1 is observed to
form upon photolysis.

12
Table 2: Product Selectivity Exhibited in micelle-like structures of polymer A.32
1 2 3 4
O Ph
O
OH
Ph
O
OH
Ph O
OHPh
O
O
Ph
Photoproduct
distribution (%)
Medium 1 2 3 4
Hexane 60 30 7 3
Polymer-A >99 0 0 0
The photo-Fries Rearrangement in Polymers
Polymers bearing aryl ester functional groups are capable of undergoing the
photo-Fries rearrangement in a manner analogous to that observed with small molecules.
Gupta, et al.36, 37 and more recently Vallon et al.
38 studied the photo-Fries rearrangement
of polycarbonates (Figure 10). The UV photo-induced rearrangement occurring in the
polycarbonate backbone has long been implicated as a source of photo-degradation of the
polymer. Furthermore it has been noted that the products of the photochemical
rearrangement (primarily hydroxyacetophenones) have a photo-stabilizing action on the
polymer resulting from an increased long wavelength absorbance (~360nm) due to the
n�π* transition of hydroxyacetophenone. Photo-stabilization occurs as incoming UV
radiation is absorbed and released through radiationless processes.39

13
CH3
CH3
O C O
On
hν
CH3
CH3
O C
HO
On
hν
CH3
H3C
OH
C
HO
On
Figure 10: Photo-Fries rearrangement of polycarbonate.38
About ten years after the first reports of the photo-Fries rearrangement in small
molecules, the photo-Fries rearrangement of polymers bearing aryl ester pendant groups
was reported. These systems differ from the doped polymer film systems studied by
Weiss in that the photo-Fries active moiety is covalently attached to the polymer
backbone. Guillet at the University of Toronto was first to report such systems in the
form of acrylic polymers.11,12 Fréchet later reported similar systems in the form of
styrenic polymers.10 More recently, Kern has continued work in this field reporting
polymers of increased chromophore complexity.13-15
Guillet’s seminal work reported the synthesis and photo-Fries rearrangement of
poly(phenyl acrylate), PPA (Table 3).11 A few years later Guillet extended this work,
studying the photo-Fries rearrangement in poly(naphthyl acrylate).12 In PPA, the phenyl
ester groups are attached to the carbon backbone. Guillet observed that these photo-Fries
active moieties rearrange upon exposure to UV light in the range of 220-340 nm to give a

14
polymer with pendant ortho and para-hydroxyacetophenone groups. The rearrangement
was confirmed by the appearance of a new band in the absorption spectrum of the
polymer at 340 nm corresponding to the n�π* excitation of the hydroxyacetophenone
group.
Table 3: Quantum Yields Observed in the Irradiation of Poly(phenyl acrylate) (PPA) and Phenyl Acetate (PA).11
O
R
O
R1 = CH3
R2 = -(CH-CH2)n-
hν
HO
R
O
R
O
OH
ortho para
Solvent φφφφortho φφφφpara φφφφo/φφφφp PA (R1) Cyclohexane 0.18 0.2 1.1
PA (R1) 1,2-Dichloroethane 0.18 0.22 1.2
PPA (R2) 1,2-Dichloroethane 0.12 0.23 1.9
PPA (R2) Solid film (air) 0.1 0.13 1.3
PPA (R2) Solid film (N2) 0.1 0.14 1.4
(all quantum yields are ±0.02)
Guillet reported the wavelength dependence of the photo-Fries rearrangement in
PPA. The highest quantum yield was obtained upon irradiation at 250 nm; no
rearrangement was observed with irradiation at 300 nm. Guillet also compared the
quantum yields for rearrangement of the polymer in the solid state and in solution (Table
3). Quantum yields were lower for the polymer in both the solid and solution states as
compared to the quantum yields of the analogous small molecules.
In 1985, Fréchet and Tessier reported the synthesis and photochemistry of poly(p-
acetoxystyrene), PAS, and poly(p-formyloxystyrene), PFS. Their intention was to use
these polymers as photoresist materials in microlithographic processing by exploiting the
difference in solubility that exists between the rearranged and unrearranged polymer.

15
Because the para position of the phenyl ring is blocked by covalent attachment to the
polymer backbone, upon irradiation, the acetate group in PAS, can only rearrange to the
ortho position on the phenyl ring (Figure 11 A). Fréchet and Tessier noted that unlike
PAS, PFS undergoes an efficient decarbonylation of the formyl group resulting in the
generation of carbon monoxide and phenol (Figure 11 B). The resulting phenol absorbs
incident irradiation to a lesser degree than the hydroxyacetophenone product from PAS.
For this reason, a complete conversion nearing 100% is observed in the photo-Fries
reaction of PFS while PAS only reaches a ultimate conversion of about 40%.10 The
ultimate conversion is defined as the maximum conversion at which further exposure to
UV light results in little or no increase on conversion.
O
CH3
O
n
OH
CH3
O
n
hν
A
O
H
O
n
OH
C
O
n
hν
B
Figure 11: Photo-Fries rearrangement of poly(p-acetoxystyrene) (A) and poly(p-formyloxystyrene) (B).10
More recently, Kern et al., revisited the photo-Fries reaction in PAS and
expanded their studies to include the olefin metathesis polymers, poly(bicyclo[2.2.1]hept-
5-ene-2-carboxylic acid, phenyl ester) (Figure 12 A) and poly(bicyclo[2.2.1]hept-5-ene-
2,3-dicarboxylic acid, diphenyl ester) (Figure 12 B).13 Kern’s observations of the photo-

16
Fries rearrangement led to a different conclusion than that of Guillet. By following
changes in the absorption spectra of PPA and comparing them to the absorption
characteristics of o- and p-hydroxyacetophenones, Guillet concluded that the major
product of the rearrangement is the para-hydroxyacetophenone.11 This is
counterintuitive to previously reported observations.17, 20, 21
Figure 12: Novel photo-Fries active polymers synthesized by Kern.13-15
Kern used FT-IR to follow the rearrangement. FT-IR spectroscopic analysis of o-
and p-hydroxyacetophenones was used to identify absorption bands from the polymer
following rearrangement. Figure 13 shows changes in the IR spectra of PAS (A) and
poly(bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, phenyl ester) (B). The ortho- and para-
hydroxyacetophenone products give rise to the IR absorption bands at 1632 and 1670cm-1
(respectively) as seen in Figure 13 B. At the same time, a decrease in the intensity of the
ester carbonyl band near 1745 cm-1 is observed (Figure 13 A and B). This IR data
confirms the predominant product in the rearrangement is indeed the ortho isomer.

17
A B
Figure 13: FT-IR spectra of PAS (A) and poly(bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, phenyl ester) (B) undergoing the photo-Fries rearrangement.13
The focus of Kern’s published work was the increase of refractive index (∆n) that
accompanies the photo-Fries rearrangement in PAS and polymers bearing aryloxy
functionality. Kern reported that photolysis of PAS converting 80% of the ester groups
to ortho-hydroxyacetophenone resulted in a ∆n of 0.05. Figure 14 shows Kern’s
refractive index data obtained from PAS in the form of Cauchy fit curves1 where the
dotted line represents data for the refractive index of the polymer following the photo-
Fries rearrangement. Kern also reported the potential utility of these polymers in
photolithographic patterning procedures by utilization of postexposure reactions to
functionalize the hydroxyl groups generated upon photo-Fries rearrangement. It was
demonstrated that following rearrangement, the exposed hydroxyl group on the
hydroxyacetophenone can be reacted with carboxylic acid chlorides, dansyl chloride, 2,4-
dinitrophenylhydrazine, or FeCl3 followed by NH4SCN.14 These post exposure reactions
1 The Cauchy equation is an empirical relationship relating refractive index to wavelength. The
relationship takes the following form: ( ) ⋅⋅⋅++=42 λλ
λCB
An where A, B and C are constants unique to
the material.

18
can be used to modify the surface properties or covalently attach desired molecules only
in areas exposed to UV energy.
Figure 14: Cauchy fit curve obtained from PAS.13
Additionally, Kern described the synthesis of three polymers (Figure 15) bearing
photo-Fries active naphthaloxy- and triarylamino moieties and studied their reactivity
under two-photon irradiation conditions.14 In the process of two-photon irradiation, aryl
ester moieties might be excited to their S1 state by simultaneous absorption of two
photons of a longer wavelength where the combined energy of both photons is sufficient
to actuate the photo-Fries rearrangement. Two-photon irradiation would allow for three-
dimensional spatial control of where the photo-Fries rearrangement occurs because
rearrangement will only occur where two distinct beams of long wavelength energy are
focused to intersect. Spatial control over the photo-Fries rearrangement would be useful
for fabricating waveguides within polymers and writing optically stored data to a polymer
medium. It is yet to be confirmed whether the change in refractive index, observed by
Kern in these experiments was the result of a two-photon induced photo-Fries
rearrangement or merely the result of a thermal reaction of unknown mechanism.

19
n n n
O O
N
A B C
O O
N
O O
Figure 15: Additional photo-Fries active polymers studied by Kern.15
Utility of the Current Research
An additional goal of this research was to determine the magnitude of refractive
index change attainable in the polymers under investigation. The rearrangement depicted
in Figure 16 exhibits a difference in refractive index between the products and reactants
giving rise to a ∆n of about 0.05 upon quantitative rearrangement of reactants to products.
Materials capable of undergoing changes in refractive index are of considerable interest
in holographic imaging systems, optical data storage devices and waveguides in the field
of photonics.38, 39 Previous approaches to making materials capable of refractive index
changes include cross-linking of cinnamoyl double bonds creating cyclobutane dimers,40
photo-chemically induced isomerization of norbornadiene moieties41 and doping
poly(methyl methacrylate) films with photo-reactive molecules.38, 39 Depending on the
system, the change in refractive index may be positive or negative and may also be
reversible.2 Refractive index changes of photo-Fries active polymers are positive and
irreversible.
2 See Appendix I for a description of systems capable of refractive index modulation.

20
O
O
H3C
hνOHOH
O
CH3 OH
O CH3
n=1.5030 n=1.5584 n=1.5577 n=1.5408
Figure 16: Refractive index change associated with photo-Fries rearrangement of acetoxybenzene.
In order to utilize refractive index changes of photo-Fries active polymers in
commercial devices the polymers must be engineered in such a way to maximize their
ability to undergo rearrangement. Two factors that can be addressed to improve the
utility of photo-Fries active polymers are altering the structure of the aryl ester such that
longer wavelengths are capable of actuating the photo-Fries rearrangement (with
limitation - vide infra) and sensitizing the rearrangement to increase conversion of ester.
Addressing these factors can increase the extent of photo-Fries rearrangement (and
subsequently ∆n) while reducing the dose of energy necessary to elicit the desired ∆n.
The structure of the photo-Fries active chromophore can be altered in such a way
that it absorbs at a longer wavelength. However, if the absorbed wavelength is not
energetic enough to cause homolytic cleavage of the acyloxy bond, the photo-Fries
rearrangement will not occur. For example, an anthryl ester absorbing at 366 nm does
not undergo photo-Fries rearrangement upon irradiation at that wavelength.42, 43 Work
published by Kern, et al. reported examples of chromophores that undergo photo-Fries
rearrangement at longer wavelengths.15 The polymer shown in Figure 17 is one such
polymer capable of undergoing rearrangement upon irradiation with 310 nm light. To
date, this is the longest wavelength reported in which the photo-Fries rearrangement has
been observed in a polymer.

21
O O
n
Figure 17: Structure of poly(naphthylene-1-yl 4-vinyl-benzoate).15
Molecular orbital calculations of sensitization of the photo-Fries rearrangement
have been reported in the literature.44 These experiments carried out by Schwetlick et al.
calculated the fluorescence and absorption maxima of several aromatic heterocycles in
order to assess their ability to sensitize or inhibit the photo-Fries rearrangement of N-
phenyl-urethane. Additionally there have been a few reports of sensitization of the
photo-Fries rearrangement which report an acceleration of the rearrangement in the
presence of carbonyl compounds such as benzophenone, acetophenone and, flavone in
addition to dye sensitizers which show less of an effect.25, 26, 45 Though not a true photo-
Fries rearrangement, photo-acid catalysis of the decarbonylation of poly(p-formyloxy
styrene) has been reported by Loong (Figure 18).46, 47
O
H
O
n
Ph3S+ AsF6
-
254nm
OH
n
Figure 18: Photo-acid catalysis of the photo-Fries rearrangement of PFS in the presence of triarylsulfonium hexafluoroarsenate.
An interesting example of sensitization was presented in a 1999 paper by Guillet
et al. They observed energy transfer from poly(sodium styrenesulfonate-co-2-

22
vinylfluorene) to naphthyl acetate (Figure 19).48 In aqueous solutions, poly(sodium
styrenesulfonate-co-2-vinylfluorene) forms micelle-like structures in which the fluorene
residues are localized within the hydrophobic core. Upon incorporation of hydrophobic,
photo-Fries-active, small molecules, like naphthyl acetate, light energy absorbed by
fluorene is subsequently transferred to naphthyl acetate molecules inducing the photo-
Fries rearrangement.
O3S
Na
n mhν
O
O
Energy Transfer
O
O S1
S1
photo-FriesRearrangement
OHOH O
OH
O
poly(sodium styrenesulfonate-co-2-vinylfluorene)
1-naphthyl acetate
Figure 19: Energy transfer observed between poly(sodium styrenesolfonate-co-2-vinylfluorene) and 1-naphthyl acetate.
The phenomenon in Figure 19 is known as the antenna effect in which the
“antenna molecule”, fluorene chromophores in this case, absorb energy and transfer it to
an adjacent antenna molecule or a proximate, non-covalently attached, second molecule.
Energy transfer occurs through the Förster energy transfer mechanism3 and therefore,
molecules must be within the Förster radius of each other in order to transfer energy.49, 50
3 See Appendix II for a description of Förster energy transfer.

23
In such a system, the concentration of the antenna molecule must be high enough to
ensure that antenna molecules are statistically within the Förster radius of the photo-Fries
active molecules. At the same time, there is an upper limit to the concentration at which
increased amounts of antenna molecule have a decreasing effect on the enhancement of
the photo-Fries rearrangement.
Preliminary experiments are reported in this thesis to show how a long-
wavelength-absorbing photo-Fries active chromophore in the presence of a sensitizer
molecule can lead to an increased conversion of the photo-Fries active chromophore from
aryl ester to hydroxyacetoarene. This was achieved by studying the photo-Fries
rearrangement of naphthalene-1-yl benzoate in the presence of fluorene as an antenna
molecule (Figure 20).
hν
O OEnergy Transfer
O O
S1
photo-FriesRearrangement
S1
Rearranged Products
Figure 20: Antenna effect in the rearrangement of naphthalene-1-yl benzoate in the presence of fluorene.

24
Experimental
Materials
Unless otherwise noted, all reagents were used as received, without further
purification.
Tetrahydrofuran (99%), 2,2′-Azobis(2-methylpropionitrile) (recrystalized from
methanol), p-toluenesulfonic acid monohydrate (98%), phenylchloroformate (99%),
allyltrimethylsilane (98%), 1-hexene (97%), anhydrous aluminum chloride (99.99%), 1-
naphthol (≥99%), triethylamine (99%) phenyl chloroformate (99%), and coumarin were
obtained from Sigma-Aldrich.
Phenyl acrylate (97%), p-acetoxystyrene (95%, stabilized with 15-25 ppm.
Phenothiazine), and n-butyl acrylate (98%, stabilized with 50 ppm 4-methoxyphenol)
were purchased from Alfa Aesar.
Toluene was purchased from EMD and dried over CaH2 purchased from Sigma-
Aldrich.
Methanol (HPLC grade) was purchased from Burdick & Jackson.
Dibenzoyl peroxide (75%), ethyl acrylate (99.5%), and fluorene (98%) were
purchased from Acros.
Petroleum ether was obtained from Mallinckrodt Chemicals.
Methacryloxyethoxy-trimethylsilane, (25-30% methylhydrosiloxane)-
(dimethylsiloxane) copolymer, and platinum-divinyl tetramethyl disiloxane complex in
xylene (2.1-2.4% Pt conc.) were purchased from Gelest.
Activated carbon (50-200 mesh) was purchased from Fisher.

25
Dichloromethane (dried over CaH2), thin layer chromatography plates (silica gel
IB-F), diatomaceous earth, and silica gel (40-140 mesh) was obtained from J. T. Baker.
Poly(vinyl butyral) was obtained from Pfaltz & Bauer, Inc.
Instrumentation
Proton NMR data was gathered using a Brucker 300 MHz. spectrometer with all
samples prepared in chloroform-d (Aldrich, 99.8 atom % D, 0.05% v/v TMS) unless
otherwise noted.
Glass transition data was gathered using a TA Instruments DSC 2010 with
samples prepared in non-hermetic aluminum pans. All Tg values are reported as Tg
midpoint temperatures.
UV-visible spectroscopic data was collected on a Hewlett-Packard 8453 Diode
Array instrument.
IR spectroscopic data was collected on a Shimadzu IR Prestige-21. UV-visible
and IR data was gathered in transmission mode from polymer films spin-cast onto IR
grade CaF2 windows (Red Optronics, 25.4 mm diameter x 3 mm thick) from 5% wt./vol.
solutions in toluene filtered through Whatman® 13mm glass microfiber GF/B syringe
filtration devices (1.0 µm pore size) unless otherwise noted.
Spin coating was done using a Model P6700 Series spin coater from Specialty
Coating Systems Inc.
Irradiation of polymer films was achieved using a 15 watt Spectroline Model XX-
15F Shortwave (254 nm) UV lamp or a UVP EL Series (302 nm) UV lamp at a distance
of 4 cm.

26
Refractive index and film thickness measurements were made using a Woollam
Variable Angle Spectroscopic Ellipsometer (VASE). VASE measurements were made
on films cast on 4” polished silicon P100 test wafers (Wafer Works Corp.) using
solutions prepared as described above unless otherwise noted.
Synthesis
OO
AIBN, Toluene
70-75°C, 20h
OO
n
Figure 21: Synthesis of poly(p-acetoxystyrene).
Poly(p-acetoxystyrene) - PAS. To a 40 mL capacity Carious tube, AIBN (100
mg, 0.6 mmol), p-acetoxystyrene (9.637 g, 59.4 mmol) and toluene (2 mL) was added.
The solution was degassed with three freeze thaw cycles by freezing the tube contents in
a dry ice/acetone bath (-78 °C). The tube was sealed and heated in an oil bath to 70-75
°C for 20 hours. The resulting solid clear mass of polymer was dissolved in 75 mL
toluene and precipitated from 800 mL methanol. The resulting polymer was air dried at
ambient temperature overnight to yield 9.05 g (94% conversion). FT-IR (cm-1) 1760
(acetate ester). Tg = 123 °C.

27
OOdibenzoyl peroxide, toluene
70-75°C, 19h
OO
n
Figure 22: Synthesis of poly(phenyl acrylate).
Poly(phenyl acrylate) - PPA. To a 40 mL capacity Carius tube, dibenzoyl
peroxide (25 mg, 0.15 mmol), phenyl acrylate (5 g, 33.8 mmol) and toluene (10 mL)
were added. The solution was degassed by purging with argon for one hour. The tube
was sealed and heated in an oil bath to 70-75 °C for 19 hours. Following the
polymerization reaction, a highly viscous clear solution was formed. The viscous
solution was diluted with 35 mL toluene and precipitated in 750 mL petroleum ether.
The resulting polymer was air dried at ambient temperature overnight to yield 4.31 g
(86% conversion). FT-IR (cm-1) 1760 (acrylate ester). Tg = 50 °C.
OO
65°C, 20h
O O
CH2
CH3
n m
OOO O
CH2
CH3
AIBN
Figure 23: Synthesis of poly(phenyl acrylate-co-ethyl acrylate).
Poly(phenyl acrylate-co-ethyl acrylate) – PA-EA. To a 40 mL capacity Carious
tube, AIBN (25 mg, 0.15 mmol), phenyl acrylate (2.50 g, 16.8 mmol) and ethyl acrylate
(1.68 g, 16.8 mmol) were added. The solution was degassed by purging with argon for
one hour. The tube was sealed and heated in an oil bath to 65 °C for 20 hours. The
resulting clear mass of solid polymer was dissolved in 90 mL toluene. The polymer did
not fully dissolve indicating the formation of cross-linked polymer. The solution was
filtered through a funnel with a plug of glass wool in the stem yielding 0.809 g insoluble

28
polymer. The soluble polymer in the remaining filtrate was precipitated in 1 L of
methanol. The resulting polymer was air dried at ambient temperature overnight then
dried for 24 hours at 40 °C in a vacuum oven to yield 2.90 g polymer (69% conversion).
FT-IR (cm-1) 1754 (phenyl acrylate ester), 1730 (ethyl acrylate ester). Tg = 34 °C. 1H-
NMR, δ (ppm) 4.1 (broad unresolved, 1.2H, CH2 of ethyl acrylate), 6.8-7.5 (broad
unresolved, 5H, aromatic protons of phenyl acrylate), other signals were not well enough
resolved for assignment. Phenyl acrylate content was 62 mol. % as determined by 1H-
NMR.
AIBN, Toluene
65°C, 19h
OO
O O
CH2
CH2
n m
CH2
CH3
OO
O O
CH2
CH2
CH2
CH3
Figure 24: Synthesis of poly(p-acetoxystyrene-co-butyl acrylate).
Poly(p-acetoxystyrene-co-butyl acrylate) – PA-BA. To a 40 mL capacity
Carious tube, AIBN (30 mg, 0.18 mmol), p-acetoxystyrene (3.16 g, 19.5 mmol), butyl
acrylate (2.5 g, 19.5 mmol) and toluene (1.5 mL) were added. The solution was degassed
by purging with argon for 45 minutes. After sealing the tube and heating in an oil bath at
65°C for 19 hours an opaque white solid was obtained. The solid was dissolved in 27 mL
toluene and precipitated from 500 mL methanol. The resulting precipitated polymer was
air dried at ambient temperature overnight then dried for 24 hours at 40 °C in a vacuum
oven to yield 4.84 g polymer (85% conversion). FT-IR (cm-1) 1763 (acetate ester), 1725
(butyl acrylate ester). Tg = 47 °C. 1H-NMR, δ (ppm) 0.9 (unresolved d, 5.8H, CH3 of

29
butyl acrylate), 6.5-7.0 (broad unresolved, 4H, aromatic protons of acetoxy styrene).
Acetoxy styrene content was 65 mol. % as determined by Tg and the Fox equation.4
OO AIBN, Toluene
65°C, 19h
O O
CH2
CH2
n m
OOO O
CH2
CH2
CH2
CH3
CH2
CH3
Figure 25: Synthesis of poly(phenyl acrylate-co-butyl acrylate).
Poly(phenyl acrylate-co-butyl acrylate) – PA-BA. To a 40 mL capacity Carius
tube, AIBN (30 mg, 0.18 mmol), phenyl acrylate (2.89 g, 19.5 mmol), butyl acrylate (2.5
g, 19.5 mmol) and toluene (1.5 mL) were added. The solution was degassed by purging
with argon for 45 minutes. The tube was sealed and heated in an oil bath at 65°C for 19
hours. Following reaction, a clear highly viscous solution resulted. The thick solution
was diluted with 27 mL toluene and precipitated from 500 mL ice-cold methanol, kept
cold by an ice bath. The resulting polymer was air dried at ambient temperature
overnight then dried for 24 hours at 40 °C in a vacuum oven to yield 4.37 g polymer
(81% conversion). FT-IR (cm-1) 1756 (phenyl acrylate ester), 1730 (butyl acrylate ester).
Tg = 6 °C. 1H-NMR, δ (ppm) 4 (broad unresolved, 3.4H, -COO-CH2 of butyl acrylate),
6.8-7.5 (broad unresolved, 10H, aromatic protons of phenyl acrylate). Phenyl acrylate
content was 54 mol. % as determined by 1H-NMR.
4 The Fox equation is; )()()(
1
bg
b
ag
a
cog T
W
T
W
T+= where Tg is the glass transition of the copolymer (co) or
homopolymer of monomer a (a) or monomer b (b) and W is the weight percent content of monomer a or monomer b.

30
O O
CH2
CH2
n m
OO
OH
O O
CH2
CH2
n m
OO
O
p-toluenesulfonic acid
THF, 90 min.
TMS
O O
CH2
CH2
OO
O
TMS
AIBN65°C, 50 min.
Figure 26: Synthesis of poly(phenyl acrylate-co-hydroxyethyl methacrylate).
Poly(phenyl acrylate-co-hydroxyethyl methacrylate) – PA-HEMA. To a
heavy walled glass tube fitted with a Teflon lined crowned cap, AIBN (20 mg, 0.12
mmol), phenyl acrylate (1.70 g, 11.5 mmol) and methacryloxyethoxy-trimethylsilane
(2.32 g, 11.5 mMol) were added. The solution was degassed with bubbling argon for 30
minutes prior to capping the tube. The tube was heated in a circulating water bath at
65°C for 50 minutes until the solution began to noticeably thicken and bubble slightly.
The thickened solution was removed from the heat and quenched by placing in ice-cold
water. The thickened solution was diluted with 20 mL tetrahydrofuran. Small portions
of polymer did not dissolve indicating the formation of cross-linked polymer. The
solution was filtered through a funnel with a plug of glass wool in the stem yielding
0.174 g insoluble polymer. The soluble polymer in the resulting filtrate was precipitated
from 400 mL n-hexane. Upon air-drying the resulting polymer at ambient temperature

31
overnight, 2.16 g (58% conversion) of poly(phenyl acrylate-co-hydroxyethyl
methacrylate) with TMS-protected hydroxyl groups was afforded.
In order to obtain deprotected poly(phenyl acrylate-co-hydroxyethyl
methacrylate), the TMS-protected polymer was dissolved in 30 mL tetrahydrofuran.
While stirring the solution, distilled water was added drop-wise until the solution reached
its clouding point. Approximately 5 mg p-toluenesulfonic acid monohydrate was added
to the solution which was then stirred at ambient temperature for an additional 90
minutes. The dissolved polymer was then precipitated into 400 mL distilled water
yielding 1.53 g of TMS deprotected polymer. FT-IR (cm-1) 1755 (phenyl acrylate ester),
1725 (hydroxyethyl methacrylate ester), 3500 (broad, hydroxyl). Tg = 112 °C. 1H-NMR,
(THF-d8) δ (ppm) 1.5 (s, 5.3H, backbone CH3 of hydroxyethyl methacrylate), 6.8-7.5
(broad unresolved, 5H, aromatic protons of phenyl acrylate). Phenyl acrylate content was
50 mol. % as determined by Tg and the Fox equation.

32
O O
CH2
CH2
n m
OO
OH
O O
CH2
CH2
n m
OO
O
p-toluenesulfonic acid
THF, 90 min.
TMS
O O
CH2
CH2
OO
O
TMS
AIBN65°C, 50 min.
Figure 27: Synthesis of poly(p-acetoxystyrene-co-hydroxyethyl methacrylate).
Poly(p-acetoxystyrene-co-hydroxyethyl methacrylate) – AS-HEMA. To a
heavy walled glass tube fitted with a Teflon lined crowned cap, AIBN (24 mg, 0.15
mmol), p-acetoxystyrene (2.65 g, 16.3 mmol) and methacryloxyethoxy-trimethylsilane
(2.205 g, 10.9 mMol) were added. The solution was degassed with bubbling argon for 30
minutes prior to capping the tube. The tube was heated in a circulating water bath at
65°C for about one hour until the solution began to noticeably thicken. The thickened
solution was removed from heat and diluted with 15 mL tetrahydrofuran. The dissolved
polymer was precipitated from 300 mL n-hexane. Upon air-drying the resulting polymer
at ambient temperature overnight, 2.54 g (52% conversion) of poly(acetoxy styrene-co-
hydroxyethyl methacrylate) with TMS-protected hydroxyl groups was afforded.
In order to obtain deprotected poly(acetoxy styrene-co-hydroxyethyl
methacrylate), the TMS-protected polymer was dissolved in 40 mL tetrahydrofuran.

33
While stirring the solution, distilled water was added drop-wise until the solution reached
its clouding point. Approximately 5 mg p-toluenesulfonic acid monohydrate was added
to the solution which was then stirred at ambient temperature for an additional 90
minutes. The dissolved polymer was then precipitated from 600 mL distilled water
yielding 2.39 g of TMS-deprotected polymer. FT-IR (cm-1) 1755 (acetate ester), 1725
(hydroxyethyl methacrylate ester), 3400 (broad, hydroxyl). Tg = 108 °C. 1H-NMR,
(THF-d8) δ (ppm) 1.5 (s, 4.1H, backbone CH3 of hydroxyethyl methacrylate), 6.5-7.0
(broad unresolved, 4H, aromatic protons of acetoxy styrene). Acetoxy styrene content
was 42 mol. % as determined by 1H-NMR.
OO
Si Si O
CH3
CH3
CH3
H
toluene, Pt catalyst
40 °C, 24h
OO
On m
Si Si O
CH3
CH3
CH3
On m
Figure 28: Synthesis of p-acetoxystyrene functionalized polysiloxane.
p-Acetoxystyrene functionalized polysiloxane. Under a stream of argon in a
three neck 50 mL round bottom flask, 3 g (25-30% methylhydrosiloxane)-
(dimethylsiloxane) copolymer (molecular weight 1600-2400 g mol-1), p-acetoxy styrene
(2.73 g, 16.88 mmol) and toluene (20 mL) were added and heated to 40 °C while
constantly stirring. One hundred microliters of platinum-divinyl tetramethyl disiloxane
complex in xylene (2.1-2.4% Pt conc.) was slowly added. The reaction was stirred for 24
hours and progress was monitored via FT-IR. After 24 hours, the reaction was checked
via 1H-NMR (CDCl3, 300 MHz.) to confirm disappearance of Si-H resonance at 4.7 ppm.
1-Hexene (1.4 mL, 0.011 moles) was added to the reaction which was stirred for another

34
48 hours to react with any remaining traces of Si-H. After cooling to room temperature,
activated carbon (1.5g) was added and stirred for 3 hours. The resulting solution was
filtered through a fine fritted glass funnel and then filtered with 0.2 µm PTFE filters. The
solvent was removed in vacuo yielding a brown oil which upon cooling below 0 °C
separated into a dark brown layer at the bottom and a light clear-whitish layer at the top.
The top layer was separated from the bottom layer and dissolved in 10 mL methylene
chloride. About 1 g diatomaceous earth was added to the solution which was stirred at
room temperature for about an hour and filtered through a fine fritted glass funnel.
Solvent was removed in vacuo from the resulting filtrate yielding a clear-whitish oil
(4.2956 g). The oil was heated to 55 °C under high vacuum (0.03-0.04µ) for 24 hours to
remove traces of acetoxy styrene. FT-IR (cm-1) 1755 (acetate ester), 1260 (Si-CH3). Tg =
-88 °C. 1H-NMR, δ (ppm) 2.25 (s, 3H, CH3 of acetoxy), 6.75-7.25 (broad unresolved,
4H, aromatic protons of acetoxy styrene). Acetoxy styrene content is 25 mol. % as
determined by 1H-NMR.

35
Phenyl 3-butenoate functionalized polysiloxane.
OSi
Cl
O CH2Cl2, AlCl3
room temp., 4hOO
1
Figure 29: Synthesis of phenyl 3-butenoate (1).
Phenyl 3-butenoate (1). Under a stream of argon in a three neck 250 mL round
bottom flask, aluminum chloride (5 g, 37.5 mmol) and 20 mL dry methylene chloride
were added under stirring to form a slurry. Phenyl chloroformate (5.34 g, 34.1 mmol)
was slowly added to the slurry. After 20 minutes of stirring, allyltrimethylsilane (4.56 g,
39.9 mmol, dissolved in 40 mL dry methylene chloride) was added drop-wise over 20
minutes. The reaction was stirred at room temperature for 4 hours with reaction progress
being monitored via TLC (9:1 - hexane:ethyl acetate). Reaction was quenched with 70
mL cold distilled water added drop-wise very slowly. After separating the aqueous and
organic layer, the aqueous layer was washed three times with 70 mL methylene chloride.
The combined organic layers contained a brown emulsion that was broken up upon the
addition of calcium chloride. The resulting clarified organic layer was concentrated via
rotary evaporator yielding a brown oil as a crude product. Compound 1 was isolated
from the crude product via distillation under reduced pressure (0.03-0.04 µ) to yield
4.5603 g (83%) of clear oil with a slight yellow tint. FT-IR (cm-1) 2980 (=CH stretch),
1284 (ester, C=O stretch), 1493 (benzene ring), 1120 (ester, C-O-C). 1H-NMR, δ (ppm)
3.34 (d, 2H, aliphatic CH2), 5.27 (m, 2H, C=CH2), 6.04 (m, 1H, C=CH-), 7.04-7.46 (m,
5H, aromatic protons).

36
OO
1
toluene, Pt catalyst
40 °C, 24h
O
OSi Si O
CH3
CH3
CH3
H
On m
Si Si O
CH3
CH3
CH3
On m
Figure 30: Synthesis of phenyl 3-butenoate functionalized polysiloxane.
Hydrosilylation of phenyl 3-butenoate. Under a stream of argon in a three neck
50 mL round bottom flask, 3 g (25-30% methylhydrosiloxane)-(dimethylsiloxane)
copolymer (molecular weight 1600-2400 g mol-1), compound 1 (2.73 g, 16.88 mmol) and
toluene (20 mL) were added and heated to 40 °C while constantly stirring. One hundred
microliters of platinum-divinyl tetramethyl disiloxane complex in xylene (2.1-2.4% Pt
conc.) was slowly added. The reaction was stirred for 24 hours and progress was
monitored via FT-IR. An additional 100 µL of catalyst was added and the reaction was
stirred for another 24 hours, then checked via FT-IR. The solution gradually turned a
cloudy white color over the course of the reaction. 1-Hexene (1.4 mL, 0.011 moles) was
added to the reaction which was stirred for another 24 hours to react with any remaining
traces of Si-H. The solvent was removed in vacuo yielding a thick tan colored oil. The
oil was dissolved in 10 mL 1:1 methylene chloride: hexane. Silica gel (3 g) was added
and stirred vigorously overnight. The silica gel was filtered off and the solvent was
removed in vacuo from the resulting filtrate yielding a clear-whitish oil with a slight tan
color remaining. The oil was heated to 50 °C under high vacuum (0.03-0.04µ) for 24
hours to remove traces of phenyl 3-butenoate resulting in 2.3475 g of white oil with a
slight gray/tan color. FT-IR (cm-1) 1750 (ester, C=O stretch), 1260 (Si-CH3). Tg =-64°C.
1H-NMR, δ (ppm) 0.08 (s, Si-CH3), 6.96-7.04 (broad singlet, 1H, para aromatic proton),

37
7.08-7.40 (m, 4H, ortho and meta aromatic protons). Phenyl 3-butenoate content was 20
mol. % as determined by 1H-NMR.
Kinetics Analysis
Kinetic analysis was carried out according to methods reported by Kern.13
Polymer films were cast on calcium fluoride windows in triplicate. Hydroxyethyl
methacrylate copolymers were cast from 5% solutions in tetrahydrofuran. Siloxane
polymers were cast from 10% solutions in toluene. Irradiations took place in a glove bag
purged with dry argon. Irradiated films were all of thicknesses that were not optically
opaque. Irradiation of hydroxyethyl methacrylate copolymers was done with dry films
and films submerged under distilled water, equilibrated to their hydrated state by leaving
them submerged overnight. Reaction progress was monitored by FT-IR and UV-visible
spectroscopy.
Variable angle spectroscopic ellipsometry (VASE) Analysis.
Polymer films cast on silicon wafers were irradiated in a glove bag purged with
dry argon. Irradiated films were all of thicknesses that were not optically opaque.
Hydroxyethyl methacrylate copolymers were cast from 5% solutions in tetrahydrofuran.
Siloxane polymers were cast from 10% solutions in toluene. Irradiation of hydroxyethyl
methacrylate copolymers was done with films submerged under distilled water,
equilibrated to their hydrated state by leaving them submerged overnight. The wafers
were sequentially masked in such a way that each third of the film was irradiated for a
different amount of time causing a different conversion of ester in each region. Siloxane
polymer films were too delicate for sequential masking so they were irradiated for a

38
given amount of time and analyzed using the VASE. The irradiation and analysis was
repeated to gather refractive index data at different extents of photo-Fries rearrangement.
Psi (Ψ) and delta (∆) data obtained from VASE analysis was gathered over the
wavelength range of 400 nm to 800 nm (data points gathered every 10 nm) at 65°, 70°
and 75° angles of incidence. The model applied to the Ψ and ∆ data consisted of an
absorbing substrate (1 mm thick silicon) with a non-absorbing Cauchy layer and a top
layer to account for surface roughness (effective medium approximation - 50% void
space). Cauchy coefficients A, B and C in addition to the thickness of the Cauchy and
surface roughness layers were fit to the experimental data obtained from analysis. Fitting
of the experimental data provided refractive index data over the range of 400-800 nm
used to construct Cauchy curves.
Sensitization of the photo-Fries rearrangement.
OH Cl O
Triethyl amine, CH2Cl2
0°C to RT, 60 min. O O
2
Figure 31: Synthesis of naphthalene-1-yl benzoate (2)
Synthesis of naphthalene-1-yl benzoate. To a 100 mL round bottom flask
purged with argon, 1-naphthol (1.75 g, 12.1 mmol) and 35 mL methylene chloride were
added while stirring. The solution was cooled to 0 °C. Triethyl amine (1.35 g, 13.31
mmol) and benzoyl chloride (1.87 g, 13.31 mmol) was added drop-wise to the solution.
The solution became turbid and yellow in color. The solution was stirred for 30 minutes

39
at 0 °C, warmed to room temperature and stirred an additional 60 minutes at room
temperature. Reaction progress was monitored by TLC (8:1 hexane:ethyl acetate). When
TLC indicated complete consumption of 1-naphthol, the reaction was quenched by the
addition of 30 mL distilled water and 30 mL methylene chloride. The resulting organic
layer was concentrated via rotary evaporator to ~30 mL. The concentrated organic layer
was washed with 30 mL aq. HCl (pH = 4-5), 30 mL distilled water, 30 mL aq. sodium
bicarbonate and finally 30 mL distilled water. Solvent was removed from the resulting
organic phase yielding a light tan – yellowish oil as crude product. The crude product
was purified via silica gel chromatography (10 cm high by 4 cm diameter column
dimensions) using a gradient elution of cyclohexane, 50:1 cyclohexane: ethyl acetate and
finally 50:2 cyclohexane: ethyl acetate. Compound 2 was isolated as 2.5441 g (85%) of a
clear light brown oil that that became a white crystalline solid upon refrigeration.
Melting point: 54-55 °C. FT-IR (ATR single reflectance diamond anvil) (cm-1) 3035
(aromatic CH), 1725 (ester C=O), 1600 (aromatic C=C), 1510 (aromatic C=C), 1260,
1253 (C-O-Ar), 1220 (C-O-C). 1H-NMR, δ (ppm) all signals in aromatic region 8.35 (d,
2H), 7.87-7.97 (m, 2H), 7.79 (d, 1H), 7.65-7.72 (m, 1H) 7.45-7.61 (m, 5H), 7.38 (d, 1H).
Photo-Fries rearrangement of naphthalene-1-yl benzoate. Films of poly(vinyl
butyral) were cast (in triplicate) onto CaF2 windows from 5% (w/v) solutions in
tetrahydrofuran filtered through 1µm syringe filters. Solutions were doped with 5% wt.
compound 2 and 0, 15 or 25% wt. fluorene. After drying at room temperature overnight,
the films were irradiated in a glove bag purged with dry argon using a 302 nm lamp.
Reaction progress was monitored via FT-IR or UV/visible spectroscopy.
Results and Discussion

40
Synthesis of Polymers
In order to conduct a systematic study of the variables that might affect the photo-
Fries rearrangement in polymer systems, vinyl, acrylic and siloxane-based polymers with
differing molar content of photo-Fries active residues (Table 3) were synthesized.5 The
polymers can be divided into four main categories; homopolymers (PAS and PPA),
hydrophobic acrylic copolymers (PA-EA, AS-BA and PA-BA), hydrophilic acrylic
copolymers (PA-HEMA and AS-HEMA), and siloxane copolymers (AS-SiO and PA-
SiO).
Table 4: Properties of polymers under investigation.
Polymer Name Abbreviation Tg (°C) Photo-Fries active monomer
content (mole %)
poly(p-acetoxy styrene) PAS 123 100
poly(phenyl acrylate) PPA 50 100
Poly(phenyl acrylate-co-ethyl acrylate) PA-EA 34 62
Poly(p-acetoxystyrene-co-butyl acrylate) AS-BA 47 65
Poly(phenyl acrylate-co-butyl acrylate) PA-BA 6 54
Poly(phenyl acrylate-co-hydroxyethyl methacrylate) PA-HEMA 112 50
Poly(p-acetoxystyrene-co-hydroxyethyl methacrylate) AS-HEMA 108 42
p-Acetoxystyrene functionalized polysiloxane. AS-SiO -88 25
Phenyl 3-butenoate functionalized polysiloxane. PA-SiO -64 20
Homopolymers were easily obtained by free radical polymerization under
conditions similar to those published by Guillet, et.al.11 and Fréchet, et.al.10 These same
conditions were successfully applied to the synthesis of hydrophobic acrylic copolymers.
Initial attempts to copolymerize hydroxyethyl methacrylate (HEMA) with photo-Fries
active monomers under free radical conditions (Figure 32), however, led to the formation
of insoluble cross-linked polymers, presumably due to the presence of ethylene glycol
dimethacrylate as a contaminant in the HEMA. Methods have, however, been published
in the literature for the synthesis of linear HEMA polymers.52,53 Hirao reported the
5 See Appendix III for the structures of the polymers under investigation.

41
synthesis of soluble HEMA by anionic polymerization of the TMS protected monomer,
2-trimethylsilyloxyethyl methacrylate, followed by hydrolysis of the polymer.54 Assadi,
et.al.55 demonstrated the successful synthesis of soluble polymer by the free-radical
polymerization of trimethylsilyloxyethyl methacrylate followed by hydrolysis to cleave
the trimethylsilyl group. In the present research, the process reported by Assadi was
adapted to copolymerize TMS-HEMA and the copolymer was deprotected to yield the
desired high molecular weight linear HEMA copolymer.
OO
or
OO
O O
OH
Insoluble Crosslinked PolymerAIBN65°C, 24h
Figure 32: Initial polymerization conditions for HEMA copolymers. In order to obtain photo-Fries active siloxane polymers, pendant groups were
attached to a commercially available siloxane polymer via platinum-catalyzed
hydrosilylation. Hydrosilylation to add olefins to alkyloxyhydrosilanes has been
extensively reported.56-60 Prior to functionalization of each polymer, hydrosilylation of
the model compound 1,3,5,7-tetramethyl-cyclotetrasiloxane (D4H) was carried out to
probe the reactivity of the olefin towards the silicon-hydrogen bond and the effectiveness

42
of the catalyst.6 Once proper conditions were determined from studies of the model
compounds synthesis of functionalized polysiloxanes was easily achieved.
Kinetics of the photo-Fries Rearrangement
The kinetics of the photo-Fries rearrangement was studied using FT-IR in
a process analogous to that described by Kern.13 Figure 33 shows an example of FT-IR
spectra obtained from PPA as a function of time exposed to 254 nm irradiation. The
conversion of reactant to products in the rearrangement was monitored by observing the
decrease of intensity in the absorption band corresponding to the carbonyl stretch of the
aryl ester at about 1760 cm-1. Copolymers containing nonfunctional methacrylate
comonomers exhibit a second absorption band near 1760 cm-1 that slightly overlaps the
aryl ester absorption being monitored. Even with this overlay, conversion can be
accurately followed by the percent decrease in intensity of the FT-IR absorption band of
the aryl ester moiety.
6 See Appendix IV for a description of these experiments.

43
Figure 33: Overlaid FT-IR Spectra of PPA at 0, 30 and 60 minutes.
New peaks in the FT-IR spectra, corresponding to the ortho and para
rearrangement products, emerge near 1635 and 1675cm-1; and the yield of the ortho and
para products was calculated, based on the mole fraction of ester consumed and the
amount of ortho and para product formed, using Beer’s law and absorbance coefficients
reported by Kern,
The kinetics of the rearrangement in polymer films was measured in triplicate to
statistically account for experimental error. Error associated with the absorbance
coefficients published by Kern is unknown and may be a source of systematic error when
treating the data in this way. Nonetheless, the results obtained from PAS are in
agreement with those reported by Kern. Based upon this precedent, the same
methodology was applied to the analysis of all homopolymers and copolymers studied in
this research.
55
60
65
70
75
80
85
90
95
100
105
15001550160016501700175018001850
cm-1
%T
0 min. 20 min. 40 min.
Ester
ortho
para
OO
n
HO
O
n
OH
O
n
ortho para

44
Research described in this thesis expands upon the work of Guillet, Fréchet and
Kern by systematically investigating the following effects; 1) constraint, 2) proximity
and 3) dielectric constant on the photo-Fries rearrangement. This has been achieved by
copolymerizing monomers bearing photo-Fries active substituents with selected
nonfunctional co-monomers. Nonfunctional co-monomers are any monomer that does
not exhibit aryl ester moieties and as a result is not capable of undergoing any established
photoreaction.
1) Constraint. Constraint has two elements, limitation of rotational degrees of
freedom by tethering the functional group pendant to or within the main chain of a high
molecular weight polymer and mobility related to free volume and the glass transition of
amorphous polymers. In PAS, there is free rotation around the both the aryloxy- and the
acyloxy- bonds of pendant aryl ester groups. In contrast, rotation around the acyloxy
bond in PPA is sterically restricted in PPA (Figure 34). One might therefore expect that
the rate of the photo-Fries rearrangement in PAS might be slightly enhanced over that in
PPA.
n
vs.
n
O O
OO
Figure 34: Comparison of constraint of PAS (left) and PPA (right).
It is also important to note the difference in tethering in Figure 34. Both PAS and
PPA exhibit the same photo-Fries active phenyl ester functional group. The phenyl ester
functional group in PPA is tethered to the polymer backbone through the carbonyl
carbon. PAS is attached to the polymer chain at the para position of the phenyl ring.

45
Accordingly, the para position is blocked and, ortho/para product selectivity cannot be
observed in PAS as it is in PPA.
The rotational degrees of freedom accessible by the photo-Fries active moiety do
not however have an affect on the kinetics. Figure 35 compares the conversion of PAS
and PPA. These polymers have the same phenyl ester moiety tethered to the polymer
backbone; however they are attached in a different way allowing only PPA to rearrange
to both an ortho and para rearrangement product. In the case of PAS, the untethered
geminal radical is an acyloxy radical while in PPA it is a larger phenoxy radical. It was
expected that the increased degree of freedom in PAS would result in a higher initial rate
of rearrangement; instead, both polymers appear to exhibit the same initial rate in
conversion over time. This observation indicates that homolytic bond scission of the
acyloxy bond is not affected by tethering or rotational degrees of freedom.
0 20 40 60 80 100 120
0
10
20
30
40
50
% C
on
vers
ion
Time (min.)
PAS
PPA
Figure 35: Conversion of PAS and PPA versus time.
Mobility associated with free volume is directly related to the glass transition.
The glass transition temperature serves as an approximation of the temperature at which

46
there is the onset of large-scale segmental motion of the polymer backbone. At
temperatures in excess of the glass transition temperature, radicals formed upon
irradiation of aryloxy groups would be expected to have a greater propensity to form para
and cage escape products, due to an increased mobility as a result of increased free
volume.
Increased mobility related to decreased glass transition temperature of the
polymers is observed to have no effect on the initial rate of rearrangement. Figure 37
shows the conversion of PAS compared to that of AS-BA. PAS has a Tg of 123 °C while
that for AS-BA is 47 °C. Despite a difference of over 70 °C, there is no significant
difference in the conversion/time plots of PAS and AS-BA. However both of these
polymers have a Tg that is greater than room temperature.
0 20 40
0
10
20
30
40
50
% C
onvers
ion
Time (min.)
PAS
AS-BA
Figure 36: Conversion of PAS and AS-BA verses time.
PPA, PA-EA and PA-BA shown in Figure 37 have Tg values of 50, 34 and 6°C,
respectively. The temperature at which photolysis was carried out was slightly above

47
room temperature (ca. 30 – 35 °C), due to heat generated by the UV lamp. As a result,
the copolymers, PA-EA and PA-BA, are undergoing photoreaction at temperatures near
or above their glass transition temperatures. Despite this difference, the initial rate of the
photo-Fries rearrangement is observed to be approximately the same in all three
polymers.
0 5 10 15 20 25 30
0
10
20
30
40
50
% C
onvers
ion
Time (min.)
PPA
PA-EA
PA-BA
Figure 37: Conversion of PPA, PA-EA and PA-BA verses time.
A decreased glass transition temperature has no effect on the initial rate but does
change the distribution of rearranged products. Figure 38 shows the yield obtained from
the rearrangement of PPA and PA-EA which have glass transition temperatures of 50 and
34 °C, respectively. Rearrangement of PPA shows a preference for the ortho rearranged
product with an ortho to para ratio (O:P ratio) of about 3. Maximum yields of 30 and
10% are achieved for the ortho and para products, respectively. In PA-EA higher yields
of 50% and 26% are obtained for the ortho and para products, respectively, with an O:P
ratio of about 2. These observations support the original hypothesis that increased

48
mobility in the polymers with lower Tg leads to increased preference for the para
rearrangement product.
0 20 40 60 80 100 120
0
5
10
15
20
25
30
35
40
45
50
55
% y
ield
Time (min.)
PPA : Ortho
PA-EA : Ortho
PPA : Para
PA-EA : Para
Figure 38: Yield of ortho and para products in PPA and PA-EA rearrangements.
The hydration state (i.e. dry compared to swollen) of a hydrophilic hydrogel
polymer might also increase the mobility of radicals within the polymer matrix. In
HEMA containing copolymers, mobility is influenced by the hydration state of the
polymer. It was hypothesized that swollen HEMA polymers would achieve a higher
ultimate conversion due to an increased mobility of radicals within the water swollen free
volume of the polymer but Figure 39 and Figure 40 show the hydration state has no
effect on the conversion (i.e., there is no significant difference in the conversion of dry
and water swollen polymer) but has a notable effect on the formation of products in the
rearrangement. This observation indicates the water swollen free volume (and difference
in dielectric constant of the local environment) has no effect on homolytic bond scission
of the acyloxy bond.
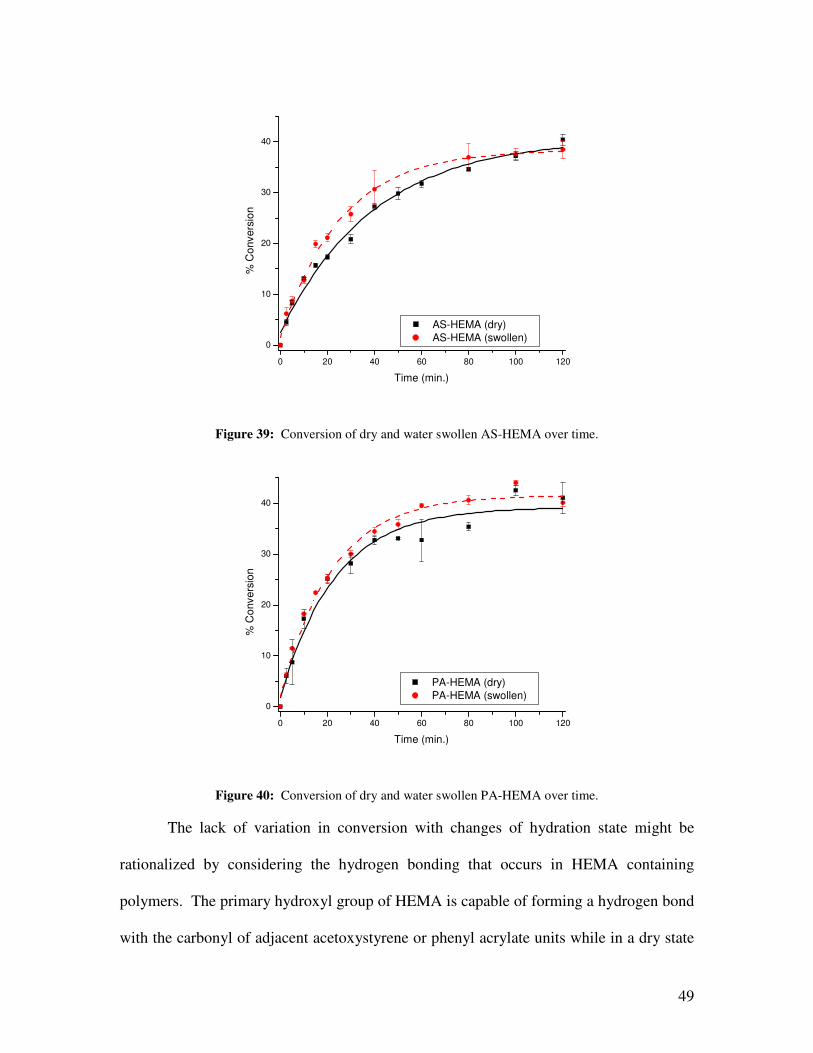
49
0 20 40 60 80 100 120
0
10
20
30
40
% C
onvers
ion
Time (min.)
AS-HEMA (dry)
AS-HEMA (swollen)
Figure 39: Conversion of dry and water swollen AS-HEMA over time.
0 20 40 60 80 100 120
0
10
20
30
40
% C
onvers
ion
Time (min.)
PA-HEMA (dry)
PA-HEMA (swollen)
Figure 40: Conversion of dry and water swollen PA-HEMA over time.
The lack of variation in conversion with changes of hydration state might be
rationalized by considering the hydrogen bonding that occurs in HEMA containing
polymers. The primary hydroxyl group of HEMA is capable of forming a hydrogen bond
with the carbonyl of adjacent acetoxystyrene or phenyl acrylate units while in a dry state

50
thus restricting the movement of untethered radicals. When the polymer becomes
swollen with water, water molecules form a network of hydrogen bonds that mimic the
hydrogen bonding which occurs in the dry polymer. The hydrogen bond network formed
by water restricts the movement of untethered radicals much like movement is restricted
in the dry polymer.
Figure 41 shows the yield of the ortho rearrangement product from AS-HEMA to
be about 20% lower in the water-swollen polymer. One might speculate that the acyloxy
radical formed in the rearrangement in AS-HEMA has increased mobility when the
polymer is hydrated, thus, allowing for increased occurrence of cage escape. The fate of
the acyloxy radical is not known. There was no significant change in the pH of the water
in which the polymer films were immersed. No other analysis was performed on the
water to determine if the radical had diffused into solution.
0 20 40 60 80 100 120
0
10
20
30
40
50
% y
ield
Time (min.)
Ortho (dry)
Ortho (swollen)
Figure 41: Yield of ortho product in AS-HEMA rearrangement.
The yield of rearrangement products obtained from PA-HEMA seen in Figure 42
also shows differences when comparing the dry and water swollen states. Here the

51
polymer is capable of undergoing rearrangement to both an ortho- and para-isomer. The
formation of ortho isomer shows no significant difference between dry and swollen states
while the para isomer exhibits a lower yield when the polymer is in a swollen state. It
was originally thought that the increased mobility brought about by the water swollen
free volume would allow for an increased formation of para rearranged product;
however, this is not the case.
0 20 40 60 80 100 120
0
10
20
30
40
50
% y
ield
Time (min.)
Ortho (dry)
Ortho (swollen)
Para (dry)
Para (swollen)
Figure 42: Yield of ortho and para products in PA-HEMA rearrangement
2) Proximity. The effect of proximity is defined as the influence of one
functional group on an adjacent or proximate functional moiety. For instance, a triad of
structurally repeating units in PAS homopolymer contains phenyl esters surrounded only
by other phenyl esters. In random or alternating copolymers of acetoxystyrene, the
phenyl esters groups are likely to be separated by non-photoactive co-monomer groups
which act as spacers and tend to isolate the photo-Fries active moieties from one another.
The relative proximity of phenyl ester moieties can be controlled by varying the mole
fraction of photo-Fries active monomer and its reactivity ratio relative to the non-

52
photoactive co-monomer. As photo-Fries active units decrease in proximity to one
another (decreased photo-Fries active monomer content) the ultimate conversion of the
rearrangement is observed to increase (Table 5).
Table 5: Ultimate Conversion observed in Polymers Under Investigation.
Polymer Photo-Fries active monomer
content (mole %)
Ultimate conversion
observed (%)
PAS 100 49 PPA 100 50
PA-EA 62 79 AS-BA 65 59 PA-BA 54 52
PA-HEMA 50 44 AS-HEMA 42 39
AS-SiO 25 91 PA-SiO 20 90
The observation of increased conversion in copolymers containing lower
concentrations of photo-Fries active monomer is may be caused by the absorption
characteristics of the hydroxyacetophenone moieties generated in the photo-Fries
rearrangement. Hydroxyacetophenones exhibit a new absorption band near 350 nm in
addition to an increased molar absorptivity near 254 nm. Since the photo-Fries
rearrangement is catalyzed by 254 nm energy, rearranged units at the film surface can
screen UV light from unrearranged units deeper in the film resulting in slower
rearrangement or no rearrangement at those depths.
This screening effect was mitigated by casting films to a thickness (3000 Å) at
which their optical density was low; i.e., UV light will pass through the entire depth of
the film, even if 100% conversion to the strongly UV-absorbing hydroxyacetophenone
were to occur. Despite this precaution, the conversion was still observed to reach a limit.
The observation of a limited conversion can be rationalized by considering the fact that

53
hydroxyacetophenones might act as long range quenchers which suppress the photo-Fries
rearrangement in their immediate vicinity (within a Förster radius). Quenching results
from energy transfer from excited aryl ester functional groups to hydroxyacetophenone
moieties which dissipate energy through non-radiative processes.37
Studies by McCourt, et al. showed that energy quenching by the rearrangement
product molecules of 2-naphthyl acetate in poly(methyl methacrylate) films gave rise to a
spatially non-random distribution of rearranged moieties.60 In other words,
rearrangement does not occur in regions of space that are within the Förster radius of a
molecule that has already rearranged. The conversion of aryl ester groups in polymer
films will thus progress towards a maximum value related to the concentration of
hydroxyacetophenone moieties which quench excited aryl esters in their vicinity. As the
photo-Fries active chromophores become more dilute in a copolymer, they are separated
to distances greater than the Förster radius. Accordingly, hydroxyacetophenone groups
generated by the photo-Fries rearrangement may screen absorbed light but cannot quench
photo-excited aryl ester groups thereby allowing for an increased ultimate conversion.
Figure 43 shows hypothetical dyads in PAS and PA-BA. Upon rearrangement of
a repeat unit in PAS, the resulting hydroxyacetophenone is capable of forming an
intramolecular hydrogen bond or forming a hydrogen bond with an adjacent, or
proximate ( a 1,3- or 1,5- interaction), aryl ester moiety. Formation of hydrogen bonds
with adjacent aryl ester moieties is limited in copolymers such as PA-BA due to
likelihood that hydroxyacetophenone groups will be spatially isolated from one another.

54
OO
O O
CH2
CH2
n m
CH2
CH3
OO OO
n
hν hν
OO OO
n
H
OO OO
n
H
OO
O O
CH2
CH2
n m
CH2
CH3
H
Figure 43: Difference in proximity demonstrated between PAS (left) and PA-BA (right).
3) Dielectric Constant. Dielectric constant (electrostatic permittivity) is
determined by the chemical characteristics (apolar, polar aprotic, polar protic, dipole
moment) of the polymer or polymer composite. By varying the chemical composition of
the polymer backbone (carbon chain or siloxane) and the relative hydrophilicity of the
polymer substituents, the dielectric constant can be substantially varied. This is
analogous to varying the solvent in which the photo-Fries rearrangement of a small
molecule is carried out. Previous experiments have been reported indicating that solvent
polarity or whether a protic/aprotic solvent is used has little effect on the photo-Fries
rearrangement.20, 21 It is presumed then that varying the dielectric constant of the
polymer medium will also have little or no effect on the photo-Fries rearrangement.

55
0 20 40 60 80 100 120
0
10
20
30
40
50
60
70
80
90
100
% C
onvers
ion
Time (min.)
PAS
AS-SiO
Figure 44: Conversion of PAS and AS-SiO.
0 20 40 60 80 100 120
0
10
20
30
40
50
60
70
80
90
100
% C
onvers
ion
Time (min.)
PPA
PA-SiO
Figure 45: Conversion of PPA and PA-SiO.
Figure 44 shows the conversion in PAS compared to AS-SiO. Figure 45 shows
the same plot comparing PPA and AS-SiO. A higher initial rate of rearrangement in
siloxane polymers is an unexpected result. Previous reports indicated that altering the

56
dielectric constant by changing the solvent has no effect on the photo-Fries
rearrangement.20, 21 While the initial rate of the photo-Fries rearrangement was found to
be greater in each siloxane compared to the homopolymers, because of the marked
difference in Tg, this increase rate cannot be unequivocally attributed the difference in
dielectric constant. It is important to note here that the siloxane polymers contain a much
lower content of photo-Fries active monomer (20-25 mol.%) resulting in a drastically
decreased proximity of aryl ester units towards one another. This decreased proximity
leads to a very high ultimate conversion of 90%. Additionally, the Tg of the siloxane
polymers are much lower than the other polymers under investigation (-88 and -64 °C,
AS-SiO and PA-SiO – respectively). It is possible that these variables may be
responsible for the difference in initial rate of rearrangement rather than the difference in
dielectric constant. Based upon comparisons of these polymers, it is not possible to
isolate the variable of dielectric constant to probe its effect on the photo-Fries
rearrangement.
Refractive Index Changes
Measurements of refractive index were made using variable angle spectroscopic
ellipsometry (VASE) according to methods reported by Kern13. Very high precision
values (≤0.001) in refractive index were obtained from this method. Results obtained
from PAS were in agreement with Kern’s published results. Just as Kern reported, a 0.05
∆n was observed in PAS.13 The experimentally obtained Cauchy fit curves seen in
Figure 46 match well with those of Kern (shown in inset plot). The curves are each at an
increasing irradiation time and thus an increased extent of conversion. The wavelength
range of Cauchy fit curves reported in this thesis is from 400-800 nm while Kern’s range

57
extends down to 350 nm. In the region of 350 to 400 nm, a drop of refractive index is
observed. This feature is the result of the new absorption band in that wavelength range
and is not visible from the curves obtained in this study but can be noted in Kern’s
results.
1.5400
1.5600
1.5800
1.6000
1.6200
1.6400
400 450 500 550 600 650 700 750 800
Wavelength (nm)
n
0 15 45 120
Figure 46: Cauchy fit curves showing ∆n of PAS with increasing irradiation time (in min) with Kern’s data shown in inset plot.
Replication of Kern’s observations provided a basis to apply the same analysis to
the other polymers under study in order to determine their respective ∆n. PPA exhibited
a ∆n of 0.07 under an equivalent conversion as PAS. The Cauchy fit curves of PPA at
increasing irradiation times are shown in Figure 47.

58
1.5500
1.5700
1.5900
1.6100
1.6300
1.6500
1.6700
400 450 500 550 600 650 700 750 800
Wavelength (nm)
n
0 5 45 120
Figure 47: Cauchy fit curves showing ∆n in PPA with increasing irradiation time (in min).
Similar Cauchy fit curves were obtained for all of the other polymers under
investigation. The curves had a maximum refractive index at 400 nm and minimum at
800 nm. Figure 48 and Figure 49 show results obtained from hydrophobic and
hydrophilic copolymers (respectively). In all cases, as the rearrangement progressed the
curve translated upwards with very little change in the general shape. The ∆n remained
relatively constant over the wavelength range.

59
1.5200
1.5300
1.5400
1.5500
1.5600
1.5700
1.5800
1.5900
1.6000
1.6100
400 450 500 550 600 650 700 750 800
Wavelength (nm)
n
0 10 20 45
Figure 48: Cauchy fit curves showing ∆n in PA-EA with increasing irradiation time (in min).
1.5300
1.5400
1.5500
1.5600
1.5700
1.5800
1.5900
400 450 500 550 600 650 700 750 800
Wavelength (nm)
n
0 15 80
Figure 49: Cauchy fit curves showing ∆n in PA-HEMA with increasing irradiation time (in min).

60
Table 6 summarizes the experimentally determined ∆n of the polymers under
investigation. Homopolymers exhibit the highest ∆n. Copolymers containing
approximately half of the photo-Fries active monomer content show about half of the ∆n
observed in the analogues homopolymer. The siloxane polymers which contain an even
lower content exhibit a proportionally lower ∆n.
Table 6: Observed ∆n in Polymers Under Investigation.
Polymer Photo-Fries active monomer
content (mole %)
Maximum
observed
conversion (%)
Expected
∆∆∆∆n
Observed ∆∆∆∆n at
400 nm
PAS 100 49 0.025 0.051 PPA 100 50 0.025 0.070
PA-EA 62 79 0.040 0.033 AS-BA 65 59 0.030 0.019 PA-BA 54 52 0.026 0.031
PA-HEMA 50 44 0.022 0.018 AS-HEMA 42 39 0.020 0.017
AS-SiO 25 91 0.046 0.014 PA-SiO 20 90 0.045 0.012
An interesting trend is observed when comparing the maximum observed
conversion to the observed ∆n particularly in the case of homopolymers. The ∆n of 0.05
for the rearrangement of acetoxybenzene occurs when the rearrangement is carried out to
completion; therefore it was hypothesized that a 50% conversion would lead to a nominal
∆n of 0.025. The homopolymers PAS and PPA exhibit a ∆n of 0.05 or more when only
half of the photo-Fries active moieties have rearranged. When the rearrangement occurs
in solution or liquid phase, the aryl ester and hydroxyacetophenone are not isolated from
each other (due to molecular motion in solution) relative to their proximity when tethered
to a polymer. Accordingly, the refractive index contributions sum in a linear fashion and
the overall change in refractive index will be proportionate to the percent conversion and
the relative concentrations of aryl ester and hydroxacetophenone in the solution. This

61
observation suggests that the contribution to the refractive index from polymer dyads or
triads wherein hydroxyacetophenone moieties flanked by aryl ester groups is essentially
different from that for isolated hydroxyacetophone groups. Such a comparison can be
seen in Figure 50 where the ∆n is higher in triads compared to dyads and single aryl ester
units was calculated by Vogel’s method.66, 67
OOOH
O
OO OO OH
O
OO OH
O
OO
HH
2
OO
HH
3
1.3850 1.4123
∆n=0.0273
1.3934 1.4247
∆n=0.0313
1.3964 1.4288
∆n=0.0324
Figure 50: Refractive indices of isolated aryl ester groups, dyads and triads calculated from group contributions.
An additional characteristic of the refractive index change that is important for
utilization of these polymers in devices is a dose dependent response. Figure 51 shows
the refractive index change of PA-EA as extent of conversion increases. A linear trend is

62
observed demonstrating that the desired degree of refractive index change can be
modulated by applying the appropriate dose of energy.
y = 0.0003x - 3E-05
R2 = 0.9968
0
0.005
0.01
0.015
0.02
0.025
0 10 20 30 40 50 60 70
Conversion (%)
∆n
Figure 51: Linear dose dependent modulation of ∆n in PA-EA.
Preliminary Sensitization Experiments
Naphthalene-1-yl benzoate (compound 2) was synthesized according to methods
published by Kern.15 This small molecule is a model compound analogous to the polymer
in Figure 17 and is capable of undergoing rearrangement upon exposure to 310 nm light.
Compound 2 was doped into a film of poly(vinyl butyral), PVB, at a concentration of 5%
by weight. Fluorene was chosen as an antenna molecule because of its absorption and
emission characteristics near 310 nm. Fluorene was of particular interest because it had
been successfully used as an antenna molecule in Guillet’s 1999 publication. Increasing
concentrations of fluorene were doped in multiple PVB films. Progress of the
rearrangement in the presence of fluorene was monitored via FT-IR.
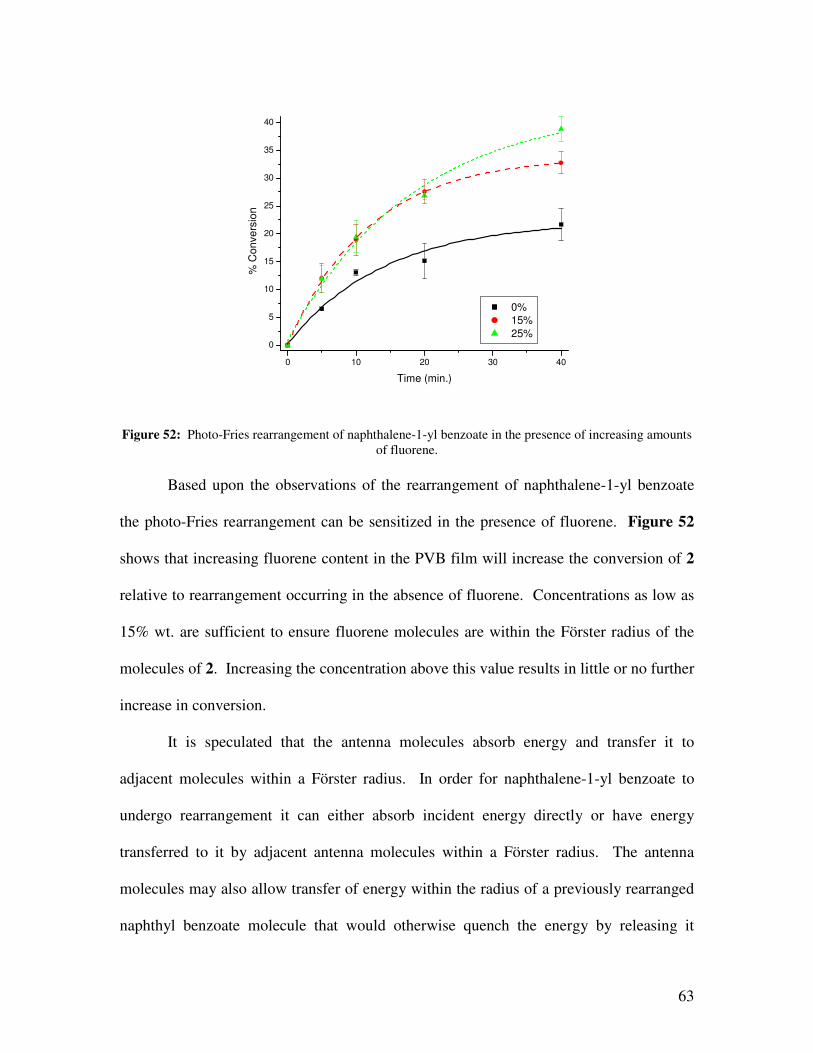
63
0 10 20 30 40
0
5
10
15
20
25
30
35
40
% C
on
ve
rsio
n
Time (min.)
0%
15%
25%
Figure 52: Photo-Fries rearrangement of naphthalene-1-yl benzoate in the presence of increasing amounts of fluorene.
Based upon the observations of the rearrangement of naphthalene-1-yl benzoate
the photo-Fries rearrangement can be sensitized in the presence of fluorene. Figure 52
shows that increasing fluorene content in the PVB film will increase the conversion of 2
relative to rearrangement occurring in the absence of fluorene. Concentrations as low as
15% wt. are sufficient to ensure fluorene molecules are within the Förster radius of the
molecules of 2. Increasing the concentration above this value results in little or no further
increase in conversion.
It is speculated that the antenna molecules absorb energy and transfer it to
adjacent molecules within a Förster radius. In order for naphthalene-1-yl benzoate to
undergo rearrangement it can either absorb incident energy directly or have energy
transferred to it by adjacent antenna molecules within a Förster radius. The antenna
molecules may also allow transfer of energy within the radius of a previously rearranged
naphthyl benzoate molecule that would otherwise quench the energy by releasing it

64
through non-radiative processes. The presence of antenna molecules statistically
increases the probability that an incoming photon of energy will trigger a rearrangement
of a naphthyl benzoate molecule.
Conclusions and Future Directions
Through the series of experiments reported in this thesis the effects of constraint,
proximity and dielectric constant on the photo-Fries rearrangement in polymers have
been investigated. The following conclusions can be drawn from this work.
1. Constraint in terms of rotational degrees of freedom has no impact on the rate of
the photo-Fries rearrangement. Constraint in terms of increased mobility due to
free volume (decreased glass transition temperature) has no effect on the initial
rate of the rearrangement but causes an increase in the yield of para rearranged
products in the photolysis of PPA copolymers.
2. Increased mobility in hydrated hydrophilic copolymers has no effect on the initial
rate of rearrangement but increased formation of cage escape products is
observed.
3. Decreasing the proximity of the photo-Fries active moieties to one another in
copolymers results in an increased ultimate conversion of the rearrangement.
4. The rate of the photo-Fries reaction in siloxane-based polymers is substantially
greater than that in carbon-chain polymers. While the dielectric constant of
siloxane polymers is much lower than that in carbon chain polymers, because of
the substantial difference in Tg of these polymers, this rate enhancement cannot be
unequivocally attributed to dielectric constant. Other factors such as decreased
proximity and lower Tg may be the cause of the differences observed.

65
5. Dose dependant changes in refractive index ranging from 0.07 in PPA
homopolymer to 0.01 in siloxane polymers containing 20 mole % of aryl ester
functional groups were measured.
The preliminary experiments in sensitization of the photo-Fries rearrangement of
small molecules active at a longer wavelength suggest the need to further develop
polymers based upon these systems. The refractive index change of naphthyl-1-yl
benzoate has yet to be evaluated. Further work must also be done to determine the
feasibility of using two-photon irradiation on long wavelength active photo-Fries
polymers. Based upon the results of the photo-Fries rearrangement in siloxane polymers,
low Tg polymers with decreased proximity of photo-Fries active moieties would allow for
high conversions to be achieved relatively quickly. Incorporation of antenna molecules
into such polymers (either covalently or non-covalently) may allow for the conversion to
be increased even further. The presence of antenna molecules within such a polymer
may also allow for an increased concentration of photo-Fries active monomer thus
increasing the total ∆n that may be realized.
Appendix
Appendix I – Existing methods of refractive index modulation.
Refractive index changes resulting from cross-linking of cinnamoyl double bonds
forming cyclobutane dimers were reported by Nagata et al.42

66
H2
CH2
C
CH3 CH3
n m
O
CH2
O
OO
O
O O
O
Polymer Strand 1
Polymer Strand 2
H2
CHC
H2
CHC
n m
O
O
O
Polymer Strand 1
Polymer Strand 2
OH
O
O
O
UV
UV
Ultraviolet induced cross-linking of cinnamoyl double bonds leads to a decrease
in refractive index of 0.015.
Refractive index changes resulting from photo-chemical induced isomerization of
norbornadiene moieties attached to calixarenes were reported by Kudo et al.43
67
R
OR
CH2
n
R=
OH
O
O
Ph
OH
O
O
Ph
hν OH
O
O
Ph
∆
Refractive index changes of 0.028 – 0.061 can be realized. Refractive index
changes are positive and can be reversed upon addition of heat.
Refractive index changes resulting from photoreaction of mesoionic sulfur-
substituted phenyloxatriazolones doped into poly(methyl methacrylate), PMMA, films
were reported by Kato et al.41
NN
O
O
NN
ON
O
NN
SN
O
NN
SN
S
NN
ON
S
The above compounds were doped into PMMA films and upon exposure to UV
light the compounds underwent photoreaction. The exact nature of the photoreaction is

68
unknown but suspected to involve a nitrene adduct with molecules in the surrounding
environment. Irreversible decreases in refractive index from 0.0008 – 0.036 were noted.
Refractive index changes resulting from photoreaction of azoaromatic
chromaphores doped into PMMA films were reported by Mendonca et al.40
N
N NH2
O2N
N
N N
O2N
N
N N
O2N
OH
Cl OH
Disperse Orange 3
Disperse Red 1
Disperse Red 13
The above dyes were doped into PMMA films and upon exposure to femtosecond
laser energy undergo photoreaction. The exact nature of the photoreaction is unknown.
A change in refractive index was demonstrated as a part of this work by constructing a
waveguide device, but the exact ∆n was not quantified.
Appendix II - Förster energy transfer.
Förster energy transfer is the transfer of energy from a molecule or functional
group in its excited state to an acceptor molecule or functional group. Förster energy
transfer is also known as resonance energy transfer (RET). RET is typically

69
characterized by an overlap in the emission spectrum of the donor and the absorption
spectrum of the acceptor (though this alone does not guarantee the occurrence of energy
transfer). RET occurs without the emission of a photon from donor to acceptor unlike
radiative energy transfer. Energy transfer occurs through long-range interactions of the
dipoles of donors and acceptors. When the donor absorbs a photon of light its dipole
begins to oscillate at a specific frequency. The oscillating dipole of the donor can then
interact with the dipole of a nearby acceptor. If the resonance frequency of the acceptor
is similar to the donor’s frequency, the acceptor will begin to oscillate as well thus
transferring energy to the acceptor.
Rate of energy transfer (kT) is affected by the relative overlap between donor
emission and acceptor absorption, quantum yield of the donor, orientation of donor and
acceptor dipoles and distance between donor and acceptor. Equation 1 shows kT as a
function of decay of donor in absence of acceptor (τD), distance between donor and
acceptor (r) and the Förster distance (R0)62. The Förster distance is the distance at which
energy transfer is 50% efficient. Equation 2 shows the relationship between efficiency
(E), the Förster distance and acceptor-donor distance. An efficiency of 50% (0.5) is
obtained if r = R0. The Förster distance is typically between 20-60 Å.
6
01
=
r
Rk
D
T τ (1)
660
60
rR
RE
+= (2)
A more detailed explanation of RET can be found in Principles of Fluorescence
Spectroscopy by Lakowicz.63
Appendix III - Structures of polymers under investigation.

70
OO
OO
nn
OO
O O
CH2
CH2
n m
CH2
CH3
O O
CH2
CH2
n m
CH2
CH3
OO
O O
CH2
CH3
n m
OO
O O
CH2
CH2
n m
OO
OH
OO
O O
CH2
CH2
n m
OH
Si O
CH3
Si O
CH3
CH3
n m
Si O
CH3
Si O
CH3
CH3
n m
OO
O
O
PAS PPA PA-EA
AS-BA
PA-BA
AS-HEMA
PA-HEMA
AS-SiO
PA-SiO
Appendix IV – Probing olefin reactivity in the hydrosilylation reaction via hydrosilylation of D4
H siloxane as a model compound.
It has been reported in the literature that the reaction of a silicon hydrogen bond
with an olefin, called hydrosilylation, is dependent on the reactivity of the olefin
substrate, reaction conditions, catalyst type and catalyst concentration.57-59, 64 As a result
the reactivity of aryl ester containing olefin substrates towards D4H siloxane was probed
before carrying out a similar reaction on a polysiloxane bearing silane bonds.

71
The initial approach towards polysiloxanes with aryl ester pendant groups
analogous in structure to PAS was to use phenol 4-(2-propenyl)-acetate (1) as an olefin
substrate. This compound was obtained according to the following synthesis.
OMe
BBr3, CH2Cl2
-78°C to RT50 min., 85%
OH
Ac2O, Et3N, 4-DMAP, CH2Cl2
0°C to RT, 20 min., 71%
O
CH3
O
1
The compound was obtained in 44% yield over two steps. The following reaction
of compound 1 was attempted but unexpectedly found to yield no product. Increasing the
temperature and amount of catalyst had no effect on the reaction.
O Si
CH3
H4
OO
O Si
CH3
4
OO
Toluene, Pt catalyst
70 °C, o/n
A similar reaction was carried out using allyl benzene as the olefin substrate.
This reaction did proceed efficiently to completion after one hour at 50 °C. There are no
known reports in the literature of difficulty in hydrosilylation of olefins containing aryl
esters. At present time, it is unclear as to why this reaction is not proceeding.
Based upon previous precedent64, a second approach was developed using p-
acetoxy styrene as the starting olefin. Reactivity of p-acetoxy styrene towards silane

72
under platinum catalyzed hydrosilylation conditions was probed by carrying out the
following reaction.
O Si
CH3
H4
OO
O Si
CH3
4
OO
Toluene, Pt catalyst
RT, 30 mins.
The reaction proved to be very facile leading to predominant α substitution across
the double bond and quantitative functionalization of silane. Based upon these
observations it was concluded p-acetoxystyrene was sufficiently reactive towards silane
and would be suitable for synthesis of polysiloxanes with aryl ester pendant groups
analogous in structure to PAS.
Reactivity of phenyl 3-butenoate towards silane under platinum catalyzed
hydrosilylation conditions was probed by carrying out the following reaction.
O Si
CH3
H4
OOO Si
CH3
4Toluene, Pt catalyst
50 °C, 2 h.
O
O
The reaction was not as facile as previous reaction with styrene. An incomplete
functionalization of silane was observed leaving some exposed Si-H bonds. Extent of

73
functionalization can easily be increased by increasing the equivalents of olefin and/or
allowing reaction to run for longer than two hours. Based upon these observations it was
concluded phenyl 3-butenoate was sufficiently reactive towards silane and would be
suitable for synthesis of polysiloxanes with aryl ester pendant groups analogous in
structure to PPA.
Appendix V – State of present work towards sensitization of the photo-Fries rearrangement.
The plan for investigation of the sensitization of the photo-Fries rearrangement
included an investigation of the use of fluorene and coumarin as sensitizing molecules.
At the time this thesis was completed, only the experiments with fluorene produced data
of sufficient quality. Experiments with coumarin yielded insufficient data. Additionally,
the reactivity of naphthalene-2-yl benzoate has yet to be probed in this system. This
molecule has been synthesized using the same conditions as the 1 substituted isomer but
starting with 2-naphthol as the alcohol.
In experiments with coumarin, a set of doped PVB films identical to those
described for fluorene was prepared with coumarin (in place of fluorene) at
concentrations of 5, 15 and 25% wt. FT-IR data was unsuccessful at monitoring the
progress of the rearrangement because the carbonyl stretching band of coumarin overlaps
that of naphthalene-1-yl benzoate. Progress of the rearrangement can also be monitored
by following the increase in absorption at 360 nm in the UV spectrum (see below);
however only single measurements were taken at each data point so no averages or error
bars could be calculated. Additionally, no data was gathered with 0% coumarin content.
When using coumarin as a sensitizing molecule it is important to maintain the
concentration at levels low enough to minimize the known dimerization of coumarin65

74
that can occur when exposed to light. It is not clear from this data where the lower
concentration limit is to insure coumarin molecules are within the Förster radius of the
naphthyl ester molecules. Further experiments must be done in order to determine the
effectiveness of coumarin as a sensitizer for the photo-Fries rearrangement.
0 10 20 30 40
0.00
0.02
0.04
0.06
0.08
0.10
0.12
Ab
so
rba
nce
Time (min.)
5%
15%
25%
The approach of doping a PVB film with desired aryl ester and varying levels of
sensitizer molecule serves as a good system to test the ability of a molecule to sensitize
the rearrangement and to probe the reactivity of a particular aryl ester. This system may
also allow for the ability of a sensitizer molecule to absorb under two-photon irradiation
conditions to be assessed. Sensitizer molecules that are capable of two-photon absorption
have yet to be determined.
Despite the utility of doped PVB films as a test system, it is still desirable to have
both aryl ester and sensitizer incorporated covalently into a polymer. Methods have been
published by Kern for the synthesis of the monomer naphthalene-1-yl vinyl-benzoate.15
Additionally, the synthesis of the monomer 2-vinylfluorene was published by Guillet.33

75
Additional chemistry for synthesis of other monomers with sensitizer structures, like
coumarin, has yet to be determined. Incorporation of these monomers into a polymer at
appropriate levels will produce a polymer capable of undergoing a sensitized photo-Fries
rearrangement.

76
References
1. Cui, C.; Weiss, R. G., Photo-Fries Rearrangements of 2-Naphthyl Acrylates as Probes of the Size and Shape of Guest Sites Afforded by Unstretched and Stretched Low-Density Polyethylene Films. A Case of Remarkable Selectivity. J. Am. Chem.
Soc. 1993, 115, (21), 9820-9821.
2. Tung, C.-H.; Ying, Y.-M., Photochemistry of Phenyl Phenylacetates Adsorbed on Pentasil and Faujasite Zeolites. J. Chem. Soc., Perkin Trans. 2 1997, 1319-1322.
3. Banu, H. S.; Pitchumani, K.; Srinivasan, C., Effect of Cyclodextrin Complexation on Photo-Fries Rearrangement of Naphthyl Esters. Tetrahedron 1999, 55, 9601-9610.
4. Tung, C.-H.; Wu, L.-Z.; Yuan, Z.-Y.; Guan, J.-Q.; Ying, Y.-M.; Wang, H.-W.; Xu, X.-H., Remarkable Product Selectivity in Photochemical Reactions Within Supramolecular Systems. Materials Science and Engineering C 1999, 10, 75-81.
5. Tung, C.-H.; Xu, X.-H., Selectivity in the Photo-Fries Reaction of Phenyl Phenylacetates Included in a Nafion Membrane. Tetrahedron Letters 1999, 40, (1), 127-130.
6. Gu, W.; Weiss, R. G., Photo-Fries Rearrangements of Phenyl Phenylacylates in Polyethylene Films. Comparison of Reactivity and Selectivity with 1-Naphthyl Phenylacylates. J. Org. Chem. 2001, 66, (5), 1775-1780.
7. Gu, W.; Bi, S.; Weiss, R. G., Photo-Fries Rearrangements of 1-naphthyl esters in the Glassy and Melted States of poly(vinyl acetate). Comparisons With Reactions in Less Polar Polymers and Low-viscosity Solvents. Photochem. Photobiol. Sci. 2002, 1, 52-59.
8. Kaanumalle, L. S.; Nithyanandhan, J.; Pattabiraman, M.; Jayaraman, N.; Ramamurthy, V., Water-Soluble Dendrimers as Photochemical Reaction Media: Chemical Behavior of Singlet and Triplet Radical Pairs Inside Dendritic Reaction Cavities. J. Am. Chem. Soc. 2004, 126, (29), 8999-9006.
9. Xu, J.; George, M.; Weiss, R. G., Photo-Fries rearrangements of 1-naphthyl (R)-2-phenylpropanoate in poly(vinyl acetate) and ethyl acetate. Influence of medium polarity and polymer relaxation on motions of singlet radical pairs. Anais de
Academia Brasileria de Ciencias 2006, 78, (1), 31-44.
10. Frechet, J. M. J.; Tessier, T. G., Poly[p-(formyloxystyrene]: Synthesis and Radiation-Induced Decarbonylation. Macromolecules 1985, 18, (3), 317-320.
11. Li, S.-K. L.; Guillet, J. E., Studies of the photo-Fries reaction in solid poly(phenyl acrylate). Macromolecules 1977, 10, (4), 840-44.
12. Merle-Aubry, L.; Holden, D. A.; Merle, Y.; Guillet, J. E., Photophysics and Photochemistry of Naphthyl Ester Polymers in Solution. Macromolecules 1980, 13, 1138-1143.

77
13. Hofler, T.; Grieber, T.; Gstrein, X.; Trimmel, G.; Jakopic, G.; Kern, W., UV Reactive Polymers for Refractive Index Modulation Based on the Photo-Fries Rearrangement. Polymer 2007, 48, 1930-1939.
14. Griesser, T.; Hofler, T.; Temmel, S.; Kern, W.; Trimmel, G., Photolithographic Patterning of Polymer Surfaces Using the Photo-Fries Rearrangement: Selective Postexposure Reactions. Chem. Mater. 2007, 19, (12), 3011-3017.
15. Daschiel, U.; Höfler, T.; Jakopic, G.; Schmidt, V.; Kern, W., Selected Polymers that Contain Aromatic Ester Units: Synthesis, Photoreactions, and Refractive Index Modulation. Macromolecular Chemistry and Physics 2007, 208, (11), 1190-1201.
16. Gillberg-LaForce, G. E.; al., e. Polymeric Direct Imaging Holographic Compositions. 5,128,223, July 7, 1992, 1992.
17. Kobsa, H., Rearrangement of Aromatic Esters by Ultraviolet Radiation. Journal of
Organic Chemistry 1962, 27, (7), 2293-2298.
18. Anderson, J. C.; Reese, C. B., Photo-Induced Fries Rearrangements. Proc. Chem.
Soc. 1960, June, 217.
19. Coppinger, G. M.; Bell, E. R., Photo-Fries Rearrangement of Aromatic Esters. Role of Steric and Electronic Factors. The Journal of Physical Chemistry 1966, 70, (11), 3479-3489.
20. Sandner, M. R.; Trecker, D. J., On the Mechanism of the Photo-Fries Reaction. Journal of the American Chemical Society 1967, 89, (22), 5725-5726.
21. Sandner, M. R.; Hedaya, E.; Trecker, D. J., Mechanistic Studies of the Photo-Fries Reaction. Journal of the American Chemical Society 1968, 90, (26), 7249-7254.
22. Jimenez, M. C.; Miranda, M. A.; Scaiano, J. C.; Tormos, R., Two-photon processes in the photo-Claisen and photo-Fries rearrangements. Direct observation if dienic ketenes generated by photolysis of transient cyclohexa-2,4-dienones. Chemical
Communications 1997, 1487-88.
23. Lochbrunner, S.; Zissler, M.; Piel, J.; Riedle, E., Real Time Observation of the Photo-Fries Rearrangement. Journal of Chemical Physics 2004, 120, (24), 11634-11639.
24. Fries, K. F., G., Ber. Dtsch. Chem. Ges. 1908, 41, 2447.
25. Crevatín, L. K.; Bonesi, S. M.; Erra-Balsells, R., Photo-Fries Rearrangement of Carbazol-2-yl Sulfonates: Efficient Tool for the Introduction of Sulfonyl Groups into Polycyclic Aromatic Compounds. Helvetica Chimica Acta 2006, 89, (6), 1147-1157.
26. Bonesi, S. M.; Crevatín, L. K.; Erra-Balsells, R., Photochemistry of 2-acyloxycarbazoles. A potential tool in the synthesis of carbazole alkaloids. Photochem. Photobiol. Sci. 2004, 3, (4), 381 - 388.
27. Park, K. K.; Lee, J. J.; Ryu, J., Photo-Fries rearrangement of N-arylsulfonamides to aminoaryl sulfone derivatives. Tetrahedron 2003, 59, (39), 7651.

78
28. Norell, J. R., Organic reactions in Liquid Hydrogen Fluoride. IV. The Fries Rearrangement of Aryl Benzoates. J. Org. Chem. 1973, 38, (10), 1924-1928.
29. Oliveira, A. M. A. G.; Oliveira-Campos, A. M. F.; Raposo, M. M. M.; Griffiths, J.; Machado, A. E. H., Fries Rearrangement of Dibenzofuran-2-yl Ethanoate Under Photochemical and Lewis-acid-catalysed Conditions. Tetrahedron 2004, 60, (29), 6145-6154.
30. Bagno, A.; Kantlehner, W.; Kress, R.; Saielli, G.; Stoyanov, E., Fries Rearrangement of Aryl Formates: A Mechanistic Study by Means of 1H, 2H and 11B NMR Spectroscopy and DFT Calculations. J. Org. Chem. 2006, 71, (25), 9331-9340.
31. Cui, C.; Wang, X.; Weiss, R. G., Investigation of the Photo-Fries Rearrangements of Two 2-Naphthyl Alkanoates by Experiment and Theory. Comparison with the Acid-Catalyzed Reactions. J. Org. Chem. 1996, 61, (6), 1962-1974.
32. Arumugam, S.; Vutukuri, D. R.; Thayumanavan, S.; Ramamurthy, V., Amphiphilic Homopolymer as a Reaction Medium in Water: Product Selectivity within Polymeric Nanopockets. J. Am. Chem. Soc. 2005, 127, (38), 13200-13206.
33. Nowakowska, M.; Storsberg, J.; Zapotoczny, S.; Guillet, J. E., Studies of the Antenna Effect in Polymer Molecules. 28. Photo-Fries Rearrangement of 1-Naphthyl Acetate in Aqueous Solutions of Poly(sodium styrenesulfonate-co-2-vinylfluorene). New J. Chem. 1999, 23, 617-623.
34. Mauritz, K. A.; Moore, R. B., State of Understanding of Nafion. Chem. Rev. 2004, 104, (10), 4535-4586.
35. Gierke, T. D.; Munn, G. E.; Wilson, F. C., The morphology in nafion perfluorinated membrane products, as determined by wide- and small-angle x-ray studies. J.
Polym. Sci., Polym. Phys. 1981, 19, (11), 1687.
36. Gupta, A.; Rembaum, A.; MoaCanin, J., Solid State Photochemistry of Polycarbonates. Macromolecules 1978, 11, (6), 1285-1288.
37. Gupta, A.; Liang, R.; Moacanin, J.; Goldbeck, R.; Kliger, D., Photochemical Processes in Polymeric Systems. 2. Photochemistry of a Polycarbonate of Bisphenol A in SOlution and in the Solid Phase. Macromolecules 1980, 13, (2), 262-267.
38. Vallon, S.; Drevillon, B.; Poncin-Epaillard, F.; Klemberg-Sapieha, J. E.; Martinu, L., Argon Plasma Treatment of Polycarbonate: In situ Spectroellipsometry Study and Polymer Characterizations. J. Vac. Sci. Technol. A 1996, 14, (6), 3194-3201.
39. Allen, N. S., Photostabilising Action of ortho-Hydroxy Aromatic Compounds: A Critical Review. Polymer Photochemistry 1983, 3, 167-187.
40. Mendonca, C. R.; Cerami, L. R.; Shih, T.; Tilghman, R. W.; Baldacchini, T.; Mazur, E., Femtosecond laser waveguide micromachining of PMMA films with azoaromatic chromophores. Opt. Express 2008, 16, (1), 200.

79
41. Yuichi Kato, K. H., Photoinduced Refractive Index Change of Polymer Films Containing Mesoionic Sulfur-Substituted Phenyloxatriazolones. Macromolecular
Chemistry and Physics 2002, 203, (16), 2290-2295.
42. Nagata, A.; Sakaguchi, T.; Ichihashi, T.; Miya, M.; Ohta, K., Refractive Index Decrease During Photocrosslinking in Photopolymers Suitable for Holographic Recording. Macromol. Rapid Commun. 1997, 18, 191-196.
43. Kudo, H.; Ueda, W.; Sejimo, K.; Mitani, K.; Nishikubo, T.; Anada, T., New Large Refractive-Index Change Materials: Synthesis and Photochemical Valence Isomerization of the Calixarene Derivatives Containing Norbornadiene Moieties. Bulletin of the Chemical Society of Japan 2004, 77, (7), 1415.
44. Veierov, D.; Bercovici, T.; Fischer, E.; Mazur, Y.; Yogev, A., Effect of Substituents and of Wavelength of Irradiation on the Photo-Fries-Rearrangement of Enol-Esters Preliminary Communication. Helvetica Chimica Acta 1975, 58, (4), 1240-1243.
45. Veierov, D.; Mazur, Y.; Fischer, E., Mechanistic studies in the photochemical Fries rearrangement of enol esters. Journal of the Chemical Society, Perkin Transactions
2: Physical Organic Chemistry 1980, 11, 1659-1664.
46. Mehlhorn, A.; Schwenzer, B.; Schwetlick, K., Mo-calculations of the energy transfer activities of organic [pi]-structures in the photo-fries rearrangement--II: Selection of sensitizers and inhibitors of the photo-fries reaction based on theoretical absorption and fluorescence data. Tetrahedron 1977, 33, (12), 1489.
47. Tsao, R.; Eto, M., Photolysis of Flutolanil and the Effect of Some Photosensitizers. Agric. Biol. Chem. 1991, 55, (3), 763-768.
48. Loong, W.-a.; Su, A.-n., Enhanced oxygen reactive ion etching resistance of diazonaphthoquinone-poly(formyloxystyrene) resist system by photoacid catalyzed photo-fries rearrangement and potassium ion treatment in aqueous solution. Microelectronic Engineering 1991, 13, (1-4), 101.
49. Loong, W.-A.; Chen, R.-H., Photoacid Catalyzed Photo-Fries Rearrangements of Poly(P-Formyloxystyrene) and Formyloxynovolac. Molecular Crystals and Liquid
Crystals 1990, 183, (1), 481 - 489.
50. Holden, D. A.; Shephard, S. E.; Guillet, J. E., Studies of the Antenna Effect in Polymer Molecules. 4. Energy Migration and Transfer in 2-Naphthyl Methacrylate-Aryl Vinyl Ketone Copolymers. Macromolecules 1982, 15, (6), 1481-1485.
51. Guillet, J. E., Polymer Photophysics and Photochemistry - An Introduction to the
Study of Photoprocesses in Macromolecules. Cambridge University Press: Cambridge, 1985; p 391.
52. Gregonis, D. E.; Russell, G. A.; Andrade, J. D.; deVisser, A. C., Preparation and properties of stereoregular poly(hydroxyethyl methacrylate) polymers and hydrogels. Polymer 1978, 19, (11), 1279.
53. Teruo Okano, M. K. I. S., The influence of hydrophilic and hydrophobic domains on water wettability of 2-hydroxyethyl methacrylate-styrene copolymers. Journal of
Applied Polymer Science 1978, 22, (2), 369-377.

80
54. Hirao, A.; Kato, H.; Yamaguchi, K.; Nakahama, S., Polymerization of monomers containing functional groups protected by trialkylsilyl groups. 5. Synthesis of poly(2-hydroxyethyl methacrylate) with a narrow molecular weight distribution by means of anionic living polymerization. Macromolecules 1986, 19, (5), 1294-1299.
55. Assadi, M. G.; Mahkam, M.; Tajrezaiy, Z., Synthesis and characterization of some organosilicon derivatives of poly 2-hydroxyethyl methacrylate with cubane as a cross-linking agent. Journal of Organometallic Chemistry 2005, 690, (21-22), 4755.
56. Michalczyk, M. J.; Farneth, W. E.; Vega, A. J., High-Temperature Stabilization of Cross-linked Siloxanes Glasses. Chemistry of Materials 1993, 5, (12), 1687-1689.
57. Malki, A. E.; Hannioui, A.; Knouzi, J.; Vaultier, M., Regioselective Hydrosilylation of Allylic Derivatives by the Siloxanes D4H and MDHM in the Presence of Platinum Catalysts. Phys. Chem. News 2004, 17, 107-112.
58. Musolf, M. C.; Speier, J. L., The Addition of Silicon Hydrides to Olefinic Double Bonds. X. Addition to Phenylalkenes. The Nuclear Magnetic Resonance Proton Spectra of (Phenylalkyl)silanes. J. Org. Chem. 1964, 29, (9), 2519-2524.
59. Mukbaniani, O.; Tatrishvili, T.; Titvinidze, G.; Mukbaniani, N., Hydrosilylation Reactions of Methylhydridesiloxane to Styrene and alpha-methylstyrene. Journal of
Applied Polymer Science 2006, 101, 388-394.
60. Yamamoto, Y.; Ayama, K. Ester Group-Containing Siloxane Compound, and Its Preperation Method. 6,107,504, August 22, 2000, 2000.
61. Wang, Z.; Holden, D. A.; McCourt, F. R. W., Generation of nonrandom chromophore distributions by the photo-Fries reaction of 2-naphthyl acetate in poly(methyl methacrylate). Macromolecules 1990, 23, (16), 3773-3779.
62. Förster, T., Intermolecular Energy Migration and Fluoresence. Ann. Phys. 1948, 2, 55-75.
63. Lakowicz, J. R., Principles of Fluorescence Spectroscopy. Second Edition ed.; Springer: 1999; p 725.
64. Lewis, L. N., On the mechanism of metal colloid catalyzed hydrosilylation: proposed explanations for electronic effects and oxygen cocatalysis. J. Am. Chem.
Soc. 1990, 112, (16), 5998-6004.
65. Kim, H. C.; Kreiling, S.; Greiner, A.; Hampp, N., Two-photon-induced cycloreversion reaction of coumarin photodimers. Chemical Physics Letters 2003, 372, (5-6), 899.
66. Krevelen, D. W. V., Properties of Polymers - Their Correlation with Chemical
Structure; Their Numerical Estimation and Prediction From Additive Group
Contributions. 3 ed.; Elsevier: New York, 1990; p 875.
66. Vogel, A. I., Cresswell, W. T. and Leicester, J. Bond Refractions for Tin, Silicon, Lead, Germanium and Mercury Compounds. J. Phys. Chem. 1954, 58, (2), 174-177.

81


















