
The Cholesteryl Ester Cycle in Macrophage Foam
9
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 255, No. 19, hue of October 10, pp. 9344-9352, 1980 Printed m U.S.A. The Cholesteryl Ester Cycle in Macrophage Foam Cells CONTINUAL HYDROLYSIS AND RE-ESTERIFICATION OF CYTOPLASMIC CHOLESTERYL ESTERS* (Received for publication, May 2, 1980, and in revised form, June 17, 1980) Michael S. Brown, Y. K. Ho, and Joseph L. Goldstein From the Devartments of Molecular Genetics and Znternal Medicine, University of Texas Health Science Center at Dallas, Dallas, Texas 75235 . Mouse peritoneal macrophages take up acetylated human low density lipoprotein by receptor-mediated endocytosis, hydrolyze its cholesteryl esters in lyso- somes, and re-esterify the cholesterol in the cytoplasm. The re-esterified cholesterol accumulates as cytoplas- mic lipid droplets that resemble the droplets seen in “foam cells” of atherosclerotic lesions. In the present studies, we have investigated the metabolism of these cytoplasmic lipid droplets by incubating mouse peri- toneal macrophages with acetylated human low den- sity lipoprotein and then withdrawing the lipoprotein and following the fate of the cytoplasmic cholesteryl esters. The results demonstrate that these esters un- dergo a continual cycle of hydrolysis and re-esterifica- tion with a half-life of about 24 h. Hydrolysis appears to be mediated by a non-lysosomal cholesteryl ester hydrolase whose activity is resistant to lysosomal in- hibitors such as chloroquine and ammonium chloride. Re-esterification is mediated by an acyl-CoA:choles- terol acyltransferase enzyme whose activity is en- hanced in extracts of cells that have accumulated cho- lesteryl esters. Progesterone, an inhibitor of macro- phage acyl-CoAxholesterol acyltransferase activity in vitro, inhibits the re-esterification of hydrolyzed cho- lesteryl esters in intact cells. As a result, progesterone produces a decline in the cellular content of cholesteryl ester and a reciprocal increase in the free cholesterol content of the cells. Inasmuch as the re-esterification reaction uses acyl-CoA derivatives that require ATP for their synthesis, the continual hydrolysis and re- esterification of cholesterol in macrophage foam cells constitutes a futile cycle that wastes ATP. Incubation of cholesteryl ester-laden macrophages with high den- sity lipoprotein disrupts the cycle by removing free cholesterol from the cell, thereby suppressing the re- esterification reaction and leading to net cholesteryl ester hydrolysis. Thus, high density lipoprotein pro- motes net cholesteryl ester hydrolysis without actually increasing the rate of the hydrolytic reaction. These findings may have relevance to the pathologic changes that occurwhen macrophages and other scavenger cells become overloaded with cytoplasmic cholesteryl ester droplets in disease states. In a variety of pathologic conditions, the cholesterol com- ponent of circulating plasma lipoproteins becomes desposited in body tissues (1-3). Much of this cholesterol accumulates as * Supported by Research Grant HL-20948 from the National In- stitutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. cholesteryl ester droplets in the cyotplasm of macrophage-like scavenger cells. Under the light microscope the droplets im- part to the cells a foamy, vacuolated appearance that accounts for their description as “foam cells.” By electron microscopy most of the cholesteryl ester droplets are not surrounded by a limiting membrane and hence are believed to be located free in the cytoplasm and not within lysosomes (4, 5). The mech- anism for the accumulation of these cytoplasmic cholesteryl esters and the reason for their lack of excretion by the mac- rophage is unknown. Recently a model system has been devised by which the major histologic and biochemical features of foam cells can be reproduced in vitro (6, 7). Macrophages derived from the mouse peritoneal cavity are incubated in monolayer culture with human plasma low density lipoprotein that has been chemically modified byacetylation. The acetyl-LDL’ binds to receptor siteson the macrophage surface whereupon it is internalized by adsorptive endocytosis and delivered to lyso- somes (6,7). Within the lysosome the cholesteryl esters of the lipoprotein are hydrolyzed, liberating free cholesterol. The free cholesterol crosses the lysosomal membrane and enters the cytoplasm where much of it is re-esterified by a micro- somal acyl-CoA:cholesterol acyltransferase. The resulting cholesteryl esters accumulate as cytoplasmic lipid droplets (7). The development of the above in vitro system has made possible a study of the metabolism of cytoplasmic cholesteryl ester droplets in macrophages. In earlier studies, we observed that the cytoplasmic cholesteryl ester droplets disappeared when the source of incoming cholesterol (i.e. the acetyl-LDL) was removed from the medium. This morphologic change was associated with a fall in the cholesteryl ester content of the cells as determined by mass measurements (7). The choles- terol released from the cholesteryl ester droplets appeared in the medium exclusively as free (Le. unesteritied) cholesterol (8). Thus, excretion of the stored cholesteryl esters required their preliminary hydrolysis. Net hydrolysis did not occur to any appreciable degree unless the culture medium contained a substance that was capable of binding the unesterified cholesterol and removing it from the cell (8). Among the most potent cholesterol-accepting substances were intact red blood cells and certain plasma lipoproteins, especially high density lipoprotein (8). In thepresence of a cholesterol acceptor such as HDL, the cytoplasmically stored cholesteryl esters were rapidly hydrolyzed, and the free cholesterol was excreted from the cell. In the absence of these agents, the cholesteryl esters ’ The abbreviations used are: LDL, low density lipoprotein; HDL, high density lipoprotein; r-[ch~lesteryl-~H-linoleate]acetyl-LDL, hep- tane-extracted acetyl-LDL reconstituted with [’HJchoZesteryl linole- ate; ACAT, acyl-CoA:cholesterol acyltransferase; DMEM, Dulbecco’s modified Eagle’s medium. 9344
Transcript of The Cholesteryl Ester Cycle in Macrophage Foam

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 255, No. 19, h u e of October 10, pp. 9344-9352, 1980 Printed m U.S.A.
The Cholesteryl Ester Cycle in Macrophage Foam Cells CONTINUAL HYDROLYSIS AND RE-ESTERIFICATION OF CYTOPLASMIC CHOLESTERYL ESTERS*
(Received for publication, May 2, 1980, and in revised form, June 17, 1980)
Michael S. Brown, Y. K. Ho, and Joseph L. Goldstein From the Devartments of Molecular Genetics and Znternal Medicine, University of Texas Health Science Center a t Dallas, Dallas, Texas 75235 .
Mouse peritoneal macrophages take up acetylated human low density lipoprotein by receptor-mediated endocytosis, hydrolyze its cholesteryl esters in lyso- somes, and re-esterify the cholesterol in the cytoplasm. The re-esterified cholesterol accumulates as cytoplas- mic lipid droplets that resemble the droplets seen in “foam cells” of atherosclerotic lesions. In the present studies, we have investigated the metabolism of these cytoplasmic lipid droplets by incubating mouse peri- toneal macrophages with acetylated human low den- sity lipoprotein and then withdrawing the lipoprotein and following the fate of the cytoplasmic cholesteryl esters. The results demonstrate that these esters un- dergo a continual cycle of hydrolysis and re-esterifica- tion with a half-life of about 24 h. Hydrolysis appears to be mediated by a non-lysosomal cholesteryl ester hydrolase whose activity is resistant to lysosomal in- hibitors such as chloroquine and ammonium chloride. Re-esterification is mediated by an acyl-CoA:choles- terol acyltransferase enzyme whose activity is en- hanced in extracts of cells that have accumulated cho- lesteryl esters. Progesterone, an inhibitor of macro- phage acyl-CoAxholesterol acyltransferase activity in vitro, inhibits the re-esterification of hydrolyzed cho- lesteryl esters in intact cells. As a result, progesterone produces a decline in the cellular content of cholesteryl ester and a reciprocal increase in the free cholesterol content of the cells. Inasmuch as the re-esterification reaction uses acyl-CoA derivatives that require ATP for their synthesis, the continual hydrolysis and re- esterification of cholesterol in macrophage foam cells constitutes a futile cycle that wastes ATP. Incubation of cholesteryl ester-laden macrophages with high den- sity lipoprotein disrupts the cycle by removing free cholesterol from the cell, thereby suppressing the re- esterification reaction and leading to net cholesteryl ester hydrolysis. Thus, high density lipoprotein pro- motes net cholesteryl ester hydrolysis without actually increasing the rate of the hydrolytic reaction. These findings may have relevance to the pathologic changes that occur when macrophages and other scavenger cells become overloaded with cytoplasmic cholesteryl ester droplets in disease states.
In a variety of pathologic conditions, the cholesterol com- ponent of circulating plasma lipoproteins becomes desposited in body tissues (1-3). Much of this cholesterol accumulates as
* Supported by Research Grant HL-20948 from the National In- stitutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
cholesteryl ester droplets in the cyotplasm of macrophage-like scavenger cells. Under the light microscope the droplets im- part to the cells a foamy, vacuolated appearance that accounts for their description as “foam cells.” By electron microscopy most of the cholesteryl ester droplets are not surrounded by a limiting membrane and hence are believed to be located free in the cytoplasm and not within lysosomes (4, 5). The mech- anism for the accumulation of these cytoplasmic cholesteryl esters and the reason for their lack of excretion by the mac- rophage is unknown.
Recently a model system has been devised by which the major histologic and biochemical features of foam cells can be reproduced in vitro (6, 7). Macrophages derived from the mouse peritoneal cavity are incubated in monolayer culture with human plasma low density lipoprotein that has been chemically modified by acetylation. The acetyl-LDL’ binds to receptor sites on the macrophage surface whereupon it is internalized by adsorptive endocytosis and delivered to lyso- somes (6 ,7) . Within the lysosome the cholesteryl esters of the lipoprotein are hydrolyzed, liberating free cholesterol. The free cholesterol crosses the lysosomal membrane and enters the cytoplasm where much of it is re-esterified by a micro- somal acyl-CoA:cholesterol acyltransferase. The resulting cholesteryl esters accumulate as cytoplasmic lipid droplets (7).
The development of the above in vitro system has made possible a study of the metabolism of cytoplasmic cholesteryl ester droplets in macrophages. In earlier studies, we observed that the cytoplasmic cholesteryl ester droplets disappeared when the source of incoming cholesterol (i.e. the acetyl-LDL) was removed from the medium. This morphologic change was associated with a fall in the cholesteryl ester content of the cells as determined by mass measurements (7). The choles- terol released from the cholesteryl ester droplets appeared in the medium exclusively as free (Le. unesteritied) cholesterol (8). Thus, excretion of the stored cholesteryl esters required their preliminary hydrolysis. Net hydrolysis did not occur to any appreciable degree unless the culture medium contained a substance that was capable of binding the unesterified cholesterol and removing it from the cell (8). Among the most potent cholesterol-accepting substances were intact red blood cells and certain plasma lipoproteins, especially high density lipoprotein (8). In the presence of a cholesterol acceptor such as HDL, the cytoplasmically stored cholesteryl esters were rapidly hydrolyzed, and the free cholesterol was excreted from the cell. In the absence of these agents, the cholesteryl esters
’ The abbreviations used are: LDL, low density lipoprotein; HDL, high density lipoprotein; r-[ch~lesteryl-~H-linoleate]acetyl-LDL, hep- tane-extracted acetyl-LDL reconstituted with [’HJchoZesteryl linole- ate; ACAT, acyl-CoA:cholesterol acyltransferase; DMEM, Dulbecco’s modified Eagle’s medium.
9344

Cholesteryl Ester Cycle in Macrophages 9345
remained within the cell indefinitely as visible cytoplasmic lipid droplets (7 , 8).
In the current experiments, we have begun to study the turnover of the stored cytoplasmic cholesteryl esters in mac- rophages and to determine why there is no net hydrolysis of these esters in the absence of a cholesterol acceptor. To answer these questions, we have used two experimental ap- proaches that permit separate measurements of the synthesis, hydrolysis, and excretion of cytoplasmic cholesteryl esters. The fust approach involves the use of acetyl-LDL that has been reconstituted with ['Hlcholesteryl linoleate, hereafter designated as r-[ch~ZesteryZ-~H-linoleate]acetyl-LDL. When macrophages are incubated with r-[~holesteryl-~H-linole- atelacetyl-LDL, the cells incorporate the hydrolyzed ['HI- cholesterol into re-esterified cholesteryl ester droplets. The presence of radiolabeled cholesterol in cytoplasmic cholesteryl esters allows measurement of initial rates of hydrolysis and excretion of these esters. The second approach involves a double label protocol in which the macrophages are f is t induced to accumulate cytoplasmic cholesteryl esters labeled with 3H in the fatty acid portion and are subsequently switched to medium containing I4C-labeled fatty acids. This protocol permits separate measurements of the hydrolysis and re-esterifcation of the cytoplasmic cholesteryl esters. These studies have revealed that stored cytoplasmic cholesteryl es- ters are constantly undergoing a cycle of hydrolysis and re- esterification. The presence of a cholesterol acceptor in the medium interrupts this cycle, promoting the net hydrolysis and excretion of the stored cholesteryl esters. This net hy- drolysis is achieved primarily through a reduction in the rate of the ACAT-mediated re-esterification reaction and not by an increase in the rate of hydrolysis per se.
EXPERIMENTAL PROCEDURES
Materials-Male and female NCS mice (25 to 30 g) were obtained from The Rockefeller University. [1,2-'H]Cholesterol (43 Ci/mmol), [l-'4C]oleic acid (56 mCi/mmol), and sodium ["'II]iodide (16 mCi/pg) were purchased from Amersham/Searle. r9.10-"H]Oleic acid (7.12 Ci/ m o l ) was obtained from New England Nuclear Corp. ['4C]Oleate and ["Hloleate were bound to bovine serum albumin as previously described (9). [3H]Cholestery2 linoleate was synthesized from [1,2- 3H]cholesterol and linoleyl chloride (10). Human albumin (crystal- lized) was purchased from Miles Biochemicals (catalogue No. 82-301- 1). Dulbecco's modified Eagle medium (catalogue No. 320-1885) and Dulbecco's phosphate-buffered saline (catalogue No. 310-4190) were purchased from Grand Island Biological Co. All other tissue culture supplies and reagents for assays were obtained from sources as previously reported (7, 10).
Lipoproteins-Human LDL (d 1.019 to 1.063 g/ml), HDLa (d 1.125 to 1.215 g/ml), and lipoprotein-deficient serum (d > 1.215 g/ml) were isolated from the plasma of individual healthy subjects by ultracen- trifugation (11). LDL was acetylated with repeated additions of acetic anhydride ( 6 ) . Acetyl-LDL radiolabeled with ["H]cholesteryl linoleate was prepared by a previously described reconstitution method in which the endogenous neutral lipids of acetyl-LDL were extracted with heptane and replaced with ["Hlcholesteryl linoleate (7, 10). The resulting reconstituted lipoprotein, designated r-[cholesteryE-"H-li- noleatelacetyl-LDL, was shown to comigrate with acetyl-LDL in agarose gel electrophoresis at pH 8.3 (7). Acetyl-LDL was radiolabeled with Iz5I as previously described (6) . The concentrations of acetyl- LDL and HDL are given in terms of the protein content of the lipoproteins.
Mouse Macrophage Monolayers-Resident peritoneal cells were harvested from mice in phosphate-buffered saline by a previously described modification (6, 7) of the procedure developed by Edelson and Cohn (12). The fluid from 20 to 40 mice ( 6 to 10 X 10" cells/ mouse) was pooled, and the cells were collected by centrifugation (400 X g, 10 min, room temperature) and washed once with 30 ml of Dulbecco's modified Eagle medium. The cells were resuspended in DMEM containing 20% (v/v) fetal calf serum, penicillin (100 units/ ml), and streptomycin (100 pg/ml) at a final concentration of 2 to 3 X lo6 cells/ml. The 1-ml aliquots of this cell suspension were dis-
pensed into plastic Petri dishes (35 mm X 10 mm) and incubated in a humidified Con (570) incubator at 37°C. After 1 to 2 h, each dish was washed three times with 2 ml of DMEM without serum to remove nonadherent cells, after which the macrophage monolayers were used for experiments (day 0).
Standard Method for Loading Macrophages with [3HJCholes- teryl Esters-On day 0, each macrophage monolayer received 1 ml of DMEM containing 2 mg/ml of human albumin and 50 p g / d of r- [ch~lesteryl-~H-linoleate]acetyl-LDL (670 to 2710 cpm/nmol of cho- lesteryl linoleate; mass ratio of cholesterol to protein, 0.60 to 0.97). and the monolayers were incubated at 37°C for 24 h. On day 1, the medium was discarded, and each monolayer was washed twice with 2 ml of DMEM containing 10% fetal calf serum followed by one wash with 2 ml of DMEM (standard wash method). Each monolayer was then incubated at 37°C for 24 h with 1 ml of DMEM containing 1 mg/ml of human albumin. On day 2, the medium was discarded, and each monolayer was washed once with 2 ml of DMEM and then used for assay of cholesteryl ester hydrolysis and excretion as indicated in the legends. AU incubations were performed at 37°C unless otherwise indicated.
Extraction of Sterols from Macrophage Monolayers and Culture Medium-Free and esterified cholesterol were extracted directly from macrophage monolayers in situ in the plastic Petri dish. Each washed monolayer was incubated with hexane/isopropyl alcohol (3:2) for 30 min at room temperature (13). The organic solvent was removed, each monolayer was rinsed briefly with 1 ml of solvent of the same composition, and the two organic solvent extracts were combined. An internal standard containing [14C]cholesterol (30 pg, IO00 cpm) was added, and the sample was further processed for gas-liquid chroma- tography or thin layer chromatography as described below. After the lipids had been extracted, the cells in the monolayer were dissolved in 1 ml of 0.2 N NaOH, and aliquots were removed for protein determination. Sterols were extracted from culture medium with chloroform/methanol according to the method of Bligh and Dyer (14) as previously described (7).
Assays-The mass of free and esterified cholesterol was measured by gas-liquid chromatography (7). The cellular content of free and esterified ["H]cholesterol derived from r-[cholesteryl-"H-linoleate]- acetyl-LDL and the excretion of the liberated ["H]cholesterol into the medium was quantified by extraction as above and thin layer chro- matography as previously described (7). The data are expressed as the amount of free or esterified [,'H]cholesterol contained within the cells or the medium per milligram of total cell protein. The amount of [l-'4C]oleate or [9,10-.'H]oleate incorporated into cholesteryl esters and triglycerides by macrophage monolayers was determined by incubation with the labeled fatty acid bound to albumin (9), followed by extraction as above and thin layer chromatography as previously described (7). The activity of acyl-CoA:cholesterol acyltransferase in cell-free extracts of macrophages was measured by a previously reported method (7). The assay was linear with time up to 60 min and with extract concentration up to 100 pg of protein/200 pl reaction volume. ACAT activity is expressed as the picomoles of cholesteryl ['4C]oleate formed/h/mg of total extract protein. The total cellular content of '''1 radioactivity derived from the binding and uptake of '"I-labeled acetyl-LDL and the amount degraded to trichloroacetic acid-soluble (non-iodide) material was measured as previously de- scribed (6). The protein content of lipoproteins and cells was deter- mined by the procedure of Lowry et al. (15) with bovine serum albumin as standard. In experiments in which two isotopic labels were used, all data were corrected for the spillover between isotope chan- nels. Unless otherwise stated, each data point in the figures represents the average of duplicate incubations.
RESULTS
Net Hydrolysis and Excretion of Cytoplasmic ChoZesteryl Esters in the Presence of HDL--In the standard protocol, mouse peritoneal macrophages were incubated in the presence of acetyl-LDL that had been reconstituted with [3H]choZes- teryZ linoleate (designated as r-[~hoZesteryZ-~H-linoleate]ace- tyl-LDL). Previous studies have shown that the macrophages take up the r-[ch~ZesteryZ-~H-linoleate]acetyl-LDL by adsorp- tive endocytosis, hydrolyze the cholesteryl esters in lysosomes, and re-esterify the cholesterol in the cytoplasm for storage as cholesteryl ester droplets (6, 7). After incubation with r-[cho- ZesteryZ-"H-linoleatelacetyl-LDL for 24 h, the cells were

9346 Cholesteryl Ester Cycle in Macrophages
washed and incubated for an additional 24 h in lipoprotein- free medium containing 1 mg/ml of albumin (which does not remove cholesterol from the cells (8)). During this period, the cells reached a state of equiIibrium in which all of the [:'H]choZesteryZ linoleate had been hydrolyzed and most of the liberated [3H]cholesterol had been re-esterified such that the content of ["H]cholesteryl esters remained reasonably con- stant. HDL was added at this point to promote the hydrolysis and excretion of the stored [3H]cholesteryl esters.
The above points are illustrated in the experiment of Fig. 1. When cells were allowed to accumulate [3H]cholesteryl esters by incubation with r-[ch~lesteryl-~H-linoleate)acetyl-LDL and were then washed and incubated in the absence of lipo- proteins, the cellular content of ['H]cholesteryl esters (closed symbols, Fig. L 4 ) and free ["H]cholesterol (closed symbols, Fig. 1B) remained relatively constant throughout the 72-h duration of the experiment. When HDL was added at zero time, the cellular content of [3H]cholesteryl esters declined rapidly, falling by more than 50% (200 nmol/mg of protein) over 24 h (Fig. lA), and the cellular content of free ['HI- cholesterol declined by about 70 nmol/mg of protein (Fig. 1B). Although most of the decline in radioactive cellular sterol represented a loss in ["Hlcholesteryl ester (200 out of 270 nmol/mg of protein), all of the excreted radioactivity re- covered in the medium was free ["H]cholesterol (250 nmol/ mg of protein) (Fig. E ) . No [3HH]cholesteryl ester was found in the medium. The response was similar when HDL was added after a delay of 24 h or 48 h. In each case, HDL produced a rapid net hydrolysis of the ~?H]cholesteryl ester and excretion of free ["H]cholesterol.
Fig. 2 shows the amount of free ~"H]cholesteroI excreted from the cells at 8 h and 24 h after the addition of HDL as a function of the concentration of HDL added to the culture medium. The amount of excreted free [:'H]cholesterol in- creased with increasing HDL concentrations and reached a maximal rate at a lipoprotein concentration between 50 and 133 pg of protein/ml.
Excretion of free [3H]cholesterol in the presence of HDL was dependent on temperature (Fig. 3). At 4°C the excretion was reduced by 95% as compared with 37°C. At 24°C a small amount of free tJH]cholesterol was excreted during the 1st h of incubation with HDL, but excretion soon stopped (Fig. 3B). A t 37OC, excretion of free VHlcholesterol continued linearly for at least 7 h. The lack of HDL-mediated excretion of free ["H]cholesterol at 4°C was associated with a lack of hydrolysis of the cellular [3H]cholesteryl esters (data not shown).
To confm that the loss of [3H]cholesteryl esters from the macrophages reflected a loss of cholesteryl ester mass and not
simply a one-€or-one exchange of labeled cellular cholesterol for unlabeled HDL cholesterol, the experiment shown in Fig. 4 was performed. Cells were loaded with r-[cholesteryl-"H- linoleatelacetyl-LDL according to the standard protocol and
t //
0 ' " " " Y 0 50 ( 0 0 1 5 0 200 250 300 1
HDL (pq protetn/ml) Hours
FIG. 2. Excretion of free [3€KJcholesterol from macrophages as a function of the concentration of HDL. A, macrophages were incubated with r-CchoZesteryZ-"H-linoleate]acetyl-LDL for 24 h, washed, and incubated for a further 24 h in the absence of lipoproteins according to the standard procedure. On day 2, each monolayer received 1 ml of DMEM containing the indicated concentration of HDL. After incubation at 37°C for either 8 h (0) or 24 h (O), the amount of iree [?H]cholesterol in the medium was measured by thin layer chromatography. B presents the data of A replotted as a function of the time of incubation with HDL. The cellular content of free and esterified [%]cholesterol at zero time was 213 and 405 nmol/ mg of ceU protein, respectively.
- 2:u 0 2 4 6 0 2 4 6 - Hours
FIG. 3. Effect of temperature on the excretion of free [3HJ- cholesterol from macrophages in the presence of HDL. Macro- phages were incubated with r-[cholesteryl-"H-linoleatelacetyt-LDL for 24 h, washed, and incubated a further 24 h in the absence of lipoproteins according to the standard procedure. On day 2, each monolayer received 1 ml of minimum Eagle's medium (without bicar- bonate) containing 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesul- fonic acid (pH 7.4) and either no HDL (A) or 200 p g / d of HDL (B). After incubation for the indicated time at the indicated temperature, the amount of free [3H]cholesterol in the medium was measured by thin layer chromatography. The cellular content of free and esterified [JH]cholesterol at zero time was 187 and 449 nmol/mg of cell protein, respectively.
r p. j3H~Cholesleryl Eslers In Cells [ 8 [3H]Cholesterol In Cells
50 c-1 - 0 ' ' ~ " " ' ' ' ~ " '
0 12 24 36 48 60 72 0 12 24 36 48 60 72
Hours Hours Hours
FIG. 1. Triggering of hydrolysis and excretion of stored (250 pg/ml) was added to individual groups of dishes at zero time, 24 [3H]cholesteryl esters by addition of HDL. Macrophages were h, or 48 h as indicated by the arrows. After incubation at 37OC in the incubated with r-[cholestery6"H-linoleate]acetyl-LDL for 24 h, absence (01 or presence of HDL (0, A, U) for the indicated time, the washed, and incubated a further 24 h in the absence of lipoproteins esterified r3H]cholesterol in the ceUs (A), the free ["H]cholesterol in according to the standard procedure. On day 2 (zero time), each the cells (B) , and the free [3H]cholesterol in the medium (C) was monolayer received 1 ml of DMEM containing no HDL (e). HDL measured by thin layer chromatography,

Cholesteryl Ester Cycle in Macrophages 9347
I I I I
0 50 0 3 IX) 2 0 0 250 0 0 2 04 06 08 10
HDLfpgpoIeln/mli HDLImgprole8nlmll
FIG. 4 (left). HDLstimulated hydrolysis of stored cholesteryl esters; comparison of isotopic measurements with mass meas- urements. Macrophages were incubated with r-[cholesteryl-,'H- linoleate]acetyl-LDL for 24 h, washed, and incubated a further 24 h in the absence of lipoproteins according to the standard procedure except that each monolayer contained twice the usual number of macrophages. On day 2, each monolayer received 1 ml of DMEM containing the indicated concentration of HDL. After incubation at 37°C for 24 h, the cellular sterols were extracted with hexane/isopro- pyl alcohol (3:2). The lipid extract was used for separate measure- ments of the mass and radioactivity of free (0, 0) and esterified (A, A) cholesterol by gas-liquid chromatography (mass measurement) (A, 0) and thin layer chromatography (TLC) (radioactivity measure- ment) (A, 0). FIG. 5 (right). HDLstimulated hydrolysis of cellular choles-
teryl ["Cloleate, but not ['4C]tiglycerides. Macrophage mono- layers were prepared as described under "Experimental Procedures." On day 0, each monolayer received 1 ml of DMEM containing 20% fetal calf serum. On day 1, the medium was replaced with 1 ml of DMEM containing 10% fetal calf serum, 50 pg/ml of acetyl-LDL, and 0.2 mM ['4C]oleate-albumin (5300 cpm/nmol). After incubation at 37°C for 24 h, each monolayer was washed by the standard method, after which the cells were incubated for 24 h with 1 ml of DMEM containing the indicated concentration of HDL. Following the second incubation, the monolayers were washed and harvested, and their content of cholesteryl ['4C]oleate (0) and ['4C]triglycerides (0) was measured by thin layer chromatography. The "100% of control" values were 216 and 114 nmol/mg of protein for cholesteryl ["C]- oleate and ['4C]triglycerides, respectively.
were subsequently incubated with different concentrations of HDL. Following extraction with hexane/isopropyl alcohol, the cell extracts were divided into two aliquots. One aliquot was used to measure the content of free [3H]cholesterol and ['H]cholesteryl ester by thin layer chromatography and scin- tillation counting. The second aliquot was used for measure- ment of the cellular mass of free and esterified cholesterol by gas-liquid chromatography. Under all circumstances, there was close agreement between the two methods used to assess cholesteryl ester content, indicating that there was no dilution of the radioactivity by exchange with the unlabeled choles- terol of HDL. The data for free cholesterol were slightly different. In the absence of HDL, the mass of free cholesterol in the cells was about 25% higher than could be accounted for by radioactive cholesterol derived from hydrolysis of r-[cho- le~teryl-~H-linoleate]acetyl-LDL. This excess mass of free cholesterol presumably represented unlabeled cholesterol that was present in the cells prior to the addition of the r-[ choles- teryL3H-linoleate]acetyl-LDL. In this experiment, the amount of free cholesterol measured either by gas-liquid chromatog- raphy or by radioactivity did not change significantly in the presence of HDL. In three other experiments conducted with a similar protocol, the cellular content of free cholesterol declined by an average of 22% when measured by mass and 26% when measured by radioactivity.
Under conditions in which HDL stimulated the net hydrol- ysis and excretion of cholesteryl esters, the lipoprotein had little effect on the hydrolysis or excretion of triglycerides (Fig. 5 ) . This was shown by incubating macrophages with unlabeled acetyl-LDL plus [ 14C]oleate. At the end of this incubation, the
cells had incorporated the radioactive fatty acid into ["CJ- triglycerides as well as into cholesteryl [14C]oleate. The sub- sequent addition of HDL led to a drop in the cellular content of cholesteryl ['4C]oleate without a significant change in the content of ['4C]triglycerides.
Hydrolysis and Re-esterification of Stored Cholesteryl Es- ters in the Absence and Presence of €€DL-The above data indicate that macrophages do not engage in net hydrolysis of cholesteryl esters unless the culture medium contains an acceptor, such as HDL, that can bind the liberated cholesterol. The failure of net hydrolysis in the absence of a cholesterol acceptor might occur as a result of one of two mechanisms: 1) the cholesteryl ester hydrolase reaction might be slow in the absence of a cholesterol acceptor, but be activated when a cholesterol acceptor is present; or 2) the cholesteryl ester
* A NO HDL E HDL. 250p~/ml
"""""
0 - Q - 0 I < I ~-0-0
0 6 12 18 24 30 0 6 12 18 24 30 Hours HOUE
FIG. 6. Hydrolysis and re-esterification of stored cholesteryl esters in macrophages incubated in the absence and presence of HDL. Macrophage monolayers were prepared as described under "Experimental Procedures." On day 0, each monolayer received 1 ml of DMEM containing 2 mg/ml of human albumin, 50 pg/ml of acetyl- LDL, and 0.2 mM ['JH]oleate-albumin (270,000 cpm/nmol). After 24 h at 37OC, each monolayer was washed by the standard method and then incubated for a further 24 h with 1 ml of DMEM containing 1 mg/ml of human albumin and 0.2 mM ['H]oleate-albumin (270,000 cpm/nmol) in the absence of lipoproteins. On day 2, each monolayer was washed in the same manner as on day 1, after which triplicate monolayers were harvested to determine the cellular content of cholesteryl r3H]oleate (zero time). The remaining monolayers re- ceived 1 ml of DMEM containing 1 mg/ml of human albumin and 0.2 mM [14C]oleate-albumin (6,040 cpm/nmol) in the absence ( A ) or presence (B) of 250 pg/ml of HDL. After the indicated interval at 37"C, triplicate monolayers were washed four times with 2.5 ml of ice-cold phosphate-buffered saline and the cellular content of choles- teryl [:'H]oleate (0) and cholesteryl [I4C]oleate (0) was determined by thin layer chromatography and double label scintillation counting. The dashed line indicates the total cholesteryl oleate content, i.e. the sum of the values for cholesteryl r3H]oleate and cholesteryl ["C]- oleate.
0 2 4 6 0 2 4 6 + HO",* HOW*
FIG. 7. HDLstimulated excretion of free [3H]cholesterol; lack of inhibition by acetyl-LDL. Macrophages were incubated with r-[~holesteryl-~H-linoleate]acetyl-LDL for 24 h, washed, and incubated for a further 24 h in the absence of lipoproteins according to the standard procedure. On day 2 (zero time), each monolayer received 1 ml of DMEM containing either no HDL (A) or 200 pg/ml of HDL ( B ) in the presence of the indicated concentration of acetyl- LDL. After incubation at 37°C for the indicated time, the amount of free [3H]cholesterol in the medium was measured by thin layer chromatography. The cellular content of free and esterified r3H]- cholesterol at zero time was 190 and 488 nmol/mg of protein, respec- tively.
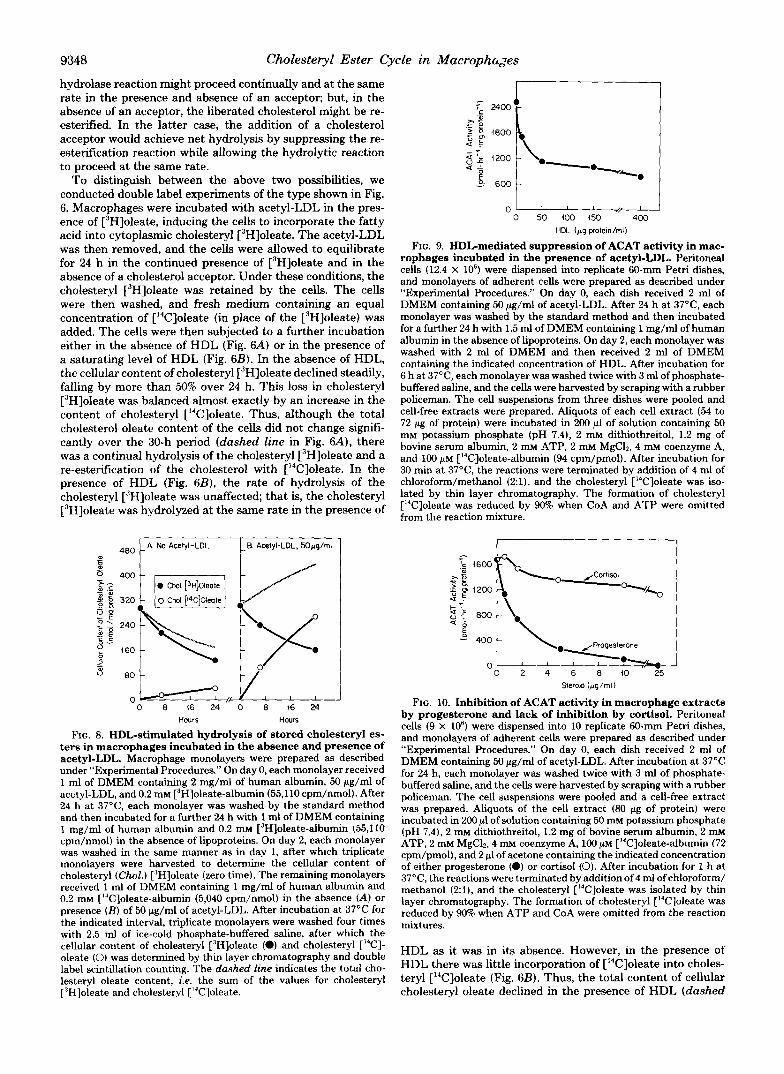
9348 Cholesteryl Ester Cycle in Macrophages
hydrolase reaction might proceed continually and at the same rate in the presence and absence of an acceptor; but, in the absence of an acceptor, the liberated cholesterol might be re- esterified. In the latter case, the addition of a cholesterol acceptor would achieve net hydrolysis by suppressing the re- esterification reaction while allowing the hydrolytic reaction to proceed at the same rate.
To distinguish between the above two possibilities, we conducted double label experiments of the type shown in Fig. 6. Macrophages were incubated with acetyl-LDL in the pres- ence of ['Hloleate, inducing the cells to incorporate the fatty acid into cytoplasmic cholesteryl ['Hloleate. The acetyl-LDL was then removed, and the cells were allowed to equilibrate for 24 h in the continued presence of r3H]oleate and in the absence of a cholesterol acceptor. Under these conditions, the cholesteryl [3H]oleate was retained by the cells. The cells were then washed, and fresh medium containing an equal concentration of ['4C]oleate (in place af the r3H]01eate) was added. The cells were then subjected to a further incubation either in the absence of HDL (Fig. 6A) or in the presence of a saturating level of HDL (Fig. 6B) . In the absence of HDL, the cellular content of cholesteryl [3H]oleate declined steadily, falling by more than 50% over 24 h. This loss in cholesteryl [3H]oleate was balanced almost exactly by an increase in the content of cholesteryl ['4C]oleate. Thus, although the total cholesterol oleate content of the cells did not change signifi- cantly over the 30-h period (dashed line in Fig. 6A), there was a continual hydrolysis of the cholesteryl [3H]oleate and a re-esterification of the cholesterol with ['4C]oleate. In the presence of HDL (Fig. 6B), the rate of hydrolysis of the cholesteryl ['Hloleate was unaffected; that is, the cholesteryl [3H ]oleate was hydrolyzed at the same rate in the presence of
0 50 100 150 400
HDL (pg prote~n/mll
FIG. 9. HDL-mediated suppression of ACAT activity in mac- rophages incubated in the presence of acetyl-LDL. Peritoneal cells (12.4 X lo6) were dispensed into replicate 60-mm Petri dishes, and monolayers of adherent cells were prepared as described under "Experimental Procedures." On day 0, each dish received 2 ml of DMEM containing 50 pg/ml of acetyl-LDL. After 24 h at 37"C, each monolayer was washed by the standard method and then incubated for a further 24 h with 1.5 ml of DMEM containing 1 mg/ml of human albumin in the absence of lipoproteins. On day 2, each monolayer was washed with 2 ml of DMEM and then received 2 ml of DMEM containing the indicated concentration of HDL. After incubation for 6 h at 37"C, each monolayer was washed twice with 3 ml of phosphate- buffered saline, and the cells were harvested by scraping with a rubber policeman. The cell suspensions from three dishes were pooled and cell-free extracts were prepared. Aliquots of each cell extract ( 5 4 to 72 pg of protein) were incubated in 200 p1 of solution containing 50 mM potassium phosphate (pH 7.4), 2 mM dithiothreitol, 1.2 mg of bovine serum albumin, 2 m~ ATP, 2 mM MgCh, 4 m~ coenzyme A, and 100 PM ['4C]oleate-albumin (94 cpm/pmol). After incubation for 30 min at 37"C, the reactions were terminated by addition of 4 ml of chloroform/methanol (2:1), and the cholesteryl [I4C]oleate was iso- lated by thin layer chromatography. The formation of cholesteryl ['4C]oleate was reduced by 90% when CoA and ATP were omitted from the reaction mixture.
1" Acetyl-LDL, 50pg/ml I
80
0 0 8 (6 24
Hours 0 8 16 24
Hours
FIG. 8. HDL-stimulated hydrolysis of stored cholesteryl es- ters in macrophages incubated in the absence and presence of acetyl-LDL. Macrophage monolayers were prepared as described under "Experimental Procedures." On day 0, each monolayer received 1 ml of DMEM containing 2 mg/ml of human albumin, 50 pg/ml of acetyl-LDL, and 0.2 mM [3H]oleate-albumin (55,110 cpm/nmol). After 24 h at 37OC, each monolayer was washed by the standard method and then incubated for a further 24 h with 1 ml of DMEM containing 1 mg/ml of human albumin and 0.2 mM ['Hloleate-albumin (55,110 cpm/nmol) in the absence of lipoproteins. On day 2, each monolayer was washed in the same manner as in day 1, after which triplicate monolayers were harvested to determine the cellular content of cholesteryl (Chol.) [3H]oleate (zero time). The remaining monolayers received 1 ml of DMEM containing 1 mg/ml of human albumin and 0.2 mM [L4C]oleate-albumin (5,040 cpm/nmol) in the absence (A) or presence ( B ) of 50 pg/ml of acetyl-LDL. After incubation at 37°C for the indicated interval, triplicate monolayers were washed four times with 2.5 ml of ice-cold phosphate-buffered saline, after which the cellular content of cholesteryl ["Hloleate (0) and cholesteryl ['*C]- oleate (0) was determined by thin layer chromatography and double label scintillation counting. The dashed line indicates the total cho- lesteryl oleate content, i.e. the sum of the values for cholesteryl ['Hloleate and cholesteryl [14C]oleate.
I
- 0 2 4 6 8 10 . 2 5 Sterod (pg/ml)
FIG. 10. Inhibition of ACAT activity in macrophage extracts by progesterone and lack of inhibition by cortisol. Peritoneal cells (9 X 10') were dispensed into 10 replicate 60-mm Petri dishes, and monolayers of adherent cells were prepared as described under "Experimental Procedures." On day 0, each dish received 2 ml of DMEM containing 50 pg/ml of acetyl-LDL. After incubation at 37°C for 24 h, each monolayer was washed twice with 3 ml of phosphate- buffered saline, and the cells were harvested by scraping with a rubber policeman. The cell suspensions were pooled and a cell-free extract was prepared. Aliquots of the cell extract (80 pg of protein) were incubated in 200 p1 of solution containing 50 mM potassium phosphate (pH 7.4), 2 mM dithiothreitol, 1.2 mg of bovine serum albumin, 2 mM ATP, 2 mM MgC12.4 m~ coenzyme A, 100 p~ ['4C]oleate-albumin (72 cpm/pmol), and 2 p1 of acetone containing the indicated concentration of either progesterone (0) or cortisol (0). After incubation for 1 h at 37"C, the reactions were terminated by addition of 4 ml of chloroform/ methanol (2:1), and the cholesteryl ["C]oleate was isolated by thin layer chromatography. The formation of cholesteryl ['4C]oleate was reduced by 90% when ATP and CoA were omitted from the reaction mixtures.
HDL as it was in its absence. However, in the presence of HDL there was little incorporation of [14C]oleate into choles- teryl ['*C]oleate (Fig. 6B) . Thus, the total content of cellular cholesteryl oleate declined in the presence of HDL (dashed

Cholesteryl Ester Cycle in Macrophages
line in Fig. 6B) because re-esterification was suppressed at a time when hydrolysis continued.
Effect of Acetyl-LDL on the Hydrolysis and Re-Esterifi- cation of Stored Cholesteryl Esters in the Absence and Pres- ence of HDL-Macrophages were loaded with r-[cholesteryZ- 3H-linoleate]acetyl-LDL by the standard protocol. The cells were then incubated in the presence of various concentrations of unlabeled acetyl-LDL and the excretion of free [‘H]choles- terol was measured. In the absence of HDL, there was little excretion of free [3H]cholesterol (Fig. 7A). The addition of acetyl-LDL stimulated this excretion slightly. In the presence of HDL there was a marked increase in the rate of excretion of free [3H]cholesterol (Fig. 7B). The addition of acetyl-LDL had no effect on this excretion. Thus, in the presence of HDL, hydrolysis and excretion of stored cholesteryl esters continued despite the presence of acetyl-LDL.
To study the effect of acetyl-LDL on the unidirectional processes of cholesteryl ester hydrolysis and re-esterification, we employed the double label oleate protocol. Fig. 8 shows an experiment in which macrophages were induced to accumu- late cholesteryl [3H]oleate by incubation with acetyl-LDL and [3H]oleate. The cells were then switched to medium contain- ing [‘%]oleate in the presence of HDL. As in the earlier experiment of Fig. 6, under these conditions there was a net loss of cholesteryl oleate that resulted from rapid hydrolysis of cholesteryl [3H]oleate that was unaccompanied by signifi- cant re-esterification with [%]oleate (Fig. 8A). When the same excretion experiment was performed in the presence of acetyl-LDL (Fig. 8B), cholesteryl [3H]oleate hydrolysis pro- ceeded at nearly the same rate as in the absence of acetyl- LDL (compare with Fig. 8A). However, in the presence of acetyl-LDL there was a progressive increase in the total cholesteryl oleate content because the acetyl-LDL markedly stimulated the esterification of [ %]oleate. The experiments of Figs. 7 and 8 indicate that acetyl-LDL causes a progressive increase in the content of cytoplasmic cholesteryl esters be- cause the cholesterol derived from acetyl-LDL stimulates the esterification reaction without affecting the hydrolysis reac- tion.
Enzymatic Basis of the Cholesterol Re-esterification Re- action-Previous studies have shown that the increase in cholesteryl ester synthesis in macrophages incubated with acetyl-LDL is associated with a marked increase in ACAT activity as measured in cell-free extracts (7). Fig. 9 shows that the opposite change takes place when increasing amounts of HDL are added to macrophages previously loaded with cho- lesteryl esters by incubation with acetyl-LDL. After the cells had been incubated with HDL for 6 h, ACAT activity, as measured in cell-free extracts, was reduced by 65%. HDL had a maximal effect at about 50 pg/ml, the same concentration at which the removal of cellular cholesteryl esters reached a maximum (Figs. 2 and 4).
To further test the conclusion that the re-esterification of stored cholesteryl esters was catalyzed by the ACAT enzyme, we employed progesterone, a steroid that is a known inhibitor of the ACAT reaction in human fibroblasts (16). Fig. 10 shows that progesterone was also a potent inhibitor of ACAT activity in homogenates of mouse peritoneal macrophages. Another Ll-carbon steroid, cortisol, did not inhibit this enzyme.
Progesterone, which inhibits the ACAT reaction in uitro, also blocked the re-esterification of stored cholesteryl esters in intact cells. Fig. 11 shows an experiment in which macro- phages were incubated with acetyl-LDL in the presence of C3H]oleate so that they formed cholesteryl [3H]oleate. As in the experiment of Fig. 6, the macrophages were allowed to equilibrate for 24 h and were then switched to medium con- taining [‘%]oleate. The cells were then further incubated in
B. Prcqestemne - 1 Owq/ml .0 2Opg/ml *A
0 6 12 18 24 30 0 6 iz 18 24 30
FIG. 11. Inhibition of the re-esterification of stored choles- teryl esters by progesterone. Macrophage monolayers were pre- Dared as described under “Experimental Procedures.” On day 0. each monolayer received 1 ml of DMEM containing 2 mg/ml of human albumin and 0.2 mu [3H]oleate-albumm (26,860 cpm/nmol). After 24 h at 37°C each monolayer was washed by the standard method and then incubated for a further 24 h with 1 ml of DMEM containing 1 ma/ml of human albumin and 0.2 mM [“Hloleate-albumin (26,800 cpm/nmol) in the absence of lipoproteins: On day 2, each monolayer was washed in the same manner as on day 1, after which triplicate monolayers were harvested to determine the cellular content of cholesteryl [3H]oleate (zero time). The remaining monolayers re- ceived 1 ml of DMEM containing 1 mg/ml of human albumin, 0.2 mM [‘4C]oleate-albumin (5,100 cpm/nmol), and 5 al of ethanol to which was added either no progesterone (A) or the indicated concentration of progesterone (B). After incubation at 37°C for the indicated inter- val, triplicate monolayers were washed four tunes with 2.5 ml of ice- cold phosphate-buffered saline, after which the cellular content of cholesteryl [3H]oleate (0) and cholesteryl [‘%Joleate (0) was deter- mined by thin layer chromatography.
the absence of a cholesterol acceptor. Fig. 11A shows that under these conditions the cells hydrolyzed the cholesteryl [3H]oleate and re-esterified the cholesterol with [‘%]oleate, as shown previously. Fig. 11B shows that the addition of progesterone did not affect the rate of hydrolysis of cholesteryl r3H]oleate. On the other hand, progesterone completely blocked the re-esterification of cholesterol with [%]oleate. In other experiments, we showed that progesterone did not in- hibit the incorporation of [‘4C]oleate into [‘4C]triglycerides under conditions in which it completely inhibited cholesteryl [‘%]oleate formation.
In the experiment of Fig. 11B the total cholesteryl ester content of the cells declined in the presence of progesterone, even though HDL was not present in the medium. To deter- mine whether the liberated [3H]cholesterol was retained by the cells or excreted, we incubated macrophages with r-[cho- ZesteryZ-3H-linoleate]acetyl-LDL and allowed the cells to ac- cumulate [3H]cholesteryl esters and free [3H]cholesterol (Ta- ble I). The cells were then incubated in the absence of HDL and either in the presence or absence of progesterone. After this incubation the cells that were maintained in the presence of progesterone had a [3H]cholesteryl ester content that was 97 nmol/mg lower than that of the cells incubated in the absence of progesterone. This decline in cellular [3H]choles- teryl esters in the progesterone-treated cells was balanced by an increase of 84 nmol/mg in the cellular content of free [3H]cholesterol. Progesterone did not increase the excretion of free [3H]cholesterol into the medium (Table I). Thus, in the absence of a cholesterol acceptor, progesterone blocks the re-esterification reaction and leads to an accumulation of free cholesterol within the cell.
Enzymatic Basis of the Cholesteryl Ester Hydrolase Re- action-To determine whether the cholesteryl esterase en- zyme that hydrolyzed the stored cytoplasmic cholesteryl es- ters was the same as the lysosomal cholesteryl esterase (also called lysosomal acid lipase) that digests the incoming choles-

9350 Cholesteryl Ester Cycle in Macrophages
TABLE I Progesterone-mediated accumulation of free [3H]cholesterol in macrophages previously loaded with [aH]cholesteryl esters
Macrophages were incubated with r-[~holesteryl-~H-linoleate]acetyl-LDL for 24 h, washed, and incubated for a further 24 h in the absence of lipoproteins according to the standard procedure. On day 2, each monolayer received 1 ml of DMEM containing 1 mg/ml of human albumin and 5 p1 of ethanol to which was added either no progesterone or 10 pg/ml of progesterone. After incubation at 37°C for 24 h, the content of free and esterified [3H]cholesterol in the cells and in the medium was measured by thin layer chromatography.
Addition to medium ["H]Cholesteryl esters Free ['Hlcholesterol
Cells Medium Cells Medium nrnol/mgprotein nrnol/rng protein
None (A) 397 (363-448)" 7.7 (6.3-9.4) 182 (163-198) 24 (21-29) Progesterone (B) 300 (294-306) -9.1 (6.5-11) 266 (259-273) 19 (17-23) Change due to progesterone -97 +1.4 +a4
(R - A) -5
'I mean and range of triplicate incubations.
A. C. 1
50 t t ,, no HDL
0 12 24 36 40 0 4 0 i 2 46
Chloroqulne (pM) Arnrnonlurn Chlorlde (rnM)
FIG. 12. Failure of chloroquine or ammonium chloride to inhibit the excretion of free [3H]cholesterol derived from the hydrolysis of stored [3H]cholesteryl esters. Macrophages were incubated with r-[~holesteryl-~H-lioleate]acetyl-LDL for 24 h, washed, and incubated for a further 24 h in the absence of lipoproteins according to the standard procedure. On day 2, the dishes were divided into two groups. In the first group (A and C), each monolayer received 1 ml of DMEM containing either no HDL (A) or 250 pg/ml of HDL (A, in the presence of the indicated concentration of either chloroquine (A) or ammonium chloride (C). After incubation at 37OC for 4 h, the content of free [3HJcholesterol in the medium was measured by thin layer chromatography. The cellular content of free and esterified ["H]cholesterol in the absence of chloroquine and ammonium chloride was 191 and 404 nmol/mg of cell protein, respec- tively. In the second group ( E and D), each monolayer received 1 ml of DMEM containing 10% human lipoprotein-deficient serum and 25 p g / d of i""Ilabeled acetyl-LDL (105,000 cpm/pg of protein) in the presence of the indicated concentration of either chloroquine ( E ) or ammonium chloride (D). After incubation for 4 h at 37"C, the total ceUular content of i251 radioactivity (0) and the amount of '2"I-labeled trichloroacetic acid-soluble (non-iodide) material in the medium (@I were determined.
teryl esters of acetyl-LDL, we performed the experiment shown in Fig. 12. Macrophages were induced to accumulate re-esterified cholesterol by incubation with r-[cholesteryl-"H- linoleatelacetyl-LDL according to the standard procedure. The cells were then washed and incubated with varying concentrations of two lysosomal enzyme inhibitors, chloro- quine (Fig. 12A) or ammonium chloride (Fig. 12C), in the presence or absence of HDL. Neither agent significantly af- fected the hydrolysis and excretion of the stored ["Hlcholes- teryl esters (Fig. 12, A and 0. To ensure that these two agents were inhibiting lysosomal enzymes, in the same experiment we incubated the macrophages with 12SI-labeled acetyl-LDL which is taken up and hydrolyzed in lysosomes. Previous
LYSOSOME 1 CYTOPLASM I I AMP
- 1 ~
Acceptu HDL. RBC. etc.
FIG. 13. Two-compartment model for cholesteryl ester me- tabolism in macrophages illustrating the cytoplasmic choles- teryl ester cycle. The salient features of this model are discussed in the text. RBC, red blood cells.
studies have shown that when the proteolytic degradation of '251-labeled LDL is blocked by chloroquine the hydrolysis of the lipoprotein's cholesteryl esters is also blocked (6, 7). In the experiment of Fig. 12, increasing concentrations of chlo- roquine inhibited the hydrolysis of the protein component of the "'I-labeled acetyl-LDL and caused a corresponding in- crease in the amount of cell-bound undegraded lipoprotein (Fig. 12B). Ammonium chloride also inhibited the degradation of the 12'I-labeled acetyl-LDL. In this case, however, there was also an inhibition of uptake of the 'Z51-labeled acetyl-LDL (Fig. 120). Thus, under conditions in which the lysosomal digestive apparatus was inhibited by chloroquine or ammo- nium chloride, the macrophages remained able to hydrolyze and excrete stored cytoplasmic ['H]cholesteryl esters.
DISCUSSION
Fig. 13 shows a two-compartment model that we propose to account for the observations in this study and earlier studies of cholesteryl ester metabolism in mouse peritoneal macro- phages (6-8, 17). Lipoprotein-bound cholesteryl esters that enter the macrophage via receptor-mediated endocytosis are delivered to lysosomes (frrst cellular compartment) where the cholesteryl esters are hydrolyzed. The liberated cholesterol leaves the lysosome and enters the cytoplasm (second cellular compartment) where it has two fates. Some of the cholesterol is immediately excreted. The remainder of the excess choles- terol is re-esterified by the ACAT enzyme and accumulates in the cytoplasm as cholesteryl ester droplets.
The major new finding in the current study is that the cholesteryl ester droplets of macrophages, despite their quies- cent appearance in electron micrographs (7) , are not meta- bolically inert. The cholesteryl esters are continually undergo- ing hydrolysis. When an acceptor for cholesterol, such as HDL, is present in the culture medium, the liberated choles- terol is excreted from the cell. When no cholesterol acceptor

Cholesteryl Ester Cycle in Macrophages 9351
is available, the liberated cholesterol is retained in the cell where it is re-esterified by the ACAT enzyme. In the absence of a cholesterol acceptor, about 50% of the stored cholesteryl esters are hydrolyzed and re-esterified each day in a type of futile cycle. Inasmuch as the ACAT enzyme uses a fatty acyl- CoA derivative that requires ATP for its synthesis, each turn of the cholesteryl ester cycle has the net effect of breaking down one molecule of ATP to AMP and pyrophosphate.
In addition to the ACAT reaction, it is theoretically possible that some of the cholesteryl ester turnover within macro- phages might represent a fatty acid exchange reaction cata- lyzed by a reversible cholesteryl ester hydrolase that does not consume ATP. Such an exchange mechanism is rendered unlikely in the current studies because: 1) crude extracts of cholesteryl ester-loaded macrophages incorporate [‘%]oleate into cholesteryl [%]oleate at appreciable rates only in the presence of CoA and ATP (Ref. 7 and Figs. 9 and 10); 2) the increased rate of synthesis of cholesteryl esters in intact macrophages incubated with acetyl-LDL is accompanied by an increase in ACAT activity as measured in cell-free extracts (7); 3) the decreased rate of cholesteryl ester formation in intact macrophages incubated with HDL is accompanied by a decrease in ACAT activity as measured in cell-free extracts (Fig. 9); and 4) progesterone, which inhibits ACAT activity in vitro (Fig. lo), inhibits cholesterol re-esterifcation in intact macrophages without affecting cholesteryl ester hydrolysis (Fig. 11B).
The mechanism by which the addition of HDL reduces ACAT activity is not yet understood. Prior experiments with human fibroblasts (9, 16, 18) as well as with macrophages (7) indicate that the ACAT activity is enhanced when the cells accumulate “excess” cholesterol (i.e. an amount that is greater than is required for membrane synthesis). Conversely, ACAT activity is reduced when the amount of excess cholesterol is reduced. It seems likely that HDL lowers ACAT activity primarily by removing cholesterol from the cell, thereby re- ducing a crucial cellular pool of free cholesterol. The reduction of this cholesterol pool may simply deprive the ACAT enzyme of its cholesterol substrate. Alternatively, the removal of cho- lesterol may have additional regulatory effects that contribute to the suppression of ACAT activity. Studies in tibroblasts suggest that an increased availability of cholesterol stimulates the ACAT by two mechanisms: it provides additional sub- strate and it somehow increases the catalytic activity of the enzyme as well (18, 19).
Whereas HDL causes net hydrolysis of cholesteryl esters in macrophages by removing cholesterol and suppressing the ACAT reaction, acetyl-LDL causes net synthesis of choles- teryl esters by delivering cholesterol and stimulating the ACAT reaction. On the basis of earlier experiments using an indirect approach, we suggested that acetyl-LDL might also inhibit the hydrolysis of cytoplasmic cholesteryl esters (7). This suggestion has not been substantiated by the more direct experiments in the present studies. As shown in Figs. 7 and 8, acetyl-LDL did not affect the hydrolysis reaction; it simply stimulated the ACAT reaction, and this caused a net increase in cellular cholesteryl esters.
The current experiments focus attention on the ACAT enzyme as a crucial determinant of cholesteryl ester retention as well as cholesteryl ester synthesis in macrophages. The ACAT activity, in turn, is determined primarily by the avail- ability of cholesterol. The experiments with progesterone suggest that a cholesterol acceptor is necessary for removal of cholesterol even when the ACAT enzyme is inhibited and the cellular content of free cholesterol is increased (Table I). The mechanisms by which HDL and other cholesterol acceptors
facilitate the removal of cellular cholesterol is now under study.
The continual hydrolysis and re-esterification of cholesteryl esters in macrophage foam cells resembles a similar process previously described for triglycerides in adipose tissue cells. The classic studies of Steinberg and Vaughan demonstrated that adipose tissue triglycerides are continually hydrolyzed and re-synthesized in a series of trans-esterification reactions that use fatty acyl-CoA derivatives (reviewed in Ref. 20). In adipose tissue the rates of hydrolysis and re-esterification can be varied independently by the presence of hormones and metabolic substrates, the net change in triglyceride being dictated by the relative rates of the two reactions. In macro- phage foam cells, we have not yet been able to find agents that regulate the hydrolytic part of the cycle; only the ACAT seems to be regulated by the agents so far tested. Another interesting parallel exists between macrophages and adipo- cytes. In adipocytes, excretion of the liberated fatty acids, like the excretion of cholesterol from macrophages, requires an acceptor substance in the medium (20). However, whereas albumin is a good acceptor for fatty acids in the adipocyte system, it is a poor acceptor for cholesterol in the macrophage system (8).
There is evidence to suggest that a cholesteryl ester cycle like that which occurs in isolated mouse peritoneal macro- phages may also take place in uiuo in foam cells of athero- sclerotic lesions. Thus, several investigators have shown an increased rate of incorporation of %-labeled oleate into cho- lesteryl [“‘Cloleate by intact aortic tissue, isolated foam cells, and cell-free preparations of cholesteryl ester-loaded athero- sclerotic aortas from man and experimental animals (reviewed in Ref. 21). If a cholesteryl ester cycle does occur in athero- sclerotic vessels, the question arises as to whether the attend- ant consumption of ATP occurring in a poorly oxygenated tissue might contribute to the eventual necrosis of foam cells in the artery wall.
Acknowledgments-We thank Gloria Brunschede, Michael Gais- bauer, Clyde Wetherill, and Wendy Womack for their excellent tech- nical assistance.
REFERENCES
1. Goodman, Dew. S. (1965) Physiol. Rev. 45, 747-839 2. Fredrickson. D. W.. Goldstein. J. L.. and Brown, M. S. (1978) in
The Metabolic Basis of Inherited Disease (Stanbury, J. B., Wyngaarden, J. B., and Fredrickson, D. S., eds) pp. 604-655, McGraw-Hill Book Co., New York
3. Mahley, R. W. (1979) Atherosclerosis Rev. 5, l-34 4. Bulklev. B. H.. Buia. L. M.. Ferrans. V. J., Bulklev, G. B., and
Rob&s, W. c. (i9j5) Arch. Pathol. 99,293-300 - 5. Buja, L. M., Kovanen, P. T., and Bilheimer, D. W. (1979) Am. J.
Pathol. 97,327-357 6. Goldstein, J. L., Ho, Y. K., Basu, S. K., and Brown, M. S. (1979)
Proc. Natl. Acad. Sri. U. S. A. 76, 333-337 7. Brown, M. S., Goldstein, J. L., Krieger, M., Ho, Y. K., and
Anderson, R. G. W. (1979) J. Cell Biol. 82.597-613 8. Ho, Y. K., Brown, M. S., and Goldstein, J. L. (1980) J. Lipid Res.
21,391-398 9. Goldstein, J. L., Dana, S. E., and Brown, M. S. (1974) Proc. Natl.
Acad. Sci. U. S. A. 71.4288-4292 10. Krieger, M., Brown, M. S., Faust, J. R., and Goldstein, J. L. (1978)
J. Biol. Chem. 253,4093-4101 11. Brown, M. S., Dana, S. E., and Goldstein, J. L. (1974) J. Biol.
Chem. 249,789-796 12. Edelson, P. J., and Cohn, Z. A. (1976) in In Vitro Methods in
Cell-Mediated and Tumor Immunity (Bloom, B. R., and David, J. R., eds) pp. 333-340, Academic Press, New York
13. Hara, A., and Radin, N. S. (1978) Anal. Biochem. 90,420-426 14. Bligh, E. G., and Dyer, W. J. (1959) Can. J. Biochem. Physiol.
37,911-917 15. Lowry, 0. H., Rosebrough, N. J., Farr, A. L., and Randall, R. J.

9352 Cholesteryl Ester Cycle in Macrophages (1951) J. Biol. Chen. 193,265-275 Chem. 250,4025-4027
Brown, M. S. (1978) Proc. Natl. Acad. Sci. U. S. A. 76, 1877- 1881
Mahley, R. W. (1980) J. Bwl. Chem. 265,1839-1848 Physiological Society, Washington, D. C.
16. Goldstein, J. L., Faust, J. R., Dygos, J. N., Chorvat, R. J., and 19. Goldstein, J. L., and Brown, M. S. (1977) Annu. Rev. Biochem.
20. Steinberg, D., and Vaughan, M. (1965) in Handbook of Physiology 17. Goldstein, J. L., Ho, Y. K., Brown, M. S., Innerarity, T. L., and (Renold, A. E., and Cahill, G. F., Jr., eds) pp 335-347, American
18. Brown, M. S., Dana, S. E., and Goldstein, J. L. (1975) J. Biol. 21. St. Clair, R. W. (1976) Atherosclerosis Rev. 1, 61-117
46,897-930










![eeri Enzyme Engineering - longdom.org · catalyze the hydrolysis of ester bonds in substrates such as vitamin esters, phospholipids, triglycerides (TGs) and cholesteryl esters [1].](https://static.fdocuments.us/doc/165x107/5e14d1ebd622ec4625428ebc/eeri-enzyme-engineering-catalyze-the-hydrolysis-of-ester-bonds-in-substrates-such.jpg)




![Surface Properties of Cholesteryl Ester Liquid Crystalmint.uthm.edu.my/images/DOCUMENTS/PMiNT_Paper_3.pdf · literature [8]. However, the chemical compounds of the cholesteryl ester](https://static.fdocuments.us/doc/165x107/5f90eb74f0ec4316ef0bf1d2/surface-properties-of-cholesteryl-ester-liquid-literature-8-however-the-chemical.jpg)


