
The Activity Spectrum of Vif from Multiple HIV-1 Subtypes against
11
The Activity Spectrum of Vif from Multiple HIV-1 Subtypes against APOBEC3G, APOBEC3F, and APOBEC3H Mawuena Binka, a Marcel Ooms, a Myeika Steward, a and Viviana Simon a,b,c Department of Microbiology, a The Global Health and Emerging Pathogens Institute, b and Division of Infectious Diseases, Department of Medicine, c Mount Sinai School of Medicine, New York, New York, USA The APOBEC3 family comprises seven cytidine deaminases (APOBEC3A [A3A] to A3H), which are expressed to various degrees in HIV-1 susceptible cells. The HIV-1 Vif protein counteracts APOBEC3 restriction by mediating its degradation by the protea- some. We hypothesized that Vif proteins from various HIV-1 subtypes differ in their abilities to counteract different APOBEC3 proteins. Seventeen Vif alleles from seven HIV-1 subtypes were tested for their abilities to degrade and counteract A3G, A3F, and A3H haplotype II (hapII). We show that most Vif alleles neutralize A3G and A3F efficiently but display differences with respect to the inhibition of A3H hapII. The majority of non-subtype B Vif alleles tested presented some activity against A3H hapII, with two subtype F Vif variants being highly effective in counteracting A3H hapII. The residues required for activity were mapped to two residues in the amino-terminal region of Vif (positions 39F and 48H). Coimmunoprecipitations showed that these two amino acids were necessary for association of Vif with A3H hapII. These findings suggest that the A3H hapII binding site in Vif is distinct from the regions important for A3G and A3F recognition and that it requires specific amino acids at positions 39 and 48. The differential Vif activity spectra, especially against A3H hapII, suggest adaptation to APOBEC3 repertoires representative of different human ancestries. Phenotypic assessment of anti-APOBEC3 activity of Vif variants against several cytidine deaminases will help reveal the requirement for successful replication in vivo and ultimately point to interventions targeting the Vif- APOBEC3 interface. A polipoprotein B mRNA-editing, catalytic polypeptide (APOBEC3) cytidine deaminases act as intracellular inhib- itors of HIV-1 replication (1, 8). The family comprises seven pro- teins (APOBEC3A [A3A] to A3H), which differ in catalytic activ- ity and susceptibility to HIV-1 Vif-mediated degradation (1, 8, 38). In the absence of a functional Vif, some APOBEC3 proteins (e.g., A3G, A3F, and A3H haplotype II [hapII]) exert restriction through editing- and nonediting-based mechanisms, thereby pre- venting efficient viral spread in the next round of infection (1, 8, 13). HIV-1 isolates harbor a Vif allele that degrades A3G and some other antiretroviral APOBEC3 proteins in a ubiquitin/ proteasome-dependent manner (1, 8, 38). A3G and A3F contain two deaminase domains while the A3H protein consists of only one catalytic domain. Moreover, A3H has several haplotypes, some of which are highly active against HIV-1 (haplotypes II, V, and VII) (5, 9, 18, 19, 29, 43, 52). Interestingly, the allele frequency of the active A3H hapII differs considerably among human ethnicities, being high in African and low in Euro- pean and Asian populations (29, 43). There remains some uncer- tainty with respect to the Vif neutralization sensitivity of these A3H protein variants (5, 10, 41, 52). The global HIV-1 epidemic is mostly driven by non-subtype B virus strains from group M (12), but our current knowledge on functional determinants of Vif-APOBEC3 interactions is almost exclusively based on the interactions of HIV-1 subtype B molecu- lar clones (LAI, HXB2, and NL4-3) with the reference A3G. These studies have generated a body of evidence indicating that the amino-terminal region of Vif is important for binding to A3G and A3F proteins (3, 16, 23, 24, 26, 34, 35, 37, 42, 44). Vif residues that are important for selective association with A3G/A3F vary: Vif residues 14 to 17 (DRMR) are important for interaction with A3F, whereas residues 40 to 44 (YRHHY) are required for A3G binding (26, 34, 35, 49). Other studies showed that Vif residues I9, K22, E45, and N48 are necessary for A3G binding, whereas Q12 is needed for A3F binding (37, 44). In addition, tryptophans at po- sitions 11 and 79 and at positions 5, 21, 38, and 89 are important for A3F and A3G binding, respectively (42). The YXXL (X stands for any residue) motif spanning positions 69 to 72 and the WX SLVK motif from positions 21 to 26 are necessary for the degra- dation of both A3G and A3F (6, 32). The C-terminal region of Vif contains the SLQYLA SOCS box (residues 145 to 151), which is important for Vif binding to elongin C in the E3 ubiquitin ligase complex (39, 50). This region is highly conserved among HIV-1 subtypes, and mutations within this domain disrupt the interaction between Vif and Elongin C, thereby abrogating anti-APOBEC3 activity (20, 36). This region also contains the zinc binding HCCH motif consisting of residues H108, C114, C133, and H139, which are necessary for Vif binding to Cullin 5, which mediates polyubiquitination of APOBEC3 pro- teins, followed by their degradation via the 26S proteasome (21, 25, 31, 47, 48). To date, the three-dimensional structure of Vif has not yet been solved by X ray or nuclear magnetic resonance (NMR), but a conformational analysis of HXB2 Vif by hydrogen exchange mass spectrometry suggested that the N-terminal Vif region is more structured than the C-terminal portion of the Vif protein (22). Taken together, our current understanding suggests that the amino-terminal region of HIV-1 Vif allows APOBEC3- Received 23 August 2011 Accepted 13 October 2011 Published ahead of print 19 October 2011 Address correspondence to Viviana Simon, [email protected]. Supplemental material for this article may be found at http://jvi.asm.org/. Copyright © 2012, American Society for Microbiology. All Rights Reserved. doi:10.1128/JVI.06082-11 0022-538X/12/$12.00 Journal of Virology p. 49 –59 jvi.asm.org 49 on January 7, 2019 by guest http://jvi.asm.org/ Downloaded from
Transcript of The Activity Spectrum of Vif from Multiple HIV-1 Subtypes against

The Activity Spectrum of Vif from Multiple HIV-1 Subtypes againstAPOBEC3G, APOBEC3F, and APOBEC3H
Mawuena Binka,a Marcel Ooms,a Myeika Steward,a and Viviana Simona,b,c
Department of Microbiology,a The Global Health and Emerging Pathogens Institute,b and Division of Infectious Diseases, Department of Medicine,c Mount Sinai School ofMedicine, New York, New York, USA
The APOBEC3 family comprises seven cytidine deaminases (APOBEC3A [A3A] to A3H), which are expressed to various degreesin HIV-1 susceptible cells. The HIV-1 Vif protein counteracts APOBEC3 restriction by mediating its degradation by the protea-some. We hypothesized that Vif proteins from various HIV-1 subtypes differ in their abilities to counteract different APOBEC3proteins. Seventeen Vif alleles from seven HIV-1 subtypes were tested for their abilities to degrade and counteract A3G, A3F, andA3H haplotype II (hapII). We show that most Vif alleles neutralize A3G and A3F efficiently but display differences with respectto the inhibition of A3H hapII. The majority of non-subtype B Vif alleles tested presented some activity against A3H hapII, withtwo subtype F Vif variants being highly effective in counteracting A3H hapII. The residues required for activity were mapped totwo residues in the amino-terminal region of Vif (positions 39F and 48H). Coimmunoprecipitations showed that these twoamino acids were necessary for association of Vif with A3H hapII. These findings suggest that the A3H hapII binding site in Vif isdistinct from the regions important for A3G and A3F recognition and that it requires specific amino acids at positions 39 and 48.The differential Vif activity spectra, especially against A3H hapII, suggest adaptation to APOBEC3 repertoires representative ofdifferent human ancestries. Phenotypic assessment of anti-APOBEC3 activity of Vif variants against several cytidine deaminaseswill help reveal the requirement for successful replication in vivo and ultimately point to interventions targeting the Vif-APOBEC3 interface.
Apolipoprotein B mRNA-editing, catalytic polypeptide(APOBEC3) cytidine deaminases act as intracellular inhib-
itors of HIV-1 replication (1, 8). The family comprises seven pro-teins (APOBEC3A [A3A] to A3H), which differ in catalytic activ-ity and susceptibility to HIV-1 Vif-mediated degradation (1, 8,38). In the absence of a functional Vif, some APOBEC3 proteins(e.g., A3G, A3F, and A3H haplotype II [hapII]) exert restrictionthrough editing- and nonediting-based mechanisms, thereby pre-venting efficient viral spread in the next round of infection (1,8, 13). HIV-1 isolates harbor a Vif allele that degrades A3G andsome other antiretroviral APOBEC3 proteins in a ubiquitin/proteasome-dependent manner (1, 8, 38).
A3G and A3F contain two deaminase domains while the A3Hprotein consists of only one catalytic domain. Moreover, A3H hasseveral haplotypes, some of which are highly active against HIV-1(haplotypes II, V, and VII) (5, 9, 18, 19, 29, 43, 52). Interestingly,the allele frequency of the active A3H hapII differs considerablyamong human ethnicities, being high in African and low in Euro-pean and Asian populations (29, 43). There remains some uncer-tainty with respect to the Vif neutralization sensitivity of theseA3H protein variants (5, 10, 41, 52).
The global HIV-1 epidemic is mostly driven by non-subtype Bvirus strains from group M (12), but our current knowledge onfunctional determinants of Vif-APOBEC3 interactions is almostexclusively based on the interactions of HIV-1 subtype B molecu-lar clones (LAI, HXB2, and NL4-3) with the reference A3G. Thesestudies have generated a body of evidence indicating that theamino-terminal region of Vif is important for binding to A3G andA3F proteins (3, 16, 23, 24, 26, 34, 35, 37, 42, 44). Vif residues thatare important for selective association with A3G/A3F vary: Vifresidues 14 to 17 (DRMR) are important for interaction with A3F,whereas residues 40 to 44 (YRHHY) are required for A3G binding(26, 34, 35, 49). Other studies showed that Vif residues I9, K22,
E45, and N48 are necessary for A3G binding, whereas Q12 isneeded for A3F binding (37, 44). In addition, tryptophans at po-sitions 11 and 79 and at positions 5, 21, 38, and 89 are importantfor A3F and A3G binding, respectively (42). The YXXL (X standsfor any residue) motif spanning positions 69 to 72 and the WXSLVK motif from positions 21 to 26 are necessary for the degra-dation of both A3G and A3F (6, 32).
The C-terminal region of Vif contains the SLQYLA SOCS box(residues 145 to 151), which is important for Vif binding toelongin C in the E3 ubiquitin ligase complex (39, 50). This regionis highly conserved among HIV-1 subtypes, and mutations withinthis domain disrupt the interaction between Vif and Elongin C,thereby abrogating anti-APOBEC3 activity (20, 36). This regionalso contains the zinc binding HCCH motif consisting of residuesH108, C114, C133, and H139, which are necessary for Vif bindingto Cullin 5, which mediates polyubiquitination of APOBEC3 pro-teins, followed by their degradation via the 26S proteasome (21,25, 31, 47, 48). To date, the three-dimensional structure of Vif hasnot yet been solved by X ray or nuclear magnetic resonance(NMR), but a conformational analysis of HXB2 Vif by hydrogenexchange mass spectrometry suggested that the N-terminal Vifregion is more structured than the C-terminal portion of the Vifprotein (22). Taken together, our current understanding suggeststhat the amino-terminal region of HIV-1 Vif allows APOBEC3-
Received 23 August 2011 Accepted 13 October 2011
Published ahead of print 19 October 2011
Address correspondence to Viviana Simon, [email protected].
Supplemental material for this article may be found at http://jvi.asm.org/.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.06082-11
0022-538X/12/$12.00 Journal of Virology p. 49–59 jvi.asm.org 49
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

specific interactions while the C-terminal region of Vif connects itto the host cell degradation machinery.
Although Vif variants have been characterized genotypically(2, 40, 45, 46), we know very little about the range of phenotypicactivity of non-subtype B Vif variants against the differentAPOBEC3 proteins. Indeed, naturally occurring single residuesubstitutions may affect the Vif/APOBEC3 phenotype, with full-length subtype B Vif alleles displaying neutralization defectsagainst A3G, A3F, or both deaminases (27, 37). A recent studycompared the anti-A3G activity of Vif alleles derived from varioussubtypes and recombinants (A, B, C, CRF01, and CRF02) andfound that subtype C Vif alleles displayed higher activity againstA3G (14). The correlates of the increased activity were mapped toseveral residues in the amino-terminal region of Vif (14), but ourunderstanding of the requirements of diverse Vif proteins tocounteract multiple APOBEC3 proteins remains incomplete.
We hypothesized that Vif alleles, especially those obtainedfrom non-B subtype isolates that adapted to replication in indi-viduals of different ancestries, may differ in their relative activitiesagainst A3G, A3F, and A3H hapII. In this study, we show thatmost Vif alleles from different HIV-1 subtypes have activityagainst A3G and A3F. In contrast, only half of the Vif alleles testedwere partially or fully active against A3H hapII. Two distinct res-idues in Vif (39F and 48H) were required for the efficient neutral-ization of A3H hapII by Vif, suggesting that the single deaminaseA3H may interact with a nonlinear domain in Vif.
MATERIALS AND METHODSCloning. Twelve viral isolates and five molecular clones from differentsubtypes were obtained through the NIH AIDS Research and ReferenceReagent Program (Table 1). Briefly, viral RNA was extracted from the viralstocks using a Qiagen QIAamp Viral RNA Mini Kit, followed by reversetranscription and amplification using primers located outside the Vif cod-ing region. The PCR fragments were cloned into pSC-A (StrataClone PCRcloning vector; Stratagene), and several clones were sequenced and man-ually aligned to the published sequences. Clones with Vif sequences iden-tical to the published sequences were used as templates for PCR amplifi-cation with Vif primers specific for the 5= and 3= regions of each variantand containing NotI and EcoRI restriction sites. PCR fragments weredigested with NotI/EcoRI, and the coding regions of the subcloned 17 Vifvariants were inserted in pCRV1 as described previously (37, 51). Site-directed mutagenesis of subtype F1 and F2 Vifs was performed usingoverlapping PCR as described previously (37). The mutated Vifs werecloned into pCRV1 vector and confirmed by sequencing. Primer se-quences are available upon request.
Amino-terminally FLAG-tagged A3G, A3F, and A3H hapII in PTR600expression plasmids have been described previously (10, 28, 30). Acarboxyl-terminal triple hemagglutinin (HA) tag was added to A3G andA3H hapII by overlapping PCR, followed by cloning into PTR600. Anuntagged version of A3H hapII was cloned into PTR600. All DNA prep-arations were sequenced to confirm the integrity of the APOBEC3 se-quences. NCBI reference sequence numbers for the APOBEC3 used areNM 021822.3 (A3G), NM 145298.5 (A3F), and FJ376614.1 (A3H hapII,splice variant SV-183).
Cell culture. HEK 293T and TZM-bl cells were grown in Dulbecco’smodified Eagle’s medium (Invitrogen) supplemented with 100 U/ml pen-icillin/streptomycin and 10% fetal bovine serum (FBS). TZM-bl cells wereprovided by J. C. Kappes and X. Wu through the AIDS Research andReference Reagent Program, Division of AIDS, NIAID, National Insti-tutes of Health, NIH Reagent program.
Infectivity assay. To determine the level of infectivity rescue achievedby each Vif variant, viral stocks were produced in the presence and ab-sence of APOBEC3. Briefly, 3.0 � 105 HEK 293T cells were cotransfected
with 500 ng of pNL4-3�Vif, 5 ng of Vif, and 50 ng of PTR600FLAG-A3G,50 ng of PTR600FLAG-A3F, or 25 ng of PTR600FLAG-A3H expressionplasmids, using 4 �g/ml polyethylenimine (PEI; Polysciences, Inc.).The total amount of DNA transfected was 1,000 ng, with pcDNA3.1 serv-ing as filler. Viral supernatants were collected at 44 to 48 h posttransfec-tion, clarified by centrifugation, and stored at �80°C. Forty-microlitersupernatants were used to infect 1 � 104 TZM-bl cells in black 96-wellplates. Infections were done in triplicate with viral supernatants fromthree independent experiments for each APOBEC3 protein tested. Infec-tivity of the virus particles in the TZM-bl cells was assessed at 44 to 48 hpostinfection using a Galacto-Star System for detecting �-galactosidaseactivity (Applied Biosystems), as described previously (10, 37).
Vif expression. A total of 3.0 � 105 HEK 293T cells were transfectedwith 200 ng of Vif expression plasmids, 100 ng of PTR600-green fluores-cent protein (GFP), and 700 ng of pcDNA (24-well plate; the total amountof DNA transfected was 1,000 ng with 4 �g/ml PEI). SDS lysis buffer (1%SDS, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA) was added tothe transfected cells 48 h later, and the clarified lysates were analyzed byWestern blotting.
Degradation assay. A total of 3.0 � 105 HEK 293T cells in 24-wellplates were cotransfected with 500 ng of pNL4-3�Vif, 100 ng of PTR600-GFP, 20 ng of pCRV1-Vif, and 100 ng of PTR600FLAG-A3G, 100 ng ofPTR600FLAG-A3F, or 50 ng of PTR600FLAG-A3H hapII expressionplasmids (total amount of DNA transfected, 1,000 ng; filler DNA,pcDNA3.1).
The degradation of tagged and untagged A3H hapII was compared bycotransfecting HEK 293T cells as described above with the following ex-pression plasmids: PTR600FLAG-A3H hapII (50 ng) and pCRV1-Vif (50ng) or PTR600-A3H hapII (untagged; 100 ng) with pCRV1-Vif (100 ng).The total amount of DNA transfected was 1,000 ng (filler DNA, pcDNA).Transfections were performed with PEI (4 �g/ml). At 48 h posttransfec-tion cells were lysed (in lysis buffer consisting of 1% SDS, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA), clarified by centrifugation, andanalyzed by Western blotting.
Western blotting. Transfected HEK 293T cell lysates were separatedon 10% bis-Tris 26-well gels (Invitrogen) and transferred by semidrytransfer onto polyvinylidene difluoride (PVDF) membranes (ThermoScientific). Membranes were blocked in 1% (wt/vol) milk solution for 1 hand incubated in primary antibody overnight. HIV-1 Vif was detectedwith a rabbit anti-Vif primary antibody (NIH catalog number 2221) at1:1,000 dilution and with goat anti-rabbit secondary antibody (1:5,000;Sigma). FLAG-tagged APOBEC3 proteins were detected with a mouseanti-FLAG primary antibody (1:5,000; Sigma), GFP was detectedwith rabbit anti-GFP primary antibody (1:2,000; Santa Cruz), andglyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected witha mouse anti-GAPDH primary antibody (1:2,000; Santa Cruz). Goat anti-rabbit or goat anti-mouse secondary antibody (both, Sigma) was at a1:5,000 dilution. Membranes were washed in wash buffer (0.1% Tween inphosphate-buffered saline [PBS]) and incubated in secondary antibodyfor 2 h. Membranes were developed with SuperSignal West Pico or Femtosubstrate (Thermo Scientific) in an LAS-3000 imaging system (FujiFilm).Only nonsaturated signals were acquired for further analysis with Image-Gauge, version 4.0, software.
Vif-A3H coimmunoprecipitation. A total of 7 � 105 HEK 293T cellswere transfected with pCRV1-Vif expression plasmids (200 ng),PTR600HA-A3H hapII (800 ng), PTR600HA-A3G (800 ng), or PTR600-GFP (800 ng) using Fugene 6 according to the manufacturer’s instruc-tions. At 24 h after transfection, clasto-lactacystin �-lactone (10 �M;Sigma) was added to the transfection to prevent potential Vif-mediatedA3H degradation. At 48 h after transfection, cells were washed with PBSand lysed on ice for 15 min in lysis buffer (1% Triton X-100 in PBSsupplemented with EDTA-free protease inhibitor cocktail [Roche]). Pro-teins were clarified by centrifugation at 4°C (8,200 � g for 10 min at 4°C)and incubated with anti-HA-coated beads (Easyview; Sigma) for 1 h at4°C. Beads were extensively washed with lysis buffer supplemented with
Binka et al.
50 jvi.asm.org Journal of Virology
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

75 mM NaCl. Bound proteins were eluted by boiling the beads in sampleloading buffer. Cell lysates and bound proteins were analyzed by Westernblotting.
A3H polyclonal serum. Two rabbits were immunized three times withthe A3H peptide Gln-Phe-Asn-Asn-Lys-Arg-Arg-Leu-Arg-Arg-Pro-Tyr-Tyr-Pro-Arg (A3H residues 12 to 26). Custom antibody production wasperformed by Pocono Rabbit Farm and Laboratory, Inc. Rabbit poly-clonal serum was used at a 1:2,000 dilution to detect transfected untaggedand tagged A3H.
Statistical analysis. GraphPad Prism, version 5, software was used toperform all statistical tests (minimum, maximum, mean, t tests, andSpearman rank correlation). P values are two sided, and values of �0.05were considered to be significant.
RESULTSGenotype and expression of selected Vif alleles. Inspection of�1,500 Vif alleles deposited at the Los Alamos HIV sequence da-tabase revealed considerable variation in Vif sequences. Somesubtype-specific motifs such as 8L (subtype C), 45DCXH48 (sub-type D), and 91QRK93 (subtype AE) were also identified. Weselected 12 viral isolates from patients representing seven HIV-1subtypes and five replication-competent full-length molecularclones from three different subtypes. The subtype-specific motifslisted above were present in the selected Vif alleles. The patientsfrom whom the viruses were originally isolated lived in variouscountries around the world, like Brazil, Rwanda, Zimbabwe, andThailand (Table 1). Analysis of the phylogenic relationships of theselected Vif alleles and their corresponding consensus sequencesconfirmed their subtypes (Fig. 1A).
At the protein sequence level, the 17 Vif alleles displayed, onaverage, 15% diversity relative to HIV subtype B molecular cloneLAI (maximum, 19%; minimum, 7%). The greatest number ofchanges were observed for Vif G1 (37/192 residues), while Vif B3(29/192 residues) was most similar to LAI Vif. Inspection of theN-terminal region revealed polymorphic positions both withinand outside the domains known to be relevant for APOBEC3-specific interactions while the HCCH Cullin 5-binding motif andthe SLQYLA Elongin C-binding motif were conserved in all 17 Vif
TABLE 1 Description of the Vif alleles used in this study
Vif name Strain name Country of origin
A1 93RW037 RwandaA2 92RW008 RwandaB-NL4-3 pNL4-3 USAB-LAI pLAI.2 USAB1 92TH026 ThailandB2 AD.MDR01 USAB3 93TH305 ThailandC1 92ZW101 ZimbabweC2 92ZW106 ZimbabweC3 pMJ4 BotswanaD1 p94UG114.1.6 UgandaD2 94KE102 KenyaAE1 p90CF402.1.8 Central African RepublicF1 93BR029 BrazilF2 93BR019 BrazilF3 93BR020 BrazilG1 HIV-1 G3 Nigeria
FIG 1 A panel of 17 Vif alleles from different subtypes. (A) Phylogenetic relationships of subtype consensus sequences and selected Vif alleles from differentsubtypes were inferred using the neighbor-joining method. Bootstrap values of 70% or higher are shown next to the branches (1,000 replicates). Distances werecomputed using the maximum composite likelihood method. All positions containing gaps and missing data were eliminated from the data set (completedeletion option). There were a total of 568 positions in the final data set. Phylogenetic analyses were conducted in MEGA4. (B) Vif variants from different subtypes areexpressed to comparable levels. pCRV1 Vif expression plasmids were transfected with GFP expression plasmid into HEK 293T cells. Lysates were separated by SDS-PAGEat 48 h posttransfection, transferred to PVDF membranes, and probed with antibodies against Vif, GFP, and GAPDH. (C) Relative Vif expression for each Vif variant isshown. Nonsaturated signals were acquired, background was subtracted, and values were normalized to GFP expression levels. NL4-3 Vif expression was set to 1. Theaverage and standard deviations of three independent experiments are shown. Error bars represent standard deviations. � denotes anti.
Activity of Vif from HIV-1 Subtypes against APOBEC3
January 2012 Volume 86 Number 1 jvi.asm.org 51
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

alleles (see Fig. S1 in the supplemental material for Vif sequencealignments).
The majority of Vif variants were similarly expressed upontransfection in 293T cells (Fig. 1B and C), with the exception of thesubtype C Vif C1, which consistently displayed a 2-fold decrease insignal intensity (Fig. 1C). This difference may reflect lower Vifprotein expression or reduced antibody recognition by the poly-clonal anti-Vif rabbit serum. All 17 Vif variants encode 192 resi-dues, but some Vif bands migrated differently during gel electro-phoresis, likely reflecting variations in protein charges (Fig. 1B,compare Vif F1 to Vif F3 or Vif G1).
Anti-APOBEC3 phenotypes of Vif alleles. The different Vifproteins were analyzed for rescue of infectivity of NL4-3 with adeletion of Vif (pNL4-3�Vif) in the presence of A3G, A3F, andA3H hapII. In addition, APOBEC3 degradation by the Vif panelwas assessed by cotransfecting the Vifs and APOBEC3, followedby Western blot analysis. Titrations of Vif expression plasmids (5ng, 100 ng, and 500 ng) and APOBEC3 plasmids (25 ng, 50 ng, and100 ng) in the presence of NL4-3�Vif were performed to selectassay conditions and plasmid ratios that allowed optimal discrim-ination of both an increase and a decrease in rescue of infectivity(data not shown). Five nanograms of Vif and 50 ng of A3G or 50ng of A3F were used in the experiments comparing the anti-A3G/A3F activities of different Vif variants. In addition, simi-lar experiments were performed with Vif-deleted HIV-1 mo-lecular clones pLAI, pMJ4 (subtype C, Vif C3), and p94UG114(subtype D, Vif D1) to exclude potential isolate and subtypeincompatibilities (data not shown). Of note, Capsid p24 pro-duction upon transfection was comparable for all viral stocks(data not shown).
Most HIV-1 Vif alleles counteract A3G and A3F restriction.The infectivity of viral stocks was quantified 48 h posttransfectionusing TZM-bl reporter cells as described previously (10, 28, 37).An empty pCRV1 plasmid and the NL4-3 Vif mutant C133S,which abrogates cullin 5 binding (C133S), were used as negativecontrols. Infectivity of NL4-3�Vif produced in the absence ofboth APOBEC3 and Vif was set to 100% relative infectivity (Fig.2A and C; see also Fig. 3A). We chose the widely used molecularclone LAI Vif variant as a reference for comparison to other Vifs(Fig. 1A), and the differences relative to LAI Vif were calculatedusing a t test (Prism GraphPad).
In the absence of Vif or in the presence of the Vif C133S mu-tant, A3G strongly reduced infectivity to 1%. The reference LAIVif rescued infectivity from A3G restriction to 60% � 5%, and themajority of Vif alleles tested were similarly or more active than LAIVif (Fig. 2A).
Four Vif variants (NL4-3, B2, C1, and G1) displayed low tomoderate activities against A3G (relative infectivity, 35% � 4.5%,36% � 2.1%, 10.3% � 3.5%, and 35% � 2.5%, respectively), andonly one Vif allele (B3) was fully defective against A3G (relativeinfectivity, 2.7% � 1.5%). Interestingly, Vif C1, C2, and C3 wereexpressed to lower levels than the other Vifs, and correcting infec-tivity data with the Vif expression levels shown in Fig. 2A and Bdemonstrated that all subtype C Vifs were relatively more activeagainst A3G.
The ability of the Vifs to degrade the APOBEC3s was deter-mined by transfecting HEK 293T cells with 20 ng of the differentVif expression plasmids and 100 ng of A3G (Fig. 2B) or A3F (Fig.2D). The rescue of infectivity correlated with the ability to degradeA3G in the producer cell, with the exception of Vif C1, which
FIG 2 Activity of Vif alleles against A3G and A3F. (A) Anti-A3G activities of the different Vif variants. Infectivity of pNL4-3�Vif cotransfected with Vif allelesand A3G was assessed at 48 h posttransfection on TZM-bl reporter cells. Data shown are representative of three independent experiments. Error bars representstandard deviations. Unpaired t tests were computed to determine whether differences between LAI Vif infectivity and a given Vif variant reached significance (�,P � 0.05; ��, P � 0.01; ���, P � 0.001 [Prism software]). (B) Degradation of A3G by the different Vif alleles. Transfected lysates were separated by SDS-PAGEat 48 h posttransfection, transferred to PVDF membranes, and probed with antibodies against FLAG, Vif, and GFP. One representative Western blot is shown.(C) Anti-A3F activities of the different Vif variants. Infectivity of pNL4-3�Vif cotransfected with Vif alleles and A3G was assessed at 48 h posttransfection onTZM-bl reporter cells. Data shown are representative of three independent experiments. Error bars represent standard deviations. Unpaired t tests werecomputed to determine whether differences between LAI Vif infectivity and a given Vif variant reached significance (�, P � 0.05; ��, P � 0.01; ���, P � 0.001[Prism software]). (D) Degradation of A3F by the different Vif alleles. Transfected lysates were separated by SDS-PAGE at 48 h posttransfection, transferred toPVDF membranes, and probed with antibodies against FLAG, Vif, and GFP. Representative Western blots are shown.
Binka et al.
52 jvi.asm.org Journal of Virology
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from
degraded A3G efficiently but failed to rescue viral infectivity to thesame degree.
A3F is less potent than A3G in restricting NL4-3�Vif comple-mented with an empty control plasmid or with the mutant VifC133S (no-Vif and Vif C133S, 10% compared to 1% with A3G).LAI Vif rescued infectivity in the presence of A3F to 64% � 5%,with all Vif alleles displaying activity against A3F. Vif variants AE1,F2, and F3 were more active than LAI Vif (relative infectivity,73% � 8.1%, 85% � 8.5%, and 71% � 6.6%, respectively). VifC1, which displayed low activity against A3G, showed averageactivity against A3F (relative infectivity, 50% � 2.0%). Interest-ingly, Vif B3, which was not active against A3G, displayed some
activity against A3F (relative infectivity, 26% � 3.1%) although itsability to degrade A3F was modest (Fig. 2D). Overall, most Vifswere able to degrade and counteract A3G and A3F restriction.
Most non-subtype B HIV-1 Vif alleles counteract A3H hapIIrestriction. More than eight naturally occurring A3H haplotypeshave been described, with A3H hapII having potent anti-HIV ac-tivity (10, 29, 43). There remains some uncertainty about the ex-tent to which A3H is partially or completely resistant to Vif neu-tralization (5, 10, 29, 41, 43, 52). We therefore tested whether thedifferent Vif alleles have activity against A3H hapII (Fig. 3A).Based on initial optimization experiments (data not shown), weused less APOBEC3 expression plasmid for the experiments with
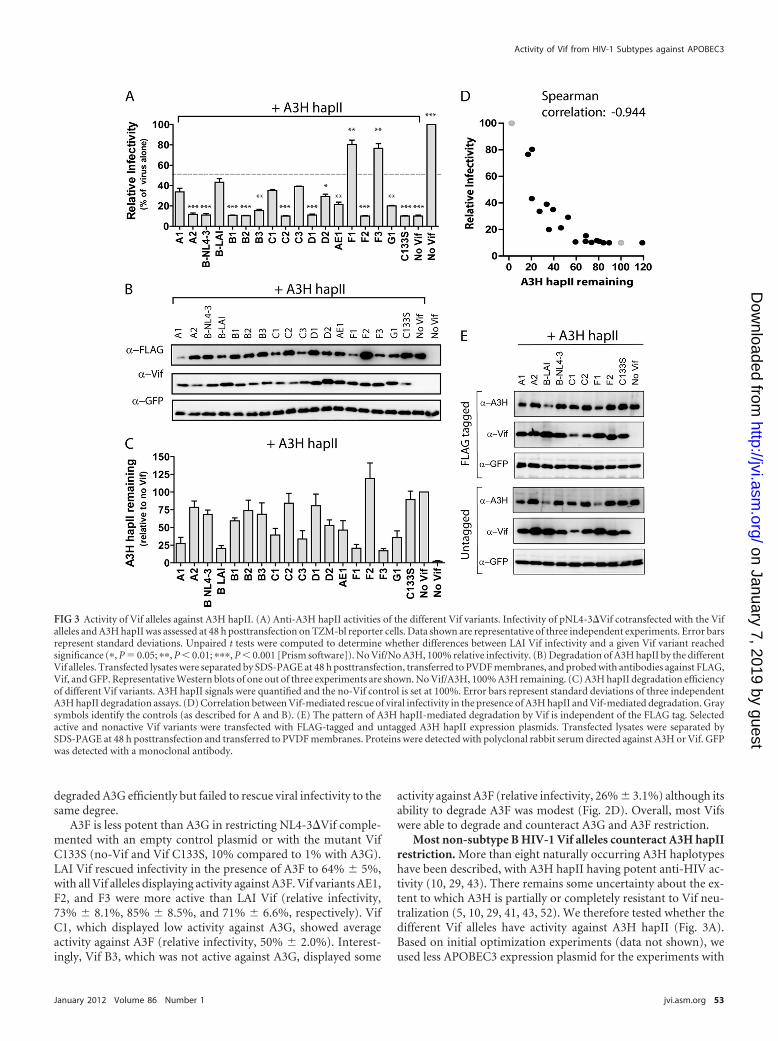
FIG 3 Activity of Vif alleles against A3H hapII. (A) Anti-A3H hapII activities of the different Vif variants. Infectivity of pNL4-3�Vif cotransfected with the Vifalleles and A3H hapII was assessed at 48 h posttransfection on TZM-bl reporter cells. Data shown are representative of three independent experiments. Error barsrepresent standard deviations. Unpaired t tests were computed to determine whether differences between LAI Vif infectivity and a given Vif variant reachedsignificance (�, P � 0.05; ��, P � 0.01; ���, P � 0.001 [Prism software]). No Vif/No A3H, 100% relative infectivity. (B) Degradation of A3H hapII by the differentVif alleles. Transfected lysates were separated by SDS-PAGE at 48 h posttransfection, transferred to PVDF membranes, and probed with antibodies against FLAG,Vif, and GFP. Representative Western blots of one out of three experiments are shown. No Vif/A3H, 100% A3H remaining. (C) A3H hapII degradation efficiencyof different Vif variants. A3H hapII signals were quantified and the no-Vif control is set at 100%. Error bars represent standard deviations of three independentA3H hapII degradation assays. (D) Correlation between Vif-mediated rescue of viral infectivity in the presence of A3H hapII and Vif-mediated degradation. Graysymbols identify the controls (as described for A and B). (E) The pattern of A3H hapII-mediated degradation by Vif is independent of the FLAG tag. Selectedactive and nonactive Vif variants were transfected with FLAG-tagged and untagged A3H hapII expression plasmids. Transfected lysates were separated bySDS-PAGE at 48 h posttransfection and transferred to PVDF membranes. Proteins were detected with polyclonal rabbit serum directed against A3H or Vif. GFPwas detected with a monoclonal antibody.
Activity of Vif from HIV-1 Subtypes against APOBEC3
January 2012 Volume 86 Number 1 jvi.asm.org 53
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

A3H hapII (25 ng of A3H hapII compared to 50 ng of A3G orA3F). Under these experimental conditions, A3H hapII reducesinfectivity to 10% � 1.0% in the absence of Vif or with the mutantVif C133S.
Interestingly, 9 out of 17 Vif variants were able to rescue infec-tivity in the presence of A3H hapII. Of the active Vifs, Vif F1 andF3 were, by far, the most efficient at counteracting A3H hapII (F1,80% � 7.5%; F3, 77% � 8.0%) (Fig. 3A), followed by LAI, twosubtype C Vif variants (C1 and C3), and one subtype A Vif variant(A1). Of note, Vif C1, which had low activity against A3G andmodest activity against A3F, was active against A3H hapII (35% �2.0%). Three out of four subtype B variants failed to counteractA3H hapII restriction, with LAI Vif being the sole exception. LAIVif rescued viral infectivity in the presence of A3H hapII to 43% �6.1%, whereas NL4-3 Vif was defective in rescuing A3H-mediatedrestriction (11% � 2.6%). This observation is in good agreementwith reports showing that Vif from LAI but not NL4-3 is activeagainst A3H hapII (18, 19).
The ability of Vif variants to degrade the A3H hapII was deter-mined by transfecting HEK 293T cells with 20 ng of the differentVif expression plasmids and 50 ng of A3H (Fig. 3B). Overall, A3HhapII was more resistant to degradation than A3G and A3F (Fig.3B, compare the no-Vif and Vif C133S control levels to the level ofA3H in the presence of other Vifs). The Vif variants LAI, F1, andF2 degraded A3H hapII efficiently (Fig. 3C, e.g., only 20% of the“no Vif” A3H hapII control remaining), whereas the other Vifvariants were partially or completely defective with respect to deg-radation in the producer cell (Fig. 3B and C). The degree of Vif-mediated rescue of viral infectivity in the presence of A3H hapIIcorrelated well with the efficiency to degrade A3H hapII (Fig. 3D).
Adding epitope tags to A3H hapII has been reported to influ-ence Vif-mediated A3H hapII degradation (18). To test whetherthe N-terminal FLAG tag to A3H hapII affects its susceptibility toVif, we analyzed 10 Vif alleles with N-terminal FLAG tags anduntagged A3H hapII for degradation. We transfected the selectedVif variants and the two A3H expression plasmids into HEK 293Tcells and used polyclonal rabbit serum raised against an A3H pep-tide to detect both A3H hapII variants in the cell lysates by West-ern blotting (Fig. 3E). The A3H hapII degradation patterns weresimilar for the N-terminally tagged and untagged A3H hapII pro-teins (Fig. 3E), indicating that the N-terminal FLAG tag did notaffect its sensitivity to Vif degradation.
Taken together, in contrast to the efficient activity of most ofthe Vifs against A3G and A3F, Vifs from different subtypes dis-played considerable differences in counteracting A3H hapII re-striction.
The spectrum of anti-APOBEC3 activities varies between Vifvariants. Since multiple APOBEC3 proteins are expressed inHIV-1 susceptible cells, we were interested in a comprehensiveassessment of Vif-mediated anti-APOBEC3 activities. Figure 4provides a heat map representation of the anti-APOBEC3 spec-trum of each Vif allele (complete rescue indicated in green andmaximum restriction shown in red). Vif variants F1 and F3showed high activity against all three deaminases tested (broad-spectrum anti-APOBEC3 activity), while Vif B3 displayed low ac-tivity against all three APOBEC3s. Interestingly, we also notedA3-specific Vif defects in which one (Vif C1 and Vif F2) or two(Vif C2) specific APOBEC3s were not neutralized. Overall, thepatterns of rescue from A3G and A3F were similar among Vifs butdid not correlate with the activities of these Vifs against A3H ha-pII. This indicates that residues in Vif that interact with A3G andA3F are likely different from those that interact with A3H hapII.
Identification of the Vif residues required for A3H hapII deg-radation. Subtype F Vif variants were efficient in counteractingA3G and A3F but differed in their abilities to block A3H hapII(Fig. 4). This apparent difference between F1 and F2 with respectto A3H hapII was used to accurately determine the Vif require-ments for interacting with A3H hapII. We focused on theN-terminal part of Vif because this region has been previouslyshown to interact with A3G and A3F. Since Vif F1 and Vif F2differ at only five residues in the N-terminal region (positions 39,48, 61, 62, and 63) (Fig. 5A), we generated a series of mutants inwhich residue 39, residue 48, or residues 61 to 63 were exchangedbetween the two Vifs (Fig. 5A). We tested the infectivity of NL4-3�Vif with these Vif mutants in the presence and absence of A3HhapII (Fig. 5B). Infectivity data showed that two residues, 39F and48H, are responsible for the difference between Vif F1 and Vif F2and that both are required for efficient anti-A3H hapII activity.Replacing either the phenylalanine (F) at position 39 with a serine(39S) or the histidine (H) at position 48 with an asparagine (48N)rendered Vif F1 inactive against A3H hapII, while the introduc-tion of both residues (F39 and H48) was required to render Vif F2active against A3H hapII (Fig. 5B). The ability of these mutants todegrade A3H hapII was also assessed (Fig. 5C) and showed that theability to rescue infectivity correlated with A3H hapII degradation(Fig. 5B and C). Interestingly, mutations at these two positionsaffected only the activity against A3H hapII, whereas the ability tocounteract A3G and A3F was unaffected (data not shown).
Before Vif can mediate the degradation of APOBEC3 by theproteasome, both proteins must first physically interact. We spec-ulated, therefore, that the Vif proteins that failed to counteractA3H hapII might be inefficiently interacting with A3H hapII. We
FIG 4 Comparison of the anti-APOBEC3 activities of different Vif alleles. A heat map representation illustrates the spectrum of infectivity obtained with eachVif variant in the presence of A3G, A3F, and A3H hapII. Normalized infectivity values are plotted. The controls for each deaminase are shown on the right sideof the panel: the level of suppression achieved (%) in the absence of Vif is depicted (red), and the infectivity (%) in the absence of APOBEC3 is set to 100 (green).Relative infectivity values are the average of three independent experiments shown in Fig. 2 and 3. Max denotes maximum.
Binka et al.
54 jvi.asm.org Journal of Virology
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

performed coimmunoprecipitation experiments by transfectingVif F1 and Vif F2 with A3H hapII or A3G in HEK 293T cells. Theproteasome inhibitor clasto-lactacystin �-lactone was added tothe medium 24 h before lysis to prevent APOBEC3 degradation.Vif F1 was more efficient in precipitating A3H hapII than Vif F2,whereas both Vif F1 and F2 associated efficiently with A3G (Fig.5D). In addition, we established that the two residues were neces-sary and sufficient to determine the ability of Vif F variants toassociate with A3H hapII by cotransfected Vifs F1 and F2 as well asmutant Vifs M4 and M3 with A3H hapII in HEK 293T cells. Vif F1and mutant M3 (with residues 39F and 48H) immunoprecipitatedwith A3H hapII, while defective Vifs F2 and mutant M4 (with 39Sand 48N) both failed to associate with A3H hapII. This indicatesthat a phenylalanine at position 39 and a histidine at position 48
determine the association between subtype F Vifs and A3H hapII(Fig. 5E). These results correlated well with the observed infectiv-ity and degradation phenotypes (Fig. 5B and C).
Alanine scanning of residues 38 to 49 in the amino-terminalregion of subtype F Vif. In addition to the polymorphic nature ofthe amino acids at position 39 and 48, changes within the regionbetween these two residues may affect the interaction between Vifand APOBEC3. Indeed, several motifs in this region have beendescribed to affect subtype B Vif function against A3G (e.g.,40YRHHY44, 45E, and 48N) (34, 37). We individually replaced allamino acids between positions 38 and 49 in Vif F1 with alaninesand analyzed their effects on rescue of infectivity in the presence ofA3G, A3F, and A3H hapII. The alanine scanning of this Vif F1region showed that distinct positions had differential effects on
FIG 5 Identification of the residues important for activity of subtype F Vif variants against A3H hapII. (A) A schematic of the Vif N-terminal mutants generatedto investigate activity against A3H hapII. Vif F1 and Vif F2 differ at positions 39, 48, and 61 to 63. (B) Infectivities of pNL4-3�Vif cotransfected with Vif F1, VifF2, and Vif mutants M1 to M8 and A3H hapII were assessed at 48 h posttransfection on TZM-bl reporter cells. Data shown are representative of threeindependent experiments. Error bars represent standard deviations of triplicate experiments. (C) Western blot depicting A3H hapII degradation in the presenceof Vif F1, Vif F2, and Vif mutants M1 to M8. Transfected lysates were separated by SDS-PAGE at 48 h posttransfection, transferred to PVDF membranes, andprobed with antibodies against FLAG, Vif, and GFP. Data shown are representative of three independent experiments. (D) Coimmunoprecipitation (Co-IP) ofsubtype F Vif variants with A3H hapII and A3G. Subtype F Vif F1, Vif F2, or empty control plasmid was cotransfected with HA-tagged A3H hapII, HA-taggedA3G, or control plasmid. APOBEC3 degradation was prevented by the addition of the proteasome inhibitor clasto-lactacystin �. Cleared lysates were incubatedwith anti-HA-coated beads and washed vigorously. Proteins were analyzed by Western blotting. (E) Coimmunoprecipitation of subtype F Vif variants with A3HhapII. Subtype F Vif F1, Vif F2, and Vif mutants M4 and M3 were cotransfected with HA-tagged A3H hapII. A3H hapII degradation was prevented by the additionof the proteasome inhibitor clasto-lactacystin �. Cleared lysates were incubated with anti-HA-coated beads and washed vigorously. Proteins were analyzed byWestern blotting.
Activity of Vif from HIV-1 Subtypes against APOBEC3
January 2012 Volume 86 Number 1 jvi.asm.org 55
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

activity against A3G, A3F, and A3H hapII (Fig. 6). Alanines atposition 38, 48, and 49 rendered Vif F1 defective against all threeAPOBEC3s (Fig. 6A, B, C, and E) but did not affect Vif expression(Fig. 6D). None of the other mutations reduced neutralization ofA3F, while other mutations showed APOBEC3-specific effects:40A completely abrogated anti-A3G and anti-A3H hapII activity,and several substitutions in this region resulted in a specific loss offunction against A3H hapII (41A, 40A, and 45A) (Fig. 6C). Posi-tion 39, one of the two positions that determined the differencebetween Vif F1 and F2 with regard to A3H degradation, was moretolerant to being mutated to an alanine (from an aromatic phe-nylalanine) than to the nucleophilic serine. Western blot analysisshowed that the efficiency in rescue of infectivity correlates withthe level of APOBEC3 degradation by the respective Vifs. Takingthese results together, alanine scanning mutagenesis of the regionbetween residues 39 and 48 in subtype F Vif suggests that theresidues necessary for A3H hapII recognition are distinct fromthose required for Vif-A3G or Vif-A3F interactions.
Next, we analyzed the sequences between positions 38 and 49of the initial subtype set of Vifs to understand why only some Vifscounteract A3H hapII. Figure 7 shows an alignment of the proteinregion of interest (positions 38 to 49) of the 17 Vif proteins testedand their respective activities against A3H hapII. Nearly all Vifsthat efficiently counteracted A3H hapII carried the 39F and 48Hcombination, whereas most of the inactive Vifs carried alternative
pairs. Only a few exceptions existed: Vif B2 contains the FH pairbut lacks anti-A3H activity, which could be caused by the uniqueglycine (G) at position 41 (41G, all other active Vifs carry 41R).This possibility is further supported by the alanine scanning re-sults, which showed the importance of position 41 for anti-A3Hfunction (Fig. 6C). Vif D2, which carries an LH pair instead of theFH pair, displayed low activity against A3H hapII. The leucine (L)at position 39 shares the same hydrophobic and nonpolar prop-erties as the phenylalanine (F), indicating that some variation atposition 39 is tolerated with amino acids of similar properties, like39A (Fig. 6C).
In summary, a 39F-48H or a 39L-48H pair is required for ac-tivity against A3H hapII but not for neutralization of A3G andA3F. In addition, amino acid changes at critical residues betweenthese positions (e.g., 40, 41, and 45) may abrogate anti-A3H hapIIfunction in the presence of the FH/LH pairs.
DISCUSSION
At least five cytidine deaminases of the APOBEC3 family restrictretroviruses if left unimpeded by Vif (1, 4, 8, 11, 38). Our knowl-edge of the anti-APOBEC3 spectra of diverse non-B subtype Vifvariants is limited to A3G (14).
In this study, we assessed whether Vifs from different HIV-1subtype strains are able to counteract specific APOBEC3 proteins(A3G, A3F, and A3H hapII) with comparable efficiencies. We
FIG 6 Characterization of the Vif/APOBEC3 interface that allows degradation of A3H hapII in the Vif F1 context. The anti-APOBEC3 activities of the 12 differentalanine mutants in Vif F1 (alanine scanning mutagenesis of the region between residues 38 and 49) are shown. Infectivity of pNL4-3�Vif cotransfected with the Vifmutants and A3G (A), A3F (B), and A3H hapII (C) was assessed 48 h posttransfection on TZM-bl reporter cells. Data shown are representative of three independentexperiments. Error bars represent standard deviations. Western blots showing A3G, A3F, and A3H hapII degradation by the mutant F1 Vifs are directly under therespective infectivity values. (D) Transfected Vif F1 mutants are expressed to comparable levels. Representative Western blots are shown for Vif expression and the GFPtransfection control. (E) Heat map summarizing the anti-A3G, -A3F, and -A3H hapII phenotypes associated with each Vif F1 alanine mutant.
Binka et al.
56 jvi.asm.org Journal of Virology
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

found that several non-B subtype Vif alleles were more efficient inneutralizing A3G, A3F, and A3H hapII than the commonly usedsubtype B Vifs. Indeed, most Vif alleles tested in this study againstA3G and A3F were at least as active as the Vif obtained from thesubtype B LAI molecular clone (Fig. 2 and 4). Moreover, the ma-jority of non-B subtypes were able to neutralize A3H hapII (Fig. 3and 7). The subtype F Vif variants F1 and F3 were especially effi-cient in neutralizing A3H hapII (�70% of the no-A3H control),while a second group of Vifs (Vif A1, C1, C3, and LAI) displayedmoderate activity against A3H hapII (e.g., �30 to 50% infectivityof the no-A3H control). Two residues, 39F and 48H, were re-quired for this activity (Fig. 5). Alanine scanning mutagenesis re-vealed that changes to other residues between positions 39 and 48may affect the anti-A3H hapII function even in the presence of therequired 39F-48H pair (Fig. 6). The observed variation in anti-A3H hapII activity of the Vifs tested in this study, however, waslargely explained by the presence/absence of this specific aminoacid pair (Fig. 7). For example, the difference in anti-A3H activitiesbetween the LAI and NL4-3 Vifs is likely due to the substitution atposition 48, but the additional differences between NL4-3 and LAIVifs at positions 47 and 50 may also affect anti-A3H activity (Fig. 7). Arecent analysis of HIV-1 Vif sequence variation in an Argentineanpediatric cohort (7) showed that approximately 70% of subtype Fstrains encoded FH at positions 39/48, while 30% carried ZH or XNcombinations (where Z indicates any amino acid other than F, and X
denotes any residue), suggesting that the majority of subtype F strainslikely counteract A3H hapII.
Our findings suggest that HIV-1 Vif variants display a spec-trum of neutralization phenotypes, which cannot be easily pre-dicted from the Vif genotype. Some Vifs failed to neutralize A3Gbut rescued infectivity in the presence of A3F or A3H hap II (e.g.,Vif C1), while others displayed activity against A3G and A3F butnot A3H hapII. We propose that the sum of these activities likelydetermines the degree to which APOBEC3 restriction is overcomein the context of given individual’s APOBEC3 composition. Morecomprehensive data sets relating Vif genotype to Vif phenotypeare needed to develop algorithms that allow prediction of anti-APOBEC3 activities.
The structure-function experiments performed in this studyimply that the Vif interface recognizing A3H hapII is distinct fromthe A3G or A3F interface. Multiple distant residues are involved,suggesting a nonlinear recognition domain. We speculate that theVif alleles lacking activity against A3H hapII were originally ob-tained from individuals who did not express active A3H variantsand, thus, did not require A3H neutralization. Interestingly, theanti-APOBEC3 activity of LAI Vif was more potent as well asbroader than that of NL4-3 Vif; the LAI Vif not only neutralizedA3G and A3F with higher efficiency but also was the only subtypeB Vif that displayed activity against A3H hapII. Endogenous A3Hexpression levels are believed to be modest, but the differentialneutralization spectra of several Vif variants of different subtypessupport the role of A3H as a restriction factor in vivo.
Some limitations of our study need to be mentioned. First, wecannot draw generalized conclusions about subtype-specific ac-tivities since the sample sizes are not sufficiently representative.Moreover, the Vif alleles used in this study were cloned fromviral isolates obtained by coculture. Future studies on the anti-APOBEC3 function should use Vif variants directly obtained fromthe plasma of patients in order to obtain information on the Vifactivity spectrum without bias introduced by passaging the viralstock in cells from a different donor. Moreover, experimental ap-proaches using overexpression assays are frequently used to probefor HIV-APOBEC3 interactions, but specific assays using primaryT lymphocytes encoding specific APOBEC3 haplotypes areneeded to further validate our observations at more physiologicalAPOBEC3 expression levels.
The APOBEC3 locus is polymorphic, with several haplotypesfor each of these deaminases (1). Proviruses with footprints of pastcytidine deamination are found in many infected patients inde-pendently of the HIV-1 subtype (1, 15, 17, 33), suggesting thatvariation in the spectra of the anti-APOBEC3 activities of HIV-1Vif and/or antiretroviral activity of Vif-resistant APOBEC3 pro-teins is important in vivo. HIV-1-infected patients with certainA3H haplotypes displayed fewer G-to-A mutations and lower viralloads (9), and it is attractive to speculate that Vif variants thatcounteract multiple APOBEC3 proteins decrease the complexityof the viral quasi-species as well as the pathogenicity of HIV-1 in agiven infected individual. A previous study found that Vif variantsfrom a patient harboring the active A3H hapII were active againstthis variant while Vif variants from patients with A3H hapI failedto degrade A3H hapII (19). This observation, together with ourfindings, suggests a more complex picture in which Vif may adaptto the APOBEC3 haplotype repertoire of the infected host. Futurestudies looking for clinical associations between APOBEC3 and
FIG 7 Correlation between Vif genotype and activity against A3H hapII. Theamino acid alignment of the 17 Vifs shows the region between residues 38 to 49in the amino-terminal portion of Vif. Vif F1 serves as reference in this align-ment. The Vifs are ranked in descending order of anti-A3H hapII activity (Fig.4). Nine Vif variants (F1 to G1) rescued viral infectivity in the presence of A3HhapII to 20% or more. These infectivity values were significantly higher (P �0.05, t test) from the no-Vif or the Vif C133S controls. The level of rescue isindicated as follows: ���, high, with infectivity �50% of the no-A3H con-trol; ��, moderate, with 50 to 30% of the no-A3H control; and �, low, with20 to 30% of the no-A3H control. Infectivity values of �20% of the no-A3Hcontrol were defined as lack of neutralization activity (—). The amino acidpairs at locations 39 and 48 are grouped as FH, ZH, and XN. Z denotes anyresidue except F, and X denotes any residue.
Activity of Vif from HIV-1 Subtypes against APOBEC3
January 2012 Volume 86 Number 1 jvi.asm.org 57
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

HIV/AIDS disease progression will have to focus on multipleAPOBEC3 deaminases in conjunction with the Vif phenotype.
ACKNOWLEDGMENTS
We thank all the members of the Simon laboratory for helpful discussions.This research was supported by National Institutes of Health grants
AI089246, AI90935, and AI064001.
REFERENCES1. Albin JS, Harris RS. 2010. Interactions of host APOBEC3 restriction
factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol.Med. 12:e4.
2. Bell CM, et al. 2007. Molecular characterization of the HIV type 1 subtypeC accessory genes vif, vpr, and vpu. AIDS Res. Hum. Retroviruses 23:322–330.
3. Conticello SG, Harris RS, Neuberger MS. 2003. The Vif protein of HIVtriggers degradation of the human antiretroviral DNA deaminaseAPOBEC3G. Curr. Biol. 13:2009 –2013.
4. Cullen BR. 2006. Role and mechanism of action of the APOBEC3 familyof antiretroviral resistance factors. J. Virol. 80:1067–1076.
5. Dang Y, et al. 2008. Human cytidine deaminase APOBEC3H restrictsHIV-1 replication. J. Biol. Chem. 283:11606 –11614.
6. Dang Y, Wang X, Zhou T, York IA, Zheng YH. 2009. Identification ofa novel WxSLVK motif in the N terminus of human immunodeficiencyvirus and simian immunodeficiency virus Vif that is critical forAPOBEC3G and APOBEC3F neutralization. J. Virol. 83:8544 – 8552.
7. De Maio FA, et al. 2011. Effect of HIV-1 Vif variability on progression topediatric AIDS and its association with APOBEC3G and CUL5 polymor-phisms. Infect. Genet. Evol. 11:1256 –1262.
8. Goila-Gaur R, Strebel K. 2008. HIV-1 Vif, APOBEC, and intrinsic im-munity. Retrovirology 5:51.
9. Gourraud PA, et al. 2011. APOBEC3H haplotypes and HIV-1 pro-viralvif DNA sequence diversity in early untreated human immunodeficiencyvirus-1 infection. Hum. Immunol. 72:207–212.
10. Harari A, Ooms M, Mulder LC, Simon V. 2009. Polymorphisms andsplice variants influence the antiretroviral activity of human APOBEC3H.J. Virol. 83:295–303.
11. Harris RS, Liddament MT. 2004. Retroviral restriction by APOBEC pro-teins. Nat. Rev. Immunol. 4:868 – 877.
12. Hemelaar J, Gouws E, Ghys PD, Osmanov S. 2011. Global trends inmolecular epidemiology of HIV-1 during 2000 –2007. AIDS 25:679 – 689.
13. Hultquist JF, et al. 2011. Human and rhesus APOBEC3D, APOBEC3F,APOBEC3G, and APOBEC3H demonstrate a conserved capacity to re-strict Vif-deficient HIV-1. J. Virol. 85:11220 –11234.
14. Iwabu Y, et al. 2010. Differential anti-APOBEC3G activity of HIV-1 Vifproteins derived from different subtypes. J. Biol. Chem. 285:35350 –35358.
15. Janini M, Rogers M, Birx DR, McCutchan FE. 2001. Human immuno-deficiency virus type 1 DNA sequences genetically damaged by hypermu-tation are often abundant in patient peripheral blood mononuclear cellsand may be generated during near-simultaneous infection and activationof CD4[sup]� T cells. J. Virol. 75:7973–7986.
16. Kao S, et al. 2003. The human immunodeficiency virus type 1 Vif proteinreduces intracellular expression and inhibits packaging of APOBEC3G(CEM15), a cellular inhibitor of virus infectivity. J. Virol. 77:11398–11407.
17. Kijak GH, et al. 2008. Variable contexts and levels of hypermutation inHIV-1 proviral genomes recovered from primary peripheral blood mono-nuclear cells. Virology 376:101–111.
18. Larue RS, Lengyel J, Jonsson SR, Andresdottir V, Harris RS. 2010.Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mam-malian host and is capable of cross-species activity. J. Virol. 84:8193– 8201.
19. LI MM, Wu LI, Emerman M. 2010. The range of human APOBEC3Hsensitivity to lentiviral Vif proteins. J. Virol. 84:88 –95.
20. Liu B, Yu X, Luo K, Yu Y, Yu XF. 2004. Influence of primate lentiviralVif and proteasome inhibitors on human immunodeficiency virus type 1virion packaging of APOBEC3G. J. Virol. 78:2072–2081.
21. Luo K, et al. 2005. Primate lentiviral virion infectivity factors are substratereceptors that assemble with cullin 5-E3 ligase through a HCCH motif tosuppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444 –11449.
22. Marcsisin SR, et al. 2011. On the solution conformation and dynamics ofthe HIV-1 viral infectivity factor. J. Mol. Biol. 410:1008 –1022.
23. Mariani R, et al. 2003. Species-specific exclusion of APOBEC3G fromHIV-1 virions by Vif. Cell 114:21–31.
24. Mehle A, et al. 2004. Vif overcomes the innate antiviral activity ofAPOBEC3G by promoting its degradation in the ubiquitin-proteasomepathway. J. Biol. Chem. 279:7792–7798.
25. Mehle A, Thomas ER, Rajendran KS, Gabuzda D. 2006. A zinc-bindingregion in Vif binds Cul5 and determines cullin selection. J. Biol. Chem.281:17259 –17265.
26. Mehle A, et al. 2007. Identification of an APOBEC3G binding site inhuman immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J. Virol. 81:13235–13241.
27. Mulder LC, Harari A, Simon V. 2008. Cytidine deamination inducedHIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 105:5501–5506.
28. Mulder LC, et al. 2010. Moderate influence of human APOBEC3F onHIV-1 replication in primary lymphocytes. J. Virol. 84:9613–9617.
29. OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. 2008. Antiret-roelement activity of APOBEC3H was lost twice in recent human evolu-tion. Cell Host Microbe 4:249 –259.
30. Ooms M, Majdak S, Seibert CW, Harari A, Simon V. 2010. The local-ization of APOBEC3H variants in HIV-1 virions determines their antiviralactivity. J. Virol. 84:7961–7969.
31. Paul I, Cui J, Maynard EL. 2006. Zinc binding to the HCCH motif ofHIV-1 virion infectivity factor induces a conformational change that me-diates protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 103:18475–18480.
32. Pery E, Rajendran KS, Brazier AJ, Gabuzda D. 2009. Regulation ofAPOBEC3 proteins by a novel YXXL motif in human immunodeficiencyvirus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol.83:2374 –2381.
33. Piantadosi A, Humes D, Chohan B, McClelland RS, Overbaugh J. 2009.Analysis of the percentage of human immunodeficiency virus type 1 se-quences that are hypermutated and markers of disease progression in alongitudinal cohort, including one individual with a partially defectiveVif. J. Virol. 83:7805–7814.
34. Russell RA, Pathak VK. 2009. Identification of Two Distinct HIV-1 Vifdeterminants critical for interactions with human APOBEC3G andAPOBEC3F. J. Virol. 83:1992–2003.
35. Schrofelbauer B, Senger T, Manning G, Landau NR. 2006. Mutationalalteration of human immunodeficiency virus type 1 Vif allows for func-tional interaction with nonhuman primate APOBEC3G. J. Virol. 80:5984 –5991.
36. Simon JH, Sheehy AM, Carpenter EA, Fouchier RA, Malim MH. 1999.Mutational analysis of the human immunodeficiency virus type 1 Vif pro-tein. J. Virol. 73:2675–2681.
37. Simon V, et al. 2005. Natural variation in Vif: differential impact onAPOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pat-hog. 1:e6.
38. Smith JL, Bu W, Burdick RC, Pathak VK. 2009. Multiple ways oftargeting APOBEC3-virion infectivity factor interactions for anti-HIV-1drug development. Trends Pharmacol. Sci. 30:638 – 646.
39. Stanley BJ, et al. 2008. Structural insight into the human immunodefi-ciency virus Vif SOCS box and its role in human E3 ubiquitin ligase as-sembly. J. Virol. 82:8656 – 8663.
40. Stephens EB, Singh DK, Pacyniak E, McCormick C. 2001. Comparisonof Vif sequences from diverse geographical isolates of HIV type 1 andSIV(cpz) identifies substitutions common to subtype C isolates and exten-sive variation in a proposed nuclear transport inhibition signal. AIDS Res.Hum. Retroviruses 17:169 –177.
41. Tan L, Sarkis PT, Wang T, Tian C, Yu XF. 2009. Sole copy of Z2-typehuman cytidine deaminase APOBEC3H has inhibitory activity againstretrotransposons and HIV-1. FASEB J. 23:279 –287.
42. Tian C, et al. 2006. Differential requirement for conserved tryptophans inhuman immunodeficiency virus type 1 Vif for the selective suppression ofAPOBEC3G and APOBEC3F. J. Virol. 80:3112–3115.
43. Wang X, et al. 2011. Analysis of human APOBEC3H haplotypes andanti-human immunodeficiency virus type 1 activity. J. Virol. 85:3142–3152.
44. Wichroski MJ, Ichiyama K, Rana TM. 2005. Analysis of HIV-1 viralinfectivity factor-mediated proteasome-dependent depletion ofAPOBEC3G: correlating function and subcellular localization. J. Biol.Chem. 280:8387– 8396.
45. Wieland U, et al. 1994. In vivo genetic variability of the HIV-1 vif gene.Virology 203:43–51.
Binka et al.
58 jvi.asm.org Journal of Virology
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

46. Wieland U, et al. 1997. Diversity of the vif gene of human immunodefi-ciency virus type 1 in Uganda. J. Gen. Virol. 78:393– 400.
47. Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu XF. 2007. Zinc chelation inhibitsHIV Vif activity and liberates antiviral function of the cytidine deaminaseAPOBEC3G. FASEB J. 21:217–222.
48. Xiao Z, et al. 2007. Characterization of a novel Cullin5 binding domain inHIV-1 Vif. J. Mol. Biol. 373:541–550.
49. Yamashita T, Doi N, Adachi A, Nomaguchi M. 2008. Growth ability insimian cells of monkey cell-tropic HIV-1 is greatly affected by downstreamregion of the vif gene. J. Med. Invest. 55:236 –240.
50. Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. 2004. Selective assembly ofHIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complexthrough a novel SOCS box and upstream cysteines. Genes Dev. 18:2867–2872.
51. Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. 2004.APOBEC3G incorporation into human immunodeficiency virus type 1particles. J. Virol. 78:12058 –12061.
52. Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. 2010. A single amino aciddifference in human APOBEC3H variants determines HIV-1 Vif sensitiv-ity. J. Virol. 84:1902–1911.
Activity of Vif from HIV-1 Subtypes against APOBEC3
January 2012 Volume 86 Number 1 jvi.asm.org 59
on January 7, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from









![Vif and Apobec3G in the innate immune response to HIV: a ... · their proposed anointing as ‘guardians of the genome’ [8,17,18]. Apobec3H, while a robust anti-viral in primates,](https://static.fdocuments.us/doc/165x107/5ed505b2ad38025d974e484f/vif-and-apobec3g-in-the-innate-immune-response-to-hiv-a-their-proposed-anointing.jpg)








