
The 2-oxoacid dehydrogenase multi-enzyme complex of the archaeon Thermoplasma...
10
The 2-oxoacid dehydrogenase multi-enzyme complex of the archaeon Thermoplasma acidophilum ) recombinant expression, assembly and characterization Caroline Heath 1 , Mareike G. Posner 1 , Hans C. Aass 1 , Abhishek Upadhyay 2 , David J. Scott 3 , David W. Hough 1 and Michael J. Danson 1 1 Centre for Extremophile Research, Department of Biology and Biochemistry, University of Bath, UK 2 Department of Biology and Biochemistry, University of Bath, UK 3 National Centre for Macromolecular Hydrodynamics, School of Biosciences, University of Nottingham, Sutton Bonington, UK In aerobic bacteria and eukaryotes, a family of 2-oxoacid dehydrogenase multi-enzyme complexes (OADHCs) functions in the pathways of central metabolism. The complexes are responsible for the oxi- dative decarboxylation of 2-oxoacids to their corre- sponding acyl-CoAs. Members of the family include the pyruvate dehydrogenase complex (PDHC), which catalyzes the conversion of pyruvate to acetyl-CoA and so links glycolysis and the citric acid cycle; the 2-oxoglutarate dehydrogenase complex (OGDHC), which catalyzes the conversion of 2-oxoglutarate to succinyl-CoA within the citric acid cycle; and the branched-chain 2-oxoacid dehydrogenase complex (BCOADHC), which oxidatively decarboxylates the branched-chain 2-oxoacids produced by the transami- nation of valine, leucine and isoleucine. The complexes comprise multiple copies of three component enzymes: 2-oxoacid decarboxylase (E1), dihydrolipoyl acyl-trans- ferase (E2) and dihydrolipoamide dehydrogenase (E3) [1–3]. E2 forms the structural core of the complex, with multiple polypeptide chains associating into octa- hedral (24-mer) or icosahedral (60-mer) configurations, depending on the particular complex and the source organism [2,4]. E1 and E3 bind noncovalently to the Keywords Archaea; metabolism; multi-enzyme complex; 2-oxoacid dehydrogenase; thermophile Correspondence M. J. Danson, Centre for Extremophile Research, Department of Biology and Biochemistry, University of Bath, Bath, BA2 7AY, UK Fax: +44 1225 386779 Tel: +44 1225 386509 E-mail: [email protected] (Received 29 June 2007, revised 23 August 2007, accepted 24 August 2007) doi:10.1111/j.1742-4658.2007.06067.x The aerobic archaea possess four closely spaced, adjacent genes that encode proteins showing significant sequence identities with the bacterial and eukaryal components comprising the 2-oxoacid dehydrogenase multi- enzyme complexes. However, catalytic activities of such complexes have never been detected in the archaea, although 2-oxoacid ferredoxin oxidore- ductases that catalyze the equivalent metabolic reactions are present. In the current paper, we clone and express the four genes from the thermophilic archaeon, Thermoplasma acidophilum, and demonstrate that the recombi- nant enzymes are active and assemble into a large (M r ¼ 5 · 10 6 ) multi- enzyme complex. The post-translational incorporation of lipoic acid into the transacylase component of the complex is demonstrated, as is the assembly of this enzyme into a 24-mer core to which the other components bind to give the functional multi-enzyme system. This assembled complex is shown to catalyze the oxidative decarboxylation of branched-chain 2-oxoacids and pyruvate to their corresponding acyl-CoA derivatives. Our data constitute the first proof that the archaea possess a functional 2-oxo- acid dehydrogenase complex. Abbreviations BCOADHC, branched-chain 2-oxoacid dehydrogenase complex; CoASH, coenzyme-A; DLS, dynamic light scattering; E1, 2-oxoacid decarboxylase; E2, dihydrolipoyl acyl-transferase; E3, dihydrolipoamide dehydrogenase; FOR, ferredoxin oxidoreductase; IPTG, isopropyl thio-b-D-galactoside; M r , relative molecular mass; OADHC, 2-oxoacid dehydrogenase complex; OGDHC, 2-oxoglutarate dehydrogenase multienzyme complex; PDHC, pyruvate dehydrogenase complex; TPP, thiamine pyrophosphate. 5406 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS
-
Upload
caroline-heath -
Category
Documents
-
view
212 -
download
0
Transcript of The 2-oxoacid dehydrogenase multi-enzyme complex of the archaeon Thermoplasma...

The 2-oxoacid dehydrogenase multi-enzyme complex ofthe archaeon Thermoplasma acidophilum ) recombinantexpression, assembly and characterizationCaroline Heath1, Mareike G. Posner1, Hans C. Aass1, Abhishek Upadhyay2, David J. Scott3,David W. Hough1 and Michael J. Danson1
1 Centre for Extremophile Research, Department of Biology and Biochemistry, University of Bath, UK
2 Department of Biology and Biochemistry, University of Bath, UK
3 National Centre for Macromolecular Hydrodynamics, School of Biosciences, University of Nottingham, Sutton Bonington, UK
In aerobic bacteria and eukaryotes, a family of
2-oxoacid dehydrogenase multi-enzyme complexes
(OADHCs) functions in the pathways of central
metabolism. The complexes are responsible for the oxi-
dative decarboxylation of 2-oxoacids to their corre-
sponding acyl-CoAs. Members of the family include
the pyruvate dehydrogenase complex (PDHC), which
catalyzes the conversion of pyruvate to acetyl-CoA
and so links glycolysis and the citric acid cycle; the
2-oxoglutarate dehydrogenase complex (OGDHC),
which catalyzes the conversion of 2-oxoglutarate to
succinyl-CoA within the citric acid cycle; and the
branched-chain 2-oxoacid dehydrogenase complex
(BCOADHC), which oxidatively decarboxylates the
branched-chain 2-oxoacids produced by the transami-
nation of valine, leucine and isoleucine. The complexes
comprise multiple copies of three component enzymes:
2-oxoacid decarboxylase (E1), dihydrolipoyl acyl-trans-
ferase (E2) and dihydrolipoamide dehydrogenase (E3)
[1–3]. E2 forms the structural core of the complex,
with multiple polypeptide chains associating into octa-
hedral (24-mer) or icosahedral (60-mer) configurations,
depending on the particular complex and the source
organism [2,4]. E1 and E3 bind noncovalently to the
Keywords
Archaea; metabolism; multi-enzyme
complex; 2-oxoacid dehydrogenase;
thermophile
Correspondence
M. J. Danson, Centre for Extremophile
Research, Department of Biology and
Biochemistry, University of Bath, Bath,
BA2 7AY, UK
Fax: +44 1225 386779
Tel: +44 1225 386509
E-mail: [email protected]
(Received 29 June 2007, revised 23 August
2007, accepted 24 August 2007)
doi:10.1111/j.1742-4658.2007.06067.x
The aerobic archaea possess four closely spaced, adjacent genes that encode
proteins showing significant sequence identities with the bacterial and
eukaryal components comprising the 2-oxoacid dehydrogenase multi-
enzyme complexes. However, catalytic activities of such complexes have
never been detected in the archaea, although 2-oxoacid ferredoxin oxidore-
ductases that catalyze the equivalent metabolic reactions are present. In the
current paper, we clone and express the four genes from the thermophilic
archaeon, Thermoplasma acidophilum, and demonstrate that the recombi-
nant enzymes are active and assemble into a large (Mr ¼ 5 · 106) multi-
enzyme complex. The post-translational incorporation of lipoic acid into
the transacylase component of the complex is demonstrated, as is the
assembly of this enzyme into a 24-mer core to which the other components
bind to give the functional multi-enzyme system. This assembled complex
is shown to catalyze the oxidative decarboxylation of branched-chain
2-oxoacids and pyruvate to their corresponding acyl-CoA derivatives. Our
data constitute the first proof that the archaea possess a functional 2-oxo-
acid dehydrogenase complex.
Abbreviations
BCOADHC, branched-chain 2-oxoacid dehydrogenase complex; CoASH, coenzyme-A; DLS, dynamic light scattering; E1, 2-oxoacid
decarboxylase; E2, dihydrolipoyl acyl-transferase; E3, dihydrolipoamide dehydrogenase; FOR, ferredoxin oxidoreductase; IPTG, isopropyl
thio-b-D-galactoside; Mr, relative molecular mass; OADHC, 2-oxoacid dehydrogenase complex; OGDHC, 2-oxoglutarate dehydrogenase
multienzyme complex; PDHC, pyruvate dehydrogenase complex; TPP, thiamine pyrophosphate.
5406 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS

E2 core. E1 may occur as a homodimer or as an a2b2hetero-tetramer, depending upon the source and the
type of complex, although in all cases E3 is a dimer of
identical subunits.
E2 also forms the catalytic core of the complex; a
lipoyl moiety, covalently attached to a lysine residue in
the lipoyl domain of the E2, serves as a swinging arm,
connecting the active sites of each enzyme and chan-
nelling substrate through the complex [2,3]. Thus, E1
catalyzes the thiamine pyrophosphate (TPP)-dependent
decarboxylation of the 2-oxoacid and the transfer
of the resulting acyl group to the lipoic acid of E2.
E2 then transfers the acyl-group to coenzyme-A
(CoASH), after which E3 serves to reoxidize the
dihydrolipoyl moiety. It does so by the reduction of
the noncovalently bound cofactor FAD, in conjunction
with a protein disulfide bond and an amino acid base,
all of which are themselves then reoxidized by NAD+
to form NADH.
In Archaea [5] and anaerobic bacteria, the equiva-
lent oxidation of 2-oxoacids is catalyzed by an un-
related, and structurally more simple, family of
2-oxoacid ferredoxin oxidoreductases (FORs). This
comprises the pyruvate FOR, the 2-oxoglutarate FOR
and the 2-oxoisovalerate FOR, which catabolize the
oxidative decarboxylation of pyruvate, 2-oxoglutarate
and the branched-chain 2-oxoacids, respectively [6–8].
The pyruvate FOR from the halophilic archaeon Halo-
bacterium halobium is an a2b2 structure [9], whereas in
Sulfolobus solfataricus and Aeropyrum pernix it is an
ab dimer, and an octamer (a2b2c2d2) in Pyrococcus
furiosus, Methanothermobacter thermoautotrophicum
and Archaeoglobus fulgidus [reviewed in 5,8]. The cata-
lytic reaction of FORs does not involve a lipoic acid
moiety or NAD+; rather, the acyl-moiety formed on
decarboxylation of the 2-oxoacid is handed on directly
to CoASH, and the reducing equivalents to ferredoxin
via the enzyme’s iron-sulfur centre [6–8].
No OADHC activity has ever been detected in any
archaeon [5]. However, detection of E3 and lipoic acid
in various archaea [10–12] led to the discovery of a
putative OADHC operon in Haloferax volcanii [13]
and our subsequent detection of similar putative ope-
rons in the genome sequences of the aerobic archaea
Thermoplasma acidophilum, S. solfataricus, Sulfolobus
acidocaldarius, A. pernix, Pyrobaculum aerophilum,
Halobacterium NRC1 and Ferroplasma acidophilum
(M. G. Posner, unpublished data).
We have previously expressed the E1a and E1bgenes of the putative OADHC from T. acidophilum in
Escherichia coli and shown the recombinant proteins
to assemble into an a2b2 enzyme that catalyzes the
decarboxylation of the branched-chain 2-oxoacids and
pyruvate [14]. In the current paper, we report the clon-
ing and expression of the E2 and E3 genes of the same
operon from T. acidophilum, and the in vitro assembly
and characterization of an active 2-oxoacid dehydro-
genase complex.
This, then, is the first evidence that the putative
OADHC operon in an archaeon encodes a 2-oxoacid
dehydrogenase multi-enzyme complex that is func-
tional in vitro and therefore may have physiological
significance.
Results
Expression and purification of the E1, E2 and E3
components
The E1 component
The E1 a2b2 recombinant enzyme was produced as
described previously, and was shown to be catalyti-
cally active with the branched-chain 2-oxoacids
4-methyl-2-oxopentanoate, 3-methyl-2-oxopentanoate
and 3-methyl-2-oxobutanoate, and with pyruvate [14].
By dynamic light scattering (DLS), its Mr was found
to be 168 000 (± 6000), consistent with the value
of 165 000 determined by gel filtration [14] and the
expected value of 157 000 from the protein
sequences.
The E2 component
The gene encoding the E2 component was PCR-ampli-
fied from T. acidophilum genomic DNA and cloned
into the pET28a expression vector, as described in
Experimental procedures. The E2 protein was then
expressed in two host systems: E. coli BL21(DE3) cells,
in medium supplemented with lipoic acid but without
isopropyl thio-b-d-galactoside (IPTG) induction, and
E. coli BL21(DE3)pLysS cells without supplementation
but with IPTG induction. Both methods yielded solu-
ble E2 protein, although the level of expression was
significantly greater in the pLysS cells. In each case,
E2 was purified to >95% homogeneity using His-
Bind affinity chromatography followed by anion
exchange chromatography.
The E2 Mr values predicted from the published gene
sequence are 46 276 for unlipoylated protein and
46 464 for the polypeptide possessing a single lipoyl
residue. Accordingly, mass spectrometric analysis
revealed that the E2 expressed in E. coli grown in
lipoic acid-supplemented medium comprised an
approximately equimolar mixture of unlipoylated
(Mr ¼ 46 273) and lipoylated (Mr ¼ 46 461) protein.
Furthermore, from MS-analysis of tryptic fragments,
C. Heath et al. 2-Oxoacid dehydrogenase complex from the Archaea
FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS 5407

lipoylation was shown to have occurred at K42, which
by sequence comparisons corresponds to the lipoylated
lysine residue in bacterial E2 components (Fig. 1).
However, the E2 protein produced by expression in
induced cells, without lipoic acid supplement to the
growth medium, was <5% lipoylated. As reported
below, consistent with these data is the observation
that only the complex assembled using the lipoylated
E2 component showed detectable catalytic activity in
the overall complex assay.
The E3 component
The gene encoding the E3 component was PCR-
amplified from T. acidophilum genomic DNA and
cloned into the pET28a expression vector, as
described in Experimental procedures. Expression of
the E3 gene was carried out in E. coli BL21(DE3)
cells. Small-scale expression trials with induced versus
uninduced host cells revealed a higher E3 activity in
the soluble cell extract from uninduced cells, and this
was confirmed by SDS ⁄PAGE. The soluble protein
was purified to >95% homogeneity by heat precipi-
tation at 60 �C and His-Bind affinity chromatogra-
phy. The absorption spectrum of the purified protein
showed peaks at 375, 450 and 475 nm that are char-
acteristic of the presence of FAD in the enzyme;
using a molar absorption coefficient for FAD of
11 300 m)1Æcm)1 at 455 nm [15], the flavin content
was calculated to be 1.0 (± 0.1) FAD per polypep-
tide (Mr ¼ 49 867). Analytical gel filtration revealed
an Mr ¼ 100 000 for the purified enzyme, and DLS
gave a similar value (117 000 ± 2000), suggesting a
dimeric structure, as has been found for the bacterial
and eukaryotic enzymes. The maximal specific activity
of the enzyme was found to be 22 (± 1) lmolÆmin)1Æmg)1, a value that is considerably higher than
the overall complex activity (see below).
Assembly of OADHC from the recombinant
components
Complex assembly
When E1, E2 and E3 were incubated at 55 �C for
10 min in 20 mm sodium phosphate buffer, pH 7.5,
containing 2 mm MgCl2 and 0.2 mm TPP, prior to
assay, whole complex activity was subsequently
detected with the branched-chain 2-oxoacids 4-methyl-
2-oxopentanoate, 3-methyl-2-oxopentanoate and
3-methyl-2-oxobutanoate, and with pyruvate. The
10-min incubation is necessary to allow E1-TPP bind-
ing [16], but no increase in rate was seen when the
incubation, after subunit mixing, was extended to 1 h
at 4 �C, 25 �C or 55 �C.With an E2 : E3 molar ratio fixed at 1 : 1 (E2 poly-
peptide to E3 dimer), titration with E1 resulted in an
increase in whole complex activity until a maximum
was reached at a molar ratio (E1 ⁄E2) of approximately
2–3 : 1 [E1 a2b2 tetramer to E2 polypeptide] (Fig. 2).
However, a reduction of the E3 ratio did not cause a
significant change in whole complex activity, and by
investigating the mixing of various amounts of the
complex components it was found that maximum
OADHC activity was achieved when the E1, E2 and
E3 subunits were mixed in a molar stoichiometry of
3 : 1 : 0.1. However, as described below, this stoichio-
metry does not equate to the amounts of the three
enzymes in the assembled complex.
B. subtilis PDHC
B. subtilis BCOADHCTp. acidophilum OADHCE. coli OGDHC
E. coli PDHC (lipoyl domain 3)
Fig. 1. Alignment of various E2 sequences around the lipoylated
lysine residue. The T. acidophilum E2 protein sequence was aligned
with those of the E2 components of the following OADHCs using
the CLUSTALW multiple sequence alignment program: Bacillus subtilis
PDHC; B. subtilis BCOADHC; E. coli OGDHC; E. coli PDHC (lipoyl
domain 3). Residues 27–53 of the T. acidophilum sequence are
shown, along with the aligned regions of the other E2 sequences,
and the lysine residue that is lipoylated in each sequence is marked
by a (*).
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.1
0 1 2 3 4 5 6 7
Molar Ratio of E1:E2
Act
ivit
y (A
U/m
in)
Fig. 2. Whole complex activity as a function of the E1 : E2 ratio.
Lipoylated E2 and E3 were mixed in a molar ratio of 1 : 1 (E2 poly-
peptide to E3 dimer), and to this mixture varying amounts of E1
were added. The mixture was then incubated at 55 �C for 10 min
in the presence of 2 mM MgCl2 and 0.2 mM TPP, following which
samples were taken and assayed for whole complex activity
(expressed as A (absorbance units).min)1) using the substrate
3-methyl-2-oxopentanoate. The molar ratio of E1 : E2 is expressed
as E1 a2b2 tetramer to E2 polypeptide.
2-Oxoacid dehydrogenase complex from the Archaea C. Heath et al.
5408 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS

OADHC activity
The whole complex activity detected with 4-methyl-2-
oxopentanoate, 3-methyl-2-oxopentanoate, 3-methyl-2-
oxobutanoate, and pyruvate, but not with 2-oxogluta-
rate, is entirely consistent with the substrate specificity
observed for the recombinant E1 enzyme in the
absence of the other complex components [14]. The
relative activities are given in Table 1, along with those
for the isolated E1 component and for the BCOADHC
from bovine kidney. Using 3-methyl-2-oxopentanoate
as substrate, the assembled T. acidophilum complex
exhibited a hyperbolic dependence of velocity on the
2-oxoacid concentration, with KM ¼ 250 (± 40) lm
and Vmax ¼ 4 (± 0.1) lmolÆmin)1Æmg)1 (E2). This spe-
cific activity is comparable to that of the Bacillus
stearothermophilus PDHC [8–10 lmo1Æmin)1Æmg)1 (E2)
[17].
Relative molecular mass of the E2 core and the
assembled complex
Analytical centrifugation and DLS were used to deter-
mine the Mr of both the E2 core and the assembled
complex.
E2 core
Sedimentation velocity analysis was carried out at
40 �C as described in Experimental procedures on
recombinant E2 that was 50% lipoylated and had been
purified by His Bind� (Novagen, Merck Biosciences
Ltd., Nottingham, UK) and then anion-exchange chro-
matography. The sedimentation coefficient distribu-
tions showed a major symmetrical peak constituting
95% of the total protein (Fig. 3). Analysis of the data
by direct fitting using the finite element solution of the
Lamm equation in sedfit [18], gave a sedimentation
coefficient (corrected to water at 20�C and infinite
dilution; s�20,w) ¼ 27 (± 1) S (Svedberg units; 1S =
10)13 seconds) and an Mr ¼ 1.1 (± 0.1) · 106. From
the E2 polypeptide Mr of 46 276, the E2 protein there-
fore comprises 23.8 polypeptides; that is, it assembles
into a core of 24 subunits possessing octahedral sym-
metry.
DLS analysis at 55 �C, the optimum growth temper-
ature of T. acidophilum, gave an E2 Mr value ¼ 1.0
(± 0.1) · 106, in good agreement with the value deter-
mined by analytical centrifugation. This E2 sample
was nonlipoylated, demonstrating that lipoylation is
not a prerequisite for assembly of the core. Interest-
ingly, the Mr of the same E2 at 25 �C, with no prior
heat treatment at 55 �C, was estimated by DLS to be
55 000, close to the sequence-predicted monomer value
(46 276); this indicates that the assembly of E2 into a
24-mer core is temperature dependent.
The assembled complex
Sedimentation velocity analysis at 40 �C of assembled
complex, created by mixing the components in a
3 : 1 : 0.1 (E1 a2b2: E2 polypeptide: E3 a2) molar
ratio, revealed three discrete protein peaks with s�20,wvalues of 50S (24% of total protein), 19S (8%) and 6S
Table 1. Substrate specificities of the T. acidophilum 2-oxoacid
dehydrogenase complex. The T. acidophilum assembled complex
was assayed as described in Experimental procedures. For
3-methyl-2-oxopentanoate, Vmax ¼ 4 lmolÆmin–1Æmg–1 (E2), and
KM ¼ 250 lM; other activities were determined at saturating sub-
strate concentrations.
2-Oxoacid substrate
Ratio of specific activities
T. acidophilum
Bovine kidney
BCOADHCb
Assembled
complex E1a
3-methyl-2-oxopentanoate 1.0 1.0 1.0
4-methyl-2-oxopentanoate 0.5 0.3 1.5
3-methyl-2-oxobutyrate 0.9 0.6 0.2
pyruvate 0.2 0.2 0.4
2-oxoglutarate 0 0 0
a Data from [14]. b Data from [25].
Fig. 3. Sedimentation coefficient distributions of the E2 protein.
Sedimentation velocity of lipoylated E2 protein was carried out at
40 �C as described in Experimental procedures. The sedimentation
velocity distribution was obtained by the c(s) method [34] using the
program SEDFIT, and the data were then directly fitted using the
finite element solution of the Lamm equation in SEDFIT [18] to give
values of the sedimentation coefficient in the buffer of sedimenta-
tion [20 mM Tris ⁄ HCl (pH 8.5), 0.4 M NaCl and 1 mM phenyl-
methanesulfonyl fluoride].
C. Heath et al. 2-Oxoacid dehydrogenase complex from the Archaea
FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS 5409
(68%), respectively. Unfortunately, attempts to fit
these data using the finite element solution of the
Lamm equation in sedfit [18] were unsuccessful, thus
failing to give Mr values for these proteins. Using the
relationship that s � (Mr)2 ⁄ 3 for globular proteins, and
incorporating the values of Mr ¼ 6.1 · 106 and
s�20,w ¼ 60S for the E. coli PDHC [19], an approxi-
mate value of Mr ¼ 4.7 · 106 was calculated for what
is assumed to be an assembled Thermoplasma complex
of 50S.
DLS is better able to deal with the polydispersity in
the sample of assembled complex. Analysis of the auto-
correlation curve using the volume distribution algo-
rithm gives an Mr of 5.0 (± 0.2) · 106 for the complex
at 55 �C, in close agreement with that estimated by sed-
imentation velocity experiments described above.
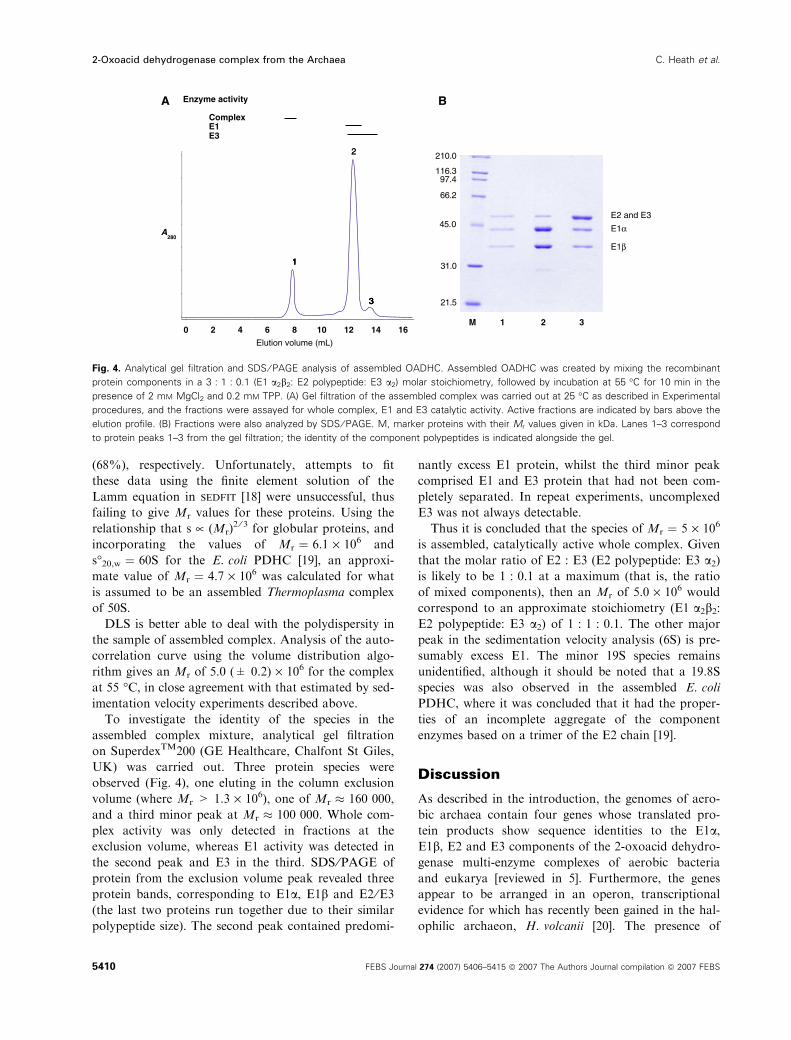
To investigate the identity of the species in the
assembled complex mixture, analytical gel filtration
on SuperdexTM200 (GE Healthcare, Chalfont St Giles,
UK) was carried out. Three protein species were
observed (Fig. 4), one eluting in the column exclusion
volume (where Mr > 1.3 · 106), one of Mr � 160 000,
and a third minor peak at Mr � 100 000. Whole com-
plex activity was only detected in fractions at the
exclusion volume, whereas E1 activity was detected in
the second peak and E3 in the third. SDS ⁄PAGE of
protein from the exclusion volume peak revealed three
protein bands, corresponding to E1a, E1b and E2 ⁄E3(the last two proteins run together due to their similar
polypeptide size). The second peak contained predomi-
nantly excess E1 protein, whilst the third minor peak
comprised E1 and E3 protein that had not been com-
pletely separated. In repeat experiments, uncomplexed
E3 was not always detectable.
Thus it is concluded that the species of Mr ¼ 5 · 106
is assembled, catalytically active whole complex. Given
that the molar ratio of E2 : E3 (E2 polypeptide: E3 a2)
is likely to be 1 : 0.1 at a maximum (that is, the ratio
of mixed components), then an Mr of 5.0 · 106 would
correspond to an approximate stoichiometry (E1 a2b2:E2 polypeptide: E3 a2) of 1 : 1 : 0.1. The other major
peak in the sedimentation velocity analysis (6S) is pre-
sumably excess E1. The minor 19S species remains
unidentified, although it should be noted that a 19.8S
species was also observed in the assembled E. coli
PDHC, where it was concluded that it had the proper-
ties of an incomplete aggregate of the component
enzymes based on a trimer of the E2 chain [19].
Discussion
As described in the introduction, the genomes of aero-
bic archaea contain four genes whose translated pro-
tein products show sequence identities to the E1a,E1b, E2 and E3 components of the 2-oxoacid dehydro-
genase multi-enzyme complexes of aerobic bacteria
and eukarya [reviewed in 5]. Furthermore, the genes
appear to be arranged in an operon, transcriptional
evidence for which has recently been gained in the hal-
ophilic archaeon, H. volcanii [20]. The presence of
0 2 4 6 8 10 12 14 16
Elution volume (mL)
1
3
1
3
1
2
3
1
2
3
Enzyme activityA BComplex E1 E3
45.0
31.0
21.5
66.2
97.4116.3
210.0
M 1 2 3
E2 and E3
E1α
E1β
A280
Fig. 4. Analytical gel filtration and SDS ⁄ PAGE analysis of assembled OADHC. Assembled OADHC was created by mixing the recombinant
protein components in a 3 : 1 : 0.1 (E1 a2b2: E2 polypeptide: E3 a2) molar stoichiometry, followed by incubation at 55 �C for 10 min in the
presence of 2 mM MgCl2 and 0.2 mM TPP. (A) Gel filtration of the assembled complex was carried out at 25 �C as described in Experimental
procedures, and the fractions were assayed for whole complex, E1 and E3 catalytic activity. Active fractions are indicated by bars above the
elution profile. (B) Fractions were also analyzed by SDS ⁄ PAGE. M, marker proteins with their Mr values given in kDa. Lanes 1–3 correspond
to protein peaks 1–3 from the gel filtration; the identity of the component polypeptides is indicated alongside the gel.
2-Oxoacid dehydrogenase complex from the Archaea C. Heath et al.
5410 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS

these genes is unexpected, as all the archaea possess
ferredoxin oxidoreductases that catalyze the equivalent
oxidation of the same 2-oxoacids as those used by the
bacterial and eukaryal dehydrogenase complexes;
moreover, activity of the 2-oxoacid dehydrogenase
complexes has never been detected in any archaeon.
The obvious question raised by these observations is
whether or not the archaeal ‘OADHC’ genes actually
encode functional proteins that assemble into a multi-
enzyme complex. The E1 component defines the sub-
strate specificity of the whole complex, and we have
previously reported heterologous expression of the
a2b2 E1 enzyme from T. acidophilum and shown it to
be catalytically active with the branched-chain 2-oxo-
acids and pyruvate [14]. In the current paper, the E2
and E3 genes have also been successfully cloned and
expressed as soluble proteins in E. coli, and their cata-
lytic activities have been demonstrated.
Generation of an active E2 enzyme requires lipoyla-
tion of a specific lysine residue. In E. coli the lipoyla-
tion of its own E2 components can occur via two
routes [21,22]. The endogenous pathway involves the
covalent attachment of a C-8 intermediate of fatty acid
biosynthesis to the target lysine on E2 by enzyme LipB;
subsequently, LipA catalyzes the incorporation of sul-
fur atoms to generate the lipoic acid moiety. However,
if lipoic acid is supplied in the growth medium, E. coli
preferentially uses its exogenous pathway, which
employs lipoate protein ligase A; this enzyme catalyzes
the adenylation of lipoic acid after uptake into the cell
and its subsequent transfer to the E2 lysine residue.
Knowing that the lipoylation process can take place
across the species barrier, albeit with varying efficien-
cies [2, and references therein], lipoylation of recombi-
nantly expressed Thermoplasma E2 was tested and
optimized. Up to 50% lipoylation was achieved when
the rate and level of expression in E. coli was slowed
down by decreasing the growth temperature and avoid-
ing induction, whilst at the same time supplementing
the growth medium with lipoic acid.
Clearly, therefore, the E. coli machinery is able to rec-
ognize the lipoyl-domain of the Thermoplasma E2
enzyme, the target lysine of which is flanked by D and V
residues, as it is in the E2 protein of the E. coli OGDHC
and of the Bacillus subtilis BCOADHC (Fig. 1). How-
ever, in addition to the identity of the neighbouring
residues, it is the exact positioning of the lysine in the
lipoyl domain that is fundamental to target lysine recog-
nition [23], implying that the fold of the Thermoplasma
enzyme has been conserved in this region.
Whilst the Thermoplasma recombinant E2 has not
been assayed for catalytic activity in isolation, assem-
bly of the whole active complex from its individually
expressed components shows that it is indeed a func-
tional enzyme. This assembly process, studied by both
analytical ultracentrifugation and DLS, has been dem-
onstrated to involve the formation of a 24-mer E2
core, which binds E1 and E3 components to give a
complex that has the same substrate specificity as that
determined for the isolated E1 enzyme; namely, it is a
branched-chain 2-oxoacid dehydrogenase complex that
is also active with pyruvate. An E2 core that comprises
an assembly of 24 polypeptide chains is consistent with
other branched-chain 2-oxoacid multi-enzyme com-
plexes from bacteria and eukarya [24,25], some of
which also have activity with pyruvate. Additionally,
the E1 subunit in those branched-chain complexes is
also an a2b2 oligomer [24,25]. What is particularly
interesting is that the assembly of the E2 core in the
Thermoplasma enzyme is temperature dependent, incu-
bation to at least 40 �C (the temperature of the ultra-
centrifugation) being required.
The data in Fig. 2 show that overall complex activ-
ity increased linearly with the ratio of E1 : E2 mixed
together, as was found with the E. coli PDHC for
example [26], until maximal activity was achieved at a
mixing ratio of 2 : 1. However, both ultracentrifuga-
tion and DLS estimate the Mr of the assembled com-
plex to be around 5 · 106, closely fitting the tentative
conclusion of an E1(a2b2) ⁄E2 ⁄E3(a2) stoichiometry of
1 : 1 : 0.1. These experiments indicate that not all the
E1 molecules may be tightly bound to the E2 core
and ⁄or that assembly might be substrate enhanced.
Whilst uncertainties remain over the exact subunit
composition of the assembled complex, the important
conclusion from our data is that the four OADHC
ORFs in the archaeon T. acidophilum encode the com-
ponents of a functional 2-oxoacid dehydrogenase
multi-enzyme complex, the first to be identified in this
domain of life. Thus, OADHCs were probably present
in the common ancestor to the Bacteria and Archaea,
and have been retained in aerobic members of each
domain. The Thermoplasma enzyme possesses catalytic
activity with branched-chain 2-oxo acids and pyruvate,
but it remains to be established whether other archaeal
OADHCs have the same or different substrate specific-
ities. However, whatever specificity is found, the physi-
ological role of these OADHCs in the archaea remains
a mystery given the presence of active 2-oxoacid FORs
that catalyze the equivalent chemical reactions.
In studies of the OADHC from the halophilic archa-
eon H. volcanii, we could find no growth substrate that
would induce the expression of OADHC activity [20],
nor was any physiological defect apparent in this
organism when the E3 gene was inactivated by inser-
tional mutagenesis [27]. Interestingly, Wanner & Soppa
C. Heath et al. 2-Oxoacid dehydrogenase complex from the Archaea
FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS 5411

[28] have found an additional gene cluster in H. volca-
nii comprising three genes that would appear to code
for OADHC E1a and E1b subunits, and an unat-
tached lipoyl domain; however, no genes for a com-
plete E2 or an E3 were present. Evidence for a
function during nitrate-respirative growth on Casami-
no acids was presented, although the metabolic sub-
strate could not be identified.
In conclusion, with respect to the archaeal four-gene
OADHC cluster that we have studied in this paper, it
is highly unlikely that the genes would have been
retained in a highly sophisticated and functional state
without a physiological role. We suggest that proteo-
mic studies on this thermoacidophile need to be insti-
tuted to reveal this role.
Experimental procedures
Materials
Bacteriological media were purchased from Sigma-Aldrich
(Poole, UK) or Fisher Scientific (Loughborough, UK).
Expression vector pET28a, E. coli expression strain
BL21(DE3), BugBuster Protein Extraction Reagent and
Benzonase� nuclease were purchased from Novagen-Merck
(Nottingham, UK). E. coli JM109 cells, pGEM-T vector,
restriction endonucleases, T4 DNA ligase and Taq polymer-
ase were purchased from Promega (Southampton, UK).
Vent DNA polymerase was from New England Biolabs
(Hitchin, UK). Lipoic acid, phenylmethanesulfonyl fluoride
and antibiotics were purchased from Sigma-Aldrich.
SDS ⁄PAGE molecular mass markers were from Bio-Rad
(Hemel Hempstead, UK).
Plasmids pET19b-E1a and pET28a-E1b, which, respec-
tively, coexpress the a and b subunits of the T. acidophilum
E1, have been described previously [14].
Bioinformatics
The putative OADHC operon was identified in the T. aci-
dophilum DSM1728 genome from the ENTREZ Nucleotide
database (http://www.3.ncbi.nlm.nih.gov). E1a: Ta1438;
E1b: Ta1437; E2: Ta1436; and E3: Ta1435.
Recombinant DNA techniques
E2 and E3 gene amplification
Preparation of genomic DNA from T. acidophilum strain
DSM 1728 has been described previously [29]. The E2 and
E3 genes were PCR-amplified from this genomic
DNA using primers that engineered restriction sites into the
5¢- and 3¢ ends of the gene products: NdeI and XhoI for the
E2 gene, and NheI and EcoRI for E3. Oligonucleotides
were as follows (restriction sequences are underlined):
E2 forward: CGCCATATGTACGAATTCAAACTGC
CAGACATAGG
E2 reverse: CCGCTCGAGTCAGATCTCGTAGAT
TATAGCGTTCGG
E3 forward: CTACGAGAGCTAGCATGTACGATGC
AATAATAATAGGTTC
E3 reverse: TTTAAAAATGGAATTCAATGAGAT
GGT.
PCR amplification was carried out using Vent polymer-
ase, and A-tails were added to the products with Taq poly-
merase. Both genes were then separately cloned into the
intermediate vector pGEM-T using T4 DNA ligase, and
the clones were amplified in E. coli strain JM109 grown in
Luria–Bertani LB media [1% (w ⁄ v) tryptone, 1% (w ⁄ v)NaCl, 0.5% (w ⁄ v) yeast extract] supplemented with carben-
icillin (50 lgÆmL)1). Plasmids were extracted using the BD
Biosciences (Palo Alto, CA) NucleoSpin Plasmid kit, and
the genes were sequenced for fidelity. The E2 and E3 genes
were then excised from pGEM-T, using the appropriate
restriction endonucleases, purified by electrophoresis in a
0.8% (w ⁄ v) agarose gel, and extracted from the gel using
the Qiagen (Hilden, Germany) Qiaex II Gel Extraction kit.
Construction of expression vectors pET28a-E2 and
pET28a-E3
Expression vector pET28a was prepared for recombinant
ligation by NdeI ⁄XhoI restriction endonuclease digestion for
E2, and NheI ⁄EcoRI digestion for E3, and purified by gel
electrophoresis and gel extraction as already described. E2
and E3 genes were separately ligated into the vectors using
T4 DNA ligase, generating the recombinant expression vec-
tors pET28a-E2 and pET28a-E3. This pET28a vector thus
introduced a 20- and a 23-amino acid sequence at the N-ter-
mini of the E2 and E3 recombinant protein products, respec-
tively, with each containing a 6-histidine tag sequence.
Expression and purification of OADHC
components
E2 expression and purification
Two different methods of expression were used. Several col-
onies of E. coli strain BL21(DE3), freshly transformed with
plasmid pET28a-E2, were picked from LB agar plates and
used to inoculate 2 L of LB medium supplemented with
kanamycin (30 lgÆmL)1) and 0.2 mm dl-lipoic acid. Incu-
bation was at 30 �C for 20 h in darkness, with no induction
by IPTG, after which cells were harvested by centrifugation
at 6000 g. Alternatively, an overnight culture (20 mL,
A600 0.6) of freshly transformed E. coli BL21(DE3)pLysS
cells was used to inoculate 1 L of LB medium supple-
mented with kanamycin (30 lgÆmL)1). After induction with
IPTG (at A600 0.6) and subsequent overnight incubation at
37 �C, cells were harvested as described before. The former
2-Oxoacid dehydrogenase complex from the Archaea C. Heath et al.
5412 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS

method allows production of recombinant E2 of which up
to 50% is lipoylated, whereas in the latter method only 5%
of the E2 is lipoylated.
Purification of the recombinant E2 was carried out at
25 �C, unless otherwise stated. Samples were analyzed by
SDS ⁄PAGE on a 10% (w ⁄ v) polyacrylamide gel at each
step of the purification, and protein concentrations were
determined from A280 values. Frozen cells were disrupted
by resuspension in BugBuster (5 mLÆg)1 wet cells) supple-
mented with Benzonase nuclease (1 lLÆmL)1), incubated on
ice with gentle agitation for 30 min, and centrifuged at
16 000 g for 20 min at 4 �C to pellet cell debris. The solu-
ble cell extract was subjected to His Bind� Resin chroma-
tography, and fractions containing E2 protein were pooled
and dialyzed overnight into 20 mm Tris ⁄HCl buffer,
pH 9.0, 10% (v ⁄ v) glycerol. The protein was then subjected
to anion exchange chromatography on an Amersham Bio-
sciences (Chalfont St Giles, UK) Akta FPLC system, using
a 5 mL Q-Sepharose Hi-Trap column equilibrated with
50 mm Tris ⁄HCl buffer, pH 8.5. Protein was eluted over a
0–0.8 m gradient of NaCl in the same buffer, at a flow rate
of 1 mLÆmin)1 over 60 min. Fractions containing E2 were
stored at 4 �C in the elution buffer supplemented with
1 mm phenylmethanesulfonyl fluoride.
E3 expression and purification
For expression of the E3 enzyme, a 20 mL overnight cul-
ture of transformed BL21(DE3) (A600 0.6) was used to
inoculate 1 L of LB medium supplemented with kanamycin
(30 lgÆmL)1). Cells were incubated at 37 �C until 5 h after
the A600 had reached 0.6 (with no induction by IPTG), and
were then collected by centrifugation. Cell disruption was
carried out as described for the purification of recombinant
E2. The soluble cell extract was subjected to heat precipita-
tion at 65 �C for 5 min, and precipitated material removed
by centrifugation at 16 000 g for 20 min at 4 �C. E3 in the
remaining soluble fraction was purified by His-Bind Resin
chromatography, dialyzed overnight into 20 mm Tris ⁄HCl
buffer, pH 8.4, and then stored at 4 �C.
Assembly of the OADHC multi-enzyme complex
OADHC was assembled in vitro by mixing together recom-
binant E1a, E1b, E2 and E3 proteins at 55 �C for 0–1 h.
Molar ratios to enzyme E2 varied from 0.5 to 6.0 (E1a2b2)
and 0.01–1.0 (E3a2). Each assembled complex was assayed
for overall complex activity as described below.
SDS-PAGE
Analysis of protein purity and determination of polypeptide
Mr values were carried out by SDS ⁄PAGE in a resolving
gel containing 10% (w ⁄ v) acrylamide [30].
Enzyme assays
E1 enzymic activity was assayed spectrophotometrically by
following the 2-oxoacid-dependent reduction of 2,6-dichlo-
rophenolindophenol (DCPIP) at 595 nm [31]. Assays were
carried out at 55 �C in 20 mm potassium phosphate
(pH 7.0), 2 mm MgCl2 and 0.2 mm TPP. Buffer and recom-
binant E1a2b2 enzyme were pre-incubated at 55 �C for
10 min; 50 lm DCPIP was then added and the assay
started by the addition of the 2-oxoacid substrate (pyru-
vate, 2-oxoglutarate, 4-methyl-2-oxopentanoate, 3-methyl-2-
oxopentanoate or 3-methyl-2-oxobutanoate).
E3 was assayed at 55 �C in 50 mm EPPS buffer (pH 8.0)
containing 0.4 mm dihydrolipoamide and 1 mm NAD+.
The reaction, in a final volume of 1 mL, was started by the
addition of enzyme, and activity was monitored by measur-
ing the production of NADH at 340 nm.
Overall complex activity was assayed at 55 �C in 50 mm
potassium phosphate buffer (pH 7.0) containing 2.5 mm
NAD+, 1 mm MgCl2, 0.2 mm TPP, 0.13 mm CoASH and
2.6 mm cysteine-HCl [32]. Buffer and assembled enzyme
complex were pre-incubated at 55 �C for 10 min to allow
binding of TPP to E1. The assay, in a final volume of
1 mL, was started by the addition of the 2-oxoacid substrate
as for the assay of E1, and OADHC activity was monitored
by measuring the production of NADH at 340 nm.
Kinetic parameters were determined by the direct linear
method of Eisenthal & Cornish-Bowden [33].
Gel filtration
Analytical gel filtration was carried out at 25 �C on the
Amersham Biosciences Akta FPLC system, using a
Superdex 200 10 ⁄ 300 GL column. Protein standards
were: b-amylase (Mr ¼ 200 000), alcohol dehydrogenase
(150 000), BSA (66 000), carbonic anhydrase (29 000) and
cytochrome c (12 400). For analysis of E3, the column was
equilibrated with 20 mm sodium phosphate (pH 7.0), 0.1 m
NaCl and 10% (v ⁄ v) glycerol; peak fractions were assayed
for E3 enzymic activity. Analysis of assembled complex was
carried out in 20 mm sodium phosphate (pH 7.0), 2 mm
MgCl2, 0.1 m NaCl and 10% (v ⁄ v) glycerol. Peak fractions
were assayed for E1, E3 and OADHC activity.
Analytical ultracentrifugation
All analytical ultracentrifugation experiments were carried
out on a Beckman XL-A analytical ultracentrifuge (Beck-
man-Coulter, CA). Sedimentation velocity experiments were
carried out at 15 000 r.p.m., and cells were scanned every
5 min at 280 nm. For sedimentation of E2, the buffer was
20 mm Tris ⁄HCl (pH 8.5), 0.4 m NaCl and 1 mm phen-
ylmethanesulfonyl fluoride; for whole complex, the buffer
was the same as that used in the analytical gel filtration
C. Heath et al. 2-Oxoacid dehydrogenase complex from the Archaea
FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS 5413

analysis, 20 mm sodium phosphate (pH 7.0), 2 mm MgCl2,
100 mm NaCl and 10% (v ⁄ v) glycerol. The set temperature
on the centrifuge was 40 �C, and the solution densities were
directly measured at this temperature using an Anton-Paar
DMA 5000 high-precision density-meter. Sedimentation
velocity distributions were obtained by the c(s) method [34]
using the program sedfit. Data were then directly fitted
using the finite element solution of the Lamm equation in
sedfit [18] to give values of the sedimentation coefficient
and of Mr.
Dynamic light scattering
All DLS measurements were performed using a Zetasizer
Nano S from Malvern Instruments Ltd. (Malvern, UK).
Prior to DLS measurements, protein solutions
(1 mgÆmL)1) in 20 mm sodium phosphate buffer, pH 7.5,
10% glycerol (v ⁄ v) and 0.1 m NaCl were filtered through
a 0.02 lm membrane filter (Whatman� Anotop 10, Fisher
Scientific, Loughborough, UK) to remove dust particles.
However, it was found necessary to filter enzyme E3
through a 0.22 lm membrane filter (Millipore, Watford,
UK). DLS measurements were carried out at 25 �C or
55 �C. Mr values were derived from the measured hydro-
dynamic radii using the Protein Utilities feature of the
dispersion technology software, version 4.10, supplied
with the instrument.
Mass spectrometry
Determination of the E2 polypeptide mass
A 100 pmol sample of recombinant E2 was injected on to
a MassPrep on-line desalting cartridge (2.1 · 10 mm)
(Waters, Milford, MA), eluted with an increasing acetoni-
trile concentration [2 vol acetonitrile + 98 vol aqueous for-
mic acid (1%, v ⁄ v) to 98 vol acetonitrile + 2 vol aqueous
formic acid (1%, v ⁄ v)] and delivered to an electrospray ion-
ization mass spectrometer (LCT, Micromass, Manchester,
UK) that had previously been calibrated using myoglobin.
An envelope of multiply charged signals was obtained and
deconvoluted using maxent1 software to give the molecular
mass of the protein.
Mapping the lipoylation of E2
Recombinant E2 (50 pmol) was dialyzed against 50 mm
ammonium bicarbonate on a VS membrane disc (Millipore)
for 30 min. Sequencing grade, modified porcine trypsin
(Promega) (60 ng) was added and the sample incubated at
37 �C for 16 h. A portion of the sample was diluted in 5%
(v ⁄ v) formic acid and the peptides separated using an Ulti-
Mate nanoLC (LC Packings, Amsterdam, the Netherlands)
equipped with a PepMap C18 trap and column. The eluant
was sprayed into a Q-Star Pulsar XL tandem mass spec-
trometer (Applied Biosystems, Foster City, CA) and ana-
lyzed in Information Dependent Acquisition mode. The
MS ⁄MS data generated were analyzed using the Mascot
search engine (Matrix Science, London, UK), with lipoyla-
tion selected as a possible lysine modification. The MS ⁄MS
spectrum corresponding to the modified peptide was also
interpreted ‘manually’ using BioAnalyst (Applied Biosys-
tems) tools.
Acknowledgements
MJD and DWH thank the US Air Force Office of Sci-
entific Research (Arlington, VA, USA) for generous
financial support. CH and MGP gratefully acknowl-
edge the receipt of Postgraduate Studentships from the
UK Biotechnology and Biological Sciences Research
Council and from the University of Bath, respectively.
We thank Dr Jean van den Elsen (University of Bath,
UK) for allowing us to use the DLS Zetasizer Nano S,
and Dr Catherine Botting, BMS Mass Spectrometry
and Proteomics Facility, University of St Andrews,
UK, for carrying out the MS analyses.
References
1 Perham RN (1991) Domains, motifs, and linkers in
2-oxo acid dehydrogenase multienzyme complexes – a
paradigm in the design of a multifunctional protein.
Biochemistry 30, 8501–8512.
2 Perham RN (2000) Swinging arms and swinging
domains in multifunctional enzymes: catalytic machines
for multistep reactions. Ann Rev Biochem 69, 961–1004.
3 Perham RN, Jones DD, Chauhan HJ & Howard MJ
(2002) Substrate channelling in 2-oxo acid dehydrogenase
multienzyme complexes. Biochem Soc Trans 30, 47–51.
4 Izard T, Ævarsson A, Allen MD, Westphal AH, Per-
ham RN, de Kok A & Hol WGJ (1999) Principles of
quasi-equivalence and Euclidean geometry govern the
assembly of cubic and dodecahedral cores of pyruvate
dehydrogenase complexes. Proc Natl Acad Sci USA 96,
1240–1245.
5 Danson MJ, Lamble HJ & Hough DW (2007) Central
metabolism. In Archaea: Molecular and Cell Biology
(Cavicchioli R, ed.). Chapter 12, pp. 260–287. ASM
Press, Washington, DC.
6 Kerscher L & Oesterhelt D (1982) Pyruvate – ferredoxin
oxidoreductase: new findings on an ancient enzyme.
Trends Biochem Sci 7, 371–374.
7 Schut GJ, Menon AL & Adams MWW (2001) 2-Keto
acid oxidoreductases from Pyrococcus furiosus and
Thermococcus litoralis. Methods Enzymol 331, 144–158.
8 Ragsdale SW (2003) Pyruvate ferredoxin oxidoreductase
and its radical intermediate. Chem Rev 103, 2333–2346.
2-Oxoacid dehydrogenase complex from the Archaea C. Heath et al.
5414 FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS

9 Plaga W, Lottspeich F & Oesterhelt D (1992) Improved
purification, crystallization and primary structure of
pyruvate: ferredoxin oxidoreductase from Halobacterium
halobium. Eur J Biochem 205, 391–397.
10 Danson MJ, Eisenthal R, Hall S, Kessell SR &
Williams DL (1984) Dihydrolipoamide dehydrogenase
from halophilic archaebacteria. Biochem J 218, 811–818.
11 Smith LD, Bungard SJ, Danson MJ & Hough DW
(1987) Dihydrolipoamide dehydrogenase from the ther-
moacidophilic archaebacterium Thermoplasma acidophi-
lum. Biochem Soc Trans 15, 1097–1097.
12 Pratt KJ, Carles C, Carne TJ, Danson MJ & Stevenson
KJ (1989) Detection of bacterial lipoic acid: a modified
gas chromatographic – mass spectrometric procedure.
Biochem J 258, 749–754.
13 Jolley KA, Maddocks DG, Gyles SL, Mullan Z,
Tang S-L, Dyall-Smith ML, Hough DW & Danson MJ
(2000) 2-Oxoacid dehydrogenase multienzyme complexes
in the halophilic Archaea? Gene sequences and protein
structural predictions. Microbiology 146, 1061–1069.
14 Heath C, Jeffries AC, Hough DW & Danson MJ (2004)
Discovery of the catalytic function of a putative 2-oxo-
acid dehydrogenase multienzyme complex in the ther-
mophilic archaeon Thermoplasma acidophilum. FEBS
Lett 577, 523–527.
15 Massey V (1960) Identity of diaphorase and lipoyl dehy-
drogenase. Biochim Biophys Acta 37, 314–322.
16 Mann S, Melero CP, Hawksley D & Leeper FJ (2004)
Inhibition of thiamin diphosphate dependent enzymes
by 3-deazathiamin diphosphate. Org Biomol Chem 2,
1732–1741.
17 Chauhan HJ, Domingo GJ, Jung HI & Perham RN
(2000) Sites of limited proteolysis in the pyruvate decar-
boxylase component of the pyruvate dehydrogenase
multienzyme complex of Bacillus stearothermophilus and
their role in catalysis. Eur J Biochem 267, 7158–7169.
18 Schuck P (1998) Sedimentation analysis of non-interact-
ing and self-associating solutes using numerical solu-
tions to the Lamm equation. Biophys J 75, 1503–1512.
19 Danson MJ, Hale G, Johnson P, Perham RN, Smith J
& Spragg P (1979) Molecular weight and symmetry of
the pyruvate dehydrogenase multienzyme complex of
Escherichia coli. J Mol Biol 129, 603–617.
20 Al-Mailem DM, Hough DW & Danson MJ (2007) The
2-oxoacid dehydrogenase complex from Haloferax vol-
canii. Extremophiles, doi: 10.1007/s00792-007-0091-0.
21 Miller JR, Busby RW, Jordan SW, Cheek J,
Henshaw TF, Ashley GW, Broderick JB, Cronan JE &
Marletta MA (2000) Escherichia coli LipA is a lipoyl
synthase: in vitro biosynthesis of lipoylated pyruvate
dehydrogenase complex from octanoyl-acyl carrier
protein. Biochemistry 39, 15166–15178.
22 Morris T, Reed K & Cronan J Jr (1995) Lipoic acid
metabolism in Escherichia coli: the lplA and lipB genes
define redundant pathways for ligation of lipoyl groups
to apoprotein. J Bacteriol 177, 1–10.
23 Wallis NG & Perham RN (1994) Structural dependence
of post-translational modification and reductive acetyla-
tion of the lipoyl domain of the pyruvate dehydrogenase
multienzyme complex. J Mol Biol 236, 209–216.
24 Ævarsson A, Seger K, Turley S, Sokatch JR & Hol WGJ
(1999) Crystal structure of 2-oxoisovalerate dehydroge-
nase and the architecture of 2-oxo acid dehydrogenase
multienzyme complexes. Nature Struct Biol 6, 785–792.
25 Pettit FH, Yeaman SJ & Reed LJ (1978) Purification
and characterisation of branched-chain a-ketoacid dehy-
drogenase complex of bovine kidney. Proc Natl Acad
Sci USA 75, 4881–4885.
26 Bates DL, Danson MJ, Hale G, Hooper EA &
Perham RN (1977) Self-assembly and catalytic activity
of the pyruvate dehydrogenase multienzyme complex of
Escherichia coli. Nature 268, 313–316.
27 Jolley KA, Rapaport E, Hough DW, Danson MJ,
Woods WG, Dyal I & Smith ML (1996) Dihydrolipoa-
mide dehydrogenase from the halophilic archaeon Halo-
ferax volcanii: homologous overexpression of the cloned
gene. J Bacteriol 178, 3044–3048.
28 Wanner C & Soppa J (2002) Functional role for a
2-oxo acid dehydrogenase in the halophilic archaeon
Haloferax volcanii. J Bacteriol 184, 3114–3121.
29 Sambrook J & Russell DW (2001) Molecular Cloning: A
Laboratory Manual, 3rd edn. Cold Spring. Harbour
Laboratory Press, Cold Spring Harbour, NY.
30 Laemmli UK (1970) Cleavage of structural proteins dur-
ing the assembly of the head of bacteriophage T4. Nat-
ure 227, 680–685.
31 Lessard IAD & Perham RN (1994) Expression in Esc-
herichia coli of genes encoding the E1a and E1b subun-
its of the pyruvate-dehydrogenase complex of Bacillus
stearothermophilus and assembly of a functional E1
Component (a2b2) in-vitro. J Biol Chem 269, 10378–
10383.
32 Domingo GJ, Chauhan HJ, Lessard IAD, Fuller C &
Perham RN (1999) Self-assembly and catalytic activity
of the pyruvate dehydrogenase multienzyme complex
from Bacillus stearothermophilus. Eur J Biochem 266,
1136–1146.
33 Eisenthal R & Cornish-Bowden A (1974) The direct
linear plot. A new graphical procedure for estimating
enzyme kinetic parameters. Biochem J 139, 715–720.
34 Schuck P (2000) Size distribution analysis of macromole-
cules by sedimentation velocity ultracentrifugation and
Lamm equation modeling. Biophys J 78, 1606–1619.
C. Heath et al. 2-Oxoacid dehydrogenase complex from the Archaea
FEBS Journal 274 (2007) 5406–5415 ª 2007 The Authors Journal compilation ª 2007 FEBS 5415


















