
TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE, … · ii TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE,...
83
TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE, ATTENUATES BEHAVIOUR DEFICITS FOLLOWING TRAUMATIC BRAIN INJURY IN RATS by Wen-Jia Zhang A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Physiology University of Toronto © Copyright by Wen-Jia Zhang (2011)
Transcript of TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE, … · ii TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE,...

TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE, ATTENUATES BEHAVIOUR DEFICITS FOLLOWING
TRAUMATIC BRAIN INJURY IN RATS
by
Wen-Jia Zhang
A thesis submitted in conformity with the requirements for the degree of Master of Science
Department of Physiology University of Toronto
© Copyright by Wen-Jia Zhang (2011)

ii
TAT-9C, A TAT-FUSION CYSTEINE-RICH PEPTIDE,
ATTENUATES BEHAVIOUR DEFICITS FOLLOWING
TRAUMATIC BRAIN INJURY IN RATS
Wen-Jia Zhang
Master of Science
Department of Physiology
University of Toronto
2011
Abstract
Peroxynitrite, a highly oxidative molecule, plays a role in neuronal cell death following traumatic
brain injury (TBI). A peptide comprised of the HIV-1 tat transduction domain fused to nine
cysteine residues (Tat-9c) was previously designed to act as an exogenous target for nitrosylation
by peroxynitrite. The present study’s aim was to explore the efficacy of Tat-9c in maintaining
neurological function following TBI. Rats treated with Tat-9c exhibited significant improvement
in performance compared to controls 24 hrs following TBI in the Beam-Walk task but not in the
Rota-Rod task. Injured animals, given the drug, show a recovery as indicated by similar
performance on the Morris Water Maze task compared to sham controls. These findings suggest
Tat-9c may constitute a potential therapy for improving motor and cognitive function following
TBI.

iii
Acknowledgments
This thesis would not have been possible without the assistance of a few individuals who
believed in me and kept me going along this adventure to the very end. Firstly, I would like to
thank Dr. Michael Tymianski for giving me this opportunity to experience and contribute to this
exiting field in neuroscience. It was also a pleasure to work alongside other esteemed scientists
and scientists-in-training in Dr. Tymianski’s lab: Ishraq Alim, Andrew Barszczyk, Douglas J
Cook, Hong Cui, Zhanxin Ji, Rongwen Li, Hong Sun, Xuijun Sun, Kinga Szydlowska, and Lucy
Teves. I would also like to thank the members of my supervisory committee, Dr. Andrew Baker,
Dr. James Eubanks, and Dr. Martin Wojtowicz for their advice and constructive criticism. For
the unconditional love and limitless hearty meals, I would like to thank my family for their
encouragements, including my vociferous parrot for keeping me awake. I am also grateful to my
friends for making sure I still had a life outside the laboratory. Finally, I owe my deepest
gratitude to Jeff Dason for his generous assistance and friendship over the years, to Rene Persaud
for destroying me on my practice dissertation so I didn’t feel as bad during the real one, and
especially to Dr. Martin Wojtowicz who has provided me with superb mentorship and excellent
teaching all the way since my naïve undergraduate years – I would not be where I am today
without the exceptional patience, support, and guidance from these extraordinary people.

iv
Table of Contents
ABSTRACT ................................................................................................................................... ii
ACKNOWLEDGMENTS ........................................................................................................... iii
TABLE OF CONTENTS ............................................................................................................ iv
LIST OF TABLES ...................................................................................................................... vii
LIST OF FIGURES ................................................................................................................... viii
LIST OF ABBREVIATIONS ..................................................................................................... ix
INTRODUCTION ......................................................................................................................... 1
1.1 Traumatic Brain Injury ......................................................................................................... 1
1.1.1 Epidemiology of Traumatic Brain Injury .................................................................. 1
1.1.2 Definition and Severity Index of Traumatic Brain Injury ......................................... 2
1.1.3 Neuropathological Classification of TBI .......................................................................... 2
1.1.4 Biomechanical Classification of TBI ......................................................................... 3
1.2 Pathophysiology of Traumatic Brain Injury ........................................................................ 4
1.2.1 Cerebral Blood Flow .................................................................................................. 4
1.2.2 Neuroinflammation .................................................................................................... 4
1.2.3 Excitotoxicity and Oxidative Stress ........................................................................... 5
1.2.3.1 Glutamate Excitotoxicity ................................................................................. 5
1.2.3.2 NMDAR-Mediated Glutamate Excitotoxicity ................................................. 6
1.2.4 Protease-Mediated Cell Death ................................................................................... 9
1.2.5 Cell Death Pathways of Secondary Injury in Summary ............................................ 9
1.3 INJURY MODELS OF TRAUMATIC BRAIN INJURY................................................. 11
1.3.1 Weight Drop Model ................................................................................................. 11
1.3.2 Cortical Control Impact (CCI) Model ...................................................................... 13

v
1.3.3 Fluid Percussion Injury (FPI) Model ....................................................................... 15
1.4 Current Research for Treatment of Traumatic Brain Injury .............................................. 17
1.4.1 Management of TBI ................................................................................................. 17
1.4.2 Neuroprotection Drugs for TBI ............................................................................... 17
1.4.3 Neuroprotection Through the Scavenging of Radical Species ................................ 20
1.5 Design of Tat-9c and Tat-9a .............................................................................................. 21
1.6 Neurobehaviour Models for Assessing TBI Severity ........................................................ 22
1.6.1 Beam-Walk .............................................................................................................. 22
1.6.2 Rota-Rod .................................................................................................................. 23
1.6.3 Morris Water Maze .................................................................................................. 24
HYPOTHESIS AND GOALS .................................................................................................... 26
2.1 Research Goals ................................................................................................................... 26
2.2 Hypothesis I ....................................................................................................................... 27
2.3 Hypothesis II ...................................................................................................................... 27
2.4 Hypothesis III ..................................................................................................................... 27
MATERIALS AND METHODS ............................................................................................... 28
3.1 Lateral Fluid Percussion Injury Model .............................................................................. 28
3.1.1 Surgical Preparation ................................................................................................. 28
3.1.2 Drug Injection .......................................................................................................... 28
3.1.3 Fluid Percussion Injury ............................................................................................ 29
3.2 Treatment Preparation ........................................................................................................ 31
3.2.1 Preparation of Tat-9c for Animal Injection ............................................................. 31
3.2.2 Preparation of Tat-9a for Animal Injection ............................................................. 31
3.2.3 Preparation of Saline for Animal Injection .............................................................. 31
3.2.4 Preparation of Sham Animals .................................................................................. 31
3.3 Implication of Various Behavioural Paradigms ................................................................. 32

vi
3.3.1 Beam-Walk Model ................................................................................................... 32
3.3.2 Rota-Rod Model ....................................................................................................... 32
3.3.3 Morris Water Maze Model ....................................................................................... 33
3.4 Statistical Analysis ............................................................................................................. 33
RESULTS .................................................................................................................................... 34
4.1 Validation of Behaviour Assays Utilized to Assess Severity of TBI................................. 34
4.1.1 Beam-Walk Model ................................................................................................... 34
4.1.2 Rota-Rod Model ....................................................................................................... 36
4.1.3 Morris Water Maze Model ....................................................................................... 38
4.2 Effects of Tat-9c on Motor Behaviour Function Following TBI ....................................... 43
4.3 Effects of Tat-9c on Memory Function Following TBI ..................................................... 47
DISCUSSION .............................................................................................................................. 50
5.1 Behaviour Assays Utilized to Assess Severity of TBI ....................................................... 50
5.2 Tat-9c Protects Motor Behaviour Function Following TBI ............................................... 51
5.3 Effects of Tat-9c on Memory Function Following TBI ..................................................... 52
5.4 Limitations of the Study ..................................................................................................... 52
5.4.1 Disadvantages of the FPI Model .............................................................................. 52
5.4.2 Limitations of Tat-9c ............................................................................................... 53
5.4.3 The Complex Nature of TBI .................................................................................... 53
5.4.3.1 Cell Death Pathway Targets ........................................................................... 53
5.4.3.2 Limitations and Variability Amongst Subjects .............................................. 53
5.5 Future Directions ................................................................................................................ 55
CONCLUSIONS ......................................................................................................................... 57
REFERENCES AND LINKS..................................................................................................... 58

vii
List of Tables
Table 1-1 Neuroprotective strategies for traumatic brain injury………………..…………..19

viii
List of Figures
Figure 1-1
Figure 1-2
Figure 1-3
Figure 1-4
Figure 1-5
Figure 1-6
Figure 1-7
Figure 1-8
Figure 3-1
Figure 3-2
Figure 4-1
Figure 4-2
Figure 4-3
Figure 4-4
Figure 4-5
Figure 4-6
Figure 4-7
Figure 4-8
Figure 4-9
Figure 4-10
Figure 4-11
NMDAR-mediated glutamate excitotoxicity
The major pathways associated with secondary injury following TBI
Representation of the weight drop model of TBI
Representation of the controlled cortical impact (CCI) model of TBI
Representation of the fluid percussion injury (FPI) model of TBI
Representation of the beam walk device
Representation of the rota-rod device
Representation of the Morris Water Maze (MWM) assay
Dorsal representation of a rat skull
Representation of the magnitude of injury
Beam-walk motor function is impaired after TBI
Rota-rod motor function is impaired after TBI
Learning ability was not affected following TBI
Swim speed was not affected following TBI
Total distance traveled was not affected following TBI
Spatial memory is impaired after TBI
Beam-walk motor deficits are reduced in Tat-9c treated animals
Number of falls is reduced in Tat-9c treated rats following TBI
No improvement in rota-rod performance by Tat-9c treated rats
Rate of learning in Tat-9c rats was no different to any other group
Effects of Tat-9c on spatial memory
8
10
12
14
16
22
23
25
29
30
35
37
39
40
41
42
44
45
46
48
49

ix
List of Abbreviations
AIF
AMPA
atm
BBB
CBF
CCI
CNS
CsA
DNA
EPO
FPI
HBO
HIV-1
MPTP
MWM
NAC
NMDA
NMDAR
nNOS
NO
ONOO-
PSD-95
TAT-9A
TAT-9C
TBI
apoptosis inducing factor
-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
standard atmospheric pressure
blood brain barrier
cerebral blood flow
controlled cortical impact
central nervous system
cyclosporin A
deoxyribonucleic acid
erythropoietin
fluid percussion injury
hyperbaric oxygen
human immunodeficiency virus – type 1
mitochondrial permeability transition pore
morris water maze
N-acetylcysteine
N-methyl-D-aspartate
NMDA receptor
neuronal nitric oxide synthase
nitric oxide
peroxynitrite
postsynaptic density protein of 95kDa
tat transduction domain fused to 9 alanine residues
tat transduction domain fused to 9 cysteine residues
traumatic brain injury

1
Chapter 1 INTRODUCTION
1.1 Traumatic Brain Injury
1.1.1 Epidemiology of Traumatic Brain Injury
Traumatic brain injury (TBI), also entitled the “Silent Epidemic,” presents severe health and
economic burden to the global community. It is known as the “Silent Epidemic” by doctors
because the uninformed public has been rattled by its devastating repercussions (Abelson-
Mitchell, 2008; Headway, 2001; The Brain Injury Association of Canada). According to the
World Health Organization, TBI will surpass many diseases and become the major cause of
death and disability worldwide by the year 2020 (Hyder et al., 2007). The Centers for Disease
Control and Prevention estimates that 1.4 million people sustain a TBI each year in the United
States alone and of those 1.4 million people, 50,000 individuals will die from their injuries and
another 235,000 will be hospitalized (Summers et al., 2009). In 2000, the Centers for Disease
Control and Prevention estimates that at least 5.3 million Americans are currently suffering from
long-term disabilities caused by TBI. As a result of TBIs, direct costs (medical costs,
rehabilitation, and treatment) and indirect costs (loss of wages and productivity) total an
estimated $60 billion in the United States in 2000. The leading causes of TBI are falls (28%),
motor vehicle-traffic crashes (20%), other accidental events (19%), and assaults (11%) while a
further 9% of all TBI cases do not have a known cause (Summers et al., 2009).
TBIs can also vary in severity ranging from mild to severe. A survey from 1995 showed that
most TBIs are classified as mild, 51%, with 21% as moderate, and 19% as severe. The rest were
unknown (Thurman et al., 1999). Overall, due to its tremendous prevalence in society, TBI
represents a major socioeconomic crisis in the world.

2
1.1.2 Definition and Severity Index of Traumatic Brain Injury
Traumatic brain injury can be characterized as a non-congenital insult to the brain from an
external mechanical force with possible permanent or temporary impairment of cognitive,
physical, and psychosocial functions (Dawodu, 2003). The severity of TBI is broken down into
three categories: mild, moderate, and severe.
A mild head injury is defined as a brief period of unconsciousness but can also have long-lasting
sequelae (Guerrero et al., 2000). People with mild head injury may suffer from memory deficits,
thought processing, and concentration (Abelson-Mitchell, 2006).
A moderate head injury is described as a loss of consciousness between 15 minutes and six hours
(Headway, 2001). The symptoms of moderate head injury include physical, psychological,
cognitive, and behavioural deficits (Abelson-Mitchell, 2006).
A severe head injury occurs when a person has been in a coma for at least six hours and
generally, the longer a patient stays in a coma, the poorer the outcome will be for physical and
cognitive function (Annoni et al., 1992).
1.1.3 Neuropathological Classification of TBI
The highly destructive characteristics of TBI occur at two distinct levels. First of all, there is an
immediate CNS tissue disruption categorized as primary injury. Primary injury is a result of the
initial mechanical impact where shearing, compression, and stretching of the brain results in
rapid neuronal death of the affected area (Nolan, 2005; Wong et al., 2005). Following primary
injury, a secondary injury mediated by several complex biochemical signaling pathways become
activated. This process may arise hours following primary injury and can continue for several
days thereafter leading to apoptosis of neurons (Nolan, 2005; Wong et al., 2005).

3
1.1.4 Biomechanical Classification of TBI
Upon impact, the mechanical load on the brain causes the brain to move either in normal but
excessively accelerated or anatomically abnormal ways, depending on the site and direction of
the load. Due to the varying ways a mechanical load can be applied to the cranium, three types of
mechanical loading responsible for TBI have been classified as impact, impulsive, and static
loading (Ommaya et al., 2002).
Impact loading involves direct contact of a solid object to the head at a given speed. Its duration
is defined as < 50 s. Due to the collision of an object with a skull, movement and deformation of
tissue occurs and deformation of the skull at the site of impact is common. Impact loading can
occur whether the head was stationary (inert) or moving (accelerated) (LaPlaca et al., 2007).
Impulse loading, on the other hand, does not involve any contact towards the head and is chiefly
a consequence of excessive acceleration or deceleration of the head. Because there is no impact,
usually it does not result in any skull deformation. The duration of impulse loading ranges
between 50 – 200 ms (Davis, 2000). An example of impulse loading occurs when a sudden brake
is applied to a motor vehicle previously driving at high speeds. The seatbelt will stop the body
from moving but also cause the head and neck to snap forwards and backwards, thereby causing
damage to the brain.
Finally, static loading is caused by excessive force applied to the head for over 200 ms. Static
loading is the least common type (Ommaya et al., 2002) and it often results in deformation of the
skull since static loading happens usually in the context of a head injury caused by compression
(Davis, 2000).
In all types of mechanical loading, a sudden change in head motion will occur. This acceleration
can elevate intracranial pressure in certain areas of the skull, create axonal injury or bleeding if
head movement ruptures neuronal or vascular tissue, and cause tissue damage as a result of stress
waves produced by the brain hitting the skull (Davis, 2000). All types of mechanical loading are
associated with primary injury.

4
1.2 Pathophysiology of Traumatic Brain Injury
The pathways involved in secondary injury leading to apoptosis of neurons can include altered
cerebral blood flow, inflammation, increased intracellular calcium and extracellular glutamate
levels as well as oxidative damage to various molecules of the cell (Liu et al., 2002; Moro et al.,
2004; Werner et al., 2007). As the secondary injury does not manifest immediately following the
primary injury, it is thought that there is potential for neuroprotection for TBI at the secondary
injury level by manipulating the signaling pathways involved (Beauchamp et al., 2008).
1.2.1 Cerebral Blood Flow
In order for the brain to function, it requires a constant supply of oxygen and nutrients; hence,
cerebral blood flow (CBF) is tightly coupled to neuronal activity and cerebral glucose
metabolism (Giza et al., 2001). The blood-brain barrier (BBB) is created by specialized vascular
endothelial cells and tight junctions to prevent the passive diffusion of electrolytes and large
proteins. The BBB helps to maintain the integrity of the cerebral environment (Greve et al.,
2009). Following TBI, the BBB can fail leading to a collapse of the cerebral environment and the
disruption of the autoregulation for CBF (Greve et al., 2009). CBF can be decreased up to 80%
after a TBI (Giza et al., 2001) as a result of various secondary injury conditions such as cerebral
edema, hemorrhage, hypoxia, inflammation, and increased intracranial pressure (Nolan, 2005).
Furthermore, disrupted CBF may also play a role in exacerbating secondary injury by providing
less oxygen and nutrients to the brain (thus compounding the stress that neurons undergo
following TBI) (Bramlett et al., 2004) as well as stimulate the release of glutamate, free radicals,
and nitric oxide (Greve et al., 2009).
1.2.2 Neuroinflammation
Neuroinflammation is the activation of the brain’s innate immune system in response to an
inflammatory challenge and is characterized by many cellular and molecular changes within the
brain such as activation of glial cells, synthesis of pro-inflammatory molecules such as
cytokines, chemokines, and prostanoids, and up-regulation of infiltrating leukocytes (Hein et al.,
2009). Both primary and secondary insults can induce a neuroinflammatory response leading to
an increase in BBB permeability, synthesis of scar tissue, and release of neurotoxic mediators

5
such as nitric oxide and thus, aggravate secondary brain damage (Hein et al, 2009; Werner et al.,
2007).
1.2.3 Excitotoxicity and Oxidative Stress
1.2.3.1 Glutamate Excitotoxicity
Glutamate is the primary excitatory neurotransmitter in the central nervous system (CNS).
Excitatory neurotransmitters cause ion channels on the cell membrane to open resulting in a net
influx of positively charged ions (cations). This net influx of cations causes the neuron to
depolarize and if sufficient levels of excitation are provided, action potentials are fired.
Glutamate toxicity, neuronal death due to overstimulation of the CNS by high concentrations of
glutamate, was first observed in the retinal layers of neonatal mice fed monosodium glutamate
(Lucas et al., 1957), but the term “glutamate excitotoxicity” was not coined until 1969 by Olney.
Glutamate acts on both ionotropic and metabotropic receptors. The ionotropic glutamate
receptors are divided into three types according to their agonists: N-methyl-D-aspartate
(NMDA), -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate. All
three channels possess various sites by which their biochemical properties can be altered. AMPA
and kainate receptors are ligand-gated ion channels that are calcium impermeable and by
triggering downstream cell death pathways, they can play a minor role in excitotoxicity
(Waxman et al., 2005). However, the NMDA receptor (NMDAR), which is activated by binding
of glutamate and glycine, is highly permeable to calcium and is the primary receptor responsible
for excitotoxicity (Olney, 1969). Following TBI, the NMDARs are over-activated by the
excessive release of glutamate and thus, the NMDARs depolarize and disrupt the membrane
potential leading to neuronal overexcitation and death through excitotoxicity (Giza et al., 2001;
Kermer et al., 1999). Glutamate also acts on metabotropic receptors to release calcium from
stores but it is its involvement with ionotropic receptors that plays an important role following
TBI (Arundine et al., 2004).

6
1.2.3.2 NMDAR-Mediated Glutamate Excitotoxicity
It was first thought in 1985 that calcium entry into neurons was the essential determinate for
glutamate excitotoxicity (Choi, 1985). However, subsequent studies confirmed that it is
specifically the path of calcium entry (mediated by NMDARs) and not the calcium load itself
that is central to the neurodegenerative process (Choi, 1987; Sattler et al., 1998; Tymianski et al.,
1993).
The neurodegenerative process initiated by calcium entry through the NMDAR is associated
with nitric oxide synthase (NOS), which generates nitric oxide (NO) leading to cell death
(Dawson et al., 1991, 1993, 1994, and 1996). Thus, the current model of NMDA-mediated
glutamate excitotoxicity was formed (Figure 1-1).
NO is a gaseous free radical that is found in all tissues, especially in high concentrations in the
brain where it plays a large role in various physiological and pathological pathways (Cherian et
al., 2004). Physiologically, NO is involved in mediating CBF as a potent vasodilator, memory
formation and synaptic plasticity, release of neurotransmitters, and intracellular signaling
(Cherian et al., 2004; Moncada et al., 2006). Pathologically, on its own, NO is a weak toxin
however it can easily react with other species containing an unpaired electron (Gibson et al.,
2005) such as superoxide. When NO and superoxide react with one another, it produces the
reactive nitrogen species known as peroxynitrite (ONOO-) through the chemical reaction: O2-
+
NO ONOO-. ONOO- was introduced 20 years ago as the principal reactive oxygen species
(ROS) involved in producing tissue injury in a variety of neurological disorders (Beckman,
1991). The lethality of ONOO- is actually induced through the potent free radicals derived from
its decomposition (Gibson et al., 2005; Singh et al., 2006) and at physiological pH, ONOO- is
highly reactive (Aulak et al., 2004). ONOO- can decompose in two ways. The first is when
ONOO- is protonated to form peroxynitrous acid (ONOOH) which then undergoes hemolytic
decomposition to form the highly reactive nitrogen dioxide radical (NO2) and OH through the
chemical reaction ONOOH NO2 + OH (Hall et al., 2010). The second reaction is when
ONOO- reacts with carbon dioxide (CO2) to form nitrosoperoxocarbonate (ONOOCO2) which
then decomposes into NO2 and the carbonate radical (CO3): (ONOOCO2 NO2 + CO3
(Hall et al., 2010). Also ONOO- can interact with lipids to form lipid peroxyl radicals (LOO)

7
(Krishnaswamy and Chi, 2006). All of the radicals derived from ONOO- (OH, NO2, CO3,
LOO) can in turn either peroxidize lipids (Radi et al., 1991), nitrate proteins (Beckham et al.,
1992; Ischiropoulos et al., 1992) or fragment DNA to cause harm (Fiskum et al., 2004; Salgo et
al., 1995a; Salgo et al., 1995b). All of the above radicals are shown to be up regulated in the first
24 hrs post TBI in rodents (Cobbs et al., 1997; Graham et al., 2000; Rao et al., 1999) and the
damaging effects of ONOO- is further validated in a fluid percussion injury (FPI) model of TBI
showing that ONOO- formation results in neuronal protein nitration and cell death (Lau et al.,
2006).
Postsynaptic density protein of 95kDa (PSD-95) has been shown to provide a structural link
between neuronal nitric oxide synthase (nNOS) and NMDARs (Sattler et al., 1999). Hence by
uncoupling the interaction between PSD-95 and the NMDAR, neuroprotection can be achieved
through the inhibition of NO production as demonstrated by Arundine et al., 2004.
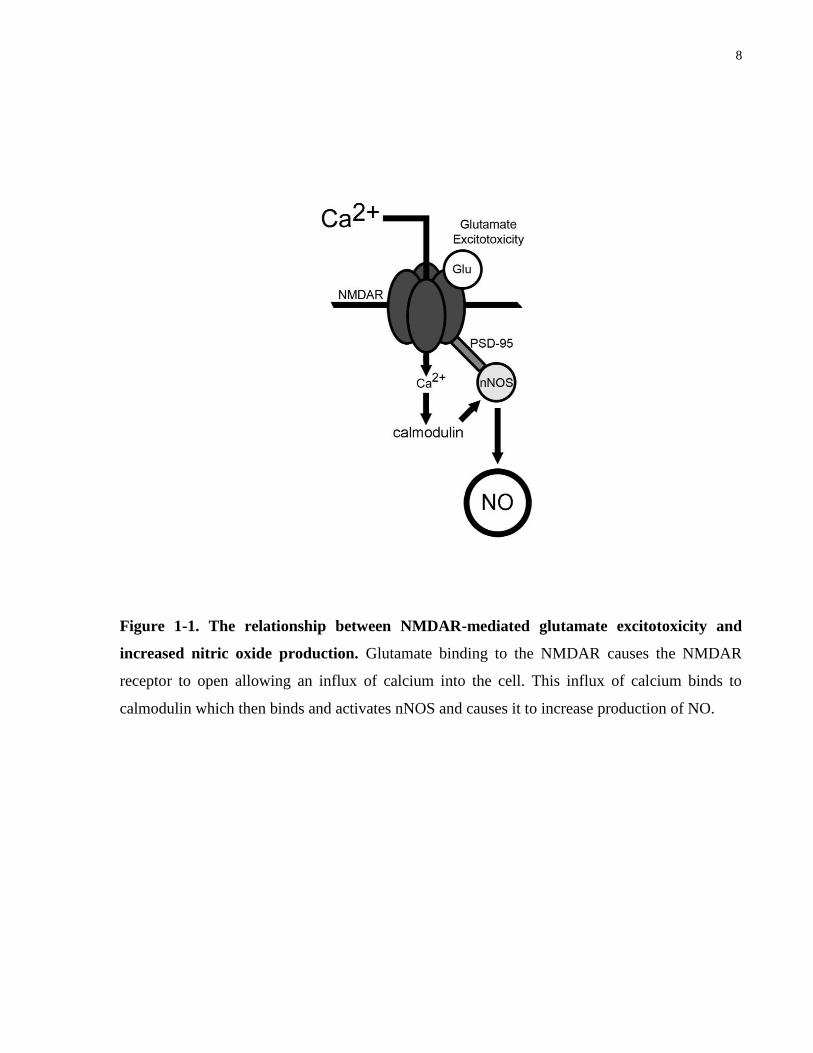
8
Figure 1-1. The relationship between NMDAR-mediated glutamate excitotoxicity and
increased nitric oxide production. Glutamate binding to the NMDAR causes the NMDAR
receptor to open allowing an influx of calcium into the cell. This influx of calcium binds to
calmodulin which then binds and activates nNOS and causes it to increase production of NO.

9
1.2.4 Protease-Mediated Cell Death: Caspases and Calpains
Caspases have been shown to be active following TBI in both cell cultures and whole animal
models (Clark et al., 2000; Keane et al., 2001; Qiu, et al., 2002; Yakovlev et al., 1997) and
calpains are a family of enzymes which have been shown to be another important mediator of
cellular injury following TBI (Greve et al., 2009).
Caspases represent a family of cysteine proteases, which fragment DNA (Wong et al., 2005). It
was first discovered to be involved in apoptosis, or programmed cell death, in the nematode C.
elegans (Wong et al., 2005). Caspases are translated as an inactive proenzyme, which can only
be activated by proteolytic digestion of the proenzyme into two subunits, which then form
together to make an active holoenzyme. Active caspases can then activate other caspases and
cause cellular cytoskeleton and genomic DNA to degrade.
Calpains are calcium-dependent cysteine proteases that, under normal physiological conditions,
have a variety of cellular regulatory functions such as cytoskeletal maintenance; however,
following TBI, they have been shown to be key enzymes targeting axonal proteins leading to
cytoskeletal breakdown and disruption of axonal transport (Greve et al., 2009). Calpains have
also been implicated in the release of endonuclease apoptosis-inducing factor (AIF) from the
mitrochondria, which causes the degradation of genomic DNA (Polster et al., 2005).
1.2.5 Cell Death Pathways of Secondary Injury in Summary
Despite the importance of CBF, neuroinflammation, excitotoxicity, reactive nitrogen species,
caspases, and calpains in TBI (Figure 1-2), there has yet to be any successful pharmacological
intervention developed for the treatment of secondary neuronal injury. One reason for this is that
various studies involving potential therapies have demonstrated histological improvement
without any behavioural protection (Clark et al., 2000). Although the histological aspect of TBI
is unquestionably valuable, in the clinical setting, the end-goal is to rescue the behavioural
deficits following TBI and therefore this study will focus on the behavioural outcomes following
an injury. Furthermore, although it is acknowledged that the cell death pathways associated with
secondary injury are extensive and complex, this study will focus on affecting the excitotoxicity
outcomes of TBI and the resulting behavioural consequences.
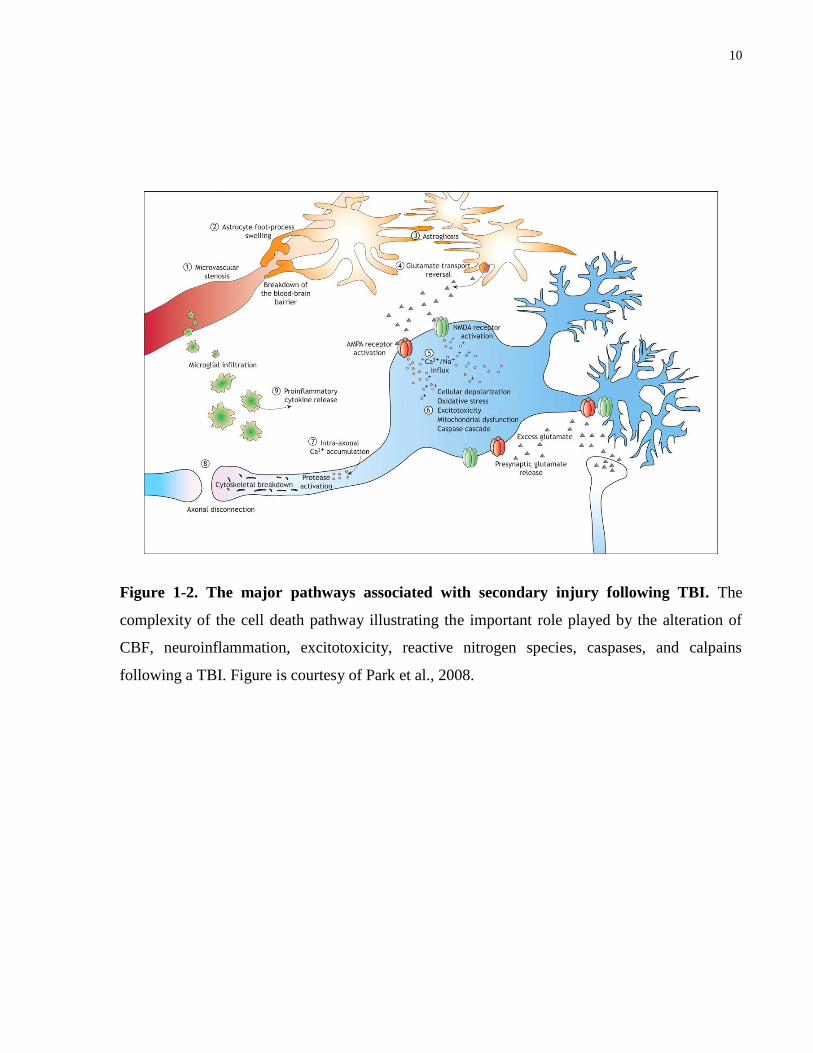
10
Figure 1-2. The major pathways associated with secondary injury following TBI. The
complexity of the cell death pathway illustrating the important role played by the alteration of
CBF, neuroinflammation, excitotoxicity, reactive nitrogen species, caspases, and calpains
following a TBI. Figure is courtesy of Park et al., 2008.

11
1.3 Injury Models of Traumatic Brain Injury
There are several experimental models of TBI that attempts to recreate the pathological
conditions through the application of mechanical forces. Examples of these models include
devices that induce impact to a intact or freely moving cranium (Nilsson et al., 1977), generate
an impact to a fixed, immobilized cranium (Gurdjian et al., 1943), cause TBI by accelerating or
rotating the skull (Gennerelli et al., 1983), and trigger TBI by rapid impact of a fluid bolus
against the intact dural surface of the brain (fluid percussion injury model). Currently the three
most commonly used animal models of TBI are the weight drop model, the controlled cortical
impact model, and the fluid percussion injury model (FPI).
1.3.1 Weight Drop Model
The weight drop model, first characterized by Feeney et al., 1981, is considered by most to be
the original TBI model (Morales et al., 2005). The weight drop model employs the gravitation
forces of a weight to deliver the impact on an anesthetized animal performed under head
restraints (Feeney et al., 1981). As illustrated in Figure 1-3, this can be performed either to the
closed cranium, but most commonly, the weight is dropped through a craniotomy directly onto
the brain (O’Connor et al., 2011). The severity of the injury is controlled by adjusting the height
and mass of the weight. One advantage of this model is that very little preparation of the animal
is required (no trephination of the skull) thus it is a fast and easy technique; however, a clear
disadvantage include the possibility of a rebound injury which can cause chances of inaccuracy
and inability to control the velocity of the injury (Morales et al., 2005).
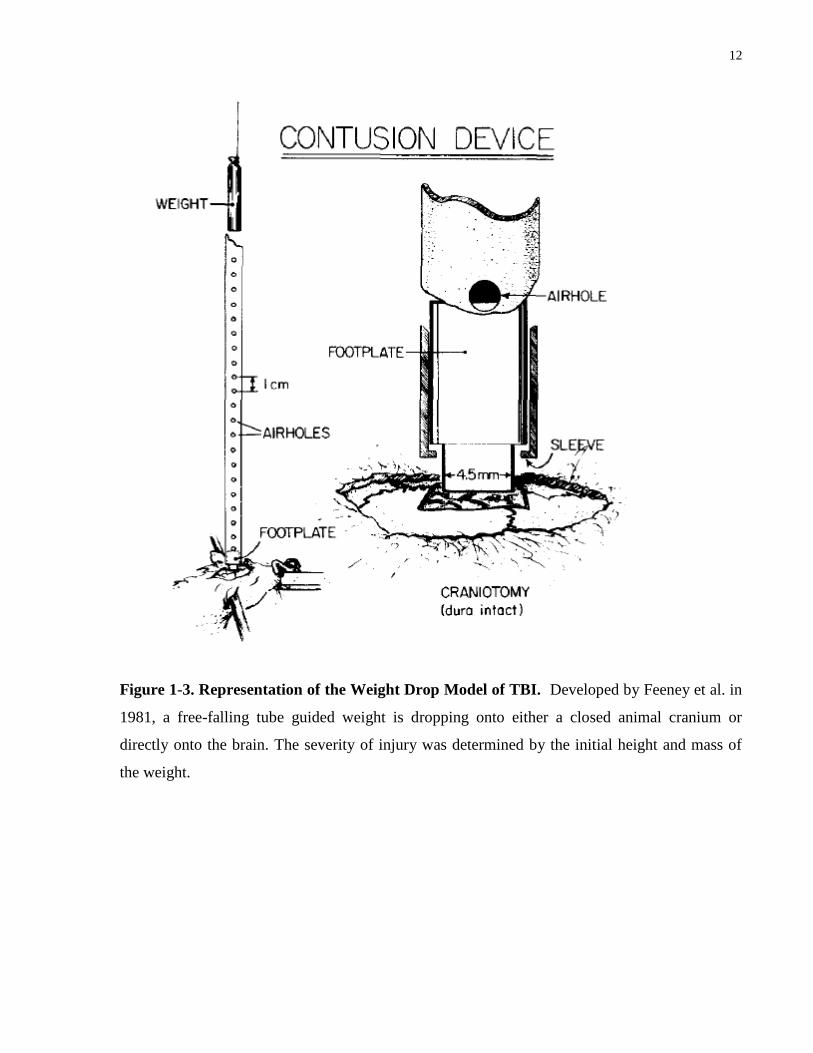
12
Figure 1-3. Representation of the Weight Drop Model of TBI. Developed by Feeney et al. in
1981, a free-falling tube guided weight is dropping onto either a closed animal cranium or
directly onto the brain. The severity of injury was determined by the initial height and mass of
the weight.

13
1.3.2 Controlled Cortical Impact (CCI) Model
The controlled cortical impact (CCI) model was first developed in the ferret (Lighthall, 1988)
and later adapted for the rat by Dixon et al. in 1991. This model uses a pneumatic impactor to
generate mechanical energy delivered to the intact dura following trephination of the exposed
skull (Morales et al., 2005). As illustrated in Figure 1-4, while the head of the animal is
restrained, pressurized air is employed as the source of the mechanical energy for loading to the
brain. The severity of the injury is controlled by the depth of cortical deformation, the duration of
the impact, also known as the dwell time, as well as the velocity. The CCI model improves upon
the weight drop model as it is able to control the velocity and depth of impact and there is also no
risk for a rebound injury to occur (O’Connor et al., 2011). It is well documented that the cerebral
hemodynamic responses in the animal such as elevated intracranial pressure, decreased blood
and cerebral perfusion pressures, histological and cellular alterations, as well as functional
deficits are all directly related to the depth and velocity parameters of the CCI making it arguably
the best model of head trauma in terms of having the best control over mechanical factors
(Cernak, 2005). Furthermore, the CCI model has been shown to reproduce the wide spectrum of
focal-type damage in TBI observed in the clinic setting. These include observed changes in brain
edema, elevated cerebral blood flow, neuroendocrine and metabolic changes, and coma as well
as skull deformation and related cortical compression (Cernak, 2005). However, one
disadvantage of the CCI model is the lack of brain-stem deformation, resulting in minimal
mortality, an aspect that does not mimic the real-life setting of TBI (Morales et al., 2005).
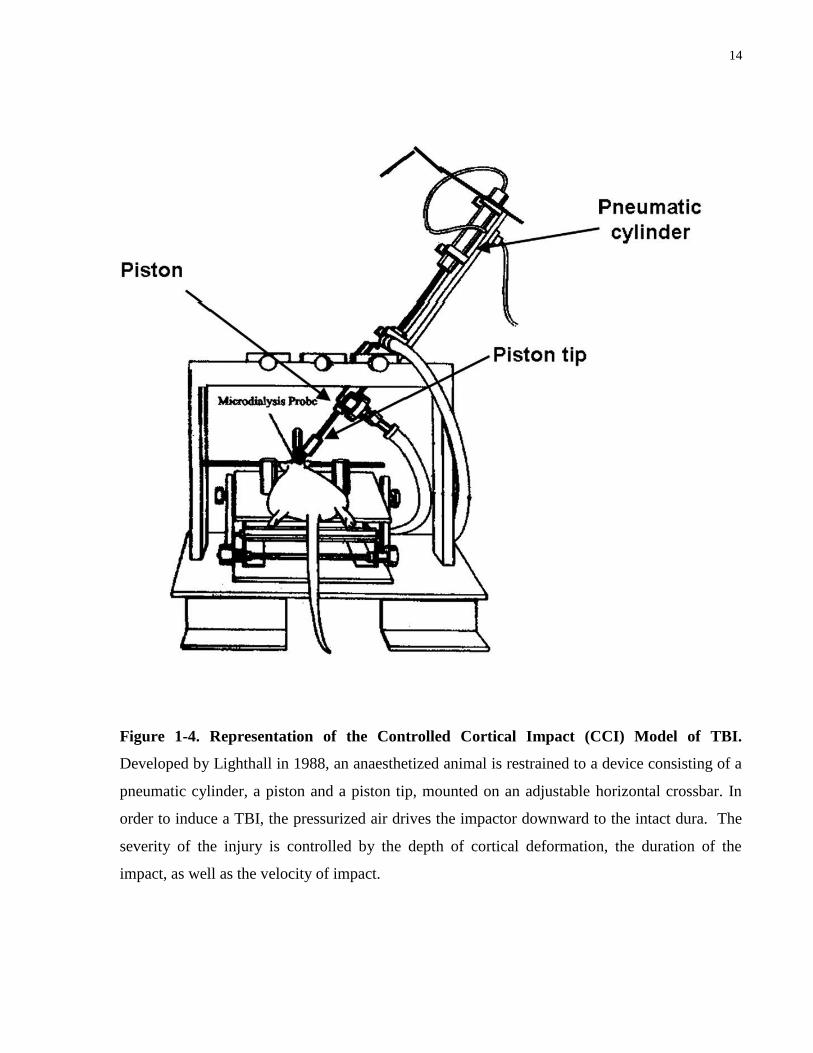
14
Figure 1-4. Representation of the Controlled Cortical Impact (CCI) Model of TBI.
Developed by Lighthall in 1988, an anaesthetized animal is restrained to a device consisting of a
pneumatic cylinder, a piston and a piston tip, mounted on an adjustable horizontal crossbar. In
order to induce a TBI, the pressurized air drives the impactor downward to the intact dura. The
severity of the injury is controlled by the depth of cortical deformation, the duration of the
impact, as well as the velocity of impact.

15
1.3.3 Fluid Percussion Injury (FPI) Model
The FPI model has since become the primary model for experimental TBI (Laurer et al., 2002).
It was initially developed for use in the cat and rabbit animal model (Hayes et al., 1987);
however in 1987, it was adapted to the rat model by McIntosh et al., 1987. Levels of injury
severity can be adjusted by changing the pendulum height, which defines the force of the fluid
pressure pulse transmitted through the saline tube and ultimately towards the exposed intact dura
of a restrained and anesthetized animal. This is illustrated in Figure 1-5. There are three severity
levels measured in standard atmospheric pressure (atm): mild (about 1.4 atm), moderate (about
2.7 atm), and severe (about 3.7 atm) (Prins et al., 1996). The neuropathological sequelae caused
by the FPI device transpires in the hippocampus, thalamus, and cortex of the brain with limited
involvement of the brainstem and contralateral hemisphere (Hicks et al., 1996; Smith et al.,
1997). The FPI model reproduces several aspects of human TBI such as focal contusion,
hemorrhages, traumatic axonal injury (Graham et al., 2000; McIntosh et al., 1989) as well as
BBB disruption, neuronal loss, and alterations in CBF (Narayan et al., 1996). It also causes acute
and chronic behavioural abnormalities similar to human TBI (Faden et al., 2001; Hamm, 2001;
Pierce et al., 1998; Sanders et al., 1999). The extensive use and productivity of the FPI model
over the past 20 years has helped refine clinical trials in human head injury and vice versa
(Thompson et al, 2005) and thus, the FPI model was chosen for this study.

16
Figure 1-5 Representation of the Fluid Percussion Injury (FPI) Model of TBI.
Developed by Hayes et al., in 1987, a pendulum from a certain height impacts the piston
containing saline, which in turn forces a fluid bolus into the sealed cranial cavity of the rat
causing a TBI. The severity of the injury is controlled by changing the pendulum height, which
defines the force of the fluid pressure.

17
1.4 Current Research for Treatment of Traumatic Brain Injury
There have been many experimental studies showing a reduction of secondary injury is possible;
however, the management of TBI is challenging due to its complexity of sequelae (Jain, 2008).
These studies include different management strategies for TBI and various drugs for
neuroprotection. However, many successes in the laboratory have yet to be translated into the
clinic setting (Zhang, 2005).
1.4.1 Management of TBI
There are many different ways to manage TBI to help reduce its physical and cognitive deficits.
The most important aspect of TBI management is the control of cerebral edema and raised
intracranial pressure (Jain, 2008). Current treatments for cerebral edema include osmotherapy
(administration of hypertonic mannitol or hypertonic saline) however they are not often
beneficial since they withdraw water from healthy brain areas as well as damaged areas (Jain,
2008). Hyperbaric oxygen (HBO) therapy (administration of oxygen at pressures greater than the
atmospheric pressure at sea level) has shown signs of being an effective treatment for cerebral
edema and assists in reducing intracranial pressure; however, this method is limited to the
availability of hyperbaric chambers (Jain, 2008).
1.4.2 Neuroprotection Drugs for TBI
There are many different neuroprotective strategies being investigated (Table 1-1). As of June
2008, approximately 150 clinical trials for TBI are listed; however, the failure rate in clinical
trials has been high (Jain, 2008). Hence, continued research efforts are required to identify and
test new neuroprotective agents as well as examine combined therapies for treating TBI. Three of
the most well known drugs designed for TBI are Citicoline, Cyclosporin A (CsA), and
Erythropoietin (Jain, 2008).
Citicoline is endogenously used for getting more choline into the brain. In a rat model of TBI,
citicoline was shown to prevent neuronal loss in the hippocampus and helped decrease cortical
contusion volume. Citicoline also improved neurological recovery in rats (Dempsey et al, 2003).
Cyclosporin A, an immunosuppressant, has been shown to improve brain function, memory, and
learning in animals models of TBI and the neuroprotective properties of CsA are found to be

18
mediated through modulation of the mitochondrial permeability transition pore (MPTP). By
inhibiting the opening of MPTP with CsA, the mitochondrial membrane potential and calcium
homeostasis is maintained and isolated in the mitochondrial, thereby preventing cell death
(Sullivan et al., 1999).
Erythropoietin (EPO) has also demonstrated neuroprotective potential in cell culture and animal
models of TBI however the exact mechanism of EPO is unclear. It has been hypothesized that
EPO protects neurons by inhibiting TBI-induced neuronal apoptosis (Liao et al., 2008) and that
EPO protects motor deficits by reducing cerebral edema (Grasso et al., 2007). Currently, it is in
phase II/III clinical trials for TBI (Jain, 2008).

19
Table 1-1. Neuroprotective strategies for traumatic brain injury. Table of many different
neuroprotective strategies currently being investigated for the treatment of TBI.

20
1.4.3 NeuroprotectionThrough the Scavenging of Radical Species
One approach to neuroprotection through the pathway illustrated in Figure 1-1 is to design a
compound that chemically scavenges for the radical species produced by ONOO-. By offering a
more attractive compound as bait, one hopes to limit the destructive actions of the free radicals.
Two compounds have been previously designed to accomplish this: polyethylene glycol-
conjugated superoxide dismutase (PEG-SOD) and Tirilazad; however, both failed to provide any
therapeutic benefit following TBI (Hall et al., 2010). The reason for PEG-SOD’s failure lies
within the compound’s large size. Being a large protein, it was found to be unlikely to penetrate
the BBB and therefore its radical scavenging effects would have been only limited to the
microvasculature (Hall et al., 2010). Tirilazad, on the other hand, fared better than PEG-SOD. In
the laboratory, it was demonstrated to be effective in multiple animal models of TBI (Dimlich et
al., 1990; Hall et al., 1988; McIntosh et al., 1992) but once it progressed into the clinical
development phase, the results of Tirilazad failed to show a significant positive effect in
moderate or severe TBI patients (Marshall et al., 1998). It is unknown why Tirilazad’s
theurapeutic effects did not translate from the laboratory to the clinical setting. Perhaps its failure
lies in its extremely specific mechanism of action - preventing lipid peroxidation by targeting
only the lipid peroxyl radical (LOO) (Hall et al., 2010). As described earlier, the free radicals
derived from ONOO- not only peroxidize lipids (Radi et al., 1991), but can also nitrate proteins
(Beckham et al., 1992; Ischiropoulos et al., 1992) and fragment DNA (Fiskum et al., 2004; Salgo
et al., 1995a; Salgo et al., 1995b). Therefore, a more effective compound would have attributes
such as having a smaller size in order to cross the BBB and to target not one but many different
free radicals resulting from ONOO-.

21
1.5 Design of Tat-9c and Tat-9a
Peptide-based therapies have been steadily gaining in popularity. Peptides can be made cell-
permeant by fusing them to the cell-membrane transduction domain of the human
immunodeficiency virus-type 1 (HIV-1) Tat protein (Fawell et al., 1994). This approach has been
used to show neuroprotective effects of disrupting the nNOS/PSD-95 interaction in in vivo
models of acute cerebral ischemia (Aarts et al., 2002). Furthermore, it has been demonstrated
that tyrosine-containing peptides can successfully scavenge peroxynitrite-derived radicals and
protect motor neuron cultures from peroxynitrite-mediated cell death (Ye et al., 2007). However,
Alvarez et al. (1999) illustrated that cysteine residues are more reactive when compared to
tyrosine. When a derivative of cysteine, N-acetylcysteine (NAC), was examined in in vivo
studies, no gross histological benefits following TBI were observed (Thomale et al., 2006); yet,
NAC was able to protect against oxidative insults in vitro (Gow et al., 2000). Based on these
findings, Tat-9c, a cysteine-rich, cell-permeant, polypeptide composed of the tat protein
conjugated to nine cysteines was developed.
The Tat-9a peptide was also synthesized as a negative control for Tat-9c. Tat-9a is composed of
nine alanines attached to the tat protein.

22
1.6 Neurobehaviour Models of Assessing TBI Severity
Many neuromotor deficits such as difficulties with coordination, posture, and steadiness of
movement following TBI have been reported in humans (Schalen et al., 1994). The Beam-Walk
and Rota-Rod tests are often used to measure neuromotor deficits following TBI in animal
models.
1.6.1 Beam-Walk
The Beam-Walk is one of the most common techniques used to assess motor function following
FPI (Dixon et al., 1991). It is used to assess the more complex components of vestibulomotor
function and coordination that a simple beam-balance test cannot detect (Hamm et al., 1994). In
the Beam-Walk test, animals are required to traverse a suspended narrow beam and enter a goal
box at the end of the beam as illustrated in Figure 1-6. Motor function is assessed by measuring
the animal’s latency to enter the goal box at the end of the beam.
Figure 1-6 Representation of the Beam Walk Device. For the Beam-Walk test, animals are
required to traverse a suspended narrow beam and enter a goal box at the end of the beam. Motor
function is assessed by measuring the animal’s latency to enter the goal box at the end of the
beam.

23
1.6.2 Rota-Rod
The Rota-Rod task was first developed by Dunham et al. in 1957. The procedure required a rat to
maintain its balance on a rod rotating at a constant speed as illustrated in Figure 1-7. Jones et al.
increased the sensitivity of the task by gradually increasing the speed of the rod while it rotated
in 1968. Hamm et al. in 1994 was the first to apply it as a test for motor deficits following TBI.
Hamm et al., also found that the Rota-Rod test is a more sensitive and efficient test at detecting
injury severity produced by a TBI than the Beam-Walk and Beam-Balance tests.
Figure 1-7 Representation of the Rota-Rod Device. For the Rota-Rod test, animals are
required to maintain its balance on a rod rotating at a constant or gradually accelerating speed.
Motor function is assessed by measuring the animal’s latency to stay balanced on top of the
rotating rod.

24
1.6.3 Morris Water Maze
Cognitive performance in the laboratory is tested through a variety of hippocampal-dependant
spatial mazes. The Morris Water Maze (MWM) was designed to assess the cognitive processes
of spatial learning and working memory (D’Hooge et al., 2001; Morris et al., 1982). As
illustrated in Figure 1-8, the MWM consists of a large circular pool filled with opaque water. A
small escape platform is hidden beneath the water and during a number of training trials, animals
learn to find the location of the platform to escape from the water (Morris et al., 1982). Basic
MWM protocols include hidden-platform acquisition training and probe trial testing (D’Hooge et
al., 2001). Hidden-platform acquisition records how long it takes for an animal to find the hidden
platform however during a probe trial, the platform is removed and the trained animal is allowed
to swim for a fixed amount of time. A probe trial tests the spatial accuracy, or memory, of the
animal (D’Hooge et al., 2001). The MWM was the first cognitive test used with the FPI model
(Smith et al., 1991) and still remains the most frequent test to assess cognitive function after FPI.

25
Figure 1-8 Representation of the Morris Water Maze (MWM) testing room and apparatus.
The MWM consists of a large circular pool filled with opaque water. A small escape platform is
hidden beneath the water and during a number of training trials, animals learn to find the location
of the platform to escape from the water. Hidden-platform acquisition records how long it takes
for an animal to find the hidden platform however, in Day 1 of training, often the animal’s swim
path and duration is longer as shown above. If the animal is capable of learning following a
number of training trials, the swim path and duration taken to the platform should be lessened as
shown above in Day 6. During a probe trial, the platform is removed and the trained animal is
allowed to swim for a fixed amount of time. A probe trial tests the spatial accuracy, or memory,
of the animal to remember where the platform is located. If the animal has memory, it should
spend most of its time swimming in the correct quadrant as shown above in Day 7. The above
illustration is courtesy of Buccafusco, 2009.

26
Chapter 2 HYPOTHESIS AND GOALS
2.1 Research Goals
Despite significant research detailing various cell death pathways involved in secondary injury
following a TBI, no successful pharmacological therapies have been fashioned that consistently
attenuate neurological deficits caused by a TBI and/or in manner where the mechanism of drug
action is clear and defined (Jain, 2008).
However, past research has determined that peroxynitrite plays a key part in neuronal cell death.
Hence, our lab has developed a peptide, Tat-9c, which we believe will be a potent nytrosylation
target for peroxynitrite and potentially prevent neuronal cell death caused by peroxynitrite.
One way of testing new drugs is to examine TBI injured animals as a whole unit instead of
fragments of their neuronal structures. Thus, the aim of this study was to explore the efficacy of
Tat-9c by observing an animal’s gain or loss of function following TBI.
In this thesis, the following hypotheses were tested:

27
2.2 Hypothesis I
The Beam-Walk, Rota-Rod, and Morris Water Maze behaviour paradigms can
discriminate the severity of a TBI implemented through a FPI device.
Since the aim of this study was to explore the efficacy of Tat-9c by observing an animal’s gain or
loss of function following FPI, various neurobehaviour paradigms needed to be validated in
order to investigate their robustness at assessing injury severity and recovery. This hypothesis
was derived from previously published data supporting the Beam-Walk, Rota-Rod, and Morris
Water Maze as effective tests for TBI (Hallam et al., 2004; Hamm et al., 1994).
2.3 Hypothesis II
There will be significant increase in motor function following FPI in rats treated with Tat-
9c compared to Tat-9a or saline treated animals.
This hypothesis was postulated based on the findings of Lau et al., 2006 who showed that
ONOO- formation results in neuronal protein nitration and cell death and the following theory
that Tat-9c will be a potent nitrosylation target for peroxynitrite; thus, Tat-9c can potentially
prevent neuronal cell death leading to a decrease in motor performance.
2.4 Hypothesis III
There will be significant improvement in memory performance following FPI in rats
administered with Tat-9c compared to Tat-9a or saline treated animals.
This hypothesis was based on our findings from our second hypothesis. It is conceived that if
Tat-9c is acting in the same manner to restore motor function, it may play the same protective
role in the hippocampus where spatial learning and memory occurs.

28
Chapter 3 MATERIALS AND METHODS
All of the following animal experiments were approved by the University Health Network
Animal Resource Center, which is fully accredited by the Canadian Council on Animal Care.
3.1 Lateral Fluid Percussion Injury Model
The lateral FPI model was performed as described in Lau et al (2006).
3.1.1 Surgical Preparation
Male Sprague-Dawley rats weighing 350g – 375g were used in this study. 24 hours prior to
injury, rats were anesthetized using 2% isofluorane. The scalp of the rat was shaved and the
incision site was swabbed with alcohol to minimize infection. A sagittal incision was made from
the bregma to the lambda. Curved hemostats were placed to retract the overlying skin. Cotton
swabs were then used to separate the connective tissue as well as to clean the area of the exposed
skull. Using a dental drill, a small indent was made in the middle of the cranium flanked by the
bregma, the lambda, the sagittal suture, and the right temporal ridge (Figure 3-1). With the indent
as a guide, a 4.8mm craniotomy was made using a trephine. A modified Leur-loc was then
attached over the craniotomy and sealed into place using Acron MC/R dental acrylic (GC
America, Alsip, IL) and Krazy glue. Once the dental acrylic cement has hardened, the Leur-loc
was then filled with saline and a small piece of gelfoam was placed inside the Leur-loc to reduce
infection. The skin was sutured over the entire setup and the animal was returned to its cage
overnight.
3.1.2 Drug Injection
The following day, animals were anesthetized using 2% isofluorane and animal weights were
recorded. To substantiate a functional effect, we first tested Tat-9c as a pretreatment. This is a
common approach for testing therapies for injury as demonstrated by Shein et al, 2009; Di
Giorgio et al, 2008; Czeiter et al, 2009). Therefore 30 minutes prior to injury, either saline or
5nmol/g of Tat-9c or Tat-9a was administered via tail vein injection over 10 minutes. Once the
injection was complete, the animals were returned to their home cages to await another 20

29
minutes before TBI was induced. All injections were performed with the experimenter blind to
the drug injected. An assistant prepared the drugs in advance to the drug injection.
3.1.3 Fluid Percussion Injury
Before a TBI was produced, animals were again anesthetized with 2% isofluorane and the
incision site from the previous day was reopened. The piece of gelfoam inside the Leur-loc was
removed and the entire area was cleansed with saline. Prior to attaching the animal to the FPI
device (Custom Design and Fabrication, Richmond, VA), the Leur-loc was again filled with
saline. Once the animal was securely attached to the device via the Leur-loc (Figure 1-5), they
were subjected to a 2.4 atm moderate injury as described previously (McIntosh et al., 1989). A
pressure transducer attached to an oscilloscope recorded the magnitude of each injury (Figure 3-
2). Following injury, sometimes it was necessary to resuscitate an animal by chest compressions.
If the animal survived, the entire Leur-loc setup with dental acrylic was removed and the wound
was again sutured.
Figure 3-1 Dorsal Representation of a Rat Skull. A small indent was made in the
middle of the cranium flanked by the bregma, the lambda, the sagittal suture, and the right
temporal ridge. This indent is where the FPI device will be connected to the rat’s skull.
30
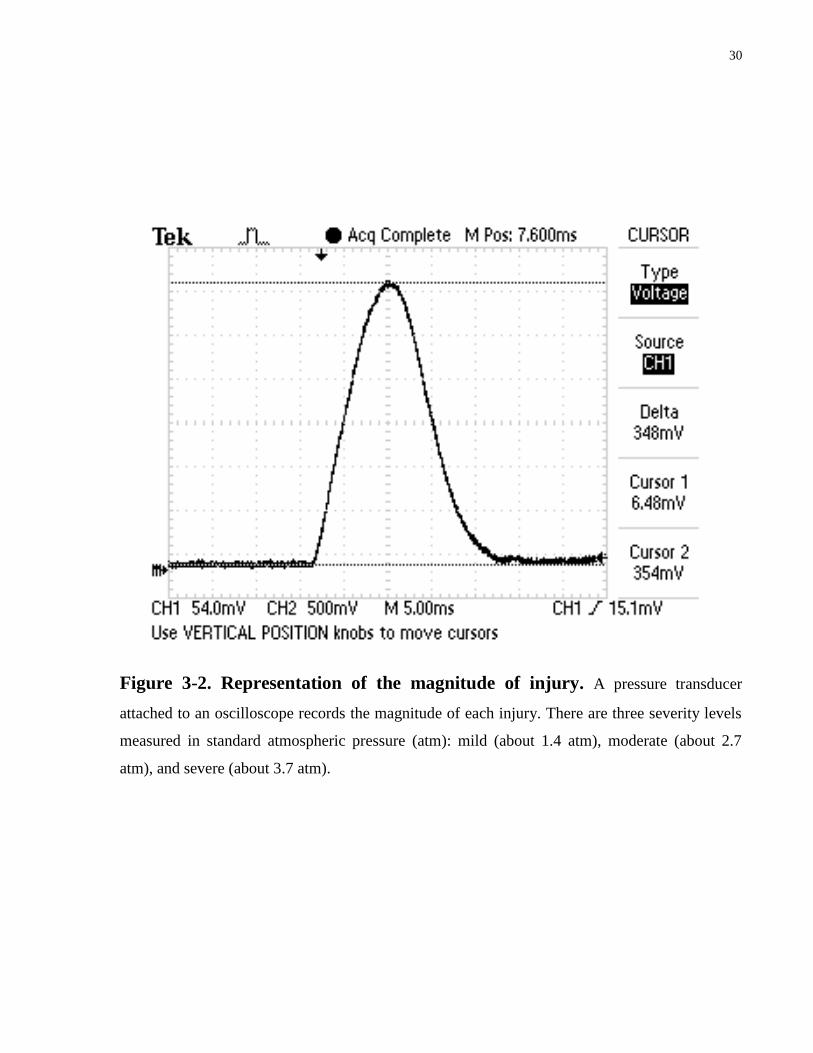
Figure 3-2. Representation of the magnitude of injury. A pressure transducer
attached to an oscilloscope records the magnitude of each injury. There are three severity levels
measured in standard atmospheric pressure (atm): mild (about 1.4 atm), moderate (about 2.7
atm), and severe (about 3.7 atm).

31
3.2 Treatment Preparation
3.2.1 Preparation of Tat-9c for Animal Injection
The Tat-9c peptide (YGRKKRRQRRRCCCCCCCCC) was synthesized at the Advanced Protein
Technology Centre (Hospital for Sick Children, Toronto, Ontario, Canada) by Dr. Nam-Chiang
Wang. The lyophilized form of the peptide was resuspended in deoxygenated normal saline
solution and the completely soluble solution was aliquoted under anoxic conditions. Aliquots
were stored at 4oC until use (5nmol/g).
3.2.2 Preparation of Tat-9a for Animal Injection
The Tat-9a peptide (YGRKKRRQRRRAAAAAAAAA) was also synthesized at the Advanced
Protein Technology Centre (Hospital for Sick Children, Toronto, Ontario, Canada) by Dr. Nam-
Chiang Wang. The lyophilized form of the peptide was resuspended in deoxygenated normal
saline solution and the completely soluble solution was aliquoted under anoxic conditions to
mirror the preparation of Tat-9c as much as possible. Tat-9a was designed to be a negative
control for Tat-9c. Aliquots were stored at 4oC until use (5nmol/g).
3.2.3 Preparation of Saline for Animal Injection
Saline solution was purchased from Baxter DIN 00786160.
3.2.4 Preparation of Sham Animals
Sham animals underwent surgical preparation for the FPI in the same manner as treatment
animals however they were not subjected to the injury. Sham animals were included in this study
to ensure that treatments such as anaesthesia or surgery will not affect the outcome parameters.
This is especially important as isoflurane, the anaesthetic approved in our protocol, has been
shown to exert some neuroprotection on the injured brain (Statler et al., 2006). Animals
remained under anesthetic during suturing and returned to their home cage to await behavioural
trials.

32
3.3 Implication of Various Behavioural Paradigms
Animals were evaluated on motor or cognitive function immediately after injury and for up to 15
days post-injury depending on the various behaviour models utilized. All the following
behaviour experiments were performed with the experimenter blinded to the treatment that the
animals received.
3.3.1 Beam-Walk Model
The Beam-Walk model consists of a 90cm x 2.5cm wooden beam suspended 60cm above a
padded table in a dark room. At one end of the beam, negative stimulus was provided through
white noise and bright light (60W light bulb). At the other end of the beam, a positive
environment was supplied by a dark cardboard box. Prior to injury, animals were trained on the
Beam-Walk by placing them at the end with the negative stimulus. Once the animals
successfully traversed the beam within 10 seconds over 3 consecutive trials, they were deemed
trained and returned to their cages. Following surgical preparation and before injury, animals
were tested on the Beam-Walk to determine their baseline values. The average latency of 3 trials
was recorded. Any animal that had difficulty crossing the beam was discarded from the
experiment as it would signify that an unintentional injury has occurred during the surgical
preparation before a FPI was given. Once an animal has been subjected to a FPI, the average
latency of 3 trials was recorded for various time points following the injury. If an animal could
not traverse the beam and fell off, they were given an automatic 60 seconds.
3.3.2 Rota-Rod Model
The Rota-Rod model consists of a rotating rod suspended 50cm over weight sensitive planks that
stop recording time if a rat falls on it. Before injury, rats were acclimatized to the setup and
trained to stay on the rotating rod for at least 2 minutes. The speed of the rod started at 2rpm and
was steadily increased by 3rpm in 10 second intervals until the animal fell completely off the
rungs of the rod. Following surgical preparation and before injury, animals were tested on the
Rota-Rod to determine their baseline values. Any animal that had difficulty staying on the
rotating beam for less than 30 seconds was discarded from the experiment as it would signify

33
that an unintentional injury has occurred during the surgical preparation before a FPI was given.
Once an animal has been subjected to a FPI, the mean duration of 3 trials per day each rat spent
on the rotating rod was recorded for various time points following the injury.
3.3.3 Morris Water Maze Model
This test was performed as described previously (Hallam et al., 2004). The water maze task was
used to measure spatial reference memory 11-15 days post-injury allowing animals who were
given a TBI enough time to recover their motor deficits in order to swim. Water temperature
levels were kept between 23-25C. Non-toxic blue paint (made by Gothic Tempera and
purchased at various art stores) was diluted into the pool until the platform was not visible.
Visual cues in different shapes (circles, rectangles, symbols, etc.) were cut out of cardboard
paper and foam material and placed around the pool in plain sight of the animal. Animals were
tested 4 trials each day for 5 consecutive days to find a hidden platform in 120 seconds. As soon
as the animal has found the platform, time was stopped. The swim path, swim speed, and latency
to find the hidden platform were measured using a computer controlled tracking system
(SMART, San Diego Instruments, San Diego, CA). If an animal was unable to find the platform
within 120 seconds, the experimenter would lead the animal towards it and allow it to stay on the
platform for an extra 30 seconds before returning it to its cage where they were thoroughly dried.
On the last day, 15 days post-injury, a final probe trial was also conducted where the platform
was removed. The rat was placed inside the water maze and allowed to swim freely for a total of
120 seconds. From the probe test, the percent of time the animal spent in the correct quadrant
was measured and recorded.
3.4 Statistical Analysis
All data were expressed as mean ± SD, except where noted. Data analysis was performed using
SigmaStat3.0; SPSS, Chicago, IL. Behaviour data for each dependent variable were analyzed
with repeated measures ANOVAs. Statistical significance was considered at the p < 0.05 level.

34
Chapter 4 HYPOTHESIS AND GOALS
4.1 Validation of Behaviour Assays Utilized to Assess Severity of TBI
To assess the impact of Tat-9c on neurobehavioural outcome following TBI, we used the Beam-
Walk, Rota-Rod, and the Morris Water Maze, which have all been previously demonstrated to
effectively measure TBI severity as mentioned above. Although the success of these behaviour
assays is well documented, it was still essential to verify whether these assays were able to
reflect the impairments caused by the TBI in this study.
4.1.1 Beam-Walk Model
The Beam-Walk model was first assessed using male Sprague-Dawley rats. All animals were
trained 24 hours before surgical preparation and tested for baseline results between the surgical
preparation and the injury time point. TBI rats were subjected to a moderate 2.4atm FPI. At 1, 2,
3, and 7 days post-injury, Beam-Walk performance was assessed (Figure 4-1). Experiment data
was collected up to one week following TBI since the Beam-Walk becomes insensitive to motor
deficits after one week. Results showed a significant difference between TBI and sham groups at
all time points post-injury which is consistent with previous literature (Hamm et al., 1994). TBI
injured animals required on average seven times longer than sham animals to traverse the beam
24 hours following TBI and also demonstrated a gradual trend towards behavioural improvement
throughout the experiment. These results suggest that the Beam-Walk is an effective model to
assess severity of TBI but only up to one week post-injury.
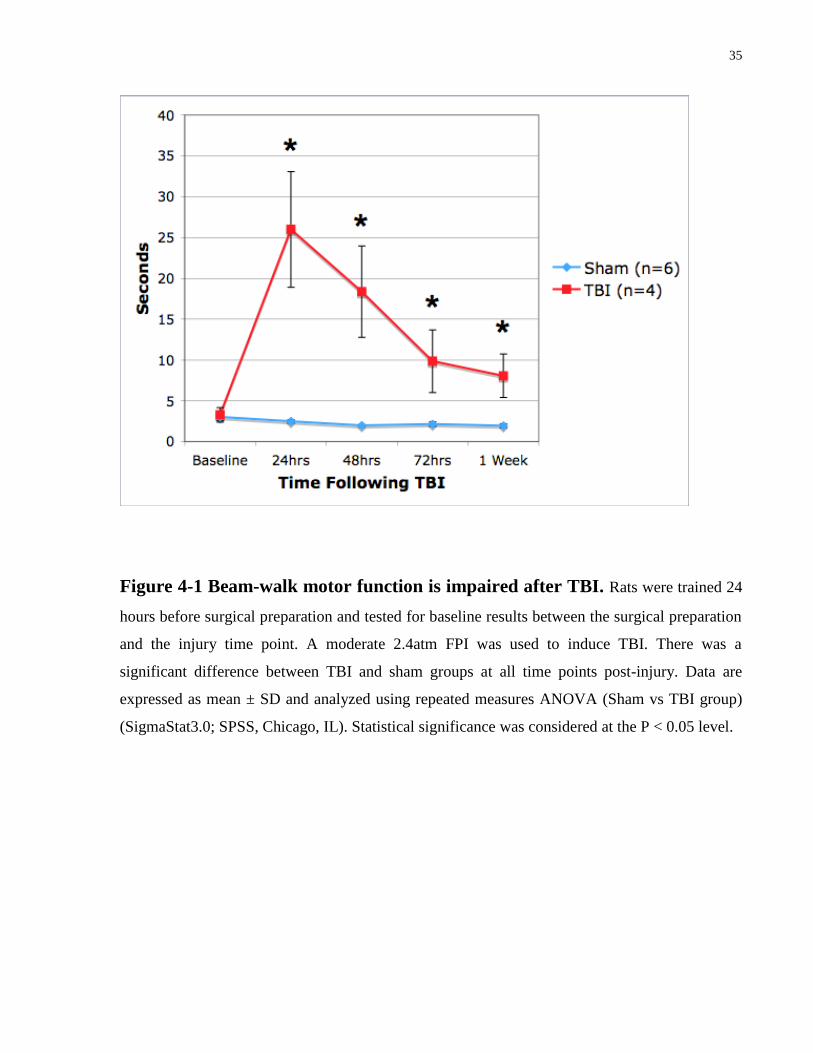
35
Figure 4-1 Beam-walk motor function is impaired after TBI. Rats were trained 24
hours before surgical preparation and tested for baseline results between the surgical preparation
and the injury time point. A moderate 2.4atm FPI was used to induce TBI. There was a
significant difference between TBI and sham groups at all time points post-injury. Data are
expressed as mean ± SD and analyzed using repeated measures ANOVA (Sham vs TBI group)
(SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.

36
4.1.2 Rota-Rod Model
The Rota-Rod model was tested in conjunction with the Beam-Walk model using the same group
of 10 male Sprague-Dawley rats. All animals were trained 24 hours before surgical preparation
and tested for baseline results between the surgical preparation and the injury time point. TBI
rats were subjected to a moderate 2.4atm FPI. At 1, 2, 3, and 7 days post-injury, Rota-Rod
performance was assessed (Figure 4-2). These time points were used to coincide with the Beam-
Walk experiments. Results showed a significant difference between TBI and sham groups at all
time points post-injury. TBI injured animals showed greatest motor deficits (about 70%
decrease) 24 hours following TBI which was then preceded by a gradual trend towards
behavioural improvement throughout the experiment. These findings also suggest that the Rota-
Rod is a more sensitive index for assessing motor impairment as the rate of motor recovery is
slower than the Beam-Walk. This also is consistent with previous research comparing the Beam-
Walk and Rota-Rod models (Hamm et al., 1994). The greatest differences between TBI and
sham animals were seen in the first three days post-injury. Hence, it was decided that future
experiments involving the Beam-Walk and Rota-Rod would only be tested up to 72 hours post-
injury.
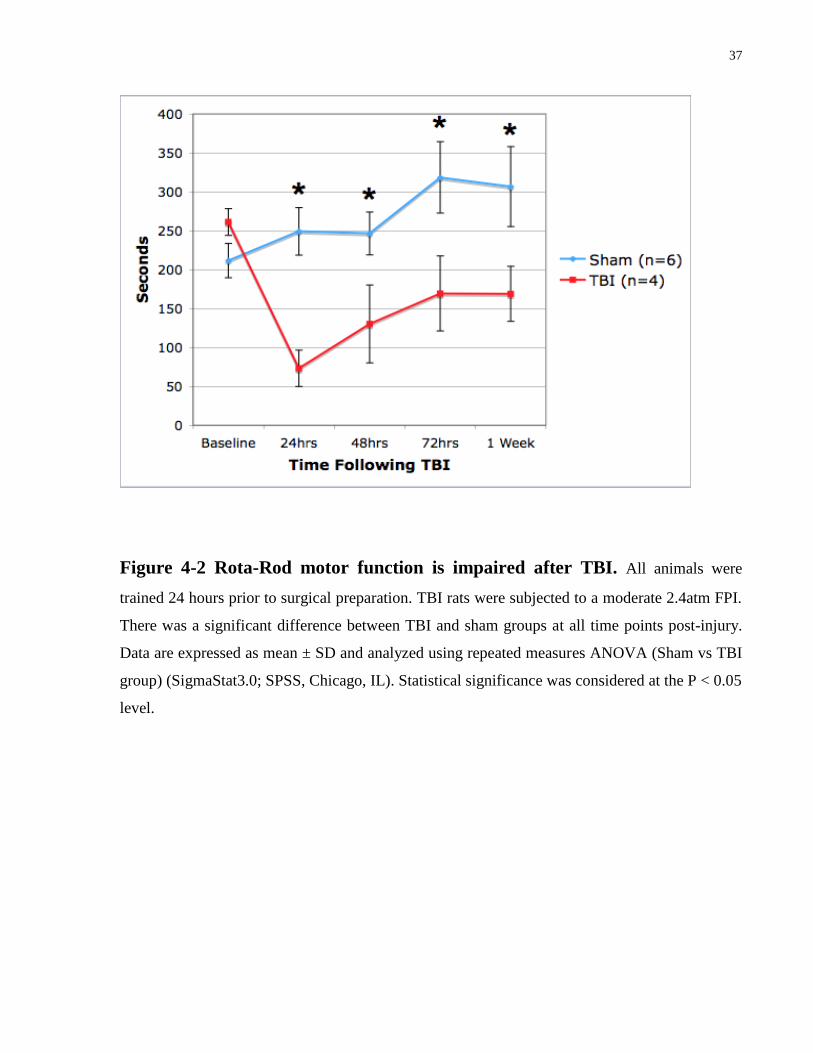
37
Figure 4-2 Rota-Rod motor function is impaired after TBI. All animals were
trained 24 hours prior to surgical preparation. TBI rats were subjected to a moderate 2.4atm FPI.
There was a significant difference between TBI and sham groups at all time points post-injury.
Data are expressed as mean ± SD and analyzed using repeated measures ANOVA (Sham vs TBI
group) (SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05
level.

38
4.1.3 Morris Water Maze Model
The Morris Water Maze Model was used to assess cognitive defects caused by TBI. The TBI
group was subjected to a moderate 2.4atm FPI and 11 days post-injury, all animals were
introduced to the Morris Water Maze. Results showed there was no significant difference
between TBI and sham groups at all time points when measuring latency to platform (Figure 4-3)
which is consistent with previous literature (Hallam et al., 2004; Reid et. al. 2008). After just
four days, all animals were able to learn the location of the hidden platform. The average swim
speed (Figure 4-4) and total distance traveled (Figure 4-5) were also not significant between the
two groups indicating that 11 days post-injury is an adequate time point to start the behaviour
experiment as the injured animals have recovered their motor function sufficiently enough to
swim. Furthermore, a final probe test was performed 15 days post-injury. Results indicate that
TBI injured animals performed significantly lower than sham animals in the memory test (Figure
4-6). In all, the results confirm that the Morris Water Maze is a practical test for showing
deficiencies in memory impairment following TBI.
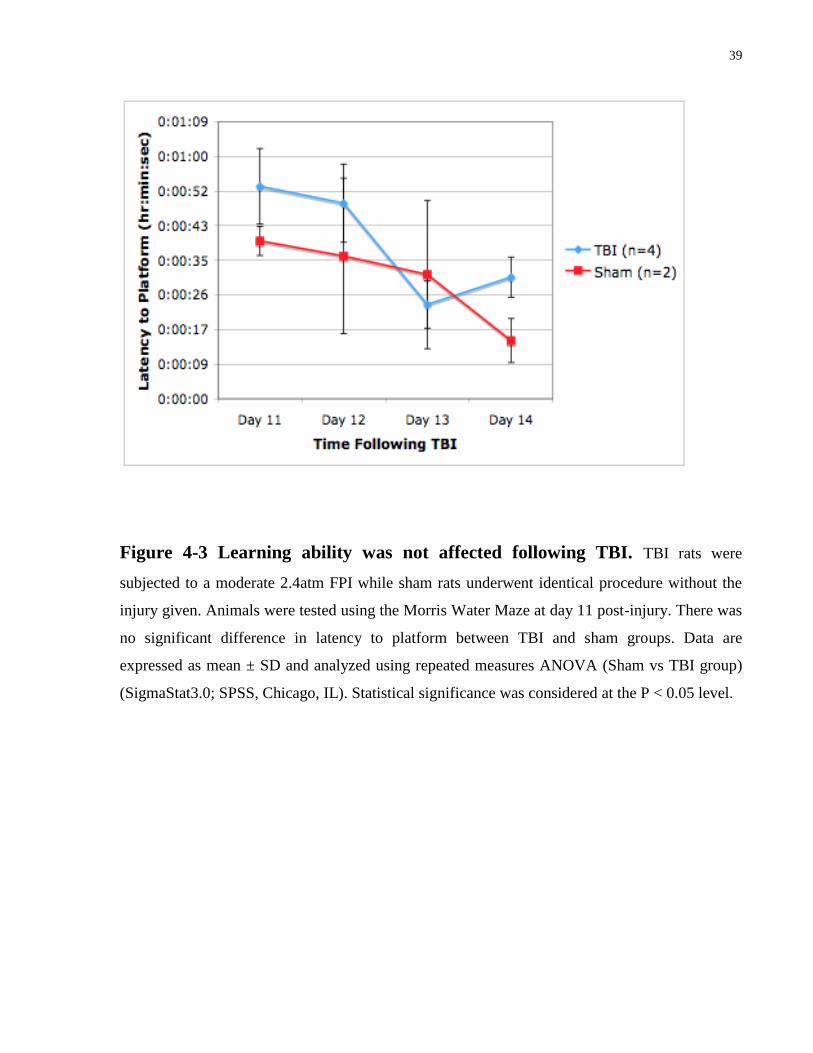
39
Figure 4-3 Learning ability was not affected following TBI. TBI rats were
subjected to a moderate 2.4atm FPI while sham rats underwent identical procedure without the
injury given. Animals were tested using the Morris Water Maze at day 11 post-injury. There was
no significant difference in latency to platform between TBI and sham groups. Data are
expressed as mean ± SD and analyzed using repeated measures ANOVA (Sham vs TBI group)
(SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.
40
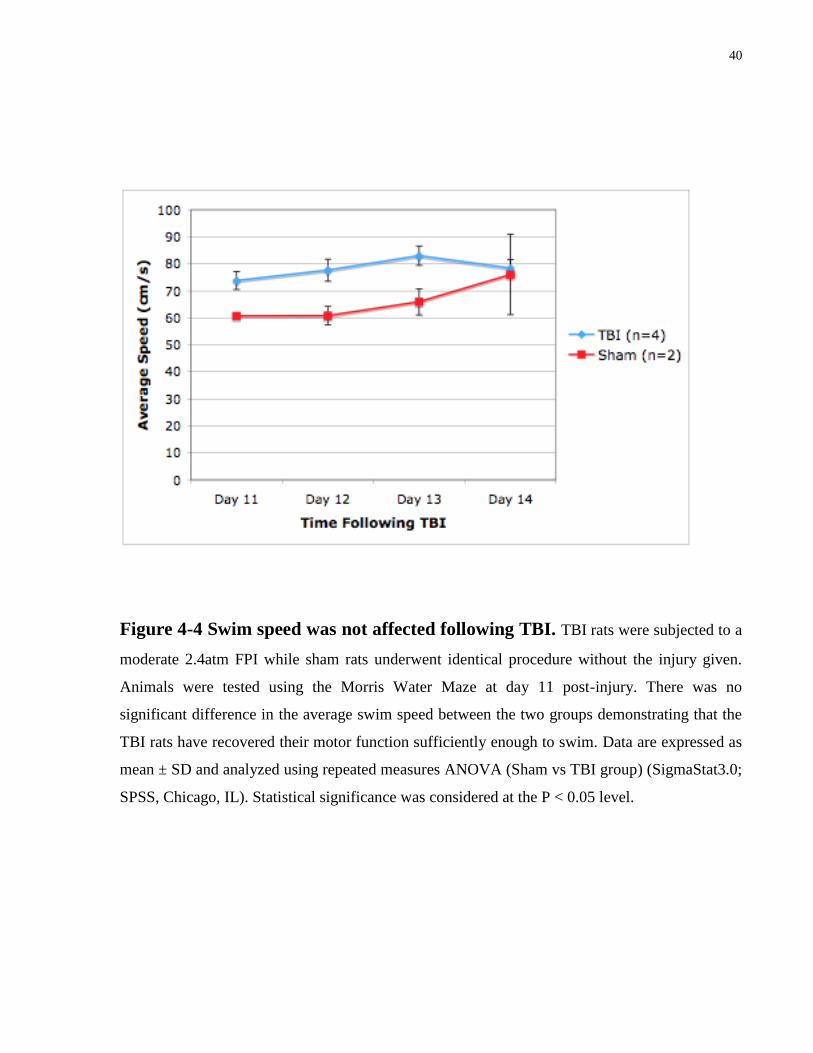
Figure 4-4 Swim speed was not affected following TBI. TBI rats were subjected to a
moderate 2.4atm FPI while sham rats underwent identical procedure without the injury given.
Animals were tested using the Morris Water Maze at day 11 post-injury. There was no
significant difference in the average swim speed between the two groups demonstrating that the
TBI rats have recovered their motor function sufficiently enough to swim. Data are expressed as
mean ± SD and analyzed using repeated measures ANOVA (Sham vs TBI group) (SigmaStat3.0;
SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.
41
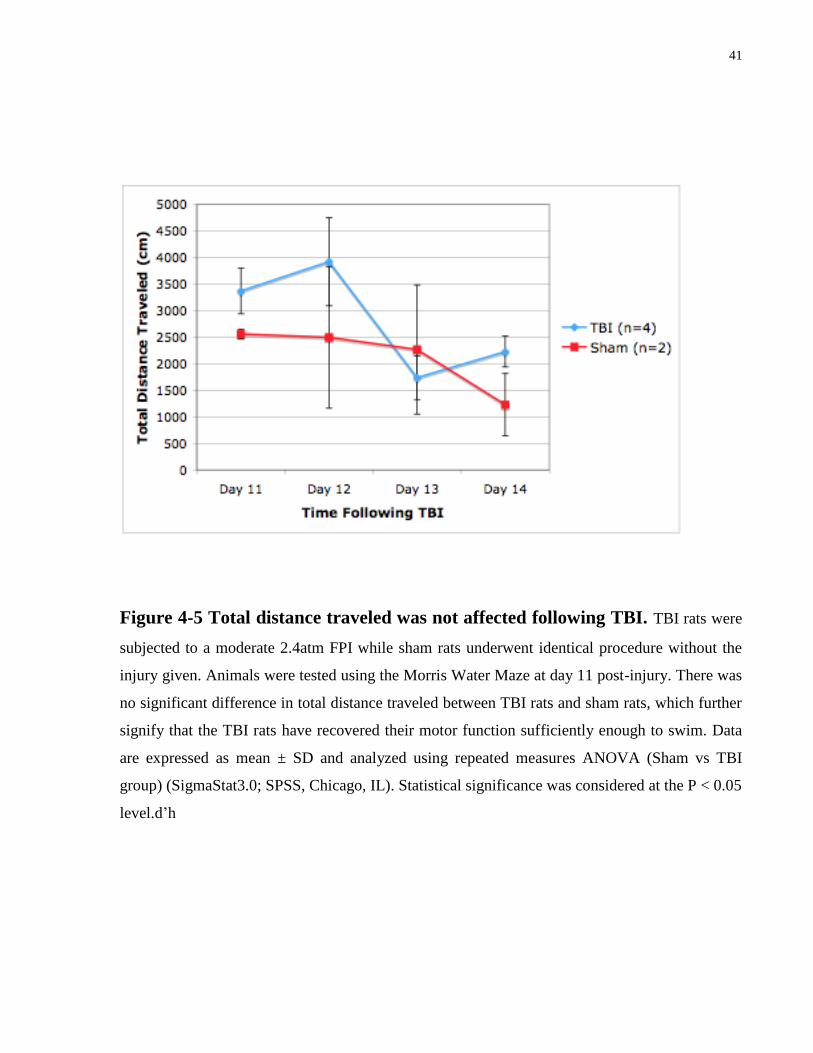
Figure 4-5 Total distance traveled was not affected following TBI. TBI rats were
subjected to a moderate 2.4atm FPI while sham rats underwent identical procedure without the
injury given. Animals were tested using the Morris Water Maze at day 11 post-injury. There was
no significant difference in total distance traveled between TBI rats and sham rats, which further
signify that the TBI rats have recovered their motor function sufficiently enough to swim. Data
are expressed as mean ± SD and analyzed using repeated measures ANOVA (Sham vs TBI
group) (SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05
level.d’h
42
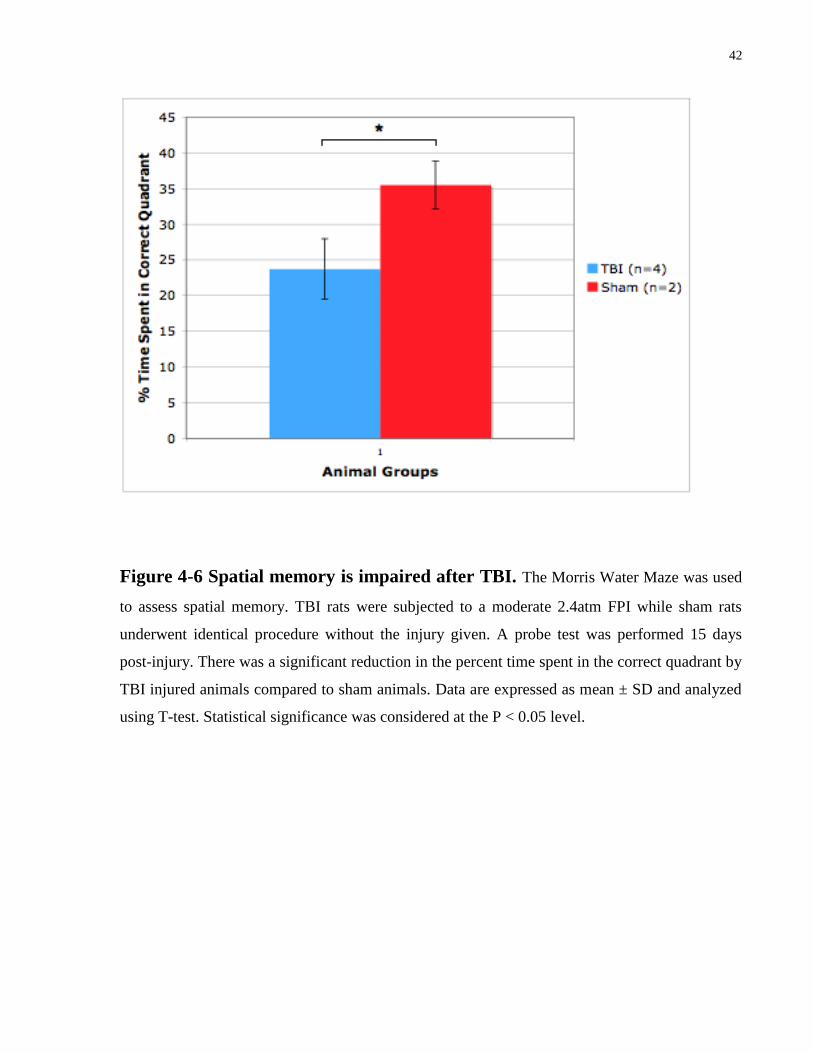
Figure 4-6 Spatial memory is impaired after TBI. The Morris Water Maze was used
to assess spatial memory. TBI rats were subjected to a moderate 2.4atm FPI while sham rats
underwent identical procedure without the injury given. A probe test was performed 15 days
post-injury. There was a significant reduction in the percent time spent in the correct quadrant by
TBI injured animals compared to sham animals. Data are expressed as mean ± SD and analyzed
using T-test. Statistical significance was considered at the P < 0.05 level.

43
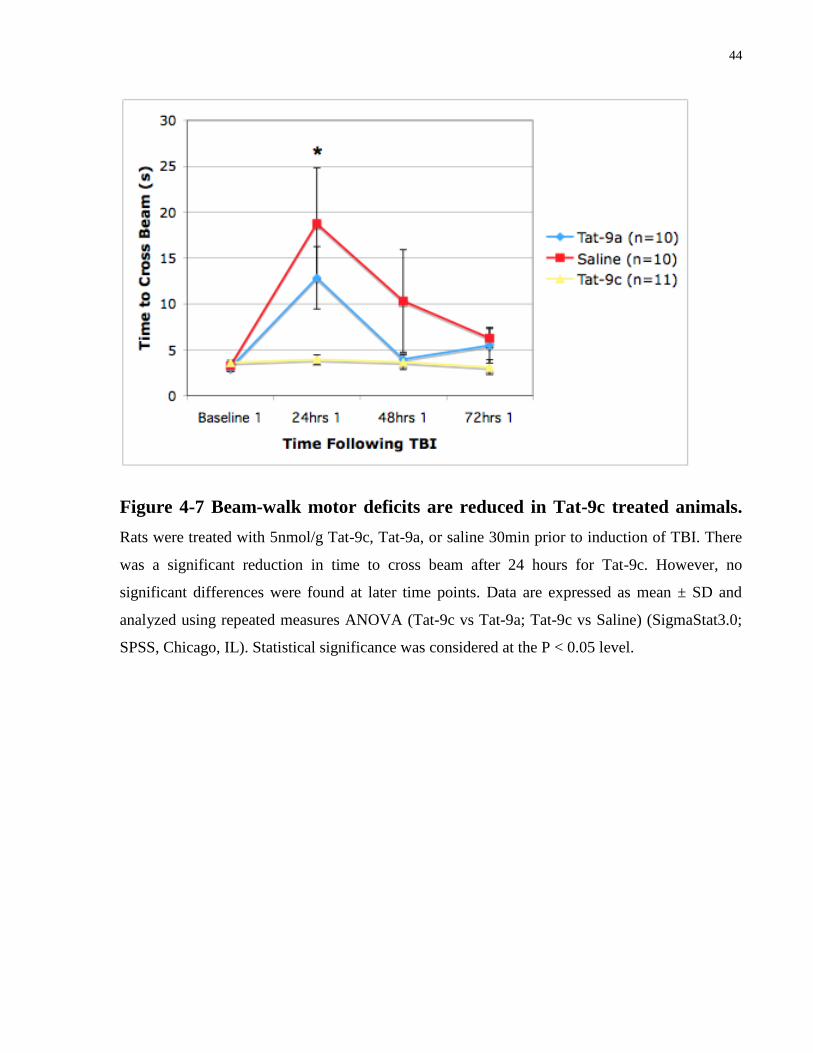
4.2 Effects of Tat-9c on Motor Behaviour Function Following TBI
To examine Tat-9c’s properties, a larger study using 20 male Sprague-Dawley rats was
conducted to further characterize behavioural differences between the treatment groups. Rats
were treated with either 5nmol/g Tat-9c, Tat-9a, or saline 30min prior to FPI. At 1, 2, and 3 days
post-injury, Beam-Walk and Rota-Rod performance was assessed. The Beam-Walk results
showed a trend towards behavioural improvement with Tat-9c but only demonstrated statistical
significance at the 24 hours post-injury time point (Figure 4-7). However, overall, Tat-9c greatly
reduced the number of falls on the Beam-Walk when compared to other treatments (Figure 4-8).
On the other hand, the Rota-Rod assay surprisingly did not find any statistical differences
between Tat-9c and the other treatment groups at all time points following TBI (Figure 4-9).
Possible explanations for this are considered in the Discussion Section: 5.2 on pg 67.
44
Figure 4-7 Beam-walk motor deficits are reduced in Tat-9c treated animals.
Rats were treated with 5nmol/g Tat-9c, Tat-9a, or saline 30min prior to induction of TBI. There
was a significant reduction in time to cross beam after 24 hours for Tat-9c. However, no
significant differences were found at later time points. Data are expressed as mean ± SD and
analyzed using repeated measures ANOVA (Tat-9c vs Tat-9a; Tat-9c vs Saline) (SigmaStat3.0;
SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.
45
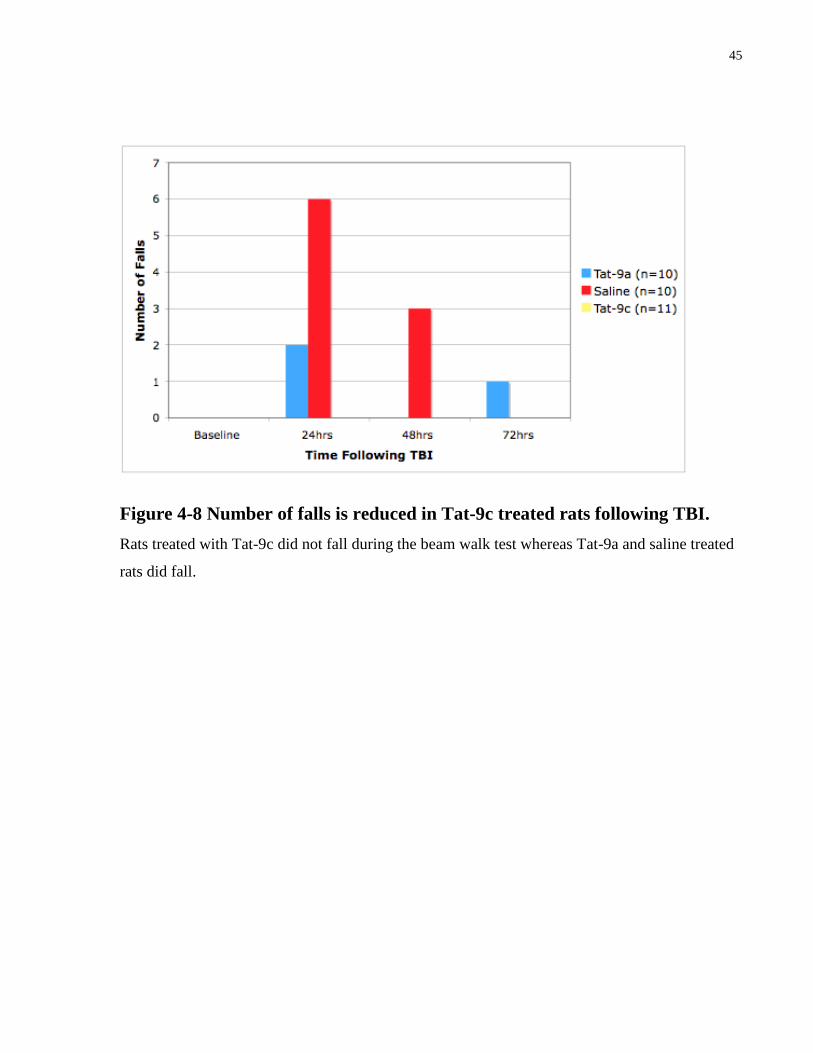
Figure 4-8 Number of falls is reduced in Tat-9c treated rats following TBI.
Rats treated with Tat-9c did not fall during the beam walk test whereas Tat-9a and saline treated
rats did fall.
46
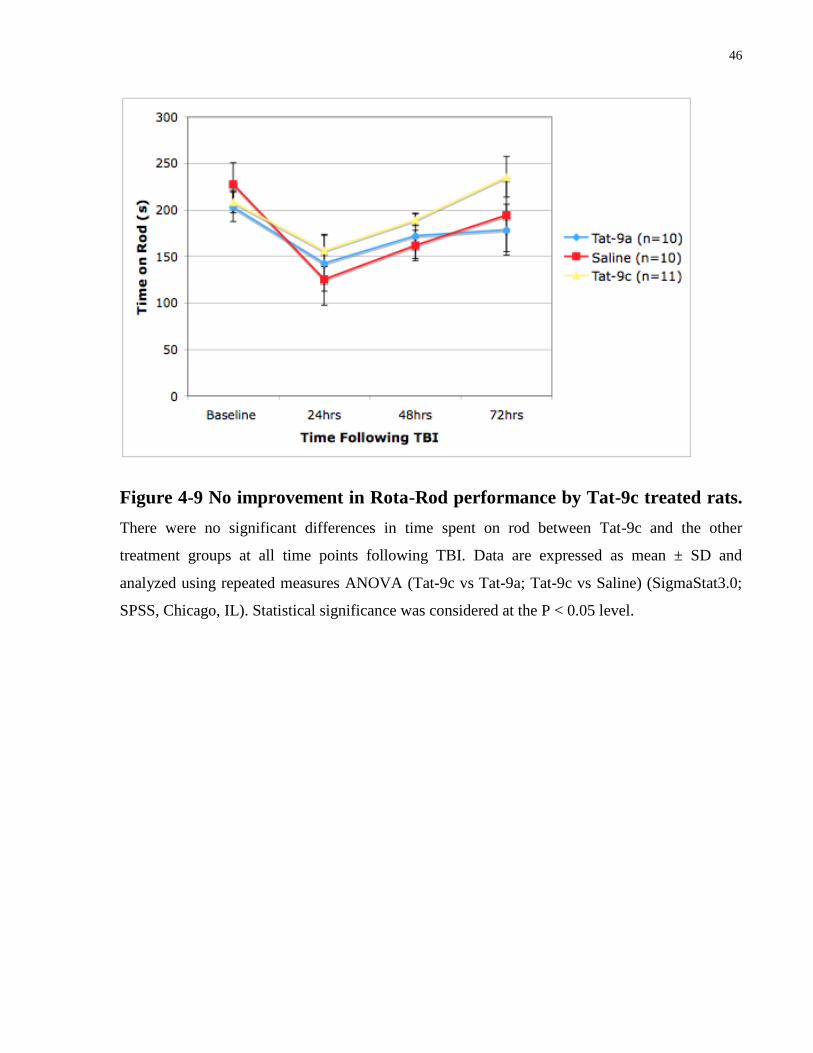
Figure 4-9 No improvement in Rota-Rod performance by Tat-9c treated rats.
There were no significant differences in time spent on rod between Tat-9c and the other
treatment groups at all time points following TBI. Data are expressed as mean ± SD and
analyzed using repeated measures ANOVA (Tat-9c vs Tat-9a; Tat-9c vs Saline) (SigmaStat3.0;
SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.

47
4.3 Effects of Tat-9c on Memory Function Following TBI
A further study involving 89 male Sprague-Dawley rats was performed to examine cognitive
behavioural differences between the treatment groups: sham control, saline control, Tat-9a, and
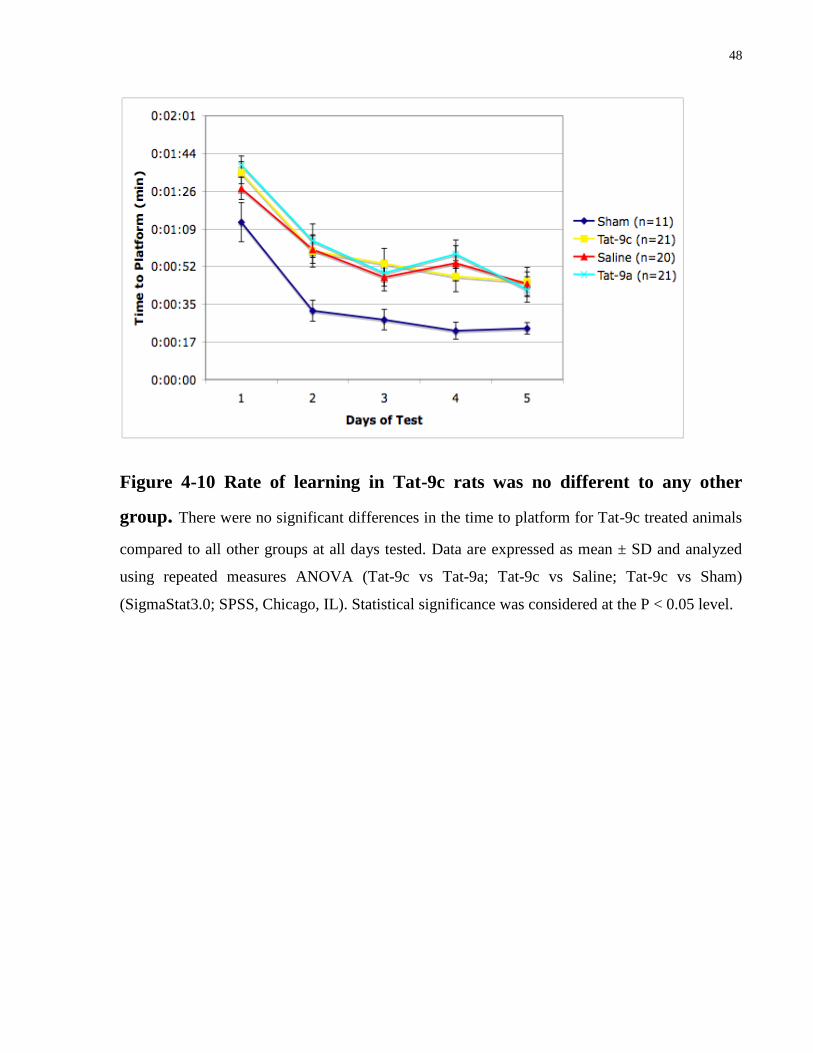
Tat-9c. Results show no significant differences in rate of learning for Tat-9c treated animals
compared to all other groups during testing days 11-15 post injury (Figure 4-10). According to
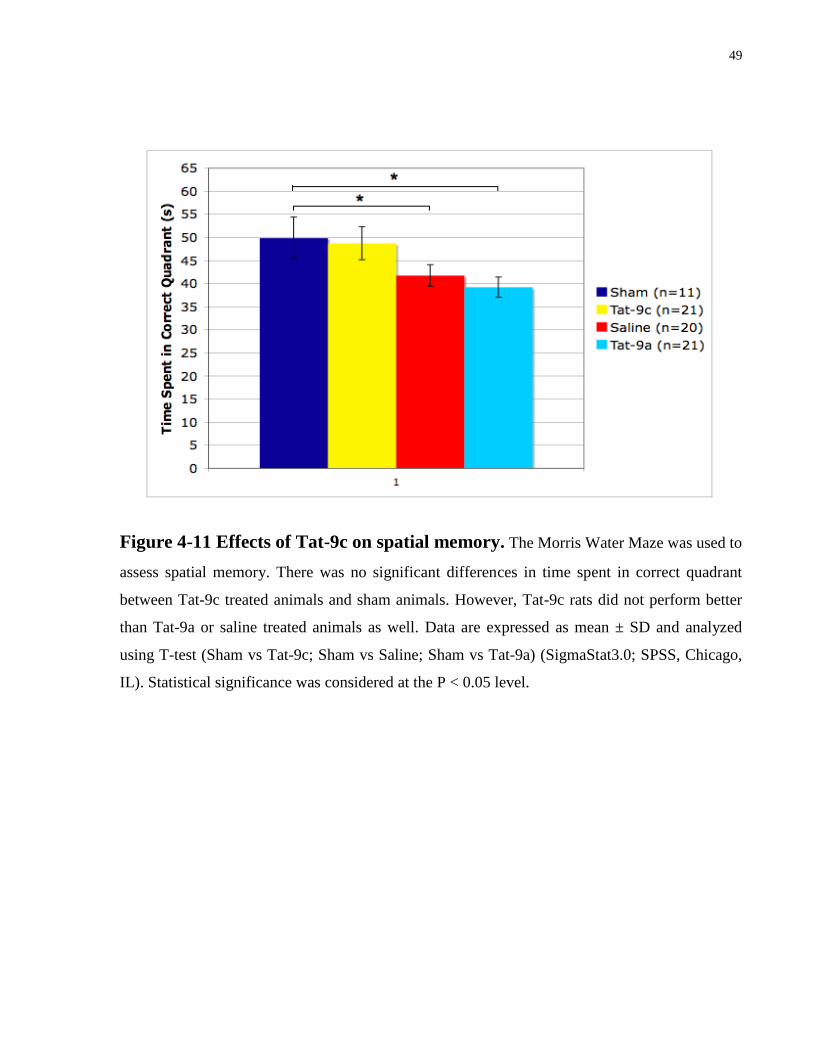
Fig. 4-11, there was also no statistical differences between Tat-9c treated animals and sham
animals in the memory probe test while Tat-9a and saline treated animals performed significantly
poorer than sham in the probe test (Figure 4-11). However, by comparing Tat-9c, Tat-9a, and
saline to each other, there were also no statistical differences. These ambiguous results are
reviewed in the Discussion Section: 5.4 on pg 70-72.
48
Figure 4-10 Rate of learning in Tat-9c rats was no different to any other
group. There were no significant differences in the time to platform for Tat-9c treated animals
compared to all other groups at all days tested. Data are expressed as mean ± SD and analyzed
using repeated measures ANOVA (Tat-9c vs Tat-9a; Tat-9c vs Saline; Tat-9c vs Sham)
(SigmaStat3.0; SPSS, Chicago, IL). Statistical significance was considered at the P < 0.05 level.
49
Figure 4-11 Effects of Tat-9c on spatial memory. The Morris Water Maze was used to
assess spatial memory. There was no significant differences in time spent in correct quadrant
between Tat-9c treated animals and sham animals. However, Tat-9c rats did not perform better
than Tat-9a or saline treated animals as well. Data are expressed as mean ± SD and analyzed
using T-test (Sham vs Tat-9c; Sham vs Saline; Sham vs Tat-9a) (SigmaStat3.0; SPSS, Chicago,
IL). Statistical significance was considered at the P < 0.05 level.

50
Chapter 5 DISCUSSION
5.1 Behaviour Assays Utilized to Assess Severity of TBI
In order to assess severity of TBI, three assays were tested: Beam-Walk, Rota-Rod, and Morris
Water Maze. All three assays were able to significantly discriminate injured animals from sham.
It was important to validate the Beam-Walk task as it was modified and consists of components
based from the original model created by Feeney et al., 1982 and the one used by Hallam et al.,
2004 who designed the Beam-Walk with four pegs as barriers. Another reason why it was
important to test the Beam-Walk and Rota-Rod test was that the methods of this study requires
the same animals to be trained and tested on the Beam-Walk and Rota-Rod together. This has not
been done previously and it was necessary to observe if there was any interference of the training
of one task with another present. The process of forgetting has long been observed and studied
and the standard school of thought is that it is caused by interference. Interference can be
proactive (forgetting caused by prior learning) or retroactive (forgetting caused by subsequent
learning) (Wixted, 2004). Because of the interference phenomenon, we needed to verify that
failure to perform any of the behavior tasks was due to the injury and not to interference.
Luckily, Figures 4-1 and 4-2 were consistent with other findings which verify that interference
did not play a role in our experiments. Another important feature in our Beam-Walk and Rota-
Rod tests was the addition of baseline testing for all animals. This allowed for the use of each
animal to be their own control and also assisted in detecting any animals that were
unintentionally injured prior to TBI. The Beam-Walk and Rota-Rod results were consistent with
finding by Hamm et al., 1994 where data demonstrated a significant difference in motor function
between TBI and sham injured animals. It was also similar in that performance of injured
animals improved gradually to baseline values throughout the time period confirming that the
Beam-Walk and Rota-Rod tasks are only effective at assessing TBI severity one week post-
injury.
The Morris Water Maze test was included in this study to investigate the cognitive function of
animals following TBI. The Morris Water Maze specifically measures new acquisition of a
spatial reference memory task. The extensive memory deficits observed by the injured group is
similar to previous studies (Smith et al., 1991; and Hallam et al., 2004), which found that TBI

51
can cause deficits in spatial memory. TBI animals appeared to be able to retain the ability to
learn. This is also consistent with reports from Reid et. al. 2008 who showed that the learning
curve for TBI and sham animals was not significantly different.
5.2 Tat-9c Protects Motor Behaviour Function Following TBI
Tat-9c appears to improve motor function in rats following TBI in the Beam-Walk study but
only at the 24 hours post-injury time point. Changes were most pronounced on the Beam-Walk
task where complex coordination with cognitive components was required to successfully
traverse the beam. Not only did the time latency improve in the Tat-9c treated group at the 24
hours time point but there was also a decreased incidence of falls from the beam at all time
points. However, it is difficult to judge the effects of Tat-9c beyond 24 hours post-injury since
the animals administered with negative controls, Tat-9a and saline, had also quickly recovered
their motor function to the point where Tat-9a animals performed identically to their baseline
values after the 48 hours post-injury time point. It was surprising to see that Tat-9a animals
showed such quick signs of recovery as Tat-9a was meant to be a negative control and should
have no positive effects. This phenomenon may be due to the tat sequence alone as it was first
discovered by Vaslin et al in 2009 that unconjugated tat demonstrated a neuroprotective effect
against mild NMDA excitotoxicity however the underlying mechanisms of Tat’s interference
with the NMDAR signaling is still unknown.
Furthermore, it was interesting to find no significant differences in the Rota-Rod test between
treatment groups. As the Rota-Rod test is described to be more sensitive and proficient at
assessing motor deficits following FPI (Hamm et al., 1994), it can be interpreted that Tat-9c was
unable to recover finer motor function required to perform the Rota-Rod test and not the Beam-
Walk test. Thus, while Tat-9c was able to rescue Beam-Walk deficits, subtler motor impairments
likely persisted and these were demonstrated using the more sensitive Rota-Rod assay. Another
explanation for the negative results could be that Tat-9c’s therapeutic effects were not distributed
evenly in the brain however no histology was done to examine this. A future experiment to
explore this would be to tag tat-9c with a fluorescent tag in order to determine its distribution
within the brain. It has been reported that locomotion induces activation of the sensorimotor
cortex however when accelerating, the prefrontal cortex and premotor cortex also becomes active
(Suzuki et al., 2004). Since the Rota-Rod test involves an accelerating rotating rod, different

52
areas of the brain were required compared to the Beam-Walk; therefore, the results could suggest
that Tat-9c plays a stronger role in different areas of the brain.
5.3 Effects of Tat-9c on Memory Function Following TBI
Tat-9c also appears to improve cognitive function in rats following TBI in the Morris Water
Maze study but only in the probe test for memory. Tat-9c had no effect on learning which is
what we expected since Tat-9c was not designed to improve existing cognitive function but to
protect against functional decline due to injury. Since TBI has already been shown to have no
effect on learning (Reid et al, 2008), we did not expect Tat-9c to play any effect either. The
results from the probe test revealed that there was no statistical differences between Tat-9c
treated animals and sham animals while Tat-9a and saline treated animals performed
significantly poorer than sham in the probe test. There were no significant differences between
the Tat-9c and Tat-9a or saline groups. Explanations for this are described below under
Limitations of the Study. Nonetheless, these results show that Tat-9c can protect memory
function following TBI but more subjects are required to be assured.
5.4 Limitations of the Study
5.4.1 Disadvantages of the FPI Model
One concern is the variability of injury presented by the lateral fluid percussion model. The FPI
model has been demonstrated to replicate many of the features associated with human TBI
(McIntosh et al., 1989, Pierce et al., 1998, Statler et al., 2001, Morales et al., 2005, Thompson et
al., 2005) and it is currently the most commonly used experimental model of TBI (Frey et al.,
2009). However, investigators utilizing this model are often confronted with two main
disadvantages: the model’s sensitivity to individual operators and its sensitivity to slight
differences in the piston’s o-rings and cylinder (Frey et al., 2009). It is absolutely necessary for
the operator to skillfully position and release the pendulum consistently throughout an
experiment.

53
5.4.2 Limitations of Tat-9c
The mechanism of action of Tat-9c provides the drug with a very limited and short therapeutic
window. The drug would need to be administered rapidly following TBI to provide even a
chance of interfering with the initial up regulation of free radical production which occurs in the
first 60 minutes post trauma (Hall et al., 2010). The attractiveness of Tat-9c as a potential
therapy for TBI is diminished due to its clinical impracticality however, when there are no
alternative therapies at present time, limited is still better than nothing at all.
5.4.3 The Complex nature of TBI
5.4.3.1 Cell Death Pathway Targets
Another reason for the lack of successful pharmacological intervention developed for TBI may
be that many proposed therapies have a mechanism of only individual specific targets on the cell
death pathway. It is becoming increasingly evident that various cell death pathways do not exist
independently of each other, and two or more of these pathways are likely active following TBI.
These interactions may either be synergistic where they assist each other in exacerbating cell
death or competitive, where they slow the progression of cell death. It is difficult to observe
these interactions in in vivo experiments however there is evidence from in-vitro studies
suggesting that these interactions may play a large role in determining the efficacy of
neuroprotective drugs (Pike et al., 2000; Mohr et al., 1997; Guttmann et al., 2001). Therefore,
designing a drug that only targets a small portion of the cell death pathways following TBI such
as Tat-9c may be limiting its therapeutic abilities and hence the observed efficacy of the drug
was not as robust as expected.
5.4.3.2 Limitations and Variability Amongst Subjects
Rodents have become the most widely experimented subject for examining TBI within the
laboratory. They are easy to handle individually while carrying out the surgery and also simple to
utilize in large numbers. The costs are relatively low compared to larger mammals and with the
availability of knockout animals, desired genes can be genetically ablated (or overexpressed with
the use of transgenic mice), making the study of a certain gene or protein of interest possible in
rodents (Morganti-Kossmann et al. 2010). However, no matter how severe a TBI can be induced

54
in rodents, seldom will they enter a state of coma that is often seen with TBI patients clinically
(Morganti-Kossmann et al. 2010). This represents one limitation to the use of rodent models as
severe disabilities of TBI in humans will not display in the rodent and any potential therapies
discovered to alleviate severe symptoms in the rodent will not translate to humans.
Another limitation is that responses to TBI varies from species to species and even within the
same strain of rodent, individual rodents have been shown to behave differently in neurological
motor and cognitive testing (Morganti-Kossmann et al. 2010) and thereby skewing any
measurements of potential therapies.

55
5.5 Future Directions
Although this study provided a crucial first step in the investigation of Tat-9c as a possible drug
therapy for TBI through in vivo models, there is still much more research that needs to be
accomplished.
The biggest question that needs to be resolved is the mechanism by which Tat-9c provides
neuroprotection. The initial hypothesized mechanism of action of Tat-9c is that it acts as an
intracellular scavenger reacting with peroxynitrite derived free radicals. However, there can be
other scenarios in which Tat-9c is attenuating behavioural deficits. For example, since Tat-9c
was designed as a polypeptide, it avoids a number of potential neurotoxic mechanisms attributed
to L-cysteine or homocysteine, which as a single amino acid, can alter NMDARs and inhibit
glutamate uptake in astrocytes (Janaky et. al., 2000).
The present study could also be enhanced by investigating the therapeutic window of Tat-9c.
Although Tat-9c showed promise when injected as a single bolus pre-injury, this application is
not very useful clinically. It is clear from the Beam-Walk results that Tat-9c’s therapeutic
potential can come in effect as early as 24 hours after TBI but it is unknown if Tat-9c can still
provide benefits when given post-injury. For example, the protection provided by Tat-9c may
simply be a result of the very efficient ROS scavenging at the time of impact. An ideal future
experiment would be to test Tat-9c’s effectiveness each hour post TBI and determine the latest
time-point it can be administered and still attenuate behavioural deficits.
In addition, as the pathophysiology of TBI is multi-faceted and complex, the combination of
other potential drug therapies with Tat-9c can also be assessed. For instance, if Tat-9c is used in
combination with anti-apoptotic drugs, the net beneficial effect may be potentiated.
Furthermore, due to the variability of disabilities seen amongst individual animals subjected to
the uniform experimental protocol, perhaps a better way to approach testing for potential drug
therapies is to first categorize the severity of injury AFTER the injury has occurred and then test
and compare the severity groups amongst each other. For example, following application of
injury, animals can then be divided between those who tolerated the infarct relatively well
compared to those who may have required chest compressions. Perhaps the results of Tat-9c
would have been more dramatic in one group over the other but if combined together, as

56
performed in this study, the results would have dampened and this may also help explain the
large error bars.
Overall, initial experiments with Tat-9c show that it appears to be a potential candidate for a TBI
drug but further experiments are required to confirm its mechanism of action and define its
therapeutic limits.

57
Chapter 6 CONCLUSIONS
One of the major findings of this study includes the validation of various behaviour paradigms to
assess severity of TBI. Without this step, further evaluation of potential drug therapies for TBI
would not be as thorough. Three assays were tested: Beam-Walk, Rota-Rod, and Morris Water
Maze. All three models were able to significantly discriminate injured animals from sham. The
Beam-Walk and Rota-Rod assays illustrated that due to innate recovery, these models were best
utilized up to one week post-injury. These findings are consistent with previous research
presented by Hamm et al (1994). The Morris Water Maze results are also consistent with those
found by Reid et al (2008) where it is demonstrated that TBI has no effect on spatial learning but
plays a crucial role in memory. Once the behaviour methodology was established, evaluation of
Tat-9c as a potential drug therapy for TBI was performed. Overall, the results demonstrate that
Tat-9c has the potential to provide early and late behavioural protection following TBI. The
Beam-Walk model highlighted the early beneficial effects of Tat-9c where the motor function of
those animals administered with Tat-9c was significantly superior than those given saline or Tat-
9a. Moreover, in the Morris Water Maze test, there was no statistical difference between Tat-9c
treated animals and sham animals in the memory probe test while Tat-9a and saline treated
animals performed significantly poorer than sham suggesting that Tat-9c can still provide late
behavioural protection as much as 15 days post-injury. These significant findings demonstrate
that TAT-9c may be a potential therapy for improving motor function and memory following
TBI.

58
References and Links
1. Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW,
and Tymianski M. (2002) Treatment of ischemic brain damage by perturbing
NMDA receptor-PSD-95 protein interactions. Science. 298(5594): 846-50.
2. Abelson-Mitchell N. (2008) Epidemiology and prevention of head injuries:
literature review. Journal of Clinical Nursing. 17(1): 46-57.
3. Alvarez B, Ferrer-Sueta G, Freeman BA, and Radi R (1999) Kinetics of
peroxynitrite reaction with amino acids and human serum albumin. J Biol Chem
274:842-848.
4. Annoni JM, Beer S, and Kesselring J. (1992) Severe traumatic brain injury –
epidemiology and outcome after 3 years. Disability and Rehabilitation. 14(1): 23-6.
5. Arundine M and Tymianski M. (2003) Molecular mechanisms of calcium-
dependent neurodegeneration in excitotoxicity. Cell Calcium. 34:325–337.
6. Arundine M, Aarts M, Lau A, and Tymianski M. (2004) Vulnerability of central
neurons to secondary insults after in vitro mechanical stretch. J. Neurosci
24(37):8106-8123.
7. Aulak KS, Koeck T, Crabb JW, and Stuehr DJ. (2004) Dynamics of protein
nitration in cells and mitochondria. Am J Physiol Heart Circ Physiol. 286: H30–
H38.

59
8. Beauchamp K, Mutlak H, Smith WR, Shohami E, and Stahel PF. (2008)
Pharmacology of traumatic brain injury: where is the “golden bullet”? Mol Med.
14(11-12):731-40.
9. Beckman JS, Ischiropoulos H, Zhu L, van der WM, Smith C, Chen J, Harrison J,
Martin JC, Tsai M (1992) Kinetics of superoxide dismutase- and iron-catalyzed
nitration of phenolics by peroxynitrite. Arch Biochem Biophys 298: 438-445.
10. Beckman JS. (1991) The double-edged role of nitric oxide in brain function and
superoxide-mediated injury. J Dev Physio 15: 53-59.
11. Bramlett HM, and Dietrich WD. (2004) Pathophysiology of cerebral ischemia and
brain trauma: similarities and differences. Journal of Cerebral Blood Flow and
Metabolism 24:133-150.
12. Buccafusco JJ (2009) Methods of Behaviour Analysis in Neuroscience, 2nd
edition.
Chapter 13: Spatial Navigation (Water Maze) Tasks.
13. Cernak I (2005) Animal Models of Head Trauma. NeuroRx 2(3): 410-422.
14. Cherian L, Hlatky R., and Robertson C.S. (2004) Nitric oxide in traumatic brain
injury. Brain Pathology 4:195-201.
15. Choi DW (1985) Glutamate neurotoxicity in cortical cell culture is calcium
dependent. Neurosci Lett 58: 293-297.
16. Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7: 369-
379.

60
17. Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J,
Loeffert JE, Nathaniel PD, Jin KL, and Graham SH (2000) Caspase-3 mediated
neuronal death after traumatic brain injury in rats. J Neurochem 74: 740-753.
18. Cobbs CS, Fenoy A, Bredt DS, and Noble LJ. (1997) Expression of nitric oxide
synthase in the cerebral microvasculature after traumatic brain injury in the rat.
Brain Res 751: 336-338.
19. Czeiter E, Buki A, Bukovics P, Farkas O, Pal J, Kovesdi E, Doczi T, Sandor J.
(2009) Calpain inhibition reduces axolemmal leakage in traumatic axonal injury.
Molecules 14(12): 5115-23.
20. D’Hooge R and De Deyn PP. (2001) Applications of the morris water maze in the
study of learning and memory. Brain Res Brain Res Rev. 36(1): 60-90.
21. Davis AE. (2000) Mechanisms of traumatic brain injury: biomechanical, structural
and cellular considerations. Critical Care Nursing Quarterly. 23(3): 1-13.
22. Dawodu ST. (2003) Traumatic Brain Injury: Definition, Epidemiology,
Pathophysiology. Available at: Http://www.emedicine.com/pmr/topic212.htm
(Accessed on 12 July 2009).
23. Dawson VL, Dawson TM, London ED, Bredt DS, and Snyder SH (1991) Nitric
oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad
Sci USA 88: 6368-6371.

61
24. Dawson VL, Dawson TM, Bartley DA, Uhl GR, and Snyder SH (1993)
Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J
Neurosci 13: 2651-2661.
25. Dawson DA, Graham DI, McCulloch J, and Macrae IM (1994) Anti-ischaemic
efficacy of a nitric oxide synthase inhibitor and a N-methyl-D-aspartate receptor
antagonist in models of transient and permanent focal cerebral ischaemia. Br J
Pharmacol 113: 247-253.
26. Dawson VL, Kizushi VM, Huang PL, Snyder SH, and Dawson TM (1996)
Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-
deficient mice. J Neurosci 16: 2479-2487.
27. Dempsey RJ, Raghavendra Rao VL (2003) Cytidinediphosphocholine treatment to
decrease traumatic brain injury-induced hippocampal neuronal death, cortical
contusion volume, and neurological dysfunction in rats. J. Neurosurg. 98: 867-73.
28. Di Giorgio AM, Hou Y, Zhao X, Zhang B, Lyeth BG, Russell MJ. (2008) Dimethyl
sulfoxide provides neuroprotection in a traumatic brain injury model. Restor Neurol
Neurosci 26(6): 501-7.
29. Dimlich RV, Tornheim PA, Kindel RM, Hall ED, Braughler JM, and McCall JM.
(1990) Effects of a 21-aminosteroid (U-74006F) on cerebral metabolites and edema
after severe experimental head trauma. Adv Neurol 52: 365-75.
30. Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, and Hayes RL (1991) A
controlled cortical impact model of traumatic brain injury in the rat. J Neurosci
Methods. 39(3):253-62.

62
31. Dunham NW and Miya TS. (1957) A note on a simple apparatus for detecting
neurological deficits in rats and mice. J Am Pharm Assoc 46(3): 208-9.
32. Faden AI, O’Leary DM, Fan L, Bao W, Mullins PG, and Movsesyan VA. (2001)
Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in
vitro and improves outcome after brain trauma. Exp. Neurol. 167: 435-444.
33. Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, and Barsoum J.
(1994) Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad
Sci USA. 91(2): 664-8.
34. Feeney, DM, Boyeson MG, Linn RT, Murray HM and Dail WG. (1981) Responses
to cortical injury: I. Methodology and local effects of contusions in the
rat. Brain Res. 211(1):67–77.
35. Fiskum G, Rosenthal RE, Vereczki V, Martin E, Hoffman GE, Chinopoulos C, and
Kowaltowski A. (2004) Protection against ischemic brain injury by inhibition of
mitochondrial oxidative stress. Journal of Bioenergetics and Biomembranes.
36(4):347-52.
36. Frey LC, Hellier J, Unkart C, Lepkin A, Howard A, Hasebroock K, Serkova N,
Liang L, Patel M, Soltesz I, and Staley K. (2009) A novel apparatus for lateral fluid
percussion injury in the rat. J Neurosci Methods 177(2): 267-72.
37. Gahm C, Holmin S, and Mathiesen T. (2000). Temporal profiles and cellular
sources of three nitric oxide synthase isoforms in the brain after experimental
contusion. Neurosurgery 46: 169-177.

63
38. Gennerelli TA, Seqawa H, and Wald U (1983) Physiological response to angular
acceleration of the head. Head Injury: Basic and Clinical Aspects. Raven Press,
New York
39. Gibson CL, Coughlan TC, and Murphy SP. (2005) Glial nitric oxide and ischemia.
Glia 50:417–426.
40. Giza CC, and Hovda DA. (2001) The neurometabolic cascade of concussion.
Journal of Athletic Training. 36(3): 228-35.
41. Gow AJ, Chen Q, Gole M, Themistocleous M, Lee VM, and Ischiropoulos H
(2000) Two distinct mechanisms of nitric oxide-mediated neuronal cell death show
thiol dependency. Am J Physiol Cell Physiol 278: C1099-C1107.
42. Graham DI, Raghupathi R, Saatman KE, Meaney D, McIntosh TK (2000) Tissue
tears in the white matter after lateral fluid percussion injury in the rat: relevance to
human brain injury. Acta Neuropathol 99: 117-124.
43. Grasso G (2007) Neuroprotection by erythropoietin administration after
experimental traumatic brain injury. Brain Res 1182: 92-105.
44. Greve MW, and Zink, BJ. (2009) Pathophysiology of Traumatic Brain Injury.
Mount Sinai Journal of Medicine. 76: 97-104.
45. Guerrero JL, Thurman DJ, and Sniezek JE. (2000) Emergency department visits
associated with traumatic brain injury: United States, 1995-1996. Brain Injury.
14(2): 181-6.

64
46. Gurdjian ES, and Webster JF. (1943) Experimental head injury with special
reference to the mechanical factors in acute trauma. Surg. Gynecol. Obstet. 76: 623-
34.
47. Guttmann RP, Baker DL, Seifert KM, Cohen AS, Coulter DA, and Lynch DR
(2001) Specific proteolysis of the NR2 subunit at multiple sites by calpain. J
Neurochem 78: 1083-1093.
48. Hall ED, Vaishnav RA, and Mustafa AG. (2010) Antioxidant therapies for
traumatic brain injury. Neurotherapeutics 7(1): 51-61.
49. Hall ED, Yonkers PA, McCall JM, and Braughler JM. (1988) Effects of the 21-
aminosteroid U74006F on experimental head injury in mice. J Neurosurg 68: 456-
61.
50. Hallam TM, Floyd CL, Folkerts MM, Lee LL, Gong QZ, Lyeth BG, Muizelaar JP,
and Berman RF. (2004) Comparison of behavioral deficits and acute neuronal
degeneration in rat lateral fluid percussion and weight-drop brain injury models. J.
Neurotrauma 21(5): 521-539.
51. Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, and Jenkins LW. (1994) The rotarod
test: an evaluation of its effectiveness in assessing motor deficits following
traumatic brain injury. J Neurotrauma 11(2): 197-96.
52. Hamm RJ (2001) Neurobehavioural assessment of outcome following traumatic
brain injury in rats: an evaluation of selected measures. J. Neurotrauma 18: 1207-
16.

65
53. Hayes RL, Stalhammer D, and Povlishock JT (1987) A new model of concussive
brain injury in the cat produced by extradural fluid volume loading. Brain Inj 1: 93-
112.
54. Headway (2001) Head Injury: The Hidden Disability. Headway, London.
55. Hein AM, and O’Banion MK. (2009) Neuroinflammation and memory: the role of
prostaglandins. Mol Neurobiol. 40(1): 15-32.
56. Hicks R, Soares H, Smith D, and McIntosh T. (1996) Temporal and spatial
characterization of neuronal injury following lateral fluid-percussion brain injury in
the rat. Acta Neuropathol. 91: 236-46.
57. Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. (2007)
The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation.
22(5): 341-53.
58. Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, and Beckman JS
(1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase.
Arch Biochem Biophys 298: 431-437.
59. Jain KK. (2008) Neuroprotection in traumatic brain injury. Drug Discovery Today
13(23-24): 1082-9.
60. Janaky R, Varga V, Hermann A, Saransaari P, and Oja SS. (2000) Mechanisms of
L-cysteine neurotoxicity. Neurochem Res 25(9-10): 1397-405.

66
61. Jones BJ and Roberts DJ. (1968) The quantitative measurement of motor inco-
ordination in naïve mice using an accelerating rotarod. J Pharm Pharmocol. 20(4):
302-4.
62. Keane RW, Kraydieh S, Lotocki G, Alonso OF, Aldana P, and Dietrich WD (2001)
Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J Cereb
Blood Flow Metab 21: 1189-1198.
63. Kermer P, Klöcker N, and Bähr M. (1999) Neuronal death after brain injury:
Models, mechanisms, and therapeutic strategies in vivo. Cell Tissue Res 298:383–
395.
64. Krishnaswamy G, and Chi DS. (2006) Mast Cells Methods and Protocols Chapter
19: Assays for Nitric Oxide Expression. Humana Press Inc pg. 246.
65. LaPlaca MC, Simon CM, Prado GR, and Cullen DK (2007) CNS injury
biomechanics and experimental models. Prog Brain Res 161: 13-26.
66. Lau A, Arundine M, Sun HS, Jones M, and Tymianski M. (2006) Inhibition of
caspase-mediated apoptosis by peroxynitrite in traumatic brain injury. J. Neurosci.
26(45):11540-11553.
67. Laurer HL, Meaney DF, Margulies SS, and McIntosh TK. (2002). Modeling brain
injury/trauma. Encyclopedia of the Human Brain. San Diego.
68. Liao ZB (2008) Recombinant human erythropoietin administration protects cortical
neurons from traumatic brain injury in rats. Eur. J. Neurol. 15: 140-9.

67
69. Lighthall JW (1988) Controlled cortical impact: A new experimental brain injury
model. J Neurotrauma. 5(1): 1-15.
70. Liu PK, Robertson CS, and Valadka A. (2002) The association between neuronal
nitric oxide synthase and neuronal sensitivity in the brain after brain injury. Ann N
Y Acad Sci. 962: 226-41.
71. Lucas DR, and Newhouse JP (1957) The toxic effect of sodium L-glutamate on the
inner layers of the retina. AMA Arch Ophthalmol 58: 193-201.
72. Marshall LF, Maas AI, Marshall SB, Bricolo A, Fearnside M, Iannotti F, Klauber
MR, Lagarrigue J, Lobato R, Persson L, Pickard JD, Piek J, Servadei F, Wellis
GN, Morris GF, Means ED, and Musch B. (1998) A multicenter trial on the
efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg 89(4): 519-
25.
73. McIntosh TK, Thomas M, Smith D, and Banbury M. (1992) The novel 21-
aminosteroid U74006F attenuates cerebral edema and improves survival after brain
injury in the rat. J Neurotrauma 9: 33-46.
74. McIntosh TK, Noble L, Andrews B, Faden AI (1987). Traumatic brain injury in the
rat: characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma
4: 119-134.
75. McIntosh TK, Vink R, and Noble L (1989) Traumatic brain injury in the rat:
characerization of a lateral fluid-percussion model. Neuroscience. 28: 233-44.
76. Mohr S, Zech B, Lapetina EG, and Brune B (1997) Inhibition of caspase-3 by S-
nitrosation and oxidation caused by nitric oxide. Biochem Biophys Res Commun
238: 387-391.
http://www.ncbi.nlm.nih.gov.myaccess.library.utoronto.ca/pubmed?term=%22Marshall%20LF%22%5BAuthor%5D
http://www.ncbi.nlm.nih.gov.myaccess.library.utoronto.ca/pubmed?term=%22Marshall%20SB%22%5BAuthor%5D

68
77. Moncada S, Bolanos JP. (2006) Nitric oxide, cell bioenergetics and
neurodegeneration. J Neurochem. 97(6): 1676-89.
78. Morales DM, Marklund N, Lebold D, Thompson HJ, Pitkanen A, Maxwell WL,
Longhi L, Laurer H, Maegele M, Neugebauer E, Graham DI, Stocchetti N, and
McIntosh TK. (2005) Experimental models of traumatic brain injury: Do we really
need to build a better mousetrap? Neuroscience 126(4): 971-989.
79. Morris RG, Garrud P, Rawlins JN, and O’Keefe J. (1982) Place navigation
impaired in rats with hippocampal lesions. Nature. 297(5868): 681-3.
80. Morganti-Kossmann MC, Yan E, and Bye N. (2010) Animal models of traumatic
brain injury: is there an optimal model to reproduce human brain injury in the
laboratory? Injury 41:S10-3.
81. Moro MA, Cardenas A, Hurtado O, Leza JC, and Lizasoain I. (2004) Role of nitric
oxide after brain ischaemia. Cell Calcium 36(3-4): 265-75.
82. Narayan RK, Wilberger JE, and Povlishock JT (1996) Neurotrauma. McGraw Hill:
New York.
83. Nilsson B, Ponten U, Voigt G. (1977) Experimental head injury in the rat. J.
Neurosurg. 47: 241-51.
84. Nolan S. (2005) Traumatic brain injury: a review. Critical Care Nursing Quarterly.
28(2): 188-194.

69
85. O’Connor WT, Smyth A, and Gilchrist MD. (2011) Animal models of traumatic
brain injury: A critical evaluation. Pharmacology & Therapeutics 130(2): 106-113.
86. Olney JW (1969) Brain lesions, obesity, and other disturbances in mice treated with
monosodium glutamate. Science 164: 719-721.
87. Ommaya AK, Goldsmith W, and Thibault L. (2002) Biomechanics and
neuropathology of adult and paediatric head injury. Br J Neurosurg. 16(2): 220-42.
88. Park E, Bell JD, and Baker AJ. (2008) Traumatic brain injury: Can the
consequences be stopped? Canadian Medical Association Journal 178(9): 1163-
1170.
89. Pierce JE, Smith DH, Trojanowski JQ, and McIntosh TK (1998). Enduring
cognitive, neurobehavioural and histopathological changes persist for up to one
year following severe experimental brain injury in rats. Neuroscience 87: 359-69.
90. Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, and Hayes RL (2000)
Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic
cell death in septo-hippocampal cell cultures. J Neurotrauma 17: 283-298.
91. Polster BM, Basanez G, Etxebarria A, Hardwick JM, and Nicholls DG (2005)
Calpain I induces cleavage and release of apoptosis-inducing factor from isolated
mitochondria. J Biol Chem 280: 6447-6454.
92. Prins ML, Lee SM, Cheng CL, Becker DP, and Hovda DA. (1996) Fluid percussion
brain injury in the developing and adult rat: a comparative study of mortality,
morphology, intracranial pressure and mean arterial blood pressure. Brain Res Dev
Brain Res. 95(2): 272-82.

70
93. Qiu J, Whalen MJ, Lowenstein P, Fiskum G, Fahy B, Darwish R, Aarabi B, Yuan J,
and Moskowitz MA (2002) Upregulation of the Fas receptor death-inducing
signaling complex after traumatic brain injury in mice and humans. J Neurosci 22:
3504-3511.
94. Radi R, Beckman JS, Bush KM, and Freeman BA (1991) Peroxynitrite-induced
membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide.
Arch Biochem Biophys 288: 481-487.
95. Rao VL, Dogan A, Bowen KK, and Dempsey RJ. (1999) Traumatic injury to rat
brain upregulates neuronal nitric oxide synthase expression and L-[3H]
nitroarginine binding. J Neurotrauma 16: 865-877.
96. Reid WM and Hamm RJ. (2008) Post-injury atomoxetine treatment improves
cognition following experimental traumatic brain injury. J Neurotrauma 25(3):
248-56.
97. Salgo MG, Bermudez E, Squadrito GL, and Pryor WA (1995a) Peroxynitrite causes
DNA damage and oxidation of thiols in rat thymocytes [corrected]. Arch Biochem
Biophys 322: 500-505.
98. Salgo MG, Stone K, Squadrito GL, Battista JR, and Pryor WA (1995b)
Peroxynitrite causes DNA nicks in plasmid pBR322. Biochem Biophys Res
Commun 210: 1025-1030.
99. Sanders MJ, Dietrich WD, and Green EJ (1999). Cognitive function following
traumatic brain injury: effects of injury severity and recovery period in a
parasagittal fluid-percussive injury model. J. Neurotrauma. 16: 915-25.

71
100. Sattler R, Charlton MP, Hafner M, and Tymianski M (1998) Distinct influx
pathways, not calcium load, determine neuronal vulnerability to calcium
neurotoxicity. J Neurochem 71: 2349-2364.
101. Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, and Tymianski M (1999)
Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by
PSD-95 protein. Science 284: 1845-1848.
102. Schalen W, Hansson L, Nordstrom G, and Nordstrom CH. (1994) Psychosocial
outcome 5-8 years after severe traumatic brain lesions and the impact of
rehabilitation services. Brain Injury 8: 49-63.
103. Shein NA, Grigoriadis N, Alexandrovich AG, Simeonidou C, Lourbopoulos A,
Polyzoidou E, Trembovler V, Mascagni P, Dinarello CA, and Shohami E. (2009)
Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional
recovery, and induces glial apoptosis following experimental traumatic brain injury.
FASEB J. 23(12): 4266-75.
104. Singh IN, Sullivan PG, Deng Y, Mbye LH, and Hall ED. (2006) Time course of
post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model
of focal traumatic brain injury: implications for neuroprotective therapy. Journal of
Cerebral Blood Flow & Metabolism. 26:1407–1418
105. Smith DH, Okiyama K, Thomas MJ, Claussen B, and McIntosh TK. (1991)
Evaluation of memory dysfunction following experimental brain injury using the
Morris water maze. J Neurotrauma 8(4): 259-69.

72
106. Smith DH, Chen XH, and Pierce JE (1997) Progressive atrophy and neuron death
for one year following brain trauma in the rat. J. Neurotruama. 14: 715-27.
107. Statler KD, Alexander H, and Vagni V. (2006) Isoflurane exerts neuroprotective
actions at or near the time of severe traumatic brain injury. Brain Res 1076: 216-24.
108. Sullivan PG, Thompson MB, and Scheff SW (1999) Cyclosporin A attenuates acute
mitochondrial dysfunction following traumatic brain injury. Experimental
Neurology 160: 226-34.
109. Summers CR, Ivins B, Schwab KA. (2009) Traumatic brain injury in the United
States: an epidemiologic overview. Mount Sinai Journal of Medicine. 76(2):105-10.
110. Suzuki M, Miyai I, Ono T, Oda I, Konishi I, Kochiyama T, and Kubota K. (2004)
Prefrontal and premotor cortices are involved in adapting walking and running
speed on the treadmill: an optical imaging study. Neuroimage. 23(3): 1020-6.
111. The Brain Injury Association of Canada. Available at www.biac-aclc.ca/
112. Thomale UW, Griebenow M, Kroppenstedt SN, Unterberg AW, and Stover JF
(2006) The effect of N-acetylcysteine on posttraumatic changes after controlled
cortical impact in rats. Intensive Care Med 32:149-155.
113. Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, and
McIntosh TK (2005) Lateral fluid percussion brain injury: a 15 year review and
evaluation. J Neurotrauma 22(1) 42-75.

73
114. Thurman D, and Guerrero J. (1999) Trends in hospitalization associated with
traumatic brain injury. JAMA. 282(10): 954-7.
115. Tymianski M, Charlton MP, Carlen PL, and Tator CH (1993) Source specificity of
early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci 13:
2085-2104.
116. Vaslin A, Rummel C, and Clarke PG. (2009) Unconjugated TAT carrier peptide
protects against excitotoxicity. Neurotox Res 15(2): 123-6.
117. Waxman EA and Lynch DR. (2005) N-methyl-D-aspartate Receptor Subtypes:
Multiple Roles in Excitotoxicity and Neurological Disease. Neuroscientist.
11(1):37–49.
118. Werner C, and Engelhard K. (2007) Pathophysiology of traumatic brain injury. Br J
Anaesth. 99(1): 4-9.
119. Wixted JT. (2004) The psychology and neuroscience of forgetting. Annu Rev
Psychol. 55: 235-69.
120. Wong J, Hoe NW, Zhiwei F and Ng I. (2005) Apoptosis and traumatic brain injury
Neurocritical Care 3(2):177–82.
121. Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, and Faden AI (1997)
Activation of CPP32-like caspases contributes to neuronal apoptosis and
neurological dysfunction after traumatic brain injury. J Neurosci 17: 7415-7424.

74
122. Ye Y, Quijano C, Robinson KM, Ricart KC, Strayer AL, Sahawneh MA, Shacka JJ,
Kirk M, Barnes S, ccavitti-Loper MA, Radi R, Beckman JS, and Estevez AG.
(2007) Prevention of peroxynitrite-induced apoptosis of motor neurons and PC12
cells by tyrosine-containing peptides. J Biol Chem 282:6324-6337.
123. Zhang X, Chen Y, Jenkins LW, Kochanek PM, and Clark RSB. (2005) Bench-to-
bedside review: Apoptosis programmed cell death triggered by traumatic brain
injury. Crit Care 9(1): 66–75.






![TAT-902S [1 650] TAT- 1 600 102S F] TAT-312V TAT-322V ...TAT-902S [1 650] TAT- 1 600 102S F] TAT-312V TAT-322V TAT-332S 1/2 1/2 1/2 1/2 1/2 1/2 I OOOX420 IOOOX500 1200X500 TAT-1 52S](https://static.fdocuments.us/doc/165x107/6125a0cefb88a6479b4afa46/tat-902s-1-650-tat-1-600-102s-f-tat-312v-tat-322v-tat-902s-1-650-tat-.jpg)











