
Systemic Lupus Erythematosus Immune Complexes Increase · PDF fileSystemic Lupus Erythematosus...
11
of May 25, 2018. This information is current as NK Cells dim Cells and CD319 on CD56 CD229 (LY-9) on Plasmacytoid Dendritic Family Members CD319 (CRACC) and Complexes Increase the Expression of SLAM Systemic Lupus Erythematosus Immune Rönnblom Maija-Leena Eloranta, Yenan T. Bryceson and Lars Niklas Hagberg, Jakob Theorell, Heinrich Schlums, ol.1301022 http://www.jimmunol.org/content/early/2013/08/16/jimmun published online 16 August 2013 J Immunol Material Supplementary 2.DC1 http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130102 average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2013 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on May 25, 2018 http://www.jimmunol.org/ Downloaded from by guest on May 25, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Systemic Lupus Erythematosus Immune Complexes Increase · PDF fileSystemic Lupus Erythematosus...

of May 25, 2018.This information is current as NK CellsdimCells and CD319 on CD56
CD229 (LY-9) on Plasmacytoid Dendritic Family Members CD319 (CRACC) andComplexes Increase the Expression of SLAM Systemic Lupus Erythematosus Immune
RönnblomMaija-Leena Eloranta, Yenan T. Bryceson and Lars Niklas Hagberg, Jakob Theorell, Heinrich Schlums,
ol.1301022http://www.jimmunol.org/content/early/2013/08/16/jimmun
published online 16 August 2013J Immunol
MaterialSupplementary
2.DC1http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130102
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2013 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Systemic Lupus Erythematosus Immune Complexes Increasethe Expression of SLAM Family Members CD319 (CRACC)and CD229 (LY-9) on Plasmacytoid Dendritic Cells andCD319 on CD56dim NK Cells
Niklas Hagberg,* Jakob Theorell,† Heinrich Schlums,† Maija-Leena Eloranta,*
Yenan T. Bryceson,† and Lars Ronnblom*
Patients with systemic lupus erythematosus (SLE) display an activated type I IFN system due to unceasing IFN-a release from
plasmacytoid dendritic cells (pDCs) stimulated by nucleic acid–containing immune complexes (ICs). NK cells strongly promote
the IFN-a production by pDCs; therefore, we investigated surface molecules that could be involved in the pDC–NK cell cross-talk.
In human PBMCs stimulated with RNA-containing ICs (RNA-ICs), the expression of the signaling lymphocyte activation molecule
(SLAM) family receptors CD319 and CD229 on pDCs and CD319 on CD56dim NK cells was selectively increased. Upregulation of
CD319 and CD229 on RNA-IC–stimulated pDCs was induced by NK cells or cytokines (e.g., GM-CSF, IL-3). IFN-a–producing
pDCs displayed a higher expression of SLAM molecules compared with IFN-a2 pDCs. With regard to signaling downstream of
SLAM receptors, pDCs expressed SHIP-1, SHP-1, SHP-2, and CSK but lacked SLAM-associated protein (SAP) and Ewing’s
sarcoma-activated transcript 2 (EAT2), indicating that these receptors may act as inhibitory receptors on pDCs. Furthermore,
pDCs from patients with SLE had decreased expression of CD319 on pDCs and CD229 on CD56dim NK cells, but RNA-IC
stimulation increased CD319 and CD229 expression. In conclusion, this study reveals that the expression of the SLAM receptors
CD319 and CD229 is regulated on pDCs and NK cells by lupus ICs and that the expression of these receptors is specifically altered
in SLE. These results, together with the observed genetic association between the SLAM locus and SLE, suggest a role for CD319
and CD229 in the SLE disease process. The Journal of Immunology, 2013, 191: 000–000.
Systemic lupus erythematosus (SLE) is an autoimmunedisease characterized by the presence of immune complexes(ICs) that deposit in tissue and cause inflammation (1). Such
SLE ICs are typically formed by autoantibodies and nucleic acid–containing autoantigens, which are released by dying cells (2). SLEICs promote tissue inflammation and immune activation throughseveral mechanisms, most importantly by complement activationand FcgR triggering of immune cells (3). In response to SLE ICs,macrophages and neutrophils produce proinflammatory cytokinesand proteolytic enzymes (4), whereas plasmacytoid dendritic cells(pDCs) produce type I IFN (5). The DNA- or RNA-containing SLE
ICs (interferogenic ICs) are internalized by pDCs through FcgRIIAand trigger TLR7 or TLR9 activation, ultimately leading to IFN-asecretion (5). A continuous activation of pDCs by interferogenicICs is a prominent feature of SLE and causes an increased ex-pression of type I IFN–induced genes (an IFN signature) in bothcirculating cells (6, 7) and affected tissues (8). An IFN signaturealso has been observed in other systemic autoimmune diseases thatexhibit interferogenic ICs (9, 10), suggesting that an activation ofthe type I IFN system may be important in the development of anautoimmune disease process. This assumption is also supported bythe observation that treatment of infectious or malignant diseaseswith IFN-a can trigger the development of several autoimmunediseases (11, 12).Previous studies showed that NK cells interact with pDCs to
potently enhance IFN-a production upon stimulation with viruses,synthetic oligonucleotides, or RNA-containing ICs (RNA-ICs)(13–15). Mechanistically, NK cells promoted RNA-IC–inducedIFN-a secretion by pDCs via soluble factors, such as MIP-1b, andvia LFA-1–dependent cell–cell interactions. In addition to IFN-a,the secretion of several cytokines and chemokines implicated inthe pathogenesis of SLE (e.g., IFN-g, IL-6, IL-8, MIP-1b, andTNF-a) was also enhanced in RNA-IC–stimulated pDC–NK cellcocultures (15). Consequently, pDC–NK cell cross-talk could beimportant in promoting sustained type I IFN production and theinflammatory response in systemic autoimmune diseases.In the current study, we aimed to identify cell surface molecules
of possible importance in the interaction between pDCs and NKcells when activated by RNA-ICs. By screening pDCs and NK cellsfor molecules that were regulated by RNA-ICs, consisting of SLE-IgG and U1snRNP particles, we identified two members of thesignaling lymphocyte activation molecule (SLAM) family that
*Section of Rheumatology, Department of Medical Sciences, Uppsala University, S-75185 Uppsala, Sweden; and †Department of Medicine, Center for Infectious Medicine,Karolinska Institute, Karolinska University Hospital Huddinge, S-141 86 Stockholm,Sweden
Received for publication April 16, 2013. Accepted for publication July 22, 2013.
This work was supported by grants from the Swedish Research Council, the SwedishRheumatism Foundation, the Torsten and Ragnar Soderberg Foundation, King GustafV’s 80-Year Foundation, Controlling Chronic Inflammatory Diseases with CombinedEfforts, the Knut and Alice Wallenberg Foundation, Clas Groschinsky’s Memorial Fund,the Jeanssons Foundation, the Ake Olsson Foundation for Hematological Research, theAke Wiberg Foundation, and the Karolinska Institute Research Foundation.
Address correspondence and reprint requests to Niklas Hagberg, Systemic Autoimmu-nity Group, Department of Medical Sciences, Clinical Research Department 3, Entrance85, 3rd Floor, S-751 85 Uppsala, Sweden. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: EAT2, Ewing’s sarcoma-activated transcript 2;HAIG, heat-aggregated IgG; IC, immune complex; MFI, median fluorescence in-tensity; pDC, plasmacytoid dendritic cell; RNA-IC, RNA-containing immune com-plex; SAP, signaling lymphocyte activation molecule–associated protein; SLAM,signaling lymphocyte activation molecule; SLE, systemic lupus erythematosus.
Copyright� 2013 by The American Association of Immunologists, Inc. 0022-1767/13/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1301022
Published August 16, 2013, doi:10.4049/jimmunol.1301022 by guest on M
ay 25, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

were strongly upregulated on pDCs and NK cells by RNA-ICs:CD319 (SLAMF7, CRACC, CS1) and CD229 (SLAMF3, LY9).In humans, the SLAM family of receptors includes seven mem-bers (CD150 [SLAMF1], CD48 [SLAMF2], CD229, CD244[SLAMF4, 2B4], CD84 [SLAMF5], CD352 [SLAMF6, NTB-A],and CD319) encoded by a locus on chromosome 1 (16). With theexception of CD48, which interacts with CD244, all of the SLAMmembers interact via homophilic interactions. The SLAM recep-tors have important immunomodulatory effects, and the SLAMlocus has been genetically associated with human SLE and mousemodels of SLE (17–21). Therefore, we investigated the mecha-nism for the regulation of CD319 and CD229 and the expressionof signaling molecules downstream of these receptors in pDCs andNK cells. Finally, the expression of all SLAM family members inpDCs and NK cells was compared between healthy individualsand patients with SLE.
Materials and MethodsCells
PBMCs were isolated from healthy blood donors or patients with SLE(Rheumatology Unit, Uppsala University Hospital) by density-gradientcentrifugation. For the comparison of SLAM expression on PBMCs fromhealthy individuals and patients with SLE, PBMCs were resuspended in 90%FCS and 10% DMSO and stored at280˚C pending flow cytometry analysis.NK cells and pDCs were isolated by negative selection, according to themanufacturer’s instructions (NK or pDC cell isolation kit; Miltenyi Biotec,Bergisch Gladbach, Germany), and monocyte-depleted PBMCs were pre-pared using CD14 MicroBeads (Miltenyi Biotec). Isolated pDCs and NKcells were routinely .95% BDCA-2+ and CD32CD56+, respectively. If nototherwise stated, cells were cultured in 96-well V-bottom plates (Nunc,Roskilde, Denmark) using macrophage serum–free medium supplementedwith HEPES (20 mM), penicillin (60 mg/ml), and streptomycin (100 mg/ml)(all from Invitrogen, Carlsbad, CA) at a concentration of 1 3 106 PBMCs/well, 50 3 103 pDCs/well, or 100 3 103 NK cells/well. In some experi-ments, cells were preincubated for 30 min with chloroquine (1 mg/ml),cycloheximide (10 mg/ml), or actinomycin D (1 mg/ml) (Sigma-Aldrich,St. Louis, MO). All cell cultures were incubated at 37˚C with 5% CO2.
Patients
All patients fulfilled at least four of the American College of Rheumatologycriteria for SLE. Patients were 50.2 6 12.0 y old. The disease activity wasdetermined by the SLE disease activity index (22); the median score was 2(range, 0–13). The study was approved by the local ethics committee atUppsala University, and informed consent was obtained from all patientsand controls.
Flow cytometry and mAbs
For flow cytometry, fluorochrome-conjugated mAbs to CD3 (HIT3a or SK7),CD14 (MfP9), CD56 (NCAM16.2), CD19 (HIB19) (all from BD Bio-sciences, San Jose, CA), CD8 (3B5; Invitrogen), CD11c (Bu15; BioLegend,San Diego, CA), BDCA-2 (AC114), and BDCA-4 (AD5-17F6) (both fromMiltenyi Biotec) were used to identify different cell types, as specified. Afluorescent, fixable dead cell marker (LIVE/DEAD near IR; Invitrogen) wasused to exclude dead cells from the analysis. Fluorochrome-conjugatedmAbs to CD69 (TP1.55.3; Beckman Coulter, Indianapolis, IN), CD150(A12[7D4]; eBioscience, San Diego, CA), CD48 (MEM102; BioLegend),CD229 (HLy9.1.25; BioLegend), CD244 (C1.7; BioLegend), CD84(CD84.1.21 [BioLegend] and 2G7 [eBioscience]), NTB-A (#292811; R&DSystems, Minneapolis, MN), and CD319 (162.1; BioLegend) were used todetermine surface expression. If not otherwise stated, pDCs were definedas CD32CD142CD562BDCA-2+, and NK cells were defined as CD32
CD142CD56+. Flow cytometry data were acquired on a FACSCanto II oran LSR Fortessa (BD Biosciences) and analyzed using FlowJo software(version 7.6 and 9.4; TreeStar, Ashland, OR).
For functional studies of pDCs, unlabeled mAbs to CD319 (162.1;BioLegend), CD229 (HLy9.1.25; BioLegend), or BDCA-2 (AC114; Mil-tenyi Biotec) were used.
Stimulation of cells
SLE IgG was purified by protein G chromatography from a patient serumcontaining autoantibodies to SmB, SmD, U1-RNPA, U1-RNP C, ribosomalPAg, histone, and dsDNA (2). U1snRNP particles were purified from HeLa
cells, as previously described (23). U1snRNP particles were used togetherwith SLE IgG at a final concentration of 2.5 mg/ml and 1 mg/ml, re-spectively. Heat-aggregated normal IgG (HAIG) was prepared by heatingOctagam (Octapharma, Stockholm, Sweden) at 63˚C for 60 min at a con-centration of 50 mg/ml and used at a final concentration of 1 mg/ml. TheTLR9 agonist ODN2216 (Cybergene, Stockholm, Sweden) was used at afinal concentration of 3 mg/ml.
For stimulation of cells with cytokines, GM-CSF (2 ng/ml; Leukine;Berlex, Montville, NJ), IFN-a (500 U/ml; Intron A; Schering-Plough,Bloomfield, NJ), IL-3 (10 ng/ml; R&D Systems), IFN-g (100 U/ml),IL-12 (10 ng/ml), IL-18 (100 ng/ml), MIP-1b (50 ng/ml), or TNF-a (10ng/ml; all from PeproTech, Rocky Hill, NJ) was used.
For receptor cross-linking of pDCs, cells were stimulated with RNA-ICsin 96-well flat-bottom ELISA plates coated with F(ab9)2 goat anti-mouseIgG Ab (10 mg/ml; Southern Biotechnology Associates, Birmingham,AL) and 10 mg/ml anti-CD319, anti-CD229, or anti–BDCA-2 or relevantisotype-control mAb.
IFN-a detection
Concentration of IFN-a in culture supernatants was determined using adissociation-enhanced lanthanide fluoroimmunoassay, as previously de-scribed (24).
For detection of intracellular IFN-a, PBMCs depleted of monocytes(1 3 106/well) were stimulated with RNA-ICs for 9 h, with the addition ofGolgiPlug (BD Biosciences) after 6 h. Cells were spun down and stainedfor surface markers, fixed in 1% paraformaldehyde, and permeabilizedwith 0.5% saponin (Sigma-Aldrich) before being stained with allophyco-cyanin-conjugated anti-human IFN-a (LT27:295; Miltenyi Biotec) andanalyzed by flow cytometry.
Western blot analysis
Isolated pDCs or NK cells were resuspended in lysis buffer (50 mMHEPES[pH 7.5], 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, and 1% Triton X-100) supplemented with protease inhibitor mixture (Pierce, Rockford, IL).After lysis, cells were centrifuged at 4˚C for 10 min at 14,000 3 g. Pro-teins were separated by SDS-PAGE (NuPAGE; Invitrogen) and transferredto a polyvinylidene difluoride membrane (Millipore, Billerica, MA) byWestern blotting. A rat anti–SLAM-associated protein (SAP) mAb (XLP1D12; Cell Signaling Technology, Danvers, MA) and HRP-conjugatedgoat anti-rat IgG (H+L) F(ab9)2 fragments (Invitrogen) were used to de-tect SAP. Rabbit polyclonal Abs to Ewing’s sarcoma-activated transcript 2(EAT2), SHIP-1, SHP-2, CSK (Proteintech Group, Chicago, IL), anda rabbit mAb to SHP-1 (EPR5519; Epitomics, Burlingame, CA), followedby HRP-conjugated goat anti-rabbit IgG (H+L) (Invitrogen), were used todetect the indicated signaling molecules.
ResultsCD319 and CD229 expression is increased on both pDCs andNK cells after RNA-IC stimulation
In a screen of receptors and ligands potentially involved in theRNA-IC–induced pDC–NK cell cross-talk, we examined expres-sion of 42 surface molecules (summarized in Supplemental TableI) on pDCs and CD56dim and CD56bright NK cells in PBMCscultured with or without RNA-ICs for 6 h.As expected, strong upregulation on pDCs and NK cells was seen
for the early activation marker CD69 (median fluorescence in-tensity [MFI] was increased 37- and 7-fold on pDCs and CD56dim
NK cells, respectively), confirming that RNA-ICs are potentactivators of both pDCs and NK cells (Fig. 1A). Remarkably, inaddition to CD69, CD319 showed a strong increase in surfaceexpression on both pDCs and CD56dim NK cells (2.7- and 2.2-foldmean increase in MFI, respectively; Fig. 1A). In contrast, RNA-ICs did not induce an increase in expression of CD69 or CD319 onB cells or T cells (Supplemental Fig. 1A).The finding of dynamic regulation of CD319 expression, a re-
ceptor belonging to the immunomodulatory SLAM receptor familyassociated with SLE, prompted us to examine the expression of allseven SLAM family members on pDCs and NK cells before andafter RNA-IC stimulation. In freshly isolated PBMCs, nearly allpDCs expressed CD48, CD229, and CD319, whereas expression ofCD84 and NTB-A was lower. CD56dim and CD56bright NK cells
2 SLE-IC REGULATION OF CD319 AND CD229
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
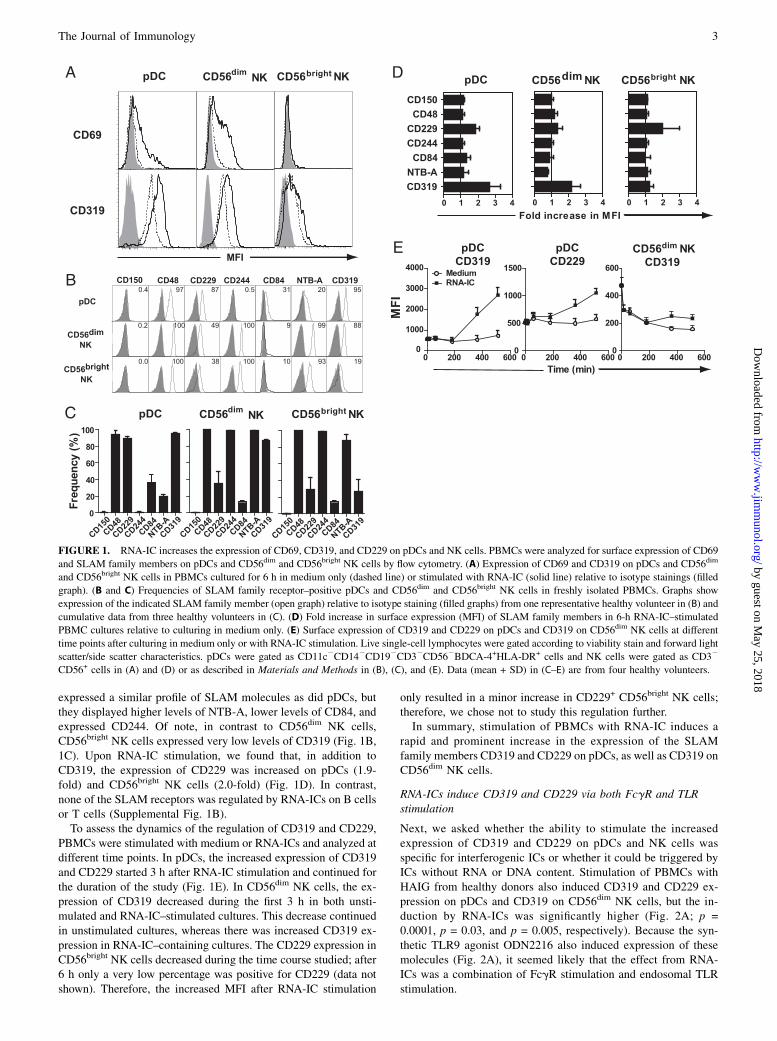
expressed a similar profile of SLAM molecules as did pDCs, butthey displayed higher levels of NTB-A, lower levels of CD84, andexpressed CD244. Of note, in contrast to CD56dim NK cells,CD56bright NK cells expressed very low levels of CD319 (Fig. 1B,1C). Upon RNA-IC stimulation, we found that, in addition toCD319, the expression of CD229 was increased on pDCs (1.9-fold) and CD56bright NK cells (2.0-fold) (Fig. 1D). In contrast,none of the SLAM receptors was regulated by RNA-ICs on B cellsor T cells (Supplemental Fig. 1B).To assess the dynamics of the regulation of CD319 and CD229,
PBMCs were stimulated with medium or RNA-ICs and analyzed atdifferent time points. In pDCs, the increased expression of CD319and CD229 started 3 h after RNA-IC stimulation and continued forthe duration of the study (Fig. 1E). In CD56dim NK cells, the ex-pression of CD319 decreased during the first 3 h in both unsti-mulated and RNA-IC–stimulated cultures. This decrease continuedin unstimulated cultures, whereas there was increased CD319 ex-pression in RNA-IC–containing cultures. The CD229 expression inCD56bright NK cells decreased during the time course studied; after6 h only a very low percentage was positive for CD229 (data notshown). Therefore, the increased MFI after RNA-IC stimulation
only resulted in a minor increase in CD229+ CD56bright NK cells;therefore, we chose not to study this regulation further.In summary, stimulation of PBMCs with RNA-IC induces a
rapid and prominent increase in the expression of the SLAMfamily members CD319 and CD229 on pDCs, as well as CD319 onCD56dim NK cells.
RNA-ICs induce CD319 and CD229 via both FcgR and TLRstimulation
Next, we asked whether the ability to stimulate the increasedexpression of CD319 and CD229 on pDCs and NK cells wasspecific for interferogenic ICs or whether it could be triggered byICs without RNA or DNA content. Stimulation of PBMCs withHAIG from healthy donors also induced CD319 and CD229 ex-pression on pDCs and CD319 on CD56dim NK cells, but the in-duction by RNA-ICs was significantly higher (Fig. 2A; p =0.0001, p = 0.03, and p = 0.005, respectively). Because the syn-thetic TLR9 agonist ODN2216 also induced expression of thesemolecules (Fig. 2A), it seemed likely that the effect from RNA-ICs was a combination of FcgR stimulation and endosomal TLRstimulation.
FIGURE 1. RNA-IC increases the expression of CD69, CD319, and CD229 on pDCs and NK cells. PBMCs were analyzed for surface expression of CD69
and SLAM family members on pDCs and CD56dim and CD56bright NK cells by flow cytometry. (A) Expression of CD69 and CD319 on pDCs and CD56dim
and CD56bright NK cells in PBMCs cultured for 6 h in medium only (dashed line) or stimulated with RNA-IC (solid line) relative to isotype stainings (filled
graph). (B and C) Frequencies of SLAM family receptor–positive pDCs and CD56dim and CD56bright NK cells in freshly isolated PBMCs. Graphs show
expression of the indicated SLAM family member (open graph) relative to isotype staining (filled graphs) from one representative healthy volunteer in (B) and
cumulative data from three healthy volunteers in (C). (D) Fold increase in surface expression (MFI) of SLAM family members in 6-h RNA-IC–stimulated
PBMC cultures relative to culturing in medium only. (E) Surface expression of CD319 and CD229 on pDCs and CD319 on CD56dim NK cells at different
time points after culturing in medium only or with RNA-IC stimulation. Live single-cell lymphocytes were gated according to viability stain and forward light
scatter/side scatter characteristics. pDCs were gated as CD11c2CD142CD192CD32CD562BDCA-4+HLA-DR+ cells and NK cells were gated as CD32
CD56+ cells in (A) and (D) or as described in Materials and Methods in (B), (C), and (E). Data (mean + SD) in (C–E) are from four healthy volunteers.
The Journal of Immunology 3
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
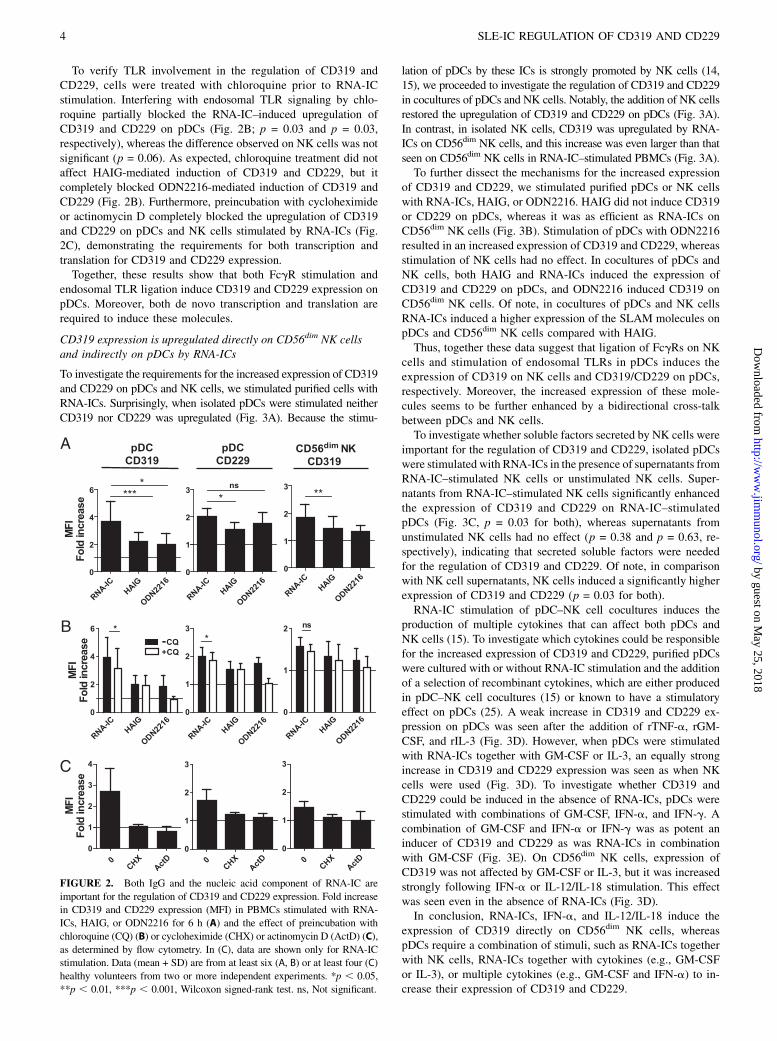
To verify TLR involvement in the regulation of CD319 andCD229, cells were treated with chloroquine prior to RNA-ICstimulation. Interfering with endosomal TLR signaling by chlo-roquine partially blocked the RNA-IC–induced upregulation ofCD319 and CD229 on pDCs (Fig. 2B; p = 0.03 and p = 0.03,respectively), whereas the difference observed on NK cells was notsignificant (p = 0.06). As expected, chloroquine treatment did notaffect HAIG-mediated induction of CD319 and CD229, but itcompletely blocked ODN2216-mediated induction of CD319 andCD229 (Fig. 2B). Furthermore, preincubation with cycloheximideor actinomycin D completely blocked the upregulation of CD319and CD229 on pDCs and NK cells stimulated by RNA-ICs (Fig.2C), demonstrating the requirements for both transcription andtranslation for CD319 and CD229 expression.Together, these results show that both FcgR stimulation and
endosomal TLR ligation induce CD319 and CD229 expression onpDCs. Moreover, both de novo transcription and translation arerequired to induce these molecules.
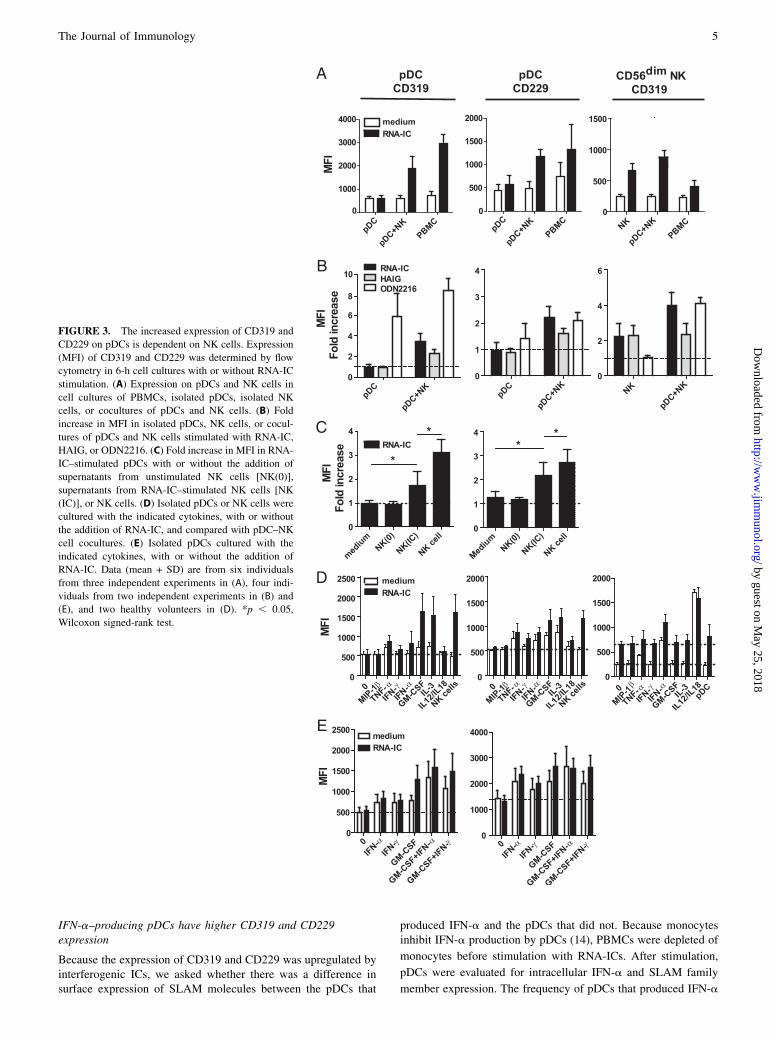
CD319 expression is upregulated directly on CD56dim NK cellsand indirectly on pDCs by RNA-ICs
To investigate the requirements for the increased expression of CD319and CD229 on pDCs and NK cells, we stimulated purified cells withRNA-ICs. Surprisingly, when isolated pDCs were stimulated neitherCD319 nor CD229 was upregulated (Fig. 3A). Because the stimu-
lation of pDCs by these ICs is strongly promoted by NK cells (14,15), we proceeded to investigate the regulation of CD319 and CD229in cocultures of pDCs and NK cells. Notably, the addition of NK cellsrestored the upregulation of CD319 and CD229 on pDCs (Fig. 3A).In contrast, in isolated NK cells, CD319 was upregulated by RNA-ICs on CD56dim NK cells, and this increase was even larger than thatseen on CD56dim NK cells in RNA-IC–stimulated PBMCs (Fig. 3A).To further dissect the mechanisms for the increased expression
of CD319 and CD229, we stimulated purified pDCs or NK cellswith RNA-ICs, HAIG, or ODN2216. HAIG did not induce CD319or CD229 on pDCs, whereas it was as efficient as RNA-ICs onCD56dim NK cells (Fig. 3B). Stimulation of pDCs with ODN2216resulted in an increased expression of CD319 and CD229, whereasstimulation of NK cells had no effect. In cocultures of pDCs andNK cells, both HAIG and RNA-ICs induced the expression ofCD319 and CD229 on pDCs, and ODN2216 induced CD319 onCD56dim NK cells. Of note, in cocultures of pDCs and NK cellsRNA-ICs induced a higher expression of the SLAM molecules onpDCs and CD56dim NK cells compared with HAIG.Thus, together these data suggest that ligation of FcgRs on NK
cells and stimulation of endosomal TLRs in pDCs induces theexpression of CD319 on NK cells and CD319/CD229 on pDCs,respectively. Moreover, the increased expression of these mole-cules seems to be further enhanced by a bidirectional cross-talkbetween pDCs and NK cells.To investigate whether soluble factors secreted by NK cells were
important for the regulation of CD319 and CD229, isolated pDCswere stimulated with RNA-ICs in the presence of supernatants fromRNA-IC–stimulated NK cells or unstimulated NK cells. Super-natants from RNA-IC–stimulated NK cells significantly enhancedthe expression of CD319 and CD229 on RNA-IC–stimulatedpDCs (Fig. 3C, p = 0.03 for both), whereas supernatants fromunstimulated NK cells had no effect (p = 0.38 and p = 0.63, re-spectively), indicating that secreted soluble factors were neededfor the regulation of CD319 and CD229. Of note, in comparisonwith NK cell supernatants, NK cells induced a significantly higherexpression of CD319 and CD229 (p = 0.03 for both).RNA-IC stimulation of pDC–NK cell cocultures induces the
production of multiple cytokines that can affect both pDCs andNK cells (15). To investigate which cytokines could be responsiblefor the increased expression of CD319 and CD229, purified pDCswere cultured with or without RNA-IC stimulation and the additionof a selection of recombinant cytokines, which are either producedin pDC–NK cell cocultures (15) or known to have a stimulatoryeffect on pDCs (25). A weak increase in CD319 and CD229 ex-pression on pDCs was seen after the addition of rTNF-a, rGM-CSF, and rIL-3 (Fig. 3D). However, when pDCs were stimulatedwith RNA-ICs together with GM-CSF or IL-3, an equally strongincrease in CD319 and CD229 expression was seen as when NKcells were used (Fig. 3D). To investigate whether CD319 andCD229 could be induced in the absence of RNA-ICs, pDCs werestimulated with combinations of GM-CSF, IFN-a, and IFN-g. Acombination of GM-CSF and IFN-a or IFN-g was as potent aninducer of CD319 and CD229 as was RNA-ICs in combinationwith GM-CSF (Fig. 3E). On CD56dim NK cells, expression ofCD319 was not affected by GM-CSF or IL-3, but it was increasedstrongly following IFN-a or IL-12/IL-18 stimulation. This effectwas seen even in the absence of RNA-ICs (Fig. 3D).In conclusion, RNA-ICs, IFN-a, and IL-12/IL-18 induce the
expression of CD319 directly on CD56dim NK cells, whereaspDCs require a combination of stimuli, such as RNA-ICs togetherwith NK cells, RNA-ICs together with cytokines (e.g., GM-CSFor IL-3), or multiple cytokines (e.g., GM-CSF and IFN-a) to in-crease their expression of CD319 and CD229.
FIGURE 2. Both IgG and the nucleic acid component of RNA-IC are
important for the regulation of CD319 and CD229 expression. Fold increase
in CD319 and CD229 expression (MFI) in PBMCs stimulated with RNA-
ICs, HAIG, or ODN2216 for 6 h (A) and the effect of preincubation with
chloroquine (CQ) (B) or cycloheximide (CHX) or actinomycin D (ActD) (C),
as determined by flow cytometry. In (C), data are shown only for RNA-IC
stimulation. Data (mean + SD) are from at least six (A, B) or at least four (C)
healthy volunteers from two or more independent experiments. *p , 0.05,
**p , 0.01, ***p , 0.001, Wilcoxon signed-rank test. ns, Not significant.
4 SLE-IC REGULATION OF CD319 AND CD229
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
IFN-a–producing pDCs have higher CD319 and CD229expression
Because the expression of CD319 and CD229 was upregulated byinterferogenic ICs, we asked whether there was a difference insurface expression of SLAM molecules between the pDCs that
produced IFN-a and the pDCs that did not. Because monocytesinhibit IFN-a production by pDCs (14), PBMCs were depleted of
monocytes before stimulation with RNA-ICs. After stimulation,
pDCs were evaluated for intracellular IFN-a and SLAM family
member expression. The frequency of pDCs that produced IFN-a
FIGURE 3. The increased expression of CD319 and
CD229 on pDCs is dependent on NK cells. Expression
(MFI) of CD319 and CD229 was determined by flow
cytometry in 6-h cell cultures with or without RNA-IC
stimulation. (A) Expression on pDCs and NK cells in
cell cultures of PBMCs, isolated pDCs, isolated NK
cells, or cocultures of pDCs and NK cells. (B) Fold
increase in MFI in isolated pDCs, NK cells, or cocul-
tures of pDCs and NK cells stimulated with RNA-IC,
HAIG, or ODN2216. (C) Fold increase in MFI in RNA-
IC–stimulated pDCs with or without the addition of
supernatants from unstimulated NK cells [NK(0)],
supernatants from RNA-IC–stimulated NK cells [NK
(IC)], or NK cells. (D) Isolated pDCs or NK cells were
cultured with the indicated cytokines, with or without
the addition of RNA-IC, and compared with pDC–NK
cell cocultures. (E) Isolated pDCs cultured with the
indicated cytokines, with or without the addition of
RNA-IC. Data (mean + SD) are from six individuals
from three independent experiments in (A), four indi-
viduals from two independent experiments in (B) and
(E), and two healthy volunteers in (D). *p , 0.05,
Wilcoxon signed-rank test.
The Journal of Immunology 5
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
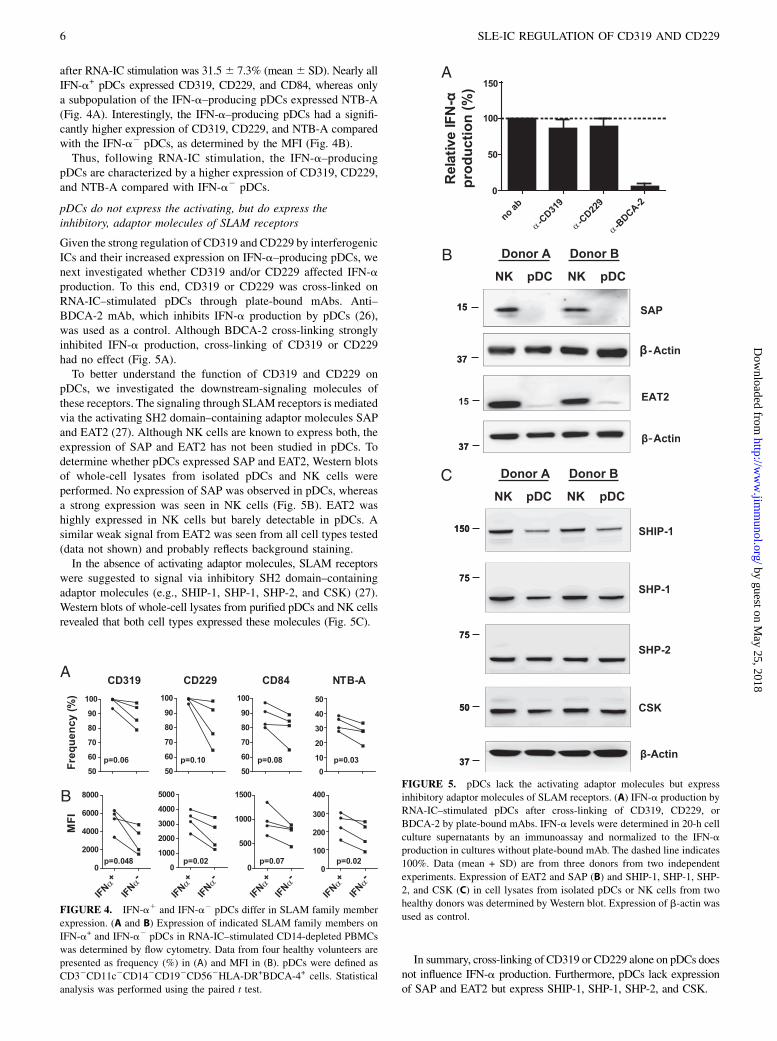
after RNA-IC stimulation was 31.56 7.3% (mean6 SD). Nearly allIFN-a+ pDCs expressed CD319, CD229, and CD84, whereas onlya subpopulation of the IFN-a–producing pDCs expressed NTB-A(Fig. 4A). Interestingly, the IFN-a–producing pDCs had a signifi-cantly higher expression of CD319, CD229, and NTB-A comparedwith the IFN-a2 pDCs, as determined by the MFI (Fig. 4B).Thus, following RNA-IC stimulation, the IFN-a–producing
pDCs are characterized by a higher expression of CD319, CD229,and NTB-A compared with IFN-a2 pDCs.
pDCs do not express the activating, but do express theinhibitory, adaptor molecules of SLAM receptors
Given the strong regulation of CD319 and CD229 by interferogenicICs and their increased expression on IFN-a–producing pDCs, wenext investigated whether CD319 and/or CD229 affected IFN-aproduction. To this end, CD319 or CD229 was cross-linked onRNA-IC–stimulated pDCs through plate-bound mAbs. Anti–BDCA-2 mAb, which inhibits IFN-a production by pDCs (26),was used as a control. Although BDCA-2 cross-linking stronglyinhibited IFN-a production, cross-linking of CD319 or CD229had no effect (Fig. 5A).To better understand the function of CD319 and CD229 on
pDCs, we investigated the downstream-signaling molecules ofthese receptors. The signaling through SLAM receptors is mediatedvia the activating SH2 domain–containing adaptor molecules SAPand EAT2 (27). Although NK cells are known to express both, theexpression of SAP and EAT2 has not been studied in pDCs. Todetermine whether pDCs expressed SAP and EAT2, Western blotsof whole-cell lysates from isolated pDCs and NK cells wereperformed. No expression of SAP was observed in pDCs, whereasa strong expression was seen in NK cells (Fig. 5B). EAT2 washighly expressed in NK cells but barely detectable in pDCs. Asimilar weak signal from EAT2 was seen from all cell types tested(data not shown) and probably reflects background staining.In the absence of activating adaptor molecules, SLAM receptors
were suggested to signal via inhibitory SH2 domain–containingadaptor molecules (e.g., SHIP-1, SHP-1, SHP-2, and CSK) (27).Western blots of whole-cell lysates from purified pDCs and NK cellsrevealed that both cell types expressed these molecules (Fig. 5C).
In summary, cross-linking of CD319 or CD229 alone on pDCs doesnot influence IFN-a production. Furthermore, pDCs lack expressionof SAP and EAT2 but express SHIP-1, SHP-1, SHP-2, and CSK.
FIGURE 4. IFN-a1 and IFN-a2 pDCs differ in SLAM family member
expression. (A and B) Expression of indicated SLAM family members on
IFN-a+ and IFN-a2 pDCs in RNA-IC–stimulated CD14-depleted PBMCs
was determined by flow cytometry. Data from four healthy volunteers are
presented as frequency (%) in (A) and MFI in (B). pDCs were defined as
CD32CD11c2CD142CD192CD562HLA-DR+BDCA-4+ cells. Statistical
analysis was performed using the paired t test.
FIGURE 5. pDCs lack the activating adaptor molecules but express
inhibitory adaptor molecules of SLAM receptors. (A) IFN-a production by
RNA-IC–stimulated pDCs after cross-linking of CD319, CD229, or
BDCA-2 by plate-bound mAbs. IFN-a levels were determined in 20-h cell
culture supernatants by an immunoassay and normalized to the IFN-a
production in cultures without plate-bound mAb. The dashed line indicates
100%. Data (mean + SD) are from three donors from two independent
experiments. Expression of EAT2 and SAP (B) and SHIP-1, SHP-1, SHP-
2, and CSK (C) in cell lysates from isolated pDCs or NK cells from two
healthy donors was determined by Western blot. Expression of b-actin was
used as control.
6 SLE-IC REGULATION OF CD319 AND CD229
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
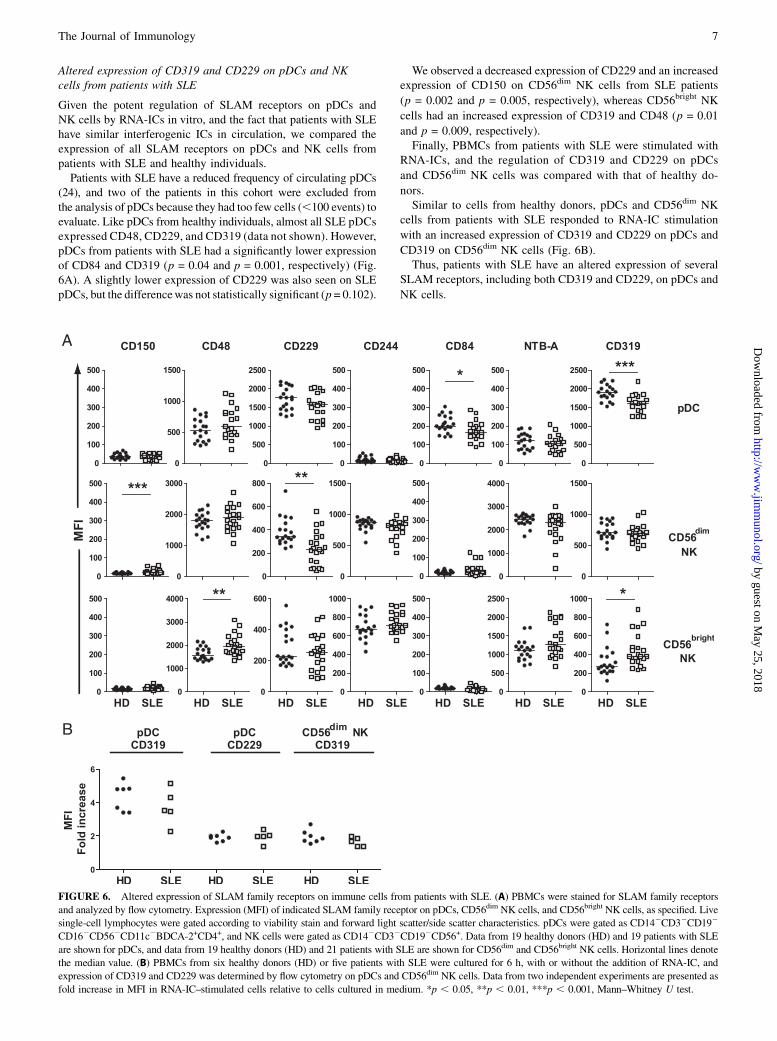
Altered expression of CD319 and CD229 on pDCs and NKcells from patients with SLE
Given the potent regulation of SLAM receptors on pDCs andNK cells by RNA-ICs in vitro, and the fact that patients with SLEhave similar interferogenic ICs in circulation, we compared theexpression of all SLAM receptors on pDCs and NK cells frompatients with SLE and healthy individuals.Patients with SLE have a reduced frequency of circulating pDCs
(24), and two of the patients in this cohort were excluded fromthe analysis of pDCs because they had too few cells (,100 events) toevaluate. Like pDCs from healthy individuals, almost all SLE pDCsexpressed CD48, CD229, and CD319 (data not shown). However,pDCs from patients with SLE had a significantly lower expressionof CD84 and CD319 (p = 0.04 and p = 0.001, respectively) (Fig.6A). A slightly lower expression of CD229 was also seen on SLEpDCs, but the differencewas not statistically significant (p = 0.102).
We observed a decreased expression of CD229 and an increasedexpression of CD150 on CD56dim NK cells from SLE patients
(p = 0.002 and p = 0.005, respectively), whereas CD56bright NK
cells had an increased expression of CD319 and CD48 (p = 0.01
and p = 0.009, respectively).Finally, PBMCs from patients with SLE were stimulated with
RNA-ICs, and the regulation of CD319 and CD229 on pDCs
and CD56dim NK cells was compared with that of healthy do-
nors.Similar to cells from healthy donors, pDCs and CD56dim NK
cells from patients with SLE responded to RNA-IC stimulation
with an increased expression of CD319 and CD229 on pDCs and
CD319 on CD56dim NK cells (Fig. 6B).Thus, patients with SLE have an altered expression of several
SLAM receptors, including both CD319 and CD229, on pDCs and
NK cells.
FIGURE 6. Altered expression of SLAM family receptors on immune cells from patients with SLE. (A) PBMCs were stained for SLAM family receptors
and analyzed by flow cytometry. Expression (MFI) of indicated SLAM family receptor on pDCs, CD56dim NK cells, and CD56bright NK cells, as specified. Live
single-cell lymphocytes were gated according to viability stain and forward light scatter/side scatter characteristics. pDCs were gated as CD142CD32CD192
CD162CD562CD11c2BDCA-2+CD4+, and NK cells were gated as CD142CD32CD192CD56+. Data from 19 healthy donors (HD) and 19 patients with SLE
are shown for pDCs, and data from 19 healthy donors (HD) and 21 patients with SLE are shown for CD56dim and CD56bright NK cells. Horizontal lines denote
the median value. (B) PBMCs from six healthy donors (HD) or five patients with SLE were cultured for 6 h, with or without the addition of RNA-IC, and
expression of CD319 and CD229 was determined by flow cytometry on pDCs and CD56dim NK cells. Data from two independent experiments are presented as
fold increase in MFI in RNA-IC–stimulated cells relative to cells cultured in medium. *p , 0.05, **p , 0.01, ***p , 0.001, Mann–Whitney U test.
The Journal of Immunology 7
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

DiscussionSLAM family receptors are important immunomodulatory re-ceptors involved in the cross-talk between a number of immune
cells (16). The SLAM locus is genetically associated with SLE
(17–21), and recent data suggest involvement of SLAM receptors
in the pathogenesis of lupus (28–30). In the current study, we
found that the expression of the SLAM family receptors CD319
and CD229 was dynamically regulated on pDCs and CD56dim NK
cells by RNA-ICs typically found in lupus. Importantly, there was
no general upregulation of all SLAM molecules on pDCs and NK
cells or an increase in SLAM family receptor expression on T and
B cells by RNA-ICs. Thus, lupus ICs seem to selectively regulate
the expression of certain SLAM molecules on two cell types,
which together have the capacity to produce large amounts of type
I IFN after activation.Interferogenic ICs, such as RNA-ICs, have the capacity to ac-
tivate both FcgRIIA and endosomal TLRs. We showed that sig-
naling induced by engagement of both the IgG component and the
nucleic acid–containing autoantigen in the RNA-ICs contributed
to the increased expression of CD319 and CD229 on pDCs and
CD319 on CD56dim NK cells. When the mechanisms behind the
regulation of CD319 and CD229 were investigated further, we
found that transcription and translation were required. Experi-
ments using isolated or mixed populations of purified pDCs and
NK cells stimulated with RNA-ICs revealed that the upregulation
of CD319 and CD229 on pDCs by RNA-ICs required NK cells.
This effect was partially mediated by soluble factors produced by
NK cells, because supernatant from RNA-IC–stimulated NK cells
increased CD319/CD229 expression. RNA-ICs in combination
with GM-CSF or IL-3, or GM-CSF together with IFN-a or IFN-g
was an equally strong inducer of CD319 and CD229 on pDCs as
NK cells and RNA-ICs. These results demonstrate that the regu-
lation of at least some SLAM molecules on pDCs is complex and
involve several pathways that need to act in concert to trigger an
increased expression. In support of this conclusion, a previous
study showed that the combination of HSV and IL-3 is necessary
to upregulate CD319 in pDCs (31). Whether the induction of
SLAM molecules is coupled to maturation of pDCs to efficient
Ag-presenting dendritic cells is unknown, but this clearly is a
possibility given the fact that both IL-3 and GM-CSF are stimuli
for pDC differentiation (25).In contrast to pDCs, purified NK cells activated by RNA-ICs
exhibited a strong increase in CD319 expression on CD56dim NK
cells. This increase was even more pronounced than the increase
in CD319 among CD56dim NK cells in PBMC cultures. Conse-
quently, inhibitory cells present in the PBMC population may
suppress the upregulation of SLAM molecules on NK cells, pos-
sibly via the release of reactive oxygen species, TNF-a, and PGE2,
which suppress NK cell function (14). We also noted that IFN-a
and IL-12/IL-18 could upregulate CD319 on CD56dim NK cells,
representing an interesting mechanism whereby inflammation may
augment NK cell recognition of, and activation by, hematopoietic
cells. Notably, RNA-ICs or proinflammatory cytokines increased
the CD319 expression on NK cells to comparable levels. Thus, it
appears that at least partially different signaling pathways lead to
CD319 expression in pDCs and NK cells. Furthermore, we noted
that IL-12/IL-18 increased the expression of CD319 on CD56bright
NK cells (data not shown). These cytokines are well-known acti-
vators of NK cells (32), and one mechanism by which both IL-12/
IL-18 and RNA-ICs might contribute to increased NK cell cyto-
toxicity is by upregulation of CD319.The pDC population is heterogeneous with respect to the ex-
pression of several molecules and the capacity to produce IFN-a.
To clarify whether there was any difference in the expression ofdifferent SLAM molecules between pDCs with and without theability to synthesize IFN-a, we stimulated the cells with RNA-ICsand investigated SLAM expression and IFN-a content among thepDCs. This experiment revealed that the IFN-a–producing pDCsdisplayed higher expression of CD319 and CD229, as well asNTB-A, compared with IFN-a2 cells. It is well known that onlyFcgRIIA-expressing pDCs have the capacity to produce IFN-a inresponse to interferogenic ICs (33, 34), and the differences inSLAM expression between IFN-a+ and IFN-a2 pDCs could bedue to the lack of FcgRIIA/TLR-7 activation of the latter pop-ulation. Thus, our observation may indicate that induction of IFN-aproduction and increased SLAM molecule expression by RNA-ICstimulation of pDCs are linked processes in the activation of pDCs,although this needs to be formally proven.On NK cells, CD319 and CD229 act as coactivating receptors,
which results in increased cytotoxicity against targets cells ex-pressing CD319 and CD229 (35, 36), whereas, on B cells, ligationof CD319 induces proliferation and cytokine production (37). Incontrast, no study of the function of SLAM receptors on pDCs hasbeen reported. When investigating the potential role of CD319 andCD229 on pDC function, we noted that cross-linking of thesereceptors by immobilized mAbs did not affect the RNA-IC–induced IFN-a secretion in isolated pDCs. This finding does notnecessarily exclude the involvement of CD319 and CD229 inregulating IFN-a production, because SLAM receptors functionas coreceptors. Thus, CD319 and CD229 may be dependent onconcomitant engagement of other activating or inhibitory recep-tors to exert their effect. Although such receptors are well char-acterized on NK cells, the identity of these receptors on pDCsremains to be determined. Blocking experiments might be a wayto circumvent the need for coligation of other receptors, but thelack of Abs that effectively block the homophilic binding ofCD319 or CD229 regrettably precludes these experiments. Giventhese circumstances, we cannot exclude a function for CD319 andCD229 in regulating IFN-a production.With the exception of CD48, the SLAM molecules contain in-
tracellular immuno-tyrosine switch motifs that bind and signal viathe SH2 domain–containing adaptor molecules SAP and EAT2(27). CD319 differs from the other SLAM family receptors in that itonly recruits EAT2 (38). SAP is expressed in T cells, NK cells,NKT cells, and perhaps some B cells, whereas EAT2 expression hasbeen described in NK cells, dendritic cells, and macrophages (27).The presence of SAP or EAT2 has not previously been investigatedin pDCs. When isolated pDCs and NK cells were examined for SAPand EAT2, no SAP and little to no EAT2 were detected in pDCs. Incontrast, and as expected, high levels of both SAP and EAT2 weredetected in NK cells. Ligation of CD319 in NK cells deficient inEAT2 was shown to abolish the activating effect of CD319 andresult in inhibition of NK cell cytotoxicity (39). Furthermore, li-gation of CD319 on CD4+ T cells or monocytes, which naturallylack EAT2 expression, inhibits Ag-induced proliferation and LPS-induced production of TNF-a, respectively (39, 40). The inhibitoryeffect of SLAM receptors in cells lacking SAP and EAT2 probablycan be explained by the fact that, in the absence of these molecules,SLAM receptor signaling couples to inhibitory adaptor proteins,such as SHIP-1, SHP-1, SHP-2, and CSK (27, 41). By Western blot,we showed that pDCs expressed these molecules, suggesting thatCD319 and CD229 act as inhibitory coreceptors on pDCs. Anotherpossibility is that these receptors promote the activation of othercells expressing the corresponding molecules. For instance, SLAMfamily receptors on NK cells enhance LFA-1–dependent conjugateformation (41); because IFN-a production is strongly promoted byLFA-1–mediated interaction between pDCs and NK cells (15),
8 SLE-IC REGULATION OF CD319 AND CD229
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

expression of CD229 and CD319 on pDCs may support the pDC–NK cell cross-talk.Finally, we investigated the expression of the SLAM molecules
on pDCs and NK cells from patients with SLE. Patients with SLEhad decreased expression of CD84 and CD319 on circulatingpDCs, which is at variance with the in vitro stimulation of PBMCsfrom healthy individuals with RNA-ICs. However, RNA-IC stimu-lation of SLE PBMCs increased the expression of CD319 andCD229 on pDCs, demonstrating that SLE pDCs retain their ca-pacity to upregulate these molecules. One explanation for thedecreased SLAM expression on circulating pDCs in SLE could bethat SLE IC-activated cells migrate from the circulation to inflamedtissues. Such redistribution from blood to tissues of pDCs thatproduce IFN-a was also described in SLE (42, 43) and mightexplain our observation. On SLE NK cells, decreased expressionof CD229 on CD56dim NK cells and increased expression ofCD319 and CD48 on CD56bright NK cells were observed. Theexact consequence of the altered expression of SLAM moleculesin SLE NK cells is unknown, but it might reflect several previousobservations. For instance, the lower expression of CD229 onCD56dim cells may contribute to the decreased cytotoxicity of SLENK cells (44, 45). In contrast, the increased expression of SLAMmolecules on CD56bright NK cells may play a role in the increasedproinflammatory cytokine production in SLE (46). Conceivablysupporting the latter interpretation is the recent observation thatpatients with SLE have increased expression of CD229 and NTB-A on T cells and that activation of CD229 or NTB-A, togetherwith CD3, promotes IL-17 production (30).In summary, our study shows that RNA-ICs regulate a subset of
SLAM molecules on pDCs and NK cells and that lupus patientshave an altered expression of CD319 and CD229 on these celltypes. Our observations are important in light of emerging datademonstrating that the SLAM family of receptors is implicated inautoimmune disease processes. Because SLAM molecules havebeen suggested as both possible biomarkers and therapeutic targetsin SLE (30), further studies of the precise function of SLAMreceptors on pDCs and their role in the pathogenesis of SLE seemhighly warranted.
AcknowledgmentsWe thank Gert Weber for kindly providing the U1snRNP complexes,
Gunnar Alm for reviewing the manuscript, and Anne Tronnberg, Charlotta
Jakobsson, and Rezvan Kiani Dehkordi for excellent technical assistance.
DisclosuresThe authors have no financial conflicts of interest.
References1. Tsokos, G. C. 2011. Systemic lupus erythematosus. N. Engl. J. Med. 365: 2110–
2121.2. Lovgren, T., M. L. Eloranta, U. Bave, G. V. Alm, and L. Ronnblom. 2004. In-
duction of interferon-alpha production in plasmacytoid dendritic cells by im-mune complexes containing nucleic acid released by necrotic or late apoptoticcells and lupus IgG. Arthritis Rheum. 50: 1861–1872.
3. Pisetsky, D. S. 2012. Antinuclear antibodies in rheumatic disease: a proposal fora function-based classification. Scand. J. Immunol. 76: 223–228.
4. Ravetch, J. V., and S. Bolland. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19:275–290.
5. Ronnblom, L., M. L. Eloranta, and G. V. Alm. 2006. The type I interferon systemin systemic lupus erythematosus. Arthritis Rheum. 54: 408–420.
6. Baechler, E. C., F. M. Batliwalla, G. Karypis, P. M. Gaffney, W. A. Ortmann,K. J. Espe, K. B. Shark, W. J. Grande, K. M. Hughes, V. Kapur, et al. 2003.Interferon-inducible gene expression signature in peripheral blood cells ofpatients with severe lupus. Proc. Natl. Acad. Sci. USA 100: 2610–2615.
7. Bennett, L., A. K. Palucka, E. Arce, V. Cantrell, J. Borvak, J. Banchereau, andV. Pascual. 2003. Interferon and granulopoiesis signatures in systemic lupuserythematosus blood. J. Exp. Med. 197: 711–723.
8. Peterson, K. S., J. F. Huang, J. Zhu, V. D’Agati, X. Liu, N. Miller,M. G. Erlander, M. R. Jackson, and R. J. Winchester. 2004. Characterization of
heterogeneity in the molecular pathogenesis of lupus nephritis from transcrip-tional profiles of laser-captured glomeruli. J. Clin. Invest. 113: 1722–1733.
9. Higgs, B. W., Z. Liu, B. White, W. Zhu, W. I. White, C. Morehouse, P. Brohawn,P. A. Kiener, L. Richman, D. Fiorentino, et al. 2011. Patients with systemic lupuserythematosus, myositis, rheumatoid arthritis and scleroderma share activationof a common type I interferon pathway. Ann. Rheum. Dis. 70: 2029–2036.
10. Ronnblom, L., and M. L. Eloranta. 2013. The interferon signature in autoim-mune diseases. Curr. Opin. Rheumatol. 25: 248–253.
11. Ronnblom, L. E., G. V. Alm, and K. E. Oberg. 1991. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann. Intern. Med. 115: 178–183.
12. Borg, F. A., and D. A. Isenberg. 2007. Syndromes and complications of inter-feron therapy. Curr. Opin. Rheumatol. 19: 61–66.
13. Della Chiesa, M., C. Romagnani, A. Thiel, L. Moretta, and A. Moretta. 2006.Multidirectional interactions are bridging human NK cells with plasmacytoidand monocyte-derived dendritic cells during innate immune responses. Blood108: 3851–3858.
14. Eloranta, M. L., T. Lovgren, D. Finke, L. Mathsson, J. Ronnelid, B. Kastner,G. V. Alm, and L. Ronnblom. 2009. Regulation of the interferon-alpha pro-duction induced by RNA-containing immune complexes in plasmacytoid den-dritic cells. Arthritis Rheum. 60: 2418–2427.
15. Hagberg, N., O. Berggren, D. Leonard, G. Weber, Y. T. Bryceson, G. V. Alm,M. L. Eloranta, and L. Ronnblom. 2011. IFN-a production by plasmacytoiddendritic cells stimulated with RNA-containing immune complexes is promotedby NK cells via MIP-1b and LFA-1. J. Immunol. 186: 5085–5094.
16. Cannons, J. L., S. G. Tangye, and P. L. Schwartzberg. 2011. SLAM familyreceptors and SAP adaptors in immunity. Annu. Rev. Immunol. 29: 665–705.
17. Kono, D. H., R. W. Burlingame, D. G. Owens, A. Kuramochi, R. S. Balderas,D. Balomenos, and A. N. Theofilopoulos. 1994. Lupus susceptibility loci in NewZealand mice. Proc. Natl. Acad. Sci. USA 91: 10168–10172.
18. Hogarth, M. B., J. H. Slingsby, P. J. Allen, E. M. Thompson, P. Chandler,K. A. Davies, E. Simpson, B. J. Morley, and M. J. Walport. 1998. Multiple lupussusceptibility loci map to chromosome 1 in BXSB mice. J. Immunol. 161: 2753–2761.
19. Tsao, B. P., R. M. Cantor, K. C. Kalunian, C. J. Chen, H. Badsha, R. Singh,D. J. Wallace, R. C. Kitridou, S. L. Chen, N. Shen, et al. 1997. Evidence forlinkage of a candidate chromosome 1 region to human systemic lupus eryth-ematosus. J. Clin. Invest. 99: 725–731.
20. Moser, K. L., B. R. Neas, J. E. Salmon, H. Yu, C. Gray-McGuire, N. Asundi,G. R. Bruner, J. Fox, J. Kelly, S. Henshall, et al. 1998. Genome scan of humansystemic lupus erythematosus: evidence for linkage on chromosome 1q inAfrican-American pedigrees. Proc. Natl. Acad. Sci. USA 95: 14869–14874.
21. Cunninghame Graham, D. S., T. J. Vyse, P. R. Fortin, A. Montpetit, Y. C. Cai,S. Lim, T. McKenzie, L. Farwell, B. Rhodes, L. Chad, et al; CaNIOS GenESInvestigators. 2008. Association of LY9 in UK and Canadian SLE families.Genes Immun. 9: 93–102.
22. Bombardier, C., D. D. Gladman, M. B. Urowitz, D. Caron, and C. H. Chang; TheCommittee on Prognosis Studies in SLE. 1992. Derivation of the SLEDAI. Adisease activity index for lupus patients. Arthritis Rheum. 35: 630–640.
23. Weber, G., S. Trowitzsch, B. Kastner, R. Luhrmann, and M. C. Wahl. 2010.Functional organization of the Sm core in the crystal structure of human U1snRNP. EMBO J. 29: 4172–4184.
24. Cederblad, B., S. Blomberg, H. Vallin, A. Perers, G. V. Alm, and L. Ronnblom.1998. Patients with systemic lupus erythematosus have reduced numbers ofcirculating natural interferon-alpha- producing cells. J. Autoimmun. 11: 465–470.
25. Ghirelli, C., R. Zollinger, and V. Soumelis. 2010. Systematic cytokine receptorprofiling reveals GM-CSF as a novel TLR-independent activator of humanplasmacytoid predendritic cells. Blood 115: 5037–5040.
26. Dzionek, A., Y. Sohma, J. Nagafune, M. Cella, M. Colonna, F. Facchetti,G. Gunther, I. Johnston, A. Lanzavecchia, T. Nagasaka, et al. 2001. BDCA-2,a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates an-tigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp.Med. 194: 1823–1834.
27. Veillette, A. 2010. SLAM-family receptors: immune regulators with or withoutSAP-family adaptors. Cold Spring Harb. Perspect. Biol. 2: a002469.
28. Kim, J. R., S. O. Mathew, R. K. Patel, R. M. Pertusi, and P. A. Mathew. 2010.Altered expression of signalling lymphocyte activation molecule (SLAM) familyreceptors CS1 (CD319) and 2B4 (CD244) in patients with systemic lupuserythematosus. Clin. Exp. Immunol. 160: 348–358.
29. Chatterjee, M., K. Kis-Toth, T. H. Thai, C. Terhorst, and G. C. Tsokos. 2011.SLAMF6-driven co-stimulation of human peripheral T cells is defective in SLET cells. Autoimmunity 44: 211–218.
30. Chatterjee, M., T. Rauen, K. Kis-Toth, V. C. Kyttaris, C. M. Hedrich, C. Terhorst,and G. C. Tsokos. 2012. Increased expression of SLAM receptors SLAMF3 andSLAMF6 in systemic lupus erythematosus T lymphocytes promotes Th17 dif-ferentiation. J. Immunol. 188: 1206–1212.
31. Schuster, P., N. Donhauser, K. Pritschet, M. Ries, S. Haupt, N. A. Kittan,K. Korn, and B. Schmidt. 2010. Co-ordinated regulation of plasmacytoid den-dritic cell surface receptors upon stimulation with herpes simplex virus type 1.Immunology 129: 234–247.
32. Fehniger, T. A., M. H. Shah, M. J. Turner, J. B. VanDeusen, S. P. Whitman,M. A. Cooper, K. Suzuki, M. Wechser, F. Goodsaid, and M. A. Caligiuri. 1999.Differential cytokine and chemokine gene expression by human NK cells fol-lowing activation with IL-18 or IL-15 in combination with IL-12: implicationsfor the innate immune response. J. Immunol. 162: 4511–4520.
33. Bave, U., M. Magnusson, M. L. Eloranta, A. Perers, G. V. Alm, andL. Ronnblom. 2003. Fc gamma RIIa is expressed on natural IFN-alpha-
The Journal of Immunology 9
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

producing cells (plasmacytoid dendritic cells) and is required for the IFN-alphaproduction induced by apoptotic cells combined with lupus IgG. J. Immunol.171: 3296–3302.
34. Means, T. K., E. Latz, F. Hayashi, M. R. Murali, D. T. Golenbock, andA. D. Luster. 2005. Human lupus autoantibody-DNA complexes activate DCsthrough cooperation of CD32 and TLR9. J. Clin. Invest. 115: 407–417.
35. Bouchon, A., M. Cella, H. L. Grierson, J. I. Cohen, and M. Colonna. 2001.Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor ofthe CD2 family. J. Immunol. 167: 5517–5521.
36. Dong, Z., M. E. Cruz-Munoz, M. C. Zhong, R. Chen, S. Latour, and A. Veillette.2009. Essential function for SAP family adaptors in the surveillance of hema-topoietic cells by natural killer cells. Nat. Immunol. 10: 973–980.
37. Lee, J. K., S. O. Mathew, S. V. Vaidya, P. R. Kumaresan, and P. A. Mathew.2007. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine ex-pression on human B lymphocytes. J. Immunol. 179: 4672–4678.
38. Tassi, I., and M. Colonna. 2005. The cytotoxicity receptor CRACC (CS-1)recruits EAT-2 and activates the PI3K and phospholipase Cgamma signalingpathways in human NK cells. J. Immunol. 175: 7996–8002.
39. Cruz-Munoz, M. E., Z. Dong, X. Shi, S. Zhang, and A. Veillette. 2009. Influenceof CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on naturalkiller cell function. Nat. Immunol. 10: 297–305.
40. Kim, J. R., N. C. Horton, S. O. Mathew, and P. A. Mathew. 2013. CS1(SLAMF7) inhibits production of proinflammatory cytokines by activatedmonocytes. Inflamm. Res. 62: 765–772.
41. Dong, Z., D. Davidson, L. A. Perez-Quintero, T. Kurosaki, W. Swat, andA. Veillette. 2012. The adaptor SAP controls NK cell activation by regulating theenzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Im-munity 36: 974–985.
42. Blomberg, S., M. L. Eloranta, B. Cederblad, K. Nordlin, G. V. Alm, andL. Ronnblom. 2001. Presence of cutaneous interferon-alpha producing cells inpatients with systemic lupus erythematosus. Lupus 10: 484–490.
43. Tucci, M., C. Quatraro, L. Lombardi, C. Pellegrino, F. Dammacco, andF. Silvestris. 2008. Glomerular accumulation of plasmacytoid dendritic cells inactive lupus nephritis: role of interleukin-18. Arthritis Rheum. 58: 251–262.
44. Hoffman, T. 1980. Natural killer function in systemic lupus erythematosus.Arthritis Rheum. 23: 30–35.
45. Hervier, B., V. Beziat, J. Haroche, A. Mathian, P. Lebon, P. Ghillani-Dalbin,L. Musset, P. Debre, Z. Amoura, and V. Vieillard. 2011. Phenotype and functionof natural killer cells in systemic lupus erythematosus: excess interferon-gproduction in patients with active disease. Arthritis Rheum. 63: 1698–1706.
46. Ronnblom, L., and K. B. Elkon. 2010. Cytokines as therapeutic targets in SLE.Nat. Rev. Rheumatol. 6: 339–347.
10 SLE-IC REGULATION OF CD319 AND CD229
by guest on May 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from














