
Synthesis and thermal responsiveness of self-assembled gold nanoclusters
4
ISSN 1359-7345 Chemical Communications www.rsc.org/chemcomm Volume 46 | Number 34 | 14 September 2010 | Pages 6189–6392 COMMUNICATION Silvija Gradečak et al. Synthesis and thermal responsiveness of self-assembled gold nanoclusters FEATURE ARTICLE Dario Braga et al. The growing world of crystal forms View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Synthesis and thermal responsiveness of self-assembled gold nanoclusters

Chemical ScienceA new global impact journal for exceptional research from across the chemical sciences
What’s different about Chemical Science, you might ask? Firstly, we know that current approaches to selecting and publishing articles aren’t always as effective as they might be. Secondly, we appreciate the challenges you face in getting your work published, noticed and recognised.
We want to change this. So Chemical Science will make it easier for you to get the recognition that your work deserves.
A hand-picked team of dynamic Associate Editors, led by Editor-in-Chief David MacMillan, will ensure that every article has a fair review. And as active researchers, they will guarantee that the journal represents the best new thinking in the chemical sciences.
Submit your next article to Chemical Science – and step into the spotlight.
Recognition starts here
0110
67
www.rsc.org/chemicalscienceRegistered Charity Number 207890
ISSN 1359-7345
Chemical Communications
www.rsc.org/chemcomm Volume 46 | Number 34 | 14 September 2010 | Pages 6189–6392
COMMUNICATIONSilvija Gradečak et al.Synthesis and thermal responsiveness of self-assembled gold nanoclusters
FEATURE ARTICLEDario Braga et al.The growing world of crystal forms
Publ
ishe
d on
10
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
War
saw
on
30/1
0/20
14 1
5:13
:18.
View Article Online / Journal Homepage / Table of Contents for this issue

6246 Chem. Commun., 2010, 46, 6246–6248 This journal is c The Royal Society of Chemistry 2010
Synthesis and thermal responsiveness of self-assembled gold
nanoclustersw
Shenqiang Ren, Sung-Keun Lim and Silvija Gradecak*
Received 10th June 2010, Accepted 14th July 2010
DOI: 10.1039/c0cc01829e
A simple and versatile approach was developed to generate
hierarchical assemblies of ultra-small gold nanocluster thin films
using the combination of galvanic reaction and a block copolymer
coordinated with gold complex. Variation of the temperature
allows effective control over the optical response of these stimuli-
responsive organic-nanocluster hybrid structures.
Noble metal-atomic clusters are composed of a small number
of atoms, with diameters typically ranging from sub-nanometre
to approximately 2 nm.1 They have molecule-like characteristics
and exhibit unique and size-dependent optical, electronic, and
magnetic properties.2,3 For example, due to their enhanced
luminescence over a broad range of absorption wavelengths,4
metal nanoclusters are ideal candidates for light-harvesting
applications.5,6 Controlled synthesis and assembly of the noble
metal nanocluster and their manipulation via external stimuli
would enable design of functional devices based on these
unique properties. Several methods of the metal cluster
synthesis have been developed so far;2,7,8 however, the design
of the functional and responsive ultra-small metal nanoclusters
has been impeded by the lack of simultaneous control over
their synthesis and spatial organization in a film-on-substrate
geometry, as well as by the challenges in incorporating external
stimuli to generate stable responsive nanocluster composites.
In this communication, we describe the templated synthesis
of gold (Au) nanoclusters embedded in copolymer matrix
using the combination of galvanic reaction and self-assembled
block copolymer (BCP). Strong selectivity and homogeneous
spatial distribution of as-synthesized Au nanoclusters in one
block was achieved by selective gold precursor loading, while
BCPs enabled thermally-responsive and fully reversible optical
behaviour of the resulting Au nanocluster composites. In
contrast to previous approaches of using BCP micelles
coordinated with a metal complex9 or using galvanic displace-
ment combined with other assembly techniques,10 both of
which result in synthesis of noble metal nanoparticles, our
method simultaneously combines these two syntheses. In this
way, we control the Au reduction rate and thus synthesize self-
assembled and size-selective nanoclusters.
BCPs are versatile platforms to achieve ordered nanoscale
hybrid materials because of their tendency to self-assemble in
periodic structures of tens of nanometres.11�13 To direct
position- and size-controlled synthesis of Au nanoclusters, in
this work we used polystyrene-b-poly(4-vinylpyridine)
(PS-b-P4VP) BCP with the molecular weights of PS and
P4VP being 19 kg mol�1 and 5.2 kg mol�1, respectively. Au
was loaded selectively into the P4VP domains by dissolving
PS-b-P4VP with HAuCl4 at a ratio of Au/pyridyl of 0.3
(PS-b-P[4VP(HAuCl4)0.3]) in 0.5% (w/w) toluene/tetrahydrofuran
(THF) solution. During this process, anionic complexes AuCl4�
bind to a protonated pyridine moiety due to electrostatic
interactions. A monolayer of the mixture was then spin-coated
on GaAs(111) substrate (Fig. 1a). Atomic force microscopy
(AFM) images of the spun-cast film revealed that no assembly
occurred in the pristine state (Fig. 1b). After THF solvent
annealing (Fig. 1c), the quasi-hexagonal assembly was
observed in the resulting thin film sample, as confirmed by
AFM imaging and the corresponding fast Fourier transform
(Fig. 1d). The sample was then immersed in 1% hydrofluoric
acid (HF) solution for 30 seconds. The HF is selectively
delivered to the semiconductor surface via the hexagonally
ordered spherical P4VP/HAuCl4 cores and the consequent
chemistry of the galvanic displacement is based upon a simple
electrochemical reaction in which sufficiently oxidizing Au(III)
ions (in the form of AuCl4�) are reduced by GaAs substrate.9
The resulting Au nanoclusters connected by P4VP chain
possess the same quasi-hexagonal packing as that of the parent
BCP (Fig. 1e and f). AFM revealed that the surface of Au
loaded films is comprised of quasi-hexagonal arrays of small
localized depressions in the individual P4VP cores (Fig. 1f).
The depressions are likely formed during the volume shrinkage
that occurs from the original HAuCl4 complex to Au metal
nanoclusters reduced by galvanic displacement at the
P4VP microdomain.
Fig. 1 Schematic illustration (upper row) and corresponding AFM
height images (lower row) of Au nanocluster composite synthesis using
BCP and galvanic reaction. Scale bar is 50 nm, identical for all AFM
images. (a and b) PS-b-P4VP doped with Au complex spun cast onto
GaAs(111). (c and d) Self-assembled thin film after solvent annealing. Inset
in (d) is the fast Fourier transform of the corresponding AFM image.
(e and f) The same sample after HF treatment. Au nanoclusters form in
P4VP domains embedded in PS matrix due to the galvanic displacement.
Massachusetts Institute of Technology, Department of MaterialsScience and Engineering, 77 Massachusetts Avenue, Cambridge,MA 02139, USA. E-mail: [email protected] Electronic supplementary information (ESI) available: Fullexperimental details, scanning transmission electron microscopy, highresolution TEM and its corresponding FFT, chemical analysis andUV-Vis absorbance spectrum. See DOI: 10.1039/c0cc01829e
COMMUNICATION www.rsc.org/chemcomm | ChemComm
Publ
ishe
d on
10
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
War
saw
on
30/1
0/20
14 1
5:13
:18.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Commun., 2010, 46, 6246–6248 6247
Plan-view transmission electron microscopy (TEM) of the
self-assembled PS-b-P4VP(HAuCl4)0.3 nanocomposite after
galvanic reaction (Fig. 2a) shows that these films are
comprised of a quasi-hexagonal arrays of circular P4VP
domains with 15 nm diameters and 25 nm spacing, which
correlates well with the AFM observations. Each domain is
uniformly filled with Au nanoclusters (ESIw, Fig. S1 and S2),
and the size of each nanocluster is significantly smaller
(B1.0 � 0.2 nm) than the corresponding P4VP domain size.
The crystalline nature and the overall diameter of as-prepared
Au nanoclusters were confirmed using high-resolution TEM
(Fig. 2a, inset). Notably, the sizes of the Au nanoclusters can
be modulated through variations in the deposition conditions,
such as immersion time and Au complex concentration.
The size distributions of Au nanoclusters increase with
P4VP(HAuCl4) loading concentration. From TEM images,
the average nanocluster sizes were measured to be 1.0 � 0.2,
1.3 � 0.2, 1.7 � 0.2 and 2.2 � 0.3 nm for loading concentrations
of 0.3, 0.5, 0.8, and 1, respectively (Fig. 2). According to the
‘‘magic numbers’’ for Au nanoclusters,14 the sizes of the
resulting sub-2-nm clusters closely match the sizes of Au
nanoclusters corresponding toB38, 75 and 146 Au atoms.15,16
In our case, the observed size distribution within a sample can
be due to the nature of our synthesis or caused by intrinsic
issues of TEM imaging. Electron beam irradiation can obfuscate
the boundary between cluster and polymer matrix during
TEM imaging and we estimate uncertainty in the size
measurement to be one or two lattice fringes, typically around
0.2 nm. In addition to the nanocluster size control, the
Au complex concentrations have a critical effect on their
spatial distribution in response to observed BCP morphological
changes (Fig. 2a–d). As HAuCl4 loading in the P4VP domain
is increased, the transition from hexagonal (Fig. 2a and b),
gyroid (Fig. 2c) to lamellar morphology (Fig. 2d) is observed
due to the modification of the copolymer block volume ratio.17
Since the Au nanoclusters are not covalently linked to the
P4VP block, bonding between the Au nanoclusters and the
pyridine can be either strengthened or weakened by external
stimuli to adjust the arrangement and architecture of the
polymer chain, and in turn transform the spatial distribution
and ordering of the embedded Au nanoclusters. This brings
about unique opportunities to create responsive materials
where external stimuli can be used to alter the spatial distribution
of Au nanoclusters and the macroscopic properties of the
nanocluster assemblies. We used thermally-responsive properties
of PS-b-P4VP BCP to change the dimensionality and overall
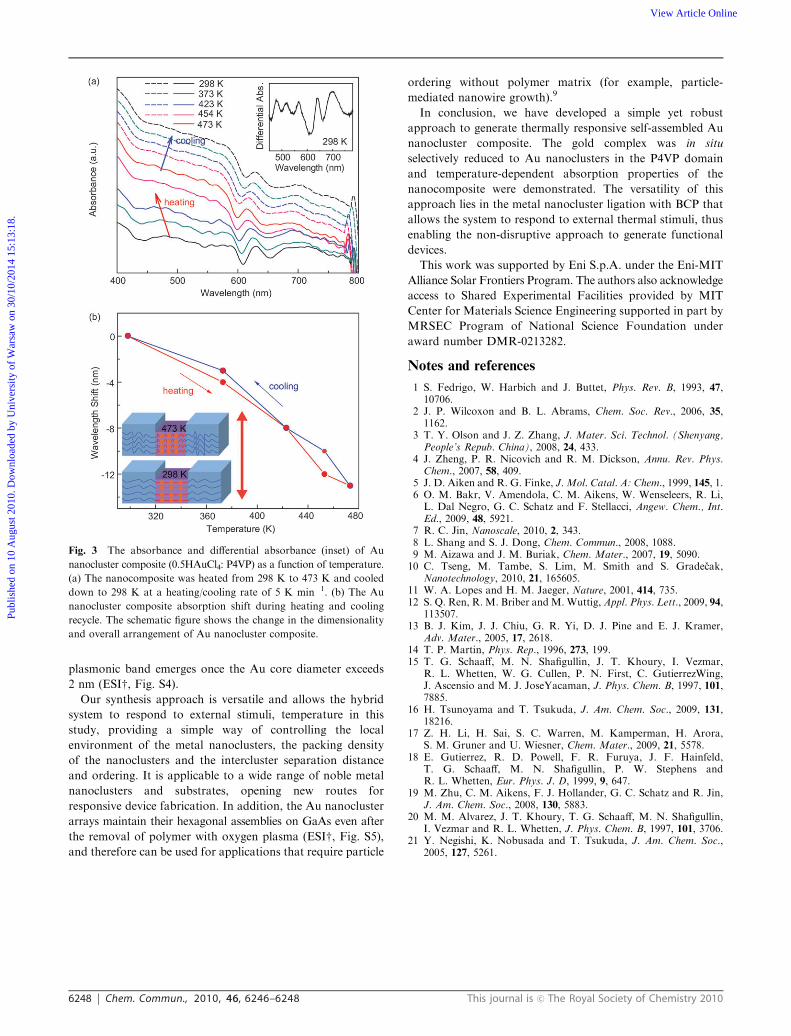
arrangements of the Au nanocluster assemblies. Fig. 3a shows
in situ thermally responsive UV-Vis spectra of the 1.3 nm Au
nanocluster composite. Distinct onsets of absorption bands at
about 480 nm, 565 nm, 640 nm, and 700 nm were observed at
room temperature (Fig. 3a, inset) and these closely match the
characteristic optical absorption features of the classical Au
nanocluster core with 75 atoms (B1.3 nm).18 They may be
interpreted as arising from intraband (between levels in quantized
sp band) or interband (d band to sp band) transitions or a
mixture of the two.19,20 An additional absorption band at
520 nm was also observed, but further studies are needed to
clarify its origin. An interesting feature was observed when the
sample was heated from 298 to 473 K: the 640 nm absorption
band blue shifted by 13 nm, and the process was reversible
upon cooling back to 298 K (Fig. 3b). When the nanocomposite
is heated above the glass transition temperature of each block,
it firstly causes extension of the PS coil-block because of its
lower glass transition temperature (373 K) than that of P4VP
(476 K). Further increase of the temperature reduces the
stiffness of the P4VP(Au nanoclusters) comb-block and leads
to a structural rearrangement of Au nanoclusters and their
relative distance. Therefore, the observed thermally responsive
absorption is likely caused by the interaction between pyridine
and Au nanoclusters and/or the inter-nanocluster distance
evolution during the heating process.21 In the cooling cycle,
a reverse process is observed. Thus, using temperature as a
switch it is possible to direct Au nanocluster distribution.
This process occurs within minutes and is fully reversible.
The optical absorption spectra of clusters synthesized using
different loading concentrations demonstrate the corresponding
nanocluster size-dependent behaviour. Ultimately, a surface
Fig. 2 Bright-field TEM images of Au nanocluster composites and
the corresponding Au nanocluster size distributions as a function
of the xHAuCl4: P4VP concentration. (a) x = 0.3; (b) x = 0.5;
(c) x = 0.8; (d) x = 1. Insets show high resolution TEM images of
representative Au nanoclusters observed within each P4VP domain.
Same scale-bars for all bright-field (50 nm) and high-resolution TEM
images (1 nm). (e) Au nanocluster size distributions correspond to
images (a)–(d).
Publ
ishe
d on
10
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
War
saw
on
30/1
0/20
14 1
5:13
:18.
View Article Online
6248 Chem. Commun., 2010, 46, 6246–6248 This journal is c The Royal Society of Chemistry 2010
plasmonic band emerges once the Au core diameter exceeds
2 nm (ESIw, Fig. S4).Our synthesis approach is versatile and allows the hybrid
system to respond to external stimuli, temperature in this
study, providing a simple way of controlling the local
environment of the metal nanoclusters, the packing density
of the nanoclusters and the intercluster separation distance
and ordering. It is applicable to a wide range of noble metal
nanoclusters and substrates, opening new routes for
responsive device fabrication. In addition, the Au nanocluster
arrays maintain their hexagonal assemblies on GaAs even after
the removal of polymer with oxygen plasma (ESIw, Fig. S5),and therefore can be used for applications that require particle
ordering without polymer matrix (for example, particle-
mediated nanowire growth).9
In conclusion, we have developed a simple yet robust
approach to generate thermally responsive self-assembled Au
nanocluster composite. The gold complex was in situ
selectively reduced to Au nanoclusters in the P4VP domain
and temperature-dependent absorption properties of the
nanocomposite were demonstrated. The versatility of this
approach lies in the metal nanocluster ligation with BCP that
allows the system to respond to external thermal stimuli, thus
enabling the non-disruptive approach to generate functional
devices.
This work was supported by Eni S.p.A. under the Eni-MIT
Alliance Solar Frontiers Program. The authors also acknowledge
access to Shared Experimental Facilities provided by MIT
Center for Materials Science Engineering supported in part by
MRSEC Program of National Science Foundation under
award number DMR-0213282.
Notes and references
1 S. Fedrigo, W. Harbich and J. Buttet, Phys. Rev. B, 1993, 47,10706.
2 J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35,1162.
3 T. Y. Olson and J. Z. Zhang, J. Mater. Sci. Technol. (Shenyang,People’s Repub. China), 2008, 24, 433.
4 J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys.Chem., 2007, 58, 409.
5 J. D. Aiken and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1.6 O. M. Bakr, V. Amendola, C. M. Aikens, W. Wenseleers, R. Li,L. Dal Negro, G. C. Schatz and F. Stellacci, Angew. Chem., Int.Ed., 2009, 48, 5921.
7 R. C. Jin, Nanoscale, 2010, 2, 343.8 L. Shang and S. J. Dong, Chem. Commun., 2008, 1088.9 M. Aizawa and J. M. Buriak, Chem. Mater., 2007, 19, 5090.10 C. Tseng, M. Tambe, S. Lim, M. Smith and S. Gradecak,
Nanotechnology, 2010, 21, 165605.11 W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735.12 S. Q. Ren, R. M. Briber andM.Wuttig, Appl. Phys. Lett., 2009, 94,
113507.13 B. J. Kim, J. J. Chiu, G. R. Yi, D. J. Pine and E. J. Kramer,
Adv. Mater., 2005, 17, 2618.14 T. P. Martin, Phys. Rep., 1996, 273, 199.15 T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar,
R. L. Whetten, W. G. Cullen, P. N. First, C. GutierrezWing,J. Ascensio and M. J. JoseYacaman, J. Phys. Chem. B, 1997, 101,7885.
16 H. Tsunoyama and T. Tsukuda, J. Am. Chem. Soc., 2009, 131,18216.
17 Z. H. Li, H. Sai, S. C. Warren, M. Kamperman, H. Arora,S. M. Gruner and U. Wiesner, Chem. Mater., 2009, 21, 5578.
18 E. Gutierrez, R. D. Powell, F. R. Furuya, J. F. Hainfeld,T. G. Schaaff, M. N. Shafigullin, P. W. Stephens andR. L. Whetten, Eur. Phys. J. D, 1999, 9, 647.
19 M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin,J. Am. Chem. Soc., 2008, 130, 5883.
20 M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin,I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706.
21 Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc.,2005, 127, 5261.
Fig. 3 The absorbance and differential absorbance (inset) of Au
nanocluster composite (0.5HAuCl4: P4VP) as a function of temperature.
(a) The nanocomposite was heated from 298 K to 473 K and cooled
down to 298 K at a heating/cooling rate of 5 K min�1. (b) The Au
nanocluster composite absorption shift during heating and cooling
recycle. The schematic figure shows the change in the dimensionality
and overall arrangement of Au nanocluster composite.
Publ
ishe
d on
10
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
War
saw
on
30/1
0/20
14 1
5:13
:18.
View Article Online


















