
Synopsis of study HPV-030 PRI (111507)
24
Personal identifiable data of investigators (investigator names) are not published in this report, as consent according to Section 4a of the German Federal Act on Data Protection is not available for any of the investigators. Synopsis of study HPV-030 PRI (111507) Pharmaceutical entrepreneur: GlaxoSmithKline GmbH & Co. KG Prinzregentenplatz 9 81675 Munich Germany
Transcript of Synopsis of study HPV-030 PRI (111507)

Personal identifiable data of investigators (investigator names) are not published in this report, as consent according to Section 4a of the German Federal Act on Data Protection is not available for any of the investigators.
Synopsis of study HPV-030 PRI (111507) Pharmaceutical entrepreneur: GlaxoSmithKline GmbH & Co. KG Prinzregentenplatz 9 81675 Munich Germany

The study summarized below may involve approved and non-approved uses, formulations or treatment regimens. The results reported for any single study may not reflect the overall results obtained during all studies involving the same product. Before prescribing any product mentioned in this Register, healthcare professionals should consult the prescribing information for the product approved in their country.
HPV-030 (111507) 1
Name of company: GlaxoSmithKline Biologicals, Rixensart, Belgium Name of finished product: GlaxoSmithKline Biologicals’ human papillomavirus (HPV) vaccine Name of active substance: HPV-16 L1 virus-like particles HPV-18 L1 virus-like particles
TABULAR FORMAT REFERRING TO PART OF THE DOSSIER
Volume:
Page:
(for national authority only)
Study No.: HPV-030 (111507) Title of the study : A phase IIIb, randomized, open, multicentre study to evaluate the immunogenicity and safety of GlaxoSmithKline Biologicals’ HPV-16/18 L1 VLP adjuvanted vaccine (Cervarix™) co-administrated with GlaxoSmithKline Biologicals’ Hepatitis B vaccine (Engerix-B™) in healthy female subjects aged 9 - 15 years Coordinating investigators: This study was conducted by three coordinating investigators in two European countries (Netherlands, Sweden). Study centre(s): This study was conducted in seven study centers: two centers in the Netherlands and five centers in Sweden. Publication (reference): Not published as of June 2010. Study period: Study initiation date (First Subject, First Visit): 09 April 2008 Study completion date for the active phase (Last Subject, Last Visit, Month 7): 28 Aug 2009
Clinical phase: IIIb
Objectives: Co-primary: • To demonstrate non-inferiority of the hepatitis B immune response at Month 7 when hepatitis B vaccine was
co-administered with HPV-16/18 vaccine at Months 0, 1 and 6 (HPV+HepB group) as compared to when hepatitis B vaccine was administered alone at Months 0, 1 and 6 (HepB group). Criterion for non-inferiority: The objective was reached if the upper limit of the 95% confidence interval (CI) for the difference in the percentage of subjects with anti-HBs antibody titres ≥ 10 mIU/mL (seroprotection) between HepB group and HPV+HepB group (HepB minus HPV+HepB) at Month 7 was below 10%.
• To demonstrate non-inferiority of the HPV immune response at Month 7 when the HPV-16/18 vaccine was co-administered with hepatitis B vaccine (HPV+HepB group) as compared to when the HPV-16/18 vaccine was administered alone (HPV group). Criteria for non-inferiority were assessed sequentially as follows: 1. Non inferiority in terms of seroconversion rates:
The objective was reached if for each antigen the upper limits of the 95% confidence interval (CI) for the difference in the percentage of subjects who had seroconverted at Month 7 in the HPV group versus the HPV+HepB group (HPV minus HPV+HepB) at Month 7 were below 5%.
2. Non-inferiority in terms of GMTs:
The objective was reached if for each antigen the upper limits of the 95% CI for the ratio of GMTs one month post Dose 3 (Month 7) were below 2. For anti-HPV-16 and anti-HPV-18, this ratio was calculated as the GMT in HPV group divided by the GMT in HPV+HepB group.
Secondary: Immunogenicity • To evaluate, one month after Dose 3 (Month 7), in the HepB vaccine recipients, the immune response against
hepatitis B with respect to seroconversion rates and GMTs.

HPV-030 (111507) 2
• To evaluate, one month after Dose 2 (Month 2), in the HPV vaccine recipients, the immune response against HPV-16 and HPV-18 with respect to seroconversion rates and GMTs.
• To evaluate, one month after Dose 2 (Month 2), in the HepB vaccine recipients, the immune response against hepatitis B with respect to seroconversion rates, seroprotection rates and GMTs.
Safety • To evaluate the incidence and intensity of solicited local symptoms reported during the 7-day period (Days 0 -
6) following each and any vaccination in all study groups. • To evaluate the incidence, intensity and relationship to vaccination of solicited general symptoms reported
during the 7-day period (Days 0 - 6) following each and any vaccination in all study groups. • To evaluate the incidence, intensity and relationship to vaccination of unsolicited adverse events (AEs)
reported during the 30-day period (Days 0 - 29) following any vaccination in all study groups. • To assess the safety of the study vaccine with respect to the nature, intensity and relationship to vaccination
of serious adverse events (SAEs) in all study groups throughout the study period. • To evaluate the occurrence of medically significant conditions throughout the study period (up to Month 12)
regardless of causal relationship to vaccination and intensity in all study groups. Study design: • A Phase IIIb, randomized, controlled, parallel-group, multicountry, multicentre study with three treatment
groups. • Treatment allocation: Internet-based randomization (1:1:1). The randomization was to be age-stratified, with
approximately the same number of subjects in the three age strata (9 years, 10 - 12 years and 13 - 15 years). • Vaccination schedules:
• Cervarix (HPV-16/18 L1 VLP with adjuvant): Three doses of vaccine administered intramuscularly, with the second and third dose given one month and six months after the first dose, respectively.
• Engerix-B (HepB): Three doses of vaccine administered intramuscularly, with the second and third dose given one month and six months after the first dose respectively.
Synopsis Table 1. Treatment groups Vaccine administered Groups Month 0 Month 1 Month 6 HPV (N = 252) HPV-16/18 HPV-16/18 HPV-16/18 HPV + HepB (N = 252) HPV-16/18 + HepB HPV-16/18 + HepB HPV-16/18 + HepB HepB (N = 252) HepB HepB HepB N = planned number of subjects Note: After completion of the safety follow-up and outside the study protocol, Engerix-B will be offered to the HPV group and Cervarix will be offered to the HepB group, if commercially available in the participating country.

HPV-030 (111507) 3
• Blinding: Open. • Control: Active. • Type of study: Self-contained. • Data collection: Remote Data entry (RDE). • Duration of the study: Approximately 12 months per subject i.e., a seven-month active phase followed by a
five-month follow-up phase for safety. • Visits: Five visits scheduled per subject at Months 0, 1, 2, 6 and 7 (with telephone contact at Month 12). • Blood samples to evaluate immunogenicity: Three blood samples, taken at Month 0, Month 2 and Month 7. • Safety and reactogenicity monitoring:
1. Solicited signs and symptoms were to be self-reported by all subjects, using a diary card, on the day of vaccination (Day 0) and on the 6 subsequent days (Days 1 - 6).
2. Unsolicited signs and symptoms were to be self-reported in all subjects within 30 days (Days 0 - 29) after each vaccination.
3. SAEs were to be reported in all subjects throughout the study period (up to Month 12).
4. Medically significant conditions were to be reported in all subjects throughout the entire study (up to Month 12) regardless of causal relationship to vaccination and intensity.
5. Pregnancies and pregnancy outcomes were to be recorded throughout the entire study period (up to Month 12) and subjects will be followed until delivery (even if delivery occurs after the end of the study).
• An interim analysis was conducted on immunogenicity data collected at one month after the second dose of vaccine (Month 2), in the subset of the first 150 subjects (approximately) with Month 2 data available (about 50 subjects per group).
• Final analysis was conducted on all immunogenicity and safety data collected up to the end of the active phase (Month 7).
• The results of the safety follow-up (up to Month 12) will be provided in a separate annex report. Number of subjects: Approximately 756 subjects (252 subjects per group) aged 9 - 15 years were to be enrolled in study centers in Europe to reach a target of 603 evaluable subjects for the analysis at Month 7 (201 evaluable subjects in each group), refer to Synopsis Table 2. In each study group, there were three age strata (9 years, 10 - 12 years and 13 - 15 years) and approximately 84 subjects were to be enrolled per stratum Synopsis Table 2. Number of subjects in the study Number of subjects Total HPV+HepB group HPV group HepB group Planned 756 252 252 252 Enrolled 744 Completed 728 246 240 242 Total Vaccinated cohort (TVC) 741 247 247 247 According to protocol (ATP) cohort for Safety 720 241 240 239 ATP cohort for Immunogenicity 676 225 222 229

HPV-030 (111507) 4
Diagnosis and criteria for inclusion: Healthy females between, and including, 9 and 15 years of age (had not attained her 16th birthday) at the time of the first vaccination. Written informed consent was obtained from each subject’s parent /legally acceptable representative (LAR) and written informed assent was obtained from each subject prior to the performance of any study-specific procedures. Subjects had to be free of obvious health problems as established by medical history and clinical examination. Subjects were not to be pregnant. Absence of pregnancy was to be verified with a urine pregnancy test. Subjects had to be of non-childbearing potential, i.e., had a current tubal ligation, hysterectomy, ovariectomy or be pre-menarcheal, or if the subject was of childbearing potential or became of child-bearing potential during the study, she was to be abstinent or to have used adequate contraception for 30 days prior to vaccination and to have agreed to continue such precautions for two months after completion of the vaccination series. Subjects were excluded from the study if they were using any investigational/non-registered product other than the study vaccine within 30 days prior to first study vaccination or were planning to use it during the study period. Subjects concurrently participating in another clinical study in which they had been or would be exposed to an investigational or non-investigational product during the study period were excluded. Also forbidden were planned administration/administration of non-routine vaccines not foreseen by the study protocol within 30 days of vaccination, chronic administration of immunosuppressants or other immune modifying drugs within 6 months prior to vaccination, previous or planned administration of an HPV or hepatitis B vaccine not foreseen by the study protocol during the study period, previous administration of MPL or adjuvant, administration of immunoglobulins or any blood products administered within 3 months prior to vaccination or planned to be administered during the study period. Subjects with a history of hepatitis B infection and subjects with known exposure to hepatitis B within 6 weeks prior to vaccination were excluded. Pregnant or breastfeeding subjects, subjects planning to or likely to become pregnant and subjects planning to discontinue contraception during the study period and up to 2 months after last vaccination were excluded. Subjects with cancer/autoimmune disease under treatment, known acute or chronic clinically significant neurologic, hepatic or renal functional abnormality, history of allergic disease or reactions likely to be exacerbated by any component of the vaccines, or any immunosuppressive or immunodeficient condition were excluded. Study vaccine, dose, mode of administration, lot no.: Vaccination schedule /site: Three doses of the adjuvanted study vaccine HPV-16/18 L1 VLP (HPV-16/18) (Cervarix) were administered intramuscularly according to a 0, 1, 6-month schedule. Vaccine composition /dose /lot number: Each dose of HPV-16/18 vaccine (Cervarix) contained HPV-16 L1 VLP, HPV-18 L1 VLP, MPL and aluminium salt. Lot number: AHPVA023D (expiry date: 30/04/2010). Reference vaccine /Comparator, dose and mode of administration, lot no.: Vaccination schedule /site: Three doses of the control hepatitis B (HepB) vaccine (Engerix-B) were administered intramuscularly according to a 0, 1, 6-month schedule. Vaccine composition /dose /lot number: Each dose of HepB vaccine (Engerix-B) contained HBs Ag (Hepatitis B surface antigen) and aluminium salt. Lot number: AHBVB407B (expiry date: 28/02/2010) and AHBVB420A (expiry date: 28/02/2010). Duration of treatment: Duration of study was approximately 12 months for each subject.

HPV-030 (111507) 5
Criteria for evaluation: Co-primary endpoints(Immunogenicity) • Anti-HBs seroprotection status at Month 7 in subjects receiving the HepB vaccine. • Anti-HPV-16/18 seroconversion status at Month 7 in subjects receiving the HPV-16/18 vaccine. • Anti-HPV-16/18 antibody titres at Month 7 in subjects receiving the HPV-16/18 vaccine. Secondary endpoints (Immunogenicity and Safety /Reactogenicity) • Anti-HBs seroconversion status and antibody titres at Month 7 in subjects receiving the HepB vaccine. • Anti-HPV-16/18 seroconversion status and antibody titres at Month 2 in subjects receiving the HPV vaccine. • Anti-HBs seroconversion and seroprotection status and antibody titres at Month 2 in subjects receiving the
HepB vaccine. • Occurrence of any and Grade 3 solicited local symptoms (injection site pain, redness and swelling) during the
7-day period (Days 0 - 6) following each and any vaccination in all study groups. • Occurrence of any, Grade 3 and causally related to vaccination solicited general symptoms during the 7-day
period (Days 0 - 6) following each vaccination in all study groups. • Occurrence of any, Grade 3 and causally related to vaccination unsolicited AEs during the 30-day period
(Days 0 - 29) following any vaccination in all study groups. • Occurrence of any, Grade 3 and causally related to vaccination SAEs in all study groups throughout the active
phase of the study (up to Month 7). • Occurrence of medically significant conditions in all study groups throughout the active phase of the study (up
to Month 7) regardless of causal relationship to vaccination and intensity. • Occurrence of any, Grade 3 and causally related to vaccination SAEs in all study groups throughout the safety
follow-up (up to Month 12) (to be presented in a separate annex report). • Occurrence of medically significant conditions in all study groups throughout the safety follow-up (up to Month
12) regardless of causal relationship to vaccination and intensity (to be presented in a separate annex report). Statistical methods: Study cohorts The Total Vaccinated cohort included all vaccinated subjects for whom data were available. The total analysis of safety included all subjects with at least one vaccine administration documented for unsolicited AEs and concomitant medication and included subjects with documented doses for solicited symptoms. The Total Vaccinated cohort for analysis of immunogenicity included vaccinated subjects for whom data concerning immunogenicity endpoint measures were available. The Total Vaccinated cohort analysis was performed per treatment actually administered. The ATP cohort for analysis of safety included all subjects who had received three doses of study/control vaccine according to their random assignment, for whom administration site of study vaccine was known and who had not received a vaccine not specified or forbidden in the protocol. The ATP cohort for analysis of immunogenicity included all evaluable subjects (i.e. those meeting all eligibility criteria, complying with the procedures defined in the protocol, with no elimination criteria. during the study) for whom data concerning immunogenicity endpoint measures were available. This included subjects for whom assay results were available for antibodies against at least one study vaccine antigen component after vaccination. The Interim Vaccinated Cohort included subjects who received at least one dose of vaccine and with a blood sample available at Month 0 and Month 2 The subset of 9 year old subjects in the Total Vaccinated cohort included all 9 year old vaccinated subjects for whom data were available. The total analysis of safety included all 9 year old subjects with at least one vaccine administration documented for unsolicited AEs and concomitant medication and included 9 year old subjects with documented doses for solicited symptoms. The 9 year old subjects in ATP cohort for analysis of immunogenicity included all evaluable 9 year old subjects (i.e. those meeting all eligibility criteria, complying with the procedures defined in the protocol, with no elimination criteria during the study) for whom data concerning immunogenicity endpoint measures were available. This included 9 year old subjects for whom assay results were available for antibodies against at least one study vaccine antigen

HPV-030 (111507) 6
component after vaccination. Analysis of demographics Demographic characteristics (age, gender, and race) of each study cohort were tabulated. The mean age (plus range and standard deviation) of the vaccinated subjects, as a whole, and per group, was calculated. The distribution of subjects enrolled among the study sites was tabulated as a whole and per group. Immunogenicity The primary analysis was based on the ATP cohort for analysis of immunogenicity. A second analysis based on the Total Vaccinated cohort was performed to supplement the ATP analysis. Between group comparisons (co-primary objectives) • The two-sided standardized asymptotic 95% CI for the group differences in the percentage of subjects with
anti-HBs antibody titres ≥10 mIU/mL at Month 7 in the HepB group and (minus) the HPV+ HepB group, was calculated. Non-inferiority of HPV+ HepB to HepB with respect to anti-HBs was demonstrated if the upper limit of this 95% CIs was below the pre-defined clinical limit of 10%.
• The two-sided standardized asymptotic 95% CI of the anti-HPV-16 and anti-HPV-18 for the group differences in the seroconversion rate at Month 7 between the HPV group and (minus) the HPV+ HepB group, was calculated. Non-inferiority of HPV+ HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 was demonstrated if the upper limits of these 95% CIs were below the pre-defined clinical limit of 5%.
• If the non-inferiority of HPV+ HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 SC rates was reached, the two-sided 95% CI of the anti-HPV-16 and anti-HPV-18 for the GMTs ratios at Month 7 between the HPV group and (divided by) the HPV+ HepB group, was calculated. Non-inferiority of HPV+ HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 was demonstrated if the upper limits of these 95% CIs were below the pre-defined clinical limit of 2.
Within group evaluations (secondary objectives) For the HPV+ HepB and HPV groups at each time point that a blood sample result was available (Months 0, 2 and 7): • Seroconversion and seropositivity rates for anti-HPV-16 and anti-HPV-18 (with exact 95% CI) were calculated
per pre-vaccination status.

HPV-030 (111507) 7
• Anti-HPV-16 and anti-HPV-18 GMTs with 95% CI and range of antibody titres were tabulated for antibodies for each antigen per pre-vaccination status.
• The distribution of antibody titres for anti-HPV-16 and anti-HPV-18 at Month 7 was displayed using reverse cumulative distribution curves for the sub-cohort of initially seronegative subjects.
For the HPV+ HepB and HepB groups at each time point that a blood sample result was available (Months 0, 2 and 7): • Seroconversion/seropositivity and seroprotection rates for anti-HBs (with exact 95% CI) were calculated in
subjects with undetectable anti-HBs and anti-HBc titres at Month 0. • Anti-HBs GMTs with 95% CI and range of antibody titres were tabulated in subjects with undetectable anti-
HBs and anti-HBc titres at Month 0. • The distribution of antibody titres for anti-HBs at Month 2 and Month 7 was displayed using reverse cumulative
distribution curves in subjects with undetectable anti-HBs and anti-HBc titres at Month 0. Analysis of safety The primary analysis was based on the Total Vaccinated cohort. A second analysis based on this ATP cohort for safety was performed to supplement the Total Vaccinated cohort analysis.
• The percentage of subjects with at least one local solicited AE, with at least one general solicited AE and with any solicited AE during the solicited follow-up period (Days 0 - 6) was tabulated with exact 95% CI after each vaccine dose and overall. The percentage of doses followed by at least one local solicited AE, by at least one general solicited AE and by any solicited AE was tabulated, over the whole vaccination course, with exact 95% CI. The same tabulation was done for Grade 3 solicited AEs, solicited AEs related to vaccination and Grade 3 solicited AEs related to vaccination.
• The percentage of subjects with at least one local (solicited or unsolicited) AE, with at least one general (solicited or unsolicited) AE and with any (solicited or unsolicited) AE during the 30-days follow-up period (Days 0 - 29) was tabulated with exact 95% CI after each vaccine dose and overall. The percentage of doses followed by at least one local (solicited or unsolicited) AE, by at least one general (solicited or unsolicited) AE and by any (solicited or unsolicited) AE was tabulated, over the whole vaccination course, with exact 95% CI. The same tabulation was done for Grade 3 solicited and unsolicited AEs, solicited and unsolicited AEs related to vaccination and Grade 3 solicited and unsolicited AEs related to vaccination.
• The percentage of subjects reporting each individual solicited local and general AE during the solicited follow-up period was tabulated with exact 95% CI. The percentage of doses followed by each individual solicited local and general AE was tabulated, per dose and over the whole vaccination course, with exact 95% CI.
• For all solicited symptoms, the same tabulation was performed for Grade 3 AEs, for AEs with relationship to vaccination (general symptoms only) and for Grade 3 AEs related to vaccination (general symptoms only).
• The median duration of solicited local and general symptoms during the solicited follow-up period was tabulated. The same tabulation was performed for Grade 3 solicited symptoms.
• The number of solicited local and general symptoms ongoing beyond the 7-day follow-up period, together with the time to resolution was described in detail.
• The proportion of subjects with at least one report of unsolicited AE classified by the Medical Dictionary for Regulatory Activities (MedDRA), whenever available, and reported up to 30 days after vaccination was tabulated with exact 95% CI. The same tabulation was performed for Grade 3 unsolicited AEs and for unsolicited AEs with a relationship to vaccination.
• The proportion of subjects with at least one report of a medically significant AE classified by MedDRA, whenever available, and reported up to 30 days after vaccination was tabulated with exact 95% CI. A similar table was produced for medically significant AEs starting Day 30 after each dose till the end of the study.
• The proportion of subjects with at least one report of New Onset of Chronic Disease classified by MedDRA, whenever available, and reported during the entire study period was tabulated with exact 95% CI.
• The proportion of subjects with at least one report of New Onset of Autoimmune Disease classified by MedDRA, whenever available, and reported during the entire study period was tabulated with exact 95% CI.
• SAEs and withdrawal due to AE(s) were described in detail. • The proportion of subjects who started to receive at least one concomitant medication during the 30-day
follow-up period after each vaccination was calculated with 95% CI. The use of antipyretics and antibiotics

HPV-030 (111507) 8
was also reported.
Interim analysis An interim analysis was performed on immunogenicity data collected at one month after the second dose of vaccine (Month 2) in the subset of the first 150 subjects (approximately) with Month 2 data available (about 50 subjects per group). The interim analysis for immunogenicity was descriptive only and included subjects who received at least one dose of vaccine and with a blood sample available at Month 0 and Month 2. • Seroconversion and seropositivity rates for anti-HPV-16 and anti-HPV-18 at Month 2 (with exact 95% CI) were
calculated per pre-vaccination status in the HPV group and the HPV+HepB group. • GMT with 95% CI and range of antibody titres was tabulated for antibodies for anti-HPV-16 and anti-HPV-18
at Month 2, per pre-vaccination status, in the HPV group and the HPV+HepB group. • Seroconversion and seropositivity rates for anti-HBs at Month 2 (with exact 95% CI) were calculated in
subjects with undetectable anti-HBs and anti-HBc titres at Month 0 in the HPV+HepB group and the HepB group.
• GMT with 95% CI and range of antibody titres were tabulated for antibodies for anti-HBs at Month 2 in subjects with undetectable anti-HBs and anti-HBc titres at Month 0 in the HPV+HepB group and the HepB group.
Since the interim analysis was limited to descriptive statistics of a secondary endpoint, no correction for multiplicity was applied. This analysis was performed on uncleaned data by an external statistician to avoid access to unblinded data by GSK staff. No stopping rule was applied. No clinical report was written at the time of the interim analysis but all produced tables supplement this final clinical report. Dissemination of results from the interim analysis was limited within GSK and only shared outside of GSK on a limited basis and under conditions of confidentiality. Summary:
ATP cohort for immunogenicity: The mean age of the subjects at study entry was 11.4 years. The majority (94.5%) of the population was of White- Caucasian/European heritage.
Demography
Total Vaccinated cohort: The mean age of the subjects at study entry was 11.4 years. The majority (94.2%) of the population was of White- Caucasian/European heritage.
The primary analysis of immunogenicity was based on the ATP cohort for immunogenicity. Analyses based on the Total Vaccinated cohort, the Interim Cohort and 9 year old subjects in the ATP cohort for immunogenicity were performed to supplement the ATP analysis.
Immunogenicity:
Immunogenicity –Co-primary objective - Non-inferiority assessment for the hepatitis B immune response between HPV + HepB and HepB groups at Month 7 The percentage of initially seronegative subjects with anti-HepB antibody concentrations ≥ 10 mIU/mL (seroprotection level) and anti-HepB GMTs in the HPV+HepB and HepB groups at one month post Dose 3 (Month 7) are presented in Synopsis Table 6. Non-inferiority of HPV+HepB to HepB with respect to anti-HepB seroprotection rate was demonstrated since the upper limit (UL) of the 95% confidence interval (CI) on the difference of seroprotection was below the pre-defined clinical limit of 10%, refer to Synopsis Table 3. Synopsis Table 3 Non-inferiority assessment of HPV+HepB group compared to HepB group with respect to anti-HBs seroprotection rate, Post Dose 3- Month 7 in subjects initially seronegative for anti-HBs and anti-HBc prior to Dose 1 (ATP Cohort for Immunogenicity)
Difference in seroprotection rate (Group 2 minus Group 1)
95 % CI Group 1 N % Group 2 N % Difference % LL UL HPV+HepB 194 97.9 HepB 181 100 HepB – (HPV+HepB) 2.06 -0.04 5.19 HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine, HepB = Engerix-B vaccine N = number of subjects with available results; % = percentage of subjects with HBs AB concentration ≥ 10 mIU/mL; 95% CI = 95% Standardized asymptotic confidence interval; LL = lower limit, UL = upper limit

HPV-030 (111507) 9
Immunogenicity –Co-primary objective - Non-inferiority assessment for the HPV-16/18 immune response between HPV + HepB and HPV groups at Month 7 The percentage of initially seronegative subjects with anti-HPV-16 antibody titres ≥ 8 EL.U/mL (seroconversion level) and anti-HPV-16 GMTs, in the HPV+HepB and HPV groups at one month post Dose 3, Month 7 are presented in Synopsis Table 7. The percentage of initially seronegative subjects with anti-HPV-18 antibody titres ≥ 7 EL.U/mL (seroconversion level) and anti-HPV-18 GMTs in the HPV+HepB and HPV groups at one month post Dose 3, Month 7 are presented in Synopsis Table 8. Non-inferiority of HPV+HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 seroconversion rates was demonstrated since the ULs of the 95% CIs on the differences of SC were below the pre-defined clinical limit of 5%, refer to Synopsis Table 4. Synopsis Table 4 Non-inferiority assessment of HPV+HepB group compared to HPV group in terms of anti-HPV-16 and anti-HPV/18 seroconversion rate, Post Dose 3, Month 7 in initially seronegative subjects (ATP Cohort for Immunogenicity)
Difference in seropositivity rate (Group 2 minus Group 1)
95 % CI Group 1 N % Group 2 N % Difference % LL UL Anti-HPV-16 HPV+HepB 207 99.0* HPV 200 100 HPV – (HPV+HepB) 0.97 -0.93 3.46 Anti-HPV-18 HPV+HepB 200 99.5 HPV 202 100 HPV – (HPV+HepB) 0.50 -1.38 2.78 HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine N = number of subjects with available results; % = percentage of subjects with Anti-HPV-16 concentration ≥ 8 ELU/ML or with Anti-HPV-18 ≥ 7 ELU/ML; 95% CI = 95% Standardized asymptotic confidence interval; LL = lower limit, UL = upper limit; * = At the time of the final analysis, inconsistent HPV antibody titers were observed at Month 7 (post Dose 3) for two subjects, in the HepB+ HPV group, when compared to their Month 2 (post Dose 2) results. After re-testing, 1 of the subjects (originally seronegative for HPV-16 at Month 7) was found to have seroconverted for HPV-16 at Month 7. A post-hoc analysis of all immunogenicity data involving anti-HPV-16 antibody titres was performed and is provided as supplemental data. Since non-inferiority of HPV+HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 seroconversion rates was demonstrated, the two-sided 95% CIs for anti-HPV-16 and anti-HPV-18 GMT ratios at Month 7 (HPV group divided by HPV+HepB group) were calculated. Non-inferiority of HPV+HepB to HPV with respect to anti-HPV-16 and anti-HPV-18 GMTs was demonstrated since the ULs of the 95% CIs on the ratios of GMTs were below the pre-defined clinical limit of 2, refer to Synopsis Table 5. Synopsis Table 5 Non-inferiority assessment of HPV+HepB group compared to HPV group in terms of anti-HPV-16 and HPV-18 GMT ratios, Post Dose 3, Month 7 in initially seronegative subjects (ATP Cohort for Immunogenicity)
GMT ratio (HPV / HPV+HepB)
HPV HPV+HepB 95% CI Anti body N GMT N GMT Value LL UL Anti-HPV-16 (ELU/ML) 200 21712.6 207 19819.8* 1.10 0.90 1.33 Anti-HPV-18 (ELU/ML) 202 8838.6 200 8835.1 1.00 0.84 1.20 HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine GMT = geometric mean antibody concentration; N = Number of subjects with pre-vaccination results available 95% CI = 95% confidence interval for the GMT ratio (Anova model - pooled variance); LL, UL = lower, upper limit ; * = At the time of the final analysis, inconsistent HPV antibody titers were observed at Month 7 (post Dose 3) for two subjects, in the HepB+ HPV group, when compared to their Month 2 (post Dose 2) results. After re-testing, 1 of the subjects (originally seronegative for HPV-16 at Month 7) was found to have seroconverted for HPV-16 at Month 7. A post-hoc analysis of all immunogenicity data involving anti-HPV-16 antibody titres was performed and is provided as supplemental data.
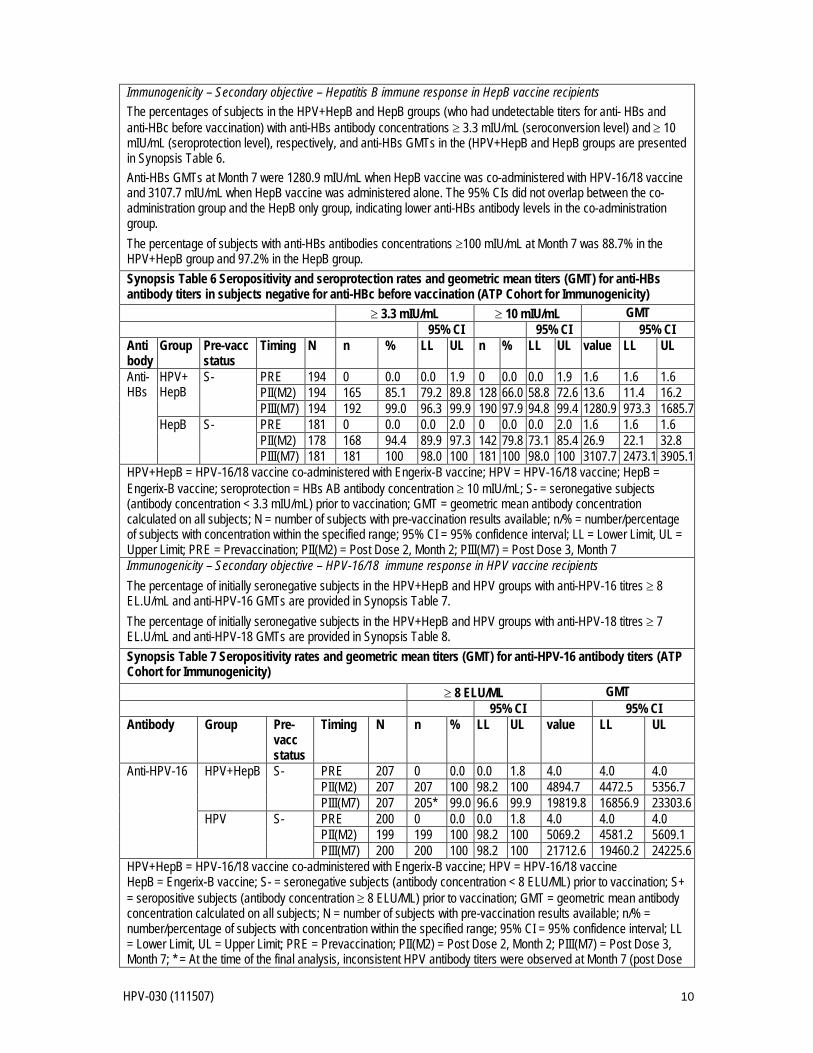
HPV-030 (111507) 10
Immunogenicity – Secondary objective – Hepatitis B immune response in HepB vaccine recipients The percentages of subjects in the HPV+HepB and HepB groups (who had undetectable titers for anti- HBs and anti-HBc before vaccination) with anti-HBs antibody concentrations ≥ 3.3 mIU/mL (seroconversion level) and ≥ 10 mIU/mL (seroprotection level), respectively, and anti-HBs GMTs in the (HPV+HepB and HepB groups are presented in Synopsis Table 6. Anti-HBs GMTs at Month 7 were 1280.9 mIU/mL when HepB vaccine was co-administered with HPV-16/18 vaccine and 3107.7 mIU/mL when HepB vaccine was administered alone. The 95% CIs did not overlap between the co-administration group and the HepB only group, indicating lower anti-HBs antibody levels in the co-administration group. The percentage of subjects with anti-HBs antibodies concentrations ≥100 mIU/mL at Month 7 was 88.7% in the HPV+HepB group and 97.2% in the HepB group. Synopsis Table 6 Seropositivity and seroprotection rates and geometric mean titers (GMT) for anti-HBs antibody titers in subjects negative for anti-HBc before vaccination (ATP Cohort for Immunogenicity)
≥ 3.3 mIU/mL ≥ 10 mIU/mL GMT 95% CI 95% CI 95% CI
Anti body
Group Pre-vacc status
Timing N n % LL UL n % LL UL value LL UL
Anti-HBs
HPV+ HepB
S- PRE 194 0 0.0 0.0 1.9 0 0.0 0.0 1.9 1.6 1.6 1.6 PII(M2) 194 165 85.1 79.2 89.8 128 66.0 58.8 72.6 13.6 11.4 16.2 PIII(M7) 194 192 99.0 96.3 99.9 190 97.9 94.8 99.4 1280.9 973.3 1685.7
HepB S- PRE 181 0 0.0 0.0 2.0 0 0.0 0.0 2.0 1.6 1.6 1.6 PII(M2) 178 168 94.4 89.9 97.3 142 79.8 73.1 85.4 26.9 22.1 32.8 PIII(M7) 181 181 100 98.0 100 181 100 98.0 100 3107.7 2473.1 3905.1
HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine; HepB = Engerix-B vaccine; seroprotection = HBs AB antibody concentration ≥ 10 mIU/mL; S- = seronegative subjects (antibody concentration < 3.3 mIU/mL) prior to vaccination; GMT = geometric mean antibody concentration calculated on all subjects; N = number of subjects with pre-vaccination results available; n/% = number/percentage of subjects with concentration within the specified range; 95% CI = 95% confidence interval; LL = Lower Limit, UL = Upper Limit; PRE = Prevaccination; PII(M2) = Post Dose 2, Month 2; PIII(M7) = Post Dose 3, Month 7 Immunogenicity – Secondary objective – HPV-16/18 immune response in HPV vaccine recipients The percentage of initially seronegative subjects in the HPV+HepB and HPV groups with anti-HPV-16 titres ≥ 8 EL.U/mL and anti-HPV-16 GMTs are provided in Synopsis Table 7. The percentage of initially seronegative subjects in the HPV+HepB and HPV groups with anti-HPV-18 titres ≥ 7 EL.U/mL and anti-HPV-18 GMTs are provided in Synopsis Table 8. Synopsis Table 7 Seropositivity rates and geometric mean titers (GMT) for anti-HPV-16 antibody titers (ATP Cohort for Immunogenicity)
≥ 8 ELU/ML GMT 95% CI 95% CI
Antibody Group Pre-vacc status
Timing N n % LL UL value LL UL
Anti-HPV-16 HPV+HepB S- PRE 207 0 0.0 0.0 1.8 4.0 4.0 4.0 PII(M2) 207 207 100 98.2 100 4894.7 4472.5 5356.7 PIII(M7) 207 205* 99.0 96.6 99.9 19819.8 16856.9 23303.6
HPV S- PRE 200 0 0.0 0.0 1.8 4.0 4.0 4.0 PII(M2) 199 199 100 98.2 100 5069.2 4581.2 5609.1 PIII(M7) 200 200 100 98.2 100 21712.6 19460.2 24225.6
HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine HepB = Engerix-B vaccine; S- = seronegative subjects (antibody concentration < 8 ELU/ML) prior to vaccination; S+ = seropositive subjects (antibody concentration ≥ 8 ELU/ML) prior to vaccination; GMT = geometric mean antibody concentration calculated on all subjects; N = number of subjects with pre-vaccination results available; n/% = number/percentage of subjects with concentration within the specified range; 95% CI = 95% confidence interval; LL = Lower Limit, UL = Upper Limit; PRE = Prevaccination; PII(M2) = Post Dose 2, Month 2; PIII(M7) = Post Dose 3, Month 7; * = At the time of the final analysis, inconsistent HPV antibody titers were observed at Month 7 (post Dose

HPV-030 (111507) 11
3) for two subjects, in the HepB+ HPV group, when compared to their Month 2 (post Dose 2) results. After re-testing, 1 of the subjects (originally seronegative for HPV-16 at Month 7) was found to have seroconverted for HPV-16 at Month 7. A post-hoc analysis of all immunogenicity data involving anti-HPV-16 antibody titres was performed and is provided as supplemental data. Synopsis Table 8 Seropositivity rates and geometric mean titers (GMT) for anti-HPV-18 antibody titers (ATP cohort for Immunogenicity)
≥ 7 ELU/ML GMT 95% CI 95% CI
Antibody Group Pre-vacc status
Timing N n % LL UL value LL UL
Anti-HPV-18 HPV+HepB S- PRE 200 0 0.0 0.0 1.8 3.5 3.5 3.5 PII(M2) 200 200 100 98.2 100 4790.4 4338.9 5288.8 PIII(M7) 200 199 99.5 97.2 100 8835.1 7636.3 10222.1
HPV S- PRE 202 0 0.0 0.0 1.8 3.5 3.5 3.5 PII(M2) 201 201 100 98.2 100 4663.8 4228.2 5144.3 PIII(M7) 202 202 100 98.2 100 8838.6 7948.5 9828.4
HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine HepB = Engerix-B vaccine; S- = seronegative subjects (antibody concentration < 7 ELU/ML) prior to vaccination; S+ =; seropositive subjects (antibody concentration ≥ 7 ELU/ML) prior to vaccination; GMT = geometric mean antibody concentration calculated on all subjects; N = number of subjects with pre-vaccination results available; n/% = number/percentage of subjects with concentration within the specified range; 95% CI = 95% confidence interval; LL = Lower Limit, UL = Upper Limit; PRE = Prevaccination; PII(M2) = Post Dose 2, Month 2; PIII(M7) = Post Dose 3, Month 7 Total Vaccinated cohort Immunogenicity data obtained from the analysis of the Total Vaccinated cohort groups were consistent with those obtained from the ATP cohort for immunogenicity. Interim Analysis at Month 2 Month 2 immunogenicity data obtained from the analysis of the Interim Vaccinated cohort groups were consistent with those obtained from the ATP cohort for immunogenicity at Month 2. 9 year old subjects in the ATP cohort for immunogenicity Immunogenicity data obtained from the analysis of the 9 year old subjects in the ATP cohort for immunogenicity were consistent with those obtained from the entire ATP cohort for immunogenicity
The primary analysis of safety was performed on the Total Vaccinated cohort. Analyses based on the ATP safety cohort were performed to supplement the Total Vaccinated cohort analysis.
Safety / Reactogenicity:
The percentage of subjects who completed the vaccination course was 100% in the HPV+HepB group, 98.0% in the HPV group and 98.8% in the HepB group. Overall compliance in returning symptom sheets was ≥99.2% in all groups Solicited local symptoms Solicited local symptoms (pain, redness and swelling) reported overall per dose for each vaccine during the 7 - day post-vaccination period are presented in Synopsis Table 9. Pain was the most common solicited local symptom. The majority of solicited local symptoms overall per dose resolved within five days and the majority of solicited grade 3 local symptoms within two days.

HPV-030 (111507) 12
Solicited general symptoms Solicited general symptoms reported overall per dose during the 7-day post-vaccination period are presented in Synopsis Table 10. The most common general symptoms were fatigue, gastrointestinal symptoms, headache and myalgia. The majority of solicited general symptoms overall per dose resolved within four days and the majority of grade 3 general symptoms within three days. No subjects experienced urticaria/rash within 30 minutes after vaccination. Synopsis Table 9 Incidence of solicited local symptoms reported during the 7-day (Days 0-6) post-vaccination period following each dose and overall (Total Vaccinated Cohort)
HPV+HepB HPV HepB 95 % CI 95 % CI 95 % CI
Symptom Product Type N n % LL UL
N n % LL UL N n % LL UL
Pain Total All 737 681 92.4 90.2 94.2 728 635 87.2 84.6 89.6 731 352 48.2 44.5 51.8 Grade 3 737 68 9.2 7.2 11.6 728 42 5.8 4.2 7.7 731 5 0.7 0.2 1.6 HepB All 737 338 45.9 42.2 49.5 731 352 48.2 44.5 51.8 Grade 3 737 5 0.7 0.2 1.6 731 5 0.7 0.2 1.6 HPV All 737 658 89.3 86.8 91.4 728 635 87.2 84.6 89.6 Grade 3 737 66 9.0 7.0 11.3 728 42 5.8 4.2 7.7 Redness Total All 737 231 31.3 28.0 34.8 728 218 29.9 26.6 33.4 731 96 13.1 10.8 15.8 (mm) [50.1 - … 737 13 1.8 0.9 3.0 728 5 0.7 0.2 1.6 731 1 0.1 0.0 0.8 HepB All 737 90 12.2 9.9 14.8 731 96 13.1 10.8 15.8 [50.1 - … 737 2 0.3 0.0 1.0 731 1 0.1 0.0 0.8 HPV All 737 213 28.9 25.7 32.3 728 218 29.9 26.6 33.4 [50.1 - … 737 11 1.5 0.7 2.7 728 5 0.7 0.2 1.6 Swelling Total All 737 222 30.1 26.8 33.6 728 172 23.6 20.6 26.9 731 66 9.0 7.1 11.3 (mm) [50.1 - … 737 17 2.3 1.3 3.7 728 14 1.9 1.1 3.2 731 1 0.1 0.0 0.8 HepB All 737 71 9.6 7.6 12.0 731 66 9.0 7.1 11.3 [50.1 - … 737 3 0.4 0.1 1.2 731 1 0.1 0.0 0.8 HPV All 737 210 28.5 25.3 31.9 728 172 23.6 20.6 26.9 [50.1 - … 737 15 2.0 1.1 3.3 728 14 1.9 1.1 3.2 HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine HPV = HPV-16/18 vaccine HepB = Engerix-B vaccine For each dose and overall/subject: N= number of subjects with at least one documented dose n/%= number/percentage of subjects reporting at least once the symptom For Overall/dose: N= number of documented doses n/%= number/percentage of doses followed by at least one type of symptom Total: n/%= number/percentage of subjects/doses with at least one local symptom whatever the number of injections. 95%CI= Exact 95% confidence interval; LL = lower limit, UL = upper limit
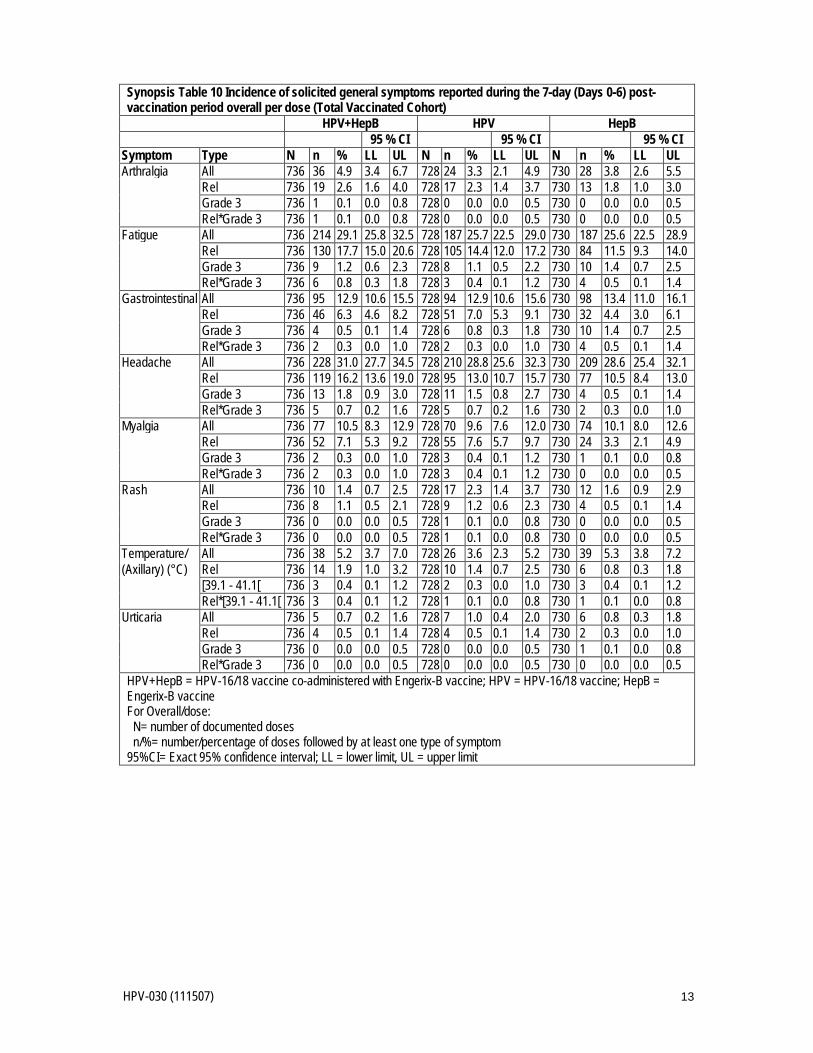
HPV-030 (111507) 13
Synopsis Table 10 Incidence of solicited general symptoms reported during the 7-day (Days 0-6) post-vaccination period overall per dose (Total Vaccinated Cohort)
HPV+HepB HPV HepB 95 % CI 95 % CI 95 % CI
Symptom Type N n % LL UL N n % LL UL N n % LL UL Arthralgia All 736 36 4.9 3.4 6.7 728 24 3.3 2.1 4.9 730 28 3.8 2.6 5.5 Rel 736 19 2.6 1.6 4.0 728 17 2.3 1.4 3.7 730 13 1.8 1.0 3.0 Grade 3 736 1 0.1 0.0 0.8 728 0 0.0 0.0 0.5 730 0 0.0 0.0 0.5 Rel*Grade 3 736 1 0.1 0.0 0.8 728 0 0.0 0.0 0.5 730 0 0.0 0.0 0.5 Fatigue All 736 214 29.1 25.8 32.5 728 187 25.7 22.5 29.0 730 187 25.6 22.5 28.9 Rel 736 130 17.7 15.0 20.6 728 105 14.4 12.0 17.2 730 84 11.5 9.3 14.0 Grade 3 736 9 1.2 0.6 2.3 728 8 1.1 0.5 2.2 730 10 1.4 0.7 2.5 Rel*Grade 3 736 6 0.8 0.3 1.8 728 3 0.4 0.1 1.2 730 4 0.5 0.1 1.4 Gastrointestinal All 736 95 12.9 10.6 15.5 728 94 12.9 10.6 15.6 730 98 13.4 11.0 16.1 Rel 736 46 6.3 4.6 8.2 728 51 7.0 5.3 9.1 730 32 4.4 3.0 6.1 Grade 3 736 4 0.5 0.1 1.4 728 6 0.8 0.3 1.8 730 10 1.4 0.7 2.5 Rel*Grade 3 736 2 0.3 0.0 1.0 728 2 0.3 0.0 1.0 730 4 0.5 0.1 1.4 Headache All 736 228 31.0 27.7 34.5 728 210 28.8 25.6 32.3 730 209 28.6 25.4 32.1 Rel 736 119 16.2 13.6 19.0 728 95 13.0 10.7 15.7 730 77 10.5 8.4 13.0 Grade 3 736 13 1.8 0.9 3.0 728 11 1.5 0.8 2.7 730 4 0.5 0.1 1.4 Rel*Grade 3 736 5 0.7 0.2 1.6 728 5 0.7 0.2 1.6 730 2 0.3 0.0 1.0 Myalgia All 736 77 10.5 8.3 12.9 728 70 9.6 7.6 12.0 730 74 10.1 8.0 12.6 Rel 736 52 7.1 5.3 9.2 728 55 7.6 5.7 9.7 730 24 3.3 2.1 4.9 Grade 3 736 2 0.3 0.0 1.0 728 3 0.4 0.1 1.2 730 1 0.1 0.0 0.8 Rel*Grade 3 736 2 0.3 0.0 1.0 728 3 0.4 0.1 1.2 730 0 0.0 0.0 0.5 Rash All 736 10 1.4 0.7 2.5 728 17 2.3 1.4 3.7 730 12 1.6 0.9 2.9 Rel 736 8 1.1 0.5 2.1 728 9 1.2 0.6 2.3 730 4 0.5 0.1 1.4 Grade 3 736 0 0.0 0.0 0.5 728 1 0.1 0.0 0.8 730 0 0.0 0.0 0.5 Rel*Grade 3 736 0 0.0 0.0 0.5 728 1 0.1 0.0 0.8 730 0 0.0 0.0 0.5 Temperature/ All 736 38 5.2 3.7 7.0 728 26 3.6 2.3 5.2 730 39 5.3 3.8 7.2 (Axillary) (°C) Rel 736 14 1.9 1.0 3.2 728 10 1.4 0.7 2.5 730 6 0.8 0.3 1.8 [39.1 - 41.1[ 736 3 0.4 0.1 1.2 728 2 0.3 0.0 1.0 730 3 0.4 0.1 1.2 Rel*[39.1 - 41.1[ 736 3 0.4 0.1 1.2 728 1 0.1 0.0 0.8 730 1 0.1 0.0 0.8 Urticaria All 736 5 0.7 0.2 1.6 728 7 1.0 0.4 2.0 730 6 0.8 0.3 1.8 Rel 736 4 0.5 0.1 1.4 728 4 0.5 0.1 1.4 730 2 0.3 0.0 1.0 Grade 3 736 0 0.0 0.0 0.5 728 0 0.0 0.0 0.5 730 1 0.1 0.0 0.8 Rel*Grade 3 736 0 0.0 0.0 0.5 728 0 0.0 0.0 0.5 730 0 0.0 0.0 0.5 HPV+HepB = HPV-16/18 vaccine co-administered with Engerix-B vaccine; HPV = HPV-16/18 vaccine; HepB = Engerix-B vaccine For Overall/dose: N= number of documented doses n/%= number/percentage of doses followed by at least one type of symptom 95%CI= Exact 95% confidence interval; LL = lower limit, UL = upper limit

HPV-030 (111507) 14
Unsolicited symptoms The percentage of doses followed by at least one unsolicited symptom classified by MedDRA Primary System Organ Class and Preferred Term within the 30-day post vaccination period was 24.3 % in the HPV+HepB group, 18.4% in the HPV group and 17.5% in the HepB group. The most common unsolicited symptoms were nasopharyngitis, headache and oropharyngeal pain. The proportion of subjects reporting at least one unsolicited grade 3 symptom during within the 30-day post vaccination period following each dose and overall was less than 3.0% in all three groups. Serious Adverse Events: By Month 7, one fatality was reported for a subject in the HPV+HepB group due to traumatic brain injury. The subject did not complete the study. Two subjects reported two non-fatal SAEs: one subject in the HPV+HepB group reported one SAE (ovarian cyst) and one subject in the HPV group reported one SAE (nephrolithiasis). The SAE of ovarian cyst resolved with sequelae. The SAE of nephrolithiasis resolved. None of the SAEs were considered to be related to the study vaccination by the investigator. Withdrawals due to adverse events/serious adverse events: In addition to the subject in the HPV+HepB group who did not complete the study due to a fatal SAE, two subjects in the HepB group withdrew from the study due to a non-serious AE by Month 7. Medically significant adverse events: By Month 7, the percentage of subjects reporting medically significant adverse events classified by MedDRA Primary System Organ Class and Preferred Term was 12.6% in the HPV+HepB group, 11.3% in the HPV group and 8.9% in the HepB group. New Onset of Chronic Diseases By Month 7, four NOCDs classified by MedDRA Preferred Term were identified in four subjects. Two subjects in the HPV+HepB group, one subject in the HPV group and one subject in the HepB group reported urticaria, based on GSK assessment. None of the NOCDs were classified as SAEs and none were assessed as possibly related to vaccination in the opinion of the investigator. None of the NOCDs was identified as a potential NOAD. NOCD assessment by the investigator was not done. Pregnancies One pregnancy for a subject in the HPV+HepB group was reported by Month 7. The subject underwent elective abortion after six weeks of pregnancy. Concomitant medication The use of concomitant medication overall per dose during the during the 30-day post-dose vaccination periods was 19.2% for the HPV+HepB group, 17.2% for the HPV group and 14.1% for the HepB group. Antipyretics were used by 14.7% of the subjects in the HPV+HepB group, 12.8% of the subjects in the HPV group and 9.6% of the subjects in the HepB group during the 30-day post vaccination period, overall per dose. No prophylactic antipyretics or antibiotics were taken by any subjects in any groups. ATP cohort of safety The safety data obtained from the analysis of the ATP cohort of safety were consistent with those obtained from the Total Vaccinated cohort. 9 year old subjects in the Total Vaccinated cohort The safety data obtained from the analysis of the 9 year old subjects in the Total Vaccinated cohort were consistent with those obtained from the entire Total Vaccinated cohort.

HPV-030 (111507) 15
Conclusions: This Phase IIIb, randomized, controlled, open, multicentre study was designed to evaluate the immunogenicity and safety of GlaxoSmithKline Biologicals’ HPV-16/18 L1 VLP adjuvanted vaccine co-administered with GlaxoSmithKline Biologicals’ hepatitis B vaccine (Engerix-B) in healthy female subjects aged 9 - 15 years. The co-primary non-inferiority objectives of the study were met:
• Non-inferiority of the hepatitis B immune response, with respect to anti-HBs seroprotection rate at Month 7, was demonstrated when the HepB vaccine was co-administered with HPV-16/18 vaccine at Month 0, 1 and 6, compared to when HepB vaccine was administered alone at Month 0, 1 and 6.
• Non-inferiority of the HPV-16/18 immune response, with respect to seroconversion rates and GMTs at Month 7, was demonstrated when HPV-16/18 vaccine was co-administered with HepB vaccine at Month 0, 1 and 6, compared to when HPV-16/18 vaccine was administered alone at Month 0, 1 and 6.
Anti-HBs GMTs at Month 7 were 1280.9 mIU/mL when HepB vaccine was co-administered with HPV-16/18 vaccine and 3107.7 mIU/mL when HepB vaccine was administered alone. The 95% CIs did not overlap between the co-administration group and the HepB only group, indicating lower anti-HBs antibody levels in the co-administration group. This finding is probably of little clinical relevance as non-inferiority between both groups was shown with regards to seroprotection levels. For hepatitis B vaccines, the most important is to achieve seroprotection at anti-HBs antibody level ≥ 10 mIU/mL, a level that is widely recognized as indicative of a protective response to hepatitis B vaccines and that is associated with induction of long-term immune memory for anti-HBs. In the present study, seroprotection in the co-administration group was reached for ≥ 97.9% of the subjects at Month 7 and non-inferiority in terms of seroprotection rate was demonstrated as one of the co-primary study objectives. Furthermore, anti-HBs antibody levels ≥ 100 mIU/mL were reached for 88.7% of the subjects at Month 7 in the co-administration group. The immune response to the hepatitis B antigen was consistent with previously published data on hepatitis B vaccine when given to adolescents [Leroux-Roels, 2000]. These data are also in line with previously reported results for co-administration of other HPV and hepatitis B vaccines (Gardasil co-administered with Recombivax HB), showing a decrease in anti-HBs GMTs in the co-administration group compared to the HB vaccine only group, with non-inferiority reached with respect to the seroprotection rate [Wheeler, 2008]. The safety and reactogenicity profile was generally similar for all HPV-16/18 vaccine recipients. One fatal SAE was reported. Two subjects reported two non-fatal SAEs, one resolved and one resolved with sequelae. None of the SAEs were considered to be related to the study vaccination by the investigator. Two subjects were withdrawn from the study due to a non-serious AE. One pregnancy which was terminated by elective abortion was reported during the study. The overall incidence of solicited local and solicited general adverse events during the 7-day follow-up period was similar for the HPV+HepB group and the HPV group. Incidence of solicited local events was higher in HPV-16/18 vaccine recipients than in HepB vaccine only recipients. Incidence of solicited general events was similar in all groups. It can be concluded that data from this study support co-administration of GSK Biologicals’ HPV-16/18 L1 adjuvanted vaccine with GSK Biologicals’ Hepatitis B vaccine (Engerix-B) in 9 - 15 years old girls. GSK Biologicals’ HPV-16/18 L1 adjuvanted vaccine was found to be immunogenic and generally well tolerated when administered to 9-15 year old subjects, alone or co-administered with Engerix-B. Date of report: 17-JUN-2010

The study summarized below may involve approved and non-approved uses, formulations or treatment regimens. The results reported for any single study may not reflect the overall results obtained during all studies involving the same product. Before prescribing any product mentioned in this Register, healthcare professionals should consult the prescribing information for the product approved in their country.
HPV-030 (111507) 1
Name of company: GlaxoSmithKline Biologicals, Rixensart, Belgium Name of finished product: GlaxoSmithKline Biologicals’ human papillomavirus (HPV) vaccine Name of active substance: HPV-16 L1 virus-like particles HPV-18 L1 virus-like particles
TABULAR FORMAT REFERRING TO PART OF THE DOSSIER
Volume:
Page:
(for national authority only)
Study No.: HPV-030 (111507) Title of the study : A phase IIIb, randomized, open, multicentre study to evaluate the immunogenicity and safety of GlaxoSmithKline Biologicals’ HPV-16/18 L1 VLP adjuvanted vaccine (Cervarix™) co-administrated with GlaxoSmithKline Biologicals’ Hepatitis B vaccine (Engerix-B™) in healthy female subjects aged 9 - 15 years Coordinating investigators: This study was conducted by three coordinating investigators in two European countries (The Netherlands, Sweden). Study centre(s): This study was conducted in seven study centers: two centers in the Netherlands and five centers in Sweden. Publication (reference): Not published as of July 2010. Study period: Study initiation date (First Subject, First Visit): 09 April 2008 Study completion date for the active phase : 28 Aug 2009 Completion date Extended Safety Follow-Up (ESFU) Last Subject, Last Safety Contact, Month 12): 08 January 2010 Database Freeze: 19 April 2010
Clinical phase: IIIb
ESFU objectives: • To assess the safety of the study vaccine with respect to the nature, intensity and relationship to vaccination
of serious adverse events (SAEs) in all study groups throughout the study period. • To evaluate the occurrence of medically significant conditions throughout the study period (up to Month 12)
regardless of causal relationship to vaccination and intensity in all study groups.
The clinical study report dated June 2010 presented analyses of safety and immunogenicity data collected up to Month 7. Safety data collected up to the Month 12 follow-up telephone contact (from Month 7 to Month 12 and from Month 0 to Month 12) are presented in this annex report. Study design: • A Phase IIIb, randomized, controlled, parallel-group, multi-country, multi-centre study with three treatment
groups. • Treatment allocation: Internet-based randomization (1:1:1). The randomization was age-stratified, with
approximately the same number of subjects in the three age strata (9 years, 10 - 12 years and 13 - 15 years). • Vaccination schedules:
• Cervarix (HPV-16/18 L1 VLP with adjuvant): Three doses of vaccine administered intramuscularly, with the second and third dose given one month and six months after the first dose, respectively.
• Engerix-B (HepB): Three doses of vaccine administered intramuscularly, with the second and third dose given one month and six months after the first dose respectively.

HPV-030 (111507) 2
Synopsis Table 1. Treatment groups Vaccine administered Groups Month 0 Month 1 Month 6 HPV (N = 252) HPV-16/18 HPV-16/18 HPV-16/18 HPV + HepB (N = 252) HPV-16/18 + HepB HPV-16/18 + HepB HPV-16/18 + HepB HepB (N = 252) HepB HepB HepB N = number of subjects planned to be enrolled. • Blinding: Open. • Control: Active. • Type of study: Self-contained. • Data collection: Remote Data entry (RDE). • Duration of the study: Approximately 12 months per subject i.e., a seven-month active phase followed by a
five-month follow-up phase for safety. • Visits: Five visits scheduled per subject at Months 0, 1, 2, 6 and 7 (with telephone contact at Month 12). • Blood samples to evaluate immunogenicity: Three blood samples taken at Month 0, Month 2 and Month 7. • Safety and reactogenicity monitoring:
(1) Solicited signs and symptoms were to be self-reported by all subjects, using a diary card, on the day of vaccination (Day 0) and on the 6 subsequent days (Days 1 - 6). (2) Unsolicited signs and symptoms were to be self-reported in all subjects within 30 days (Days 0 - 29) after each vaccination. (3) SAEs were to be reported in all subjects throughout the study period (up to Month 12). (4) Medically significant conditions were to be reported in all subjects throughout the entire study (up to Month 12) regardless of causal relationship to vaccination and intensity. (5) Pregnancies and pregnancy outcomes were to be recorded throughout the entire study period (up to Month 12) and subjects will be followed until delivery (even if delivery occurs after the end of the study).
• An interim analysis was conducted on immunogenicity data collected at one month after the second dose of vaccine (Month 2), in the subset of the first 150 subjects (approximately) with Month 2 data available (about 50 subjects per group).
• Final analysis was conducted on all immunogenicity and safety data collected up to the end of the active phase (Month 7).
The results of the safety follow-up (up to Month 12) are provided in this annex report. Number of subjects: Approximately 756 subjects (252 subjects per group) aged 9 - 15 years were to be enrolled in study centres in Europe to reach a target of 603 evaluable subjects for the analysis at Month 7 (201 evaluable subjects in each group), refer to Synopsis Table 2. In each study group, there were three age strata (9 years, 10 - 12 years and 13 - 15 years) and approximately 84 subjects were to be enrolled per stratum.
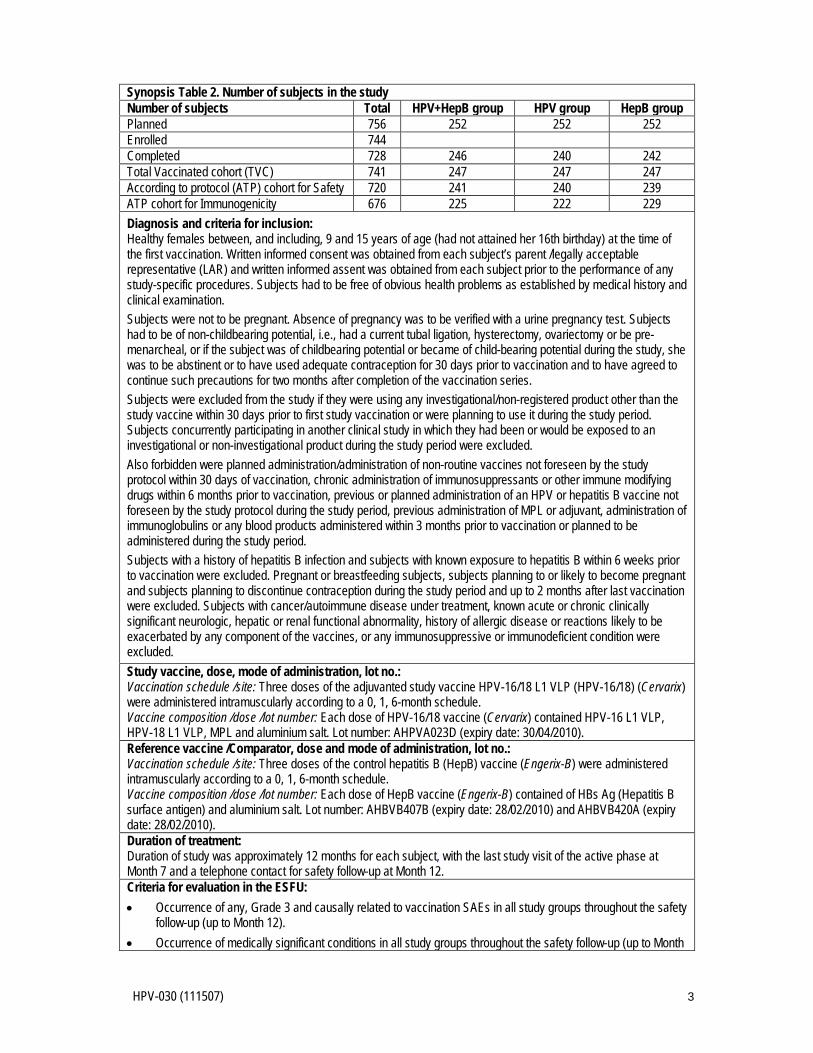
HPV-030 (111507) 3
Synopsis Table 2. Number of subjects in the study Number of subjects Total HPV+HepB group HPV group HepB group Planned 756 252 252 252 Enrolled 744 Completed 728 246 240 242 Total Vaccinated cohort (TVC) 741 247 247 247 According to protocol (ATP) cohort for Safety 720 241 240 239 ATP cohort for Immunogenicity 676 225 222 229 Diagnosis and criteria for inclusion: Healthy females between, and including, 9 and 15 years of age (had not attained her 16th birthday) at the time of the first vaccination. Written informed consent was obtained from each subject’s parent /legally acceptable representative (LAR) and written informed assent was obtained from each subject prior to the performance of any study-specific procedures. Subjects had to be free of obvious health problems as established by medical history and clinical examination. Subjects were not to be pregnant. Absence of pregnancy was to be verified with a urine pregnancy test. Subjects had to be of non-childbearing potential, i.e., had a current tubal ligation, hysterectomy, ovariectomy or be pre-menarcheal, or if the subject was of childbearing potential or became of child-bearing potential during the study, she was to be abstinent or to have used adequate contraception for 30 days prior to vaccination and to have agreed to continue such precautions for two months after completion of the vaccination series. Subjects were excluded from the study if they were using any investigational/non-registered product other than the study vaccine within 30 days prior to first study vaccination or were planning to use it during the study period. Subjects concurrently participating in another clinical study in which they had been or would be exposed to an investigational or non-investigational product during the study period were excluded. Also forbidden were planned administration/administration of non-routine vaccines not foreseen by the study protocol within 30 days of vaccination, chronic administration of immunosuppressants or other immune modifying drugs within 6 months prior to vaccination, previous or planned administration of an HPV or hepatitis B vaccine not foreseen by the study protocol during the study period, previous administration of MPL or adjuvant, administration of immunoglobulins or any blood products administered within 3 months prior to vaccination or planned to be administered during the study period. Subjects with a history of hepatitis B infection and subjects with known exposure to hepatitis B within 6 weeks prior to vaccination were excluded. Pregnant or breastfeeding subjects, subjects planning to or likely to become pregnant and subjects planning to discontinue contraception during the study period and up to 2 months after last vaccination were excluded. Subjects with cancer/autoimmune disease under treatment, known acute or chronic clinically significant neurologic, hepatic or renal functional abnormality, history of allergic disease or reactions likely to be exacerbated by any component of the vaccines, or any immunosuppressive or immunodeficient condition were excluded. Study vaccine, dose, mode of administration, lot no.: Vaccination schedule /site: Three doses of the adjuvanted study vaccine HPV-16/18 L1 VLP (HPV-16/18) (Cervarix) were administered intramuscularly according to a 0, 1, 6-month schedule. Vaccine composition /dose /lot number: Each dose of HPV-16/18 vaccine (Cervarix) contained HPV-16 L1 VLP, HPV-18 L1 VLP, MPL and aluminium salt. Lot number: AHPVA023D (expiry date: 30/04/2010). Reference vaccine /Comparator, dose and mode of administration, lot no.: Vaccination schedule /site: Three doses of the control hepatitis B (HepB) vaccine (Engerix-B) were administered intramuscularly according to a 0, 1, 6-month schedule. Vaccine composition /dose /lot number: Each dose of HepB vaccine (Engerix-B) contained of HBs Ag (Hepatitis B surface antigen) and aluminium salt. Lot number: AHBVB407B (expiry date: 28/02/2010) and AHBVB420A (expiry date: 28/02/2010). Duration of treatment: Duration of study was approximately 12 months for each subject, with the last study visit of the active phase at Month 7 and a telephone contact for safety follow-up at Month 12. Criteria for evaluation in the ESFU: • Occurrence of any, Grade 3 and causally related to vaccination SAEs in all study groups throughout the safety
follow-up (up to Month 12). • Occurrence of medically significant conditions in all study groups throughout the safety follow-up (up to Month

HPV-030 (111507) 4
12) regardless of causal relationship to vaccination and intensity. Statistical methods: Study cohorts The Total Vaccinated cohort included all vaccinated subjects for whom data were available. The total analysis of safety included all subjects with at least one vaccine administration documented for unsolicited AEs and concomitant medication and included subjects with documented doses for solicited symptoms. The Total Vaccinated cohort analysis was performed per treatment actually administered. The ESFU Vaccinated cohort included all subjects with at least one vaccine administration documented that could be contacted at the safety follow-up telephone contact at Month 12. The ESFU cohort analysis was performed per treatment actually administered The subset of 9 year old subjects in the Total Vaccinated cohort included all 9 year old vaccinated subjects for whom data were available. The total analysis of safety included all 9 year old subjects with at least one vaccine administration documented for unsolicited AEs and concomitant medication and included 9 year old subjects with documented doses for solicited symptoms. The subset of 9 year old subjects in the ESFU Vaccinated cohort included all 9 year old subjects with at least one vaccine administration documented that could be contacted at the safety follow-up telephone contact at Month 12. The ESFU cohort analysis was performed per treatment actually administered Analysis of demographics Demographic characteristics (age, gender, race) of the study cohorts during the active phase were tabulated in the study report dated June 2010. The mean age (plus range and standard deviation) of the vaccinated subjects, as a whole and per group, was calculated. The distribution of subjects enrolled among the study sites was tabulated as a whole and per group.
Analysis of safety The primary analysis of safety was performed on the ESFU Vaccinated cohort for data reported during the ESFU period (Month 7 to Month 12). In addition, safety data reported during the entire study period (Month 0 to Month 12) were analysed on the Total Vaccinated cohort and the ESFU Vaccinated cohort. The safety analyses were performed per vaccine group, overall in subjects aged 9 - 15 years and in subjects aged 9 years. • The proportion of subjects with at least one report of a medically significant AE classified by MedDRA,
whenever available, and reported up to 30 days after vaccination was tabulated with exact 95% CI. A similar table was produced for medically significant AEs starting Day 30 after each dose till the end of the study.
• The proportion of subjects with at least one report of New Onset of Chronic Disease classified by MedDRA, whenever available, and reported during the entire study period was tabulated with exact 95% CI.
• The proportion of subjects with at least one report of New Onset of Autoimmune Disease classified by MedDRA, whenever available, and reported during the entire study period was tabulated with exact 95% CI.
• Pregnancies and pregnancy outcomes, SAEs and withdrawal due to AE(s) were described in detail. Summary: Overall, 744 subjects were enrolled in the study and 741 subjects were vaccinated: 247 subjects in each of the HPV+HepB group, the HPV group and the HepB group. Of these subjects, 726 subjects completed the ESFU: 245 subjects in the HPV+HepB group, 240 subjects in the HPV group and 241 subjects in the HepB group. Fifteen subjects could not be contacted during the ESFU period and were therefore not included in the ESFU analyses. In the subset of 9 year old subjects, 239 subjects were enrolled in the study. All of them were vaccinated: 79 subjects in the HPV+HepB group, 80 subjects in the HPV group and 80 subjects in the HepB group. Of these subjects, 234 subjects completed the ESFU: 79 subjects in the HPV+HepB group, 77 subjects in the HPV group and 78 subjects in the HepB group. Five subjects could not be contacted during the ESFU period and were therefore not included in the ESFU analyses.
Month 7 to Month 12 safety analysis (ESFU Vaccinated cohort) Safety
Serious adverse events: During the ESFU period (Month 7 to Month 12), three subjects reported three SAEs: one subject in the HPV+HepB group reported one SAE (appendicitis), one subject in the HPV group reported one SAE (forearm fracture) and one subject in the HepB group reported one SAE (ankle fracture). All SAEs were resolved. None of the SAEs were considered by the investigator to be related to study vaccination. In the subset of 9 year old subjects, no SAEs were reported.

HPV-030 (111507) 5
Withdrawals due to adverse events/serious adverse events: No subjects were withdrawn due to AEs or SAEs. Medically significant adverse events: The percentage of subjects reporting medically significant adverse events (AEs) (at least one symptom) classified by MedDRA Primary System Organ Class and Preferred Term was 0% in the HPV+HepB group, 0.8% in the HPV group and 0.8% in the HepB group. In the subset of 9 year old subjects, the percentage of subjects reporting medically significant adverse events (at least one symptom) classified by MedDRA Primary System Organ Class and Preferred Term was 0% in the HPV+HepB group, 1.3% in the HPV group and 0% in the HepB group. New onset of chronic diseases: One NOCD classified by MedDRA Primary System Organ Class and Preferred Term was identified, based on GSK assessment in one subject in the HPV group who reported urticaria). The NOCD was not classified as an SAE and was not assessed as possibly related to vaccination in the opinion of the investigator. The NOCD was not identified as a potential NOAD. NOCD assessment by the investigator was not done. Pregnancies: No pregnancies were reported during the ESFU period. Month 0 to Month 12 safety analysis (Total Vaccinated cohort) Serious Adverse Events: During the entire study period, one fatality was reported for a subject in the HPV+HepB group due to traumatic brain injury due to train accident in the Total Vaccinated cohort. This fatal SAE was not considered to be related to the study vaccination by the investigator. Five subjects reported five non-fatal SAEs: two subjects in the HPV+HepB group reported two SAEs (ovarian cyst and appendicitis), two subjects in the HPV group reported two SAEs (nephrolithiasis and forearm fracture) and one subject in the HepB group reported one SAE (ankle fracture). The SAE of ovarian cyst resolved with sequelae. All other non-fatal SAEs resolved. None of the non-fatal SAEs were considered to be related to the study vaccination by the investigator. In the subset of 9 year old subjects, no SAEs were reported. Withdrawals due to adverse events/serious adverse events: In addition to the subject in the HPV+HepB group who did not complete the study due to a fatal SAE, two subjects in the HepB group withdrew from the study due to a non-serious AE during the active phase. No subjects were withdrawn from the study due to SAEs or AEs during the ESFU period. Medically significant adverse events: During the entire study period, the percentage of subjects reporting medically significant AEs classified by MedDRA Primary System Organ Class and Preferred Term was 12.6% in the HPV+HepB group, 12.1% in the HPV group and 9.3% in the HepB group in the Total Vaccinated cohort. In the subset of 9 year old subjects, the percentage of subjects reporting medically significant AEs (at least one symptom) classified by MedDRA Primary System Organ Class and Preferred Term was 10.1% in the HPV+HepB group, 10.0% in the HPV group and 13.8% in the HepB group. New Onset of Chronic Diseases During the entire study period, five NOCDs classified by MedDRA term were identified in five subjects. Two subjects in the HPV+HepB group, two subjects in the HPV group and one subject in the HepB group all reported urticaria, based on GSK assessment in the Total Vaccinated cohort. None of these NOCDs were classified as SAEs and none were assessed as possibly related to vaccination in the opinion of the investigator. None of the NOCDs were identified as potential NOADs. NOCD assessment by the investigator was not done.
In the subset of 9 year old subjects, two subjects in the HPV group reported two NOCDs of urticaria. Pregnancies One pregnancy for a subject in the HPV+HepB group was reported during the entire study period. The subject underwent elective abortion after six weeks of pregnancy.

HPV-030 (111507) 6
Conclusions: This Phase IIIb, randomized, controlled, open, multi-centre study was designed to evaluate the immunogenicity and safety of GlaxoSmithKline Biologicals’ HPV-16/18 L1 VLP adjuvanted vaccine (Cervarix) co-administered with GlaxoSmithKline Biologicals’ hepatitis B vaccine (Engerix-B) in healthy female subjects aged 9 - 15 years. The final analysis of the HPV-030 study at Month 7 demonstrated non-inferiority of the hepatitis B immune response, with respect to anti-HBs seroprotection rate, as well as non-inferiority of the HPV-16/18 immune response, with respect to seroconversion rates and GMTs, when HPV-16/18 and HepB vaccine were co-administered, compared to when either vaccine was administered alone. The safety and reactogenicity profile was generally similar for all HPV-16/18 vaccine recipients. Overall, it was concluded that data from this study support co-administration of GSK Biologicals’ HPV-16/18 L1 adjuvanted vaccine (Cervarix) with GSK Biologicals’ Hepatitis B vaccine (Engerix-B) in 9 - 15 years old girls. GSK Biologicals’ HPV-16/18 L1 adjuvanted vaccine (Cervarix) was found to be immunogenic and generally well tolerated when administered to 9-15 year old subjects, alone or co-administered with Engerix-B. The objective of the extended safety follow-up (ESFU) was to assess the safety of the HPV-16/18 L1 adjuvanted vaccine up to six months after the last vaccine dose (Month 12) with respect to medically significant AEs, NOCDs, SAEs and pregnancies. During the entire study period, one fatal SAE was reported. This fatal SAE was not considered to be related to the study vaccination by the investigator. Five subjects reported five non-fatal SAEs, four resolved and one resolved with sequelae. None of the SAEs were considered to be related to the study vaccination by the investigator. Two subjects were withdrawn from the study due to a non-serious AE. One pregnancy which was terminated by elective abortion was reported during the study. In conclusion, in 9 - 15 year old girls, the HPV-16/18 L1 adjuvanted vaccine (Cervarix) and GSK Biologicals’ Hepatitis B vaccine (Engerix-B) appeared to be generally well tolerated up to six months after the last vaccine dose when given alone or when co-administered. Date of annex report 1: 14-JUL-2010

Appendix 1 to Synopsis for study HPV-030 PRI (111507) Overview of Protocol Amendments No protocol amendments were submitted for this study.

Appendix 2 to Synopsis for study HPV-030 PRI (111507) List of study sites
Country Name
Centre Status
Clinical Site Institution Name
Clinical Site Department Name
Clinical Site Street Clinical Site ZIP Code
Clinical Site City
Netherlands Concluded
-Closed UMC St. Radboud Not Defined Geert Grooteplein-
Zuid 10 6525 GA NIJMEGEN
Netherlands Concluded-Closed
GGD Not Defined Schiedamsedijk 95 3011 EN ROTTERDAM
Sweden Concluded-Closed
Ladulaas Kliniska Studier
Kungsfors Center Industrigatan 2 SE-511 62
SKENE
Sweden Concluded-Closed
Center för läkemedelsstudier
Not Defined Föreningsgatan 26, vån 3
SE-211 52
MALMÖ
Sweden Concluded-Closed
Vrinnevisjukhuset Barn- och ungdomsmedicinska kliniken
Vrinnevisjukhuset SE-601 82
NORRKÖPING
Sweden Concluded-Closed
Universitetssjuk-huset Linköping
Allergicentrum Universitetssjuk-huset Linköping
SE-581 85
LINKÖPING
Sweden Concluded-Closed
Kvartersakuten - Bothnia Research Center
Not Defined Timmermansgatan 26
SE-972 31
LULEÅ


















