
Surface functionalization of cellulose by grafting oligoether chains
8
Materials Chemistry and Physics 120 (2010) 438–445 Contents lists available at ScienceDirect Materials Chemistry and Physics journal homepage: www.elsevier.com/locate/matchemphys Surface functionalization of cellulose by grafting oligoether chains El hadji Babacar Ly a , Julien Bras a , Patrizia Sadocco b , Mohamed Naceur Belgacem a,∗ , Alain Dufresne a , Wim Thielemans a,b,1 a Laboratoire de Génie des Procédés Papetiers (UMR 5518 CNRS-CTP-INPG), Institut National Polytechnique de Grenoble, 461 Rue de la papeterie, 38402 St Martin d’Hères, France b Sperimentale Carta, Cartoni e Paste per Carta, Piazza Leonardo da Vinci 16, 20133 Milano, Italy article info Article history: Received 22 August 2009 Received in revised form 7 November 2009 Accepted 22 November 2009 Keywords: Cellulose fibres Surface chemical modification Oligoethers Contact angle X-ray photoelectron spectroscopy Biodegradability abstract Two cellulosic substrates (Whatman paper and wood fibres) were chemically modified using different oligoether chains; poly(ethylene) (POE), poly(propylene) (PPG) and poly(tetrahydrofuran) (PTHF) glycols with different lengths were first converted into mono-NCO-terminating macromolecules to allow direct grafting to the cellulose substrates. This step was achieved by reacting the chosen oligoether with 2,4- toluene diisocyanate. The prepared macromolecular grafts were then coupled with the cellulose surface and the resulting treated substrates were fully characterized by contact angle measurements, elemental analysis, scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). Thus, all the techniques implemented showed clear evidence of successful grafting, namely: (i) when using PPG grafts, the polar contribution to the surface energy decreased from approximately 25 to virtually 0 mJ m −2 and the wettability by water decreased, as the water contact angle shifted from around 40 to above 90 ◦ ; (ii) nitrogen atoms were detected by elemental analysis and XPS; (iii) the aliphatic carbon contents increased from 11 to about 39–50%, depending on the oligoether used; and (iv) small spheres having about 100 nm diameter were detected by SEM. Moreover, the grafted fibres were submitted to biodegradation tests which showed that they conserved their biodegradable character, although with a slower biodegradation rate. The novelty of the present paper is the direct grafting of the polymeric matrix onto the fibre surface thanks to a new modification strategy involving the use of a diisocyanate as a mediator between the matrix and the reinforcing elements. The covalently linked polymeric chains constituting the matrix could melt under heating, thus, yielding the interdiffusion of the macromolecular grafts and forming the composite. © 2009 Elsevier B.V. All rights reserved. 1. Introduction The exploitation of natural fibres, and particularly lignocellu- losic fibres, as an alternative to inorganic materials to reinforce polymeric matrices has been reviewed in several recent investi- gations [1–5]. The main driving forces of such tendency is that cellulose fibres provide several potential advantages, namely: (i) their low density, which gives the possibility of preparing lighter composites; (ii) their bio-renewable character; and (iii) their ubiq- uitous availability at low cost and in a variety of forms. Moreover, cellulose-based composites present the additional advantage that they can be valorised as a fuel at the end of their life cycle much ∗ Corresponding author. Tel.: +33 4 76 82 69 62; fax: +33 4 76 82 69 33. E-mail addresses: [email protected], [email protected] (M.N. Belgacem). 1 Current address: Driving Innovation in Chemistry and Chemical Engineering (DICE), School of Chemistry and Process and Environmental Research Division- Faculty of Engineering, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. more easily than those made from glass-fibres and thermo-set resins which leave molten glass residue in the recovery boilers. Cellulose fibres possess nevertheless a major drawback. They are highly hydrophilic, and water take up, even from atmo- spheric moisture, reduces drastically their dimensional stability and strength properties. Indeed, cellulose-containing composite materials are highly sensitive to moisture uptake and release depending on the surrounding atmospheric conditions, which leads to mechanical failure. Moreover, the mechanical properties depend strongly on the quality of the fibre/matrix interface. Very often, the matrices used are hydrophobic (polyolefins) and the interfacial properties between such polymeric matrices and cellulose fibres are very poor. For these two reasons, cellulose fibres are, generally, subjected to surface modifications in order to achieve maximum compatibility, or even better fibre–matrix covalent bridges, and therefore good adhesion. The treatment strategies and reactions applied to the surface modification of cellulose have been recently reviewed and included etherification, esterification, urethane and siloxanes formation, and treatments using lignin [1,4–7]. More recently, new ways of cellulose grafting via click chemistry were also reported [8–10]. 0254-0584/$ – see front matter © 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.matchemphys.2009.11.032
-
Upload
el-hadji-babacar-ly -
Category
Documents
-
view
236 -
download
8
Transcript of Surface functionalization of cellulose by grafting oligoether chains

S
EAa
b
a
ARRA
KCSOCXB
1
lpgctcuct
N
(F2
0d
Materials Chemistry and Physics 120 (2010) 438–445
Contents lists available at ScienceDirect
Materials Chemistry and Physics
journa l homepage: www.e lsev ier .com/ locate /matchemphys
urface functionalization of cellulose by grafting oligoether chains
l hadji Babacar Lya, Julien Brasa, Patrizia Sadoccob, Mohamed Naceur Belgacema,∗,lain Dufresnea, Wim Thielemansa,b,1
Laboratoire de Génie des Procédés Papetiers (UMR 5518 CNRS-CTP-INPG), Institut National Polytechnique de Grenoble, 461 Rue de la papeterie, 38402 St Martin d’Hères, FranceSperimentale Carta, Cartoni e Paste per Carta, Piazza Leonardo da Vinci 16, 20133 Milano, Italy
r t i c l e i n f o
rticle history:eceived 22 August 2009eceived in revised form 7 November 2009ccepted 22 November 2009
eywords:ellulose fibresurface chemical modificationligoethersontact angle-ray photoelectron spectroscopyiodegradability
a b s t r a c t
Two cellulosic substrates (Whatman paper and wood fibres) were chemically modified using differentoligoether chains; poly(ethylene) (POE), poly(propylene) (PPG) and poly(tetrahydrofuran) (PTHF) glycolswith different lengths were first converted into mono-NCO-terminating macromolecules to allow directgrafting to the cellulose substrates. This step was achieved by reacting the chosen oligoether with 2,4-toluene diisocyanate. The prepared macromolecular grafts were then coupled with the cellulose surfaceand the resulting treated substrates were fully characterized by contact angle measurements, elementalanalysis, scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). Thus, all thetechniques implemented showed clear evidence of successful grafting, namely: (i) when using PPG grafts,the polar contribution to the surface energy decreased from approximately 25 to virtually 0 mJ m−2 andthe wettability by water decreased, as the water contact angle shifted from around 40 to above 90◦; (ii)nitrogen atoms were detected by elemental analysis and XPS; (iii) the aliphatic carbon contents increasedfrom 11 to about 39–50%, depending on the oligoether used; and (iv) small spheres having about 100 nm
diameter were detected by SEM. Moreover, the grafted fibres were submitted to biodegradation testswhich showed that they conserved their biodegradable character, although with a slower biodegradationrate. The novelty of the present paper is the direct grafting of the polymeric matrix onto the fibre surfacethanks to a new modification strategy involving the use of a diisocyanate as a mediator between thematrix and the reinforcing elements. The covalently linked polymeric chains constituting the matrixcould melt under heating, thus, yielding the interdiffusion of the macromolecular grafts and forming the composite.. Introduction
The exploitation of natural fibres, and particularly lignocellu-osic fibres, as an alternative to inorganic materials to reinforceolymeric matrices has been reviewed in several recent investi-ations [1–5]. The main driving forces of such tendency is thatellulose fibres provide several potential advantages, namely: (i)heir low density, which gives the possibility of preparing lighter
omposites; (ii) their bio-renewable character; and (iii) their ubiq-itous availability at low cost and in a variety of forms. Moreover,ellulose-based composites present the additional advantage thathey can be valorised as a fuel at the end of their life cycle much∗ Corresponding author. Tel.: +33 4 76 82 69 62; fax: +33 4 76 82 69 33.E-mail addresses: [email protected],
[email protected] (M.N. Belgacem).1 Current address: Driving Innovation in Chemistry and Chemical Engineering
DICE), School of Chemistry and Process and Environmental Research Division-aculty of Engineering, University of Nottingham, University Park, Nottingham, NG7RD, UK.
254-0584/$ – see front matter © 2009 Elsevier B.V. All rights reserved.oi:10.1016/j.matchemphys.2009.11.032
© 2009 Elsevier B.V. All rights reserved.
more easily than those made from glass-fibres and thermo-setresins which leave molten glass residue in the recovery boilers.
Cellulose fibres possess nevertheless a major drawback. Theyare highly hydrophilic, and water take up, even from atmo-spheric moisture, reduces drastically their dimensional stabilityand strength properties. Indeed, cellulose-containing compositematerials are highly sensitive to moisture uptake and releasedepending on the surrounding atmospheric conditions, which leadsto mechanical failure. Moreover, the mechanical properties dependstrongly on the quality of the fibre/matrix interface. Very often,the matrices used are hydrophobic (polyolefins) and the interfacialproperties between such polymeric matrices and cellulose fibresare very poor. For these two reasons, cellulose fibres are, generally,subjected to surface modifications in order to achieve maximumcompatibility, or even better fibre–matrix covalent bridges, andtherefore good adhesion. The treatment strategies and reactions
applied to the surface modification of cellulose have been recentlyreviewed and included etherification, esterification, urethane andsiloxanes formation, and treatments using lignin [1,4–7]. Morerecently, new ways of cellulose grafting via click chemistry werealso reported [8–10].
stry an
triiurusrdt
(
dttecd
TT
E.h.B. Ly et al. / Materials Chemi
Very recent studies (limited to those published in 2009) relatedo the development of the present topic [11–20] have also appearedeporting a variety of strategies associated with the surface mod-fication of cellulose. They are mostly based on polymer graftingn order to modify the surface properties of cellulose fibers, callingpon the use of the following approaches: “grafting from” free-adical polymerization, “grafting onto” thanks to esterification orrethane formation reactions and layer-by-layer adsorption. In fact,everal very recent papers were reported dealing with the prepa-ation and the characterization of a variety of cellulose or celluloseerivatives-based substrates. The main breakthroughs achieved inhese papers could be drawn as follows:
(i) Preparation of biodegradable starch grafted cellulose fibers[11].
(ii) Grafting of cellulose fibres and/or cellulose acetate films byoligomeric polycaprolactone [12,13].
(iii) Surface-initiated atom transfer radical polymerization (i.e.,“grafting from”) of polystyrene or poly(ethyl acrylate) chainsonto cellulose nanocrystals or fibres [14,15].
(iv) Controlled surface modification of cellulose fibers by amino-derivatives using N,N′-carbonyldiimidazole, as activator [16].
(v) Cellulose fibers self-reinforced composites preparation bytheir partial oxypropylation [17,18].
(vi) The use of ionic liquid homogeneous esterification ofcellulose with succinic anhydride in the presence of n-bromosuccinimide, as a catalyst [19].
vii) The preparation of l-lysine aminocarboxylic modified cellu-lose [20].
Among these treatment strategies, very few propose to graftirectly the polymeric matrix onto the fibre surface. The innova-
iveness of the present study was then to graft the matrix directlyo the surface of cellulose fibres thanks to a new reaction strat-gy involving the use of a diisocyanate as a linking agent. Theovalently linked matrix could melt and the composite would beirectly achieved through interdiffusion of the polymer grafts. Thisable 1he oligoethers used in this work and their characteristics.
Name Abbreviation Chemical formula
Polyethylene glycol PEG
Polyethylene glycol monomethylether PEGME
Polypropylene glycol PPG
Polypropylene glycol monobutylether PPGMBE
Polytetrahydrofuran PTHF
Phenyl isocyanate PI
Tolylene 2,4-diisocyanate TDI
d Physics 120 (2010) 438–445 439
study describes the new way of cellulose fibre grafting for differ-ent oligoethers and the surface properties obtained like surfaceenergy and biodegradability. The grafted fibres could represent anew promising route to obtain a composite material where thethermoplastic matrix is directly linked on the cellulose fibre sur-face.
2. Materials and methods
2.1. Materials
Whatman paper (WP) (purchased from Whatman Co.) and bleached softwoodkraft pulps (BSK) (an industrial product supplied by the pulp and papermakingindustry) were used as received. WP was used as reference material, because ofits high purity and availability in the form of paper sheets, a suitable substrate forcontact angle measurements. Moreover, WP can be used as such in composite for-mulations processed by impregnation of fibermats. Therefore, in such a context, theinvestigation of such a substrate could be relevant.
The oligoethers as well as phenyl isocyanate (PI) and 2,4-toluene diisocyanate(TDI) used in this work were purchased from Sigma–Aldrich Inc. and used asreceived. Their chemical structures as well as the molecular weights investigatedare shown in Table 1. Dibutyltin dilaureate, DBTL (95%) was also obtained fromSigma–Aldrich and used to catalyze the urethane formation reaction. Anhydroustoluene, tetrahydrofuran and methylene chloride were of the highest purity avail-able and purchased from Sigma–Aldrich. They were used as received.
2.2. Grafted fibres synthesis
The grafting reactions were carried out in three steps, as sketched in Scheme 1,forming the following products: oligoether + PI: adduct I, adduct I + TDI: adduct II andadduct II + cellulose. These reactions were performed in anhydrous toluene, in orderto avoid the hydrolysis of the NCO functions and prevent swelling of the cellulosefibres to limit the reaction to the fibre surface. The first step consisted of introducing30 ml of the solvent (toluene or tetrahydrofuran) into a three-necked round-bottomflask under nitrogen atmosphere. Then, 1 equiv. of PI is added (see Table 2) and thereaction medium heated to 90 ◦C, where it was kept for 4 h. The second step involvedthe addition of the right amount of TDI (Table 2) and the heating of the reactionmixture at 90 ◦C for 24 h, in the presence of 0.1 ml DBTL. The third stage of grafting
is started by the introduction of 0.5 g of cellulose fibres into a three-necked flaskfollowed by the addition of 10 equiv. of the previously prepared grafting agent. Theresulting suspension was maintained at 90 ◦C and the reaction was left for 72 h.At the end of the grafting reactions, the modified fibres were recovered byfiltration over an extraction thimble and soxhlet-extracted for 6 h using methy-lene chloride, in order to remove the physically adsorbed unbound materials. The
Molecular weight (g mol−1) Density Source CAS no.
2000 1 Sigma 25322-68-3
2000 1 Sigma 9004-74-4
2000, 2500, 3000, 3300, 4000 1 Sigma 25322-69-4
2500 1 Sigma 9003-13-8
2900 0.97 Sigma 25190-06-1
119 1.096 Sigma 103-71-9
174 1.21 Sigma 584-84-9

440 E.h.B. Ly et al. / Materials Chemistry and Physics 120 (2010) 438–445
es
2
1wtP
ocasTtttfupcra
Vm
aKsc
TT
Table 3Molecule probes for contact angle measurements and their characteristics.
Liquid �DL �P
L �TL
Water 21.8 51 72.8Ethylene glycol 29.0 19 48Formamide 37.6 20.4 58
Scheme 1.
xtracted fibres were then dried for 48 h at 105 ◦C and stored under a dry atmo-phere.
.3. Characterization
FTIR spectra were obtained by preparing pellets of dried KBr powder containing% (w/w) of the investigated solid samples. Liquid polymers and others moleculesere analyzed by depositing a drop of liquid in between two NaCl pellets. Six-
een scans were taken between 4000 and 400 cm−1, each 4 cm−1, on a PerkinElmerARAGON 1000 FTIR spectrometer equipped with spectrum software.
Static and dynamic measurements of contact angle were carried out using vari-us liquids. The apparatus used was a DataPhysics instrument, equipped with a CDDamera, which allowed both the determination of contact angle at equilibrium, withprecision of ±1◦ , and the kinetics of its evolution, by taking up to 1000 images per
econd, starting within a few tens of milliseconds after the deposition of the drop.he contact angles at equilibrium of pure liquids having different polarity were usedo calculate the dispersive and polar contributions to the surface energy according tohe approach proposed by Owens and Wendt [21]. The probes used to carry out con-act angle measurements were n-hexadecane, �-bromonaphthalene, diidomethane,ormamide and water. The first four molecules were high purity commercial prod-cts, whereas water was twice distilled in our laboratory. The total, dispersive andolar components of these liquids are listed in Table 3. WP was used as such forontact angle measurements, whereas BSK fibres were converted to film-like mate-ial by pressing a disk under 1 ton pressure. This method allows the preparation ofsmoother and less porous surface compared with WP.
Elemental analysis was carried out at the Laboratoire Centrale d’Analyses deernaison. This technique is based on atomic absorption of the investigated ele-ents.
X-ray photoelectron spectroscopy (XPS) experiments were recorded on a XR3E2pparatus (Vacuum Generators, U.K.). It was equipped with monochromated Mg� X-ray source (1253.6 eV) and operated at 15 kV under a current of 20 mA. Theamples were previously dried for a week and then placed in an ultrahigh vacuumhamber (10−8 mbar) with electron collection by a hemispherical analyzer at an
able 2he amount of PI and TDI to be used to prepare adducts 1 and 2.
Oligoether Mw (g mol−1) DP wPIa (mg) wTDI
a (mg)
PEG 2000 45 595 870PEGME 2000 45 – 870
PPG
2000 34 595 8702500 42 476 6963000 51 397 5803300 56 360 5284000 69 298 435
PPGMBE 2500 42 – 696PTHF 2900 40 410 600
a The amount of PI or TDI to react with 10 g of the corresponding oligoether.
n-Hexadecane 27.5 0 27.8�-Bromonaphthalene 43.5 0.9 44.4Diiodomethane 48.5 2.3 50.8
angle of 90◦ . The signal decomposition was performed using Spectrum NT, and theC–H signal was used as a reference peak at 285.0 kV.
A ZEISS-ULTRA55 field emission scanning electron microscope (FESEM) with anacceleration voltage of 15 kV, was used to analyze grafted and non-grafted fibres.Each sample was coated with a gold thin layer.
The biodegradability assessments were carried out applying the standard testmethod (ISO 14855-2005) for the evaluation of the complete decomposition of thematerials to CO2 and water by the action of compost microorganisms [22]. Two tech-nical standards (EN14955-2006: Plastic evaluation of compostability – test schemeand specifications and ISO/FDIS 17088-2007: Specifications for compostable plas-tics) are available. They both claim that ultimate biodegradability of a given materialin compost should yield a minimum value of 90% within 90–180 testing days for thematerial to be considered biodegradable.
The samples were mixed with mature compost (inoculum) in a ratio equal to1:12. In order to allow the testing of small quantities of sample, the composting mix-ture volume was increased through the addition of vermiculite, as an inert materialand acting as water sink. Therefore, the reactor composting mixture was, as follows:36 g of vermiculite plus 36 g of mature compost. The water content of the vermi-culite/compost mixture was adjusted to 34%. A reactor having 500 ml capacity wasemployed. It was equipped with inlet and outlet tubes for the aeration with com-pressed humidified air. CO2 was previously removed from the inlet air by adsorptionon soda lime. The samples (3 g for each reactor) were tested in duplicate includingblank experiments (only compost and vermiculite without the sample). The test wasconducted in an incubator at 58 ± 2 ◦C. The mature compost inoculum was obtainedfrom a composting plant treating 70% of municipal garden green waste and 30% ofmunicipal organic waste. The CO2 production was evaluated by the % of CO2 contentin the outlet reactors air measured by an infrared gas analyzer (NDIR, EcoControlEC400).
The percentage of biodegradation was expressed as the % of CO2 production withrespect to the theoretical CO2 content of the sample, which was deduced from thetotal organic carbon content value and from the amount of sample added to eachreactor.
3. Results and discussions
As discussed before, the grafting reactions were carried out inthree steps (Scheme 1). The first adduct was obtained by reactingone the chosen oligo-diol with PI, in order to block one of the twoOH functions and to leave the second one, for further reaction. Thisstep was not performed when dealing with a mono-functionalizedoligoether, namely: PEGME and PPGBE.
The FTIR spectra corresponding to the polyether, before andafter the first reaction step (reaction with PI) showed the follow-ing main differences. As expected, before treatment, the presence ofbands at 3494, 2868, 2970 and 1110 cm−1, associated to OH, CO andC–H from CH2 and CH3, respectively, was detected in all the spec-tra. The addition of PI gave rise to the appearance of 2 new peaks at2260 and 1732 cm−1, attributed to the presence of the NCO func-tion and the formation of urethane moieties, respectively. The latterpeak confirms the occurrence of the reaction. The reaction was leftto occur and monitored by FTIR spectroscopy, which allowed thedetermination of the end of the reaction, when the peak associ-ated with PI (2260 cm−1) disappeared completely and the signalcorresponding to urethane formation (1732 cm−1) remained con-stant. Other peaks appeared in the modified PPG spectrum, namely:those at around 3300, 730 and 694 cm−1, corresponding to aro-
matic ring from PI. The reaction between polyol and PI was overafter approximately 4 h.The addition of TDI to adduct I yielded the formation of adductII, as shown in Fig. 1, which gives rise to the following features inthe FTIR spectrum:
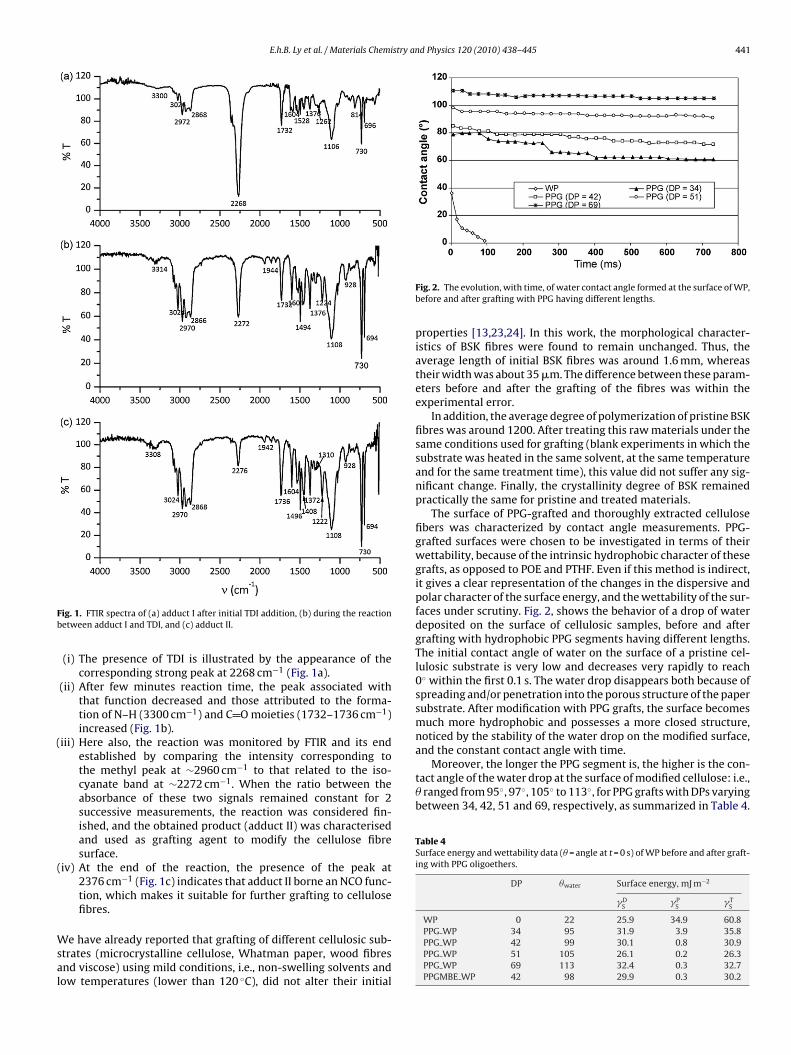
E.h.B. Ly et al. / Materials Chemistry and Physics 120 (2010) 438–445 441
Fb
(
(
Wsal
Moreover, the longer the PPG segment is, the higher is the con-tact angle of the water drop at the surface of modified cellulose: i.e.,� ranged from 95◦, 97◦, 105◦ to 113◦, for PPG grafts with DPs varyingbetween 34, 42, 51 and 69, respectively, as summarized in Table 4.
Table 4Surface energy and wettability data (� = angle at t = 0 s) of WP before and after graft-ing with PPG oligoethers.
DP �water Surface energy, mJ m−2
�DS �P
S �TS
WP 0 22 25.9 34.9 60.8
ig. 1. FTIR spectra of (a) adduct I after initial TDI addition, (b) during the reactionetween adduct I and TDI, and (c) adduct II.
(i) The presence of TDI is illustrated by the appearance of thecorresponding strong peak at 2268 cm−1 (Fig. 1a).
(ii) After few minutes reaction time, the peak associated withthat function decreased and those attributed to the forma-tion of N–H (3300 cm−1) and C O moieties (1732–1736 cm−1)increased (Fig. 1b).
iii) Here also, the reaction was monitored by FTIR and its endestablished by comparing the intensity corresponding tothe methyl peak at ∼2960 cm−1 to that related to the iso-cyanate band at ∼2272 cm−1. When the ratio between theabsorbance of these two signals remained constant for 2successive measurements, the reaction was considered fin-ished, and the obtained product (adduct II) was characterisedand used as grafting agent to modify the cellulose fibresurface.
iv) At the end of the reaction, the presence of the peak at2376 cm−1 (Fig. 1c) indicates that adduct II borne an NCO func-tion, which makes it suitable for further grafting to cellulosefibres.
e have already reported that grafting of different cellulosic sub-trates (microcrystalline cellulose, Whatman paper, wood fibresnd viscose) using mild conditions, i.e., non-swelling solvents andow temperatures (lower than 120 ◦C), did not alter their initial
Fig. 2. The evolution, with time, of water contact angle formed at the surface of WP,before and after grafting with PPG having different lengths.
properties [13,23,24]. In this work, the morphological character-istics of BSK fibres were found to remain unchanged. Thus, theaverage length of initial BSK fibres was around 1.6 mm, whereastheir width was about 35 �m. The difference between these param-eters before and after the grafting of the fibres was within theexperimental error.
In addition, the average degree of polymerization of pristine BSKfibres was around 1200. After treating this raw materials under thesame conditions used for grafting (blank experiments in which thesubstrate was heated in the same solvent, at the same temperatureand for the same treatment time), this value did not suffer any sig-nificant change. Finally, the crystallinity degree of BSK remainedpractically the same for pristine and treated materials.
The surface of PPG-grafted and thoroughly extracted cellulosefibers was characterized by contact angle measurements. PPG-grafted surfaces were chosen to be investigated in terms of theirwettability, because of the intrinsic hydrophobic character of thesegrafts, as opposed to POE and PTHF. Even if this method is indirect,it gives a clear representation of the changes in the dispersive andpolar character of the surface energy, and the wettability of the sur-faces under scrutiny. Fig. 2, shows the behavior of a drop of waterdeposited on the surface of cellulosic samples, before and aftergrafting with hydrophobic PPG segments having different lengths.The initial contact angle of water on the surface of a pristine cel-lulosic substrate is very low and decreases very rapidly to reach0◦ within the first 0.1 s. The water drop disappears both because ofspreading and/or penetration into the porous structure of the papersubstrate. After modification with PPG grafts, the surface becomesmuch more hydrophobic and possesses a more closed structure,noticed by the stability of the water drop on the modified surface,and the constant contact angle with time.
PPG WP 34 95 31.9 3.9 35.8PPG WP 42 99 30.1 0.8 30.9PPG WP 51 105 26.1 0.2 26.3PPG WP 69 113 32.4 0.3 32.7PPGMBE WP 42 98 29.9 0.3 30.2

442 E.h.B. Ly et al. / Materials Chemistry and Physics 120 (2010) 438–445
Fig. 3. The correlation between the DPs of PPG coupling segments and the watercontact angle formed at the surface of WP, before and after grafting.
Table 5Elemental analysis of WP, before and after grafting with oligoethers.
DP C H O N
AlfF
cvnArs0wevs
ti
WP – 42.7 6.7 50.9 –PPG WP 69 48.5 6.9 37.7 3.8PEG WP 45 46.4 7.1 46.0 0.5
linear correlation with good regression coefficient was estab-ished between the DP of PPG grafts and the water contact angleormed at the respective modified cellulose surface, as shown inig. 3.
These data are confirmed by the values of dispersive and polarontributions to the surface energy of these substrates. In fact,irgin model cellulose (WP) displayed dispersive and polar compo-ents of the surface energy of about 25 and 35 mJ m−2, respectively.fter modification with PPG grafts, the dispersive componentemained roughly constant, while the polar contribution to theurface energy decreased drastically and reached values close tomJ m−2. Here also, lowest polar component of the surface energyas reached when using the longest PPG segment. The mono-alkyl
ther derivative (PPGMBE with a DP of 42) was also found to reduce
ery efficiently the hydrophilic and polar character of the celluloseurface.Elemental analysis (Table 5) shows an increase in the propor-ion of the amount of carbon atoms, compared with that detectedn the initial substrate and the detection of nitrogen atoms. These
Fig. 4. SEM micrographs of the surface of WP (a) b
Fig. 5. Full XPS spectra of (a) virgin WP, and (b) PPG3300-grafted, (c) PEGMBE2500-grafted, (d) PEGME2000-grafted and (e) PTHF2900-grafted WP.
data confirm those obtained by FTIR spectroscopy and confirm theoccurrence of the grafting.
For some modified fibres, SEM analysis detected surface changesafter modification, as shown in Fig. 4 for the PTHF-grafted sample.After grafting, the fibres seem to be covered by individual spheri-cal particles of 100 nm size. Unfortunately these particles did notcover the fibre surface uniformly. Thus, SEM data of the paper sam-ples thoroughly purified by solvent soxhlet extraction, confirms theoccurrence of the grafting, but revealed that it is not uniform, atleast in terms of the thickness of the grafted layer. In fact, sincecontact angle measurements revealed a good hydrophobization, itcan be thought that the full surface was grafted, but the polymergrafts density could vary from one location to another. The paper
areas grafted with a very small amount of hydrophobic polymergrafts are not detectable by SEM, but they can be grafted enough togive very good response towards a drop of water.The XPS data of WP before and after grafting are summarisedin Table 6 and shown in Figs. 5 and 6. The full spectra (Fig. 5) for
efore and (b) after grafting with PTHF2900.
E.h.B. Ly et al. / Materials Chemistry and Physics 120 (2010) 438–445 443
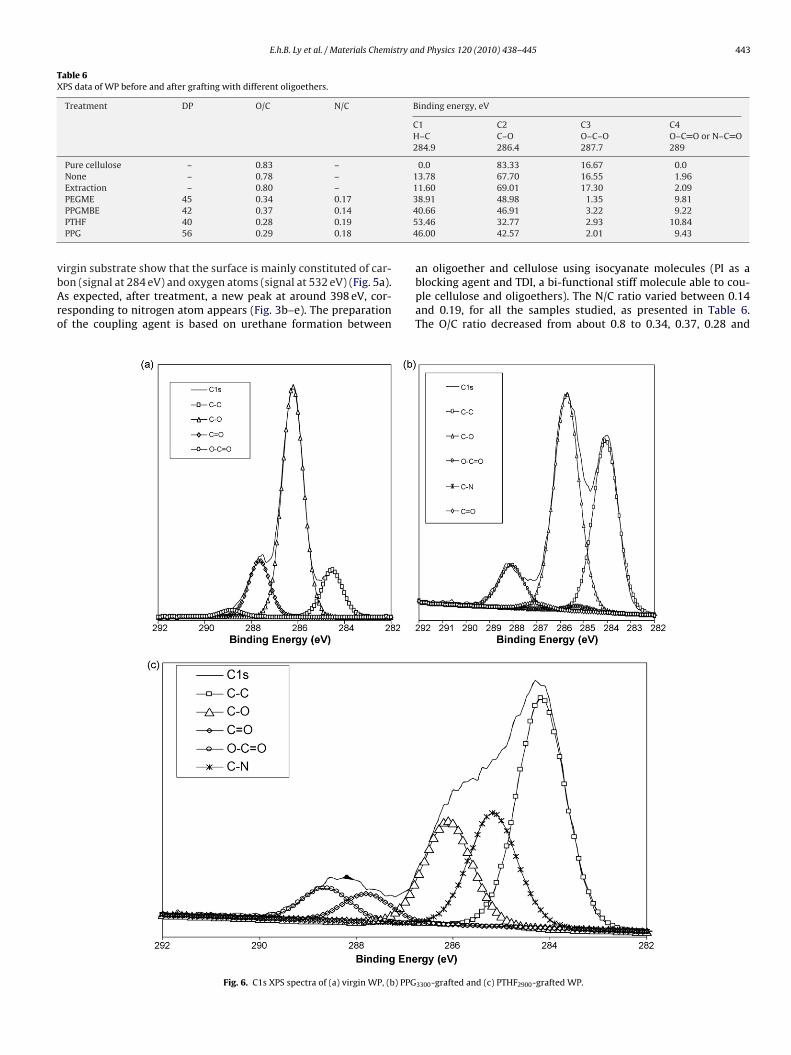
Table 6XPS data of WP before and after grafting with different oligoethers.
Treatment DP O/C N/C Binding energy, eV
C1 C2 C3 C4H–C C–O O–C–O O–C O or N–C O284.9 286.4 287.7 289
Pure cellulose – 0.83 – 0.0 83.33 16.67 0.0None – 0.78 – 13.78 67.70 16.55 1.96Extraction – 0.80 – 11.60 69.01 17.30 2.09
3454
vbAro
PEGME 45 0.34 0.17PPGMBE 42 0.37 0.14PTHF 40 0.28 0.19PPG 56 0.29 0.18
irgin substrate show that the surface is mainly constituted of car-
on (signal at 284 eV) and oxygen atoms (signal at 532 eV) (Fig. 5a).s expected, after treatment, a new peak at around 398 eV, cor-esponding to nitrogen atom appears (Fig. 3b–e). The preparationf the coupling agent is based on urethane formation betweenFig. 6. C1s XPS spectra of (a) virgin WP, (b) PPG
8.91 48.98 1.35 9.810.66 46.91 3.22 9.223.46 32.77 2.93 10.846.00 42.57 2.01 9.43
an oligoether and cellulose using isocyanate molecules (PI as a
blocking agent and TDI, a bi-functional stiff molecule able to cou-ple cellulose and oligoethers). The N/C ratio varied between 0.14and 0.19, for all the samples studied, as presented in Table 6.The O/C ratio decreased from about 0.8 to 0.34, 0.37, 0.28 and3300-grafted and (c) PTHF2900-grafted WP.

444 E.h.B. Ly et al. / Materials Chemistry a
F
0Wsb
asiaaep2aC[ewpt(
Tcoc5tcsbIs
agBrd3dnagPag
[[[
[
[[[
[
[
[
[20] R. Manríquez, F.A. López-Dellamary, J. Frydel, T. Emmler, H. Breitzke, G.
ig. 7. Biodegradation curves of BSK and PPG3300-grafted and PTHF2900-grafted BSK.
.29, for PEGME2000-, PEGMBE2500-, PTHF2900- and PPG3300-treatedP, respectively (Table 6). This is in agreement with the chemical
tructure of the used oligoethers, which have an O/C ratio varyingetween 0.33 and 0.20.
The C1s was deconvoluted in order to quantify the relativebundance of carbon atom types. As discussed in our previoustudies, theoretically speaking, pure cellulose exhibits two peaksn its deconvoluted C1s XPS spectra [4,25,26], namely: (i) C–Ot 286.7 eV, associated to alcohols and ethers groups (peak C2),nd (ii) O–C–O at 288.3 attributed to acetal and carbonyl moi-ties (peak C3). The XPS analysis of pristine WP displays theresence of four peaks (C1, C2, C3 and C4), at 285.0, 286.7,88.3 and 289.0 eV, and corresponding to C1 (C–H), C2 (C–O)nd C3 (O–C–O and/or C O), respectively. Thus, the unexpected1 peak is associated to non-oxidized alkane-type carbon atoms4,25–29] and attributed to impurities associated with the pres-nce of residual lignin, extractive substances and fatty acids,hereas the weak C4 signal is assigned to carboxylic functionsresent in hemicellulose macromolecules. After soxhlet extrac-ion with methylene chloride, the spectrum of WP did not changeTable 6).
The C1s spectra of the PPG- and PTHF-treated WP (Fig. 6 andable 6) revealed a major difference related to the C1 and C4 peaks,ompared to that of initial WP (Fig. 6a). Moreover, the appearancef a new peak associated with C–N links was observed (Fig. 6b and). In fact, the intensity of C1 increased from about 11 to 39, 41,3 and 46%, for PEGME2000-, PEGMBE2500-, PTHF2900- and PPG3300-reated WP, respectively (Table 6). This is consistent with thehemical structure of the used oligoethers, which contain aliphaticequences. The intensity of the C3 peak decreased significantlyecause none of the grafting molecules contains such a moiety.
nstead, the intensity of the C4 signal increased because the graftingegments bear urethane groups.
Biodegradability tests were performed on grafted BSK fibresnd the obtained results show different behaviors between non-rafted and grafted fibres, as shown in Fig. 7. Thus, for non-graftedSK the biodegradation starts slowly but immediately increasedegularly to reach more than 80% of biodegradation after 20ays of composting. The full biodegradation was attained after0 days. The grafting seems to slow down the initial degra-ation rate by a factor of 2, independently from the chemicalature of the grafted segments. Moreover, the modification yieldedn induction period in the biodegradation kinetics, since the
rafted fibres started to biodegrade after 5 and 7 days, forPG- and PTHF-grafted BSK, respectively. This can probably bettributed to the higher hydrophobic character and length of PTHFrafts, as already confirmed by the wettability measurements.[[
nd Physics 120 (2010) 438–445
The induction period is probably associated with the removalof the grafted macromolecular layers before attacking cellulosicsubstrates.
In all cases, the ultimate biodegradation reached an averagevalue of 90% within 60 days biodegradation. According to the twotechnical standard (EN14955-2006 and ISO/FDIS 17088-2007), allthe materials studied here can be considered as biodegradablematerials, thus allowing to conclude that grafting with aliphaticoligoethers enhances substantially the hydrophobic character ofthe treated fibres, but did not alter their intrinsic ability to undergobiological degradation, although it somehow increases the degra-dation time. This is further evidence of the fact that the treatment islocated mainly at the surface of the fibres, which yields significantchanges in the surface properties but did not alter substantially thebulk properties.
4. Conclusion
Cellulose fibres, a renewable raw material, can be success-fully modified with functionalized oligoethers yielding hydrophilicand biodegradable substrates. Thus, the judicious choice of thepolar/non-polar balance, as well as, the length of the macromolec-ular segments of the coupling agent gave rise to a variety ofinteresting modified fibres, which should now be tested in differentareas of applications, such as flexible packaging films or cellulose-reinforced hydro-stable composite materials.
Acknowledgements
This research forms an integral part of the SUSTAINPACK project,funded by the European Community under the 6th Framework Pro-gram. Wim Thielemans also greatly acknowledges the EuropeanCommunity for his Marie Curie Intra-European Fellowship (MEIF-CT-2005-025125).
References
[1] M.N. Belgacem, A. Gandini, Surface modification of cellulose fibres, in: A. Bel-gacem, Gandini (Eds.), Monomers, Polymers and Composites from RenewableResources, Elsevier, Amsterdam, 2008, pp. 385–400 (chapter 18).
[2] A special issue of 14 publications devoted to cellulose-based composite mate-rials and entitled eco-composites, Compos. Sci. Technol. 63 (2004).
[3] A special issue devoted to cellulose-based composites, Compos. Interface 12(2005).
[4] M.N. Belgacem, A. Gandini, Compos. Interface 12 (2005) 41.[5] M.N. Belgacem, A. Gandini, Natural fibre-surface modification and charac-
terisation, in: T. Sabu, L. Pothan (Eds.), Cellulose Fibre Reinforced PolymerComposites, Old City Publishing, Philadelphia, 2009, pp. 14–46 (chapter 2).
[6] W. Thielemans, R.P. Wool, Polym. Compos. 26 (2005) 695.[7] W. Thielemans, R.P. Wool, Compos. Part A: Appl. Sci. Manuf. 35 (2004) 327.[8] W.H. Binder, R. Sachsenhofer, Macromol. Rapid Commun. 28 (2007) 15.[9] J. Hafrén, W. Zou, A. Corbova, Macromol. Rapid. Commun. 27 (2006) 1362.10] M. Krouit, J. Bras, M.N. Belgacem, Eur. Polym. J. 44 (2008) 4074.11] D. Song, Y. Zhao, C. Dong, Y. Deng, J. Appl. Polym. Sci. 113 (2009) 3019.12] S. Klébert, L. Nagy, A. Domjan, B. Pukánszky, J. Appl. Polym. Sci. 113 (2009)
3255.13] O. Paquet, M. Krouit, J. Bras, W. Thielemans, M.N. Belgacem, Acta Mater. (2009),
doi:10.1016/j.actamat.2009.09.057.14] G. Morandi, L. Heath, W. Thielemans, Langmuir 25 (2009) 8280.15] G. Zampano, M. Bertoldo, S. Bronco, Carbohydr. Polym. 75 (2009) 22.16] S. Alila, A.M. Ferraria, A.M. Botelho do Rego, S. Boufi, Carbohydr. Polym. 77
(2009) 553.17] A.J. de Menezes, D. Pasquini, A.A.d.S. Curvelo, A. Gandini, Carbohydr. Polym. 76
(2009) 437.18] A.J. de Menezes, D. Pasquini, A.A.d.S. Curvelo, A. Gandini, Cellulose 16 (2009)
239.19] L. Chuan-Fu, Z. Ai-Ping, L. Wei-Ying, Y. Feng-Xia, S. Run-Cang, J. Agric. Food
Chem. 57 (2009) 1814.
Buntkowsky, H.-H. Limbach, I.G. Shenderovich, J. Phys. Chem. B 113 (2009)934.
21] D.K. Owens, R.C. Wendt, J. Appl. Polym. Sci. 13 (1969) 1741.22] ISO 14855-2005, Determination of the ultimate aerobic biodegradability of
plastic materials under controlled composting conditions.

stry an
[[
[
E.h.B. Ly et al. / Materials Chemi
23] J.A. Trejo-O’Reilly, J.Y. Cavaillé, A. Gandini, Cellulose 4 (1997) 305.24] J.A. Trejo-O’Reilly, J.Y. Cavaillé, N.M. Belgacem, A. Gandini, J. Adhes. 67 (1998)
359.25] A. Ahmed, A. Adnot, J.L. Grandmaison, S. Kaliaguine, J. Doucet, Cellulose Chem.
Technol. 21 (1987) 483.
[[[[
d Physics 120 (2010) 438–445 445
26] G.M. Dorris, D. Gray, Cellulose Chem. Technol. 12 (1978) 9.27] G.M. Dorris, D. Gray, Cellulose Chem. Technol. 12 (1978) 721.28] D. Gray, Cellulose Chem. Technol. 12 (1978) 735.29] M.N. Belgacem, G. Czeremuszkin, S. Sapieha, A. Gandini, Cellulose 2 (1995)
145.


















