
Suppression of Growth of Renal Carcinoma Cells by the von...
5
(CANCER RESEARCH 55. 4804-4807, November 1, 1995) Advances in Brief Suppression of Growth of Renal Carcinoma Cells by the von Hippel-Lindau Tumor Suppressor Gene Fan Chen, Takeshi Kishida, Fuh-Mei Duh, Paul Renbaum, Mary L. Orcutt, Laura Schmidt, and Berton /liar1 Biological Carcinogenesis and Development Program, SA1C Frederick [F. C., F~M. D., L. S.}, Laboratory of Immunobiologv, National Cancer Institute ¡T.K., P. R., M. L. O., ft Z.ÃOE, Frederick, Maryland 21702 Abstract Clear cell renal carcinomas are most frequently characterized by loss of function of both copies of the von Hippel-Lindau (VHL) disease gene, suggesting that the \ III gene product plays an important role in regulat ing renal cell proliferation. To directly assess the function of the Mil gene product, we transfected the wild-type \ III gene into two renal carcinoma cell lines that lacked normal expression of the gene. Expression of the wild-type \ III gene led to a dramatic suppression of growth in two renal carcinoma cell lines, A498 and UMRC6 in vitro, as measured by colony formation and direct cell counting. Transfection of a naturally occurring mutant \ III gene (nucleotide 713 G to A, Arg to Gin) did not lead to growth suppression of these renal carcinoma cells, nor did transfection of the wild-type VHL gene into two non-renal tumor cell lines that expressed the endogenous wild-type \ III gene. Expression constructs, which in cluded the first ATG at nucleotide 214, were sufficient to produce the strongest growth suppression. These experiments provide direct evidence that the \ III gene product functions to suppress the growth of renal carcinoma cells and also provide a model for mapping the domains of the VHL protein important in suppressing tumor growth. Introduction von Hippel-Lindau disease is an inherited disorder characterized by a predisposition to develop tumors in the eyes, brain, spinal cord, kidney, adrenal gland, and pancreas (1). The VHL gene, located at 3p25-26, was recently cloned and shown to be mutated in the germ- line of affected family members, as well as in sporadic RCC,~ he- mangioblastomas, and pheochromocytomas (2-7). Studies of sporadic RCC have frequently shown loss of heterozygosity of chromosome 3p, accompanied by mutational inactivation of the other VHL alÃ-ele, strongly suggesting that the VHL gene plays an important role in tumor growth suppression (2, 4, 5). The central issue surrounding tumor suppressor genes concerns their biological function in negative growth regulation (8). Both the p53 and the retinoblastoma suscepti bility genes demonstrated dramatic growth suppression when they were introduced into tumor cells with inactivated endogenous genes (9-11). Similar effects with the WTI, APC, and pI6 genes have also been reported recently (12-14). These studies confirm the hypothesis that wild-type tumor suppressor genes function as suppressors of neoplastic growth (8). Inactivation of both copies of the wild-type gene leads to a pathway of uncontrolled cell growth (8-14). We tested several VHL gene constructs for the ability to suppress growth by transfecting these genes into RCC lines that lacked endog enous wild-type VHL genes. Introducing VHL gene constructs, includ ing the first ATG codon extending to the stop codon (without the VHL 3' untranslated region), consistently showed growth suppression of Received 8/17/95; accepled 9/21/95. The costs of publication of this article were defrayed in part hy the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1To whom requests for reprints should be addressed, at Laboratory of Immunobiology, Building 560, Room 12-71, NCI-FCRDC, Frederick, MD 21702. •¿ The abbreviations used are: RCC, renal cell carcinoma; ORF. open reading frame. RCC lines lacking the endogenous VHL gene but had no effect on two non-RCC lines that expressed wild-type VHL. This work provides direct evidence that the VHL gene can suppress the growth of human RCC. Materials and Methods VHL Gene Expression Constructs. Various VHL gene sequences were cloned into the mammalian expression vector pCR3 (Invitrogen, San Diego, CA) under the control of the constitutive cytomegalovirus promoter and containing the neomycin resistance gene as a selection marker. The construct VHL-gp7 started from nucleotide 190 with an introduced ATG codon and extended to nucleotide 852, the end of the ORF of the VHL cDNA sequence (2). VHL-H1 started from nucleotide 214, the first in-frame ATG site within the VHL cDNA sequence, and extended to nucleotide 852. VHL-H2 started from nucleotide 373, the second in-frame ATG site, and extended to nucleotide 852. A mutant derived from VHL-H1 was generated by making aGtoA substitution at nucleotide 713, resulting in an amino acid change from Arg to Gin, and was designated as VHL-Hl-713 mut. This 713 mutation is a naturally occurring hot spot mutation identified in many VHL families who were affected by RCC, pheochromocytoma, and other tumors (3, 5, 6). A control construct containing the VHL-H1 sequence in reverse orientation was desig nated as VHL-Hl-antisense. All constructs were confirmed by sequencing. Cell Lines. Human RCC lines A498 and UMRC6 were grown in medium RPMI 1640 with 10% PCS (GIBCO-BRL, Bethesda, MD) as described pre viously (15, 16). A498 has a 4-base deletion at nucleotides 639-642 of the VHL gene (5), and UMRC6 has a 10-base deletion at nucleotides 717-726 (5).3 In addition, chromosome 3p loss was reported in both cases (5). A human ovarian cancer cell line SKOV3 and a human lung cancer cell line NC1-H23 were also used for this study. Both cell lines expressed the VHL gene, and no VHL mutation/deletion was identified in either of them (5). Transfections. Transfections were performed using the cationic lipid, Li- pofectamine (GIBCO-BRL), with cells at 60-70% confluence, as described by the manufacturer. Preliminary tests were performed to determine the amount of lipofectamine and DNA required to produce the highest transfection efficiency. For colony counting, A498 line in T-25 cm flasks, 20 /xl/3.5 jig (Lipo- fectamine/DNA), was used (Table 1). For A498 and UMRC6 lines in 3.5-cm plates, 6.0 fil/1.0 ng (Lipofectamine/DNA) was used. For SKOV3 and NCI- H23 lines in 3.5-cm plates, 1.2 fil/0.2 /xg (Lipofectamine/DNA) was used. Geneticin (G418; GIBCO-BRL) selection started 48 h after transfection. G418 concentrations for each cell line were: A498, 500 fig/ml; UMRC6, 250 /¿g/ml; SKOV3, 1000 /iig/ml; and NCI-H23, 1000 tig/ml. G418-resistant colonies were counted after 18 days of G418 selection, whereas individual G418- resistant cells were counted after 8 days of G418 selection. RNA Isolation and RT-PCR. Total RNA was isolated from cultured G418-resistant cells using RNazol (Tel-Test, Friendswood, TX). Endogenous and exogenous VHL mRNA were amplified by differential RT-PCR. The first cDNA strand was synthesized using AMV reverse transcriptase (Promega, Madison, WI) for endogenous VHL, with the antisense primer 8TM13F located in the VHL 3' untranslated region (nucleotides 921-940, 5'-GGA AGO AAG GAA CCA GTC CTG TAT C-3'), and then amplified by PCR with the sense primer pCR3Sl (nucleotides 666-695, 5'-CAC ACT GCC ACT GTA TAC TCT GAA AGA GCG -3') and a nested antisense primer, 6b (nucleotides 902-930, 5'-TAC CAT CAA AAG CTG AGA TGA AAC ACT GTA AGT- 1M. H. Wei, unpublished data. 4804 on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Transcript of Suppression of Growth of Renal Carcinoma Cells by the von...

(CANCER RESEARCH 55. 4804-4807, November 1, 1995)
Advances in Brief
Suppression of Growth of Renal Carcinoma Cells by the von Hippel-Lindau Tumor
Suppressor Gene
Fan Chen, Takeshi Kishida, Fuh-Mei Duh, Paul Renbaum, Mary L. Orcutt, Laura Schmidt, and Berton /liar1
Biological Carcinogenesis and Development Program, SA1C Frederick [F. C., F~M. D., L. S.}, Laboratory of Immunobiologv, National Cancer Institute ¡T.K., P. R., M. L. O.,
ft Z.Ì,Frederick, Maryland 21702
Abstract
Clear cell renal carcinomas are most frequently characterized by loss offunction of both copies of the von Hippel-Lindau (VHL) disease gene,
suggesting that the \ III gene product plays an important role in regulating renal cell proliferation. To directly assess the function of the Mil geneproduct, we transfected the wild-type \ III gene into two renal carcinoma
cell lines that lacked normal expression of the gene. Expression of thewild-type \ III gene led to a dramatic suppression of growth in two renal
carcinoma cell lines, A498 and UMRC6 in vitro, as measured by colonyformation and direct cell counting. Transfection of a naturally occurringmutant \ III gene (nucleotide 713 G to A, Arg to Gin) did not lead togrowth suppression of these renal carcinoma cells, nor did transfection ofthe wild-type VHL gene into two non-renal tumor cell lines that expressedthe endogenous wild-type \ III gene. Expression constructs, which in
cluded the first ATG at nucleotide 214, were sufficient to produce thestrongest growth suppression. These experiments provide direct evidencethat the \ III gene product functions to suppress the growth of renalcarcinoma cells and also provide a model for mapping the domains of theVHL protein important in suppressing tumor growth.
Introduction
von Hippel-Lindau disease is an inherited disorder characterized by
a predisposition to develop tumors in the eyes, brain, spinal cord,kidney, adrenal gland, and pancreas (1). The VHL gene, located at3p25-26, was recently cloned and shown to be mutated in the germ-line of affected family members, as well as in sporadic RCC,~ he-
mangioblastomas, and pheochromocytomas (2-7). Studies of sporadic
RCC have frequently shown loss of heterozygosity of chromosome3p, accompanied by mutational inactivation of the other VHL alÃele,strongly suggesting that the VHL gene plays an important role intumor growth suppression (2, 4, 5). The central issue surroundingtumor suppressor genes concerns their biological function in negativegrowth regulation (8). Both the p53 and the retinoblastoma susceptibility genes demonstrated dramatic growth suppression when theywere introduced into tumor cells with inactivated endogenous genes(9-11). Similar effects with the WTI, APC, and pI6 genes have alsobeen reported recently (12-14). These studies confirm the hypothesisthat wild-type tumor suppressor genes function as suppressors ofneoplastic growth (8). Inactivation of both copies of the wild-typegene leads to a pathway of uncontrolled cell growth (8-14).
We tested several VHL gene constructs for the ability to suppressgrowth by transfecting these genes into RCC lines that lacked endogenous wild-type VHL genes. Introducing VHL gene constructs, includ
ing the first ATG codon extending to the stop codon (without the VHL3' untranslated region), consistently showed growth suppression of
Received 8/17/95; accepled 9/21/95.The costs of publication of this article were defrayed in part hy the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1To whom requests for reprints should be addressed, at Laboratory of Immunobiology,Building 560, Room 12-71, NCI-FCRDC, Frederick, MD 21702.
•¿�The abbreviations used are: RCC, renal cell carcinoma; ORF. open reading frame.
RCC lines lacking the endogenous VHL gene but had no effect on twonon-RCC lines that expressed wild-type VHL. This work provides
direct evidence that the VHL gene can suppress the growth of humanRCC.
Materials and Methods
VHL Gene Expression Constructs. Various VHL gene sequences werecloned into the mammalian expression vector pCR3 (Invitrogen, San Diego,CA) under the control of the constitutive cytomegalovirus promoter andcontaining the neomycin resistance gene as a selection marker. The constructVHL-gp7 started from nucleotide 190 with an introduced ATG codon and
extended to nucleotide 852, the end of the ORF of the VHL cDNA sequence(2). VHL-H1 started from nucleotide 214, the first in-frame ATG site withinthe VHL cDNA sequence, and extended to nucleotide 852. VHL-H2 startedfrom nucleotide 373, the second in-frame ATG site, and extended to nucleotide852. A mutant derived from VHL-H1 was generated by making a G to A
substitution at nucleotide 713, resulting in an amino acid change from Arg toGin, and was designated as VHL-Hl-713 mut. This 713 mutation is a naturally
occurring hot spot mutation identified in many VHL families who wereaffected by RCC, pheochromocytoma, and other tumors (3, 5, 6). A controlconstruct containing the VHL-H1 sequence in reverse orientation was designated as VHL-Hl-antisense. All constructs were confirmed by sequencing.
Cell Lines. Human RCC lines A498 and UMRC6 were grown in mediumRPMI 1640 with 10% PCS (GIBCO-BRL, Bethesda, MD) as described previously (15, 16). A498 has a 4-base deletion at nucleotides 639-642 of theVHL gene (5), and UMRC6 has a 10-base deletion at nucleotides 717-726 (5).3
In addition, chromosome 3p loss was reported in both cases (5). A humanovarian cancer cell line SKOV3 and a human lung cancer cell line NC1-H23
were also used for this study. Both cell lines expressed the VHL gene, and noVHL mutation/deletion was identified in either of them (5).
Transfections. Transfections were performed using the cationic lipid, Li-pofectamine (GIBCO-BRL), with cells at 60-70% confluence, as described by
the manufacturer. Preliminary tests were performed to determine the amount oflipofectamine and DNA required to produce the highest transfection efficiency.For colony counting, A498 line in T-25 cm flasks, 20 /xl/3.5 jig (Lipo-fectamine/DNA), was used (Table 1). For A498 and UMRC6 lines in 3.5-cmplates, 6.0 fil/1.0 ng (Lipofectamine/DNA) was used. For SKOV3 and NCI-H23 lines in 3.5-cm plates, 1.2 fil/0.2 /xg (Lipofectamine/DNA) was used.Geneticin (G418; GIBCO-BRL) selection started 48 h after transfection. G418
concentrations for each cell line were: A498, 500 fig/ml; UMRC6, 250 /¿g/ml;SKOV3, 1000 /iig/ml; and NCI-H23, 1000 tig/ml. G418-resistant colonieswere counted after 18 days of G418 selection, whereas individual G418-
resistant cells were counted after 8 days of G418 selection.RNA Isolation and RT-PCR. Total RNA was isolated from cultured
G418-resistant cells using RNazol (Tel-Test, Friendswood, TX). Endogenousand exogenous VHL mRNA were amplified by differential RT-PCR. The first
cDNA strand was synthesized using AMV reverse transcriptase (Promega,Madison, WI) for endogenous VHL, with the antisense primer 8TM13F locatedin the VHL 3' untranslated region (nucleotides 921-940, 5'-GGA AGO AAGGAA CCA GTC CTG TAT C-3'), and then amplified by PCR with the senseprimer pCR3Sl (nucleotides 666-695, 5'-CAC ACT GCC ACT GTA TACTCT GAA AGA GCG -3') and a nested antisense primer, 6b (nucleotides902-930, 5'-TAC CAT CAA AAG CTG AGA TGA AAC ACT GTA AGT-
1M. H. Wei, unpublished data.
4804
on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

GROWTH SUPPRESSION OF RCC CELLS BY THE VHL GENE
Table 1 Colony formation of A498 cells
Transfectedconstructs"VHL-Hl-anlisenseVHL-gp7VHL-H1VHL-H2VHL-m-713
mutExp.l50,
69*2e233Exp.268,690023Exp.3131,167262242Exp.420,201,41,030,23Exp.531,353.
117,64,
1345,41Exp.6107,
14929,2726,2580,74145,
102" Transfection procedures were described in "Materials and Methods." After 18 days
of G418 selection, cells were stained by méthylèneblue and counted under a microscope.A group of cells that numbered more than 20 was counted as a colon.
' The number (if colonies observed.
' The VHL-gp7 group and VHL-H1 group are statistically significantly different from
the VHL-Hl-antisense groups (P < 0.001, l lest); the VHL-H2 group is also significantlydifferent from VHL-Hl-anlisense group (P < 0.01). There is no difference between theVHL-gp7 and VHI.-H1 groups.
3'). For exogenous VHL, the first cDNA strand was synthesized with theantisense primer VHLBGHAS2 located in the vector's BGH-.V untranslatedregion (nucleotides 868-897 of pCR3, 5'-CAG TGG GAG TGG CAC CTTCCA GGG TCA AGO -3') and then amplified by PCR with the sense primer
pCR3Sl and a nested antisense primer VHLBGHAS1 from the vector (nucleotides 802-831 of pCR3, 5'-AAC TAG AAG OCA CAO T CG AGO CTGTAT ACT-3'). The use of these primers enabled us to specifically amplify and
identify cither the endogenous (274 bp) or the exogenous (306 bp) VHLtranscripts.
Results
The cloned VHL cDNA contains two in-frame ATG codons with an
ORF extending upstream from the first ATG, making the actualtranslation initiation site uncertain (2). To examine growth suppression activity, we prepared three wild-type VHL constructs with different NH,-termini, an antisense control, and a mutant VHL construct
beginning at the first ATG codon and containing the 713 (A to G, Argto Gin) hot spot mutation (Refs. 3 and 5; see "Materials and Methods"). To determine whether these constructs could drive synthesis of
the VHL proteins, each of the expression vectors was transientlytransfected into COS-7 cells and assayed by probing a Western blot
with polyclonal antibodies against the VHL protein. Specific expression products of appropriate molecular weights were detected for eachof the transfected constructs 48 h after transfection. For VHL-gp7, M,
33,000, 31,000 and 17,000 expression products were seen. ForVHL-H1 and VHL-H1-713 mut, M, 31,000 and 17,000 proteins weredetected, and for VHL-H2, a single band of M, 17,000 protein wasdetected. Untransfected COS-7 cells and the transfected antisense
construct did not produce detectable VHL proteins (data not shown).We introduced the VHL expression constructs into two RCC lines
lacking normal VHL expression to examine their effects on cellproliferation. Six experiments were performed in which the VHLexpression constructs were transfected into the A498 line (Table 1).Following 18 days of G418 selection, cells were stained with méthylèneblue (Fisher Scientific. Pittsburgh, PA), and colonies containingmore than 20 cells were counted (Table 1). Transfection of theVHL-gp7 and VHL-H 1 constructs into A498 cells resulted in 5-20-
fold fewer colonies than cells transfected with either the antisensecontrol or VHL-H\-7I3 mut constructs. Transfection of the VHL-H2construct into A498 cells resulted in only a 2-fold reduction ofG418-resistant colonies.
Similar results were also observed in another RCC line, UMRC6.These cells however, grow in a diffuse manner, making it difficult toquantitate growth suppression by colony formation, and instead, after8 days of G418 selection, cells were stained and photographed. Fig. 1shows a magnified photograph of UMRC6 cells following transfection with each of the expression constructs and G418 selection. Again,both VHL-gp7 and VHL-H 1 induced strong suppressive effects, whilethe VHL-H2 constructs induced a milder effect, when compared to theantisense control or the VHL-Hl-7/.f mut. In contrast to the growth
suppression observed with these RCC lines, the human ovarian carcinoma cell line SKOV3 did not respond to any of the transfectedVHL constructs (Fig. 1).
To further quantitate the growth suppression induced by the VHLgene product, individual cells were counted following transfection and8 days of G418 selection and compared with the VHL antisensecontrol. As seen in Fig. 2, the growth of the A498 line was suppressed
"'•'•»'"V-•'•"'•¿�
r. ¿: - f: \\ ....V--. •¿�;'•x*•'.- •¿�../•-•»••¿�-.;- -'-,
.- «
B
3Fig. 1. Microscopic pictures of the effects of the VHL gene expression in UMRC6 and SKOV3 lines. Pictures were taken 10 days after G418 selection. A. UMRC6 line; B. SKOV3
line. Expression constructs /. VHL-Hl-anlisense; 2, VHL-gp7; 3, VHL-H 1; 4. VHL-H2; 5, VHL-H1-7/J muÃ.
4805
on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
GROWTH SUPPRESSION OF RCC CELLS BY THE VHL GENE
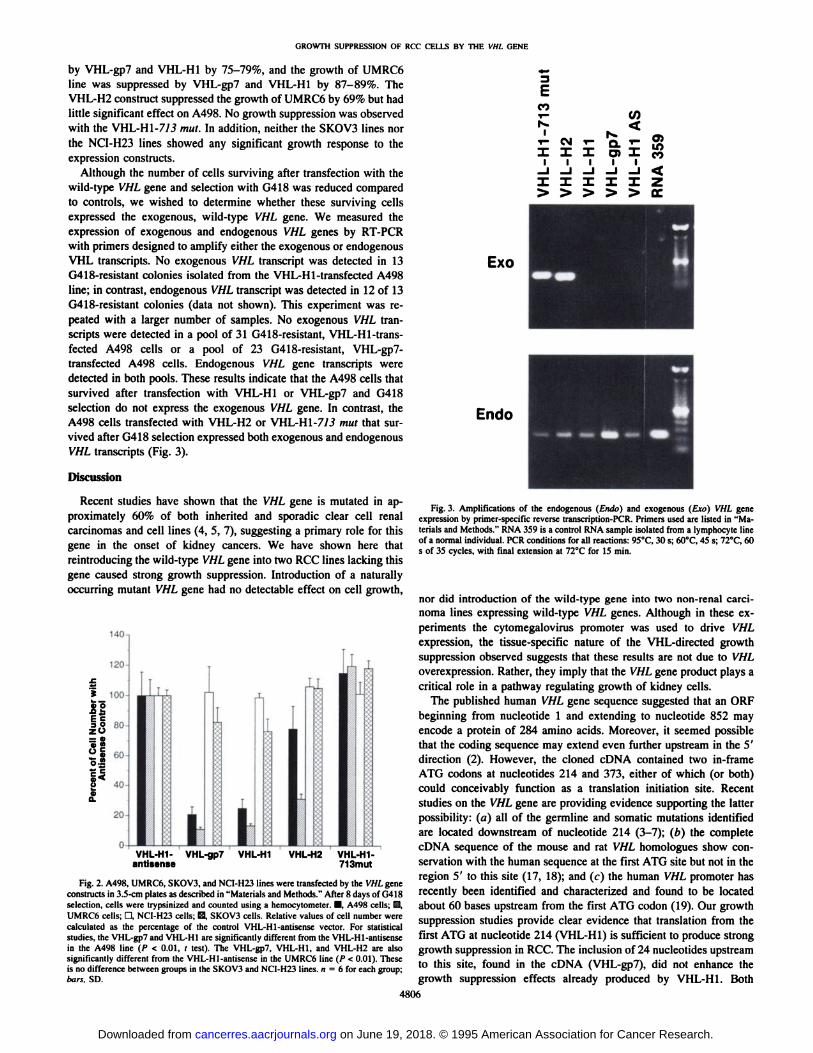
by VHL-gp7 and VHL-H1 by 75-79%, and the growth of UMRC6line was suppressed by VHL-gp7 and VHL-H1 by 87-89%. TheVHL-H2 construct suppressed the growth of UMRC6 by 69% but had
little significant effect on A498. No growth suppression was observedwith the VHL-H1-7/3 mut. In addition, neither the SKOV3 lines northe NCI-H23 lines showed any significant growth response to the
expression constructs.Although the number of cells surviving after transfection with the
wild-type VHL gene and selection with G418 was reduced compared
to controls, we wished to determine whether these surviving cellsexpressed the exogenous, wild-type VHL gene. We measured theexpression of exogenous and endogenous VHL genes by RT-PCR
with primers designed to amplify either the exogenous or endogenousVHL transcripts. No exogenous VHL transcript was detected in 13G418-resistant colonies isolated from the VHL-Hl-transfected A498
line; in contrast, endogenous VHL transcript was detected in 12 of 13G418-resistant colonies (data not shown). This experiment was re
peated with a larger number of samples. No exogenous VHL transcripts were detected in a pool of 31 G418-resistant, VHL-Hl-transfected A498 cells or a pool of 23 G418-resistant, VHL-gp7-
transfected A498 cells. Endogenous VHL gene transcripts weredetected in both pools. These results indicate that the A498 cells thatsurvived after transfection with VHL-H1 or VHL-gp7 and G418
selection do not express the exogenous VHL gene. In contrast, theA498 cells transfected with VHL-H2 or VHL-H1-7/3 mut that sur
vived after G418 selection expressed both exogenous and endogenousVHL transcripts (Fig. 3).
Discussion
Recent studies have shown that the VHL gene is mutated in approximately 60% of both inherited and sporadic clear cell renalcarcinomas and cell lines (4, 5, 7), suggesting a primary role for thisgene in the onset of kidney cancers. We have shown here thatreintroducing the wild-type VHL gene into two RCC lines lacking this
gene caused strong growth suppression. Introduction of a naturallyoccurring mutant VHL gene had no detectable effect on cell growth,
BO
T T I
VHL-H1- VHL-gp7 VHL-H1 VHL-H2 VHL-H1-antisense 713mut
Fig. 2. A498, UMRC6, SKOV3, and NCI-H23 lines were transfected by the VHL geneconstructs in 3.5-cm plates as described in "Materials and Methods." After 8 days of G418
selection, cells were trypsinized and counted using a hemocytometer. •¿�.A498 cells; &,UMRC6 cells; d, NCI-H23 cells; S, SKOV3 cells. Relative values of cell number werecalculated as the percentage of the control VHL-H1-antisense vector. For statisticalstudies, the VHL-gp7 and VHL-H1 arc significantly different from the VHL-Hl-antisensein the A498 line (/> < 0.01, I tesi). The VHL-gp7. VHL-H1. and VHL-H2 are alsosignificantly different from Ihe VHL-Hl-antisense in the UMRC6 line (P < 0.01). Theseis no difference between groups in the SKOV3 and NCI-H23 lines. «= 6 for each group;bars. SD.
Exo
Endo
Fig. 3. Amplifications of the endogenous (Endo) and exogenous (Ero) VHL geneexpression by primer-specific reverse Iranscription-PCR. Primers used are listed in "Materials and Methods." RNA 359 is a control RNA sample isolated from a lymphocyte lineof a normal individual. PCR conditions for all reactions: 95°C,30 s; 60°C,45 s; 72°C,60s of 35 cycles, with final extension at 72°Cfor 15 min.
nor did introduction of the wild-type gene into two non-renal carcinoma lines expressing wild-type VHL genes. Although in these ex
periments the cytomegalovirus promoter was used to drive VHLexpression, the tissue-specific nature of the VHL-directed growth
suppression observed suggests that these results are not due to VHLoverexpression. Rather, they imply that the VHL gene product plays acritical role in a pathway regulating growth of kidney cells.
The published human VHL gene sequence suggested that an ORFbeginning from nucleotide 1 and extending to nucleotide 852 mayencode a protein of 284 amino acids. Moreover, it seemed possiblethat the coding sequence may extend even further upstream in the 5'
direction (2). However, the cloned cDNA contained two in-frame
ATG codons at nucleotides 214 and 373, either of which (or both)could conceivably function as a translation initiation site. Recentstudies on the VHL gene are providing evidence supporting the latterpossibility: (a) all of the germline and somatic mutations identifiedare located downstream of nucleotide 214 (3-7); (h) the complete
cDNA sequence of the mouse and rat VHL homologues show conservation with the human sequence at the first ATG site but not in theregion 5' to this site (17, 18); and (c) the human VHL promoter has
recently been identified and characterized and found to be locatedabout 60 bases upstream from the first ATG codon (19). Our growthsuppression studies provide clear evidence that translation from thefirst ATG at nucleotide 214 (VHL-H1) is sufficient to produce strong
growth suppression in RCC. The inclusion of 24 nucleotides upstreamto this site, found in the cDNA (VHL-gp7), did not enhance thegrowth suppression effects already produced by VHL-H1. Both
4806
on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

GROWTH SUPPRESSION OF RCC CELLS BY THE VHl. GENE
constructs induced similar growth suppression in RCC lines. Webelieve that the results of this study, combined with those mentionedabove, strongly suggest that the full-length functional VHL gene
product begins at the first ATG at nucleotide 214 and extends to theTAA stop cudon at nucleotide 852. The biological significance of thetranslation product beginning with the second ATG is unclear. Thisconstruct (VHL-H2) induced a partial reduction in the growth of
UMRC6 and A498 (Fig. 2). The transcripts of this gene constructwere detectable in rapidly growing cells (Fig. 3).
The differential RT-PCR experiments analyzed the cells that sur
vived after transfection with different recombinant constructs andG418 selection. The A498 cells transfected with VHL-gp7 andVHL-H1 showed only the endogenous VHL transcripts. The A498cells transfected with the VHL-H2 or VHL-H1-7/.? mut constructs
showed both the endogenous and exogenous VHL messages. Thesedata suggest that wild-type VHL expression in these lines is incom
patible with rapid cell growth.A single nucleotide change (G to A) at nucleotide 713, resulting in
a single amino acid change (Arg to Gin), eliminated the growthsuppressive effect of the VHL gene. This naturally occurring mutationhas been identified as a hot spot mutation, present in many unrelatedVHL families, as well as sporadic RCC (3, 5, 6). Patients carrying thismutation in their germline are affected by pheochromocytoma andRCC, a VHL type 2 disease by previous classification (3). Thismutation has been shown recently to inhibit the binding by the VHLprotein of two low molecular weight cellular proteins ( 18).4 These two
in vitro assays for VHL protein function will be valuable in studyingthe biological and biochemical aspects of the VHL gene function.
The ability to suppress the growth of tumor cells is the mostimportant aspect in understanding the biological function of the VHLgene. Although the exact intracellular mechanism of this suppressionstill remains unclear, the loss of V7/Z.-directed growth suppression,
either by mutation or by transcriptional inactivation (20), seems to bea crucial event to the development of RCC. The demonstration of thegrowth suppressive effect of the VHL gene in cultured RCC cellsprovides direct evidence that the VHL gene plays an essential role inthe tumorigenesis of RCC.
Acknowledgments
We thank C. Riggs for his help in the statistical work. Dr. T. Stackhouse forVHL antibodies, and Dr. M. H. Wang for many helpful discussions in thecourse of this work.
References1. Mclmon, K. L, and Rosen. S. W. Lindau's disease: review of the literature and the
study of a large kindred. Am. J. Med., 36: 595-607, 1964.2. Latif, F., Tory, K., Gnarra, J., Yao, M., Duh, F-M., Orcutl, M. L., Stackhouse. T.,
Bkuzmin. I.. Modi. W., Geil. L.. Schmidt, L., Zhou, F., Li, H., Wei., M. H., Chen, F.,Glenn. G., Choke, P.. Walther, M. M., Weng, Y.. Duan, D-S. R.. Dean. M.. Glavac.D.. Richards. F. M., Crossey, P. A., Ferguson-Smith. M. A.. Paslier. D. E.. Chuma-
4 T. Kishida, unpublished data.
kov, 1., Cohen, D.. Chinault, A. C.. Mäher.E.. Linchan. W. M., Zhar. B., and Lerman.M. I. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science(Washington DC). 260: 1317-1320. 1993.
3. Chen, F., Kishida, T., Yao, M., Hustad, T., Glavac, D., Dean, M., Gnarra, J. G.,Orcutt, M. L., Duh, F. M. Glenn, G., Green, J., Hsia, E., Larmeli, J., Li, Maio. Wei,M. H., Schmidt. 1... Tory. K.. Kuzmin, I., Stackhouse. Y., Latif. F.. Lineham. M.,Lcrman. M. I., and Zhar, B. Germline mutations in the von Hippel-Lindau diseasetumor suppressor gene: correlation with phcnotype. Hum. Mutât.,5: 66-72, 1995.
4. Whaley, J. M., Gahlich, J., Galhert, L., Hsia, E., Lamiell, J. M., Green, J. S., Collins.D., Neumann, H. P. H., Laidlaw, J.. Li, F. P., Klein-Szanto, A. J. P., Seizinger, B. R.,and Kiev, N. Germ-line mutations in the von Hippel-Lindau tumor suppressor genearc similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma.Am. J. Hum. Genet., 55: 1092-1102. 1994.
5. Gnarra, J. R.. Tory, K., Weng, Y., Schmidt, L., Wei, M. H., Latif, F.. Liu, F., Chen,F., Duh. F-M., Lubensky, I., Duan, D. R.. Florence, C., Pozzatti. R., Wallher. M. M..
Bander, N. H., Grossman, H. B.. Brauch, H., Pomer. S., Brook, J. D., Isaacs, W. B.,Lerman, M. I.. Zbar, B., and Linchan. W. M. Mutations of the VHL tumor suppressorgene in renal carcinoma. Nat. Genet., 7: 85-90. 1994.
6. Crossey, P. A., Richards, F. M., Foster, K.. Green, J. S., Prowsc. A.. Latif, F.. Lerman,M. I., Zbar, B., Affara, N. A., Ferguson-Smith. A. F.. and Mäher.E. R. Identificationof intragenic mutation in the von Hippel-Lindau disease tumor suppressor gene andcorrelation with disease phenotype. Hum. Mol. Genet.. 3: 1303-130X, 1994.
7. Shuin, T., Kondo. K., Torigoe, S., Kishida, T., Kubota Y., Hosaka, M.. Nagashima,Y., Kitamura, H.. Latif, F.. Zbar. B.. Lerman, M. I., and Yao, M. Frequent somaticmutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor genein primary human renal cell carcinomas. Cancer Res.. 54: 2852-2855, 1994.
8. Knudson, A. G. Hereditary cancer, oncogenes. and antioncogenes. Cancer Res., 45:1437-1443, 1985.
9. Chen, P-L.. Chen, Y., Bookstein, R., and Lee. W-H. Genetic mechanism of tumorsuppression by the human p53 gene. Science (Washington DC), 250: 1576-1580,
1990.10. Huang, H-J. S., Yce, J-K., Shew, J-Y., Chen. P-L.. Bookstein. R., Friedmann, T., Lee,
E. Y-H. P., and Lee, W-H. Suppression of the neoplastic phenotype by replacement of theKB gene in human cancer cells. Science (Washington DC). 242: 1563-1566, 1988
11. Baker, S. J., Markowitz, S., Fearon, E. R., Willson. J.. and Vogelstein. B. Suppressionof human colorectal carcinoma cell growth by wild-type p53. Science (WashingtonDC), 250: 912-914. 1990.
12. Haber, D. A., Park, S., Maheswaran. S., Englert, C., Re, G. G., Hazen-Martin, D. L.,Sens. D. A., and Garvin, A. J. WTl-mediated growth suppression of Wilms tumorcells expressing a WT1 splicing variant. Science (Washington DC), 262: 2057-2059,
1993.13. Arap, W., Nishikawa, R., Furnari, F. B., Cavenee, W. K., and Huang, H-J. S.
Replacement of the plf>/CDKN2 gene suppresses human glioma cell growth. CancerRes., 55: 1351-1354, 1995.
14. Groden, J., Joslyn, G.. Samowilz, W., Jones, D., Bhaltacharyya. N.. and Spirio, L.Response of colon cancer cell lines to the introduction of APC, a colon-specific tumorsuppressor gene. Cancer Res., 55: 1531-1539, 1995.
15. Giard, D. J., Aaronson, S. A., Todaro, G. J., Arnstein, P., Kersey, J. H., Dosik. H., andParks, W. P. In \-iirn cultivation of human tumors: establishment of cell lines derivedfrom a series of solid tumors. J. Nail. Cancer. Inst., 51: 1417-1423. 1973.
16. Grossman, H. B., Wedemeyer. G., and Ren, L. Human renal cell carcinoma: characterization of five new cell lines. J. Surg. Oncol., 28: 237-244, 1985.
17. Gao, J.. Naglich, J. G., Laidlaw. J., Whaley, J. M., Seizinger, B. R., and Kley, N.Cloning and characterization of a mouse gene with homology to the human vonHippel-Lindau disease tumor suppressor gene: implication for the potential organization of the human von Hippel-Lindau disease gene. Cancer Res., 55: 743-747,
1995.18. Duan, D. R.. Humphrey, J. S., Chen, D. Y., Weng. Y., Sugegawa, J., Lee, S.. Gnarra,
J. G., Linehan, W. M.. and Klausner. R. D. Characterization of the VHL tumorsuppressor gene product: localization, complex formation, and the effect of naturalinactivating mutations. Prix:. Nati. Acad. Sci. USA, 91: 6459-6463, 1995.
19. Kuzmin, I.. Duh. F. M., Latif, F., Geil, L., Zbar, B., and Lerman, M. I. Identificationof the promotor of the human von Hippel-Lindau disease tumor suppressor gene.Oncogene, IO: 2185-2194, 1995.
20. Herman J. G.. Lalif. F.. Weng, Y.. Lerman. M. I., Zbar, B., Liu, S.. Samid. D., Duan,D-H. R.. Ganrra. J. R.. Linehan, W. M., and Baylin. S. T. Silencing of the VHLtumor-suppressor gene by DNA methylalion in renal carcinoma. Proc. Nail. Acad.Sci. USA, 91: 9700-9704, 1994.
4807
on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1995;55:4804-4807. Cancer Res Fan Chen, Takeshi Kishida, Fuh-Mei Duh, et al. Hippel-Lindau Tumor Suppressor GeneSuppression of Growth of Renal Carcinoma Cells by the von
Updated version
http://cancerres.aacrjournals.org/content/55/21/4804
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/55/21/4804To request permission to re-use all or part of this article, use this link
on June 19, 2018. © 1995 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from


















