
Supporting Information - Universiteit van Amsterdam
54
Supporting Information Mechanistic Insight into the Catalytic Promiscuity of Amine Dehydrogenases : Asymmetric Synthesis of Secondary and Primary Amines Vasilis Tseliou, Marcelo F. Masman, Wesley Bçhmer, Tanja Knaus, and Francesco G. Mutti* [a] cbic_201800626_sm_miscellaneous_information.pdf cbic_201800626_sm_Video.avi
Transcript of Supporting Information - Universiteit van Amsterdam

Supporting Information
Mechanistic Insight into the Catalytic Promiscuity ofAmine Dehydrogenases: Asymmetric Synthesis ofSecondary and Primary AminesVasilis Tseliou, Marcelo F. Masman, Wesley Bçhmer, Tanja Knaus, and Francesco G. Mutti*[a]
cbic_201800626_sm_miscellaneous_information.pdfcbic_201800626_sm_Video.avi

S1
Table of Contents
Abbreviations 3
List of substrates, amine donors and obtained products 3
General Information 4
Biocatalytic transformations 4
4.1. Screening of a panel of carbonyl compounds (ketones and aldehydes) with different amine donors for
the reductive amination catalysed by Ch1‐AmDH and Rs‐AmDH ....................................................................... 4
4.2. Reductive amination of 1 using different concentrations of catalyst .................................................... 8
4.3. Reductive amination of 1 at different concentrations of methylamine (b) ............................................ 9
Computational molecular modelling 10
5.1. Rhodococcus sp. amine dehydrogenase (Rs‐AmDH) ............................................................................ 10
5.2. Chimeric amine dehydrogenase (Ch1‐AmDH) ...................................................................................... 11
Experimental studies on the reaction mechanism 14
6.1. Disproving in‐situ generation of ammonia by enzymatic oxidative deamination of amine donor (1st
option in Scheme 2, main manuscript). ............................................................................................................ 14
6.2. Testing for possible elimination of cyclopropanol (2nd option in Scheme 2, main manuscript) ........... 15
6.3. Testing for possible NAD+/NADH redox‐mediated formal aminotransferase activity (3rd option in
Scheme 2, main manuscript) ............................................................................................................................ 16
6.4. Study of oxidative deamination on racemic N‐(1‐(4'‐fluorophenyl)propan‐2‐yl)cyclopropanamine (rac‐
1d) catalysed by Rs‐AmDH ............................................................................................................................... 18
Synthesis of secondary α‐chiral amines as reference compounds 20
7.1. Procedure for the protection of amines employing a tert‐butyloxycarbonyl group ............................. 20
Synthesis of tert‐butyl (R)‐N‐[1‐(4‐fluorophenyl)propan‐2‐yl]carbamate. ................................................... 20
Synthesis of tert‐butyl (R)‐hexan‐2‐ylcarbamate. ........................................................................................ 21
7.2. Procedure for the (m)ethylation of protected amines ......................................................................... 22
Synthesis of tert‐butyl (R)‐ethyl(1‐(4‐fluorophenyl)propan‐2‐yl)carbamate. .............................................. 22
Synthesis of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)(methyl)carbamate. ......................................... 23
Synthesis of tert‐butyl (R)‐hexan‐2‐yl(methyl)carbamate. .......................................................................... 24
7.3. Procedure for the deprotection of amines with a tert‐butyloxycarbonyl group .................................. 25
Synthesis of (R)‐methyl(1‐methyl‐2‐(4‐fluorophenyl)ethyl)amine. .............................................................. 25
Synthesis of (R)‐N‐[2‐(4‐fluorophenyl)‐1‐methylethyl]ethylamine. ............................................................. 26
Synthesis of (R)‐N‐methylhexan‐2‐amine. ................................................................................................... 27
7.4. Procedure for the reductive amination of 4’‐fluorophenylacetone to rac‐N‐(1‐(4‐fluorophenyl)
propan‐2‐yl) cyclopropanamine. ...................................................................................................................... 28
Analytical methods for the determination of conversions and of the absolute configurations 29
8.1. GC‐FID Method for the determination of the conversions ................................................................... 29
8.2. GC‐FID Method for the determination of the absolute configuration of primary amines ................... 29
8.3. GC‐MS Method for the identification of the products .......................................................................... 29
8.4. GC‐MS Method for the determination of the absolute configuration of secondary amines ................ 29

S2
Retention times 30
GC‐FID and GC‐MS chromatograms 31
References 53

S3
Abbreviations
AmDH amine dehydrogenase (variant)
Rs‐AmDH amine dehydrogenase variant originated from the L‐phenylalanine dehydrogenase from
Rhodoccoccus species.[1]
Ch1‐AmDH chimeric amine dehydrogenase generated through domain shuffling of Bb‐PhAmDH variant
and L‐AmDH variant.[1b, 2]
Cb‐FDH formate dehydrogenase from Candida boidinii
ee enantiomeric excess
ω‐TA ω‐transaminase
DCM dichloromethane
N.d. not detected
N.m. not measured
List of substrates, amine donors and obtained products
OF
6
O O
1
O
2 3
5 O
O
7
O
8
O11 O12
O
9
O
10
O4
O
13
Figure S1. List of prochiral ketones and aldehydes tested in this study.
NH2b NH2 NH2
NH2
NH2
NH2
NH2HN
HN
HN
c f
j
d e
ig h k
NH2
l
Figure S2. List of amine donors tested in this study

S4
NH2F
6b
1a
HN5eHN
HNF
1b HNF
1cHN
F
1d
NH2 HN
6cHN
6fHN
2a 2b
NH2 HN
3a 3b
6gN
6hN
HN4b
Figure S3. Products obtained in this study
General Information
All chemicals were purchased in highest grade available without further purification from Sigma Aldrich, Alfa
Aesar, Acros Organics, Fluka or TCI. Nicotinamide cofactor (NAD+) was purchased from Melford Biolaboratories
(Chelsworth, Ipswich, UK). The Ni2+ affinity columns (HisTrap FF, 5 mL) were purchased from GE Healthcare Bio‐
Sciences (Munich, Germany). Cyclopropanol was purchased by BOC sciences (USA)
Enantiopure (S) and (R)‐configured amines 1a‐3a were synthesized by stereoselective amination using
established enzymatic methods as reported in literature (employing commercially available
stereocomplementary ω‐transaminases ATA‐113, ATA‐117 from Codexis, Redwood City, California, US).[3]
Enantiopure secondary (S) and (R)‐configured amines 1b, 2b and racemic amine 1d were synthesized chemically
as described in section 7.
The amine dehydrogenases Rs‐AmDH and Ch1‐AmDH as well as Cb‐FDH were expressed and purified as
described previously.[1b]
Biocatalytic transformations
4.1. Screening of a panel of carbonyl compounds (ketones and aldehydes) with
different amine donors for the reductive amination catalysed by Ch1‐AmDH
and Rs‐AmDH
To identify the products, in the initial set of experiments, all performed biocatalytic reactions were analysed by
GC‐MS. In the cases that the product could be identified, conversions were determined by GC‐FID and further
validated with authentic reference compounds. A summary of the screening can be found in Table S1. A detailed
list of GC‐traces obtained by GC‐MS and GC‐FID measurements of the samples after reductive amination are
shown in section 10.

S5
Table S1. GC‐MS analysis of the biocatalytic reductive amination reactions performed in this study.
Substrate Amine donor AmDH Secondary amine formation [%]
“Promiscuous “ Primary amine formation [%]
1
Methylamine (b)
Ch1‐AmDH 19 30
Rs‐AmDH 2 N.d.
Ethylamine (c)
Ch1‐AmDH 1 7
Rs‐AmDH 8 35
Cyclopropylamine (d)
Ch1‐AmDH 4 17
Rs‐AmDH 36 14
Propargylamine (e)
Ch1‐AmDH N.d. 44
Rs‐AmDH N.d. 66
N‐propylamine (f)
Ch1‐AmDH 3 N.d.
Rs‐AmDH N.d. N.d.
Dimethylamine (g)
Ch1‐AmDH N.d. N.d.
Rs‐AmDH N.d. N.d
Ethyl‐methylamine (h)
Ch1‐AmDH N.d. N.d
Rs‐AmDH N.d. N.d
Allylamine (i)
Ch1‐AmDH N.d. 11
Rs‐AmDH N.d. 14
Iso‐propylamine (j)
Ch1‐AmDH N.d. N.d.
Rs‐AmDH <1 4
Pyrrolidine (k)
Ch1‐AmDH N.d. N.d.
Rs‐AmDH N.d. N.d.
Aniline (l) Ch1‐AmDH N.d. N.d.
Rs‐AmDH N.d. N.d.
2
Methylamine (b)
Rs‐AmDH N.d. <1
Ch1‐AmDH 6 13
Ethylamine (c)
Rs‐AmDH N.d. 5
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Propargylamine (e)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. 13
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Dimethylamine (g)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethyl‐methylamine (h)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. <1
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Pyrrolidine (k)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
3
Methylamine (b)
Rs‐AmDH N.d. 6
Ch1‐AmDH 18 10
Ethylamine (c)
Rs‐AmDH N.d. 3
Ch1‐AmDH N.d. 9
Cyclopropylamine (d)
Rs‐AmDH N.d. 11
Ch1‐AmDH N.d. 3
Propargylamine (e)
Rs‐AmDH N.d. 13
Ch1‐AmDH N.d. 26
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. 4

S6
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
4
Methylamine (b)
Rs‐AmDH 24 N.d.
Ch1‐AmDH N.d. N.d.
Ethylamine (c)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d) Rs‐AmDH N.d. N.d.
Propargylamine (e) Rs‐AmDH N.d. N.d.
N‐propylamine (f)
Rs‐AmDH 3 N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i) Rs‐AmDH 8 N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
5
Methylamine (b)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethylamine (c)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH <1 N.d.
Ch1‐AmDH 2 N.d.
Propargylamine (e)
Rs‐AmDH 4 N.d.
Ch1‐AmDH 12 N.d.
N‐propylamine (f)
Rs‐AmDH 7 N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
6
Methylamine (b)
Rs‐AmDH 36 N.d.
Ch1‐AmDH 14 N.d.
Ethylamine (c)
Rs‐AmDH 22 N.d.
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Propargylamine (e)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
N‐propylamine (f)
Rs‐AmDH 14 N.d.
Ch1‐AmDH N.d. N.d.
Dimethylamine (g)
Rs‐AmDH 3 N.d.
Ch1‐AmDH 2 N.d.
Ethyl‐methylamine (h)
Rs‐AmDH 8 N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Pyrrolidine (k)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
7
Methylamine (b)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH 2 N.d.
Ethylamine (c)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.

S7
Cyclopropylamine (d)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Propargylamine (e)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
8
Methylamine (b)
Rs‐AmDH 9 4
Ch1‐AmDH N.d. 2
Ethylamine (c)
Rs‐AmDH N.d. <1
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH 10 N.d.
Ch1‐AmDH N.d. N.d.
Propargylamine (e)
Rs‐AmDH N.d. <1
Ch1‐AmDH N.d. <1
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Dimethylamine (g)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethyl‐methylamine (h)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Pyrrolidine (k)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
9
Methylamine (b) Rs‐AmDH 2 N.d.
Ethylamine (c) Rs‐AmDH N.d. <1
Cyclopropylamine (d) Rs‐AmDH 27 N.d.
Propargylamine (e) Rs‐AmDH 34 N.d.
N‐propylamine (f) Rs‐AmDH N.d. N.d.
Allylamine (i) Rs‐AmDH 9 N.d.
Iso‐propylamine (j) Rs‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
10
Methylamine (b) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethylamine (c) Rs‐AmDH N.d. N.d.
Cyclopropylamine (d) Rs‐AmDH N.d. N.d.
Propargylamine (e) Rs‐AmDH N.d. N.d.
N‐propylamine (f) Rs‐AmDH N.d. N.d.
Allylamine (i) Rs‐AmDH N.d. N.d.
Iso‐propylamine (j) Rs‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
11
Methylamine (b)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethylamine (c)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.

S8
Propargylamine (e)
Rs‐AmDH <1 N.d.
Ch1‐AmDH <1 N.d.
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
12
Methylamine (b)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Dimethylamine (g)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethyl‐methylamine (h)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Pyrrolidine (k)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
13
Methylamine (b)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethylamine (c)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Cyclopropylamine (d)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Propargylamine (e)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
N‐propylamine (f)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Dimethylamine (g)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Ethyl‐methylamine (h)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Allylamine (i)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Iso‐propylamine (j)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Pyrrolidine (k)
Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d.
Aniline (l) Rs‐AmDH N.d. N.d.
Ch1‐AmDH N.d. N.d. Reaction conditions: 0.5 mL reaction volume; substrate 10 mM; Rs‐AmDH 102.8 μΜ or Ch1‐AmDH 91.8 μM; Cb‐FDH 23.5 μΜ; NAD+ 1mM,
48 h, 170 rpm.
N.d.: not detected (the expected product, under assay conditions)
4.2. Reductive amination of 1 using different concentrations of catalyst
We investigated the influence of the enzyme loading into the reaction mixture for the reductive amination of 1
with b, catalysed by Ch1‐AmDH. In all reactions, 1 (10 mM) was used in presence of catalytic amount of NAD+ (1
mM) and Cb‐FDH (23.5 μΜ) for recycling of the nicotinamide cofactor. Reactions were run at 30 oC for 48 h and
the work up was performed as described in the main paper. In these experiments, the concentration of the
amine donor (b) was fixed to 1 M, whereas the final concentration of Ch1‐AmDH was varied from 4.5 μΜ to 91.8
μΜ. The results are summarized in Table S2 and Figure 1A (main manuscript).

S9
Table S2. Reductive amination of 1 with methylamine b (1M, pH 8.5) using different final concentrations of Ch1‐AmDH.
Conversions depicted here are the average value obtained from three independent experiments with standard deviation.
4.3. Reductive amination of 1 at different concentrations of methylamine (b)
The reductive amination of substrate 1 with b as amine donor catalysed by Ch1‐AmDH was further investigated
by varying the methylamine concentration. In particular, the reductive amination was carried out at the final
concentrations of: 0.2 M; 0.4 M; 0.6 M; 0.8 M; 1 M; 2 M; 3 M; 4 M; 5M and 6M of amine donor b as buffer
system while keeping the pH value constant at 8.5 (adjusted with formic acid) and the enzyme loading at 91.8
μΜ. In all reactions, 10 mM of substrate 1 was used as well as catalytic amount of NAD+ (1 mM). Cb‐FDH (23.5
μΜ) was also added for recycling of the nicotinamide cofactor. Reactions were run at 30 oC for 48 h and the work
up was performed as described in the main paper. Conversions as well as ee values are reported in Table S3 and
Fig. 1B (main manuscript).
Table S3. Reductive amination of 1 using Ch1‐AmDH at varied concentrations of methylamine buffer (b, pH: 8.5)
Conversions depicted here is the average value obtained from three independent experiments with standard deviation.
Enzyme [µM] Secondary Amine [%] Primary Amine [%]
4.5 0.5 ± 0.29 0.4 ± 0.33
9 0.8 ± 0.34 1.0 ± 0.49
22.9 2.4 ± 0.62 2.7 ± 0.47
45.5 10.1 ± 1.42 7.1 ± 0.63
91.8 14.7 ± 0.65 7.9 ± 0.42
Methylamine [M][M] 1b 1a
Conv. [%]a ee [%] Conv. [%]a ee [%]
0.2 3.1 ± 0.68 N.m. 1.1 ± 0.03 N.m.
0.4 6.4 ± 1.11 N.m. 2.9 ± 0.23 N.m.
0.6 10.0 ± 0.82 N.m. 4.9 ± 0.32 N.m.
0.8 13.3 ± 1.04 N.m. 6.9 ± 0.57 N.m.
1 14.7 ± 0.65 71.7 ± 1.05 (R) 7.9 ± 0.42 >99 % (R)
2 19.1 ± 0.53 71.3 ± 0.51 (R) 10.3 ± 0.13 >99 % (R)
3 25.4 ± 0.58 69.7 ± 1.91 (R) 11.1 ± 0.24 >99 % (R)
4 33.4 ± 0.26 67.6 ± 0.59 (R) 10.6 ± 0.02 >99 % (R)
5 38.7 ± 1.15 64.8 ± 0.70 (R) 8.3 ± 0.19 >99 % (R)
6 39.4 ± 0.44 63.9 ± 0.49 (R) 6.1 ± 0.16 >99 % (R)

S10
Computational molecular modelling
5.1. Rhodococcus sp. amine dehydrogenase (Rs‐AmDH)
Figure S4. Enzyme‐substrate calculated binding poses in the active site of Rs‐AmDH for the imine intermediate formed during
the reaction of: A) 4’‐fluoro‐phenylacetone (1) with ammonia (a); B) and C) 4’‐fluoro‐phenylacetone (1) with ethylamine (c)
in two different binding poses. Similarly, D) acetophenone (10) with ammonia (a); E) and F) acetophenone (10) with
ethylamine (c) in two different binding poses. G) 4‐phenyl‐2‐butanone (9) with ammonia (a); H) and I) 4‐phenyl‐2‐butanone
(9) with ethylamine (c) in two different binding poses.
A B C
D E F
G H I

S11
Figure S5. The best enzyme‐substrate calculated binding poses in the active site of Rs‐AmDH for the imine intermediate
formed during the reaction of: A) hexanal (4) with methylamine (b), B) heptanal (5) with methylamine (b), C)
phenylacetaldehyde (6) with methylamine (b), D) phenylacetaldehyde (6) with ethylamine (c) and E) phenylacetaldehyde (6)
with n‐propylamine (f).
5.2. Chimeric amine dehydrogenase (Ch1‐AmDH)
Table S4: Homology model generation of Ch1‐AmDH. The full amino acid sequence was explored. The multimeric state, the
selected templates, the excluded templates, and the number of models generated per run are shown. The template that was
selected as main contributor to the hybrid model is demarked in bold font. Moreover, the accuracy of the generated models
is reported by the use of Z‐scores.[4] The overall Z‐scores for all models have been calculated as the weighted averages of the
individual Z‐scores (Dihedrals, Packing 1D, and Packing 3D) using the formula Overall = 0.145*Dihedrals + 0.390*Packing1D
+ 0.465*Packing3D.
run Multimeric
state Templates Cofactor Ligand
Number of
models
Overall Z‐score
(Quality)a
Overall Z‐score
(Quality)b
exploratory monomer 1LEH[5], 3VPX[6], 1C1X[7], 1C1D [7], 1BXG [8]
none none 23 ‐0.290 (Good)
ND
production monomer 1C1D [7] NADH L‐Phe 10 ‐0.520 (Good)
‐0.520 (Good)
ND: Not determined a. Z‐score and quality of the model after hybrid model generation. b. Best Z‐score and quality of the model after molecular dynamic refinement.
A B C
D E

S12
Figure S6: Interatomic distance between the hydride atom of NADH and the pro‐chiral carbon atom of the imine
intermediates vs. simulation time. The measured distance vs. time per each simulation run is shown. The average value is
shown with black traces within a grey area that indicates the standard deviation amongst simulation runs. The simulation
run 5 (denoted with an asterisk) of substrate 1a* was not considered for the average calculation due to fact that the substrate
moved away from the active site. The threshold distance is indicated with a dotted black line at 3.0 Å.

S13
Figure S7: Dihedral angle χ versus time of the simulation. The measured angle vs. time per each simulation run is shown. The
average value is shown with black traces within a grey area that indicates the standard deviation amongst simulation runs.
The simulation run 5 of substrate 1a* was not considered for the average calculation due to fact that the substrate moved
away from the active site. The average values of dihedral angle for the formation of R‐ and S‐products are also shown as
dashed lines.

S14
Experimental studies on the reaction mechanism
6.1. Disproving in‐situ generation of ammonia by enzymatic oxidative deamination
of amine donor (1st option in Scheme 2, main manuscript).
To demonstrate that free ammonia is not generated as a result of the oxidative deamination of the amine donor
(e.g. cyclopropylamine, d) catalysed by Rs‐AmDH, we incubated cyclopropylamine (50 mM) in the presence of
Rs‐AmDH (102.8 μΜ) and stoichiometric NAD+ (60 mM). The reactions were performed in duplicates for 1 h, 2 h
and 7 h. After the reported times, reactions were quenched with KOH and extracted with ethyl acetate
supplemented with toluene (10 mM) as internal standard. For each set of reactions the average ratio between
the GC areas of cyclopropylamine and toluene was calculated. A blank experiment was also performed in
triplicate by incubating cyclopropylamine (50 mM) in the reaction buffer (phosphate buffer pH 8.5). After basic
extraction with ethyl‐acetate containing the internal standard (toluene, 10 mM), the ratios between
cyclopropylamine and toluene areas (d/t) were calculated. Comparison among the values (d/t) obtained from
the reactions performed with those obtained from the blank reactions revealed that cyclopropylamine was not
consumed by the enzyme over time. Therefore, a possible scenario in which free ammonia is generated by the
oxidative deamination of cyclopropylamine by RsAmDH and subsequently used by the enzyme to produce the
primary amine can be excluded.
NH2 RsAmDH
KPi pH 8.5+ NAD+ O
+ NADH + NH3
Reaction time: 1h, 2h, 7h at 30oC
Internal stadard (10 mM) used with extraction solvent (ethyl acetate):
d
Reaction:
Blank:NH2
d
KPi pH 8.5+ , extraction with ethyl acetate contanining
t
as internal stadard (10 mM):
t
Table S5. Analytical quantification of the consumption of cyclopropylamine over time
Replicate Compound GC Area GC Area
Blank Reaction
1 h 2 h 7 h
1st
d 31.6 24.3 27.6 31.9
t 68.4 75.7 72.4 68.1
d/t 0.462 0.321 0.381 0.469
2nd
d 28.2 26.8 29.6 32.9
t 71.8 73.2 70.4 67.2
d/t 0.393 0.366 0.420 0.488
3nd
d 22.2
t 77.8
d/t 0.285
Average d/t 0.380± 0.089 0.343 ± 0.023 0.401 ± 0.019 0.478 ± 0.010

S15
6.2. Testing for possible elimination of cyclopropanol (2nd option in Scheme 2, main
manuscript)
We investigated a possible elimination of cyclopropanol as a result of a nucleophilic attack of a water molecule
on the iminium intermediate 1d*, which is generated during the reductive amination catalysed by Rs‐AmDH
using substrate 1 with amino donor d. Hence, the reference compound (cyclopropanol, 50 mM) was injected
into GC‐MS to have reference GC trace and MS. Incubation of cyclopropanol in the reaction buffer (30oC, 48 h,
170 rpm) followed by extraction in MTBE (1 x 600 μL) revealed that cyclopropanol is stable under the reaction
conditions. Extraction of the reaction performed with RsAmDH (102.8 μM) in cylopropylamine buffer (d, 1M, pH
8.5) supplemented with NAD+ (1 mM) using Cb‐FDH (23.5 μΜ) as recycling system did not result in any
detectable amount of cyclopropanol in the reaction mixture.
GCMS chromatograph of MTBE
GCMS chromatograph of cyclopropanol in MTBE (analytical reference)
GCMS chromatograph after incubation of cyclopropanol in buffer and extraction in MTBE (control experiment)
GCMS chromatograph of the reaction catalysed by RsAmDH with 1 and d
1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
(x10,000,000)TIC
1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
(x10,000,000)TIC
1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
(x10,000,000)TIC
1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
(x10,000,000)TIC
Cyclopropanol
Cyclopropanol

S16
Overlay of the cyclopropanol (Black) and reaction with RsAmDH (mageda) chromatographs
Mass spectrum of cyclopropanol
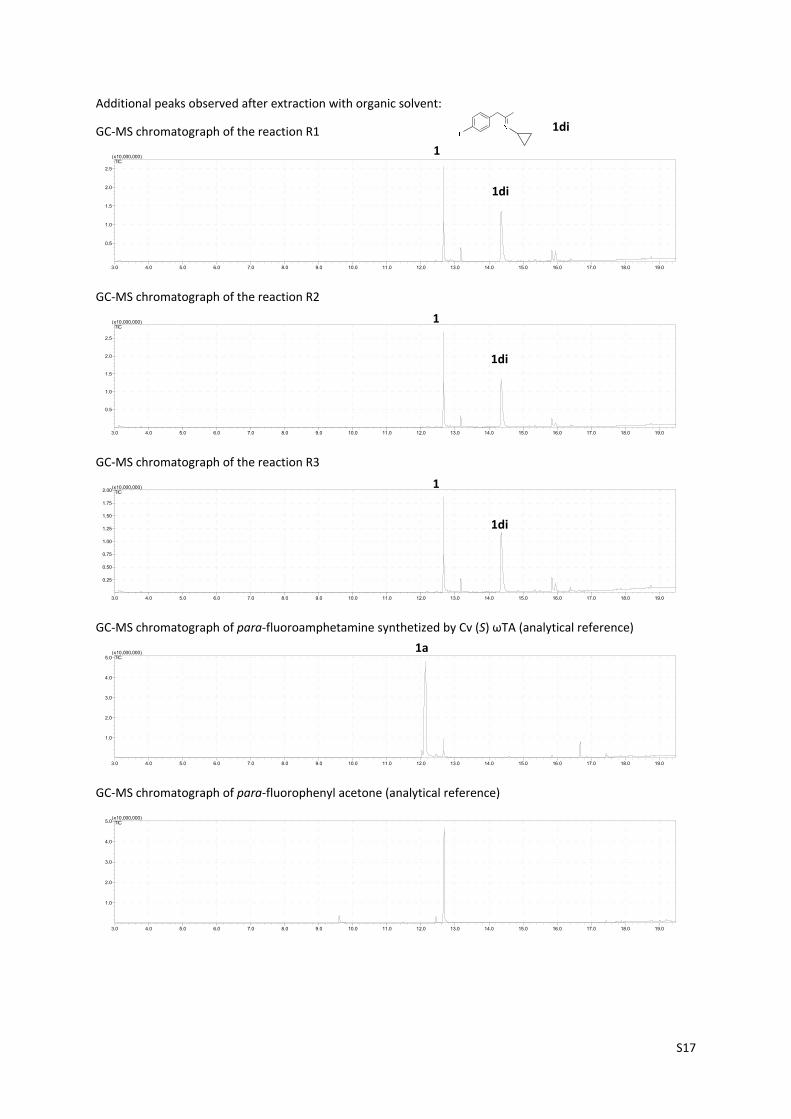
6.3. Testing for possible NAD+/NADH redox‐mediated formal aminotransferase
activity (3rd option in Scheme 2, main manuscript)
In order to rule out any possible “classical” transaminase activity, we incubated 4'‐fluorophenylacetone 1 (10
mM) in cyclopropylamine buffer (d, 1 M pH 8.5) with Rs‐AmDH in the presence and absence of PLP (0.5 mM) for
48 h at 30 oC (See below R1‐R3). The reaction with PLP was also performed without the AmDH in order to assess
spontaneous chemical transamination. As expected no primary amine product was obtained in any of the
reactions performed. The identical spectra in all cases, showed only the starting compound present in the
reaction mixture after 48 h as well as the secondary imine adduct that is spontaneously formed during extraction
with organic solvent.
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
1.0
2.0
3.0
4.0
5.0
6.0
7.0
8.0
(x10,000,000)
5.0 10.0 15.0 20.0 25.0 30.0 35.0 40.0 45.0 50.0 55.0 60.00
10
20
30
40
50
%
5729
27
39
18
41
15 55
2 368 12 24 45 52 60
Cyclopropanol
1a1
1d
1di
1 1a d
S17
Additional peaks observed after extraction with organic solvent:
GC‐MS chromatograph of the reaction R1
GC‐MS chromatograph of the reaction R2
GC‐MS chromatograph of the reaction R3
GC‐MS chromatograph of para‐fluoroamphetamine synthetized by Cv (S) ωΤΑ (analytical reference)
GC‐MS chromatograph of para‐fluorophenyl acetone (analytical reference)
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.5
1.0
1.5
2.0
2.5
(x10,000,000)TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.5
1.0
1.5
2.0
2.5
(x10,000,000)TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00(x10,000,000)
TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
1.0
2.0
3.0
4.0
5.0(x10,000,000)
TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
1.0
2.0
3.0
4.0
5.0(x10,000,000)
TIC
1di
1di
1
1di
1
1di
1
1a
S18
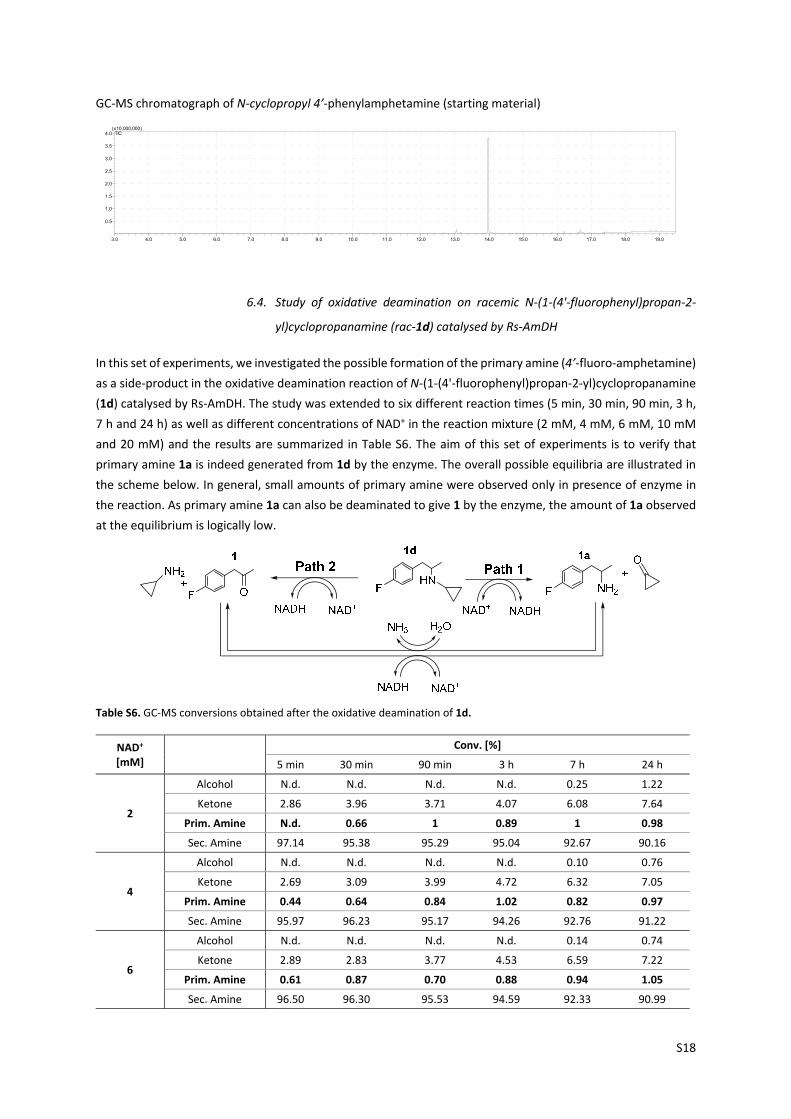
GC‐MS chromatograph of N‐cyclopropyl 4’‐phenylamphetamine (starting material)
6.4. Study of oxidative deamination on racemic N‐(1‐(4'‐fluorophenyl)propan‐2‐
yl)cyclopropanamine (rac‐1d) catalysed by Rs‐AmDH
In this set of experiments, we investigated the possible formation of the primary amine (4’‐fluoro‐amphetamine)
as a side‐product in the oxidative deamination reaction of N‐(1‐(4'‐fluorophenyl)propan‐2‐yl)cyclopropanamine
(1d) catalysed by Rs‐AmDH. The study was extended to six different reaction times (5 min, 30 min, 90 min, 3 h,
7 h and 24 h) as well as different concentrations of NAD+ in the reaction mixture (2 mM, 4 mM, 6 mM, 10 mM
and 20 mM) and the results are summarized in Table S6. The aim of this set of experiments is to verify that
primary amine 1a is indeed generated from 1d by the enzyme. The overall possible equilibria are illustrated in
the scheme below. In general, small amounts of primary amine were observed only in presence of enzyme in
the reaction. As primary amine 1a can also be deaminated to give 1 by the enzyme, the amount of 1a observed
at the equilibrium is logically low.
Table S6. GC‐MS conversions obtained after the oxidative deamination of 1d.
NAD+ [mM]
Conv. [%]
5 min 30 min 90 min 3 h 7 h 24 h
2
Alcohol N.d. N.d. N.d. N.d. 0.25 1.22
Ketone 2.86 3.96 3.71 4.07 6.08 7.64
Prim. Amine N.d. 0.66 1 0.89 1 0.98
Sec. Amine 97.14 95.38 95.29 95.04 92.67 90.16
4
Alcohol N.d. N.d. N.d. N.d. 0.10 0.76
Ketone 2.69 3.09 3.99 4.72 6.32 7.05
Prim. Amine 0.44 0.64 0.84 1.02 0.82 0.97
Sec. Amine 95.97 96.23 95.17 94.26 92.76 91.22
6
Alcohol N.d. N.d. N.d. N.d. 0.14 0.74
Ketone 2.89 2.83 3.77 4.53 6.59 7.22
Prim. Amine 0.61 0.87 0.70 0.88 0.94 1.05
Sec. Amine 96.50 96.30 95.53 94.59 92.33 90.99
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0 18.0 19.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0(x10,000,000)
TIC

S19
10
Alcohol N.d. N.d. N.d. N.d. N.d. 0.24
Ketone 2.75 3.21 3.64 4.34 5.77 6.29
Prim. Amine 0.52 1.09 0.98 0.86 0.75 1.12
Sec. Amine 96.73 95.70 95.38 94.80 93.48 92.35
20
Alcohol N.d. N.d. N.d. N.d. N.d. 0.24
Ketone 2.88 3.03 2.96 4.30 5.43 5.50
Prim. Amine 0.82 1.16 1.11 1.08 0.84 1.04
Sec. Amine 96.30 95.81 95.93 94.62 93.73 93.22
We have also verified the possible enantiodiscrimination of Rs‐AmDH in an experiment of kinetic resolution on
rac‐1d. For the efficient regeneration of the catalytic NAD+ coenzyme, the H2O forming NAD‐oxidase from
Streptococcus mutans (NOx) was applied. The enantiomeric excess of the remaining starting material 1d was
monitored during the time. Results are summarised in Table S7.
Notably, in this case, amine 1a could not be observed because 1a is rapidly and irreversibly deaminated to 1
under the applied reaction conditions. However, an enantioenriched mixture of 1d starting material was
generated indicating that the enzyme can differentiate between the two enantiomers of 1d.
Table S7. Determination of conversion into 1 and of the ee of the remaining 1d after different reaction times
Experimental conditions: 0.5 mL final volume in Eppendorf tubes, buffer strength: 50 mM Kpi, pH 8.5, T: 30 °C, 170 rpm on orbital shaker,
[1d]: 20 mM, [NAD+]: 1 mM, [NOx] = 5 µM, [RsAmDH] = 102.8 μM; n.m. = not measured
Time Conversion Enantiomeric ratio (er) ee
into 1 [%] remaining 1d [%] [%]
0h n.m. 50:50 0
1h n.m. 48:52 4
2h n.m. 46:54 8
48h, 1st sample 25 43:57 14
48h, 2nd sample 30 42:58 16

S20
Synthesis of secondary α‐chiral amines as reference compounds
7.1. Procedure for the protection of amines employing a tert‐butyloxycarbonyl
group
Synthesis of tert‐butyl (R)‐N‐[1‐(4‐fluorophenyl)propan‐2‐yl]carbamate.
To a solution of (R)‐1‐(4‐fluorophenyl)‐2‐aminopropane (206.6 mg, 1.35 mmol) in dry dichloromethane (9 mL,
previously dried on 3 Å molecular sieves), triethylamine (200 µL, 1.43 mmol) and di‐tert‐butyl dicarbonate (322.6
mg, 1.48 mmol) were added at 0 °C (ice bath) under N2. The mixture was stirred for 2 h and then quenched with
water. The water layer was extracted with dichloromethane (3 times). The combined organic phase was dried
with magnesium sulphate. Evaporation of the solvent yielded a yellow oil that was purified by column
chromatography (material: silica 60; column internal diameter: 5 cm; high of silica bed: 30 mm; eluent; 85:15, v
v‐1, EtOAc : petroleum ether). The product crystallized out of solution as a white solid (321.3 mg, 1.27 mmol, 94
%). Rf = 0.59 in EtOAc : petroleum ether (85:15).
Figure S8. 1H‐NMR of tert‐butyl (R)‐N‐[1‐(4‐fluorophenyl)propan‐2‐yl]carbamate (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer tert‐butyl (S)‐N‐[1‐(4‐
fluorophenyl)propan‐2‐yl]carbamate. Analogous NMR was obtained as for the R‐enantiomer.

S21
Synthesis of tert‐butyl (R)‐hexan‐2‐ylcarbamate.
To a solution of (R)‐aminohexane (0.7 mL, 5.22 mmol) in dry dichloromethane (35 mL, previously dried on 3 Å
molecular sieves), triethylamine (770 µL, 5.52 mmol) and di‐tert‐butyl dicarbonate (1.2 g, 5.50 mmol) were
added at 0 °C (ice bath) under N2. The mixture was stirred for 2 h and then quenched with water. The water
layer was extracted with dichloromethane (3 times). The combined organic phase was dried with magnesium
sulphate. Evaporation of the solvent yielded a colorless liquid that was purified by column chromatography
(material: silica 60; column internal diameter: 5 cm; high of silica bed: 30 mm; eluent; 1:1, v v‐1, dichloromethane
: petroleum ether). The product obtained as a colorless oil (1.00 g, 4.96 mmol, 95 %). Rf = 0.20 in EtOAc :
petroleum ether (1:1).
Figure S9. 1H‐NMR of tert‐butyl (R)‐hexan‐2‐ylcarbamate (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer tert‐butyl (S)‐hexan‐2‐ylcarbamate.
Analogous NMR was obtained as for the R‐enantiomer.
S22
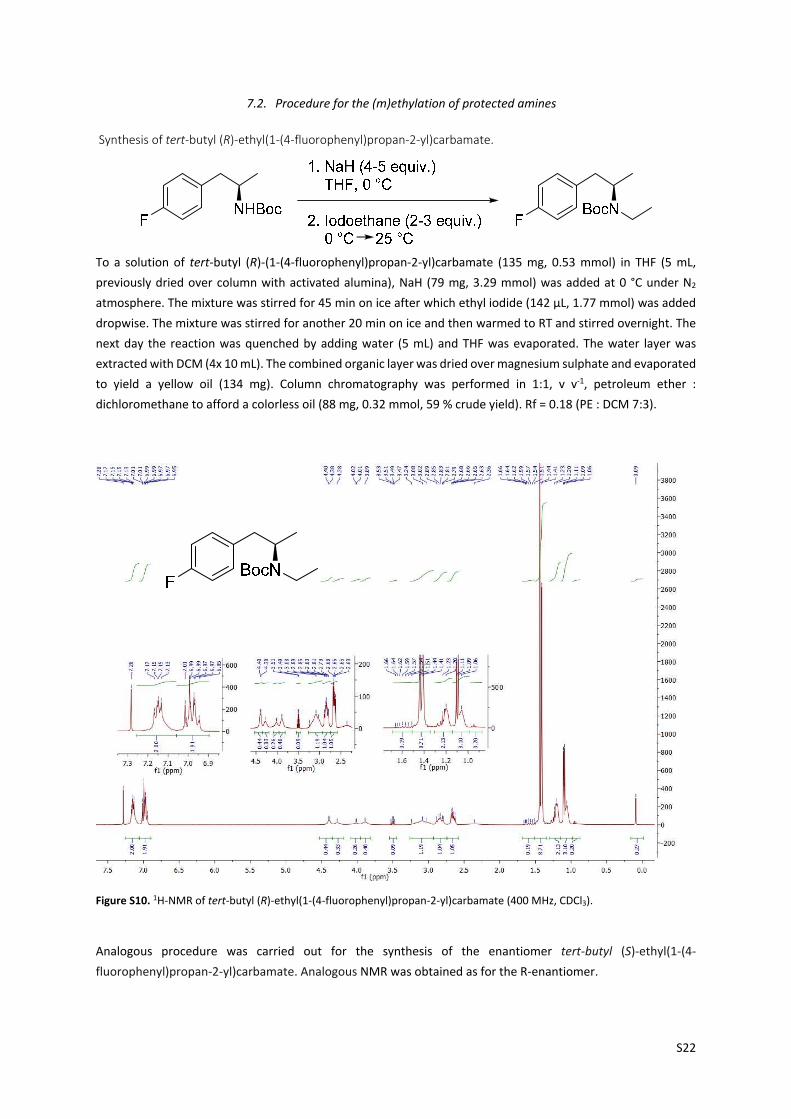
7.2. Procedure for the (m)ethylation of protected amines
Synthesis of tert‐butyl (R)‐ethyl(1‐(4‐fluorophenyl)propan‐2‐yl)carbamate.
To a solution of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)carbamate (135 mg, 0.53 mmol) in THF (5 mL,
previously dried over column with activated alumina), NaH (79 mg, 3.29 mmol) was added at 0 °C under N2
atmosphere. The mixture was stirred for 45 min on ice after which ethyl iodide (142 µL, 1.77 mmol) was added
dropwise. The mixture was stirred for another 20 min on ice and then warmed to RT and stirred overnight. The
next day the reaction was quenched by adding water (5 mL) and THF was evaporated. The water layer was
extracted with DCM (4x 10 mL). The combined organic layer was dried over magnesium sulphate and evaporated
to yield a yellow oil (134 mg). Column chromatography was performed in 1:1, v v‐1, petroleum ether :
dichloromethane to afford a colorless oil (88 mg, 0.32 mmol, 59 % crude yield). Rf = 0.18 (PE : DCM 7:3).
Figure S10. 1H‐NMR of tert‐butyl (R)‐ethyl(1‐(4‐fluorophenyl)propan‐2‐yl)carbamate (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer tert‐butyl (S)‐ethyl(1‐(4‐
fluorophenyl)propan‐2‐yl)carbamate. Analogous NMR was obtained as for the R‐enantiomer.
S23
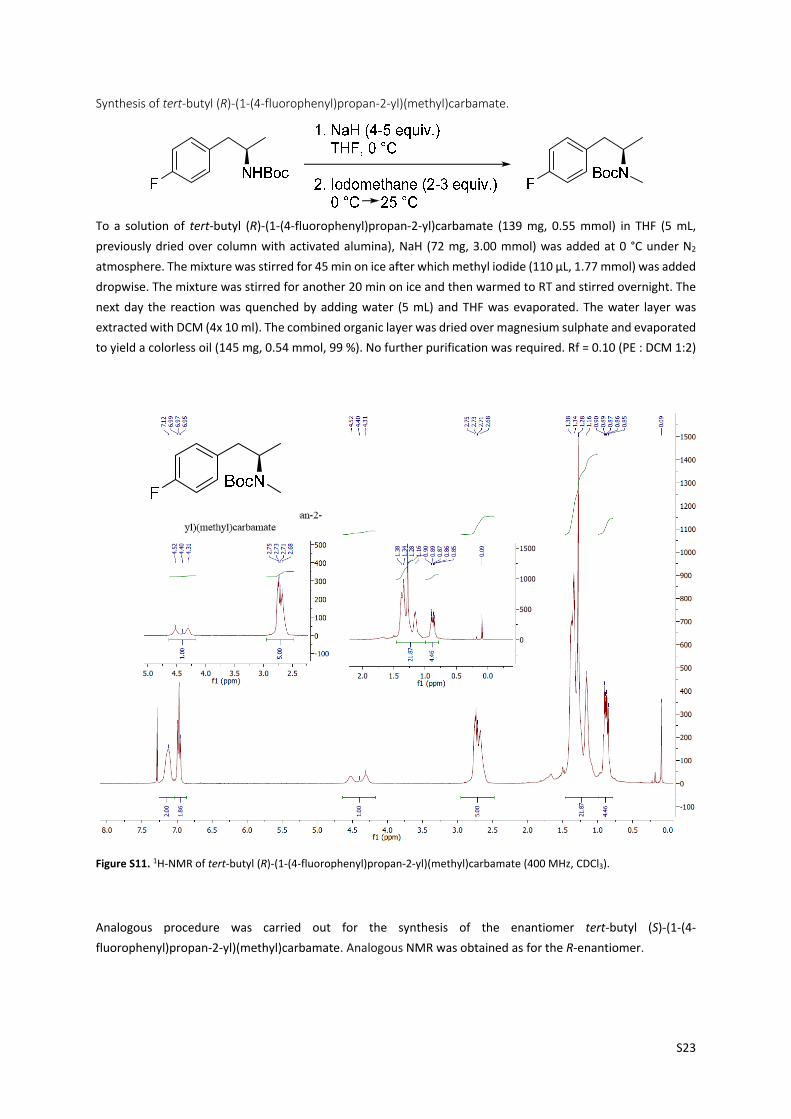
Synthesis of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)(methyl)carbamate.
To a solution of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)carbamate (139 mg, 0.55 mmol) in THF (5 mL,
previously dried over column with activated alumina), NaH (72 mg, 3.00 mmol) was added at 0 °C under N2
atmosphere. The mixture was stirred for 45 min on ice after which methyl iodide (110 µL, 1.77 mmol) was added
dropwise. The mixture was stirred for another 20 min on ice and then warmed to RT and stirred overnight. The
next day the reaction was quenched by adding water (5 mL) and THF was evaporated. The water layer was
extracted with DCM (4x 10 ml). The combined organic layer was dried over magnesium sulphate and evaporated
to yield a colorless oil (145 mg, 0.54 mmol, 99 %). No further purification was required. Rf = 0.10 (PE : DCM 1:2)
Figure S11. 1H‐NMR of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)(methyl)carbamate (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer tert‐butyl (S)‐(1‐(4‐
fluorophenyl)propan‐2‐yl)(methyl)carbamate. Analogous NMR was obtained as for the R‐enantiomer.
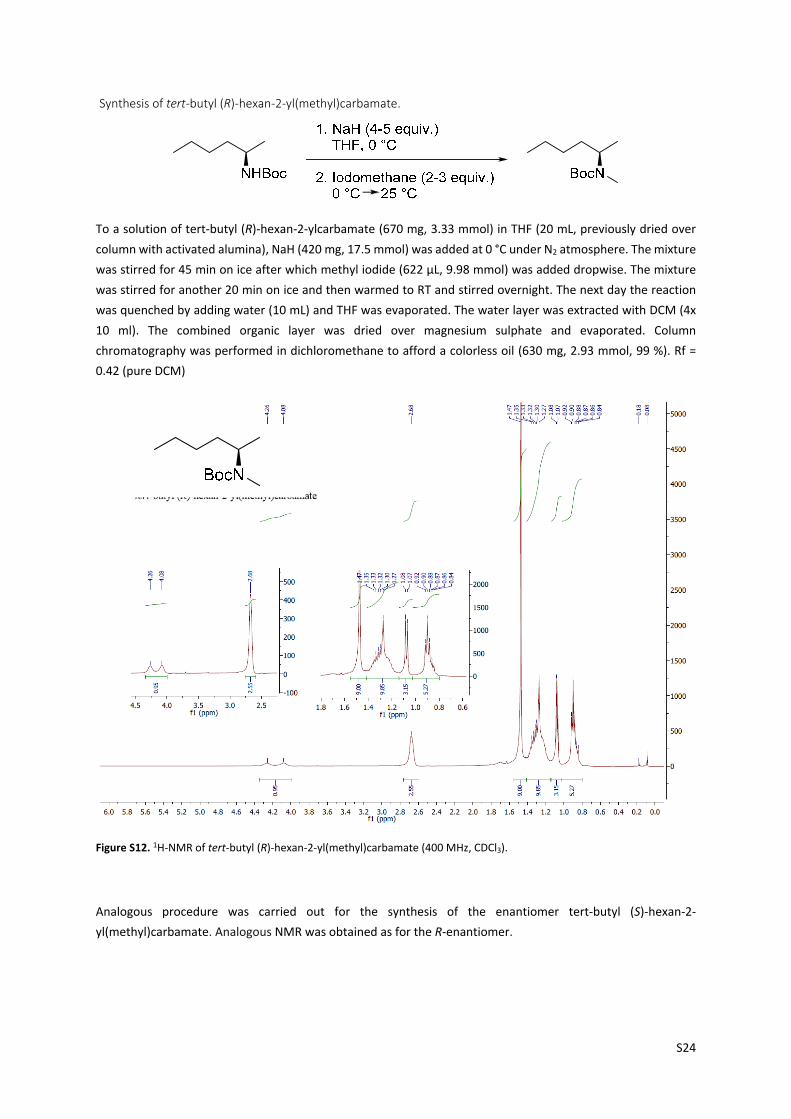
S24
Synthesis of tert‐butyl (R)‐hexan‐2‐yl(methyl)carbamate.
To a solution of tert‐butyl (R)‐hexan‐2‐ylcarbamate (670 mg, 3.33 mmol) in THF (20 mL, previously dried over
column with activated alumina), NaH (420 mg, 17.5 mmol) was added at 0 °C under N2 atmosphere. The mixture
was stirred for 45 min on ice after which methyl iodide (622 µL, 9.98 mmol) was added dropwise. The mixture
was stirred for another 20 min on ice and then warmed to RT and stirred overnight. The next day the reaction
was quenched by adding water (10 mL) and THF was evaporated. The water layer was extracted with DCM (4x
10 ml). The combined organic layer was dried over magnesium sulphate and evaporated. Column
chromatography was performed in dichloromethane to afford a colorless oil (630 mg, 2.93 mmol, 99 %). Rf =
0.42 (pure DCM)
Figure S12. 1H‐NMR of tert‐butyl (R)‐hexan‐2‐yl(methyl)carbamate (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer tert‐butyl (S)‐hexan‐2‐
yl(methyl)carbamate. Analogous NMR was obtained as for the R‐enantiomer.

S25
7.3. Procedure for the deprotection of amines with a tert‐butyloxycarbonyl group
Synthesis of (R)‐methyl(1‐methyl‐2‐(4‐fluorophenyl)ethyl)amine.
To a solution of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)(methyl)carbamate (143 mg, 0.53 mmol) in
dichloromethane (5 mL, distilled over calcium hydride), trifluoro acetic acid (5 mL, 65.3 mmol) was added at 0
°C under N2 atmosphere. The mixture was stirred for 2 h on ice and then warmed to RT. When the reaction was
complete, the solvent was evaporated. The crude was quenched with NaHCO3 (10 mL, saturated) and the water
layer was extracted with methyl tert‐butyl ether (10 mL). The organic layer was washed with brine, dried over
magnesium sulphate, and evaporated to yield a pink/brown oil (37 mg, 0.13 mmol, 25 % after work‐up).
Figure S13. 1H‐NMR of (R)‐methyl(1‐methyl‐2‐(4‐fluorophenyl)ethyl)amine containing trace of impurity (marked with “i”
originating from Boc‐anhydride) (400 MHz, CDCl3). Further attempt to purify was unsuccessful. Still, the purity of the final
compound was well sufficient for our purpose (analytical reference compound for GC‐FID and GC‐MS).
Analogous procedure was carried out for the synthesis of the enantiomer (S)‐methyl(1‐methyl‐2‐(4‐
fluorophenyl)ethyl)amine. Analogous NMR was obtained as for the R‐enantiomer.
i
i

S26
Synthesis of (R)‐N‐[2‐(4‐fluorophenyl)‐1‐methylethyl]ethylamine.
To a solution of tert‐butyl (R)‐(1‐(4‐fluorophenyl)propan‐2‐yl)(ethyl)carbamate (88 mg, 0.31 mmol) in
dichloromethane (5 mL, distilled over calcium hydride), trifluoro acetic acid (5 mL, 65.3 mmol) was added at 0
°C under N2 atmosphere. The mixture was stirred for 2 h on ice and then warmed to RT. When the reaction was
complete, the solvent was evaporated. The crude was quenched with NaHCO3 (10 mL, saturated) and the water
layer was extracted with methyl tert‐butyl ether (10 mL). The organic layer was washed with brine, dried over
MgSO4, and evaporated to yield a pink/brown oil (16 mg, 0.09 mmol, 28 % yield after work‐up). Rf = 0.40 in DCM
: MeOH : NH4OH (95 : 5 : 0.5).
Figure S14. 1H‐NMR of (R)‐N‐[2‐(4‐fluorophenyl)‐1‐methylethyl]ethylamine (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer (S)‐N‐[2‐(4‐fluorophenyl)‐1‐
methylethyl]ethylamine. Analogous NMR was obtained as for the R‐enantiomer.

S27
Synthesis of (R)‐N‐methylhexan‐2‐amine.
To a solution of tert‐butyl (R)‐hexan‐2‐yl(methyl)carbamate (0.34 mL, 1.38 mmol) in dichloromethane (5 mL,
distilled over calcium hydride), trifluoro acetic acid (5 mL, 65.3 mmol) was added at 0 °C under N2 atmosphere.
The mixture was stirred for 2 h on ice and then warmed to RT. When the reaction was complete, the solvent
was evaporated. The crude was quenched with NaHCO3 (10 mL, saturated) and the water layer was extracted
with methyl tert‐butyl ether (10 mL). The organic layer was washed with brine, dried over magnesium sulphate,
and evaporated to yield a pink/brown oil (150 mg, 0.97 mmol, 70 % yield after work‐up).
Figure S15. 1H‐NMR of (R)‐N‐methylhexan‐2‐amine (400 MHz, CDCl3).
Analogous procedure was carried out for the synthesis of the enantiomer (S)‐N‐methylhexan‐2‐amine.
Analogous NMR was obtained as for the R‐enantiomer.
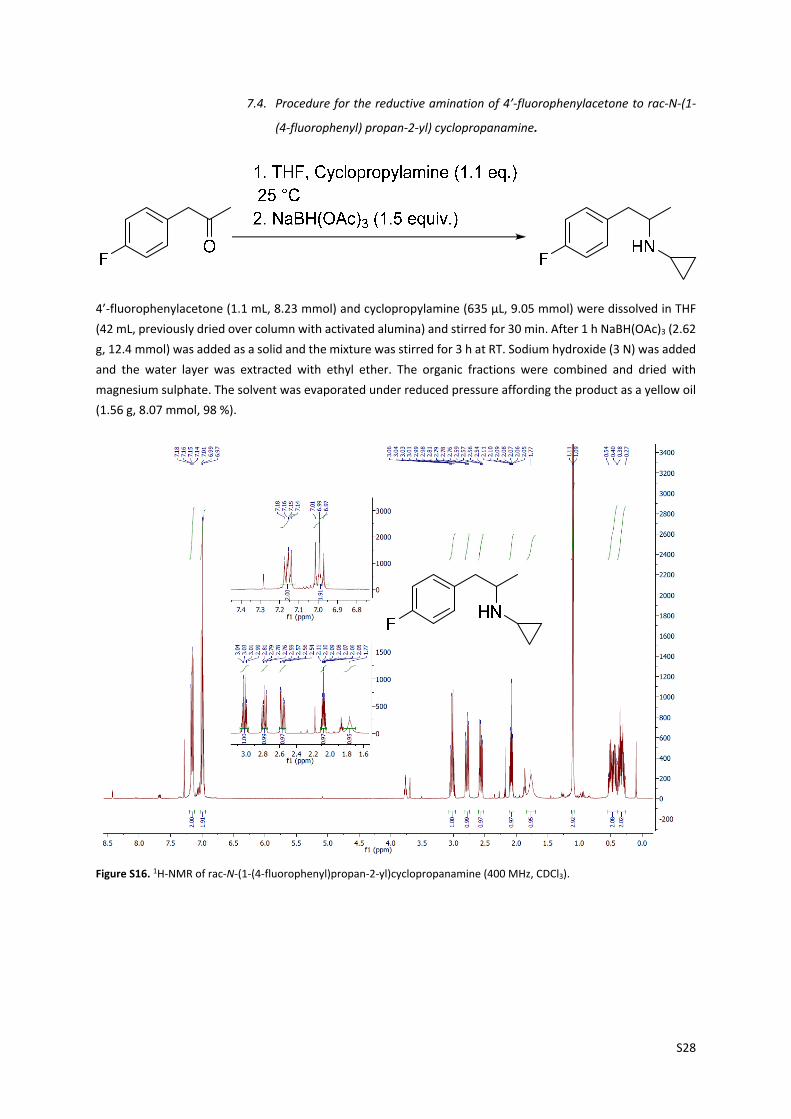
S28
7.4. Procedure for the reductive amination of 4’‐fluorophenylacetone to rac‐N‐(1‐
(4‐fluorophenyl) propan‐2‐yl) cyclopropanamine.
4’‐fluorophenylacetone (1.1 mL, 8.23 mmol) and cyclopropylamine (635 µL, 9.05 mmol) were dissolved in THF
(42 mL, previously dried over column with activated alumina) and stirred for 30 min. After 1 h NaBH(OAc)3 (2.62
g, 12.4 mmol) was added as a solid and the mixture was stirred for 3 h at RT. Sodium hydroxide (3 N) was added
and the water layer was extracted with ethyl ether. The organic fractions were combined and dried with
magnesium sulphate. The solvent was evaporated under reduced pressure affording the product as a yellow oil
(1.56 g, 8.07 mmol, 98 %).
Figure S16. 1H‐NMR of rac‐N‐(1‐(4‐fluorophenyl)propan‐2‐yl)cyclopropanamine (400 MHz, CDCl3).

S29
Analytical methods for the determination of conversions and of the absolute configurations
8.1. GC‐FID Method for the determination of the conversions
The conversions for the reductive amination of the ketones were determined by GC using a 7890A GC system
(Agilent Technologies), equipped with a FID detector using H2 as carrier gas and a DB‐1701 column from Agilent
(30 m, 250 μm, 0.25 μm).
DB1701‐30m‐B: constant pressure 6.9 psi, split ratio 40:1, T injector 250 °C. Temperature Program: T initial 60
°C, hold 6.5 min, gradient 20 °C/min up to 100 °C; hold 1 min, gradient 20 °C/min up to 280 °C; hold 1 min.
8.2. GC‐FID Method for the determination of the absolute configuration of primary
amines
After the extraction of the obtained amines, a solution of DMAP (50 mg) in 1 mL of acetic anhydride was
prepared. In total, 50 μL of this solution were added to 600 μL mixture of dichloromethane which contained the
obtained amines (extract). The mixtures were shaken at 25 oC for 30 min. After that, 500 μL of water was added
and shaken for another 30 min at 25 oC. The samples were centrifuged for 10 min at 14.800 rpm and the organic
phases were dried with magnesium sulfate prior to injection in a Chrompack Chiracel Dex‐CB column (length 25
m, internal diameter 0.32 mm)
DEX‐CB‐Method‐A: (Standard‐Long): Constant Flow: 1.4 mL/min, split ratio 40:1, T injector 200 °C. Temperature
Program: T initial 100 °C, hold 2 min, gradient 1°C/min up to 130 °C; hold 5 min, gradient 10 °C/min up to 170
°C; hold 10 min, gradient 10 °C/min up to 180 °C; hold 1 min
8.3. GC‐MS Method for the identification of the products
Amines were identified using a QP2010SE GCMS system (Shimadzu), with He as carrier gas using the same DB‐
1701 column from Agilent (30 m, 250 μm, 0.25 μm) as mentioned above.
DB1701‐30m‐Method‐B: GC program parameters: Linear Velocity: 38.3 cm/sec, pressure 65.4 KPa, Flow: 1.10
mL/min, split ratio 40:1, T injector 250 °C. Temperature Program: T initial 60 °C, hold 6.5 min, gradient 20 °C/min
up to 100 °C; hold 1 min, gradient 20 °C/min up to 280 °C; hold 1 min. MS program, parameters: Ion Source
Temperature: 200 °C, Detector Voltage: 0.1 Kv, Start Time: 3 min, End Time: 19.5 min, Start m/z: 43, End m/z:
600.
8.4. GC‐MS Method for the determination of the absolute configuration of
secondary amines
The absolute configuration of the secondary amines were determined by QP2010SE GCMS system (Shimadzu)
equipped with a Hydrodex‐β‐TBDAC column from Macherey‐Nagel (L: 50m, OD: 0.40 mm, ID: 0.25 mm)
Hydrodex‐β‐TBDAC‐Method 1. GC program parameters: Linear Velocity: 30 cm/sec, pressure 133 KPa, Flow: 1.08
mL/min, split ratio 40:1, T injector 220 °C. Temperature Program: T initial 100 °C, hold 2 min, gradient 1 °C/min
up to 220 °C; hold 2 min. MS program, parameters: Ion Source Temperature: 200 °C, Detector Voltage: 0.1 Kv,
Start Time: 5 min, End Time: 124 min, Start m/z: 43, End m/z: 600.
S30
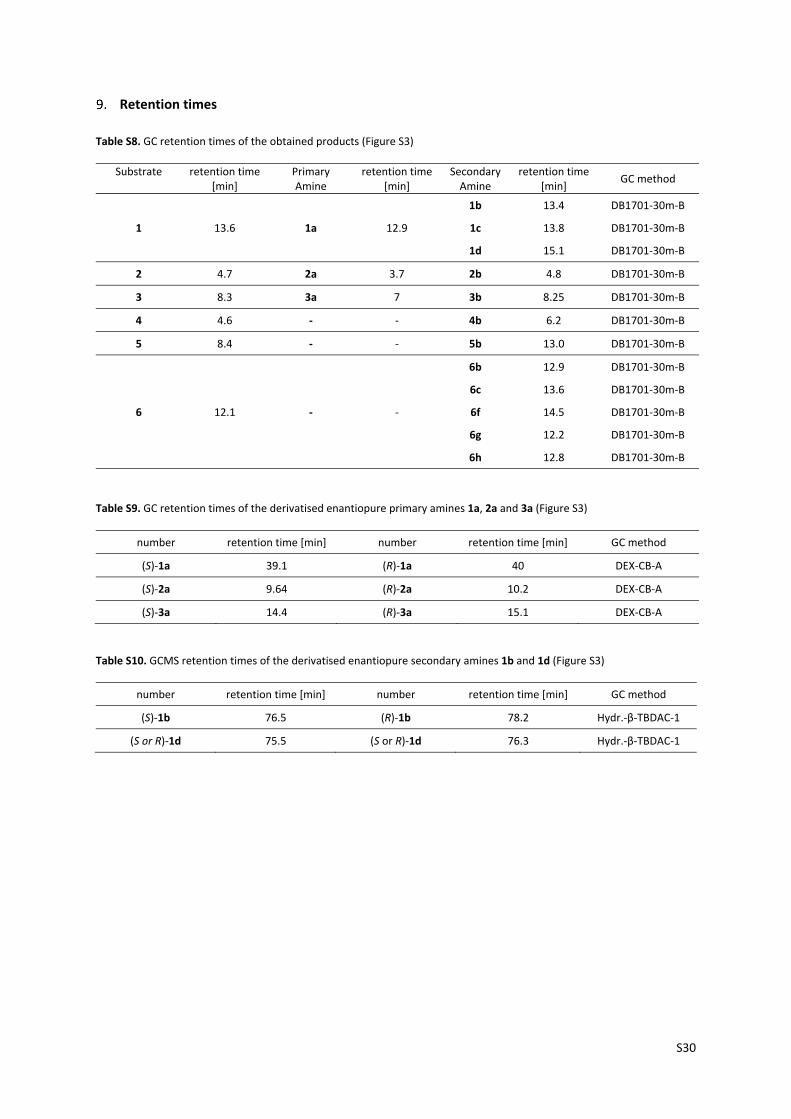
Retention times
Table S8. GC retention times of the obtained products (Figure S3)
Substrate
retention time [min]
Primary Amine
retention time [min]
Secondary Amine
retention time [min]
GC method
1
1a
1b 13.4 DB1701‐30m‐B
13.6 12.9 1c 13.8 DB1701‐30m‐B
1d 15.1 DB1701‐30m‐B
2 4.7 2a 3.7 2b 4.8 DB1701‐30m‐B
3 8.3 3a 7 3b 8.25 DB1701‐30m‐B
4 4.6 ‐ ‐ 4b 6.2 DB1701‐30m‐B
5 8.4 ‐ ‐ 5b 13.0 DB1701‐30m‐B
6
‐
6b 12.9 DB1701‐30m‐B
6c 13.6 DB1701‐30m‐B
12.1 ‐ 6f 14.5 DB1701‐30m‐B
6g 12.2 DB1701‐30m‐B
6h 12.8 DB1701‐30m‐B
Table S9. GC retention times of the derivatised enantiopure primary amines 1a, 2a and 3a (Figure S3)
number retention time [min] number retention time [min] GC method
(S)‐1a 39.1 (R)‐1a 40 DEX‐CB‐A
(S)‐2a 9.64 (R)‐2a 10.2 DEX‐CB‐A
(S)‐3a 14.4 (R)‐3a 15.1 DEX‐CB‐A
Table S10. GCMS retention times of the derivatised enantiopure secondary amines 1b and 1d (Figure S3)
number retention time [min] number retention time [min] GC method
(S)‐1b 76.5 (R)‐1b 78.2 Hydr.‐β‐TBDAC‐1
(S or R)‐1d 75.5 (S or R)‐1d 76.3 Hydr.‐β‐TBDAC‐1

S31
GC‐FID and GC‐MS chromatograms
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
(x10,000,000)TIC
GC‐FID chromatogram of the reaction
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
1
2
3
4
5
6
7
%44
109
83
57 69107 13863
11891152 207162 178 191
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
1
2
3
4
5
6
7
%58
109
56
83
43
15276 13510796 166122 148 207168 193185
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
1
2
3
4
5
6
7
%56
109
83
16563 133
10350 89 148123207197177184
217
Additional peak observed after extraction in organic solvent:1bi
1b 1a
GC‐MS chromatogram of the reaction with enzyme
1a
1b
1
1bi
GC‐MS chromatograph of the primary amine
GC‐MS chromatograph of the secondary amine
GC‐MS chromatograph of the imine (generated via mixing ketone with methylamine in organic solvent)
1a
1b
1
1bi
1
1a
1b
1
1bi
1bi
1b
1a
S32
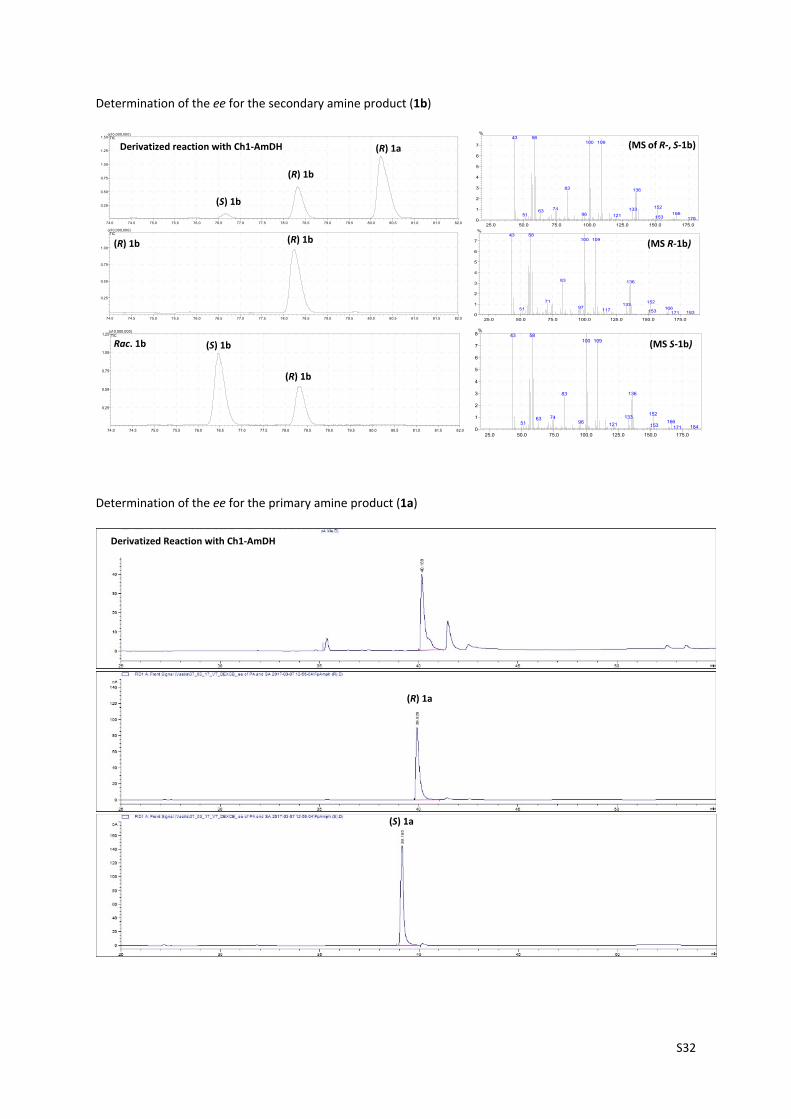
Determination of the ee for the secondary amine product (1b)
Determination of the ee for the primary amine product (1a)
74.0 74.5 75.0 75.5 76.0 76.5 77.0 77.5 78.0 78.5 79.0 79.5 80.0 80.5 81.0 81.5 82.0
0.25
0.50
0.75
1.00
(x10,000,000)TIC
74.0 74.5 75.0 75.5 76.0 76.5 77.0 77.5 78.0 78.5 79.0 79.5 80.0 80.5 81.0 81.5 82.0
0.25
0.50
0.75
1.00
1.25
1.50(x10,000,000)TIC
74.0 74.5 75.0 75.5 76.0 76.5 77.0 77.5 78.0 78.5 79.0 79.5 80.0 80.5 81.0 81.5 82.0
0.25
0.50
0.75
1.00
1.25(x10,000,000)TIC
Derivatized reaction with Ch1‐AmDH
(R) 1b
Rac. 1b
25.0 50.0 75.0 100.0 125.0 150.0 175.00
1
2
3
4
5
6
7
%58
10043
109
83 136
71 152133
97 16651 117 153 183171
25.0 50.0 75.0 100.0 125.0 150.0 175.00
1
2
3
4
5
6
7
8%
58100
43109
13683
1521337463
1669651 121 153 184171
25.0 50.0 75.0 100.0 125.0 150.0 175.00
1
2
3
4
5
6
7
%58
10043
109
83 136
15274 133631669651 121 153 178
(MS R‐1b)
(MS of R‐, S‐1b)
(R) 1b
(S) 1b
(R) 1a
(R) 1b
(R) 1b
(S) 1b
Derivatized Reaction with Ch1‐AmDH
(R) 1a
(S) 1a
(MS S‐1b)

S33
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
1
2
3
4
5
6
7
%44
109
83
57 69107 13863
11891152 207162 178 191
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.00
5
10
15
20
25
30%
43109
15283
5763
8975 133101 121 147 191165 177
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
1.0
2.0
3.0
4.0
5.0
(x10,000,000)TIC
50.0 75.0 100.0 125.0 150.0 175.00
1
2
3
4
5
6
7
%7244
109
73
8356
57
137 166101 11596 121 180150
Additional peak observed after extraction in organic solvent:
1 1c 1a
1ci
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
1
1a
1c 1ci
1a
1
1ci1c
GC‐MS chromatograph of the primary amine
GC‐MS chromatograph of the secondary amine
GC‐MS chromatograph of the ketone
1a
1
1c
1a
1
1c

S34
Determination of the ee of the primary Amine product (1a)
(R) 1a
(S) 1a
Reaction with Ch1‐AmDH
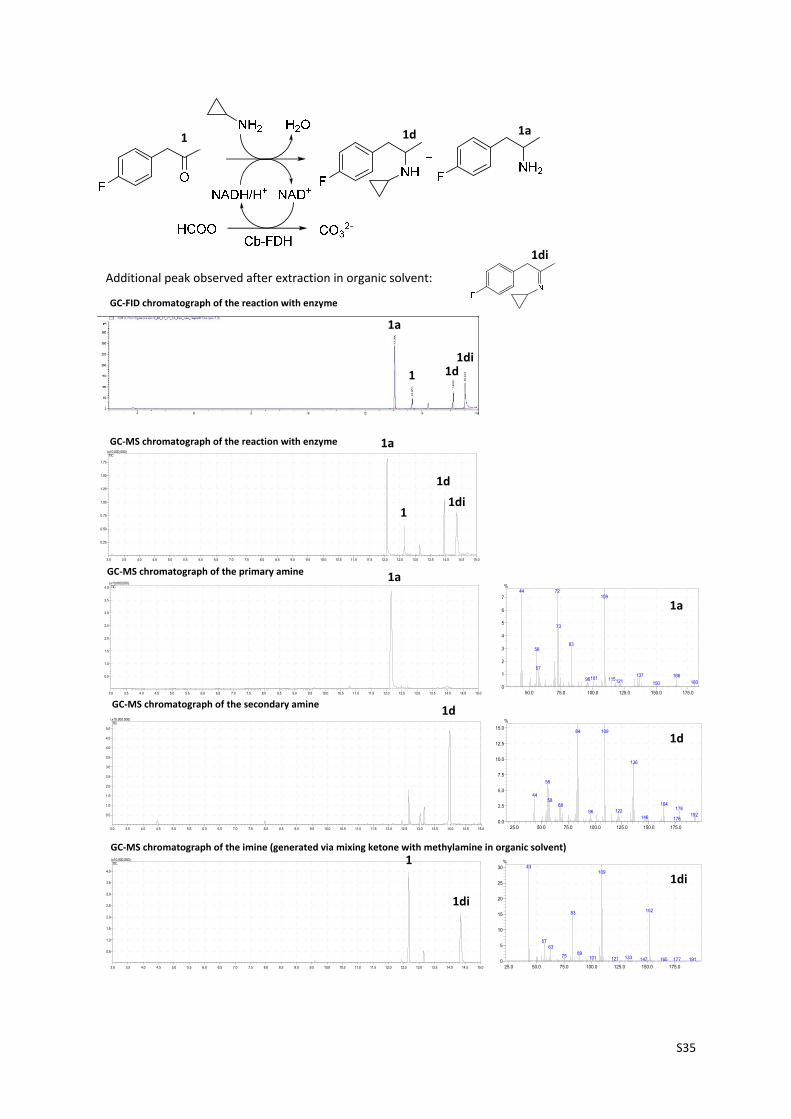
S35
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0 14.5 15.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
(x10,000,000)TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.00.0
2.5
5.0
7.5
10.0
12.5
15.0
%
84 109
136
56
4458
1646817812296
192146 176
50.0 75.0 100.0 125.0 150.0 175.00
1
2
3
4
5
6
7
%7244
109
73
8356
57
137 166101 11596 121 180150
25.0 50.0 75.0 100.0 125.0 150.0 175.00
5
10
15
20
25
30%
43109
15283
5763
8975 133101 121 147 191165 177
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0 14.5 15.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0 14.5 15.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0 14.5 15.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0(x10,000,000)TIC
Additional peak observed after extraction in organic solvent:
1 1d 1a
1di
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the secondary amine
GC‐MS chromatograph of the primary amine
GC‐MS chromatograph of the imine (generated via mixing ketone with methylamine in organic solvent)
1
1a
1d1di
1a
1
1d
1di
1a
1d
1
1di
1a
1d
1di
S36
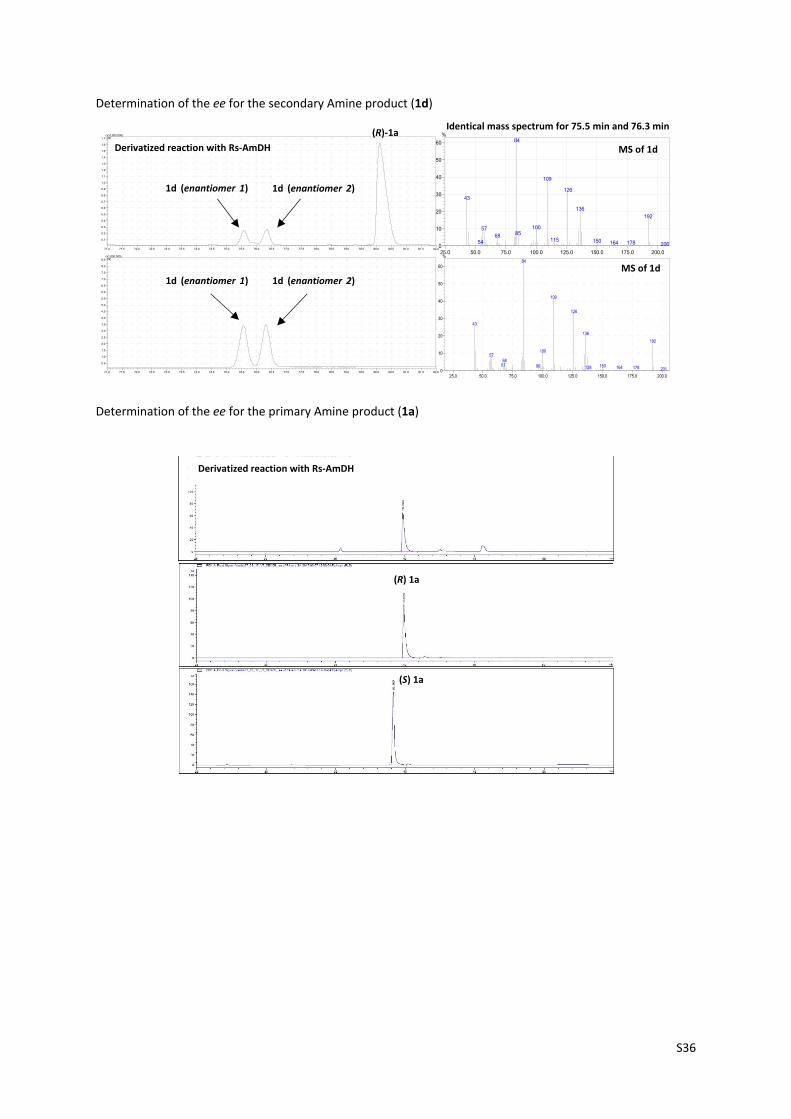
Determination of the ee for the secondary Amine product (1d)
Determination of the ee for the primary Amine product (1a)
71.0 71.5 72.0 72.5 73.0 73.5 74.0 74.5 75.0 75.5 76.0 76.5 77.0 77.5 78.0 78.5 79.0 79.5 80.0 80.5 81.0 81.5 82.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
1.3
1.4
1.5
1.6
1.7(x10,000,000)
TIC
71.0 71.5 72.0 72.5 73.0 73.5 74.0 74.5 75.0 75.5 76.0 76.5 77.0 77.5 78.0 78.5 79.0 79.5 80.0 80.5 81.0 81.5 82.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5(x1,000,000)
TIC
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
10
20
30
40
50
60
%84
109
126
43
136
192
10057
6867 15096 138 164 178 201
25.0 50.0 75.0 100.0 125.0 150.0 175.0 200.00
10
20
30
40
50
60
%84
109
126
43
136192
100578568
115 15054 164 178 206
Derivatized reaction with Rs‐AmDH
MS of 1d
1d (enantiomer 1)
Identical mass spectrum for 75.5 min and 76.3 min
MS of 1d
(R)‐1a
Derivatized reaction with Rs‐AmDH
(S) 1a
(R) 1a
1d (enantiomer 2)
1d (enantiomer 1)
1d (enantiomer 2)
S37
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75(x10,000,000)TIC
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0
0.25
0.50
0.75
1.00
1.25
1.50(x10,000,000)TIC
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
(x10,000,000)TIC
3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0 14.5 15.0
0.25
0.50
0.75
1.00
(x10,000,000)TIC
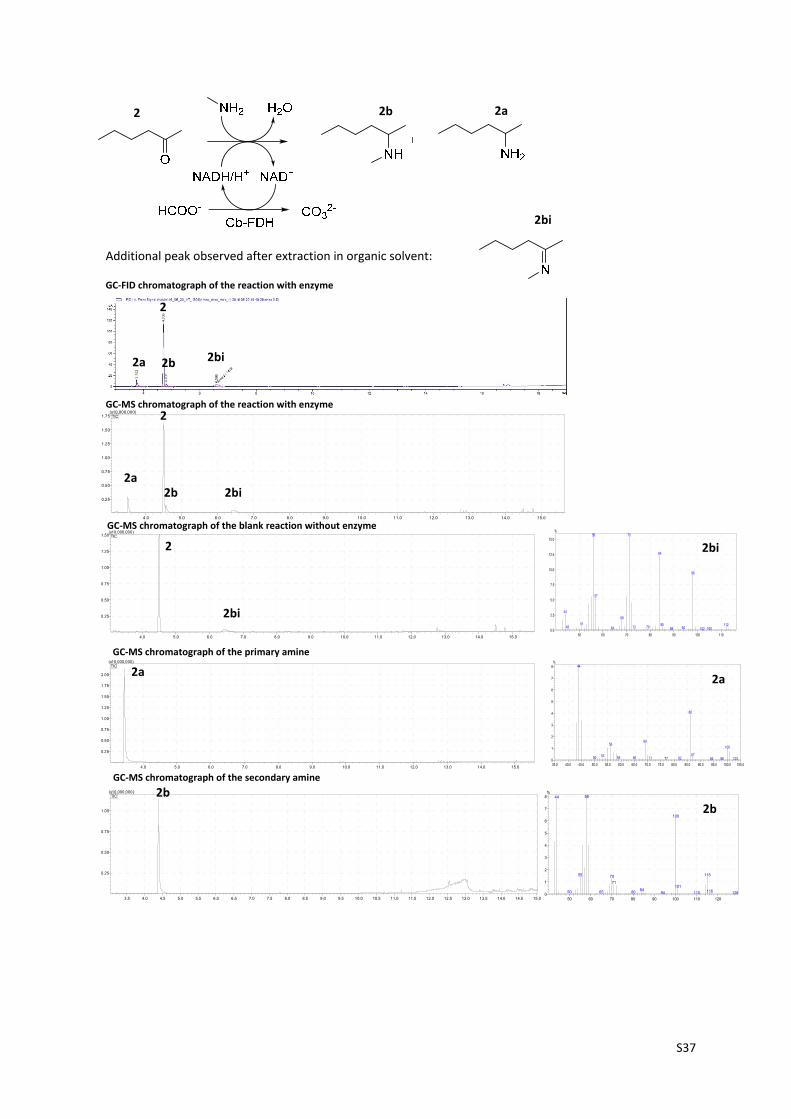
2 2b 2a
Additional peak observed after extraction in organic solvent:
2bi
2
2a 2b 2bi
2
2a2b 2bi
2a
2b
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the secondary amine
GC‐MS chromatograph of the primary amine
GC‐MS chromatograph of the blank reaction without enzyme
2
2bi
50 60 70 80 90 100 1100.0
2.5
5.0
7.5
10.0
12.5
15.0
%7156
84
98
57
44
68
51 85 11245 7973 9464 10589 102
2bi
35.0 40.0 45.0 50.0 55.0 60.0 65.0 70.0 75.0 80.0 85.0 90.0 95.0 100.0 105.00
1
2
3
4
5
6
7
8%
44
86
6956
100
875350 59 65 71 8277 9894 103
2a
50 60 70 80 90 100 110 1200
1
2
3
4
5
6
7
8%
5844
100
11555 70
71101
84 11650 65 80 94 110 128
2b
S38
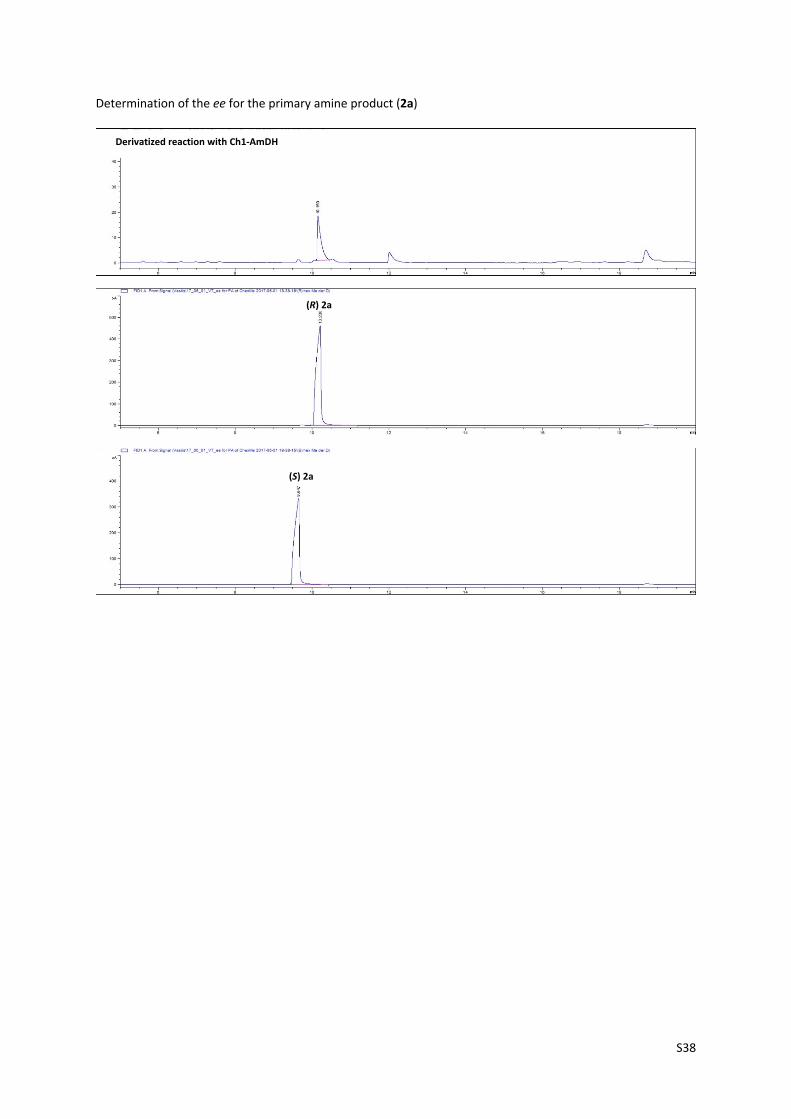
Determination of the ee for the primary amine product (2a)
Derivatized reaction with Ch1‐AmDH
(R) 2a
(S) 2a

S39
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
(x10,000,000)TIC
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
(x10,000,000)TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0
0.5
1.0
1.5
2.0
2.5
3.0(x10,000,000)TIC
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0 13.0 14.0 15.0 16.0 17.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
(x10,000,000)TIC
3 3b 3a
Additional peak observed after extraction in organic solvent:
3bi
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the blank reaction without enzyme
GC‐MS chromatograph of the primary amine
GC‐MS chromatograph of the ketone
3
3b3a
3bi
3
3b
3a 3bi
3
3bi
3a
3
50 60 70 80 90 100 110 1200.0
2.5
5.0
7.5
10.0
12.5
15.0
%
7156
84
43
9872 11257
12681 9651 66 11010391 117
3bi
50 60 70 80 90 100 110 1200.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
%44
100
55
11458 708353 69
8165 9777 8646 91119
109105
3a
40 50 60 70 80 90 100 110 120 1300
5
10
15
20
25
30%
43 58
71
114
9985
53 11545 8160 69 10091 109 124 131
3

S40
Determination of the ee for the primary amine product (3a)
Derivatized reaction with ch1‐AmDH
(R) 2a
(S) 2a

S41
4 4b
Additional peak observed after extraction in organic solvent:
Additional peak observed as reaction byproduct:(generated by Cb‐FDH)
4bi
4bo
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0
0.25
0.50
0.75
1.00
1.25
(x10,000,000)TIC
44b4bi
4bo
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of blank reaction without enzyme
GC‐MS chromatograph of the substrate
3.0 4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0
0.5
1.0
1.5
2.0
2.5(x10,000,000)
TIC
44bi
4.0 5.0 6.0 7.0 8.0 9.0 10.0 11.0 12.0
0.5
1.0
1.5
2.0
2.5
(x10,000,000)TIC 4

S42
Mass spectrum of the peak at 6.2 min (secondary amine product 4b)
50 60 70 80 90 100 110 120
0
1
2
3
%44
115
58
55
7053 1008669
7865 11246 96 10591 119

S43
Additional peak observed after extraction in organic solvent:
5 5e
5ei
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5
0.25
0.50
0.75
1.00
1.25
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50
2.75
3.00
(x10,000,000)TIC
GC‐FID chromatograph of the reaction with enzyme
GC‐FID chromatograph of the blank reaction without enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the aldehyde and imine (generating via mixing the aldehyde and the amine donor in organic solvent)
5
5e
5ei
5
5ei
5
5ei
5e
5
5ei
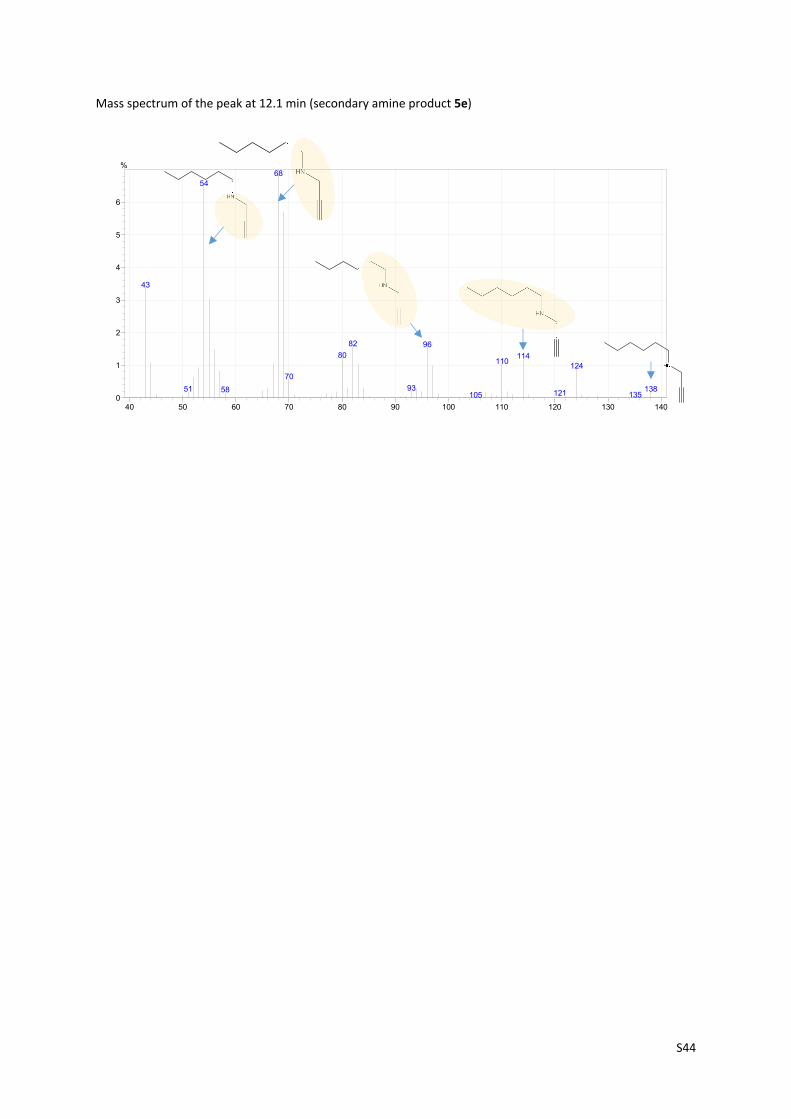
S44
Mass spectrum of the peak at 12.1 min (secondary amine product 5e)
40 50 60 70 80 90 100 110 120 130 1400
1
2
3
4
5
6
%68
54
43
82 9680 114
110124
70
9351 13858 121 135105

S45
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5(x1,000,000)
TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0(x1,000,000)
TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50(x10,000,000)
TIC
Additional peak observed after extraction in organic solvent:
6 6b
6bi
66bi6b
6bi
6
6b
6bi
6
6
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the blank reaction without enzyme
GC‐MS chromatograph of the substrate
S46
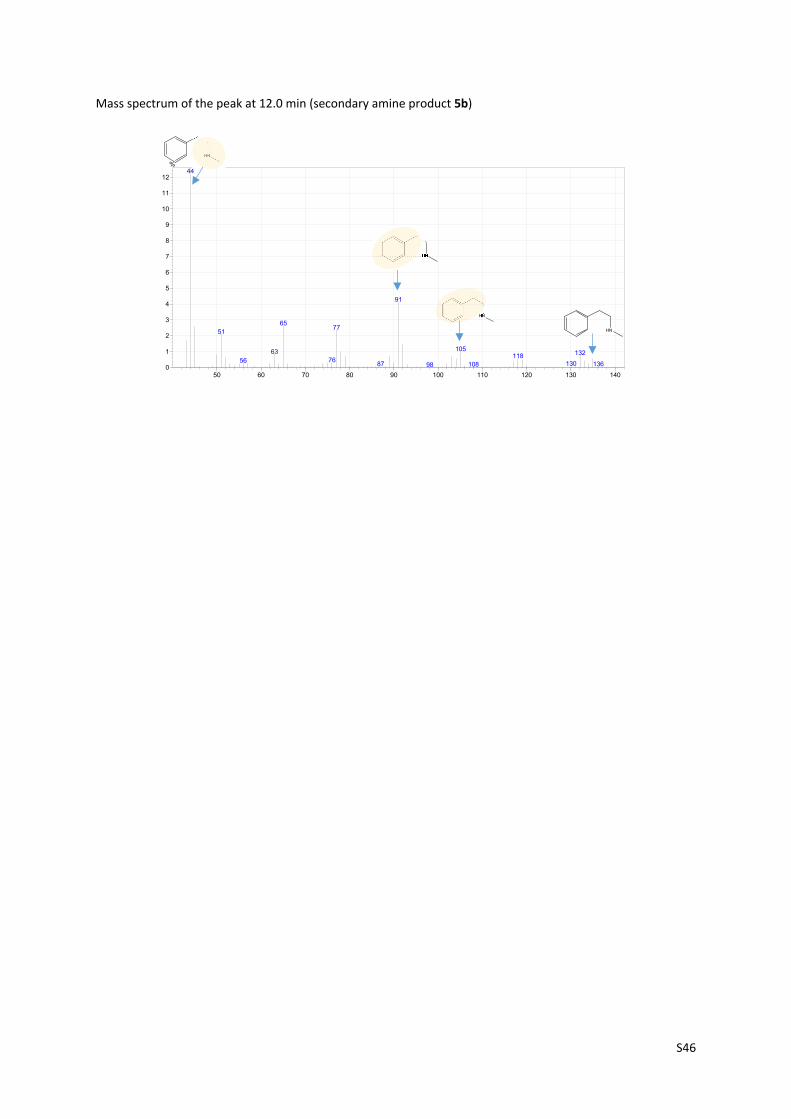
Mass spectrum of the peak at 12.0 min (secondary amine product 5b)
50 60 70 80 90 100 110 120 130 1400
1
2
3
4
5
6
7
8
9
10
11
12
%44
91
6577
51
10563 1321187656 13087 13610898
HN
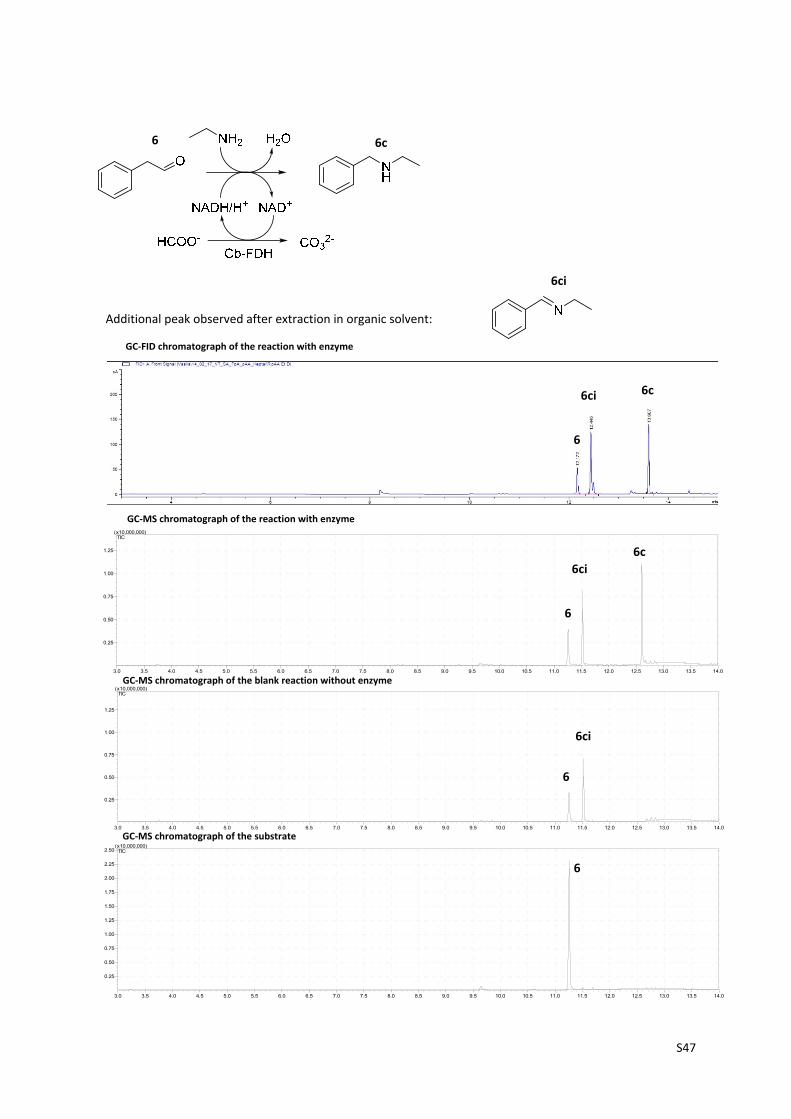
S47
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.25
0.50
0.75
1.00
1.25
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.25
0.50
0.75
1.00
1.25
(x10,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5 14.0
0.25
0.50
0.75
1.00
1.25
1.50
1.75
2.00
2.25
2.50(x10,000,000)
TIC
Additional peak observed after extraction in organic solvent:
6 6c
6ci
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the blank reaction without enzyme
GC‐MS chromatograph of the substrate
6
6ci 6c
6c
6
6ci
6ci
6
6
S48
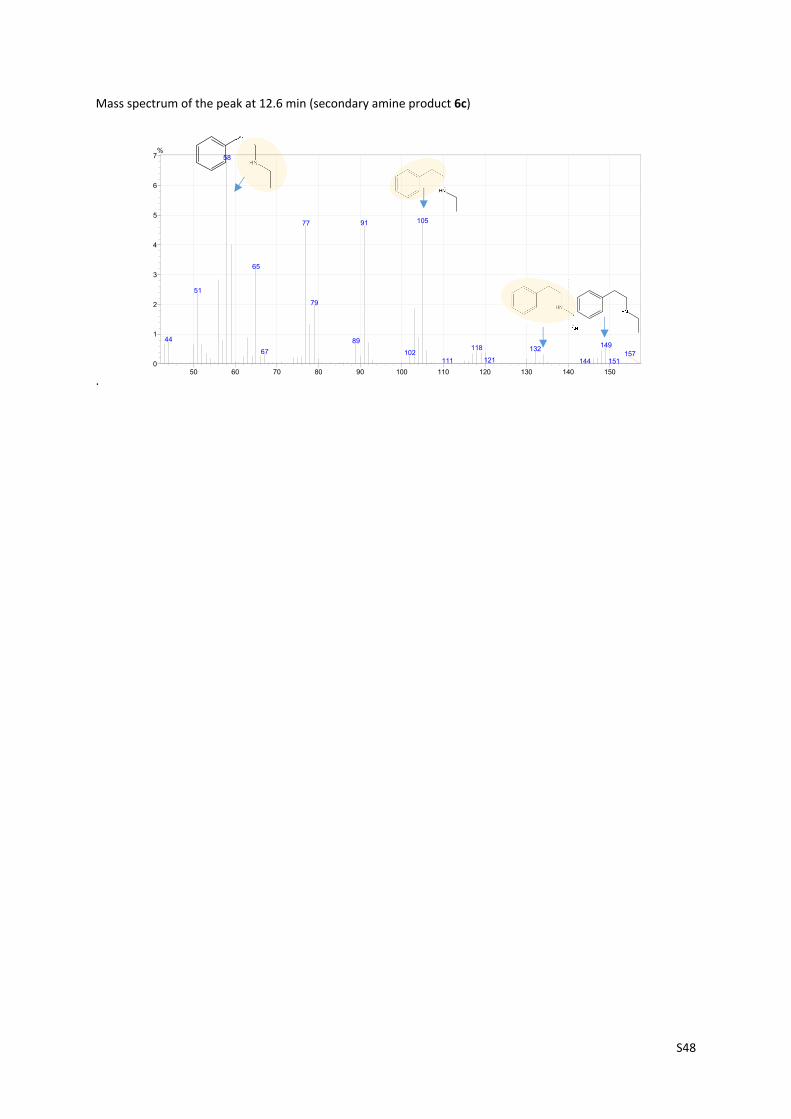
Mass spectrum of the peak at 12.6 min (secondary amine product 6c)
.
50 60 70 80 90 100 110 120 130 140 1500
1
2
3
4
5
6
7%
58
1059177
65
51
79
44 89149118 13267 102
121111 144 151157
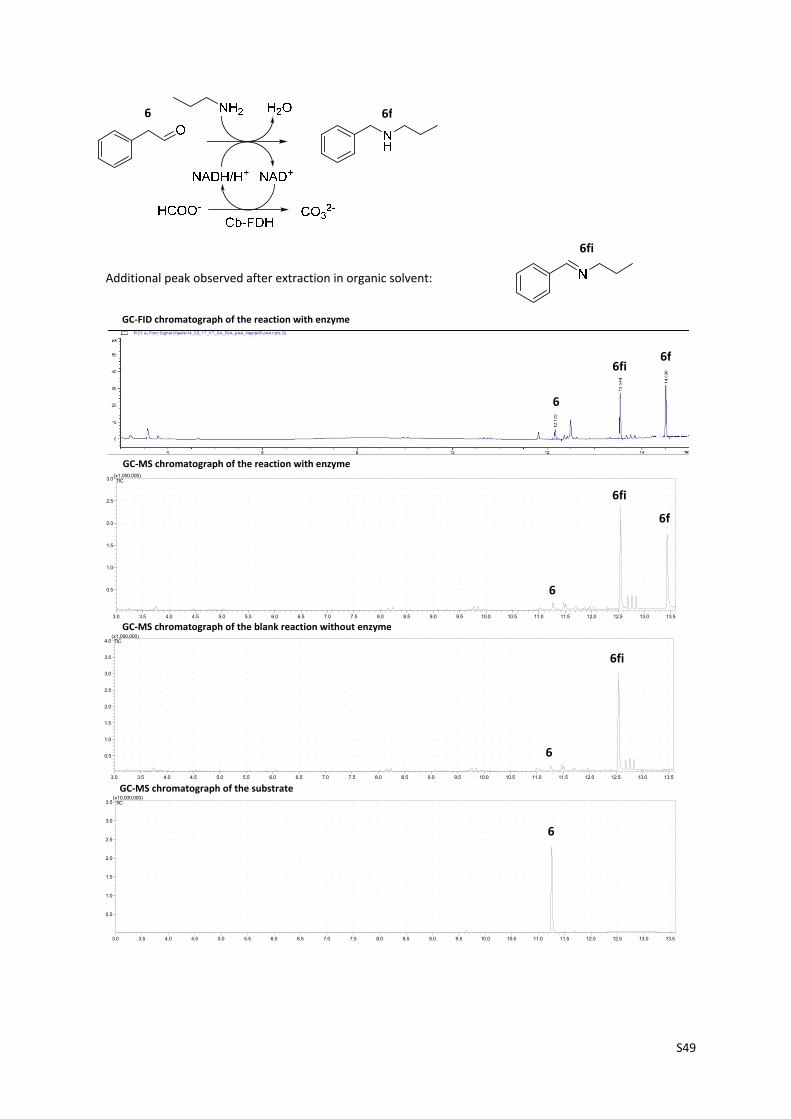
S49
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5
0.5
1.0
1.5
2.0
2.5
3.0(x1,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0(x1,000,000)TIC
3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 8.5 9.0 9.5 10.0 10.5 11.0 11.5 12.0 12.5 13.0 13.5
0.5
1.0
1.5
2.0
2.5
3.0
3.5(x10,000,000)
TIC
Additional peak observed after extraction in organic solvent:
6 6f
6fi
6
6fi6f
6
6fi
6f
6
6fi
6
GC‐FID chromatograph of the reaction with enzyme
GC‐MS chromatograph of the reaction with enzyme
GC‐MS chromatograph of the blank reaction without enzyme
GC‐MS chromatograph of the substrate

S50
Mass spectrum of the peak at 13.4 min (secondary amine product 6f)
40 50 60 70 80 90 100 110 120 130 140 150 160 1700.0
2.5
5.0
7.5
10.0
12.5
%72
105
43
77
91
65134
51
56 12016310283 130115 138 152146 173169

S51
6
6
6g
6g
6
GC‐FID chromatograph of the reaction with enzyme
GC‐FID chromatograph of the blank reaction without enzyme
GC‐FID chromatograph of the substrate
6
Mass spectrum of the peak at 12.90 (secondary amine 6g)
50 60 70 80 90 100 110 120 130 1400
1
2
3
4
5
6
%58
77
10544 51 65 9157 79
133102 11771 10885 97139
125

S52
6 6h
6
GC chromatograph of the reaction with enzyme
GC chromatograph of the blank reaction without enzyme
GC chromatograph of the substrate
Mass spectrum of the peak at 12.90 (secondary amine 6h)
6
6
6h
50.0 75.0 100.0 125.0 150.00
1
2
3
4
5
6
7%
7244
43
10577
57
10385 9151 65
111
13297 125118
146134

S53
References
[1] a) L. J. Ye, H. H. Toh, Y. Yang, J. P. Adams, R. Snajdrova, Z. Li, ACS Catal. 2015, 5, 1119-1122; b) T. Knaus, W. Böhmer, F. G. Mutti, Green Chem. 2017, 19, 453-463.
[2] B. R. Bommarius, M. Schürmann, A. S. Bommarius, Chem. Commun. 2014 50, 14953-14955.
[3] D. Koszelewski, I. Lavandera, D. Clay, D. Rozzell, W. Kroutil, Adv. Synth. Catal. 2008, 350, 2761-2766.
[4] a) R. A. Laskowski, M. W. MacArthur, D. S. Moss, J. M. Thornton, J. Appl. Crystallogr. 1993, 283-291; b) R. W. Hooft, G. Vriend, C. Sander, E. E. Abola, Nature 1996, 381, 272-272.
[5] P. J. Baker, A. P. Turnbull, S. E. Sedelnikova, T. J. Stillman, D. W. Rice, Structure 1995, 3, 693-705.
[6] Y. Zhao, T. Wakamatsu, K. Doi, H. Sakuraba, T. Ohshima, J. Mol. Catal. B: Enzym. 2012, 83, 65-72.
[7] N. M. W. Brunhuber, J. B. Thoden, J. S. Blanchard, J. L. Vanhooke, Biochemistry 2000, 39, 9174-9187.
[8] J. L. Vanhooke, J. B. Thoden, N. M. W. Brunhuber, J. S. Blanchard, H. M. Holden, Biochemistry 1999, 38, 2326-2339.


















