
Structural elucidation of transmembrane transporter protein bilitranslocase: Conformational analysis...
11
Structural elucidation of transmembrane transporter protein bilitranslocase: Conformational analysis of the second transmembrane region TM2 by molecular dynamics and NMR spectroscopy Amrita Roy Choudhury a , Andrej Perdih a , Špela Župerl a , Emilia Sikorska b , Tom Solmajer a , Stefan Jurga c,d , Igor Zhukov e,1 , Marjana Novič a, ⁎ a National Institute of Chemistry, Ljubljana, Slovenia b Faculty of Chemistry, University of Gdańsk, Gdańsk, Poland c NanoBioMedical Centre, Adam Mickiewicz University, Poznan, Poland d Department of Macromolecular Physics, Faculty of Physics, Adam Mickiewicz University, Poznan, Poland e Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland abstract article info Article history: Received 15 March 2013 Received in revised form 31 May 2013 Accepted 6 June 2013 Available online 15 June 2013 Keywords: Transmembrane peptides Bilitranslocase Drug target Molecular dynamic simulations NMR spectroscopy Membrane proteins represent about a third of the gene products in most organisms, as revealed by the genome sequencing projects. They account for up to two thirds of known drugable targets, which emphasizes their critical pharmaceutical importance. Here we present a study on bilitranslocase (BTL) (TCDB 2.A.65), a membrane protein primarily involved in the transport of bilirubin from blood to liver cells. Bilitranslocase has also been identified as a potential membrane transporter for cellular uptake of several drugs and due to its implication in drug uptake, it is extremely important to advance the knowledge about its 3D structure. However, at present, only a limited knowl- edge is available beyond the primary structure of BTL. It has been recently confirmed experimentally that one of the four computationally predicted transmembrane segments of bilitranslocase, TM3, has a helical structure with hydrophilic amino acid residues oriented towards one side, which is typical for transmembrane domains of mem- brane proteins. In this study we confirmed by the use of multidimensional NMR spectroscopy that the second transmembrane segment, TM2, also appears in a form of α-helix. The stability of this polypeptide chain was ver- ified by molecular dynamics (MD) simulation in dipalmitoyl phosphatidyl choline (DPPC) and in sodium dodecyl sulfate (SDS) micelles. The two α-helices, TM2 corroborated in this study, and TM3 confirmed in our previous in- vestigation, provide reasonable building blocks of a potential transmembrane channel for transport of bilirubin and small hydrophilic molecules, including pharmaceutically active compounds. © 2013 Elsevier B.V. All rights reserved. 1. Introduction Membrane proteins attract attention of scientists because of their important and intriguing role in membrane trafficking, thus being also extensively studied drug targets [1,2]. Crossing of the membrane for hydrophilic molecules is enabled through an ion channel or mem- brane embedded transporter protein. Many ion-channel diseases are associated with the selectivity and gating, which allow for organized ionic permeation while maintaining the barrier property of the mem- brane [3]. Discernibly, the 3D structure of a protein is mandatory for all detailed studies of membrane transporters. There are a plethora of approaches to resolve the protein secondary structure, including NMR spectroscopy [4–6], X-ray crystallography and electron micros- copy [7–9], or single molecule manipulation and detection methods, such as atomic force microscopy (AMF) [10] and fluorescence reso- nance energy transfer (FRET) [11,12]. However, the membrane proteins are extremely difficult to prepare for experimental measurements and only a small portion of structurally resolved proteins in the PDB data- base (less than 5 %) are membrane proteins [13,14]. Biochimica et Biophysica Acta 1828 (2013) 2609–2619 Abbreviation: BTL, bilitranslocase protein; TM2, peptide representing the second (positions 73–95) transmembrane region of BTL protein; TM2A, peptide representing the second (positions 75–94) transmembrane segment of BTL protein; TM2B, peptide representing a prolonged second transmembrane segment (positions 73–99) of BTL protein; TM3, peptide represented third transmembrane region (residues 220–237) of BTL protein; HSQC, Heteronuclear Single Quantum Correlation spectroscopy; TAV, time-averaged distance restraints; RFD, radial distribution function; CD, circular di- chroism; SDS, sodium dodecyl sulfate; DPC, dodecylphosphocholine; DPPC, dipalmitoyl phosphatidyl choline; DSS, sodium 2,2-dimethyl-2-silapentane-5-sulfonate; DLPC, 1,2-dilauroyl-sn-glycero-3-phosphocholine; DMPC, 1,2-dimyristoyl-sn-glycero-3- phosphocholine; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; MD, molecular dynamics; CPNN, counter-propagation neural network; AMF, atomic force micros- copy; FRET, fluorescence resonance energy transfer; SD, steepest descent; ABNR, Adopted Basis Newton–Raphson method ⁎ Corresponding author at: National Institute of Chemistry, Hajdrihova 19, 1001 Ljubljana, Slovenia. Tel.: +386 1 4760 253; fax: +386 1 4760 300. E-mail addresses: [email protected] (A. Roy Choudhury), [email protected] (A. Perdih), [email protected] (Š. Župerl), [email protected] (E. Sikorska), [email protected] (T. Solmajer), [email protected] (S. Jurga), [email protected] (M. Novič). 1 Corresponding for NMR data: [email protected], Institute of Biochemistry and Bio- physics, Polish Academy of Sciences, ul. Pawińskiego 5a, 02–106 Warsaw, Poland, Tel./fax: +48 22 592 2038. 0005-2736/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.bbamem.2013.06.006 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamem
Transcript of Structural elucidation of transmembrane transporter protein bilitranslocase: Conformational analysis...

Biochimica et Biophysica Acta 1828 (2013) 2609–2619
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bbamem
Structural elucidation of transmembrane transporter proteinbilitranslocase: Conformational analysis of the second transmembraneregion TM2 by molecular dynamics and NMR spectroscopy
Amrita Roy Choudhury a, Andrej Perdih a, Špela Župerl a, Emilia Sikorska b, Tom Solmajer a, Stefan Jurga c,d,Igor Zhukov e,1, Marjana Novič a,⁎a National Institute of Chemistry, Ljubljana, Sloveniab Faculty of Chemistry, University of Gdańsk, Gdańsk, Polandc NanoBioMedical Centre, Adam Mickiewicz University, Poznan, Polandd Department of Macromolecular Physics, Faculty of Physics, Adam Mickiewicz University, Poznan, Polande Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland
Abbreviation: BTL, bilitranslocase protein; TM2, pe(positions 73–95) transmembrane region of BTL proteinthe second (positions 75–94) transmembrane segmentrepresenting a prolonged second transmembrane segmprotein; TM3, peptide represented third transmembraof BTL protein; HSQC, Heteronuclear Single Quantum Ctime-averaged distance restraints; RFD, radial distribuchroism; SDS, sodium dodecyl sulfate; DPC, dodecylphosphosphatidyl choline; DSS, sodium 2,2-dimethyl-2-s1,2-dilauroyl-sn-glycero-3-phosphocholine; DMPC,phosphocholine; DOPC, 1,2-dioleoyl-sn-glycero-3-phdynamics; CPNN, counter-propagation neural networcopy; FRET, fluorescence resonance energy transfer;Adopted Basis Newton–Raphson method⁎ Corresponding author at: National Institute of Chemistr
Slovenia. Tel.: +386 1 4760 253; fax: +386 1 4760 300.E-mail addresses: [email protected] (A. Ro
[email protected] (A. Perdih), [email protected] (Š. Žu(E. Sikorska), [email protected] (T. Solmajer), [email protected]@ki.si (M. Novič).
1 Corresponding for NMR data: [email protected], Instphysics, Polish Academy of Sciences, ul. PawińskiegoTel./fax: +48 22 592 2038.
0005-2736/$ – see front matter © 2013 Elsevier B.V. Allhttp://dx.doi.org/10.1016/j.bbamem.2013.06.006
a b s t r a c t
a r t i c l e i n f oArticle history:Received 15 March 2013Received in revised form 31 May 2013Accepted 6 June 2013Available online 15 June 2013
Keywords:Transmembrane peptidesBilitranslocaseDrug targetMolecular dynamic simulationsNMR spectroscopy
Membrane proteins represent about a third of the gene products in most organisms, as revealed by the genomesequencing projects. They account for up to two thirds of known drugable targets, which emphasizes their criticalpharmaceutical importance. Herewe present a study on bilitranslocase (BTL) (TCDB 2.A.65), amembrane proteinprimarily involved in the transport of bilirubin fromblood to liver cells. Bilitranslocase has also been identifiedas apotentialmembrane transporter for cellular uptake of several drugs and due to its implication in drug uptake, it isextremely important to advance the knowledge about its 3D structure. However, at present, only a limited knowl-edge is available beyond the primary structure of BTL. It has been recently confirmed experimentally that one ofthe four computationally predicted transmembrane segments of bilitranslocase, TM3, has a helical structure withhydrophilic amino acid residues oriented towards one side,which is typical for transmembrane domains ofmem-brane proteins. In this study we confirmed by the use of multidimensional NMR spectroscopy that the secondtransmembrane segment, TM2, also appears in a form of α-helix. The stability of this polypeptide chain was ver-ified bymolecular dynamics (MD) simulation in dipalmitoyl phosphatidyl choline (DPPC) and in sodium dodecylsulfate (SDS)micelles. The twoα-helices, TM2 corroborated in this study, and TM3 confirmed in our previous in-vestigation, provide reasonable building blocks of a potential transmembrane channel for transport of bilirubinand small hydrophilic molecules, including pharmaceutically active compounds.
© 2013 Elsevier B.V. All rights reserved.
ptide representing the second; TM2A, peptide representingof BTL protein; TM2B, peptideent (positions 73–99) of BTLne region (residues 220–237)orrelation spectroscopy; TAV,tion function; CD, circular di-phocholine; DPPC, dipalmitoylilapentane-5-sulfonate; DLPC,1,2-dimyristoyl-sn-glycero-3-osphocholine; MD, moleculark; AMF, atomic force micros-SD, steepest descent; ABNR,
y, Hajdrihova 19, 1001 Ljubljana,
y Choudhury),perl), [email protected] (S. Jurga),
itute of Biochemistry and Bio-5a, 02–106 Warsaw, Poland,
rights reserved.
1. Introduction
Membrane proteins attract attention of scientists because of theirimportant and intriguing role in membrane trafficking, thus beingalso extensively studied drug targets [1,2]. Crossing of the membranefor hydrophilic molecules is enabled through an ion channel or mem-brane embedded transporter protein. Many ion-channel diseases areassociated with the selectivity and gating, which allow for organizedionic permeation while maintaining the barrier property of the mem-brane [3]. Discernibly, the 3D structure of a protein is mandatory forall detailed studies of membrane transporters. There are a plethoraof approaches to resolve the protein secondary structure, includingNMR spectroscopy [4–6], X-ray crystallography and electron micros-copy [7–9], or single molecule manipulation and detection methods,such as atomic force microscopy (AMF) [10] and fluorescence reso-nance energy transfer (FRET) [11,12]. However, themembrane proteinsare extremely difficult to prepare for experimental measurements andonly a small portion of structurally resolved proteins in the PDB data-base (less than 5 %) are membrane proteins [13,14].

2610 A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
In this study we present an attempt to resolve the transmembranedomains of bilitranslocase (BTL), a plasma membrane protein func-tioning as organic anion carrier. It is involved in several biologicalprocesses in different organs; the most studied role of BTL is thetransport of bilirubin from blood to liver cells [15–20], but it wasalso detected in the heart or gastrointestinal tract [21–25]. In vitro ex-periments have confirmed that BTL has an active role in the transportof many organic molecules through the cell membrane [23,25,26]. Re-cently it has been shown [27] that carrier-mediated and active uptakeof pharmaceutical drugs may be more common than is usually as-sumed. We believe that BTL could be considered as a potential drugcarrier and should thus be considered in rational drug discovery anddevelopment. For this reason alone it would be very important to re-solve the 3D structure of BTL. The primary sequence of the 340 aminoacids long protein BTL has been available for some time [28]; howev-er, its 3D structure is not yet solved. The comparison with other struc-turally resolved proteins is not supportive, because BTL shows nohomology with known proteins. As the tertiary and the quaternarystructures of BTL remain unknown, the composition, arrangement,and structure of the transporting channel is yet to be studied in detail.However, it is assumed that the BTL polypeptide chain consists of fourmembrane spanning segments, which presumably show α helicalconformation [29]. Evidence from both computational and experi-mental studies [30–32] supports the hypothesis. The structure ofone of these helices, the third transmembrane domain (TM3) hasbeen previously determined experimentally in our group [33] andsupported by molecular dynamics (MD) simulations [34–38].
Antibody studies have revealed the residues 65–75 to containthe bilirubin binding motif [19,20,29,31]. This region is located imme-diately before the transmembrane region TM2, thus indicating the in-volvement of TM2 in forming the transporting channel. Furthermore,inhibition of transport by antibody raised against residues 235–246shows the transmembrane region TM3 (220–238) to be also impor-tant in transporting mechanism of the protein [29,31]. Therefore, adetailed study of both transmembrane regions TM2 and TM3 wereperformed considering their functional importance. The transmem-brane region TM3 was reported previously and its helical structureis found to be stable [33]. In the present work, we have continuedthe molecular dynamic simulations and NMR studies on transmem-brane segments TM2, TM2A and TM2B (BTL residues 73–95, 75–94,and 73–99, respectively) to determine their structure in a membranelike environment. In addition, CD measurements for TM2B and TM3were also performed.
2. Materials and methods
2.1. Synthetic TM2B and TM3 peptides
Synthetic peptides used in this study were purchased fromCASLO Laboratory, Denmark (www.caslo.com). The peptides TM2Band TM3 correspond to bilitranslocase (UniProt O88750) regionsSer73–Leu99 and Gly220–Tyr238, respectively. They were synthe-sized as lyophilized trifluoroacetate salts. The synthetic sequencesof TM2B and TM3 were SSFCLFVATLQSPFSAGVSGLCKAILLKKKKand GSVQCAGLISLPIAIEFTKKKK, respectively, with purity higherthan 93.8%. The sequences include the predicted transmembraneregions, along with a few amino acid residues beyond the trans-membrane region termini in case of TM2B. Due to extreme hydro-phobicity there are four lysines (a Lys tag KKKK) added at theC-terminus to avoid problems during synthesis, purification andNMR sample preparation. The four Lys tags do not replace any ofthe amino acid at the C-termini of the TM2B or TM3 sequence;they are just added to the desired AA sequence. Throughout of fur-ther discussion we shall refer to the peptide chain Ser73 to Leu99 asa prolonged second transmembrane segment TM2B.
2.2. Transmembrane region prediction of BTL using chemometrics model
We have done the initial prediction of the transmembrane regionsof BTL using the first version of the alpha transmembrane regionprediction model [32]. The alpha transmembrane region predictiontool is a data-driven model based on a counter-propagation neuralnetwork (CPNN) classifier built using transmembrane and non-transmembrane protein segments, which are characterized mathe-matically using amino acid adjacency matrix. The developed classifiershows 90.75% of overall prediction accuracy when it is challengedwith external validation set.
The 340 residues long BTL sequence was segmented into 329overlapping peptides, each of which was 20 amino acids long. TheCPNN classifier then predicted each of the segments as either trans-membrane or non-transmembrane. Based on this CPNN classification,the transmembrane regions of BTL were predicted.
2.3. Statistics based scoring function for refinement of N- and C-terminiof BTL transmembrane segments
In order to improve the prediction of the transmembrane regions,especially the transmembrane region termini, we have incorporatedstatistical data in the enhanced prediction model [33]. Position specif-ic amino acid preference was calculated for the terminal positionsof experimentally known transmembrane regions in a large non-redundant data set derived from Protein Data Bank. The developedamino acid preference patterns were used to score the probabletransmembrane region termini. We considered all possible terminalresidue combinations from the initial prediction made by the CPNNclassifier. Each combination was then scored using the amino acidpreference data. Terminal positions with preferred amino acidswere awarded with positive scores, whereas presence of statisticallyavoided amino acids was penalized. One of the top 3 scoring seg-ments meeting a particular length and position criteria was reportedas the final transmembrane region. As the prediction was based onstatistical data, the termini of the predicted transmembrane regionsof BTL are considered to be more statistically favorable and plausiblycloser to experimental observations.
2.4. Preparation of the predicted BTL transmembrane α-helix TM2 for themolecular dynamic simulation
Molecular systems were constructed, simulated and analyzedusing CHARMM modeling environment [39] by analogous methodol-ogy as used in our previous study of studying the stability of the TM3segment [33]. The initial conformation for the amino acid sequencethat was predicted to encompass the transmembrane region TM2(BTL residues: 73–95) SSFCLFVATLQSPFSAGVSGLCK from the initialchemometrics prediction model (described in Section 2.2) and anoth-er conformation of the recently developed (Section 2.3) predictionmodel incorporating statistical data TM2A (BTL residues: 75–94)FCLFVATLQSPFSAGVSGLC were generated by available CHARMM to-pology and structural libraries [40–43]. An α-helical conformationfor each linear sequence was generated by constraining the backbonetorsion angles φ and ψ to the values of −57° and −47° for eachamino acid backbone torsion angle [44]. Analogously as in our previ-ous study [33], additional amino acids corresponding to the residueslocated prior to the start (71–72 BTL residues HL for TM2 systemand 73–74 BTL residues SS from the TM2A system) and subsequentto (96–97 BTL residues AI for TM2 system and 95–96 BTL residuesKA from the TM2A system) the end of the transmembrane helix onthe BTL sequences were added on the C-terminal and N-terminalend of TM2 and TM2A. These additional amino acid residues werenot constrained to the alpha helical conformation.
CHARMM-GUIMembrane builderwas utilized for the construction ofthe membrane-lipid system [45]. The α-helices TM2 and TM2A were

2611A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
oriented along the Z-axis. Dipalmitoylphosphatidylcholine (DPPC) lipidmolecules were selected to represent the lipid bilayer [46]. A rectangularbox consisted of two layers of lipids along with 12 Å broad layers ofwater molecules above and below the lipid surface. CHARMM-GUI wasalso used to construct the individual parts of this complex system. Eachα-helix was inserted into the membrane using the insertion method,where a protein is inserted into a pre-equilibrated lipid bilayer [39,45].The final molecular systems comprised 18,707 (TM2 system) and16,750 atoms (TM2A system), containing 81 (TM2 system) and 72(TM2A system) DPPC molecules respectively.
2.5. Molecular dynamics simulation procedure
Molecular dynamics (MD) calculationswere performed by CHARMMmolecular modeling suite [39]. CHARMM parameter and topology files(version 27) for proteins and lipids were utilized to specify the forcefield parameters of the protein and lipid DPPC molecules [40–43]. Equil-ibration of themembranewas performed using the analogous scheme asin our previous study [33] resulting in a total of 375 ps simulation time.The systemwas primaryminimized for 500 steps using steepest descent(SD) method followed by 500 steps of the modified Adopted BasisNewton–Raphson (ABNR) method.
The equilibration of the system was performed in 6 consecutivesteps with the use of several force constants (See Table S1 of theSupporting material for the definition and description of the usedforce constraints at each equilibration step). In the first two equilibra-tion steps (1–2) the system was simulated for 25 ps by Langevin dy-namics using 1 fs time step. Further four equilibration steps (3–6)were performedwith the standardMD procedure by leapfrog integra-tion scheme. To reduce the possible problemwith the numerical inte-gration, 1 fs time-step was used in the third step with the totalequilibration time of 25 ps. In the next stages of the integrationscheme, 2 fs step along with SHAKE algorithm was applied. The sim-ulation times for steps 4–6 were 100 ps long [45].
Production trajectories were generated using a leapfrog integra-tion scheme and 2 fs simulation step using SHAKE algorithm. 20 nslong MD simulation was performed for both TM2 and TM2A trajecto-ries. Results of the MD simulations were visualized and analyzedusing VMD [47,48] and Gnuplot program [49].
2.6. CD measurements
CD spectra were recorded for TM2 and TM3 fragments of BTL pro-tein using Jasco J-815 spectropolarimeter at 25 °C. Experiments wereperformed over the 185–260 nm range, every spectrumwas repeatedthree times to increase signal-to-noise ratio. All measurements wereprovided on 0.15 mg/ml peptide solutions using a 2 cm/min scanspeed. pH values of solutions were controlled just before start theexperiments and varied between 6.5 and 7.0 in aqueous solution,with addition of 20 mM SDS, 20 mM DPC, and 20 mM DPC:SDSmicelle with molar ratio 5:1. Final spectra were corrected bysubtracting the background and analyzed as mean molar ellipticityΘ (degree × cm2 × dmol−1) vs. wavelength λ (nm). The content ofthe secondary structure was calculated from the spectra using aCONTINLL method [50].
The analysis is based on the approximation that CD spectrumcould be presented as linear combination of secondary structure com-ponents (Eq. (1)):
CDλ ¼ ∑ f kCkλð Þ ð1Þ
where fk is the fraction of each secondary structure, and Ckλ compo-nent is the ellipticity of kth secondary structural element at eachwavelength. In the case of TM2 analysis, the Ckλ component was de-termined from the reference database containing 37 soluble and 13membrane proteins (SMP50) with known secondary structures.
2.7. NMR spectroscopy
The NMR sample was obtained by making a solution of about1 mM TM2B fragment in 90%/10% H2O/D2O containing about 32 mgof deuterated sodium dodecyl sulfate micelles (SDS-d25) (Sigma Al-drich). The SDS-d25:TM2 ratio was kept approximately as 40:1which is exceeded the CMC (8.3 mM) at pH 6.5. An ultrasound bathwas used for nearly 20 min to increase homogeneity of the complex.
All NMR data sets were acquired at 303 K using two Agilent VNMRS800 NMR spectrometers operated at 18.8 T (1H resonance frequency799.81 or 799.94 MHz). Both spectrometers were equipped with fourchannels, gradient unit along z-axis, DirectDrive console and 1H/13C/15Nprobe-head with inverse detection. Additionally, some of NMRexperiments were performed at the same temperature on VarianUnity + 500 NMR spectrometer (1H resonance frequency 500.61 MHz)equippedwith three channels, z-gradient unit and 1H/13C/15N probeheadwith inverse detection. 2D 1H–1H TOCSY spectra were recorded withmixing times 15 and 80 ms using MLEV-17 pulse scheme for spinlock[51]. In order to eliminate effect spin diffusion, 2D 1H–1H NOESY datasets were acquired with 80, 120 and 150 ms mixing times. 13C and 15Nchemical shifts were assigned in base heteronuclear 2D 1H–15N HSQCand 1H–13C HSQC experiments performed on natural abundance of 13Cand 15N isotopes. All chemical shifts were referenced with respectto external sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) usingΞ = 0.251449530 andΞ = 0.101329118 ratios for indirectly referenced13C and 15N resonances, respectively [52]. All recorded NMR data wereprocessed by NMRPipe software [53] and analyzed with the Sparky pro-gram [54].
2.8. Structure calculation of TM2 peptide
3D structure calculations were performed with CYANA 2.1 [55] soft-ware package. The calculation protocol initialized with 200 structurescreated with randomly chosen torsion angles. The 250 nontrivial (139intraresidual, 78 sequential, and 33 medium range) 1H–1H distanceconstraints were evaluated from 2D homonuclear NOESY spectrum ac-quired with mixing times 120 ms which were applied for structure so-lution (Table 1). The calibration of peaks volume to distance constraintswas done with the CALIBA followed with simulated annealing proce-dure included 10,000 steps of molecular dynamic in torsion anglespace [55].
2.9. Construction of TM2 peptide in SDS micelle and molecular dynamicsimulations
The initial construction of a single molecule of SDS was done onthe basis of previously published parameters [56,57]. After minimiza-tion in vacuum, a starting model containing 60 monomers of SDS wasplaced in a cubic box enlarged by about 8 Å in each direction that in-cluded 7126 water molecule [58]. To reach an equilibrium density ofthe entire system, a 1 ns NTP MD simulation was carried out.
The simulations with the program AMBER 11 [59] were started bypositioning the TM2 fragment into the box with its center of mass co-inciding with SDS micelle. Total charge +5 of the entire system wereneutralized using chloride ions. To remove the initial bad contactbetween peptide and SDS micelle core and preventing water penetra-tion during equilibration the peptide and bulk water were kept underweak harmonic constraints for initial 20,000 steps of minimization(the steepest descent method). The entire system was minimizedfor 20,000 steps without any constraints. After that, the wholecomplex was subjected to molecular dynamic simulations withtime-averaged (TAV) distance restraints derived from NMR spectros-copy with the force constants for interproton connectivities equalto 50 kcal/(mol × Å2). The geometry of all peptide bonds waskept trans-, as follows from NMR data, with the force constantf = 50 kcal/(mol × rad2). MD simulations were performed with a

Table 1Statistic of distance constraints used for high-resolution 3D structure calculations andquality ensemble of 10 NMR derived structures of TM2B peptide on the last steps ofMD simulations with TAV.
Total number of NOE restraints 250Intraresidual (|i − j| = 0) 139Sequential (|i − j| = 1) 78Medium-range (|i − j| ≤ 5) 33Long-range (|i − j| > 5) 0
Structure Z-score1-st generation packing quality 3.97 ± 1.592-nd generation packing quality 3.85 ± 1.36Ramachandran plot appearance −0.74 ± 0.48χ1/χ2 rotamer normality −2.58 ± 0.63
RMS Z-scoresBond lengths 1.200 ± 0.004Bond angles 0.718 ± 0.015Side chains planarity 0.384 ± 0.079Improper dihedral distribution 1.077 ± 0.046
Ramachandran plot summary for residues 75…99 (%)Backbone atoms 0.30 ± 0.08All heavy atoms 0.45 ± 0.08
2612 A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
time step 2 fs and 9.0 Å cutoff radius. The coordinates wererecorded at 4 ps (2000-th step) each and 10 conformationspresented the last steps of MD simulation were chosen for finalstructure analysis.
In order to assess the influence of the parameters needed to startthe molecular dynamic simulations, a different protocol was tested.The 80 SDS molecules together with one molecule of TM2 peptidewere positioned in the water box with a volume corresponding tothe SDS concentration in NMR experiments (170 mM). The simula-tions with previously evaluated distance constraints (TAV protocol)were performed for 17 ns long trajectories. The evaluated structuraldata were compared with those obtained by the initially appliedprotocol.
3. Results and discussion
3.1. BTL transmembrane regions predicted by the chemometrics models
The initial prediction of the transmembrane regions of BTL wasperformed using the developed alpha transmembrane region predic-tion model as described previously [32]. We considered only longstretches of 10 or more consecutive overlapping segments predictedas transmembrane for the final prediction of the transmembrane re-gions, and further analyzed them. Four such stretches of consecutivesegments were predicted as transmembrane in case of BTL thatspan over residues 16–53 (19 segments), 63–103 (20 segments),213–246 (15 segments), and 250–285 (17 segments). As the proteinsegments are overlapping, instead of reporting these whole stretchesas transmembrane regions, only the central residues of these trans-membrane segment stretches were considered. This was to makesure that the residues in predicted transmembrane regions werecommon to more segments predicted as transmembrane than asnon-transmembrane. The initial transmembrane regions of BTL,TM1, TM2, TM3, and TM4, were accordingly predicted to be at resi-dues 24–45, 73–95, 221–238, and 258–277, respectively [32].
In the next step, based on the initial prediction, the transmem-brane regions were predicted utilizing statistical data on amino acidpreference. The incorporation of statistical data helps in fine-tuningthe transmembrane region termini, and makes the predictions morestatistically plausible. Instead of reporting the central residues ofthe predicted transmembrane stretch as the final transmembrane re-gion, the peptide sequences, defined by all the possible combinationsof the termini of the constituent overlapping segments forming a
transmembrane stretch, are considered as candidates for the trans-membrane region. Each peptide sequence, thus formed, was thenscored using the statistically generated amino acid preference pat-terns. The transmembrane region was predicted such that it wasbounded by best scoring combination of terminal residues. In caseof transmembrane region TM2 of BTL, the initially predicted region73–95 had a lower statistical score than the top scoring region,which was found to be at 75–94 (TM2A). The other three transmem-brane regions predicted are TM1A 24–48, TM3A 220–238, and TM4A254–276. Therefore, in both the initial and improved predictions, thecentral residues of the transmembrane regions remain same withonly variation at the terminal residues. The predicted transmembraneregion is therefore essentially similar in both cases, with TM2A beingmore statistically favored.
3.2. Assessment of stability of the predicted BTL transmembrane helix 2by the MD simulation in the DPPC membrane
MD simulation was performed for BTL transmembrane helix TM2(residues 73–95) and its variant TM2A (residues 75–94) to investi-gate if the BTL sequences could adopt a stable helical conformation.In order to enable a comparison with our previous study [33] thesame protocol of the data analysis was applied.
The structure of the starting transmembrane α-helices TM2 andTM2A is shown in Fig. 1. Two additional amino acids were added onthe C-terminal and the N-terminal end of each of the α helices inorder to enable both ends of the α helix to explore more conforma-tional space and soften the boundary. The water molecules mimickedextracellular and intracellular compartments. Despite reasonablylong 20 ns MD simulation runs a caveat must be stated that this sim-ulation time does not enable a full coverage of the conformationalspace for an unambiguous quantitative stability assessment [33].The produced trajectories were visually inspected for overall conforma-tional changes and in both systems under study (TM2 and TM2A) theα-helical conformation was retained. High resolution animations forboth TM2 and TM2A systems are provided (see Supp. Mat., Movie S1and Movie S2).
Representative snapshots of the α-helices TM2 and TM2A fromthe molecular dynamics (MD) simulation in the lipid membrane arepresented in Fig. 1. Their secondary structure was visualized usingSTRIDE algorithm within VMD software suite [48]. The conformation-al behavior in time showed comparable overall structure. Both se-quences TM2 and TM2A displayed preserved α-helical secondaryconformation. The presence of proline in the predicted transmem-brane sequence (Pro85) resulted in an expected kink formation inboth helices due to the sterical interference with the backbone ofthe preceding turn. These observations are in line with literaturedata regarding the secondary structure behavior of helices in mem-brane environment [60,61]. Also similar effect was observed in ourprevious study of TM3 where a Pro231 in the transmembraneα-helix showed analogous behavior [33]. Thus, from the qualitativeperspective stable helix was retained for both helices within theperformed MD simulation times.
Subsequently, RMSD values for all backbone atoms initially gener-ated in the α helical positions were calculated. The RMSD values werecalculated by aligning all conformations of the individual α helix tothe last MD-generated α-helix conformation [33]. For the structuralalignment, backbone atoms (C', CA, N) were used for those residuesthat were predicted to form the α-helix by the chemometrics predic-tions [32]. Obtained RMSD parameters corroborated that α-helix con-formations of both TM2 and TM2A did not undergo substantialchanges during the MD simulations. The average RMSD for helixTM2 was 0.72 Å with a standard deviation of 0.29 Å, and for thehelix TM2A the average RMSD value was 0.59 Å with the standard de-viation of 0.21 Å. Also RMSD graphs were plotted for both simulatedsystems as a function of the simulation time and are schematically
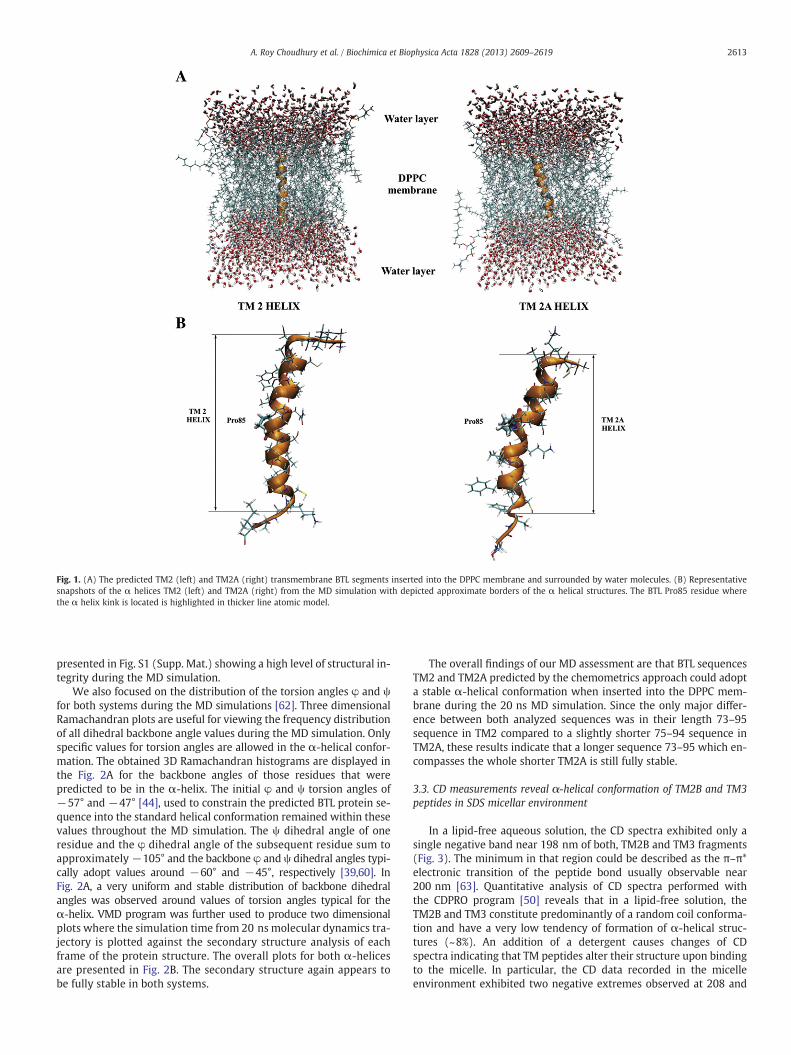
Fig. 1. (A) The predicted TM2 (left) and TM2A (right) transmembrane BTL segments inserted into the DPPC membrane and surrounded by water molecules. (B) Representativesnapshots of the α helices TM2 (left) and TM2A (right) from the MD simulation with depicted approximate borders of the α helical structures. The BTL Pro85 residue wherethe α helix kink is located is highlighted in thicker line atomic model.
2613A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
presented in Fig. S1 (Supp. Mat.) showing a high level of structural in-tegrity during the MD simulation.
We also focused on the distribution of the torsion angles φ and ψfor both systems during the MD simulations [62]. Three dimensionalRamachandran plots are useful for viewing the frequency distributionof all dihedral backbone angle values during the MD simulation. Onlyspecific values for torsion angles are allowed in the α-helical confor-mation. The obtained 3D Ramachandran histograms are displayed inthe Fig. 2A for the backbone angles of those residues that werepredicted to be in the α-helix. The initial φ and ψ torsion angles of−57° and −47° [44], used to constrain the predicted BTL protein se-quence into the standard helical conformation remained within thesevalues throughout the MD simulation. The ψ dihedral angle of oneresidue and the φ dihedral angle of the subsequent residue sum toapproximately −105° and the backbone φ and ψ dihedral angles typi-cally adopt values around −60° and −45°, respectively [39,60]. InFig. 2A, a very uniform and stable distribution of backbone dihedralangles was observed around values of torsion angles typical for theα-helix. VMD program was further used to produce two dimensionalplots where the simulation time from 20 ns molecular dynamics tra-jectory is plotted against the secondary structure analysis of eachframe of the protein structure. The overall plots for both α-helicesare presented in Fig. 2B. The secondary structure again appears tobe fully stable in both systems.
The overall findings of our MD assessment are that BTL sequencesTM2 and TM2A predicted by the chemometrics approach could adopta stable α-helical conformation when inserted into the DPPC mem-brane during the 20 ns MD simulation. Since the only major differ-ence between both analyzed sequences was in their length 73–95sequence in TM2 compared to a slightly shorter 75–94 sequence inTM2A, these results indicate that a longer sequence 73–95 which en-compasses the whole shorter TM2A is still fully stable.
3.3. CD measurements reveal α-helical conformation of TM2B and TM3peptides in SDS micellar environment
In a lipid-free aqueous solution, the CD spectra exhibited only asingle negative band near 198 nm of both, TM2B and TM3 fragments(Fig. 3). The minimum in that region could be described as the π–π*electronic transition of the peptide bond usually observable near200 nm [63]. Quantitative analysis of CD spectra performed withthe CDPRO program [50] reveals that in a lipid-free solution, theTM2B and TM3 constitute predominantly of a random coil conforma-tion and have a very low tendency of formation of α-helical struc-tures (~8%). An addition of a detergent causes changes of CDspectra indicating that TM peptides alter their structure upon bindingto the micelle. In particular, the CD data recorded in the micelleenvironment exhibited two negative extremes observed at 208 and

Fig. 2. (A) Three dimensional Ramachandran histograms for the backbone torsion angles ϕ and ψ which were predicted to be a part of the transmembrane helix (generated forresidues: 73–95 TM2 helix and 75–94 TM2A helix), (B) Two dimensional plots of the secondary structure analysis of each frame of the protein structure against simulation timefrom the MD simulation. Purple color depict the α helix structure (green indicated the turn structure and blue depicted the 310 helical structure) Residue numbers on the y-axisare from 1–27 or 1–24 which corresponds to 73–95 or 75–94 residues of the BTL sequence.
2614 A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
220 nm, which is characteristic for α-helices. Quantitative analysisindicates that the helical structure content varies from 32% to 42 %in micelle-bound TM2B which is higher than that in previously stud-ied TM3 peptide, where the α-helical conformation does not exceed25% in any of the studied micelle-bound state. Moreover, the leveland/or stabilization of the helical fold in the TM2B and TM3 appearto depend on the nature of the polar head of the lipids. The CD spectraof TM2B segment in DPC exhibited higher population of α-helical
conformation than in SDS, whereas in case of TM3 fragment, theopposite effect is detected. The binding of peptides to a micelle is acomplex dynamic process controlled by two major non-covalent in-teractions: electrostatic interactions between the polar head groupson the micelle surface and the “charged” side chains of the peptideand hydrophobic interactions between the hydrophobic side chainsof the peptide and the hydrophobic core of the micelle [64]. Thus,the dissimilarity in helicity in TM2B and TM3 peptides in different
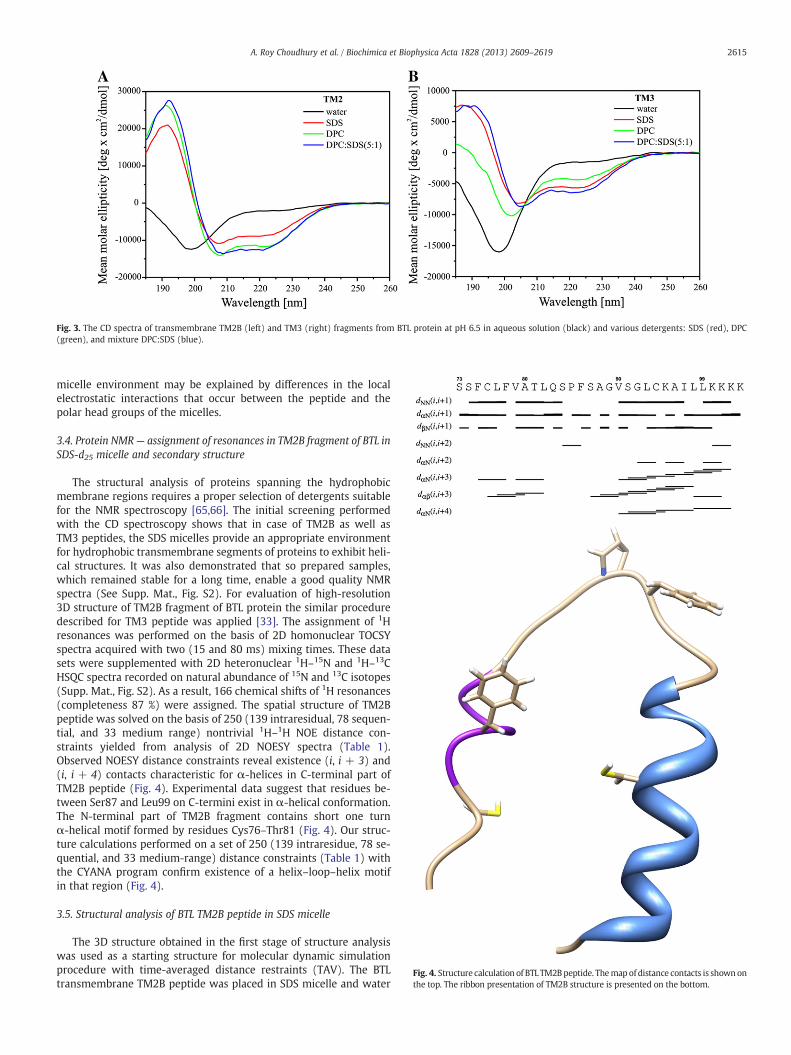
Fig. 3. The CD spectra of transmembrane TM2B (left) and TM3 (right) fragments from BTL protein at pH 6.5 in aqueous solution (black) and various detergents: SDS (red), DPC(green), and mixture DPC:SDS (blue).
Fig. 4. Structure calculation of BTL TM2Bpeptide. Themapof distance contacts is shownonthe top. The ribbon presentation of TM2B structure is presented on the bottom.
2615A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
micelle environment may be explained by differences in the localelectrostatic interactions that occur between the peptide and thepolar head groups of the micelles.
3.4. Protein NMR— assignment of resonances in TM2B fragment of BTL inSDS-d25 micelle and secondary structure
The structural analysis of proteins spanning the hydrophobicmembrane regions requires a proper selection of detergents suitablefor the NMR spectroscopy [65,66]. The initial screening performedwith the CD spectroscopy shows that in case of TM2B as well asTM3 peptides, the SDS micelles provide an appropriate environmentfor hydrophobic transmembrane segments of proteins to exhibit heli-cal structures. It was also demonstrated that so prepared samples,which remained stable for a long time, enable a good quality NMRspectra (See Supp. Mat., Fig. S2). For evaluation of high-resolution3D structure of TM2B fragment of BTL protein the similar proceduredescribed for TM3 peptide was applied [33]. The assignment of 1Hresonances was performed on the basis of 2D homonuclear TOCSYspectra acquired with two (15 and 80 ms) mixing times. These datasets were supplemented with 2D heteronuclear 1H–15N and 1H–13CHSQC spectra recorded on natural abundance of 15N and 13C isotopes(Supp. Mat., Fig. S2). As a result, 166 chemical shifts of 1H resonances(completeness 87 %) were assigned. The spatial structure of TM2Bpeptide was solved on the basis of 250 (139 intraresidual, 78 sequen-tial, and 33 medium range) nontrivial 1H–1H NOE distance con-straints yielded from analysis of 2D NOESY spectra (Table 1).Observed NOESY distance constraints reveal existence (i, i + 3) and(i, i + 4) contacts characteristic for α-helices in C-terminal part ofTM2B peptide (Fig. 4). Experimental data suggest that residues be-tween Ser87 and Leu99 on C-termini exist in α-helical conformation.The N-terminal part of TM2B fragment contains short one turnα-helical motif formed by residues Cys76–Thr81 (Fig. 4). Our struc-ture calculations performed on a set of 250 (139 intraresidue, 78 se-quential, and 33 medium-range) distance constraints (Table 1) withthe CYANA program confirm existence of a helix–loop–helix motifin that region (Fig. 4).
3.5. Structural analysis of BTL TM2B peptide in SDS micelle
The 3D structure obtained in the first stage of structure analysiswas used as a starting structure for molecular dynamic simulationprocedure with time-averaged distance restraints (TAV). The BTLtransmembrane TM2B peptide was placed in SDS micelle and water

2616 A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
box and subjected to MD simulations with AMBER 11 workpackage[59]. The protocol of creation micelle and all other parameters usedfor simulations were described in detail in our previous publication[33]. Due to a long time needed for stabilization of the position ofTM2B peptide inside the SDS micelle, the time of MD trajectory wasextended to 10 ns (Supp. Mat., Fig. S3), which is two times longerthan the TM3 peptide simulations. All experimental data used forstructure evaluation and quality analysis of solved 3D structure withWhatIf program are presented in Table 1.
The 3D structure of BTL transmembrane TM2B peptide evaluatedafter 10 ns MD simulations appears as helix–loop–helix motifwith two α-helices buried inside hydrophobic part of SDS micelle(Fig. 5). The N- and C-terminal α-helices contain residues Cys76–Leu82 and Phe86–Leu99, respectively. The Pro85 in loop region is inconformation trans- and the interhelical angle between N- andC-terminal α-helices is 80 ± 2.5° (Fig. 5). The residues Gln83–Pro85that constitute the central loop are located on the outer part of SDSmicelle and accessible for solvent (Fig. 5). Due to expected strong in-teractions of C-terminal Lys-Tag with the micelle surface, the MD sim-ulations were performed with another protocol excluded ‘size effect’of the media (described in Section 2.8). The calculations exhibiteda similar fold of TM2B fragment in SDS detergent, characterizedwith exposed central loop and interhelical angle between N- andC-terminal helices of nearly 80° (See Supp. Mat., Fig. S4).
The NMR studies revealed the transmembrane region TM2B to bein helix–loop–helix motif. This finding is not uncommon in view ofthe recent extensive study of transmembrane proline-containing pep-tides in various lipid environments (DLPC (1,2-dilauroyl-sn-glycero-3-phosphocholine), DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine)and DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) lipid bilayers ofdifferent thicknesses) using solid stateNMR [61,67]. Significantly, regard-less of lipid identity the segmental directions of peptide segment on ei-ther side of the proline adopt significant tilt angles (from 0° to 70°)with respect to the lipid membrane normal. The effect of the missingintra-helical NH…O = C hydrogen bond between the proline (whichhas no NH group) and the amino acid four residues earlier in theα-helical sequence causing the distortion was further enhanced by theeffect of globular micelle structure (see graphic abstract). This is coupledto the fact that the peptide is buried in the hydrophobic SDS micelle andthe effect of the aqueous phase surrounding the micelle cannot be ruled
Fig. 5. High-resolution 3D structure of transmembrane TM2B fragment of BTL protein in SDS11 program suite. The structure presents as helix–loop–helix motif with Pro85 as a central
out in a simple way. Thus, the peptide tilted shape appears to be plausi-ble. It awaits further study before we canmake a rationale about its pos-sible role in the BTL transmembrane functional properties.
The two helices were found at positions Cys76–Thr81 and Ser87–Leu99, and the complete transmembrane region consisted of residuesCys76–Leu99. Even the five residues (Lys95–Leu99) that extend be-yond the C-terminal of the transmembrane region TM2A (Phe75–Cys94) predicted by the model, show helical structure, which is stillin accordance with the prediction results (Fig. 6). It has to be statedthat in order to facilitate the synthesis and purification the peptide se-quencewas amended by addition of a Lys Tag containing four lysine res-idues (KKKK) at the C terminal. Interestingly, the structure of this tail isalso helical (Fig. 5), however, such observation can be discarded as anartifact of little importance for the main purpose of the present study.
As the final transmembrane regions were predicted using statisti-cal amino acid preference pattern, the region Phe75–Cys94 was foundto be more statistically favorable with higher scores for the N- andC-terminal positions. However, both regions Phe75–Cys94 (fromfinal prediction model) and Cys76–Leu99 (from NMR studies) camefrom the transmembrane region stretch Gly65–Gln103 as predictedby CPNN classifier. As explained before (Sections 2.2, 2.3 and 3.1),all the transmembrane regions from a single transmembrane stretchhave the same conserved core with different combinations of termi-nal residues, and all have the equal probability to represent the actualtransmembrane fragment. Therefore, the BTL segments Phe75–Cys94or Cys76–Leu99, sharing the same conserved core residues, have anequal probability to form the transmembrane region.
Residues from TM2B fragment exposed to solution could be select-ed with Radial Distribution Function (RDF) (Fig. S5) evaluated withthe ptraj program from AMBER 11 software suite on the basis of thehigh-resolution 3D structure. A detail analysis of extracted data ex-hibits Ser73, Ser74, Cys76, Ala80, Gln83, Pro85, Ala88, and Ser91 asresidues with high solubility of side chains (Fig. 6A). As followsfrom the list, those residues constitute mostly N-termini and the cen-tral loop of the helix–loop–helix motif which are more accessible tosolvent due to their location in the outer region of the SDS micelle.In comparison with the previously determined high-resolution 3Dstructure of TM3 fragment in SDS media, a stronger kink is observedin the helix–loop–helix motif of the TM2B segment. Also the solventaccessible surface appears to be more evenly shaped (Fig. 6B).
micelle obtained after 10 ns molecular dynamic simulations using TAV with the AMBERresidue.
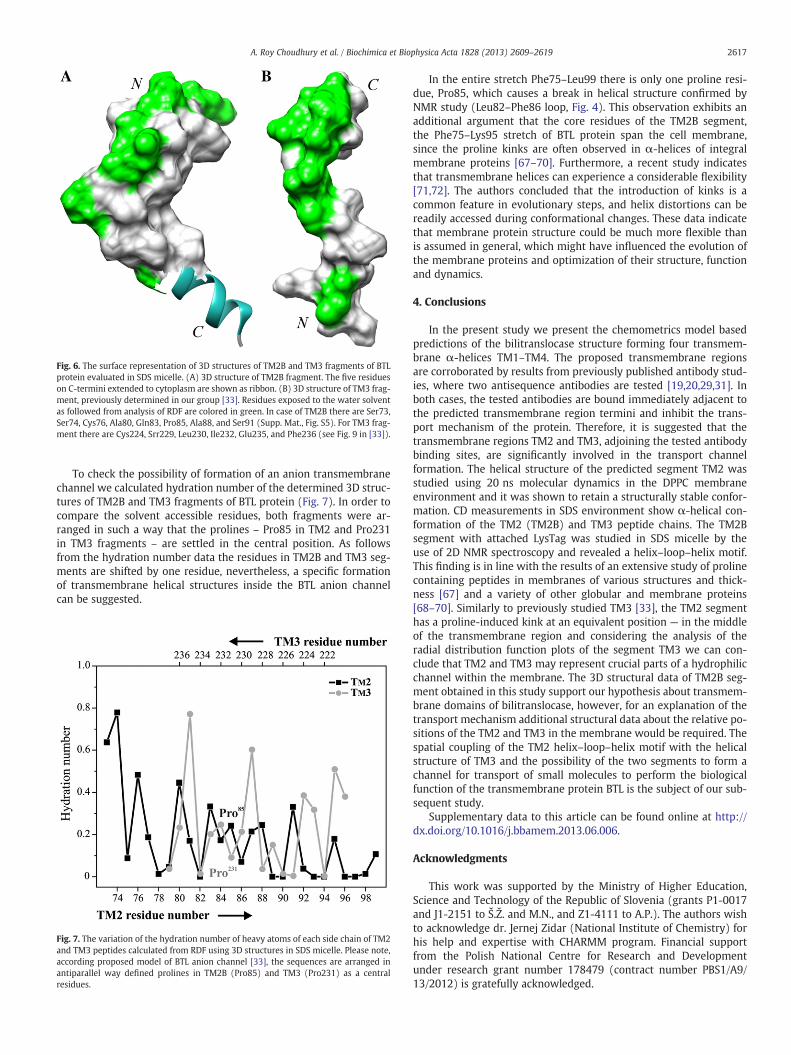
Fig. 6. The surface representation of 3D structures of TM2B and TM3 fragments of BTLprotein evaluated in SDS micelle. (A) 3D structure of TM2B fragment. The five residueson C-termini extended to cytoplasm are shown as ribbon. (B) 3D structure of TM3 frag-ment, previously determined in our group [33]. Residues exposed to the water solventas followed from analysis of RDF are colored in green. In case of TM2B there are Ser73,Ser74, Cys76, Ala80, Gln83, Pro85, Ala88, and Ser91 (Supp. Mat., Fig. S5). For TM3 frag-ment there are Cys224, Srr229, Leu230, Ile232, Glu235, and Phe236 (see Fig. 9 in [33]).
2617A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
To check the possibility of formation of an anion transmembranechannel we calculated hydration number of the determined 3D struc-tures of TM2B and TM3 fragments of BTL protein (Fig. 7). In order tocompare the solvent accessible residues, both fragments were ar-ranged in such a way that the prolines – Pro85 in TM2 and Pro231in TM3 fragments – are settled in the central position. As followsfrom the hydration number data the residues in TM2B and TM3 seg-ments are shifted by one residue, nevertheless, a specific formationof transmembrane helical structures inside the BTL anion channelcan be suggested.
Fig. 7. The variation of the hydration number of heavy atoms of each side chain of TM2and TM3 peptides calculated from RDF using 3D structures in SDS micelle. Please note,according proposed model of BTL anion channel [33], the sequences are arranged inantiparallel way defined prolines in TM2B (Pro85) and TM3 (Pro231) as a centralresidues.
In the entire stretch Phe75–Leu99 there is only one proline resi-due, Pro85, which causes a break in helical structure confirmed byNMR study (Leu82–Phe86 loop, Fig. 4). This observation exhibits anadditional argument that the core residues of the TM2B segment,the Phe75–Lys95 stretch of BTL protein span the cell membrane,since the proline kinks are often observed in α-helices of integralmembrane proteins [67–70]. Furthermore, a recent study indicatesthat transmembrane helices can experience a considerable flexibility[71,72]. The authors concluded that the introduction of kinks is acommon feature in evolutionary steps, and helix distortions can bereadily accessed during conformational changes. These data indicatethat membrane protein structure could be much more flexible thanis assumed in general, which might have influenced the evolution ofthe membrane proteins and optimization of their structure, functionand dynamics.
4. Conclusions
In the present study we present the chemometrics model basedpredictions of the bilitranslocase structure forming four transmem-brane α-helices TM1–TM4. The proposed transmembrane regionsare corroborated by results from previously published antibody stud-ies, where two antisequence antibodies are tested [19,20,29,31]. Inboth cases, the tested antibodies are bound immediately adjacent tothe predicted transmembrane region termini and inhibit the trans-port mechanism of the protein. Therefore, it is suggested that thetransmembrane regions TM2 and TM3, adjoining the tested antibodybinding sites, are significantly involved in the transport channelformation. The helical structure of the predicted segment TM2 wasstudied using 20 ns molecular dynamics in the DPPC membraneenvironment and it was shown to retain a structurally stable confor-mation. CD measurements in SDS environment show α-helical con-formation of the TM2 (TM2B) and TM3 peptide chains. The TM2Bsegment with attached LysTag was studied in SDS micelle by theuse of 2D NMR spectroscopy and revealed a helix–loop–helix motif.This finding is in line with the results of an extensive study of prolinecontaining peptides in membranes of various structures and thick-ness [67] and a variety of other globular and membrane proteins[68–70]. Similarly to previously studied TM3 [33], the TM2 segmenthas a proline-induced kink at an equivalent position — in the middleof the transmembrane region and considering the analysis of theradial distribution function plots of the segment TM3 we can con-clude that TM2 and TM3 may represent crucial parts of a hydrophilicchannel within the membrane. The 3D structural data of TM2B seg-ment obtained in this study support our hypothesis about transmem-brane domains of bilitranslocase, however, for an explanation of thetransport mechanism additional structural data about the relative po-sitions of the TM2 and TM3 in the membrane would be required. Thespatial coupling of the TM2 helix–loop–helix motif with the helicalstructure of TM3 and the possibility of the two segments to form achannel for transport of small molecules to perform the biologicalfunction of the transmembrane protein BTL is the subject of our sub-sequent study.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamem.2013.06.006.
Acknowledgments
This work was supported by the Ministry of Higher Education,Science and Technology of the Republic of Slovenia (grants P1-0017and J1-2151 to Š.Ž. and M.N., and Z1-4111 to A.P.). The authors wishto acknowledge dr. Jernej Zidar (National Institute of Chemistry) forhis help and expertise with CHARMM program. Financial supportfrom the Polish National Centre for Research and Developmentunder research grant number 178479 (contract number PBS1/A9/13/2012) is gratefully acknowledged.

2618 A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
References
[1] The Welcome Trust Case Control Consortium, Genome-wide association study of14,000 cases of seven common diseases and 3,000 shared controls, Nature 447(2007) 661–678.
[2] S.R. Pfeffer, Unsolved mysteries in membrane traffic, Annu. Rev. Biochem. 76(2007) 5.1–5.17.
[3] M.A. Zaydman, J.R. Silva, J. Cui, Ion channel associated diseases: overview of mo-lecular mechanisms, Chem. Rev. 112 (2012) 6319–6333.
[4] S.J. Opella, F.M. Marassi, Structure determination of membrane proteins by NMRspectroscopy, Chem. Rev. 104 (2004) 3587–3606.
[5] H.J. Kim, S.C. Howell, W.D. Van Horn, Y.H. Jeon, C.R. Sanders, Recent advances inthe application of solution NMR spectroscopy to multi-span integral membraneproteins, Prog. Nucl. Magn. Reson. Spectrosc. 55 (2009) 335–360.
[6] R. Linser, M. Dasari, U. Fink, P. Schmieder, J.M. Lopez del Amo, S. Marcovic, M.Hiller, H. Oschkinat, D. Oesterheld, B. Reif, Proton-detected solid-state NMRspectroscopy of fibrillar and membrane proteins, Angew. Chem. Int. Ed. Engl. 50(2011) 4508–4512.
[7] J.R. Helliwell, How to solve protein structures with an X-ray laser, Science 339(2013) 146–147.
[8] S. Boutet, L. Lomb, G.J. Williams, T.R.M. Barends, A. Aquila, R.B. Doak, U.Weierstall, D.P. DePonte, J. Steinbrener, R.L. Shoeman, M. Messerschmidt, A.Barty, T.A. White, S. Kassemeyer, R.A. Kirian, M.M. Seibert, P.A. Montanez, C.Kenney, R. Herbst, P. Hart, J. Pines, G. Haller, S.M. Gruner, H.T. Philipp, M.W.Tate, M. Hromalik, L.J. Koerner, N. van Bakel, J. Morse, W. Ghonsalves, D.Arnlund, M.J. Bogan, C. Caleman, R. Fromme, C.Y. Hampton, M.S. Hunter, L.C.Johansson, G. Katona, C. Kupitz, M. Liang, A.V. Martin, K. Nass, L. Redecke, F.Stellato, N. Timneanu, D. Wang, N.A. Zatsepin, D. Schafer, J. Defever, R. Neutze,P. Fromme, J.C.H. Spence, H.N. Chapman, I. Schlichting, High-resolution proteinstructure determination by serial femtosecond crystallography, Science 337(2012) 362–364.
[9] L. Redecke, K. Nass, D.P. DePonte, T.A. White, D. Rehders, A. Barty, F. Stellato, M.Liang, T.R.M. Barends, S. Boutet, G.J. Williams, M. Messerschmidt, M.M. Seibert,A. Aquila, D. Arnlund, S. Bajt, T. Barth, M.J. Bogan, C. Caleman, T.C. Chao, R.B.Doak, H. Fleckenstein, M. Frank, R. Fromme, L. Galli, I. Grotjohann, M.S. Hunter,L.C. Johansson, S. Kassemeyer, G. Katona, R.A. Kirian, R. Koopmann, C. Kupitz, L.Lomb, A.V. Martin, S. Mogk, R. Neutze, R.L. Shoeman, J. Steinbrener, N.Timneanu, D. Wang, U. Weierstall, N.A. Zatsepin, J.C.H. Spence, P. Fromme, I.Schlichting, M. Duszenko, C. Betzel, H.N. Chapman, Natively inhibited Trypanosomabrucei cathepsin B structure determined by using an X-ray laser, Science 339 (2013)227–230.
[10] D.J. Müller, Y.F. Dufrêne, Atomic force microscopy as a multifunctional moleculartoolbox in nanobiotechnology, Nat. Nanotechnol. 3 (2008) 261–269.
[11] G.W. Gordon, G. Berry, X.H. Liang, B. Levine, B. Herman, Quantitative fluorescenceresonance energy transfermeasurements usingfluorescencemicroscopy, Biophys. J. 74(1998) 2702–2713.
[12] R.B. Sekar, A. Periasamy, Fluorescence resonance energy transfer (FRET) micros-copy imaging of live cell protein localizations, J. Cell. Biol. 160 (2003) 629–633.
[13] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N.Shindyalov, P.E. Bourne, The Protein Data Bank, Nucl. Acids. Res. 28 (2000)235–242, (Available: http://www.pdb.org/pdb/home/home.do. Accessed 22 De-cember 2011).
[14] G.E. Tusnady, Z. Dosztanyi, I. Simon, Transmembrane proteins in the Protein DataBank: identification and classification, Bioinformatics 20 (2004) 2964–2972,(Available: http://pdbtm.enzim.hu/. Accessed 22 December 2011).
[15] S. Passamonti, L. Battiston, G.L. Sottocasa, Arylsulfonylation of bilitranslocase inplasma membranes from rat liver enables to discriminate between natural andartificial substrates, Biochim. Biophys. Acta 1323 (1977) 130–136.
[16] S. Passamonti, G.L. Sottocasa, The quinoid structure is the molecular requirementfor recognition of phthaleins by the organic anion carrier at the sinusoidal plasmamembrane level in the liver, Biochim. Biophys. Acta 943 (1988) 119–125.
[17] L. Battiston, S. Passamonti, A. Macagno, G.L. Sottocasa, The bilirubin-binding motifof bilitranslocase and its relation to conserved motifs in ancient biliproteins,Biochem. Biophys. Res. Commun. 247 (1998) 687–692.
[18] A.M. Torres, G.C. Lunazzi, W. Stremmel, C. Tiribelli, Bilitranslocase andsulfobromophthalein/bilirubin-binding protein are both involved in the hepaticuptake of organic anions, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 8136–8139.
[19] A. Brandoni, G. Di Giusto, R. Franca, S. Passamonti, A.M. Torres, Expression of kid-ney and liver bilitranslocase in response to acute biliary obstruction, Nephron.Physiol. 114 (2010) 35–40.
[20] S. Passamonti, M. Terdoslavich, R. Franca, A. Vanzo, F. Tramer, E. Braidot, E.Petrussa, A. Vianello, Bioavailability of flavonoids: a review of their membranetransport and the function of bilitranslocase in animal and plant organisms,Curr. Drug. Metab. 10 (2009) 369–394.
[21] L. Ziberna, M. Lunder, F. Tramer, G. Drevensek, S. Passamonti, The endothelialplasma membrane transporter bilitranslocase mediates rat aortic vasodilation in-duced by anthocyanins, Nutr. Metab. Cardiovasc. 23 (2011) 68–74.
[22] L. Ziberna, F. Tramer, S. Moze, U. Vrhovsek, F. Mattivi, S. Passamonti, Transportand bioactivity of cyanidin 3-glucoside into the vascular endothelium, FreeRadic. Bio. Med. 52 (2012) 1750–1759.
[23] V. Nicolin, V. Grill, F. Micali, P. Narducci, S. Passamonti, Immunolocalisationof bilitranslocase in mucosecretory and parietal cells of the rat gastric mucosa,J. Mol. Histol. 36 (2005) 45–50.
[24] A. Maestro, M. Terdoslavich, A. Vanzo, A. Kuku, F. Tramer, V. Nicolin, F. Micali, G.Decorti, S. Passamonti, Expression of bilitranslocase in the vascular endotheliumand its function as a flavonoid transporter, Cardiovasc. Res. 85 (2010) 175–183.
[25] A. Vanzo, M. Terdoslavich, A. Brandoni, A.M. Torres, U. Vrhovsek, S. Passamonti,Uptake of grape anthocyanins into the rat kidney and the involvement ofbilitranslocase, Mol. Nutr. Food Res. 52 (2008) 1106–1116.
[26] A. Karawajczyk, V. Drgan, N. Medic, G. Oboh, S. Passamonti, M. Novič, Propertiesof flavonoids influencing the binding to bilitranslocase investigated by neuralnetwork modeling, Biochem. Pharmacol. 73 (2007) 308–320.
[27] Š. Župerl, S. Fornasaro, M. Novič, S. Passamonti, Experimental determination andprediction of bilitranslocase transport activity, Anal. Chim. Acta 705 (2011)322–333.
[28] G. Lunazzi, C. Tiribelli, B. Gazzin, G.L. Sottocasa, Further studies on bilitranslocase,a plasma membrane protein involved in hepatic organic anion uptake, Biochim.Biophys. Acta Biomembr. 685 (1982) 117–122.
[29] S. Passamonti, M. Terdoslavich, A. Margon, A. Cocolo, N. Medic, F. Micali, G.Decorti, M. Franko, Uptake of bilirubin into HepG2 cells assayed by thermal lensspectroscopy, FEBS J. 272 (2005) 5522–5535.
[30] S. Passamonti, L. Battiston, G.L. Sottocasa, On the mechanism of bilitranslocasetransport inactivation by phenylmethylsulphonyl fluoride, Mol. Membr. Biol. 16(1999) 167–172.
[31] S. Passamonti, A. Cocolo, E. Braidot, E. Petrussa, C. Peresson, N. Medic, F. Macri, A.Vianello, Characterization of electrogenic bromosulfophthalein transport in car-nation petal microsomes and its inhibition by antibodies against bilitranslocase,FEBS J. 272 (2005) 3282–3296.
[32] A. Roy Choudhury, M. Novič, Data-driven model for the prediction of proteintransmembrane regions, SAR QSAR Environ. Res. 20 (2009) 741–754.
[33] A. Perdih, A. Roy Choudhury, Š. Župerl, E. Sikorska, I. Zhukov, T. Solmajer, M.Novič, Structural analysis of a peptide fragment of transmembrane transporterprotein bilitranslocase, PLoS One 7 (2012) e38967.
[34] A. Elofsson, G. von Heijne, Membrane protein structure: prediction versus reality,Annu. Rev. Biochem. 76 (2007) 125–140.
[35] A. Perdih, M. Kotnik, M. Hodoscek, T. Solmajer, Targeted molecular dynamics sim-ulation studies of binding and conformational changes in E. coliMurD, Proteins 68(2007) 243–254.
[36] J.M. Shea, C.L. Brooks III, From folding theories to folding proteins: a review andassessment of simulation studies of protein folding and unfolding, Annu. Rev.Phys. Chem. 52 (2001) 499–535.
[37] L. Bu, C.L. Brooks III, De novo prediction of the structures of M. tuberculosis mem-brane proteins, J. Am. Chem. Soc. 130 (2008) 5384–5385.
[38] T.H. Duong, E.L. Mehler, H. Weinstein, Molecular dynamics simulation of mem-branes and a transmembrane helix, J. Comput. Phys. 151 (1999) 358–387.
[39] B.R. Brooks, C.L. Brooks III, A.D. MacKerell Jr., L. Nilsson, B. Roux, Y. Won, G.Archontis, C. Bartels, S. Boresch, L. Caves, Q. Cui, A.R. Dinner, S. Fischer, J. Gao,M. Hodoscek, K. Kuczera, T. Lazaridis, J. Ma, E. Paci, R.W. Pastor, R.J. Petrella, C.B.Post, M. Schaefer, B. Tidor, R.M. Venable, H.L. Woodcock, X. Wu, D.M. York, M.Karplus, CHARMM: the biomolecular simulation program, J. Comput. Chem. 30(2009) 1545–1615.
[40] A.D. MacKerell Jr., D. Bashford, M. Bellott, R.L. Dunbrack Jr., J.D. Evanseck, M.J.Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K.Kuczera, F.T.K. Lau, C. Mattos, S. Michnick, T. Ngo, D.T. Nguyen, B. Prodhom,W.E. Reiher III, B. Roux, M. Schlenkrich, J.C. Smith, R. Stote, J. Straub, M.Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, M. Karplus, All-atom empirical poten-tial for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B 102(1998) 3586–3616.
[41] A.D. MacKerell Jr., M. Feig, C.L. Brooks III, Extending the treatment of backboneenergetics in protein force fields: limitations of gas-phase quantum mechanicsin reproducing protein conformational distributions in molecular dynamics sim-ulations, J. Comput. Chem. 25 (2004) 1400–1415.
[42] N. Foloppe, A.D. MacKerell Jr., All-atom empirical force field for nucleic acids:I. Parameter optimization based on small molecule and condensed phase macro-molecular target data, J. Comput. Chem. 25 (2004) 86–104.
[43] A.D. MacKerell Jr., N.K. Banavali, All-atom empirical force field for nucleic acids:II. Application to molecular dynamics simulations of DNA and RNA in solution,J. Comput. Chem. 25 (2004) 105–120.
[44] C. Li, P. Gao, H. Qin, R. Chase, P.L. Gorkov, W.W. Brey, T.A. Cross, Uniformly alignedfull-length membrane proteins in liquid crystalline bilayers for structural charac-terization, J. Am. Chem. Soc. 129 (2007) 5304–5305.
[45] S. Jo, T. Kim, V.G. Iyer, W. Im, CHARMM-GUI: a web-based graphical user interfacefor CHARMM, J. Comput. Chem. 29 (2008) 1859–1865.
[46] J. Zidar, F. Merzel, M. Hodošček, K. Rebolj, K. Sepčić, P. Maček, D. Janežič,Liquid-ordered phase formation in cholesterol/sphingomyelin bilayers: all-atommolecular dynamics simulations, J. Phys. Chem. B 113 (2009) 15795–15802.
[47] W. Humphrey, A. Dalke, K. Schulten, VMD: visual molecular dynamics, J. Mol.Graph. 14 (1996) 33–38.
[48] D. Frishman, P. Argos, Knowledge-based protein secondary structure assignment,Proteins 23 (1995) 566–579.
[49] Gnuplot software, http://www.gnuplot.info, (accessed 30 September 2012).[50] N. Sreerama, R.W. Woody, Estimation of protein secondary structure from circu-
lar dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methodswith an expanded reference set, Anal. Biochem. 287 (2000) 252–260.
[51] A. Bax, D.G. Davis, MLEV-17 based two-dimensional homonuclear magnetizationtransfer spectroscopy, J. Magn. Reson. 65 (1985) 355–360.
[52] D.S. Wishart, C.G. Bigam, J. Yao, F. Abilgaad, H.J. Dyson, E. Oldfield, J.L. Markley,B.D. Sykes, 1H, 13C, and 15N chemical shifts referencing in biomolecular NMR, J.Biomol. NMR 6 (1995) 135–140.
[53] F. Delaglio, S. Grzesiek, G.W. Vuister, G. Zhu, J. Pfeifer, A. Bax, NMRPipe: amultidimensional spectral processing system based on UNIX pipes, J. Biomol.NMR 6 (1995) 277–293.

2619A. Roy Choudhury et al. / Biochimica et Biophysica Acta 1828 (2013) 2609–2619
[54] T.D. Goddard, D.G. Kneller, SPARKY 3, University of California, San Francisco, 2013.[http://www.csb.yale.edu/userguides/datamanip/sparky/sparky_descrip.html].
[55] P. Guntert, C. Mumenthaler, K. Wuthrich, Torsion angle dynamics for NMRstructure calculation with the new program DYANA, J. Mol. Biol. 273 (1997)283–298.
[56] K.J. Schweighofer, U. Essman, M. Berkowitz, Simulation of sodium dodecyl sulfateat the water-vapor and water-carbon tetrachloride interfaces at low surface cov-erage, J. Phys. Chem. B 101 (1997) 3793–3799.
[57] S. Rodziewicz-Motowidlo, E. Sikorska, M. Oleszczuk, C. Czaplewski, Conformationalstudies of vasopressin and mesotocin using NMR spectroscopy and molecularmodelling methods, Part II: Studies in the SDS micelle, J. Pept. Sci. 14 (2008) 85–96.
[58] C.D. Bruce, M.L. Berkowitz, L. Perera, M.D.E. Forbes, Molecular dynamics simula-tion of sodium dodecyl sulfate micelle in water: micellar structural characteristicsand counterion distribution, J. Phys. Chem. B 106 (2002) 3788–3793.
[59] D.A. Case, T.A. Darden, T.E. Cheatham, C.L. Simmerling, J. Wang, R.E. Duke, R. Luo,R.C. Walker, W. Zhang, K.M. Merz, B. Roberts, B. Wang, S. Hayik, A. Roitberg, G.Seabra, I. Kolossváry, K.F. Wong, F. Paesani, J. Vanicek, X. Wu, S.R. Brozell, T.Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D.R. Roe, D.H.Mathews, M.G. Seetin, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko,P.A. Kollman, AMBER 11, University of California, San Francisco, 2010.
[60] J.S. Richardson, The anatomy and taxonomy of proteins, Adv. Protein Chem. 34(1981) 167–339.
[61] R. Smith, F. Separovic, T.J. Milne, A. Whittaker, F.M. Bennett, B.A. Cornell, A.Makriyannis, Structure and orientation of the pore-forming peptide melittin, inlipid bilayers, J. Mol. Biol. 241 (1994) 456–466.
[62] A. Perdih, T. Solmajer, MurD ligase from Escherichia coli: C-terminal domain clos-ing motion, Comput. Theor. Chem. 979 (2012) 73–81.
[63] S.M. Kelly, N.C. Price, The use of circular dichroism in the investigation of proteinstructure and function, Curr. Protein Pept. Sci. 1 (2000) 349–384.
[64] R.P. Hicks, E. Mones, H. Kim, B.W. Koser, D.A. Nichols, A.K. Bhattacharjee, Compar-ison of the conformation and electrostatic surface properties of magainin pep-tides bound to sodium dodecyl sulfate and dodecylphosphocholine micelles,Biopolymers 68 (2003) 459–470.
[65] L. Columbus, J. Lipfert, K. Jambunathan, D.A. Fox, A.Y.L. Sim, S. Doniach, S.A. Lesley,Mixing and matching detergents for membrane protein NMR structure determi-nation, J. Am. Chem. Soc. 131 (2009) 7320–7326.
[66] C.M. Franzin, X.-M. Gong, K. Thai, J. Yu, F.M. Marassi, NMR of membrane proteinsin micelles and bilayers: the FXYD family proteins, Methods 41 (2007) 398–408.
[67] J.M. Rankenberg, V.V. Vostrikov, C.D. DuVall, D.V. Greathouse, R.E. Koeppe II, C.V.Grant, S.J. Opella, Proline kink angle distributions for GWALP23 in lipid bilayers ofdifferent thicknesses, Biochemistry 51 (2012) 3554–3564.
[68] G. von Heijne, Proline kinks in transmembrane alpha-helices, J. Mol. Biol. 218(1991) 499–503.
[69] P. Chakrabarti, S. Chakrabarti, C\H … O hydrogen bond involving proline residuesin α-helices, J. Mol. Biol. 284 (1998) 867–873.
[70] S. Tuzi, A. Naito, H. Saitô, Local protein structure and dynamics at kinked trans-membrane α-helices of [1-13C]Pro-labeled bacteriorhodopsin as revealed bysite-directed solid-state 13C NMR, J. Mol. Struct. 654 (2003) 205–214.
[71] Z. Cao, J.U. Bowie, Shifting hydrogen bonds may produce flexible transmembranehelices, PNAS 109 (2012) 8121–8126.
[72] M.R.R. de Planque, D.T.S. Rijkers, J.I. Fletcher, R.M.J. Liskamp, F. Separovic, TheαM1 segment of the nicotinic acetylcholine receptor exhibits conformationalflexibility in a membrane environment, Biochim. Biophys. Acta Biomembr. 1665(2004) 40–47.





![Modul Audit Energi [TM2]](https://static.fdocuments.us/doc/165x107/563dbb55550346aa9aac3c3b/modul-audit-energi-tm2.jpg)












