
stereochemistry - PKU · Stereochemistry, Jian Pei, College of Chemistry, Peking University,...
106
1 Stereochemistry Stereochemistry 裴 裴 坚 坚 北京大学化学与分子工程学院 北京大学化学与分子工程学院 2006 2006 年春 年春 1985-1995, S. B., S. M., Ph. D Peking University 1995-1998, Postdoctoral, National University of Singapore 1998-2000, Postdoctoral, UCSB 2000-2001, Associate Research Fellow, Research Fellow Institute of Materials Research and Engineering Singapore 2001.4 - Associate Professor, Professor Peking University Jian Pei Jian Pei Stereochemistry, Jian Pei, College of Chemistry, Peking University, The universe is dissymmetrical…… ----- Louis Pasteur Stereochemistry, Jian Pei, College of Chemistry, Peking University, Why Preface Introduction Chapter 1 Basic Stereochemical Concepts and Vocabularies Chapter 2 Asymmetric Organic Reaction and the Classification Chapter 3 Type 1 Reactions: Reactions in Which no New Chiral Centres Are Created Chapter 4 Type 2 Reactions: Chapter 5 Type 3 Reactions: Chapter 6 Type 4 Reactions: Contents Stereochemistry, Jian Pei, College of Chemistry, Peking University, Introduction Introduction Stereochemistry, Jian Pei, College of Chemistry, Peking University, History 1. 1801, Haüy: quartz crystals exhibited hemihedral phenomena; 2. 1809, Malaus: quartz crystals induced the polarization of light; 3. 1812, Biot: quartz plate rotated the plane of polarized light …; 4. 1815, Biot: difference between the rotation caused by…; 5. 1822, Herschel: a correlation between hemihedralism and optical rotation; 6. 1846, Pasteur: tartaric acid; 7. 1848, Pasteur: separated enantiomorphous crystals of sodium ammonium salts of tartaric acid; 8. 1874, J. H. van’t Hoff and J. A. Le Bel: Tetrahedron carbon A clockwise direction: dextrorotatory molecule: (+) or d; A counterclockwise direction: levorotatory molecule: (-) or l; Enantiomers of a given molecule have specific rotations with the same magnitude but in oppsite directions. Stereochemistry, Jian Pei, College of Chemistry, Peking University, Introduction Introduction
Transcript of stereochemistry - PKU · Stereochemistry, Jian Pei, College of Chemistry, Peking University,...

1
StereochemistryStereochemistry
裴裴 坚坚
北京大学化学与分子工程学院北京大学化学与分子工程学院
2006 2006 年春年春
1985-1995, S. B., S. M., Ph. D
Peking University
1995-1998, Postdoctoral, National University of Singapore1998-2000, Postdoctoral, UCSB2000-2001, Associate Research Fellow,
Research FellowInstitute of Materials Research and EngineeringSingapore
2001.4 - Associate Professor, ProfessorPeking University
Jian PeiJian Pei
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The universe is dissymmetrical……
----- Louis Pasteur
The universe is dissymmetrical……
----- Louis Pasteur
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Why
Preface IntroductionChapter 1 Basic Stereochemical Concepts
and VocabulariesChapter 2 Asymmetric Organic Reaction and
the ClassificationChapter 3 Type 1 Reactions: Reactions in Which
no New Chiral Centres Are CreatedChapter 4 Type 2 Reactions:Chapter 5 Type 3 Reactions:Chapter 6 Type 4 Reactions:
Preface IntroductionChapter 1 Basic Stereochemical Concepts
and VocabulariesChapter 2 Asymmetric Organic Reaction and
the ClassificationChapter 3 Type 1 Reactions: Reactions in Which
no New Chiral Centres Are CreatedChapter 4 Type 2 Reactions:Chapter 5 Type 3 Reactions:Chapter 6 Type 4 Reactions:
Contents
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
IntroductionIntroduction
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
History1. 1801, Haüy: quartz crystals exhibited hemihedral phenomena;2. 1809, Malaus: quartz crystals induced the polarization of light;3. 1812, Biot: quartz plate rotated the plane of polarized light …;4. 1815, Biot: difference between the rotation caused by…;5. 1822, Herschel: a correlation between hemihedralism and
optical rotation;6. 1846, Pasteur: tartaric acid;7. 1848, Pasteur: separated enantiomorphous crystals of sodium
ammonium salts of tartaric acid;8. 1874, J. H. van’t Hoff and J. A. Le Bel: Tetrahedron carbon
1. 1801, Haüy: quartz crystals exhibited hemihedral phenomena;2. 1809, Malaus: quartz crystals induced the polarization of light;3. 1812, Biot: quartz plate rotated the plane of polarized light …;4. 1815, Biot: difference between the rotation caused by…;5. 1822, Herschel: a correlation between hemihedralism and
optical rotation;6. 1846, Pasteur: tartaric acid;7. 1848, Pasteur: separated enantiomorphous crystals of sodium
ammonium salts of tartaric acid;8. 1874, J. H. van’t Hoff and J. A. Le Bel: Tetrahedron carbon
A clockwise direction: dextrorotatory molecule: (+) or d; A counterclockwise direction: levorotatory molecule: (-) or l;Enantiomers of a given molecule have specific rotations with the same magnitude but in oppsite directions.
A clockwise direction: dextrorotatory molecule: (+) or d; A counterclockwise direction: levorotatory molecule: (-) or l;Enantiomers of a given molecule have specific rotations with the same magnitude but in oppsite directions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
IntroductionIntroduction

2
Emil Fischer:
COOH
Bu-tH
CONH2
COOCH3
Bu-tH
CONH2
CH2N2
HNO2
COOCH3
Bu-tH
COOH H2NNH2
CONHNH2
Bu-tH
COOHHNO2
CON3
Bu-tH
COOHCONH2
Bu-tH
COOH
(+)-1 [ ]Dα 20= + 50
(-)-1 [ ]Dα 20= -45
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
IntroductionIntroduction
CA
B
D
E
CD
B
A
E
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
IntroductionIntroduction
Because of their similarity in structure, diastereoisomers often have very similar chemical and physical properties. Consequently, separation of diastereoisomer mixtures will usually be more difficult than separation of constitutional isomers. Enantiopure materials have been important for the synthesis of natural products since the latter almost always are found in nature as a single enantiomers.In some cases, one enantiomer may be completely inactive; in others this enantiomer may be active in another deleterioussense, leading to undesirable side-effects of the drug.
Because of their similarity in structure, diastereoisomers often have very similar chemical and physical properties. Consequently, separation of diastereoisomer mixtures will usually be more difficult than separation of constitutional isomers. Enantiopure materials have been important for the synthesis of natural products since the latter almost always are found in nature as a single enantiomers.In some cases, one enantiomer may be completely inactive; in others this enantiomer may be active in another deleterioussense, leading to undesirable side-effects of the drug.
SignificanceSignificance
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HO
HO COOH
HH2N
(L)-dopa
HO
HO COOH
NH2H
(D)-dopa
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SignificanceSignificance
NMe
HOH
Me
(-)-benzomorphia(ease pain, unhabituational)
(+)-benzomorphia( faintly pain-easing, habituational)
MeN
OH
H
Me
止痛,不成瘾 弱止痛,成瘾
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SignificanceSignificance
(-)-benzopyriyldiol(strong carcinogenicity)
HO
O
OH(+)-benzopyriyldiol(no carcinogenicity)
OH
O
OH
强致癌性 无致癌性
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SignificanceSignificance

3
SNN
N
O
ONHC(CH3)3
HO
(R)-timolol (adrenergic)
SNN
N
O
ONHC(CH3)3
HO
(S)-timolol (ineffective)
肾上腺素能阻断剂 无活性
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SignificanceSignificance
Basic Stereochemical Concepts and Vocabularies
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chapter 1Chapter 1
OutlineOutline
1.1. Selectivity1.2. Chirality1.3. Determining Enantiomer
Composition1.4 General Strategies for Asymmetric
Synthesis
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chemoselectivity
Regioselectivity
Diastereoselectivity
EnatioselectivityStereoselectivity
1.1. 1.1. SelectivitySelectivity
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChemoselectivityChemoselectivity
Chemoselctivity is the preferential reaction of one functional group over another under the reaction conditions employed.
MeOOCR
O
NaBH4
MeOOCR
H OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HO OHMeOOCR
O
HOH2CR
O
MeOOCR
OO LiAlH4
Na2SO4 (aq.) H+/H2O
Function of the Protecting GroupFunction of the Protecting Group
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

4
RegioselectivityRegioselectivity
Regioselctivity is the preferential formation of one isomer of the product in a reaction in which one (or less commonly two) other isomer (s) may also be formed.
Regioselctivity is the preferential formation of one isomer of the product in a reaction in which one (or less commonly two) other isomer (s) may also be formed.
HBrH2O
BrHBrperoxide
CH2Br
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Regioisomer is formed in a reaction may depend critically on the conditions under which it is carried out.
Regioisomer is formed in a reaction may depend critically on the conditions under which it is carried out.
Same Mechanisms
R
R'
R
R'
R R
Br
R
R
base R
R
R
R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioisomerRegioisomer
OH
R'
R R'
HO RR
R
O
R' R'R'
R
O
R R'
O
R R'
H+
Nu--O
R
R'
Nu
R
Nu
O-
R'
Control of regioselectivity in these cases is less easy although not impervious to changes in reaction conditions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivityTwo or more identical but distinguishable functional
groups in one molecule:
COOH
COOH
HO CH3OH
H+O
H+
LiAlH4 Na2SO4(aq)CH2OH
CH2OH
HO
O
H+
CH2OHO
O
CH2OH
O
O
10 %
NaH, DMFPhCH2Br
CH2OBnO
OH+
CH2OBnHO
HO
MeSO2ClPyr
Base CH2OBnO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity
StereoselectivityStereoselectivity
Stereoisomers are isomers which have the same atoms and bonds in common but different arrangementsin space.
Stereoisomers are isomers which have the same atoms and bonds in common but different arrangementsin space.
Chirality or Handedness
Come from the Greek word Chier, which means hand in English
Chirality or Handedness
Come from the Greek word Chier, which means hand in English
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Conformational isomers: nonisolableEnantiomers: a special sort of stereoisomer where the two molecules in question have a mirror-image or enantiomeric relationship.
Diastereoisomers:Stereoisomers which do not have an enantiomeric relationship.A chiral molecule containing at least two chiral elements.Some achiral molecules can exist as diastereoisomers:The E/Z stereoisomers of alkenes are not enantiomers andtherefore diastereoisomers.
Conformational isomers: nonisolableEnantiomers: a special sort of stereoisomer where the two molecules in question have a mirror-image or enantiomeric relationship.
Diastereoisomers:Stereoisomers which do not have an enantiomeric relationship.A chiral molecule containing at least two chiral elements.Some achiral molecules can exist as diastereoisomers:The E/Z stereoisomers of alkenes are not enantiomers andtherefore diastereoisomers.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
StereoisomersStereoisomers

5
Enatioselectivity: in a reaction, is either the preferential formation of one enantiomer of the product over the other or the preferential reaction of one enantiomer of the (usually racemic) starting material over the other.
Diastereoselectivity: is the preferential formation in a reaction of one diastereoisomer of the product over the other.
Note: in a diastereoselective reaction
The product may be either as a single enantiomer or as a racemate, or excess of one enantiomer over the other between these extremes.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
StereoselectivityStereoselectivity
c
y
b
xz-
a+ a cb
zyx
b
x
c
y
a
z
enant.
c
y
b
xz-
a+
Completely diastereoselective, but the product is bound to be racemic.
a cb
zyx
a cb
y xz-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
StereoselectivityStereoselectivity
Stereoselectivity:
Diastereoselectivity
Enatioselectivity
CompleteHigh (> 90 %)Moderatelow
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
StereoselectivityStereoselectivity
Enatioselectivity:normally quoted as the enantiomeric excess (e.e.), which is the mole fraction of the major enantiomer expressed as a percentage , i.e. with an excess of the R or S form.
e.e. =mole fraction R - mole fraction S
mole fraction R + mole fraction S * 100 % = α[ ]obs
α[ ]max* 100 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
EnatioselectivityEnatioselectivity
d. r.: diastereoisomeric ratiod. r.: diastereoisomeric ratio
Diastereoselectivity:usually given as the diastereoisomeric excess (d.e.), which is the mole fraction of the major diastereoisomer D1 in a mixture of two diastereoisomers D1 and D2 against usually expressed as a percentage:
d.e. =mole fraction D1 - mole fraction D2
mole fraction D1 + mole fraction D2* 100 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DiastereoselectivityDiastereoselectivity
Stereospecificity: a term reserved for the circumstance in which stereo-differentiated reactants give stereo-differentiated products and/or show different reactivity.
Stereospecificity: a term reserved for the circumstance in which stereo-differentiated reactants give stereo-differentiated products and/or show different reactivity.
ax
bcNu-
aNu
bcNu
a
cb x
a
cb
Nu-
y x
z
b a
cNu-
ba
yx y x
z
a b
cNu-
ab
yx
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
StereospecificityStereospecificity

6
1.2 Chirality1.2 Chirality
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chirality is a fundamental properties of many three-dimensional objects. An object is chiral if it cannot be superimposed on its mirror image. There is one property in which chiral compounds differ from achiral compounds and in which enantiomers differ from each other. This property is the direction in which rotate plane-polarized light, and this is called optical rotation or optical activity.
Chirality is a fundamental properties of many three-dimensional objects. An object is chiral if it cannot be superimposed on its mirror image. There is one property in which chiral compounds differ from achiral compounds and in which enantiomers differ from each other. This property is the direction in which rotate plane-polarized light, and this is called optical rotation or optical activity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality
C N P S S
O
AsymmetryAsymmetry
1. Compounds with an asymmetric carbon atom: chiral center;2. Compounds with another quaternary covalent chiral center
binding to four different groups that occupy the four corners of a tetrahedron: Si, Ge, N, Mn, Cu, Bi, and Zn;
3. Compounds with trivalent asymmetric atoms:
1. Compounds with an asymmetric carbon atom: chiral center;2. Compounds with another quaternary covalent chiral center
binding to four different groups that occupy the four corners of a tetrahedron: Si, Ge, N, Mn, Cu, Bi, and Zn;
3. Compounds with trivalent asymmetric atoms:
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
N
H
CH3
Cl
N
H
CH3ClO
N
Ph
CH3Ph
In a three-membered heterocyclic ring
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AsymmetryAsymmetry
N
N
Me
Me
The bridgehead structure
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AsymmetryAsymmetry
ChiralA molecule which is non-superimposable upon its mirror image.
ChiralityArises from the absence of some symmetry elements.
The chiral element is most usually a chiral centre or, as it is sometimes called stereogenic centre in the molecule.
The configuration at this chiral centre is the arrangement in space of the substituents responsible for its chirality.
The chirality of a molecule may be alternatively ascribed to the presence of a chiral axis.
ChiralA molecule which is non-superimposable upon its mirror image.
ChiralityArises from the absence of some symmetry elements.
The chiral element is most usually a chiral centre or, as it is sometimes called stereogenic centre in the molecule.
The configuration at this chiral centre is the arrangement in space of the substituents responsible for its chirality.
The chirality of a molecule may be alternatively ascribed to the presence of a chiral axis.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality

7
a
b
c
d
a
bd b
a
d
cc
enant.
One chiral center
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality
a bc
xy z
Two chiral centers
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality
a cb
zyx a cb
yxz
a cb
zyx
b
x
c
y
a
zenant.
b
x
a
y
c
z
c ab
zyx
enant.
Stereoisomers
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality FischerFischer’’s Conventions Convention
Glyceraldehyde as the standard
C OHH
CHO
CH2OH
C HHO
CHO
CH2OH
C
OHH
HO C
HHO
HO
CH2OH
CHO
CH2OH
OHH
CH2OH
CHO
CH2OH
HHO
Advantages:1. Enables the systematic
stereochemical presentation of a large number of natural of natural products;
2. Still useful for carbohydratesor amino acids today.
Limitations:Do not resemble the model reference compound glyceraldehyde;difficult to correlate the terpene compounds with glyceraldehye
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The CahnCahn--IngoldIngold--PrelogPrelog ConventionConvention
In the system, atoms or groups bonded to the chiral center are prioritized based on the sequence rules.1) An atom having a higher atomic number has priority over
one with a lower atomic number; for isotopic atoms, the isotope with a higher mass precedes the one with the lowermass;
2) If two or more of the atoms directly bonded to the asym-metric atom is identical, the atoms attached to them will compared according to the same sequence rule.
In the system, atoms or groups bonded to the chiral center are prioritized based on the sequence rules.1) An atom having a higher atomic number has priority over
one with a lower atomic number; for isotopic atoms, the isotope with a higher mass precedes the one with the lowermass;
2) If two or more of the atoms directly bonded to the asym-metric atom is identical, the atoms attached to them will compared according to the same sequence rule.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The CahnCahn--IngoldIngold--PrelogPrelog ConventionConvention
3) For multiple bonds, a doubly or triply bonded atom is duplicated or triplicated with the atom to which it is connected.
4) For vinyl groups, a group having the (Z)-configuration precedes the same groups having the (E) configuration, andan (R)-group have precedence over an (S)-group for
pseudochiral centres.
3) For multiple bonds, a doubly or triply bonded atom is duplicated or triplicated with the atom to which it is connected.
4) For vinyl groups, a group having the (Z)-configuration precedes the same groups having the (E) configuration, andan (R)-group have precedence over an (S)-group for
pseudochiral centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

8
CH2OH
OHH
CHO
D-Glyceraldehyde in Fischer's convention(R)-glyceraldehyde in the Cahn-Ingold-Prelog convention
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The CahnCahn--IngoldIngold--PrelogPrelog ConventionConvention
A equals C
A
A
C
A equals C
A
A
A
C
C
C
C
CC
CC
CHCH
CH
HC
CH
Cequals
R originating from the Latin word rectus, which means right in English;S originating from the Latin word sinister, which means left in English.
R originating from the Latin word rectus, which means right in English;S originating from the Latin word sinister, which means left in English.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The CahnCahn--IngoldIngold--PrelogPrelog ConventionConvention
CenterCenter--ChiralChiral
x
z wy
x, y, z and w different substituents
The system Cxyzw has no symmetry when x, y, z, and w are different groups, and this system is referred to as a central chiral system.
The system Cxyzw has no symmetry when x, y, z, and w are different groups, and this system is referred to as a central chiral system.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
a
b
c
d
if a>b>c>d, R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CenterCenter--ChiralChiral
Axial Axial ChrialityChriality
OHOH
R
For a system with four groups arranged out of the plane in paired about an axis, the system is asymmetric when the groups on each side of the axis are different. Such a system is referred to as an axial chiral system.
For a system with four groups arranged out of the plane in paired about an axis, the system is asymmetric when the groups on each side of the axis are different. Such a system is referred to as an axial chiral system.
NO2
COOH
Me
OMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
C C C
C C Cd
ca
bif a > b, c > d, (R)
c
d
a
bif a>b, c > d, (S)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Axial Axial ChrialityChriality

9
N
N
O
CH2COOHPhH2Cc d
a b
BrCMe2OH
Br
But
Br
Br
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Axial Axial ChrialityChriality
CH3
HH
HOOC
H
H3C COOH
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Axial Axial ChrialityChriality
OO
old, obsolete method(R)-configuration
OO
Currently used method(S)-configuration
12
3 4 4
3
2
1
For chiral spirocyclic compounds
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Axial Axial ChrialityChriality Planar Planar ChrialityChriality
Br
CH2
Planar chirality arises from the desymmetrization of a symmetric plane in such a way that chirality depends on a distinction between the two sides of the plane and on the pattern of the three determining groups.
Planar chirality arises from the desymmetrization of a symmetric plane in such a way that chirality depends on a distinction between the two sides of the plane and on the pattern of the three determining groups.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral Plane “Pilot Atom”or Descriptor
Br
O
CH2Pilot atom
12
3 CH2H2C
CH2H2C
Br
Pilot atom
12
3pS pS
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Planar Planar ChrialityChriality
Metallocene molecules:Replace the η6-π bond by six σ single bonds.
According to the CIP rules, treated as a central chiral system.
Metallocene molecules:Replace the η6-π bond by six σ single bonds.
According to the CIP rules, treated as a central chiral system.
CH3
CHO
Cr(CO)3
CH3
CHO
Cr(CO)3
the most preferred atom
1
2
3
4S
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Planar Planar ChrialityChriality

10
Helical Helical ChrialityChriality
Helicity is a special case of chirality in which molecules are shaped as a right- or left-handed spiral like a screw or spiral stairs. The configurations are designed M and P, respectively, According to the helical direction.
Helicity is a special case of chirality in which molecules are shaped as a right- or left-handed spiral like a screw or spiral stairs. The configurations are designed M and P, respectively, According to the helical direction.
M: counterclockwise orientation P: clockwise helix
M
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Octahedral StructuresOctahedral Structures
Extention of the sequence rule makes it possible to arrange an octahedral structure in such a way that the ligands are placed octahedrally in an order of preference.
Extention of the sequence rule makes it possible to arrange an octahedral structure in such a way that the ligands are placed octahedrally in an order of preference.
2 5
3 4
1
C
6
1
4 3
5 2C
Observer's place
S R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Pseudochiral CentresPseudochiral Centres
A Cabcd system is called a pseudochiral cnetre when a/b are pair of enantiomeric groups and c/d are different from a/b as well as different from each other.
A Cabcd system is called a pseudochiral cnetre when a/b are pair of enantiomeric groups and c/d are different from a/b as well as different from each other.
CH2OH
OHH
OHH
H OH
CH2OH
CH2OH
OHH
HHO
H OH
CH2OH
pseudochiral centre
s rNH
MeMe
Molecules that belong to Cnor Dn point groups are also chiral
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HO
CH3
Symmetric face
perchiral carbon atom
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Pseudochiral CentresPseudochiral Centres
Compounds with other atoms
N
N
Me
Me
N
NMe
Me
N
MeO
N
Me O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality
OOSS OSS
O
N P
NMe2
HO
O
As
Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality

11
PC6H4OHPh
S
R R'R''
S
OR'RO
S
R'RO
S
S
O
OStereochemistry, Jian Pei, College of Chemistry, Peking University,
ChiralityChirality Double Bond Double Bond Prochirality Prochirality
Enantiofaces
H
Me H
Me
1. OsO4
2. H2O +HO OH
H MeMe H HO OH
H MeMe H
OEt
H1. MeMgI2. NH4Cl
resi Et
H Me
OH Et
H OH
Me+
NPh
Me R 1. LiAlH4
2. Na2SO4 (aq) Ph
Me H
NHR Ph
Me NHR
H+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
c
a bRe Si
b a
c
a b
cRe Si
O
RHRe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
enantioface or enantiotopic faces
A colckwise ordering of the substituents on the double bond defines the face views as re, an anticlockwise ordering as si.
In the reaction with a prochiral double bond, an achiral reagent cannot distinguish between enantiofaces and the product, therefore, is always racemic.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
Prochirality is not an inherent property of the double bond alone but only arises in its combination with reagent.
H
Me H
Me
Br2 +
Br+
H MeMe H Br+
H MeMe H
Br
Br
H
Me
Me
H
Identical or homotopic faces
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
The difference between the carbonyl group and carbon-carbon double bond:A configured prochiral double bond can give rise to contiguous chiral centres from attack on its two enantiofaces whereas a non-configured prochiral double bond can give rise to only one.
The difference between the carbonyl group and carbon-carbon double bond:A configured prochiral double bond can give rise to contiguous chiral centres from attack on its two enantiofaces whereas a non-configured prochiral double bond can give rise to only one.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality

12
H
Me H
Me
1. OsO4
2. H2O+
HO OH
H MeMe H HO OH
H MeMe H
OEt
H1. MeMgI2. NH4Cl
resi Et
H Me
OH Et
H OH
Me+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
N
O2N
t-Bu
RCO3HN
t-BuH
O
O2N
The carbon-nitrogen double bond may also be configured but the addition product does not normally have an assignable configuration …..
The carbon-nitrogen double bond may also be configured but the addition product does not normally have an assignable configuration …..
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
MeOOC
COOMe
Me
MeOOCCOOMe
MeH
Me H
COOMe
COOMe
MeOOC
COOMeMe Me
H
MeOOC
COOMe
Enant.
The possession of enantiofaces isn’t restricted to isolated double bonds.
H Me
COOMe
COOMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality Prochiral Prochiral ((ProstereogenicProstereogenic) ) CentresCentres
H3COH
HHpro-R
pro-S
H3COH
HD
H3COH
DH
entantiotopic
CH2COOH
CH2COOH
H
HOMeOH/H+
CH2COOH
CH2COOMe
H
HO
CH2COOMe
CH2COOH
H
HO
pro-S
pro-R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Identification of the individual enantiotopic atoms or groups uses an obvious extension of Cahn-Ingold-Prelog convention:One or other of two atoms or groups is assigned priority over the other (e. g. by substituting D for H or 18O for 16O), but the priority of the remaining two substituents is not affected.
The resulting configuration (R or S) defines the atom or group promoted as pro-R or pro-S.
Identification of the individual enantiotopic atoms or groups uses an obvious extension of Cahn-Ingold-Prelog convention:One or other of two atoms or groups is assigned priority over the other (e. g. by substituting D for H or 18O for 16O), but the priority of the remaining two substituents is not affected.
The resulting configuration (R or S) defines the atom or group promoted as pro-R or pro-S.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality
HOOC COOHHH
HOOC COOHHD
HOOC COOHDH
Two enantiotopic atoms or groups in a molecule are not necessarily substituents on the same carbon atom:the two hydrogen atoms in the meso-diacid are also enantiotopic since…
Two enantiotopic atoms or groups in a molecule are not necessarily substituents on the same carbon atom:the two hydrogen atoms in the meso-diacid are also enantiotopic since…
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Bond Double Bond Prochirality Prochirality

13
Prochirality Prochirality ------DiastereofacesDiastereofaces
a
cb
[O]
a
cb
Oa
cb
O
diastereoisomers
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
- Δ G≠ = RTlnkΔ Δ G≠ = - [Δ G≠(1) - Δ G≠(2)] = RTln(k1 / k2)
If k1 / k2 = 100, Δ Δ G≠ = 11.5 kJ / mol
- Δ G≠ = RTlnkΔ Δ G≠ = - [Δ G≠(1) - Δ G≠(2)] = RTln(k1 / k2)
If k1 / k2 = 100, Δ Δ G≠ = 11.5 kJ / mol
a
cb [O]
a
cb
[O]
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Prochirality Prochirality ------DiastereofacesDiastereofaces
DiastereotopicDiastereotopic Atoms or GroupsAtoms or Groups
diastereoisomers
a
cb
z HH
a
cb
z HD
a
cb
z DH
diastereotopic
Br
COOHF
Me
Cl
MeBr
COOHF
Ph
Cl
Ph
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HOOC COOHH OH
H H HOOC COOHH OH
D H
HOOC COOHH OH
H D
a ac
RBRBRA RA
b
2 × RA and 2 × RB
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DiastereotopicDiastereotopic Atoms or GroupsAtoms or Groups
Diastereotopic atoms or groups also are not necessarily bound to the same atom.
Diastereotopic atoms or groups also are not necessarily bound to the same atom.
Br
F
Me
H
H
Me
Br
F
Me
H
H
Et
Br
F
Et
H
H
Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DiastereotopicDiastereotopic Atoms or GroupsAtoms or Groups Homotopic Homotopic Faces and GroupsFaces and Groups
COOMeCOOMe
OR1
H
OR1
HH
COOMe
OR1
H
OR1
H COOMeR
COOMe
OR1
H
OR1
H COOMe
H
R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

14
HOH
H H
H HH H
H
H
HH
H
H
OH OH
Me MePhPh
OH OH
Ph MePhMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Homotopic Homotopic Faces and GroupsFaces and Groups Reaction of Reaction of DiastereofacesDiastereofaces
a
cb [O]
a
cb
O
a
cb
OR R R R
S+
It has been pointed out above that, in principle, an achiral reagent can distinguish between two diastereofaces of a double bond.
It has been pointed out above that, in principle, an achiral reagent can distinguish between two diastereofaces of a double bond.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric induction
(R) + (S)
either or
(R,R) + (S,S) (R,S) + (S,R)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of DiastereofacesDiastereofaces
Intermolecular Asymmetric Introduction
Enantiofaces react at the same rate with achiral reagents since the two transition state energies involved are identical.
Enantiofaces react at the same rate with achiral reagents since the two transition state energies involved are identical.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces
For one enantioface to react in preference to the other they must be converted into diastereofaces.
For one enantioface to react in preference to the other they must be converted into diastereofaces.
However, it is not mandatory that the two faces of the alkene are diastereofaces from the outset of the reaction.However, it is not mandatory that the two faces of the
alkene are diastereofaces from the outset of the reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces
G
Reaction coordinate
H H
ba
x y*
y* x
H Ha b
y* x
a bHH
y* x
a bHH
Ha b
y* x
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces

15
2
BHMe tBu
Me tBuH HB H
2
NaOH, H2O2, MeOH
Me tBuH H
HO H
R 76 %, 60 % ee
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces
G
Reaction coordinate
b
a
x y
y x
ba
y* x
a b
y* x
a b
a
y* x
bS*
S*
S*
S*
S*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces
OHPh
Me
OPh
Me
Me2NNMe2
OMe
OMe
hv, 52%
OH
OH
MePh
MePh
Ph
MeO
OHPh
Me
OHMe
Ph
OHMe
Ph
meso-isomer
8.3 % e.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces
a
cb
[O]
a
cb
Oa
cb
O
diastereoisomers
Sharpless Sharpless OxidationOxidation
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
G
Reaction coordinate
b
a
x y*
b
a
b
ax y*
x y*
ba
x
y*
ba
xy*
x
ba
ba
x
more lessy*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reaction of Reaction of EnantiofacesEnantiofaces Asymmetric InductionAsymmetric Induction
G
Reaction coordinate
b
a
bH
x y*
bax
H b
y*
bax
b H
y*
bax
H b
y*
bax
b H
y*
b
bHa
xb
Hba
x
y*
more less
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
in Reaction of Enantiotopic Atoms or Groups

16
1.3. Determining Enantiomer Composition
1.3. Determining Enantiomer Composition
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Measuring Specific RotationMeasuring Specific Rotation
optical purityOne of the terms for describing enantiomer composition
Refers to the ratio of observed specific rotation to the maximumor absolute specific rotation of a pure enantiomer sample
Drawbacks:1) The specific rotation of the pure enantiomer;2) Affected by numerous factors;3) A large quantity of the sample;4) Chemically pure sample for measurement.
Many enantiomer compositions determined by this method in earlier years were incorrect
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The NMR MethodThe NMR Method
1. Measured in a chiral solvent or with a chiral solvating agent
2,2,2-trifluoro-1-phenylethanol, 19F NMR in (-)-α-phenylethylamine
O
C3F7
O
Eu3
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The NMR MethodThe NMR Method
2. Measured with a chiral chemical shift reagent 2. Measured with a chiral chemical shift reagent
H
OH
S
H1. Me3OBF42. NaOH(10 %)
OH
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
3. Chiral Derivatizing Agents3. Chiral Derivatizing Agents
COOH
CF3
OCH3
COOH
CF3
OCH3
(R)-Mosher's Acid (S)-Mosher's Acid
CH2NH2
CF3
OCH3
CH2NH2
CF3
OCH3
(R)-Mosher's Amine (S)-Mosher's Amine
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The NMR MethodThe NMR Method
4. Other New Agents4. Other New Agents
HO
HO
COOR
COOR
PCl3 +COOR
COOROP
OCl R*OH
2 min
COOR
COOROP
OR*O
HO
31P NMR 1.5 ppm 0.7 ppm 1.5 ppm
MeOH
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The NMR MethodThe NMR Method

17
4. Other New Agents4. Other New Agents
NP
N
CH3
CH3
N
CH3
CH3
+ R*OH S8N
PN
CH3
CH3
OR*
S
PO
OO
ClP
N
OPh
Z
Y
CH3
OOP
O Cl
NP
N
CH3
CH3
OR*
Z = S, O,Y = Cl, NHR*, OR*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The NMR MethodThe NMR MethodDetermining the enantiomer composition of
chiral glycol or cyclic ketonesDetermining the enantiomer composition of
chiral glycol or cyclic ketones
O
Pr
+
R
HO OH*
PrOO
R*
PrOO
R*
*
O
R
+
Ph
H2N NH2
Ph
*R
NHHN
PhPh13C NMR or chiral HPLC
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Measurement of the EnantiomerMeasurement of the Enantiomer
1) Chromatographic methods using chiral colummnsa. Gas chromatographyb. Liquid chromatography
2) Capillary Electrophoresis with enantioselective supporting electrolytes
1) Chromatographic methods using chiral colummnsa. Gas chromatographyb. Liquid chromatography
2) Capillary Electrophoresis with enantioselective supporting electrolytes
Other MethodsOther Methods
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Determining Absolute ConfigurationDetermining Absolute Configuration
1) X-Ray Diffraction Methods2) Chiroptical Methods3) The Chemical Interrelation Method4) Prelog’s Method5) Horeau’s Method6) NMR
1) X-Ray Diffraction Methods2) Chiroptical Methods3) The Chemical Interrelation Method4) Prelog’s Method5) Horeau’s Method6) NMR
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
General Strategies for Asymmetric SynthesisGeneral Strategies for Asymmetric Synthesis
1. Chiron Approaches
2. Acyclic Diastereoselective Approaches
3. Double asymmetric Synthesis
1. Chiron Approaches
2. Acyclic Diastereoselective Approaches
3. Double asymmetric Synthesis
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiron ApproachesChiron Approaches
A great number of natural compounds have been employed as chiral starting materials for asymmetric syntheses.
A great number of natural compounds have been employed as chiral starting materials for asymmetric syntheses.
O
O
Me
Me
HO
MeHO
O
CHO
OH
HO
CHO
CH2OH
OH
OH
HO
OH
CHO(+)-exobrevicomin
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

18
H2NNH
N COOH
OH NH2 O CH3
COOH
H2N
OH
CH2NH2
.
.CH2OH
HO
OH
CHO
HO
OH
Naturally occurring chiral compounds provide an enormous range and diversity of possible starting materials.
Natural CarbohydratesAmino Acids
Naturally occurring chiral compounds provide an enormous range and diversity of possible starting materials.
Natural CarbohydratesAmino Acids
negamycin
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiron ApproachesChiron Approaches AcyclicAcyclic Diastereoselective ApproachesDiastereoselective Approaches
1. Substrate-controlled methods;2. Auxiliary-controlled methods;3. Reagent-controlled methods;4. Catalyst-controlled methods.
1. Substrate-controlled methods;2. Auxiliary-controlled methods;3. Reagent-controlled methods;4. Catalyst-controlled methods.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled MethodsControlled Methods
The substrate-controlled reaction is called the first generation of asymmetric synthesis. It is based on intramolecular contact witha stereogenic unit that already exists in the chiral substrate.
The substrate-controlled reaction is called the first generation of asymmetric synthesis. It is based on intramolecular contact witha stereogenic unit that already exists in the chiral substrate.
S* P*R
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AuxiliaryAuxiliary--controlled methodscontrolled methods
The auxiliary-controlled reaction is referred to as the second generation of asymmetric synthesis. This approach is similarto the first generation method in which the asymmetric control is achieved intramolcularly by a chiral group in the substrate.
The auxiliary-controlled reaction is referred to as the second generation of asymmetric synthesis. This approach is similarto the first generation method in which the asymmetric control is achieved intramolcularly by a chiral group in the substrate.
S + A* P-A*R P*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ReagentReagent--Controlled MethodsControlled Methods
The reagent-controlled reaction is called the third generation of asymmetric synthesis. It is based on an achiral substrate directly converted to the chiral product using a chiral reagent..
The reagent-controlled reaction is called the third generation of asymmetric synthesis. It is based on an achiral substrate directly converted to the chiral product using a chiral reagent..
S R*P*
S* R*P*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CatalystCatalyst--Controlled Methods.Controlled Methods.
The most significant advance in asymmetric synthesis in the pastthree decades has been the application of chiral catalysts to induce the conversion of achiral substrates to chiral products
The most significant advance in asymmetric synthesis in the pastthree decades has been the application of chiral catalysts to induce the conversion of achiral substrates to chiral products
S Cat*
P*
S Cat / L*
P*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

19
Double Asymmetric SynthesisDouble Asymmetric Synthesis
Pioneered : Horeau, A. 1968;Reviewed: Masamune, S. 1985.
The idea involves the asymmetric reaction of an enantiomerically pure substrate and an enantiomerically pure reagent. There are also reagent-controlled reactions and substrate-controlled reactions in this category.
*A C(x) I *A
*B C(y)
II*A *C *C *B
(*Cn)-C(z)
III
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HO
O
O
OCH3
H
HO
Re
Si
O OXR
O OXR
1:4.5
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric SynthesisDouble Asymmetric Synthesis
O HCH2Ph
OM
Re
Si
O
YR
OX
O
YR
OX
1:8
OCOCH2Ph
O HCH2Ph
OM
Si
Re
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric SynthesisDouble Asymmetric Synthesis
O HCH2Ph
OM
Re
Si
O
YR
OXR
O
YR
OXR
1:40
O HCH2Ph
OM
Si
Re
O
O
OCH3
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric SynthesisDouble Asymmetric Synthesis
QuestionsQuestions
Asymmetric and Dissymmetric
D / L and d / l
Racemic, meso, racemization, and scalemic
syn / anti and erythro / threo
Asymmetric and Dissymmetric
D / L and d / l
Racemic, meso, racemization, and scalemic
syn / anti and erythro / threo
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Prochirality:
Pro-R and Pro-S:
Optical activity, optical isomer, and optical purity
Re and Si
Prochirality:
Pro-R and Pro-S:
Optical activity, optical isomer, and optical purity
Re and Si
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
QuestionsQuestions

20
x y
a
b
c
b
a
b
a
b
a
b
a
b
a
b
a
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
QuestionsQuestions
Asymmetric Organic Reactions and the Classification
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chapter 2Chapter 2
OutlineOutline
2.1. Common Classification2.2. Stereoselective Synthesis of Molecules
Having Two Chiral Centres2.3. New Classification
Classification of Stereochemical Reactions
2.1. Common Classification2.2. Stereoselective Synthesis of Molecules
Having Two Chiral Centres2.3. New Classification
Classification of Stereochemical Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.1. 2.1. Common ClassificationCommon Classification
2.1.1. α-Alkylation and Catalytic Alkylation of Carbonyl Compounds;
2.1.2. Aldol and Related Reactions;2.1.3. Asymmetric Oxidations;2.1.4. Asymmetric Diels-Alder and Other
Cyclization Reactions;2.1.5. Asymmetric Catalytic Hydrogenation and
Other Reduction Reactions;2.1.6. Enzymatic Reactions and Miscellaneous
Asymmetric Syntheses
2.1.1. α-Alkylation and Catalytic Alkylation of Carbonyl Compounds;
2.1.2. Aldol and Related Reactions;2.1.3. Asymmetric Oxidations;2.1.4. Asymmetric Diels-Alder and Other
Cyclization Reactions;2.1.5. Asymmetric Catalytic Hydrogenation and
Other Reduction Reactions;2.1.6. Enzymatic Reactions and Miscellaneous
Asymmetric Syntheses
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
αα--AlkylationAlkylation and Catalytic and Catalytic AlkylationAlkylationof Carbonyl Compoundsof Carbonyl Compounds
1) Preparation of Quaternary Carbon Centres;
2) Preparation of α-Amino Acids;
3) Nucleophilic Substitution of Chiral Centres;
4) Chiral Catalyst-induced Aldehyde Alkylation
5) Catalytic Asymmetric Addition of Dialkylzinc to Ketones;
6) Asymmetric Cyanohydrination
7) Asymmetric α-Hydroxyphosphonylation
1) Preparation of Quaternary Carbon Centres;
2) Preparation of α-Amino Acids;
3) Nucleophilic Substitution of Chiral Centres;
4) Chiral Catalyst-induced Aldehyde Alkylation
5) Catalytic Asymmetric Addition of Dialkylzinc to Ketones;
6) Asymmetric Cyanohydrination
7) Asymmetric α-Hydroxyphosphonylation
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Preparation of Quaternary Carbon Preparation of Quaternary Carbon CentresCentres
NH2
OHR
HOOC
ON
O
O
R
1. LDA, THF
2. R'XN
O
O
R
R'
bucyclic lactamR = Me, Ph
endo : exo = 9-30 : 1for R = Ph1 -1.5 : 1 for R = Me
1. LDA, THF
2. R''XN
O
O
R
R''
R'
75-95 % de for R = Ph90-93 % de for R = Me
H2SO4 / BuOH
(R = Ph) Ph CO2Bu
O R'' R'
1. Red-Al2. Bu4NH2PO4 Me CHO
O R'' R' HO-
O
R'R''
-OH从下面进攻,由-iPr的构型诱导
R′从下面进攻,由-iPr和R的构型共同
诱导
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
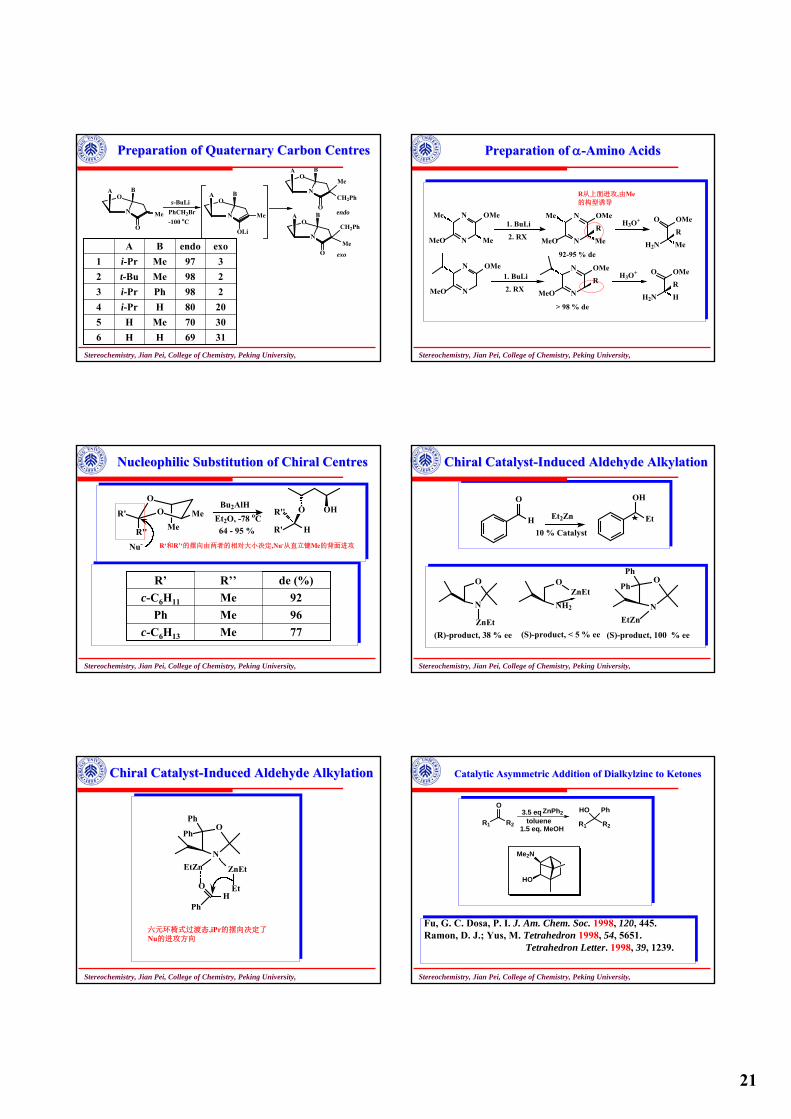
21
N
OA B
O
Me
s-BuLiPhCH2Br-100 oC
N
OA B
OLi
Me
N
OA B
O
Me
CH2Ph
N
OA B
O
CH2Ph
Me
endo
exo
3169HH63070MeH52080Hi-Pr4298Phi-Pr3298Met-Bu2397Mei-Pr1
exoendoBA
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Preparation of Quaternary Carbon Preparation of Quaternary Carbon CentresCentres Preparation of Preparation of αα--Amino AcidsAmino Acids
N
N
MeO
OMe
Me
Me1. BuLi
2. RX N
N
MeO
OMe
Me
Me
R
92-95 % de
H3O+
H2N
OMe
Me
R
O
N
N
MeO
OMe1. BuLi
2. RX N
N
MeO
OMe
R
> 98 % de
H3O+
H2N
OMe
H
R
O
R从上面进攻,由Me的构型诱导
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
NucleophilicNucleophilic Substitution of Chiral Substitution of Chiral CentresCentres
Me
O
R'
R''
O Me
Nu-
Bu2AlHEt2O, -78 oC64 - 95 % R' H
OR'' OH
77Mec-C6H13
96MePh92Mec-C6H11
de (%)R’’R’
R‘和R’‘的摆向由两者的相对大小决定,Nu-从直立键Me的背面进攻
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral CatalystChiral Catalyst--Induced Aldehyde Induced Aldehyde AlkylationAlkylation
H
O
Et2Zn
10 % Catalyst
Et
OH
N
O
ZnEt(R)-product, 38 % ee
NH2
OZnEt
(S)-product, < 5 % ee
N
OPh
Ph
EtZn
(S)-product, 100 % ee
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
N
OPh
Ph
EtZn ZnEt
EtOH
Ph
六元环椅式过渡态,iPr的摆向决定了Nu的进攻方向
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral CatalystChiral Catalyst--Induced Aldehyde Induced Aldehyde AlkylationAlkylation Catalytic Asymmetric Addition of Catalytic Asymmetric Addition of DialkylzincDialkylzinc to to KetonesKetones
R2R1
OZnPh2
toluene1.5 eq. MeOH
3.5 eqR2R1
HO Ph
Me2N
HO
Fu, G. C. Dosa, P. I. J. Am. Chem. Soc. 1998, 120, 445.Ramon, D. J.; Yus, M. Tetrahedron 1998, 54, 5651.
Tetrahedron Letter. 1998, 39, 1239.
Fu, G. C. Dosa, P. I. J. Am. Chem. Soc. 1998, 120, 445.Ramon, D. J.; Yus, M. Tetrahedron 1998, 54, 5651.
Tetrahedron Letter. 1998, 39, 1239.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

22
Catalytic Asymmetric Addition of Catalytic Asymmetric Addition of DialkylzincDialkylzinc to to KetonesKetones
RPh
O ZnR'2toluene
Ti(OPri)RPh
HO R'
OH
H
SO2
NH
e.e. > 89 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.1.2. 2.1.2. AldolAldol and Related Reactionsand Related Reactions
1) Substrate-Controlled Aldol Reactions;
2) Reagent-Controlled Aldol Reactions;
3) Chiral Catalyst-Controlled Asymmetric Aldol Reactions;
4) Double Asymmetric Aldol Reactions;
5) Asymmetric Allylation Reaction;
6) Asymmetric Allylation and Alkylation of Imines
7) Henry Reactions
1) Substrate-Controlled Aldol Reactions;
2) Reagent-Controlled Aldol Reactions;
3) Chiral Catalyst-Controlled Asymmetric Aldol Reactions;
4) Double Asymmetric Aldol Reactions;
5) Asymmetric Allylation Reaction;
6) Asymmetric Allylation and Alkylation of Imines
7) Henry Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled Controlled AldolAldol ReactionsReactions
ON
O
RO
ON
O
RO
Ph
Oxazolidones as chiral Auxiliaries
ON
O
ROB(Bun)2
NOR
OB(Bun)2O
Z-enolate
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ON
O
R
OB(Bun)2
ON
O
RO
R2BOTf
i-Pr2NEt
R'CHO
O
BOH
R'R
Bun
Bun
N
O
O
ON
O
O
R'R
OH
MeO
O
R'R
OHMeONa
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled Controlled AldolAldol ReactionsReactions
ON
O
ROB(Bun)2
PhO
N
O
RO
Ph R2BOTf
i-Pr2NEt
R'CHO
ON
O
O
R'R
OH
PhMeO
O
R'R
OHMeONa
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled Controlled AldolAldol ReactionsReactions
ON
O
OB(Bun)2
OHCO
N
O
O OH
ON
O
O OH
1.75 : 1
无手性辅基诱导选择性较低
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled Controlled AldolAldol ReactionsReactions

23
ON
O
ROB(Bun)2
Ph OHCO
N
O
O OHPh
ON
O
O OH
Ph
600 : 1
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SubstrateSubstrate--Controlled Controlled AldolAldol ReactionsReactions PyrrolidinesPyrrolidines as chiral Auxiliariesas chiral Auxiliaries
NH
MOMO
MOMO
N
MOMO
MOMO
O
N
MOMO
MOMO
OM
RCHON
MOMO
MOMO
O OH
HO
O OH
非对映选择性类似Evans助剂
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
H
R
N
MOMO
OMOMH
Me OZrLn
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
PyrrolidinesPyrrolidines as chiral Auxiliariesas chiral Auxiliaries
N
MOMO
MOMO
RO
N
MOMO
MOMO
O
R'R
O
1. BuLi
2. R'COCl
Zn(BH4)2
KBEt3H
N
MOMO
MOMO
O
R'R
OH
N
MOMO
MOMO
O
R'R
OH
syn
anti
构型相反
构型相反
两手性中心构型相反
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
PyrrolidinesPyrrolidines as chiral Auxiliariesas chiral Auxiliaries
AminoalcoholsAminoalcohols as Chiral Auxiliariesas Chiral Auxiliaries
R*O OTMS
RCHOTiCl4
Me2N
TiOOPh
H H
ClCl
Cl
OH
Me
Me2N
TiOOH
Ph H
ClCl
Cl
OH
Me
anti-product 77% syn-product 23 %
E-enolate得到anti-product
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AcylsultamAcylsultam Systems as Chiral AuxiliariesSystems as Chiral Auxiliaries
O
N(Ar)SO2Ph
OTBS
i-PrCHO
TiCl4 R*O
O
Me
OH
+ syn
O 2S
N
O
E t2B O Tfi-P r2N E t
S
N
O B E t2
R C H OTiC l4
R *N R
O HO
R *N R
O HO
a b
anti: syn = 93 :7
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

24
93/0/70.5TiCl4
97/0/31TiCl4
98/0/22TiCl4
78/0/222Et2BOTf27/37/362Et2AlCl
b/a/others
Mole equiv. of Lewis acid / EtCHO
Lewis acid
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AcylsultamAcylsultam Systems as Chiral AuxiliariesSystems as Chiral Auxiliaries αα--SillylSillyl KetonesKetones
H3C
TBS
O
H3CO
R
OH
d.e. 92-98 %, e.e. > 98 %
H3C
TBS
OBBu2RCHO H3C
O
R
OH
TBS
d.e. 92-98 %, e.e. > 98 %
HBF4
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
R1 OR2
H
OM
R4
R3
R1 R4R2
OOH
R3
O
BO
R3
R4L
LHR1H
HR2
R1 R4R2
OOH
R3
O
BO
H
R4L
LHR1HR3
R2
R1 R4R2
OOH
R3
O
BO L
LH
R2
HR1R3
H
R4
R1 R4R2
OOH
R3
O
BO L
LH
R2
HR1H
R3
R4
R1 R4R2
OOH
R3
L
Re-attack;(Z)-enolate
Re-attack;(E)-enolate
Si-attack;(Z)-enolate
Si-attack;(E)-enolate
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
αα--SillylSillyl KetonesKetones ReagentReagent--Controlled Controlled AldolAldol ReactionsReactions
Aldol Condensations Induced by Chiral Boron Compounds
BOTf
BOTf
BN
O
Me
Ph
OMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BOTf
O
N i-Pr2NEt2
B
O
N
1. RCHO2. 3N H2SO43. CH2N2
RCOOMe
OH
Me
RCOOMe
Me
OHsyn anti
RCHOn-PrCHO
c-HexCHO
n-BuCHO
e.e. % for anti7784
79
anti : syn
91: 995 : 594 : 6
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ReagentReagent--Controlled Controlled AldolAldol ReactionsReactions
O
BN L
L
Me
O
H
R
O
BN L
LO
R
H
Me
H和R互换
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ReagentReagent--Controlled Controlled AldolAldol ReactionsReactions

25
AldolAldol Reactions by CoreyReactions by Corey’’s Reagentss Reagents
Corey’s Reagents
BNN
Ph Ph
SO2ArArSO2
Br
a: Ar: p-CH3C6H4
b: Ar: p-NO2C6H4
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AldolAldol Reactions by CoreyReactions by Corey’’s Reagentss Reagents
C6H5S
Oa
RCHO C6H5S
O
R
HO H
R = Ph, 91 % e.e.R = i-Pr, 83 % e.e.
C6H5S
Ob
RCHO C6H5S
O
R
HO H
H
R = Ph, 95 % e.e.R = i-Pr, 97 % e.e.
syn/anti = 98.3 : 1.7 syn/anti = 94.5 : 5.5
O
BOH
RR'
H
C6H5S N
N
Ph
Ph
ArSO2
SO2Ar
构型相反
Ph的构型促使ArSO2向内
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AldolAldol Reactions by Miscellaneous ReagentsReactions by Miscellaneous Reagents
R1 R2
OH O
R1 R2
OH O
BOTf
OLi
OBut
TiDAGOO
ODAG
OBut
RCHO
R OBut
OH O
OO
O
O
OOH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Asymmetric AldolAldol Reactions by Chiral catalyst Reactions by Chiral catalyst
Mukaiyama’s Reactions
R'
ORRCHOL. A. R'
O
R
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
NS
S O
Sn(OTf)2
NEt
N
Me
N NS
S OSnTfO
N N
RCHONS
S O OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Asymmetric AldolAldol Reactions by Chiral catalyst Reactions by Chiral catalyst
CHOTMSBnO
OPh
OTMSNCH3
NH
Sn(OTf)2SnO
OPhTMS
OH
OH
O
H27C13 OH
OH
NHBOC
87 % syn/anti = 97 : 3 91 % e.e. for syn
10 steps with 15 % overall yieldD- erythro-sphingosine (without BOC)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Asymmetric AldolAldol Reactions by Chiral catalyst Reactions by Chiral catalyst
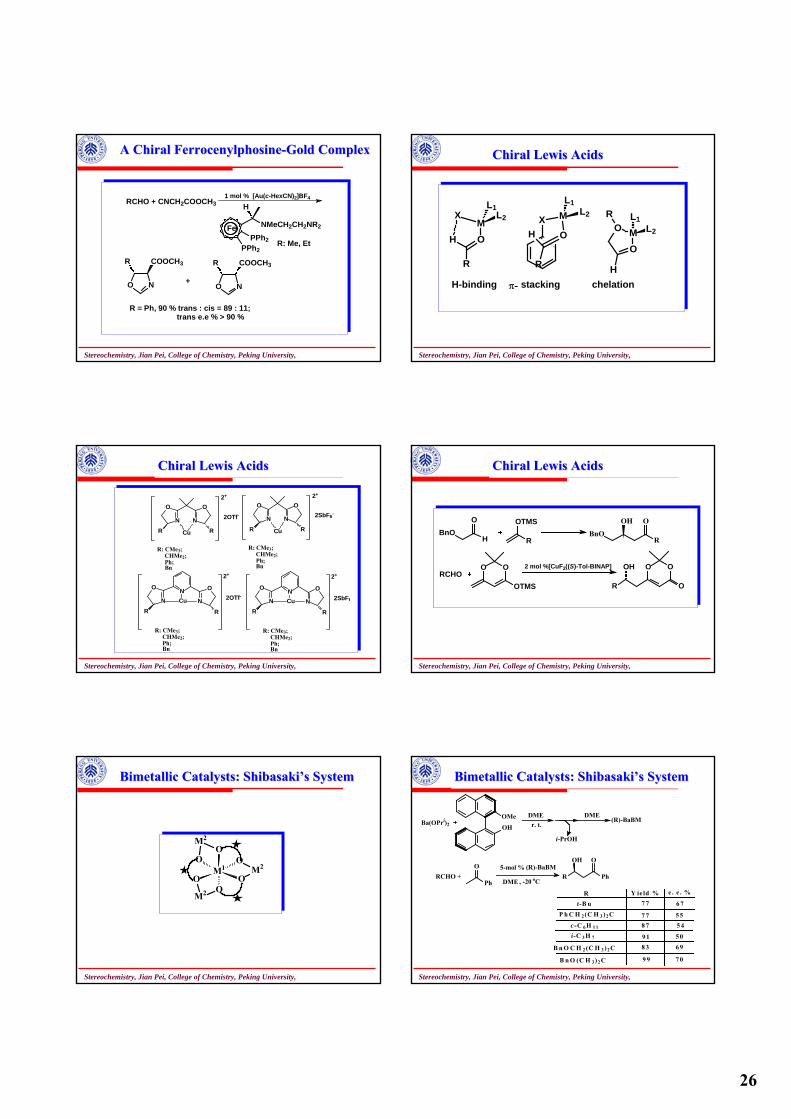
26
A Chiral A Chiral FerrocenylphosineFerrocenylphosine--Gold ComplexGold Complex
PPh2PPh2
NMeCH2CH2NR2
H
Fe
RCHO + CNCH2COOCH31 mol % [Au(c-HexCN)2]BF4
R: Me, Et
NO
R COOCH3
+ NO
R COOCH3
R = Ph, 90 % trans : cis = 89 : 11; trans e.e % > 90 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral Lewis AcidsChiral Lewis Acids
H
R
OM
XL1
L2
R
OX M
H
L1L2
OMO
H
R L1L2
H-binding stacking chelationπ-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
NN
OO
R RCu
2+
2OTf-
R: CMe3; CHMe2; Ph; Bn
NN
OO
R RCu
2+
2SbF6-
R: CMe3; CHMe2; Ph; Bn
2+
2OTf-
R: CMe3; CHMe2; Ph; Bn
N
O
R
N
O
R
NCu
2+
2SbF6
R: CMe3; CHMe2; Ph; Bn
N
O
R
N
O
R
NCu
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral Lewis AcidsChiral Lewis Acids
BnOH
O OTMS
RBnO
R
OH O
RCHOOO
OTMS
2 mol %[CuF2((S)-Tol-BINAP] OO
OR
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral Lewis AcidsChiral Lewis Acids
Bimetallic Catalysts: Bimetallic Catalysts: ShibasakiShibasaki’’s Systems System
O
M1
O
O O
OOM2
M2
M2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Ba(OPri)2 OH
OMe DMEr. t.
i-PrOH
DME(R)-BaBM
RCHO + Ph
O (R)-BaBM5-mol %
DME , -20 oCPh
O
R
OH
R Y ie ld % e . e . %
t-B u 7 7 6 7P h C H 2(C H 3 )2C
c -C 6H 1 1
i-C 3 H 7
B n O C H 2 (C H 3 )2C
B n O (C H 3)2 C
7 78 7
9 18 3
9 9
5 55 4
5 06 9
7 0
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Bimetallic Catalysts: Bimetallic Catalysts: ShibasakiShibasaki’’s Systems System

27
Double Asymmetric Double Asymmetric AldolAldol ReactionsReactions
MeOOC CHO
O
SPh
B MeOOCSPh
OH O
MeOOCSPh
OH O
3:2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
R: PhCH2OCH2CH2: OB(n-C4H9); 28:1OB(C-C5H9); 100:1
R: (CH3)CH: OB(n-C4H9); >100:1OB(C-C5H9); no reaction
R: PhCH2OCH2CH2: OB(n-C4H9); 28:1OB(C-C5H9); 100:1
R: (CH3)CH: OB(n-C4H9); >100:1OB(C-C5H9); no reaction
O
t-BuMe2SiO H
B
R'CHO
R'
OH O
OSiMeBut
R'
OH O
OSiMeBut
R: PhCH2OCH2CH2; a:b = 16:1 (CH3)2CH > 100:1
a
b
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric Double Asymmetric AldolAldol ReactionsReactions
BO
Me SCEt3
MeOOC CHO
O
O
COR
4,5-anti-matched case: 200 : 1
BO
Me SCEt3
MeOOC CHO
O
O
COR
4,5-syn-mismatched case: 55 : 1
O
BO
H
CH3R1H
Me
Et3CS
H
O
BO
H
SCEt3HR1H3C
H3CH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric Double Asymmetric AldolAldol ReactionsReactions
O
t-BuMe2SiO H
B
MeOOC CHO MeOOCR*
OH O
MeOOCR*
OH O
matched pair a:b > 100:1
a
b
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric Double Asymmetric AldolAldol ReactionsReactions
O
t-BuMe2SiO H
B
MeOOC CHO MeOOCR*
OH O
MeOOCR*
OH O
mismatched pair a:b = 1:30
a
b
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric Double Asymmetric AldolAldol ReactionsReactions Asymmetric Asymmetric AllylationAllylation ReactionsReactions
Me M
M
Me RCHOR
OH
MeR
OH
Me
M: SiMe3, SnBu3, BR2, AlR2, MaX, Li, CrX2, TiCp2X, ZrCp2X, InBr
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

28
The Roush ReactionThe Roush Reaction
B O
O
COOR
COORB O
O
COOR
COOR B O
O
COOR
COOR
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
B O
O
COOR
COOR
B O
O
COOR
COOR
RCHO4 A molecular sieves R
OH
RCHO4 A molecular sieves R
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The Roush ReactionThe Roush Reaction
BO O
OCOOR
ORO
H
R'Me
Hfavored
O
B O
O
ORO
Me
H
H
R'
OR
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The Roush ReactionThe Roush Reaction The Corey ReactionThe Corey Reaction
BNN
Ts Ts
Br
Ph Ph
BNN
Ts Ts
Ph Ph
BNN
Ts Ts
Ph Ph
RCHO
O
BN
N
Ts
Ts
Ph
Ph
R
H R
HO H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BNN
Ts Ts
Ph Ph
X
RCHO
O
BN
N
Ts
Ts
Ph
Ph
R
HX R
HO HX
X: Br, Cl
Catalytic Asymmetric Catalytic Asymmetric AllylationAllylation ReactionsReactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
R
HO HX
R
HO H
R
HO H
OO HR
H2C
R
H OCH2OCH3TMS
R
H OCH2OCH3
HO
R
H OCH2OCH3O
R
H OHO
R
H OCH2OCH3
SnBu3
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
AllylicAllylic CompoundsCompounds

29
Asymmetric Asymmetric AllylationAllylation and and AlkylationAlkylation of Iminesof Imines
H
NTMS B
21.
2. H3O+
H NH2
BNN
Ts Ts
Me Ph
H
NTMS
N
BN
N
Me
PhTs
Ph
HTMS
Ts Ph
NH2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Cat.R
PdCl
PdCl
R
N
R1 H
R2
SnBu3R1
NHR2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Asymmetric AllylationAllylation and and AlkylationAlkylation of Iminesof Imines
ON
PhBu3Sn LA
O
H
H
NLA
Ph
SnBu3
O
N Ph
H
H H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Asymmetric AllylationAllylation and and AlkylationAlkylation of Iminesof Imines The Henry ReactionThe Henry Reaction
OLiOLi
LaCl3NaOHH2O
optically active La catalyst
CHO CH3NO210 mol% NO2
OH
O CHO
CH3NO2La-(R)-BINOL
O NO2
OH
80 %, 92 % e.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.1.3. 2.1.3. Asymmetric OxidationsAsymmetric Oxidations
R2
R1
R3
OH
"O"(S,S)-D-(-)tartrate(unnatural)
(R,R)-L-(+)tartrate(unnatural)
"O"
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Characteristics• Simplicity: all the ingredients are inexpensive and
commercially available;• Reliability: It succeeds with most allylic alcohols,
although bulky substituents at R are deleterious;• High optical purity: optical purity of the product is generally
> 90 % e.e. and usually > 95 %;• Predictable absolute stereochemistry:•Relative insensitivity to preexisting chiral centers;•Versatility of 2,3-epoxy alcohols as intermediates.
Characteristics• Simplicity: all the ingredients are inexpensive and
commercially available;• Reliability: It succeeds with most allylic alcohols,
although bulky substituents at R are deleterious;• High optical purity: optical purity of the product is generally
> 90 % e.e. and usually > 95 %;• Predictable absolute stereochemistry:•Relative insensitivity to preexisting chiral centers;•Versatility of 2,3-epoxy alcohols as intermediates.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Sharpless EpoxidationSharpless Epoxidation ReactionsReactions

30
MechanismMechanism
TiOO
O
OO
OTiOO
O
O
OTiOO
O
OOTi
OO
O
O
HO
TiOO
O
O
HO
O
OOH
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OO OH
Ti(OPri)4
t-BuOOH
OO OH
O
OO OH
O+
no catalyst: a : b = 2.3 :1(+)-DET: mismatched a : b = 1 : 22;(-)-DET, matched a : b = 90 : 1
a b
Double Asymmetric Induced Double Asymmetric Induced EpoxidationEpoxidation ReactionsReactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OH
OO (+)-DET
OH
OO
OOH
OO
O
30 : 1
OH
OO
(-)-DET
OH
OO
O OH
OO
O
3 : 2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Double Asymmetric Induced Double Asymmetric Induced EpoxidationEpoxidation ReactionsReactions
R'
+R''
O
L. A.R'
O
R''
exo
endo
exo
endo
Asymmetric DielsAsymmetric Diels--Alder ReactionsAlder Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chiral Chiral DienophilesDienophiles
O
OR*
R*
O
NR2*
O
Chiral Dienes
O
O
Ph
H OMe
There are few reports about chiral auxiliary-attached dienecomponents due, in part, tothe difficulty of the preparation of the modified dienes
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Dipolar Asymmetric Dipolar CycloadditionCycloaddition
HNSO2
COCl
NSO2
ORCNO
NSO2
O
N OR
L-selectrideOH
N OR
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

31
R
NMe
O-
H
H
X
Cr(CO)3
H
R
NMe
O-
H
X
H
Cr(CO)3
H
endo-transition state (A) to cis
exo-transition state (A) to trans
N+
H
O- MeTMS
Cr(CO)3
Ph
CAN
R
TMS ONX
H3C
3R 5S
TMS
N+
H
O- Me
Ph
CAN
TMS
ONX
H3C
3S 5R
Cr(CO)3
Cerium ammonium nitrate
(-)-
(+)-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Asymmetric Dipolar Asymmetric Dipolar CycloadditionCycloaddition 2.1.5. 2.1.5. Asymmetric Catalytic Reduction ReactionsAsymmetric Catalytic Reduction Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.1.6. 2.1.6. Enzymatic Reactions and Enzymatic Reactions and
Miscellaneous Asymmetric SynthesesMiscellaneous Asymmetric Syntheses
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
Racemic: relative configurationEnantiopure: absolute configuration
cb
a
p
q
ab
yxp
x z
y
p
ra
qx
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
To maximize the yield of compound 1 with, and to minimize the yiled of its diastereoisomer 1’, four method are empolyedTo maximize the yield of compound 1 with, and to minimize the yiled of its diastereoisomer 1’, four method are empolyed
cb
ap
x z
y
cb
ap
xy
z1 1'
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
Method A:Starting materials having chiral centres in 1 already in place
Method A:Starting materials having chiral centres in 1 already in place
OH
OH
HO
HO
HO
OH
HOHO H
OH
OH
OH
OHH
O
ZnCl2
H
OH
OHH
OO
O
O
1. MeSO2Cl, Pyr2. NaI, DMF, Zn
H HO
O
O
O1. Pd/C, H22. HOAc, H2O
HOHO H
OH
OHH
Tetraol
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres

32
x z
y
cb
a1
cb
a
x z
y
px z
y
cb
a
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
tBuOOCNH
COOH
R 1H
HN COOM e
H R 2
DCC
tBuOOCNH
CO
R 1H
H 2N COOM e
H R 2
+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
Method B:Starting materials having chiral centres in 1 already in place
Method B:Starting materials having chiral centres in 1 already in place
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
px
LG
z
cb
ay-
p
xzc
b
a y
p
xzc
b
a COR
mCPBA p
xzc
b
a OCOR
pc
b
a
zO
R2 R1
pc
b
a
zR2
R1
CHO
pc
b
a
zR2
LGR1
x-
pc
b
a
z
R2
R1
x
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres
Method C:Creating both chiral centres in a reaction
Method C:Creating both chiral centres in a reaction
HO OH
b ya x
b y
xa
OsO4
+
H OH
b ya x
HO OH
b yxa
Br2
+H OH
b yxa
Br
yxBr
ba+Br
b a Br
yx
1. R2BH2. NaOH, H2O2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentresMethod D:Using a parent chiral centre in a precursor to control the configuration of newly created chiral centre
Method D:Using a parent chiral centre in a precursor to control the configuration of newly created chiral centre
a
b cp
y'
x z
a
b cp
y
zx
1' 1
a
b cp
y
xx
1''
x z
a
b cp
y
zx
1
a
b cp
1'' a
b cp
y
zx
1y'
zx
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
2.2 2.2 Molecules with Two Chiral Molecules with Two Chiral CentresCentres

33
2.3 2.3 Classification of Classification of StereochemicalStereochemical Reactions Reactions
1) Type 0 Reactions; 2) Type 1 Reactions;3) Type 2 Reactions;4) Type 3 Reactions;5) Type 4 Reactions
1) Type 0 Reactions; 2) Type 1 Reactions;3) Type 2 Reactions;4) Type 3 Reactions;5) Type 4 Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reactions without no selectivityIn stereoselective synthesis, these reactions are carried out onsubstrates having one or more chiral centres, no new chiral centres are created and all bonds remain unbroken to the chiral centres which survive the reaction
Reactions without no selectivityIn stereoselective synthesis, these reactions are carried out onsubstrates having one or more chiral centres, no new chiral centres are created and all bonds remain unbroken to the chiral centres which survive the reaction
O
HSPh
1. PhMgBr
2. NH4Cl HPhHO
SPh
1. Me3O+BF4-
2. NaOH HPh
O
C10H21MgBr CuI
HPhHO
H23C11OH23C11
H O
Type 0 Reactions Type 0 Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Reactions with either complete or retention of configuration at an existing chiral centre;
Reactions with either complete or retention of configuration at an existing chiral centre;
aX
bcNu- Nu
a
cb
inversiona
SnR3
bc
R1Li aLi
bc
R2Xa
R2
bc
overall retention
Type 1 ReactionsType 1 Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 ReactionsType 2 Reactions
Type 2 reactions involve the diastereoselective formation of a product containing at least two chiral centres from the reaction of one or more prochiral double bondscontained in one or more achiral starting materials using achiral reagents.
Cycoladdtions,Electrocyclic reactions
Sigmatropic rearrangementsAdditions to double bonds in which two chiral centres are created
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
x
y
a
b
[3,3]sigmatrpoicrearrangement
bx
y
a
+ enantiomer
y
ab
xcontotatory electrocyclic reaction
a
by
x
b
ax
yenantiomer
xa
b yRCO3H O
b ya x
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 ReactionsType 2 Reactions
Reactions involve the stereoselective formation of one or more additional chiral centres from the reaction of one or more prochiral double bonds contained in one or more achiral starting materials using achiral reagents;
Type 3 ReactionsType 3 Reactions
Cycloaddition reactions;Electrocyclic reactionsSigmatropic rearrangementsAddition reactions to double bonds
Cycloaddition reactions;Electrocyclic reactionsSigmatropic rearrangementsAddition reactions to double bonds
Difference with Type 2:One and more existing chiral centres in the starting material, the reagents
or some part of the reaction ensemble.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

34
2
BH t-BuMe
2
B H
Me Bu-tHH
NaOH
H2O2/MeOH
H
Me Bu-tHH
HO
R
99 % e.e.
76 %, 60 % e.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 3 ReactionsType 3 Reactions
R1
R2 R3
OH(+)-diethyl tartrateCH2Cl2
Ti(OiPr)4, t-BuO2H(-)-diethyl tartrate
O OH
R3R2
R1
90 % e.e.
O
OH
R3R2
R1
90 % e.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 3 ReactionsType 3 Reactions
Reactions are those which require the exercise of astereocontrol but do not involve the formation of
tetrahedral centres or other chiral elements;
i. Elimination from substrate containing two contiguous chiral centres;
ii. Addition to alkynes or allenes;iii. Substitution on a (usually already configured) double bond;iv. Sigmatropic rearrangement in which formation of
configured double bonds accompanies the loss of chiral centres;
v. Union of two functional groups with direct formation of the double bond;
vi. Addition-elimination using a molecule containing a double bond;
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 4 ReactionsType 4 Reactions
x
y
Oa
b
x
ya
bO
type II
x
y
Oa
b
c
d
x
ya
bO
c
d type III
O
c
d
O
c
d type IV
How to define the reaction having one chiral How to define the reaction having one chiral centrescentres??
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
QuestionsQuestions
ON
O
ROB(Bun)2
PhO
N
O
RO
Ph R2BOTf
i-Pr2NEt
R'CHO
ON
O
O
R'R
OH
PhMeO
O
R'R
OHMeONa
Please give the reasonable transition state of this reaction:
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 Reactions:Reactions in Which no New Chiral
Centres Are Created
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chapter 3Chapter 3

35
OutlineOutline
1. Commercial Enantiopure Starting Materials;
2. Second-Rank Starting Materials;
3. Synthesis of Single Diastereoisomers Using Type 0 Reactions;
4. Bicyclic Compounds as Sources of Chiral Centres;
5. Readily Available Fissionable Bicyclic Compounds;
6. Fissionable Bridges;
7. Readily Available Fissionable Tricyclic Compounds
1. Commercial Enantiopure Starting Materials;
2. Second-Rank Starting Materials;
3. Synthesis of Single Diastereoisomers Using Type 0 Reactions;
4. Bicyclic Compounds as Sources of Chiral Centres;
5. Readily Available Fissionable Bicyclic Compounds;
6. Fissionable Bridges;
7. Readily Available Fissionable Tricyclic Compounds
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Why?Why?
This type reaction necessary in this case will convert functional groups in the starting material into those required in the product.
This type reaction necessary in this case will convert functional groups in the starting material into those required in the product.
Starting materials:Readily available enantiopure having one, two, or more chiral centres.
Starting materials:Readily available enantiopure having one, two, or more chiral centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Since Type 0 reactions do not require stereoselectivity, it might be assumed that a synthesis consisting of only Type 0 reactions would not constitute a stereoselective one.
Since Type 0 reactions do not require stereoselectivity, it might be assumed that a synthesis consisting of only Type 0 reactions would not constitute a stereoselective one.
Stereoselective synthesis is now widely taken to be the synthesisof a single stereoisomer of the product by whatever means which include only Type 0 reactions if the chiral centres of the productare already in place in the starting materials.
Stereoselective synthesis is now widely taken to be the synthesisof a single stereoisomer of the product by whatever means which include only Type 0 reactions if the chiral centres of the productare already in place in the starting materials.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Why?Why?
The Type 0 reactions may also include the removal of superfluous chiral centres.The application of Type 0 reactions to a limited number of readily available enantiopure starting materials having one, two or more chiral centres is the means by which a much greater number of enantiopure products are made available.
The Type 0 reactions may also include the removal of superfluous chiral centres.The application of Type 0 reactions to a limited number of readily available enantiopure starting materials having one, two or more chiral centres is the means by which a much greater number of enantiopure products are made available.
These enantiopure products will often in turn be used for further transformations or they may be synthetic
targets themselves.
These enantiopure products will often in turn be used for further transformations or they may be synthetic
targets themselves.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 Reactions
Syntheses consisting of only Type 0 reactions will require a
knowledge of the availability of starting materials having
chiral cnetres in place with the correct relative or absolute
configuration for those required in the product.
Syntheses consisting of only Type 0 reactions will require a
knowledge of the availability of starting materials having
chiral cnetres in place with the correct relative or absolute
configuration for those required in the product.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 Reactions
NH
HO
NH2COOH
H
HOOC
(S)-glutamic acid
HNO2
OH
HOOCO 1. EtOH, H+
2. NaBH4
OH
HOH2CO 1. TsCl, Pyr.
2. NaN3, DMF OHO
N3
H2, Pd/C
NH
OHO BH3/THF
NH
HO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 Reactions

36
O
HSPh
1. PhMgBr
2. NH4Cl HPhHO
SPh
1. Me3O+BF4-
2. NaOH HPh
O
C10H21MgBr CuI
HPhHO
H23C11OH23C11
H O
OH23C11
H O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 ReactionsHO
PhS
O Baker's yeast
OH
HHO
SPh
1. TsCl, Pyr2. NaOH H
O
SPh
SPh
OH
1. C10H21MgBr
2. H+
SPh
HO C11H23H
1. Me3O+BF4-
2. NaOH
O
C11H23H
HO C11H23H
O
O
10 % H2SO4
C11H23H
OHO
PCC
C11H23H
OO
O
O
CuMgBr)2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 Reactions
O
O H
HO
HO HO
CH2
CH2
CH2
H OH
HHO
CH2COCH3
CH3CH2OH
H OH
OHH
HHO
H OH
CHO
exo-Brevicomin
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 0 ReactionsType 0 Reactions
Glucose Me2CO
ZnCl2
O
OO
OH
HH
O
O 1. NaH, PhCH2Cl
2. H+, H2O O
OO
OBn
HH
HO
HO
1. MsCl, Pyr.2. I-
3. MeOH, HClO
OH
OBn
HH
OMe
1. NaH2. CS2
3. MeIO
OBn
HH
OMe
O
S
SMe
SnBu3OOBn
HH
OMeBu3SnH
OOBn
HH
OMe
1. AcOH, H2O2. Ph3PCHCOCH3
OHOBn
HH
O 1. H2, R-Ni
2. H2, Pd/C O
O
The BartonThe Barton--McCombieMcCombie MethodMethod
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OHOBn
HH
O
HOO
HO H
O
O H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The BartonThe Barton--McCombieMcCombie MethodMethod
Readily available starting materials enantio- and diastereoisomerically pure.Readily available starting materials enantio- and diastereoisomerically pure.
What constitutes a readily available starting material?What constitutes a readily available starting material?
S
NO
H2N H
COOH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Readily available starting materialsReadily available starting materials

37
SecondSecond--Rank Starting MaterialsRank Starting Materials
O
H2O2
NaOH
O
OPhSNa
O
O-
PhS
PhS
ONa
RIR
O
PhS1. mCPBA
2.
R
O
H+, H2O orNaOH, H2O
O
反式消除
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
O
O3HOOC
HOOC
O
on Ca salt
Br2
OBr
BrNaOMe COOMe
NaOHCOOMe
SecondSecond--Rank Starting MaterialsRank Starting Materials
Trans稳定
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SecondSecond--Rank Starting MaterialsRank Starting Materials
O
1. HCl2. NaOH
COOH
Citronellic acid
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
O Br2, HOAc O
BrBr2ClSO3H
O
Br
Br
ZnHBr
O
Br
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SecondSecond--Rank Starting MaterialsRank Starting Materials
O
Br
H+
Br
OH
Br
OH
H BrOH
(2,3)-exo Me shift
H BrOH
-H+
H BrOH
Br2
H BrOH
Br+
(2,3)-exo Me shift
H BrOH
Br
H BrOH
Br
H Br
Br
O
Br
Br
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SecondSecond--Rank Starting MaterialsRank Starting Materials
O
Br
KI, DMFO
I
NaCH(COOMe)2
O
MeOOC
NH2OHN
MeOOC
OH
1. TsCl, pyr
2. CF3COOH COOMe
CNH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SecondSecond--Rank Starting MaterialsRank Starting Materials

38
O
Br
Br2, ClSO3HBr2, 95 % O
Br
Br
O
Br
Br
Br
Zn HBr
75 %O
Br
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SecondSecond--Rank Starting MaterialsRank Starting Materials Synthesis of Single DiastereoisomersSynthesis of Single Diastereoisomers
O O
OH
H
Ph
OH
OH
H
Ph
COOH
OH
HOH
COOH
Ph
CH2OH
OH
HOH
COOH
OH
HO O
O O
OH
HO
OH
HO OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OH
OHHO
R-Ni
OH
HO OH
2equiv. tBuPh2SiClTHF, imidaz.
PCC
OSiPh2But
tBuPh2SiO O
mCPBA MeOHH+
PCC PhCH2+PPh3Cl-
BuLi
Bu4N+F- AcOH
O
OH
H
Ph
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Synthesis of Single DiastereoisomersSynthesis of Single Diastereoisomers BicyclicBicyclic Compounds as Sources of Chiral Compounds as Sources of Chiral CentresCentres
Unsubstituted bicyclic compounds can serve as usefulsources of monocyclic compounds whose relative or absolute configuration at two chiral centres is defined.
This requires that one of the bridges in the bicyclic compound can be cleaved without affecting the configuration at either chiral centre originating from the bridgehead positions.
Unsubstituted bicyclic compounds can serve as usefulsources of monocyclic compounds whose relative or absolute configuration at two chiral centres is defined.
This requires that one of the bridges in the bicyclic compound can be cleaved without affecting the configuration at either chiral centre originating from the bridgehead positions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
O3
COOH
COOH
H
HCOOH
COOH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BicyclicBicyclic Compounds as Sources of Chiral Compounds as Sources of Chiral CentresCentres The Schreiber ModificationThe Schreiber Modification
O3, MeOHNaHCO3
O3, MeOHpTsOH
CHOOH
CHO
OMe
Ac2OEt3N COOMe
CHO
CHOOH
CH(OMe)2
OMe
NaHCO3
Me2S CHO
CH(OMe)2
Et3NAc2O COOMe
CH(OMe)2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
the Ozonolysis Reaction

39
COOH
NH2
COOH
CONH2
O
O
O
Completely Stereoselective ConversionCompletely Stereoselective Conversion
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CC--C to CC to C--N BondsN Bonds
Readily Available Fissionable Readily Available Fissionable BicyclicBicyclic CompoundsCompounds
The use of made of bicyclic compounds for the preparation of disubstituted monocyclic compounds of the defined relative configuration by Type 0 reactions will depend upon their availability and on the ease with which one ring can be cleaved.
The use of made of bicyclic compounds for the preparation of disubstituted monocyclic compounds of the defined relative configuration by Type 0 reactions will depend upon their availability and on the ease with which one ring can be cleaved.
A large number of bicyclic compounds are readily available by a variety of Type 2 cycloadditions including the
Diles-Alder reaction.
A large number of bicyclic compounds are readily available by a variety of Type 2 cycloadditions including the
Diles-Alder reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Readily Available Fissionable Readily Available Fissionable BicyclicBicyclic CompoundsCompounds
p q
x
a
p q
x
a
rp
qx
a
rp
qx
a
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
Any reaction which leads to bond breaking can be harnessed to bring about cleavage of a ring provided
that the atoms of the functional group involved can be incorporated into a ring.
Any reaction which leads to bond breaking can be harnessed to bring about cleavage of a ring provided
that the atoms of the functional group involved can be incorporated into a ring.
Lactones are particularly useful fissionable rings because they are cleaved under mild conditions to give two chemo-
differentiated functional groups.
Lactones are particularly useful fissionable rings because they are cleaved under mild conditions to give two chemo-
differentiated functional groups.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
OmCPBA O
O
MeOH
H
OH
COOMeDIBAL
O
OH
Ph3PCHCO2Me
H
OH
CO2Me
取代多的R基优先迁移
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
O
OO
OBnO
1. LiCH2PO(OMe)2
2. H2O
O
OO
BnOOH
CH2P(OMe)2
O
NaOMe, MeOH
OO
BnO OH
CH2P(OMe)2
O
O
CrO3pyr
OO
BnO O
CH2P(OMe)2
O
O
K2CO3
18-crown-6
OO
BnOO
Wittg Reaction冠醚可以增强试剂的碱性
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges

40
hv, O2MeOH, thiorea O
O
OH
OH
H
HOH
OH
NCOPh
O O
NCOPh Al/HgNHCOPh
HO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
OAc
OAc
NN
MeO2CCO2Me
OAc
OAcN
NCO2Me
MeO2C
NHMeOH
NH2
OH
MeO
HO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
O
O
O
1. LDA2. TMSCl
O
O
OTMS
1. O32. CrO3
CO2H
COOH
O
O
SN
O
H
H
ArR-Ni
SNHAr
OH
H
H
R-Ni NHArOH
H
H
酮的α-H酸性大于酯
过量的还原剂还原硫醚
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
O
O
1. PhSCl2. HOCH2CH2OH
H+
O
PhS
ClO
O
1. mCPBA2. DBUCHCl3
O
PhSO2 O
O
MeLi
O
PhSO2 O
O
O
OPhSO2
MeOH
AcOCN
O
成砜可以增强α-H的酸性
Li和O的络合作用使Me从上面进攻
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Fissionable BridgesFissionable Bridges
Readily available Fissionable Readily available Fissionable TricyclicTricyclic CompoundsCompounds
The use of tricyclic compounds for the synthesis of bicyclic/monocyclic compounds is widespread.
The use of tricyclic compounds for the synthesis of bicyclic/monocyclic compounds is widespread.
The small number of commercially available tricyclic compounds of any complexity means that assembly of the required tricyclic
compound will almost always be necessary with substitution on one or more bridges appropriate for their cleavage as required.
The small number of commercially available tricyclic compounds of any complexity means that assembly of the required tricyclic
compound will almost always be necessary with substitution on one or more bridges appropriate for their cleavage as required.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Readily available Fissionable Readily available Fissionable TricyclicTricyclic CompoundsCompounds
Br2Br
Br
KOHdioxane
Br
O- O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

41
N
Cl
dioxaneH2O
NH2O N
CHO
NaBH4 Ac2O, pyr
AcN
H
H
H
CH2OAc
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Readily available Fissionable Readily available Fissionable TricyclicTricyclic CompoundsCompounds
O
CF3CO3HNa2HPO4
O
O
LiAlH4
OHCH2OH
Ph3CClpyr
CrO3 NH2OHH
H
H
CH2OCPh3NHO
AcN
H
H
H
CH2OAcTsClpyr
Al2O3 LiAlH4 HClCHCl3
Ac2Opyr
保护一级醇
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Readily available Fissionable Readily available Fissionable TricyclicTricyclic CompoundsCompounds
SummarySummary
Synthesis using Type 0 reactions relies heavily on the availabilityand recognition of appropriate starting materials in which the chiral centre(s) in the target molecule must already be present.
Synthesis using Type 0 reactions relies heavily on the availabilityand recognition of appropriate starting materials in which the chiral centre(s) in the target molecule must already be present.
Although the most useful starting materials containing two chiral centres in Type 0 synthesis are those which are single enantiomers, racemic compounds may also be of use in the synthesis of target molecules in racemic form.
Although the most useful starting materials containing two chiral centres in Type 0 synthesis are those which are single enantiomers, racemic compounds may also be of use in the synthesis of target molecules in racemic form.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The number of cheap enantiopure compounds which are commercially available is small and their variety is limited. However, in the hands of a competent chemist working in a well-found laboratory this small number of compounds can
provide access to a much larger number of second-rankstarting materials, most of which are not commercially available.
The number of cheap enantiopure compounds which are commercially available is small and their variety is limited. However, in the hands of a competent chemist working in a well-found laboratory this small number of compounds can
provide access to a much larger number of second-rankstarting materials, most of which are not commercially available.
Bicyclic compounds are fertile sources of monocyclic and acyclic molecules having at least two chiral centres of defined relative or absolute configuration. The two chiral centres present at the bridgehead or ring junction positions of these bicyclic compounds are conserved in Type 0 reactions which open one or both rings.
Bicyclic compounds are fertile sources of monocyclic and acyclic molecules having at least two chiral centres of defined relative or absolute configuration. The two chiral centres present at the bridgehead or ring junction positions of these bicyclic compounds are conserved in Type 0 reactions which open one or both rings.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Awareness of the existence of second-rank starting materialsand retrieval of methods for their preparation can be accomplished by chemical literature searching methods including computer-assisted searches. Ultimately, however, there is no substitute for familiarity with as wide a rangeof organic chemistry as possible and particularly with the chemistry of the compounds.
Awareness of the existence of second-rank starting materialsand retrieval of methods for their preparation can be accomplished by chemical literature searching methods including computer-assisted searches. Ultimately, however, there is no substitute for familiarity with as wide a rangeof organic chemistry as possible and particularly with the chemistry of the compounds.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Type 1 Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chapter 4Chapter 4

42
Those Proceeding With Either Inversion or With Rentention of Configuration at a Single Chiral
Centre
Part 1Part 1
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OutlineOutline
4.1. Inversion of Configuration;4.2. Stereoelectronic Control;4.3. Transition-state Geometry;4.4. Limitations of the SN2 Reaction in
Stereoselective Synthesis;4.5. Double Inversion --- Retention 4.6. 1,2-Rearrangements with Inversion of
Configuration at the Migration Terminus4.7. 1,2-Rearrangements with Retention of
Configuration at The Migration Terminus4.8. Substitution with Retention of Configuration
via Configurationally Stable Carbanions 4.9. Summary
4.1. Inversion of Configuration;4.2. Stereoelectronic Control;4.3. Transition-state Geometry;4.4. Limitations of the SN2 Reaction in
Stereoselective Synthesis;4.5. Double Inversion --- Retention 4.6. 1,2-Rearrangements with Inversion of
Configuration at the Migration Terminus4.7. 1,2-Rearrangements with Retention of
Configuration at The Migration Terminus4.8. Substitution with Retention of Configuration
via Configurationally Stable Carbanions 4.9. Summary
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
bc
a
Nu-
bc
a
NuX X Nua
bc
σ∗-orbital
4.1. 4.1. Inversion of ConfigurationInversion of Configuration
The bond changes which accompany the SN2 reaction take place concertedly: as the Nu-C bond is forming so the C-X bond is breaking and a single transition state separates the starting materials and products.
The bond changes which accompany the SN2 reaction take place concertedly: as the Nu-C bond is forming so the C-X bond is breaking and a single transition state separates the starting materials and products.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
In Type 1 reactions, at least one bond to a chiral centre is broken and a new bond is formed completely stereoselectively.
In Type 1 reactions, at least one bond to a chiral centre is broken and a new bond is formed completely stereoselectively.
In concerted reactions such as the SN2 type, the orbitalswhich are involved in making new bonds have a defined spatial relationship to those orbitals which arise from the breaking of existing bonds.
In concerted reactions such as the SN2 type, the orbitalswhich are involved in making new bonds have a defined spatial relationship to those orbitals which arise from the breaking of existing bonds.
4.2. 4.2. StereoelectronicStereoelectronic ControlControl
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Scheme illustrates the inversion of configuration which characterises an SN2 reaction
4.2. 4.2. StereoelectronicStereoelectronic ControlControl
In SN2 reaction, the part of the σ∗-orbital shown is directed at the angle of 180o to orbitals comprising the C-X σ bond undergoing cleaving. Bond formation, therefore, which occurs by overlap of this empty σ∗ orbital with the filled orbital of the nucleophile, takes place with inversion of configuration.
In SN2 reaction, the part of the σ∗-orbital shown is directed at the angle of 180o to orbitals comprising the C-X σ bond undergoing cleaving. Bond formation, therefore, which occurs by overlap of this empty σ∗ orbital with the filled orbital of the nucleophile, takes place with inversion of configuration.
bc
a
Nu-
bc
a
NuX X Nua
bc
σ∗-orbital
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
When, as in the SN2 reaction, a specific spatial relationship exists between bonds made and broken, the reaction is said to proceed under stereoelectronic control.
When, as in the SN2 reaction, a specific spatial relationship exists between bonds made and broken, the reaction is said to proceed under stereoelectronic control.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.2. 4.2. StereoelectronicStereoelectronic ControlControl

43
4.3. 4.3. TransitionTransition--state Geometrystate Geometry
In SN2 reaction, the transition state is assumed to be as shown in scheme in which C-a, C-b and C-c bonds lie in a plane and the Nu-C and C-X bonds are colinear.
In SN2 reaction, the transition state is assumed to be as shown in scheme in which C-a, C-b and C-c bonds lie in a plane and the Nu-C and C-X bonds are colinear.
This transition-state geometry (TSG) is an idealized representation for most SN2 reactions. If, for example, C-X bond breaking runs ahead of Nu-C bond making, or vice versa, then this will result in a transition state in which C-a, C-b and C-c bonds deviate from coplanarity.
This transition-state geometry (TSG) is an idealized representation for most SN2 reactions. If, for example, C-X bond breaking runs ahead of Nu-C bond making, or vice versa, then this will result in a transition state in which C-a, C-b and C-c bonds deviate from coplanarity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
Since the SN2 reaction is sensitive to steric effects, ideally this reaction requires attack by a non-bulky and highly polarisable nucleophile on a sterically unencumbered sp3-hybridised carbon atom bearing a good leaving group.
Since the SN2 reaction is sensitive to steric effects, ideally this reaction requires attack by a non-bulky and highly polarisable nucleophile on a sterically unencumbered sp3-hybridised carbon atom bearing a good leaving group.
The most widely used leaving group:Halides, OZ groups (esters of the hydroxyl group with strong acids:tosylates, mesylates, triflates
The best nucleophiles:CN-, N3
-, RS-, I-, etc
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
The stereoelectronic control present in the SN2 results in inversion of configuration in the product.
The stereoelectronic control present in the SN2 results in inversion of configuration in the product.
The complete stereoselectivity is impervious to the presence of the other chiral centres elsewhere in the molecule
The complete stereoselectivity is impervious to the presence of the other chiral centres elsewhere in the molecule
The SN1 reaction can occasionally also be highly stereoselective if the carboncation through which it proceeds is captured by a nucleophile predominantly from one face.
The SN1 reaction can occasionally also be highly stereoselective if the carboncation through which it proceeds is captured by a nucleophile predominantly from one face.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OOMe
Me
H
H H
C6H13Me2AlOAr O
OMe
Me
HH H
C6H13
Al Me
MeOAr
H+ OHOMe
Me
HH
H
C6H13Me
70 %, > 99 % retention, Ar = C6F5
The SN1 reaction can occasionally also be highly stereoselective if the carboncation through which it proceeds is captured by a nucleophile predominantly from one face.
The SN1 reaction can occasionally also be highly stereoselective if the carboncation through which it proceeds is captured by a nucleophile predominantly from one face.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
If the latter does occur, it is invariably the result of the molecular environment in which the carboncation is generated as in ion-pair formation: unlike the SN2 reaction, the SN1 reaction is not an inherently stereoselective (stereospecific) reaction.
If the latter does occur, it is invariably the result of the molecular environment in which the carboncation is generated as in ion-pair formation: unlike the SN2 reaction, the SN1 reaction is not an inherently stereoselective (stereospecific) reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
Limitation the Usefulness of Limitation the Usefulness of Intermolecular SIntermolecular SNN2 Reactions2 Reactions
First, attack at tertiary centres is so sterically retarded that reaction, if it occurs at all, results in elimination and
alkene formation rather than substitution.
First, attack at tertiary centres is so sterically retarded that reaction, if it occurs at all, results in elimination and
alkene formation rather than substitution.
HX
Nu-+ NuH + X-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

44
Second, although primary carbon centres are well suited for SN2 substitution, inversion of configuration is only apparent
when deuterium (or trituim) substitution is present.
Second, although primary carbon centres are well suited for SN2 substitution, inversion of configuration is only apparent
when deuterium (or trituim) substitution is present.
Nu- XR
HDNu
R
HD
+ X-
Nu- XR
HHNu
R
HH
+ X-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Limitation the Usefulness of Limitation the Usefulness of Intermolecular SIntermolecular SNN2 Reactions2 Reactions
Third, attempted SN2 substitution at secondary centres is also often blighted by competitive elimination especially
when the nucleophile has any basic character.
Third, attempted SN2 substitution at secondary centres is also often blighted by competitive elimination especially
when the nucleophile has any basic character.
MeMeOHOTs
+MeO2C Ph
CO2MeMeMeOH
Ph
CO2Me
CO2Me
OO
H
( )-sarracenin
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Limitation the Usefulness of Limitation the Usefulness of Intermolecular SIntermolecular SNN2 Reactions2 Reactions
OR1
R2109oO
119o
In epoxides, the angle between the two exocyclic bonds is widened as a consequence of the higher s-character which these bonds contain.
This increased s-character is itself a consequence of the higher p-character which is required for formation of the ring bonds to accommodate the geometry of the small ring.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
Widening of the bond angles allows easier ingress of the nucleophiles.
Complexation of the epoxide oxygen can assist in the ring opening such that the substitution has both
‘push’ form the nucleophile and ‘pull’ from the leaving group.
O
B F 3 .E t 2 OB u L i, -7 8 o C
+ O-B F 3
B u -
O H
B u
H OB u
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
The diminished basicity and increased nucleophilicity of carbonions when complexed with copper facilitates substitution at secondary centres and particularly those on epoxide rings.
The diminished basicity and increased nucleophilicity of carbonions when complexed with copper facilitates substitution at secondary centres and particularly those on epoxide rings.
Bu COOEt
OTfMe2CuLi, -70 oC Bu COOEt
Me
Me2CuLi
OO O
MeOBn
OO
MeOBn
OHMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
The epoxide is opened at the less hindered position highly regio-selectivity (11:1) and also completely stereoselectively.The epoxide is opened at the less hindered position highly
regio-selectivity (11:1) and also completely stereoselectively.
Me2CuLi
OO O
MeOBn
OO
MeOBn
OHMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis

45
O
Me TMSHH
Li2CuCN(CH2TMS)2
2TMSCH2Li + CuCNMe
TMSHO
CH2TMS
H
H
Epoxide ring opening by nucleophiles does not necessarily occur at the sterically less hindered. In this case attack of the nucleophile is believed to take place initially by addition to the silicon substituent and is thence directed to the more hindered position on the ring.
Epoxide ring opening by nucleophiles does not necessarily occur at the sterically less hindered. In this case attack of the nucleophile is believed to take place initially by addition to the silicon substituent and is thence directed to the more hindered position on the ring.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
SN2 substitution at secondary centres is also facilitated when steric congestion in the transition state is reduced in other ways.
This is the case when one or more of the substituents on Cα in the substrate is sp2- or sp-hybridised, i.e. a vinyl or ethynyl group.
A heteroatom substituent (C, N, O) is smaller by virtue of the lone pair(s) (rather than substituents) that it bears. Unfortunately, these unsaturated substituents and heteroatoms may also facilitate SN1 substitution (C+ stabilisation) with its only occasionally complete stereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
CO2Me
H
N3
OHRH
O
H CO2MeHR
NaN3HO
N3H
CO2MeH
R +
Ring opening of the enantiopure epoxide with azide ion followed by reaction of two products with triphenylphosphine results in the formation of aziridine. In spite of the fact that the ring opening is not regiospecific, only a single enantiopureaziridine is obtained.
Ring opening of the enantiopure epoxide with azide ion followed by reaction of two products with triphenylphosphine results in the formation of aziridine. In spite of the fact that the ring opening is not regiospecific, only a single enantiopureaziridine is obtained.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
O
H CO2MeHR
NaN3HO
N3H
CO2MeH
RPh3P
DMF, 80 oC
HO
NH
CO2MeH
R PPh3
NHP
O
HH CO2MeR
Ph3PO
CO2Me
H
H
R-N H
Ph3
NH
CO2MeHR H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
CO2Me
H
N3
OHRH
O
H CO2MeHR
NaN3 Ph3PDMF, 80 oC
NH
CO2MeHR H
CO2Me
H
N
OHRH
Ph3P
H HR CO2Me
OPPh3
NCO2Me
H
HN-
ORH
PPh3
从两边进攻的最终产物相同
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
RO- also as the nucleophiles
Like carbanions, oxyanions (RO-) are also basic and the classic method for replacement of one C-O bond with inversion. This transformation can now be accomplished in better yield by making use of the Mitsunobu reaction followed by the hydrolysis.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis

46
H
H
HO
HTsCl, pyr.
H
H
TsO
H
HO-, DMF
H
H
HO
H
HO-
H
H
PhCOO
H
Ph3P, EtO2CNNCO2Et (DEAD)
PhCOOH, THF
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
The mechanism of the Mitsunobu reactionsThe mechanism of the Mitsunobu reactions
Widely used for converting a secondary alcohol directly into its inverted ester
Widely used for converting a secondary alcohol directly into its inverted ester
EtO2CNNCO2Et (DEAD) Ph3P EtO2CN NCO2Et
PPh3
R1COOH
EtO2CN NHCO2Et
PPh3
R2OH EtO2CN NHCO2Et
PPh3
OR2R1COO-H+
R1COOR2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
This reaction can also be used for the construction of large-ring compounds as in the closure of compounds to the ring system.
This reaction can also be used for the construction of large-ring compounds as in the closure of compounds to the ring system.
S
SH11C5
OTHP1. BuLi2. O
Br3.
LiO
O
Li
4. MeOH, THF, H3O+
H11C5
OH
S
S
OH
O OH
Ph3PDEAD
H11C5
O
S
S
OH
O
只适用于醛
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
EtOOCCOOEt
OHO1. , H+;
2. LiAlH4;3. TsCl, pyr;4. NaBr, DMSO;5. HOAc, H2O
BrH2CCH2Br
OH
KOH
CH2BrO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
OO
TsO OBn
1. KNO2, DMF2. H2O
OO
HO OBn
Other method for C-O to C-O conversion of configuration
Other methods for C-O to O-C conversion with inversion of configuration which minimise yields of alkene by-products:Some of these methods were developed for prostaglandin synthesis, where elimination to give alkene was a particular problem with existing methods.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
tBuOTs
KNO2, DMSODME18-crown-6
tBu
OH
tBuOH
KNO2, DMSODME18-crown-6
tBu
OMs
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
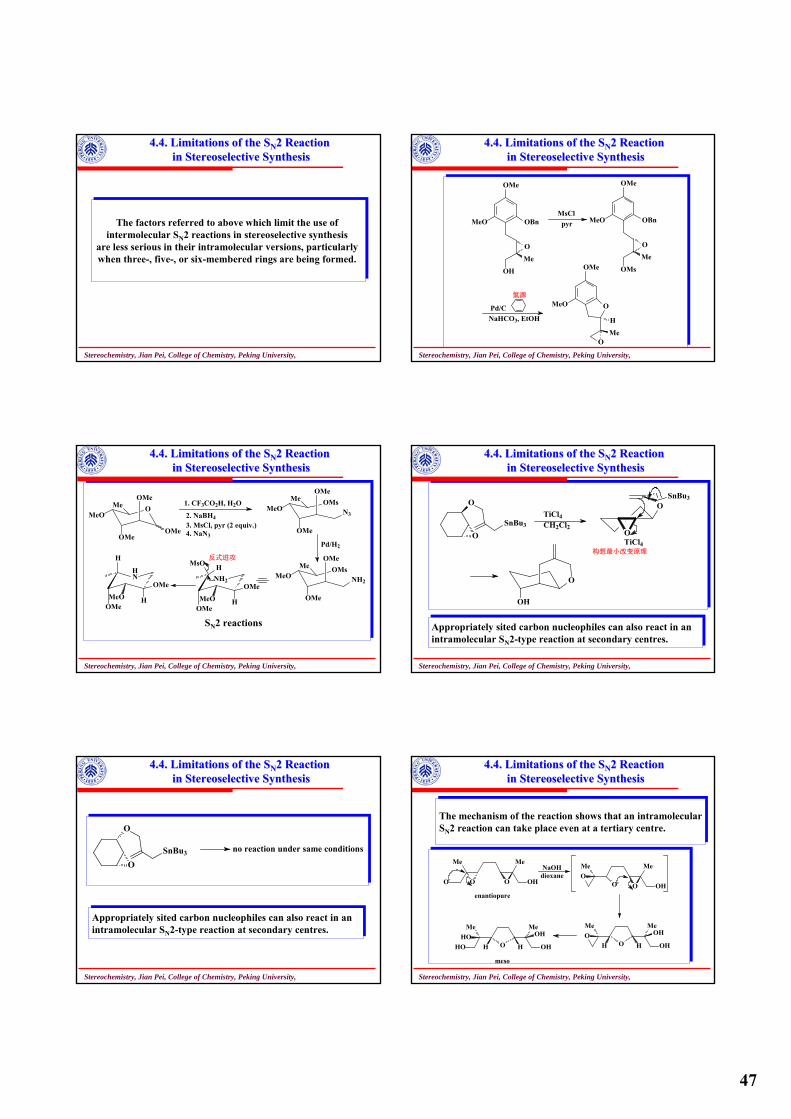
47
The factors referred to above which limit the use of intermolecular SN2 reactions in stereoselective synthesis
are less serious in their intramolecular versions, particularly when three-, five-, or six-membered rings are being formed.
The factors referred to above which limit the use of intermolecular SN2 reactions in stereoselective synthesis
are less serious in their intramolecular versions, particularly when three-, five-, or six-membered rings are being formed.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
OMe
MeO OBn
OH
O
Me
MsClpyr
OMe
MeO OBn
OMs
O
Me
Pd/CNaHCO3, EtOH
OMe
MeO O
O
H
Me
氢源
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
OMeO
OMe
MeOMe
OMe
1. CF3CO2H, H2O
2. NaBH43. MsCl, pyr (2 equiv.)4. NaN3
OMsMeO
OMe
MeOMe
N3
Pd/H2
OMsMeO
OMe
MeOMe
NH2NH2
MeO H
OMe
MsO H
OMe
N
MeO H
OMe
H
OMe
H
SN2 reactions
反式进攻
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
O
O
SnBu3
TiCl4CH2Cl2
OTiCl4
OSnBu3
O
OH
Appropriately sited carbon nucleophiles can also react in an intramolecular SN2-type reaction at secondary centres.
Appropriately sited carbon nucleophiles can also react in an intramolecular SN2-type reaction at secondary centres.
构想最小改变原理
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
O
O
SnBu3 no reaction under same conditions
Appropriately sited carbon nucleophiles can also react in an intramolecular SN2-type reaction at secondary centres.
Appropriately sited carbon nucleophiles can also react in an intramolecular SN2-type reaction at secondary centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
OHO- O O
MeMeNaOH
dioxaneOHO
MeMeO
O-
OH
MeMeO
OH H
OH
OH
MeMeHO
OH H
OH
HO
enantiopure
meso
The mechanism of the reaction shows that an intramolecular SN2 reaction can take place even at a tertiary centre.
The mechanism of the reaction shows that an intramolecular SN2 reaction can take place even at a tertiary centre.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis

48
TsOCO2H
OH
OH
NaOEtEtOH
COO-
O-
O
O
HH
HO
O-
O
O
O
HHO
H
HO
Ac2OO
O
HAcO
H
AcO
Treatment of the enantiopure acid with sodium ethoxide followed by acetylation gave diacetate, whose relative and absolute configurations suggest that a Payne rearrangement is involved and inversion at both adjacent chiral centres has occurred.
Treatment of the enantiopure acid with sodium ethoxide followed by acetylation gave diacetate, whose relative and absolute configurations suggest that a Payne rearrangement is involved and inversion at both adjacent chiral centres has occurred.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.4. 4.4. Limitations of the SLimitations of the SNN2 Reaction 2 Reaction in Stereoselective Synthesisin Stereoselective Synthesis
Limitation the Usefulness of Limitation the Usefulness of Intermolecular SIntermolecular SNN2 Reactions2 Reactions
1. Attack at teriary centres is so sterically retarded that reaction, if it occurs at all, results in elimination and alkene formation rather than substitution.
2. Although primary carbon centres are well suited for SN2 substitution, inversion of configuration is only apparent when deuterium (or trituim) substitution is present.
3. Attempted SN2 substitution at secondary centres is also often blighted by competitive elimination especially when the nucleophile has any basic character.
1. Attack at teriary centres is so sterically retarded that reaction, if it occurs at all, results in elimination and alkene formation rather than substitution.
2. Although primary carbon centres are well suited for SN2 substitution, inversion of configuration is only apparent when deuterium (or trituim) substitution is present.
3. Attempted SN2 substitution at secondary centres is also often blighted by competitive elimination especially when the nucleophile has any basic character.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
COOHNH2H
HOOC 1 M HClHNO2
COOHN2
+HC
HO
O
C
C
O
HOOH
OO
OH
HOOC
An intramolecular SN2 reaction with inversion and formation of an unstable α–lactone is followed by a second intramolecular substitution with inversion.
An intramolecular SN2 reaction with inversion and formation of an unstable α–lactone is followed by a second intramolecular substitution with inversion.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
COOHMeH
H NH2
COOHMeH
H OHHNO2
1. Mitsunobu
2. hydrolysisCOOH
MeH
HO H
Three inversions and overall just inversion.Three inversions and overall just inversion.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
Double inversion commonly results from neighboring group participation of which α- and then γ-lactone formation.
Double inversion commonly results from neighboring group participation of which α- and then γ-lactone formation.
NOBn
H
H
Me
COOMe
HI
Bu3NMeCN, H2O N
OBnH
H
Me
O
H OMe
NOBn
H
H
Me O OH
91 %
NMeH COOMe
I
HBnO
6 %
I-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
NOBn
H
H
Me
O
H OMe NMe
H COOMe
I
HBnO
I-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention

49
MeO
MeONMe
HO
H
(CO)3Cr
HBF4CH2Cl2
< - 20oC
MeO
MeONMe
H
(CO)3Cr
MeO
MeO
NMe
H
(CO)3Cr
O2hvMeO
MeO
NMe
H
Cyclisation of this compound lacking the chromium tricarbonyl ligand gave rise to the product with only 6 % e.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
OTsHOAc H
H
-OAc
OAc
a-OAc
ba
b
AcOAcO
H H
AcO- AcO-a b
The first inversion is the result of the intramolecular necleophilic displacement mediated by partial delocalization of the bond with the formation of a non-classical carbocation. The second substitution path a takes place by attack on this non-classical ion by the solvent acetic acid with the partially formed bond acting as a leaving group.
The first inversion is the result of the intramolecular necleophilic displacement mediated by partial delocalization of the bond with the formation of a non-classical carbocation. The second substitution path a takes place by attack on this non-classical ion by the solvent acetic acid with the partially formed bond acting as a leaving group.Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
R R
X
PdL2PdL2
R
H
R
H
Nu- R R
Nu
TMS Ph
OCOOMe
R PH
MeOOC COOMe
Pd(PBu3)n- CO2
Ph
H
TMS
HPd
Bu3P OMe
Net retention of configuration can also be accomplished at allylic secondary carbon centres by carbon nucleophiles using palladiumchemistry.
Net retention of configuration can also be accomplished at allylic secondary carbon centres by carbon nucleophiles using palladiumchemistry.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
HO OAcPd(PPh3)4
HO
Pd+
Nu- HO Nu
Nu: CH(CO2Me)2 80 % PhS 86 %
N 74 %
O
O
Retention here is the result of two substitutions and although neither is the SN2 reaction, the overall stereochemistry is that which would obtain if this were the case.
Retention here is the result of two substitutions and although neither is the SN2 reaction, the overall stereochemistry is that which would obtain if this were the case.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.5. 4.5. Double Inversion Double Inversion ------ Retention Retention
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
The migration of a substituent from one carbon to an adjacent one is frequently accompanied by stereochemical changes.
The migration of a substituent from one carbon to an adjacent one is frequently accompanied by stereochemical changes.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The stereoelctronic requirementfor this rearrangement is anantiperiplanar disposition of the bonds to migrating group and leaving group as shown in both transitions.
The stereoelctronic requirementfor this rearrangement is anantiperiplanar disposition of the bonds to migrating group and leaving group as shown in both transitions.
OHc
a
OH
b
d
H+OH2
+
c
a
O
b
d
H
dO
ca
b
OH2+
d
a
O
b
c
H
cO
d ab
Loss of one chiral centre.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus

50
Pinacol rearrangement of acyclic diols is seldom regioselective although completely stereoselective.
A small rotation around the C-C bond in which migration of d is stereoelectronically preferred.
Protonation and ionisation of the other hydroxyl accompanied by migration of a or b can lead to other regioisomers.
Pinacol rearrangement of acyclic diols is seldom regioselective although completely stereoselective.
A small rotation around the C-C bond in which migration of d is stereoelectronically preferred.
Protonation and ionisation of the other hydroxyl accompanied by migration of a or b can lead to other regioisomers.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
OHc
a
OH
b
d
H+OH2
+
c
a
O
b
d
H
dO
ca
b
OH2+
d
a
O
b
c
H
cO
d ab
Regioselectivity may be accomplished if migration of c is easier than d if it has a higher migratory aptitude.
Regioselectivity may be accomplished if migration of c is easier than d if it has a higher migratory aptitude.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
O
O HO
MeMsO H
O H
Bu
Et3Al, CH2Cl2, - 42 oC
H
Me
BuO
1. BuCCLi, THF2. LiAlH4, THF3. pyrH+OTs-
4. MsCl, Et3N
> 99 % e. e.
Solved by selective mesylation of the secondary hydroxyl; this thenbecomes then better leaving group. Exclusive migration of the vinyl group occurs with retention of double bond configuration in the alkene and inversion configuration at the migration terminus.
Solved by selective mesylation of the secondary hydroxyl; this thenbecomes then better leaving group. Exclusive migration of the vinyl group occurs with retention of double bond configuration in the alkene and inversion configuration at the migration terminus.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
OHc
a
OH
b
d
H+OH2
+
c
a
O
b
d
H
dO
ca
b
OH2+
d
a
O
b
c
H
cO
d ab
The migration of c or d in the rearrangement is promoted and assisted by the pull of the developing carbocation but the migration may be assisted as much by a push from a substituent on the migrationorigin.
The migration of c or d in the rearrangement is promoted and assisted by the pull of the developing carbocation but the migration may be assisted as much by a push from a substituent on the migrationorigin.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
OOBn
H
H
C8H17O
1. CH2 CH2MgBr2. TMSCl, imidazole
OOBn
H
H
H17C8O
SiMe3
Cl-
Ti
H17C8 OBn
OHO
(iPrO)2TiCl2-78oC to -40oC
Driving force:The formation of the strong Si-Cl bond;The conversion of the resulting alkoxide anion into a carbonyl group long with relief of ring strain from opening of the epoxide.
Driving force:The formation of the strong Si-Cl bond;The conversion of the resulting alkoxide anion into a carbonyl group long with relief of ring strain from opening of the epoxide.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus
Commercially available enantiopure compounds containing a C-O bond at a chiral centre are more abundant than analogues containing C-C bonds.It is noteworthy, therefore, that the overall conversions in above two schemes fashion C-C bonds at the expense of C-Obonds and this is acoomplished diastereoselectively in spite of the fact that the substrates undergoing reaction are not necessarily single diastereoisomers.
Commercially available enantiopure compounds containing a C-O bond at a chiral centre are more abundant than analogues containing C-C bonds.It is noteworthy, therefore, that the overall conversions in above two schemes fashion C-C bonds at the expense of C-Obonds and this is acoomplished diastereoselectively in spite of the fact that the substrates undergoing reaction are not necessarily single diastereoisomers.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus

51
It is noted previously that intermolecular SN2 substitution using carbon-centred nucleophiles at secondary and tertiary centres is not generally synthetically useful.Above two scheme provide at least a partial solution to this problem: the nucleophile is added initially to a carbonyl group flanking the chiral centre and this is followed by intramolecular transfer to the chiral centre.
It is noted previously that intermolecular SN2 substitution using carbon-centred nucleophiles at secondary and tertiary centres is not generally synthetically useful.Above two scheme provide at least a partial solution to this problem: the nucleophile is added initially to a carbonyl group flanking the chiral centre and this is followed by intramolecular transfer to the chiral centre.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.6. 1,24.6. 1,2--Rearrangements with Inversion of Rearrangements with Inversion of Configuration at The Migration TerminusConfiguration at The Migration Terminus WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
c
a
ed
bed
c abx
c
a
ed
b reaction with Nu-
(Type III reaction?)loss of +ve charged fragment(regiocontrol: Type IV reaction?)
daughter carboncation by further rearrangementX-
The 1,2-shift is a rearrangement in which a group c migrates with its σ-bonded pair of electrons to an electron-deficient centre.
The 1,2-shift is a rearrangement in which a group c migrates with its σ-bonded pair of electrons to an electron-deficient centre.
A
B
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
In general, a new more stable carboncation (B) is formed from that (A) formally derived by loss of a leaving group X. It is likely that the migration of c is already underway as X departs and that the carbocation (A) is not ‘fully developed’. A stable product is formed from (B) either by loss of a positively charged fragment (usually a proton) or by reaction with a necleophile.
In general, a new more stable carboncation (B) is formed from that (A) formally derived by loss of a leaving group X. It is likely that the migration of c is already underway as X departs and that the carbocation (A) is not ‘fully developed’. A stable product is formed from (B) either by loss of a positively charged fragment (usually a proton) or by reaction with a necleophile.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
Alternatively, further rearrangement can occur and this daughter carbocation can be stabilised in either of the above ways. In any event, further stereo- or regiocontrol may be required if a single product is to be formed
Alternatively, further rearrangement can occur and this daughter carbocation can be stabilised in either of the above ways. In any event, further stereo- or regiocontrol may be required if a single product is to be formed
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
The stereoelectronic requirement in these Wagner-Meerwein rearrangement is just the same as that obtaining in above two Schemes with the migrating groups acting as the nucleophile in an intramolecular SN2-like substitution of the leaving group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
TsO O-
O
O
CH3O-
O
O
CH2
H
O
OOH
Me
H
H
The most common and useful 1,2-shifts of this type are those that are constrained to take place in conformationally restricted molecules such that, stereoelectronically, one group is better disposed for migration over others, even though their inherent migratory aptitudes might be similar.
The most common and useful 1,2-shifts of this type are those that are constrained to take place in conformationally restricted molecules such that, stereoelectronically, one group is better disposed for migration over others, even though their inherent migratory aptitudes might be similar.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions

52
O
Br
H+
Br
OH
Br
OH
H BrOH
(2,3)-exo Me shift
H BrOH
-H+
H BrOH
Br2
H BrOH
Br+
(2,3)-exo Me shift
H BrOH
Br
H BrOH
Br
H Br
Br
O
Br
Br
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
Wagner-Meerwein rearrangements dominate the carbocation-mediated chemistry of strained bicyclic systems and, in particular, that of substituted bicyclo[2.2.1]heptanes including camphor).
Wagner-Meerwein rearrangements dominate the carbocation-mediated chemistry of strained bicyclic systems and, in particular, that of substituted bicyclo[2.2.1]heptanes including camphor).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
WagnerWagner--MeerweinMeerwein Rearrangement ReactionsRearrangement Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Me
AgSbF6toluene
Cl-
N
O
H64 %
SiO
H H
MeN
Cl SiO
HH
MeN
H
Rearrangement ReactionsRearrangement Reactions
Neighboring group participation (NGP) can also result in rearrangement as in the conversion of this compound to the morphinane ring system; here there is NGP by nitrogen in formation of the aziridinium ion and NGP by the double bond in formation of addition ring and in both steps there is inversion of configuration.
Neighboring group participation (NGP) can also result in rearrangement as in the conversion of this compound to the morphinane ring system; here there is NGP by nitrogen in formation of the aziridinium ion and NGP by the double bond in formation of addition ring and in both steps there is inversion of configuration.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Rearrangement ReactionsRearrangement Reactions
OH
HO
H
H
MeSPh
H+ OHH
H
MeSPh
OHHMe
H SPhO
H
Me
SPh
H
Sulphur readily enters into NGP and migration of the sulphur substituent is often the result, particularly when a more stablecarbocation is generated thereby.
Sulphur readily enters into NGP and migration of the sulphur substituent is often the result, particularly when a more stablecarbocation is generated thereby.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Rearrangement ReactionsRearrangement Reactions 4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
1,2-Rearrangement are not limited to those in which carbon is the migrating terminus.
1,2-Rearrangement are not limited to those in which carbon is the migrating terminus.
R1 R2
O HOO R
O
O
OR
O
O
HR1
R2
R1 OR2
O
The ease with which the O-O bond is broken, for example, facilitates migration from carbon to oxygen in the Baeyer-Villiger reactions.
The ease with which the O-O bond is broken, for example, facilitates migration from carbon to oxygen in the Baeyer-Villiger reactions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

53
R1 R2
O HOO R
O
O
OR
O
O
HR1
R2
R1 OR2
O
It is likely that this reaction includes an SN2 attack on oxygen by the migrating group R2 but this cannot be proved from inversion of configuration since oxygen cannot be a chiral centre.
It is likely that this reaction includes an SN2 attack on oxygen by the migrating group R2 but this cannot be proved from inversion of configuration since oxygen cannot be a chiral centre.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
RCH2
Obc a
RCOOOHbc a
O
RCH2
O
However, if the migrating group R2 is chiral then the rearrangement will be a Type I reaction since a bond to a chiral centre is broken and a new one is made.
However, if the migrating group R2 is chiral then the rearrangement will be a Type I reaction since a bond to a chiral centre is broken and a new one is made.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
Two factors happily conspire together to make the Baeyer-Villiger a valuable reaction in stereoselective synthesis.
1. The more substituted bond to the carbonyl group is the one that migrates to oxygen:tertiary alkyl > secondary alkyl > primary alkyl > methyl;
2. If the migrating α-carbon is chiral, the new carbon oxygenbond is formed with retention of configuration.
Two factors happily conspire together to make the Baeyer-Villiger a valuable reaction in stereoselective synthesis.
1. The more substituted bond to the carbonyl group is the one that migrates to oxygen:tertiary alkyl > secondary alkyl > primary alkyl > methyl;
2. If the migrating α-carbon is chiral, the new carbon oxygenbond is formed with retention of configuration.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
The reluctance of methyl to migrate in the Baeyer-Villiger reaction is used in the Criegee sequence for conversion of a γ-lactone into a 1,3-diol.
The reluctance of methyl to migrate in the Baeyer-Villiger reaction is used in the Criegee sequence for conversion of a γ-lactone into a 1,3-diol.
O
H
O
MeLiOH
H
OMe
1. CH3CO3H2. Ac2O, DMAP3. LiAlH4
4. H+
OH
H
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
a
c
b
d
R2BHa b
dc
BR2
HH2O2, OH-
a b
dc
BR2
H
OOH
a b
dc
OBR2
H
HO-a b
dc
OH
H
Hydroboration of an alkene followed by transformation of the C-C bond into a C-O bond is an invaluable route to alcohol.
Hydroboration of an alkene followed by transformation of the C-C bond into a C-O bond is an invaluable route to alcohol.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
SiMe(OEt)2
30 % H2O2
KHF2, DMF OH
SiF3
DMFOH
mCPBA
100 % exo
95 % endo
Replacement of C-Si bonds by C-O bonds can now be reliable accomplished with retention of configuration at carbon.
Replacement of C-Si bonds by C-O bonds can now be reliable accomplished with retention of configuration at carbon.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus

54
Ph
O
(PhMe2Si)2CuLi
Ph
O
SiMe2Ph
HBF4
Ph
O
SiMe2
H
F-
Ph
O
SiMe2
FPh
O
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
SiR
ArCOOO- Si
R
OO Ar
O
SiOR
ROH
These rearrangements are triggered by weakness of the O-O bond and mechanisms resembling that aboveScheme are operative.
These rearrangements are triggered by weakness of the O-O bond and mechanisms resembling that aboveScheme are operative.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
NH2
O
cb
aKOH, Br2
Hofmann N
O
cb
a
Br
Cl
O
cb
aN3
-
CurtiusN
O
cb
a
N2
Nc
b
a CO
H2ONH2
cb
a
OH
O
cb
a
HN3 Schmidt Lossen
N
O
cb
a
OAc cb
a
NH
O
OAc
base
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
Migration from carbon to nitrogen occurs in the Hofmann, Schmidt, Curtius and Lossen rearrangements, all of which proceed via an intermediate isocyanate formed with retention of configuration in the migrating group.
Migration from carbon to nitrogen occurs in the Hofmann, Schmidt, Curtius and Lossen rearrangements, all of which proceed via an intermediate isocyanate formed with retention of configuration in the migrating group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.7. 1,24.7. 1,2--Rearrangements with Retention of Rearrangements with Retention of Configuration at The Migration TerminusConfiguration at The Migration Terminus
In stabilized carbanions, e.g. enolates, the carbanion is planar and achiral. Simple carbations are usually tetrahedral but have low barriers to invcersion. Consequently, the formation of a carbanion from an enantiopure precursor followed by reaction with an electrophile will in general give a racemic product because the carbanion is not configura-tionally stable.
In stabilized carbanions, e.g. enolates, the carbanion is planar and achiral. Simple carbations are usually tetrahedral but have low barriers to invcersion. Consequently, the formation of a carbanion from an enantiopure precursor followed by reaction with an electrophile will in general give a racemic product because the carbanion is not configura-tionally stable.
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
a
Hbc base a
bc fast
abc E+ a
Ebc
enantiopure racemicE+: electrophile
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
The situation here is analogous to that of tetrahedral sp3-hybridised (pyramidal) trivalent nitrogen, where inversion is so fast as to preclude the possibility of isolating enantiomers.
Na
bc
fastN
a
bc
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

55
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
One way in which the inversion barrier in sp3-hybridised amines can be raised is by having one or more of the substituents a, b or c as heteroatoms, e.g. O, N or halogen. The presence of non-bonding electron pairs on the heteroatom and/or its σ electron-withdrawing character raises the energy of the transition state for inversion relative to the ground state as compared with simple trialkylamines.
One way in which the inversion barrier in sp3-hybridised amines can be raised is by having one or more of the substituents a, b or c as heteroatoms, e.g. O, N or halogen. The presence of non-bonding electron pairs on the heteroatom and/or its σ electron-withdrawing character raises the energy of the transition state for inversion relative to the ground state as compared with simple trialkylamines.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
Me H
HOPh
SnBu3
OMe BuLi, -78 oCTHF
Bu3SnI Me H
HOPh
Li
OMe
Me2CO
Me H
HOPh
OMeMe
MeHO
It was shown by Still and Sreekumar that the same inversion-retarding effect of heteroatom effect of heteroatom substitution could be applied to carbonions.
It was shown by Still and Sreekumar that the same inversion-retarding effect of heteroatom effect of heteroatom substitution could be applied to carbonions.
transmetalation
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
1. BuLi2. CO23. H+
iPr SnR3
OEtOH
(S)-BINAL-H iPr SnR3
H OH iPr2NEtBOMCl iPr SnR3
H OBOM
iPr COOH
H OBOM
93 %98 % e.e.
CH2N2
iPr COOCH3
H OBOM1. HNO2
2. CH2N23. BOMCl
iPr COOH
H NH2
O
OAl
H
OEt
Li+Converted into the hydroxy acid derivative with complete retention of configuration.
Converted into the hydroxy acid derivative with complete retention of configuration.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
H
xy
bX
a
H H
xy
bH
a
From what has been said previously regarding the limitations ofthe SN2 reaction, it might appear that with abcX as a secondary centre there is little prospect of being able to carry out the transformation to form two adjacent carbon chiral centres of defined absolute configuration.
From what has been said previously regarding the limitations ofthe SN2 reaction, it might appear that with abcX as a secondary centre there is little prospect of being able to carry out the transformation to form two adjacent carbon chiral centres of defined absolute configuration.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
B
RH
O
OCl
Nu-B
RH
O
OClNu
ZnCl2Nu
B
RH
OO
Secondary α–chloroboronates undergo substitution by nucleophiles with clean inversion of configuration via rearrangement of intermediate boronate anions.
Secondary α–chloroboronates undergo substitution by nucleophiles with clean inversion of configuration via rearrangement of intermediate boronate anions.
Another example of SN2 substitution via intramolecular rearrangement
Another example of SN2 substitution via intramolecular rearrangement
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
B
iPrH
O
OCl
iPrH
O
MeO
1. THF, -100 oC2. ZnCl2, MeCN
68 %
iPrH
O
MeO
B
iPrH
OO
1. Bu3SnLi2. H2O2,NaOH3. ClCH2OMe4. BuLi
1. H2O2, NaOH
2. H+
iPrH
HO
OH
iPrH
Substitution with tributyltin: inversion;Replacement of boron hydroxyl: retention of configuration.
Substitution with tributyltin: inversion;Replacement of boron hydroxyl: retention of configuration.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

56
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
The scope of this unprecedented synthesis of a molecular containing two adjacent chiral centres is considerable, particularly since the configurations of the centres can be selected by appropriate choice of the enantiomer of the starting materials.
The scope of this unprecedented synthesis of a molecular containing two adjacent chiral centres is considerable, particularly since the configurations of the centres can be selected by appropriate choice of the enantiomer of the starting materials.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
increased s-character
increasedp-character
Carbanions which are generated on three-membered rings also have enhanced configuration stability. The hybridisation of their ring atoms provides a greater degree of p-character in the overlapping hybrid orbitals forming the bent banana bonds of the ring; This allows for a narrower angle than the 109o between normal sp3-bybrid orbitals.
Carbanions which are generated on three-membered rings also have enhanced configuration stability. The hybridisation of their ring atoms provides a greater degree of p-character in the overlapping hybrid orbitals forming the bent banana bonds of the ring; This allows for a narrower angle than the 109o between normal sp3-bybrid orbitals.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
At the transition state for inversion of the carbanion, the ring bonds at this carbon are the result of overlap of sp2-hybrid orbitals having an increased angle of 120o
between them.The net result is that the energy of the transition state for this carbanion inversion is raised relative to that of a carbanion in an acyclic substrate which can accommodate normal sp2-bond angles without the same increase in strain.
At the transition state for inversion of the carbanion, the ring bonds at this carbon are the result of overlap of sp2-hybrid orbitals having an increased angle of 120o
between them.The net result is that the energy of the transition state for this carbanion inversion is raised relative to that of a carbanion in an acyclic substrate which can accommodate normal sp2-bond angles without the same increase in strain.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
X X X
The same rationale accounts for the increased inversion barriersat nitrogen in aziridines by compared with cyclic amines.
The same rationale accounts for the increased inversion barriersat nitrogen in aziridines by compared with cyclic amines.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
Ph
Ph
Me
COPhNaNH2
toluene
Ph
Ph98 % e. e.
H2O Ph
Ph H96 % e. e.
OD
H
D
Ph3Si
BuLi
-78 oCH2OO
D
H
Ph3Si
OD
H
H
Ph3Si
1. LDA, -75 oC NH
PhSPh
O
2. BuLi3. D2O
ND
PhSPh
O
Haller Bauer cleavage
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
Examples of carbanion generation and reaction with an electrophile with retention of configuration in a cyclopropane, an epoxide and an aziridine.
Examples of carbanion generation and reaction with an electrophile with retention of configuration in a cyclopropane, an epoxide and an aziridine.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

57
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
1. LDA, -75 oC NH
PhSPh
O
2. BuLi3. D2O
ND
PhSPh
O
The optical rotation of the deuterated product was almost identical with that of the starting material, showing that carbanion and nitrogen inversion had not occurred.
The optical rotation of the deuterated product was almost identical with that of the starting material, showing that carbanion and nitrogen inversion had not occurred.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4. 8. 4. 8. Substitution with Retention of Configuration Substitution with Retention of Configuration via Configurationally Stable via Configurationally Stable Carbanions Carbanions
Finally, the reaction of many metal-carbon bonds, with transfer of a ligand from the metal to carbon, takes place with retention of configuration.Transmetallation reactions, in which one metal-carbon bond is replaced by another metal-carbon bond, also usually proceed with retention of configuration at carbon.
Finally, the reaction of many metal-carbon bonds, with transfer of a ligand from the metal to carbon, takes place with retention of configuration.Transmetallation reactions, in which one metal-carbon bond is replaced by another metal-carbon bond, also usually proceed with retention of configuration at carbon.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
There are limitations to the use of SN2 reactions in stereoselective synthesis.Tertiary substrates do not react intermolecularly and the reactions of secondary substrates are unsatisfactory withnucleophiles of even modest bulk, including most carbanions. In intramolecular reactions, however, SN2 substitution is practicable, sometimes even at tertiary centres.
There are limitations to the use of SN2 reactions in stereoselective synthesis.Tertiary substrates do not react intermolecularly and the reactions of secondary substrates are unsatisfactory withnucleophiles of even modest bulk, including most carbanions. In intramolecular reactions, however, SN2 substitution is practicable, sometimes even at tertiary centres.
Double inversion ( = retention) of configuration is common in many reactions in which a neighboring group participates.Net retention of configuration can also be accomplished using palladium chemistry.
Double inversion ( = retention) of configuration is common in many reactions in which a neighboring group participates.Net retention of configuration can also be accomplished using palladium chemistry.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
1,2-Migration of C-C bonds to sp3-hybridised centres with loss of a leaving group commonly proceeds with inversion at the migration terminus with the migrating bond behaving as the nucleophile in an SN2-type substitution.
1,2-Migration of C-C bonds to sp3-hybridised centres with loss of a leaving group commonly proceeds with inversion at the migration terminus with the migrating bond behaving as the nucleophile in an SN2-type substitution.
Concerted 1,2-migration with retention of configuration in the migrating sp3 carbon takes place in the Baeyer-Villiger and relatedreactions where migration of a C-C, C-B or C-Si bond takesplace to give a C-O bond. Similar 1,2-migrations from carbonto nitrogen with retention of configuration in the migrating group take place in the Hofmann, Curtius and related reactions.
Concerted 1,2-migration with retention of configuration in the migrating sp3 carbon takes place in the Baeyer-Villiger and relatedreactions where migration of a C-C, C-B or C-Si bond takesplace to give a C-O bond. Similar 1,2-migrations from carbonto nitrogen with retention of configuration in the migrating group take place in the Hofmann, Curtius and related reactions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Pyramidal carbanions substituted with oxygen, nitrogen or halogens have retarded rates of inversion and, at sufficiently low temperatures. The carbonion is configurationally stable and can be generated and related with retention of configuration. Carbanions generated on three-membered rings are also configurationally stable at lower temperatures.
Pyramidal carbanions substituted with oxygen, nitrogen or halogens have retarded rates of inversion and, at sufficiently low temperatures. The carbonion is configurationally stable and can be generated and related with retention of configuration. Carbanions generated on three-membered rings are also configurationally stable at lower temperatures.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Simple Chirality Transfer Reactions:
Those in Which a Single Chiral Centre is Transferred with
Concomitant Migration of one or More Double Bonds
Part 2Part 2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

58
OutlineOutline
4.10. 1,3-Simple Chirality Transfer via the SE2’ Reaction; 4.11. 1,3-Simple Chirality Transfer via the SN2’ Reaction;4.12. Simple Chirality Transfer via Concerted
Sigmatropic Rearrangement;4.13. 1,3-Simple Chirality Transfer by [2,3]
Sigmatropic Rearrangement;4.14. Simple Chirality Transfer via [3,3]
Sigmatropic Rearrangement;4.15. [1,5] Sigmatropic Rearrangement;4.16. Simple Chirality Transfer in the
Ene (Retroene) Reaction 4.17. Summary
4.10. 1,3-Simple Chirality Transfer via the SE2’ Reaction; 4.11. 1,3-Simple Chirality Transfer via the SN2’ Reaction;4.12. Simple Chirality Transfer via Concerted
Sigmatropic Rearrangement;4.13. 1,3-Simple Chirality Transfer by [2,3]
Sigmatropic Rearrangement;4.14. Simple Chirality Transfer via [3,3]
Sigmatropic Rearrangement;4.15. [1,5] Sigmatropic Rearrangement;4.16. Simple Chirality Transfer in the
Ene (Retroene) Reaction 4.17. Summary
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 1 reactions discussed in the previous part are those in which either inversion or retention of configuration takes place at a chiral centre.
Type 1 reactions discussed in the previous part are those in which either inversion or retention of configuration takes place at a chiral centre.
In Part 2, all reactions involve simple transfer of chirality or, more precisely, those which take place with the loss of the existing chiral centres, the migration of at least one configur-ation double bond and the creation of a new chiral centre elsewhere in the molecule.
In Part 2, all reactions involve simple transfer of chirality or, more precisely, those which take place with the loss of the existing chiral centres, the migration of at least one configur-ation double bond and the creation of a new chiral centre elsewhere in the molecule.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 1 reactionsType 1 reactions
4.10. 1,34.10. 1,3--Simple Chirality TransferSimple Chirality Transfer
via the SE2’ ReactionThe reaction of allylsilanes with electrophiles is an SE2’ reaction:Loss of the silyl group and reaction with electrophiles takes place in an anti fashion with 1,3-transfer of chirality.
via the SE2’ ReactionThe reaction of allylsilanes with electrophiles is an SE2’ reaction:Loss of the silyl group and reaction with electrophiles takes place in an anti fashion with 1,3-transfer of chirality.
d
c
SiMe3
ba
d
c SiMe3
ab
E+
dc
E
a
b
dc
E b
a
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.10. 1,34.10. 1,3--Simple Chirality TransferSimple Chirality Transfer
OMe3Si
O
ClCl
1. PhSCl2. NaF, MeOH 65 %
OO
ClClPhS
Note: reaction takes place from only one of the two conformationsNote: reaction takes place from only one of the two conformations
Me
H H
PhSiMe3
E+ Me
H H
PhE
85 % e. e. E = tBu or CH2OH, 86 % e. e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
A small number of examples are known in which the inherent anti-bias of the SE2’ is overridden by other factors and synattack of the electrophile and loss of trialkylsilyl group is the stereochemical outcome.
A small number of examples are known in which the inherent anti-bias of the SE2’ is overridden by other factors and synattack of the electrophile and loss of trialkylsilyl group is the stereochemical outcome.
Allylstannanes react with electrophiles in an SE2’ reaction with the same anti preference as allysilanes
Allylstannanes react with electrophiles in an SE2’ reaction with the same anti preference as allysilanes
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.10. 1,34.10. 1,3--Simple Chirality TransferSimple Chirality Transfer 4.11. 1,34.11. 1,3--Simple Chirality TransferSimple Chirality Transfer
X
Nu-
Nu
X-
via the SN2’ ReactionAn SN2’ reaction is the nucleophilic attack at the terminal sp2 centre of an ally system with migration of the double bond and expulsion of a leaving group from the allylic position.
via the SN2’ ReactionAn SN2’ reaction is the nucleophilic attack at the terminal sp2 centre of an ally system with migration of the double bond and expulsion of a leaving group from the allylic position.
With appropriate substitution on the allyl system, this reaction also results in 1,3-transfer of chirality.
With appropriate substitution on the allyl system, this reaction also results in 1,3-transfer of chirality.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

59
Nu
X
Nu
X
The stereoelectronic requirement for the SN2’ reactions arises from the necessity for the developing p-orbital formed the breaking C-X bond to overlap with the adjacent p-orbital of the existing π-bond to form the new π-bond in the product.
The stereoelectronic requirement for the SN2’ reactions arises from the necessity for the developing p-orbital formed the breaking C-X bond to overlap with the adjacent p-orbital of the existing π-bond to form the new π-bond in the product.
anti syn
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
This stereoelectronic requirement can be satisfied by attack of the nucleophile in either of two orientations relative to the leaving group (syn or anti).
This stereoelectronic requirement can be satisfied by attack of the nucleophile in either of two orientations relative to the leaving group (syn or anti).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
a cb
d
X
Nu-
ab
Nu
c d
ad
b
c
X ab
Nu dc
anti-derived
In an enantiopure acyclic substrate having a double bond of defined configuration, there are two conformations from which both syn and anti reaction can proceed.
In an enantiopure acyclic substrate having a double bond of defined configuration, there are two conformations from which both syn and anti reaction can proceed.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
ad
b
c
X
Nu-
ab
Nu
dc
a cb
d
XNu-
ab
Nuc d
syn-derived
The products from each of these conformations have opposite configurations at both the chiral centre and the double bond.
The products from each of these conformations have opposite configurations at both the chiral centre and the double bond.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
In general, the SN2’ reaction is not highly stereoselective; attack of the nucleophile may be preferentially syn or antito some degree depending on the nucleophile, the leaving group and whether the substitution is inter- or intramolecular.
In general, the SN2’ reaction is not highly stereoselective; attack of the nucleophile may be preferentially syn or antito some degree depending on the nucleophile, the leaving group and whether the substitution is inter- or intramolecular.
The application of copper and palladium chemistry to the SN2’reaction has greatly increased the reliability of its stereochemical outcome.
The application of copper and palladium chemistry to the SN2’reaction has greatly increased the reliability of its stereochemical outcome.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
H
OHO
HOBn
1. Me2CuLi, THF, Et2O2. H2O
HO
MeH
OBnOH)(
H
OO
HOBn
Li
Me-
H
OH
Me
Me
OH
HOBn
H OH
MeH
OBn
HO
Me
88 %
12 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction

60
Although the reaction has high anti stereoselectivity, two diastereoisomers are still obtained (albeit indisparate amounts) because two conformations of the starting material react competitively.
Although the reaction has high anti stereoselectivity, two diastereoisomers are still obtained (albeit indisparate amounts) because two conformations of the starting material react competitively.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
H CO2Me
H
OTs
HtBuMe2SiO
H
Me
Me-
H
CO2MeHH
tBuMe2SiOH
Me
Me
Me2Cu(CN)Li2BF3, THF, -78 oC
96 %> 99 : 1
In favorable cases, cuprate-mediated SN2’ reactions on acyclic substrates can be highly stereoselective, but this is more likely when one of the options of anti reaction is removed by conformational restraints, e.g. when the allylic system is incorporated in a ring.
In favorable cases, cuprate-mediated SN2’ reactions on acyclic substrates can be highly stereoselective, but this is more likely when one of the options of anti reaction is removed by conformational restraints, e.g. when the allylic system is incorporated in a ring.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
Me
OAcMe
OAc
Me(CN)CuLi
Me
Me
Me
Me1.2 % 98.8 %94 % 5.9 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
CuCN
R
OAc
HH
CuCN
R
H
R
(6) (7)
CuCNR
HH R
HH
R
(8) SN2 product SN2' product
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
In these reactions, a π-complex of the alkene with copper isbelieved to be converted into a σ-complex with loss of the
leaving group followed by metal-to-carbon transfer of the ligand R (with retention of configuration at carbon).
In these reactions, a π-complex of the alkene with copper isbelieved to be converted into a σ-complex with loss of the
leaving group followed by metal-to-carbon transfer of the ligand R (with retention of configuration at carbon).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
Control of the stereochemistry, therefore, is brought about by the π-complex formation which, in an otherwise unbiased five-or six-membered ring system, will be form the side opposite to the acetoxy group and hence lead to the anti mode of substitution.
Control of the stereochemistry, therefore, is brought about by the π-complex formation which, in an otherwise unbiased five-or six-membered ring system, will be form the side opposite to the acetoxy group and hence lead to the anti mode of substitution.
However, when this face is hindered, even the copper-catalysed SN2’ reaction can revert to the syn mode.
However, when this face is hindered, even the copper-catalysed SN2’ reaction can revert to the syn mode.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction

61
It is noteworthy that the formation of a π-allyl complex is apparently not favored when the other lignand on copper is cyanide, since the product from transfer of R to the carbon which originally bore the acetoxy group– an SN2 reaction – is at best a very minor one.
It is noteworthy that the formation of a π-allyl complex is apparently not favored when the other lignand on copper is cyanide, since the product from transfer of R to the carbon which originally bore the acetoxy group– an SN2 reaction – is at best a very minor one.
However, using a dialkylcuprate, R2CuLi, this is the major pathway and the regiospecificity of substitution of the SN2’reaction is lost.
However, using a dialkylcuprate, R2CuLi, this is the major pathway and the regiospecificity of substitution of the SN2’reaction is lost.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
It has been suggested that the tendency for cuprates to bring about SN2’ reactions with anti stereochemistry can be ascribed to the ability of the filled and diffuse d-orbital of the nucleophilic copper atom to interact with both π*- and σ*-orbitals of the substrate in a bidentate fashion.
It has been suggested that the tendency for cuprates to bring about SN2’ reactions with anti stereochemistry can be ascribed to the ability of the filled and diffuse d-orbital of the nucleophilic copper atom to interact with both π*- and σ*-orbitals of the substrate in a bidentate fashion.
Cu
L
R
X
d (Cu)
C C
σ* C-X
π*
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
H
Me
OPhNH
O
1. MeLi2. CuI3. MeLi
H
Me
ON
OPh
MeCu-
-CO2
H
Me
N
Ph
MeCu- Me
Me
H
Me
Me
Exceptionally, synaddition in these cuprate-mediated SN2’ substitutions can be effected by coordinations of the cuprate to the leaving group, thus directing addition of the copper to the synface of the allyl group.
Exceptionally, synaddition in these cuprate-mediated SN2’ substitutions can be effected by coordinations of the cuprate to the leaving group, thus directing addition of the copper to the synface of the allyl group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
Pd
OHOCOArH
OCOArH
OH
(Ph3P)4PdEt3N, MeCN
OHHH
O H H
OH
HPd)(
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
By contrast, SN2’ reactions which are brought about by involve-ment of palladium invariably proceed with anti formation of π-allyl complexes followed by anti attack of the nucleophile leading to an overall syn stereochemistry.
By contrast, SN2’ reactions which are brought about by involve-ment of palladium invariably proceed with anti formation of π-allyl complexes followed by anti attack of the nucleophile leading to an overall syn stereochemistry.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
Palladium is generally used in only catalytic amounts. Whereas in copper-mediated SN2’ reactions the cuprate is the source of the nucleophile, in palladium-catalysed SN2’ reactions the nucleophile comes from elsewhere.
Palladium is generally used in only catalytic amounts. Whereas in copper-mediated SN2’ reactions the cuprate is the source of the nucleophile, in palladium-catalysed SN2’ reactions the nucleophile comes from elsewhere.
The nucleophile attacks intramolecularly to form a five-membered ring: there can be no attack at other terminus of the system in intermediate because this would require a trans-double bond to contained in a seven-membered ring.
The nucleophile attacks intramolecularly to form a five-membered ring: there can be no attack at other terminus of the system in intermediate because this would require a trans-double bond to contained in a seven-membered ring.
The alternative transition state for five-membered ring formation is disfavoured because of A1,3-strain.
The alternative transition state for five-membered ring formation is disfavoured because of A1,3-strain.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction

62
OHMe O
HMe
H
(Ph3P)4Pd, THF COO-HMe H
MeH
Pd+
90 %-CH(CO2Me)2
HC(CO2Me)2
HMe
HMe
COO-
OH
HMe
HMe
H
O
)(
In these palladium-catalysed reactions, complete stereo- and regioselectivity using intermolecular attack on acyclic substrates by nucleophiles has also been achieved using the lactone.
In these palladium-catalysed reactions, complete stereo- and regioselectivity using intermolecular attack on acyclic substrates by nucleophiles has also been achieved using the lactone.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
OHMe O
HMe
H
(Ph3P)4Pd, THF COO-HMe H
MeH
Pd+
90 %-CH(CO2Me)2
HC(CO2Me)2
HMe
HMe
COO-
OH
HMe
HMe
H
O
)(
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
The complete stereoselectivity here reflects the lower concentration of the alternative conformer from which ionisation could occur and/or its sluggish reaction withthe bulky complexed palladium.
The complete stereoselectivity here reflects the lower concentration of the alternative conformer from which ionisation could occur and/or its sluggish reaction withthe bulky complexed palladium.
The regioselectivity in this reaction is thought to be the result of charge repulsion between the incoming nucleophile and the carboxylate anion.
The regioselectivity in this reaction is thought to be the result of charge repulsion between the incoming nucleophile and the carboxylate anion.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
O
1. (iPrO)3P, Pd(OAc)2
N
N
N
N
NH2
Li
2. , DMSO, THF
3. ClCO2Me, pyr
MeO2CO N
N
NN
NHCO2Me
LiCH(NO2)SO2Ph(dba)3Pd2, PPh3, CHCl3
N
N
NN
NHCO2MeNO2
PhO2S
N
N
NN
NH2HO
HO OH
(+)-aristeromycin
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
OH
OH
OH
HO
HO2C
O
NH
NCO2Me
(Ph3P)4PdTHF N NCO2Me
HH
HO
The π-allylpalladium species is trapped intramolecularly by anitrogen nucleophile with net intention of configuration at theepoxide carbon atom, i.e. overall this is not equivalent to an SN2’reaction.
The π-allylpalladium species is trapped intramolecularly by anitrogen nucleophile with net intention of configuration at theepoxide carbon atom, i.e. overall this is not equivalent to an SN2’reaction.Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction
nBu
H
H
AlMe2
+O
O
1. (Ph3P)4Pd, THF
2. H2O, H+
HOOC
tBu
Whereas attack of a nucleophile on the π- allylpalladium intermediate is usually anti, attack involving aryl or vinyl zinc halides or vinylalanes is syn; in these cases the nucleophile is transferred first to the palladium(transmetallation) and thence to the syn face.
Whereas attack of a nucleophile on the π- allylpalladium intermediate is usually anti, attack involving aryl or vinyl zinc halides or vinylalanes is syn; in these cases the nucleophile is transferred first to the palladium(transmetallation) and thence to the syn face.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction

63
OO
(Ph3P)4Pd, THFPhZnCl
-O2C
Pd+Ph3P PPh3
-O2C
Pd+Ph PPh3
H+
OHO
H
Ph
H
racemic, 94 %
Ph亲核性较弱,易转移到Pd上
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The SThe SNN22’’ ReactionReaction 4.12. 4.12. Simple Chirality Transfer via Concerted Simple Chirality Transfer via Concerted SigmatropicSigmatropic RearrangementRearrangement
-Y+X
Y
X
H H
In a sigmatropic rearrangement, the breaking of an existing σ-bond is accompanied by the making of a new σ-bond elsewhere in the same molecule; consequential shifting of one or more double bonds is generally required.
In a sigmatropic rearrangement, the breaking of an existing σ-bond is accompanied by the making of a new σ-bond elsewhere in the same molecule; consequential shifting of one or more double bonds is generally required.
[3,3]
[2,3]
[1,5]
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Stereoelectronic control in these sigmatropic rearrangement is a consequence of transition-state geometries (TSG) in which, as always, overlap of the appropriate interacting orbitals(including those form any intervening π-bonds) is maximised.
Stereoelectronic control in these sigmatropic rearrangement is a consequence of transition-state geometries (TSG) in which, as always, overlap of the appropriate interacting orbitals(including those form any intervening π-bonds) is maximised.
Sigmatropic rearrangements are one of the important sub-classes of pericyclic reaction first recognised by Woodward and Hoffman. Pericyclic reactions are single-step reactions which proceed via cyclic transition states in which bonds are being made and broken concertedly.
Sigmatropic rearrangements are one of the important sub-classes of pericyclic reaction first recognised by Woodward and Hoffman. Pericyclic reactions are single-step reactions which proceed via cyclic transition states in which bonds are being made and broken concertedly.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.12. 4.12. Simple Chirality Transfer via Concerted Simple Chirality Transfer via Concerted SigmatropicSigmatropic RearrangementRearrangement
Concertedness in these pericyclic reactions is allowed only when the orbitals which overlap have the correct symmetry. If the symmetry is not appropriate, the reaction may still proceed but via a non-concerted pathway involving more than a single transition state.
Concertedness in these pericyclic reactions is allowed only when the orbitals which overlap have the correct symmetry. If the symmetry is not appropriate, the reaction may still proceed but via a non-concerted pathway involving more than a single transition state.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.12. 4.12. Simple Chirality Transfer via Concerted Simple Chirality Transfer via Concerted SigmatropicSigmatropic RearrangementRearrangement
Concertedness in these reactions is often accompanied by high stereoselectivity; non-concerted reactions are less likely to be highly stereoselective.
Concertedness in these reactions is often accompanied by high stereoselectivity; non-concerted reactions are less likely to be highly stereoselective.
A feature of pericyclic reactions is that their stereochemical course can usually be predictly when the demands of stereoelectronic control (including correct symmetry of the overlapping orbitals) are considered. Such predictions can be made by using rules devised by Woodward and Hoffman or by using the frontier molecular orbital midification (FMO) of those rules devised by Fukui.
A feature of pericyclic reactions is that their stereochemical course can usually be predictly when the demands of stereoelectronic control (including correct symmetry of the overlapping orbitals) are considered. Such predictions can be made by using rules devised by Woodward and Hoffman or by using the frontier molecular orbital midification (FMO) of those rules devised by Fukui.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.12. 4.12. Simple Chirality Transfer via Concerted Simple Chirality Transfer via Concerted SigmatropicSigmatropic RearrangementRearrangement
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
OS
Ar CH2-
O O-O-
+SAr
sulphoxide sulphenate ester [2,3] Wittig rearrangement
S+S
CH2-
sulphonium ylide allyl sulphide
N+N
CH2-
[2,3] Stevens rearrangement
In acyclic system
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

64
Both the Wittig and Stevens rearrangements are prefaced by [2.3] to distinguish them from their [1,2] (non-concerted) variants.
Both the Wittig and Stevens rearrangements are prefaced by [2.3] to distinguish them from their [1,2] (non-concerted) variants.
Depending on the substitution present, the [2,3] sigmatropic rearrangement may in the net creation of chiral centres in Type 2 or 3 reaction as illustrated for Wittig rearrangement.
Depending on the substitution present, the [2,3] sigmatropic rearrangement may in the net creation of chiral centres in Type 2 or 3 reaction as illustrated for Wittig rearrangement.
O
Me
-O
racemic(b) Type 2
O Me
SiMe3
Me
-O
MeMe
SiMe3(c) Type 3
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
CH2-
O
H
H HMe O-
(a) simple chirality transfer (Type 3)
We shall consider only those arrangement in which chirality is transferred, i.e. there is not gain in the number of chiral centres. For this to be so the double bond must be configured and both it and the existing chiral centre must be present in the pericyclic array.
We shall consider only those arrangement in which chirality is transferred, i.e. there is not gain in the number of chiral centres. For this to be so the double bond must be configured and both it and the existing chiral centre must be present in the pericyclic array.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
Y-
X
ba
cd e
YbX-
c
d
e
a
Y-
X
ba
c e
d
YbX
c
a
e
d
In [2,3]rearrangements involving simple chirality transfer there are only two positions for the chiral centre: either at the allylic or homoallylic position.
In [2,3]rearrangements involving simple chirality transfer there are only two positions for the chiral centre: either at the allylic or homoallylic position.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
σ-Bond breaking
σ-Bond forming
Maximised overlap of the orbitals involved in the [2,3] rearrangement can be accommodated.
Maximised overlap of the orbitals involved in the [2,3] rearrangement can be accommodated.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
(A) (D)
(B)(C)
HOMOLUMO
same phase
σ-Bond breaking
σ-Bond forming
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
A formalism by which the symmetry of the orbitals involved in this rearrangement can be deduced:The bond which is breaking is located (A) and the two radical species obtained by a (hypothetical) homolytic cleavage of this bond are identified (B)
A formalism by which the symmetry of the orbitals involved in this rearrangement can be deduced:The bond which is breaking is located (A) and the two radical species obtained by a (hypothetical) homolytic cleavage of this bond are identified (B)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement

65
Consideration of the highest occupied molecular orbitals (HOMOs) of these two radical species (C) shows that lobesrequired to overlap to form the new σ-bond have the same phase (the lobes of the orbitals produced from the breaking bond must have the same phase) and thus concertedrearrangement is allowed, giving the product (D).
Consideration of the highest occupied molecular orbitals (HOMOs) of these two radical species (C) shows that lobesrequired to overlap to form the new σ-bond have the same phase (the lobes of the orbitals produced from the breaking bond must have the same phase) and thus concertedrearrangement is allowed, giving the product (D).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
O O-
The envelope conformation shown for the transition state geometry in previous Scheme is not necessarily that through which all [2,3] sigmatropic rearrangements pass.
The envelope conformation shown for the transition state geometry in previous Scheme is not necessarily that through which all [2,3] sigmatropic rearrangements pass.
The stereochemistry of some [2,3] Wittig rearrangements is better accommodate using an alternative “envelope”.
The stereochemistry of some [2,3] Wittig rearrangements is better accommodate using an alternative “envelope”.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
There is a syn relationship between the C-C bond made and the C-O bond broken. Alternatively, one can say that the CH2O unit is transferred across one face of allyl system(superafacial migration)
There is a syn relationship between the C-C bond made and the C-O bond broken. Alternatively, one can say that the CH2O unit is transferred across one face of allyl system(superafacial migration)
Oa
bH
Rc
a
b H
Rc
O-
c
b
aH
R
O
()
cH
R b
aO-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
In an acyclic system, there are always two conformation (non-envelope), through which the rearrangement can proceed when a chiral centre is present in the starting material.
In an acyclic system, there are always two conformation (non-envelope), through which the rearrangement can proceed when a chiral centre is present in the starting material.
Oa
bH
Rc
a
b H
Rc
O-
c
b
aH
R
O
()
cH
R b
aO-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
O
Me
(R)-Alpine borateOH
Me
1. H2, Lindlar (Type 4)
2. NaH, ICH2SnMe3
OSnMe3
HMe
(R)-Alpine borateB
A particularly valuable experimental by transmetallation.A particularly valuable experimental by transmetallation.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
OSnMe3
HMe
BuLi-78 oC
OLi
HMe
Me
HH
OLi
H
Me H
OLi
H
H+OH
Me H
Me
HH
OLi ()
H
Me
HLiO OLi
Me H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement

66
The preference for rearrangement via (A) rather than (B) is strong when the R group is large and when b is largerThan hydrogen.
The preference for rearrangement via (A) rather than (B) is strong when the R group is large and when b is largerThan hydrogen.
However, this selectivity would be expected to decline when the allylic position bearing R is also sunstituted with a group R1 of a similar bulk. Preferential reaction via (B) can be accomplished if c is a bulky group (methyl or larger) and b is a hydrogen as in next sample.
However, this selectivity would be expected to decline when the allylic position bearing R is also sunstituted with a group R1 of a similar bulk. Preferential reaction via (B) can be accomplished if c is a bulky group (methyl or larger) and b is a hydrogen as in next sample.
A
B
Oa
bH
Rc
a
b H
Rc
O-
c
b
aH
R
O
()
cH
R b
aO-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
Bu
OH
Me
1. KH2. Bu3SnCH3I
Bu
O
Me
SnBu3
BuLi
OH
HBu
HMe
Li
H
H Bu
HMe
OLi
Me
H
HBu
H
OLi
()H+
BuMe
OH
The interaction between the butyl and methyl groups indicated in (A) is sufficient to direct all of the rearrange-ment via conformer (B).
The interaction between the butyl and methyl groups indicated in (A) is sufficient to direct all of the rearrange-ment via conformer (B).
B
A
A和B的相对含量取决于R,c的邻交叉作用大,还是R,b(a)的1,3-strain大
A和B的相对含量取决于R,c的邻交叉作用大,还是R,b(a)的1,3-strain大
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
Allyl Sulphoxide-allyl sulphenate ester interconversion.Allyl Sulphoxide-allyl sulphenate ester interconversion.
The alternative location of the chiral centre in a [2,3] rearrange-ment is at the homoallylic position. This can be the case with sulphoxides since the sulphur is pyramidal and normally configuratioanally stable, i. e. the lone pair of electrons on sulphur does not invert at a measurable rate at r. t..
The alternative location of the chiral centre in a [2,3] rearrange-ment is at the homoallylic position. This can be the case with sulphoxides since the sulphur is pyramidal and normally configuratioanally stable, i. e. the lone pair of electrons on sulphur does not invert at a measurable rate at r. t..
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
SOAr H
H
S-O
Ar
OH
H
S
Ar
OH
H
SAr
H
H
S-O
Ar
Enantiopure allylic sulphoxides racemise much more rapidly than those lacking an allyl group.
Enantiopure allylic sulphoxides racemise much more rapidly than those lacking an allyl group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
H
H Me
HArSO
SOAr
Me
H
H
H
OSArMe
H
SAr
Me
H
SOAr
H
Me
O-
Me
H
S-O
Ar
[2,3] Sigmatropic rearrangement provides a route for the inversion configuration of the allyl double bond in allyl sulphoxides.
[2,3] Sigmatropic rearrangement provides a route for the inversion configuration of the allyl double bond in allyl sulphoxides.
77 % 23 %
OO
两个构型不一致
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
+
H O
R
H
ArS
C5H11
HH
R
H
O
C5H11
HArS
H
H
O
C6H11
HArS
R
H
H
C5H11
R
HArS
O-
H
C5H11
H
H
RArS
O-
C5H11
HH
R
HS+
Ar O-
If an allylic sulphoxide contains a configured double bond and a chiral centre at the allylic position, equilibration via sulphenate inverts the configuration of the double bond and the chiral centre,in which the chirality at sulphur is not specified.
If an allylic sulphoxide contains a configured double bond and a chiral centre at the allylic position, equilibration via sulphenate inverts the configuration of the double bond and the chiral centre,in which the chirality at sulphur is not specified. Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement

67
If a sulphenate esterisprepared from a chiral allylic alcohol Containing a configured double bond, the [2,3] sigmatropic re-arrangement sets up an equilibrium between two sulphenates.
If a sulphenate esterisprepared from a chiral allylic alcohol Containing a configured double bond, the [2,3] sigmatropic re-arrangement sets up an equilibrium between two sulphenates.
R C5H11
HHO 1. ArSCl, Et3N
2. P(OMe)3, MeOH
R C5H11
HArSO
H
C5H11
H
H
H
RS O-
ArC5H11O-S
R
H
Ar H
H
C5H11OS
R
H
Ar H
H
R C5H11OH
H H
Ar: p-MeC6H4
R: O
(CH2)6CO2Me
THPO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
MeH
OH
H
1. KH2. ICH2SnMe3
3. BuLi, -78 oC4. MeOH, -78 oC
Me
H
HOHIn Cyclic Systems
Reduction of the conformational freedom of a system undergoing [2,3] rearrangement by its incorporation into a cyclic system will usually lead to increased stereoselectivity.
Reduction of the conformational freedom of a system undergoing [2,3] rearrangement by its incorporation into a cyclic system will usually lead to increased stereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
Whereas the sulphoxide-sulphenate ester equilibrium usually lies on the side of the former, the selenoxide-selenate equilibrium favours the latter. The resulting selenate ester is readily hydrolysed to the alcohol.
Whereas the sulphoxide-sulphenate ester equilibrium usually lies on the side of the former, the selenoxide-selenate equilibrium favours the latter. The resulting selenate ester is readily hydrolysed to the alcohol.
OH
HO OMe
OO
ArSeCNBu3P
OArSe
H OMe
OO
H2O2pyr, CH2Cl2
O
OMe
OO
OH
H
Ar: o-NO2C6H4
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
S
H
1. OTf
2. LDA S
H
SH
[2,3] Rearrangement of an allyl sulphonium ylide is exemplified by the conversion of (A) to (B). Since this rearrangement regenerates an allylic thioether, it may be carried out iteratively to provide larger ring compounds.
[2,3] Rearrangement of an allyl sulphonium ylide is exemplified by the conversion of (A) to (B). Since this rearrangement regenerates an allylic thioether, it may be carried out iteratively to provide larger ring compounds.
A
B
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.13. 1,34.13. 1,3--Simple Chirality Transfer by [2,3] Simple Chirality Transfer by [2,3] SigmatropicSigmatropic RearrangementRearrangement
X X X
X X X
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
The transition-state geometry for [3,3] sigmatropic rearrangement can be either chair- or boat-shaped. The transition-state geometry for [3,3] sigmatropic rearrangement can be either chair- or boat-shaped.
Like the [2,3] rearrangement, the concertedness of the [3,3] rearrangement is allowed since the symmetry of the orbitals of the two allyl radicals produced by (hypothetical) homolytic cleavage of the σ-bond allows for in-phase overlap of the terminal lobes to form a stable new - bond.
Like the [2,3] rearrangement, the concertedness of the [3,3] rearrangement is allowed since the symmetry of the orbitals of the two allyl radicals produced by (hypothetical) homolytic cleavage of the σ-bond allows for in-phase overlap of the terminal lobes to form a stable new - bond.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The all-carbon [3,3] rearrangement – the Cope rearrangement –is useful in stereoselective synthesis but we shall illustrate chirality transfer by particular reference to the Claisen rearrangement since this reaction has been more widely studied and used in stereoselective synthesis.
The all-carbon [3,3] rearrangement – the Cope rearrangement –is useful in stereoselective synthesis but we shall illustrate chirality transfer by particular reference to the Claisen rearrangement since this reaction has been more widely studied and used in stereoselective synthesis.
The Claisen rearrangement is closely related to the [2,3] Wittig rearrangement and, a sin the latter, a C-C bond is made at the expense of the C-O bond which is broken on a single face of the allyl system.
The Claisen rearrangement is closely related to the [2,3] Wittig rearrangement and, a sin the latter, a C-C bond is made at the expense of the C-O bond which is broken on a single face of the allyl system.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement

68
Acyclic Cases
Oa
H
H
b Oa
H
H
b
O
H
b
H
a
O
H
b
a
H
[1,4]chirality transfer
[1,3]chirality transfer
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
For chirality transfer using [3,3] sigmatropic rearrangement, there must be at least one chiral centre present in the pericyclic array and one configured double bond, and since in the Claisen rearrangement these is only one sp3-hydridised carbon, this must be the chiral centre. The chirality transfer in Claisen reactions is 1,4 or 1,3 depending on whether the configured double bond is present in the vinyl or allyl ether mioety.
For chirality transfer using [3,3] sigmatropic rearrangement, there must be at least one chiral centre present in the pericyclic array and one configured double bond, and since in the Claisen rearrangement these is only one sp3-hydridised carbon, this must be the chiral centre. The chirality transfer in Claisen reactions is 1,4 or 1,3 depending on whether the configured double bond is present in the vinyl or allyl ether mioety.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Oa
H
Hb Oa
H
H
b
Ob
Ha
H
Oa
H
bH
b
HO
a
H
diastereoisomers
a
H
OH
b
b
H
a
HO
Hb
Oa
H
Oa
H
H
b
b
H
H
O
a
diastereoisomers
enantiomers
enantiomers
There are four possible transition-state geometries through 1,4-chirality transfer can be effected using the single stereo-isomer of the vinyl ether. The product from two chairTSGs are diastereoisomers as are those from the two boat TSGs; the two diastereoisomers formed by the chair TSGs are enantiomers of those formed by the boat TSGs.
There are four possible transition-state geometries through 1,4-chirality transfer can be effected using the single stereo-isomer of the vinyl ether. The product from two chairTSGs are diastereoisomers as are those from the two boat TSGs; the two diastereoisomers formed by the chair TSGs are enantiomers of those formed by the boat TSGs.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Oa
H
Hb
c
Oa
H
b
H
c )(
The great use of the Claisen rearrangement in synthesis derives from a number of factors including: (a) the ease of preparation of substituted allyl vinyl ethers; (b) the equilibrium position which favours the carbonyl
compound;(c) the usefulness of products of the rearrangement for further
manipulation
The great use of the Claisen rearrangement in synthesis derives from a number of factors including: (a) the ease of preparation of substituted allyl vinyl ethers; (b) the equilibrium position which favours the carbonyl
compound;(c) the usefulness of products of the rearrangement for further
manipulation
(A) (B)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
OH
R
Me
R
H
MeOO
H
R
Me
O
R
H
Me
R = Et: 9 : 1R = iPr: 13 : 1
Two important consequences of this preferencefor TSG (A).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Its value in stereoselective synthesis is the results not only of a clear preference either for chair transition state (usual for acyclic substrates) or for the boat (common for cyclic substrates), but also that chair TSG in which b is equatorial (A) rather than axial (B).
Its value in stereoselective synthesis is the results not only of a clear preference either for chair transition state (usual for acyclic substrates) or for the boat (common for cyclic substrates), but also that chair TSG in which b is equatorial (A) rather than axial (B).
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
)(O
H
Et
Me
Me Et
H
MeO
Me
O
H
Et
Me
Me
O
Et
H
Me
Me
> 99 % < 1%
1. There will be increasingly selective formation of an E-double bond as the 1,3-diaxial-type interaction in TSG (B) becomes more serious.
1. There will be increasingly selective formation of an E-double bond as the 1,3-diaxial-type interaction in TSG (B) becomes more serious.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement

69
R
OH
MeO OMe
Me NMe2
OMe
Me NMeMeO-
R
O
MeO NMe2
R
O
NMe2
O
R
NMe2
Relevant to the nature of c is the reaction by which the vinyl ether in the starting materials is formed. Two valuable modifications of the Claisen rearrangement in situ and in both, the bulk of the c group leads to enhanced diastereoselectivity.
Relevant to the nature of c is the reaction by which the vinyl ether in the starting materials is formed. Two valuable modifications of the Claisen rearrangement in situ and in both, the bulk of the c group leads to enhanced diastereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
R
OH
R
O
EtO
R
O
OEt
, 138 oCMeC(OEt)3
EtCO2H
A closely related procedure uses an orthoester as the source of the vinyl ether carbons and the product is an ester, a weak acid is usually present here as a catalyst.
A closely related procedure uses an orthoester as the source of the vinyl ether carbons and the product is an ester, a weak acid is usually present here as a catalyst.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
R
O
O
1. LDA2. R'3SiCl
O
OSiR'3 H
R
O
R
H
OSiR'3
O
OSiR'3
R
Not only does the trialkylsilyloxy group improve the stereoselectivity [large diaxial repulsion between R and OSiR1
3 in the alternative chair TSG (down)] but it also facilitates the rearrangement such that it proceeds at or close to ambient temperature.
Not only does the trialkylsilyloxy group improve the stereoselectivity [large diaxial repulsion between R and OSiR1
3 in the alternative chair TSG (down)] but it also facilitates the rearrangement such that it proceeds at or close to ambient temperature.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
O
H
Me
Me
Me2N
O Me
NMe2
Me
H
Me Me
HHOC(OMe)2
NMe2
Me
xylene, 17 h
2. Use of an enantiopure allyl vinyl ether will result in a product of predictable configuration at its sp3-chiral centre.
2. Use of an enantiopure allyl vinyl ether will result in a product of predictable configuration at its sp3-chiral centre.
90 % enantioselectivity
Two important consequences of this preference for TSG (A).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Enantiopurified secondary allyl alcohols required for enantioselective Claisen rearrangement are accessible by enantioselective reduction of acetylenic ketones with Alpineborane or by kinetic resolution using the enantioselective Sharpless epoxidation.
Enantiopurified secondary allyl alcohols required for enantioselective Claisen rearrangement are accessible by enantioselective reduction of acetylenic ketones with Alpineborane or by kinetic resolution using the enantioselective Sharpless epoxidation.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Use of sterically hindered Al catalysts.
The bulky Al catalyst, by complexing to the ether oxygen, forces the isobutyl group to occupy the pseudo-axial position, if the the oxygen is more sp2- than sp3-hybridised.
The bulky Al catalyst, by complexing to the ether oxygen, forces the isobutyl group to occupy the pseudo-axial position, if the the oxygen is more sp2- than sp3-hybridised.
OiBu
MeBr
But
But
OAl
Me
O
tBu
tBu
Br
OMe
Bui
Al
OMe
AlBui )(
OMe
BuiH
_
OMe
Bui
H
78 % e. e.
84 : 16
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement

70
O b
OSiR3
aO
H
ba
O-
R3SiClO
H
ba
OSiR3
O
H
ba
O
LDA
O
H
b
O-
a
O b
OSiR3
a
R3SiClO
H
b
OSiR3
a
A difficulty in practice in bringing about 1,4-chirality transfer in Claisen rearrangement is the generation of a configurationally homo-geneous vinyl ether double bond in the vinyl allyl ether undergoing rearrangement.
High selectivity 9 : 1
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
OBnO
OH
Me
Li
LDA
OBnO
O
Me3SiCl
OBnO
OTMSH
Me1. H2O2. CH2N2
OBnO
OMeH
Me
If the substituent a contains achelating atom (O, S or N), the configurational purity of the silyl ketene acetal is often assured because the chelation in the enolate demands that the double bond be contained in a ring.
If the substituent a contains achelating atom (O, S or N), the configurational purity of the silyl ketene acetal is often assured because the chelation in the enolate demands that the double bond be contained in a ring.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
OO
chair
H
MeO
boat
H
MeO
H
[3,3]Sigmatropic Rearrangement Involving Rings.[3,3]Sigmatropic Rearrangement Involving Rings.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
If the allyl unit in the Claisen rearrangement is confined to a ring with eight or less members, the four TSGs available in an acyclic case are reduced to two. Only one face of the allyl double bond can enter into the rearrangement and the configuration of the new chiral centre on the ring is predictable irrespectiveof whether a chair or boat TSG is involved.
If the allyl unit in the Claisen rearrangement is confined to a ring with eight or less members, the four TSGs available in an acyclic case are reduced to two. Only one face of the allyl double bond can enter into the rearrangement and the configuration of the new chiral centre on the ring is predictable irrespectiveof whether a chair or boat TSG is involved.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
H
R1O
HR2
R4
R3
O
R2
H
R1
R4
R3)( H
R1O
R2
R4
R3
A substituent R2 on the vinyl ether double bond and cissubstituents R3, R4 on the ring are likely to destabilise the chair more than the boat form, and it is in this case that the boat-derived stereostructure is most likely to be found for the product.
A substituent R2 on the vinyl ether double bond and cissubstituents R3, R4 on the ring are likely to destabilise the chair more than the boat form, and it is in this case that the boat-derived stereostructure is most likely to be found for the product.
Boat-derived product
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
O
H
HOH
O
Me
Me
1. LDA
2. TMSCl O
H
HOTMSH
TMSO
Me
Me
OTMSO
Me
TMSO
Me
PhCH3reflux
TMSOO
OTMS
Me
Me H
H
HO
Me
MeMeO2C
When the vinyl ether moiety is confined to aring of eight or less atoms, the only possibleTSG is the boat form. The Ireland-Claisenrearrangement is capable of bringing about the formation of a quaternary chiral centre.
When the vinyl ether moiety is confined to aring of eight or less atoms, the only possibleTSG is the boat form. The Ireland-Claisenrearrangement is capable of bringing about the formation of a quaternary chiral centre.
A quaternary chiral centre:in this case, directly bonded to two tertiary chiral centres.
A quaternary chiral centre:in this case, directly bonded to two tertiary chiral centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement

71
OH
OH O -
H
H
H +
H
HO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Both the Claisen and Cope rearrangements may lead to products in which one of the double bonds in the first-formed product is part an enolate which is converted into a ketone in the work-up. In the retro-synthetic analysis of the product as a target molecule, vinylcarbinol does not immediately suggest itself as a precursor– the synthetic potential of the [3,3] rearrangement is disguised. This disguised results from need to draw the enolised form of the product in an unfovoured boat conformation for the possibility of the reverse sigmatropic rearrangement to become evident
Both the Claisen and Cope rearrangements may lead to products in which one of the double bonds in the first-formed product is part an enolate which is converted into a ketone in the work-up. In the retro-synthetic analysis of the product as a target molecule, vinylcarbinol does not immediately suggest itself as a precursor– the synthetic potential of the [3,3] rearrangement is disguised. This disguised results from need to draw the enolised form of the product in an unfovoured boat conformation for the possibility of the reverse sigmatropic rearrangement to become evident
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Nevertheless, blowing the cover of a disguised reverse [3,3] sigmatropic rearrangement may be rewarding, particularly since the anionic ‘oxy-Cope’ rearrangement can be carried out under very mild conditions. Evans and Golob showed that conversion of alcohol into its alkoxide brought about an increase of up to 1017-fold in the rate of the Cope rearrange-ment. Thus, whereas thermal rearrangement of the starting material was previously carried out at 300 oC, the potassium alkoxide, in the presence of a crown ether, rearrangement below room temperature.
Nevertheless, blowing the cover of a disguised reverse [3,3] sigmatropic rearrangement may be rewarding, particularly since the anionic ‘oxy-Cope’ rearrangement can be carried out under very mild conditions. Evans and Golob showed that conversion of alcohol into its alkoxide brought about an increase of up to 1017-fold in the rate of the Cope rearrange-ment. Thus, whereas thermal rearrangement of the starting material was previously carried out at 300 oC, the potassium alkoxide, in the presence of a crown ether, rearrangement below room temperature.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Much use has been made of the mild conditions required for this anionic oxy-Cope rearrangement, particularly since the anionic oxygen has been found to have a preference for the ‘equatorial’ position which is greater than that of a hydroxygroup.
Much use has been made of the mild conditions required for this anionic oxy-Cope rearrangement, particularly since the anionic oxygen has been found to have a preference for the ‘equatorial’ position which is greater than that of a hydroxygroup.
Me Me
MeOH KH, 18-crown-6
DME, reflux
Me
MeMe
O-
H+
MeMe
HO
Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
R1 R2
OH
1. NaH
2. CF3CN3. H+, H2O
R1 R2
OHN
CF3
xylenereflux
R1 R2
NHCOCF3
Some examples of chirality transfer in [3,3] sigmatropic rearrangements other than the Claisen rearrangement:
Some examples of chirality transfer in [3,3] sigmatropic rearrangements other than the Claisen rearrangement:
S
S
MeCO3H S
S
O-
S
S
O-
S
SO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement
Me Me
S
Cl3CCOCl
Zn/Cu, Et2O
Cl2C C OS
O-
Cl
Cl
Me
Me
Cl
Cl
Me
Me
S
O
67 %, 81 e. e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.14. 4.14. Simple Chirality Transfer via [3,3] Simple Chirality Transfer via [3,3] SigmatropicSigmatropic RearrangementRearrangement

72
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
ba R
cd
ba
cd
R
ba
cd
R
ba
dc
R
ba
dc
R
ba
R
dc
Thermal [1,5] sigmatropic rearrangement of the C-R bond takes place suprafacially with retention of configuration in the migrating group R. The hypothetical homolytic cleavage of the C-R bond results in a pentadienyl radical whose terminal lobes have correct phase for this suprafacial rearrangement to occur.
Thermal [1,5] sigmatropic rearrangement of the C-R bond takes place suprafacially with retention of configuration in the migrating group R. The hypothetical homolytic cleavage of the C-R bond results in a pentadienyl radical whose terminal lobes have correct phase for this suprafacial rearrangement to occur.Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ba R
cd
ba
cd
R
ba
cd
R
ba
dc
R
ba
dc
R
ba
R
dc
Thus, given the configurations of the terminal double bond and the chiral centre in the starting material, the transition state geometry allows one to predictconfidently the relationship between the configuration of the new chiral centre and that of the double bond in the product, although there remains the question of whether path (above) will be favoured over (down) or vice versa.
Thus, given the configurations of the terminal double bond and the chiral centre in the starting material, the transition state geometry allows one to predictconfidently the relationship between the configuration of the new chiral centre and that of the double bond in the product, although there remains the question of whether path (above) will be favoured over (down) or vice versa.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
ba R
cd
ba
cd
R
ba
cd
R
ba
dc
R
ba
dc
R
ba
R
dc
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
In practice, the majority of [1,5] shifts have been those of hydrogen although other groups (CHO, PhCH3, SiMe3 and CH3CO) have been shown to have higher migratory aptitudes than hydrogen. Because of the difficulty in attainingthe s-cis-diene conformation depicted in an acyclic substrate, [1,5] shifts are most commonly found in dienyl systems which are contained in a ring and especially in cyclopentadienyl, cyclohexadienyl, cycloheptadienyl or cyclohepta-trienyl systems. The ambiguity as to the stereochemical outcome which exists in the acyclic case is also removed since superficial migration is possible only to one face in these cyclic systems.
In practice, the majority of [1,5] shifts have been those of hydrogen although other groups (CHO, PhCH3, SiMe3 and CH3CO) have been shown to have higher migratory aptitudes than hydrogen. Because of the difficulty in attainingthe s-cis-diene conformation depicted in an acyclic substrate, [1,5] shifts are most commonly found in dienyl systems which are contained in a ring and especially in cyclopentadienyl, cyclohexadienyl, cycloheptadienyl or cyclohepta-trienyl systems. The ambiguity as to the stereochemical outcome which exists in the acyclic case is also removed since superficial migration is possible only to one face in these cyclic systems.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
R2
HR1
R2
R1
H
H
R2
R1
H
Unfortunately, it is often difficult in practice in these case to limit the [1,5] shift to a single migration and serial [1,5] H rearrangements often result. In cyclopentadienyl systems, facile [1,5] sigmatropic rearrangement usually means that isomeriacally pure mono- or disubstituted derivatives are unobtained. From a synthetic point of view this is most unfortunate because it limits the use of substituted cyclopentadienes as dienes in Diels-Alder cycloadditions: a mixture of interconverting dienes will usually result in an intractable mixture of isomeric adducts.
Unfortunately, it is often difficult in practice in these case to limit the [1,5] shift to a single migration and serial [1,5] H rearrangements often result. In cyclopentadienyl systems, facile [1,5] sigmatropic rearrangement usually means that isomeriacally pure mono- or disubstituted derivatives are unobtained. From a synthetic point of view this is most unfortunate because it limits the use of substituted cyclopentadienes as dienes in Diels-Alder cycloadditions: a mixture of interconverting dienes will usually result in an intractable mixture of isomeric adducts.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
hydroquinoneBu3N, 190 oC
moreceasily redu1. H2/Pd2. O3
3. DMSwork-up
HOHC
O
Possibly the most useful [1,5] sigmatropic rearrangements are those used to in tandem reactions: the diene from one or more [1,5] shifts is trapped, often by an intramolecular pericyclic reaction.
Possibly the most useful [1,5] sigmatropic rearrangements are those used to in tandem reactions: the diene from one or more [1,5] shifts is trapped, often by an intramolecular pericyclic reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement

73
ba H
cd
H H
cd
ba
H
H H
cdb
a H
More amenable to control are homo-[1,5] sigmatropic shifts in which a cyclopropane or other three-membered ring takes the place of one of the double bonds; the product is now non-conjugated and therefore cannot undergo a further [1,5] rearrangement. The migrating group here is again usually a hydrogen and the energetically favorable cleavage of the strainedthree-membered ring ensures that reaction is irreversible.
More amenable to control are homo-[1,5] sigmatropic shifts in which a cyclopropane or other three-membered ring takes the place of one of the double bonds; the product is now non-conjugated and therefore cannot undergo a further [1,5] rearrangement. The migrating group here is again usually a hydrogen and the energetically favorable cleavage of the strainedthree-membered ring ensures that reaction is irreversible.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
OMe
MeH
H
H
98 oCCope
OMe
Me
OMe
Me
140 oC(homo-[1,5])
O
Me
Me
OMe
HH
H
cis-cyclopropane via a Cope rearrangementcis-cyclopropane via a Cope rearrangement
trans-cyclopropane via the homo-[1,5] sigmatropic rearrangementtrans-cyclopropane via the homo-[1,5] sigmatropic rearrangement
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.15. [1,5] 4.15. [1,5] SigmatropicSigmatropic RearrangementRearrangement
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction
The simplest all-carbon ene reaction
This reaction has features in common with both the Diels-Alder reaction and the [1,5] sigmatropic rearrangement of hydrogen, but the transition-state geometry (TSG) which results from overlap or orbitals is more like that of the [2,3] sigmatropic rearrangement.
This reaction has features in common with both the Diels-Alder reaction and the [1,5] sigmatropic rearrangement of hydrogen, but the transition-state geometry (TSG) which results from overlap or orbitals is more like that of the [2,3] sigmatropic rearrangement.
H
ene
H
'retro-ene'
'ene'
enophile
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The ene reaction and its reverse, the retroene reaction, are pericyclic reactions: bonds are made and broken concertedly in a cyclic transition state involving six electrons and the symmetry of the orbitals involved is appropriate for this to occur.
The ene reaction and its reverse, the retroene reaction, are pericyclic reactions: bonds are made and broken concertedly in a cyclic transition state involving six electrons and the symmetry of the orbitals involved is appropriate for this to occur.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction
b
Hd
c
ab
Hdc
a
H
dc
a b
H
dc
a
b
diastereoisomers
Since there is a single sp3-centre available in ene reaction ensemble, simple chirality transfer requires this to be the chiral centre and one of two double bonds to be configured.
Since there is a single sp3-centre available in ene reaction ensemble, simple chirality transfer requires this to be the chiral centre and one of two double bonds to be configured.
Two transition state to diastereosiomeric productsTwo transition state to diastereosiomeric products
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction
CO2MeMeO2C
S
D
HiPr
H
Ph
NN
D
H
iPrH
Ph
NN CO2Me
CO2Me
Both diastereoisomeric products are formed in the reaction in which the S/R ratio at the new chiral centre in the the product is the same as the D/H ratio at the vinyl position.
Both diastereoisomeric products are formed in the reaction in which the S/R ratio at the new chiral centre in the the product is the same as the D/H ratio at the vinyl position.
NN
D
iPrH
H Ph
NN
MeO2C
MeO2C
D
iPrH
H
Ph
CO2MeMeO2C
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction

74
OSiME2But
SEt
Me H+ PhCHO Me2AlCl
CH2Cl2, -78 oC PhOSiMe2But
SEt
Me
OH
Me
HH
H
tBuMe2SiO
O
EtS
Al-
PhH
MeEtS
Ph HOH
tBuMe2SiO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction
Some of the the most useful stereoselective ene/retroene reactions have one or more carbon atoms in the ene or enophile components replaced by heteroatoms. Thus the role of the enophile is often taken on by N=N, C=O, O=O, etc., in stead of C=C or C≡C. With a carbonyl group as the enophile, the ene reaction can be catalysed by Lewis acids, e.g. SnCl4 or R3Al, which coordinate with the oxygen of the carbonyl group and greatly reduce the temperature at which the reaction will take place.
Some of the the most useful stereoselective ene/retroene reactions have one or more carbon atoms in the ene or enophile components replaced by heteroatoms. Thus the role of the enophile is often taken on by N=N, C=O, O=O, etc., in stead of C=C or C≡C. With a carbonyl group as the enophile, the ene reaction can be catalysed by Lewis acids, e.g. SnCl4 or R3Al, which coordinate with the oxygen of the carbonyl group and greatly reduce the temperature at which the reaction will take place.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
4.16. 4.16. Simple Chirality Transfer in the Simple Chirality Transfer in the EneEne ((RetroeneRetroene) Reaction) Reaction
SummarySummary
Simple chirality transfer reactions are those in which a chiral centre in the starting material is lost and a new chiral centre is created elsewhere in the molecule; this transformation is mediated by the shift of one or more double bonds, at least one of which is configured, to a new position in the product.Diastereoselectivity in these reactions is more likely when all or part of the system undergoing the chiraltity transfer is in-corporated into a cyclic system, although the substitution present in the starting material can sometimes give rise to high diastereoselectivity even in acyclic cases.
Simple chirality transfer reactions are those in which a chiral centre in the starting material is lost and a new chiral centre is created elsewhere in the molecule; this transformation is mediated by the shift of one or more double bonds, at least one of which is configured, to a new position in the product.Diastereoselectivity in these reactions is more likely when all or part of the system undergoing the chiraltity transfer is in-corporated into a cyclic system, although the substitution present in the starting material can sometimes give rise to high diastereoselectivity even in acyclic cases.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The reversibility of the allylic sulphoxide-sulphenate ester [2,3]sigmatropic rearrangement allows the interconversion of diastereoisomeric sulphenate esters and hence provides a means of inverting the configuration in an enantiopure allylicalcohol at both the double bond and the chiral centre simultaneously.
The reversibility of the allylic sulphoxide-sulphenate ester [2,3]sigmatropic rearrangement allows the interconversion of diastereoisomeric sulphenate esters and hence provides a means of inverting the configuration in an enantiopure allylicalcohol at both the double bond and the chiral centre simultaneously.
The [3,3] sigmatropic rearrangement is a most valuable simple chirality transfer reaction because of the usual preference in an acyclic system for a chair-shaped transition state with the bulkier substituent located in an equatorial position. Using the Ireland-Claisen rearrangement requires methods for diatereoselective enolate generation. The oxy-Cope [3,3] sigmatropic rearrangement can proceed at even lower temperatures than the Ireland-Claisenrearrangement.
For the [1,5] sigmatropic rearrangement to be useful in synthesis,Ways must be found of circumventing the serial [1,5] sigmatropic rearrangements which otherwise tend to occur in cyclic dienes.
The [3,3] sigmatropic rearrangement is a most valuable simple chirality transfer reaction because of the usual preference in an acyclic system for a chair-shaped transition state with the bulkier substituent located in an equatorial position. Using the Ireland-Claisen rearrangement requires methods for diatereoselective enolate generation. The oxy-Cope [3,3] sigmatropic rearrangement can proceed at even lower temperatures than the Ireland-Claisenrearrangement.
For the [1,5] sigmatropic rearrangement to be useful in synthesis,Ways must be found of circumventing the serial [1,5] sigmatropic rearrangements which otherwise tend to occur in cyclic dienes.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Type 2 Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Chapter 5Chapter 5

75
Part 1Part 1
Simple Diastereoselectivity in 1,2-Addition to Alkenes,
Diels-Alder and 1,3-Dipolar Cycloadditions and The Ene Reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OutlineOutline
1. General;2. Relative Configuration in Type 2 Reactions;3. Epoxidaton, Aziridination and Cyclopropanation;4. Bromination and Similar additions to Alkenes;5. Hydroboration-Oxidation;6. Diels-Alder Reactions;7. Type 2 1,3-dipolar cycloadditions8. The Ene reaction9. Summary
1. General;2. Relative Configuration in Type 2 Reactions;3. Epoxidaton, Aziridination and Cyclopropanation;4. Bromination and Similar additions to Alkenes;5. Hydroboration-Oxidation;6. Diels-Alder Reactions;7. Type 2 1,3-dipolar cycloadditions8. The Ene reaction9. Summary
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
In Type 2 reactions, two or more chiral centres are formed simultaneously from one or more precursors, at least one of which is prochiral but neither of which is chiral; if a reagent is used, this is achiral.
In Type 2 reactions, two or more chiral centres are formed simultaneously from one or more precursors, at least one of which is prochiral but neither of which is chiral; if a reagent is used, this is achiral.
Type 2 ReactionsType 2 Reactions
The largest number of highly diastereoselective Type 2 reactionsare those in which the prochiral precursors are double bonds, usually C=C, C=O, C=N or C=C-C=C. These reactions include:1) Those which result in the addition of X, X-X or X-Y across a
configured double bond;2) Many pericycle reactions including electrocyclic reactions,
cycloadditions, simatropic rearrangements and cheletropic additions.
The largest number of highly diastereoselective Type 2 reactionsare those in which the prochiral precursors are double bonds, usually C=C, C=O, C=N or C=C-C=C. These reactions include:1) Those which result in the addition of X, X-X or X-Y across a
configured double bond;2) Many pericycle reactions including electrocyclic reactions,
cycloadditions, simatropic rearrangements and cheletropic additions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 ReactionsType 2 ReactionsType 2 Reactions
Almost all of the products from these Type 2 reactions are either cyclic or are formed via cyclic transition states and thediastereoselectivity which arises is referred to as simplediastereoselectivity.
Almost all of the products from these Type 2 reactions are either cyclic or are formed via cyclic transition states and thediastereoselectivity which arises is referred to as simplediastereoselectivity.
R R
a b+
x
y
'addition' a
bR
R
xy
R R
a b+
- RR' a
bR
R'
xy
R' R'
x y R = R' = Hoxidation
Two chiral centres are formed from addition of a precursor having a prochiral centre to a prochiral double bond.
Two chiral centres are formed from addition of a precursor having a prochiral centre to a prochiral double bond.
from the combination of two precursors each having prochiral centres.
from the combination of two precursors each having prochiral centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Relative Configuration in Type 2 ReactionsRelative Configuration in Type 2 Reactions
The absence of chirality in the precursor(s) in Type 2 reactionshas two important consequences:1) The products are always racemic and therefore any
stereoselectivity in Type 2 reactions will be diastereoselectivity;2) There can be no asymmetric introduction.
The absence of chirality in the precursor(s) in Type 2 reactionshas two important consequences:1) The products are always racemic and therefore any
stereoselectivity in Type 2 reactions will be diastereoselectivity;2) There can be no asymmetric introduction.
It is this latter consequence which distinguishes Type 2 from Type 3 reactions:Type 2 reactions become Type 3 (or Type 2/3) when one of the components contains a chiral centres.
It is this latter consequence which distinguishes Type 2 from Type 3 reactions:Type 2 reactions become Type 3 (or Type 2/3) when one of the components contains a chiral centres.
A characteristic of many Type 2 reactions which proceed via cyclic transition states is that they show inherent diastereoselectivity (stereospecificity).
A characteristic of many Type 2 reactions which proceed via cyclic transition states is that they show inherent diastereoselectivity (stereospecificity).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
This arises from the single well defined transition state geometries (TSGs) through which the reactions proceed. The relative configuration at the two more chiral centres that are formed derives from the TSG and from the configuration(s) of the prochiral precursor(s).
This arises from the single well defined transition state geometries (TSGs) through which the reactions proceed. The relative configuration at the two more chiral centres that are formed derives from the TSG and from the configuration(s) of the prochiral precursor(s).
Other type 2 reactions (e.g. the aldol reaction) lack this inherent diastereoselectivity. Nevertheless, such reactions can occasionally exhibit high diastereoselectivity because of the particular combination of substituents on their reacting components. The occasional diastereoselectivity which results in Type 2 versions of these reactions will clearly be less reliable than inherent diastereoselectivity.
Other type 2 reactions (e.g. the aldol reaction) lack this inherent diastereoselectivity. Nevertheless, such reactions can occasionally exhibit high diastereoselectivity because of the particular combination of substituents on their reacting components. The occasional diastereoselectivity which results in Type 2 versions of these reactions will clearly be less reliable than inherent diastereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Relative Configuration in Type 2 ReactionsRelative Configuration in Type 2 Reactions

76
For some Type 2 reactions proceeding vi cyclic transition states(e.g. Diels-Alder reactions), the diastereoselectivity may be inherent or occasional or both, depending on the substitution ofthe reacting components.
For some Type 2 reactions proceeding vi cyclic transition states(e.g. Diels-Alder reactions), the diastereoselectivity may be inherent or occasional or both, depending on the substitution ofthe reacting components.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Relative Configuration in Type 2 ReactionsRelative Configuration in Type 2 Reactions EpoxidatonEpoxidaton, , AziridinationAziridination and and CyclopropanationCyclopropanation
In the Bartlett mechanisms for the epoxidation of alkenes using peroxyacids, both bonds to peroxy oxygen are formed from the same face of the configured double bond in a single step.
In the Bartlett mechanisms for the epoxidation of alkenes using peroxyacids, both bonds to peroxy oxygen are formed from the same face of the configured double bond in a single step.
a y
b x
OR
OOH
O
a yb x
O
a yb x
a y
b x
OR
OO H
Both faces of the alkene are equally likely to react in this way leading to a racemic product.
Both faces of the alkene are equally likely to react in this way leading to a racemic product.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The syn addition of oxygen to the double bond results in the inherent diastereoselectivity of the reaction and is a feature of all peroxyacid epoxidations of configured double bonds.
The syn addition of oxygen to the double bond results in the inherent diastereoselectivity of the reaction and is a feature of all peroxyacid epoxidations of configured double bonds.
The same syn addition is found in the formation of aziridines from alkenes using the aziridinating agent.The same syn addition is found in the formation of aziridines from alkenes using the aziridinating agent.
N
N
R O
NH2
Pd(OAc)4
N
N
R O
NHOAc
Me
Me
Me H
H Me
OMe
ONH
X
N
Me HH Me
X
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
EpoxidatonEpoxidaton, , AziridinationAziridination and and CyclopropanationCyclopropanation
The same syn addition is found in the cyclopropanation of alkenes using a carbenoid (the Simmons-Smith reaction).
The same syn addition is found in the cyclopropanation of alkenes using a carbenoid (the Simmons-Smith reaction).
Me MeH HMe Me
H H+ ICH2ZnI
Me Me
H HC
IZn I
H H
Type 2 aziridination and cyclopropanation of alkenes can also be brought about by cheletropic addition of singlet nitrenes and carbenes, respectively.
Type 2 aziridination and cyclopropanation of alkenes can also be brought about by cheletropic addition of singlet nitrenes and carbenes, respectively.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
EpoxidatonEpoxidaton, , AziridinationAziridination and and CyclopropanationCyclopropanation
BrominationBromination and Similar additions to Alkenesand Similar additions to Alkenes
Bromination of a configured double bond is similar to epoxidation in that a cyclic bromonium ion is formed by syn addition to each face of the double bond. However, the bromonium ion undergoes spontaneous ring opening by bromide anion in an SN2 fashion leading to overall anti addition of bromide.
Bromination of a configured double bond is similar to epoxidation in that a cyclic bromonium ion is formed by syn addition to each face of the double bond. However, the bromonium ion undergoes spontaneous ring opening by bromide anion in an SN2 fashion leading to overall anti addition of bromide.
Br2 Br-
a y
b x
Br+
a yb x
Br+
a yb x
Bra
y
b
xBr
Bra
y
b
xBr
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Ring opening of the bromonium ion is regiospecific with attack of bromide at the a,b-substituted carbon, although in fact the same racemic dibromide would have been obtained had the bromide attacked in the opposite regio-sense, at the x,y-substituted carbon (in the same SN2 fashion).
Ring opening of the bromonium ion is regiospecific with attack of bromide at the a,b-substituted carbon, although in fact the same racemic dibromide would have been obtained had the bromide attacked in the opposite regio-sense, at the x,y-substituted carbon (in the same SN2 fashion).
Br2, M+Nu-
Nu-a y
b xBr+
a yb x
Bra
y
b
xNu
Nua
y
b
xBr+ mirror image
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BrominationBromination and Similar additions to Alkenesand Similar additions to Alkenes

77
Ring opening of the bromonium ion may be brought about by nucleophiles other than bromide if these are present (including the solvent itself) and in these cases there will be the possibility of regioisomer formation.
Ring opening of the bromonium ion may be brought about by nucleophiles other than bromide if these are present (including the solvent itself) and in these cases there will be the possibility of regioisomer formation.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BrominationBromination and Similar additions to Alkenesand Similar additions to Alkenes
Bromonium ion formation and ring opening in a stereo-defined way, if not always in a regio-defined way, is analogous to a number of other anti additions to alkenes which proceed via ring opening of –onium ions including episulphonium and selenenium as well as other halonium ions of which the iodonium ion is the most important.
Bromonium ion formation and ring opening in a stereo-defined way, if not always in a regio-defined way, is analogous to a number of other anti additions to alkenes which proceed via ring opening of –onium ions including episulphonium and selenenium as well as other halonium ions of which the iodonium ion is the most important.
S
RSe
R
O
R
The oxonium ion formed by reaction of an epoxide with an electrophile usually a proton or Lewis acid like the bromonium ion, then undergoes rapid SN2 ring opening by nucleophiles.These overall anti additions are more valuable because most other highly diastereoselective Type 2 additions to double bondstake place in a syn fashion.
The oxonium ion formed by reaction of an epoxide with an electrophile usually a proton or Lewis acid like the bromonium ion, then undergoes rapid SN2 ring opening by nucleophiles.These overall anti additions are more valuable because most other highly diastereoselective Type 2 additions to double bondstake place in a syn fashion.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
BrominationBromination and Similar additions to Alkenesand Similar additions to Alkenes
HydroborationHydroboration--OxidationOxidation
Although the product from hydrogenation of an alkene is not cyclic, the transition state is regarded as being cyclic in character.
Although the product from hydrogenation of an alkene is not cyclic, the transition state is regarded as being cyclic in character.
R2BH
a y
b x
Type 2
a yb x
H BR2
a yb x
H BR2
H2O2, NaOH
Type 1
a yb x
H OH
The overall addition of water to the double bond is syn since replacement of the boron-carbon bond by oxygen-carbon proceeds with complete retention of configuration at carbon. Complete regioselectivity has been assumed with the boron adding to the x,y-substituted alkene carbon. This regioselectivity is normally determined by the bulk of the substituents a,b or x,y on the alkene, with the boron adding to the less substituted carbon.
The overall addition of water to the double bond is syn since replacement of the boron-carbon bond by oxygen-carbon proceeds with complete retention of configuration at carbon. Complete regioselectivity has been assumed with the boron adding to the x,y-substituted alkene carbon. This regioselectivity is normally determined by the bulk of the substituents a,b or x,y on the alkene, with the boron adding to the less substituted carbon.
区域选择性:B加到位阻小的C上
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HydroxylationHydroxylation
Epoxidation of a configured double bond followed by treatment of the epoxide with aqueous acid brings about anti hydroxylation (also referred as dihydroxylation).
Epoxidation of a configured double bond followed by treatment of the epoxide with aqueous acid brings about anti hydroxylation (also referred as dihydroxylation).
O
a yb x
O
a yb x
H
OH H
-H+
OHa
y
b
xHO
+mirror image
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
A stereochemically complementary method for hydroxylation of alkene giving the product of syn addition uses osmium tetraoxide followed by cleavage of the osmate ester. It is possible that the osmate ester is formed by rearrangement of the four-membered ring species. The same syn-hydroxylation can be accomplished using potassium permanganate; in this case a cyclic manganate ester is an intermediate.
A stereochemically complementary method for hydroxylation of alkene giving the product of syn addition uses osmium tetraoxide followed by cleavage of the osmate ester. It is possible that the osmate ester is formed by rearrangement of the four-membered ring species. The same syn-hydroxylation can be accomplished using potassium permanganate; in this case a cyclic manganate ester is an intermediate.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HydroxylationHydroxylation
a y
b x
a yb x
OOs
O
O O
OsO4
a yb x
OsOO O
O
NaHCO3
a yb x
OHHO
a y
b x KMnO4
a yb x
OMn
O
O O- H2O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HydroxylationHydroxylation

78
The relative configuration shown for the two chiral centres is a consequence of the conrotatory mode of ring closure. This conrotatory ring closure is dictated by the symmetry of the highest occupied molecular orbital (HOMO), ψ2; only by rotating around the axes shown in the same sense does in-phase overlap of the p-orbitals on C-1 and C-4 (Frontier Molecular Orbital Theory).
The relative configuration shown for the two chiral centres is a consequence of the conrotatory mode of ring closure. This conrotatory ring closure is dictated by the symmetry of the highest occupied molecular orbital (HOMO), ψ2; only by rotating around the axes shown in the same sense does in-phase overlap of the p-orbitals on C-1 and C-4 (Frontier Molecular Orbital Theory).
Ring closure of the same butadiene by photochemical means via the HOMO of first excited single (ψ3) gives a cyclobutene having the alternative relative configuration since the orbital symmetry-directed cyclisation in this case is disrotatory.
Ring closure of the same butadiene by photochemical means via the HOMO of first excited single (ψ3) gives a cyclobutene having the alternative relative configuration since the orbital symmetry-directed cyclisation in this case is disrotatory.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HydroxylationHydroxylation Type 2 Type 2 PericyclicPericyclic ReactionsReactions
Electrocyclic ReactionsIn Type 2 4π→2π thermal electrocyclic ring closure, two configured double bonds are converted into two adjacent chiral centres.
In Type 2 4π→2π thermal electrocyclic ring closure, two configured double bonds are converted into two adjacent chiral centres.
ba x
y
b
a x
y a
b y
x
enantiomers
ba x
y
conrotatory
conrotatory
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ba x
y
b
a y
x a
b x
y
enantiomers
ba x
y
disrotatory
disrotatory
hv
hv
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Type 2 PericyclicPericyclic ReactionsReactions CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity
In the Type 2 reactions, two adjacent chiral centres are formed in the product. In cycloadditions, more than two chiral centres may be formed and, when only two are formed, they are not necessarily on adjacent carbon atoms.
In the Type 2 reactions, two adjacent chiral centres are formed in the product. In cycloadditions, more than two chiral centres may be formed and, when only two are formed, they are not necessarily on adjacent carbon atoms.
ac
a+ y
xH
H y H
a a
xH+ mirror image
The singlet carbene adds in completely syn fashion to the configured alkene, i.e. the trans relationship of x and y in the alkene is retained in the cyclopropane product
The singlet carbene adds in completely syn fashion to the configured alkene, i.e. the trans relationship of x and y in the alkene is retained in the cyclopropane product
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ac
a+ H
xH
y H y
a a
xH+ mirror image
Since addition to the corresponding cis-alkene gives only the product in which x and y are cis, this cycopropanation by singlet carbenes is stereospecific. This complete syn diastereoselectivity is inherent in the carbene cycloaddition mechanism and, in fact, is used to determine the spin state of the carbene since triplet carbenes do not normally show such complete diastereoselectivity.
Since addition to the corresponding cis-alkene gives only the product in which x and y are cis, this cycopropanation by singlet carbenes is stereospecific. This complete syn diastereoselectivity is inherent in the carbene cycloaddition mechanism and, in fact, is used to determine the spin state of the carbene since triplet carbenes do not normally show such complete diastereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity
y H
b a
xHa
cb
+ Hx
H
y H y
a b
xH
+ mirror image
If, however, the carbene bears two different substituents, then a third chiral centre is formed in the product.
If, however, the carbene bears two different substituents, then a third chiral centre is formed in the product.
Although both cyclopropanes have x and y trans (inherent diastereoselectivity), prediction of the relative configuration of the third (a,b-substituted) chiral centre is far from safe and both diastereoisomers will in general be formed, albeirt in unequal amounts.
Although both cyclopropanes have x and y trans (inherent diastereoselectivity), prediction of the relative configuration of the third (a,b-substituted) chiral centre is far from safe and both diastereoisomers will in general be formed, albeirt in unequal amounts.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity

79
The inherently diastereoselective part of the carbene addition is the result of concerted addition of the electron-deficient carbene to either enantioface of the alkene, a process analogous to epoxidation and giving rise to racemic cyclopropane.
The inherently diastereoselective part of the carbene addition is the result of concerted addition of the electron-deficient carbene to either enantioface of the alkene, a process analogous to epoxidation and giving rise to racemic cyclopropane.
ac
a+ y
xH
H y H
a a
xH+ mirror image
a y
b x
OR
OOH
O
a yb x
O
a yb xa y
b x
OR
OO H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity
Using a carbene bearing two different substituents, however, the two enantiofaces of the alkenes become diastereofaces when associated withthe carbene in the transition state. As a result, the two transition states
are diastereoisomeric and give rise to two racemic diastereoisomericproducts; in each case the inherent diastereoselectivity referred to above is still manifested.
Using a carbene bearing two different substituents, however, the two enantiofaces of the alkenes become diastereofaces when associated withthe carbene in the transition state. As a result, the two transition states
are diastereoisomeric and give rise to two racemic diastereoisomericproducts; in each case the inherent diastereoselectivity referred to above is still manifested.
x H
b a
yH
C
x H
a b
yH
C
x H
b a
yH
x H
a b
yHHx
ab
H y
Hx
ba
H y
Hx
ab
H y
Hx
ba
H y
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity
Whether the creation of the third chiral centre at the erstwhile carbenecarbon results in a single diastereoisomers in the cyclopropanation(occasional diastereoselectivity) depends on the relative energies of these two compounds, which in turn will depend on the steric and/or electronic interactions between a and x (or y) and y (or x) in these transition states. These interactions can lead to to a single (relative) configuration at the third chiral centre (most often when metal-coordinated carbenes or carbenoids are used). However, complete diastereoselectivityin this sense is not inherent to the cyclopropanation.
Whether the creation of the third chiral centre at the erstwhile carbenecarbon results in a single diastereoisomers in the cyclopropanation(occasional diastereoselectivity) depends on the relative energies of these two compounds, which in turn will depend on the steric and/or electronic interactions between a and x (or y) and y (or x) in these transition states. These interactions can lead to to a single (relative) configuration at the third chiral centre (most often when metal-coordinated carbenes or carbenoids are used). However, complete diastereoselectivityin this sense is not inherent to the cyclopropanation.
The division of the diastereoselectivity obtaining in cyclo-propanation into inherent and occasional is typical of many of the Type 2 cycloadditions which follow.
The division of the diastereoselectivity obtaining in cyclo-propanation into inherent and occasional is typical of many of the Type 2 cycloadditions which follow.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
CycloadditionsCycloadditions -- Inherent and Occasional Inherent and Occasional DiastereoselectivityDiastereoselectivity DielsDiels--Alder ReactionsAlder Reactions
The transition-state geometry for the Diels-Alder reaction is that shown below, where both components add suprafacially. This is the best way in which the frontier molecular orbitals of the diene and dienophile can overlap in-phase.
The transition-state geometry for the Diels-Alder reaction is that shown below, where both components add suprafacially. This is the best way in which the frontier molecular orbitals of the diene and dienophile can overlap in-phase.
HOMO
LUMO HOMO
LUMO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
In the Type 2 Diels-Alder reactions, both the diene and the dieno-phile are achiral and if a catalyst is used, this is also achiral. Diels-Alder reactions in which chiral centres are present in the sub-stiuents on the diene, or catalyst are Type 3 or Type 2/3 and will be considered later.
In the Type 2 Diels-Alder reactions, both the diene and the dieno-phile are achiral and if a catalyst is used, this is also achiral. Diels-Alder reactions in which chiral centres are present in the sub-stiuents on the diene, or catalyst are Type 3 or Type 2/3 and will be considered later.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HOMO
LUMO HOMO
LUMO
As the transition-state geometry shows above, syn addtion takes place on one face of dienophile by the diene and reciprocally on one face of the diene by the dienophile. This mutual (stereospecific) syn addition of diene and dienophile is an inherent stereochemical characteristic of the Diels-Alder reactions.
As the transition-state geometry shows above, syn addtion takes place on one face of dienophile by the diene and reciprocally on one face of the diene by the dienophile. This mutual (stereospecific) syn addition of diene and dienophile is an inherent stereochemical characteristic of the Diels-Alder reactions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions

80
Ha
a HH
yx
H
2 3
Hy
xH
Ha
a H
23
Hy
xH
H
a
a
H23
a
xa y
1
2
5 5'
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
It is reflected in the stereostructure of the product cyclohexene (5), in which four chiral centres have been created in the reaction of diene (1) and dienophile (2). Thus in (5), the trans disposition of x and y in the dienophile (2), and of the substituents a (relative to the C2-C3 bond) of the diene (1), has been retained.
It is reflected in the stereostructure of the product cyclohexene (5), in which four chiral centres have been created in the reaction of diene (1) and dienophile (2). Thus in (5), the trans disposition of x and y in the dienophile (2), and of the substituents a (relative to the C2-C3 bond) of the diene (1), has been retained.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
Ha
a HH
yx
H
2 3
Hy
xH
Ha
a H
23
Hy
xH
Ha
a
H2
3
or Hy
Hx
H
a
a
H23
Hy
xH
H
a
a
H23
a
xa y
1
23 4
5 5'
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
There is now little doubt that the Diels-Alder reactions can proceed via a continuum of transition states in terms of the degree to which the new –bonds between the diene and dienophileare made. However, complete syn diastereoselectivity in addtionsto the dienophile is a stereochemical ballmark of all Diels-Alder reactions and means, for example, that for the reaction the cycolhexene diastereoisomers (3) and (4) are not obtained.
There is now little doubt that the Diels-Alder reactions can proceed via a continuum of transition states in terms of the degree to which the new –bonds between the diene and dienophileare made. However, complete syn diastereoselectivity in addtionsto the dienophile is a stereochemical ballmark of all Diels-Alder reactions and means, for example, that for the reaction the cycolhexene diastereoisomers (3) and (4) are not obtained.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
The enantiomeric product and the transition state which leads to it are drawn as the mirror images of thosshown in the previous scheme. Reacting opposite face of both dieneand dienophile to those used in the previous scheme will also lead to enantiomers. In the remainder of this chapter, the products from Type 2 reactions will usually be drawn as a single enantiomers but will in fact always have been formed as racemates.
The enantiomeric product and the transition state which leads to it are drawn as the mirror images of thosshown in the previous scheme. Reacting opposite face of both dieneand dienophile to those used in the previous scheme will also lead to enantiomers. In the remainder of this chapter, the products from Type 2 reactions will usually be drawn as a single enantiomers but will in fact always have been formed as racemates.
yx
H
H
aHH a
yx
H
H
a
HHa
a
H H
aH
H
y
x
xH
a
Hy
a
H
H
enantiomers
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
In this transition state, y locates on the same side as the diene (the endo position) and x on the opposite side (the exo position) and the product is therefore (5): in the transition state in anotherscheme, the location of x and y is revised, x being endo and y exo. In this scheme, the inherent syn addition of diene to dienophileand dienophile to diene still obtains.
In this transition state, y locates on the same side as the diene (the endo position) and x on the opposite side (the exo position) and the product is therefore (5): in the transition state in anotherscheme, the location of x and y is revised, x being endo and y exo. In this scheme, the inherent syn addition of diene to dienophileand dienophile to diene still obtains.
y
HH
x
Ha
a H
23
y
HH
x
H
a
a
H23
a
xa y
5 5'
endo
exo exo,endo是针对某个取代基而言的
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions

81
Comparison of the two cyclohexene products reveals that these are indeed diastereoisomers and not enantiomers or regiomers. Where one or other or both of these diastereoisomers is produceddepends on the preference of x (or y) for the endo (or exo) position. This exo/endo diastereoselectivity of the Diels-Alder reaction is occasional, although in ether case the inherent synaddition to the dienophile and diene still prevails.
Comparison of the two cyclohexene products reveals that these are indeed diastereoisomers and not enantiomers or regiomers. Where one or other or both of these diastereoisomers is produceddepends on the preference of x (or y) for the endo (or exo) position. This exo/endo diastereoselectivity of the Diels-Alder reaction is occasional, although in ether case the inherent synaddition to the dienophile and diene still prevails.
yx
H
H
a
HHa
y
HH
x
H
a
a
H23
5
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
The situation at this point is very similar to that described earlier for addition of an unsymmetrical carbene to a figured alkenes: diastereoselectivity is in part inherent and in part occasional. However, there is an additional complication in the Diels-Alder reaction when both the diene and the dienophile are unsymmetrically substituted. This is the problem not of diastereoselectivity but of regioselectivity.
The situation at this point is very similar to that described earlier for addition of an unsymmetrical carbene to a figured alkenes: diastereoselectivity is in part inherent and in part occasional. However, there is an additional complication in the Diels-Alder reaction when both the diene and the dienophile are unsymmetrically substituted. This is the problem not of diastereoselectivity but of regioselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
Hx
y
H
Hb
a HH
xy
H
H
b
a
Hb
ya x
xH
Hy
Hb
a Hx
HH
y
H
b
a
Hb
ya x
Hy
xH
Hb
a HH
yx
H
H
b
a
Hb
xa y
y
HH
x
Hb
a Hy
HH
x
H
b
a
Hb
xa y
diastereoisomers
diastereoisomers
regioisomers
Thus in the reaction of the unsymmetrical diene and dienophile there are four possible products (each a racemate; only one enantiomers of each is shown).
Thus in the reaction of the unsymmetrical diene and dienophile there are four possible products (each a racemate; only one enantiomers of each is shown).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
Consequently, for the formation of a single product, the Type 2 Diels-Alder reactions must be completely diastereoselective in the exo/endo sense and also completely regioselective; syn addition to the diene and the dienophile can be taken for granted.
Consequently, for the formation of a single product, the Type 2 Diels-Alder reactions must be completely diastereoselective in the exo/endo sense and also completely regioselective; syn addition to the diene and the dienophile can be taken for granted.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
DielsDiels--Alder ReactionsAlder Reactions
RegioselectivityRegioselectivity in the Dielsin the Diels--Alder ReactionsAlder Reactions
Frontier molecular orbital theory can be used to predict the regioselectivity of Diels-Alder reactions by
(a) Identifying the HOMO and LUMO of diene and dienophilewhich are closer in energy.
(b) Containing the orbital coefficients on the terminal atoms of the diene and dienophile in this HOMO-LUMO combination.
(c) Matching the larger coefficient at the 1 and 4 positions of the diene with the larger coefficient at the 1 and 2 positions of the dieneophile.
Frontier molecular orbital theory can be used to predict the regioselectivity of Diels-Alder reactions by
(a) Identifying the HOMO and LUMO of diene and dienophilewhich are closer in energy.
(b) Containing the orbital coefficients on the terminal atoms of the diene and dienophile in this HOMO-LUMO combination.
(c) Matching the larger coefficient at the 1 and 4 positions of the diene with the larger coefficient at the 1 and 2 positions of the dieneophile.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ax
HOMO LUMO
preferrred
a
x
a
x
'ortho'
'meta'
a
x1
4
5
6
Where the size of the circle represents the magnitude of the coefficients on the interacting lobes, formation of the ‘ortho” isomer is favored.
Where the size of the circle represents the magnitude of the coefficients on the interacting lobes, formation of the ‘ortho” isomer is favored.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in the Dielsin the Diels--Alder ReactionsAlder Reactions

82
In practice, formation of a single regioisomer in the sense is often the case when a is a strongly electron-donating substituents and x a strongly electron-withdrawing substituent. This regioselectivity is that which would result if, in the transition state, formation of the 4,5-bond has run slightly ahead of that of the 1,6-bond leading to stabilisation by a and x of the fractional charges thereby generated.
In practice, formation of a single regioisomer in the sense is often the case when a is a strongly electron-donating substituents and x a strongly electron-withdrawing substituent. This regioselectivity is that which would result if, in the transition state, formation of the 4,5-bond has run slightly ahead of that of the 1,6-bond leading to stabilisation by a and x of the fractional charges thereby generated.
ax
HOMO LUMO
preferrred
a
x
a
x
'ortho'
'meta'
a
x1
4
5
6
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in the Dielsin the Diels--Alder ReactionsAlder Reactions
The regioselectivity of the Diels-Alder reactions may be affected by a number of factors including catalysis, pressure and solvent, but for the many reactions which are not very regioselective, one reliable solution is to tether the diene and dienophile together in such a way that the reaction -now intramolecular-can form only one regioisomer.
The regioselectivity of the Diels-Alder reactions may be affected by a number of factors including catalysis, pressure and solvent, but for the many reactions which are not very regioselective, one reliable solution is to tether the diene and dienophile together in such a way that the reaction -now intramolecular-can form only one regioisomer.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in the Dielsin the Diels--Alder ReactionsAlder Reactions
From our previous discussion, exo/endo diastereoselectivity is determined by the preference of the dienophile substituentsfor exo or endo positions in the cycloaddition transition state.
From our previous discussion, exo/endo diastereoselectivity is determined by the preference of the dienophile substituentsfor exo or endo positions in the cycloaddition transition state.
Exo/endo-Diastereoselectivity (occasional diastereoselectivity) in the Diels-Alder reaction
Exo/endo-Diastereoselectivity (occasional diastereoselectivity) in the Diels-Alder reaction
RO
O+
O
O
R
O
O
RH
H
R : OMe, 72 % CO2Me, 65 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
There are a few Diels-Alder reactions which show complete exo diastereoselectivity, usually as a result of steric repulsion in the endo transition state.
There are a few Diels-Alder reactions which show complete exo diastereoselectivity, usually as a result of steric repulsion in the endo transition state.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
H
H
b
a
H
R
O
b
CORa
aH
b
HO
R
HOMO (diene)
LUMO (dienophile)
attractivesecondary interaction
primary interaction
endo Selectivity is found when the dienophile bears single carbonyl group. This is thought to result from an attractive secondary orbital interaction in the endo transition state between orbitals on the carbonyl group and the diene although other explanations have been suggested. In frontier orbital terms, this attractive secondary orbital interaction can be ascribed to overlap of orbitals of matching symmetry on the carbonyl carbon and the diene C-2 in the dominant HOMO-LUMO pair.
endo Selectivity is found when the dienophile bears single carbonyl group. This is thought to result from an attractive secondary orbital interaction in the endo transition state between orbitals on the carbonyl group and the diene although other explanations have been suggested. In frontier orbital terms, this attractive secondary orbital interaction can be ascribed to overlap of orbitals of matching symmetry on the carbonyl carbon and the diene C-2 in the dominant HOMO-LUMO pair.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
The dienophile reacts as a 2π system but is, in fact, part of a 4 πsystem. The LUMO of this 4π system is that it has the appropriate symmetry for both primary and secondary orbital overlap with the HOMO of the 4π system of the diene.
The dienophile reacts as a 2π system but is, in fact, part of a 4 πsystem. The LUMO of this 4π system is that it has the appropriate symmetry for both primary and secondary orbital overlap with the HOMO of the 4π system of the diene.
O
O
O
O
OH
HOO
O
OH
Hno
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity

83
Attractive secondary interactions of this type are the bias of the Alder endo rule which is more usually applied to bicyclicsystems, e.g. the formation of the endo isomer from reaction of cyclopentadiene and maleic anhydride.
Attractive secondary interactions of this type are the bias of the Alder endo rule which is more usually applied to bicyclicsystems, e.g. the formation of the endo isomer from reaction of cyclopentadiene and maleic anhydride.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
The same endo selectivity is responsible for the stereostructure of the product from reaction of the acyclic diene and maleic anhydride.
The same endo selectivity is responsible for the stereostructure of the product from reaction of the acyclic diene and maleic anhydride.
O
O
O
O
OH
HOO
O
OH
Hno
OOO
OMe
TMSO
O
O
O
HOMe
H H
TMSO
O
OH
HOH
OMe
TMSO H+
O
H
H
O
OO
OMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Complete endo selectivity in Diels-Alder reaction is the expectationrather that the rule. Thus the reactions of cyclopentadiene with various carbonyl-substituted dienophile show that the endo (carbonyl)-substituted diastereoiomers is not the exclusive product nor even sometimes the major product. It appears that a methyl group has its own attractive secondary interaction withthe diene and can compete with the carbonyl group for the endo position.
Complete endo selectivity in Diels-Alder reaction is the expectationrather that the rule. Thus the reactions of cyclopentadiene with various carbonyl-substituted dienophile show that the endo (carbonyl)-substituted diastereoiomers is not the exclusive product nor even sometimes the major product. It appears that a methyl group has its own attractive secondary interaction withthe diene and can compete with the carbonyl group for the endo position.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Me CO2Me
CO2Me+
CO2Me
CO2Me
Me CO2Me
Me
CO2Me
75 : 2594 : 6 (AlCl3)
CO2Me+
CO2Me
HCO2Me
H
78 : 2295 : 5 (AlCl3)
CO2MeMe+
CO2Me
MeCO2Me
Me
31 : 6960 : 40 (AlCl3)
Me
CO2Me
+
CO2Me
H
Me
CO2Me
H
Me
54 : 4694 : 6 (AlCl3)
endo (carbonyl) Diastereoselectivity is much improved by catalysis with Lewis acid although, in the case of methyl methacrylate, this only changes the endo/exo ratio from 31:69 to 60:40.
endo (carbonyl) Diastereoselectivity is much improved by catalysis with Lewis acid although, in the case of methyl methacrylate, this only changes the endo/exo ratio from 31:69 to 60:40.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
NHCOCl3
+
CHO110 oC, 36 h
46 %
NHCOCl3CHO
NHCOCl3CHO
3 : 1
SPh
+
CO2Me 130 oC, 20 h
90 %
CO2Me
SPh
CO2Me
SPh
1 : 1
Incomplete endo selectivity is not restricted to Diels-Alder reactions with cyclopentadiene as the diene, as the example using acyclic dienes, only the cis-subsituted products are endo-derived. Not surprisingly, endo selectivity increases when dienophiles such as maleic anhydride are used in which both alkene carbons are substituted with carbonyl groups, and there are cis.
Incomplete endo selectivity is not restricted to Diels-Alder reactions with cyclopentadiene as the diene, as the example using acyclic dienes, only the cis-subsituted products are endo-derived. Not surprisingly, endo selectivity increases when dienophiles such as maleic anhydride are used in which both alkene carbons are substituted with carbonyl groups, and there are cis.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity

84
Increased diastereoselectivity may also be accompanied by increased regioselectivity and both can be accounted for in frontier orbital terms by an augmented secondary interaction.
Increased diastereoselectivity may also be accompanied by increased regioselectivity and both can be accounted for in frontier orbital terms by an augmented secondary interaction.
aH
b
HO
R
HOMO (diene)
LUMO (dienophile)
increase in orbital coefficient leading to greatersecondary interaction
without catalysis
aH
b
HOE+
R
with catalysis by E+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
endo Diastereoselectivity for many Diels-Alder reactions along with regioselectivity and rate constants can be markedly increased by changes in other experimental conditions and particularly by using water as the solvent, by addition of metalsalts to organic solvents (e.g. LiClO4 in diethyl ether), and by an increase in pressure.
endo Diastereoselectivity for many Diels-Alder reactions along with regioselectivity and rate constants can be markedly increased by changes in other experimental conditions and particularly by using water as the solvent, by addition of metalsalts to organic solvents (e.g. LiClO4 in diethyl ether), and by an increase in pressure.
+
OMe
COMe
COMe+
in cyclopentadiene 3.9 : 1in H2O 21.4 : 1in H2O, LiCl 28 : 1
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Thus, endo/exo selectivity in the reaction of cyclopentadieneand methyl vinyl ketone is increased in water and is increased further using water containing dissolved lithium chloride.
Thus, endo/exo selectivity in the reaction of cyclopentadieneand methyl vinyl ketone is increased in water and is increased further using water containing dissolved lithium chloride.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
It is thought that, as a result of hydrophobic reactions with water, the reactants are aggregated and subjected to an internal pressure in the solvent cavities that they occupy. Thiseffect is magnified in the presence of lithium chloride.
It is thought that, as a result of hydrophobic reactions with water, the reactants are aggregated and subjected to an internal pressure in the solvent cavities that they occupy. Thiseffect is magnified in the presence of lithium chloride.
LiClO4, Et2O+
OMeO
CO2Me
CO2Me+
8 : 1 4 : 1
RT, 4 h94 %
H2O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Lithium perchlorate dissolves in diethyl ether, in the reaction of cyclopentadiene and methyl acrylate in this solvent, the exo/endo ratio is increased over that obtained using water. The origin of this increased selectivity is thought to be the result of lithium functioning as a Lewis acid.
Lithium perchlorate dissolves in diethyl ether, in the reaction of cyclopentadiene and methyl acrylate in this solvent, the exo/endo ratio is increased over that obtained using water. The origin of this increased selectivity is thought to be the result of lithium functioning as a Lewis acid.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Diels-Alder reactions using furans as dienes are potentially useful because the furans are often readily available and oxygen bridge in the bicyclo[2.2.1] adducts is easily broken. However, the reaction of simple furans in this reation is often reversible and the equilibrium may lie on the side of the furan and dienophile rather than adduct.
Diels-Alder reactions using furans as dienes are potentially useful because the furans are often readily available and oxygen bridge in the bicyclo[2.2.1] adducts is easily broken. However, the reaction of simple furans in this reation is often reversible and the equilibrium may lie on the side of the furan and dienophile rather than adduct.
O
O
O+
O
O
O
H
H
O O
OH
H+
20 kbar
1 bar
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity

85
A further consequence of the reversibility is that, when the equilibrium does favor the adduct, a thermodynmicallyformed mixture of endo and exo products may be obtained. Occasional diastereoselectivity under these conditions is, in general, less likely than in kinetically controlled irreversibleDiels-Alder reactions
A further consequence of the reversibility is that, when the equilibrium does favor the adduct, a thermodynmicallyformed mixture of endo and exo products may be obtained. Occasional diastereoselectivity under these conditions is, in general, less likely than in kinetically controlled irreversibleDiels-Alder reactions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
Characteristic of Diels-Alder reactions is a high negative volume of activation (from –25 to –45 cm3 for intermolecular reactions). The application of high pressures to these cycloadditions (8-20 kbar), therefore, increases the rate of reaction. Using simple furans, the application of high pressure often brings about Diels-Alder reactions in greater yields than can be achieved at atmospheric pressure and the products are kinetically formed. Formation of the endo diastereoisomer is usually favored but high diastereoselectivity is in common.
Characteristic of Diels-Alder reactions is a high negative volume of activation (from –25 to –45 cm3 for intermolecular reactions). The application of high pressures to these cycloadditions (8-20 kbar), therefore, increases the rate of reaction. Using simple furans, the application of high pressure often brings about Diels-Alder reactions in greater yields than can be achieved at atmospheric pressure and the products are kinetically formed. Formation of the endo diastereoisomer is usually favored but high diastereoselectivity is in common.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
The Diels-Alder adducts, formed at high pressure, may start to dissociate at atmospheric pressure. If these adducts are required for synthetic purposes, therefore, they must be reacted directly in a way which eliminates this possibility of cycloreversion.
The Diels-Alder adducts, formed at high pressure, may start to dissociate at atmospheric pressure. If these adducts are required for synthetic purposes, therefore, they must be reacted directly in a way which eliminates this possibility of cycloreversion.
O
O
O+
O
O
O
H
H
O O
OH
H+
20 kbar
1 bar
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
ExoExo//endoendo--DiastereoselectivityDiastereoselectivity
The problem of both incomplete regioselectivity and endo/exodiastereoselectivity in the intermolecular Diels-Alder reactions arise because of the ability of diene and dienophile to react via different transition states of comparable energies.
The problem of both incomplete regioselectivity and endo/exodiastereoselectivity in the intermolecular Diels-Alder reactions arise because of the ability of diene and dienophile to react via different transition states of comparable energies.
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
Complete control of the regioselectivity in the Diels-Alder reaction can be exercised by linking the diene and dienophiletogether using a tether of limited length, i.e. comprised of a limited number of atoms. In practice, the tethers most often used contain three or four atoms with the diene linked at C-1 or C-4.
Complete control of the regioselectivity in the Diels-Alder reaction can be exercised by linking the diene and dienophiletogether using a tether of limited length, i.e. comprised of a limited number of atoms. In practice, the tethers most often used contain three or four atoms with the diene linked at C-1 or C-4.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
These intramolecular Diels-Alder (IMDA) reactions lead, in the all-carbon cases, to hydroindene or hydronaphthalenesystems.
These intramolecular Diels-Alder (IMDA) reactions lead, in the all-carbon cases, to hydroindene or hydronaphthalenesystems.
(CH2)n (CH2)n
It is important to be able to represent the transition states for these IMDA reactions adequately so that interactions of substituents on the diene, dienophile and tether can be appreciated.
It is important to be able to represent the transition states for these IMDA reactions adequately so that interactions of substituents on the diene, dienophile and tether can be appreciated.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
For the hydroindene system, the approach of diene and dienophile can be depicted as in the intermolecular versions.
For the hydroindene system, the approach of diene and dienophile can be depicted as in the intermolecular versions.
H HH
H
H
trans
H H
H
H
H
cis
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions

86
For the hydronaphthalene system it is often more appropriate to draw the corresponding transition states such that the chair-like perspective of the tether becomes apparent.
For the hydronaphthalene system it is often more appropriate to draw the corresponding transition states such that the chair-like perspective of the tether becomes apparent.
H
H
H
H
H
H
trans
H
H
H
H
H
H
cis
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
A single regioisomer is formed in these IMDA reactions (when n≤4) because the alternative bicyclo[n.3.1] products would be considerably more strained and the transition states leading to them would be correspondingly raised in energy. Although suitable tethering of the diene and dienophile will dictate the regioselectivity of the Diels-Alder reaction, it does not necessarily the exo/endo diastereoselectivity; either cis or trans-fused bicyclic compounds can be formed.
A single regioisomer is formed in these IMDA reactions (when n≤4) because the alternative bicyclo[n.3.1] products would be considerably more strained and the transition states leading to them would be correspondingly raised in energy. Although suitable tethering of the diene and dienophile will dictate the regioselectivity of the Diels-Alder reaction, it does not necessarily the exo/endo diastereoselectivity; either cis or trans-fused bicyclic compounds can be formed.
(CH2)n
H
H
n = 3, 4
(CH2)n
H
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
However, when a cycloaddition such as the Diels-Alder reaction is carried out intramolecularly, particularly with a tether of limited length, new steric and electronic interactions may come into play by comparison with the intermolecular version of the reaction. In general the difference between exo and endotransition-state energies will be augmented by such interactions and higher occasional diastereoselectivity will result.
However, when a cycloaddition such as the Diels-Alder reaction is carried out intramolecularly, particularly with a tether of limited length, new steric and electronic interactions may come into play by comparison with the intermolecular version of the reaction. In general the difference between exo and endotransition-state energies will be augmented by such interactions and higher occasional diastereoselectivity will result.
An addition benefit of intramolecularity is entropy derived: the probability of the diene and dienophile coming together is increased if they are appropriately linked. This will manifest itself in reaction at a lower temperature than is needed for analogous intermolecular reactions and, consequently, by and large, to greater exo/endo selectivity.
An addition benefit of intramolecularity is entropy derived: the probability of the diene and dienophile coming together is increased if they are appropriately linked. This will manifest itself in reaction at a lower temperature than is needed for analogous intermolecular reactions and, consequently, by and large, to greater exo/endo selectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
Secondary orbital interactions can favour endo diastereoselectivitywith a carbonyl-bonded substituent on a dienophile and that this often feeble interaction can be augmented by complexation of the carbonyl group with a Lewis acid. This same increase in endodiastereoselectivity can be brought about in an acid-catalysedIMDA reaction.
Secondary orbital interactions can favour endo diastereoselectivitywith a carbonyl-bonded substituent on a dienophile and that this often feeble interaction can be augmented by complexation of the carbonyl group with a Lewis acid. This same increase in endodiastereoselectivity can be brought about in an acid-catalysedIMDA reaction.
aH
b
HO
R
HOMO (diene)
LUMO (dienophile)
increase in orbital coefficient leading to greatersecondary interaction
without catalysis
aH
b
HOE+
R
with catalysis by E+
H
H
b
a
H
R
O
aH
b
HO
R
HOMO (diene)
LUMO (dienophile)
attractivesecondary interaction
primary interaction
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
The triene is cyclised thermally to cis- and trans- hydroindenes (A) and (B) in a 28:72 ratio, whereas in the presence of ethylaluminium dichloride as a catalyst, only the trans-fused product is obtained, in 80 % yield.
The triene is cyclised thermally to cis- and trans- hydroindenes (A) and (B) in a 28:72 ratio, whereas in the presence of ethylaluminium dichloride as a catalyst, only the trans-fused product is obtained, in 80 % yield.
80 %
CO2Me
EtAlCl223 oC, 48 h
O
MeO
Al-
H
H
H
O
MeO
H
H
HH
O
MeO
H
H
H
150 oC, 40 h72 %72 parts28 parts
HH
H
CO2Me
HH
H
CO2Me
H
五元环的张力决定选择性
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
The effect of a catalyst cannot, however, always be relied upon;the corresponding triene containing the cis-α,β-unsaturated ester likewise gives a mixture of diastereoisomers but their ratio is almost unaffected by carrying out the reaction in the presence of Lewis acids such as EtAlCl2.
The effect of a catalyst cannot, however, always be relied upon;the corresponding triene containing the cis-α,β-unsaturated ester likewise gives a mixture of diastereoisomers but their ratio is almost unaffected by carrying out the reaction in the presence of Lewis acids such as EtAlCl2.
MeO2C
HCO2Me
H
HCO2Me
H
+
180 oC, 5 h 67 : 33
63 : 37Et2AlCl, 23 oC, 40 h
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions

87
Higher endo diastereoselectivity appears to be more common in the formation of cis-hydronaphthalenes than cis-hydroindenes; possibly the four atom tether gives rise to a marginally looser transition states which allows more efficient secondary orbital interaction (a). The Lewis acid-catalysed reaction in (b) also gives the endo (carbonyl) adduct.
Higher endo diastereoselectivity appears to be more common in the formation of cis-hydronaphthalenes than cis-hydroindenes; possibly the four atom tether gives rise to a marginally looser transition states which allows more efficient secondary orbital interaction (a). The Lewis acid-catalysed reaction in (b) also gives the endo (carbonyl) adduct.
OSiEt3
H
HO
H
H
O
KF, MeOH, 0 oC
H
HO
(a)
六元环的柔性相对较大,endo选择性Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
O
O
O
H
HO
KF, MeOH, 0 oC(b)
H
O
H
HOAl 67 %
H
O
H
HO
E-diene
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
RHH
R
H
H
+
)(
H
H
R
H
H
R H)(R = H, 220 oC, 48 : 52R = Me, 160 oC, 94 : 6
trans cis
B
A
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
The exo/endo ratio in these IMDA reactions can be influenced by steric interactions between substituents in the diene and the tether (including ‘axial’ hydrogens). The steric interaction between an alkyl group R and the axial hydrogen shown in (A) is greater than that between R and the axial hydrogen shown in (B), so exo becomes the preferred mode of addition.
The exo/endo ratio in these IMDA reactions can be influenced by steric interactions between substituents in the diene and the tether (including ‘axial’ hydrogens). The steric interaction between an alkyl group R and the axial hydrogen shown in (A) is greater than that between R and the axial hydrogen shown in (B), so exo becomes the preferred mode of addition.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
The presence of a heteroatom in the tether can also affect the exo/endo ratio. Complete diastereoselectivity arises from the preference for endo transition state (A) in which overlap of the urethane and diene systems is largely conserved and the A1,3-strain present in (A) is absent.
The presence of a heteroatom in the tether can also affect the exo/endo ratio. Complete diastereoselectivity arises from the preference for endo transition state (A) in which overlap of the urethane and diene systems is largely conserved and the A1,3-strain present in (A) is absent.
N
CO2Me
190 oC, 16 h N
MeO OH
H N
CO2MeH
H
endo
N
MeO OH)(
A
B
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
Heating the triene, whose diene component is linked to the dienophile via a non-terminal carbon atom, gave the oxabicyclo[4.3.1], which contains a bridgehead double bond. The tightness of the transition state here ensures that the reaction is completely diastereoselective (exo-ethoxycarbonyl) and, of course, completely regioselective.
Heating the triene, whose diene component is linked to the dienophile via a non-terminal carbon atom, gave the oxabicyclo[4.3.1], which contains a bridgehead double bond. The tightness of the transition state here ensures that the reaction is completely diastereoselective (exo-ethoxycarbonyl) and, of course, completely regioselective.
MeO O
HH
EtO2C
180 oC78 %
MeO O
HH
EtO2C 1. Pt/H22. NaOEt, EtOH
MeO
HH
EtO2C
CO2Et
?
MeOR
EtO2C
HH
EtO2C
(A)
(B)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions

88
Reduction of the double bond in (A) takes place exclusively fromthe exo face and ring opening give (B). To appreciate the selectivity inherent in this synthesis of (B) one need only consider the problems that would arise in its attempted synthesis starting with the intermolecular Diels-Alder reactions.
Reduction of the double bond in (A) takes place exclusively fromthe exo face and ring opening give (B). To appreciate the selectivity inherent in this synthesis of (B) one need only consider the problems that would arise in its attempted synthesis starting with the intermolecular Diels-Alder reactions.
Linking together the two partners in acycloaddition reaction by a tether designed to be easily broken is valuable for the stereo-selective synthesis of both cyclic and acyclic target molecules.
Linking together the two partners in acycloaddition reaction by a tether designed to be easily broken is valuable for the stereo-selective synthesis of both cyclic and acyclic target molecules.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Intramolecular DielsType 2 Intramolecular Diels--Alder (IMDA) ReactionsAlder (IMDA) Reactions
Replacement of one or more atoms in the all-carbon Diels-Alder reaction by heteroatoms gives rise to six-membered-ring hetereocycles. The double bonds of hetereoatom-containing dienophiles include C=S, C=N, C=O, N=O, O=O, N=S=O and N=N and hetereoatom-containing dienes.
Replacement of one or more atoms in the all-carbon Diels-Alder reaction by heteroatoms gives rise to six-membered-ring hetereocycles. The double bonds of hetereoatom-containing dienophiles include C=S, C=N, C=O, N=O, O=O, N=S=O and N=N and hetereoatom-containing dienes.
HetereocyclesHetereocycles via Dielsvia Diels--Alder reactionsAlder reactions
O NRN
S
Of particular interest in stereoselective synthesis are the reactions of electron-rich dienes with carbonyl groups (usually aldehydes) as the dienophile. These reactions are catalysed by Lewis acids, e.g. zinc chloride or europium salts, and are highly endo diastereoselective.
Of particular interest in stereoselective synthesis are the reactions of electron-rich dienes with carbonyl groups (usually aldehydes) as the dienophile. These reactions are catalysed by Lewis acids, e.g. zinc chloride or europium salts, and are highly endo diastereoselective.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
66 %TMSO
Me
OMe
H
OMeHMe H
Eu(fod)3
TMSOMe
OMe
H
Me
OMeH
H
Et3N, MeOH
O
Me
MeMe
OMeO
TFA, Et2O
O
Me
MeMe
O
Eu(fod)3, 0.5-5mol%
TMSOMe
OMe
MeCH3CHO
CHCl3
O
OCF3
tBu
fod
Eu试剂促使Me占endo
Me占e键,孤对电子占a键
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HetereocyclesHetereocycles via Dielsvia Diels--Alder reactionsAlder reactions
The occasional diastereoselectivity is dominated by the large bulk of the europium with its associated ligands; its coordination to the carbonyl group anti to the methyl and its exo placement in the transition state to minimise steric effects lead to an endoplacement of the aldehyde methyl group.
The occasional diastereoselectivity is dominated by the large bulk of the europium with its associated ligands; its coordination to the carbonyl group anti to the methyl and its exo placement in the transition state to minimise steric effects lead to an endoplacement of the aldehyde methyl group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HetereocyclesHetereocycles via Dielsvia Diels--Alder reactionsAlder reactions
The 2-methoxy substituent in the adduct can be retained or eliminated, depending on the work-up, and the products are related to natural sugars.
The 2-methoxy substituent in the adduct can be retained or eliminated, depending on the work-up, and the products are related to natural sugars.
The ability of other metal ions (Ti4+, Mg2+) to chelate α- and β-alkoxyaldehydes allows diastereoselective cycloaddtion of aldehydes with chiral centres α to the carbonyl group.
The ability of other metal ions (Ti4+, Mg2+) to chelate α- and β-alkoxyaldehydes allows diastereoselective cycloaddtion of aldehydes with chiral centres α to the carbonyl group.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
HetereocyclesHetereocycles via Dielsvia Diels--Alder reactionsAlder reactions Type 2 1,3Type 2 1,3--dipolar dipolar cycloadditionscycloadditions
Like Diels-Alder reactions, 1,3-dipolar cycloadditions involve 4π + 2π concerted reaction of a 1,3-dipolar species (the 4πcomponent) and a dipolarophile (the 2π component).
Like Diels-Alder reactions, 1,3-dipolar cycloadditions involve 4π + 2π concerted reaction of a 1,3-dipolar species (the 4πcomponent) and a dipolarophile (the 2π component).
1,3-Dipoles can be divided into two classes:
1. Those in which three atoms comprising the 1,3-dipole are
linear;
2. Those in which they are not;
1,3-Dipoles can be divided into two classes:
1. Those in which three atoms comprising the 1,3-dipole are
linear;
2. Those in which they are not;
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

89
azomethine ylide
diazoalkane
nitroneazide
carbonyl ylide
nitrile ylide
ozonenitrile oxideBentlinear
C N O-+
C N C+ _
N N N+ _
N N C+ _
Table 1. Example of 1,3-dipolesTable 1. Example of 1,3-dipoles
O OO
- -
C CO
-+
C ON
-+
C CN
-+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 1,3Type 2 1,3--dipolar dipolar cycloadditionscycloadditions
Since the products of these cycloadditions are five-membered rings in which the 1,3-dipolar residue must be bent, one might expect the linear type to be less reactive than those which are already bent. This, however, does not seem to be important; the energy required to bend a linear dipole in the transition state is apparently relatively small.
Since the products of these cycloadditions are five-membered rings in which the 1,3-dipolar residue must be bent, one might expect the linear type to be less reactive than those which are already bent. This, however, does not seem to be important; the energy required to bend a linear dipole in the transition state is apparently relatively small.
a cb
-+ + x y cb
ax y
cb
ay x
+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 1,3Type 2 1,3--dipolar dipolar cycloadditionscycloadditions
As shown in this scheme, with an unsymmetrical 1,3-dipole and dipolarophile, the possibility of regioisomers arises. The major regioisomer formed in this case can be predicted using the same procedure applied to the Diels-Alder reaction-the HOMO/ LUMO pair closer in energy is identified and, within this pair, those terminal atoms on the dipole and dipolarophile bearing the largest orbital coefficients became bonded.
As shown in this scheme, with an unsymmetrical 1,3-dipole and dipolarophile, the possibility of regioisomers arises. The major regioisomer formed in this case can be predicted using the same procedure applied to the Diels-Alder reaction-the HOMO/ LUMO pair closer in energy is identified and, within this pair, those terminal atoms on the dipole and dipolarophile bearing the largest orbital coefficients became bonded.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 1,3Type 2 1,3--dipolar dipolar cycloadditionscycloadditions
Steric effects may control the sense of regioselectivity; nitrile oxides give adducts in which the oxygen atom becomes bonded to the more hindered end of the dipolarophile in the major regioisomer.
Steric effects may control the sense of regioselectivity; nitrile oxides give adducts in which the oxygen atom becomes bonded to the more hindered end of the dipolarophile in the major regioisomer.
+ NOtBu
Me Ph
tBu
Me
N+
Ph
O-
N
Ph
ClHO
Et3N
Some linear 1,3-dipoles, e.g. nitrile oxides, are not prochiral in these cycloadditions and so the only chiral centres in the product are those derived from syn addition to the dipolarophile.
Some linear 1,3-dipoles, e.g. nitrile oxides, are not prochiral in these cycloadditions and so the only chiral centres in the product are those derived from syn addition to the dipolarophile.
Steric effect大基团和O相邻
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 1,3Type 2 1,3--dipolar dipolar cycloadditionscycloadditions
Nitrile oxides are unstable and must be generated in situ. In the reaction in next scheme, this carried out by thermolysis of the dimer (A), which is in equilibrium with a small concentration of the monomer (B). The relatively high temperature allows cycloaddition of (B) not only to 1,2-disubstituted alkenes but also to trisubstituted alkenes, which are normally less reactive.
Nitrile oxides are unstable and must be generated in situ. In the reaction in next scheme, this carried out by thermolysis of the dimer (A), which is in equilibrium with a small concentration of the monomer (B). The relatively high temperature allows cycloaddition of (B) not only to 1,2-disubstituted alkenes but also to trisubstituted alkenes, which are normally less reactive.
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
ON
N
O-
OTMS
OTMS+135-165 oC
OTMSN
O-OTMS
O2N
PhN C OC6H6
Me
H
Me
MeO2C
A
2.2 equiv.
OTMSN
O-
Me
H
Me
MeO2C
NO OTMS
HMeO2CMe Me
B
脱水试剂, PhNH2 CO2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

90
For prochiral 1,3-dipole, there can be prochiral syn addition to 1,3-dipole and dipolarophile, as in the Diels-Alder reaction (stereospecificity, inherent diastereoselectivity). However, unlike the Diels-Alder reaction in which the diene is invariably configurationally stable, the barrier to configurational inter-conversion (stereomutation) in bent 1,3-dipoles may be low as in, for example, azomethine ylides.
For prochiral 1,3-dipole, there can be prochiral syn addition to 1,3-dipole and dipolarophile, as in the Diels-Alder reaction (stereospecificity, inherent diastereoselectivity). However, unlike the Diels-Alder reaction in which the diene is invariably configurationally stable, the barrier to configurational inter-conversion (stereomutation) in bent 1,3-dipoles may be low as in, for example, azomethine ylides.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Na
b y
x
R
-+ -a
b
N
y
x
R
+ -a
b
N
x
y
R
+
N
R
xab y N
R
yab x
Na
b x
y
R
-+
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Na
b y
x
R
-+ -a
b
N
y
x
R
+ -a
b
N
x
y
R
+
N
R
xab y N
R
yab x
Na
b x
y
R
-+
A B
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Consequently, a mixture of diastereoisomeric cycloadductsmay be obtained (with each of the stereoisomeric 1,3-dipolar species A and B reacting with complete syn selectivity). Using unreactive dipolarophiles, this may be the result even if the 1,3-dipolar species was initially generated as a single diastereoisomer.
Consequently, a mixture of diastereoisomeric cycloadductsmay be obtained (with each of the stereoisomeric 1,3-dipolar species A and B reacting with complete syn selectivity). Using unreactive dipolarophiles, this may be the result even if the 1,3-dipolar species was initially generated as a single diastereoisomer.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
However, for cycloaddition of prochiral 1,3-dipoles which react via a single configuration with a prochiral dipolarophile the consequence are analogous to those in the Diels-Alder reaction; four (racemic) products may be formed when regioisomers are also considered.
However, for cycloaddition of prochiral 1,3-dipoles which react via a single configuration with a prochiral dipolarophile the consequence are analogous to those in the Diels-Alder reaction; four (racemic) products may be formed when regioisomers are also considered.
endo/exo (occasional) Diastereoselectivity in 1,3-dipolar cycloaddition is, as in the Diels-Alder reaction, determined by the preference of the dipolarophile substituents for the endo or exo position. However, it appears that secondary orbital interactions which usually favour placement of, e.g. carbonyl substituents in the endo position in the Diels-Alder reactions, are much weaker in the corresponding 1,3-dipolar cycloaddition, if they are present at all.
endo/exo (occasional) Diastereoselectivity in 1,3-dipolar cycloaddition is, as in the Diels-Alder reaction, determined by the preference of the dipolarophile substituents for the endo or exo position. However, it appears that secondary orbital interactions which usually favour placement of, e.g. carbonyl substituents in the endo position in the Diels-Alder reactions, are much weaker in the corresponding 1,3-dipolar cycloaddition, if they are present at all.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Like nitrile oxides, azomethine ylides must be also be generated in situ and some of the different ways in which this can be done are shown.
Like nitrile oxides, azomethine ylides must be also be generated in situ and some of the different ways in which this can be done are shown.
(a) aziridine ring opening(a) aziridine ring opening
ArNH
MeO2C
ArNH
MeO2C
+
_
OO
O ArN
OO
O
H
MeO2CHH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions

91
(b) α-amino acid decarboxylation(b) α-amino acid decarboxylation
N CO2HH
+
O
O
OMeOH
OO
OH2+
N-O2C
OO
N+_
CO2MeHN
O N CO2Me_
+HN
O NCO2Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
(c) 1,2-prototropy
PhCHN
CO2Me
CHPh 105 oCC C
NPh
H Ph
CO2Me
H
-+ NPh
OO
NPhO
OHN
H
H
HPh
MeO2C Ph
(d) fluorosesilylation
N
Me
CH2SiMe3
AgF
N
Me
CH2-
Ag
+
MeO2C CO2Me
N
Me
CO2Me
CO2Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Examples of diastereoselective nitrone and carbonyl ylide 1,3-dipolar cycloadditions.
Examples of diastereoselective nitrone and carbonyl ylide 1,3-dipolar cycloadditions.
N
O-
+ + Phtoluenereflux
+
H
Ph
NH
O-
NO
HCH2CH2Ph
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
CHO
O
N2
Rh2(OAc)2CHO
O
+ _CHO
O+
_
O OO
O
OO
O
OH
H
CH3 O
O
CH3
O
O
OH
H
+_
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SetereochemistrySetereochemistry of 1,3of 1,3--Dipolar Dipolar CycloadditionsCycloadditions
Intramolecular 1,3Intramolecular 1,3--Dipolar Dipolar CycloadditionsCycloadditions
The weakened endo-directing secondary orbital interaction in the transition state for 1,3-dipolar cycloaddition means that selectivity in the endo sense is reduced and may easily be overridden by other effects leading to exo diastereoselectivity. High regioselectivity also cannot be relied upon and in any case, where it obtains, the alternative regioisomer may be the one that is actually desired.
The weakened endo-directing secondary orbital interaction in the transition state for 1,3-dipolar cycloaddition means that selectivity in the endo sense is reduced and may easily be overridden by other effects leading to exo diastereoselectivity. High regioselectivity also cannot be relied upon and in any case, where it obtains, the alternative regioisomer may be the one that is actually desired.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Intramolecularity in 1,3-dipolar cycloadditions is an especially valuable device for directing the regiochemical course of the reaction, and often results in enhanced occasional (exo/endo) selectivity also. The latter is usually the result of the limited length of the tether, coupled with non-bonded interactions of substituents on the tether and the 1,3-dipole or dipolarophile. Electronic effects resulting from enforced orientations of substituents may also be introduced or magnified in these intramolecular 1,3-dipolar cycloadditions.
Intramolecularity in 1,3-dipolar cycloadditions is an especially valuable device for directing the regiochemical course of the reaction, and often results in enhanced occasional (exo/endo) selectivity also. The latter is usually the result of the limited length of the tether, coupled with non-bonded interactions of substituents on the tether and the 1,3-dipole or dipolarophile. Electronic effects resulting from enforced orientations of substituents may also be introduced or magnified in these intramolecular 1,3-dipolar cycloadditions.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Intramolecular 1,3Intramolecular 1,3--Dipolar Dipolar CycloadditionsCycloadditions

92
Some examples of completely diastereoselective and regiospecificintramolecular 1,3-dipolar cycloadditions:
Some examples of completely diastereoselective and regiospecificintramolecular 1,3-dipolar cycloadditions:
MeO
OMe
CHOMeNHCH2CO2Et
toluene MeO
OMe
H
NCO2Et
HHMe
_+
NMeO
MeO
H
H
CO2Et
H
Me
89 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Intramolecular 1,3Intramolecular 1,3--Dipolar Dipolar CycloadditionsCycloadditions
OS
ArNHOH
S
H
ArNO-
H+
S
H
ArNO
H
R-Ni
74 %
Me
H
H
MeArNH
OH
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Intramolecular 1,3Intramolecular 1,3--Dipolar Dipolar CycloadditionsCycloadditions
H H
diene
dienophile
H H
enophile
The The EneEne ReactionsReactions
The ene and the Diels-Alder reactions are related pericyclicreactions which frequently co-occur in the reaction of a dienebearing an allylic C-H bond (an enecomponent) with dienophile.
The ene and the Diels-Alder reactions are related pericyclicreactions which frequently co-occur in the reaction of a dienebearing an allylic C-H bond (an enecomponent) with dienophile.
A
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
The inherent diastereoselectivity (stereospecificity) in the enereaction is evident from inspection of (A): both bonds are made syn to the enophile (the configuration of the enophile is retained in the product) and the bond made to the double bond of the enecomponent is syn to the hydrogen which is transferred.
The inherent diastereoselectivity (stereospecificity) in the enereaction is evident from inspection of (A): both bonds are made syn to the enophile (the configuration of the enophile is retained in the product) and the bond made to the double bond of the enecomponent is syn to the hydrogen which is transferred.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The role of the enophile can be assumed by the same double bonds that can function as dienophiles in the Diels-Alder reaction, e.g. C=O, C=N, C=S, N=N, N=O as well as C=C. Preparatively, aldehydes are particularly useful as enophilesbecause their reactivity is greatly increased by complexationwith Lewis acids, allowing them to be used at low temperatures. The products using aldehydes as enophiles are homoallylic alcohols, i.e. the ene reaction brings about the same transformation as is accomplished by using allylmetaladdition to the aldehyde.
The role of the enophile can be assumed by the same double bonds that can function as dienophiles in the Diels-Alder reaction, e.g. C=O, C=N, C=S, N=N, N=O as well as C=C. Preparatively, aldehydes are particularly useful as enophilesbecause their reactivity is greatly increased by complexationwith Lewis acids, allowing them to be used at low temperatures. The products using aldehydes as enophiles are homoallylic alcohols, i.e. the ene reaction brings about the same transformation as is accomplished by using allylmetaladdition to the aldehyde.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
H
OH
R
H
O
HR
ene using aldehyde
M
OH
R
M
O
HR
ene using aldehyde
H+
H
O
HR
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions

93
O
HH
HH
H H
HH
H
H
HH
HH
H
HO
43 o
39 o
The transition-state geometries of the lowest energy for the enereactions of propene with ethylene and of propene with formaldehyde have been calculated by Loncharich and Houkand found to be very similar. For the carbonyl-ene reaction, the substituents on carbons 1 and 1’ are staggered, as illustrated in the Newman projection along with the (forming) 1-1’ carbon-carbon formation.
The transition-state geometries of the lowest energy for the enereactions of propene with ethylene and of propene with formaldehyde have been calculated by Loncharich and Houkand found to be very similar. For the carbonyl-ene reaction, the substituents on carbons 1 and 1’ are staggered, as illustrated in the Newman projection along with the (forming) 1-1’ carbon-carbon formation.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
The most important stereochemical features of the Type 2 enereaction by reference to the Lewis acid-catalysed reactions of aldehydes with allyl systems shall be illustrated. With some combinations of substituted ene/enophile the mechanism of the catalysed reaction may involve ion-pair intermediates, but this does not appear to be important for the reactions discussed below.
The most important stereochemical features of the Type 2 enereaction by reference to the Lewis acid-catalysed reactions of aldehydes with allyl systems shall be illustrated. With some combinations of substituted ene/enophile the mechanism of the catalysed reaction may involve ion-pair intermediates, but this does not appear to be important for the reactions discussed below.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
O
HR
H
H H
Ha
b
ALn R
ba
HOH
Ra
HO
b
H
O
RH
H
H H
H
ab
ALn H
ba
ROH
Ra
H
b
OH
A endo
B exo
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
In the reaction of a 1,1-disubstituted propene with an aldehyde, two transition states, exo and endo, are possible. In these transition states, the carbonyl oxygen is assumed to be sp2-hybridised with the Lewis acid ALn, complexed to the lone pair syn to the aldehyde hydrogen to minimise steric effects.
In the reaction of a 1,1-disubstituted propene with an aldehyde, two transition states, exo and endo, are possible. In these transition states, the carbonyl oxygen is assumed to be sp2-hybridised with the Lewis acid ALn, complexed to the lone pair syn to the aldehyde hydrogen to minimise steric effects.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
The major factors which control the occasional (exo/endo) diastereoselectivity in these ene reactions are (i) stericinteractions between R and a in (A) and between R and b in (B) and (ii) steric interactions between the Lewis acid ALn and substituents on the ene component.
The major factors which control the occasional (exo/endo) diastereoselectivity in these ene reactions are (i) stericinteractions between R and a in (A) and between R and b in (B) and (ii) steric interactions between the Lewis acid ALn and substituents on the ene component.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
O
HMeO2C
H
H H
H
HMe
AlMe2OTf
CO2MeH
HO
Me
H
O
CO2MeH
H
H H
H
HMe
TfOMe2lAH
MeH
CO2MeOH
CO2MeH
H
Me
OH
-78 oC
CH2Cl2
-78 oCCH2Cl2
MeO2C
MeH
HOH
+
9 : 91
65 %
(A)(B)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions

94
Methyl glyoxylate reacts with an excess of (Z)-but-2-ene in the presence of AlMe2OTf to give a 9:91 ratio of diastereoisomers resulting from endo and exo addition, respectively. This is rationalised on the basis of greater steric interaction between substituents on the coordinated aluminium and the ene methyl group in (A) than between the methoxycarbonyl group and the same methyl group in (B).
Methyl glyoxylate reacts with an excess of (Z)-but-2-ene in the presence of AlMe2OTf to give a 9:91 ratio of diastereoisomers resulting from endo and exo addition, respectively. This is rationalised on the basis of greater steric interaction between substituents on the coordinated aluminium and the ene methyl group in (A) than between the methoxycarbonyl group and the same methyl group in (B).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
Catalysis of the ene reaction of alkyl glyoxylates by tin(IV) chloride is different because the tin atom is able to complex with the oxygens of both carbonyl groups. Reaction with (E)-but-2-ene gives mainly trans-diastereoisomers (A) because of greater repulsion between the Lewis acid and the methyl group in (B). Cis and trans refer to the relative positions of the substituents(Me, OH) with the lonest chain drawn in the zig-zagconformation; the hydrogens at these chiral centres are omitted for clarity.
Catalysis of the ene reaction of alkyl glyoxylates by tin(IV) chloride is different because the tin atom is able to complex with the oxygens of both carbonyl groups. Reaction with (E)-but-2-ene gives mainly trans-diastereoisomers (A) because of greater repulsion between the Lewis acid and the methyl group in (B). Cis and trans refer to the relative positions of the substituents(Me, OH) with the lonest chain drawn in the zig-zagconformation; the hydrogens at these chiral centres are omitted for clarity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
OH
H
H H
H
MeH
SnCl4O
MeO
CO2MeMe
HO
H
H
H
HMe
CO2MeOH
CO2MeMe
H
H
OH
-78 oC
CH2Cl2
-78 oCCH2Cl2
MeO2C
MeH
HOH
A
O
H
H H
H
MeH
H
O
MeO
Cl4Sn
C B A : B = 92 : 8
构型相反
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
The same trans isomer is the major diastereoisomer obtained when (Z)-but-2-ene is used, although in this case the diastereoselectively is not high; probably there is some interaction between the olefinic proton and the substituents on the tin.
The same trans isomer is the major diastereoisomer obtained when (Z)-but-2-ene is used, although in this case the diastereoselectively is not high; probably there is some interaction between the olefinic proton and the substituents on the tin.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
CO2MeH
HO
Me
H
-78 oCCH2Cl2
MeO2C
MeH
HOH
O
H
H H
H
HMe
H
O
MeO
Cl4Sn
trans : cis = 72 : 28
On the other hand, this 2-trimethylsilyl substituent brings about a reversal of the sense of diastereoselectivity using the 1,2-cis-dimethyl isomer.
On the other hand, this 2-trimethylsilyl substituent brings about a reversal of the sense of diastereoselectivity using the 1,2-cis-dimethyl isomer.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions
TMS
trans:cis = 98:2
OH
TMS
H H
H
MeH
SnCl4O
MeO
H
HMe
CO2MeOH
TMS
CO2MeMe
H
H
OH
-78 oC
CH2Cl2
A
The presence of an additional substituent on the enecomponent can further increase the diastereoselectivity.
The presence of an additional substituent on the enecomponent can further increase the diastereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions

95
TMS
trans:cis = 7:93
OH
TMS
H H
H
HMe
SnCl4O
MeO
H
MeH
CO2MeOH
TMS
CO2MeH
H
Me
OH
-78 oC
CH2Cl2
A
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The The EneEne ReactionsReactions RegioselectivityRegioselectivity in The in The EneEne ReactionsReactions
Two major drawbacks to the use of the intermolecular ene reaction in synthesis have been the high temperatures previously needed for reaction and the lack of regioselectivity generally exhibited in such cases. The discovery that Lewis acid catalysis enables the intermolecular carbonyl ene reaction to be accomplished at temperatures as low as –78 oC with beneficial effects on the diastereoselectivity has stimulated efforts to bring about regioselectivity in the reaction.
Two major drawbacks to the use of the intermolecular ene reaction in synthesis have been the high temperatures previously needed for reaction and the lack of regioselectivity generally exhibited in such cases. The discovery that Lewis acid catalysis enables the intermolecular carbonyl ene reaction to be accomplished at temperatures as low as –78 oC with beneficial effects on the diastereoselectivity has stimulated efforts to bring about regioselectivity in the reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Me
RO+
O
H CO2Me
SnCl4 -78 oC
CH2Cl2RO
OH
CO2Me
R = Si(CH3)(t-Bu), CH2Ph, Ph
As usual, this can be accomplished by carrying out the reaction intramolecularly, but work by Nakaihas shown that, in an intermolecular reaction, an allylic oxygen or even a homoallylic oxygen can retard hydrogen transfer from this allyllic position, thereby favouring transfer from alternative allylic position.
As usual, this can be accomplished by carrying out the reaction intramolecularly, but work by Nakaihas shown that, in an intermolecular reaction, an allylic oxygen or even a homoallylic oxygen can retard hydrogen transfer from this allyllic position, thereby favouring transfer from alternative allylic position.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in The in The EneEne ReactionsReactions
CH2
RO Se+
H
R
O-RO
Hnot
H
OR
The effect seems to be partly steric and partly electronic on origin and is reminiscent of the complete regioselectivitywhich obtains in the syn elimination reactions of β-alkoxyalkylselenoxides.
The effect seems to be partly steric and partly electronic on origin and is reminiscent of the complete regioselectivitywhich obtains in the syn elimination reactions of β-alkoxyalkylselenoxides.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in The in The EneEne ReactionsReactions
trans 99%
OH
H
Me H
HH
SnCl4O
MeO
SiO
H
H
CO2MeOH
Me
SiO CO2Me
H OHMe
SiO
-78 oC
CH2Cl2
A
Thus a Type 2 ene reaction using the silyloxy-substituted (E)-but-2-ene with methyl glyoxylate and tin(IV) chloride is both highly regio- and diastereoselective.
Thus a Type 2 ene reaction using the silyloxy-substituted (E)-but-2-ene with methyl glyoxylate and tin(IV) chloride is both highly regio- and diastereoselective.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in The in The EneEne ReactionsReactions
This scheme also shows that when the hydrogen which is transferred in the ene reaction is derived from a secondary centre (as opposed to a methyl group in the schemes above), a configured double bond is formed, which, in the absence of a substituent at the 2-position on the ene component, is the parent ene compenent.
This scheme also shows that when the hydrogen which is transferred in the ene reaction is derived from a secondary centre (as opposed to a methyl group in the schemes above), a configured double bond is formed, which, in the absence of a substituent at the 2-position on the ene component, is the parent ene compenent.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
RegioselectivityRegioselectivity in The in The EneEne ReactionsReactions

96
SummarySummary
The Type 2 reactions considered in this chapter are those which are in part stereospecific (inherently diasteroselectivite). Thus, syn additions of a chiral regents XY to configured double bonds give single daistereoisomers as a result of the mechanism of the reaction in which both chiral centres are created more or less simultaneously: overall anti additions result from Type 1 reactions on these syn addition products.
The Type 2 reactions considered in this chapter are those which are in part stereospecific (inherently diasteroselectivite). Thus, syn additions of a chiral regents XY to configured double bonds give single daistereoisomers as a result of the mechanism of the reaction in which both chiral centres are created more or less simultaneously: overall anti additions result from Type 1 reactions on these syn addition products.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Likewise, cycloaddition reactions, e.g. the Diels-Alder reaction, also contain an inherently dastereoselective element arising from the reciprocal syn (suprafacial) addition of the components. However, these cycloaddition reactions may also be diastereoselective in another sense (exo/endo for the Diels-Alder reaction) which is occasional, I.e. depends on the substitution of the interacting components and, unlike the inherent diastereoselectivity, will not be complete in most cases.
Likewise, cycloaddition reactions, e.g. the Diels-Alder reaction, also contain an inherently dastereoselective element arising from the reciprocal syn (suprafacial) addition of the components. However, these cycloaddition reactions may also be diastereoselective in another sense (exo/endo for the Diels-Alder reaction) which is occasional, I.e. depends on the substitution of the interacting components and, unlike the inherent diastereoselectivity, will not be complete in most cases.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Part 2Part 2
Type 2 Reactions:
Occasional Diastereoselectivity
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
OutlineOutline
1. Type 2 2π + 2π cycloadditions;2. Type 2 photochemical 1,3-cycloaddition of arenes;3. Photochemistry in crystals;4. Other 2π + 2π cycloadditions;5. Other cycloadditions;6. 1,4-Cheletropic additions to 1,3-dienes;7. Other 1,4-additions to 1,3-dienes;8. [3,3] Sigmatropic rearrangements;9. [2,3] Sigmatropic rearrangements10. Summary
1. Type 2 2π + 2π cycloadditions;2. Type 2 photochemical 1,3-cycloaddition of arenes;3. Photochemistry in crystals;4. Other 2π + 2π cycloadditions;5. Other cycloadditions;6. 1,4-Cheletropic additions to 1,3-dienes;7. Other 1,4-additions to 1,3-dienes;8. [3,3] Sigmatropic rearrangements;9. [2,3] Sigmatropic rearrangements10. Summary
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
In this part, Type 2 reactions will be considered whose diastereoselectivity is in the main brought about by the particular substitution on the interacting components, i.e. it is occasional rather than inherent in the mechanism of the reaction.
In this part, Type 2 reactions will be considered whose diastereoselectivity is in the main brought about by the particular substitution on the interacting components, i.e. it is occasional rather than inherent in the mechanism of the reaction.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 ReactionsType 2 Reactions
From orbital symmetry consideration, concerted cycloaddition of two alkenes to give a cyclobutane is allowed suprafacially-suprafacially, i.e. syn with respect to both alkenes, provided that one reacts via its singlet excited state. Unfortunately, in practice there are number of factors which conspire to reduce the theoretical diastereoselective promise of this reaction, at least with acyclic alkenes.
1) the tendency for alkenes to undergo photochemical trans/cis (E/Z) diastereoisomerism.
2) in many intermolecular reactions, the photoexcited state involved is a triplet, from which concerted cycloaddition is precluded because of the necessity for spin inversion.
From orbital symmetry consideration, concerted cycloaddition of two alkenes to give a cyclobutane is allowed suprafacially-suprafacially, i.e. syn with respect to both alkenes, provided that one reacts via its singlet excited state. Unfortunately, in practice there are number of factors which conspire to reduce the theoretical diastereoselective promise of this reaction, at least with acyclic alkenes.
1) the tendency for alkenes to undergo photochemical trans/cis (E/Z) diastereoisomerism.
2) in many intermolecular reactions, the photoexcited state involved is a triplet, from which concerted cycloaddition is precluded because of the necessity for spin inversion.
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

97
In general, therefore, photochemical 2π + 2π cycloadditionsdo not have an inherent diastereoselectivity of the kind that is present in, e.g., Diels-Alder additions. However, if appropriate constraints are applied, this cycloaddition can display high occasional diastereoselectivity. Thus, photochemical E/Z diastereoisomerism can be eliminated by incorporation of the alkene into a ring of sufficiently small size (five-membered or less). By carrying out the reaction intramolecularly, there is a greater chance of reaction occurring via a singlet state and regioselectivity is more likely to be obtained using a tether of an appropriate length.
In general, therefore, photochemical 2π + 2π cycloadditionsdo not have an inherent diastereoselectivity of the kind that is present in, e.g., Diels-Alder additions. However, if appropriate constraints are applied, this cycloaddition can display high occasional diastereoselectivity. Thus, photochemical E/Z diastereoisomerism can be eliminated by incorporation of the alkene into a ring of sufficiently small size (five-membered or less). By carrying out the reaction intramolecularly, there is a greater chance of reaction occurring via a singlet state and regioselectivity is more likely to be obtained using a tether of an appropriate length.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
When both alkenes are contained in rings (n ≤ 5) the cycloadditionis invariably syn on both of them although exo and endo products may still be formed as in the photochemical dimerisation of maleicanhydride. With unsymmetrical cyclic alkenes, head-to-head or head-to-tail dimerisation (regioisomerism) is also possible (in addition to exo/endo addition).
When both alkenes are contained in rings (n ≤ 5) the cycloadditionis invariably syn on both of them although exo and endo products may still be formed as in the photochemical dimerisation of maleicanhydride. With unsymmetrical cyclic alkenes, head-to-head or head-to-tail dimerisation (regioisomerism) is also possible (in addition to exo/endo addition).
O
O
O
hv
O
O
O
O
O
O
H
H
HO
O
O
O
O
O
H
HH
+
endo exo
O
hv+
O
OOO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Most stereoselective synthesis which make use of photochemical 2π + 2πreactions take advantage of the E/Z configurational stability of cyclic alkenes. The use of α,β-unsaturated carbonyl compounds with their longer wavelength absorption allows their selective excitation in the presence of the alkenes.
Most stereoselective synthesis which make use of photochemical 2π + 2πreactions take advantage of the E/Z configurational stability of cyclic alkenes. The use of α,β-unsaturated carbonyl compounds with their longer wavelength absorption allows their selective excitation in the presence of the alkenes.
CN OEt
+hv
95 %
CN
H
OEt
OOSiMe2
tBuMe hv, Pyrex, hexane68 %
OOSiMe2
tBu
Me H+HO
O
Me
Retro-aldol reactionand a aldol reaction
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Ring opening of the strained cyclobutane intermediate or product is also a pervasive feature of syntheses involving these 2π + 2πreactions photochemical cycloadditions as in the de Mayo reaction.
Ring opening of the strained cyclobutane intermediate or product is also a pervasive feature of syntheses involving these 2π + 2πreactions photochemical cycloadditions as in the de Mayo reaction.
Me
Me
+O
OH
Me
hv, pentaneMe
Me
OH
Me
O
CHO
Me
MeMe
O
Me
MeO
Retro-aldol
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
The direct creation of two adjacent quaternary chiral centres diastereoselectivity, by syn addition to the alkene C=C bond with the formation of two C-C bonds, is a conversion for which there are few other methods available. Any lack of diastereoselectivityin addition to the enol ether moiety is immaterial since retroaldolisation destroys the chirality at these two centres
The direct creation of two adjacent quaternary chiral centres diastereoselectivity, by syn addition to the alkene C=C bond with the formation of two C-C bonds, is a conversion for which there are few other methods available. Any lack of diastereoselectivityin addition to the enol ether moiety is immaterial since retroaldolisation destroys the chirality at these two centres
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Although photostimulated 2π + 2π reactions involving cyclohexenes are not always reliably syn, the strain present in the trans-fused diastereoisomer (1) allows its easy conversion into the cis-fused form (2) by base-catalyzed equilibration.
Although photostimulated 2π + 2π reactions involving cyclohexenes are not always reliably syn, the strain present in the trans-fused diastereoisomer (1) allows its easy conversion into the cis-fused form (2) by base-catalyzed equilibration.
O
+ hv
O
H
H
(1) 4 parts
O
H
H
(2) 1 parts
+
dil. base
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions

98
CO2Me+
O
Me Me
Mehv
85 %
isophorone
OH
Me
CO2Me
K Selectride76 %
H
Me
O
O
1. Li/NH3, tBuOH
2. Oxidation92%
H
H
H
OO
K-Selectride = K(C2H5CH)3B
CH3
Uses a reductive cleavage of the photochemically-derived cyclobutane (3) as a model for a synthesis of (±)-10-epijuneol.
Uses a reductive cleavage of the photochemically-derived cyclobutane (3) as a model for a synthesis of (±)-10-epijuneol.(3)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Lineatin (8) is an aggregation pheromone of the female ambrosia beetle Trypodendron lineatum, a pest causing damage to sawn timber. The obvious disconnection of the target molecule is to the retroacetalisation product (9). However, White used the less obvious retrosynthetic step to give (7) as the target.
Lineatin (8) is an aggregation pheromone of the female ambrosia beetle Trypodendron lineatum, a pest causing damage to sawn timber. The obvious disconnection of the target molecule is to the retroacetalisation product (9). However, White used the less obvious retrosynthetic step to give (7) as the target.
O
O
(8)
CHOOH
OH
(9)
NaH/Et2O67 %
O
OH
HMeTsO
(7)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Synthesis of (7) uses 2π + 2π photoaddition of ethyne to (4) in a key step and also makes good use of the shape of unsaturated [4.2.0] skeleton in directing the hydroboration of (5) and in DIBAL reduction of (6) (after tosylation). Intramolecular SN2 displacement by the hemiacetal hydroxyl group gives (±) lineatin.
Synthesis of (7) uses 2π + 2π photoaddition of ethyne to (4) in a key step and also makes good use of the shape of unsaturated [4.2.0] skeleton in directing the hydroboration of (5) and in DIBAL reduction of (6) (after tosylation). Intramolecular SN2 displacement by the hemiacetal hydroxyl group gives (±) lineatin.
O
OH
HMeTsO
(7)
O
O
Me
hv, HC CH, MeCN O
OH
(4)
1. MeLi2. PCC (via hemiacetal)
OMe
O
O
OH
HMeHO
(5)
1. B2H6
2. H2O2, OH-1. TsCl, pyr2. DIBAL
(6)Me的空阻使双键的加成具选择性
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Photochemical 2π + 2π photoaddition of cyclic alkenes with carbonyl groups (the Paterno-Büchi reaction) have been widely studied. Using aliphatic aldehydes, singlet excited states are involved and higher diastereoselectivity results than with, for example, aromatic ketones.
Photochemical 2π + 2π photoaddition of cyclic alkenes with carbonyl groups (the Paterno-Büchi reaction) have been widely studied. Using aliphatic aldehydes, singlet excited states are involved and higher diastereoselectivity results than with, for example, aromatic ketones.
O+ n-C8H17CHO hv
O C8H17
O
HH
OO
H
H
C8H17
O
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions
The strain in four-membered (oxetane) ring of the the product is used to advantage. (Type 0) Hydrolysis of the oxetane-acetal unit in (10) leads to a single aldol diastereoisomers (11). The shape of the bicyclo[3.2.0] system allows completely diastereoselective hydrogenation of (10) (Type 3) from the more accessible face; after hydrolysis, the product is (12).
The strain in four-membered (oxetane) ring of the the product is used to advantage. (Type 0) Hydrolysis of the oxetane-acetal unit in (10) leads to a single aldol diastereoisomers (11). The shape of the bicyclo[3.2.0] system allows completely diastereoselective hydrogenation of (10) (Type 3) from the more accessible face; after hydrolysis, the product is (12).
OMe Me+ EtCHO hv
O Et
O
HMe
MeHCl
0.01 NMe
Et
O
OH
MeO(10) (11)
H2, Rh/Al2O3
O Et
O
HMeH
Me
H2O
H+Me
Et
OH
OH
MeO(12)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 2Type 2 2ππ + 2+ 2ππ CycloadditionsCycloadditions Type 2 Photochemical 1,3Type 2 Photochemical 1,3--CycloadditionCycloaddition of of ArenesArenes
1,3-Cycloaddition of benzene and substituted benzenes to alkenes under the influence of light can be a reaction of high diastereoselectivity. syn Addition to the alkene is accompanied by complete endo diastereoselectivity (methyl groups cis to the double bond).
1,3-Cycloaddition of benzene and substituted benzenes to alkenes under the influence of light can be a reaction of high diastereoselectivity. syn Addition to the alkene is accompanied by complete endo diastereoselectivity (methyl groups cis to the double bond).
Me
Me
Me
Me
MeMe
hv
H
H
H H
MeMe
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

99
The reaction with substituted benzenes is also often highly regioselective.
The reaction with substituted benzenes is also often highly regioselective.
OMe OMeOMe
hv
OMe
H
H H
H
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Photochemical 1,3Type 2 Photochemical 1,3--CycloadditionCycloaddition of of ArenesArenes
HMe
Me
hv
HMe
Me
HMe
COOH
H
H
(-)-retigeranic acid
This photochemical 1,3-cycloaddition, and particularly the intramolecular version of it, has been brilliantly in syntheses of a number of natural products, which is part of a synthesis of (-)-retigeranic acid.
This photochemical 1,3-cycloaddition, and particularly the intramolecular version of it, has been brilliantly in syntheses of a number of natural products, which is part of a synthesis of (-)-retigeranic acid.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Type 2 Photochemical 1,3Type 2 Photochemical 1,3--CycloadditionCycloaddition of of ArenesArenes
Photochemistry in CrystalsPhotochemistry in Crystals
One of the advantages of photochemistry is that it can be carried out on compounds in the crystalline state as well as in solution.
One of the advantages of photochemistry is that it can be carried out on compounds in the crystalline state as well as in solution.
In a crystal, molecules are stacked in a highly ordered way, usually in the same conformation, and there is little variation in the atomic distances separating nearest neighbors. For intermolecular reactions to occur, therefore, the separation between two molecules and the relative orientation of the reacting functional groups must be appropriate. When this is the case, completely diastereoselective reaction may ensue as in the photochemical dimerisation of hexadienoic acid and its derivatives.
In a crystal, molecules are stacked in a highly ordered way, usually in the same conformation, and there is little variation in the atomic distances separating nearest neighbors. For intermolecular reactions to occur, therefore, the separation between two molecules and the relative orientation of the reacting functional groups must be appropriate. When this is the case, completely diastereoselective reaction may ensue as in the photochemical dimerisation of hexadienoic acid and its derivatives.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Photochemical reaction (sensitized) of these compounds in solution gives mixtures of diastereoisomers.
Photochemical reaction (sensitized) of these compounds in solution gives mixtures of diastereoisomers.
R1
R2
H H
R1
R2
H H
R1
R2
H H
R1
R2
H H
.> 290 nm, hv (solid)Pyrex R1
R2
H H
R1
R2
H H
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Photochemistry in CrystalsPhotochemistry in Crystals
O
O
O
hv
O
O
O
O
O
O
H
H
HO
O
O
O
O
O
H
HH
+
endo exo
Likewise, the photolysis of maleic anhydride when carried out in the crystalline, gives only the endo diastereoisomers.
Likewise, the photolysis of maleic anhydride when carried out in the crystalline, gives only the endo diastereoisomers.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Photochemistry in CrystalsPhotochemistry in Crystals Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Orbital symmetry considerations not only predict concerted suprafacial-suprafacial cycloaddition of two alkenes when one is in its excited state (2πs + 2πs) but also concerted cycloaddition of two ground state alkenes with one alkene acting antarafacially.
Orbital symmetry considerations not only predict concerted suprafacial-suprafacial cycloaddition of two alkenes when one is in its excited state (2πs + 2πs) but also concerted cycloaddition of two ground state alkenes with one alkene acting antarafacially.
In practice, alkenes fail to undergo concerted 2πs + 2πa cycloadditions, presumably because steric interactions between substituents on the alkene sp2-carbons and other strain factors are too severe.
In practice, alkenes fail to undergo concerted 2πs + 2πa cycloadditions, presumably because steric interactions between substituents on the alkene sp2-carbons and other strain factors are too severe.
HOMOLUMO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

100
Reduction of this steric interaction would be expected if the substituents at one terminus of the alkene were absent which would be the case using a ketene. Ketenes do react with alkenes to give cyclobutanones but it is not clear that this cycloadditionis a simple 2πs + 2πa with the ketene acting antarafacially. This is because the orthogonal π*-orbital of the carbonyl group in the ketene may be involved, making the reaction a 2π + 2π + 2π type.
Reduction of this steric interaction would be expected if the substituents at one terminus of the alkene were absent which would be the case using a ketene. Ketenes do react with alkenes to give cyclobutanones but it is not clear that this cycloadditionis a simple 2πs + 2πa with the ketene acting antarafacially. This is because the orthogonal π*-orbital of the carbonyl group in the ketene may be involved, making the reaction a 2π + 2π + 2π type.
HR3
R2H R1
HOMOLUMO
O
R2
H
R1H
R3
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Although ketene-alkene cycloadditions are inherently syndiastereoselective (syn stereospecific) in addition to the alkenethere is evidence to suggest that in the transition state, formation of the bond to the electron-deficient ketene carbonyl has run ahead of that between the other two carbons, i.e. the reaction has dipolar character.
Although ketene-alkene cycloadditions are inherently syndiastereoselective (syn stereospecific) in addition to the alkenethere is evidence to suggest that in the transition state, formation of the bond to the electron-deficient ketene carbonyl has run ahead of that between the other two carbons, i.e. the reaction has dipolar character.
O D
HPh
Ph Ph
H
C OPh
Ph
H
Ph H
D
H
Ph D
HO H
DPh
Ph Ph
H
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
δ+
The transition state of lowest energy in the cycloaddition of ethene and ketene is that shown in next Figure, which illustrates that whilst the C1-C3 bond is almost fully the C2-C4 bond is far less developed.
The transition state of lowest energy in the cycloaddition of ethene and ketene is that shown in next Figure, which illustrates that whilst the C1-C3 bond is almost fully the C2-C4 bond is far less developed.
H HC
O
H
H
H H1
2
43
δ-δ-
Asynchronous bonding in the transition state helps to explain the regiochemistry of this reaction; the alkene carbon less able to sustain the partial positive charge becomes bonded to the ketene carbonyl carbon.
Asynchronous bonding in the transition state helps to explain the regiochemistry of this reaction; the alkene carbon less able to sustain the partial positive charge becomes bonded to the ketene carbonyl carbon.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
H RC
O
H
H
H H
+
-O
R
C CH
PhO+ 16 h, CHCl3, 20 oC C
O
C
HPh
C
O
C
HPh
(13)(14)
(15)H H
HPh O
26 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Antarafacial addition of the ketene as show in the figure also explains a subtle stereochemical feature of these 2πs + 2πacycloaddition which leads to the larger ketene substituentsbeing located in the more sterically crowed environment in the product. Thus, addition of phenylketene to cyclopentadienegives as excess of the bicyclo[3.2.0]heptenone diastereoisomer (15) in which the phenyl group is endo.
Antarafacial addition of the ketene as show in the figure also explains a subtle stereochemical feature of these 2πs + 2πacycloaddition which leads to the larger ketene substituentsbeing located in the more sterically crowed environment in the product. Thus, addition of phenylketene to cyclopentadienegives as excess of the bicyclo[3.2.0]heptenone diastereoisomer (15) in which the phenyl group is endo.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
This stereochemical outcome is a consequence of the preferred approach of the ketene in (13), in which the hydrogen of the ketene is directed towards the diene, coupled with the required direction of twisting around the ketene C=C bond as shown in (14). Like the Diels-Alder reaction, therefore, the diastereoselectivity of the 2πs + 2πaaddition of an alkene and a ketene can have an inherent component (syn addition to the alkene) and an occasional component (endo/exo addition)
This stereochemical outcome is a consequence of the preferred approach of the ketene in (13), in which the hydrogen of the ketene is directed towards the diene, coupled with the required direction of twisting around the ketene C=C bond as shown in (14). Like the Diels-Alder reaction, therefore, the diastereoselectivity of the 2πs + 2πaaddition of an alkene and a ketene can have an inherent component (syn addition to the alkene) and an occasional component (endo/exo addition)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions

101
The endo diastereoselectivity which is found in this scheme cannot be expected in all cases, particularly where the ketene is disubstituted with groups of a similar bulk.
The endo diastereoselectivity which is found in this scheme cannot be expected in all cases, particularly where the ketene is disubstituted with groups of a similar bulk.
C CH
PhO+ 16 h, CHCl3, 20 oC C
O
C
HPh
C
O
C
HPh
(13)(14)
(15)H H
HPh O
26 %
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Intramolecular ketene-alkene cycloadditionshave been used to synthesis four-membered ring-containing products, e.g. β-trans-bergotamene (16)
Intramolecular ketene-alkene cycloadditionshave been used to synthesis four-membered ring-containing products, e.g. β-trans-bergotamene (16)
CO2H
O
1. Ph3=CH2
2. H2O(40 %)
CO2H
CH2
(COCl)2
COCl
CH2
NHiPr2
tolueneH
CO
O
1. NH2NH2
2. KOtBu, DMSO
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions
Keteniminium salts can also play the role of the antarafacialcomponent in 2πs + 2πa cycloadditions.
Keteniminium salts can also play the role of the antarafacialcomponent in 2πs + 2πa cycloadditions.
O
N(CF3SO2)2O
collidine, ClCH2CH2Cl H
C N+
H
C N+
H
N+H2OCCl4
H
O
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 2Other 2ππ + 2+ 2ππ CycloadditionsCycloadditions Other Other CycloadditionsCycloadditions
In recent years, considerable effort has been directed towards developing cycloaddition reactions which would give carbocyclicfive-membered rings with the facility and stereoselectivity with which the Diels-Alder reaction gives six membered rings. This effort has been necessary because of the dearth of available all-carbon 1,3-dipoles and their generally inefficient reactions with alkenes to give five-membered rings. An all-carbon 1,3-dipole which might be expected to have enhanced stability would be one in which a methyl group was present at C-2.
In recent years, considerable effort has been directed towards developing cycloaddition reactions which would give carbocyclicfive-membered rings with the facility and stereoselectivity with which the Diels-Alder reaction gives six membered rings. This effort has been necessary because of the dearth of available all-carbon 1,3-dipoles and their generally inefficient reactions with alkenes to give five-membered rings. An all-carbon 1,3-dipole which might be expected to have enhanced stability would be one in which a methyl group was present at C-2.
+ -N N
hv
or
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
N
N
H
MeO2C
H
H
MeCNreflux
H
MeO2C
H
H
MeO2C
H
H
H
H H
H
CO2MeH H
HO
( + )-hirsutene-
The part is biradicaloid in its behaviour rather than dipolar and whilst addition to alkenes to give five membered rings occurs, diastero-and regioselectivityare high only when the reaction is intramolecular, the tricyclic skeleton of hirsutene is formed.
The part is biradicaloid in its behaviour rather than dipolar and whilst addition to alkenes to give five membered rings occurs, diastero-and regioselectivityare high only when the reaction is intramolecular, the tricyclic skeleton of hirsutene is formed.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other Other CycloadditionsCycloadditions
Trost has successfully used palladium complexes of the 1,3-dipole in cycloaddition reactions. These are generated in situ, e.g. by palladium-induced decomposition of 2-(trimethylsilyl-methyl)allyl acetate.
Trost has successfully used palladium complexes of the 1,3-dipole in cycloaddition reactions. These are generated in situ, e.g. by palladium-induced decomposition of 2-(trimethylsilyl-methyl)allyl acetate.
SiMe3
OAc+ PdL4
SiMe3
PdL4+
-OAc
PdL4+
-
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other Other CycloadditionsCycloadditions

102
These trimethylenemethane intermediates cycloadd efficiently to double bonds which are conjugated with at least one electron-withdrawing group.
These trimethylenemethane intermediates cycloadd efficiently to double bonds which are conjugated with at least one electron-withdrawing group.
SiMe3
OAc+
R EWG(Ph3P)4Pd
3-9 mol %
EWG
R
Pd(PPh3)2+
-
R
EWGEWG = CN, SO2Ph, CO2R'
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other Other CycloadditionsCycloadditions
Although high diastereoselectivity is obtained to trans-alkenes, the configuration of cis-alkenes is incompletely retained in the product. It appears, therefore, that the cycloaddition is not concerted and that formation of one bond is complete before any formation of the second bond has taken place allowing intervention of some C-C bond rotation around the original double bond of the cis-alkene.
Although high diastereoselectivity is obtained to trans-alkenes, the configuration of cis-alkenes is incompletely retained in the product. It appears, therefore, that the cycloaddition is not concerted and that formation of one bond is complete before any formation of the second bond has taken place allowing intervention of some C-C bond rotation around the original double bond of the cis-alkene.
The diastereoselectivity of this cycloaddition, therefore, is occasional. Of course, configurational stability of cis-alkenes is assured by containing them in an eight-membered or smaller ring. In these cases, addition of the complexed 1,3-dipole is highly syn selective.
The diastereoselectivity of this cycloaddition, therefore, is occasional. Of course, configurational stability of cis-alkenes is assured by containing them in an eight-membered or smaller ring. In these cases, addition of the complexed 1,3-dipole is highly syn selective.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other Other CycloadditionsCycloadditions
Ar SiMe3
OCO2Me+
Ph
CO2Me
CO2Me
Pd(OAc)2, 5 %
(iPrO)3P, toluene
Ar
Ph CO2MeCO2Me
O3
O
Ar
Ph CO2MeCO2Me
Ar = p-MeOC6H4
A substituted version of the palladium-complexed 1,3-dipole is generated, the cycloaddition is highly diastereoselective when R is electron-withdrawing (CN, COEt), giving a trans relationship between R and Ph; the diastereoselectivityis less when R is electron donating. However, if the exocyclic methylene group in the product is removed by ozonolysis, thermodynamic equilibration then generates the trans diastereoisomer (21), the first steps in a synthesis of rocaglamide.
A substituted version of the palladium-complexed 1,3-dipole is generated, the cycloaddition is highly diastereoselective when R is electron-withdrawing (CN, COEt), giving a trans relationship between R and Ph; the diastereoselectivityis less when R is electron donating. However, if the exocyclic methylene group in the product is removed by ozonolysis, thermodynamic equilibration then generates the trans diastereoisomer (21), the first steps in a synthesis of rocaglamide.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other Other CycloadditionsCycloadditions 1,41,4--CheletropicCheletropic Additions to 1,3Additions to 1,3--DienesDienes
Addition of sulphur dioxide across a 1,3-diene is a cheletropicaddition in which two new bonds to sulphur are made concertedly and the diene reacts suprafacially.
Addition of sulphur dioxide across a 1,3-diene is a cheletropicaddition in which two new bonds to sulphur are made concertedly and the diene reacts suprafacially.
In this reaction, therefore, using a diene having two prochiraldouble bonds, two chiral centres are created of defined relative configuration (inherent diastereoselectivity). Unfortunately, there is a dearth of molecules containing an atom (S in SO2) having the filled and empty orbitals necessary for cheletropic addition (siglet carbenes do not, in general react with dienes by 1,4-addition: they add 1,2 to one of the double bonds).
In this reaction, therefore, using a diene having two prochiraldouble bonds, two chiral centres are created of defined relative configuration (inherent diastereoselectivity). Unfortunately, there is a dearth of molecules containing an atom (S in SO2) having the filled and empty orbitals necessary for cheletropic addition (siglet carbenes do not, in general react with dienes by 1,4-addition: they add 1,2 to one of the double bonds).
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The reaction is , in practice, more often encountered in the reverse elimination mode, i.e. as a route to configurationally defined dienes from the corresponding dihydrothiophenedioxides (22) and (23), prepared by other means.
The reaction is , in practice, more often encountered in the reverse elimination mode, i.e. as a route to configurationally defined dienes from the corresponding dihydrothiophenedioxides (22) and (23), prepared by other means.
a b+ SO2
Sa bHH
O2(22)
ab
+ SO2Sa H
bH
O2(23)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
1,41,4--CheletropicCheletropic Additions to 1,3Additions to 1,3--DienesDienes Other 1,4Other 1,4--Additions to 1,3Additions to 1,3--DienesDienes
Cyclic and acyclic dienes can be converted completely diastereo-selectively into 1,4-addition products using palladium chemistry. Thus cyclohexa-1,3-dienes is converted into cis-1-acetoxy-4-chlorocyclohex-2-ene (24), cis-1,4-diacetoxycyclohex-2-ene (25) or trans-1,4-diacetoxycyclohex-2-ene (26), depending on the conditions.
Cyclic and acyclic dienes can be converted completely diastereo-selectively into 1,4-addition products using palladium chemistry. Thus cyclohexa-1,3-dienes is converted into cis-1-acetoxy-4-chlorocyclohex-2-ene (24), cis-1,4-diacetoxycyclohex-2-ene (25) or trans-1,4-diacetoxycyclohex-2-ene (26), depending on the conditions.
Cl OAc
89 %, > 98 % cis(24)
benzoquinonePd(OAc)2 (5 mol%)
HOAcLiClLiOAc MnO2, benzoquinone,
Pd(OAc)2 (5 mol%)LiOAc, HOAc
AcO OAc
93 %, > 91 % cis(26)
AcO OAc
93 %, > 91 % cis(25)
MnO2, benzoquinone,Pd(OAc)2 (5 mol%)LiOAc, HOAc
LiCl
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

103
The mechanism suggested for cis-1,4-acetoxychlorinationinvolves coordination of the diene to palladium followed by anti addition of acetate to give a π-allyl complex (27). Coordination by benzoquinoneis followed by external anti attack of chloride to give overall syn addition. Similar mechanisms account for the formation of (25) and (26) with the acetoxy group being intramolecularly delivered from palladium in the latter case.
The mechanism suggested for cis-1,4-acetoxychlorinationinvolves coordination of the diene to palladium followed by anti addition of acetate to give a π-allyl complex (27). Coordination by benzoquinoneis followed by external anti attack of chloride to give overall syn addition. Similar mechanisms account for the formation of (25) and (26) with the acetoxy group being intramolecularly delivered from palladium in the latter case.
(24)
Cl OAc
PdCl42-
Pd2+
Cl Cl
OAc-
OAc
PdCl/2
OO
OAc
PdCl
O
O
Cl-
HOAc, LiCl
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 1,4Other 1,4--Additions to 1,3Additions to 1,3--DienesDienes
These products of 1,4-addition to conjugated dienes are versatile starting materials for nucleophilic substitution reactions with retention or inversion of configuration.
These products of 1,4-addition to conjugated dienes are versatile starting materials for nucleophilic substitution reactions with retention or inversion of configuration.
Cl OAcOAc
(MeO2C)2CH
NaCH(CO2Me)2
Pd(0)NaCH(CO2Me)2
> 98 % trans
(MeO2C)2CH OAc
> 98 % cis
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 1,4Other 1,4--Additions to 1,3Additions to 1,3--DienesDienes
An application of this tandem 1,4-addition-nucleophilicsubstitution in a synthesis of (28), a sex pheromone of the carpenter bee.
An application of this tandem 1,4-addition-nucleophilicsubstitution in a synthesis of (28), a sex pheromone of the carpenter bee.
benzoquinone
HOAc, Pd(OAc)2LiCl, LiOAc
Cl
H OAc
H
1. PhSO2CHNO2Na+
Pd(PPh3)42. NaOH
H OH
H
NO2PhSO2
1. Rh(PPh3)3Cl, H2
2. dihydropyran, H+H
NO2PhSO2
OTHP
H
1. KMnO4, NaOH2. H+/H2O
CO2H
H OTHP
Hspontaneous O
O
(28)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Other 1,4Other 1,4--Additions to 1,3Additions to 1,3--DienesDienes [3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements
Simple diastereoselectivity in [3,3] sigmatropic rearrangements results in the transformation of two configured double bonds, separated by two saturated atoms, into two adjacent chiral centres.
Simple diastereoselectivity in [3,3] sigmatropic rearrangements results in the transformation of two configured double bonds, separated by two saturated atoms, into two adjacent chiral centres.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements
Oa
b
x
yOa
H
x
y
Oa
b
x
yO
a
b
x
y
diastereoisomers
Oa
b x
y
enantiomers
enantiomers
Ob
x
a
y
Oa
y
b
x
Oa
b x
yO
b
y
a
x
Oy
x b
a Oy
x b
aO
a
x
b
y
(29)
(30)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
The chair transition state is usually sufficiently lower in energy than the boat for formation of (29) to be favored over formationof (30) by a factor of 20:1 or more.
A change in configuration at one double bond would lead, after rearrangement via a chair transition state, to (30). Accordingly, the use made of Type 2 [3,3] rearrangements to prepare two adjacent chiral centres will depend on the availability of 1,5-dienes, configurationally defined at both double bonds.
The chair transition state is usually sufficiently lower in energy than the boat for formation of (29) to be favored over formationof (30) by a factor of 20:1 or more.
A change in configuration at one double bond would lead, after rearrangement via a chair transition state, to (30). Accordingly, the use made of Type 2 [3,3] rearrangements to prepare two adjacent chiral centres will depend on the availability of 1,5-dienes, configurationally defined at both double bonds.
For the much-used Claisen rearrangement of allyl vinyl ethers, the more difficult bond to obtain configurationally defined is, in general, that of the vinyl ether because of the dearth of methods for their diastereoselective synthesis.
For the much-used Claisen rearrangement of allyl vinyl ethers, the more difficult bond to obtain configurationally defined is, in general, that of the vinyl ether because of the dearth of methods for their diastereoselective synthesis.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements

104
The Ireland modification of the Claisen rearrangement is a valuable variant, not least because of the much lower temperature required. Diastereoselectivity in this variant requires the generation and silylation of configurationally, defined enolate in a molecule already containing a configurationally pure allyl double bond.
The Ireland modification of the Claisen rearrangement is a valuable variant, not least because of the much lower temperature required. Diastereoselectivity in this variant requires the generation and silylation of configurationally, defined enolate in a molecule already containing a configurationally pure allyl double bond.
O
O
Me
Me1. LDA, THF2. tBuMe2SiCl
OSiMe2tBu
OH
Me
H
MeH+
H
Me
OH
OH
Me
H
Me
OH
OMe
H
87
:
13(31)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements
The (E)-enolates are generated under kinetically controlled conditions and the major diastereoisomer of the product is formed via the conformations (31) and (32), respectively. The less than complete diastereoselectivityseems more likely to result from incomplete configurational homogeneity of the silyl ketene acetal double bonds in (31) and (32) than from their rearrangements proceeding in part via boat transition states.
The (E)-enolates are generated under kinetically controlled conditions and the major diastereoisomer of the product is formed via the conformations (31) and (32), respectively. The less than complete diastereoselectivityseems more likely to result from incomplete configurational homogeneity of the silyl ketene acetal double bonds in (31) and (32) than from their rearrangements proceeding in part via boat transition states.
O
O
MeMe 1. LDA, THF
2. tBuMe2SiCl
OSiMe2tBu
OH
Me
Me
HH+
H
Me
OH
OH
Me
H
Me
OH
OMe
H
11
:
89(32)
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements
The use of dipolar aprotic cosolvents (HMPA or DMPU) favors the formation of enolates having the alternative configuration to those shown and thus products with the opposite sense of diastereoselectivity.
The use of dipolar aprotic cosolvents (HMPA or DMPU) favors the formation of enolates having the alternative configuration to those shown and thus products with the opposite sense of diastereoselectivity.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[3,3] [3,3] SigmatropicSigmatropic rearrangementsrearrangements
R1
H
H
OR2
H
R1
H
H
O
R2
H
H+
R2
R1
OH
R1
H
H
O
H
R2
R1
H
H
OR2
H
H+
R2
R1
OH
(33)
(33')
2
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
Simple diastereoselectivity (Type 2) in [2,3] rearrangements has been investigated in some detail by Nakai and Mikami. It is suggested that in the Wittig rearrangement the diastereoselectivity is controlled by the balance between two factors. Thus, using the envelope conformation (33), there is an eclipsing interaction between R2 and the hydrogen at C-2, whereas in conformation (33’), there is a gauche interaction between R1 and R2.
Simple diastereoselectivity (Type 2) in [2,3] rearrangements has been investigated in some detail by Nakai and Mikami. It is suggested that in the Wittig rearrangement the diastereoselectivity is controlled by the balance between two factors. Thus, using the envelope conformation (33), there is an eclipsing interaction between R2 and the hydrogen at C-2, whereas in conformation (33’), there is a gauche interaction between R1 and R2.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Using an E-configured alkene, these two interactions appear to be comparable and diastereoselectivity is not high, although the balance can be tipped by, for example, introducing a bulky substituent at C-2. In this case, reaction from conformation (34) is favored.
Using an E-configured alkene, these two interactions appear to be comparable and diastereoselectivity is not high, although the balance can be tipped by, for example, introducing a bulky substituent at C-2. In this case, reaction from conformation (34) is favored.
Me
H
TMS
O
H
Me
H+
(34)
MeH
TMS
OH
Me
MeH
TMS
O
H
Me
Me
TMS OH
Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
For Z-configured alkenes, the stereoselectivity is higher and favors the formation of the cis diastereoisomer with, for example, a preference for reaction from conformation (35).
For Z-configured alkenes, the stereoselectivity is higher and favors the formation of the cis diastereoisomer with, for example, a preference for reaction from conformation (35).
H
Me
H
OPh
H
H+
(35)
HMe
H
O
Ph
HH
Me
H
O
H
Ph
Ph
OH
Me
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements

105
Remarkably high diastereoselectivity with both (E)- and (Z)-alkenes is found using ethynyl groups as substituents on the oxymethylene group in the Wittig rearrangement.
Remarkably high diastereoselectivity with both (E)- and (Z)-alkenes is found using ethynyl groups as substituents on the oxymethylene group in the Wittig rearrangement.
O
Rexo
Rendo
Li
(36)
2
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
Calculations by Houk suggest that the preferred transition state geometry for the Wittig rearrangement is the alternative envelope shown in (36), but this was only located when the lithium ion was included in the calculation. The preference of an ethylnyl group form the exo position in this envelope does not arise, according to Houk’s calculations, from avoidance of an eclipsing interaction between the endo-ethylnyl group and the C2-H.
Calculations by Houk suggest that the preferred transition state geometry for the Wittig rearrangement is the alternative envelope shown in (36), but this was only located when the lithium ion was included in the calculation. The preference of an ethylnyl group form the exo position in this envelope does not arise, according to Houk’s calculations, from avoidance of an eclipsing interaction between the endo-ethylnyl group and the C2-H.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
Whether the envelope conformation suggested by Nakai and Mikami or that in (36) is preferred for the transition state of the [2,3] sigmatropic rearrangement remains an open question.
Whether the envelope conformation suggested by Nakai and Mikami or that in (36) is preferred for the transition state of the [2,3] sigmatropic rearrangement remains an open question.
OH
Me
R
BuLi, THF, -85 oC
Me
H
OH
H
H
R
H+
MeH
H
O
H
R
R = H, TMS : 99 % d.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
OMe
H
R
BuLi, THF, -85 oC
H
H
OH
H
R
Me
H+
HMe
H
O
H
R
R = H, TMS : 100 % d.e.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
[2,3] [2,3] SigmatropicSigmatropic rearrangementsrearrangements
SummarySummary
Type 2 photochemical 2π + 2π cycloadditions involving alkenes are normally reliably diastereoselective only when the photo-stimulated alkene contain in a ring of less than six members.
Type 2 photochemical 2π + 2π cycloadditions involving alkenes are normally reliably diastereoselective only when the photo-stimulated alkene contain in a ring of less than six members.
Cyclobutanes and oxetanes resulting from such 2π + 2πcycloadditions are useful because of the ease with which their ring opening can be accomplished.
Cyclobutanes and oxetanes resulting from such 2π + 2πcycloadditions are useful because of the ease with which their ring opening can be accomplished.
Type 2 thermal cycloaddition of ketenes and alkenes can be highly diastereoselective since the ketene can apparently function as the 2πa component in a 2πa + 2πs cycloaddition.
Type 2 thermal cycloaddition of ketenes and alkenes can be highly diastereoselective since the ketene can apparently function as the 2πa component in a 2πa + 2πs cycloaddition.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
SummarySummary
Cycloaddition of (substituted) trimethylenemethanepalladiumcomplexes with alkenes gives five-membered rings, but the two carbon-carbon bonds are not formed simultaneously and the reaction is not inherently diastereoselective.
Cycloaddition of (substituted) trimethylenemethanepalladiumcomplexes with alkenes gives five-membered rings, but the two carbon-carbon bonds are not formed simultaneously and the reaction is not inherently diastereoselective.
Some highly diastereoselective 1,4-additions to 1,3-dienes can be accomplished using palladium chemistry.
Some highly diastereoselective 1,4-additions to 1,3-dienes can be accomplished using palladium chemistry.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,

106
SummarySummary
Type 2 [3,3] sigmatropic rearrangements are usually highly diastereoselective and proceed via chair transition states, but depend on the availability of 1,5-dienes with defined configurations for two double bonds.
Type 2 [3,3] sigmatropic rearrangements are usually highly diastereoselective and proceed via chair transition states, but depend on the availability of 1,5-dienes with defined configurations for two double bonds.
Some Type 2 [2,3] Wittig rearrangements are highly diastereoselective, particularly those with an ethynyl substituent on the oxymethylene group and an E- or Z-substituted double bond.
Some Type 2 [2,3] Wittig rearrangements are highly diastereoselective, particularly those with an ethynyl substituent on the oxymethylene group and an E- or Z-substituted double bond.
Stereochemistry, Jian Pei, College of Chemistry, Peking University,
Thanks for Thanks for Your Attention!!!Your Attention!!!












![Stereochemistry of the Brivaracetam Diastereoisomers · Chirality 1 Stereochemistry of the Brivaracetam Diastereoisomers Shi Qiu,[a] Kourosch Abbaspour Tehrani,[a] Sergey Sergeyev,[a]](https://static.fdocuments.us/doc/165x107/5f0f368a7e708231d4430bfa/stereochemistry-of-the-brivaracetam-diastereoisomers-chirality-1-stereochemistry.jpg)





