
Stabilizing biocatalysts.pdf
32
6534 Chem. Soc. Rev., 2013, 42, 6534--6565 This journ al is c The Royal Society of Chemistry 2013 Cite this: Chem. Soc. Rev., 2013, 42, 6534 Stabilizing biocatalysts Andreas S. Bommarius* a and Marie ´tou F. Paye b The area of biocatalysis itself is in rapid development, fueled by both an enhanced repertoire of protein engineering tools and an increasing list of solved problems. Biocatalysts, however, are delicate materials that hover close to the thermodynamic limit of stability. In many cases, they need to be stabilized to survive a range of challenges regarding temperature, pH value, salt type and concentration, co-solvents, as well as shear and surface forces. Biocatalysts may be delicate proteins, however, once stabilized, they are efficiently active enzymes. Kinetic stability must be achieved to a level satisfactory for large-scale proc ess appli catio n. Kine tic stability evokes resis tance to degr adati on and maintained or incr eased catalytic efficiency of the enzyme in which the desired reaction is accomplished at an increased rate. Howev er, beyon d these limitatio ns, stabl e bioca talys ts can be oper ated at high er temperat ures or co-solvent concentrations, with ensuing reduction in microbial contamination, better solubility, as well as in many cases more favorabl e equi lib ri um, and can serve as more effec ti ve templates for comb inatorial and data -driv en prote in engin eerin g. To incr ease thermodyn amic and kinet ic stabi lity , immo biliz ation , prot ein engin eerin g, and medium engin eerin g of bioca talys ts are available, the main focus of this work. In the case of protein engineering, there are three main approaches to enhancing the stability of protein biocatalysts: (i) rational design, based on knowledge of the 3D-structure and the catalytic mechanism, (ii) combinatorial design, requiring a protocol to generate diversity at the genetic level, a large, often high throughput, screening capacity to distinguish ‘hits’ from ‘misses’, and (iii) data- driven design, fueled by the increased availability of nucleotide and amino acid sequences of equivalent functionality. a School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Parker H. Petit Institute of Bioengineering and Bioscience, 315 Ferst Drive, Atlanta, GA 30332-0363, USA. E-mail: [email protected]; Fax: +1 404-894-2291 b School of Chemistry and Biochemistry, Georgia Institute of Technology, 901 Atlantic Drive, Atlanta, GA 30332-0400, USA Andreas S. Bommarius Andreas (Andy) S. Bommarius is a pro fes sor of Che mic al and Biomolecular Engineering as w el l of C he mi st r y a nd B io - chemistry at the Georgia Institute of Technology in Atlanta, GA, USA. He received hi s di pl oma in Ch emis tr y in 1984 at the Technical University of Munich , Ge rmany and hi s Chemical Engi ne er ing BS and PhD degrees in 1982 and 1989 at MIT, Cambridge, MA, USA. From 1990–2000, he led the Laboratory of Enzyme Catalysis at Degussa (now Evonik) in Wol fgan g, Ger many. At Geo rgi a Tec h since 2000 , his res ear ch inter ests cover green chemi stry and biomo lecul ar engin eerin g, specifically biocatalyst development and protein stability studies. Marie ´tou F. Paye Marie ´tou F. Paye obtained her BA de gr ee in Bi oche mi stry an d a minor in French from Middlebury College in Middlebu ry, Vermont, USA. She was awarded a Bill and Melinda Gates Schola rship for her un de rg ra du ate and gr aduate stud ies . At the Geor gia Inst itut e of Technology in Atlanta, GA, she works toward s her Ph D in the Sc ho ol of Chemistry and Bi o- ch emis tr y in the Bommar ius group, focusi ng on novel synt heses towards beta -lac tam antibiotics. Recei ved 18th April 2013 DOI: 10.1039/c3cs60137d www.rsc.org/csr Chem Soc Rev REVIEW ARTICLE P u b l i s h e d o n 2 7 J u n e 2 0 1 3 . D o w n l o a d e d b y O p e n U n i v e r s i t y o n 0 8 / 0 7 / 2 0 1 3 1 4 : 1 7 : 3 5 . View Article Online View Journal | View Issue
-
Upload
george-sebastian-antony -
Category
Documents
-
view
242 -
download
0
Transcript of Stabilizing biocatalysts.pdf

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 1/32
6534 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
Cite this: Chem. Soc. Rev., 2013,
42, 6534
Stabilizing biocatalystsAndreas S. Bommarius*a and Marietou F. Payeb
The area of biocatalysis itself is in rapid development, fueled by both an enhanced repertoire of protein
engineering tools and an increasing list of solved problems. Biocatalysts, however, are delicate materials
that hover close to the thermodynamic limit of stability. In many cases, they need to be stabilized to
survive a range of challenges regarding temperature, pH value, salt type and concentration, co-solvents,
as well as shear and surface forces. Biocatalysts may be delicate proteins, however, once stabilized, they
are efficiently active enzymes. Kinetic stability must be achieved to a level satisfactory for large-scale
process application. Kinetic stability evokes resistance to degradation and maintained or increased
catalytic efficiency of the enzyme in which the desired reaction is accomplished at an increased rate.
However, beyond these limitations, stable biocatalysts can be operated at higher temperatures or
co-solvent concentrations, with ensuing reduction in microbial contamination, better solubility, as well
as in many cases more favorable equilibrium, and can serve as more effective templates for
combinatorial and data-driven protein engineering. To increase thermodynamic and kinetic stability,
immobilization, protein engineering, and medium engineering of biocatalysts are available, the main
focus of this work. In the case of protein engineering, there are three main approaches to enhancing
the stability of protein biocatalysts: (i) rational design, based on knowledge of the 3D-structure and the
catalytic mechanism, (ii) combinatorial design, requiring a protocol to generate diversity at the genetic
level, a large, often high throughput, screening capacity to distinguish ‘hits’ from ‘misses’, and (iii) data-
driven design, fueled by the increased availability of nucleotide and amino acid sequences of equivalent
functionality.
a School of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Parker H. Petit Institute of Bioengineering and Bioscience, 315 Ferst Drive,
Atlanta, GA 30332-0363, USA. E-mail: [email protected]; Fax: +1 404-894-2291b School of Chemistry and Biochemistry, Georgia Institute of Technology, 901 Atlantic Drive, Atlanta, GA 30332-0400, USA
Andreas S. Bommarius
Andreas (Andy) S. Bommarius
is a professor of Chemical and
Biomolecular Engineering as
well of Chemistry and Bio-
chemistry at the Georgia
Institute of Technology in
Atlanta, GA, USA. He received
his diploma in Chemistry in1984 at the Technical University
of Munich, Germany and his
Chemical Engineering BS and
PhD degrees in 1982 and 1989
at MIT, Cambridge, MA, USA.
From 1990–2000, he led the
Laboratory of Enzyme Catalysis at Degussa (now Evonik) in
Wolfgang, Germany. At Georgia Tech since 2000, his research
interests cover green chemistry and biomolecular engineering,
specifically biocatalyst development and protein stability studies.
Marietou F. Paye
Marietou F. Paye obtained her BA
degree in Biochemistry and a
minor in French from Middlebury
College in Middlebury, Vermont,
USA. She was awarded a Bill and
Melinda Gates Scholarship for her
undergraduate and graduate
studies. At the Georgia Instituteof Technology in Atlanta, GA, she
works towards her PhD in the
School of Chemistry and Bio-
chemistry in the Bommarius
group, focusing on novel
syntheses towards beta-lactam
antibiotics.
Received 18th April 2013
DOI: 10.1039/c3cs60137d
www.rsc.org/csr
Chem Soc Rev
REVIEW ARTICLE View Article OnlineView Journal | View Issue

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 2/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6535
1. Need for biocatalyst stabilization
Justification for current review. The area of biocatalysis itself is in
rapid development, fueled by both an enhanced repertoire of
protein engineering tools (for more reviews, see citations1,2)
and an increasing list of solved problems, such as the synthesis
of the side chains of statins such as Lipitors and Crestors,3 as
well as the active pharmaceutical ingredients (APIs) of Lyricas
(pregabalin)4 and Januvias/Janumet s (sitagliptin)5 (Fig. 1) thepathway from glucose to 1,3-propanediol,6 and the process to
acrylamide and nicotinamide from their respective nitriles. In
parallel, the subject of stabilizing biocatalysts has developed
alongside and certainly deserves a renewed look. In addition to
this review, readers are referred to the overview of enzyme
stability by Polizzi et al.,7 the focus on stabilization through the
surrounding medium by Bommarius and Broering,8 and lastly
the works of Eijsink on both rational design9 and directed
evolution for comprehensive reviews on the topic of stabilizing
biocatalysts.10
Advantages of biocatalysts. Biocatalysis, defined as reactions
catalyzed by macromolecules such as isolated enzymes and
whole cells, has shown to be favorable in several ways.14–16
Biocatalysts are able to catalyze diverse sets of reactions includ-
ing: (a) formation or hydrolysis of peptides, esters, and amides
by hydrolases, (b) enantio- and regioselective oxidation of
alcohols, and reductions of ketones & double bonds by oxido-
reductases, (c) enantio, chemo-, and regioselective reactions by
transferases and isomerases, (d) and formation of C–C bonds
by lyases.16–20 Essentially, biocatalysis offers industrial-scale
synthesis of compounds within a controllable environment.This biochemical method of synthesis relieves many of the
disadvantages presented in the production of complex macro-
molecules such as lack of reactive specificity and in the use of
chemocatalysts such as metals during organic synthesis.16,19,21
Additionally, the shift towards green chemistry, the need for the
use of biodegradable materials, mild reaction conditions,
controlled reactions conditions, and reduced cost and
manpower have rendered biocatalysis increasingly timely in
industrial settings.15,16,19,22,23 Biocatalysis encompasses a wide
range of chemical reactions, including redox reactions and
carbon–carbon formations, as illustrated by the following two
examples.
Fig. 1 (a) Structure of enzymes studied and evolved to catalyze the production of the side chains of pharmaceutically available medicines. 12-Oxophyto-
dienoate reductase—synthesis of Lyricas (pregabalin);11 PDB: 3HGR.12 (b) D-Amino acid aminotransferase—synthesis of Januvias/Janumets (sitagliptin);5
PDB: 3LQS.13
Review Article Chem Soc Rev
View Article Online
7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 3/32
6536 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
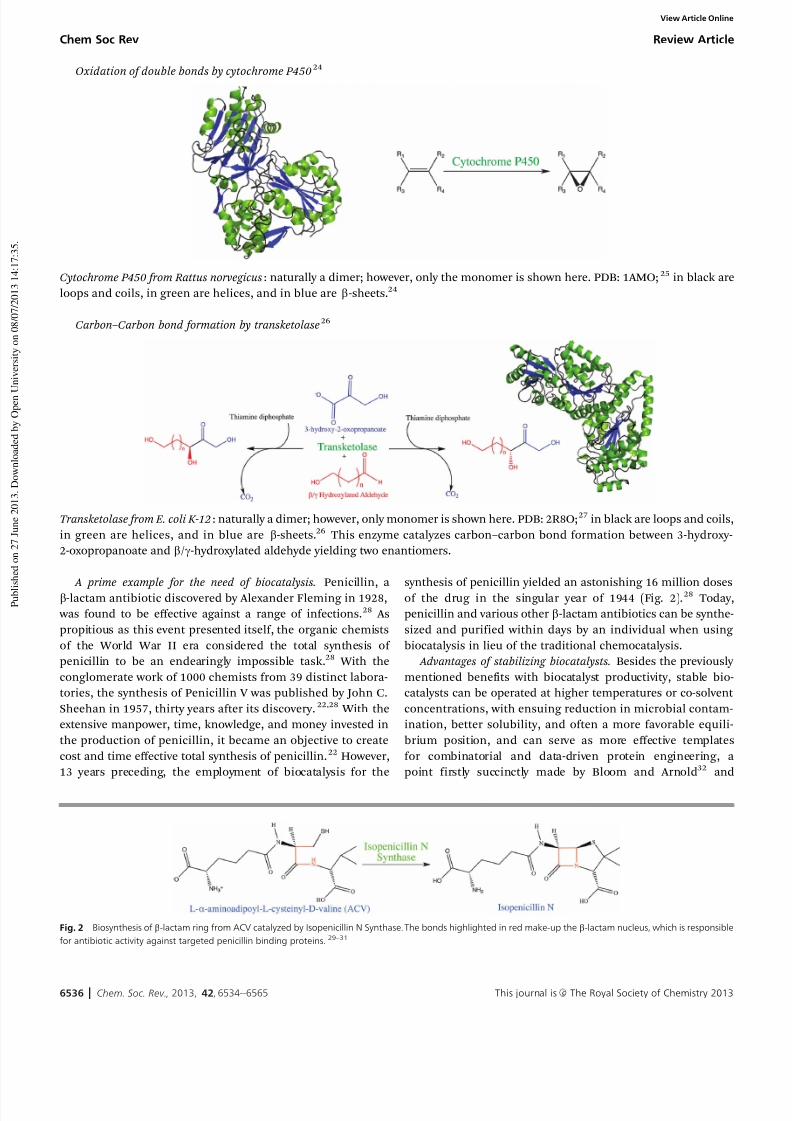
Oxidation of double bonds by cytochrome P45024
Cytochrome P450 from Rattus norvegicus: naturally a dimer; however, only the monomer is shown here. PDB: 1AMO; 25 in black are
loops and coils, in green are helices, and in blue are b-sheets.24
Carbon–Carbon bond formation by transketolase26
Transketolase from E. coli K-12: naturally a dimer; however, only monomer is shown here. PDB: 2R8O;27 in black are loops and coils,
in green are helices, and in blue are b-sheets.26 This enzyme catalyzes carbon–carbon bond formation between 3-hydroxy-
2-oxopropanoate and b/g-hydroxylated aldehyde yielding two enantiomers.
A prime example for the need of biocatalysis. Penicillin, a
b-lactam antibiotic discovered by Alexander Fleming in 1928,
was found to be effective against a range of infections.28 As
propitious as this event presented itself, the organic chemists
of the World War II era considered the total synthesis of
penicillin to be an endearingly impossible task.28 With the
conglomerate work of 1000 chemists from 39 distinct labora-
tories, the synthesis of Penicillin V was published by John C.
Sheehan in 1957, thirty years after its discovery.22,28 With the
extensive manpower, time, knowledge, and money invested in
the production of penicillin, it became an objective to create
cost and time effective total synthesis of penicillin.22 However,
13 years preceding, the employment of biocatalysis for the
synthesis of penicillin yielded an astonishing 16 million doses
of the drug in the singular year of 1944 (Fig. 2).28 Today,
penicillin and various other b-lactam antibiotics can be synthe-
sized and purified within days by an individual when using
biocatalysis in lieu of the traditional chemocatalysis.
Advantages of stabilizing biocatalysts. Besides the previously
mentioned benefits with biocatalyst productivity, stable bio-
catalysts can be operated at higher temperatures or co-solvent
concentrations, with ensuing reduction in microbial contam-
ination, better solubility, and often a more favorable equili-
brium position, and can serve as more effective templates
for combinatorial and data-driven protein engineering, a
point firstly succinctly made by Bloom and Arnold32 and
Fig. 2 Biosynthesis of b-lactam ring from ACV catalyzed by Isopenicillin N Synthase. The bonds highlighted in red make-up the b-lactam nucleus, which is responsible
for antibiotic activity against targeted penicillin binding proteins.29–31
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 4/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6537
recently discussed as a key step in the development of indus-
trial biocatalysts.33
1.1 Enzymes are delicate materials, an issue of
thermodynamics
Although enzymes designed for industrial settings are opti-
mized for efficient synthesis of compounds, the route to
obtaining these stable biocatalysts is often challenging since
naturally enzymes have not evolved for industrial environ-ments.17,34,35 Biocatalysts have to be adapted to synthesis
requirements, i.e. they must be engineered to maintain proper
folding for catalytic efficiency under ambient conditions of
temperature and pressure in the presence of high substrate
and product concentrations.36,37 Thermodynamic stability
(T m, see Section 2.1) is a required component for industrial
applications; this important parameter concerns the folding
and unfolding process enzyme.38 For example, enzymes
involved in the production of organic compounds such as
ethanol for fuel must be thermodynamically stabilized in
solution with high concentration of organic solvent to prevent
misfolding. Enzymes such as DhA of the haloalkane dehalo-genase family from Rhodococcus rhodochrous are often used for
the synthesis of enantiopure organic compounds for organic
synthesis.39,40 DhA, however, with 1,2-dibromomethane as its
natural substrate, is unstable in solvent with high concen-
tration of organic materials such as DMSO.39 This destabilizing
phenomenon was eliminated by introducing bulkier and
mostly hydrophobic amino acids in DhA’s access tunnel, which
prevented the hydrophilic organic solvent from entering the
active site. Therefore, there were more of the natural substrate
and less of the deactivating DMSO in the active site, which
yielded kinetic stability.39 Similarly, a-amino ester hydrolases
have been shown to have great potential for industrial synthesis
of b-lactam antibiotics when compared to the industrially available penicillin G acylase.41 However, these hydrolases are
thermally instable as they are easily degraded into smaller peptides.
Their rapid degradation at just slightly above ambient temperatures
requires further stabilization of the enzyme (Fig. 3).42
Thermodynamic instability of enzymes can be partly attrib-
uted to the lack of rigidity within the tertiary structure. Such
lack of rigidity often is caused by a large fractions of less stable
and very flexible random coils or a-helices less stabilized due
to lower ionic interactions, in contrast to the more stable
b-sheets.38 The added rigidity through protein engineering,
must not, however, reduce the flexibility of the enzyme neces-
sary for catalysis. Subsequently, even in reactions occurring inorganic solvents, water plays a significant role in maintaining
the entropic stability of the protein structure, requiring a care-
ful balance of water in the organic medium.43 Additionally, the
role of salts, pH, ionic strengths, temperature, and chaperones
may need to be explored when stabilizing a biocatalyst.44,45
Multimeric enzymes often dissociate at unfavorable pH values
or ionic strengths, which in case of obligate multimers inacti-
vates the enzyme. As more parameters are to be optimized, the
more challenging it becomes to achieve thermodynamic stability.
However, these steps of stabilization generally must be determined
before attempting to achieve increased kinetics stability of
the biocatalyst.
1.2 Stable biocatalysts are a prerequisite for active
biocatalysts, an issue of kinetics
Biocatalysts may be delicate proteins, however, once stabilized,
they are enzymes efficient in catalyzing their respective reactions.36
Kinetic stability (k d, t 1/2, see Section 2.2) must be achieved to a
level satisfactory for large-scale process application. Kinetic
stability evokes resistance to degradation and maintained or
increased reaction efficiency of the enzyme. Nature has been
shown to be a master of evolving enzymes to kinetically and
thermodynamically adapt to their environments as shown by
psychrophiles, mesophiles, thermophiles, and hyperthermo-philes whose orthologous enzymes evolved to have mutated
amino acids that allow for distinct molecular interactions.48,49
The theory of neutral drift states that there are several types of
mutations that occur within an enzyme over time: (a) positive
mutations that drifted to maintain functionality (advantageous
mutations), (b) mutations with no effect on functionality
(neutral mutations), and (c) ‘‘negative’’ mutations that drifted
to alter the function of the enzyme (i.e. deleterious mutations).32,50,51
Neutral drift in the literature is not linked to thermal stabili-
zation. Nonetheless, neutrally drifted and thermostabilized
templates have higher tolerance for mutations; they are able to
tolerate considerable number of changes towards new properties. When attempting to stabilize an enzyme, all three types of
mutations are encountered, increasing the challenge of protein
engineering. In the laboratories, this process of neutral drift is
achieved by introducing random mutations through error-
prone PCR or DNA shuffling, and selecting those variants with
wild-type activity.51–53 However, the application of neutral drift
and finding of purifying mutations is very dependent on an
effective high-throughput screening or selection assay to obtain
the library of advantageous mutations. However, moving
toward a kinetically stabilized template based on neutral drift
Fig. 3 Both of the above enzymes belong to the a/b-hydrolases family
of enzymes. (a) A psychrozyme named a-amino ester hydrolase (AEH) from
Xanthomonus citri (PDB 1MPX) and (b) hyper-thermozyme from Archaeon
archaeoglobus fulgidus named carboxylesterase (PDB 1JJI).46 AEH from
Xanthomonus campestris, an orthologs, has a half-life of 30 minutes at 26.8 1C.42
It is a very fragile enzyme whose tertiary structure is composed of large amounts
of random coils. Carboxylesterase has significantly less random coils in its
structure and compared to its less thermostable counterparts, this enzyme has
shorter loops or random coils.47 Both enzymes are tetrameric.
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 5/32
6538 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
may results in libraries of enzymes with altered substrate
specificity. Depending on the precise screening method,
neutral drift might result in active and stable variants, or just
optimize the finding of active but not necessarily more stable
variants. ‘Neutral’ mutations in and around the active site may
result in increased promiscuity.32,51 In addition to improved
enzymes’ half-lives, improved thermostability has been linked to
enzymes’ increased tolerability for destabilizing mutations.32,51–53
Focus of current article. To increase thermodynamic and
kinetic stability, various methods including immobilization,
protein engineering, and medium engineering of biocatalysts
are to be considered.54 In the case of protein engineering, there
are three main approaches to enhancing the stability of protein
biocatalysts (Fig. 4): (i) rational design, based on knowledge of
the 3D-structure and the catalytic mechanism, (ii) combinatorial
design, requiring a protocol to generate diversity at the genetic
level, a large, often high throughput, screening capacity todistinguish ‘hits’ from ‘misses’, and (iii) data-driven design,
fueled by the increased availability of nucleotide and amino
acid sequences of equivalent functionality.
2. Different concepts of protein biocatalyst
stability
2.1 Three types of criteria are used to judge stability
The thermodynamic stability of a protein is indicated by the
melting temperature, T m (K or 1C), of the protein or the Gibbs
free energy of unfolding, DG (kJ mol
1
) (Fig. 5). For a two-stateunfolding process (eqn (1)) at the melting temperature, T m,
the equilibrium constant K is equal to unity. Thus, DGu = 0 at
T m, eqn (3).
Biocatalysts also exhibit a degree of kinetic stability at a given
temperature and salt concentration, reported as half-life t1/2(h or d) or, preferably, as an intrinsic ( i.e. mechanism-based)
deactivation rate constant k d (h1 or d1). As elsewhere in
kinetics, such intrinsic rate constants gave to be carefully
distinguished from observed rate constants. For enzymes with
a first-order rate of deactivation, the observed deactivation rateconstant, k d,obs is linked to the observable half-life:
t1=2 ¼lnð0:5Þ
kd;obs
(4)
As shown in eqn (11), the observed deactivation rate constant,
k d,obs depends on the unfolding equilibrium and thus not just
on kinetics.
Process stability or operating stability indicates the active
lifetime of a biocatalyst active during the process of the reac-
tion. Two criteria have been employed to characterize process
stability, the total turnover number TTN (eqn (5)) and the
productivity number PN or consumption number (eqn (6)).The more defined and more generalizable dimension of merit
is the dimensionless total turnover number TTN, which scales
the amount (mass) of product formed per unit mass of (bio)-
catalyst and which thus indicates the average number of
catalytic events per active site during the entire lifetime of the
biocatalysts. For enzymes under prevalent process conditions
(for details, see Sections 2.4 and 3.3), the TTN can be deter-
mined via eqn (5):
TTN ¼ ½Product
½ðBioÞcatalyst 0s active site¼kcat;obs
kd;obs
(5)
PN ¼ mass of product
; kg
mass of prepared catalyst (6)
The observed specific catalytic activity k cat,obs is based on the
active, i.e. native, enzyme concentration [N] at given tempera-
ture and time after starting the experiment or production run
(see Rogers and Bommarius).55 Too often, experimenters
assume that [N] equals the initial enzyme concentration [E]0but that is valid only at or close to initial conditions and at
temperatures far below the melting temperature.
Good productivity numbers (PNs) and total turnover num-
bers (TTNs) of more than 10 000 to 100 000 are not uncommon.
2.2 Thermodynamic stability
It is well established that the deactivation of a biocatalyst by
thermal or chemical means may proceed via multiple inter-
mediate states.55,100 Generally, any significant conformational
change to the native fold (hence catalytically active) of an
enzyme can be expected to eliminate activity. If one or more
of these alternate conformations are thermodynamically stable,
then there may exist in the system multiple enzyme species,
each with a varied degree of tertiary and/or secondary structure;
only one of the species is assumed to remain catalytically active.
Upon exposure to the system conditions, equilibrium in the
Fig. 4 Three modes of protein stabilization: (i) protein engineering, (ii) immo-
bilization, and (iii) formulation (medium engineering).
Fig. 5 N = native state; U = Unfolded state; K is the equilibrium constant; DG is
the Gibbs free energy.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 6/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6539
vast majority of cases, especially when not involving dissocia-
tion of multimeric enzymes, is rapidly established between all
of these stable states, according to Fig. 6.In Fig. 6, the native, folded state N is shown in equilibrium
with n stable intermediate states. It is implied that the ‘‘early’’
intermediate states (n = 1, 2. . .) represent smaller departures
from the native state; thus, they have lower energy barriers than
intermediates further to the right. Each of the equilibrium
constants K n, where K n = [I]n/[I]n1, may be modeled as a van’t
Hoff equilibrium according to eqn (8), in which D H n is the
enthalpy change at the transition state conformation, T n is the
characteristic temperature at which K n equals unity (analogous
to a melting temperature T m) and R is the universal gas
constant.
lnK n ¼DH
nR
1
T n
1
T (7)
At least one of the transitions, usually the last one, In1 to In,
leads to complete unfolding of the protein. The temperature of
this transition commonly is referred to as the melting tempera-
ture, T m, observable via several techniques (see Chapter 3.1). If
the temperature difference between T n and T n+1 is sufficiently
large, several transitions can be observed within one run.
Due to the rapid establishment of equilibrium, the apparent
initial catalytic activity in the system will be that of [N]0, the
initial concentration of active enzyme. This value must be
distinguished from [E]0, the concentration of total enzyme
initially charged to the system, in terms of all equilibriumconstants K n. The model in Fig. 6 generates the initial condition
shown in eqn (8):
½N0 ¼ ½E0
1 þPn j ¼1
P j i ¼1
K i
(8)
This relationship has two important ramifications for the reporting
of enzyme specific activity under a given set of conditions:55
– The starting enzyme activity is not [E]0 but [N]0, owing to
the instantaneous equilibration of the enzyme population, and
– An incremental increase in temperature causes a doubling
of the apparent specific activity, it must be noted that a smaller
fraction of the original enzyme is contributing to the observed
activity (a fraction of the enzymes is now inactive due to the
shift in equilibrium). Hence, the magnitude of increase of the
intrinsic specific activity, according to the above correction
factor, would be expected to be:
2 1 þXn j ¼1
X j i ¼1
K i
! (9)
In many cases, techniques such as differential scanning calori-
metry (DSC), differential scanning fluorimetry (DSF), circular
dichroism (CD) spectroscopy, or ANS fluorescence (see Section 3)
may be used to determine the number of intermediate states
involved in the deactivation of the biocatalyst, thereby removing
unnecessary degrees of freedom from the above model.
2.3 Kinetic stability
In addition to the loss of activity through a rapid shift in
thermodynamic equilibrium, one must consider the entropically-
driven and time-dependent denaturation of a biocatalyst in
parallel to catalytic events. From the general case set forth in
Fig. 7, it can be assumed that any of the stable intermediates
(or the native state) may deteriorate to the permanently unfolded state, D, and that each may do so according to a
different kinetic rate constant (states which are already ‘‘loosened’’
in terms of tertiary structure may deteriorate more readily). The
rate law for denaturation is assumed to be first order, which
leads to the following time-dependent model:
Each of the first-order deactivation rate constants may be
modeled by transition state theory according to eqn (10), where
DGn is the Gibbs free energy change upon denaturation, k B is
the Boltzmann constant, h is Planck’s constant, R is the
universal gas constant, and T is temperature, the underlying
independent variable that dictates the value of the rate constant
and, therefore, the speed of time-dependent denaturation at
any given temperature.
kd;n ¼K BT
h e
DGn
RT (10)
It was shown previously that the population of native enzyme at
time t , therefore, depends on both the equilibria between
available intermediate states and the first-order deactivation
rate constants for each of those states.55 Eqn (11) shows the
expression for [N](t ) in terms of the model in Fig. 7 and the
initial value of [N]0 given in eqn (3).
½NðtÞ ¼ ½N0e
kD;0þ
Pn
j ¼1
kD; j
Q j
i ¼1
K i
1þPn j ¼1
Q j i ¼1
K i
0BB@
1CCAt
(11)
With regard to process evaluation, the expression, in the
exponential multiplied to t , is essentially a weighed average of
the first-order rate constants among all of the species present in
the system. This weighted average is the rate constant, which
would be observed in an isothermal deactivation and will
hereby be abbreviated k d,obs.
½NðtÞ ¼ ½N0e kd;obsð Þt (12)
Fig. 7 Time-dependent denaturation from multiple equilibrium intermediates.
Fig. 6 General scheme for unfolding via multiple intermediate states.
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 7/32
6540 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
2.4 Process stability
Process or operational stability relates the kinetic stability of a
(bio)catalyst to its catalytic output throughout the (bio)-
catalyst’s lifetime. A typical process is run isothermally and at
saturating concentrations of substrate. At such conditions,
V max,t = k cat [N]t = k cat,obs[N]0. The product yield (y), in moles,
during the lifetime of the biocatalyst is then equaled:55
Z 10
V max;t dt (13)
With eqn (12):
y ¼ kcat;obs½N0
Z 10
ekd;obst dt (14)
which simplifies to:
y ¼ kcat;obs½N0
1
kd;obs
(15)
The TTN scales product yield to the initial biocatalyst concen-tration [N]0:
TTN ¼ y
½N0
¼kcat;obs
kd;obs
(16)
Use of eqn (12) assumes first-order deactivation kinetics of
the biocatalyst. Under thermal stress or in the presence of
chaotropes, this assumption is justified in the overwhelming
number of cases. First-order deactivation cannot be assumed if
aggregation (a higher-order process) is the method of deactiva-
tion.56 At a constant TTN, a very active but rather unstable
catalyst is equivalent to a slower catalyst with improved stability
over time.
3. Measuring stability of biocatalysts
3.1 Thermodynamic stability: DSC, DSF, CD, and fluorescence
Thermodynamic instability typically registers conformational
change of a protein from native state N to an unfolded state U
resembling a random coil. This change is modeled as reversi-
ble, N2 U, though by far not every protein unfolding event is
reversible. Upon unfolding, hydrophobic residues are exposed
to the solvent, allowing the application of fluorescence to
record a strong increase in emission upon unfolding. Similarly,
Circular dichroism (CD) uses circularly polarized light to
measure changes in ellipticity upon the disappearance of secondary structure elements, i.e. a-helices and b-sheets.
Additional methods of tracking thermodynamic instability
include differential scanning calorimetry (DSC) and differential
scanning fluorimetry (DSF). DSC measures the change in heat
during the unfolding of the protein. However, it has been a
popular method for non-high throughput studies. DSF, in
contrast, allows the analysis of samples in well plates. DSF
can be conducted with any instrument that allows accurate
measurement and ramping of temperature, such as a real-time
PCR machine. Major conformational changes are picked up via
fluorescence interactions with a dye probe molecule, such as
SYBER orange. The fitting procedure to calculate the midpoint
of unfolding, i.e. the melting temperature (T m) is similar to the
procedure used in fluorescence experiment. The procedure has
been described in detail by Niesen et al .57 Differential scanning
fluorimetry (DSF) has been compared with differential static
light scattering (DSLS) and isothermal denaturation (ITD) for
applications in protein stability and aggregation measure-
ments.58 DSF has also been applied to screen for ionic liquidsolvents suitable for stabilizing proteins.59
3.2 Kinetic stability: half-life, s1/2
Measuring residual activity over time at given conditions of
temperature, pH value, & salt and solvent composition allows
one to follow biocatalyst deactivation and will result in an
activity-time plot that can be fitted to an empirical rate law.
In most cases, i.e. if unfolding or chemical degradation is the
determining event, a first-order rate law will be observed with
respect to biocatalyst concentration, allowing one to extract an
observed first-order deactivation rate constant , k d,obs (h1 or d1).
Dominance of aggregation or autohydrolysis in case of proteases leads to a concentration-dependent behavior that
often can be fit via a second-order rate law.56 If a first- or
second-order plot does not fit the data, the apparent reaction
order can be obtained via a Levenspiel plot of ln(dimensionless
rate) vs. ln(1-degree of conversion) or ln(d X /dt ) vs. ln(1 X ), the
slope of which equals the apparent reaction order.60 However,
in the authors’ own experience, the accessible range of bio-
catalyst concentration often does not lend itself to accurate
Levenspiel plots.
An empirical fit of residual activity over time does not reveal
any details about the mechanism of deactivation. Consequently,
the rate constant is an observed one; to obtain an intrinsic
deactivation rate constant , k d, a mechanistic representation of the events leading to deactivation is required. For the Lumry–
Eyring mechanism N2 U- D,100 the observed and intrinsic
rate constants, k d,obs and k d, respectively, are linked via:
kd;obs ¼ kd
1 þ K (17)
K ¼½N
½U (18)
with eqn (18) as the folding equilibrium constant.61 The crucial
difference between k d,obs and k d is to be noted when simplifying
eqn (17) in different temperature regimes.(i) High T , or T > T m: then [U] c [N], thus K { 1, and
k d,obsE k d(ii) Moderate T , or T o T m: then [U] o [N], thus K > 1, and
k d,obsE k d/(1 + K )
(iii) Low T , or T { T m: then [U] { [N], thus K c 1, and
k d,obsE k d/ K
Extreme care is required when attempting to determine k dby calculating k d,obs.
Experimentally, it is often more convenient to express bio-
catalyst stability in terms of the half-life t1/2 (h or d), i.e. the
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 8/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6541
time at which 50% of the initial activity is left. For enzymes that
deactivate according to a first-order rate law, the observed
deactivation rate constant k d,obs is linked to the observable
half-life by a t1/2 = (ln 0.5)/k d,obs, eqn (4). In case of measuring
T 3050 or T 6050 (see Section 3.5), the desired temperatures are those
at which the half-lives are 30 or 60 minutes, respectively.
3.3 Process stability: total turnover number TTN
The dimensionless TTN scales the observed catalytic constant k cat,obs to the observed deactivation rate constant k d,obs (for
first-order deactivation processes), as shown in eqn (10).55
Eqn (10) is valid (i) at saturation of all substrates, i.e. if k cat and not just an apparent k cat,app can be measured, (ii) if the
native enzyme concentration at time t , [N]t , is known, (iii) under
isothermal conditions, i.e. both k cat,obs and k d,obs have to be
measured at the same temperature, most often a temperature
meaningful for a process, and (iv) at the same pH value as well
as salt and co-solvent composition.
There are two other methods to determine TTN:
I. If substrate can be supplied over the lifetime of a bio-
catalyst, which usually but not always requires the use of acontinuous reactor, conversion can be followed over time and
the amount of product compared to the amount of initial
(bio)catalyst employed;
II. If temperature deactivation dominates, the biocatalyst
can be exposed to increasing thermal stress by ramping up the
temperature, measuring the instantaneous conversion, again
often as output of a continuous reactor, and fitting the time–
temperature–conversion data to a kinetic model of both activa-
tion and deactivation. This method has been demonstrated on
the example of pen G acylase by Rogers et al.62
In applied biocatalysis, such as in many industrial situa-
tions, where the purity of biocatalyst often is not known,
biocatalyst stability is expressed as an enzyme consumptionnumber (ECN):
ECN ¼ 1
PN¼
amount of enzymes spent ðgÞ
mass of product ðkg or lbÞ (19)
PN is defined in eqn (6).
The value of ECN depends on process parameters such as
temperature, pH value, & concentrations of substrate(s) and
product(s). With the molar masses of enzyme and product, ECN
and TTN can be interconverted:
TTN / 1
ECN
MWenzyme
MWproduct
(20)
It should be emphasized that TTN is not a completely suitable
quantity for the evaluation of operating stability because the
number of moles of biocatalyst is not a suitable reference for
the complexity and cost of its manufacture. However, the values
for both numerator and denominator in eqn (20) are usually
known and can be expressed in monetary terms. To assess the
application of a biocatalyst, the contribution of the biocatalyst
to the overall cost can be assessed readily.
An empirical value for the amount of enzyme required to
generate a certain amount of product can be gained via observing
conversion over time in a continuous stirred tank reactor (CSTR).
The relevant parameter for studies of operating stability of
enzymes is the product of active enzyme concentration, [E]active,
and residence time, t; [E]activet. In a continuous stirred tank
reactor (CSTR), the quantities [E]active and t are linked by the
following equation:
½Eactivet
½S0
¼ x
rðxÞ
(21)
where [S0] denotes the substrate concentration in the feed,
assumed to be constant, x is the degree of conversion, and
r (x) the conversion-dependent reaction rate.63–65 A stable
enzyme leads to a constant degree of conversion x. How-
ever, an unstable enzyme will lead to a decreasing degree of
conversion over time. To keep up the initial degree of conver-
sion and thus the output of product per unit time, either the
residence time t can be increased or, fresh enzyme can be
added, increasing [E]active. Thus, over time, the amount
of [E]active required per unit mass of product can be determined,
providing a hands-on empirical measure of total turnover
number.
3.4 Quick stability criteria: T 50 and C 50
Often, the measurement of the half-life or the deactivation rate
constant is not desirable because it would take too long to
deactivate a biocatalyst, or because a library would create too
many data points at once when following deactivation behavior
of many variants simultaneously. In such cases, T 50 (in the case
of temperature-dependent deactivation) or C 50 (in the case of
chaotrope-dependent deactivation) are rapid and useful indi-
cators of deactivation, often over 60 min, resulting in T 6050 or C 6050data. T 6050 is the temperature at which the measured residual
activity after 60 min is 50% of its initial value. The quantity isneither a thermodynamic nor a kinetic one but encompasses a
mixture of both.
By definition, T 6050 is the temperature at which [N](t )/[N]0 =
0.5, where [N] is the concentration of native, folded (hence
catalytically active) biocatalyst present in the system. Combin-
ing this fact with the result in eqn (12) for t = 3600 s gives the
following:
ln(0.5) = (k d,obs)(3600) (22)
This leads to the idea that, at T 6050, the observed first-order
rate constant of the biocatalyst takes on a particular value:
k d,obs = 1.925 10
4 s
1 (23)
As shown in eqn (11), k d,obs is a quantity which depends on
the thermodynamic equilibrium between intermediate states as
well as the propensity of each state to undergo time-dependent
denaturation. If information about mechanism or number of
intermediates is known, a priori , then a restricted model can be
imposed and the T 6050 can be calculated directly. To illustrate, it
will be assumed that a biocatalyst follows a deactivation
mechanism akin to the Lumry–Eyring mechanism: N2 U2 D.
For such a system, k d,obs at the T 6050 can be expressed as a
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 9/32
6542 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
function of individual deactivation rate constants and the
equilibrium constant between N and U:
kd;obs ¼ kd;1K 1
1 þ K 1
¼ 1:925 104 (24)
The above equation may be rewritten by expressing the
deactivation rate constants and the equilibrium constant in
terms of their transition state and van’t Hoff definitions,
respectively:
1:925 104 ¼
kBT
h e
DG1
RT
e
DH 1R
1T 1
1T
!
1 þ e
DH 1R
1T 1
1T
0BBBBB@
1CCCCCA (25)
Assuming typical parameters for unfolding and
deactivation,62,66,67
D H 1 = 108 kJ mol1
DG1 = 98 kJ mol1 T 1 = 325 K
the value of T , which is equivalent to T 6050, is then found by an
iteration with the desired level of convergence (here, to match
the value of k d,obs within 1 107) to be T 6050 = 319.1 K. So even
at 6 K below the melting temperature T m of 325 K, the enzyme is
stable only for an hour.
3.5 Additional stability indicators: T opt or T max
T opt for enzyme reactions: Peterson et al.67 define a parameter
T eq, an equivalent of the melting temperature, T m. Peterson
demonstrates that the optimum temperature, T opt , for an
enzyme reaction, i.e. the temperature at which a maximum
rate is observed that is determined from dV max
/dT with an
analytical expression, is very close to the observable maximum
in a plot of rate as a function of temperature:
T opt E T m(1 aT ) (26)
a ¼ R
DH mln
DH m
DGacat
1
(27)
Since typically, 5 105o ao 104, a T m{ 1; and thus
T opt is approximately equal to T m with a difference between T opt and T m of less than 0.018 1C. This fact had been surmised by
many practitioners but had never been supported with experi-
mental results. While the significance of T opt may seem
obvious to anyone trying to optimize the rate of an enzymaticreaction, Peterson et al. point out the significance of the
melting temperature T m next to T opt : the magnitude of T mreflects thermostability, as does the deactivation rate constant
k d, and also reflects evolutionary adaptation as the melting
temperatures of proteins often correlates with the preferred
habitat of the organism harboring the proteins.
Sometimes the T opt is referred to as T max ; while T opt commonly is utilized in connection with impending unfolding
upon further heating and T max can occur at temperatures
significantly below the melting temperature, the difference
between T max and T opt often is rather semantic. T max for enzyme
reactions: Burkholderia cepacia lipase immobilized on porous
ceramic particles, lipase PS-C II (Amano Enzyme Inc.), gave an
enantiopure product at 40–120 1C in the kinetic resolution of
1,1-diphenyl-2-propanol, with the highest conversion (39%) at
80–90 1C.68
4. Modes of stabilizing biocatalysts:immobilization, medium engineering,
protein engineering
4.1 Mode 1: immobilization
Overview. Stabilization of biocatalysts through immobilization
have been employed by industries since the 1960s for several
reasons.69 Immobilization of biocatalyst is a method by which
an enzyme is bound to a solid support through covalent or ionic
interactions, or via entrapment or crosslinking, to eliminate
diffusion of the enzyme in solution. Site-directed mutagenesis
can be employed to introduce amino acids that would facilitate
these desired types of interactions in the desired regions of the
protein. Immobilization allows for the recyclability of bio-catalysts as well for creation of robustly bound enzymes.45
Additionally, immobilization can be used in reactions requiring
biphasic systems where the product of the reaction is immedi-
ately and automatically isolated from the reaction phase. In the
case of using enzymes as biosensors, immobilization provides
the optimal control of the enzyme’s orientation as needed by
the system.34 For biodiesel production, immobilization of
enzymes yields an environmentally friendly process since
operating temperature tends to be lower while purification of
products is eased.70 Most importantly, however, the method
of immobilization can be used to stabilize enzymes both
thermodynamically and kinetically as well as change theirfunctionality.71 This phenomenon was shown by both Grazu
et al. and Cecchini et al. where multipoint covalent immobiliza-
tion of penicillin G acylase resulted in several stable variants of
the enzyme (Fig. 8).72,73 Both groups showed that by combining
site-directed mutagenesis and immobilization of enzyme in
Fig. 8 Grazu et al. showed that multipoint covalent immobilization can stabilize
the protein depending on the locations vis-a-vis the active site.72
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 10/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6543
specific regions, generally far from the active site, a stable
variant with improved reaction specificity can be created. Using
this method, an astonishing kinetic stability of penicillin G
acylase was shown by the significant reduction of the undesired
secondary hydrolysis.73
Integrative chemistry. Integrative chemistry is a newly emer-
ging concept where the designs of complex hierarchal struc-
tures inspired by nature are constructed with interdisciplinary
tools including those of chemistry.74,75 Brun et al. used inte-grative chemistry-based rational design to create the first lipase
hybrid macrocellular biocatalyst, triacylglycerol acyl hydro-
lase.74 Immobilization was used as a method of stabilization
and also to create recyclable reaction materials. Lipases from
Candida rugosa (CRl) and Thermomyces lanuginosus (TLl) were
used in their study; the former enzyme is known for its high
substrate specificity and reactivity and the latter for its high
reactivity in transesterification reactions involving the produc-
tion of biodiesel. Both enzymes were stabilized with porous and
(3-glycidyloxypropyl) trimethoxysilane (Glymo)-functionalized
silica-based hybrid macrocellular monolithic foams that are
capable of stabilizing the interface between water and oil; thissystem was dubbed Si(HIPE).
Surface display. Another example of immobilization concerns
recombinant whole-cell biocatalysis with the enzyme expressed
at the surface of the cells. The final result is an active enzyme
anchored at the cell surface.76 Kim et al. used aga2 protein
fused to epoxide hydrolase to anchor the protein to the surface
of the Rosetta strain of E. coli . Using this method, the authors
eliminated isolation and purification of epoxide hydrolase,
which allows for the maximum use of the enzyme during the
initial period of high activity right after protein synthesis and
folding. In addition, the enzyme, while in the cell, is stabilized
by multiple factors that may be unknown to the experimenter.
Kim et al. also found that the enzyme had an improved K M value of 2.6 mM with this process.
Fusion protein. Immobilization of a fusion protein of a
cellulose-binding domain (CBD) and the catalytic domain of
horseradish peroxidase (HRP) onto cellulose beads preserved
both binding and catalytic function and resulted in six-fold
enhanced stability of the construct in buffer. Cellulose-bound
CBD-enzyme was used for the first time in aqueous-organic
solvents.77 With increasing THF, acetone, acetonitrile, or ethanol
concentration, activity decreased but stayed above the level of
soluble enzyme forms. The soluble CBD-HRP fusion protein
featured lower activity than native HRP but higher thermal and
solvent stability. The immobilized fusion protein was found tobe more stable in aqueous acetone of any composition.
Single-walled carbon nanotubes (SWNTs). Single-walled carbon
nanotubes (SWNTs) were found to stabilize enzymes against
high temperatures and organic solvents than flat surfaces. The
curvature of SWNTs, which suppresses unfavorable protein–
protein lateral interactions, is the cause of the stabilization, as
was found through both experiments and theoretical analysis.
Curvature-based stabilization should not be limited to SWNTs
but should be a general property of nanomaterials.78 Similarly,
highly curved surface of C60 fullerenes enhances the stability of
soybean peroxidase at 95 1C. The half-life at that temperature of
soybean peroxidase adsorbed onto fullerenes was measured to be
2 h, compared to 25 min when adsorbed on graphite flakes, a flat
support, and 10 min for the native, soluble enzyme.79 This
phenomenon is not limited to fullerenes, but can also be extended
to other nanoscale supports including silica and gold nano-
particles, or highly active and stable polymer-nanocomposite films.
4.1.1. More examples of stabilization through immobilization.
Case I. Penicillin G acylase (PGA) was stabilized through variousmeasures: (i) insertion of a cysteine, each in different regions of
the lysine-rich enzyme surface and oxidation of the sulfhydryl
sidechain groups to form disulfide bonds, which stabilize the
enzyme via freezing of configurational entropy, (ii) subsequent
immobilization on disulfide/epoxide supports, and (iii) a series
of point mutations to increase resistance against heat and
organic solvents. The best immobilized PGA variant was
300 000 times more stable than the soluble wild-type, as judged
by a combination of increased temperature and half life, and
thus very suitable for various reactions of interest in fine
chemistry and pharma, including penicillin G hydrolysis,
enantioselective hydrolysis and synthesis reactions. Immobili-zation on disulfide-epoxide (Eupergit) and glyoxyl-agarose
supports rendered biocatalysts of comparable stability. The
stereochemistry of the PGA variants did not vary significantly
from that of the wild-type.80
Case II. PGA has also been encapsulated into a rigid poly-
vinylalcohol matrix, termed Lentikats, in the form of cross-
linked enzyme aggregates (CLEAs) to compensate for the
inadequate mechanical properties of CLEAs. While encapsula-
tion in Lentikats decreased CLEA activity by 40%, its improved
stability more than compensated for the decreased activity.
Partitioning studies of the dioxane solvent into the Lentikats
found that the solvent concentration inside the Lentikats was
lower than in the bulk and lent credence to the hypothesis that the hydrophilic environment in the matrix is primarily respon-
sible for increased solvent stability; indeed, thermal stability
was comparable to the corresponding CLEA. The PGA-CLEA
Lentikats with their favorable substrate and product partitioning
yielded a significantly higher reaction rate as well as enhanced
yield in the thermodynamically controlled synthesis of cephalo-
sporin C (ceph C) from phenylacetic acid and 7-amino-
deacetoxycephalosporanic acid (7-ADCA).81
Case III. In the next example, immobilization of formate
dehydrogenase (FDH) on highly activated glyoxyl agarose, in
contrast to other matrices, increased its stability against several
distorting influences, such as extreme pH value, high tempera-ture, presence of organic solvent, or contact with hydrophobic
interfaces as a consequence of stirring. Biocatalyst optimized
with respect to activation of the support, time and temperature
of immobilization retaining half the initial activity was stabi-
lized 50-fold at high temperatures and neutral pH value and at
10 1C with pH 4.5. At acidic pH value, where inactivation is
preceded by subunit dissociation, immobilization under such
optimized conditions stabilizes the quaternary structure of
dimeric FDH and stabilizes the glyoxyl-agarose-FDH by hun-
dreds of times over the wild-type.82
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 11/32
6544 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
Case IV. Brun et al. had created a significantly improved
efficiency of CRl in the synthesis of butyloleate ester, which is
used as a lubricant in biodiesel.73 Stabilized CRl showed an
overall 26 fold improvement in reactivity. In addition, the enzyme
was recycled 19 times during which it maintained 100% catalytic
efficiency! TLl was shown to also have increased catalytic effi-
ciency of 80% hydrolysis of triester of olive oil in non-aqueous
medium after four rounds of recycling and up to 65% in 8 rounds.
4.2 Mode 2: medium engineering
A second method of stabilization of proteins relies in formulating
favorable buffer conditions. Here, both salt concentration and
salt type are important.
Salt concentration. The concepts for the quantitative descrip-
tion of the influence of salt concentration have been developed
since the 1880s but mainly in the 1930s. In 1889, Setschenow
published his law on the dependence of the logarithm of
protein solubility on salt concentration.83
log P0
P ¼ ks½Csalt
(28)
with [P]0 and [P] the protein concentrations in presence and
absence of salt, respectively, and the k s the Setschenow coeffi-
cient. Soon after, it became clear that not the ion concentration
[C] but the ionic strength, I , was the appropriate descriptor of
salt concentration. In the 1920s, Cohn and Edsall developed
their law for fractional crystallization of proteins, which was
published in 1943:84
I ¼1
2
XZ 2mi ; in mol L1
log E 0ð Þ ¼ log S 0ð Þ K sI
(29)
Debye and H +uckel treated a point charge in a medium of
uniform dielectric constant e and based on this concept devel-
oped the Debye–H +uckel law (eqn (30)) and, at high ionic
strength, the extended Debye–H +uckel law (eqn (31))85 written
here for proteins and salts:
log( y) = A|Z proteinZ salt | I 1/2 (30)
log( y) = [ A|Z proteinZ salt | I 1/2] + [ AA*|Z proteinZ salt | I ] (31)
with [E] = enzyme solubility [g/L], b = protein solubility at zero
ionic strength I (log[E]0), and K s denoting the salting-out
constant. Except for utilizing I and [C]salt , eqn (28) and (29)
are mathematically equivalent.
Debye and H +uckel treated a point charge in a medium of
uniform dielectric constant e and based on this concept
developed the Debye–H +uckel law (eqn (30)) and, at high ionic
strength, the extended Debye–H+uckel law (eqn (31)).85 To
make use of the Debye–H +uckel law for enzyme deactivation,
we apply transition state theory, which predicts that k d =
k d,0(gproteingsalt /ga), so eqn (32) results:
log(k d) = log(k d,0) A|Z proteinZ salt | I
1/2
(32)log(k d) = log(k d,0) [ A|Z proteinZ salt | I 1/2] + [ AA*|Z proteinZ salt | I ]
(33)
Surprisingly few enzymes have been analyzed with respect to
the influence of salt concentration on stability or activity,
whether with the (extended) Debye–H+uckel model, or other-
wise. Examples include glucose dehydrogenase86 and formate
dehydrogenase.87
Salt type: influence of ions on biological molecules: the Hofmeister
series. The type of salt in most instances is even much more
important than its concentration. The significance of ion type
for protein solubility and precipitation has been known sincethe 1880s, when a pharmacist, Franz Hofmeister, observed
the dependence of the precipitation of hen-egg white lysozyme
on the type of salt 83 and qualitatively ranked the influence
of ions.
The non-specific influence of ions on many biophysical
phenomena, such as salting-in and salting-out of proteins,83,88
melting of gels,89 DNA helix-coil-transitions,90 and ion
channel formation,91 has been described qualitatively by the
Hofmeister series (1888),88 which classifies and ranks ions as chao-
tropes (water-structure breaker) or kosmotropes (water-structure
former).
Fig. 9 ranks some common anions and cations in terms of
chaotropicity and kosmotropicity, starting with the strong chaotropes Cs+ and thiocyanate (SCN) via the indifferent Cl
and Na+ ions to the strongly kosmotropic phosphate and Mg 2+
ions. In the case of well-folded enzymes, addition of destabilizing
(chaotropic) ions tends to lead to increased unfolding or
deactivation, whereas addition of stabilizing (kosmotropic)
ions tends to support preservation of activity.
Two of the most relevant of the few attempts towards a
quantification of the Hofmeister series are the dipole-cavity
model and the ion hydration model .
The dipole-cavity model , treating the protein as a dipole in a
high ionic strength aqueous solution (Kirkwood model),92
attributes the influence of ionic strength on a protein to
Fig. 9 The Hofmeister series.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 12/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6545
electrostatic (DGelectrostatic) and surface energy (DGcavity ) forces93
and arrives at the following eqn (34):
DG0 ¼ DGcavity þ DGelectrostatic
¼ A BI 0:5
1 þ CI 0:5 ðL OgÞI (34)
( A, B, C and D are constants, L and Og represent the
electrostatic and hydrophobic terms, respectively, and g is the
surface tension of the solvent.) As the surface tension g can be
written as g = g0 + Ds, with Ds as the surface tension increment
attributable to the salt, the Debye term becomes unimportant
at high salt concentrations and eqn (34) simplifies to eqn (35),
written here for rate constants:
log(k ) = log(k 0) (L ODs) I (35)
Eqn (35) resembles the Setschenow (1889)83 (eqn (28))
and the Cohn–Edsall eqn (29) (1943)84 for the solubility
of proteins.
The ion hydration model 84,94–97 describes the influence of salt
as an interaction of chaotropes and kosmotropes with layers of
hydration on the surface of the respective ion, captured by the Jones–Dole equation:98
Z
Z0
¼ 1 þ Ac0:5 þ Bc (36)
Z and Z0 are the viscosities in salt-containing and salt-free
medium, respectively; again, the Debye–H+uckel term, Ac0.5,
becomes insignificant at high salt concentration (c E 1 M),
so that relative viscosity Z / Z0 depends linearly on ion–solvent
interactions exerted by chaotropes (with a Jones–Dole B coeffi-
cient o 0) or kosmotropes ( B > 0). B values are approximately
additive for individual ion species and are available from the
literature. Usually, only anions have to be considered, as they are more strongly hydrated than cations.99
The observed kinetic deactivation constants, k d,obs, for the
deactivation reaction of native protein (N) to some deactivated
state (or ensemble of states) (D), eqn (37), strongly correlate
with the Jones–Dole B-viscosity coefficient 98 of the anion of
chaotropic salts ( B o 0) and show little variation in solutions of
kosmotropic salts ( B > 0).100 As B-viscosity coefficients are
indicative of ion hydration,95,101,102 this observation suggests
that ion hydration strongly influences a salt’s effect on a
protein and that B-viscosity coefficients may be useful for
predicting salt effects on protein deactivation.
N !kd;obsD
Specifically, the model should predict invariance of k d,obs with kosmotropic B-viscosity coefficients as well as the observed
logarithmic dependence of k d,obs with chaotropic B-viscosity
coefficients. This bimodal behavior suggests that the observeddeactivation might be the result of competing, parallel pro-
cesses, rather than unfolding and irreversible processes in
series, as implied by the Lumry–Eyring model.103,104 In addi-
tion, it would be convenient to incorporate dependencies of
k d,obs on water activity (i.e. salt concentration) as well as
temperature into the model so that salt-induced deactivation
can be predicted under a variety of solution conditions.
As seen in Fig. 10, a three-parameter exponential relation-
ship with the form of eqn (37) is able to model these
characteristics:
k d,obs = [k p + k c exp{o B}] (37)
The theoretical curves of eqn (38) generated using assumed
values for k p, k c and o shown in Fig. 10 bear striking resem-
blance to the experimental log (k d,obs) vs. B-viscosity coefficient
data and suggest that the model might be used to describe salt-
induced protein deactivation.
The overall model from salt-induced protein deactivation
gives:104
kd;obs ¼ kp þ kc exp Bo0 þ o0 ln aw
RT
(38)
Provided k p, k c, o0, and o 0 are known for a protein–solvent
combination, the model presented can be used to predict thedeactivation constants of proteins in varying salt solutions at
varying water activities and temperatures. Table 1 summarizes
the model parameters obtained for HL-ADH, a-chymotrypsin,
and mRFP.100
More recently, the dichotic behavior of the deactivation or
misfolding rate constant as a function of the Jones–Dole
B-viscosity coefficient has been extended to the misfolding of
yeast prions105 and monoclonal antibodies.106 In the following
section, several other examples of the model will be presented.
Fig. 10 Correlation of deactivation rate constants of HL-ADH (60 1C), a-chymotrypsin (50 1C), and mRFP (80 1C) with B-viscosity coefficients. All deactivations occurred
at pH 7.0. Deactivation rate constants were measured in salt solutions with water activities of aw = 0.95 (K), 0.97 (O), and 0.99 (.). Deactivation constants appear to
vary linearly with chaotropic B-viscosity coefficient (B o 0) but are relatively unaffected by kosmotropic salts (B > 0). Taken from Broering.100
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 13/32
6546 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
4.2.1. Examples of stabilization of biocatalysts through
medium engineering. Case I. To understand in details the
mechanism of action of trehalose, a known exceptional stabi-
lizer of proteins retaining enzyme activity in solution as well as
in freeze-dried state, a thorough investigation of its effect on
the thermal stability of solutions of RNase A was conducted
(Fig. 11).107 A 2 M trehalose solution raised T m of RNase A by as
much as 18 1C and the Gibbs free enthalpy (DG) by 20.1 kJ mol1
at pH 2.5, while decreasing the heat capacity of protein dena-
turation (DC p) in trehalose solutions. Such findings point
toward a general stabilization mechanism due to the elevationand broadening of the RNase A stability curve (DG vs. T ). Four to
seven molecules of trehalose were found to be excluded from
the protein surface upon denaturation at 1.5 M sugar. Investi-
gation of stability as a function of pH value suggested that
while the charge of RNase A can contribute significantly to its
stability, trehalose acts as a universal stabilizer of protein
conformation due to its exceptional effect on the properties
of solvent water around the protein.107
Case II. The irreversible thermo-inactivation of savinase at
70 1C due to autodigestion limits its industrial utilization in
detergents. The effect of sorbitol and trehalose on the stability
of savinase revealed that presence of either osmolyte prevented
autolysis of savinase at 70 1C and increased the protein’sstructural and kinetic stability.109
Case III. The apparent melting temperature, T m, of papain
shifted from a control value of 831 to a value of >90 1C in
presence of 40% sorbitol, as measured by fluorescence spectro-
scopy and differential scanning calorimetry (DSC); maximum
stabilization was seen in case of 30% sorbitol. Transitions of
both domains of papain shifted to higher temperatures. Activity
measurements with papain indicate several-fold increase in the
protein’s thermal stability in sorbitol, sucrose, xylose, or glycerol.
Partial specific volume measurements of papain in presence of these co-solvents revealed that the preferential interaction para-
meter (x3) was negative in all co-solvents and that maximal
hydration was observed in the case of glycerol where the
preferential interaction parameter was 0.165 g g 1. These results
suggest that papain is thermally stabilized in presence of polyol
co-solvents as a result of preferential hydration.110
Case IV. To investigate the effect of polyols on thermostability
and the anomalous surface tension behavior of glycerol, a
highly thermolabile two-domain protein, yeast hexokinase A,
has been monitored via differential scanning calorimetry (DSC)
and loss of activity over time. Upon addition of polyols, the DT m
for domain 1 was larger than DT m for domain 2, increasingly proportionally with the number of hydroxyl groups in polyols,
with sorbitol as the best stabilizer against both thermal stress
and chemical denaturation by urea. The increase in T m (DT m),
the retention of activity, and the increase in the surface tension
of polyol solutions correlate very well, except glycerol. However,
the DT m values show a linear correlation with apparent
molal heat capacity and volume of aqueous polyol solutions,
including glycerol. These results indicate that interfacial proper-
ties do not always correlate well with stabilizing effects, while
bulk solution properties contribute significantly to protein
stabilization. Various weak binding and exclusion effects of the
osmolytes scaled by the effects of water form a subtle balance
and influence stabilization. These effects must be understoodfor rational design of stable protein formulations.111
Table 1 Values for the three-parameter model given by eqn (28) using protein-
specific values for k p and k c. Dimensions of k p and k c forchymotrypsin are (mM min)1.
Taken from Broering et al.104
HLADH Chymotrypsin mRFP
k p (min1) 6.85 104 52.1 2.13 103
k c (min1) 3.13 104 40.8 8.03 104
o , a w = 0.95 (M) 115 4.0 27.8 13.2 61.2 2.9o , a w = 0.97 (M) 96.7 7.0 33.1 3.6 44.8 1.6o , a w = 0.99 (M) 70.0 3.1 23.2 3.2 31.6 2.2
Fig. 11 View of RNase A from Bos taurus at three different angles. PDB: 2E3W.108 The disaccharide is trehalose, which was shown to stabilize RNase A.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 14/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6547
Case V. The hydrolytic activity of penicillin V acylase (PVA,
EC 3.5.1.11) can be enhanced by adding monophasic organic
solvents but this addition also in many cases decreases the
stability of the enzyme (Fig. 12). Streptomyces lavendulae PVA’s
threshold monophasic organic solvent concentration, at
which half of the initial activity in purely aqueous systems is
recovered, in water–organic monophasic systems correlateslinearly with the free enthalpy of denaturation at 40 1C. Solvents
with log( P ) values o1.8 exert protective effects, those with log
P values >1.8 have a denaturing effect; both effects are
exacerbated at higher solvent concentrations. Deactivation rate
constants of PVA at 40 1C can be predicted in any water–organic
monophasic system.112,113
4.3 Mode 3: protein engineering
Protein engineering has been employed since the 1980s,
starting in the laundry detergent industry 115,116 This method
of stabilization differs from immobilization in that it requires
changes to the primary structure of the enzyme to confer thedesired features. Quite often, however, protein engineering is
used in conjunction with immobilization to stabilize proteins
as shown by Cecchini et al. Over the years, as described by the
theory of neutral drift, nature has evolved to select for certain
existing mutations (neutral, deleterious, or advantageous)
within orthologous proteins, which tend to suit their respective
host organisms.117,118 The laboratory settings attempt to mimic
nature, a process known as laboratory evolution.119 However,
rather than selecting for already present mutations within a sea
of bacteria medium, researchers or protein engineers study
orthologous enzymes and use other methods to introduce new
mutations for creating the desired kinetically and thermo-
dynamically enhanced protein variants. Three methods of
protein engineering are often used: rational design, combinatorial
design, and data-driven protein design.
4.3.1. Rational protein design. Rational design of bio-
catalysts is one of the most commonly used methods of proteinengineering primarily because of the efflux of available data
on protein sequence, structures, and more. The ideology of
rational design is based on studying available structures and
sequences of already known stable proteins; based on the
interactions of their respective amino acids, which are assumed
to participate in stabilization, a new variant of the protein of
interest is created through site-directed mutagenesis.120
Respectively, this method requires an extensive knowledge of
the protein of interest.1,121 However, public databases are now
rich of protein structures, although not all proteins are found
within that database and not all proteins have stable orthologs,
i.e. a-amino ester hydrolase.122
In addition, it is often difficult to determine which of these mutations found in the ortho-
logous enzymes were selected as neutral, deleterious or advan-
tages through evolution.118 Even if those mutations were
determined, the complex interactions between the primary,
secondary, and tertiary structures are often difficult to predict
or fully account for with increasing size of the protein.123 Two
theories are currently given as to how nature evolves these
enzymes: (a) the neo-Darwinian hypothesis that states that
positive mutations have already been installed in the primary
sequence of the protein and that neutral and deleterious
Fig. 12 Penicillin V Acylase from Lysinibacillus sphaericus. PDB: 3PVA.114
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 15/32
6548 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
mutations are then added over time; (b) the competing hypo-
thesis is that which states that neutral mutations confer flexibility
for evolution at the addition of deleterious or advantageous
mutations, which are selected under certain conditions.117
However, both theories agree that the sole examination of
protein structures and sequence will not always yield the
desired answers. Thus, other methods are used in addition to
rational design. Combinatorial design, discussed in the follow-
ing section, is used to account for some of the structuralinteractions within an enzyme.124
4.3.2. Examples of rational biocatalyst design. Case I. The
stability of prolipase from Rhizopus oryzae (proROL) toward
lipid oxidation products such as aldehydes was insufficient for
use in the oleochemical industry. To increase stability, 6 Lys
and all 6 His residues except for the catalytic histidine out of 22
amino acid residues (15 Lys and 7 His), prone to react with
aldehydes, were subjected to saturation mutagenesis. Active
variants were prescreened by an activity staining method on
agar plates to quickly and reliably identify mutants within the
resulting libraries that lead to more stable variants. Active
mutants were expressed in E. coli in a 96-well microtiter plateformat and challenged with octanal as a model-deactivating
agent. The most stable histidine variant (H201S) increased
stability by 60%, or even 100% in combination with a lysine
variant (H201S/K168I). Interestingly, the variants still displayed
essentially wild-type catalytic activity.125
Case II. LipC from Pseudomonas aeruginosa is a cold-adapted
and cold-acting lipase, highly salt- and heavy metal-tolerant,
with different substrate specificity at higher temperatures; all
these properties are biotechnologically and environmentally
relevant, where often activity at low temperature is sought.
However, low thermostability of LipC prevents its application
in long-term processes. Specific sites on the sequence were
picked for modification according to the highest flexibility onthe 3D model structure to increase LipC thermostability but
without decreasing cold-adapted properties. Eight mutant
libraries plus two point mutations were constructed and more
than 3000 mutant clones screened for successful LipC variants:
a LipC double variant with conserved cold-adapted properties
and 7-fold increased thermostability over the wild-type LipC
was found and has the desired properties for use in processes at
low temperatures (4–20 1C).126
Case III. Large-scale synthetic applications of cyclohexane
monooxygenase (CHMO) are hampered, however, by the
instability of the enzyme mostly owing to oxidation of cysteine
and methionine residues. Exhaustive mutation of all methio-nine and cysteine residues in the wild-type CHMO revealed that
oxidation-labile residues are mostly either surface-exposed or
located within the active site. Combinatorial mutation of two
methionine residues identified for thermostabilization but
buried within the folded protein led to two variants with either
oxidative or thermal stability, while keeping specific activity
and enantioselectivity. The most oxidation-stable variant
retained nearly 40% of its activity after incubation with H2O2
(0.2 M), whereas wild-type activity was completely lost at 5 mM
H2O2. Therefore, oxidation-stable CHMO variants might be
required for subsequent thermostabilization, so that lab-
evolved thermostability in CHMO might be masked by a high
degree of oxidation instability.127
Case IV. In the case of pharmaceutical industry, the use of
rational design to stabilize biocatalysts for the production of
organic precursors is becoming a widely valued approach.
However, during the synthesis of many active pharmaceutical
ingredients, the use of cofactors such as NADH and NADPH,
(simplified notation: NAD(P)H) is widely common. Sincethe cost of these co-factors are expensive, Park et al. have
engineered an NAD(P)H oxidase biocatalyst named NoxV from
Lactobacillus plantarum, which is able to regenerate the
NAD(P)H cofactor using oxygen with water as the byproduct.128
They showed that the enzyme has inproved Michaelis–Menten
kinetics with improved stability when compared to other
NAD(P)H oxidases.128 Based on structural comparison of NoxV
L. plantarum to NAD(P)H oxidase of L. sanfranciscensis, Park
et al. found that site-directed mutagenesis of NoxV in the
binding pocket with single mutation L179R and double muta-
tions L179R/G178R would yield two enzyme variants capable of
accepting NADH and NAD(P)H while maintaining the enzyme’sspecific activity.128 There are few other cases where rational
design is used to create an oxidase that is capable of accepting
various types of cofactors for the use of the synthesis of
pharmaceutically viable compounds.129
Case V. Often, high throughput screening can require lots
of resources and, in addition, may yield less than 0.001%
of desired variants and more than 33% chance of creating
deleterious variants.130 Ito et al., using rational design, trans-
formed a mesophilic cellulase, cellobiohydrolase II (CBH2),
into a thermostable enzyme.130 Ito et al. revealed 45 distinct
amino acids not conserved using primary structure alignment
of five thermophilic fungal CBH2 and Phanaerochaete chrysosporium
CBH2. They were able to introduce 15 additive mutations ontothe wild type, which conferred thermostability and improved
kinetics stability. Ito et al. also showed the efficacy of using
sequence homology to avoid the creation of random library that
may yield no desirable outcome while requiring extensive usage
of high throughput materials.130
Case VI. Tackling the issue of organic solvent and bio-
catalysts, Park et al. studied and devised a method to create
organic solvent-stable Candida antarctica lipase (CalB) (Fig. 13).
Enzymes involved in organic synthesis must be stabilized in the
presence of their substrates, products, and solvents. Use of
Fig. 13 Lipase CalB from Candida antarctica. PDB: 3ICV.132
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 16/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6549
organic solvents in reactions presents many benefits to indus-
try because they allow for sterile reaction conditions since
microbes are not able to grow in organic mediums, hydro-
phobic substrates can be accommodated, and the equilibrium
can be shifted towards product production.131 Park et al. were
able to create sets of rules for stabilizing enzyme in organic
medium based on previous literatures on directed evolution of
enzymes in hydrophilic solvents, and based on their study of
and results from CalB stabilization. One guidance recommendsmutating amino acids from the surface of the proteins; the
chosen surface residues must be capable of either shortening
the lengths of or increasing the number H-bonds between the
proteins hydrophilic organic solvent; increasing or strengthening
H-bonds helps prevent unfolding when water molecules are
removed from the protein. Three mutants were created: N97Q,
N264Q, and D265E, all of which had stability in organic solvent
when compared to the wild type.131 Their half-lives were
increased up to 1.5 fold while activity was increased up to
17%. Park et al. demonstrated that by understanding solvent–
enzyme interactions and using rational design, one could
construct variants that have improved functionality and stability in organic solvents.
Case VII. Similarly, Badoei-Dalfard et al. performed two
mutations on Salinivibrio zinc-metalloprotease (SVP) to
increase active site polarity and therefore maintain its hydra-
tion while the enzyme is in organic solvents such as DMF and
methanol.133 The two mutations in the active sites of SVP are
A195E and G203D. Additionally, to stabilize a surface loop, they
performed A268P. For A195E, Badoei-Dalfard et al. created a
variant with improved kinetics of 11%, 26%, 32%, and 41% in
methanol, DMF, isopropanol, and n-propanol, respectively.
Additionally, with this mutation, rate of irreversible deactivation
was reduced by 31–33% in DMF and n-propanol. Although with
G203P the improvements in kinetics stability were similar tothose of A195E, its rate of inactivation was higher by 17% and
23.1% in DMF and n-propanol, respectively.133
Both Park et al. and Badoei-Dalfard et al. introduce muta-
tions using acidic amino acids Q and E to confer stability of
enzyme in organic solvents.133 One used these amino acids to
stabilize hydrophilic interactions with organic solvent to avoid
unfolding due to removal of water while the other’s goal was to
maintain water within the active sites while in organic solvents.
These methods of rational design for enzyme in organic solvents
maybe found effective in various other enzymes. However, it is
important to note that both groups have proposed seemingly
opposite hypotheses while yielding the same results: stable
enzymes. However, the goal of Park et al. was to remove water
molecules from the surface and increase hydrophilic inter-
actions with the organic solvent, while Badoei-Dalfard et al.strive to keep water molecules within the active site when in
organic solvent. Therefore, both groups were able to confer
stability using one method: rational design revolving around
the strengthened H-bonds.
4.3.3. Combinatorial protein design. Although it is nearly
impossible to account for all the possible interactions within an
enzyme and to satisfactorily determine the role of every evolu-
tionary mutation, combinatorial protein engineering is able to
assess the suitability of a mutation based on energetic
features.124 This computational method coupled with labora-
tory assays, often high-throughput assays, can help find
mutants with the desired enzymatic functions.
124
When setsof possible mutants are determined, often, high-throughput
systems are required to eliminate undesired variants since this
approach can yield 104–1012 variants based on the number of
amino acids being considered for mutations.123 Although it
seems to be a laborious process, many valuable industrial
catalysts have been discovered through this method. One of
the most advantageous features of using combinatorial protein
design is that extensive knowledge of the protein structure is
not always required. Primary sequence is the main tool for
determining the various combinations of mutations that can be
implemented to yield a satisfactory biocatalyst using computa-
tional algorithm and consideration of the energy landscape.
4.3.4. Examples of combinatorial protein design. Case I. A drug produced by Merck to combat asthma is Singulair or
montelukast sodium.134 This molecule is sensitive to many
chemical processes including hydrogenations. One of the
compounds used during its synthesis is ()- B-chlorodiisopino-
campheylborane, () DIP-Cl, to create the one stereocenter in
the molecule (Fig. 14). () DIP-Cl is, however, corrosive and
Fig. 14 From Liang et al.; the synthesis of Singulair via () DIP-Cl as shown by Liang et al. Produced through the reduction of molecule 1, (E )-methyl 2-(3-(3-(2-(7-
chloroquinolin-2-yl)vinyl)phenyl)-3-oxopropyl) benzoate, Singulair has one stereocenter as shown by molecule two labeled ( S), stating the chirality.
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 17/32
6550 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
moisture sensitive as well as failing to be environmentally
friendly. Thus, Liang et al. set out to create a biocatalyst that
can replace () DIP-Cl. KetoREDuctase (KRED) is the enzymethey mutated using error prone PCR. The mutated enzyme
is able to catalyze the reduction of an asymmetric ( E )-methyl
2-(3-(3-(2-(7-chloroquinolin-2-yl)vinyl)phenyl)-3-oxopropyl) benzoate
to its (S)-alcohol (Fig. 15). The enantioselectivity of the enzyme
was increased to more than 99.9%, a ‘self-driven’ process due to
the precipitation of the product out of the reaction solution.134
In addition, the enzyme is water soluble and stable in 70%
organic solvents at 45 1C with a loading of 100 g L1.134 This
property yields a still active enzyme because layers of hydration
are present at the enzyme’s active site; therefore, the required
environment for catalysis is maintained while in organic–water
solution; the hydrophobic organic solvent is water miscible and
organic substrate, which would otherwise be insoluble in water, will become accessible to the enzyme under these conditions.
Additionally, the enzyme uses NAD or NADPH as a cofactor,
which is also regenerated during the reaction by KRED. This
feature is highly advantageous for industrial applications since
these cofactors can be highly costly.
Case II. Similar to enzymes used in therapeutic practices,
phytases, also known as myo-inositol hexakisphosphate phos-
phohydrolases, are enzymes supplemented in animal feed to
help increase phosphorous absorption while decreasing its
secretion.135 The positive attributes of this enzymes included
optimum in acidic pH, pepsin digestion resistance, and efficiency
in metabolizing phylate. However, to create a thermostable variant of the enzyme, directed evolution through error-prone
PCR was used and yielded a variant with an increased 6–7 1C
in T m. Additionally, thermostability was improved to 10 minutes
at 80 1C, keeping in mind that the enzyme’s environment
in-body with temperature of B37 1C! At pH 3.5, the catalytic
efficiency was improved by 56% and 152% for the two superior
variants, K46E and K65E/K97M/S209G. The mutations that
rendered these stabilizing features were found to strengthened
local interactions within the protein.135 Error-prone PCR can
yield a myriad of variants, some of which are the desired stable
proteins as shown by the example from Kim and Lie.135 While a
process that is easily introduced into any lab, selecting the
stable variants can be laborious.
Case III. To improve the half-life of wild-type b-glucosidase
from P. polymyxa of 15 minutes at 35 1C and no detectable
activity at 55 1C, some random variants that enhanced thermal
resistance of the enzyme were combined to achieve a half-life of
12 min at 65 1C, with retention of kinetic parameters. Analysis
of the putative causative agents for the increased thermalresistance were (i) the formation of an extra salt bridge, (ii)
the replacement of an Asn residue exposed to the solvent, (iii)
stabilization of the hydrophobic core due to tighter packing,
and stabilization of the quaternary structure of the protein.136
Case IV. A xylanase for animal feed as well as pulp and paper
applications was sought by Diversa (San Diego, CA) that could
withstand very high temperatures, above 90 1C. In the first
round, all possible 19 amino acid substitutions at each residue
position were generated by Diversa’s own gene site saturation
mutagenesis (GSSM) and screened for activity after a tempera-
ture challenge. After identifying nine mutations that were
responsible for enhanced thermostability, all 512 possiblecombinatorial variants of the 9 mutations (= 29) were then
generated and screened for improved thermal tolerance. A total
of 11 variants were found with melting temperatures T mincreased from 61 1C to as high as 96 1C! Thus, the consecutive
application of two evolution strategies enabled the rapid
discovery of the enzyme variant with the highest degree of
fitness, i.e. greater thermal tolerance at constant activity at
comparable temperatures.137
4.3.5. Data-driven protein design. Data-driven protein
design is similar to rational design in that it is based on
acquired information on the protein of interest or on ortho-
logous or paralogous proteins. The difference between the two
methods is that data-driven protein design’s predictions aremore accurate. Additionally, data-driven protein design is
advantageous over combinatorial protein design because it
yields a smaller size of protein library that is to be screened
while still allowing for ‘error-prone PCR’ types of muta-
tions.138,139 Unlike rational design, data-driven’s predictions
are obtained from experiments that monitor the fold or unfolding
sequence of the protein to determine ‘‘weak’’ spot, in the case
of rendering a protein more thermostable.140 Once that infor-
mation is determined, site-directed mutagenesis is performed.
By using this method, high-throughput assays are rarely
needed, which reduces the cost and labor.
4.3.6. Examples of data-driven protein design. Case I. Data-driven design has been used by Hirose et al. to improve
solubility of proteins. Biocatalysts are often tagged to ease
purification or to improve stability.141 However, these tags
can also have detrimental effects on the protein’s structure
and functions.141 Thus, Hirose designed data-driven tags, or
DDTs, by considered the effect of hydrophobic and hydrophilic
tags. Unsurprisingly, their experiments showed that hydro-
philic tags increased solubility of proteins. They also showed
tags conferring solubility were negatively charged while those
that made protein less soluble were positively charged with
Fig. 15 Liang et al. depicted the reaction catalyzed by KRED. In the place of
()DIP-Cl is KRED. The enzyme regenerates thecofactors NAD(P)Hin thepresence
of isopropanol, which is oxidized into acetone.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 18/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6551
arginine rich sequence.141 In addition, while the authors did
not focus on any specific biocatalyst, in fact they do not
explicitly mention any of them, they showed that data-driven
protein design could be used to create stable biocatalyst and to
also help determine satisfactory tags, proteins or peptides that
can help with the purification of a specific protein.
Case II. The kinetic stability of chitinase B from Serratiamarcescens (ChiB) (Fig. 16) was stabilized using semi-automated
design methods through rigidifying mutations such as Gly - Ala
and Xxx - Pro.142 Of the 15 stabilizing single variants, those
with significant stabilization clustered in one surface-exposed
region of the enzyme, with the double variant G188A/A234P
showing tenfold increase in half-life at 57 1C and a 4.2 1C
increase in the apparent T m. Likely, the mutations led to
entropic stabilization of ChiB, with partial unfolding in the
critical region in question, which triggered thermal deactiva-
tion coupled with aggregation.
Case III. In an attempt to further elucidate protein stability,
Rathi et al. used data-driven process to understand what allowsthermal stability or instability (Fig. 18).139 Citrate synthases
(CS) from five distinct host organisms each with optimal
growth temperature (T p) of 37 1C, 59 1C, 75 1C, 87 1C, and
100 1C were chosen for the study. Their sequences were
compared and their structures analyzed to find areas within
the proteins that would be more susceptible to mutations.
The Ensemble-based Computer Network Analysis (CNS) was
modified to take into account the idea that increased hydro-
phobic interactions, hydrogen bonds, and salt bridges result in
a more thermostable protein. Using two of their data sets
shown in Fig. 17, Rathi et al.139 showed that the thermal
unfolding of CS happens in stepwise manners where the
structurally stable enzyme (Fig. 17II(a)) which has dimer inter-
actions between F, G, M, and L, decomposes into a transition
state in which there is strong core interactions between G, I, L,
and M regions of the enzyme, corresponding to 330–360 K or an
enthalpy (H) of about 1 (Fig. 17II(b) and (c)). Next, with highertemperatures, the enzyme breaks down at the core region into
final smaller rigid structures connected by flexible links
(Fig. 17II(d) and (e)).
Additionally, Rathi showed that the thermal stability and
unfolding mechanisms of the CS orthologs had no correlation
while the thermostability of these orthologs showed to have a
significant correlation with the T m or T p of the host organisms
( R2 = 0.88). They also found that each orthologs had different
sets of weak spots, which are defined as the region of tertiary
structures whose interactions are disturbed at the T p. Sequence
analysis showed that these regions of weak spots were mutated
in the higher stable orthologs. In all, using data-driven strate-gies, they showed that by understanding the thermostability
rather than thermal unfolding, one could predict amino acids
involved in stabilization of thermozyme orthologs.
Case IV, V. One of the most popular drug problems is cocaine
addiction. Much research has been devoted to developing drugs
that combat cocaine addictions and one of the avenues is the
engineering of cocaine esterase (CocE) and butyrylcholinesterase
(BChE), a/b hydrolases.145,146 CocE is an enzyme found in
bacteria, i.e. Rhodococcus sp strain MB1, that use cocaine as
their sole carbon source.147 The enzyme hydrolyzes cocaine into
Fig. 16 Chitinase B from Serratia marcescens. PDB: 2WK2.143 Mechanism of Chitin hydrolysis by Chitinase B was adapted from Tews et al.144
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 19/32
6552 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
benzoic acid and Ecgonine methyl ester. On the other hand,
BChE is a human protein that catalyzes the hydrolysis of
acetylcholine into acetate and choline (Fig. 18).148 Several
stable variants of both enzymes have been generated using
site-directed mutagenesis based on rational design.73,74 How-
ever, Huang et al. and Zhen et al. were able to design thermo-
stable variants of CocE and BChE, respectively, based on data
obtained from computer simulations.149,150
Fig. 17 (I) Rathi et al.139 showed that a cartoon of CS (top figures I-A and I-B, 901 rotation of I-A) in which each letter points to the section of the protein it represents.
(II) Bottom figure shows the melting curve of CS, which happens in a step wise manner. Structure (a) represents a stable form of the protein with all interactions intact;
(b) and (c) are intermediate while (d) and (e) are inactive and lost significant local interactions.
Fig. 18 (a) Cocaine (molecule 1) hydrolyzed by butyrylcholinesterase (BChE), human liver carboxylesterase (hCE-1/2), or Cocaine Esterase (CocE).147 (b) Deacetylation
of choline esterase, a reaction catalyzed by BChE.148
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 20/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6553
In the case of CocE, Huang et al. performed molecular
dynamics simulations on wild type CocE, which has a half-life
of 11 minutes at 37 1C. Based on literatures and simulations,
Huang et al. determined that the lack of rigidity within the
active site is responsible for instability of the enzyme. The data
suggested that mutation L169K would yield a thermostable
enzyme, which was confirmed by wet-lab experiments with a
determined half-life of 570 minutes at 37 1C, equaling to more
than 50-fold or 5000% increase in stability! A finding that canbe used towards the stabilizations of other a/b hydrolases such
as a-amino ester hydrolase and platelet-activating factor acetyl-
hydrolase, a lipase from Streptomyces exfoliates.147,151
While Huang et al. focused on thermal stability of CocE,
Zheng et al. tackled the kinetic stability of BChE metabolism of
cocaine since the therapeutic enzyme must be of superior
thermostability and catalytic efficiency. Zheng et al. focused
on understanding the mechanism of catalysis with the goal to
stabilize the transition state and improve the kinetics of the
rate-limiting step.149 The group produced a quadruple variant
of BChE, A199S/S287G/A328W/Y332G, which has a 456-fold
improvement in catalytic efficiency, a promising outcome foranti-cocaine addiction therapeutics developments!149
Both Huang et al. and Zheng et al. showed that by increasing
rigidity of the active site without disrupting the structure of the
enzyme and by understanding the catalytic mechanism of the
biocatalysts, one can stabilize the key parameters through
the mutation of one or more amino acids. The key is to stabilize
the active site and the binding of the intermediate substrate
through charge–charge interactions, hydrophobic interactions,
etc. These methods of computational studies with data-driven
approach can be applied in the design of myriad of drugs, and
other industrial biocatalysts.149,150
Stabilization based on major algorithms. Case VI. Moleculardynamics (MD) simulations were employed to identify flexible
regions in haloalkane dehalogenase (DhlA), which can serve as
a target for enhancing stability via introduction of a disulfide
bond. MD simulations of the a/b-hydrolase fold protein DhlA
showed high mobility in a helix-loop-helix region in the five
a-helix cap domain, involving residues 184–211. A disulfide
cross-link was engineered between residue 201 of this flexible
region and residue 16 of the main domain. In ‘wet-lab’ experi-
ments, the mutant enzyme showed substantial changes in
both thermal and urea denaturation. The apparent transition
temperature T m,app of the oxidized form of the variant increased
from 47.5 to 52.5 1C, whereas the T m,app of the reduced variant decreased by more than 8 1C compared to the wild-type enzyme;
denaturation with urea followed a similar trend. The Gibbs free
energy of unfolding was decreased by 0.43 kcal mol1 in the
variant compared to the wild-type enzyme, also indicating that
the helix-loop-helix region was involved early in the unfolding
process. Thus, MD simulations can identify mobile protein
domains that can be targeted successfully for stabilization via
disulfide cross-linkage.152
Case VII. To increase the efficiency of data-driven protein
engineering based on iterative saturation mutagenesis (ISM),
adaptive substituent reordering algorithm (ASRA) was intro-
duced. ISM presented as an alternative to traditional quantita-
tive structure–activity relationship (QSAR) methods for
identifying potential protein mutants with desired properties
from minimal sampling of focused libraries. ASRA delivers
predictions without explicit knowledge of the structure–
property relationships by identifying the underlying regularity
of the protein property landscape. In a proof-of-principle study,
ASRA identified all or most of the best enantioselective variantsof Aspergillus niger epoxide hydrolase.153
B-FIT concept. Case VIII. Iterative saturation mutagenesis
(ISM) was applied to thermally stabilized lipase A (LipA) from
Bacillus subtilis (Fig. 19), i.e. sites in LipA were subjected to
saturation mutagenesis and the best hit of any given library is
then used as a template for randomization at other sites, and
the procedure is repeated until the desired improvement level
of the chosen property has been achieved. The randomization
sites in LipA were chosen on the basis of the highest B-factors
available from X-ray crystallography data of the wild-type (WT)
LipA, termed the B-FIT approach. After five rounds, this proce-dure resulted in pentuple variants with melting temperatures
T m increased by 45 1C that also showed improved reversibility
after heating to temperatures above 65 1C and subsequent
cooling.
The fitness-pathway landscape constructed from every
possible sequence of the five point mutations, i.e. 5! = 120
pathways, from the WT LipA to the thermostabilized variant XI,
was analyzed for epistatic interactions. The analysis revealed
significant cooperative effects between distal residues, which
are especially pronounced in the final steps towards variant XI.
MD simulations uncovered the stepwise formation of an exten-
sive H-bond/salt-bridge network on the surface of the enzyme.
Most importantly, while all five point mutations are necessary for high thermostability, only one participates directly in the
extended network. The authors suggest that evolution and then
identification of structural and functional amino acid networks
deserve attention.154
Combined thermal deactivation profiles, CD and NMR
experiments, as well as X-ray structure analyses on the three
best LipA variants and the wild-type yielded variations of
surface amino acid residues that counteracted propensity to
precipitate at elevated temperatures. Thus, not increased
conformational stability (higher T m) of the evolved variants
but rather reduction of precipitation of unfolding inter-
mediates seem to be responsible for improved thermal stressbehavior.155
Case IX. Three positions on the surface of Pseudomonas
fluorescens aryl esterase (Fig. 20) were selected according to
high flexibility as discerned from the B-factor iterative test
(B-FIT) principle and targeted first via site-saturation muta-
genesis and then via restricted libraries utilizing only consensus-
based mutations to increase its thermostability. Restricted
libraries reduce the library size significantly while ensuring a
high hit rate. The best variants demonstrate significantly
improved thermostability, 8 1C higher T m compared with the
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 21/32
6554 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
wild type without compromising specific activity, whereas
subsequent iterative saturation mutagenesis (ISM) even gave a
variant with a 9 1C increased T m, again with unchanged
catalytic properties.156
Case X. The B-FIT concept was applied to increase thethermal robustness of the epoxide hydrolase from Aspergillus
niger . Several rounds of ISM resulted in the best variant show-
ing a 21 1C increase in the T 6050 value, an 80-fold improvement in
half-life at 60 1C, and a 184 kJ mol1 improvement in the energy
of deactivation; seven other variants yielded lesser but still
respectable improvements. The best variants during the ISM
process surprisingly were obtained from neutral or even infer-
ior parent variants, from a library with few or no improved
variants. Tapping into such libraries apparently avoids dead
ends in evolution, i.e. local minima in the fitness landscape.158
4.3.7. Stabilization via the consensus concept. The concept
of stabilization via the consensus method determines the most
prevalent amino acid at a given position of an alignment of
orthologous proteins. The resulting sequence of most prevalent
amino acids is termed consensus sequence. Lehmann et al.159–161
and Amin et al.162 later applied the consensus approach to
generate a stable fungal phytase and b-lactamase, respectively.
Watanabe et al.163 used a phylogenetic approach to identify
ancestral amino acids that ultimately conferred thermal stabi-
lity to a 3-isopropylmalate dehydrogenase. Binz et al.164 used a
consensus approach to develop a conserved scaffold (i.e.,residues important for maintaining repeat structure) where
potential interacting amino acids where randomized en route
to improving interactions between repeating units of the
ankyrin protein. The result is an increase in expression, solu-
bility, and stability of the ankyrin repeat protein. DiTursi
et al.165 used Bayesian sequence-based algorithms on serine
protease sequences to identify a stabilizing motif in subtilisin
E, improving its melting temperature by 13 1C. In cases of low
level of sequence identity and/or few available amino acid
sequences, however, the consensus residue of a specific
position cannot be found easily or not at all. Thus, the
structure-guided consensus approach was developed, whichuses structural information (in addition to other criteria, see
below) to reduce the number of residues in a given protein to be
mutated and checked for stabilization, and demonstrated
thermostabilization on penicillin G acylase (PGA) in 47%
(10 of 21) of investigated single variants (Fig. 21).166 Towards
this goal, there were only 8 sequences ranging from 32.5 to
87.7% and 28.6–87.6% amino acid identity for the a- and
b-chains, respectively. Moreover, PGA is a large, heterodimeric
entity with a complicated maturation process to the active
protein (23 kDa and 63 kDa in a- and b-subunit, respectively)
Fig. 20 Aryl Esterase from Pseudomonas fluorescens. PDB: 3T4U.157
Fig. 19 LipA from Bacillus subtilis. PDB: 1ISP.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 22/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6555
and thus difficult to access through rational or combinatorial
design. In the present work, the structure-guided consensus
approach was extended towards a simple, broadly applicable,
and highly successful approach on an important, homo-
tetrameric biocatalyst with cofactor-dependent catalysis, glucose
dehydrogenase.
In summary, it was demonstrated that the stabilizing effect
of consensus mutations in multiple natural and diverse
variants of the same functionality. Additionally, it was demon-
strated that the structure-guided consensus concept is a very successful and widely applicable technique for generating
thermostable proteins160,166,168—further demonstrated in the
improvement in the stability of GDH. The stability of the
his-tagged B. subtilis GDH was improved from B20 minutes
at 25 1C toB3.5 days at 65 1C, a 106-fold improvement.169 Using
this approach it was possible to identify residues critical to
stability with considerably less effort than traditional directed
molecular evolution: of order 10 vs. order 104. The number of
investigated variants can be tuned by the experimentalist; the
two most influential factors are the amino acid identity, range
of the selected sequences, and the chosen consensus level (here
50%). In contrast to directed evolution, where key amino acidsare first identified via high-throughput screening (HTS), each
variation from the parent sequence has to be incorporated into
the parent gene, and the variant purified and characterized. In
the case of difficult and/or expensive substrates, not amenable
to HTS, a small number of variants can be characterized
directly on the substrate of interest, a strong advantage
(‘‘you get what you screen for’’).170 The success of the struc-
ture-guided consensus concept depends on the diversity of
available sequences. The number of consensus candidates
can then be reduced with available information, such as
(i) data on stability from thermostable or labile homologs,171,172
(ii) crystallographic structures, including B-factor analysis173
and Ramachandran plots,174,175 and (iii) information about the
deactivation mechanism, such as crucial subunit interactions,
which might prefer surface residues. Often, use of all available
data and tools optimizes output while minimizing effort. As
discussed, if the search was limited to the subunit–subunit
interface, the success rate would have increased from 46% to
86%. The development of thermostable enzymes is of interest
to the specialty chemical and pharma industries but also to theprotein engineer, since recent work has shown that evolvability
is linked to stability.176,177 This finding supports the idea that
protein engineering projects will benefit from starting with a
stable protein scaffold. Thus, straightforward, time-saving tech-
niques—such as the structure-guided consensus concept—is
projected to prove invaluable to protein engineering efforts.
4.3.8. Stabilization via SCHEMA. The discovery of exons
and introns gave birth to one evolutionary theory, which states
that proteins have evolved through nonhomologous gene
recombination.178 In 1978, W. Gilbert proposed that RNA
splicing presents a faster opportunity for novel proteins to be
created from older ones when compared to rare single point mutations.179,180 This concept essentially lead to the idea that
heteromeric proteins may have evolved from the recombination
of the exons of separates genetic material whether through
horizontal or vertical gene transfer.178,181 The structure of the
individual domains from the exons of these proteins would
therefore be independently stables. Enzyme stabilization
through rational design, combinatorial design, or data-driven
design can require an extensive amount of work in an expansive
time frame. Laboratory directed evolution uses combinatorial
shuffling of DNA to create stable enzymes in a timely fashion.178,182
Fig. 21 Penicillin G Acylase from E. Coli. PDB: 1GM9.167
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 23/32
6556 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
This method of stabilization is dubbed SCHEMA.183,184
‘Schema’ is the original computer algorithm, which searched
for genetic materials whose crossovers would yield favorable
interactions. While ‘schema’ focused on genetic materials, Voigt
et al. created SCHEMA, which applied the same algorithmic
concept onto protein sequence; the program attempts to identify
structural domains from distinct proteins that must have origi-
nated from one ancestor, relating back to Gilbert’s hypothesis.179,184
Additionally, by scoring the interactions between amino acidsof two parent proteins, SCHEMA can help determine which
interactions, when removed, would not cause the instability
of the parent proteins’ structures and would yield a stable
chimeric protein from the recombination of those parent
proteins.183,185 Voigt calculated the disruption, E ab, of hybrid
protein, from the recombination of fragment a of parent
protein 1 and fragment b of parent protein 2 and where
when residue i and j are within a specified distance, C ij = 1
and if not, C ij = 0. P ij simply accounts for the possibilities of no
disruption.184 This method of stabilization is more efficient
that error-prone PCR used in combinatorial design since large
amounts of amino acids are changes simultaneously to create anew hybrid protein with desired features.185
E ab ¼Xi 2a
Xi 2b
cij Pij (39)
4.3.9. Examples of stabilization via SCHEMA. Case I.
SCHEMA has already been employed in research to create
biocatalysts more stable than their already available industrial
counterparts.186–188 In an attempt to understand the stability of
arginase, which is an enzyme that catalyzes the hydrolysis of
L-arginine into ornithine, Romero et al. used SCHEMA. Arginases
(Fig. 22) are potential therapeutic agents for L-arginine auxo-
troph tumors and hyperargininemea. Thus, making them stable
is valuable.189 Recombination of Arginase I and II showed that pI
of the proteins is very significant for long-term thermostability.
The higher the pI of the arginase, the lower the kinetic stability
of the chimera.189 Essentially, SCHEMA can be used to not only
create novel and stable proteins but to understand how evolution
have chosen certain amino acids, as stated in the neutral drift
theory, to yield suitable enzymes for the host organism.
Case II. Similarly, Production of biofuels and biomass
requires a source of sugar, which is commonly obtained fromthe breakdown of cellulose by cellulase.186–188 Heinzelman
et al. have shown that by using structure-guided recombination
of three fungal class II cellobiohydrolases (CBH II), several
cellulases with thermostability of 7 1C to 15 1C higher along
with increased pH profile all with increased activity were
created.187,188 that a single mutation, C313S, was responsible
for the stabilization; stable variants were created when this
mutation is implemented in the parent proteins, Hypocrea
jecorina and H. insolens CBH II, and in a distant protein with
only 55–56% identical features, Phanerochaete chrysosporium
CBH II.187 These findings demonstrate that results from
structure-guided recombination SCHEMA can inform us of mutations that were selected through evolution to improve
thermodynamics and kinetics stability of orthologous enzymes.
Case III. Often, the disadvantage of biodegradation of bio-
mass is the thermostability of the cellulase. A thermostable
cellulase can be efficient at higher temperature and have longer
half-life, which both equals to greater amount of biodegrada-
tion.190 Heinzelman et al. showed that SCHEMA can sometimes
produce combinations that require high throughput screen-
ings.190 They were able to develop a method for finding stable
blocks of amino acids; a thermostable recombinant cellobio-
hydrolase class I (CBH I) was isolated from 390625 (58) possible
chimeras. Heinzelman et al. begun by analyzing the parent
Fig. 22 Arginase from Themus thermophilus. PDB: 2EF4.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 24/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6557
enzymes and the singular units, which were considered for
recombination. They identified 36–40 stable blocks that con-
densed into 16 thermostable CBH Is, which were more stable
than the most thermostable CBH I (Talaromyces emersonii ) at
70 1C.190 SCHEMA can be used to create novel stable recombi-
nant with improved or diversified functions. By studying and
choosing thermostable blocks of amino acids that make up the
chimera and/or by incorporating various catalytic domains, one
can make a desirable recombinant protein with diverse functionscan be created and implemented in industrial applications.
Case IV. Industrial processing of biomass relies on the
mixture of various cellulases and glycoside hydrolases with
diverse functions.191 Thus, Smith et al. demonstrated that
understanding structural differences within a family of protein
could help interpret their evolution as it relates to their
specificity as well as create myriads of protein with divergent
functions. Thus, using structure-guided recombination, Smith
et al. were able to increase the library of examined Cel48
(Fig. 23) from 13 to 73 by using the catalytic domains of three
glycoside hydrolase family 48 bacterial cellulases (Cel48). The
three parent enzymes include Clostridium cellulolyticum CelF,Clostridium stercorarium CelY, and Clostridium thermocoellum
CelS and yield 60 recombinants with various functionalities
governed by pH, temperature, stability, and specific activity
with crystalline cellulose. Similar to Heinzelman et al., Smith
et al. studied the individual parent protein fragments and found
that the stability of each fragments is additive in a recombinant,
based on their linear models of sequence property.191
Case V. One of the most important tools when working with
proteins is protein tags. Tags can be used for stabilization or
purification of a protein of interest. Avidin and steptavidin
(Fig. 24) are two of the most common tags used for protein
purification. Either enzymes can be linked to a protein of
interest and during purification, they are able to bind to
anchored biotins from which they will only be released at the
addition of a certain salt concentration and ionic strength. The
reverse of that scenario is also applicable: a protein modified
with biotin can bind to an anchored avidin. Maatta et al.
produced a thermostable avidin called chimeric avidin by
recombining avidin and avidin-related protein 4.194 When the
recombinant protein was studied in organic solvents and
elevated temperatures, it showed to have superior stability in
extreme pHs starting from pH 1 to 13 as well as improvedstability in organic solvents such as DMF and methanol; avidin
was stable at pH 13 while streptavidin was stable at pH 1.
Additionally, the binding energy of chimeric avidin is energe-
tically favorable with an enthalpy of 112.6 kJ mol1, which is
5% higher than that of avidin. Maatta et al. were able to show
that the conferred stabilizations are due to increased stability of
the tetrameric structures because of increased interactions
between the bridging amino acids.194 Maatta et al. developed
a tag that could be used for extended levels of purification due
to tag’s stability in very extreme conditions.
Case VI. While the examples given thus far use SCHEMA to
create one or two stable variants of their target protein, Otey et al. used this method to create a library of 6561 of which are
3000 distinct properly folded variants of cytochrome P450
(Fig. 25).198 Each variant have different level of stability and
capability to incorporate a heme cofactor and peroxygenases
activity; some of the properly folded enzymes are inactive. The
recombination of three cytochromes P450s (CYP102A,
CYP102A2, and CYP102A3) at seven locations yielded variants
that are more than 73% dissimilar to any of the known
sequences of cytochromes P450, which is an astonishing 4500
sequences. Comparing the sequence of correctly folded variants
to the mis/unfolded variants and inactive yet well folded
recombinants can give insights as to what regions are necessary
to maintain native fold and stability along with understanding
Fig. 23 Cel48F from Clostridium cellulolyticum. PDB: 1F9D.192 Reaction was adapted from the project of the month for the School of Theoretical Chemistry and
Biology, Royal Institute of Technology.193
Review Article Chem Soc Rev
View Article Online
7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 25/32
6558 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
what confers catalytic specificity and efficiency. This paperpresent a fine example of studying how enzymes have been
evolved and determining what amino acids are deleterious,
neutral, or advantageous using SCHEMA.198
Case VII. The low thermostability of sucrose phosphorylase
from Bifidobacterium adolescentis is a significant drawback for
applications in glycosylations. After substituting the most
flexible residues with the consensus amino acids, & based on
careful inspection of the crystal structure, introducing residues
that promotes electrostatic, the half-life of a hexuple variant at
the industrially relevant temperature of 60 1C more than
doubled over that of the wild type. Although mostly at low
temperatures, the hexuple variant also was more stable against organic solvents.199
4.5 Improvement of process stability
The operational stability of an immobilized lipase from
Thermomyces lanuginosa, commercially available as lipozyme
TL IM was investigated in the interesterification of two lipid
blends, blend A, a 55: 25: 20 wt% mixture of palm stearin
(POS), palm kernel oil (PK), and sunflower oil, and blend B,
formed by a 55: 35: 10 wt% mixture of POS, PK, and highly
concentrated (n 3)-polyunsaturated fatty acids (PUFAs).
The continuous packed-bed bioreactor operated in neat mediumat 70 1C at a residence time of 15 min for 580 h (blend A) and
390 h (blend B), respectively. The inactivation of the biocatalyst
followed a first-order deactivation model, with estimated half-
lives of 135 h and 77 h for blends A and B, respectively. The
higher levels of oxidation-prone PUFAs in blend B may explain
the lower lipase stability compared to blend A.200
5. Case studies
5.1 Record-setting stabilizations
In an especially successful example of thermal stabilization
through the application of two different methods of combina-torial protein engineering, gene site saturation mutagenesis
(GSSM) and exhaustive mutation among nine sites with a
xylanase gene, Palackal et al.137 improved the melting tempera-
ture T m of the xylanase from 61 1C to as high as 96 1C!
Among data-driven protein engineering works, two examples
stood out:
With the help of the B-FIT approach, Reetz et al.154 found
that the melting temperature T m of a pentavariant of lipase
A (LipA) from Bacillus subtilis, was increased by 45 1C, accom-
panied by improved reversibility. Simulations lend credence to
Fig. 24 (a) Avidin from Gallus gallus PDB: 1VYO;195 (b) Steptavidin from Streptomyces avidinii; PDB: 3T6F.196 (c) Avidin–biotin complex from PDB: 2AVI.197
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 26/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6559
the notion that an extended network of charges and H-bonds is
responsible for the enhanced thermal stability.154
Lastly, the structure-guided consensus approach yielded a
vastly stabilized glucose dehydrogenase (GDH). After analyzing 24 single variants identified by this approach, Vazquez-
Figueroa et al.169 succeeded to stabilize the his-tagged B. subtilis
GDH from a half-life of B20 minutes at 25 1C to B3.5 days at
65 1C, a 106-fold improvement.
5.2 Simultaneous stabilization against heat and other stresses
such as organic solvents
Whether stabilization against one stress, i.e. increasing
temperature, also helps to stabilize against other stresses such
as those stemming from organic solvents, interfaces, or high
concentrations of chaotropes or salts in general, remains an
important hypothesis of protein stability. As results disproving
this hypothesis often tend not to be disseminated, any report
confirming simultaneous stabilization must be read with caution,as generalizability might not be assured.
5.2.1. Examples of simultaneous enzyme stabilization.
Case I. Cowan et al.201,202 showed a positive correlation between
thermal stability and tolerance to organic solvents in naturally
occurring homologs of several different thermophilic proteins.
Hao and Berry demonstrated how a thermostable fructose
bisphosphate aldolase found via directed evolution also featured
increased stability in organic solvents.203
Case II. Mutants of the lipase from Bacillus subtilis, pre-
viously engineered for enhanced thermostability using directed
Fig. 25 (a) Otey et al.198 show a cartoon depiction of the recombinant protein composed of 8 distinct protein fragments. (b) Depiction of how the numerous
recombinants were derived from the shuffling of the 7 distinct interaction regions.
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 27/32
6560 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
evolution based on the B-FIT method, show significantly
increased tolerance to hostile organic solvents.204
Case III. A thermophilic Old Yellow Enzyme (‘TOYE’) (Fig. 26)
isolated from Thermoanaerobacter pseudethanolicus E39 is stable
at high temperatures (T m > 701) and simultaneously features
increased resistance to denaturation in water-miscible organic
solvents compared to a closely related mesophilic OldYellow Enzyme family member, pentaerythritol tetranitrate
reductase.205 According to sedimentation velocity and multi-
angle laser light scattering (MALS) measurements, TOYE adapts
higher-order oligomeric states, such as octamers and dodeca-
mers in solution, under stress compared to a tetrameric state at
ambient conditions, likely a major reason for increased stability.
Case IV. Penicillin V acylase (EC 3.5.1.11) from Streptomyces
lavendulae showed both enhanced activity and thermal stability
in mixed water–glycerol and water–glycol solvents. Both activity
and thermal stability increased proportionally to the amount of
added glycerol and glycol co-solvents up to a critical concen-
tration of these co-solvents, with further addition leading to
gradual protein deactivation. Thus, reaction conditions that allow simultaneously enhanced activity and stability in the
S. lavendulae Pen V acylase-catalyzed hydrolysis of penicillin V
could be defined.113
Case V. Thermolysin-like protease from Bacillus stearothermo-
philus was engineered for extreme thermostability via intro-
duction of a disulfide bond G8C/N60C (double variant, DV) plus
six other amino acid substitutions in the exposed loop region,
amino acids 56–69, to generate a protein dubbed ‘Boilysin,
BLN’. Both DV and BLN were probed for stability against water-
miscible organic solvents and detergents. The solvent concen-
trations at which 50% of initial enzyme activity were irreversibly
lost, C 50, decreased in the order methanol > 2-propanol >dimethylsulfoxide > dioxane > acetonitrile > DMF > acetone.
However, the C 50 values were remarkably higher for the BLN
and DV thermostable variants than for the wild-type enzyme,
pointing to a similar mechanism for irreversible thermal and
solvent deactivation. However, the differences of the C 50 values
of DV and BLN were not significant, while the corresponding
T 50 values of DM and BLN differed by 10 K. 207 Detergents
affected DV and BLN equally, inactivating (SDS, sulfobetaine)
or strongly activating (CTAB, CHAPS), and depending in degree
on detergent concentration but always destabilizing; Triton
X-100 and Tween 20 started inactivating beyond a threshold
concentration. Thus, the mechanism of detergent inactivation
seems to differ from that of thermal inactivation.208
Case VI. Encapsulation in a carboxymethylcellulose (CMC)-
Ca alginate liquid membrane optimized at 4% CMC, 1% CaCl2,
and 3% alginate led to >60% activity retention of the cysteine
protease cathepsin B purified from goat brain during fivebatches. The optimum pH value, maximum temperature T max ,
and apparent K m value of the free and immobilized enzyme
were measured to be 6.0, 50 1C, and 1.52 mM, & 6.0–6.5, 55 1C,
and 2.3 mM, respectively. Microencapsulation significantly
stabilized the enzyme against pH, thermal, and also solvent
instability, opening up the potential for use in catalyzing
transesterifications or transamidations.209
Case VII. Formate dehydrogenase (FDH) from Candida
boidinii , as cofactor-regeneration enzyme, had been found to
be much less stable when regenerating NAD+ to NADH than
the production enzyme, an amino acid dehydrogenase, when
reacting NADH to NAD+. Already in 2000, variants with free Cys
residues mutated to Ser or Ala were found to stabilize FDH.210 Asthe variants C23S and C23S/C262A were found to be advanta-
geous even in the absence of oxygen, other stresses were tested.
The two changes were found also to stabilize FDH against
interfaces with nitrogen bubbles, shear stress in Couette
viscometers, and shear stress and cavitation in gear pumps.87
Case VIII. Thermostable glucose dehydrogenase (GDH) var-
iants developed via an amino acid sequence-based consensus
method also showed improved stability in solutions with high
concentrations of either kosmotropic or chaotropic salts as well
as water-miscible organic solvents. Only the most stable variants
showed little deactivation dependence on salt-types along the
Hofmeister series and on salt concentration. Kinetic stability,expressed by the deactivation rate constant k d,obs, did not always
correlate with thermodynamic stability of variants, as measured
by melting temperature, T m. However, a strong correlation
( R2 > 0.95) between temperature stability and organic solvent
stability was found when plotting T 6050 versus C 6050 values.211
6. Conclusions
Nowadays, a vast set of tools exists to stabilize biocatalysts,
consisting mostly of immobilization, medium engineering, and
Fig. 26 Old yellow enzyme from Thermoanaerobacter pseudoethanolicus. PDB: 3KRU.205 Reaction was adapted from Brenna et al.206
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 28/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6561
protein engineering. When measuring enzyme stability, thermo-
dynamic stability, measured by the melting temperature T m,
has to be differentiated from both kinetic stability, measured by
the deactivation rate constant k d or half-life t, or operational
stability, also termed process stability, measured by the total
turnover number TTN, which scales enzyme activity over kinetic
stability. Protein engineering has been found to increase T m by
35–45 1C. Through data-driven protein engineering, kinetic
stabilization of several 100 000-fold has been achieved. TTNhas been found to improve by the same order of magnitude. In
contrast to stabilization against detergents or other specific
chemical entities, which often feature interactions with specific
moieties on the enzyme surface, stabilization against one stress
factor, such as high temperature, was in many cases found to
stabilize against others, such as high concentrations of chao-
tropes as well as high shear through pumping, stirring, or
cavitation. Given the vast number of successful cases of enzyme
stabilization, almost any well-folded protein should be amen-
able to such procedures.
References
1 U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol.,
2001, 5, 137–143.
2 S. Lutz, Curr. Opin. Biotechnol., 2010, 21, 734–743.
3 S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang,
J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem.,
2010, 12, 81–86.
4 C. A. Martinez, S. Hu, Y. Dumond, J. Tao, P. Kelleher and
L. Tully, Org. Process Res. Dev., 2008, 12, 392–398.
5 C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore,
S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J.
Fleitz, J. Brands, P. N. Devine, G. W. Huisman andG. J. Hughes, Science, 2010, 329, 305–309.
6 C. E. Nakamura and G. M. Whited, Curr. Opin. Biotechnol.,
2003, 14, 454–459.
7 K. M. Polizzi, A. S. Bommarius, J. M. Broering and
J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11,
220–225.
8 S. Bommarius and J. M. Broering, Biocatal. Biotransform.,
2005, 23, 125–139.
9 V. G. H. Eijsink, A. Bjork, S. Gaseidnes, R. Sirevag,
B. Synstad, B. van den Burg and G. Vriend, J. Biotechnol.,
2004, 113, 105–120.
10 V. G. H. Eijsink, S. Gaseidnes, T. V. Borchert and B. vanden Burg, Biomol. Eng., 2005, 22, 21–30.
11 C. K. Winkler, D. Clay, S. Davies, P. O’Neill, P. McDaid,
S. Debarge, J. Steflik, M. Karmilowicz, J. W. Wong and
K. Faber, J. Org. Chem., 2013, 78, 1525–1533.
12 C. Breithaupt, R. Kurzbauer, F. Schaller, A. Stintzi,
A. Schaller, R. Huber, P. Macheroux and T. Clausen,
J. Mol. Biol., 2009, 392, 1266–1277.
13 B. W. Lepore, D. Liu, Y. Peng, M. Fu, C. Yasuda,
J. M. Manning, R. B. Silverman and D. Ringe, Biochemistry,
2010, 49, 3138–3147.
14 M. Winn, J. M. Foulkes, S. Perni, M. J. H. Simmons,
T. W. Overton and R. J. M. Goss, Catal. Sci. Technol.,
2012, 2, 1544–1547.
15 B. Rosche, X. Z. Li, B. Hauer, A. Schmid and K. Buehler,
Trends Biotechnol., 2009, 27, 636–643.
16 T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38,
3117–3132.
17 M. Schrewe, M. K. Julsing, B. Buhler and A. Schmid, Chem.
Soc. Rev., 2013, DOI: 10.1039/C3CS60011D.18 J. E. Leresche and H.-P. Meyer, Org. Process Res. Dev., 2006,
10, 572–580.
19 J. A. Littlechild, J. Guy, S. Connelly, L. Mallett, S. Waddell,
C. A. Rye, K. Line and M. Isupov, Biochem. Soc. Trans.,
2007, 35, 1558–1563.
20 F. Secundo, Chem. Soc. Rev., 2013, DOI: 10.1039/C3CS35495D.
21 M. T. Reetz and S. Wu, J. Am. Chem. Soc., 2009, 131,
15424–15432.
22 T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc.
Rev., 2009, 38, 3010–3021.
23 M. Wang, T. Si and H. Zhao, Bioresour. Technol., 2012, 115,
117–125.24 D. Wistuba and V. Schurig, Angew. Chem., Int. Ed. Engl.,
1986, 25, 1032–1034.
25 M. Wang, D. L. Roberts, R. Paschke, T. M. Shea,
B. S. S. Masters and J.-J. P. Kim, Proc. Natl. Acad. Sci.
U. S. A., 1997, 94, 8411–8416.
26 D. Yi, T. Devamani, J. Abdoul-Zabar, F. Charmantray,
V. Helaine, L. Hecquet and W.-D. Fessner, ChemBioChem,
2012, 13, 2290–2300.
27 P. Asztalos, C. Parthier, R. Golbik, M. Kleinschmidt,
G. Hubner, M. S. Weiss, R. Friedemann, G. Wille and
K. Tittmann, Biochemistry, 2007, 46, 12037–12052.
28 T. S. Institute, Penicillin, http://www.scripps.edu/nicolaou/
pdfs/Sample_Chapter13.pdf.29 K. S. Goo and T. S. Sim, Curr. Comput.-Aided Drug Des.,
2011, 7, 53–80.
30 P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata,
C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997,
387, 827–830.
31 S. S. Weber, R. A. L. Bovenberg and A. J. M. Driessen,
Biotechnol. J., 2012, 7, 225–236.
32 J. D. Bloom and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A.,
2009, 106, 9995–10000.
33 A. S. Bommarius, J. K. Blum and M. J. Abrahamson, Curr.
Opin. Chem. Biol., 2011, 15, 194–200.
34 K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb.Technol., 2011, 48, 107–122.
35 J. L. Foo, C. B. Ching, M. W. Chang and S. S. J. Leong,
Biotechnol. Adv., 2012, 30, 541–549.
36 S. Robic, CBE Life Sci. Educ., 2010, 9, 189–195.
37 R. Burioni, D. Cassi, F. Cecconi and A. Vulpiani, Proteins:
Struct., Funct., Bioinf., 2004, 55, 529–535.
38 W. F. Li, X. X. Zhou and P. Lu, Biotechnol. Adv., 2005, 23,
271–281.
39 T. Koudelakova, R. Chaloupkova, J. Brezovsky, Z. Prokop,
E. Sebestova, M. Hesseler, M. Khabiri, M. Plevaka,
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 29/32
6562 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
D. Kulik, I. K. Smatanova, P. Rezacova, R. Ettrich,
U. T. Bornscheuer and J. Damborsky, Angew. Chem., Int.
Ed., 2013, 52, 1959–1963.
40 T. Koudelakova, S. Bidmanova, P. Dvorak, A. Pavelka,
R. Chaloupkova, Z. Prokop and J. Damborsky, Biotechnol.
J., 2013, 8, 32–45.
41 J. K. Blum and A. S. Bommarius, J. Mol. Catal. B: Enzym.,
2010, 67, 21–28.
42 J. K. Blum, M. D. Ricketts and A. S. Bommarius, J. Biotechnol., 2012, 160, 214–221.
43 Y. Harano, Entropy, 2012, 14, 1443–1468.
44 J. L. England and G. Haran, in Annual Review of Physical
Chemistry, ed. S. R. Leone, P. S. Cremer, J. T. Groves and
M. A. Johnson, 2011, vol. 62, pp. 257–277.
45 R. Singh, M. Tiwari, R. Singh and J.-K. Lee, Int. J. Mol. Sci.,
2013, 14, 1232–1277.
46 T. R. M. Barends, J. J. Polderman-Tijmes, P. A. Jekel,
C. M. H. Hensgens, E. J. de Vries, D. B. Janssen and
B. W. Dijkstra, J. Biol. Chem., 2003, 278, 23076–23084.
47 G. De Simone, V. Menchise, G. Manco, L. Mandrich,
N. Sorrentino, D. Lang, M. Rossi and C. Pedone, J. Mol. Biol., 2001, 314, 507–518.
48 A. Cipolla, F. Delbrassine, J.-L. Da Lage and G. Feller,
Biochimie, 2012, 94, 1943–1950.
49 P. L. Wintrode and P. H. Arnold, in Advances in Protein
Chemistry, Vol 55: Evolutionary Protein Design, ed. F. H.
Arnold, 2001, vol. 55, pp. 161–225.
50 M. Camps, A. Herman, E. Loh and L. A. Loeb, Crit. Rev.
Biochem. Mol. Biol., 2007, 42, 313–326.
51 S. Bershtein, K. Goldin and D. S. Tawfik, J. Mol. Biol., 2008,
379, 1029–1044.
52 S. Bershtein and D. S. Tawfik, Curr. Opin. Chem. Biol., 2008,
12, 151–158.
53 S. G. Peisajovich and D. S. Tawfik, Nat. Med., 2007, 4,991–994.
54 D. P. C. de Barros, P. Fernandes, J. M. S. Cabral and
L. P. Fonseca, J. Chem. Technol. Biotechnol., 2010, 85,
1553–1560.
55 T. A. Rogers and A. S. Bommarius, Chem. Eng. Sci., 2010,
65, 2118–2124.
56 P. Johnson and T. L. Whateley, Biochem. J., 1981, 193,
285–294.
57 F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protocols,
2007, 2, 2212–2221.
58 G. A. Senisterra and P. J. Finerty, Mol. BioSyst., 2009, 5,
217–223.59 J. V. Rodrigues, V. Prosinecki, I. Marrucho, L. P. N. Rebelo
and C. M. Gomes, Phys. Chem. Chem. Phys., 2011, 13,
13614–13616.
60 O. Levenspiel, Ind. Eng. Chem. Res., 1999, 38, 4140–4143.
61 R. O. Jenkins, Appl. Organomet. Chem., 2004, 18, 373.
62 T. A. Rogers, R. M. Daniel and A. S. Bommarius,
ChemCatChem, 2009, 1, 131–137.
63 K. D. A. S. Bommarius, U. Groeger and C. Wandrey,
Membrane Bioreactors for the Production of Enantiomerically
Pure a-Amino Acids, Wiley, Chichester, England, 1992.
64 A. S. Bommarius, K. Drauz, H. Klenk and C. Wandrey, in
Enzyme Engineering Xi , ed. D. S. Clark and D. A. Estell,
1992, vol. 672, pp. 126–136.
65 C. Wandrey, habilitation thesis, TH Hannover, Germany,
1977.
66 R. M. Daniel, M. J. Danson and R. Eisenthal, Trends
Biochem. Sci., 2001, 26, 223–225.
67 M. E. Peterson, R. Eisenthal, M. J. Danson, A. Spence and
R. M. Daniel, J. Biol. Chem., 2004, 279, 20717–20722.68 T. Ema, M. Kageyama, T. Korenaga and T. Sakai, Tetra-
hedron: Asymmetry, 2003, 14, 3943–3947.
69 N. End and K. U. Schoning, Immobilized Catalysts, 2004,
242, 273–317.
70 B. H. Zhang, Y. Q. Weng, H. Xu and Z. P. Mao, Appl.
Microbiol. Biotechnol., 2012, 93, 61–70.
71 D. A. Cowan and R. Fernandez-Lafuente, Enzyme Microb.
Technol., 2011, 49, 326–346.
72 V. Grazu, F. Lopez-Gallego, T. Montes, O. Abian,
R. Gonzalez, J. A. Hermoso, J. L. Garcıa, C. Mateo and
J. M. Guisan, Process Biochem., 2010, 45, 390–398.
73 D. A. Cecchini, R. Pavesi, S. Sanna, S. Daly, R. Xaiz,M. Pregnolato and M. Terreni, Appl. Microbiol. Biotechnol.,
2012, 95, 1491–1500.
74 N. Brun, A. B. Garcia, H. Deleuze, M. F. Achard, C. Sanchez,
F. Durand, V. Oestreicher and R. Backov, Chem. Mater.,
2010, 22, 4555–4562.
75 R. Backov, Soft Matter , 2006, 2, 452–464.
76 H. S. Kim, S. J. Lee and E. Y. Lee, J. Mol. Catal. B: Enzym.,
2006, 43, 2–8.
77 A. Fishman, I. Levy, U. Cogan and O. Shoseyov, J. Mol.
Catal. B: Enzym., 2002, 18, 121–131.
78 P. Asuri, S. S. Karajanagi, H. C. Yang, T. J. Yim, R. S. Kane
and J. S. Dordick, Langmuir , 2006, 22, 5833–5836.
79 P. Asuri, S. S. Karajanagi, A. A. Vertegel, J. S. Dordick andR. S. Kane, J. Nanosci. Nanotechnol., 2007, 7, 1675–1678.
80 V. G. S. G. Bonavia, J. Manuel, R. F. Lafuente, O. A. Franco,
T. M. Fernandez, C. M. Gonzalez, R. G. Garcia, J. A. Hermoso
Domingues and J. L. Garcia Lopez, WO 2007138148 A1
20071206, 2007.
81 L. Wilson, A. Illanes, B. C. C. Pessela, O. Abian,
R. Fernandez-Lafuente and J. M. Guisan, Biotechnol.
Bioeng., 2004, 86, 558–562.
82 J. M. Bolivar, L. Wilson, S. A. Ferrarotti, R. Fernandez-
Lafuente, J. M. Guisan and C. Mateo, Biomacromolecules,
2006, 7, 669–673.
83 J. Z. Setschenow, Z. Phys. Chem., 1889, 4, 117–125.84 E. J. Cohn, Proteins, amino acids and peptides as ions and
dipolar ions, Reinhold Publishing Corporation, New York,
1943.
85 J. Hine, Physical Organic Chemistry: Second Edition,
McGraw-Hill, New York, 1962.
86 C. H. Wong, D. G. Drueckhammer and H. M. Sweers, J. Am.
Chem. Soc., 1985, 107, 4028–4031.
87 A. S. Bommarius and A. Karau, Biotechnol. Prog., 2005, 21,
1663–1672.
88 F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 30/32
This journal is c The Royal Society of Chemistry 2013 Chem. Soc. Rev., 2013, 42, 6534--6565 6563
89 H. V. H. Peter and K.-Y. Wong, Science, 1964, 145, 577–580.
90 O. Sinanogl and S. Abdulnur, Fed. Proc., 1965, 24, S12–S23.
91 P. A. Grigorjev and S. M. Bezrukov, Biophys. J., 1994, 67,
2265–2271.
92 J. G. Kirkwood, Proteins, amino acids and peptides as ions
and dipolar ions, Reinhold Publishing Corporation,
New York, 1943.
93 W. Melander and C. Horvath, Arch. Biochem. Biophys.,
1977, 183, 200–215.94 K. D. Collins, Proc. Natl. Acad. Sci. U. S. A., 1995, 92,
5553–5557.
95 K. D. Collins, Biophys. J., 1997, 72, 65–76.
96 K. D. Collins, Methods, 2004, 34, 300–311.
97 K. D. Collins and M. W. Washabaugh, Q. Rev. Biophys.,
1985, 18, 323–422.
98 G. Jones and M. Dole, J. Am. Chem. Soc., 1929, 51, 2950–2964.
99 J. M. Broering and A. S. Bommarius, Biochem. Soc. Trans.,
2007, 35, 1602–1605.
100 J. M. Broering and A. S. Bommarius, J. Phys. Chem. B, 2005,
109, 20612–20619.
101 K. D. Collins, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5553–5557.102 M. W. Washabaugh and K. D. Collins, Q. Rev. Biophys.,
1985, 18, 323–422.
103 R. Lumry and H. Eyring, J. Phys. Chem., 1954, 58, 110–120.
104 J. M. Broering and A. S. Bommarius, J. Phys. Chem. B, 2008,
112, 12768–12775.
105 V. Yeh, J. M. Broering, A. Romanyuk, B. X. Chen,
Y. O. Chernoff and A. S. Bommarius, Protein Sci., 2010,
19, 47–56.
106 M. Hall, J. Rubin, S. H. Behrens and A. S. Bommarius,
J. Biotechnol., 2011, 155, 370–376.
107 J. K. Kaushik and R. Bhat, J. Biol. Chem., 2003, 278,
26458–26465.
108 D. J. Boerema, V. A. Tereshko and S. B. H. Kent, Pept. Sci.,2008, 90, 278–286.
109 A. Nasiripourdori, H. Naderi-Manesh, B. Ranjbar and
K. Khajeh, Int. J. Biol. Macromol., 2009, 44, 311–315.
110 H. A. Sathish, P. R. Kumar and V. Prakash, Int. J. Biol.
Macromol., 2007, 41, 383–390.
111 A. Tiwari and R. Bhat, Biophys. Chem., 2006, 124, 90–99.
112 M. Arroyo, R. Torres-Guzman, I. de la Mata, M. P. Castillon
and C. Acebal, Enzyme Microb. Technol., 2000, 27, 122–126.
113 M. Arroyo, R. Torres-Guzman, I. de la Mata, M. P. Castillon
and C. Acebal, Biotechnol. Prog., 2000, 16, 368–371.
114 C. G. Suresh, A. V. Pundle, H. SivaRaman, K. N. Rao,
J. A. Brannigan, C. E. McVey, C. S. Verma, Z. Dauter,E. J. Dodson and G. G. Dodson, Nat. Struct. Biol., 1999, 6,
414–416.
115 D. A. Estell, T. P. Graycar, J. V. Miller, D. B. Powers,
J. P. Burnier, P. G. Ng and J. A. Wells, Science, 1986, 233,
659–663.
116 J. A. Wells, D. B. Powers, R. R. Bott, T. P. Graycar and D. A.
Estell, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1219–1223.
117 J. P. Colletier, A. Aleksandrov, N. Coquelle, S. Mraihi,
E. Mendoza-Barbera, M. Field and D. Madern, Mol. Biol.
Evol., 2012, 29, 1683–1694.
118 M. Soskine and D. S. Tawfik, Nat. Rev. Genet., 2010, 11,
572–582.
119 F. H. Arnold, P. L. Wintrode, K. Miyazaki and
A. Gershenson, Trends Biochem. Sci., 2001, 26, 100–106.
120 H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science,
2003, 299, 1694–1697.
121 A. A.-A.-F. Amara, Pak. J. Pharm. Sci., 2013, 26, 217–232.
122 R. Kourist, H. Jochens, S. Bartsch, R. Kuipers, S. K. Padhi,
M. Gall, D. Bottcher, H. J. Joosten and U. T. Bornscheuer,ChemBioChem, 2010, 11, 1635–1643.
123 J. G. Saven, Curr. Opin. Struct. Biol., 2002, 12, 453–458.
124 S. G. Kang and J. G. Saven, Curr. Opin. Chem. Biol., 2007,
11, 329–334.
125 M. Di Lorenzo, A. Hidalgo, R. Molina, J. A. Hermoso,
D. Pirozzi and U. T. Bornscheuer, Appl. Environ. Microbiol.,
2007, 73, 7291–7299.
126 S. Cesarini, C. Bofill, F. I. J. Pastor, M. T. Reetz and P. Diaz,
Process Biochem., 2012, 47, 2064–2071.
127 D. J. Opperman and M. T. Reetz, ChemBioChem, 2010, 11,
2589–2596.
128 J. T. Park, J.-I. Hirano, V. Thangavel, B. R. Riebel and A. S.Bommarius, J. Mol. Catal. B: Enzym., 2011, 71, 159–165.
129 M. Katzberg, N. Skorupa-Parachin, M.-F. Gorwa-Grauslund
and M. Bertau, Int. J. Mol. Sci., 2010, 11, 1735–1758.
130 Y. Ito, A. Ikeuchi and C. Imamura, Protein Eng., Des. Sel.,
2013, 26, 73–79.
131 H. Park, J. Joo, K. Park and Y. Yoo, Biotechnol. Bioprocess
Eng., 2012, 17, 722–728.
132 Z. Qian, J. R. Horton, X. Cheng and S. Lutz, J. Mol. Biol.,
2009, 393, 191–201.
133 A. Badoei-Dalfard, K. Khajeh, S. M. Asghari, B. Ranjbar and
H. R. Karbalaei-Heidari, J. Biochem., 2010, 148, 231–238.
134 J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff,
N. Trinh, D. A. Kochrekar, R. Nair Cherat and G. G. Pai,Org. Process Res. Dev., 2009, 14, 193–198.
135 M.-S. Kim and X. Lei, Appl. Microbiol. Biotechnol., 2008, 79,
69–75.
136 G. Gonzalez-Blasco, J. Sanz-Aparicio, B. Gonzalez,
J. A. Hermoso and J. Polaina, J. Biol. Chem., 2000, 275,
13708–13712.
137 N. Palackal, Y. Brennan, W. N. Callen, P. Dupree, G. Frey,
F. Goubet, G. P. Hazlewood, S. Healey, Y. E. Kang,
K. A. Kretz, E. Lee, X. Q. Tan, G. L. Tomlinson,
J. Verruto, V. W. K. Wong, E. J. Mathur, J. M. Short,
D. E. Robertson and B. A. Steer, Protein Sci., 2004, 13,
494–503.138 J. F. Chaparro-Riggers, K. M. Polizzi and A. S. Bommarius,
Biotechnol. J., 2007, 2, 180–191.
139 P. C. Rathi, S. Radestock and H. Gohlke, J. Biotechnol.,
2012, 159, 135–144.
140 S. Radestock and H. Gohlke, Eng. Life Sci., 2008, 8, 507–522.
141 S. Hirose, Y. Kawamura, M. Mori, K. Yokota, T. Noguchi
and N. Goshima, New Biotechnol., 2011, 28, 225–231.
142 B. Synstad, S. Gaseidnes, D. M. F. van Aalten, G. Vriend,
J. E. Nielsen and V. G. H. Eijsink, Eur. J. Biochem., 2004,
271, 253–262.
Review Article Chem Soc Rev
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 31/32
6564 Chem. Soc. Rev., 2013, 42, 6534--6565 This journal is c The Royal Society of Chemistry 2013
143 J. M. Macdonald, C. A. Tarling, E. J. Taylor, R. J. Dennis,
D. S. Myers, S. Knapp, G. J. Davies and S. G. Withers,
Angew. Chem., Int. Ed., 2010, 49, 2599–2602.
144 I. Tews, A. C. T. vanScheltinga, A. Perrakis, K. S. Wilson
and B. W. Dijkstra, J. Am. Chem. Soc., 1997, 119, 7954–7959.
145 D. Gao, D. L. Narasimhan, J. Macdonald, R. Brim,
M.-C. Ko, D. W. Landry, J. H. Woods, R. K. Sunahara and
C.-G. Zhan, Mol. Pharmacol., 2009, 75, 318–323.
146 J. M. Turner, N. A. Larsen, A. Basran, C. F. Barbas,N. C. Bruce, I. A. Wilson and R. A. Lerner, Biochemistry,
2002, 41, 12297–12307.
147 N. A. Larsen, J. M. Turner, J. Stevens, S. J. Rosser,
A. Basran, R. A. Lerner, N. C. Bruce and I. A. Wilson, Nat.
Struct. Biol., 2002, 9, 17–21.
148 C. J. Rogers, L. M. Eubanks, T. J. Dickerson and
K. D. Janda, J. Am. Chem. Soc., 2006, 128, 15364–15365.
149 F. Zheng and C.-G. Zhan, J. Comput. Aided Mol. Des., 2008,
22, 661–671.
150 X. Huang, D. Gao and C.-G. Zhan, Org. Biomol. Chem.,
2011, 9, 4138–4143.
151 Y. Wei, L. Swenson, C. Castro, U. Derewenda, W. Minor,H. Arai, J. Aoki, K. Inoue, L. Servin-Gonzalez and
Z. S. Derewenda, Structure, 1998, 6, 511–519.
152 M. G. Pikkemaat, A. B. M. Linssen, H. J. C. Berendsen and
D. B. Janssen, Protein Eng., 2002, 15, 185–192.
153 X. J. Feng, J. Sanchis, M. T. Reetz and H. Rabitz,
Chem.–Eur. J., 2012, 18, 5646–5654.
154 M. T. Reetz, P. Soni, J. P. Acevedo and J. Sanchis, Angew.
Chem., Int. Ed., 2009, 48, 8268–8272.
155 W. Augustyniak, A. A. Brzezinska, T. Pijning, H. Wienk,
R. Boelens, B. W. Dijkstra and M. T. Reetz, Protein Sci.,
2012, 21, 487–497.
156 H. Jochens, D. Aerts and U. T. Bornscheuer, Protein Eng.,
Des. Sel., 2010, 23, 903–909.157 K. Kawasaki, H. Kondo, M. Suzuki, S. Ohgiya and S. Tsuda,
Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58,
1168–1174.
158 Y. Gumulya and M. T. Reetz, ChemBioChem, 2011, 12,
2502–2510.
159 M. Lehmann, C. Loch, A. Middendorf, D. Studer,
S. F. Lassen, L. Pasamontes, A. van Loon and M. Wyss,
Protein Eng., 2002, 15, 403–411.
160 M. Lehmann, L. Pasamontes, S. F. Lassen and M. Wyss,
Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000,
1543, 408–415.
161 M. Lehmann and M. Wyss, Curr. Opin. Biotechnol., 2001,12, 371–375.
162 N. Amin, A. D. Liu, S. Ramer, W. Aehle, D. Meijer,
M. Metin, S. Wong, P. Gualfetti and V. Schellenberger,
Protein Eng., Des. Sel., 2004, 17, 787–793.
163 K. Watanabe, T. Ohkuri, S. Yokobori and A. Yamagishi,
J. Mol. Biol., 2006, 355, 664–674.
164 H. K. Binz, M. T. Stumpp, P. Forrer, P. Amstutz and
A. Pluckthun, J. Mol. Biol., 2003, 332, 489–503.
165 M. K. DiTursi, S. J. Kwon, P. J. Reeder and J. S. Dordick,
Protein Eng., Des. Sel., 2006, 19, 517–524.
166 K. M. Polizzi, J. F. Chaparro-Riggers, E. Vazquez-Figueroa
and A. S. Bommarius, Biotechnol. J., 2006, 1, 531–536.
167 C. E. McVey, M. A. Walsh, G. G. Dodson, K. S. Wilson and
J. A. Brannigan, J. Mol. Biol., 2001, 313, 139–150.
168 B. Steipe, B. Schiller, A. Pluckthun and S. Steinbacher,
J. Mol. Biol., 1994, 240, 188–192.
169 E. Vazquez-Figueroa, J. Chaparro-Riggers and A. S. Bommarius,
ChemBioChem, 2007, 8, 2295–2301.
170 F. H. Arnold and G. Georgiou, Directed Enzyme Evolution:Screening and Selection Methods, 1st edn, Humana Press,
Totowa, New Jersey, 2003.
171 J. Gomes and W. Steiner, Food Technol. Biotechnol., 2004,
42, 223–235.
172 M. Podar and A. L. Reysenbach, Curr. Opin. Biotechnol.,
2006, 17, 250–255.
173 M. T. Reetz, J. D Carballeira and A. Vogel, Angew. Chem.,
Int. Ed., 2006, 45, 7745–7751.
174 K. Gunasekaran, C. Ramakrishnan and P. Balaram, J. Mol.
Biol., 1996, 264, 191–198.
175 A. E. Serov, E. R. Odintzeva, I. V. Uporov and V. I. Tishkov,
Biochemistry, 2005, 70, 804–808.176 W. Besenmatter, P. Kast and D. Hilvert, Proteins: Struct.,
Funct., Bioinf., 2007, 66, 500–506.
177 J. D. Bloom, S. T. Labthavikul, C. R. Otey and F. H. Arnold,
Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5869–5874.
178 L. Riechmann and G. Winter, Proc. Natl. Acad. Sci. U. S. A.,
2000, 97, 10068–10073.
179 W. Gilbert, Nature, 1978, 271, 501.
180 W. F. Doolittle, Nature, 1978, 272, 581–582.
181 C. C. F. Blake, Nature, 1978, 273, 267.
182 L. D. Bogarad and M. W. Deem, Proc. Natl. Acad. Sci. U. S. A.,
1999, 96, 2591–2595.
183 J. J. Silberg, J. B. Endelman and F. H. Arnold, Protein Eng.,
2004, 388, 35–42.184 C. A. Voigt, C. Martinez, Z. G. Wang, S. L. Mayo and F. H.
Arnold, Nat. Struct. Biol., 2002, 9, 553–558.
185 L. Yuan, I. Kurek, J. English and R. Keenan, Microbiol. Mol.
Biol. Rev., 2005, 69, 373–392.
186 N. D. Clarke, Curr. Opin. Struct. Biol., 2010, 20, 527–532.
187 P. Heinzelman, C. D. Snow, M. A. Smith, X. L. Yu,
A. Kannan, K. Boulware, A. Villalobos, S. Govindarajan,
J. Minshull and F. H. Arnold, J. Biol. Chem., 2009, 284,
26229–26233.
188 P. Heinzelman, C. D. Snow, I. Wu, C. Nguyen, A. Villalobos,
S. Govindarajan, J. Minshull and F. H. Arnold, Proc. Natl.
Acad. Sci. U. S. A., 2009, 106, 5610–5615.189 P. A. Romero, E. Stone, C. Lamb, L. Chantranupong,
A. Krause, A. E. Miklos, R. A. Hughes, B. Fechtel,
A. D. Ellington, F. H. Arnold and G. Georgiou, ACS Synth.
Biol., 2012, 1, 221–228.
190 P. Heinzelman, R. Komor, A. Kanaan, P. Romero, X. Yu,
S. Mohler, C. Snow and F. Arnold, Protein Eng., Des. Sel.,
2010, 23, 871–880.
191 M. A. Smith, A. Rentmeister, C. D. Snow, T. Wu,
M. F. Farrow, F. Mingardon and F. H. Arnold, FEBS J.,
2012, 279, 4453–4465.
Chem Soc Rev Review Article
View Article Online

7/26/2019 Stabilizing biocatalysts.pdf
http://slidepdf.com/reader/full/stabilizing-biocatalystspdf 32/32
192 G. Parsiegla, C. Reverbel-Leroy, C. Tardif, J. P. Belaich,
H. Driguez and R. Haser, Biochemistry, 2000, 39, 11238–11246.
193 T. C. A. Biology, Royal Institute of Technology School of
Biotechnology, 2008.
194 J. A. E. Maatta, Y. Eisenberg-Domovich, H. R. Nordlund,
R. Hayouka, M. S. Kulomaa, O. Livnah and V. P. Hytonen,
Biotechnol. Bioeng., 2011, 108, 481–490.
195 S. Repo, T. A. Paldanius, V. P. Hytonen, T. K. M. Nyholm,
K. K. Halling, J. Huuskonen, O. T. Pentikainen,K. Rissanen, J. P. Slotte, T. T. Airenne, T. A. Salminen,
M. S. Kulomaa and M. S. Johnson, Chem. Biol., 2006, 13,
1029–1039.
196 L. Baugh, I. Le Trong, D. S. Cerutti, N. Mehta, S. Gulich, P. S.
Stayton, R. E. Stenkamp and T. P. Lybrand, Biochemistry,
2011, 51, 597–607.
197 O. Livnah, E. A. Bayer, M. Wilchek and J. L. Sussman, Proc.
Natl. Acad. Sci. U. S. A., 1993, 90, 5076–5080.
198 C. R. Otey, M. Landwehr, J. B. Endelman, K. Hiraga,
J. D. Bloom and F. H. Arnold, PLoS Biol., 2006, 4, e112.
199 A. Cerdobbel, K. De Winter, D. Aerts, R. Kuipers,
H.-J. Joosten, W. Soetaert and T. Desmet, Protein Eng., Des. Sel., 2011, 24, 829–834.
200 N. M. Osorio, M. M. R. da Fonseca and S. Ferreira-Dias,
Eur. J. Lipid Sci. Technol., 2006, 108, 545–553.
201 D. A. Cowan, Comp. Biochem. Physiol., A: Mol. Integr.
Physiol., 1997, 118, 429–438.
202 R. K. Owusu and D. A. Cowan, Enzyme Microb. Technol.,
1989, 11, 568–574.
203 J. J. Hao and A. Berry, Protein Eng., Des. Sel., 2004, 17,
689–697.
204 M. T. Reetz, P. Soni, L. Fernandez, Y. Gumulya and
J. D. Carballeira, Chem. Commun., 2010, 46, 8657–8658.
205 B. V. Adalbjornsson, H. S. Toogood, A. Fryszkowska,C. R. Pudney, T. A. Jowitt, D. Leys and N. S. Scrutton,
ChemBioChem, 2010, 11, 197–207.
206 E. Brenna, F. G. Gatti, D. Monti, F. Parmeggiani and
S. Serra, Adv. Synth. Catal., 2012, 354, 105–112.
207 B. Van den Burg, A. de Kreij, P. Van der Veek, J. Mansfeld
and G. Venema, Biotechnol. Appl. Biochem., 1999, 30, 35–40.
208 J. Mansfeld and R. Ulbrich-Hofmann, Biotechnol. Bioeng.,
2007, 97, 672–679.
209 S. Sharma, A. Mittal, V. K. Gupta and H. Singh, Enzyme
Microb. Technol., 2007, 40, 337–342.
210 H. Slusarczyk, S. Felber, M. R. Kula and M. Pohl, Eur. J.
Biochem., 2000, 267, 1280–1289.211 E. Vazquez-Figueroa, V. Yeh, J. M. Broering, J. F. Chaparro-
Riggers and A. S. Bommarius, Protein Eng., Des. Sel., 2008,
21, 673–680.
Review Article Chem Soc Rev
View Article Online


















