
Stabilization of Lithium Transition Metal Silicates in the...
7
Stabilization of Lithium Transition Metal Silicates in the Olivine Structure Xiaoqi Sun, † Rajesh Tripathi, ‡ Guerman Popov, † Mahalingam Balasubramanian, § and Linda F. Nazar* ,† † Department of Chemistry and the Waterloo Institute of Nanotechnology, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada § Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, United States * S Supporting Information ABSTRACT: While olivine LiFePO 4 shows amongst the best electrochemical properties of Li-ion positive electrodes with respect to rate behavior owing to facile Li + migration pathways in the framework, replacing the [PO 4 ] 3− polyanion with a silicate [SiO 4 ] 4− moiety in olivine is desirable. This could allow additional alkali content and hence electron transfer, and increase the capacity. Herein we explore the possibility of a strategy toward new cathode materials and demonstrate the first stabilization of a lithium transition metal silicate (as a pure silicate) in the olivine structure type. Using LiInSiO 4 and LiScSiO 4 as the parent materials, transition metal (Mn, Fe, Co) substitutions on the In/Sc site were investigated by computational modeling via atomic scale simulation. Transition metal substitution was found to be only favorable for Co, a finding confirmed by the successful solid state synthesis of olivine Li x In y Co 2‑x‑y SiO 4 . Stabilization of the structure was achieved by entropy provided by cation disorder. ■ INTRODUCTION Lithium ion batteries are widely utilized as energy storage devices due to their high energy density, low cost, and small carbon footprint. 1−6 However, further increase in energy density and safety of these batteries is needed in order to meet future electrochemical energy storage goals. In this regard, the discovery and synthesis of new positive electrode materials have led to significant achievements in the last few decades. Ever since Goodenough and co-workers proposed the use of LiFePO 4 as a positive electrode material, 7 polyanion-based compounds have gained much attention. 8−12 Olivine LiFePO 4 has been the most successful among these. Owing to facile Li- ion conduction along one-dimensional (1D) pathways arising from ordering of the Li + and Fe 2+ on the two different octahedral sites in the lattice, 13−15 and formation of a Li solid solution on partial states of charge/discharge at a moderate overpotential, 16,17 LiFePO 4 exhibits some of the highest charge−discharge rates achieved in a Li-ion battery. 18−20 Nonetheless, the specific energy of this material is rather modest. The electrochemical de-/intercalation of Li takes place at 3.45 V (vs Li), and the maximum specific capacity is only 170 mAh g −1 . Compounds based on PO 4 3− as well as other polyanion groups such as BO 3 3− , SiO 4 4− , SO 4 2− , and SO 4 F 3− have been synthesized and examined for their Li-ion intercalation properties. 21−29 Although lithium iron borates and fluorosulfates reversibly de-/intercalate lithium, in neither case does the specific energy improve much over the phosphates. 21,26,29 Silicates, on the other hand, are a good alternative owing to the higher charge on the SiO 4 4− polyanion, which requires additional alkali capacity to balance the charge. 30−33 Thus, in addition to silicon’s abundance and non-toxic properties, the SiO 4 4− anion has a high charge to weight ratio which leads to higher specific energy storage capacity (at least theoretically). In the first and most widely studied silicate based positive electrode material, Li 2 FeSiO 4 , two lithium ions are available for extraction, based on the Fe 2+ / Fe 3+ and Fe 3+ /Fe 4+ redox couples. 24,34−37 However, the latter couple is at too high a voltage to be accessible, and thus only one lithium ion undergoes de-/intercalation. In addition, Li 2 FeSiO 4 undergoes an irreversible phase transformation on the first cycle, leading to a decrease of the discharge voltage from 3.1 to 2.8 V, and hence a drop in the energy density. 38 In order to use lithium transition metal silicates as potential high capacity electrodes for long-term applications, these materials must be synthesized in more robust framework types. Once such framework is olivine, which is also adopted by many natural silicate minerals with a general formula of M I M II SiO 4 (M I ,M II : cations). It comprises hexagonal close packed oxygen anions with the two M I and M II cations being octahedrally coordinated in sites 1 and 2, and Si 4+ occupying 1/8 of the tetrahedral sites (Figure 1). Examples include materials where both M I and M II cations are in their +2 state, such as MgFeSiO 4 (where Mg and Fe are fully disordered over site 1 and site 2) and Mn 2 SiO 4 . 39−42 However, in cases where the cations either have +1 or +3 charges such as in Li 1+ Sc 3+ SiO 4 and Li 1+ In 3+ SiO 4 , their charge difference results in perfect cation ordering over the two octahedral M1 and M2 sites. This Received: June 7, 2017 Published: July 28, 2017 Article pubs.acs.org/IC © 2017 American Chemical Society 9931 DOI: 10.1021/acs.inorgchem.7b01453 Inorg. Chem. 2017, 56, 9931−9937
Transcript of Stabilization of Lithium Transition Metal Silicates in the...

Stabilization of Lithium Transition Metal Silicates in the OlivineStructureXiaoqi Sun,† Rajesh Tripathi,‡ Guerman Popov,† Mahalingam Balasubramanian,§ and Linda F. Nazar*,†
†Department of Chemistry and the Waterloo Institute of Nanotechnology, University of Waterloo, Waterloo, Ontario N2L 3G1,Canada§Advanced Photon Source, Argonne National Laboratory, Argonne, Illinois 60439, United States
*S Supporting Information
ABSTRACT: While olivine LiFePO4 shows amongst the best electrochemicalproperties of Li-ion positive electrodes with respect to rate behavior owing to facileLi+ migration pathways in the framework, replacing the [PO4]
3− polyanion with asilicate [SiO4]
4− moiety in olivine is desirable. This could allow additional alkalicontent and hence electron transfer, and increase the capacity. Herein we explorethe possibility of a strategy toward new cathode materials and demonstrate the firststabilization of a lithium transition metal silicate (as a pure silicate) in the olivinestructure type. Using LiInSiO4 and LiScSiO4 as the parent materials, transitionmetal (Mn, Fe, Co) substitutions on the In/Sc site were investigated bycomputational modeling via atomic scale simulation. Transition metal substitutionwas found to be only favorable for Co, a finding confirmed by the successful solidstate synthesis of olivine LixInyCo2‑x‑ySiO4. Stabilization of the structure wasachieved by entropy provided by cation disorder.
■ INTRODUCTION
Lithium ion batteries are widely utilized as energy storagedevices due to their high energy density, low cost, and smallcarbon footprint.1−6 However, further increase in energydensity and safety of these batteries is needed in order tomeet future electrochemical energy storage goals. In this regard,the discovery and synthesis of new positive electrode materialshave led to significant achievements in the last few decades.Ever since Goodenough and co-workers proposed the use ofLiFePO4 as a positive electrode material,7 polyanion-basedcompounds have gained much attention.8−12 Olivine LiFePO4has been the most successful among these. Owing to facile Li-ion conduction along one-dimensional (1D) pathways arisingfrom ordering of the Li+ and Fe2+ on the two differentoctahedral sites in the lattice,13−15 and formation of a Li solidsolution on partial states of charge/discharge at a moderateoverpotential,16,17 LiFePO4 exhibits some of the highestcharge−discharge rates achieved in a Li-ion battery.18−20
Nonetheless, the specific energy of this material is rathermodest. The electrochemical de-/intercalation of Li takes placeat 3.45 V (vs Li), and the maximum specific capacity is only 170mAh g−1. Compounds based on PO4
3− as well as otherpolyanion groups such as BO3
3−, SiO44−, SO4
2−, and SO4F3−
have been synthesized and examined for their Li-ionintercalation properties.21−29 Although lithium iron boratesand fluorosulfates reversibly de-/intercalate lithium, in neithercase does the specific energy improve much over thephosphates.21,26,29 Silicates, on the other hand, are a goodalternative owing to the higher charge on the SiO4
4− polyanion,which requires additional alkali capacity to balance the
charge.30−33 Thus, in addition to silicon’s abundance andnon-toxic properties, the SiO4
4− anion has a high charge toweight ratio which leads to higher specific energy storagecapacity (at least theoretically). In the first and most widelystudied silicate based positive electrode material, Li2FeSiO4,two lithium ions are available for extraction, based on the Fe2+/Fe3+ and Fe3+/Fe4+ redox couples.24,34−37 However, the lattercouple is at too high a voltage to be accessible, and thus onlyone lithium ion undergoes de-/intercalation. In addition,Li2FeSiO4 undergoes an irreversible phase transformation onthe first cycle, leading to a decrease of the discharge voltagefrom 3.1 to 2.8 V, and hence a drop in the energy density.38 Inorder to use lithium transition metal silicates as potential highcapacity electrodes for long-term applications, these materialsmust be synthesized in more robust framework types.Once such framework is olivine, which is also adopted by
many natural silicate minerals with a general formula ofMIMIISiO4 (MI, MII: cations). It comprises hexagonal closepacked oxygen anions with the two MI and MII cations beingoctahedrally coordinated in sites 1 and 2, and Si4+ occupying1/8 of the tetrahedral sites (Figure 1). Examples includematerials where both MI and MII cations are in their +2 state,such as MgFeSiO4 (where Mg and Fe are fully disordered oversite 1 and site 2) and Mn2SiO4.
39−42 However, in cases wherethe cations either have +1 or +3 charges such as in Li1+Sc3+SiO4and Li1+In3+SiO4, their charge difference results in perfectcation ordering over the two octahedral M1 and M2 sites. This
Received: June 7, 2017Published: July 28, 2017
Article
pubs.acs.org/IC
© 2017 American Chemical Society 9931 DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937

makes these materials good potential Li-ion cathodes if atransition metal can be substituted for Sc or In. The voltage forLi extraction from LiMSiO4 (M = Mn, Fe, Co, Ni) is predictedto be around 5 V,43 which approaches the upper voltage limit oftypical electrolytes. Many attempts have been focused onsynthesizing these materials in the olivine polymorph.According to a computational study,44 olivine type LiFeSiO4has a lower free energy than the material arising from extractionof one lithium ion from Li2FeSiO4; however, in this report, theauthor’s attempts to directly synthesize olivine LiMSiO4 (M =Fe, Mn) resulted in the formation of many other products, withpyroxene (LiMSi2O6) being the major phase. While cleverstrategies to carry out subsitution of the anionic frameworknamely to partially replace P by Si in LiFePO4resulted in as o l i d s o l u t i o n b e twe en L iF ePO4 and F e 2 S iO 4(Li1−xFe1+xP1−xSixO4; 0 < x < 1), Fe2+ was found to partiallyoccupy both the M1 and M2 sites.45 The immobile anddisordered Fe2+ cation in the structure blocks Li-ion pathwaysand renders the material electrochemically inactive for x > 0.2.Herein, we study the possibility of stabilizing lithium
transition metal in the olivine framework. We investigateLiInSiO4 and LiScSiO4 as parent structures to attempt tostabilize two series of olivine silicate compounds of generalformula LiIn1−xMxSiO4 and LiSc1−xMxSiO4 (M = Mn3+, Fe3+ orCo3+). We used atomistic scale simulation to explore theenergetics of transition metal cation substitution behavior. Onthe basis of these results, solid-state synthesis of selectedmaterials and combined neutron and synchrotron refinementsof those materials were conducted to determine occupancies inthe two olivine cation sites, M1 and M2. Whereas substitutionin the scandium olivine was not successful, we find that a highdegree of substitution was obtained with cobalt substitution forindium, althoughlike the Si/P substitution described abovefor LiFePO4 this results in cobalt mixing over the two sitesand reduction to the Co2+ oxidation state.
■ EXPERIMENTAL SECTIONAtomistic Scale Simulation Methods. This study used well
established modeling techniques which are detailed elsewhere,46 andhence only a general description will be given here. Interactionsbetween ions in the olivine structures consist of a long-rangeCoulombic term and a short-range component representingelectron−electron repulsion and van der Waals interactions. Theshort-range interactions were modeled using the two-body Bucking-ham potential (Supporting Information, Table S1). An additional
three-body term was used for the SiO44− units to account for the angle
dependent nature of O−Si−O bonds, as previously used for othersulfate and phosphate units.14,47 The well-known shell model wasemployed to account for the polarizability effects of charged defects onthe electronic charge clouds.48 The In−O interatomic potential wastaken from the study of Fisher et al.49 on Ba2In2O5 and the Sc−Ointeraction was adopted and refined from a publication of Minervini etal.50 on Sc doping in Ce2O3. Other atomic potentials such as O−Oand Li−O were taken from another work of Fisher et al.,51 while Si−Opotentials adopted from the work of Siertka et al.52 were found to bethe best for reproducing the olivine structural parameters of LiInSiO4and LiScSiO4. The three body potential model used for the SiO4
4− unitwas the same as that used for PO4
3− by Islam et al.14 Many differentvalues for three body terms of SiO4
4− have been reported in theliterature, and the value used in the current work was very close to thatused by Sanders et al.53 for SiO2. Interatomic potential values to studythe doping energy of M3+ ions (M-O potential) were taken from thework of Woodely et al. and Braithwaite et al.54,55 As argued previously,these interatomic potential methods are assessed primarily by theirability to reproduce observed crystal properties. Indeed, they are foundto work well, even for compounds where there is undoubtedly a degreeof covalency such as aluminophosphates.56
The lattice relaxation about defects (such as Li vacancies) andmigrating ions was calculated by an implementation of the Mott-Littleton scheme incorporated in the GULP code.57 This methodpartitions a crystal lattice into two regions, where ions in the innerregion immediately surrounding the defect (on the order of >700ions) are relaxed explicitly. Relaxations of such a large number of ionsare important for charge defects that introduce long-range electrostaticperturbations and are not easily treated by electronic structuremethods. The outer region extends to infinity, with the outer latticerelaxations treated by quasi-continuum methods.
Synthesis and Characterization. Cobalt substituted LiInSiO4samples were synthesized by solid state reaction of Li2CO3 (Sigma-Aldrich, 99.4%), cobalt(III) acetylacetonate (Co(C5H7O2)3, Sigma-Aldrich, 99.99%), indium acetate (In(C2H3O2)3, Alfa, 99.99%), andSiO2 (Alfa, 99.8%). Precursors were ball milled at 250 rpm for 10 h,heated at a 5 °C/min rate to 900 °C and annealed for 12 h, followedby 10 °C/min rate cooling to 700 °C and a temperature hold for 24 h.All steps were carried out under pure O2. Iron/manganese, and thenonsubstituted LiInSiO4 samples were obtained by a similar procedure(using Fe2O3 and Mn2O3 as the iron and manganese precursors,respectively), with heat treatment performed at 900 °C for 12 h underair, followed by cooling the material to room temperature at a rate ofapproximately 10 °C/min, as dictated by the natural cooling of thefurnace.
For cobalt substituted samples, time-of-flight neutron diffractionexperiments were performed at 100 K on the POWGEN powderdiffractometer at the Oak Ridge National Laboratory, Tennessee, USA,and powder X-ray diffraction was carried out at 100 K on beamline 11-BM at the Advanced Photon Source (APS) at the Argonne NationalLaboratory, Illinois, USA, with an average wavelength of 0.4137 Å.Diffraction data processing was carried out using GSAS andEXPGUI.58,59 Cobalt K-edge X-ray absorption near edge spectroscopymeasurements were carried out at the sector 20-BM at the APS using aSi(111) crystal monochromator. Simultaneous cobalt metallic foil wasrun with the samples for energy calibration. The energy scale wascalibrated by defining the first derivative maxima of Co foil as 7709 eV.The spectra were normalized to unity absorption well above the edgeafter a linear background subtraction with Demeter software.60 Forother samples, powder X-ray diffraction was carried out with aPANalytical Empyrean diffractometer operating with Cu Kα radiation.
■ RESULTS AND DISCUSSION
Atomistic Scale Simulation of Transition Substitutioninto Olivine Silicate. Atomistic scale simulation methods havebeen successfully used to calculate relative substitution energiesof different dopants in a given structure for olivine LiFePO4and many other technologically important compounds.51,61 In
Figure 1. Crystal structure of olivine. The MIO6 and MIIO6 octahedraare shown in green and purple, respectively, while the SiO4 tetrahedraare shown in blue.
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9932
the present work, we use this method to reproduce the crystalstructures of olivine LiInSiO4 and LiScSiO4 based on theinteratomic potential parameters that have been previouslyreported (Table S1). Table 1 compares the values obtained bythese calculations to those reported experimentally usingdiffraction based methods,39,40 showing a maximum differencefor any lattice parameter of 0.9%. Similar differences are alsofound for calculated and experimentally obtained In−O, Sc−O,and Si−O bond lengths (Table S2). Such small deviationbetween calculated and experimental results confirms thevalidity of the interatomic potentials that were used in furthercalculations in this work. The following two formulationssimilar to the scheme used by Islam et al. previously forcalculating substitution energies in LiMPO4
51were used tocalculate the energy required for doping Mn3+, Fe3+, and Co3+
into LiInSiO4 and LiScSiO4 (equations in Kroger Vinknotation):
(1) Dopant occupies M2 site in the olivine structure:(a) M3+ (M = Mn, Fe, Co) in LiInSiO4
+ → +2In M O 2M In Ox xIn 2 3 In 2 3 (1a)
(b) M3+ (M = Mn, Fe, Co) in LiScSiO4
+ → +2Sc M O 2M Sc Ox xSc 2 3 Sc 2 3 (1b)
(2) Dopant occupies M1 site in the olivine structure(formation of an antisite defect):(a) M3+ (M = Mn, Fe, Co) in LiInSiO4
+ +
→ + ′′ +•• i
2In 2Li M O
2M 2L In O
x xIn Li 2 3
Li In 2 3 (2a)
(b) M3+ (M = Mn, Fe, Co) in LiScSiO4
+
+ +
→ + ′′••
2Sc 2Li M O
2M 2Li Sc O
x xSc Li 2 3
Li Sc 2 3 (2b)
Equations 1a and 2a (or 1b and 2b) represent two extremecases of the dopant ion completely occupying either site M1 orsite M2 in the olivine structure. The energy of formation of thedefect represented by eq 1a can be calculated as follows:
= + −E E E E21a In O M M Ox2 3 In 2 3 (3)
and that represented by eq 2a:
= + + −••E E E E E2 22a In O M Li M O2 3 Li In//
2 3 (4)
where EM2O3and EIn2O3
represent respective lattice energies andEdefect represents energy of formation of the isolated defect.Antisite defects are created together and tend to associate. Thistendency was taken into account by introducing these twodefects in the structure simultaneously. This strategy ofcalculating the energy of formation of associated defects waspreviously utilized for olivine LiMPO4 compounds.The difference in energy of formation of defects represented
by eqs 1a and 2a denotes the tendency of the substituted ion tobe disordered over the two cationic sites in the olivinestructure. Less difference in the energy will lead to equaldistribution of cations over the two sites and therefore highercationic disorder. This energy can be calculated as
= − = + −′′••E E E E E Eorder 2a 1a M Li MxLi In In (5)
Table 2 presents values of Eorder for Mn3+, Fe3+, and Co3+
substitution in olivine LiInSiO4 and LiScSiO4. Positive values ofthe Eorder indicate all substituents prefer site M2 over site M1,although Eorder decreases from Mn3+ to Fe3+ to Co3+. This trendis apparent for both parent compounds. Because the value forCo3+ substitution in LiInSiO4 is lowest, it suggests that Co
3+
could be significantly disordered in LiIn1−xCoxSiO4, whereasMn3+ to Fe3+ may not. Although values calculated here applyonly when a low degree of substituents are used (assumption ofnoninteracting defects), these trends in Eorder are expected tohold true even for high degrees of substitution.
Experimental Investigation into Cation Substitution.Cation substitution behavior was examined experimentally bysolid state synthesis, where a 1:1 molar ratio of the MI (Mn/Fe/Co) and MII (In/Sc) precursors was heated at 900 °C.Unfortunately, the XRD pattern of LiScSiO4 exhibited noevidence of any transition metal incorporation (data notshown), whereas substitution was successful in LiInSiO4, basedon the shift in the d-spacings. Figure 2 compares the XRDpatterns of the products obtained. The pattern for non-substituted LiInSiO4 is shown in the bottom trace of Figure 2for comparison. Although some In2O3 impurity is present, theolivine structure is clearly the main phase. Comparison of theXRD patterns of the pristine and substituted materials showsthat upon the addition of Mn in the precursors (InMn-11) littlechange is observed in the olivine phase of the product, and asignificant amount of Mn2O3 is present, indicating substitutionwas not successful. The reflections of the In−Fe sample (InFe-11), on the other hand, are shifted to the right of LiInSiO4,indicating a decrease in the unit cell parameter that could arisefrom the partial substitution of In3+ by the smaller Fe3+ ion.
Table 1. Comparison of Calculated and Experimentally Obtained Lattice Parameters of LiInSiO4 and LiScSiO4 Olivine Silicates
LiInSiO4 LiScSiO4
lattice parameters calc expt % diff calc expt % diff
a (Å) 4.8073 4.8448 −0.77 4.7905 4.8168 −0.55b (Å) 10.5683 10.5043 0.61 10.5250 10.4317 0.89c (Å) 6.0323 6.0634 −0.51 5.9621 5.9650 −0.05
Table 2. Eorder for Various Substituents in Olivine Type LiInSiO4 and LiScSiO4
LiInSiO4 LiScSiO4
energy (eV) Mn3+ Fe3+ Co3+ Mn3+ Fe3+ Co3+
E (M on M2) −7.2040 −7.1056 −10.8549 −6.7010 −6.6066 −10.3824E (M on M1) −6.2218 −6.1685 −10.4045 −4.5218 −4.4654 −8.9013E (order) 0.9822 0.9371 0.4504 2.1792 2.1412 1.4811
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9933
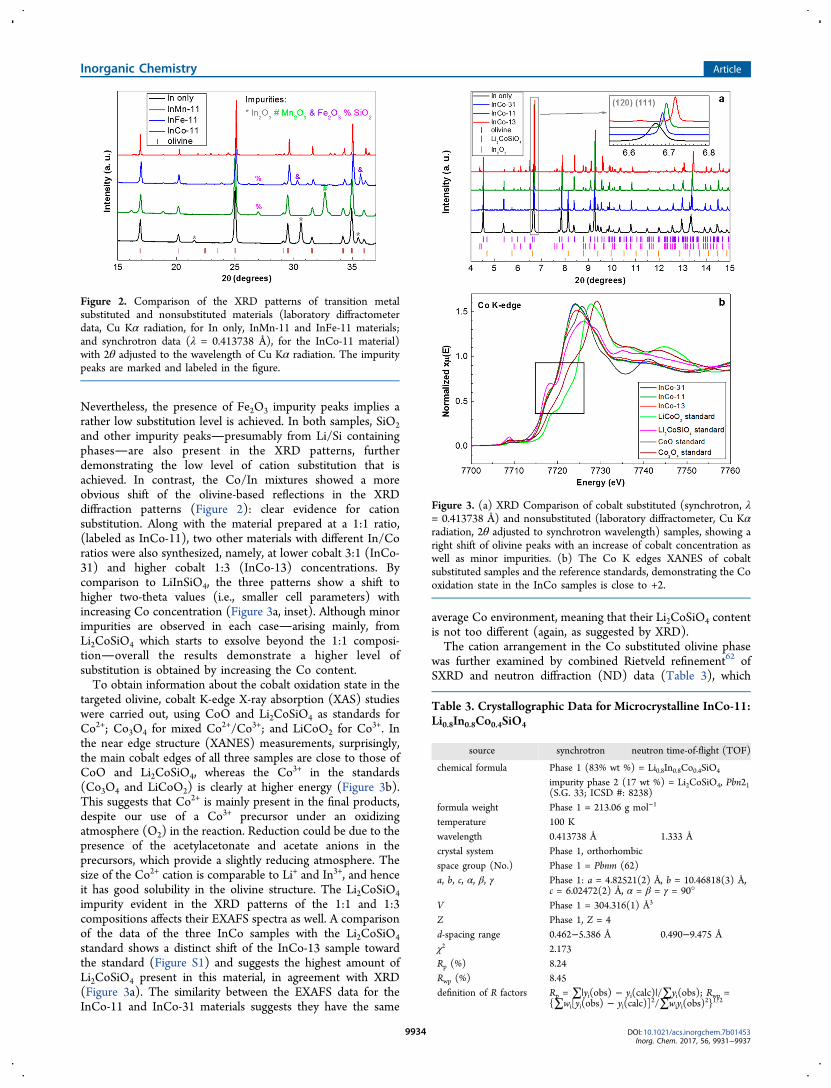
Nevertheless, the presence of Fe2O3 impurity peaks implies arather low substitution level is achieved. In both samples, SiO2and other impurity peakspresumably from Li/Si containingphasesare also present in the XRD patterns, furtherdemonstrating the low level of cation substitution that isachieved. In contrast, the Co/In mixtures showed a moreobvious shift of the olivine-based reflections in the XRDdiffraction patterns (Figure 2): clear evidence for cationsubstitution. Along with the material prepared at a 1:1 ratio,(labeled as InCo-11), two other materials with different In/Coratios were also synthesized, namely, at lower cobalt 3:1 (InCo-31) and higher cobalt 1:3 (InCo-13) concentrations. Bycomparison to LiInSiO4, the three patterns show a shift tohigher two-theta values (i.e., smaller cell parameters) withincreasing Co concentration (Figure 3a, inset). Although minorimpurities are observed in each casearising mainly, fromLi2CoSiO4 which starts to exsolve beyond the 1:1 composi-tionoverall the results demonstrate a higher level ofsubstitution is obtained by increasing the Co content.To obtain information about the cobalt oxidation state in the
targeted olivine, cobalt K-edge X-ray absorption (XAS) studieswere carried out, using CoO and Li2CoSiO4 as standards forCo2+; Co3O4 for mixed Co2+/Co3+; and LiCoO2 for Co
3+. Inthe near edge structure (XANES) measurements, surprisingly,the main cobalt edges of all three samples are close to those ofCoO and Li2CoSiO4, whereas the Co3+ in the standards(Co3O4 and LiCoO2) is clearly at higher energy (Figure 3b).This suggests that Co2+ is mainly present in the final products,despite our use of a Co3+ precursor under an oxidizingatmosphere (O2) in the reaction. Reduction could be due to thepresence of the acetylacetonate and acetate anions in theprecursors, which provide a slightly reducing atmosphere. Thesize of the Co2+ cation is comparable to Li+ and In3+, and henceit has good solubility in the olivine structure. The Li2CoSiO4impurity evident in the XRD patterns of the 1:1 and 1:3compositions affects their EXAFS spectra as well. A comparisonof the data of the three InCo samples with the Li2CoSiO4standard shows a distinct shift of the InCo-13 sample towardthe standard (Figure S1) and suggests the highest amount ofLi2CoSiO4 present in this material, in agreement with XRD(Figure 3a). The similarity between the EXAFS data for theInCo-11 and InCo-31 materials suggests they have the same
average Co environment, meaning that their Li2CoSiO4 contentis not too different (again, as suggested by XRD).The cation arrangement in the Co substituted olivine phase
was further examined by combined Rietveld refinement62 ofSXRD and neutron diffraction (ND) data (Table 3), which
Figure 2. Comparison of the XRD patterns of transition metalsubstituted and nonsubstituted materials (laboratory diffractometerdata, Cu Kα radiation, for In only, InMn-11 and InFe-11 materials;and synchrotron data (λ = 0.413738 Å), for the InCo-11 material)with 2θ adjusted to the wavelength of Cu Kα radiation. The impuritypeaks are marked and labeled in the figure.
Figure 3. (a) XRD Comparison of cobalt substituted (synchrotron, λ= 0.413738 Å) and nonsubstituted (laboratory diffractometer, Cu Kαradiation, 2θ adjusted to synchrotron wavelength) samples, showing aright shift of olivine peaks with an increase of cobalt concentration aswell as minor impurities. (b) The Co K edges XANES of cobaltsubstituted samples and the reference standards, demonstrating the Cooxidation state in the InCo samples is close to +2.
Table 3. Crystallographic Data for Microcrystalline InCo-11:Li0.8In0.8Co0.4SiO4
source synchrotron neutron time-of-flight (TOF)
chemical formula Phase 1 (83% wt %) = Li0.8In0.8Co0.4SiO4
impurity phase 2 (17 wt %) = Li2CoSiO4, Pbn21(S.G. 33; ICSD #: 8238)
formula weight Phase 1 = 213.06 g mol−1
temperature 100 Kwavelength 0.413738 Å 1.333 Åcrystal system Phase 1, orthorhombicspace group (No.) Phase 1 = Pbnm (62)a, b, c, α, β, γ Phase 1: a = 4.82521(2) Å, b = 10.46818(3) Å,
c = 6.02472(2) Å, α = β = γ = 90°V Phase 1 = 304.316(1) Å3
Z Phase 1, Z = 4d-spacing range 0.462−5.386 Å 0.490−9.475 Åχ2 2.173Rp (%) 8.24Rwp (%) 8.45definition of R factors Rp = ∑|yi(obs) − yi(calc)|/∑yi(obs); Rwp =
{∑wi[yi(obs) − yi(calc)]2/∑wiyi(obs)
2}1/2
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9934
provides information on site occupancies in solid lattices.63,64
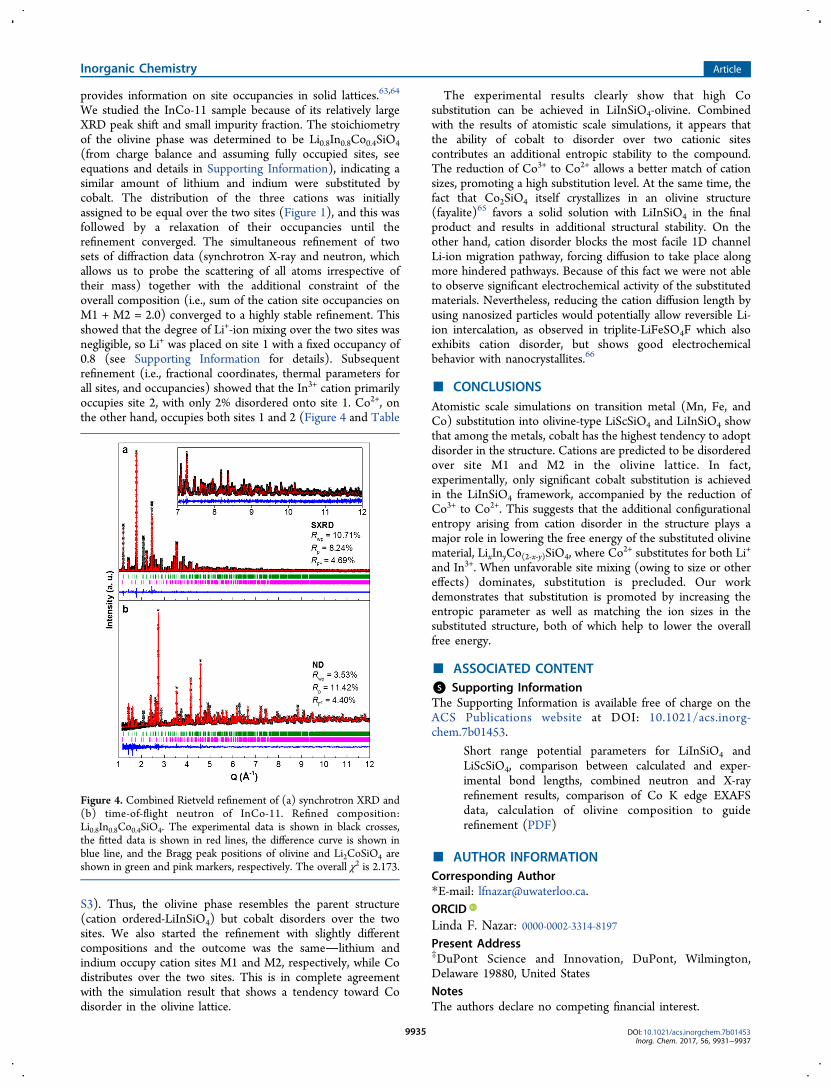
We studied the InCo-11 sample because of its relatively largeXRD peak shift and small impurity fraction. The stoichiometryof the olivine phase was determined to be Li0.8In0.8Co0.4SiO4(from charge balance and assuming fully occupied sites, seeequations and details in Supporting Information), indicating asimilar amount of lithium and indium were substituted bycobalt. The distribution of the three cations was initiallyassigned to be equal over the two sites (Figure 1), and this wasfollowed by a relaxation of their occupancies until therefinement converged. The simultaneous refinement of twosets of diffraction data (synchrotron X-ray and neutron, whichallows us to probe the scattering of all atoms irrespective oftheir mass) together with the additional constraint of theoverall composition (i.e., sum of the cation site occupancies onM1 + M2 = 2.0) converged to a highly stable refinement. Thisshowed that the degree of Li+-ion mixing over the two sites wasnegligible, so Li+ was placed on site 1 with a fixed occupancy of0.8 (see Supporting Information for details). Subsequentrefinement (i.e., fractional coordinates, thermal parameters forall sites, and occupancies) showed that the In3+ cation primarilyoccupies site 2, with only 2% disordered onto site 1. Co2+, onthe other hand, occupies both sites 1 and 2 (Figure 4 and Table
S3). Thus, the olivine phase resembles the parent structure(cation ordered-LiInSiO4) but cobalt disorders over the twosites. We also started the refinement with slightly differentcompositions and the outcome was the samelithium andindium occupy cation sites M1 and M2, respectively, while Codistributes over the two sites. This is in complete agreementwith the simulation result that shows a tendency toward Codisorder in the olivine lattice.
The experimental results clearly show that high Cosubstitution can be achieved in LiInSiO4-olivine. Combinedwith the results of atomistic scale simulations, it appears thatthe ability of cobalt to disorder over two cationic sitescontributes an additional entropic stability to the compound.The reduction of Co3+ to Co2+ allows a better match of cationsizes, promoting a high substitution level. At the same time, thefact that Co2SiO4 itself crystallizes in an olivine structure(fayalite)65 favors a solid solution with LiInSiO4 in the finalproduct and results in additional structural stability. On theother hand, cation disorder blocks the most facile 1D channelLi-ion migration pathway, forcing diffusion to take place alongmore hindered pathways. Because of this fact we were not ableto observe significant electrochemical activity of the substitutedmaterials. Nevertheless, reducing the cation diffusion length byusing nanosized particles would potentially allow reversible Li-ion intercalation, as observed in triplite-LiFeSO4F which alsoexhibits cation disorder, but shows good electrochemicalbehavior with nanocrystallites.66
■ CONCLUSIONSAtomistic scale simulations on transition metal (Mn, Fe, andCo) substitution into olivine-type LiScSiO4 and LiInSiO4 showthat among the metals, cobalt has the highest tendency to adoptdisorder in the structure. Cations are predicted to be disorderedover site M1 and M2 in the olivine lattice. In fact,experimentally, only significant cobalt substitution is achievedin the LiInSiO4 framework, accompanied by the reduction ofCo3+ to Co2+. This suggests that the additional configurationalentropy arising from cation disorder in the structure plays amajor role in lowering the free energy of the substituted olivinematerial, LixInyCo(2‑x‑y)SiO4, where Co
2+ substitutes for both Li+
and In3+. When unfavorable site mixing (owing to size or othereffects) dominates, substitution is precluded. Our workdemonstrates that substitution is promoted by increasing theentropic parameter as well as matching the ion sizes in thesubstituted structure, both of which help to lower the overallfree energy.
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acs.inorg-chem.7b01453.
Short range potential parameters for LiInSiO4 andLiScSiO4, comparison between calculated and exper-imental bond lengths, combined neutron and X-rayrefinement results, comparison of Co K edge EXAFSdata, calculation of olivine composition to guiderefinement (PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] F. Nazar: 0000-0002-3314-8197Present Address‡DuPont Science and Innovation, DuPont, Wilmington,Delaware 19880, United StatesNotesThe authors declare no competing financial interest.
Figure 4. Combined Rietveld refinement of (a) synchrotron XRD and(b) time-of-flight neutron of InCo-11. Refined composition:Li0.8In0.8Co0.4SiO4. The experimental data is shown in black crosses,the fitted data is shown in red lines, the difference curve is shown inblue line, and the Bragg peak positions of olivine and Li2CoSiO4 areshown in green and pink markers, respectively. The overall χ2 is 2.173.
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9935

■ ACKNOWLEDGMENTS
L.F.N. gratefully acknowledges support of the Natural Scienceand Engineering Council of Canada through funding from theDiscovery Grant and Canada Research Chair programs. Theauthors would like to thank Dr. Ashfia Huq for carrying outneutron diffraction measurements and Dr. Marine Cuisinier forher help in analyzing the XANES data. This research usedresources of the Advanced Photon Source, an Office of ScienceUser Facility operated for the U.S. Department of Energy(DOE) Office of Science by Argonne National Laboratory, andwas supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.
■ REFERENCES(1) Tarascon, J.-M.; Armand, M. Issues and Challenges FacingRechargeable Lithium Batteries. Nature 2001, 414, 359−367.(2) Armand, M.; Tarascon, J.-M. Building Batter Batteries. Nature2008, 451, 652−657.(3) Scrosati, B.; Garche, J. Lithium Batteries: Status, Prospects andFuture. J. Power Sources 2010, 195, 2419−2430.(4) Goodenough, J. B.; Kim, Y. Challenges for Rechargeable LiBatteries. Chem. Mater. 2010, 22, 587−603.(5) Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D.Challenges in the Development of Advanced Li-Ion Batteries: AReview. Energy Environ. Sci. 2011, 4, 3243−3262.(6) Goodenough, J. B.; Park, K.-S. The Li-Ion Rechargeable Battery:A Perspective. J. Am. Chem. Soc. 2013, 135, 1167−1176.(7) Padhi, A. K.; Nanjundaswamy, K. S.; Goodenough, J. B. Phospho-Olivines as Positive-Electrode Materials for Rechargeable LithiumBatteries. J. Electrochem. Soc. 1997, 144, 1188−1194.(8) Whittingham, W. S. Lithium Batteries and Cathode Materials.Chem. Rev. 2004, 104, 4271−4302.(9) Ellis, B. L.; Lee, K. T.; Nazar, L. F. Positive Electrode Materialsfor Li-Ion and Li-Batteries. Chem. Mater. 2010, 22, 691−714.(10) Gong, Z.; Yang, Y. Recent Advances in the Research ofPolyanion-Type Cathode Materials for Li-Ion Batteries. EnergyEnviron. Sci. 2011, 4, 3223−3242.(11) Cheng, F.; Liang, J.; Tao, Z.; Chen, J. Functional Materials forRechargeable Batteries. Adv. Mater. 2011, 23, 1695−1715.(12) Whittingham, M. S. Ultimate Limits to Intercalation Reactionsfor Lithium Batteries. Chem. Rev. 2014, 114, 11414−11443.(13) Morgan, D.; Van der Ven, A.; Ceder, G. Li Conductivity inLixMPO4 (M = Mn, Fe, Co, Ni) Olivine Materials. Electrochem. Solid-State Lett. 2004, 7, A30−A32.(14) Islam, M. S.; Driscoll, D. J.; Fisher, C. A. J.; Slater, P. R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in theLiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17,5085−5092.(15) Nishimura, S.-I.; Kobayashi, G.; Ohoyama, K.; Kanno, R.;Yashima, M.; Yamada, A. Experimental Visualization of LithiumDiffusion in LixFePO4. Nat. Mater. 2008, 7, 707−711.(16) Malik, R.; Zhou, F.; Ceder, G. Kinetics of non-equilibriumlithium incorporation in LiFePO4. Nat. Mater. 2011, 10, 587−590.(17) Liu, H.; Stobridge, F. C.; Borkiewicz, O. J.; Wiaderek, K. M.;Chapman, K. W.; Chupas, P. J.; Grey, C. P. Capturing MetastableStructures During High-rate Cycling of LiFePO4 NanoparticleElectrodes. Science 2014, 344, 1252817−1252823.(18) Huang, H.; Yin, S.-C.; Nazar, L. F. Approaching TheoreticalCapacity of LiFePO4 at Room Temperature at High Rates.Electrochem. Solid-State Lett. 2001, 4, A170−A172.(19) Kang, B.; Ceder, G. Battery Materials for Ultrafast Charging andDischaring. Nature 2009, 458, 190−193.(20) Wu, X.-L.; Jiang, L.-Y.; Cao, F.-F.; Guo, Y.-G.; Wan, L.-J.LiFePO4 Nanoparticles Embedded in a Nanoporous Carbon Matrix:Superior Cathode Material for Electrochemical Energy-StorageDevices. Adv. Mater. 2009, 21, 2710−2714.
(21) Yamada, A.; Chung, S. C. Crystal Chemistry of the Olivine-TypeLi(MnyFe1‑y)PO4 and (MnyFe1‑y)PO4 as possible 4 V CathodeMaterials for Lithium Batteries. J. Electrochem. Soc. 2001, 148,A960−A967.(22) Huang, Y.; Chernova, N. A.; Yin, Q.; Wang, Q.; Quackenbush,N. F.; Leskes, M.; Fang, J.; Omenya, F.; Zhang, R.; Wahila, M. J.;Piper, L. F. J.; Zhou, G.; Grey, C. P.; Whittingham, M. S. WhatHappens to LiMnPO4 upon Chemical Delithiation? Inorg. Chem. 2016,55, 4335−4343.(23) Legagneur, V.; An, Y.; Mosbah, A.; Portal, R.; Le Gal La Salle,A.; Verbaere, A.; Guyomard, D.; Piffard, Y. LiMBO3 (M = Mn, Fe,Co): Synthesis, Crystal Structure and Lithium Deinsertion/InsertionProperties. Solid State Ionics 2001, 139, 37−46.(24) Le Roux, B.; Bourbon, C.; Lebedev, O. I.; Colin, J. F.; Pralong,V. Synthesis and Characterization of the LiMnBO3-LiCoBO3 SolidSolution and Its Use as a Lithium-Ion Cathode Materials. Inorg. Chem.2015, 54, 5273−5279.(25) Nyten, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.;Thomas, J. O. Electrochemical Performance of Li2FeSiO4 as a New Li-Battery Cathode Material. Electrochem. Commun. 2005, 7, 156−160.(26) Rousse, G.; Tarascon, J.-M. Sulfate-Based PolyanionicCompounds for Li-Ion Batteries: Synthesis, Crystal Chemistry andElectrochemistry Aspects. Chem. Mater. 2014, 26, 394−406.(27) Lander, L.; Reynaud, M.; Rodriguez-Carvajal, J.; Tarascon, J. M.;Rousse, G. Magnetic Structures of Orthorhombic Li2M(SO4)2 (M =Co, Fe) and LixFe(SO4)2 (x = 1, 1.5) Phases. Inorg. Chem. 2016, 55,11760−11769.(28) Recham, N.; Chotard, J.-N.; Dupont, L.; Delacourt, C.; Walker,W.; Armand, M.; Tarascon, J.-M. A 3.6 V Lithium-BasedFluorosulphate Insertion Positive Electrode for Lithium-Ion Batteries.Nat. Mater. 2010, 9, 68−74.(29) Barpanda, P.; Ati, M.; Melot, B. C.; Rousse, G.; Chotard, J.-N.;Doublet, M.-L.; Sougrati, M. T.; Corr, S. A.; Jumas, J.-C.; Tarascon, J.-M. Nat. Mater. 2011, 10, 772−779.(30) Dominko, R.; Bele, M.; Gaberscek, M.; Meden, A.; Remskar, M.;Jamnik, J. Structure and Electrochemical Performance of Li2MnSiO4
and Li2FeSiO4 as Potential Li-Battery Cathode Materials. Electrochem.Commun. 2006, 8, 217−222.(31) Lyness, C.; Delobel, B.; Armstrong, A. R.; Bruce, P. The LithiumIntercalation Compound Li2CoSiO4 and Its Behavior as a PositiveElectrode for Lithium Batteries. Chem. Commun. 2007, 4890−4892.(32) Muraliganth, T.; Stroukoff, K. R.; Manthiram, A. Microwave-Solvothermal Synthesis of Nanostructured Li2MSiO4/C (M = Mn andFe) Cathodes for Lithium-Ion Batteries. Chem. Mater. 2010, 22,5754−5761.(33) He, G.; Popov, G.; Nazar, L. F. Hydrothermal Synthesis andElectrochemical Properties of Li2CoSiO4/C Nanospheres. Chem.Mater. 2013, 25, 1024−1031.(34) Nyten, A.; Kamali, S.; Haggstrom, L.; Gustafsson, T.; Thomas, J.O. The Lithium Extraction/Insertion Mechanism in Li2FeSiO4. J.Mater. Chem. 2006, 16, 2266−2272.(35) Gong, Z. L.; Li, Y. X.; He, G. N.; Li, J.; Yang, Y. NanostructuredLi2FeSiO4 Electrode Material Synthesized through Hydrothermal-Assisted Sol-Gel Process. Electrochem. Solid-State Lett. 2008, 11, A60−A63.(36) Sirisopanaporn, C.; Masquelier, C.; Bruce, P. G.; Armstrong, A.R.; Dominko, R. Dependence of Li2FeSiO4 Electrochemistry onStructure. J. Am. Chem. Soc. 2011, 133, 1263−1265.(37) Armstrong, A. R.; Kuganathan, N.; Islam, M. S.; Bruce, P. G.Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes forLithium Batteries. J. Am. Chem. Soc. 2011, 133, 13031−13035.(38) Saracibar, A.; Van der Ven, A.; Arroyo-de Dompablo, M. E.Crystal Structure, Energetics, and Electrochemistry of Li2FeSiO4
Polymorphs from First Principles Calculations. Chem. Mater. 2012,24, 495−503.(39) Hazen, R. M.; Downs, R. T.; Finger, L. W. High-PressureCrystal Chemistry of LiScSiO4: An Olivine with Nearly IsotropicCompreesion. Am. Mineral. 1996, 81, 327−334.
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9936

(40) Redhammer, G. J.; Roth, G. LiInSiO4: A New Monovalent-Trivalent Olivine. Acta Crystallogr., Sect. C: Cryst. Struct. Commun.2003, 59, i38−i40.(41) Redfern, S. A. T.; Artioli, G.; Rinaldi, R.; Henderson, C. M. B.;Knight, K. S.; Wood, B. J. Octahedral Cation Ordering in Olivine atHigh Temperature. II: An in Situ Neutron Powder Diffraction Studyon Synthetic MgFeSiO4. Phys. Chem. Miner. 2000, 27, 630−637.(42) Fujino, K.; Sasaki, S.; Takeuchi, Y.; Sadanaga, R. X-RayDetermination of Electron Distribution in Forsterite, Fayalite andTephroite. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem.1981, 37, 513−518.(43) Zhou, F.; Cococcioni, M.; Kang, K.; Ceder, G. The LiIntercalation Potential of LiMPO4 and LiMSiO4 Olivines with M = Fe,Mn, Co, Ni. Electrochem. Commun. 2004, 6, 1144−1148.(44) Arroyo y de Dompablo, M. E.; Gallardo-Amores, J. M.; García-Martínez, J.; Moran, E.; Tarascon, J.-M.; Armand, M. Is It Possible toPrepare Olivine-Type LiFeSiO4? A Joint Computational andExperimental Investigation. Solid State Ionics 2008, 179, 1758−1762.(45) Recham, N.; Casas-Cabanas, M.; Cabana, J.; Grey, C. P.; Jumas,J.-C.; Dupont, L.; Armand, M.; Tarascon, J.-M. Formation of aComplete Solid Solution between the Triphylite and Fayalite OlivineStructures. Chem. Mater. 2008, 20, 6798−6809.(46) Catlow, C. R. A. Computer Modeling in Inorganic Crystallography;Academic Press: San Diego, U.S.A, 1997.(47) Tripathi, R.; Gardiner, G. R.; Islam, M. S.; Nazar, L. F. Alkali-Ion Conduction Paths in LiFeSO4F and NaFeSO4F Tavorite-TypeCathode Materials. Chem. Mater. 2011, 23, 2278−2284.(48) Dick, B. G.; Overhauser, A. W. Theory of the DielectricConstants of Alkali Halide Crystals. Phys. Rev. 1958, 112, 90−103.(49) Fisher, C. A. J.; Islam, M. S. Defect, Protons and Conductivity inBrownmillerite-Structured Ba2In2O5. Solid State Ionics 1999, 118, 355−363.(50) Minervini, L.; Zacate, M. O.; Grimes, R. W. Defect ClusterFormation in M2O3-Doped CeO2. Solid State Ionics 1999, 116, 339−349.(51) Fisher, C. A.; Hart Prieto, M. V.; Islam, M. S. Lithium BatteryMaterials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into DefectAssociation, Transport Mechanisms, and Doping Behavior. Chem.Mater. 2008, 20, 5907−5915.(52) Sierka, M.; Sauer, J. Structure and Reactivity of a Silica andZeolite Catalysts by a Combined Quantum Mechanics−Shell-ModelPotential Approach Based on DFT. Faraday Discuss. 1997, 106, 41−62.(53) Sanders, M. J.; Leslie, M. J.; Catlow, C. R. A. InteratomicPotentials for SiO2. J. Chem. Soc., Chem. Commun. 1984, 1271−1273.(54) Woodley, S. M.; Catlow, C. R. A.; Piszora, P.; Stempin, K.;Wolska, E. Computer Modeling Study of the Lithium Ion Distributionin Quaternary Li-Mn-Fe-O Spinels. J. Solid State Chem. 2000, 153,310−316.(55) Braithwaite, J. S.; Catlow, C. R. A.; Harding, J. H.; Gale, J. D. AComputational Study of the High Voltage LixCoyMn4‑yO8 CathodeMaterial. Phys. Chem. Chem. Phys. 2000, 2, 3841−3846.(56) Henson, N. J.; Hay, P. J.; Redondo, A. Computational Studies ofCobalt-Substituted Aluminophosphates. J. Phys. Chem. A 2000, 104,2423−2431.(57) Gale, J. D.; Rohl, A. L. The General Utility Lattice Program(GULP). Mol. Simul. 2003, 29, 291−341.(58) Larson, A. C.; Von Dreele, R. B. General Structure AnalysisSystem (GSAS); Los Alamos National Laboratory Report, , LAUR 86−748; Los Alamos National Laboratory: Los Alamos, NM, 2000.(59) Toby, B. H. EXPGUI, a Graphical User Interface for GSAS. J.Appl. Crystallogr. 2001, 34, 210−213.(60) Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS:Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J.Synchrotron Radiat. 2005, 12, 537−541.(61) Bush, T. S.; Catlow, C. R. A.; Chadwick, A. V.; Cole, M.;Geatches, R. M.; Greaves, G. N.; Tomlinson, S. M. Studies of CationDopant Sites in Metal Oxides by EXAFS and Computer-SimulationTechniques. J. Mater. Chem. 1992, 2, 309−316.
(62) Rietveld, H. M. A Profile Refinement Method for Nuclear andMagnetic Structures. J. Appl. Crystallogr. 1969, 2, 65−71.(63) Murphy, D. T.; Schmid, S.; Hester, J. R.; Blanchard, P. E. R.;Miiller, W. Coordination Site Disorder in Spinel-Type LiMnTiO4.Inorg. Chem. 2015, 54, 4636−4643.(64) Biendicho, J. J.; Hsiao, K. C.; Hull, S.; West, A. R. Investigationof Antisite Defect Formation and Chemical Expansion in LiNiPO4 byin Situ Neutron Diffraction. Inorg. Chem. 2017, 56, 3657−3662.(65) Tamada, O.; Fujino, K.; Sasaki, S. Structures and ElectronDistribution of α-Co2SiO4 and α-Ni2SiO4 (Olivine Structure). ActaCrystallogr., Sect. B: Struct. Sci. 1983, 39, 692−697.(66) Tripathi, R.; Popov, G.; Sun, X.; Ryan, D. H.; Nazar, L. F. Ultra-Rapid Microwave Synthesis of Triplite LiFeSO4F. J. Mater. Chem. A2013, 1, 2990−2994.
Inorganic Chemistry Article
DOI: 10.1021/acs.inorgchem.7b01453Inorg. Chem. 2017, 56, 9931−9937
9937


















