SUPERSATURATION NUCLEATION CRYSTAL GROWTH CRYSTAL Non equilibrium Equilibrium.
Solvent fluctuations in the solvation shell determinethe activation barrier for crystal growth ratesAnish V. Dighea and Meenesh R. Singha,1
aDepartment of Chemical Engineering, University of Illinois at Chicago, Chicago, IL 60607
Edited by Alexis T. Bell, University of California, Berkeley, CA, and approved October 16, 2019 (received for review June 21, 2019)
Solution crystallization is a common technique to grow advanced,functional crystalline materials. Supersaturation, temperature, andsolvent composition are known to influence the growth rates andthereby properties of crystalline materials; however, a satisfactoryexplanation of how these factors affect the activation barrier forgrowth rates has not been developed. We report here that theseeffects can be attributed to a previously unrecognized consequenceof solvent fluctuations in the solvation shell of solute moleculesattaching to the crystal surface. With increasing supersaturation, theaverage hydration number of the glutamic acid molecule decreasesand can reach an asymptotic limit corresponding to the number ofadsorption sites on the molecule. The hydration number of theglutamic acid molecule also fluctuates due to the rapid exchange ofsolvent in the solvation shell and local variation in the supersatu-ration. These rapid fluctuations allow quasi-equilibrium betweenfully solvated and partially desolvated states of molecules, whichcan be used to construct a double-well potential and thereby toidentify the transition state and the required activation barrier. Thepartially desolvated molecules are not stable and can attach spon-taneously to the crystal surface. The activation barrier versushydration number follows the Evans–Polanyi relation. The predictedabsolute growth rates of the α-glutamic acid crystal at lower super-saturations are in reasonable agreement with the experimentalobservations.
solution crystallization | solvation dynamics | molecular dynamics |growth rate | organic crystals
Crystallization is one of the most common processes used forthe separation of molecules from solution and for the syn-thesis of crystalline materials for various applications such as ca-talysis, gas separation, and energy conversion (1, 2). The underlyingprocesses of crystallization, namely nucleation and growth, governthe structure, morphology, and size of crystals, which impart spe-cific properties to the crystalline materials (3–5). Supersatura-tion, defined as the chemical potential of solution relative to thestate when solution is in equilibrium with the crystals, is generallyregarded as the driving force for crystallization processes. Severalempirical and semiempirical kinetic models have been developedbased on this hypothesis and validated experimentally to relatenucleation and growth rates to supersaturation (2, 6–8). Examplesof empirical models for nucleation rate include classical nucleationtheory (9), 2-step nucleation theory (10), and probabilistic theory(9) and for growth rates are the Bravais–Friedel–Donnay–Harker(BFDH) method (11), the Hartman–Perdok theory (12–14), theBurton–Cabrera–Frank (BCF) model (15), the birth and spreadmodel (16), the bulk diffusion model (17), the thermochemicalmodel (18), and MONTY (19). While the majority of these modelsrely on parameters estimated from the experimental data (16), theother models are limited to only the prediction of relative rates orgrowth shapes (20, 21). The molecular simulation methods such asmolecular dynamics (MD) (22), kinetic Monte Carlo (kMC) (23,24), and hybrid MD-kMC (25) have also been applied to simulatethe growth of the crystal. However, limitations due to variation insupersaturation (22), absence of desolvation kinetics (22, 24), in-fluence of diffusion-limited growth (23), and computationalcomplexity (25) have made it difficult to obtain intrinsic growth
rates from such molecular simulations. Currently, there are notheoretical models that can predict intrinsic rates of nucleationand face-specific growth from the first principles. The presentstate is due to a limited consideration of the underlying physicsrelating supersaturation to the rates of crystallization and in-tegrating them into the existing models. The key physical processesin crystallization include self-assembly of molecules in the solution(nucleation) as well as on the facet of the crystal (growth), both ofwhich are influenced by solvents, additives, pH, and temperatureof a supersaturated solution (26, 27). The modeling of the rates ofnucleation and growth requires detailed consideration of such self-assembly processes to estimate the transition state and the activationbarrier.Fig. 1 shows a typical process of solvent-directed self-assembly
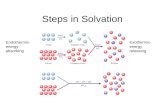
of small molecules on a growing facet of a crystal. This processinitiates in a supersaturated solution where the solute moleculesin the solution diffuse toward the crystal surface due to thechemical potential gradient. As the solvated molecules movetoward the crystal surface, the intermolecular interaction betweensolute molecules increases, causing the solvation layer to deplete.The partially solvated (or desolvated) solute molecules then in-tegrate spontaneously into the “kink” sites on the crystal surface byshedding the remaining solvation layer. The integration step iskinetically irreversible as crystals only grow and do not dissolve in asupersaturated solution. The partially solvated state of the mole-cule can be considered as a transition state, which determines theactivation barrier and hence the rate of integration of solvatedmolecules into the kink sites of the crystal surface (28). The for-mation of the partially solvated state is the rate-limiting step, whichis energetically uphill such that the fully and partially solvated
Significance
Solution crystallization is a widely used technique to growcrystalline materials such as pharmaceuticals, agrochemicals,catalysts, semiconductors, and metal–organic frameworks. Thehighly probabilistic nature and complex molecular processesspanning multiple lengths and time scales have prevented fullmechanistic understanding of crystallization processes for overa century. Here we show fluctuations in the solvation shell arekey molecular events that explain and unify the empirical ob-servations laid down since the inception of crystallization re-search. The activation barrier of the growth processes obtainedfrom these molecular fluctuations can be used to estimategrowth rates that show reasonable agreement with the ex-perimental observations, hinting at the rise of additional andefficient strategies to control solution crystallization.
Author contributions: M.R.S. designed research; A.V.D. performed research; A.V.D. andM.R.S. analyzed data; and A.V.D. and M.R.S. wrote the paper.
The authors declare no competing interest.
This article is a PNAS Direct Submission.
Published under the PNAS license.1To whom correspondence may be addressed. Email: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplemental.
First published November 11, 2019.
23954–23959 | PNAS | November 26, 2019 | vol. 116 | no. 48 www.pnas.org/cgi/doi/10.1073/pnas.1910691116
Dow
nloa
ded
by g
uest
on
June
2, 2
021
http://crossmark.crossref.org/dialog/?doi=10.1073/pnas.1910691116&domain=pdfhttps://www.pnas.org/site/aboutpnas/licenses.xhtmlmailto:[email protected]://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/cgi/doi/10.1073/pnas.1910691116
states of the molecule are in dynamic equilibrium. Similar argu-ments may be applied to the process of nucleation, where thefluctuations in supersaturations (7) lead to the instantaneous for-mation of partially solvated molecules which aggregate spontane-ously to form subcritical clusters. The desolvation of the ion/moleculeshas been previously identified as a governing step for crystalgrowth (29, 30), where the formation of the partially desolvatedion/molecule is the rate-limiting step (31). Joswiak et al. (28)have also determined the partially desolvated state of Na+/Cl− ionsto predict the activation barrier for the integration of ions into thekink sites of the NaCl crystal. However, the estimation of the ac-tivation barrier for solvated molecules to integrate or attach to thecrystal surface and thereby the prediction of absolute growth ratesremain the outstanding issues in crystallization.Two different approaches have been used to predict growth
rates from molecular simulations: 1) hybrid MD-kMC simulationsand 2) rare event simulations. The first method involves MDsimulation of the solid–liquid interface in supersaturated solu-tion to determine the rate of addition of solute to the solid in-terface and hence obtain the rate constants, which are then used astransition probabilities in kMC simulation to predict growth ratesand growth shapes of crystals (22, 25). These approaches rely oneither using simplified force fields for ideal particles or approxi-mating the rate constant for nonequilibrium processes by countingevents in the equilibrium simulations. The hybrid MD-kMC sim-ulation has been implemented to predict either the growth shapes(25) or the rate constants (22), but not the growth rates. The rareevent simulations use biased-MD methods by applying an exter-nal force to the simulation box and use methods such as um-brella sampling that can sample the crystallization coordinatewhile maintaining the information on conformational changesduring the attachment of molecules and calculating the energybarrier (28). Due to the stochastic nature of crystal growth, theidentification of the sampled path and the associated energy bar-rier that leads to the growth of the crystal is not trivial. The otherrare event simulation method is forward flux sampling, which re-quires prior information on the terminal states such as the solvatedmolecule in the bulk and the desolvated molecule at the kink siteto determine blind pathways. The complexity and longer compu-tational time involved in such rare event simulations have limitedits application to systems such as ionic crystals for prediction of therate constants for ion attachments (28), clathrate hydrates to un-derstand nucleation (32), and biomolecules to study conformational
changes (33). None of these molecular simulation methodshave been extended to predict intrinsic, absolute growth ratesof organic crystals from first principles. Here we present anMD simulation method to estimate the energy barrier for partialdesolvation of solute molecules before their integration into thekink sites. The average configuration of the solvation layer ofsolute molecules in a supersaturated solution was used to constructa double-well potential, similar to the method of Shustorovich (34,35), that yields the transition state as well as the activation barrier.The growth rates were estimated using the established thermo-chemical models (21) that account for the mechanism of growth,energetics of kink sites, and the face-specific growth factors.
TheoryMultiscale computations were performed to obtain the activationbarrier for attachment of glutamic acid molecules on the facets ofthe α-glutamic acid crystal and to calculate face-specific growthrates. The computation scheme was organized in 3 steps: 1)MD simulations of the supersaturated solution to characterizethe solvation shell dynamics; 2) molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) calculation to obtain thebinding energy of water molecules in the solvation shell, followedby the estimation of the activation barrier using the double-wellapproach; and 3) Hartman–Perdok simulation to obtain the sliceenergy of the crystal face and the estimation of absolute face-specific growth rates using mechanistic growth rate models. Theoverview of the computational approaches and the details of thecalculations performed are provided in SI Appendix.
MD Simulations. GROMACS code (36) was used to perform themolecular dynamics simulations. A dodecahedron box of fixedvolume 216.48 nm3 was set up and centered around the origin.To study the molecular events at high supersaturation, some ofthe water molecules were randomly replaced with solute molecules(22). Four different systems of supersaturation ratios, 1.2, 1.5, 2,and 2.5, were set up on the basis of experimentally measuredsolubility of α-glutamic acid crystals in water (37–39). The systemswere simulated using the Optimized Potential for Liquid Simulations–All Atom (OPLS-AA) force field for 5 ns with a time step of2 fs. The frames and energies were obtained at every 1 ps.Before the production run in the isothermal–isobaric (NPT) en-semble, systems were equilibrated for 1 ns each in canonical (NVT)and NPT ensembles, making the total simulation time equal to 7 ns
Fig. 1. Mechanism of integration of molecules on the growing face of a crystal. The solvation shell of the glutamic acid molecule in the supersaturatedsolution (GLU(l)) depletes as it moves toward the crystal surface. This partially solvated molecule (GLU
‡(l)) then integrates spontaneously to the kink site on the
crystal surface. The potential energy diagram shows the fully solvated GLU(l) in equilibrium with the energetically uphill partially solvated GLU‡(l), which then
integrates irreversibly to the kink site on the crystal surface (GLU(s)).
Dighe and Singh PNAS | November 26, 2019 | vol. 116 | no. 48 | 23955
ENGINEE
RING
Dow
nloa
ded
by g
uest
on
June
2, 2
021
https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplemental
(40). After the simulations, the distances between glutamic acidmolecules were obtained and the radial distribution function(RDF) was plotted for molecules that spend the highest time inclose vicinity to the other glutamic acid molecules. The RDF wasplotted with the center of mass of the individual glutamic acidmolecule as a reference point to analyze the distribution of watermolecules around itself. The first peak of the RDF was used toobtain the solvation shell thickness at each supersaturation.Further, to obtain the energy of interaction between the glutamicacid molecule and each water molecule in the solvation shell, theMM-PBSA method was applied to decompose the energy con-tribution on a per-molecule basis using the Poisson–Boltzmannalgorithm (41). The additional details of molecular simulationsincluding setting up of the simulation box, the MD simulationprocedure, calculation of the number of water molecules in thesolvation shell (hydration number) and solvation shell thickness,plotting of the RDF, and calculation of binding energy of watermolecules in the solvation shell are provided in SI Appendix.
Calculation of Activation Barrier and Growth Rates. Correlating thedistance between glutamic acid molecules and the hydrationnumber allows classification of the structure of the solvatedglutamic acid molecule at a certain time in the trajectory as ei-ther fully solvated or partially desolvated (SI Appendix). A framein the trajectory was classified as “fully solvated” if the distancebetween one glutamic acid molecule and other glutamic acidmolecule was greater than twice the solvation shell thickness. Allof the other frames, where distance was shorter than twice thesolvation shell thickness, were classified as “partially desolvated.”Since all of the simulations were performed without any externalforce, frames showing the extreme cases in each scenario wereconsidered and were assumed to be in quasi-equilibrium. Thestructure with the highest hydration number of all of the fullysolvated structures and the structure with the lowest hydrationnumber of all of the partially desolvated structures were furtheranalyzed to calculate the activation barrier for attachment. Thefully solvated structure is denoted as state A of glutamic acid, thepartially desolvated structure as state B, and the fully desolvatedstructure integrated into the lattice as state C. The transition ofstate B to state C is difficult to estimate using the molecularsimulation approaches while keeping the supersaturation constant.However, the energy of the state C can be approximated from thelattice energy of the α-glutamic acid crystal (SI Appendix). Toobtain the activation barrier for the transition of state A to state B,the interaction energy profiles or potential wells of 2 configura-tions representing state A and state B were obtained as a functionof distance, and were superimposed to obtain the double-wellpotential. The potential well for the state B was separated by theamount of energy required to remove the respective number ofwater molecules from the state A. The intersection point of 2potential wells determines the transition state and the height ofthe intersection gives the activation barrier. The schematic detailsof this approach are provided in SI Appendix. The intermolecularpotentials of the OPLS-AA force field with Lorentz–Berthelotmixing rules were used to calculate the potential wells.The calculation for face-specific growth rates (42) was per-
formed in 3 steps: 1) Hartman–Perdok simulations to obtain stableflat faces (F-faces) and their attachment energies, 2) estimation ofspiral morphology (step height and interstep distance) on F-faces,and 3) prediction of absolute growth rate using the mechanisticgrowth rate model. The details of growth rate calculations usingthe estimated activation barriers are described in SI Appendix.
Results and DiscussionFig. 2A shows the variation in the RDFs of water moleculesaround a glutamic acid molecule with increasing supersaturationfrom 1.2 to 2.5. The height of the RDF peaks decreases with in-creasing supersaturation due to dehydration for the primary and
secondary solvation shells. Such a phenomenon of dehydration ofthe solvation shell has been observed for simple ions, which is aneffect of higher interionic interactions in concentrated solutions(43). Fig. 2B shows a similar behavior of decrease in the hydrationnumber of glutamic acid molecules with increasing supersatura-tion, which is attributed to increasing interactions between glu-tamic acid molecules. At a sufficiently higher supersaturation, theaverage hydration number may reach an asymptotic limit corre-sponding to the number of adsorption sites on a molecule (43).The solvation shell also imposes a physical barrier for attachmentof solute molecules to either the crystal face or the nucleus. Thisphysical barrier can be anisotropic as the hydration numbers aredifferent around each functional group of the molecule. It can beseen that the effect of supersaturation is to lower the averagehydration number and thereby reduce the physical barrier forcrystallization. However, the rates of crystallization are governedby fluctuations in the solvation shell that lead to desolvationand integration of molecules to the crystal surface at a fixedsupersaturation.Fig. 3 shows the decrease in the average hydration number
(solid blue line) of a glutamic acid molecule with decreasing in-termolecular distance (solid red line) between glutamic acid mol-ecules at a supersaturation of 2.5. The method to identify thesolvation shell and the dynamics of the hydration number at dif-ferent supersaturations are given in SI Appendix. The lower aver-age hydration number is observed when the relative distancebetween molecules is lower and the intermolecular interactions aremaximum. The hydration number not only varies with the relative
Fig. 2. (A) RDFs of water molecules at different supersaturations aroundthe center of mass of a glutamic acid molecule. (B) Variation in the averagehydration number of the primary solvation shell obtained from RDFs withincreasing supersaturation.
23956 | www.pnas.org/cgi/doi/10.1073/pnas.1910691116 Dighe and Singh
Dow
nloa
ded
by g
uest
on
June
2, 2
021
https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/cgi/doi/10.1073/pnas.1910691116
motion of glutamic acid molecules but also fluctuates (solid blackline) due to the random motion of solvent molecules. The varia-tion of hydration number over a longer time scale (solid blue line)is mostly due to variations in relative distances of glutamic acidmolecules, which are dictated by local fluctuations in concentrationor supersaturation. However, the fluctuations of hydration numberat a much shorter time scale (solid black line) are due to the rapidexchange of water molecules in the solvation shell, which is gov-erned by the bulk temperature. It can be seen that both temper-ature and supersaturation affect the variation in the hydrationnumber and thereby the physical barrier for crystallization. Thefully solvated and partially desolvated states of the molecule can bedetermined based on the highest and the lowest hydration number,respectively, as shown in Fig. 3. Since the fluctuations in hydrationnumber due to temperature are much faster, the fully solvated andpartially desolvated states can be considered in quasi-equilibrium.According to the transition state theory shown in Fig. 1, theequilibrium between fully solvated and partially desolvated mole-cules determines the activation barrier.Fig. 4A shows the energy profile for the interaction between
glutamic acid molecules with increasing crystallization coordi-nate. The normalized crystallization coordinate spans the relativedistance between molecules as they travel from the bulk solution(value 0) to the crystal face (value 1). The conformation of mol-ecules does not change with increasing crystallization coordinatefor small molecules such as glutamic acid, which was confirmedfrom molecular simulations. As the crystallization coordinate in-creases, the interaction between fully solvated molecules increases,which leads to desolvation of the primary shell and formation ofpartially desolvated molecules before integrating into the crystalface. The energy required to remove water molecules from thesolvation shell is higher for the water molecules in the vicinity ofthe glutamic acid molecule (SI Appendix). The energy of transition,where the fully solvated molecules switch to a partially solvatedconfiguration, can be obtained from the double-well diagram (SIAppendix) (34, 35). The activation energy for forming partiallydesolvated molecules decreases from 26 kJ·mol−1 to 17 kJ·mol−1
with increasing supersaturation from 1.2 to 2.5. Since the transitionstate is attained long before the integration of molecules to thecrystal face, the activation energy is less sensitive to the relativeorientation of molecules. The energy profiles for glutamic acid
molecules oriented in [110] and [111] crystallographic directionsand attaching to [110] and [111] crystal faces, respectively, areshown in SI Appendix. Since the integration of partially desolvatedmolecules to the crystal face is an irreversible process, it does notaffect the activation barrier and the growth rate. A hypotheticalpathway from the transition state to the crystal state is shown inFig. 4A (dashed black line).The heat of integration is the energy difference between the
initial state (crystallization coordinate = 0.00, fully solvated mol-ecules in solution) and the final state (crystallization coordinate =1.00, molecules in the crystal lattice), which is also the sum of theenergy of the crystal face (also referred to as a slice energy) andthe hydration energy. Since the energy of the crystal face is con-stant, the heat of integration is proportional to the hydration en-ergy or the hydration number. Fig. 4B shows a linear relationshipbetween the activation barrier and the hydration number of fullysolvated glutamic acid molecules at different supersaturations.This linear relationship is similar to the Evans–Polanyi relationshipbetween the activation barrier and the heat of reaction.Fig. 5 A and B shows the experimentally measured (37) and
the predicted growth rates of [111] and [011] faces, respectively,of the α-glutamic acid crystal. The experimental growth rates
Fig. 3. Dynamics of hydration number of the glutamic acid molecule at asupersaturation of 2.5. The running average of the hydration number (solidblue line) decreases with decrease in the intermolecular distance of theglutamic acid molecule (solid red line), which is governed by fluctuations inconcentration. The instantaneous fluctuations in the hydration number (solidblack line) are due to exchange of water molecules in the solvation shell, whichincreases with increase in temperature. The shorter time-scale dynamics ofhydration number are dictated by temperature, whereas the longer time-scaledynamics are governed by local fluctuations in supersaturation.
Fig. 4. (A) Energy profiles for the integration of glutamic acid molecules ona crystal face at different supersaturations. The crystallization coordinate rep-resents relative distances between molecules as they move from the bulk phase(value 0) to the crystal surface (value 1). The transitions of the fully solvatedmolecule to the partially desolvated molecule shown as solid curves determinethe activation barrier. The dashed lines represent the hypothetical energypathway for integration of partially desolvated molecules on the crystal sur-face. (B) A linear relationship between the activation barrier to form partiallysolvated molecules and the hydration number of fully solvated molecules fordifferent supersaturations, which follows the Evans–Polanyi principle.
Dighe and Singh PNAS | November 26, 2019 | vol. 116 | no. 48 | 23957
ENGINEE
RING
Dow
nloa
ded
by g
uest
on
June
2, 2
021
https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplemental
obtained for the supersaturation range of 1.1 to 1.7 were extra-polated based on the fitting equations provided in ref. 37 up to thesupersaturation of 2.5 to extend the comparison between simu-lated and experimental data. The growth rates were calculatedusing the supersaturation-dependent activation barriers (shown inFig. 4A) in the established growth rate model. The predicted val-ues of growth rates show a good agreement with the experimentalgrowth rates for lower supersaturations. The predicted and experi-mental steady-state morphologies of the α-glutamic acid crystal arealso in good agreement (SI Appendix). For higher supersaturation,the predicted growth rates are higher than the experimental growthrates, which can be due to either overestimation of activation
barriers or change in growth mechanism from spiral to 2D nu-cleation with increasing supersaturation. The overestimation ofthe activation barriers might be due to the absence of crystalsurface in the MD simulation. The higher interactions of fullysolvated molecules near the crystal surface can result in higherdesolvation and lower hydration number of partially desolvatedmolecules and thereby lower activation barrier. However, thesolvation dynamics obtained from such MD simulations involvingthe crystal–solution interface are often affected by the continuousdepletion of supersaturation (22). Fig. 5C shows the deviation ofpredicted growth rates from the experimental growth rates for[111] and [011] faces of the α-glutamic acid crystal. The extrapo-lated experimental growth rates for supersaturation of 2.5 areoutliers as the rough growth is most likely the mechanism at sucha high supersaturation, which can increase the predicted growthrate by an order of magnitude. Therefore, the predicted growthrates deviate significantly from the experimental growth rates at asupersaturation of 2.5.
ConclusionSupersaturation is generally regarded as a driver for crystallization.Here we show that the actual driving force for crystallizationcorresponds to the energy required to desolvate molecules, whichin turn is a function of supersaturation. The hydration number andsolvation shell can be considered as the energy barrier and physicalbarrier, respectively, for solute molecules to attach to the crystalsurface. It is possible to control the solvation shell and hydrationnumber of specific functional groups in a solute molecule by ma-nipulating the solvent composition such that attachment of solutemolecules in a certain crystallographic direction is favored. Theaverage hydration number of a solute molecule in the solution canbe reduced by increasing its concentration at a constant temper-ature or increasing the supersaturation. By reducing the averagehydration number, the energy barrier to desolvate the solutemolecules can be lowered.The removal of solvent molecules from the fully solvated mole-
cules is an energetically uphill process. When a critical number ofsolvent molecules are removed, the partially desolvated moleculecan spontaneously desolvate and attach to a crystal surface via en-ergetically favorable noncovalent interactions. The partiallydesolvated state has been previously identified as a transition statefor molecular attachment processes (29, 31). The energy re-quired to desolvate a fully solvated molecule to form a partiallydesolvated molecule can be considered as the activation barrierfor crystal growth. This activation barrier is a function of su-persaturation, temperature, and solvent composition, which canbe determined from the fluctuations in the solvation shell. Thefluctuations in the hydration number are driven by 1) rapidsolvent exchange in the solvation shell, which is a function oftemperature, and 2) local variation in concentration, which is afunction of supersaturation. These fluctuations cause in-stantaneous formation of fully and partially solvated moleculesthat are in quasi-equilibrium. The transition state and activa-tion barrier can be obtained from the construction of a double-well potential using the average configuration of fully solvatedand partially desolvated molecules. The activation barrier waslinearly dependent on the hydration number, which is similar tothe Evans–Polanyi relationship. Since the activation barrier is di-rectly proportional to the average hydration number and the hy-dration number is inversely proportional to the supersaturation, itcan be seen how the activation barrier for crystal growth decreaseswith increasing supersaturation.The supersaturation-dependent activation barriers can be used
in the transition state theory to obtain rate constants for growth,which can be supplied to the mechanistic growth model to pre-dict absolute growth rates. The predicted values of growth rateswere in reasonable agreement with the experimental growth ratesof the α-glutamic acid crystal for lower supersaturations, which
Fig. 5. Comparison between experimentally measured growth rates (solidtriangles) and ab initio predicted growth rates (solid squares) for (A) the[111] face and (B) the [011] face of the α-glutamic acid crystal as a function ofsupersaturation. The extrapolated experimental growth rates are shown asopen triangles (37) and the fitted simulation data are shown as a solid blackline. (C) Deviation of the predicted growth rates from the experimentalgrowth rates for a family of faces (011) and (111).
23958 | www.pnas.org/cgi/doi/10.1073/pnas.1910691116 Dighe and Singh
Dow
nloa
ded
by g
uest
on
June
2, 2
021
https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplementalhttps://www.pnas.org/cgi/doi/10.1073/pnas.1910691116
confirms that the predicted supersaturation-dependent activationbarriers are physically meaningful. The larger disagreement be-tween experiment and theory at higher supersaturation can be dueto either the variation in the growth mechanism or overestimationof activation barriers. The prediction of the activation barrier canbe improved by considering a crystal–solution interface at a con-stant supersaturation in MD simulations. These results confirmthat the solvent fluctuation in the solvation shell is a key processthat governs the solution-based synthesis of crystalline materials.
Data Availability. All the computational data generated and as-sociated simulation details are provided in the main text andSI Appendix.
ACKNOWLEDGMENTS. This material is based on the work performed in theMaterials and Systems Engineering Laboratory at the University of Illinois atChicago. A.V.D. thanks Ms. Sneha M. Dixit from Max Planck Institute ofColloids and Interfaces for stimulating discussion on molecular simulations.We are grateful to our funding source, the US National Science Foundation(Award CBET-1706921).
1. A. S. Myerson, Handbook of Industrial Crystallization (Butterworth-Heinemann, 2002).2. D. Ramkrishna, M. R. Singh, Population balance modeling: Current status and future
prospects. Annu. Rev. Chem. Biomol. Eng. 5, 123–146 (2014).3. M. R. Singh, D. Ramkrishna, A comprehensive approach to predicting crystal morphology
distributions with population balances. Cryst. Growth Des. 13, 1397–1411 (2013).4. M. R. Singh et al., Image-analysis-based method for 3D crystal morphology measurement
and polymorph identification using confocal microscopy. Cryst. Growth Des. 12, 3735–3748 (2012).
5. A. V. Dighe, R. Y. Nemade, M. R. Singh, Modeling and simulation of crystallization ofmetal–organic frameworks. Processes 7, 527 (2019).
6. M. R. Singh, P. Verma, H.-H. Tung, S. Bordawekar, D. Ramkrishna, Screening crystalmorphologies from crystal structure. Cryst. Growth Des. 13, 1390–1396 (2013).
7. M. R. Singh, D. Ramkrishna, Dispersions in crystal nucleation and growth rates: Im-plications of fluctuation in supersaturation. Chem. Eng. Sci. 107, 102–113 (2014).
8. M. R. Singh et al., Measurement of polar plots of crystal dissolution rates using hot-stage microscopy. Some further insights into dissolution morphologies. Cryst. GrowthDes. 14, 5647–5661 (2014).
9. D. Kashchiev, Nucleation (Butterworth-Heinemann, 2000).10. D. Kashchiev, P. G. Vekilov, A. B. Kolomeisky, Kinetics of two-step nucleation of
crystals. J. Chem. Phys. 122, 244706 (2005).11. J. Donnay, D. Harker, A new law of crystal morphology extending the law of Bravais.
Am. Mineral. 22, 446–467 (1937).12. P. Hartman, W. Perdok, On the relations between structure and morphology of crystals.
I. Acta Crystallogr. 8, 49–52 (1955).13. P. Hartman, W. Perdok, On the relations between structure and morphology of crystals.
II. Acta Crystallogr. 8, 521–524 (1955).14. P. Hartman, W. Perdok, On the relations between structure and morphology of crystals.
III. Acta Crystallogr. 8, 525–529 (1955).15. W. Burton, N. Cabrera, F. Frank, The growth of crystals and the equilibrium structure
of their surfaces. Philos. Trans. R. Soc. Lond. A 243, 299–358 (1951).16. M. Ohara, R. C. Reid,Modeling Crystal Growth Rates from Solution (Prentice-Hall, 1973).17. A. Chernov, The kinetics of the growth forms of crystals. Sov. Phys. Crystallogr. 7, 728–
730 (1963).18. D. Winn, M. F. Doherty, Modeling crystal shapes of organic materials grown from
solution. AIChE J. 46, 1348–1367 (2000).19. S. Boerrigter et al., MONTY: Monte Carlo crystal growth on any crystal structure in any
crystallographic orientation; application to fats. J. Phys. Chem. A 108, 5894–5902 (2004).20. C. J. Tilbury, M. F. Doherty, Modeling layered crystal growth at increasing supersat-
uration by connecting growth regimes. AIChE J. 63, 1338–1352 (2017).21. M. A. Lovette, M. F. Doherty, Predictive modeling of supersaturation-dependent
crystal shapes. Cryst. Growth Des. 12, 656–669 (2012).22. A. M. Reilly, H. Briesen, Modeling crystal growth from solution with molecular dynamics
simulations: Approaches to transition rate constants. J. Chem. Phys. 136, 034704 (2012).23. M. Rak, M. Izdebski, A. Brozi, Kinetic Monte Carlo study of crystal growth from solution.
Comput. Phys. Commun. 138, 250–263 (2001).
24. H. Huitema, B. Van Hengstum, J. van der Eerden, Simulation of crystal growth fromLennard-Jones solutions. J. Chem. Phys. 111, 10248–10260 (1999).
25. S. Piana, M. Reyhani, J. D. Gale, Simulating micrometre-scale crystal growth fromsolution. Nature 438, 70–73 (2005).
26. G. M. Whitesides, B. Grzybowski, Self-assembly at all scales. Science 295, 2418–2421(2002).
27. G. C. Sosso et al., Crystal nucleation in liquids: Open questions and future challengesin molecular dynamics simulations. Chem. Rev. 116, 7078–7116 (2016).
28. M. N. Joswiak, M. F. Doherty, B. Peters, Ion dissolution mechanism and kinetics at kinksites on NaCl surfaces. Proc. Natl. Acad. Sci. U.S.A. 115, 656–661 (2018).
29. S. Piana, F. Jones, J. D. Gale, Assisted desolvation as a key kinetic step for crystalgrowth. J. Am. Chem. Soc. 128, 13568–13574 (2006).
30. M. Kowacz, C. Putnis, A. Putnis, The effect of cation:anion ratio in solution on themechanism of barite growth at constant supersaturation: Role of the desolvationprocess on the growth kinetics. Geochim. Cosmochim. Acta 71, 5168–5179 (2007).
31. A. E. Hofmann, I. C. Bourg, D. J. DePaolo, Ion desolvation as a mechanism for kineticisotope fractionation in aqueous systems. Proc. Natl. Acad. Sci. U.S.A. 109, 18689–18694 (2012).
32. R. S. DeFever, S. Sarupria, Nucleation mechanism of clathrate hydrates of water-soluble guest molecules. J. Chem. Phys. 147, 204503 (2017).
33. R. S. DeFever, S. Sarupria, Contour forward flux sampling: Sampling rare events alongmultiple collective variables. J. Chem. Phys. 150, 024103 (2019).
34. E. Shustorovich, Metal-Surface Reaction Energetics: Theory and Applications to Het-erogeneous Catalysis, Chemisorption, and Surface Diffusion (VCH Publishers, Inc.,New York, NY, 1991).
35. E. Shustorovich, Energetics of metal-surface reactions: Back-of-the-envelope theoreticalmodeling. J. Mol. Catal. 54, 301–311 (1989).
36. M. J. Abraham et al., GROMACS: High performance molecular simulations throughmulti-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
37. M. Kitamura, T. Ishizu, Growth kinetics and morphological change of polymorphs ofL-glutamic acid. J. Cryst. Growth 209, 138–145 (2000).
38. N. C. Kee, R. B. Tan, R. D. Braatz, Selective crystallization of the metastable α-form ofL-glutamic acid using concentration feedback control. Cryst. Growth Des. 9, 3044–3051 (2009).
39. T. Ono, H. J. M. Kramer, J. H. ter Horst, P. J. Jansens, Process modeling of the polymorphictransformation of L-glutamic acid. Cryst. Growth Des. 4, 1161–1167 (2004).
40. M. P. Allen, D. J. Tildesley, Computer Simulation of Liquids (Oxford University Press,2017).
41. R. Salomon‐Ferrer, D. A. Case, R. C. Walker, An overview of the Amber biomolecularsimulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210 (2013).
42. Z. B. Kuvadia, M. F. Doherty, Spiral growth model for faceted crystals of non-centrosymmetric organic molecules grown from solution. Cryst. Growth Des. 11,2780–2802 (2011).
43. Y. Marcus, Concentration dependence of ionic hydration numbers. J. Phys. Chem. B118, 10471–10476 (2014).
Dighe and Singh PNAS | November 26, 2019 | vol. 116 | no. 48 | 23959
ENGINEE
RING
Dow
nloa
ded
by g
uest
on
June
2, 2
021
https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.1910691116/-/DCSupplemental