
Soluble, dimeric HLA DR4-peptide chimeras: An approach for detection and immunoregulation of human...
14
Soluble, dimeric HLA DR4-peptide chimeras: An approach for detection and immunoregulation of human type-1 diabetes Ioana Preda 1 , Robert C. McEvoy 2 , Marvin Lin 1 , Constantin A. Bona 1 , Robert Rapaport 3 , Teodor D. Brumeanu 4 and Sofia Casares 1 1 Department of Microbiology, Mount Sinai School of Medicine, New York, NY, USA 2 McNeely Diabetes Center, Children's Hospitals and Clinics, St. Paul, MN, USA 3 Department of Pediatrics, Mount Sinai School of Medicine, New York, NY, USA 4 Department of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD, USA Still there are no effective methods to predict or cure type 1 diabetes (T1D) in humans. Soluble, dimeric MHC class II-peptide (DEF) chimeras have potential for both early diagnosis and immunospecific therapy. DEF chimeras prevent and reverse diabetes in mice by stimulating antigen-specific type 1 T regulatory cell (Tr1)-like cells. We also showed that diabetes could be predicted by changes in the phenotype of autoreactive CD4 T cells in peripheral blood. Herein, we demonstrated that human DEF (HLA- DR*0401/Fcc1) chimeras expressing peptides of b-cell antigens stimulate Tr1-like cells in blood of patients with T1D, non-diabetic relatives, and controls. Furthermore, the specific and stable binding of DEF chimeras to cognate TCR and CD4 coreceptor allowed quantification and phenotyping of autoreactive CD4 T cells in non-stimulated blood by FACS. Our results indicate that (1) autoreactive CD4 T cells to GAD65 autoantigen are commonly present in humans expressing diabetes-susceptible HLA-DR*0401 mole- cules; (2) these autoreactive T cells undergo avidity maturation upon encountering the self antigen early in life; (3) the disease is associated with an imbalance between autoreactive CD4 + CD25 + and CD4 + CD69 + T cells specific for GAD65. Based on this, we propose a model to explain the kinetics of autoreactive CD4 T cells in blood during the natural history of T1D. Introduction Type 1 diabetes (T1D) is an autoimmune disease mediated by T cells. Susceptibility to T1D is genetically controlled by alleles of the major histocompatibility complex (MHC) class II genes and non-MHC back- ground genes. In humans, HLA-DRB1*0401, HLA- DRB1*0301, HLA-DQB1*0302, and HLA-DQA1*0301 confer high-risk susceptibility, whereas other alleles such as HLA-DRB1*0403, HLA-DQB1*0602, and HLA- DQA1*0102 are negatively associated with the disease and may even confer resistance [1]. The association between MHC class II molecules and T1D indicates a critical role of CD4 T cells in the pathogenesis, since MHC II molecules are required for education of thymic T cell precursors and priming of CD4 T cells in the periphery. MHC class II molecules are heterodimers consisting of two noncovalently associated a and b chains Clinical immunology Correspondence: Dr. Sofia Casares, One Gustave L. Levy Place, Box 1124, New York, NY 10029, USA Fax: +1-212-5341684 e-mail: [email protected] Received 9/3/05 Revised 17/6/05 Accepted 25/7/05 [DOI 10.1002/eji.200526158] Key words: Type 1 diabetes Autoreactive CD4 T cells MHC II- Peptide complexes Homeostasis Abbreviations: DEF: dimeric MHC class II-peptide GADA: anti-GAD65 antibody hCD4: human CD4 IAA: anti- insulin antibody T1D: type 1 diabetes TcH: T cell hybridoma Tr1: type 1 T regulatory cell Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–2775 2762 f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de
-
Upload
ioana-preda -
Category
Documents
-
view
217 -
download
4
Transcript of Soluble, dimeric HLA DR4-peptide chimeras: An approach for detection and immunoregulation of human...

Soluble, dimeric HLA DR4-peptide chimeras:An approach for detection and immunoregulation ofhuman type-1 diabetes
Ioana Preda1, Robert C. McEvoy2, Marvin Lin1, Constantin A. Bona1,Robert Rapaport3, Teodor D. Brumeanu4 and Sofia Casares1
1 Department of Microbiology, Mount Sinai School of Medicine, New York, NY, USA2 McNeely Diabetes Center, Children's Hospitals and Clinics, St. Paul, MN, USA3 Department of Pediatrics, Mount Sinai School of Medicine, New York, NY, USA4 Department of Medicine, Uniformed Services University of the Health Sciences,Bethesda, MD, USA
Still there are no effective methods to predict or cure type 1 diabetes (T1D) in humans.Soluble, dimeric MHC class II-peptide (DEF) chimeras have potential for both earlydiagnosis and immunospecific therapy. DEF chimeras prevent and reverse diabetes inmice by stimulating antigen-specific type 1 T regulatory cell (Tr1)-like cells. We alsoshowed that diabetes could be predicted by changes in the phenotype of autoreactiveCD4 T cells in peripheral blood. Herein, we demonstrated that human DEF (HLA-DR*0401/Fcc1) chimeras expressing peptides of b-cell antigens stimulate Tr1-like cellsin blood of patients with T1D, non-diabetic relatives, and controls. Furthermore, thespecific and stable binding of DEF chimeras to cognate TCR and CD4 coreceptor allowedquantification and phenotyping of autoreactive CD4 T cells in non-stimulated blood byFACS. Our results indicate that (1) autoreactive CD4 T cells to GAD65 autoantigen arecommonly present in humans expressing diabetes-susceptible HLA-DR*0401 mole-cules; (2) these autoreactive T cells undergo avidity maturation upon encountering theself antigen early in life; (3) the disease is associated with an imbalance betweenautoreactive CD4+CD25+ and CD4+CD69+ T cells specific for GAD65. Based on this,we propose a model to explain the kinetics of autoreactive CD4 T cells in blood duringthe natural history of T1D.
Introduction
Type 1 diabetes (T1D) is an autoimmune diseasemediated by T cells. Susceptibility to T1D is geneticallycontrolled by alleles of the major histocompatibility
complex (MHC) class II genes and non-MHC back-ground genes. In humans, HLA-DRB1*0401, HLA-DRB1*0301, HLA-DQB1*0302, and HLA-DQA1*0301confer high-risk susceptibility, whereas other allelessuch as HLA-DRB1*0403, HLA-DQB1*0602, and HLA-DQA1*0102 are negatively associated with the diseaseand may even confer resistance [1]. The associationbetween MHC class II molecules and T1D indicates acritical role of CD4 T cells in the pathogenesis, sinceMHC II molecules are required for education of thymicT cell precursors and priming of CD4 T cells in theperiphery.
MHC class II molecules are heterodimers consistingof two noncovalently associated a and b chains
Clinical immunology
Correspondence: Dr. Sofia Casares, One Gustave L. Levy Place,Box 1124, New York, NY 10029, USAFax: +1-212-5341684e-mail: [email protected]
Received 9/3/05Revised 17/6/05
Accepted 25/7/05
[DOI 10.1002/eji.200526158]
Key words:Type 1 diabetes
� Autoreactive CD4T cells � MHC II-
Peptide complexes� Homeostasis
Abbreviations: DEF: dimeric MHC class II-peptide �GADA: anti-GAD65 antibody � hCD4: human CD4 � IAA: anti-insulin antibody � T1D: type 1 diabetes � TcH:T cell hybridoma �Tr1: type 1 T regulatory cell
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752762
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

expressed on antigen-presenting cells (APC). Theassembly of a1-b1 domains forms a groove for bindingof peptides that are presented to the specific T cells.Antigen presentation by APC expressing costimulatorymolecules fosters differentiation of T cell effectors, butthe lack of costimulation induces T cell tolerance [2].Thus, an attractive approach to treat T cell-mediatedautoimmune diseases is to use genetically engineeredMHC-peptide chimeras. We have generated the firstsoluble, dimeric MHC class II-peptide (DEF) chimerabuilt on a self-Ig scaffold [3], and demonstrated that itprevents and reverses diabetes in mice by stimulation ofantigen-specific IL-10-secreting type 1 T regulatory cell(Tr1)-like cells [4]. The therapeutic effects of DEF-likereagents have been also demonstrated in other animalmodels of autoimmune diabetes [5] and rheumatoidarthritis [6]. Here, we showed that human DEFchimeras, made of HLA-DR*0401 dimers on a humanIg-Fcc1 scaffold and expressing epitopes of GAD65 andpro-insulin, are endowed with immunoregulatoryproperties and that they stimulate antigen-specific IL-10-secreting Tr1-like cells in blood of patients with T1D,their non-diabetic relatives and unrelated controls. TheIL-10 response to DEF in vitro also indicated the presenceof autoreactive CD4 T cells in blood.
Autoantibodies are considered as a surrogate markerfor T1D, but with limited prediction. Since T1D ismediated by autoreactive T cells, analyzing these cells inperipheral blood is a good approach that surely will helpto increase the accuracy of diabetes prediction. Indeed,Trudeau et al. [7] could predict diabetes in mice bytracing autoreactive CD8 T cells in the blood. We havealso shown that diabetes could be predicted by tracingautoreactive CD4 T cells in blood [8]. Our mechanisticstudies further indicated that disease progression ismarked by an imbalance between autoreactiveCD4+CD25+ and CD4+CD69+ T cells in the pancreasand that such an event is mirrored in the phenotype ofautoreactive CD4 T cells in the blood. Once the T cellimbalance occurs, the autoimmune attack on thepancreas follows a “hit-and-run” kinetics consisting ofwaves of recruitment of autoreactive (CD4+CD69+)T cells from the blood.
Using the humanDEF chimeras, we have investigatedthe frequency and phenotype of autoreactive (GAD65)CD4 T cells in blood of patients with T1D, their relativesand controls. Our results indicate that autoreactiveT cells are commonly present in those expressing theHLA-DR*0401 molecule. Like we found in mice, diseaseprogression is associated with an imbalance betweenautoreactive CD4+CD25+ and CD4+CD69+ T cells, andthe disappearance of autoreactive T cells from the bloodcorrelated with the clinical onset of T1D.
Results
Structural integrity of human DEF chimeras
The human DEF chimeras are dimers of HLA-DR*0401built on an Ig-Fcc1 scaffold and expressing epitopes ofGAD65 (GAD-DEF) or pro-insulin (pINS-DEF) (Fig. 1A).The structural integrity of the hFcc1 component of DEFwas revealed by the ability to bind to Fc receptorsexpressed on INF-c-activated monocytes (Fig. 1B).
To assess the binding specificity of these chimericmolecules to cognate T cells, we used short-term T cellclones generated from PBMC stimulated with eitherGAD-DEF or pINS-DEFand IL-15. The specificity of theseT cell clones was demonstrated by their proliferativeresponse upon restimulation with the nominal syntheticpeptide (Fig. 1C, D, lower right panels). The GAD-DEFchimera allowed FACS detection of GAD65-specificT cells, but not of pINS-specific T cells (Fig. 1C, D,lower left panels). The binding specificity of the pINS-DEF chimera to cognate T cells was also proved by FACS(Fig. 1C, D, upper left panels). The control DEFgenerated, which expresses the Borrelia burgdorfiepitope Osp A163–175 peptide [9] did not bind to eithercells (Fig. 1C, D, upper right panels). Neither GAD-DEF,pINS-DEF or control DEF allowed FACS detection oftetanus toxoid-specific T cells (data not shown).
Since mouse DEF chimeras bind to both cognate TCRand CD4 coreceptor [10, 11], we next determined therole of CD4 interaction in stabilizing the binding ofhuman DEF to the specific T cells. For this, a mouse 33.1T cell hybridoma (TcH) recognizing the GAD65271–285peptide in the context of HLA-DR*0401 molecules [12]was transfected with the human CD4 (hCD4) gene orempty plasmid. The GAD-DEF chimera showed to bindwith higher avidity to hCD4-transfected 33.1 T cells(Fig. 1E, left panels). The pINS-DEF chimera failed todetect such cells by FACS (Fig. 1E, right panels).Furthermore, we also found that cross-linking of CD3/TCR complexes by anti-CD3mAb increased the avidity ofGAD-DEF for hCD4-transfected T cells (Fig. 1F).
These results indicate that DEF chimeras preserve thestructural integrity of MHC-peptide and Fcc1 compo-nents and that they are suitable reagents for theidentification of specific T cells by FACS.
The DEF chimeras stimulate IL-10-secretingTr1-like cells
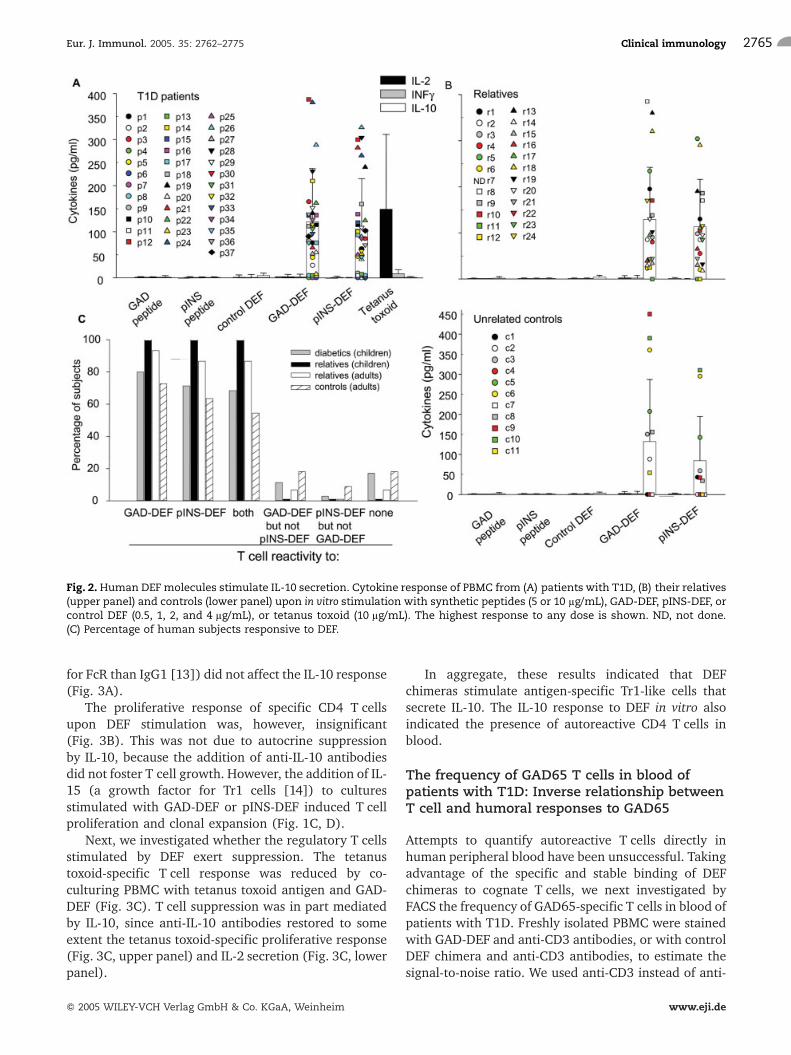
A mouse DEF chimera prevented and reversed diabetesby stimulation of antigen-specific regulatory CD4T cells, in particular Tr1-like cells that secrete IL-10[4]. Thus, we investigated whether the human DEF arealso endowed with immunoregulatory properties on thespecific T cells. As shown in Fig. 2A and B, GAD-DEF and
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2763
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

pINS-DEF stimulated IL-10 secretion in PBMC ofpatients with T1D, their first-degree relatives, andunrelated controls expressing HLA-DR*0401 molecules.No other cytokines, i.e. IL-2, INF-c (Fig. 2A, B), IL-4,TGF-b, or TGF-a (data not shown), were detected in thecultures. The IL-10 response was stimulated by the selfpeptide bound to the MHC groove of the specific DEF,since the control DEF chimera as well as the GAD65 andpINS synthetic peptides failed to stimulate IL-10(Fig. 2A, B). The secretion of IL-10 was inhibited bymonensin (data not shown), a monovalent cationophoreknown to prevent the transport of proteins from Golgi tomembrane. In contrast to the IL-10 response induced bythe specific DEF, stimulation with tetanus toxoidinduced a dominant IL-2 response (Fig. 2A). The
percentages of patients with T1D, relatives, and controlsresponding to GAD-DEF and/or pINS-DEF stimulationare shown in Fig. 2C.
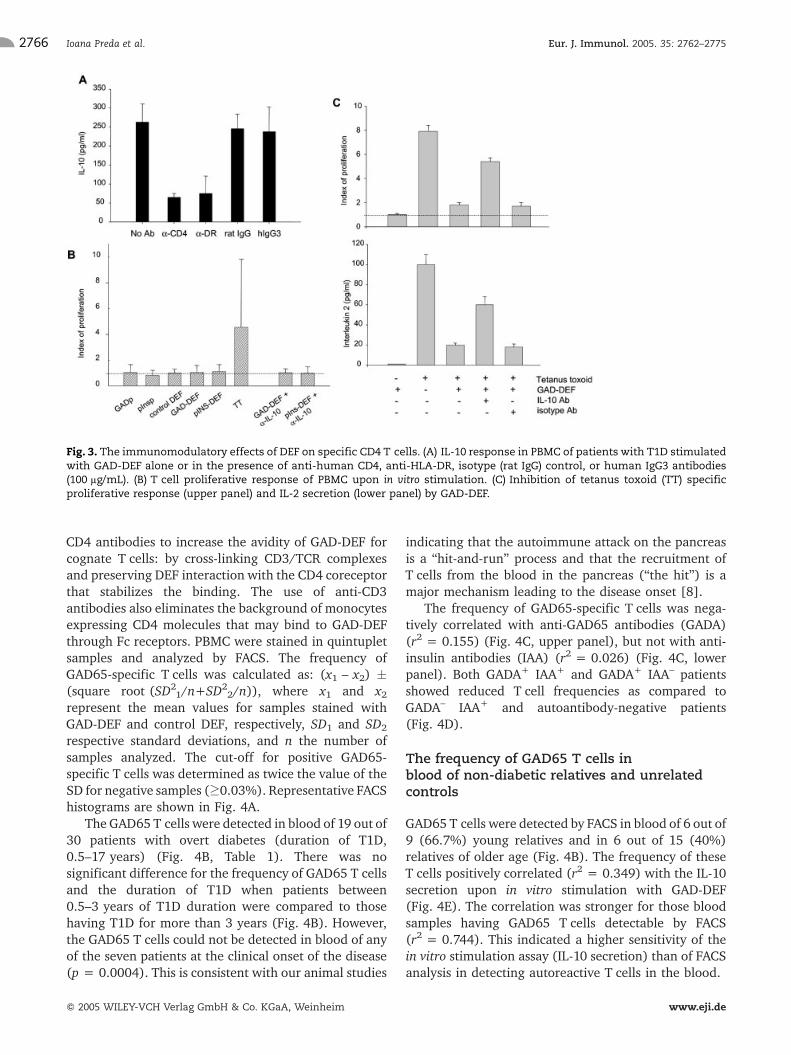
IL-10 was secreted by DEF-stimulated CD4 T cellssince anti-CD4 and anti-HLA-DR antibodies wereinhibitory (Fig. 3A). Furthermore, depletion of T cellswith anti-CD3 antibody-coated magnetic beads (effi-ciency of CD3 T cell depletion >95% as measured byFACS) resulted in more than 95% inhibition of IL-10secretion, which indicated that CD4 T cells rather thanCD4-expressing monocytes were stimulated by thespecific DEF to secrete IL-10. On the other hand, thebinding of the Fcc1 component of DEF to Fc receptors onAPC was not required to stimulate the CD4 T cells, sinceinhibition with IgG3 (which has a higher binding avidity
Fig. 1. Structural integrity of DEF chimeras. (A) Schematic representation of GAD-DEF and pINS-DEF. (B) Binding of GAD-DEF to Fcreceptors (FcR) expressed on INF-c-activated monocytes. (C) Binding of GAD-DEF to GAD65271–285-specific T cells. PBMC werestimulated with GAD-DEF and IL-15 and then restimulated with GAD65271–285 synthetic peptide (lower right panel). Cells wereanalyzed by FACS using DEF chimeras. (D) Binding of pINS-DEF to pINS73–90-specific T cells. (E) 33.1 TcH was transfected withplasmid control (upper panels) or with plasmid encoding for hCD4 (lower panels) and analyzed by FACS. GAD-DEF bound withhigher avidity and to a higher number of 33.1 TcH cells transfected with hCD4 (inner histograms). MFI, mean fluorescenceintensity. (F) Co-staining with anti-mouse CD3 antibody increased the binding avidity of GAD-DEF for hCD4-tranfected TcH cells.
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752764
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de
for FcR than IgG1 [13]) did not affect the IL-10 response(Fig. 3A).
The proliferative response of specific CD4 T cellsupon DEF stimulation was, however, insignificant(Fig. 3B). This was not due to autocrine suppressionby IL-10, because the addition of anti-IL-10 antibodiesdid not foster T cell growth. However, the addition of IL-15 (a growth factor for Tr1 cells [14]) to culturesstimulated with GAD-DEF or pINS-DEF induced T cellproliferation and clonal expansion (Fig. 1C, D).
Next, we investigated whether the regulatory T cellsstimulated by DEF exert suppression. The tetanustoxoid-specific T cell response was reduced by co-culturing PBMC with tetanus toxoid antigen and GAD-DEF (Fig. 3C). T cell suppression was in part mediatedby IL-10, since anti-IL-10 antibodies restored to someextent the tetanus toxoid-specific proliferative response(Fig. 3C, upper panel) and IL-2 secretion (Fig. 3C, lowerpanel).
In aggregate, these results indicated that DEFchimeras stimulate antigen-specific Tr1-like cells thatsecrete IL-10. The IL-10 response to DEF in vitro alsoindicated the presence of autoreactive CD4 T cells inblood.
The frequency of GAD65 T cells in blood ofpatients with T1D: Inverse relationship betweenT cell and humoral responses to GAD65
Attempts to quantify autoreactive T cells directly inhuman peripheral blood have been unsuccessful. Takingadvantage of the specific and stable binding of DEFchimeras to cognate T cells, we next investigated byFACS the frequency of GAD65-specific T cells in blood ofpatients with T1D. Freshly isolated PBMC were stainedwith GAD-DEF and anti-CD3 antibodies, or with controlDEF chimera and anti-CD3 antibodies, to estimate thesignal-to-noise ratio. We used anti-CD3 instead of anti-
Fig. 2. Human DEF molecules stimulate IL-10 secretion. Cytokine response of PBMC from (A) patients with T1D, (B) their relatives(upper panel) and controls (lower panel) upon in vitro stimulation with synthetic peptides (5 or 10 lg/mL), GAD-DEF, pINS-DEF, orcontrol DEF (0.5, 1, 2, and 4 lg/mL), or tetanus toxoid (10 lg/mL). The highest response to any dose is shown. ND, not done.(C) Percentage of human subjects responsive to DEF.
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2765
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de
CD4 antibodies to increase the avidity of GAD-DEF forcognate T cells: by cross-linking CD3/TCR complexesand preserving DEF interaction with the CD4 coreceptorthat stabilizes the binding. The use of anti-CD3antibodies also eliminates the background of monocytesexpressing CD4 molecules that may bind to GAD-DEFthrough Fc receptors. PBMC were stained in quintupletsamples and analyzed by FACS. The frequency ofGAD65-specific T cells was calculated as: (x1 – x2) �(square root (SD2
1/n+SD22/n)), where x1 and x2
represent the mean values for samples stained withGAD-DEF and control DEF, respectively, SD1 and SD2
respective standard deviations, and n the number ofsamples analyzed. The cut-off for positive GAD65-specific T cells was determined as twice the value of theSD for negative samples (�0.03%). Representative FACShistograms are shown in Fig. 4A.
The GAD65 T cells were detected in blood of 19 out of30 patients with overt diabetes (duration of T1D,0.5–17 years) (Fig. 4B, Table 1). There was nosignificant difference for the frequency of GAD65 T cellsand the duration of T1D when patients between0.5–3 years of T1D duration were compared to thosehaving T1D for more than 3 years (Fig. 4B). However,the GAD65 T cells could not be detected in blood of anyof the seven patients at the clinical onset of the disease(p = 0.0004). This is consistent with our animal studies
indicating that the autoimmune attack on the pancreasis a “hit-and-run” process and that the recruitment ofT cells from the blood in the pancreas (“the hit”) is amajor mechanism leading to the disease onset [8].
The frequency of GAD65-specific T cells was nega-tively correlated with anti-GAD65 antibodies (GADA)(r2 = 0.155) (Fig. 4C, upper panel), but not with anti-insulin antibodies (IAA) (r2 = 0.026) (Fig. 4C, lowerpanel). Both GADA+ IAA+ and GADA+ IAA– patientsshowed reduced T cell frequencies as compared toGADA– IAA+ and autoantibody-negative patients(Fig. 4D).
The frequency of GAD65 T cells inblood of non-diabetic relatives and unrelatedcontrols
GAD65 T cells were detected by FACS in blood of 6 out of9 (66.7%) young relatives and in 6 out of 15 (40%)relatives of older age (Fig. 4B). The frequency of theseT cells positively correlated (r2 = 0.349) with the IL-10secretion upon in vitro stimulation with GAD-DEF(Fig. 4E). The correlation was stronger for those bloodsamples having GAD65 T cells detectable by FACS(r2 = 0.744). This indicated a higher sensitivity of thein vitro stimulation assay (IL-10 secretion) than of FACSanalysis in detecting autoreactive T cells in the blood.
Fig. 3. The immunomodulatory effects of DEF on specific CD4 T cells. (A) IL-10 response in PBMC of patients with T1D stimulatedwith GAD-DEF alone or in the presence of anti-human CD4, anti-HLA-DR, isotype (rat IgG) control, or human IgG3 antibodies(100 lg/mL). (B) T cell proliferative response of PBMC upon in vitro stimulation. (C) Inhibition of tetanus toxoid (TT) specificproliferative response (upper panel) and IL-2 secretion (lower panel) by GAD-DEF.
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752766
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

T cells that have encountered antigen display 20–50-fold higher avidities for MHC-peptide complexes thanresting T cells, as demonstrated by Fahmy et al. [15] andAmrani et al. [16] using recombinant MHC-peptidecomplexes and FACS analysis. Thus, the lack of GAD65T cells in blood as measured by FACS using the GAD-DEFreagent could not rule out the presence of low-avidityT cells, which were indeed detected through the IL-10surrogatemarker (Fig.4E, inner left panel). Thiswasnextaddressed by in vitro stimulation T cell assays. PBMClacking GAD65 T cells by FACS were stimulated withGAD65 synthetic peptide or GAD-DEF to induce aviditymaturation, and then re-analyzed by FACS. After 12 h ofstimulation, theGAD65-specific T cells could be detectedbyFACS(Fig.4F,middlepanels).ThoseT cells stimulatedwith GAD-DEF, but not those stimulated with synthetic
peptide, were positive for intracellular IL-10 (Fig. 4F,right panels). This is consistent with our previous datashowing that DEF chimeras immunoregulate specificCD4T cells toward an IL-10-secreting phenotype (Fig. 2).Therefore,bystimulatingT cells in vitro followedbyFACSanalysis, the high-avidity GAD65 T cells were detected inrelatives (6 out of 7), controls (3 out of 3) and patients atthe onset (3 out of 3) that were negative by FACS in theabsence of stimulation. This was the consequence ofaviditymaturationofT cells rather thanofoutgrowthof apopulation of T cell precursors, because PBMC werestimulated for a shorter period (12 h) than that requiredfor the G1-S phase transition in human T cells (19.5 h)[17]. Furthermore, we also showed that stimulationwiththese antigens for longer periods (i.e. 4 days) did induceT cell proliferation (Fig. 3B).
Fig. 4. Frequency of GAD65-specific T cells in blood: Cross-sectional analysis. (A) Representative dot plots of PBMC stained withanti-CD3 antibodies and control DEF (left panels) or GAD-DEF (right panel) and analyzed by FACS. (B) Frequency of GAD65-specificT cells in PBMC of patients with T1D, their first-degree relatives, and healthy controls. (C) Relation between the frequency ofGAD65-specific T cells in patients with T1D and GADA (upper panel) or IAA (lower panel). (D) Averaged GAD65 T cell frequency inpatients with T1D having GADA and/or IAA. (E) Correlation between the frequency of GAD65 T cells in blood of relatives asmeasured by FACS and the levels of IL-10 secretionupon in vitro stimulationwith GAD-DEF. (F) Percentage of GAD+CD3+ T cells (leftpanels) and intracellular cytokines (right panels) in PBMC stimulated with GAD65271–285 peptide or GAD-DEF.
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2767
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

Table 1. Patients with T1D (p) and their first-degree relatives (r)a)
ID Gender Age years T1D durationyears
Other autoimm.disorders
HLA-DR DR4subtype
GADAU/mL
IAAU/mL
P1 M 17 2.5 none DR3/DR4 B1*0401 <4 <1P2 F 19 8 none DR3/DR4 B1*0401 84.9 <1P3 F 9 0.5 vitiligo thyroid DR3/DR4 B1*0401 54.7 43P4 F 17 12 none DR3/DR4 B1*0401 48.1 <1P5 F 15 5 none DR4/DR14 B1*0401 <4 12.7P6 F 7 2.5 celiac thyroid DR4/DR4 B1*0401 102 22.4P7 F 18 8 none DR1/DR4 B1*0401 <4 <1P8 M 20 8 none DR4/DR7 B1*0401 <4 <1P9 M 13 0.7 none DR4/DR4 B1*0401 <4 8.1p10 M 14 2 thyroid DR15/DR4 B1*0401 <4 47.3p11 F 19 1.2 none DR4/DR4 B1*0401 85.6 22.4p12 F 10 5.5 none DR3/DR4 B1*0401 <4 43.1p13 M 13 9 thyroid DR3/DR4 B1*0401 7.1 1.6p14 M 15 5.5 none DR3/DR4 B1*0401 <4 <1p15 F 17 5.5 none DR3/DR4 B1*0401 121 <1p16 M 16 8 none DR4/DR8 B1*0401 <4 16.4p17 F 19 15 none DR3/DR4 B1*0401 121 5.4p18 F 12 6 none DR4/DR8 B1*0401 <4 <1P19 M 8 1 none DR3/DR4 B1*0401 <4 5.1p20 M 9 5.5 none DR4/DR13 B1*0401 <4 <1p21 M 15 1.2 none DR1/DR4 B1*0401 42.4 <1p22 F 6 2.5 none DR4/DR13 B1*0401 <4 46.5p23 F 12 2 none DR3/DR4 B1*0401 <4 48.3p24 M 10 6 thyroid DR3/DR4 B1*0401 <4 20p25 M 16 13 none DR3/DR4 B1*0401 <4 6.4p26 F 20 17 none DR3/DR4 B1*0401 <4 56.5p27 M 13 12.5 none DR4/DR4 B1*0401 <4 2.5p28 M 10 2 none DR4/DR13 B1*0401 <4 66.4p29 M 13 1 none DR3/DR4 B1*0401 <4 13p30 F 14 12 none DR3/DR4 B1*0401 <4 12p31 M 10 onset none DR3/DR4 B1*0401 <4 26p32 F 11 onset none DR4/DR11 B1*0401 132 43p33 F 15 onset none DR3/DR4 B1*0401 10 <1p34 M 12 onset none DR4/DR4 B1*0401 <4 <1p35 F 8 onset none DR4/DR8 B1*0401 30 <1p36 M 9 onset none DR4/DR13 B1*0401 ND NDp37 M 10 onset none DR3/DR4 B1*0401 ND NDR1 F 9 non-diabetic none DR4/DR11 B1*0401 <4 <1R2 M 11 non-diabetic none DR4/DR4 B1*0401 <4 <1R3 M 7 non-diabetic none DR3/DR4 B1*0401 <4 <1R4 F 15 non-diabetic none DR4/DR13 B1*0401 <4 <1R5 M 2 non-diabetic none DR4/DR13 B1*0401 <4 <1R6 F 4 non-diabetic none DR4/DR4 B1*0401 <4 <1R7 F 19 non-diabetic none DR3/DR4 B1*0401 <4 <1R8 M 7 non-diabetic none DR3/DR4 B1*0401 <4 <1R9 F 10 non-diabetic none DR4/DR11 B1*0401 28.1 51.8r10 F 47 non-diabetic none DR4/DR7 B1*0401 <4 <1r11 F 33 non-diabetic none DR4/DR7 B1*0401 <4 <1r12 F 45 non-diabetic none DR15/DR4 B1*0401 <4 <1r13 F 36 non-diabetic none DR3/DR4 B1*0401 <4 <1r14 F 38 non-diabetic none DR3/DR4 B1*0401 <4 <1r15 M 43 non-diabetic none DR4/DR7 B1*0401 <4 <1r16 M 45 non-diabetic none DR4/DR13 B1*0401 <4 <1r17 M 35 non-diabetic none DR4/DR7 B1*0401 <4 <1r18 M 44 non-diabetic none DR4/DR7 B1*0401 <4 <1r19 M 41 non-diabetic none DR1/DR4 B1*0401 <4 <1r20 M 44 non-diabetic none DR4/DR4 B1*0401/B1*0404 <4 <1r21 M 46 non-diabetic none DR4/DR14 B1*0401 <4 <1r22 M 41 non-diabetic none DR3/DR4 B1*0401 <4 <1r23 M 46 non-diabetic none DR3/DR4 B1*0401 <4 <1r24 F 39 non-diabetic none DR4/DR7 B1*0401 <4 <1
a) Titers of autoantibodiesweremeasured by radioimmunoassay. The limits of sensitivity are 4U/mL for GADAand1U/mL for IAA.
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752768
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

In aggregate, we showed that the GAD-DEF chimeraallows FACS detection of high-avidity (antigen-experi-enced) but not low-avidity autoreactive T cells. Conse-quently, our results indicated that antigen encounteringand avidity maturation of GAD65 T cells occurs early inlife of humans expressing HLA-DR*0401 molecules,
since high-avidity T cells were commonly present inblood of very young autoantibody-negative relatives(Fig. 4B, Table 1). The presence of high-avidity GAD65T cells, preceding the appearance of autoantibodies,may predispose but does not suffice for progression todiabetes.
Fig. 5. Frequency of GAD65-specific T cells in blood: Longitudinal analysis. Frequency of GAD65-specific T cells in blood of(A) patients with T1D, (B) relatives of young age, and (C) adult relatives, as measured by FACS. Values represent means � SD ofGAD65 T cells. Titers of GADA are indicated over the plots.
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2769
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

The “hit-and-run” kinetics of GAD65 T cellsin blood
The longitudinal analyses in patients with T1D showedthat the high-avidity GAD65 T cells followed a kineticscharacterized in some cases by their transient disap-pearance from the blood (Fig. 5A). The same T cellkinetics was detected in blood of the majority (7 out of8) of the young relatives (Fig. 5B). Of note, in one ofthese young relatives, the onset of diabetes occurredwhen the high-avidity GAD65 T cells had disappearedfrom the blood (Fig. 5B; r9). This case explains the cross-sectional data showing the absence of high-avidityGAD65 T cells in patients at the clinical onset of thedisease (Fig. 4B), and further supports the concept thatthe autoimmune attack on the pancreas is a “hit-and-run” process mediated by the recruitment of autoreac-tive T cells from the blood.
The inverse relationship between the GAD65 T cellsand GAD65 autoantibodies found in the cross-sectionalanalyses (Fig. 4C, upper panel) was also consistent inthe longitudinal analyses. In two patients (Fig. 5A; p3,p6) and one autoantibody-positive relative (Fig. 5B; r9),the decrease in the frequency of GAD65 T cells wasassociated with higher anti-GAD65 antibody titers.
Within the group of adult relatives, the multiple “hit”T cell kinetics was only detected in 4 out of 11individuals (36.3%). The majority of them (7 out of11, 63.6%) showed an absence of high-avidity GAD65-specific T cells in longitudinal analysis (Fig. 5C). Thelatter is in agreement with NOD studies indicating thathigh-avidity autoreactive T cells were absent in blood ofnon-progressor mice while these T cells were detected inblood of mice that progressed toward diabetes [7]. Thus,our data showing the presence of high-avidity GAD65T cells in young relatives but their absence in themajority of the older ones suggested that a mechanismfor disease resistance may associate with the long-termdisappearance of high-avidity GAD65 T cells, presum-ably by cessation of the autoimmune process in thepancreas.
The phenotype of GAD65 T cells: a biomarkerfor T1D
While the kinetics of GAD65 T cells in blood did notallow distinguishing between patients with T1D andtheir young relatives, the phenotype of these T cells wasclearly different for both groups. Patients with T1Dshowed a higher frequency of CD69+GAD65 thanCD25+GAD65 T cells as opposed to their non-diabeticrelatives and controls that showed a higher frequency ofCD25+GAD65 T cells (Fig. 6A, upper panel, B). Themajority of high-avidity GAD65 T cells expressed eitherCD69 or CD25 (Fig. 6C), delineating two populations of
CD69+CD25– and CD69–CD25+ T cells specific forGAD65. In contrast to the phenotypic differences inthe GAD65 T cells, those T cells with specificity otherthan GAD65 peptide (CD3+GAD65–) had, however, asimilar phenotype in diabetic patients, their relatives,and controls (Fig. 6A, lower panel). In longitudinalanalysis, the GAD65 T cells in blood of patients with T1Dalso showed persistent higher expression of CD69(Fig. 7A), whereas in their autoantibody-negativerelatives, the GAD65 T cells showed a persistent higherexpression of CD25 (Fig. 7B). Together, these resultsindicated that T1D is associated with an imbalancebetween CD4+CD25+ and CD4+CD69+ T cells specificfor GAD65.
The phenotypic imbalance in the GAD65 T cellsprecedes the disease onset: a case report
Prior to the clinical onset of T1D (4 months), a youngautoantibody-positive relative developed an imbalancein the CD4+CD25+ and CD4+CD69+ T cells specific forGAD65 T cells (Fig. 7C). As indicated earlier, the clinicalonset occurred when the high-avidity GAD65 T cells haddisappeared from the blood (Fig. 5B; r9). This isconsistent with our animal studies indicating that oncethe autoreactive T cell imbalance has occurred, therecruitment of autoreactive CD4+CD69+ T cells fromthe blood into the pancreas is a major mechanismleading to the disease onset [8]. This 10-year-old relativewas autoantibody positive since the age of 2 years, andhence the phenotype imbalance in the GAD65 T cellswas a closer marker for the disease onset than theautoantibodies.
The level of glycosylated hemoglobin (HbA1c) inerythrocytes is directly proportional to the concentra-tion of glucose in blood, and it is used as a marker forlong-term blood glucose control. At the time the GAD65T cell imbalance occurred, the HbA1c was in the normalrange (5.5%). However 4 months later, at the onset ofT1D, the HbA1c was significantly increased (8.1%),suggesting a relationship between the phenotypicimbalance in the GAD65 T cells and a period of reducedb-cell function and glucose intolerance. Thus, our resultsstrongly suggest that disease progression can bemonitored through the phenotype of GAD65 T cells inblood.
Discussion
We have previously shown that a murine DEF chimeraexhibits high stability and long life in serum, i.e. 50 h[18], and immunoregulates peptide-specific CD4 T cellstoward an IL-10-secreting Tr1-like phenotype by nega-tive regulation of the STAT4 pathway involved in Th1
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752770
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

differentiation [4, 10, 11]. Furthermore, the mouse DEFprevented and reversed diabetes [4]. Here, we haveshown that human DEF chimeras are also endowed withimmonoregulatory properties, since they stimulateantigen-specific IL-10-secreting Tr1-like cells. The Tr1cells were first described by Groux et al. [19] as aregulatory T cell subset able to suppress autoimmunecolitis. Regardless of their antigenic specificity, the Tr1cells suppress local T and B cell responses through thesecretion of IL-10 and inhibitory cell-cell interactions[19–22]. IL-10 induces a profound and long-lastinganergy by a dual mechanism: suppression of cytokineproduction and proliferation of T cells [21] and down-regulation of costimulatory molecules on APC [22]. Thebeneficial effect of Tr1 cells in Th1-mediated auto-immune diseases has been demonstrated in clinicaltrials. Thus, treatment with INF-b in multiple sclerosisincreased IL-10 production which favorably altered thecourse of disease [23]. The ability of human DEFchimeras to stimulate IL-10-secreting Tr1-like cellsmakes them a potential therapeutic in T1D.
Furthermore, the IL-10 response upon stimulation ofT cells with DEF in vitro proved to be a marker for thepresence of autoreactive CD4 T cells in blood. We foundthatautoreactiveCD4T cells toGAD65andpro-insulinarecommonly present in the blood of those expressing HLA-DR*0401molecules.This is consistentwithanimal studiesindicating that the expression of diabetes-related MHCclass II molecules, but not background genes, suffices forthe positive selection of autoreactive thymic T cellprecursors and their export to the periphery [24, 25].
The DEF chimeras also allowed FACS quantificationof high-avidity GAD65 T cells in non-stimulated blood.Standard biotin-streptavidin MHC class II-peptide tetra-mers do not bind well to CD4 [26], and although theyhave four potential binding sites to interact with TCR,their rigid structure allows only a bivalent binding to asingle T cell [27]. The lower avidity of tetramers forT cells may explain why these reagents could not detectautoreactive T cells in non-stimulated blood [28].
Studies derived from T cell workshops reportedfrequencies of autoreactive T cell precursors in the
Fig. 6. The phenotype of GAD65 T cells in blood of patients with T1D and non-diabetic controls. (A) Percentage of CD69 and CD25expression on GAD65-specific T cells (GAD+CD3+, upper panel). (*) Phenotype of re-emerging high-avidity GAD65 T cells. Valuesrepresent means � SD. The phenotype of T cells with antigenic specificity other than GAD65271–285 (GAD65–CD3+) is shown in thelower panel. (B) FACShistograms showing expression of CD69 andCD25 onGAD+CD3+ T cells in PBMCof a patientwith T1D (upperpanels) and a non-diabetic relative (lower panels). (C) Co-stainingwith CD25-allophycocyanin and CD69-PE revealed two differentpopulations of CD25+CD69– and CD25–CD69+ GAD65-specific T cells.
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2771
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

range of 10–125/105, although these numbers might beunderestimated due to the presence of regulatory T cellsthat exert in vitro T cell suppression [29–35]. UsingFACS analysis, we found slightly higher frequencies forhigh-avidity GAD65 T cells (40–600/105). The fre-quency of GAD65 T cells negatively correlated withGADA autoantibodies, supporting the original observa-tion by Harrison et al. [36] using in vitro T cell assays.Autoantibodies are not pathogenic [37, 38], but theirpresence in blood is a reporter for ongoing autoimmu-nity [39]. Therefore, the inverse relationship betweenT cell and humoral responses in blood needs to becarefully analyzed. Using a mouse model of T1D, weshowed that peripheral blood is a major reservoir forautoreactive T cells and that the autoimmune attack inthe pancreas is a “hit-and-run” process mediated by thepancreatic recruitment of T cells from blood [8].Consequently, the disappearance of autoreactive T cellsfrom the blood as they are recruited to the pancreas
(“the hit”) may well result in autoreactive B cellactivation and autoantibody production. This mechan-ism could explain the inverse kinetics between thefrequency of GAD65 T cells and anti-GAD65 antibodiesin the blood. No relationship was found between theGAD65 T cells and IAA antibodies in patients with T1Dthat may account for disturbances in the anti-insulinhumoral responses in the face of insulin therapy.
Our studies also indicated that autoreactive CD4T cells undergo avidity maturation upon encounteringthe antigen early in life, since high-avidity T cells weredetected in blood of very young autoantibody-negativerelatives. The partitioning of TCR and CD4 in the plasmamembrane lipid rafts determines the avidity of T cells forMHC-peptide complexes. On the naive and restingT cells, CD4 is located in lipid rafts, whereas CD3/TCRcomplexes are mostly located in non-lipid rafts [40, 41].After interaction with antigen, the CD3/TCR complextranslocates into lipid rafts [42] and the physical
Fig. 7. The phenotype of GAD65 T cells in blood: a biomarker for disease progression. CD69 and CD25 expression on high-avidityGAD65-specific T cells in blood of (A) patients with T1D and (B) autoantibody-negative relatives. (C) A phenotypic imbalance inGAD65-specific T cells was detected in a young relative before the clinical onset of T1D. (D) A proposed model for explaining theimmunokinetics of GAD65 CD4 T cells in blood during the natural history of T1D.
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752772
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

proximity of CD3/TCR and CD4 molecules increases theavidity of T cells for the MHC-peptide complexes. Thisprocess, known as “avidity maturation” of T cells [15], isa keyevent required but not sufficient for the progressiontoward diabetes, as demonstrated in NODmice [16]. Theinitial source of b-cell antigens for avidity maturation ofT cells could be derived from a physiological wave of b-cell apoptosis that occurs in humans shortly after birth[43], similarly to what was found in NOD mice [44].
The presence of high-avidity GAD65 T cells in bloodof those expressing diabetes-susceptible HLA-DR*0401molecules may predispose, but may not suffice, forprogression toward diabetes. Like we previously foundin mice [8], T1D in humans is preceded by an imbalancebetween autoreactive CD4+CD25+ and CD4+CD69+
T cells. Regulatory CD4+CD25+ T cells are critical forthe physiological control of autoimmunity, and theirextinction associates with the development of auto-immune diseases, including T1D [45]. In humans,CD4+CD25+ T cells in peripheral blood are regulatoryas well, since they suppress the in vitro expansion ofautoreactive T cells [29, 32]. The mechanisms under-lying the extinction of CD4+CD25+ T cells and diseaseprogression are currently unknown.
A proposed model that may explain the kinetics ofautoreactive CD4 T cells in blood during the naturalhistory of T1D is illustrated in Fig. 7D. Antigenencountering and avidity maturation of autoreactiveT cells occurs early in life, presumably driven byphysiological events such as pancreatic remodelingand b-cell apoptosis. The autoimmune process issustained by waves of pancreatic T cell recruitment(“the hit”), a process under the control of regulatoryCD4+CD25+ T cells. The hallmark for disease progres-sion is the imbalance between regulatory CD4+CD25+
and CD4+CD69+ T cells (Fig. 7, upper panel). Environ-mental factors, i.e. viruses, foods, and drugs, may act asdisease triggers by inducing disturbances between theautoreactive T cell subsets. On the other hand, over-coming danger signals and immune dysregulation lead,over time, to the cessation of the autoimmune processmanifested by a long-term disappearance of high-avidityautoreactive T cells in blood (Fig. 7, lower panel).
In summary, we showed that DEF chimeras aresuitable reagents for FACS identification as well asimmunoregulation of autoreactive CD4 T cells. Accord-ingly, this approach may potentially serve for both earlydiagnosis and immunospecific therapy in T1D.
Materials and methods
DEF chimeras
The GAD-DEF and pINS-DEF chimeras are made of theextracellular domains of HLA-DR*0401 dimerized through
human Ig-Fcc1 (hFcc1). The human GAD65271–285 peptide, orthe pro-insulin (pINS)73–90 peptide, was covalently linked atthe N terminus of the B1*0401 chain. The genetic engineeringof human DEF used a strategy similar to ours previouslydescribed for mouse DEF [3]. Genes encoding for HLA-DRB1*0401 and hinge-Fcc1 were obtained by RT-PCR fromBoleth cells and human IgG1 hybridoma, ZM-1 (ATCC),respectively. Leader/peptide/linker/DRB1/Fcc1 gene wascloned under the polH promoter of the p2Bac vector(Invitrogen, San Diego, CA). The gene coding for theextracellular domain of DRA*0101 was cloned under thep10 promoter of P2Bac. Recombinant baculoviruses wereobtained as described [3]. The DEF chimeras were produced inbaculovirus-infected insect SF9 cells and purified from the cellculture supernatant by affinity chromatography using anti-human Igc1-Sepharose 4B columns. The soluble, dimeric DEFhave a molecular size of 185,000 kDa and they wererecognized by HLA-DR and hIgG1 antibodies in Western blots(data not shown).
Human subjects, HLA-DR typing, and autoantibodies
Patients with T1D and their first-degree relatives (Table 1)were recruited by informed consent at the Diabetes Centers,Children's Hospitals and Clinics, St. Paul, MN, andMount SinaiHospital, New York, NY. Assent consent was also obtained forpediatric subjects. HLA-DR*0401 non-diabetic controls withno family history of T1D were recruited at the Memorial BloodBank, Children's Hospitals and Clinics, St. Paul, MN. For HLA-DR typing, we used Dynal Reli SSO HLA-DRB and Classic SSPDRB1*04 kits (Dynal Biotech, Lafayette Hill, PA). Autoanti-bodies were determined by radioimmunoassay kits (Kronus,Boise, ID).
T cell responses
PBMC (2 � 105) in RPMI supplemented with 10% heat-inactivated human AB serum (Sigma Chemicals) were culturedwith antigens for 4 days. T cell proliferative response wasmeasured by thymidine incorporation assay (3[H]TdR) asdescribed [4]. For inhibitory experiments, anti-hCD4, anti-HLA-DR, anti-rat IgG (isotype), anti-human IgG3 or anti-human IL-10 (100 lg/mL) antibodies or monensin (2 lg/mL)(PharMingen) were added to cultures. Quantification ofcytokines was carried out by ELISA kits (Biosource Int.,Camarillo, CA). Depletion of T cells in PBMC was carried outusing anti-human CD3 antibody-coated magnetic beads,following the manufacturer's instructions (Dynal Biotech).The efficiency of T cell depletion was assessed by FACS usinganti CD3-FITC antibodies (PharMingen), and those samplesshowing more than 95% CD3 T cell depletion were used forfurther experiments.
T cell clones and T cell hybridomas (TcH)
PBMCwere cultured with GAD-DEF or pINS-DEF (3 lg/mL) inthe presence of human IL-15 (50 ng/mL) (PharMingen) for12 days. Cells were restimulated with synthetic peptides(10 lg/mL) in the presence of irradiated syngeneic PBMC.The hCD4 gene was cloned under the CMV promoter of the
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2773
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

pcDNA3 vector (Invitrogen). The 33.1 TcH was transfectedwith hCD4/pcDNA3 or pcDNA3 vector using DMRIE-C(Invitrogen). Cells were harvested 24 h after transfectionand analyzed by FACS for binding to DEF.
Flow cytometry
PBMC in PBS/1% BSA/0.1% sodium azide were incubatedwith DEF chimeras (15 lg/mL) for 1 h at room temperature,and anti-human CD3-PerCP antibody (PharMingen) wasadded for 30 min at 4�C. Cells were washed with PBS/1%BSA/0.1% sodium azide and incubated with goat F(ab')2 anti-human IgG-FITC (Southern Biotech.) for 30 min at 4�C. Tophenotype T cells in blood, PBMC were incubated with DEFchimeras as described, followed by the addition of anti-humanCD3-PerCP antibody, and either anti-human CD69-PE or anti-human CD25-PE antibodies (PharMingen). Stainings werecarried out in quintuplets. For each quintuplet, 500,000 eventswere analyzed in the gated mononuclear fraction. For DEFbinding to Fc receptors, PBMC were incubated in non-supplemented DMEM with 100 U/mL recombinant humanINF-c for 24 h. Cells were stained with DEF chimeras or anti-human IgG1 (20 lg/mL) and F(ab')2 rat anti-human IgG-FITC.Binding to FcR was analyzed in the gated monocyte cellpopulation based on forward scatter/side scatter (FSC/SSC).
Statistical analysis
Correlations were analyzed using simple regression test. Formultiple comparisons, the Kolmogorov-Smirnov test was used.
Acknowledgements: This work was supported by JDRF(1–2002–66, 1–2005–1151) and NIH/DK66421 grants toS.C., and NIH/DK61927 to T.D.B. We thank Dr. LindaWicker at the University of Cambridge for providing the33.1 TcH.
References
1 Todd, J. A. and Wicker, L. S., Genetic protection from the inflammatorydisease type 1 diabetes in humans and animal models. Immunity 2001. 15:387–395.
2 Schwartz, R. H., A cell culture model for T lymphocyte clonal anergy.Science 1990. 248: 1349–1356.
3 Casares, S., Bona, C. A. and Brumeanu, T. D., Engineering andcharacterization of a murine MHC class II-immunoglobulin chimeraexpressing an immunodominant CD4 T viral epitope. Protein Eng. 1997.10: 1295–1301.
4 Casares, S., Hurtado, A., McEvoy, R. C., Sarukhan, A., von Boehmer, H.and Brumeanu, T. D., Down-regulation of diabetogenic CD4+ T cells by asoluble, dimeric peptide-MHC class II chimera. Nat. Immunol 2002. 3:383–391.
5 Masteller, E. L., Warner, M. R., Ferlin, W., Judkowski, V., Wilson, D.,Glaichenhaus, N. and Bluestone, J. A., Peptide-MHC class II dimers astherapeutics to modulate antigen-specific T cell responses in autoimmunediabetes. J. Immunol. 2003. 171: 5587–5595.
6 Zuo, L., Cullen, C. M., DeLay, M. L., Thornton, S., Myers, L. K., Rosloniec,E. F., Boivin, G. P. and Hirsch, R., A single-chain class II MHC-IgG3 fusionprotein inhibits autoimmune arthritis by induction of antigen-specifichyporesponsiveness. J. Immunol. 2002. 168: 2554–2559.
7 Trudeau, J. D., Kelly-Smith, C., Verchere, C. B., Elliott, J. F., Dutz, J. P.,Finegood, D. T., Santamaria, P. and Tan, R., Prediction of spontaneousautoimmune diabetes in NOD mice by quantification of autoreactive T cellsin peripheral blood. J. Clin. Invest. 2003. 111: 217–223.
8 George, S. K., Preda, I., Avagyan, S., McEvoy, R. C., Rapaport, R.,Brumeanu, T. D., Casares S., Immunokinetics of autoreactive CD4 T cells inblood: a reporter for the “hit-and-run” autoimmune attack on pancreas anddiabetes progression. J. Autoimmun. 2004. 23: 151–160.
9 Gebe, J. A., Falk, B. A., Rock, K. A., Kochik, S. A., Heninger, A. K.,Reijonen, H., Kwok, W. W. and Nepom, G. T., Low-avidity recognition byCD4+ T cells directed to self-antigens. Eur. J. Immunol. 2003. 33: 1409–1417.
10 Casares, S., Zong, C. S., Radu, D. L., Miller, A., Bona, C. A. andBrumeanu, T.D., Antigen-specific signaling by a soluble, dimeric peptide/major histocompatibility complex class II/Fc chimera leading toT helper celltype 2 differentiation. J. Exp. Med. 1999. 190: 543–553.
11 Thomas, S., Kumar, R., Preda-Pais, A., Casares, S. and Brumeanu, T. D.,A model for antigen-specific T-cell anergy: displacement of CD4-p56(lck)signalosome from the lipid rafts by a soluble, dimeric peptide-MHC class IIchimera. J. Immunol. 2003. 170: 5981–5992.
12 Wicker, L. S., Chen, S. L., Nepom, G. T., Elliott, J. F., Freed, D. C., Bansal,A., Zheng, S. et al., Naturally processed T cell epitopes from humanglutamic acid decarboxylase identified using mice transgenic for the type 1diabetes-associated human MHC class II allele DRB1*0401. J. Clin. Invest.1996. 98: 2597–2603.
13 Jefferis, R. and Lund, J., Interaction sites on human IgG-Fc for FcgammaR:current models. Immunol. Lett. 2002. 82: 57–65.
14 Roncarolo, M. G., Bacchetta, R., Bordignon, C., Narula, S., Levings, M.K., Type 1 T regulatory cells. Immunol. Rev. 2001. 182: 68–79.
15 Fahmy, T. M., Bieler, J. G., Edidin, M. and Schneck, P., Increased TCRavidity after T cell activation: a mechanism for sensing low-density antigen.Immunity 2001. 14: 135–143.
16 Amrani, A., Verdaguer, J., Serra, P., Tafuro, S., Tan, R. and Santamaria,P., Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 2000. 406: 739–742.
17 Deenick, E. K., Gett, A. V. and Hodgkin, P. D., Stochastic model of T cellproliferation: a calculus revealing IL-2 regulation of precursor frequencies,cell cycle time, and survival. J. Immunol 2003. 170: 4963–4972.
18 Brumeanu, T. D., Bona, C. A. and Casares, S., T-cell tolerance andautoimmune diabetes. Int. Rev. Immunol. 2001 . 20: 301–331.
19 Groux, H., O'Garra, A., Bigler, M., Rouleau, M., Antoneuko, S., de Vries,J. E. and Roncarolo, M. G., A CD4+ T cell subset inhibits antigen-specificT cell responses and prevents colitis. Nature 1997. 389: 737–742.
20 Levings, M. K., Sangregorio, R., Galbiati, F., Squadrone, S., de WaalMalefyt, R. and Roncarolo, M. G., INF-a and IL-10 induce thedifferentiation of human type 1 T regulatory cells. J. Immunol 2001.166: 5530–5539.
21 Groux, H., Bigler, M., de Vries, J. E. and Roncarolo, M. G., Interleukin-10induces a long-term antigen-specific anergic stage in human CD4 T cells. J.Exp. Med. 1996. 184: 19–29.
22 Cavani, A., Nasorri, F., Prezzi, C., Sebastiani, S., Albanesi, C. andGirolomono, G., Human CD4+ T lymphocytes with remarkable regulatoryfunctions on dendritic cells and nickel-specific Th1 immune responses. J.Invest. Dermatol. 2000. 114: 295–302.
23 Ozenci, V., Kouwenhoven, M., Huang, Y. M., Kivisak, P. and Link, H.,Multiple sclerosis is associated with an imbalance between tumour necrosisfactor-alpha (TNF-alpha)- and IL-10-secreting blood cells that is corrected byinterferon-beta (IFN-beta) treatment. Clin. Exp. Immunol. 2000. 20:147–153.
24 Stratmann, T., Martin-Orozco, N., Mallet-Designe, V., Poirot, L.,McGavern, D., Losyev, G., Dobbs, C. M. et al., Susceptible MHC alleles,not background genes, select an autoimmune T cell reactivity. J. Clin.Invest. 2003. 112: 902–914.
25 Jang, M. H., Seth, N. P. and Wucherpfennig, K. W., Ex vivo analysis ofthymic CD4 T cells in nonobese diabetic mice with tetramers generatedfrom I-A(g7)/class II-associated invariant chain peptide precursors. J.Immunol. 2003. 171: 4175–4186.
Ioana Preda et al. Eur. J. Immunol. 2005. 35: 2762–27752774
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de

26 Crawford, F., Kozono, H., White, J., Marrack, P. and Kappler, J.,Detection of antigen-specific T cells with multivalent soluble class II MHCcovalent peptide chimeras. Immunity 1998. 8: 675–682.
27 Hackett, C. J., Sharma, O. K., Frontiers in peptide-MHC class II multimertechnology. Nat. Immunol. 2002. 3: 887–889.
28 Reijonen, H., Novak, E. J., Kochik, S., Heninger, A., Liu, A. W., Kwok, W.W. and Nepom, G. T., Detection of GAD65-specific T-cells by majorhistocompatibility complex class II tetramers in type 1 diabetic patients andat-risk subjects. Diabetes 2002. 51: 1375–1382.
29 Danke, N. A., Koelle, D. M., Yee, C., Beheray, S. and Kwok, W. W.,Autoreactive T cells in healthy individuals. J. Immunol. 2004. 72:5967–5972.
30 Schloot, N. C., Meierhoff, G., Karlsson Faresjo, M., Ott, P., Putnam, A.,Lehmann, P., Gottlieb, P. et al., Comparison of cytokine ELISpot assayformats for the detection of islet antigen autoreactive T cells. Report for thethird immunology of diabetes society T-cell workshop. J. Autoimmun.2003. 21: 365–376.
31 Naik, R. G., Beckers, C., Wentwoord, R., Frenken, A., Duinkerken, G.,Brooks-Worrell, B., Schloot, N. C., Palmer, J. P. and Roep, B. O.,Precursor frequency of T-cells reactive to insulin in recent onset type 1diabetes mellitus. J. Autoimmun. 2004. 23: 55–61.
32 Arif, S., Tree, T. I., Astill, T. P., Tremble, J. M., Bishop, A. J., Dayan, C. M.,Roep, B. O. and Peakman, M., Autoreactive T cell responses showproinflammatory polarization in diabetes but a regulatory phenotype inhealth. J. Clin. Invest. 2004. 113: 451–463.
33 Atkinson, M. A., Response of peripheral-blood mononuclear cells toglutamate decarboxylase in insulin-dependent diabetes. Lancet 1992. 339:458–459.
34 Alleva, D. G., Crowe, P. D., Jin, L., Kwok, W. W., Ling, N., Gottschalk, M.,Coulou, P. J. et al., A disease-associated cellular immune response in type 1diabetics to an immunodominant epitope of insulin. J. Clin. Invest. 2001.107: 173–180.
35 Viglietta, V., Kent, S. C., Orban, T. and Hafler, D. A., GAD65-reactiveT cells are activated in patients with autoimmune type 1a diabetes. J. Clin.Invest. 2002. 109: 895–903.
36 Harrison, L. C., Honeyman, M. C., DeAizpurua, H. J., Schmidli, R. S.,Colman, P. G., Tait, B. D. and Cram, D. S., Inverse relation betweenhumoral and cellular immunity to glutamic acid decarboxylase in subjects atrisk of insulin-dependent diabetes. Lancet 1993. 329: 1365–1369.
37 Bendelac, A., Boitard, C., Bedossa, P., Bazin, H., Bach, J. F. and Carnaud,C., Adoptive transfer does not require recruitment of host B lymphocytes. J.Immunol. 1988. 8: 2625–2628.
38 Martin, S., Wolf-Eichbaum, D., Duinkerken, G., Scherbaum, W. A., Kolb,H., Noordzij, J. G. and Roep, B. O., Development of type 1 diabetes despitesevere hereditary B-cell deficiency. N. Engl. J. Med. 2001. 345: 1036–1040.
39 Devendra, D., Yu, L. and Eisenbarth, G. S., Endocrine autoantibodies. Clin.Lab. Med. 2004. 24: 275–303.
40 Parolini, I., Sargiacomo, M., Lisanti, M. P. and Peschle, C., Signaltransduction and glycophosphatidylinositol-linked proteins (lyn, lck, CD4,CD45, G proteins, and CD55) selectively localize in Triton-insoluble plasmamembrane domains of human leukemic cell lines and normal granulocytes.Blood 1996. 87: 3783–3794.
41 Janes, P. W., Ley, S. C., Magee, A. I. and Kabouridis, P. S., The role of lipidrafts in T cell antigen receptor (TCR) signaling. Semin. Immunol. 2000. 12:23–34.
42 Simons, K. and Ikonen, E., Functional rafts in cell membranes. Nature1997. 387: 569–572.
43 Kassem, S. A., Ariel, I., Thornton, P. S., Scheimberg, I. and Glaser, B., b-cell proliferation and apoptosis in the developing normal human pancreasand in hyperinsulinism of infancy. Diabetes 2000. 49: 1325–1333.
44 Trudeau, J. D., Dutz, J. P., Arany, E., Hill, D. J., Fieldus, W. E. andFinegood, D. T., Neonatal b-cell apoptosis: a trigger for autoimmunediabetes? Diabetes 2000. 49: 1–7.
45 Bach, J. F., Regulatory T cells under scrutiny. Nat. Rev. Immunol. 2003.3:189–198.
Eur. J. Immunol. 2005. 35: 2762–2775 Clinical immunology 2775
f 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji.de


















