
Slide 1 of 21 Robert Packard, President [email protected] Supplier Auditing & Remote Auditing.
Upload
lesley-perryCategory
view
216download
1
Slide 1 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Combining Product Risk Management &
Design Controls
ISO14971
ISO 13485,
Clause 7.3
Slide 2 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Design Controls
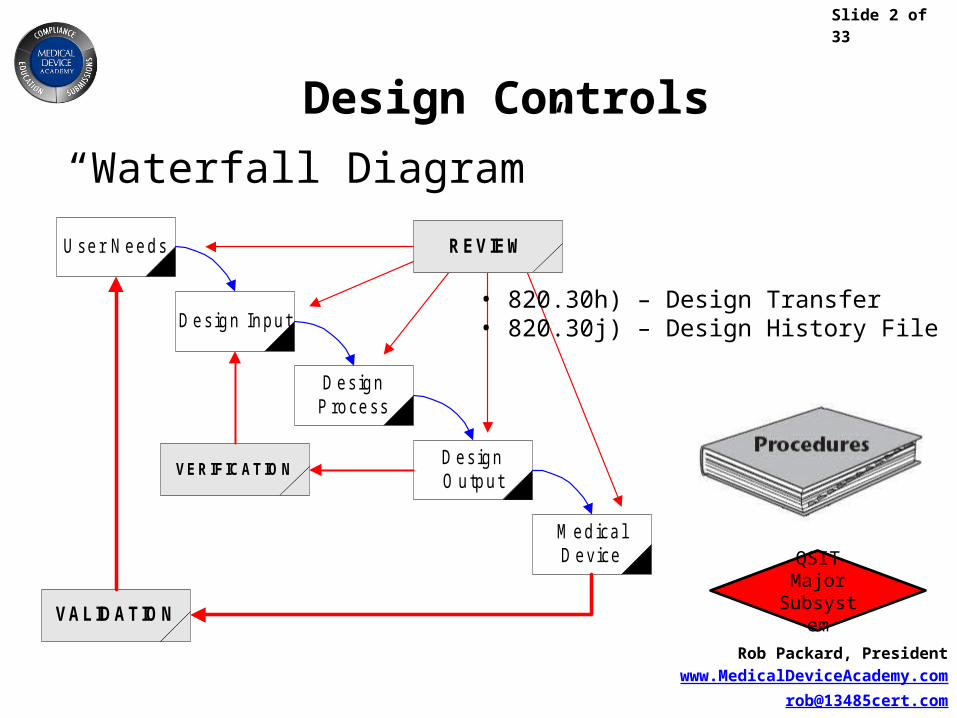
User Needs
Design Input
DesignProcess
DesignO utput
M edica lDevice
VALIDAT IO N
VERIFICAT IO N
REVIEW
“Waterfall Diagram”
• 820.30h) – Design Transfer• 820.30j) – Design History File (DHF)
QSIT Major Subsystem
Slide 3 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
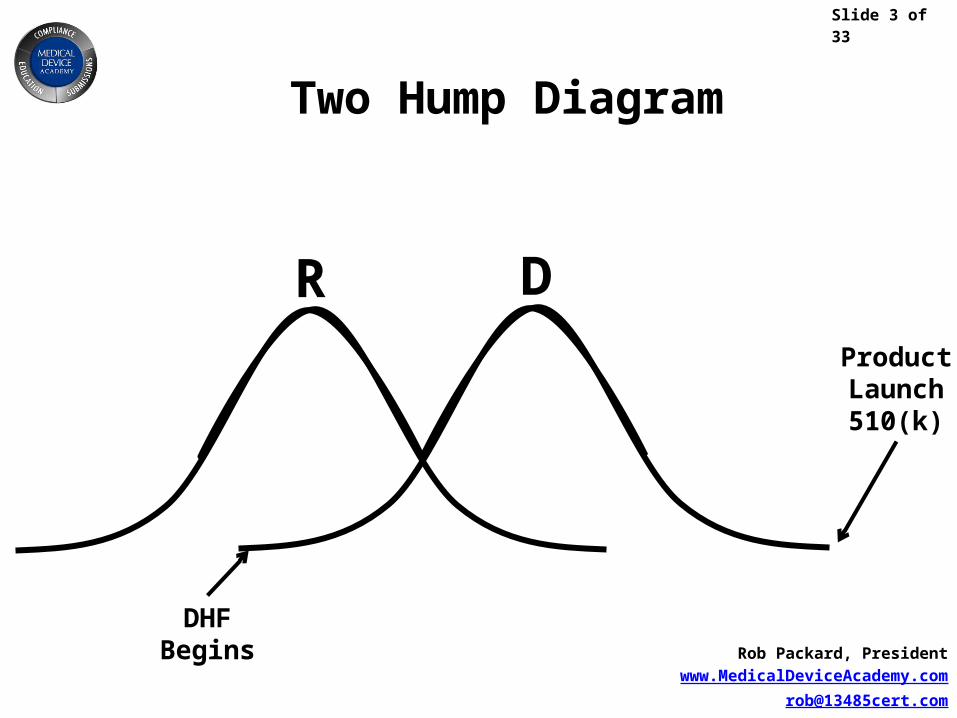
Two Hump Diagram
DHFBegins
ProductLaunch510(k)
DR
Slide 4 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
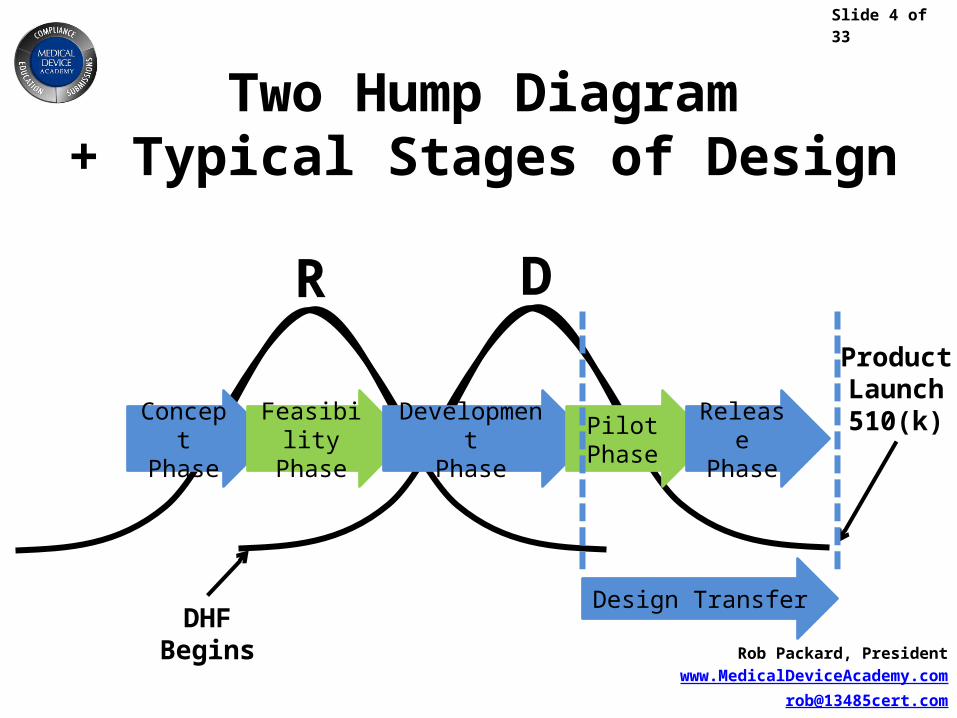
Two Hump Diagram+ Typical Stages of Design
DHFBegins
ProductLaunch510(k)
Design Transfer
ConceptPhase
FeasibilityPhase
DevelopmentPhase
PilotPhase
ReleasePhase
DR

Slide 5 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
When to Start a DHF?• After the design plan has been developed
• Upon approval of the design project
• It’s not uncommon to start a DHF late and to retroactively assemble the file from documents that were already created in team meetings.

Slide 6 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
DHF Contents• Design Plan• User Needs• Design Inputs• Design Outputs – including labeling• Verification Protocols & Reports• Validation Protocols & Reports• Process Validation & work instructions• Design Review Meeting Minutes• IOVV / Design Requirements Matrix• Risk Management File• Initial PMS Plan• Clinical Data Summary and/or Clinical Evaluation Report• Initial DMR/TF Index• Regulatory Approval
Organize Chronologicall
y

Slide 7 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
5 or 6 Phase Gates
1. 7.3.1 – Design Plan Approval
2. 7.3.2 – Design Inputs Approval
3. 7.3.3 – Design Outputs Approval
4. 7.3.5 & 7.3.6 / 1st in Humans Study Approval
5. Pilot Launch Approval
6. Commercial Launch Approval
Each Gate is a Design Review Meeting
Clause 7.4
ISO 13485 Clauses Referenced

Slide 8 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Design Reviews• ISO 13485, Clause 7.3.4• Reviews shall…
– Be systematic and according to design plan– Evaluate ability of design & development to meet
requirements– Identify problems and propose necessary actions– Include participants representing various functions of the
design team from that stage– Results and necessary actions shall be recorded– Include a reviewer not involved in that stage (21 CFR
820.30e)

Slide 9 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Design Planning• ISO 13485, Clause 7.3.1• The plan shall…
– Define stages of design & development– Define the review, verification, validation & transfer
activities for each stage– Define responsibilities & authorities
• Management shall manage interfaces• Document planning output and update as
needed
ConceptPhase

Slide 10 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Design Inputs
User Need Typical Design Input Better Design Input
Biocompatible Material = Teflon ISO 10993-1
Easy to Use Pictorial User Interface IEC 62366-1
Won't Puncture Nylon Reinforced Pouch ISTA 2A
Connects with Monitor RS232 Connection Protocol P123

Slide 11 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Input OutputVerification Validation
item User Needs Design Inputs Design Outputs Verification Test Method Validation Test Method 12345678910
Also called a Design Requirements Matrix

Slide 12 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Management / Design Controls
• Clause 7.3.2e) of ISO 13485 states that Risk management shall be an Input into Design & Development
• Clause 6.3 of ISO 14971 requires verification of effectiveness of risk controls
• Clause 6.7 of ISO 14971 requires verification of completeness of risk controls

Slide 13 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Management FileISO 14971, Clause 3.5 • File for each medical device • The risk management file shall provide
traceability for each identified hazard to:– the risk analysis– the risk evaluation– the implementation and verification of the risk control
measures– the assessment of the acceptability of any residual
risk(s)

Slide 14 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Management Activities Needed
• Risk Management Plan• Hazard Identification• Risk Assessment• Risk Control Option Analysis• Risk Control Effectiveness Verification• Risk / Benefit Analysis• Risk Management Report

Slide 15 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Management PlanISO 14971, Clause 3.4 (See Annex F for Example)• The plan shall include at least the following:
– the scope of the planned risk management activities, identifying and describing the medical device and the life-cycle phases for which each element of the plan is applicable
– assignment of responsibilities and authorities– requirements for review of risk management activities– criteria for risk acceptability, based on the manufacturer's policy for determining acceptable risk,
including criteria for accepting risks when the probability of occurrence of harm cannot be estimated
– verification activities– activities related to collection and review of relevant production and post-production information
• For each risk management plan the manufacturer should choose appropriate risk acceptability criteria
– May implement a matrix indicating which combinations of probability of harm and severity of harm are acceptable or unacceptable
• The risk management plan is part of the risk management file– Record of the changes shall be maintained in the risk management file

Slide 16 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Hazard IdentificationISO 14971, Clause 4.3• Documentation on known and foreseeable
hazards associated with the medical device in both normal and fault conditions
• Maintained in the risk management file• Annex C, Annex E, Previous Risk Analysis & TPLC
Database (http://bit.ly/FDATPLC)

Slide 17 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Assessment ToolsISO 14971, Annex G • Preliminary Hazard Analysis• Fault Tree Analysis (FTA)• Failure Mode & Effects Analysis (FMEA)• Hazard & Operability Study (HAZOP)• Hazard Analysis and Critical Control Point
(HACCP)

Slide 18 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
dFMEA
Function / ItemPotential Failure
ModePotential Failure
Effects
Severity
Probability
of Occurrence
Current Risk Controls
Verification Method
RPN
(
SxO
)
Planned Actions Assigned toUpdated Risk
Controls
Updated S
Updated O
Re-
verification
Method
Updated RPN
Cutting / Spring
Internal spring does not product adequate needle set.
Surgeon not able to cut and remove vitreous in order to allow proper access to retina for repair.
2 3
Specifiction for internal spring force.
Bench testing of parts outside design specification.
6 0
Cutting / Spring
Spring does not allow full closure of cutting port.
Surgeon not able to cut and remove vitreous in order to allow proper access to retina for repair.
2 3
Specification for intenral spring maximum length.
Bench testing of parts outside design specification.
6 0
Cutting / Spring
Spring does not fully open cutting port.
Surgeon not able to cut and remove vitreous in order to allow proper access to retina for repair.
2 3
Specification for intenral spring minimum length.
Bench testing of parts outside design specification.
6 0
Aspiration / Tubing
Vitreous is pulled into cutting port too quickly.
Elevated aspiration rate.
2 3
Specification for aspiration tubing inner diameter.
Bench testing of parts outside design specification.
6 0
Aspiration / O-ring and Tubing
Interface
O-ring does not seal propertly.
Insufficient aspiration rate.
2 3
Specification o-ring and tubing outer diameter.
Bench testing of parts outside design specification.
6 0

Slide 19 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Problems with dFMEA• Bottom-up approach doesn’t facilitate complaint
investigations• RPN scores encourage prioritization of risk
controls that deviates from MDD (EN ISO 14971:2012)
• No traceability to specific IFU Warnings & Precautions
Slide 20 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
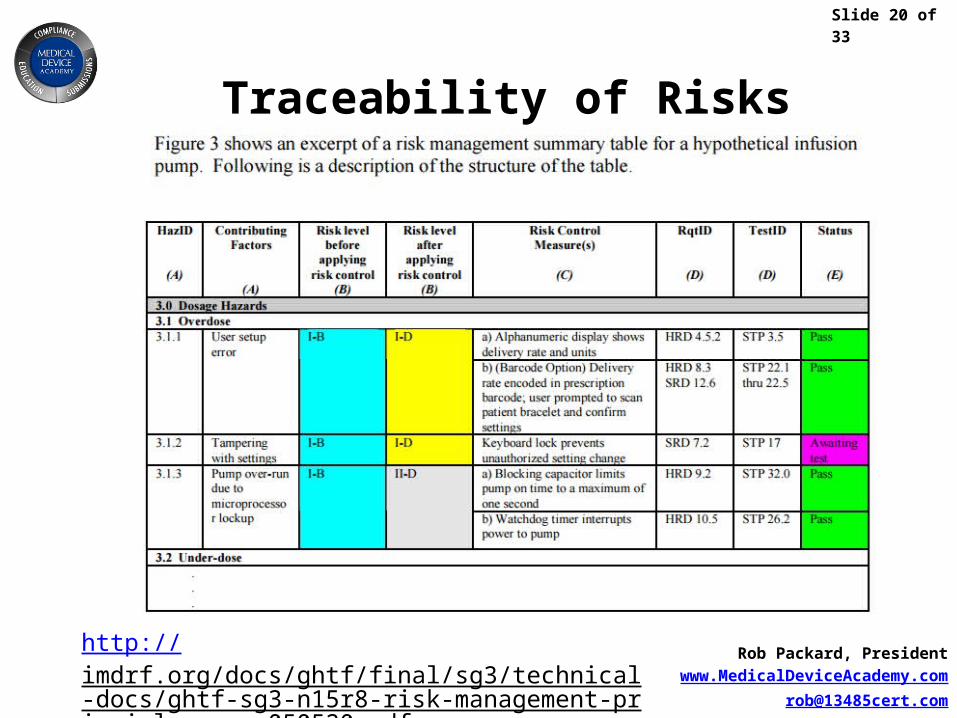
Traceability of Risks
http://imdrf.org/docs/ghtf/final/sg3/technical-docs/ghtf-sg3-n15r8-risk-management-principles-qms-050520.pdf
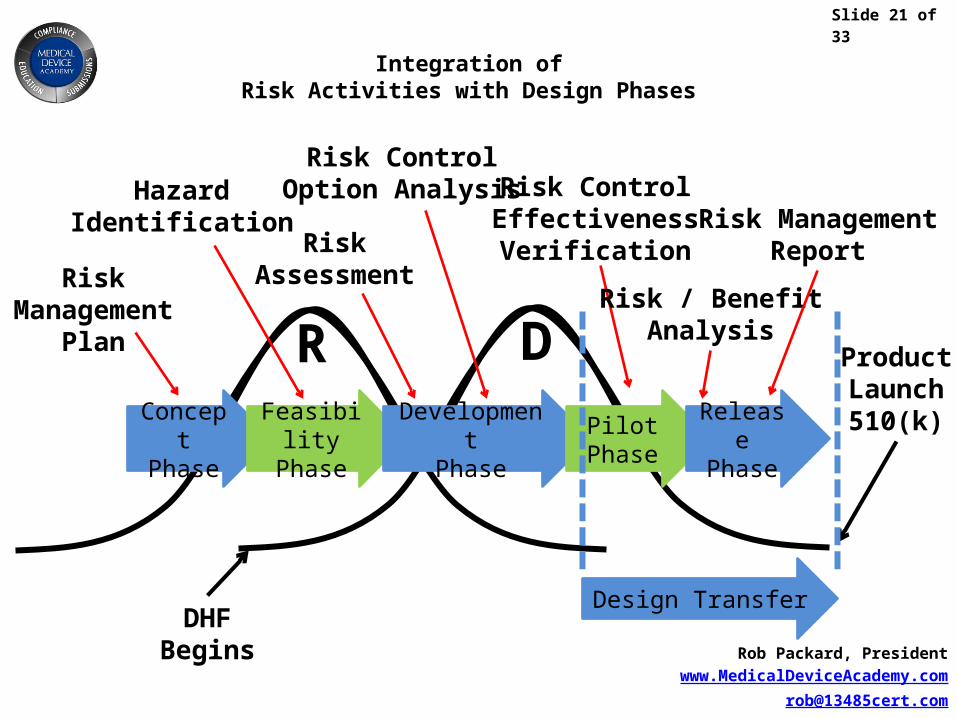
Slide 21 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Integration ofRisk Activities with Design Phases
DHFBegins
ProductLaunch510(k)
Design Transfer
ConceptPhase
FeasibilityPhase
DevelopmentPhase
PilotPhase
ReleasePhase
HazardIdentification
Risk ControlOption Analysis
RiskAssessment
Risk ControlEffectivenessVerification
RiskManagement
Plan
Risk / BenefitAnalysis
Risk ManagementReport
DR
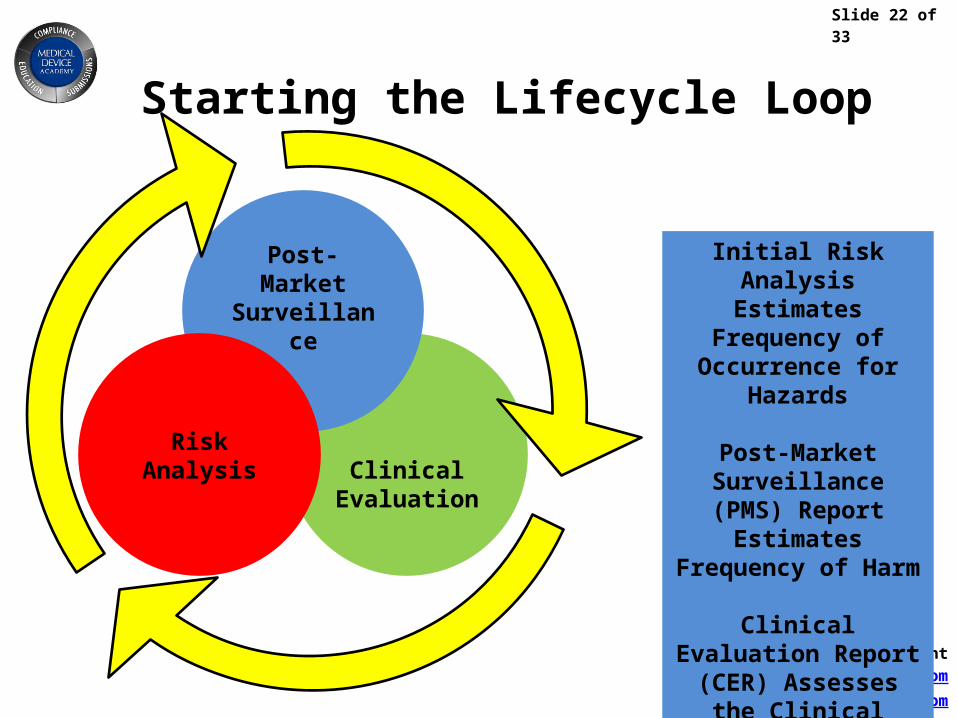
Slide 22 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Starting the Lifecycle Loop
ClinicalEvaluation
Post-Market Surveillance
RiskAnalysis
Initial Risk Analysis Estimates Frequency of Occurrence for Hazards
Post-Market Surveillance (PMS) Report Estimates
Frequency of Harm
Clinical Evaluation Report (CER) Assesses
the Clinical Risk/Benefit of the New Product

Slide 23 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Combined IOVV & Risk Traceability
Item No.
User NeedHazard Identified
Design Input
Risk Control Method(s)
Severity of Harm
Probability of Occurrence (P1)
Design Output
Verification Reference (Protocol/Report)
Validation Reference (Protocol/Report)
Probability of Occurrence Resulting in Harm (P2)
Residual Risk in IFU
PMCF Study (Protocol/Report)
123456789101112
http://medicaldeviceacademy.com/design-control-proceduresforms/

Slide 24 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Initiating Combined Matrix• Perform Hazard Identification• Work Backward to User Needs• Fill in 1st Two Columns• These are the first steps of the design process
that often lead up to creating a Regulatory Pathway Summary Document & Design / Risk Management Plan

Slide 25 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Preparation forApproval of Design Inputs
• Identify Applicable Harmonized Standards• Identify Performance Testing
– Predicate Devices 510(k) Summary– Special Controls Guidance Documents– Search ISO Standards– Are their internal specifications?

Slide 26 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Perform Risk Assessment• Identify Possible Risk Control Methods• Severity of Harm• Probability of Occurrence (P1)• Preliminary Screening against Design Inputs• Reiterate Risk Controls & Design Specifications• Finalize Design Specifications (Approve Outputs)

Slide 27 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
V&V• Verification of Risk Control Effectiveness• Preliminary Estimation of P2 based upon
literature (Need to perform literature search…Clinical Evaluation Report)
• Results of Clinical Evaluation and Risk / Benefit Analysis should determine:– if Clinical Study is Needed– If Post-Market Clinical Follow-up Study is Needed

Slide 28 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Draft IFU• Identify Residual Risks
– Warnings– Precautions– Contraindications
• This is critical for Adverse Events, Investigator Brochures and Litigation of Injuries or Death

Slide 29 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Draft PMS Plan• Post-Market Surveillance (PMS) Plan
– Needs to collect data for any residual risks– Should verify estimated risks– Should identify any missing hazards– Should identify incorrect severity of harm– Should include justification of no PMCF Study or…
• PMCF Protocol• PMCF Report
– Should include updated Risk Management Plan

Slide 30 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Risk Management Report• This is a Summary Technical Document (STED)
that summarizes all the risk management activities and references the document control numbers for the Risk Management Documents
• This should also include the dates of activities and personnel involved
• The frequency of review and update of the risk management documentation should be included

Slide 31 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Other Training Webinars
http://medicaldeviceacademy.com/implementing-risk-management-process-compliant-iso-149712007-address-seven-deviations-identified-en-iso-149712012/
http://medicaldeviceacademy.com/design-controls-implementation-21-cfr-820-30/
User Needs
Design Input
DesignProcess
DesignO utput
M edica lDevice
VALIDAT IO N
VERIFICAT IO N
REVIEW

Slide 32 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Q & A

Slide 33 of 33
Rob Packard, Presidentwww.MedicalDeviceAcademy.com
Do You Need Help with Design Controls or Risk Management?
Rob Packard
+1.802.281.4381
rob13485


















