
Short reports - adc.bmj.com filefollowed by bronchopneumonia. There was lympho-penia, neutropenia,...
5
486 Short reports TABLE Affected relatives of duodenal ulcer patients Relative No. of cases Father 7 Mother 2 Father and mother 1 Father and grandfather 2 Father, grandfather, and uncle 1 Father and 2 uncles 2 Mother, maternal grandfather, and maternal uncle 1 Brother, maternal grandparents, and maternal uncle 1 Brother 1 Sister 1 Maternal grandfather, matemal aunt, and grandmother (father's side) 1 Grandfather 2 2 uncles 1 of familial pattem in the understanding of this disease. A positive family history of peptic ulcer is an important feature and is one of the basic characteristics of a duodenal ulcer in children. Summary The families of 37 children with the established diagnosis of duodenal ulcer were studied to assess the incidence of peptic ulcer in the relatives of these children. 23 children had a positive family history of peptic ulcer: 19 children had one or other parent or sib affected with the disease. Positive family history is an important characteristic of duodenal ulcer in children. REFERENCES Robb, J. D. A., Orszulok, T. J., and Odling-Smee, G. W. (1972). Duodenal ulcers in children. Archives of Disease in Childhood, 47,688. P. PURI* and EDNA BOYD Children's Research Centre, Our Lady's Hospital for Sick Children, Crumlin, Dublin, Eire. *Correspondence to Dr. P. Puri, The Hospital for Sick Children, Great Ormond Street., London WC1N 3JH. Combined immunodeficiency with hyper-y-globulinaemia Infants with major defects in cell-mediated immunity and hypo-y-globulinaemia (severe com- bined immunodeficiency) usually die in the first few years of life from infections unless they receive a successful bone marrow transplant. Nezelof et al. (1964) described an infant with a major defect in cell-mediated immunity and normal serum immunoglobulin levels who died at 16 months from a lung infection. This paper des- cribes a similar infant with absent cell-mediated immunity and hyper-y-globulinaemia in whom we were unable to show any functional antibody. Case report A male was born at term, of normal delivery, birth- weight 3 - 6 kg, after an uneventful first pregnancy. At 10 weeks he presented with failure to thrive (weight) 4-5 kg), loose stools, and intermittent cough soon followed by bronchopneumonia. There was lympho- penia, neutropenia, and eosinophilia (lymphocytes 208/mm3, neutrophils 1440/mm3, eosinophils 800/mm3). Serum immunoglobulins were: IgG 200, IgA 20, IgM 35 mg/100 ml. Pneumonia responded slowly to anti- biotics but lymphopenia persisted (lymphocytes 70 to 389 mm3). Tonsils and lymph nodes were not clini- cally detectable and the thymus was not visible on chest x-ray. At 4 months he was readmitted with fever, loose stools, and continued poor weight gain. Stool culture grew Pseudomonas aeruginosa. Sweat sodium was 11 mmol/l and chloride 12 mmol/l. Jejunal biposy was normal and contained plasma cells. Tests for malabsor- ption were negative. Over the next 8 months he developed several upper respiratory infections, pneumococcal pneumonia, and an intractable oral Candida infection. At 12 months he weighed 8-3 kg and at 14 months was admitted to Northwick Park Hospital for further investigations of his immune status. On examination he was wasted, height and weight being well below the 3rd centile. Lateral x-ray of the nasopharynx did not show the adenoidal pad. No other significant abnormalities were found. Initial laboratory investigations. Haemoglobin 11 - 7 g/dl, neutrophils 7300/mm3, lymphocytes 291 /mm3, eosinophils 1168/mm3. Examination of the bone mar- row was normal apart from absence of iron stores. Serum immunoglobulins in mg/100 ml with 95% normal adult range in parentheses: IgG 400 (646-1484), IgA 41 (73-552), IgM 114 (50-261), IgD 5 units/ml (5-50), IgE 3 units/ml (25-373). Blood group 0, no isohaemag- glutinins detected. There were no 'natural' serum hae- magglutinating antibodies to a pooled polysaccharide antigen prepared from six common commensal sero- types of Esch. coli. Serum precipitins to Haemophilus influenzae, pneumococcus, Staphylococcus aureus, Pseudo- monas aeruginosa, Klebsiella sp., and Candida albicans were not detected. He was immunized with 1 2 ml TAB vaccine (Burroughs Wellcome) and 0 - 5 ml tetanus toxoid (Lister Institute), with a booster after 8 weeks. No antibodies were detected to either immunization up to 2 months later (Widal < 1: 10, H and O, tetanus anti- toxin <1:10 IU/ml). Intradermal injection of 0.1 ml 1% C. albicans (Bencard) failed to produce a reaction within 72 hours. Contact sensitivity to dinitrochloro- benzene (5% in acetone sensitizing dose and challenge with 0 1 % after 3 weeks) was negative. An area on the on 13 March 2019 by guest. Protected by copyright. http://adc.bmj.com/ Arch Dis Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. Downloaded from
Transcript of Short reports - adc.bmj.com filefollowed by bronchopneumonia. There was lympho-penia, neutropenia,...

486 Short reportsTABLE
Affected relatives of duodenal ulcer patients
Relative No. of cases
Father 7Mother 2Father and mother 1Father and grandfather 2Father, grandfather, and uncle 1Father and 2 uncles 2Mother, maternal grandfather, and maternal uncle 1Brother, maternal grandparents, and maternal uncle 1Brother 1Sister 1Maternal grandfather, matemal aunt, and grandmother
(father's side) 1Grandfather 22 uncles 1
of familial pattem in the understanding of thisdisease. A positive family history of peptic ulceris an important feature and is one of the basiccharacteristics of a duodenal ulcer in children.
SummaryThe families of 37 children with the established
diagnosis of duodenal ulcer were studied to assessthe incidence of peptic ulcer in the relatives ofthese children. 23 children had a positive familyhistory of peptic ulcer: 19 children had one or otherparent or sib affected with the disease. Positivefamily history is an important characteristic ofduodenal ulcer in children.
REFERENCES
Robb, J. D. A., Orszulok, T. J., and Odling-Smee, G. W. (1972).Duodenal ulcers in children. Archives of Disease in Childhood,47,688.
P. PURI* and EDNA BOYDChildren's Research Centre, Our Lady's Hospitalfor Sick Children, Crumlin, Dublin, Eire.
*Correspondence to Dr. P. Puri, The Hospital for Sick Children,Great Ormond Street., London WC1N 3JH.
Combined immunodeficiency withhyper-y-globulinaemia
Infants with major defects in cell-mediatedimmunity and hypo-y-globulinaemia (severe com-bined immunodeficiency) usually die in the firstfew years of life from infections unless they receivea successful bone marrow transplant. Nezelofet al. (1964) described an infant with a majordefect in cell-mediated immunity and normal
serum immunoglobulin levels who died at 16months from a lung infection. This paper des-cribes a similar infant with absent cell-mediatedimmunity and hyper-y-globulinaemia in whom wewere unable to show any functional antibody.
Case reportA male was born at term, of normal delivery, birth-
weight 3 - 6 kg, after an uneventful first pregnancy. At10 weeks he presented with failure to thrive (weight)4-5 kg), loose stools, and intermittent cough soonfollowed by bronchopneumonia. There was lympho-penia, neutropenia, and eosinophilia (lymphocytes208/mm3, neutrophils 1440/mm3, eosinophils 800/mm3).Serum immunoglobulins were: IgG 200, IgA 20, IgM35 mg/100 ml. Pneumonia responded slowly to anti-biotics but lymphopenia persisted (lymphocytes 70 to389 mm3). Tonsils and lymph nodes were not clini-cally detectable and the thymus was not visible on chestx-ray. At 4 months he was readmitted with fever, loosestools, and continued poor weight gain. Stool culturegrew Pseudomonas aeruginosa. Sweat sodium was 11mmol/l and chloride 12 mmol/l. Jejunal biposy wasnormal and contained plasma cells. Tests for malabsor-ption were negative.
Over the next 8 months he developed several upperrespiratory infections, pneumococcal pneumonia, andan intractable oral Candida infection. At 12 months heweighed 8-3 kg and at 14 months was admitted toNorthwick Park Hospital for further investigations ofhis immune status. On examination he was wasted,height and weight being well below the 3rd centile.Lateral x-ray of the nasopharynx did not show theadenoidal pad. No other significant abnormalitieswere found.
Initial laboratory investigations. Haemoglobin11 - 7 g/dl, neutrophils 7300/mm3, lymphocytes 291 /mm3,eosinophils 1168/mm3. Examination of the bone mar-row was normal apart from absence of iron stores.Serum immunoglobulins in mg/100 ml with 95%normal adult range in parentheses: IgG 400 (646-1484),IgA 41 (73-552), IgM 114 (50-261), IgD 5 units/ml (5-50),IgE 3 units/ml (25-373). Blood group 0, no isohaemag-glutinins detected. There were no 'natural' serum hae-magglutinating antibodies to a pooled polysaccharideantigen prepared from six common commensal sero-types of Esch. coli. Serum precipitins to Haemophilusinfluenzae, pneumococcus, Staphylococcus aureus, Pseudo-monas aeruginosa, Klebsiella sp., and Candida albicanswere not detected. He was immunized with 1 2 mlTAB vaccine (Burroughs Wellcome) and 0 - 5 ml tetanustoxoid (Lister Institute), with a booster after 8 weeks.No antibodies were detected to either immunization upto 2 months later (Widal < 1: 10, H and O, tetanus anti-toxin <1:10 IU/ml). Intradermal injection of 0.1 ml1% C. albicans (Bencard) failed to produce a reactionwithin 72 hours. Contact sensitivity to dinitrochloro-benzene (5% in acetone sensitizing dose and challengewith 0 1% after 3 weeks) was negative. An area on the
on 13 March 2019 by guest. P
rotected by copyright.http://adc.bm
j.com/
Arch D
is Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. D
ownloaded from
Short iXeportsleft thigh was painted with 2% picryl chloride and a
draining lymph node was removed 5 days later from thegroin. Histological examination of this node showedmarked depletion of lymphocytes in all areas; there were
no lymphoid follicles or germinal centres. Peripheralblood lymphocytes were incubated for 3 days with puri-fied phytohaemaglutinin (PHA, Burroughs Wellcome)and the degree of lymphoblast transformation measuredby incorporation of radioactive 14C thymidine. Therewas no increase in isotope uptake in the stimulatedcultures (unstimulated 317 counts/min per 5 x 105cells; PHA stimulated 294 counts/min per 5 x 105 cells).There was also no response with pokeweed and con-
canavallin A mitogens.
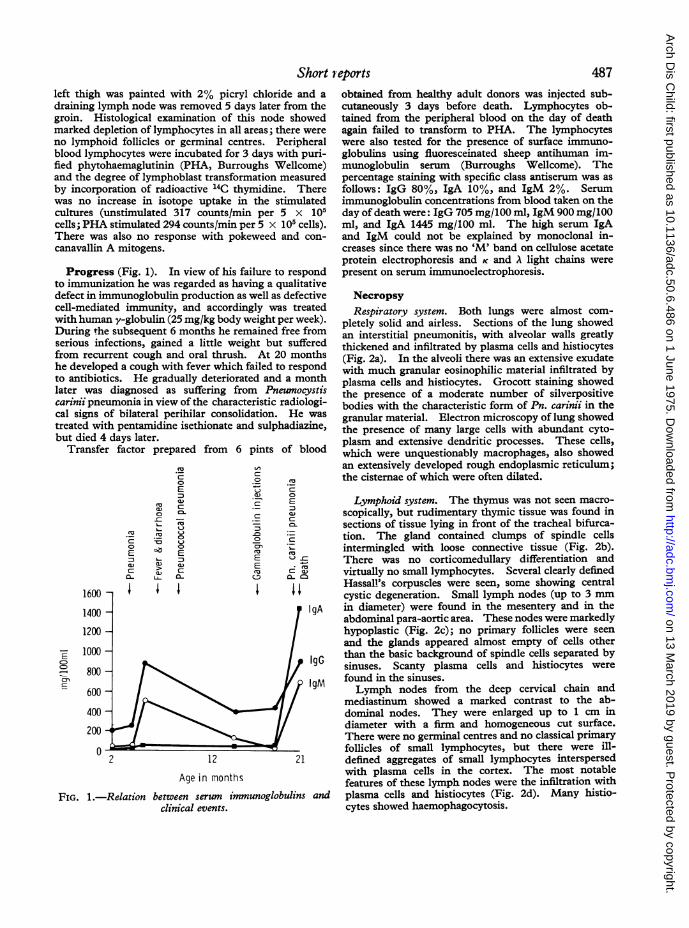
Progress (Fig. 1). In view of his failure to respondto immunization he was regarded as having a qualitativedefect in immunoglobulin production as well as defectivecell-mediated immunity, and accordingly was treatedwith human y-globulin (25 mg/kg body weight per week).During the subsequent 6 months he remained free fromserious infections, gained a little weight but sufferedfrom recurrent cough and oral thrush. At 20 monthshe developed a cough with fever which failed to respondto antibiotics. He gradually deteriorated and a monthlater was diagnosed as suffering from Pneumocystiscarinii pneumonia in view of the characteristic radiologi-cal signs of bilateral perihilar consolidation. He was
treated with pentamidine isethionate and sulphadiazine,but died 4 days later.
Transfer factor prepared from 6 pints of blood
aL)0
._Z
a)
Oa)
0
EI
0-
1600 -
1400 -
1200 -
1000 -
800 -
600 -
400 -
200
02
c-
E
3a)c
C
0~
u
a)c
V)C
0
an
EE
m
.TC
E
a)
C
C:
C a)m
gA
IgG
1gM
12 2
Age in months
FIG. 1.-Relation between serum immunoglobulins andclinical events.
obtained from healthy adult donors was injected sub-cutaneously 3 days before death. Lymphocytes ob-tained from the peripheral blood on the day of deathagain failed to transform to PHA. The lymphocyteswere also tested for the presence of surface immuno-globulins using fluoresceinated sheep antihuman im-munoglobulin serum (Burroughs Wellcome). Thepercentage staining with specific class antiserum was as
follows: IgG 80%, IgA 10%, and IgM 2%. Serumimmunoglobulin concentrations from blood taken on theday of death were: IgG 705 mg/100 ml, IgM 900 mg/100ml, and IgA 1445 mg/100 ml. The high serum IgAand IgM could not be explained by monoclonal in-creases since there was no 'M' band on cellulose acetateprotein electrophoresis and K and A light chains were
present on serum immunoelectrophoresis.
NecropsyRespiratory system. Both lungs were almost com-
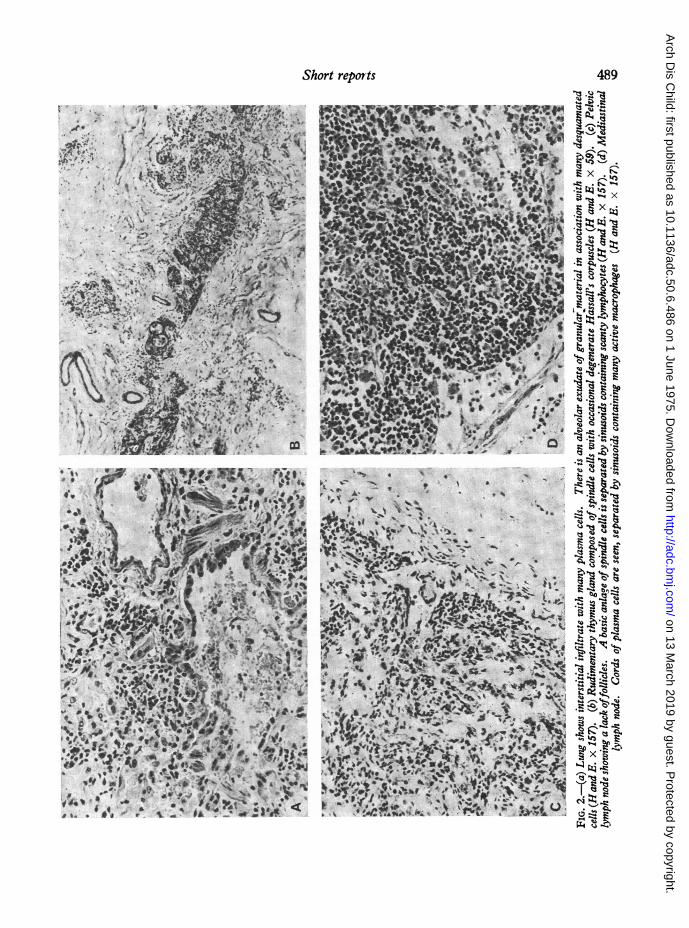
pletely solid and airless. Sections of the lung showedan interstitial pneumonitis, with alveolar walls greatlythickened and infiltrated by plasma cells and histiocytes(Fig. 2a). In the alveoli there was an extensive exudatewith much granular eosinophilic material infiltrated byplasma cells and histiocytes. Grocott staining showedthe presence of a moderate number of silverpositivebodies with the characteristic form of Pn. carinii in thegranular material. Electron microscopy of lung showedthe presence of many large cells with abundant cyto-plasm and extensive dendritic processes. These cells,which were unquestionably macrophages, also showedan extensively developed rough endoplasmic reticulum;the cistemae of which were often dilated.
Lymphoid system. The thymus was not seen macro-
scopically, but rudimentary thymic tissue was found insections of tissue lying in front of the tracheal bifurca-tion. The gland contained clumps of spindle cellsintermingled with loose connective tissue (Fig. 2b).There was no corticomedullary differentiation andvirtually no small lymphocytes. Several clearly definedHassall's corpuscles were seen, some showing centralcystic degeneration. Small lymph nodes (up to 3 mmin diameter) were found in the mesentery and in theabdominal para-aortic area. These nodes were markedlyhypoplastic (Fig. 2c); no primary follicles were seen
and the glands appeared almost empty of cells otherthan the basic background of spindle cells separated bysinuses. Scanty plasma cells and histiocytes were
found in the sinuses.Lymph nodes from the deep cervical chain and
mediastinum showed a marked contrast to the ab-dominal nodes. They were enlarged up to 1 cm indiameter with a firm and homogeneous cut surface.There were no germinal centres and no classical primaryfollicles of small lymphocytes, but there were ill-defined aggregates of small lymphocytes interspersedwith plasma cells in the cortex. The most notablefeatures of these lymph nodes were the infiltration with
plasma cells and histiocytes (Fig. 2d). Many histio-cytes showed haemophagocytosis.
487
kg
on 13 March 2019 by guest. P
rotected by copyright.http://adc.bm
j.com/
Arch D
is Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. D
ownloaded from

488 Short reportsThe spleen weighed 12 g and was unusually firm.
There was almost complete absence of lymphocyticaggregates about the splenic arterioles and elsewhere.The trabecular framework of the red pulp showedmarked fibrosis.The tonsils and adenoids were not seen in multiple
blocks taken through the area. Bone marrow showedactive haemopoiesis with plasma cells being present inlarge numbers.
Gastrointestinal system. No lymphoid aggregateswere found in the small or large gut. The appendixcontained no lymphoid follicles, but there were amoderate number of lymphocytic cells and a feweosinophils in the lamina propria. Some plasma cellswere identified in the lamina propria of the small gutand colon. All other organ systems were essentiallynormal. In particular, three normal parathyroidglands were identified histologically.
DiscussionThe clinical course, normal serum immuno-
globulin levels, gross deficiency in cell-mediatedimmunity, and the presence of enlarged mediastinallymph nodes containing plasma cells parallels thecase described by Nezelof et al. (1964). TheWHO Committee on Primary Immunodeficiency(Fudenberg et al., 1971) have classified such casesas part of the spectrum of severe combined im-munodeficiency (SCID). However, it has re-cently been suggested by Lawlor et al. (1974) thatthis condition warrants a separate classification.
It is likely that the rise in serum immunoglobulinswas related to the Pn. carinii infection, and intenseantigenic stimulation from the lungs probablyaccounts for the plasma cell infiltrate of the media-stinal lymph nodes. The hyper-y-globulinaemia,particularly the rise in IgA, was clearly out ofproportion to that seen in normal individuals afterinfection. Gelfand et al. (1973) described aninfant with SCID who developed a grossly raisedserum IgM, which they suggested was related totransfer factor therapy. This case shows thatsuch increases can occur spontaneously since it isinconceivable that the transfer factor given in thiscase caused the extreme rise of serum IgA within2 days. Furthermore, similar infants have beendescribed with very high levels of serum IgE(Kikkawa et al., 1973) and IgD (Rubenstein, Speck,and Jeannet, 1971).The normal limitation of antibody production
may depend upon the presence of special lympho-cytes which suppress the response. Experimentsin mice have shown that such suppressor lympho-cytes occur after cell mediated (Zembala andAsherson, 1973) and humoral immune reactions(Gershon and Kondo, 1971). Furthermore, mice
thymectomized at birth may develop markedlyraised levels of serum IgA (Humphrey, Parrott, andEast, 1964). It is therefore possible that theexcessive production of IgA in our patient wasrelated to the absence of suppressor T lymphocytes.
It has been suggested that SCID is due to alymphoid stem cell defect, but this is unlikely inour patient in view of the presence of lymphocytesbearing surface immunoglobulin (B cells) andplasma cells. However, it is conceivable that theseB cells were of maternal origin and that the eosino-philia and failure to thrive were manifestations of achronic graft versus host reaction.The recognition of this subgroup of SCID is
important since we may be able to learn moreabout the regulation of the immune response fromsuch cases. Clinically, it is useful to underlinethat the presence of normal or raised serum im-munoglobulins does not rule out a severe andlethal immunodeficiency. The accepted treatmentfor SCID is a bone marrow graft, preferably from ahistocompatible and mixed lymphocyte reactionnegative sib. The treatment of choice in thisrare subgroup is not clear, but it would seemrational to attempt a thymus graft initially, followedby a bone-marrow graft if this is unsuccessful.
SummaryAn infant is described with absence of cell
mediated immunity and apparent failure to producefunctional antibody despite raised serum im-munoglobulin concentrations. A marked rise inserum IgA occurred during the terminal phase ofhis illness. We draw attention to this subgroupof severe combined immunodeficiency where thereis a tendency to spontaneously develop hyper-y-globulinaemia.
We thank Drs. A. Howard, G. Loewi, M. Papamichail,and Professor J. Pepys, Judith Davies, Jill Boler, MayaShohat, and Margaret North for help with the investiga-tions. We are also grateful for assistance from theClinical Laboratory staff of the Plymouth General andNorthwick Park Hospitals.
REFERENCESFudenberg H., Good, R. A., Goodman, H. C., Hitzig, W., Kunkel,
H. G., Roitt, I. M., Rosen, F. S., Rowe, D. S., Seligmann, M.,and Soothill, J. R. (1971). Primary immunodeficiencies-re-port of a W.H.O. committee, Pediatrics, 47, 927.
Gelfand, E. W., Baumal, R., Huber, J., Crookston, M. C., andShumak, K. H. (1973). Polyclonal gammopathy and lympho-proliferation after transfer factor in severe combined im-munodeficiency disease. New England3journal ofMedicine, 289,1385.
Gershon, R. K., and Kondo, K. (1971). Infectious immunologicaltolerance. Immunology, 21, 930.
Humphrey, J. H., Parrott, D. M. V., and East, J. (1964). Studieson globulin and antibody production in mice thymectomized atbirth. Immunology, 7,419.
on 13 March 2019 by guest. P
rotected by copyright.http://adc.bm
j.com/
Arch D
is Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. D
ownloaded from
Short repor-ts 489
~4 72
x x
:i5t!b$wQ8St. .Q0 °2
t 3 $-
X _ qX
.l44t
FSz
+X.*w
on 13 March 2019 by guest. P
rotected by copyright.http://adc.bm
j.com/
Arch D
is Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. D
ownloaded from

490 Short reportsKikkawa, Y., Kamimura, K., Hamajima, T., Sekiguchi, T., Kawai,
T., Takenaka, M. and Tada, T. (1973). Thymic alymphoplasiawith hyper-IgE-globulinemia. Pediatrics, 51, 690.
Lawlor, G. J., Ammann, A. J., Wright, W. C., La Franchi, S. H.,Bilstrom, D., and Stiehm, E. R. (1974). Syndrome of cellularimmunodeficiency with immunoglobulins. Journal of Pedi-attics, 84, 183.
Nezelof, C., Jammet, M. L., Lortholary, P., Labrune, B., andLamy, M. (1964). L'hypoplasie hereditaire du thymus.Archives Francaises de P'diatrie, 21, 897.
Rubinstein, A., Speck, B., and Jeannet, M. (1971). Successfulbone-marrow transplantation in a lymphopenic immunologicdeficiency syndrome. New England Journal of Medicine 285,1399.
Zembala, M., and Asherson, G. L. (1973). Depression of the T cellphenomenon of contact sensitivity by T cells from unrespon-sive mice. Nature, 244,227.
A. D. B. WEBSTER,* G. SLAVIN, M. K. STRELLING,and G. L. ASHERSONClinical Research Centre, Northwick Park Hospital,Harrow, Middlesex; and Department of Paediatrics,Plymouth General Hospital, Devon.
*Coffespondence to Dr. A. D. B. Webster, Northwick ParkHospital, C. R. C. Harrow, Middx. HAL 3UJ.
Trial of artificial diet in treatmentof cystic fibrosis of pancreas
The major clinical features of cystic fibrosis(CF) of the pancreas include, with rare exceptions,malabsorption, malnutrition, and growth failure.Standard treatment of this disease includes the useof a high calorie, high protein diet and pancreaticenzyme supplements to improve intestinal absorp-tion. Even with such treatment nutrition andgrowth may be impaired, and undernutrition mayfurther predispose to respiratory infections.
TABLEPatients who took the diet alone
I ~~~~~~~Pretrialyear
Caae Age at Clinical Cheat -no. Sex entry grade x-ray Height g
(m~~~~~~~~~~~andheightvelocity
1 F 5 1 Clear
2 F 22 1 Clear Not available
3 F 23 1 Clear Not available
4 F 13 2 Persistent bila- Not availableteral lowerlobe changes
Darby and Seakins (1971) described the use of adietary amino acid supplement in treatment of CFbut noted no benefit to their patients. Morerecently, Allen, Mason, and Moss (1973) havereported encouraging results from the use of anartificial diet in the treatment of CF, with evidenceof improvement in growth and clinical status.We report our experience with the use of an
artificial diet in a group of patients with CF.
Patients and methods
Twelve patients with CF, whose ages ranged from4 months to 12 years and who had all been previouslytreated with normal diet plus pancreatic enzyme sup-plements, were started on an artificial diet similar tothat described by Allen et al. (1973). This consistedof Albumaid (a beef protein hydrolysate) and Caloreen(a glucose polymer), prescribed in sufficient quantitiesto provide 100% of the patient's protein and calorierequirements. It was given in water, flavoured to tastewith tomato or fruit juice, or in the case of infantsdissolved in sufficient water to provide the total dailyfluid intake. Additional eating was discouraged, butadditional fluids were permitted. All patients weregiven daily 10 ml medium chain triglyceride oil, Ketovitetablets, and liquid, and a standard mineral supplement.Pancreatic enzymes were only given when any normalfood was taken. Normal treatment, including physio-therapy and antibiotics in the presence of chest infection,was continued throughout the period of the trial. Thetrial period was for a full year in each case. If thepatients found difficulty in keeping to the diet, thefollow-up and observations described below werecontinued for a full year and advice and encouragementto continue with the diet were offered at each visit.At the beginning of the trial patients were examined
clinically and classified into grades 1 or 2 using thecriteria similar to those recently described by Mearns(1972) for assessing respiratory status. Patients without
on 13 March 2019 by guest. P
rotected by copyright.http://adc.bm
j.com/
Arch D
is Child: first published as 10.1136/adc.50.6.486 on 1 June 1975. D
ownloaded from


















