SCD (2)
52
Sickle Cell Disease, Epidemiology,Genetic History, complication and Management Dr. Richard B. Fynn Dormaa Presby Hospital
-
Upload
anastasiafynn -
Category
Documents
-
view
3 -
download
0
description
scdx
Transcript of SCD (2)

Sickle Cell Disease, Epidemiology,Genetic History,complication and Management
Dr. Richard B. Fynn Dormaa Presby Hospital
Dept. of Child Health

What Is Sickle Cell Disease?
• An inherited disease of red blood cells
• Affects hemoglobin
• Polymerization of hemoglobin leads to a cascade of effects decreasing blood flow
• Tissue hypoxia causes acute and chronic damage

Prevalence/Incidence of SCD
In African-Americans the incidence of SCD is 1 in 375 for HbSS, 1 in 835 for HbSC and 1 in 1,667 for Sickle beta-thalassemia. In addition, 1 in 12 African-Americans are carriers for the disorder
• In other U.S. populations, the prevalence of sickle cell disease is 1 in 58,000 Caucasians; 1 in 1,100 Hispanics (eastern states); 1 in 32,000 Hispanics (western states); 1 in 11,500 Asians; and 1 in 2,700 Native Americans

Sickle Cell Disease SCD Genotype
• Sickle cell anemia (SS)• Sickle Hb C disease (SC)• Sickle S beta plus
(Sβ+ thalassemia )• Sickle Beta zero
(Sβ° thalassemia)
• 65%• 25%• 8%
• 2%
Genotype Genotype prevalence

Who Is At Risk?The disease originated in at least 4 places in Africa, Mediterranean countries (such as Turkey, Greece, and Italy), and in the Indian/Saudi Arabian subcontinent. It exists in all countries of Africa and in areas where Africans have migrated.
It is most common in West and Central Africa where as many as 25% of the people have sickle cell trait and 1-2% of all babies are born with a form of the disease.
In the United States with an estimated population of over 270 million, about 1,000 babies are born with sickle cell disease each year.
In contrast, Nigeria, with an estimated 1997 population of 90 million, 45,000-90,000 babies with sickle cell disease are born each year.

Who Is At Risk?• Most common in
people whose families come from Africa, South or Central America (especially Panama), Caribbean islands, Mediterranean countries (such as Turkey, Greece, and Italy), India, and Saudi Arabia.

Some Genetic History
The error in the hemoglobin gene results from a genetic mutation that occurred many thousands of years ago in people in parts of Africa, the Mediterranean basin, the Middle East, and India.
A deadly form of malaria was very common at that time
Malaria epidemics caused the death of many In areas where malaria was a problem, children who inherited one
sickle hemoglobin gene and who, therefore, carried the sickle cell trait - had a survival advantage.
Unlike the children who had normal hemoglobin genes, they survived the malaria epidemics they grew up, had their own children, and passed on the gene- for sickle hemoglobin.

As populations migrated, the sickle cell-mutation spread to other Mediterranean areas, further into the Middle East and eventually into the Western Hemisphere.
In the United States and other countries where malaria is not a problem, the sickle hemoglobin gene no longer provides a survival advantage.
Instead, it may be a serious threat to the carrier's children, who may inherit two abnormal sickle hemoglobin genes and have sickle cell anemia.
History

Inheritance of Sickle Cell Anemia
If one parent has sickle cell trait (HbAS) and the other does not carry the sickle hemoglobin at all (HbAA) then none of the children will have sickle cell anemia.
There is a one in two (50%) chance that any given child will get one copy of the HbAS gene and therefore have the sickle cell trait.
It is equally likely that any given child will get two HbAA genes and be completely unaffected.

Inheritance of Sickle Cell Anemia
If both parents have sickle cell trait (HbAS) there is a one in four (25%) chance that any given child could be born with sickle cell anemia.
There is also a one in four chance that any given child could be completely unaffected.
There is a one in two (50%) chance that any given child will get the sickle cell trait.

Inheritance of Sickle Cell Anemia
If one parent has sickle cell trait (HbAS) and the other has sickle cell anaemia (HbSS) there is a one in two (50%) chance that any given child will get sickle cell trait and a one in two (50%) chance that any given child will get sickle cell anemia.
No children will be completely unaffected.

Inheritance of Sickle Cell Anemia
If one parent has sickle cell anaemia (HbSS) and the other is completely unaffected (HbAA) then all the children will have sickle cell trait.
None will have sickle cell anemia.
The parent who has sickle cell anemia (HbSS) can only pass the sickle hemoglobin gene to each of their children.
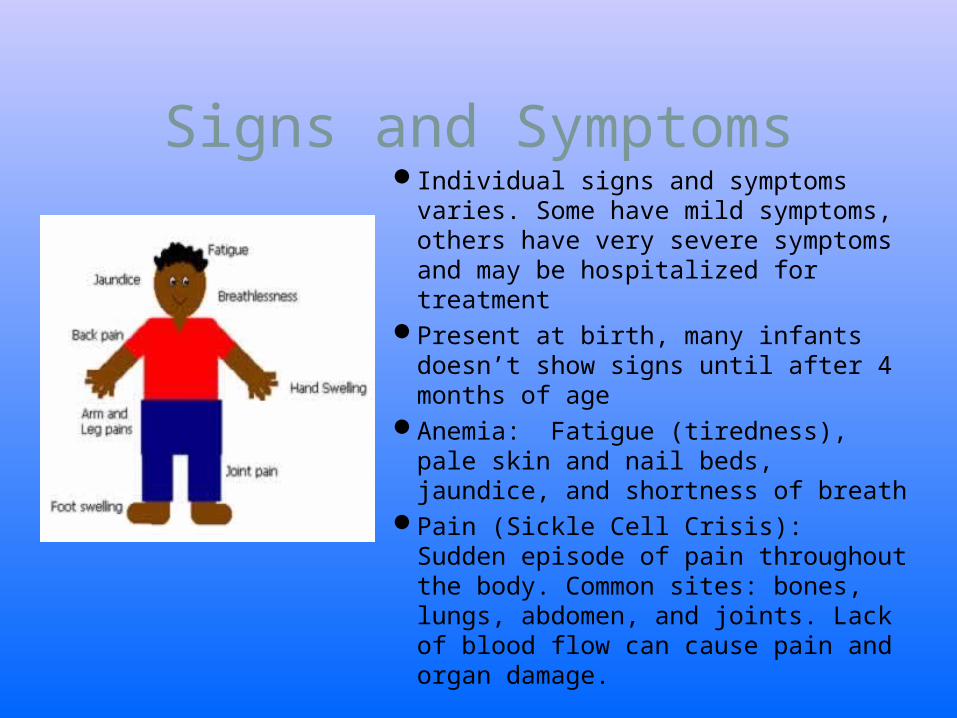
Signs and SymptomsIndividual signs and symptoms varies.
Some have mild symptoms, others have very severe symptoms and may be hospitalized for treatment
Present at birth, many infants doesn’t show signs until after 4 months of age
Anemia: Fatigue (tiredness), pale skin and nail beds, jaundice, and shortness of breath
Pain (Sickle Cell Crisis): Sudden episode of pain throughout the body. Common sites: bones, lungs, abdomen, and joints. Lack of blood flow can cause pain and organ damage.

• First described in Chicago in 1910 by James Herrick as an inherited condition that results in a decrease in the ability of red blood cells to carry oxygen throughout the body
Sickle red blood cells become hard and irregularly shaped (resembling a sickle)Become clogged in the small blood vessels and therefore do not deliver oxygen to the tissues.Lack of tissue oxygenation can cause excruciating pain, damage to body organs and even death.

Normal Vs. Sickle Red Cells
Normal• Disc-Shaped• Deformable• Life span of 120 days
Sickle• Sickle-Shaped• Rigid • Lives for 20 days or
less

Complications
• Fever/infection• Acute chest syndrome• Eye trauma (hyphema) • Priapism• Stroke• Splenic sequestration• Severe pain

Acute Chest Syndrome
Clinically:
Acute onset of fever, respiratory symptoms, new infiltrate on chest x-ray
Causes– Infection
– Fat emboli
– Lung infarct
Since you cannot distinguish between acute chest syndrome and pneumonia clinically there is no change in treatment.
A leading cause of death in sickle cell disease

Stroke
• Historically 8 to 10% of children with SS
• “Silent Stroke” in 22% of children with hemoglobin SS
Any acute neurologic symptom other than mild headache, even if transient, requires urgent evaluation.
Treatment: Chronic transfusion therapy to maintain sickle hemoglobin at or below 30%
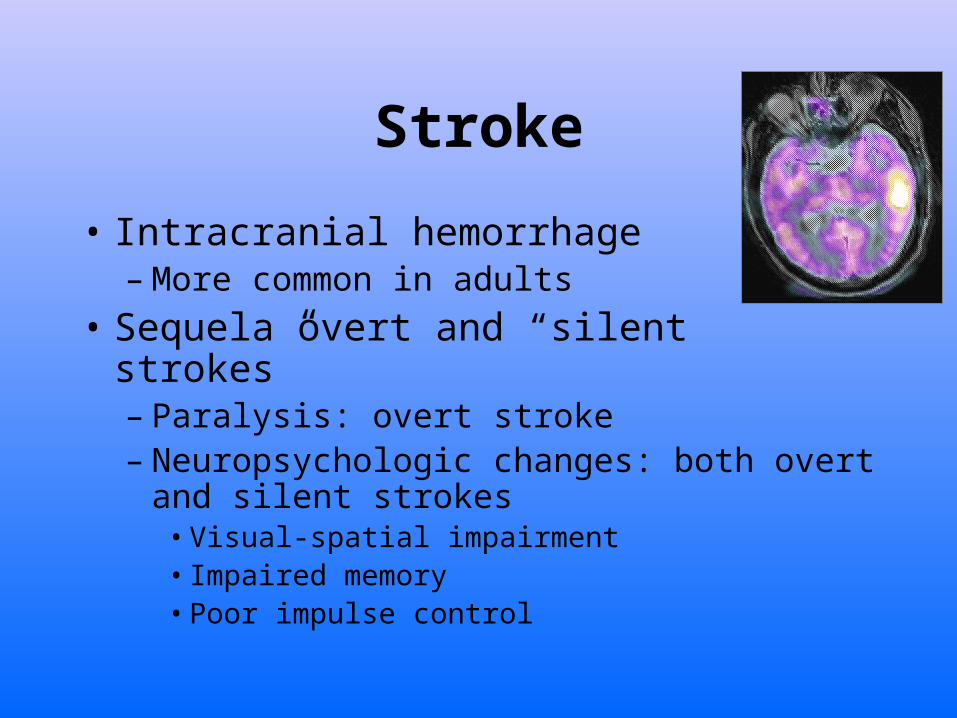
Stroke
• Intracranial hemorrhage– More common in adults
• Sequela overt and “silent strokes” – Paralysis: overt stroke– Neuropsychologic changes: both overt and
silent strokes • Visual-spatial impairment• Impaired memory• Poor impulse control

Splenic Sequestration
• Sudden trapping of blood within the spleen
• Usually occurs in infants under 2 years of age with SS
• Spleen enlarged on physical exam, may not be associated with fever, pain, respiratory, or other symptoms
• Circulatory collapse and death can occur in less than thirty minutes
•Recurrence very common (50%)•Associated with high mortality (20%)

Splenic Sequestration
• Hemoglobin SS– Incidence increased: 6 and 36 months
• Overall incidence about 15%
• Hemoglobin SC– Incidence increased: 2 and 17 years
• Mean age 8.9 years
• Can occur in adolescence and adulthood
• Incidence about 5%

Hand Foot Syndrome - Dactylitis
• Early complication of sickle cell disease
• Highest incidence 6 months to 2 years
• Painful swelling of hands and feet
• Treatment involves fluids and pain medication
• Fevers treated as medical emergency

Renal Disease
• Renal findings– Decreased ability to concentrate urine– Decreased ability to excrete potassium– Inability to lower urine pH normally– Hematuria / papillary necrosis
• Risk factors for progressive renal failure– Anemia, proteinuria, hematuria

Renal Disease
• Proteinuria/Nephrotic syndrome• 40% of SCD patients with nephrotic syndrome
develop end-stage renal disease • Occurs in ~ 20% of all patients • Occurs in 4.5% of all pediatric patients- increased
in hemoglobin SS to 6.5%– Increased incidence with age– Increased with anemia, increased MCV, and increased
leukocyte count
• Renal failure common in adults

Gall Bladder and Liver
• Gall stones and biliary sludge– Monitor by ultrasound every 1-2 years
• Cholestasis– May progress, leading to bleeding disorders or
liver failure
• Iron overload– Due to chronic transfusions
• Chronic hepatitis

Hemolysis and Vaso-occlusion
Vaso-occlusion:Occurs when the rigid sickle shaped cells fail to move through the small blood vessels, blocking local blood flow to a microscopic region of tissue. Amplified many times, these episodes produce tissue hypoxia. The result is pain, and often damage to organs.
Hemolysis:The anemia in SCD is caused by red cell destruction, or hemolysis, and the degree of anemia varies widely between patients. The production of red cells by the bone marrow increases dramatically, but is unable to keep pace with the destruction.

Bone Disease Diagnosis and Treatment
• Avascular necrosis of hips and shoulders– Index of suspicion
• Persistent hip or shoulder pain
• Plain film or MRI
• Treatment– Conservative
• NSAID’s and 6 weeks of rest off affected limb
• Physical therapy

Chronic Complications
• Anemia/Jaundice
• Brain Damage/Stroke
• Kidney failure
• Decreased lung function
• Eye disease (bleeding, retinal detachment)
• Leg ulcers
• Chronic pain management

Anemia – Jaundice
• Common and starting in the first year of life
• Decreased lifespan of sickle red cells– Hemolysis
– Anemia
– Hyperbilirubinemia
– Reticulocytosis

Leg Ulcers
• Occurs in about 25% of all hemoglobin SS patients
• Predominantly males– Incidence increased with
• Age
• Decreased hemoglobin
– Incidence decreased with alpha thalassemia
• Recurrence rate is ~ 75%

Chronic Pain
• Pain lasting >3 to 6 months
• Patients should receive comprehensive psychologic and clinical assessment
• Treatment– Analgesics– Hydroxyurea– Relaxation techniques– Physical and occupational therapy

Fever and Infection
• Fever > 38.5° C (101°F)
is an EMERGENCY• Basic laboratory
evaluation: – CBC with differential and
reticulocyte count, blood, urine, and throat cultures, urinalysis, chest x-ray
• Indications for hospitalization & IV antibiotics:-Child appears ill-Any temperature > 40°C-Abnormal laboratory values
• Start IV antibiotics IMMEDIATELY if child appears ill or temperature > 40°C (DO NOT WAIT FOR LABS)

Management
• Health maintenance
• Infection prevention
• Pain management
• Sickle emergencies
• Chronic disease management

Health Maintenance• Frequent visits: every 3 to 6 months• Immunizations
– Routine immunizations– Hib- 6 months and older– 23 valent Pneumovax at five years
• Penicillin prophylaxis beginning no later than two months
• Nutrition and fluids– Folate supplementation is controversial

Health Maintenance
• Physical exam with attention to:– Growth and development, jaundice, liver/spleen
size, heart murmur of anemia, malocclusion from increased bone marrow activity, delayed puberty
• Lab evaluations: – CBC with differential and reticulocyte count,
urinalysis, renal & liver function

Health Maintenance
Special studies• Brain- Transcranial doppler ultrasonography,
MRI/MRA• Lungs- Pulmonary function tests, Echo
cardiogram for pulmonary hypertension• Neurologic- neuropsychological testing

Current Recommendations
• Penicillin Prophylaxis: SS, SºThalassemia– 2 months to 3 years: 125 mg PO BID– Over 3 years: 250 mg PO BID
• When to discontinue is controversial
• Penicillin Prophylaxis: SC and S+ Thalassemia– SC warrants penicillin prophylaxis similar to SS– S+ Thalassemia: penicillin prophylaxis can be safely
discontinued at 5 years• Routine use in infants and children is controversial
• Special Circumstances– History of repeated sepsis, surgical splenectomy

Eye Examination
• Retinal vessel disease– Incidence 33% in
hemoglobin SC
– Incidence 3% in SS
• Annual evaluation after age 10 years by ophthalmologist– Laser photocoagulation
for vessel disease
Sea Fan
Salmon Patch: SC

Priapism
Treatment is difficult– Opioid pain medication– Intravenous fluids– Aspiration and irrigation of the
corpus cavernosum– Surgery– Blood Transfusions
• Impotence with severe disease or recurrent episodes
Urethra Corpus cavernosum
Commonly occurs in children and adolescents with SS or SC

Treatments For Splenic Sequestion
• Intravenous fluids– Maintain vascular volume
• Cautious blood transfusion– Treat anemia, sequestered
blood can be released from spleen
• Spleen removal or splenectomy– If indicated

Pain Management
Acute pain• Hand-foot syndrome (dactylitis)• Painful episodes: vasoocculsion• Splenic sequestration• Acute chest syndrome• Cholelithiasis• Priapism• Avascular necrosis• Right upper quadrant syndrome

Pain ManagementPain is an emergencyPain is an emergencyHospital evaluation:• Hydration: 1.5 times maintenance unless
acute chest syndrome suspected• Assess pain level and treat
– Do not withhold opioids– Frequently reassess pain control
• Assess for cause of pain/complications

Pain Management
Mild-moderate pain• Acetaminophen
– Hepatotoxic
• Non-steroidal anti-inflammatory agents (NSAIDs)
-Contraindicated in patients with gastritis/ulcers and renal failure
-Monitor renal function if used chronically

Pain Management
• Moderate-severe pain– Opioids are first-line treatment– Morphine sulfate or hydromorphone – Meperidine NOT recommended
• (Metabolite causes seizures & renal toxicity)
• Moderate or less severe pain– Acetaminophen or NSAID's in combination with
opioids– Other adjuvant medications (sedatives, anxiolytics)
• May increase efficacy of analgesics

Treatment
• For respiratory distress– Antibiotic coverage– Supplemental oxygen– Partial exchange transfusion
• For splenic sequestration– Repletion of intravascular volume– Severe anemia, transfuse

Treatment
• For suspicion of stroke– Exchange transfusion
• For priapism– Analgesia, hydration– Partial exchange transfusion

Treatment
• Outpatient– Vaccinations
• Pneumococcal, meningococcal, influenza vaccines
– Penicillin prophylaxis– Folic acid therapy– Hydroxyurea for severe symptoms
• Consideration for BMT for severe cases

Adolescents and Transition of Care
• Young adults (>20 years) with frequent pain crises at greatest risk for early death
• Barriers to care for young adults– Lack of adult SCD providers
– Loss of medical coverage
– Developmental (level of independence, denial of chronic illness)
– Ineffective coping skills (passive versus active)

Adolescents and Transition of Care
• Develop explicit plan for transition
• Team approach- pediatric and adult providers, social work, school/vocational staff, support groups
• Plan gradual transition (start 1 year before)
• Continue communication between pediatric & adult providers after transition

Genetic Counseling
• Who should receive counseling?-Parents of newborns with sickle disorders or traits-Pregnant women/ prenatal counseling
• What is the purpose of counseling? -Education-Informed decision-making
• Content should include: -Genetic basis, chances of disease or trait (potential pregnancy outcome), disease-related health problems, variability/unpredictability of disease, family planning, average life span

Thank You

www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf
Nelson’s Book of Medicine
eMedicine
References