
Scaling up genomic analysis with ADAM
32
Scaling up genomic analysis with ADAM Frank Austin Nothaft, UC Berkeley AMPLab [email protected], @fnothaft 11/20/2014
-
Upload
fnothaft -
Category
Engineering
-
view
635 -
download
1
Transcript of Scaling up genomic analysis with ADAM

Scaling up genomic analysis with ADAM
Frank Austin Nothaft, UC Berkeley AMPLab [email protected], @fnothaft
11/20/2014

Credit: Matt Massie & NHGRI

The Sequencing Abstraction
• Humans have 46 chromosomes and each chromosome looks like a long strong
• We get randomly distributed substrings, and want to reassemble original, whole string
It was the best of times, it was the worst of times…
It was the the best oftimes, it was
the worst ofworst of times
Metaphor borrowed from Michael Schatz
best of timeswas the worst

Genomics = Big Data
• Sequencing run produces >100 GB of raw data
• Want to process 1,000’s of samples at once to improve statistical power
• Current pipelines take about a week to run and are not horizontally scalable

How do we process a genome?

What’s our goal?
• Human genome is 3.3B letters long, but our reads are only 50-250 letters long
• Sequence of the average human genome is known
• Insight: Each human genome only differs at 1 in 1000 positions, so we can align short reads to average genome, and compute diff

Align Reads
It was the best of times, it was the worst of times…
It was the the best oftimes, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was the
the best oftimes, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst of
worst of times
best of timeswas the worst

Align Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of
times, it was
the worst ofworst of times
best of timeswas the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of times, it was
the worst ofworst of times
best of timeswas the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of times, it was
the worst ofworst of times
was the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of times, it was
the worst ofworst of times
was the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of times, it was the worst of
worst of times
was the worst

Assemble Reads
It was the best of times, it was the worst of times…
It was thethe best of times, it was the worst ofworst of timeswas the worst

Overall Pipeline Structure
From “GATK Best Practices”, https://www.broadinstitute.org/gatk/guide/best-practices

Overall Pipeline Structure
From “GATK Best Practices”, https://www.broadinstitute.org/gatk/guide/best-practices
The stages take ~100 hours; ADAM works here
End to end pipeline takes ~120 hours

Making Genomics Horizontally Scalable

Key Observations• Current genomics pipelines are I/O limited
• Most genomics algorithms can be formulated as either data/graph parallel computation
• Genomics is heavy on iteration/pipelining, data access pattern is write once, read many times
• High coverage, whole genome (>220 GB) will become main dataset for human genetics

ADAM Principles
• Use schema as “narrow waist”
• Columnar data representation + in-memory computing eliminates disk bandwidth bottleneck
• Minimize data movement: send code to data
ApplicationTransformations
Physical StorageDisk
Data DistributionParallel FS/Sharding
Materialized DataColumnar Storage
Evidence AccessMapReduce/DBMS
PresentationEnriched Models
SchemaData Models

Data Independence• Many current genomics systems require data to be
stored and processed in sorted order
• This is an abstraction inversion!
• Narrow waist at schema forces processing to be abstract from data, data to be abstract from disk
• Do tricks at the processing level (fast coordinate-system joins) to give necessary programming abstractions

Data Format
• Genomics algorithms frequently access global metadata
• Schema is fully denormalized, allows O(1) access to metadata
• Make all fields nullable to allow for arbitrary column projections
• Avro enables literate programming
record AlignmentRecord { union { null, Contig } contig = null; union { null, long } start = null; union { null, long } end = null; union { null, int } mapq = null; union { null, string } readName = null; union { null, string } sequence = null; union { null, string } mateReference = null; union { null, long } mateAlignmentStart = null; union { null, string } cigar = null; union { null, string } qual = null; union { null, string } recordGroupName = null; union { int, null } basesTrimmedFromStart = 0; union { int, null } basesTrimmedFromEnd = 0; union { boolean, null } readPaired = false; union { boolean, null } properPair = false; union { boolean, null } readMapped = false; union { boolean, null } mateMapped = false; union { boolean, null } firstOfPair = false; union { boolean, null } secondOfPair = false; union { boolean, null } failedVendorQualityChecks = false; union { boolean, null } duplicateRead = false; union { boolean, null } readNegativeStrand = false; union { boolean, null } mateNegativeStrand = false; union { boolean, null } primaryAlignment = false; union { boolean, null } secondaryAlignment = false; union { boolean, null } supplementaryAlignment = false; union { null, string } mismatchingPositions = null; union { null, string } origQual = null; union { null, string } attributes = null; union { null, string } recordGroupSequencingCenter = null; union { null, string } recordGroupDescription = null; union { null, long } recordGroupRunDateEpoch = null; union { null, string } recordGroupFlowOrder = null; union { null, string } recordGroupKeySequence = null; union { null, string } recordGroupLibrary = null; union { null, int } recordGroupPredictedMedianInsertSize = null; union { null, string } recordGroupPlatform = null; union { null, string } recordGroupPlatformUnit = null; union { null, string } recordGroupSample = null; union { null, Contig } mateContig = null;}
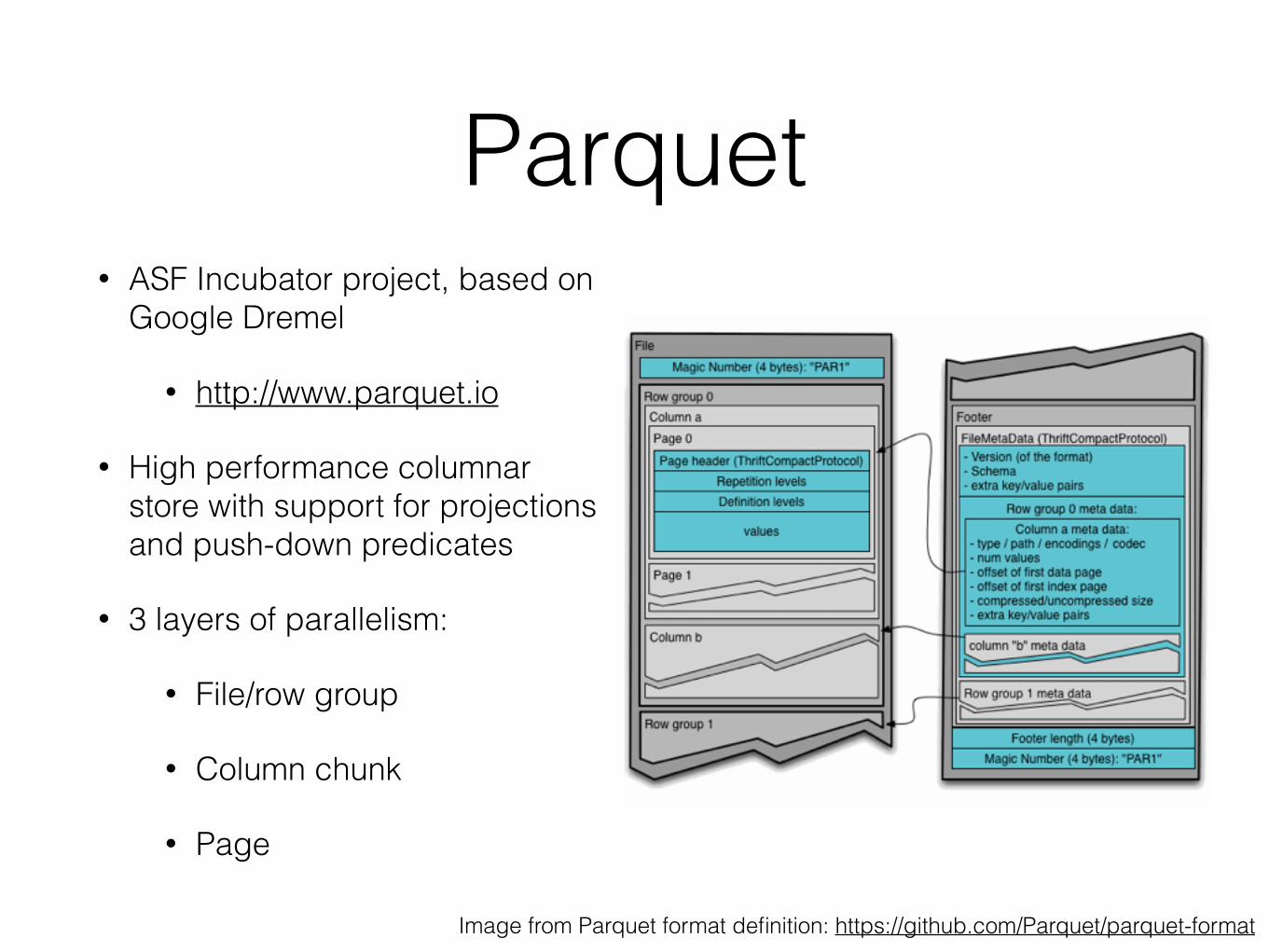
Parquet• ASF Incubator project, based on
Google Dremel
• http://www.parquet.io
• High performance columnar store with support for projections and push-down predicates
• 3 layers of parallelism:
• File/row group
• Column chunk
• Page
Image from Parquet format definition: https://github.com/Parquet/parquet-format

Access to Remote Data• For genomics, we often have a really huge dataset
which we only want to analyze part of
• This dataset might be stored in S3/equivalent block store
• Minimize data movement by allowing Parquet to support predicate pushdown/projections into S3
• Work is in progress, found at https://github.com/bigdatagenomics/adam/tree/multi-loader
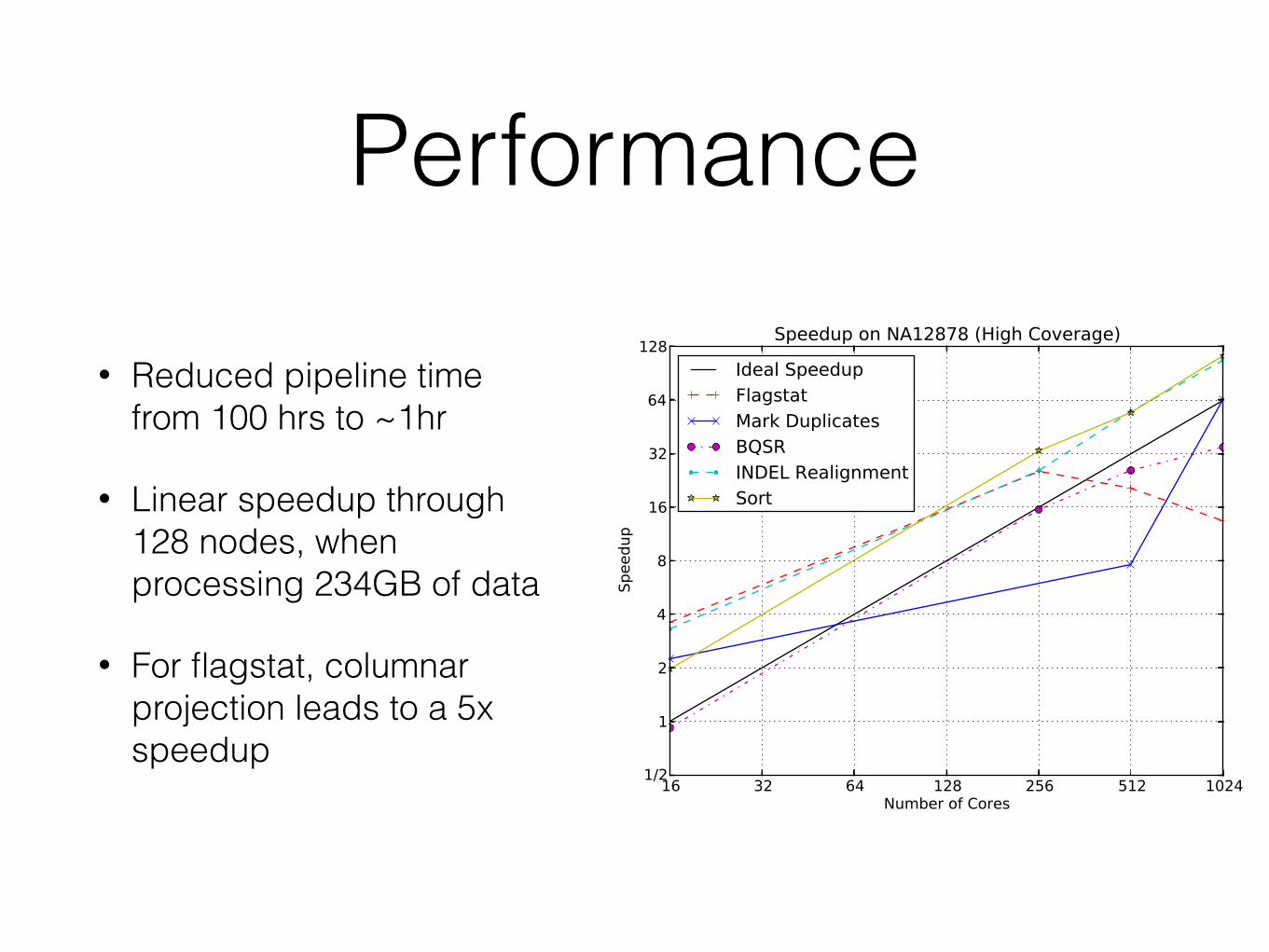
Performance
• Reduced pipeline time from 100 hrs to ~1hr
• Linear speedup through 128 nodes, when processing 234GB of data
• For flagstat, columnar projection leads to a 5x speedup

ADAM Status
• Apache 2 licensed OSS
• 25 contributors across 10 institutions
• Pushing for production 1.0 release towards end of year
• Working with GA4GH to use concepts from ADAM to improve broader genomics data management techniques

Acknowledgements• UC Berkeley: Matt Massie, André Schumacher, Jey Kottalam, Christos
Kozanitis, Dave Patterson, Anthony Joseph
• Mt. Sinai: Arun Ahuja, Neal Sidhwaney, Ryan Williams, Michael Linderman, Jeff Hammerbacher
• GenomeBridge: Timothy Danford, Carl Yeksigian
• The Broad Institute: Chris Hartl
• Cloudera: Uri Laserson
• Microsoft Research: Jeremy Elson, Ravi Pandya
• And other open source contributors, including Michael Heuer, Neil Ferguson, Andy Petrella, Xavier Tordoir!


















