
Novel hexavalent GITR agonists stimulate T cells and enhance memory formation

Article
RuvbL1 and RuvbL2 enhance aggresome formationand disaggregate amyloid fibrilsNava Zaarur1, Xiaobin Xu2, Patrick Lestienne3, Anatoli B Meriin1, Mark McComb2,
Catherine E Costello1,2, Gary P Newnam4, Rakhee Ganti4, Nina V Romanova5,
Maruda Shanmugasundaram6, Sara TN Silva7, Tiago M Bandeiras8, Pedro M Matias7,8,
Kirill S Lobachev4, Igor K Lednev6, Yury O Chernoff4,5,* & Michael Y Sherman1,**
Abstract
The aggresome is an organelle that recruits aggregated proteinsfor storage and degradation. We performed an siRNA screen forproteins involved in aggresome formation and identified novelmammalian AAA+ protein disaggregases RuvbL1 and RuvbL2.Depletion of RuvbL1 or RuvbL2 suppressed aggresome formationand caused buildup of multiple cytoplasmic aggregates. Similarly,downregulation of RuvbL orthologs in yeast suppressed theformation of an aggresome-like body and enhanced the aggregatetoxicity. In contrast, their overproduction enhanced the resistanceto proteotoxic stress independently of chaperone Hsp104.Mammalian RuvbL associated with the aggresome, and theaggresome substrate synphilin-1 interacted directly with theRuvbL1 barrel-like structure near the opening of the central chan-nel. Importantly, polypeptides with unfolded structures andamyloid fibrils stimulated the ATPase activity of RuvbL. Finally,disassembly of protein aggregates was promoted by RuvbL. Thesedata indicate that RuvbL complexes serve as chaperones in proteindisaggregation.
Keywords aggresome; amyloid; disaggregation; RuvbL
Subject Categories Protein Biosynthesis & Quality Control
DOI 10.15252/embj.201591245 | Received 10 February 2015 | Revised 9 July
2015 | Accepted 13 July 2015
Introduction
Molecular chaperones and the ubiquitin-proteasome system (UPS)
promote refolding and degradation of abnormal polypeptides.
However, in aging and disease, these systems fail to repair or
destroy abnormal polypeptides, which then tend to form small cyto-
plasmic aggregates that cause cell toxicity, leading to various
protein misfolding disorders (Sherman & Goldberg, 2001; Meriin &
Sherman, 2005). A special cellular machinery has evolved to trans-
port such aggregates to the centrosome, forming an organelle called
the aggresome (Johnston et al, 1998; Chung et al, 2001; Webb et al,
2004; Corboy et al, 2005). The aggresome serves as a storage
compartment for protein aggregates and may be actively involved in
their refolding and proteasomal or autophagic degradation. It has
been proposed that the aggresome represents a protective cellular
response to the buildup of aggregating abnormal polypeptides that
occurs when chaperones and the UPS fail to handle abnormal
species (Tanaka et al, 2004; Olzmann et al, 2008), for example
during aging or disease.
A number of factors have been implicated in aggresome forma-
tion, including a microtubule-associated histone deacetylase,
HDAC6, PLIC, ataxin 3, Hsp70, or Bag3 (Kawaguchi et al, 2003;
Burnett & Pittman, 2005; Heir et al, 2006; Marx et al, 2007;
Gamerdinger et al, 2011; Zhang & Qian, 2011). Despite these findings,
the basic molecular mechanisms of aggresome formation are poorly
understood.
Here, we developed an unbiased, full-genome siRNA screen for
factors involved in aggresome formation in mammalian cells and
identified more than 100 knockdowns that significantly affect aggre-
some formation. We have focused on a pair of homologous proteins,
RuvbL1 and RuvbL2. These proteins belong to the AAA+ (adenosine
triphosphatases associated with diverse cellular activities) super-
family of ATPases. AAA+ proteins usually form hexameric or dode-
cameric ring structures and are characterized by the presence of the
AAA+ module, which contains the highly conserved Walker A and
1 Department of Biochemistry, Boston University School of Medicine, Boston, MA, USA2 Center for Biomedical Mass Spectrometry, Boston University School of Medicine, Boston, MA, USA3 INSERM U 1053, University of Bordeaux Segalen, Bordeaux, France4 School of Biology, Georgia Institute of Technology, Atlanta, GA, USA5 Laboratory of Amyloid Biology and Institute of Translational Biomedicine, St. Petersburg State University, St. Petersburg, Russia6 Department of Chemistry, University at Albany, State University of New York, Albany, NY, USA7 Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal8 Instituto de Biologia Experimental e Tecnológica, Oeiras, Portugal
*Corresponding author. Tel: +1 404 894 1157; E-mail: [email protected]**Corresponding author. Tel: +1 617 638 5971; E-mail: [email protected]
ª 2015 The Authors The EMBO Journal 1
Published online: August 24, 2015

Walker B motifs responsible for nucleotide binding and hydrolysis,
respectively (Walker et al, 1982). Some AAA+ proteins, for exam-
ple, Hsp104 or ClpB (Doyle & Wickner, 2009; Winkler et al, 2012;
Clare & Saibil, 2013), serve as major molecular chaperones which
can promote disaggregation of protein aggregates, frequently work-
ing together with Hsp70 and Hsp40. Yeast Hsp104 is also involved
in formation of quality control protein deposits (Erjavec et al, 2007)
and controls the fragmentation and propagation of endogenous
amyloids, termed yeast prions (Chernoff et al, 1995). The genes
coding for the chaperones of the Hsp104/ClpB family, while present
in all kingdoms including bacteria, plants, various protists, and
fungi, are absent from the nuclear genomes of Metazoa. Though it
was demonstrated that the chaperone system Hsp70-Hsp40-Hsp110
can promote protein disaggregation in mammalian cells (Bukau
et al, 2006; Winkler et al, 2012; Rampelt et al, 2012; Mattoo &
Goloubinoff, 2013; Torrente & Shorter, 2014), the contribution of this
triad to the overall protein disaggregation has not been defined, and
an AAA+ disaggregase in mammalian cells may have been missed.
RuvbL1 and RuvbL2 proteins share sequence similarity to the
bacterial RuvB helicase (~30%) (Tsaneva et al, 1993; Putnam et al,
2001; Yamada et al, 2001). This similarity suggested that mamma-
lian RuvbL may also be a DNA helicase. In line with this notion,
RuvbL1 was found to be associated with the human replication
protein (RP)A3 (Qiu et al, 1998). Furthermore, DNA helicase activ-
ity of RuvbL complex was demonstrated in in vitro experiments
upon deletion of its auto-inhibitory domain II (Gorynia et al, 2011).
However, a number of publications suggest that RuvbL proteins
may have additional functions unrelated to helicase, since they are
involved in multiple protein complexes, for example, those includ-
ing TATA-binding protein (TBP) (Kanemaki et al, 1997), the large
RNA polymerase II holoenzyme (Qiu et al, 1998), chromatin remod-
eling factors (Shen et al, 2000; Jonsson et al, 2001; Jin et al, 2005;
Bakshi et al, 2006; Choi et al, 2009), certain transcription factors
(Jonsson et al, 2001; Ohdate et al, 2003), telomerase (Venteicher
et al, 2008), or phosphatidylinositol 3-kinase-related protein kinases
(Izumi et al, 2010). Furthermore, several studies have demonstrated
a role for RuvbL proteins in assembly of complexes, suggesting that
they may have chaperone-like activity (Machado-Pinilla et al,
2013). Here, we demonstrate that RuvbL functions as a general
molecular chaperone in protein quality control and facilitate
disaggregation of protein aggregates and amyloids.
Results
siRNA screening for genes involved in aggresome formation
To understand the molecular mechanisms underlying multiple steps
in the process of aggresome formation, we developed a high content
cell-based whole-genome siRNA screen (Fig 1A). We utilized two
reporters for aggresomes, RFP-fused ubiquitin (RFP-Ub), labeling
endogenous ubiquitinated proteins, and synphilin-1 tagged
with GFP (Syn-GFP), both of which were previously shown to
accumulate in aggresomes following treatment with MG132 or other
proteasome inhibitors (Engelender et al, 1999; O’Farrell et al, 2001;
Tanaka et al, 2004; Zaarur et al, 2008).
In the screen, we used HeLa cells co-expressing RFP-Ub and Syn-
GFP (Fig 1B). The entire siRNA library of the human genome was
screened. For each gene, we used a SMARTpool, that is a mix of
four different targeting siRNAs (Fig 1A). After siRNA transfections,
cells were treated with MG132 for 5 h, and the presence of aggre-
somes in different wells was evaluated by high-density microscopy
followed by image analysis (see Materials and Methods). We scored
various phenotypes, including: (i) the absence of an aggresome, (ii)
large multiple aggregates, and (iii) a smaller aggresome. We identi-
fied 425 hits that inhibited aggresome formation in more than 50%
of the cells in at least two out of three replicates. To validate the
hits, we rescreened the hit siRNA pools, by adding each siRNA oligo
from the hit SMARTpool individually in a separate well. Different
oligos from the same SMARTpool were dispensed into distant wells
to avoid local effects. Only hits that showed the inhibition of aggre-
some formation by at least two different oligos (out of 4) in all three
replicates was considered validated. By this approach, we elimi-
nated off-target effects. Images of all hit wells were rechecked
manually to confirm the results of the computer-based image analy-
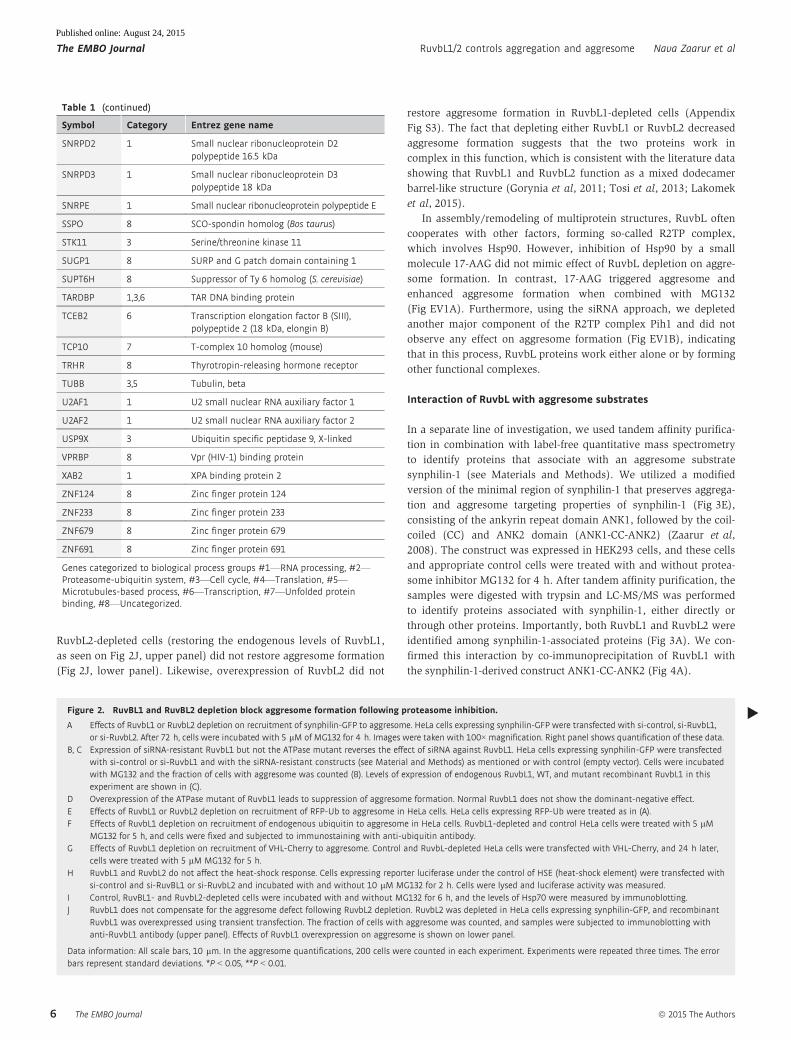
sis. Our validation procedure reduced the list to 164 hits (Table 1),
among which 29 gene knockdowns demonstrated aggresome inhibi-
tion with all four siRNA sequences.
We used a gene functional classification tool from DAVID bio-
informatics software/database (http://david.abcc.ncifcrf.gov), to
classify the validated hit genes. Using the gene ontology category-
biological process, we identified seven main groups in our gene list
(Fig 1C). The largest functional group (24% of the validated genes)
was composed of genes involved in RNA processing. The second
largest group was involved in the cell cycle (13%). Of note, almost
half of this functional group was made up of genes involved in the
proteasome-ubiquitin system.
Surprisingly, a relatively low fraction of the identified genes was
involved in cytoskeleton dynamics or binding unfolded proteins.
Among genes involved in cytoskeleton-dependent transport, we
detected several major components of the dynein motor complex,
including DYNC1H1 (dynein, cytoplasmic 1, heavy chain 1),
DNC1I2 (dynein, cytoplasmic 1, intermediate chain 2), PAFAH1B1
(dynein motor regulator, Lis1), TUBB (tubulin, beta), and ACTR1A
(ARP1, actin-related protein 1 homolog A, centractin alpha)
(Fig 1D). The presence of these “expected” genes in our list
supported the overall validity of the screen and, in addition, identi-
fied specific isoforms of components of the dynein motor complex
involved in aggresome formation.
Depletion of RuvbL1 or RuvbL2 suppresses aggresome formationin mammalian cells
RuvbL1 and RuvbL2, also known as pontin and reptin, respectively,
were two of the 29 genes in which all four siRNA sequences we
tested suppressed aggresome formation (Appendix Fig S1). Their
activity was therefore not due to off-target effects. These proteins
belong to a diverse protein family of AAA+ ATPases, which among
other proteins include molecular chaperones, like Hsp104 or ClpB
(Doyle & Wickner, 2009; Winkler et al, 2012; Clare & Saibil, 2013).
Several reports suggested that RuvbL associates with a number of
multi-protein complexes, such as telomerase, certain RNPs, mTOR,
and related kinases, and serves in their assembly, thus suggesting a
putative chaperone function (Nano & Houry, 2013). We therefore
assessed the role of RuvbL in protein aggregation and aggresome
formation.
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
2
Published online: August 24, 2015

We manually repeated tests to validate the effects of RuvbL
knockdowns using higher microscope magnification. HeLa cells
expressing Syn-GFP were depleted of either RuvbL1 or 2 using
individual siRNA, and the formation of aggresomes following
proteasome inhibition was assessed as in Zaarur et al (2008).
siRNAs against RuvbL1 or RuvbL2 suppress aggresome formation
by 60–70% (Fig 2A). A similar effect was observed with MCF10A
cells (Appendix Fig S2). Effect of RuvbL1 depletion was reversed by
expressing the siRNA-resistant version of recombinant RuvbL1
(Fig 2B and C). Interestingly, expression of the siRNA-resistant
mutant that carries two ATPase-inactivating mutations (A908G and
C915T) was ineffective (Fig 2B and C). Furthermore, unlike overex-
pression of normal RuvbL1, overexpression of the ATPase mutant
RuvbL1 partially inhibited aggresome formation (Fig 2D) and
therefore had a dominant-negative effect, further indicating that the
ATPase activity of RuvbL1 plays an important role in aggresome
formation.
Suppression of aggresome formation by depletion of RuvbL1
or RuvbL2 was also seen with RFP-Ub-decorated endogenous
polypeptides (Fig 2E). Of note, RuvbL1 depletion also reduced
recruitment of endogenous ubiquitin to the aggresome (Fig 2F). To
test whether these effects could be generalized, we conducted a
similar experiment with a different aggresome substrate, VHL-RFP;
recruitment of this polypeptide to the aggresome was strongly
suppressed by depletion of RuvbL1 (Fig 2G). Overall, these experi-
ments indicate that both RuvbL proteins are critical for aggresome
formation by a wide variety of substrates, including misfolded
polypeptides.
In its function in aggresome formation, RuvbL may act indirectly
by regulating expression of major molecular chaperones. We moni-
tored the activity of the heat-shock transcription factor Hsf1 using a
luciferase reporter (Kim et al, 2012) upon depletion of RuvbL1 or
RuvbL2. HeLa cells were infected with lentivirus encoding luciferase
under the control of HSE element in the promoter, and levels of luci-
ferase were measured in control and RuvbL-depleted cells. No
significant difference was observed (Fig 2H), indicating that RuvbL
does not play a role in heat-shock response. Similarly, depletion of
RuvbL proteins did not alter expression levels of the major chaper-
one Hsp70 (Fig 2I). Also, the effects of RuvbL depletion could not
be explained by hypothetical influence of RuvbL on protein degrada-
tion, since they could be seen upon inhibition of both proteasome
and autophagy (see, for example, Fig 6D).
Depletion of RuvbL2 leads to dramatic downregulation of
RuvbL1 (Fig 2J). Mutual regulation of RuvbL1 and RuvbL2 expres-
sion has been reported previously (Venteicher et al, 2008; Izumi
et al, 2010). Notably, overexpression of FLAG-tagged RuvbL1 in the
A
C D
B
Figure 1. High-throughput siRNA screening for aggresome formation.
A A scheme of the siRNA screening procedure as described in Materials and Methods.B Positive and negative controls from the screen. HeLa cells expressing synphilin-GFP and RFP-Ub were transfected with si-control (using anti-luciferase sequence) and
treated with 10 lM MG132 or left untreated. Scale bar, 10 lm.C Validated hits are categorized by gene ontology category-biological process, using “DAVID” bioinformatics resource (http://david.abcc.ncifcrf.gov/).D Defects in aggresome formation caused by siRNA against genes involved in microtubular transport. All cells were treated with 10 lM MG132. Scale bar, 10 lm.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
3
Published online: August 24, 2015
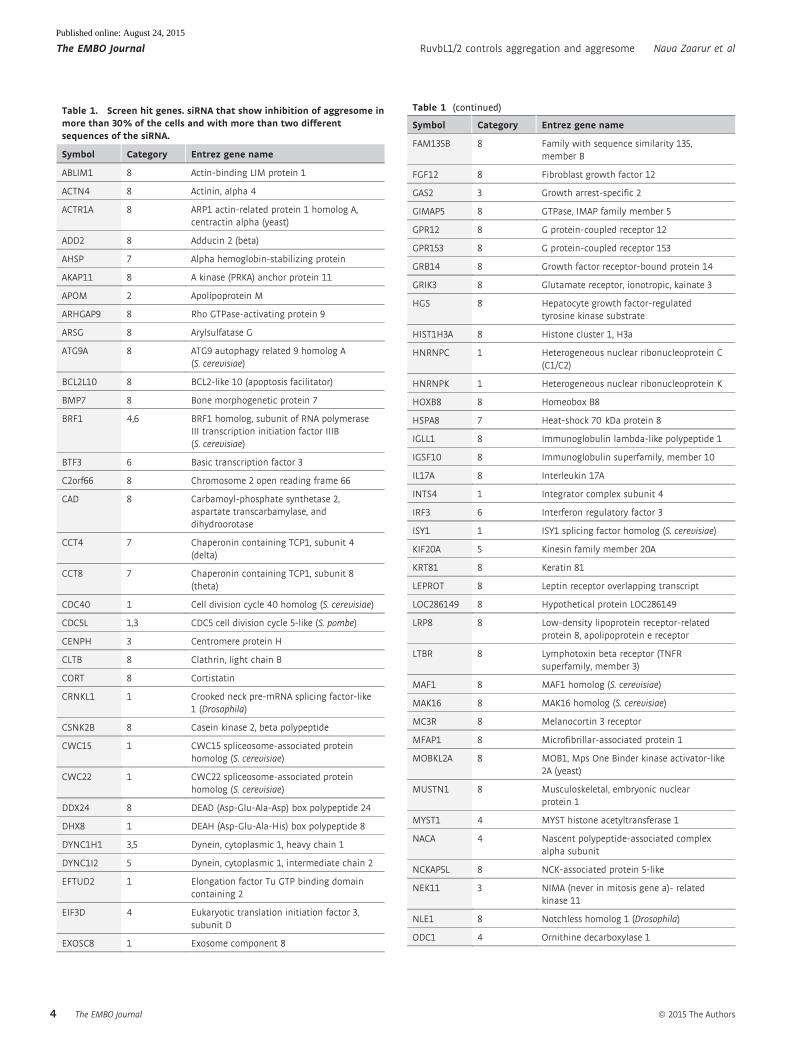
Table 1. Screen hit genes. siRNA that show inhibition of aggresome inmore than 30% of the cells and with more than two differentsequences of the siRNA.
Symbol Category Entrez gene name
ABLIM1 8 Actin-binding LIM protein 1
ACTN4 8 Actinin, alpha 4
ACTR1A 8 ARP1 actin-related protein 1 homolog A,centractin alpha (yeast)
ADD2 8 Adducin 2 (beta)
AHSP 7 Alpha hemoglobin-stabilizing protein
AKAP11 8 A kinase (PRKA) anchor protein 11
APOM 2 Apolipoprotein M
ARHGAP9 8 Rho GTPase-activating protein 9
ARSG 8 Arylsulfatase G
ATG9A 8 ATG9 autophagy related 9 homolog A(S. cerevisiae)
BCL2L10 8 BCL2-like 10 (apoptosis facilitator)
BMP7 8 Bone morphogenetic protein 7
BRF1 4,6 BRF1 homolog, subunit of RNA polymeraseIII transcription initiation factor IIIB(S. cerevisiae)
BTF3 6 Basic transcription factor 3
C2orf66 8 Chromosome 2 open reading frame 66
CAD 8 Carbamoyl-phosphate synthetase 2,aspartate transcarbamylase, anddihydroorotase
CCT4 7 Chaperonin containing TCP1, subunit 4(delta)
CCT8 7 Chaperonin containing TCP1, subunit 8(theta)
CDC40 1 Cell division cycle 40 homolog (S. cerevisiae)
CDC5L 1,3 CDC5 cell division cycle 5-like (S. pombe)
CENPH 3 Centromere protein H
CLTB 8 Clathrin, light chain B
CORT 8 Cortistatin
CRNKL1 1 Crooked neck pre-mRNA splicing factor-like1 (Drosophila)
CSNK2B 8 Casein kinase 2, beta polypeptide
CWC15 1 CWC15 spliceosome-associated proteinhomolog (S. cerevisiae)
CWC22 1 CWC22 spliceosome-associated proteinhomolog (S. cerevisiae)
DDX24 8 DEAD (Asp-Glu-Ala-Asp) box polypeptide 24
DHX8 1 DEAH (Asp-Glu-Ala-His) box polypeptide 8
DYNC1H1 3,5 Dynein, cytoplasmic 1, heavy chain 1
DYNC1I2 5 Dynein, cytoplasmic 1, intermediate chain 2
EFTUD2 1 Elongation factor Tu GTP binding domaincontaining 2
EIF3D 4 Eukaryotic translation initiation factor 3,subunit D
EXOSC8 1 Exosome component 8
Table 1 (continued)
Symbol Category Entrez gene name
FAM135B 8 Family with sequence similarity 135,member B
FGF12 8 Fibroblast growth factor 12
GAS2 3 Growth arrest-specific 2
GIMAP5 8 GTPase, IMAP family member 5
GPR12 8 G protein-coupled receptor 12
GPR153 8 G protein-coupled receptor 153
GRB14 8 Growth factor receptor-bound protein 14
GRIK3 8 Glutamate receptor, ionotropic, kainate 3
HGS 8 Hepatocyte growth factor-regulatedtyrosine kinase substrate
HIST1H3A 8 Histone cluster 1, H3a
HNRNPC 1 Heterogeneous nuclear ribonucleoprotein C(C1/C2)
HNRNPK 1 Heterogeneous nuclear ribonucleoprotein K
HOXB8 8 Homeobox B8
HSPA8 7 Heat-shock 70 kDa protein 8
IGLL1 8 Immunoglobulin lambda-like polypeptide 1
IGSF10 8 Immunoglobulin superfamily, member 10
IL17A 8 Interleukin 17A
INTS4 1 Integrator complex subunit 4
IRF3 6 Interferon regulatory factor 3
ISY1 1 ISY1 splicing factor homolog (S. cerevisiae)
KIF20A 5 Kinesin family member 20A
KRT81 8 Keratin 81
LEPROT 8 Leptin receptor overlapping transcript
LOC286149 8 Hypothetical protein LOC286149
LRP8 8 Low-density lipoprotein receptor-relatedprotein 8, apolipoprotein e receptor
LTBR 8 Lymphotoxin beta receptor (TNFRsuperfamily, member 3)
MAF1 8 MAF1 homolog (S. cerevisiae)
MAK16 8 MAK16 homolog (S. cerevisiae)
MC3R 8 Melanocortin 3 receptor
MFAP1 8 Microfibrillar-associated protein 1
MOBKL2A 8 MOB1, Mps One Binder kinase activator-like2A (yeast)
MUSTN1 8 Musculoskeletal, embryonic nuclearprotein 1
MYST1 4 MYST histone acetyltransferase 1
NACA 4 Nascent polypeptide-associated complexalpha subunit
NCKAP5L 8 NCK-associated protein 5-like
NEK11 3 NIMA (never in mitosis gene a)- relatedkinase 11
NLE1 8 Notchless homolog 1 (Drosophila)
ODC1 4 Ornithine decarboxylase 1
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
4
Published online: August 24, 2015
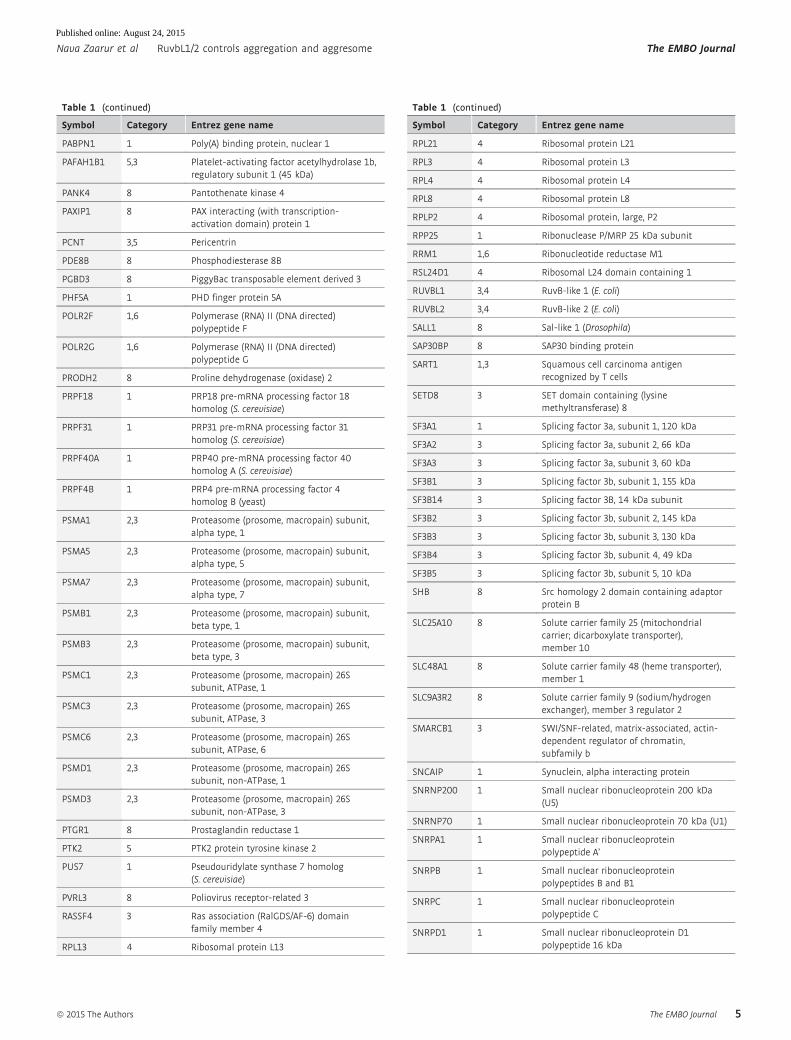
Table 1 (continued)
Symbol Category Entrez gene name
PABPN1 1 Poly(A) binding protein, nuclear 1
PAFAH1B1 5,3 Platelet-activating factor acetylhydrolase 1b,regulatory subunit 1 (45 kDa)
PANK4 8 Pantothenate kinase 4
PAXIP1 8 PAX interacting (with transcription-activation domain) protein 1
PCNT 3,5 Pericentrin
PDE8B 8 Phosphodiesterase 8B
PGBD3 8 PiggyBac transposable element derived 3
PHF5A 1 PHD finger protein 5A
POLR2F 1,6 Polymerase (RNA) II (DNA directed)polypeptide F
POLR2G 1,6 Polymerase (RNA) II (DNA directed)polypeptide G
PRODH2 8 Proline dehydrogenase (oxidase) 2
PRPF18 1 PRP18 pre-mRNA processing factor 18homolog (S. cerevisiae)
PRPF31 1 PRP31 pre-mRNA processing factor 31homolog (S. cerevisiae)
PRPF40A 1 PRP40 pre-mRNA processing factor 40homolog A (S. cerevisiae)
PRPF4B 1 PRP4 pre-mRNA processing factor 4homolog B (yeast)
PSMA1 2,3 Proteasome (prosome, macropain) subunit,alpha type, 1
PSMA5 2,3 Proteasome (prosome, macropain) subunit,alpha type, 5
PSMA7 2,3 Proteasome (prosome, macropain) subunit,alpha type, 7
PSMB1 2,3 Proteasome (prosome, macropain) subunit,beta type, 1
PSMB3 2,3 Proteasome (prosome, macropain) subunit,beta type, 3
PSMC1 2,3 Proteasome (prosome, macropain) 26Ssubunit, ATPase, 1
PSMC3 2,3 Proteasome (prosome, macropain) 26Ssubunit, ATPase, 3
PSMC6 2,3 Proteasome (prosome, macropain) 26Ssubunit, ATPase, 6
PSMD1 2,3 Proteasome (prosome, macropain) 26Ssubunit, non-ATPase, 1
PSMD3 2,3 Proteasome (prosome, macropain) 26Ssubunit, non-ATPase, 3
PTGR1 8 Prostaglandin reductase 1
PTK2 5 PTK2 protein tyrosine kinase 2
PUS7 1 Pseudouridylate synthase 7 homolog(S. cerevisiae)
PVRL3 8 Poliovirus receptor-related 3
RASSF4 3 Ras association (RalGDS/AF-6) domainfamily member 4
RPL13 4 Ribosomal protein L13
Table 1 (continued)
Symbol Category Entrez gene name
RPL21 4 Ribosomal protein L21
RPL3 4 Ribosomal protein L3
RPL4 4 Ribosomal protein L4
RPL8 4 Ribosomal protein L8
RPLP2 4 Ribosomal protein, large, P2
RPP25 1 Ribonuclease P/MRP 25 kDa subunit
RRM1 1,6 Ribonucleotide reductase M1
RSL24D1 4 Ribosomal L24 domain containing 1
RUVBL1 3,4 RuvB-like 1 (E. coli)
RUVBL2 3,4 RuvB-like 2 (E. coli)
SALL1 8 Sal-like 1 (Drosophila)
SAP30BP 8 SAP30 binding protein
SART1 1,3 Squamous cell carcinoma antigenrecognized by T cells
SETD8 3 SET domain containing (lysinemethyltransferase) 8
SF3A1 1 Splicing factor 3a, subunit 1, 120 kDa
SF3A2 3 Splicing factor 3a, subunit 2, 66 kDa
SF3A3 3 Splicing factor 3a, subunit 3, 60 kDa
SF3B1 3 Splicing factor 3b, subunit 1, 155 kDa
SF3B14 3 Splicing factor 3B, 14 kDa subunit
SF3B2 3 Splicing factor 3b, subunit 2, 145 kDa
SF3B3 3 Splicing factor 3b, subunit 3, 130 kDa
SF3B4 3 Splicing factor 3b, subunit 4, 49 kDa
SF3B5 3 Splicing factor 3b, subunit 5, 10 kDa
SHB 8 Src homology 2 domain containing adaptorprotein B
SLC25A10 8 Solute carrier family 25 (mitochondrialcarrier; dicarboxylate transporter),member 10
SLC48A1 8 Solute carrier family 48 (heme transporter),member 1
SLC9A3R2 8 Solute carrier family 9 (sodium/hydrogenexchanger), member 3 regulator 2
SMARCB1 3 SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin,subfamily b
SNCAIP 1 Synuclein, alpha interacting protein
SNRNP200 1 Small nuclear ribonucleoprotein 200 kDa(U5)
SNRNP70 1 Small nuclear ribonucleoprotein 70 kDa (U1)
SNRPA1 1 Small nuclear ribonucleoproteinpolypeptide A’
SNRPB 1 Small nuclear ribonucleoproteinpolypeptides B and B1
SNRPC 1 Small nuclear ribonucleoproteinpolypeptide C
SNRPD1 1 Small nuclear ribonucleoprotein D1polypeptide 16 kDa
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
5
Published online: August 24, 2015
RuvbL2-depleted cells (restoring the endogenous levels of RuvbL1,
as seen on Fig 2J, upper panel) did not restore aggresome formation
(Fig 2J, lower panel). Likewise, overexpression of RuvbL2 did not
restore aggresome formation in RuvbL1-depleted cells (Appendix
Fig S3). The fact that depleting either RuvbL1 or RuvbL2 decreased
aggresome formation suggests that the two proteins work in
complex in this function, which is consistent with the literature data
showing that RuvbL1 and RuvbL2 function as a mixed dodecamer
barrel-like structure (Gorynia et al, 2011; Tosi et al, 2013; Lakomek
et al, 2015).
In assembly/remodeling of multiprotein structures, RuvbL often
cooperates with other factors, forming so-called R2TP complex,
which involves Hsp90. However, inhibition of Hsp90 by a small
molecule 17-AAG did not mimic effect of RuvbL depletion on aggre-
some formation. In contrast, 17-AAG triggered aggresome and
enhanced aggresome formation when combined with MG132
(Fig EV1A). Furthermore, using the siRNA approach, we depleted
another major component of the R2TP complex Pih1 and did not
observe any effect on aggresome formation (Fig EV1B), indicating
that in this process, RuvbL proteins work either alone or by forming
other functional complexes.
Interaction of RuvbL with aggresome substrates
In a separate line of investigation, we used tandem affinity purifica-
tion in combination with label-free quantitative mass spectrometry
to identify proteins that associate with an aggresome substrate
synphilin-1 (see Materials and Methods). We utilized a modified
version of the minimal region of synphilin-1 that preserves aggrega-
tion and aggresome targeting properties of synphilin-1 (Fig 3E),
consisting of the ankyrin repeat domain ANK1, followed by the coil-
coiled (CC) and ANK2 domain (ANK1-CC-ANK2) (Zaarur et al,
2008). The construct was expressed in HEK293 cells, and these cells
and appropriate control cells were treated with and without protea-
some inhibitor MG132 for 4 h. After tandem affinity purification, the
samples were digested with trypsin and LC-MS/MS was performed
to identify proteins associated with synphilin-1, either directly or
through other proteins. Importantly, both RuvbL1 and RuvbL2 were
identified among synphilin-1-associated proteins (Fig 3A). We con-
firmed this interaction by co-immunoprecipitation of RuvbL1 with
the synphilin-1-derived construct ANK1-CC-ANK2 (Fig 4A).
Table 1 (continued)
Symbol Category Entrez gene name
SNRPD2 1 Small nuclear ribonucleoprotein D2polypeptide 16.5 kDa
SNRPD3 1 Small nuclear ribonucleoprotein D3polypeptide 18 kDa
SNRPE 1 Small nuclear ribonucleoprotein polypeptide E
SSPO 8 SCO-spondin homolog (Bos taurus)
STK11 3 Serine/threonine kinase 11
SUGP1 8 SURP and G patch domain containing 1
SUPT6H 8 Suppressor of Ty 6 homolog (S. cerevisiae)
TARDBP 1,3,6 TAR DNA binding protein
TCEB2 6 Transcription elongation factor B (SIII),polypeptide 2 (18 kDa, elongin B)
TCP10 7 T-complex 10 homolog (mouse)
TRHR 8 Thyrotropin-releasing hormone receptor
TUBB 3,5 Tubulin, beta
U2AF1 1 U2 small nuclear RNA auxiliary factor 1
U2AF2 1 U2 small nuclear RNA auxiliary factor 2
USP9X 3 Ubiquitin specific peptidase 9, X-linked
VPRBP 8 Vpr (HIV-1) binding protein
XAB2 1 XPA binding protein 2
ZNF124 8 Zinc finger protein 124
ZNF233 8 Zinc finger protein 233
ZNF679 8 Zinc finger protein 679
ZNF691 8 Zinc finger protein 691
Genes categorized to biological process groups #1—RNA processing, #2—Proteasome-ubiquitin system, #3—Cell cycle, #4—Translation, #5—Microtubules-based process, #6—Transcription, #7—Unfolded proteinbinding, #8—Uncategorized.
▸Figure 2. RuvBL1 and RuvBL2 depletion block aggresome formation following proteasome inhibition.
A Effects of RuvbL1 or RuvbL2 depletion on recruitment of synphilin-GFP to aggresome. HeLa cells expressing synphilin-GFP were transfected with si-control, si-RuvbL1,or si-RuvbL2. After 72 h, cells were incubated with 5 lM of MG132 for 4 h. Images were taken with 100×magnification. Right panel shows quantification of these data.
B, C Expression of siRNA-resistant RuvbL1 but not the ATPase mutant reverses the effect of siRNA against RuvbL1. HeLa cells expressing synphilin-GFP were transfectedwith si-control or si-RuvbL1 and with the siRNA-resistant constructs (see Material and Methods) as mentioned or with control (empty vector). Cells were incubatedwith MG132 and the fraction of cells with aggresome was counted (B). Levels of expression of endogenous RuvbL1, WT, and mutant recombinant RuvbL1 in thisexperiment are shown in (C).
D Overexpression of the ATPase mutant of RuvbL1 leads to suppression of aggresome formation. Normal RuvbL1 does not show the dominant-negative effect.E Effects of RuvbL1 or RuvbL2 depletion on recruitment of RFP-Ub to aggresome in HeLa cells. HeLa cells expressing RFP-Ub were treated as in (A).F Effects of RuvbL1 depletion on recruitment of endogenous ubiquitin to aggresome in HeLa cells. RuvbL1-depleted and control HeLa cells were treated with 5 lM
MG132 for 5 h, and cells were fixed and subjected to immunostaining with anti-ubiquitin antibody.G Effects of RuvbL1 depletion on recruitment of VHL-Cherry to aggresome. Control and RuvbL-depleted HeLa cells were transfected with VHL-Cherry, and 24 h later,
cells were treated with 5 lM MG132 for 5 h.H RuvbL1 and RuvbL2 do not affect the heat-shock response. Cells expressing reporter luciferase under the control of HSE (heat-shock element) were transfected with
si-control and si-RuvBL1 or si-RuvbL2 and incubated with and without 10 lM MG132 for 2 h. Cells were lysed and luciferase activity was measured.I Control, RuvBL1- and RuvbL2-depleted cells were incubated with and without MG132 for 6 h, and the levels of Hsp70 were measured by immunoblotting.J RuvbL1 does not compensate for the aggresome defect following RuvbL2 depletion. RuvbL2 was depleted in HeLa cells expressing synphilin-GFP, and recombinant
RuvbL1 was overexpressed using transient transfection. The fraction of cells with aggresome was counted, and samples were subjected to immunoblotting withanti-RuvbL1 antibody (upper panel). Effects of RuvbL1 overexpression on aggresome is shown on lower panel.
Data information: All scale bars, 10 lm. In the aggresome quantifications, 200 cells were counted in each experiment. Experiments were repeated three times. The errorbars represent standard deviations. *P < 0.05, **P < 0.01.
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
6
Published online: August 24, 2015

A
B
E
F
H
I
J
G
C D
Figure 2.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
7
Published online: August 24, 2015

To identify direct interactors with synphilin-1, we combined
mass spectrometry with chemical crosslinking (Fig 3B). ANK1-CC-
ANK2 with associated proteins was isolated from HEK293 cells, as
described above, and crosslinked on the affinity beads using BS3-
H12/D12 (bissulfosuccinimidylsuberate). The cross-linked proteins
were eluted from the affinity beads, digested by trypsin, and
analyzed by LC-MS/MS. The crosslinking analysis indicated that
RuvbL1 directly interacts with synphilin-1, and located the interac-
tion site, since synphilin-1K663 was linked to RuvbL1K372 (Fig 3C).
This was the only site of crosslinking between these two proteins.
We mapped the site involved in this interaction on the 3D structure
of RuvbL1 and determined that RuvbL1 interacts with synphilin
through one of its side chains, located on the surface of the ATPase
core and in proximity to the central channel (Fig 3D). Interestingly,
K663 residue critical for interaction with RuvbL1 is located in the
ankyrin repeat domain (ANK2) of synphilin-1 (Fig 3E, see arrow),
which is responsible for its aggregation (Zaarur et al, 2008). We
further built upon the uncovered structural information by testing
whether interaction with the substrate is important for the effect of
RuvbL on aggresome formation.
Functional importance of interaction of RuvbL with substratesfor aggresome formation
Previously, we demonstrated that the CC and ANK2 domains of
synphilin-1 are responsible for aggregation of this protein, while the
adjacent ANK1 domain is necessary for its aggresome targeting
(Zaarur et al, 2008). Accordingly, deletion of either CC or ANK2
from the ANK1-CC-ANK2 construct led to significantly reduced
aggregation. Yet, due to the presence of the ANK1 domain, either
deletion construct could be targeted to the aggresome, though signif-
icantly less efficiently than constructs that have all three domains.
Since the RuvbL binding site is located in the ANK2 region (see
above), first, we confirmed that deletion of the ANK2 region indeed
eliminated the association of RuvbL1 with the construct (Fig 4A).
Next, we asked whether the deletion construct lacking this region
and thus unable to bind to RuvbL remains sensitive to its depletion.
To test the role of synphilin-RuvbL interaction in aggresome
formation, HEK293 cells expressing ANK1-CC-ANK2-GFP and
ANK1-CC-GFP constructs were depleted of RuvbL1 and treated with
MG132 for 7 h. Upon the depletion, the efficiency of aggresome
formation by the full-length and ANK1-CC-ANK2-GFP constructs
dropped from 85 to 25%. On the other hand, the ANK1-CC-GFP
construct formed aggresomes in about 25% of control cells, and
RuvbL1 depletion did not significantly affect this fraction (Fig 4B).
Therefore, deletion of the RuvbL-interacting region from synphilin-1
decreased aggresome formation and made it insensitive to RuvbL,
demonstrating that interaction between synphilin-1 and RuvbL is
critical for efficient aggresome targeting of synphilin-1. These data
further support direct role of RuvbL proteins in the assembly and/or
disassembly of the aggresome.
We further tested whether RuvbL localizes to the aggresome.
FLAG-tagged RuvbL1 or HA-tagged RuvbL2 was co-expressed with
synphilin-GFP in HeLa cells, and the cells were treated with
MG132 for 2 h. In contrast to the diffuse distribution of RuvbL
proteins throughout the cytoplasm and nucleus in naı̈ve cells
(Appendix Fig S4), both RuvbL1 and RuvbL2 were recruited to
aggresomes following proteasome inhibition (Fig 4C), which was
consistent with their involvement in aggresome assembly and/or
disassembly. Furthermore, when transport of aggregates to the
aggresome was blocked by nocodazole, RuvbL localized to multi-
ple aggregates formed throughout the cytosol (Fig 4D), suggesting
that it is recruited to protein aggregates in the process of aggre-
some formation.
Direct association with aggresome and direct effect on aggresome
assembly or disassembly suggests that RuvbL may play a role in
protein aggregation or disaggregation. Indeed, microscopic observa-
tion of mammalian cells depleted of RuvbL and expressing either
Syn-GFP or VHL-GFP upon proteasome inhibition revealed that
many cells formed multiple small cytoplasmic aggregates that were
not recruited to aggresome (see Fig 2). To test for the effects of
RuvbL on the extent of protein aggregation, control and RuvbL1- or
RuvbL2-depleted HeLa cells expressing Syn-GFP were treated
with MG132. Cell lysates were fractionated by centrifugation at
13,000 g, and amounts of Syn-GFP in pellets and supernatants were
measured by immunoblotting. In the RuvbL1-depleted cells (both in
the presence and in the absence of MG132), the presence of Syn-
GFP in the pellet relative to supernatant was increased by about
two-fold (Fig 4E). Therefore, suppression of aggresome formation in
the RuvbL-depleted cells probably results from excessive protein
aggregation. These aggregates are either so large or so numerous
that they overwhelm the aggresome machinery. Such excessive
aggregation could result from a reduced ability of cells to refold or
disaggregate abnormal proteins. Taking into consideration the fact
that RuvbL proteins belong to the AAA+ protein family and that they
are involved in assembly of many protein complexes, these data
Figure 3. RuvbL1 and RuvBL2 interact with synphilin-1 directly.
A His-ANK1-CC-ANK2-GFP was expressed in HEK293 cells and isolated using cobalt affinity column, followed by anti-GFP antibody affinity column. Samples weretrypsin-digested and subjected to LC-MS/MS analysis. Results were analyzed, identified, and quantified using Progenesis LCMS and Mascot. The following RuvbL1peptides were identified: Y405-K418 (shown), and A318-R333, E47-K57, V358-K372, M61-K76, T401-K418, K60-K76, R118-K125, V91-K108, and T109-R123 (not shown).RuvbL2 peptides: V428-R437 (shown), A314-R329, G29-R39, T164-K183, Q40-R52, T115-R124, and S438-K455 (not shown).
B Work flow of determination of synphilin-1 binary interactions using isotopically labeled cross-linking and mass spectrometry.C MS2 spectra of the cross-linked peptide between synphilin-1 and RuvbL1. The orange peaks show the MS2 spectrum of the BS3-H12 cross-linked peptide, while
purple peaks show the MS2 spectrum of the BS3-D12 cross-linked peptide. The isotopically tagged cross-linkers were used to facilitate the cross-linked peptideidentification. The a peptide chain is the peptide R660-R666 from SNCAP (synphilin-1), and the b peptide chain is the peptide T363-K376 from RUVB1 (RuvbL1). Theions are classed as b ions when the charges are retained on the N-terminal fragment, and the ions are classed as y ions when the charged are retained on C-terminalfragment. Fragment ions without the cross-linker are labeled in green, and fragment ions with the cross-linker are labeled in red. The cross-linking sites weredetermined as the K663 residue on synphilin-1 and the K372 residue on RuvbL1.
D The cross-linking site on RuvbL1K372 is located at the surface of the barrel structure. The image was re-constructed from human RuvbL1 crystal structure (Matiaset al, 2006) using Protein Workshop (www.rcsb.org).
E The cross-linked site on synphilin-1 K663 is located at its ANK2 domain.
▸
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
8
Published online: August 24, 2015

suggest that RuvbL may serve a chaperone function in the assembly
or disassembly of protein aggregates.
A
C
E
B
D
Figure 3.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
9
Published online: August 24, 2015

Protein-stimulated ATPase activity of RuvbL
ATPase activity of the chaperones can be stimulated by their
polypeptide substrates, usually measured with model polypeptides,
such as casein (Woo et al, 1992; Schirmer & Lindquist, 1997).
Accordingly, we tested whether ATPase of RuvbL could be stimu-
lated by these substrates. Dodecamer RuvbL complex consisting of
two heterohexameric rings with alternating RuvbL1 and RuvbL2
monomers was purified as previously described (Gorynia et al,
2011) and used in the in vitro ATPase assay. Indeed, casein signifi-
cantly stimulated the ATPase activity of the RuvbL complex in a
dose-dependent manner (Fig 5A).
It was recently shown that domain II of both RuvbL1 and
RuvbL2 is auto-inhibitory, and its deletion enhances the ATPase
activity (Gorynia et al, 2011). Here, we tested whether deletion of
domain II affects the protein stimulation of the ATPase activity.
Accordingly, we compared casein stimulation of the ATPase of
normal and mutant RuvbL complex with deletion of the domain II
in both RuvbL1 and RuvbL2. As seen in Fig 5A, removal of the
auto-inhibitory domain II indeed enhances the stimulation of the
ATPase activity by casein. Interestingly, the protein stimulation of
the ATPase activity could also be observed with monomeric
RuvbL1 or RuvbL2 that was purified as previously described
(Gorynia et al, 2011) (Fig 5B), suggesting that interaction with
A B
C
D
E
Figure 4. Association of RuvbL with substrates is important for aggresome formation.
A Deletion of the ANK2 domain blocks association of synphilin-1 with RuvbL1. HEK293 cells were transfected with Flag-RuvbL1 and either with GFP (Cont.), full-lengthsynphilin-GFP (FL), ANK1-CC-ANK2-GFP, or ANK1-CC-GFP constructs. Cells were lysed and subjected to immunoprecipitation with anti-GFP antibody. AssociatedRuvbL1 was detected by immunoblotting with anti-RuvBL1 antibody.
B Deletion of the ANK2 domain relieves the RuvbL1 dependence of the recruitment to aggresome. HEK293 cells si-control and si-RuvbL1 were transfected with theindicated synphilin-1 deletion constructs and were treated with MG132 for 3 h. Fraction of cells with aggresome was evaluated; error bars represent standarddeviation of three repeats. **P < 0.01.
C RuvbL1 and RuvbL2 colocalize with aggresomes. HeLa cells expressing synphilin-GFP were transfected either with Flag-RuvBL1 or HA-RuvBL2. Cells were incubatedwith MG132 for 6 h, treated with 0.5% Triton X-100 for 20 s and immediately fixed. The cells were immunostained with anti-HA and anti-Flag antibodies. Scale bar:10 lm.
D RuvbL1 co-localizes with protein aggregates when transport to aggresome is blocked by nocodazole. HeLa cells transfected with Flag-RuvbL1 were incubated with5 lM MG132 and 10 lM nocodazole for 2 h and then fixed and immunostained with anti-Flag antibody.
E Effect of RuvBL1 depletion on synphilin-1 aggregation. RuvBL1-depleted and control cells were incubated with and without MG132 for 4 h. Soluble proteins andaggregates were separated by centrifugation for 15 min at 13,000 g. Pellets were 3.5 times more concentrated compared to the supernatant. The samples wereblotted with anti-GFP antibody (upper panel). Results of a typical experiment are shown. In three independent experiments, bands were quantified with ImageJ, andthe ratio of synphilin in pellets and total fractions was calculated (lower panel). Error bars represent standard deviations of the repeats.
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
10
Published online: August 24, 2015

substrate proteins may not require an assembly of the heterodode-
cameric structure.
Insulin is another polypeptide that stimulated the ATPase activity
of Hsp104 (Woo et al, 1992; Schirmer & Lindquist, 1997), and simi-
larly, it stimulated the ATPase of the RuvbL complex (Fig 5C).
Insulin is an amyloidogenic protein that can effectively form
amyloid fibrils under low pH conditions (Groenning et al, 2009).
Importantly, insulin fibrils have shown significantly stronger stimu-
latory effects on the RuvbL ATPase activity compared to soluble
insulin (Fig 5C).
Further, we compared effects of an amyloidogenic peptide
Ab1–42 on the ATPase activity of the RuvbL complex. The peptide
monomer significantly stimulated the ATPase activity (Fig 5D).
Importantly, the preformed Ab amyloid fibrils stimulated the
ATPase activity much stronger (almost four-fold). It should be
noted, that during the time of measurement of the ATPase activity,
the Ab monomer has partially polymerized (Appendix Fig S5, see
also Fig 6A). Therefore, the ATPase stimulation by the monomer is
overestimated, and the difference between effects of the amyloid
and the monomer could be even stronger. Thus, both soluble and
aggregated proteins can interact with RuvbL complex and stimulate
its ATPase activity, and amyloid fibrils have stronger effects
compared with the unaggregated forms of the corresponding
polypeptides. These data are consistent with the chaperone function
of RuvbL and further indicate that direct interaction of substrates
with RuvbL1 is functionally important.
RuvbL-stimulated remodeling of protein aggregates
To address effects of RuvbL on dynamics of protein aggregation, we
assessed how purified dodecamer RuvbL complex affects formation
of Ab fibrils by measuring thioflavin fluorescence, which is
enhanced upon binding to b-sheets, formed within aggregates. The
first step of Ab amyloidogenesis, seeding of fibrils, is a slow process,
A
C
B
D
Figure 5. Protein-stimulated ATPase activity of RuvBL.
A Effect of casein on the ATPase activity of the normal and mutant (deletion of domain II) RuvbL complexes.B Casein can stimulate the ATPase activities of monomeric RuvbL1 and RuvbL2.C Effect of soluble insulin and insulin fibrils on the ATPase activity of the RuvbL complex (see Materials and Methods).D Effect of 1–42 Ab peptide and Ab fibrils on the ATPase activity of the RuvbL complex.
Data information: All experiments were repeated at least three times; error bars represent standard deviations of the repeats.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
11
Published online: August 24, 2015

A
C
D
E
F G
B
Figure 6.
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
12
Published online: August 24, 2015

which is followed by a faster process of fibril growth. Addition of
RuvbL without ATP significantly delayed fibril seeding and reduced
the rate of fibril growth (Fig 6A). This effect could not be through
interaction of RuvbL with Ab monomers, since there was almost a
2,000× molar excess of monomers compared to the RuvbL complex.
It is likely that in the absence of ATP, RuvbL binds to oligomeric
seeds and fibrils and prevents their growth. Addition of ATP
reverses this effect of seeding suppression or fibril growth (Fig 6A
and B), suggesting that in the presence of ATP either interaction of
RuvbL with the seeds is reduced or seed multiplication is promoted
(both outcomes were seen with other chaperones). These scenarios
agree with the data pointing to direct interaction between RuvbL
and Ab. Of note, we could not investigate effects of RuvbL on
insulin fibril formation, since this process takes place at low pH
conditions, inactivating RuvbL.
Further, we tested whether RuvbL cooperates with Hsp70 and
Hsp40 (analogous to Hsp70/Hsp40/Hsp104 chaperone system) in
suppression of Ab fibril formation. In the absence of RuvbL, the
Hsp70/Hsp40 chaperone pair also delayed seeding and reduced the
rate of fibril growth. Addition of RuvbL did not further suppress fib-
ril formation (Fig EV2A). Therefore, in this reaction, RuvbL does
not cooperate with Hsp70/Hsp40, and furthermore because of the
lack of additivity, probably these chaperones interact with the same
sites on the seeds to suppress Ab polymerization. Also, stimulation
of ATPase activity of Hsp70/Hsp40 and RuvbL by casein is not addi-
tive (Fig EV2B).
To address activity of RuvbL in protein disaggregation, we used
preformed insulin fibrils (experiments with Ab fibrils were
complicated by instability of thioflavin fluorescence and fibril
heterogeneity). We evaluated fragmentation of insulin fibrils by
atomic force microscopy. The chaperone-dependent deconstruction
of insulin fibrils is known to start with fibril swelling, followed by
disintegration into shorter fragments or amorphous aggregates of
irregular size (Kurouski et al, 2012, 2013; Min et al, 2014). In
control samples without RuvbL complexes, fibrils remained intact
throughout the incubation period (Fig 6C). Incubation with RuvbL
in the absence of ATP led to significant swelling of fibrils indicating
association of RuvbL with the fibrils or partial loss of structure.
Importantly, incubation with RuvbL in the presence of ATP led to
conversion of the insulin fibrils into amorphous aggregates of irreg-
ular size, although some remaining swollen fibrils were still seen
(Fig 6C). Therefore, as with Ab, RuvbL interacted with insulin
fibrils even in the absence of ATP, and ATP addition stimulated
fibril remodeling.
To address the disaggregation effects of RuvbL in vivo, we
followed disappearance of the preformed cellular aggregates after
washout of the proteasome inhibitor. In order to exclude the auto-
phagic component in the disappearance of aggregates, the experiment
was done in the presence of the autophagy inhibitor bafilomycin
(see Materials and Methods). Under these conditions, the recovery
of aggregates was most likely due to remodeling followed by disag-
gregation, and then by either refolding or proteasome degradation
of solubilized protein molecules. An additional complexity of this
experiment is that unlike control cells that form single aggresome in
response to MG132, RuvbL-depleted cells form multiple aggregates
(see above). Therefore, to follow disappearance of comparable
aggregate structures, we blocked aggresome formation by the micro-
tubule inhibitor nocodazole. Control and RuvbL1-depleted cells
were incubated with nocodazole and MG132 for 2 h, which resulted
in formation of multiple aggregates in both cultures (Fig 6D). Then
MG132 and nocodazole were washed out, and disappearance of the
aggregates was followed microscopically. In control cells, upon
recovery from the inhibitors, multiple aggregates proceeded to
aggresome within 1–2 h in almost every cell. These aggresomes
then gradually disappeared within the next 2 h. However, the fate
of aggregates in the RuvbL-depleted cells was different: upon
removal of MG132 and nocodazole, aggresomes appeared much
slower than in control in only about 30% of cells, while multiple
aggregates remained in the rest of cells. Importantly, the disappear-
ance of aggregates from cells was not seen at least in the course of
5 h following the removal of MG132 (Fig 6D and E). Similar
suppression of disaggregation in RuvbL-depleted cells was seen with
ubiquitinated proteins (they co-localized with synphilin aggregates)
(Fig EV4).
To exclude aggregates recruitment into aggresome upon removal
of MG132 and nocodazole, we followed their disappearance in cells
depleted of dynein subunit DYNC1H1. In our screen, this depletion
resulted in strong aggresome suppression and accumulation of small
aggregates (Fig 1D). This alternative way to inhibit aggresome is
much less toxic than continued incubation with nocodazole after
removal of MG132. Accordingly, cells were transfected with
DYNC1H1 siRNA with or without RuvbL1 siRNA. Aggregates were
◀ Figure 6. RuvBL1/2 complex suppresses formation and promotes deconstruction of protein aggregates.
A RuvbL suppresses 1–42Ab fibrillation. RuvbL complex was incubated with Ab peptide and thioflavin with or without ATP for 8 h, and fluorescence was read every6 min.
B Seeding time, Tlag, and fibrillation rate, K1/2, were calculated by fitting sigmoidal equation.C Deconstruction of insulin fibril by the RuvbL complex was performed using atomic force microscopy (see Materials and Methods). AFM images showing swelling of
amyloid fibrils by RuvbL without ATP and further deconstruction in the presence of ATP. Scale bar, 500 nm.D, E Effects of RuvbL on aggregates during recovery from proteasome inhibition. Control and RuvBL1-depleted HeLa cells expressing synphilin-GFP were incubated
with 10 lM nocodazole and 5 lM MG132. After 2 h, cells were washed four times with PBS and incubated with 10 lM emetine and 1 lM bafilomycin for theindicated time periods. Time “0” is the time of the start of recovery. Scale bar, 10 lm. At indicated times, the cells with aggresome, multiple aggregates, anddiffused synphilin were counted and the quantification of the experiment is shown in (D). Cells with aggregates are cells with either aggresome or multipleaggregates.
F Effect of RuvbL on recovery of aggregates in the absence of aggresome. HeLa cells expressing synphilin-GFP were transfected with si-control and si-DYNC1H1 orwith si-RuvbL1 and si-DYNC1H1. After 72 h, cells were incubated with 10 lM MG132 for 2 h, followed by the washout of the inhibitor. Time “0” is the time of thestart of recovery in the presence of 10 lM emetine and 1 lM bafilomycin. For quantification, percentages of cells with aggregates were evaluated.
G Depletion of RuvbL1 sensitizes to proteotoxic stress. Cells were transfected with control siRNA or si-RuvbL1 and incubated with MG132 or AZC for 5 h followed by10 h of recovery. Viability of cells was measured by CellTiter 96 (Promega). The experiment was done in triplicates, error bars represent standard deviations. *P < 0.05.
Data information: In (E) and (F), 200 cells were counted in three different experiments. Error bars represent standard deviations of the repeats.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
13
Published online: August 24, 2015

induced with MG132, and the inhibitor was removed to allow for
the disappearance of aggregates. This experiment demonstrated that
RuvbL1 depletion significantly reduced the rate of aggregate disap-
pearance in cells (Fig 6F), and thus, RuvbL is a chaperone involved
in disassembly of protein aggregates.
The role of RuvbL in the aggregate disassembly suggests that it
may provide protection from proteotoxic stresses. Accordingly,
depletion of RuvbL may make cells more sensitive to treatments that
cause proteotoxicity, like inhibition of the proteasome or incubation
with amino acid analogs, which incorporate into polypeptides and
prevent normal folding. To study effects of RuvbL on cell physiol-
ogy, we treated a neuron-related cell line SY5Y with 10 lM MG132
or 10 lM of a proline analog azetidine-2-carboxylate (AZC) for 5 h,
washed out these reagents, and assessed cell viability after 10-h
recovery. These treatments were very mild and did not cause toxic-
ity in control cells, while in RuvbL1-depleted cells, a significant toxi-
city was seen (Fig 6G). Therefore, in line with its chaperone
function, endogenous RuvbL plays a role in survival of proteotoxic
stress.
RuvbL orthologs influence aggregate toxicity and formation ofaggresome-like structures in yeast
We tested whether yeast orthologs of RuvbL1 and 2 (respectively,
Rvb1 and Rvb2) are involved in the formation of aggresome-like
structures in yeast. To study protein aggregation, we chose a widely
used model of polyglutamine-containing polypeptide 103QP, which
we developed in the past (Meriin et al, 2002; Wang et al, 2009).
The 103QP construct includes exon 1 of the human huntingtin
protein with the expanded polyQ region, corresponding to the
severe form of Huntington’s disease. Due to the presence of
the proline-rich (P) region immediately following the polyQ region,
the 103QP protein is sequestered into an aggresome-like body
(Wang et al, 2009), a process similar to the recruitment of amyloid
aggregates into the IPOD compartment (Kaganovich et al, 2008).
First, we tested whether Rvb1 and Rvb2 proteins colocalize with
the aggresome-like body in yeast. Accordingly, we employed strains
in which endogenous RVB1 and RVB2 genes were fused with the
GFP ORF. These strains have been transformed with the plasmid
bearing 103QP-RFP. Indeed, both Rvb1-GFP and Rvb2-GFP localized
at single bodies in a significant fraction of yeast cells, and these
bodies colocalized with 103QP-RFP aggresome-like structures in
more than half of the cells (Fig 7A). These data indicated that, like
their mammalian counterparts, Rvb1 and Rvb2 proteins are local-
ized to aggresome-like bodies in yeast, which is consistent with their
role in aggresome formation.
To investigate the effects of Rvb1 and Rvb2 on formation of the
aggresome-like bodies, we depleted either of these proteins in yeast.
Since both RVB1 and RVB2 are essential for cell viability, we have
employed yeast strains in which a single copy of either gene is
placed under the control of the tetracycline-regulated promoter
(PTET). In these strains, expression of a particular gene can be down-
regulated by adding the tetracycline-related antibiotic doxycycline.
We used it at concentrations below 100 ng/ml to allow partial
depletion of the corresponding proteins without growth inhibition
(Appendix Fig S6). Microscopic observation showed that downregu-
lation of either RVB1 or RVB2 gene led to a significant reduction in
the proportion of cells containing large aggresome-like structures
with multiple aggregates being formed instead (Fig 7B and C). This
result indicates that similar to their mammalian counterparts, Rvb1
and Rvb2 proteins are involved in targeting misfolded proteins to
aggresome-like structures in yeast.
Previously, we demonstrated that the 103QP polypeptide usually
does not cause toxicity in yeast strains. However, either alteration of
protein sequestration patterns of 103QP (Wang et al, 2009; Gong
et al, 2012) or inhibition of the recruitment of 103QP to aggresome-
like bodies (Wang et al, 2009; Gong et al, 2012) triggered its toxicity.
In line with these findings, partial downregulation of either RVB1 or
RVB2, which suppressed targeting of 103QP to the aggresome-like
body, resulted in high toxicity of 103QP (Fig 7D). Notably, this
toxicity was caused by the expanded polyglutamine domain, since a
short polyglutamine polypeptide 25QP remained nontoxic. There-
fore, Rvb proteins appear to specifically control toxicity of poly-
peptides with expanded polyglutamine via regulation of their
aggregation status. Furthermore, consistent with their role
in proteostasis, mild downregulation of Rvb1 or Rvb2 increased
the sensitivity of yeast cells to the antibiotic hygromycin, which
causes accumulation of misfolded endogenous proteins in the cell
(Fig 7E).
According to the current paradigm, heat shock of cells causes
massive misfolding and aggregation of proteins, leading to cell
death, and the Hsp104 chaperone is the major factor counteracting
Figure 7. Rvb1 and Rvb2 influence protein aggregation and proteotoxicity in yeast.
A Rvb1-GFP and Rvb2-GFP colocalize with the aggresome-like structures in yeast. The Rvb1-GFP or Rvb2-GFP was co-expressed with 103QP-RFP.B Underexpression of Rvb1 or Rvb2 in the presence of moderate (in this case, 25 ng/ml) concentrations of doxycycline affects formation of the aggresome-like body in
cells expressing 103QP. 103QP was induced in the galactose medium for 6 h. Data obtained in a representative experiment are shown. Experiments were repeated 5times with variable concentrations of doxycycline. Numbers varied among the experiments, but effects of RVB downregulation relative to wild-type were similar.
C Appearance of the aggresome-like body and multiple aggregates.D Effects of RVB1 or RVB2 downregulation on sensitivity to protein aggregation. The 103QP construct becomes toxic to the PTET-RVB1 and PTET-RVB2 strains in the
presence of moderate concentrations (75 ng/ml) of doxycycline, which are not sufficient to inhibit growth in the presence of 25QP control. Growth inhibition by103QP is more severe in the PTET-RVB1 strain, compared to PTET-RVB2. All polyQ constructs were expressed from the galactose-inducible (PGAL) promoter.
E Downregulation of PTET-RVB1 or PTET-RVB2 by 25 ng/ml of doxycycline (Doxy) causes sensitivity to low concentration (20 lg/ml) of hygromycin (Hyg); in the sameconditions, growth of the wild-type (WT) strain is not inhibited.
F Exponential yeast cultures grown at 25°C and bearing multicopy plasmids with either RVB1 or RVB2 gene under their own promoters are more resistant to heat shockat 50°C, compared to the culture of the same strain bearing the control plasmid. Yeast cultures were plated onto �Ura medium, in order to monitor only plasmid-containing cells. Experiments were performed in triplicates.
G Excess Rvb1 or Rvb2 partially compensates for thermotolerance in the hsp104 deletion (hsp104Ä) mutant. Data shown are for the strains of BY series; major resultswere also reproduced for the strains of GT81 series (for strain description, see Appendix Supplementary Methods). Experiments were performed in triplicates.
Data information: Error bars represent standard deviations of the repeats.
▸
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
14
Published online: August 24, 2015

heat-shock-induced aggregation and cell death in yeast (Glover &
Lindquist, 1998). Hsp104 protein levels are extremely low in
exponentially growing cells at a normal temperature (25 or 30°C),
resulting in high sensitivity to heat shock (50°C). However,
overexpression of recombinant Hsp104 leads to thermotolerance.
Considering that Rvb may serve a chaperone function similar to
Hsp104, we asked whether Rvb overexpression might provide ther-
motolerance to exponential cells containing low levels of Hsp104.
Indeed, introduction of multicopy plasmids bearing RVB1 or RVB2
increased resistance to 50°C heat shock of yeast exponential cells
growing at 25°C (Fig 7F). Effect of RVB1 was the strongest (about
150-fold after 10-min exposure) and could be detected even when
expressed from a single-copy plasmid (data not shown).
Moreover, excess Rvb was able to reverse reduced thermotoler-
ance of the hsp104D strain (Fig 7G). Effects of Rvb1 or Rvb2
overexpression were similar to effects of restoration of Hsp104 on a
single-copy plasmid. These data indicate that Rvb proteins can
modulate thermotolerance independently of Hsp104. Therefore, Rvb
proteins expressed at high levels can at least partially substitute for
the Hsp104 function in thermotolerance. Together with data in
mammalian cells described above, these results support the function
for RuvbL/Rvb in protein homeostasis.
A
B
D
E F G
C
Figure 7.
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
15
Published online: August 24, 2015

Discussion
Here, we have developed a siRNA screen to identify factors involved
in aggresome formation, and RuvbL1 and RuvbL2 came up as strong
hits in the screen. Depleting both proteins reduced the fraction of
cells containing aggresomes and increased the fraction of cells with
multiple aggregates. This effect was observed with two aggregation-
prone polypeptides: synphilin-1 and VHL, as well as with endoge-
nous ubiquitylated proteins. These data suggested that (i) RuvbL
proteins play a role in control of protein aggregation, potentially as
a novel molecular chaperone and (ii) aggregates that are formed in
the absence of RuvbL are either too large to be transported to the
aggresome, or their overabundance overwhelms the aggresome
machinery. Interestingly, a chaperone function in assembly of
certain protein complexes has been proposed for mammalian RuvbL
(Nano & Houry, 2013). For example, they were suggested to partici-
pate in assembly of mTOR-like kinases, certain RNPs, telo-
merase, RNA polymerase (Venteicher et al, 2008; Izumi et al, 2011;
Horejsi et al, 2013; Kim et al, 2013; Machado-Pinilla et al, 2013),
and other complexes. Here, we report that RuvbL appears to be a
major player in general protein homeostasis.
The Hsp104 family members exist in bacteria, yeast, and plants,
where they facilitate protein disaggregation. However, they are not
found in metazoan (except mitochondria). The protein disaggregat-
ing activity existing in metazoa was attributed to the Hsp110-Hsp70-
DnaJ system, suggesting that there may be no AAA+ chaperone in
this taxonomy group that has functional analogy to Hsp104.
Indeed, mammalian Hsp110, Hsp70 (Hsc70 or Hsp70), and Hsp40
(Hdj1) were able to dissolve large protein aggregates and recover
natively folded proteins (Rampelt et al, 2012; Winkler et al, 2012;
Mattoo & Goloubinoff, 2013; Torrente & Shorter, 2014). On the other
hand, unlike Hsp104/Hsp70/Hsp40, this chaperone system failed to
disaggregate amyloids (Shorter, 2011). Our findings suggest that
RuvbL may constitute the missing AAA+ disaggregase in mamma-
lian and other metazoan cells.
Surprisingly, RuvbL expression was not induced under stressful
conditions such as heat shock or proteasome inhibition, both of
which lead to the buildup of abnormal protein species. Nor did
RuvbL depletion activate Hsf1 or cause accumulation of Hsps, two
other stress-related processes. Possibly, RuvbL becomes critical for
homeostasis only under a specific set of stressful conditions.
Previously, we demonstrated that in yeast, the model polyglu-
tamine-containing polypeptide 103QP (which corresponds to the
disease-causing exon 1 of huntingtin) accumulates in an aggresome-
like body (Wang et al, 2009), which may be the same as IPOD
compartment (Kaganovich et al, 2008). A similar polypeptide 103Q
missing the proline-rich region that follows the polyQ track could
not be recruited to this body, but instead formed multiple aggregates
in the cytoplasm that were toxic to yeast cells (Meriin et al, 2002,
2003). The formation of these multiple aggregates was dependent
on the presence of endogenous prions controlled by the AAA+ chap-
erone Hsp104, while recruitment of 103QP to the aggresome-like
body was not (Meriin et al, 2002; Wang et al, 2009). This current
study reports that yeast orthologs of RuvbL play a role in recruit-
ment of 103QP to the aggresome-like body, and downregulation of
these orthologs, which prevents the recruitment, leads to strong
103QP toxicity. These data indicate that though RuvbL may have
similar function in amyloid disassembly as Hsp104, these different
chaperones play distinct roles in recruitment to different aggregate
compartments.
A complex of data presented here strongly suggests that RuvbL
may serve as a chaperone in protein homeostasis. It includes accu-
mulation of protein aggregates upon RuvbL depletion, delayed
protein disaggregation in RuvbL-depleted cells, localization of
RuvbL to aggresomes, direct interaction of RuvbL with the aggre-
some substrate synphilin-1, stimulation of the ATPase activity of
RuvbL by misfolded proteins, and fragmentation of amyloid fibrils
in vitro. Furthermore, Ruvbl has a barrel-like structure with an
internal channel, showing certain structural similarity to common
chaperones, for example, ClpB or Hsp104 family members.
In protein disaggregation, Hsp104 cooperates with Hsp70 and
Hsp40. Here, we tested whether Hsp70/Hsp40 can similarly cooper-
ate with the RuvbL complex. A classical Hsp70/Hsp40/Hsp104
substrate luciferase could not be tested with RuvbL because of the
specificity of the latter. Indeed, RuvbL depletion did not affect luci-
ferase refolding in cells (Fig EV3), and purified RuvbL did not
stimulate luciferase refolding in vitro, indicating that luciferase is
not a RuvbL substrate. Therefore, we switched to proteins that can
interact with RuvbL. In these experiments, Hsp70/Hsp40 did not
promote ATPase activity of RuvbL-stimulated by casein (not
shown). Furthermore, Hsp70/Hsp40 did not show synergy or addi-
tivity with RuvbL in suppression of formation of Ab amyloid fibrils
(Appendix Fig S5). Finally, inhibition of Hsp70-Hsp40 interaction
with a small molecule myricetin did not affect disaggregation of
aggresome or synphilin-1 aggregates (Fig EV5). Together, these data
suggest that RuvbL does not cooperate with Hsp70/Hsp40 in its
chaperone function.
Interestingly, a function for mammalian RuvbL has been previ-
ously proposed in the assembly of certain protein complexes (Nano
& Houry, 2013), including mTOR-like kinases, certain RNPs, telo-
merase, and RNA polymerase (Venteicher et al, 2008; Izumi et al,
2011; Horejsi et al, 2013; Kim et al, 2013; Machado-Pinilla et al,
2013). In these cases, RuvbL often functions as part of the so-called
R2TP complex, which involves proteins Pih1 and Tah1 (Nano &
Houry, 2013), which connect it to Hsp90 and prefoldin (Boulon et al,
2012a,b; Mita et al, 2013). However, Hsp90 is unlikely to cooperate
with RuvbL, at least in aggresome formation, since inhibition of
Hsp90 does not mimic effects of RuvbL depletion of aggresome
(Fig EV1A). Furthermore, we did not see any effect of Pih1 depletion
on aggregation or aggresome formation (Fig EV1B). Prefoldin, on
the other hand, may be involved in delivery of substrates to the
RuvbL complex, similarly to its function in delivery of substrates to a
distinct chaperone TRiC (Hartl & Hayer-Hartl, 2002).
While DNA helicase activity of Rvb in bacteria was demonstrated
long ago, similar activity of mammalian RuvbL has not been found
until recently. Indeed, recent studies of mixed RuvbL1/2 dodecamer
structure indicated that the channel has a relatively monotonous
distribution of negative and positive charges, which was suggested
to facilitate interaction with single-stranded DNA molecules in order
to promote DNA helicase activity (Gorynia et al, 2011). On the other
hand, the charge distribution in the channel is also consistent with
binding of unfolded polypeptide chains that may be necessary for the
chaperone activity of RuvbL. We would like to emphasize, however,
that alternatively RuvbL may promote protein disaggregation not by
threading polypeptide chains through the channel, but rather by
interacting with hydrophobic surfaces or other patches on
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
16
Published online: August 24, 2015

aggregated proteins. Such interactions may be sufficient to shift the
equilibrium so that these polypeptides leave aggregates.
Materials and Methods
Reagents and antibodies
MG132, nocodazole, and bafilomycin were purchased from Biomol
(Farmingdale, NY, USA); Protease inhibitors tablets were from
Roche; Ni-NTA (Ni-nitrolotriacetate) superflow cartridge (1 × 5 ml)
was from Qiagen; MBP (Maltose Binding Protein) TrapTM HP
(1 × 1 ml) and Superdex G200 HR column (1 × 30 cm) were from
GE healthcare; Imidazole buffer solution was from Sigma.
Anti-tubulin antibodies (Abs) were from GenScript (Piscataway,
NJ, USA); anti-multi-ubiquitin Abs (FK2)—from Enzo Life Sciences
(Farmingdale, NY, USA); anti-RuvbL1 and anti-Flag Abs—from
Sigma-Aldrich (St. Louis, MO, USA); anti-HA Abs—from Cell Signal-
ing (Danvers, MA, USA); anti-GFP Abs—from Clontech (Mountain
View, CA, USA); and anti-actin Abs—from Santa Cruz (Santa Cruz,
CA, USA).
Constructs
The retroviral expression constructs with C-terminally tagged
synphilin-1 (Syn-GFP) and with mRFP-Ub were described before
(Zaarur et al, 2008, 2014). Synphilin deletion constructs ANK1-CC-
ANK2 and ANK1-CC were described before (Zaarur et al, 2008). For
tandem affinity purification, we employed the pEGFPN1 plasmid
with cloned a construct ANK1-CC-ANK2 with a His-tag at the
N-terminus and a GFP-tag at the C-terminus, as described previously
(Zaarur et al, 2008). VHL plasmid was a gift from Daniel Kaganovich
(Hebrew University, Israel). FLAG-RuvbL1 and HA-RuvbL2 were a
gift from Anindya Dutta (University of Virginia, US). For the rescue
experiment, the constructs of WT RuvbL1 and the RuvbL1 ATPase
mutant (A908G and C915T) were cloned into pCXIP and silence
mutations of A498C and C501A were introduced in both of them.
The pET21-N-ter-His6-TIP48 and pET15-His6-TIP49 plasmids coding
for RuvbL2 and RuvbL1, respectively, were a gift from I. Tsaneva.
Cells cultures, growth, and transfection
HeLa, HEK93, and SY5Y cells were grown in Dulbecco’s modified
Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum
or heat-inactivated serum (for HEK93), at 37°C in an atmosphere
of 5% CO2. For fluorescence microscopy, cells were grown on
Lab-Tek� Chambered Coverglasses (NUNC) pretreated with poly-
L-lysine (Sigma). For transient plasmid transfection, we used
Lipofectamine 2000 reagent (Invitrogen), and for siRNA transfec-
tion, we used RNAiMAX reagent (Invitrogen).
siRNA screening
High-throughput siRNA screening was done using the screening
facility of the Institute of Chemistry and Cell Biology at Harvard
Medical School, ICCB. We screened 21,121 siRNA pools of four
different sequences for each gene, from the Dharmacon human
siGENOME Smart Pool, in triplicate. HeLa cells that express
Syn-GFP and RFP-Ub were reverse-transfected with the siRNA
pool. A 10-ll mixture of the lipid, Lipofectamine RNAiMAX (Invit-
rogen), diluted 1:20 with Optimem (Gibco), was dispensed auto-
matically to a 384-well black clear bottom plate (Corning 3712,
Corning, NY) using Matrix WellMate (Thermo Scientific). Immedi-
ately after, 2 ll of siRNAs were transferred from Dharmacon
library plate to the final concentration of 50 nM. As a negative
control, we used anti-luciferase siRNA, and for transfection effi-
ciency control, we used siRNAs against GFP (to assess disappear-
ance of Syn-GFP fluorescence) and against PLK1 (polo-like kinase 1;
depletion of which caused cell death). Each plate contained nega-
tive control and transfection control wells. Plates were briefly
centrifuged. After 30 min, 500 cells per well, diluted with 40 llDMEM, were dispensed to each well. Plates were kept in the incu-
bator for 72 h, for complete depletion. MG132 solution was added
to the wells at final concentration of 10 lM. Following 5 h of incu-
bation, cells were fixed with 4% formaldehyde and stain with
Hoechst 33342 (Sigma). Each plate was run in triplicate. Auto-
mated images using an ImageExpress Micro microscope (Molecular
Device) at 20× magnification at three wavelengths (for FITC, Texas
Red and DAPI) were taken for four fields per well. The percent of
synphilin and ubiquitin aggresome was determined by image anal-
ysis software written by Tiao Xie (ICCB) in Matlab. The screening
assay was very robust and yielded a z factor of 0.67. Because of
the diverse phenotype of aggresomes, confirmation of the hit was
done manually, by viewing the images.
Cell lysis and analysis
Cells were lysed with lysis buffer (40 mM Hepes, pH 7.5, 50 mM
KCl, 1% Triton X-100, 2 mM DTT, 1 mM Na3VO4, 50 mM b-glyc-erophosphate, 50 mM NaF, 5 mM EDTA, 5 mM EGTA, 1 mM
PMSF, protease inhibitor cocktail (Roche, Switzerland)). Samples
were adjusted to have equal amount of total protein and subjected
to electrophoresis followed by immunoblotting.
Immunostaining and microscopy
For immunostaining, cells were fixed for 10 min with 4% formalde-
hyde and washed with PBS. Then the cells were blocked for 1 h
with 3% BSA, and incubated at room temperature for 2 h with
primary antibody. Following a wash with PBS, cells were incubated
for an hour with Alexa Fluor 594 donkey anti-mouse IgG or anti-
rabbit (1:500) (Molecular Probes, Eugene, OR). After a final wash,
cells were analyzed with a fluorescence microscope, Axiovert 200
(Carl Zeiss, Germany) using a 100× oil objective. To assess the frac-
tion of cells with aggresome, at least 200 cells were counted in each
well. Live fluorescence microscopy of yeast cells containing the GFP
and/or RFP tags was performed under the fluorescence microscope
Olympus BX41, using a 100× oil objective.
Immunoprecipitation
Cells were grown on 60-cm plates and transfected with the
mentioned construct. After 24 h, cells were lysed with 30 mM NaCl,
10 mM Hepes, 0.5% Triton X-100, 5% glycine, 1.5 mM MgCl2, and
protease inhibitor cocktail (Roche, Switzerland). Sample from cell
lysate was kept as the “total”. For pull down, anti-GFP-Tag-Agarose
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
17
Published online: August 24, 2015

(MBL, Woburn, MA) was used, following the manufacturer’s
protocol.
Luciferase assay
Cells were infected with HSE-Luc lentivirus and GFP retrovirus.
Three days after selection, cells were heat-shocked at 45°C for
15 min followed by 3 h of incubation at 37°C. After the incubation
period, the medium was aspirated, cells were washed twice with
PBS, lysed, and transferred into 96-well plate. 50 ll of luciferase
assay reagent (Promega) was injected into each well, and lumines-
cence was read with a luminometer. In parallel, lysates were plated
into black 96-well plates and GFP fluorescence was read by the
same luminometer. All measurements were done in triplicate, and
the assays were repeated three times.
ATPase activity assay
The ATPase assay was adapted from a previously described protocol
(Chang et al, 2008). Briefly, Malachite Green reagent was prepared
by mixing 2:1:1:2 the following solutions: 0.081% malachite green,
2.3% polyvinyl alcohol, 5.7% ammonium heptamolybdate tetrahy-
drate in 6 M HCl, and H2O. 1 lM RuvbL and protein substrates at the
indicated concentrations were diluted in buffer containing 0.017%
Triton X-100, 100 mM Tris–HCl, 20 mM KCl, and 6 mM MgCl2 at
pH7.4, in the total volume of 15 ll, and dispensed into wells of
96-well white plate. The reaction started by addition of 10 ll of
2.5 mM ATP and kept at 37°C for 2 h. 80 ll of the malachite green
reagent was added to the reaction followed by 10 ll of 32% sodium
citrate to quench the reaction. After 15 min of incubation at 37°C,
the plate was read at 620 nm. Each reaction was run in triplicates.
Fibril preparation, deconstruction assay, and atomicforce microscopy
Amyloid fibrils prepared from bovine insulin (I5500, Sigma-Aldrich)
were subjected to ATPase stimulation and deconstruction assays
(Kurouski et al, 2012, 2013). Insulin was pre-incubated at 70°C and
pH 2.4 at a concentration of 60 mg/ml for 24 h to prepare fibrils.
The fibrillation process was stopped by bringing the sample to room
temperature and centrifuging twice at 16,000 g to remove the unfib-
rillated fraction. The gelatinous phase containing fibrils was then
resuspended in HCl (pH 2.4).
Samples for fibril deconstruction assays were prepared by mixing
RuvbL preparations and fibrils in a ~1:20 protein molar ratio in the
presence of 0.1 mg/ml creatine kinase, 20 mM phosphocreatine,
10 mM MgCl2, 75 mM KCl, and 20 mM HEPES buffer pH 7.4 with
or without 5 mM ATP. The reaction mixtures were incubated at
30°C for 2 h. In the control reaction, RuvbL was replaced with an
equal volume of the buffer at 30°C for 2 h followed by imaging.
Samples for atomic force microscopy (AFM) imaging were prepared
by diluting the end product of the reactions in a 1:6 volumetric ratio
by 20 mM HEPES buffer pH 7.4 and depositing on freshly cleaved
mica disks (TED PELLA, Inc., Redding, CA) for 2 min, followed by
washing with milliQ water to remove unbound materials as well as
salt, blotting, and drying under flowing air. The samples were then
imaged in tapping mode using a SmartSPMTM-1000 atomic force
microscope from AIST-NT, Inc. (Novato, CA) and gold-coated PPP-
NCHAu AFM probes (NanosensorsTM, Neuchatel, Switzerland) with
a spring constant of 42 N/m and resonance frequency of ~270 kHz.
Images were processed using the AIST-NT SPM Control Software
v.3.5.61 (AIST-NT, Inc.).
Ab fibrillation inhibition assay
Ab1–42 peptide was solubilized with 0.15 mM NaOH to final
concentration of 0.3 mM. For the experiment, the peptide was
diluted to final concentration of 10 lM with PBS buffer containing
5 mM MgCl2, 50 mM KCl, 1 mM DTT, and 20 lM thioflavin with
and without 10 mM ATP. Protein of interest mixed in the reaction,
0.1 lM RuvbL complex, or/ and HSPs (0.2 lM HSC70 and 0.2 lMDJB1). The reaction was set in 96-well black plate, transparent
bottom, and incubated at 37°C for 12 h. Fluorescence was measured
every 6 min by plate reader.
Tandem affinity purification, label-free quantitative mass spec-
trometry, chemical cross-linking mass spectrometry, yeast strains
and plasmids, yeast cultivation procedures, and thermotolerance
assays are in the Appendix Supplementary Methods.
Expanded View for this article is available online:
http://emboj.embopress.org
AcknowledgementsWe thank Daniel Kaganovich, Anindya Dutta, Erin Fenderson and I. Tsaneva for
generous gifts of strains, plasmids and reagents, and Natalie Saini for the
strain and plasmid constructions (see Appendix Supplementary Methods). This
research was supported by the US National Institutes of Health under grants
and contracts: P41 GM104603 and HHSN268201000031C (CEC), R01
GM086890 (MYS) and R01GM093294 (YOC), by the National Science Founda-
tion under awards CHE-1152752 (IKL) and MCB-0818122 (KSL), and by
Fundação para a Ciência e Tecnologia (Portugal) through the grants PTDC/
BBB-BEP/1724/2012 (STNS, TMB and PMM) and PEst-OE/EQB/LA0004/2011
(PMM). Yeast work by NVR and YOC was funded by the grant Russian Science
Foundation from 14-50-00069. The St. Petersburg State University support
from projects 1.50.2218.2013 (YOC) and IAS 1.42.1284.2014 (travel support to
NVR) contributed to the preparation of this paper.
Author contributionsNZ performed the main body of experiments, designed experiments, and
analyzed data. PL and PMM provided purified proteins and technical expertise.
XX and MM performed experiments with mass spectrometry. CEC provided
expertise in mass spectrometry. ABM performed experiments with cell culture.
GPN, RG, and NVR performed experiments with yeast. MS performed experi-
ments with atomic force microscopy. IKL provided technical expertise in atomic
force microscopy. STNS and TMB purified proteins. KSL directed constructions
of strains and plasmids. YOC and MYS designed research and analyzed data.
Conflict of interestThe authors declare that they have no conflict of interest.
References
Bakshi R, Mehta AK, Sharma R, Maiti S, Pasha S, Brahmachari V (2006)
Characterization of a human SWI2/SNF2 like protein hINO80:
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
18
Published online: August 24, 2015

demonstration of catalytic and DNA binding activity. Biochem Biophys Res
Commun 339: 313 – 320
Boulon S, Bertrand E, Pradet-Balade B (2012a) HSP90 and the R2TP co-
chaperone complex: building multi-protein machineries essential for cell
growth and gene expression. RNA Biol 9: 148 – 154
Boulon S, Pradet-Balade B, Verheggen C, Molle D, Boireau S, Georgieva M,
Azzag K, Robert MC, Ahmad Y, Neel H, Lamond AI, Bertrand E (2012b)
HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the
cytoplasmic assembly of RNA polymerase II. Mol Cell 39: 912 – 924
Bukau B, Weissman J, Horwich A (2006) Molecular Chaperones and Protein
Quality Control. Cell 125: 443 – 451
Burnett BG, Pittman RN (2005) The polyglutamine neurodegenerative protein
ataxin 3 regulates aggresome formation. Proc Natl Acad Sci USA 102:
4330 – 4335
Chang L, Bertelsen EB, Wisen S, Larsen EM, Zuiderweg ER, Gestwicki JE
(2008) High-throughput screen for small molecules that modulate the
ATPase activity of the molecular chaperone DnaK. Anal Biochem 372:
167 – 176
Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW (1995)
Role of the chaperone protein Hsp104 in propagation of the yeast prion-
like factor [psi+]. Science 268: 880 – 884
Choi J, Heo K, An W (2009) Cooperative action of TIP48 and TIP49 in H2A.Z
exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res
37: 5993 – 6007
Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL,
Dawson TM (2001) Parkin ubiquitinates the alpha-synuclein-interacting
protein, synphilin-1: implications for Lewy-body formation in Parkinson
disease. Nat Med 7: 1144 – 1150
Clare DK, Saibil HR (2013) ATP-driven molecular chaperone machines.
Biopolymers 99: 846 – 859
Corboy MJ, Thomas PJ, Wigley WC (2005) Aggresome formation. Methods Mol
Biol 301: 305 – 327
Doyle SM, Wickner S (2009) Hsp104 and ClpB: protein disaggregating
machines. Trends Biochem Sci 34: 40 – 48
Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ,
Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM,
Ross CA (1999) Synphilin-1 associates with alpha-synuclein and promotes
the formation of cytosolic inclusions. Nat Genet 22: 110 – 114
Erjavec N, Larsson L, Grantham J, Nystrom T (2007) Accelerated aging and
failure to segregate damaged proteins in Sir2 mutants can be suppressed
by overproducing the protein aggregation-remodeling factor Hsp104p.
Genes Dev 21: 2410 – 2421
Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C (2011) BAG3
mediates chaperone-based aggresome-targeting and selective autophagy
of misfolded proteins. EMBO Rep 12: 149 – 156
Glover JR, Lindquist S (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone
system that rescues previously aggregated proteins. Cell 94: 73 – 82
Gong H, Romanova NV, Allen KD, Chandramowlishwaran P, Gokhale K,
Newnam GP, Mieczkowski P, Sherman MY, Chernoff YO (2012)
Polyglutamine toxicity is controlled by prion composition and gene
dosage in yeast. PLoS Genet 8: e1002634
Gorynia S, Bandeiras TM, Pinho FG, McVey CE, Vonrhein C, Round A, Svergun
DI, Donner P, Matias PM, Carrondo MA (2011) Structural and functional
insights into a dodecameric molecular machine - the RuvBL1/RuvBL2
complex. J Struct Biol 176: 279 – 291
Groenning M, Frokjaer S, Vestergaard B (2009) Formation mechanism of
insulin fibrils and structural aspects of the insulin fibrillation process. Curr
Protein Pept Sci 10: 509 – 528
Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from
nascent chain to folded protein. Science 295: 1852 – 1858
Heir R, Ablasou C, Dumontier E, Elliott M, Fagotto-Kaufmann C, Bedford FK
(2006) The UBL domain of PLIC-1 regulates aggresome formation. EMBO
Rep 7: 1252 – 1258
Horejsi Z, Takai H, Adelman CA, Collis SJ, Flynn H, Maslen S, Skehel JM, de Lange
T, Boulton SJ (2013) CK2 phospho-dependent binding of R2TP complex to
TEL2 is essential for mTOR and SMG1 stability. Mol Cell 39: 839 – 850
Izumi N, Yamashita A, Iwamatsu A, Kurata R, Nakamura H, Saari B, Hirano H,
Anderson P, Ohno S (2010) AAA+ proteins RUVBL1 and RUVBL2 coordinate
PIKK activity and function in nonsense-mediated mRNA decay. Sci Signal
3: ra27
Izumi N, Yamashita A, Ohno S (2011) Integrated regulation of PIKK-mediated
stress responses by AAA+ proteins RUVBL1 and RUVBL2. Nucleus 3: 29 – 43
Jin J, Cai Y, Yao T, Gottschalk AJ, Florens L, Swanson SK, Gutierrez JL, Coleman
MK, Workman JL, Mushegian A, Washburn MP, Conaway RC, Conaway JW
(2005) A mammalian chromatin remodeling complex with similarities to
the yeast INO80 complex. J Biol Chem 280: 41207 – 41212
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to
misfolded proteins. J Cell Biol 143: 1883 – 1898
Jonsson ZO, Dhar SK, Narlikar GJ, Auty R, Wagle N, Pellman D, Pratt RE,
Kingston R, Dutta A (2001) Rvb1p and Rvb2p are essential components of
a chromatin remodeling complex that regulates transcription of over 5%
of yeast genes. J Biol Chem 276: 16279 – 16288
Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between
two distinct quality control compartments. Nature 454: 1088 – 1095
Kanemaki M, Makino Y, Yoshida T, Kishimoto T, Koga A, Yamamoto K,
Yamamoto M, Moncollin V, Egly JM, Muramatsu M, Tamura T (1997)
Molecular cloning of a rat 49-kDa TBP-interacting protein (TIP49) that is
highly homologous to the bacterial RuvB. Biochem Biophys Res Commun
235: 64 – 68
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The
deacetylase HDAC6 regulates aggresome formation and cell viability in
response to misfolded protein stress. Cell 115: 727 – 738
Kim G, Meriin AB, Gabai VL, Christians E, Benjamin I, Wilson A, Wolozin B,
Sherman MY (2012) The heat shock transcription factor Hsf1 is
downregulated in DNA damage-associated senescence, contributing to the
maintenance of senescence phenotype. Aging Cell 11: 617 – 627
Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY,
Erikson RL, Cantley LC, Choo AY, Blenis J (2013) Metabolic stress controls
mTORC1 lysosomal localization and dimerization by regulating the TTT-
RUVBL1/2 complex. Mol Cell 49: 172 – 185
Kurouski D, Luo H, Sereda V, Robb FT, Lednev IK (2012) Deconstruction
of stable cross-Beta fibrillar structures into toxic and nontoxic
products using a mutated archaeal chaperonin. ACS Chem Biol 8:
2095 – 2101
Kurouski D, Luo H, Sereda V, Robb FT, Lednev IK (2013) Rapid degradation
kinetics of amyloid fibrils under mild conditions by an archaeal
chaperonin. Biochem Biophys Res Commun 422: 97 – 102
Lakomek K, Stoehr G, Tosi A, Schmailzl M, Hopfner KP (2015) Structural basis
for dodecameric assembly states and conformational plasticity of the full-
length AAA+ ATPases Rvb1. Rvb2. Structure 23: 483 – 495
Machado-Pinilla R, Liger D, Leulliot N, Meier UT (2013) Mechanism of the
AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18:
1833 – 1845
Marx FP, Soehn AS, Berg D, Melle C, Schiesling C, Lang M, Kautzmann S,
Strauss KM, Franck T, Engelender S, Pahnke J, Dawson S, von Eggeling F,
Schulz JB, Riess O, Kruger R (2007) The proteasomal subunit S6 ATPase is
ª 2015 The Authors The EMBO Journal
Nava Zaarur et al RuvbL1/2 controls aggregation and aggresome The EMBO Journal
19
Published online: August 24, 2015

a novel synphilin-1 interacting protein–implications for Parkinson’s
disease. FASEB J 21: 1759 – 1767
Matias PM, Gorynia S, Donner P, Carrondo MA (2006) Crystal structure of the
human AAA+ protein RuvBL1. J Biol Chem 281: 38918 – 38929
Mattoo RU, Goloubinoff P (2013) Molecular chaperones are nanomachines
that catalytically unfold misfolded and alternatively folded proteins. Cell
Mol Life Sci 71: 3311 – 3325
Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY (2002)
Huntington toxicity in yeast model depends on polyglutamine aggregation
mediated by a prion-like protein Rnq1. J Cell Biol 157: 997 – 1004
Meriin AB, Zhang X, Miliaras NB, Kazantsev A, Chernoff YO, McCaffery JM,
Wendland B, Sherman MY (2003) Aggregation of expanded polyglutamine
domain in yeast leads to defects in endocytosis. Mol Cell Biol 23:
7554 – 7565
Meriin AB, Sherman MY (2005) Role of molecular chaperones in
neurodegenerative disorders. Int J Hyperthermia 21: 403 – 419
Min W, Angileri F, Luo H, Lauria A, Shanmugasundaram M, Almerico AM,
Cappello F, de Macario EC, Lednev IK, Macario AJ, Robb FT (2014) A
human CCT5 gene mutation causing distal neuropathy impairs
hexadecamer assembly in an archaeal model. Sci Rep 4: 6688
Mita P, Savas JN, Ha S, Djouder N, Yates JR III, Logan SK (2013) Analysis of
URI nuclear interaction with RPB5 and components of the R2TP/prefoldin-
like complex. PLoS ONE 8: e63879
Nano N, Houry WA (2013) Chaperone-like activity of the AAA+ proteins Rvb1
and Rvb2 in the assembly of various complexes. Philos Trans R Soc Lond B
Biol Sci 368: 20110399
O’Farrell C, Murphy DD, Petrucelli L, Singleton AB, Hussey J, Farrer M, Hardy J,
Dickson DW, Cookson MR (2001) Transfected synphilin-1 forms
cytoplasmic inclusions in HEK293 cells. Brain Res Mol Brain Res 97:
94 – 102
Ohdate H, Lim CR, Kokubo T, Matsubara K, Kimata Y, Kohno K (2003)
Impairment of the DNA binding activity of the TATA-binding protein
renders the transcriptional function of Rvb2p/Tih2p, the yeast RuvB-like
protein, essential for cell growth. J Biol Chem 278: 14647 – 14656
Olzmann JA, Li L, Chin LS (2008) Aggresome formation and neurodegenerative
diseases: therapeutic implications. Curr Med Chem 15: 47 – 60
Putnam CD, Clancy SB, Tsuruta H, Gonzalez S, Wetmur JG, Tainer JA (2001)
Structure and mechanism of the RuvB Holliday junction branch migration
motor. J Mol Biol 311: 297 – 310
Qiu XB, Lin YL, Thome KC, Pian P, Schlegel BP, Weremowicz S, Parvin JD,
Dutta A (1998) An eukaryotic RuvB-like protein (RUVBL1) essential for
growth. J Biol Chem 273: 27786 – 27793
Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI,
Bukau B (2012) Metazoan Hsp70 machines use Hsp110 to power protein
disaggregation. EMBO J 31: 4221 – 4235
Schirmer EC, Lindquist S (1997) Interactions of the chaperone Hsp104 with yeast
Sup35 and mammalian PrP. Proc Natl Acad Sci USA 94: 13932 – 13937
Shen X, Mizuguchi G, Hamiche A, Wu C (2000) A chromatin remodelling
complex involved in transcription and DNA processing. Nature 406:
541 – 544
Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins:
a cell biologist thinks about neurodegenerative diseases. Neuron 29:
15 – 32
Shorter J (2011) The mammalian disaggregase machinery: Hsp110 synergizes
with Hsp70 and Hsp40 to catalyze protein disaggregation and
reactivation in a cell-free system. PLoS ONE 6: e26319
Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004)
Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective.
J Biol Chem 279: 4625 – 4631
Torrente MP, Shorter J (2014) The metazoan protein disaggregase and
amyloid depolymerase system: Hsp110, Hsp70, Hsp40, and small heat
shock proteins. Prion 7: 457 – 463
Tosi A, Haas C, Herzog F, Gilmozzi A, Berninghausen O, Ungewickell C,
Gerhold CB, Lakomek K, Aebersold R, Beckmann R, Hopfner KP (2013)
Structure and subunit topology of the INO80 chromatin remodeler and its
nucleosome complex. Cell 154: 1207 – 1219
Tsaneva IR, Muller B, West SC (1993) RuvA and RuvB proteins of Escherichia coli
exhibit DNA helicase activity in vitro. Proc Natl Acad Sci USA 90: 1315 – 1319
Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE (2008)
Identification of ATPases pontin and reptin as telomerase components
essential for holoenzyme assembly. Cell 132: 945 – 957
Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences
in the alpha- and beta-subunits of ATP synthase, myosin, kinases and
other ATP-requiring enzymes and a common nucleotide binding fold.
EMBO J 1: 945 – 951
Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman
MY (2009) Abnormal proteins can form aggresome in yeast: aggresome-
targeting signals and components of the machinery. FASEB J 23: 451 – 463
Webb JL, Ravikumar B, Rubinsztein DC (2004) Microtubule disruption inhibits
autophagosome-lysosome fusion: implications for studying the roles of
aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 36: 2541 – 2550
Winkler J, Tyedmers J, Bukau B, Mogk A (2012) Chaperone networks in protein
disaggregation and prion propagation. J Struct Biol 179: 152 – 160
Woo KM, Kim KI, Goldberg AL, Ha DB, Chung CH (1992) The heat-shock
protein ClpB in Escherichia coli is a protein-activated ATPase. J Biol Chem
267: 20429 – 20434
Yamada K, Kunishima N, Mayanagi K, Ohnishi T, Nishino T, Iwasaki H,
Shinagawa H, Morikawa K (2001) Crystal structure of the Holliday
junction migration motor protein RuvB from Thermus thermophilus HB8.
Proc Natl Acad Sci USA 98: 1442 – 1447
Zaarur N, Meriin AB, Gabai VL, Sherman MY (2008) Triggering aggresome
formation. Dissecting aggresome-targeting and aggregation signals in
synphilin 1. J Biol Chem 283: 27575 – 27584
Zaarur N, Meriin AB, Bejarano E, Xu X, Gabai VL, Cuervo AM, Sherman MY
(2014) Proteasome failure promotes positioning of lysosomes around the
aggresome via local block of microtubule-dependent transport. Mol Cell
Biol 34: 1336 – 1348
Zhang X, Qian SB (2011) Chaperone-mediated hierarchical control in
targeting misfolded proteins to aggresomes. Mol Biol Cell 22:
3277 – 3288
The EMBO Journal ª 2015 The Authors
The EMBO Journal RuvbL1/2 controls aggregation and aggresome Nava Zaarur et al
20
Published online: August 24, 2015


















