
RSC Article Template (Version 3.1) - Welcome to …doras.dcu.ie/16550/2/doras_draft.doc · Web...
45
Electronic structure calculations and physicochemical experiments quantify the competitive liquid ion association and probe stabilisation effects for nitrobenzospiropyran in phosphonium-based ionic liquids. Simon Coleman 1 , Dermot Diamond 1,2 , Damien Thompson 3* and Robert Byrne 2** 1 Biomedical Diagnostics Institute, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland 2 CLARITY: Centre for Sensor Web Technologies, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland 3 Tyndall National Institute, University College Cork, Cork, Ireland *Corresponding author: [email protected] **Corresponding author: [email protected] Abstract Liquid ion-pair (LIP) formation in ionic liquids (ILs) has been examined using a series of electronic structure calculations that measure the relative extents of ion pairing and probe stabilisation for the photochromic dye nitrobenzospiropyran ( BSP) in phosphonium- based ILs with both [Cl] - and [NTf 2 ] - anions. Physicochemical properties of the liquids were measured experimentally by adding BSP and monitoring its photochromic properties within each liquid. Taken together, the computed complexation energies and measured spectroscopic properties support recent Walden plots of 1
Transcript of RSC Article Template (Version 3.1) - Welcome to …doras.dcu.ie/16550/2/doras_draft.doc · Web...

Electronic structure calculations and physicochemical experiments quantify the competitive liquid ion association and probe stabilisation effects for nitrobenzospiropyran in phosphonium-based ionic liquids.
Simon Coleman1, Dermot Diamond1,2, Damien Thompson3* and Robert Byrne2**
1 Biomedical Diagnostics Institute, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland2 CLARITY: Centre for Sensor Web Technologies, National Centre for Sensor Research, Dublin City University, Dublin 9, Ireland3 Tyndall National Institute, University College Cork, Cork, Ireland*Corresponding author: [email protected]**Corresponding author: [email protected]
Abstract
Liquid ion-pair (LIP) formation in ionic liquids (ILs) has been examined using a series of
electronic structure calculations that measure the relative extents of ion pairing and probe
stabilisation for the photochromic dye nitrobenzospiropyran (BSP) in phosphonium-based ILs with
both [Cl]- and [NTf2]- anions. Physicochemical properties of the liquids were measured
experimentally by adding BSP and monitoring its photochromic properties within each liquid.
Taken together, the computed complexation energies and measured spectroscopic properties
support recent Walden plots of conductivity-viscosity behaviour obtained for the same ILs and
reveal the atom-scale structure and energetics that underly the observed transport properties. LIP
formation was significantly stronger in [P6,6,6,14][Cl] resulting in effectively neutral liquid
components. This was reflected in the measured ΔS‡ and Ea values of -96 J.K-1.mol-1 and +72
kcal.mol-1 respectively. By contrast, the measured ΔS‡ and Ea values in [P6,6,6,14][NTf2] of -15 J.K-
1.mol-1 and +85 kcal.mol-1 respectively, together with the lower calculated complexation energies,
reflect the lower driving force towards LIP formation with the weaker anion. Furthermore,
activation barriers to cation diffusion promote LIP relative to competing probe stabilisation
interactions. The transport properties of ILs may thus be controlled by a prudent choice of IL
1

molecular structure, with both the polar hydrophilic headgroup and non-polar hydrophobic tail of
the constituent ions serving as distinct targets for rational design.
Introduction
2

Liquid ion pair (LIP) formation is a ubiquitous feature of ionic liquids (IL) and its extent influences the
viscosity and transport properties of the IL solvent. ILs, in contrast with conventional molecular
solvents, can form persistent, relatively long-lived nanostructured domains, which mediates their
properties or “ionicity” and endows favourable properties upon ILs that make them more suitable than
molecular solvents for many applications. Exploitable IL properties can include high thermal stability,
negligible vapour pressure and wide electrochemical window.1-4 The liquid nanostructured domains are
dynamic and composed of interchangeable IL components, yet can persist over long time and length
scales. More discrete structuring, at the level of individual cation and anion pairs, may play an
important role as precursor to the formation of nanostructures and will influence their atom-scale
features. The routine implementation of ionic liquids (ILs) as “designer” solvents with user specified
properties is therefore an attractive prospect with respect to many applications in the chemical
sciences, but their effectiveness will depend to a large degree on harnessing the properties that afford
these liquids their unique properties. These fundamental properties originate from nano-scale structural
features mentioned above, which are not easily characterised by experiment alone, but provide the
basis for unique macroscopic effects, known as the “ionic liquid effect” and “ionicity”, that seem to be
observed solely in ILs.5 Since ILs consist entirely of ions whose unfavourable packing allows them to
form molten salts at conditions near ambient (under 100 ºC for room temperature ILs), the effects
observed are clearly related to this set of inherent properties. Applications in fields as diverse as
recyclable solvents,6 catalysis,7 electrochemistry,8 synthesis9 and elemental analysis10 are all made
possible by IL-mediated interactions which do not exist in conventional molecular solvents. To
determine the extent to which these “ionicity” effects exist within ILs, the physico-chemical properties
must be measured, which in turn allows for systematic isolation of the unique and critical properties
that govern these effects. Recent studies of ILs have concluded that evaluation of ILs under the
assumption that they behave similarly to molecular solvents is inaccurate and does not encompass the
3

majority of the liquid properties.11, 12 ILs are now described as complex media which contain distinct
regions which each have specific properties and combine to form the bulk properties of the liquid
system. An example of this is the approximation of solvent polarity within ILs. Polarity is a
macroscopic property that assumes the entire solvent system acts as a homogeneous medium (like a
conventional organic solvent) rather than a medium that may possess distinct nano-structured regions
with dramatically different characteristics (like ILs).13, 14 Therefore, it is understandable that while
traditional polarity probes such as Reichardt’s dye 30 can provide information about the polarity of
organic solvents, this approach fails with the more complex ILs. This is because such probe dyes with
permanent dipoles will preferentially locate in localised polar nano-domains within ILs and thus report
similar findings despite the wide variations in the characteristics of the surrounding non-polar
domains. Hence the observed bulk properties of the ILs are often significantly at variance with
expectations arising from these polarity estimations. For this reason, until recently, ILs were believed
to have almost uniform polarity similar to that of ethanol or acetonitrile. Our recent studies have
shown this estimation to be incorrect by the use of more sophisticated spirocyclic solvatochromic
probes that can be switched photonically between polar and non-polar states, enabling the probe
molecule to migrate spontaneously between polar/non-polar IL nano-structured domains.11, 12 The
resulting thermodynamic and kinetic parameters confirmed that ILs have a range of properties which
cannot be characterised via traditional polarity measurements employed for conventional molecular
solvents.
4

The solvatochromic compound nitrobenzospiropyran (BSP) forms the zwitterionic isomer merocyanine
(MC) when exposed to UV irradiation which allows its use in the measurement of solvent interactions
similar to those measured using Reichardt’s dye 30. However, the MC form is thermodynamically
unstable and normally relaxes back to its closed, uncharged spiro (SP) form. Thus, the resulting
changes in conformational/charge state make this probe far more applicable to IL characterisation, due
to its ability to interact with distinct regions within the liquids, unlike previous dyes employed. The
BSP photoswitching mechanism involves UV-triggered irradiation (typically around 365-375 nm)
cleavage of the sp3 hybridised carbon, Cspiro of the indoline ring. Figure 1a shows the structure of
spirocyclic compounds and the photoswitchable spiropyran compound in its BSP and MC states. The
MC isomer arises from cis-trans isomerisation of the molecule that creates a planar, zwitterionic
molecule with charge delocalised across the molecule, as shown in (Figure 1a). The photoswitchable
system is a reversible, first order process, wherein irradiation with white light or thermal reversion can
result in ring closure to the more stable BSP form. Thermal reversion is the process whereby MC
spontaneously uses heat from its surroundings to receive sufficient energy for the reorientation to occur
in the absence of visible light. In addition to photoswitching, the MC isomer is found to be sensitive to
its molecular environment, with both specific and non-specific solvent interactions mediating the
compound’s equilibrium between both isomers.15-18 Generally, for BSP in molecular solvents, rate
constants increase with decreasing polarity.19, 20 This is consistent with trends in estimated Kamlet-Taft
parameters, which indicate that decreasing levels of hydrogen bonding between solvent and BSP
results in reduced stabilization of the MC form.11
5

6
(b)
(a)
N O NO2
A B
UV
/ Vis N O
NO2
Spiropyran (BSP) Merocyanine (MCBSP)
N O NO2
A B
UV
/ Vis N O
NO2
Spiropyran (BSP) Merocyanine (MCBSP)

Figure 1. (a) Structure of spirocyclic compounds. Indoline (A) and pyran (B) ring and its photo-switchable states, BSP and MC. Panel (b) shows computed BSP and MC structures, overlaid with Molecular Electrostatic Potential (MEP) surfaces generated as described in the text and with surfaces coloured according to regions of net charge as marked in the scale bar from -0.10 to +0.07 atomic units (c) UV-Vis spectra of spiropyran in its open (SP) and closed (MC) forms. Inset shows thermal dependence of spiropyran thermal relaxation from which thermodynamic parameters may be derived.
We recently reported the photo- and solvatochromic properties of BSP in ILs containing the [NTf2]-
anion and showed that the kinetics and thermodynamics of the BSP-MC equilibrium was sensitive to
the nature of the cation20. Cation-based interactions inhibit the MC conversion back to the BSP isomer,
and may include through-space orbital interactions, that may be specific to the [C2mIm]+ cation16, as
well as more general electrostatic interactions. Contemporaneous studies suggest that ILs may form
ordered systems resembling pseudo-crystalline systems based on stacking of mutual charges
(aggregation) or ordered association of cation species to surrounding anions and vice versa.21-23 Lopes
7
0
0.5
1
1.5
2
2.5
3
400 500 600 700
wavelength (nm)
abso
rban
ce (a
.u)
SPMC
0.75
0.8
0.85
0.9
0.95
1
0 5 10 15 20 25 30
time (s)
abso
rban
ce (a
.u)
20°C
30°C
40°C
50°C
(c)

et al examined the formation of nanostructured domains in ILs containing the imidazolium cation and
found that certain imidazolium cations in ILs can be divided into two specific regions: a polar head
group where the ion charge resides and a non-polar region where alkyl side-chains extend into space. 24
The association of charged regions by electrostatic interactions is believed to form polar domains
within the liquids while non-polar domains are formed by steric packing of alkyl chains. Similar
structuring has also been reported for more extensively alkylated systems such as quaternary
phosphonium and ammonium based ILs.23, 25
To date, the majority of liquid-structure studies have focused on the bulk properties of ILs and
examined how the formation of domains influences the overall transport properties of the liquids.
However, very short-range, specific intra-molecular interactions that occur between the anion and
cation may be crucial to the unique effects found within ILs and these atom- and nano-scale
interactions may underlie the formation of the IL-specific bulk properties. The present study exploits
this link between the atomic, nano and macro scales for the characterisation of IL bulk properties using
BSP. Although thermal relaxation has provided insight into the proposed nano-structuring of the
liquids, minor inconsistencies have been observed which could not be explained from expected
solvent-solute interactions. When ILs are examined at a molecular level, the concept of liquid ion-pairs
(LIPs) becomes an important parameter in understanding the properties of ILs, and may explain the
origin of such thermodynamic inconsistencies. Specific intra-molecular interactions may provide subtle
but important effects upon solute introduction that serve to elucidate further the structuring of the
liquid domains. The MC isomer of the probe molecule provides a zwitterionic system that can interact
with the opposing charges found on each ion of the ILs. As thermal relaxation occurs, ion interactions
at each charge site in the molecule would be expected to stabilise the zwitterion. However, the
formation of ion-pairs in the liquid itself may offset probe stabilisation, resulting in a competition, or
8

“tug of war”, between inter (MC-ion) and intra (ion-ion) interactions. The ability of the ILs to form
tightly-bound LIPs may therefore reduce the ability of the liquid to interact with solutes and thus
mediate the ionicity and overall effects observed. Since MC is sensitive to such interactions, the
thermodynamic and kinetic parameters can quantify such liquid properties and explain the processes at
work.
To further understand these three-way effects observed between BSP and the IL ions, electronic
structure calculations were used to quantify the competitive probe-ion and ion-ion interactions. The
calculated interaction strengths ΔE for each individual ion’s interaction with both MC and its solvent
counterion provide a quantitative indication of the level of interaction occurring in each case. This
allowed for the extent of the competition between probe-ion and ion-ion interactions ΔΔE to be
quantified and compared to the observed physicochemical properties. Following the findings of
previous studies,26, 27 two phosphonium based ILs were chosen based on their Walden plot values to
determine the effects of ion-pair formation upon the properties of the ILs.
Trihexyltertadecylphosphonium Chloride; [P6,6,6,14][Cl] and Trihexyltertadecylphosphonium
bistrifluoro(sulfonyl)imide; [P6,6,6,14][NTf2] were chosen due to the large contrast in anticipated effects
due to the more localised charge on [Cl]-, meaning [P6,6,6,14][NTf2] is expected to be more weakly bound
and with more ion mobility. The theoretical models were used to determine if the formation of the
charged MC form could compete with the charge site of the cation and anion. Preservation of the IL
nanostructure, with LIP formation predominating over solute stabilisation, is essential to maintain the
transport properties within the liquids and avoid significant local fluctuations; on the other hand,
solvent-mediated biasing of photo-switchable equilibria could be a useful sensor design feature.
9

Experimental
Electronic structure calculations: IL and ion-probe electronic structures were obtained for the systems
listed in Table 1 using the Gaussian03 program28 with the B3LYP hybrid HF-DFT functional29 and 6-
311++G** basis sets. Stable geometries were obtained via nuclear relaxation to root mean square
(RMS) atomic forces and displacements below 0.0003 and 0.0012 a.u. respectively, followed by
electronic structure determination using the gfoldprint and POP=FULL keywords to generate output
files formatted for molecular electrostatic potential (MEP) visualisation using MOLEKEL unix version
4.3.32 All atomic charges were computed using a natural population analysis (NPA).30 The basis set
used is larger than in previous studies of IL structures and also uses the complete [P6,6,6,14]+ side-chains
which were previously truncated to four carbon length chains to reduce calculation costs.26 Some
calculations were repeated using the MP2 method to include electron correlation effects.31 The MP2
method is far more computationally demanding than B3LYP and so we restricted its use to the two
smallest complexes, namely MC:[Cl]- and MC:[NTf2]-. As shown in Supporting Information, while the
more detailed MP2 method provides as expected closer van der Waals’s contacts via the more explicit,
and possibly overestimated treatment of dispersion forces and so stronger ΔE values,33 the general
features of the probe-ion complex geometries are preserved and the anion-dependent complexation
energy difference ΔΔE is similar for both methods.
Physicochemical experiments: Trihexyl,tetradecyl Phosphonium Chloride (Cytec industries,
Niagara, Canada) and Trihexyl,tetradecyl Phosphonium bistrifluoro(sulfonyl)imide (Sigma
Aldrich) were purified using previously reported techniques 34 and were stored under argon to
exclude uptake of water. Spectrometric studies were carried out using a Cary 50 UV-Vis
spectrometer (JVA Analytical, Dublin, Ireland) with temperature controller and fibre optic
reflectance probe accessory. Samples were irradiated with UV light at 375nm using an in-house
10

fabricated array of UV LEDs (Roithner Lasertechnik, Vienna, Austria). Reichardts dye 30 (Sigma-
Aldrich chemicals) and 6-Nitro-1’,3’,3’-trimethylspiro[2H-1-benzopyran-2,2’-indolin] 1’,3’-
Dihdro-1’,3’,3’-trimethyl-6-nitrospiro (BSP) (Sigma-Aldrich chemicals) were used as purchased
with no further purification.
11

Results and Discussion
Computed properties for the range of IL and ion-probe complexes considered in the present study
are given in table 1 below. Comparison of the inter-ion distances in each IL pair with the ion-probe
distances, together with the computed complexation energies, provides a quantitative analysis of
the strength of ion pairing for intra-IL interactions and the competitive arrangement of the IL ions
around the zwitterionic MC probe molecule.
Table 1. Computed IL and ion-probe electronic structures.
Complex Close contact distances ΔE Δq (Å) (eV) (a.u)
[P6,6,6,14]+ : [Cl]- 2.4, 2.4, 2.7 (HCH2 : Cl); 3.8 (P : Cl) -3.54 -0.09[P6,6,6,14]+ : [NTf2]- 2.2 (HCH2 : N); 2.3, 2.4 (HCH2 : O=S) -2.89 -0.04
2.6 (HCH2 : F); 4.1 (P : N)[Cl]- : MC 2.5 (Cl : HN-CH3); 2.8 (Cl : HCH3) -0.98 -0.05
3.3 (Cl : N)[NTf2]- : MC 2.4 (O=S : HCH3); 2.6 (O=S : HC6H6) -0.73 -0.03
2.9 (N : HN-CH3); 3.6 (NNTf2 : NMC)[P6,6,6,14]+ : MC 2.4, 2.4, 2.7 (HCH2 : OPh); 3.6 (P : OPh) -0.48 +0.01
[P6,6,6,14]+ : MC : [Cl]- 2.5, 2.5, 2.9 (HCH2 : Cl); 4.0 (P : Cl) -0.58 -0.022.4 (Cl : HN-CH3); N/A (Cl : HCH3); 4.3 (Cl : NMC)
2.4, 2.7 (HCH2 : OPh); 3.9 (P : OPh)
Geometries and electronic structures calculated with B3LYP/6-311++G** model chemistry, as described in the text. E for
the two-species systems is the binding energy of the complex, as computed from the self-consistent field (SCF) energies of
the complex relative to the isolated species. For the final entry, the three-species full IL-probe complex, E is the energy of
the three-species complex minus the energies of the IL complex and the isolated MC. More negative values indicate more
favourable binding. q is the degree of charge transfer between species, as measured from the total charge of each species
in the complex and isolated systems. Negative q values generally indicate net transfer of electron density from the anion
to the cation or probe, except in the fifth entry, where the positive q indicates transfer of electron density from the probe
to the cation, and the sixth (final) entry where the negative q is the net electron transfer to MC, which is coupled with a
transfer of -0.09 a.u from Cl- to [P6,6,6,14]+, the same as for the IL in the absence of the probe (first entry).
12

IL ion-ion complexation: Computed structures of the anion:[P6,6,6,14]+ ion pairs indicate that [Cl]-
forms a tighter pair, lying 0.3Ǻ closer to the phosphorus centre than is the case for the nitrogen
centre of [NTf2]- (Table 1). For [P6,6,6,14][Cl] the tetrahedral orientation of the P-C carbons allows
for a clean approach of [Cl]- to the positive phosphorus centre. As [Cl]- approaches the P+ centre it
interacts also with CH2 hydrogens, resulting in a cradle-like structure with the chloride sitting in
the centre (Figure 2). The complex is characterised by moderate hydrogen bond type interactions
between [Cl]- and CH2 hydrogens and longer-range interactions between [Cl] - and the P+ centre.35
The chloride-phosphorus interaction, while longer range, is not believed to be of lesser importance
than the chloride-side-chain interactions. The computed dipole moment of 12 D along the Cl---P
axis illustrates how the Cl---P electrostatic interaction drives complexation; the computed
separation of 3.8 Ǻ at the upper limit of dipole-dipole interactions.
13
(a)

Figure 2. Complexation geometries with close contacts (the corresponding distances are given in Table 1) marked by dashed lines for net neutral phosphonium-based ILs, panel (a) with Cl- and (b) with [NTf2]-; carbon atoms are green, hydrogens grey, phosphorus atoms are blue, chlorines magenta, nitrogens cyan, oxygens red, sulphurs yellow and fluorine atoms are brown. Also shown are computed Molecular Electrostatic Potential (MEP) surfaces generated as described in the text and with surfaces coloured according to regions of net charge as marked in the scale bars, with the scale set in each case according to the largest net negative and net positive sites in each complex.
Replacing [Cl]- with the alternative [NTf2]- anion gives significantly weaker ion pairing, the IL
complexation strength decreases from -3.5 to -2.9 eV (Table 1), giving an anion-dependent liquid
ion pairing energy difference ΔΔE of approximately -0.6 eV. The negative charge on [NTf 2]- is
more delocalised resulting in an overall reduction in the strength and therefore effectiveness of the
charge-charge interaction, compared with the single-atom [Cl] - anion (Figure 2). The significant
net negative charge on the oxygens in particular allows [NTf 2]- to form more, but weaker,
intermolecular interactions with the phosphonium, principally via the alkyl side-chains (Figure 2).
Similar to the phosphonium chloride IL described above, moderate contacts of approximately 2.4
Ǻ are computed for the [P6,6,6,14][NTf2] complex. The similarity in intermolecular distances may
therefore be attributed to the cation, with the alkyl side-chains of the cation regulating the
14
(b)

complexation distance in both ion pairs. The increased size of the [NTf 2]- anion restricts the
approach to the phosphorus charge centre of the cation with a calculated N -:P+ distance of 4.1Ǻ.
At this distance, little or no electronic interaction can occur as reflected in the relatively low
binding energy and negligible degree of electron transfer (Table 1). In common with [P 6,6,6,14][Cl],
longer-range electrostatic interactions drive complexation, as reflected in the computed dipole
moment of 17 D parallel to the P---N axis of [P6,6,6,14][NTf2], with shorter-range polar interactions
between the anion and the phosphonium alkyl chains mediating the interaction. These calculations
give a greater understanding as to why [P6,6,6,14][Cl] (330 oC) has a lower thermal stability than that
of [P6,6,6,14][NTf2] (420 oC).36-38 Considering the structure of [P6,6,6,14][Cl] in Figure 2(a), the Cl- ion
is located in a favourable position (2.4 Å) to extract a hydrogen atom from one of the alkyl chains
thus promoting a Hoffman like elimination mechanism for thermal decomposition at lower
temperatures than that of the sterically-constrained (Figure 2b) [P6,6,6,14][NTf2].39
Having characterised the ion pair interaction within the ILs, interactions with the open, charged
merocyanine (MC) form of BSP (Figure 1a) were calculated, to probe whether the zwitterionic
MC isomer can compete for IL ion pair interaction sites and so possibly disrupt the IL structure
upon BSP → MC photoswitching.
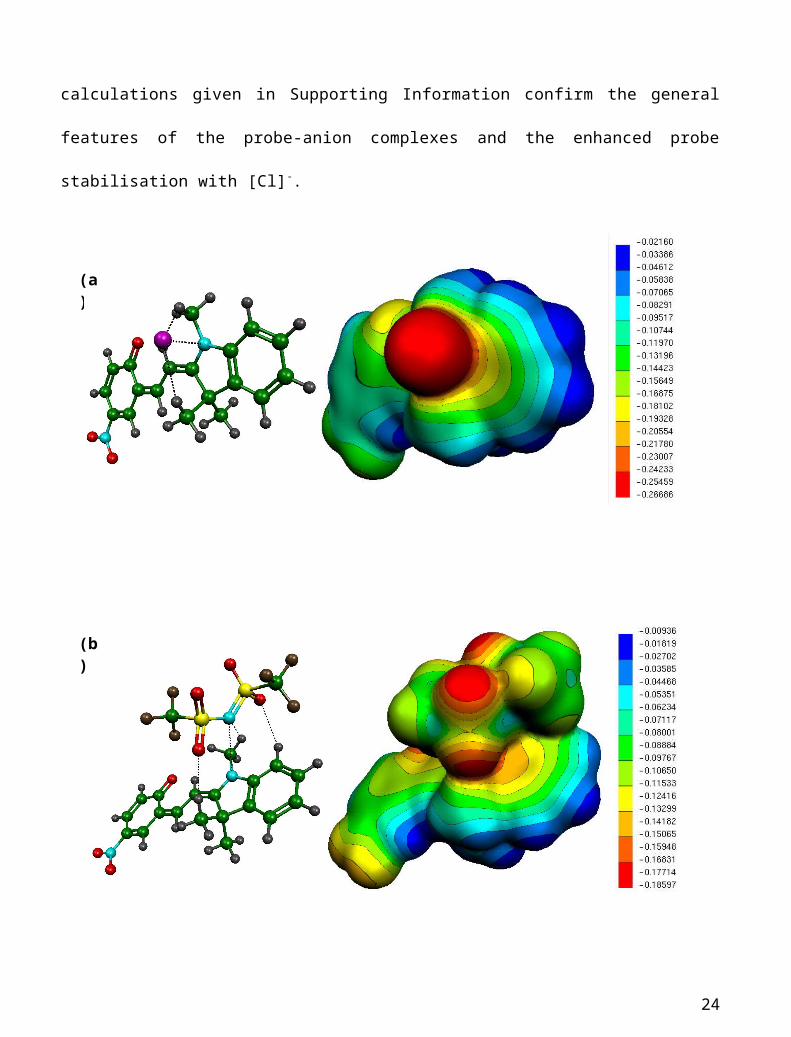
Probe-anion complexation: In common with ion pairing in the IL, Cl - interacts more strongly than
[NTf2]-, in this case [Cl]- sits 0.2Ǻ closer than [NTf2]- to the N+-CH3 site on MC (Table 1), and the
anions coordinate in a similar way to that observed for the interaction of the anions with the
phosphonium cation in the IL pairs. The methyl hydrogens on the indoline ring play a significant
role in the coordination of the anions, with contacts at ~2.4 Ǻ as the ion associates with the
positive N+-CH3 centre. The coordination of the anion with the methyl groups mediates the overall
15

interaction and stabilisation of the MC-anion couple (Figure 3). Natural population analysis (NPA)
30 of atomic charges gives a net charge of +0.28 on the methyl group of N +-CH3 compared with an
average of +0.07 on the other two methyl groups, and so it is the ability of the more compact [Cl] -
anion to make a strong contact with the N+-CH3 centre that provides the enhanced probe
stabilisation with [Cl]- compared to [NTf2]-. Reference MP2 calculations given in Supporting
Information confirm the general features of the probe-anion complexes and the enhanced probe
stabilisation with [Cl]-.
16
(a)
(b)

Figure 3. Complexation geometries with close contacts (given in Table 1) marked by dashed lines for charged ion-MC complexes, panel (a) the anionic complex with Cl - and (b) the anionic complex with [NTf2] - and (c) the cationic complex with [P6,6,6,14]+; carbon atoms are green, hydrogens grey, phosphorus atoms are blue, chlorines magenta, nitrogens cyan, oxygens red, sulphurs yellow and fluorine atoms are brown. Also shown are computed Molecular Electrostatic Potential (MEP) surfaces generated as described in the text and with surfaces coloured according to regions of net charge as marked in the scale bars, with the scale set in each case according to the largest net negative and net positive sites in each complex.
Probe-cation complexation: The MC:[P6,6,6,14]+ complex features contact distances similar to those
in [Cl]-:[P6,6,6,14]+ (Table 1), with the O(Ph) bound to the MC phenyl ring acting as the negatively-
charged centre. The strength of the interaction is weaker than for [Cl] -:[P6,6,6,14]+, with negligible
electron transfer (Table 1) and relatively weak stabilisation due to the decreased, compared with
[Cl]-, nucleophilicity and negative charge (-0.34 0.02 a.u. averaging over all systems) of the MC
OPh site; the phosphonium methyl H contacts to the MC OPh centre of 2.4 to 2.7 Ǻ are significantly
weaker than the intramolecular stabilisation due to the O---H contact of 2.2 Ǻ within MC.
MC:cation interactions are also generally weaker than MC:anion, due to the decreased localisation
of net charge at the MC positive centre (Figure 1b).
17
(c)

Probe-anion-cation complexation: The final, and computationally most demanding, system
calculated in this study is the three-species system [P6,6,6,14]+:MC:[Cl]- with the probe MC molecule
complexed simultaneously by both IL ions. The computed structure of the complex is shown in
figure 4 and summarised table 1; in general all inter-molecular contacts are weakened in the three-
species complex compared with the corresponding two-species systems, particularly for ion-MC
contacts and to a lesser extent for ion-ion contacts, giving an effective complexation energy for the
full IL-probe system of -0.58 eV (Table 1), computed from the electronic energies of the three-
species complex relative to the IL ion pair and the isolated probe molecule. The moderate binding
energy, negligible electron transfer to MC and weakened intermolecular contacts all indicate that,
while the zwitterionic probe is electrostatically stabilised by the presence of the IL pair, MC is not
sufficiently polar to break the IL pair.
Figure 4. Complexation geometries with close contacts (given in Table 1) marked by dashed lines for the net neutral complex of MC coordinated by both ions of the phosphonium chloride IL; hydrogens are omitted for clarity (and so some contact lines appear truncated), carbon atoms are green, phosphorus atoms are blue, chlorines magenta, nitrogens cyan and oxygens red. Also shown is the computed Molecular Electrostatic Potential (MEP) surface generated as described in the text and with the surface coloured according to regions of net charge as marked in the scale bar.
18

This is supported by reference calculations that replaced the large phosphonium with the
computationally more amenable imidazolium cation emim+ and allowed computation of the
influence of the IL cation on the full IL stabilisation of MC. While the detailed electronic
structures will be reported as part of a larger study of emim-based ILs, the computed emim +:MC:
[Cl]- complexation energy of -0.64 eV is very similar to the phosphonium value of -0.58 eV. This
suggests that, while the nature of both the cation and the anion can have a dramatic effect on inter-
ion IL binding energies and to a lesser extent individual ion-MC interactions (Table 1), the net
electrostatic stabilisation available to MC may not strongly depend on the nature of the IL polar
headgroups. That is, a similar small amount of charge-stabilisation will be available to MC
following inter-ion IL stabilisation, and it is the size of the non-polar region that dictates the
amount of stabilisation that actually occurs.
Headgroup stabilisation vs Tail disordering: Comparison of probe stabilisation energies with
known barriers to alkyl chain diffusion is instructive and indicates that the electrostatic
stabilisation of the probe by the polar IL headgroups is severely penalised by the long non-polar
tails of the quaternary phosphonium ion, the (putative) electrostatic interaction too weak to
overcome the chain diffusion activation barrier. From previous experimental and simulation
studies we may estimate the barrier to alkyl chain diffusion as 0.2 kcal/mol per CH 2 group.40, 41 For
[P6,6,6,14]+, this corresponds to a diffusion barrier of 6.4 kcal/mol per cation, which is just less than
half the electrostatic interaction (-0.58 eV, or -13.4 kcal/mol).
Phosphonium-based ILs form a tightly-woven hydrophobic “gel”, as described below, and so the
tight electrostatic coupling at the probe shown in figure 4 would require significant disruption of
the mesh and movement/reorientation of a large number of alkyl chains. The required relaxation
19

would propagate radially in three dimensions, and so we may anticipate that, for systems with such
large non-polar regions, the IL nanostructuring will preclude significant MC stabilisation. The
hydrophobic "gel" hypothesis above is supported by crystallographic studies based upon a similar
structure ([P10,10,10,10][Br]) that show significant structuring based upon side-chain interaction42
while structural analysis of imidazolium IL structures found that the differences between solid and
liquid structure results in only a small 10-15% expansion in the volume of the system. 43 Since
nano-scale structuring appears to be a common characteristic of all ILs, small expansions may be
expected within phosphonium ILs, preserving pseudo-crystalline ordering and thus rigidity within
the liquids.
To summarise the insights obtained from the electronic structure calculations, the intra-IL ion-ion
binding energy (ΔE) and extent of charge transfer (Δq) were both found to be stronger for [P 6,6,6,14]
[Cl] than for [P6,6,6,14][NTf2]. Computed binding energies are approximately 0.5 eV higher for the
chloride based IL. This value implies that the chloride based IL ion pairing interaction is much
stronger, as indicated by the Walden plots in previous studies.26, 27 Similarly, increased Δq values
show a greater interaction for [P6,6,6,14][Cl] than for [P6,6,6,14][NTf2], though both Δq values are < 0.1
a.u. (Table 1), indicating that non-covalent interactions dominate the ion-ion pairing in
phosphonium-based ILs. Computed binding energies are significantly lower for MC-ion
interactions compared to that of IL ion-ion interactions. [Cl] - again provides better stabilisation
compared with [NTf2]-, with a ΔE value of -0.98eV for MC:[Cl]- compared with -0.73 eV for MC-
[NTf2]-. Both MC-anion interactions are approximately three to four times weaker than the
phosphonium-anion pairing. In addition, the differences in the energies ΔΔE for MC as a function
of the anion are approximately half that calculated for the ILs. The zwitterionic MC probe is thus
less sensitive than the IL cation to the nature of the anion. Most importantly, from the range of E
20
(b)

values, -0.7 to -1.0 eV for MC-anion compared with -2.9 to -3.5 eV for the IL pair, it is clear that
while MC coordinates the IL anion, it is unable to break the IL pair. Similarly for the MC-cation
interaction, with MC-[P6,6,6,14]+ complexation is less than one sixth the strength of the anion --
[P6,6,6,14]+ IL pairing. While IL-MC through-space interactions can stabilise MC in emim-based IL
solvents16, no such covalent interactions were found for the phosphonium-based ILs, that is, no
high-lying occupied orbitals have significant inter-molecular character. Finally, the moderate
complexation energy calculated for MC with both IL ions simultaneously is not expected to be
sufficient to overcome the required disruption of the extended alkyl chain network in the long-tail
phosphonium-based ILs, and so the IL nano-structuring is preserved; this hypothesis is supported
by the calculated electronic structures, existing diffusion and Walden plot data and the
experiments described below.
Measured polarity and solvatochromic effects: The electronic structures described above serve to
rationalise some known features of IL structures and IL-probe interactions as described above,
together with some new physicochemical experiments performed as part of this study to determine
the thermal relaxation of MC. Taken together, the calculations and measurements help elucidate
the processes at work within the ILs. BSP was added to each IL and irradiated to examine the
immediate environment presented to the MC form in each liquid (Table 2). It is found that both the
[P6,6,6,14][NTf2] and [P6,6,6,14][Cl] liquids presented similar non-polar environments with a λmax shift
of 574nm while ET(30) values were found to vary in each liquid with values of 46.1 kcal.mol -1 and
43.8 kcal.mol-1 for [P6,6,6,14][NTf2] and [P6,6,6,14][Cl] respectively. Such ET(30) values were expected
since the stronger the ion pair, the more the binary system begins to resemble a single neutral
solvent molecule. The bulkier [NTf2]- anion resulted in less rigid associations between ion pairs
and so presented more ‘ideal’ ionic systems with the individual ion charge becoming more
21

apparent than that of the [Cl] - system. The result of this is that Reichardt’s Dye 30 senses a more
polar environment and thus the increase in values. While the solvatochromic shifts in the same
general region of ~570 nm may be attributed to the common phosphonium cation, the lack of
variation may be attributed to the dependence of the interaction of the anion with the MC. The
calculated strong interactions for [P6,6,6,6,14]+ with [Cl]- results in the [Cl]- becomminig somewhat
imbedded between the alkyl chains of the phosphonium cation (figure 2a). As a result the anion
becomes difficult for the MC to interact with and thus produce a stabilisation and solvatochromic
effect. Although the [NTf2]- is a bulkier anion and does not reside as close to the phosphonium
cation as the [Cl]- anion, the more diffuse nature of the ion charge and the lack of any hydrogen
bond donor sites removes the ability to stabilise the MC phenolate oxygen and the bulky nature of
the anion restricts interaction with the positive region of the MC.
Measured thermodynamic and kinetic parameters: Samples were irradiated with sample in-situ
within the cuvette holder of the spectrometer using an in-house fabricated UV LED array to induce
ring opening and MC formation. The cuvette holder was thermostatted to ensure accurate
reproducible sample heating. Upon removal of the light source , thermal relaxation first order decay
curves were examined using equation (1).
(1)
The rates of thermal relaxation were recorded at 298K and summarised in Table 2. The thermal
relaxation of [P6,6,6,14][Cl] is almost half that of [P6,6,6,14][NTf2] at 1.0 s-1 and 1.9 s-1 respectively. As
with the solvatochromic parameters previously discussed, the calculated ability of the chloride ion
to associate closer to the positive charge of the MC serves to explain the observed enhanced
22

stability (increased lifetime) of the open form. Similarly, the charge of the chloride ion is much
more concentrated because it is a single atom. In the case of the [NTf 2]- molecule the charge is
more delocalised and so the point strength of the negative charge is diminished and dispersed and
so provides less counter-charge to the positive site of the MC zwitterion. Diminished electrostatic
stabilisation therefore results in faster thermal relaxation of MC to its ground non-zwitterionic
state. Additionally, the enhanced liquid ion pairing with [Cl] - may provide a more rigid IL network
around the probe moleclue which would in turn restrict the thermal relaxation of the MC to its
closed form.
To test this hypothesis, thermodynamic parameters were derived using equations (2) (3) and (4),
providing the activation energy (Ea), entropy of activation (ΔS‡), enthalpy of activation (ΔH‡),
Gibbs free energy of activation (ΔG‡) and the equilibrium constant for activation (K‡) of each
system (Table 1) using the Eyring equation for transition state theory.44
(2) (3)
(4)
where, k = rate constantR = gas constantT = temperatureA = pre-exponential factorh = Planck’s constantkb = Boltzmann constant
The resulting entropies of activation ΔS‡ of -96.13J.K-1.mol-1 and -14.59 J.K-1.mol-1 for [P6,6,6,14][Cl]
and [P6,6,6,14][NTf2] respectively indicate that the chloride based IL does indeed present a more
rigid solvent system than that of [NTf2] - based ILs, consistent with the arguments previously put
23
forward by Fraser et al during their initial investigation into the formation of liquid ion pairing. 26
Under this hypothesis, the small size of the chloride ion drives the ion pairing that structures the
IL. The quaternary nature of the cations coupled with the length of the side-chains, e.g. [P 6,6,6,14]+,
results in the formation of a hydrophobic mesh dotted with paired charged headgroups, resulting in
a rigid liquid structure. MC is a bulky molecule which requires significant reorientation during
thermal relaxation and so its conformational space may become restricted within this rigid liquid
structure, prolonging the lifetime of the zwitterionic isomer. The bulkier [NTf2]- anion means that
ion pairing is weaker (Table 1) and so the formation of a more ‘ideal’ IL occurs, with reduced
rigidity in the liquid structure, as reflected in the lower magnitudes of the measured ΔS‡ values
and lower k values, resulting in a faster thermal relaxation of the MC to its closed form.
Similarly, lower enthalpies of activation ΔH‡ (22.63 kJ.mol-1 difference) implied that for chloride
based ILs the level of internal energy transferred by the relaxation process at the transition state of
the MC is less than that of [NTf2]-. Such reductions would be expected in the more rigid chloride
system where less interaction through solvent reorientation occurs between MC and the ions in
solution. Activation energies Ea support the enthalpic and entropic parameters, with less energy
required for the thermal relaxation of the MC to its ground state in chloride based ILs.
24

Table 2: Thermodynamic and kinetic parameters of BSP in ionic liquids. Reference values from Fraser et al.26
ET(30) λmax k20 S.D Ea ΔS‡ ΔH‡ ΔG‡20 K‡ ΔW26 ΔEdisp26
(kcal.mol-1) (nm) (x10-3s-1) ±(x10-5) (kcal.mol-1) (J.K-1.mole-1) (kJ.mol-1) (kJ.mol-1) (x10-5) (kJ.mol-1)
[P6,6,6,14][Cl] 43.8 574 1 1.04 71.51 -96.13 59.76 87.93 2.47 1.4 -46.00
[P6,6,6,14][NTf2] 46.1 574 1.9 1.39 84.94 -14.59 82.39 86.66 7.28 0.7 0.00
25

Conclusions
The discovery of nano-structuring within ionic liquids (ILs) has added new depth to both the
complexity of characterisation of IL solvent properties and the interpretation of the impact of this
structuring upon the unique effects observed in ILs. In this paper, we investigated the formation of
even smaller scale, atom-centered inter-ion interactions that mediate the formation of the nano-
structures and contribute to the observed physical (viscosity) and transport properties (conductivity)
within the liquids. The ability to influence structuring, and thus control the transport properties, could
allow for the creation of novel liquids with more predictable properties. In the present study computed
electronic structures suggest that non-covalent ion-ion electrostatic and steric interactions promote the
formation of liquid ion pairs in phosphonium-based ILs. When ions have dense concentrated charges
(for example, the chloride anion) tightly bound liquid ion pair systems are generated leading to liquids
that exhibit atypical, off-Walden plot, IL properties. In contrast, larger molecular anions such as
[NTf2]- have a more diffuse charge distribution, which gives weaker liquid ion pairing and so a more
typical IL system. In addition to this, the larger size of the anion compared to that of atomic anions
such as chloride would also result in weaker interactions with the phosphonium cation. The
experimental measurement of physical properties in the liquids via addition of the photo-chromic
spriropyran probe molecule yielded thermodynamic and kinetic data that supported the theoretical
models. Taken together the simulations and experiments indicate that the zwitterionic form of the
probe is unable to significantly disrupt IL nanostructuring in phosphonium-based ILs. The moderate
calculated IL-probe complexation energy is not expected to surmount the barrier to disruption of the
extended alkyl chain network, a hypothesis supported by the calculated electronic structures, the new
physicochemical experiments and known diffusion and Walden plot behaviour.
We may anticipate that rational engineering will allow the use of systems that incorporate the
balance between polar and non-polar interactions into their molecular design. For example, IL
26

nanostructuring may be ruptured by (a) the introduction of more polar headgroups that would
allow switching from the moderate to strong headgroup-mediated probe stabilisation coupled with
(b) shorter non-polar tails to switch from the large to moderate hydrophobic tail-mediated
diffusion barrier. While route (a) could possibly be controlled dynamically via in situ
electrochemical shifts, the IL hydrophobic tails, route (b), may serve as the first point for rational
design of IL solvents that strongly influence molecular photo-chromism, given that the
electrostatic interactions that stabilise the zwitterionic probe also promote IL nanostructuring (in a
three-site anion-zwitterion-cation “tug-of-war”). Consequently, tuning of charged sites will require
careful consideration of all ion-ion and IL-probe interactions.
In summary then, while the ‘ionicity’ of ILs can quickly become compromised by the formation of
solvent-solute interactions, a key design feature of next-generation photo-rheological systems will be
the incorporation of ionic materials that can make and break multi-site interactions, in a controllable
manner via switchable polar and/or non-polar interactions, to create probe-sensitive changes in
conductivity and viscosity.
Supporting Information Available: Anion-MC complex geometries and electronic structures calculated
with the MP2 method. This material is available free of charge via the Internet at http://pubs.acs.org.
27

Acknowledgements
We wish to acknowledge support for this research from the Biomedical diagnostics Institute (BDI)
and CLARITY, funded by Science Foundation Ireland (SFI) under Grant Nos. 05/CE3/B754 and
07/CE/I1147. Robert Byrne acknowledges Al Robertson (Cytec Canada Inc) for helpful
discussions and supply of ionic liquids. Damien Thompson acknowledges SFI for computing
resources at Tyndall National Institute and SFI/ Higher Education Authority for computing time at
the Irish Centre for High-End Computing (ICHEC).
References
1. Daibin, K.; Satoshi, U.; Robin, H.-B.; Shaik , M. Z.; Michael, G., Organic Dye-Sensitized Ionic Liquid Based Solar Cells: Remarkable Enhancement in Performance through Molecular Design of Indoline Sensitizers13. Angewandte Chemie International Edition 2008, 47, (10), 1923-1927.
2. Singh, P. K.; Kim, K.-W.; Rhee, H.-W., Development and characterization of ionic liquid doped solid polymer electrolyte membranes for better efficiency. Synthetic Metals 2009, 159, (15-16), 1538-1541.
3. Stracke, M. P.; Ebeling, G.; Cataluna, R.; Dupont, J., Hydrogen-Storage Materials Based on Imidazolium Ionic Liquids. Energy & Fuels 2007, 21, (3), 1695-1698.
4. Kalhor, H. R.; Kamizi, M.; Akbari, J.; Heydari, A., Inhibition of Amyloid Formation by Ionic Liquids: Ionic Liquids Affecting Intermediate Oligomers. Biomacromolecules 2009, 10, (9), 2468-2475.
5. Hallett, J. P.; Liotta, C. L.; Ranieri, G.; Welton, T., In Search of an "Ionic Liquid Effect". ECS Transactions 2009, 16, (49), 81-87.6. Hemeon, I.; Barnett, N. W.; Gathergood, N.; Scammells, P. J.; Singer, R. D., Manganese Dioxide Allylic and Benzylic Oxidation Reactions in
Ionic Liquids. Australian Journal of Chemistry 2004, 57, 125-128.7. Gordon, C. M., New developments in catalysis using ionic liquids. Applied Catalysis A: General 2001, 222, (1-2), 101-117.8. Lewandowski, A.; Swiderska, A., Electrochemical capacitors with polymer electrolytes based on ionic liquids. Solid State Ionics 2003, 161, (3-
4), 243-249.9. Crowhurst, L.; Falcone, R.; Lancaster, N. L.; Llopis-Mestre, V.; Welton, T., Using Kamlet-Taft Solvent Descriptors To Explain the Reactivity
of Anionic Nucleophiles in Ionic Liquids. J. Org. Chem. 2006, 71, (23), 8847-8853.10. Rodrigues, F.; do Nascimento, G. M.; Santos, P. S., Studies of ionic liquid solutions by soft X-ray absorption spectroscopy. Journal of Electron
Spectroscopy and Related Phenomena 2007, 155, (1-3), 148-154.11. Coleman, S. P.; Byrne, R.; Minkovska, S.; Diamond, D., Thermal reversion of Spirooxazine in ionic liquids containing the [NTf2]- anion.
Physical Chemistry Chemical Physics 2009, 11, (27), 5608-5614.12. Byrne, R.; Coleman, S.; Fraser, K. J.; Raduta, A.; MacFarlane, D. R.; Diamond, D., Photochromism of nitrobenzospiropyran in phosphonium
based ionic liquids. Physical Chemistry Chemical Physics 2009, 11, (33), 7286-7291.13. Reichardt, C., Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, (8), 2319-2358.14. Figueras, J., Hydrogen bonding, solvent polarity, and the visible spectrum of phenol blue and its derivativatives. J. Am. Chem. Soc. 1971, 93,
(13), 3255-3263.15. Jeliazkova, B. G.; Minkovska, S.; Deligeorgiev, T., Effect of complexation on the photochromism of 5'-(benzothiazol-2-
yl)spiroindolinonaphthooxazines in polar solvents. Journal of Photochemistry and Photobiology A: Chemistry 2005, 171, (2), 153-160.16. Ipe, B. I.; Mahima, S.; Thomas, K. G., Light-Induced Modulation of Self-Assembly on Spiropyran-Capped Gold Nanoparticles: A Potential
System for the Controlled Release of Amino Acid Derivatives. Journal of the American Chemical Society 2003, 125, (24), 7174-7175.17. Atabekyan, L. S., The Kinetics of Photocoloration of Spiropyrans upon Complexation. High Energy Chemistry 2002, 36, (6), 397-404.18. Chibisov, A. K.; Görner, H., Complexes of spiropyran-derived merocyanines with metal ions: relaxation kinetics, photochemistry and solvent
effects. Chemical Physics 1998, 237, (3), 425-442.19. Minkovska, S.; Jeliazkova, B.; Borisova, E.; Avramov, L.; Deligeorgiev, T., Substituent and solvent effect on the photochromic properties of a
series of spiroindolinonaphthooxazines. Journal of Photochemistry and Photobiology A: Chemistry 2004, 163, (1-2), 121-126.20. Byrne, R.; Fraser, K. J.; Izgorodina, E.; MacFarlane, D. R.; Forsyth, M.; Diamond, D., Photo- and solvatochromic properties of
nitrobenzospiropyran in ionic liquids containing the [NTf2]- anion. Physical Chemistry Chemical Physics 2008, 10, (38), 5919-5924.21. Consorti, C. S.; Suarez, P. A. Z.; de Souza, R. F.; Burrow, R. A.; Farrar, D. H.; Lough, A. J.; Loh, W.; da Silva, L. H. M.; Dupont, J.,
Identification of 1,3-Dialkylimidazolium Salt Supramolecular Aggregates in Solution. The Journal of Physical Chemistry B 2005, 109, (10), 4341-4349.
22. Iwata, K.; Okajima, H.; Saha, S.; Hamaguchi, H.-o., Local Structure Formation in Alkyl-imidazolium-Based Ionic Liquids as Revealed by Linear and Nonlinear Raman Spectroscopy. Accounts of Chemical Research 2007, 40, (11), 1174-1181.
23. Pott, T.; Meleard, P., New insight into the nanostructure of ionic liquids: a small angle X-ray scattering (SAXS) study on liquid tri-alkyl-methyl-ammonium bis(trifluoromethanesulfonyl)amides and their mixtures. Physical Chemistry Chemical Physics 2009, 11, (26), 5469-5475.
28

24. Canongia Lopes, J. N. A.; Padua, A. A. H., Nanostructural Organization in Ionic Liquids. The Journal of Physical Chemistry B 2006, 110, (7), 3330-3335.
25. Byrne, R.; Coleman, S.; Gallagher, S.; Diamond, D., Designer molecular probes for phosphonium ionic liquids. Physical Chemistry Chemical Physics 2010, 12, (8), 1895-1904.
26. Fraser, K. J.; Izgorodina, E. I.; Forsyth, M.; Scott, J. L.; MacFarlane, D. R., Liquids intermediate between "molecular" and "ionic" liquids: Liquid Ion Pairs? Chemical Communications 2007, (37), 3817-3819.
27. MacFarlane, D. R.; Forsyth, M.; Izgorodina, E. I.; Abbott, A. P.; Annat, G.; Fraser, K., On the concept of ionicity in ionic liquids. Physical Chemistry Chemical Physics 2009, 11, (25), 4962-4967.
28. M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; J. A. Montgomery, J.; T. Vreven; K. N. Kudin; J. C. Burant; J. M. Millam; S. S. Iyengar; J. Tomasi; V. Barone; B. Mennucci; M. Cossi; G. Scalmani; N. Rega; G. A. Petersson; H. Nakatsuji; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; M. Klene; X. Li; J. E. Knox; H. P. Hratchian; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; P. Y. Ayala; K. Morokuma; G. A. Voth; P. Salvador; J. J. Dannenberg; V. G. Zakrzewski; S. Dapprich; A. D. Daniels; M. C. Strain; O. Farkas; D. K. Malick; A. D. Rabuck; K. Raghavachari; J. B. Foresman; J. V. Ortiz; Q. Cui; A. G. Baboul; S. Clifford; J. Cioslowski; B. B. Stefanov; G. Liu; A. Liashenko; P. Piskorz; I. Komaromi; R. L. Martin; D. J. Fox; T. Keith; M. A. Al-Laham; C. Y. Peng; A. Nanayakkara; M. Challacombe; P. M. W. Gill; B. Johnson; W. Chen; M. W. Wong; C. Gonzalez; Pople, J. A. Gaussian 03 Revision C.02, Gaussian Inc: Wallingford, CT, 2004.
29. Becke, A. D., Density-functional thermochemistry. III. The role of exact exchange. The Journal of Chemical Physics 1993, 98, (7), 5648-5652.30. Reed, A. E.; Weinstock, R. B.; Weinhold, F., Natural population analysis. The Journal of Chemical Physics 1985, 83, (2), 735-746.31. Izgorodina, E. I.; Bernard, U. L.; MacFarlane, D. R., Ion-Pair Binding Energies of Ionic Liquids: Can DFT Compete with Ab Initio-Based
Methods? The Journal of Physical Chemistry A 2009, 113, (25), 7064-7072.32. Flükiger, P. F. Molekel: Molecular Visualisation Software, 4.3; University of Geneva Geneva.33. Kaplan, I. G., Intermolecular Interactions: Physical Picture, Computational Methods and Model Potentials. John Wiley & Sons Ltd:
Chichester, 2006.34. Burrell, A. K.; Sesto, R. E. D.; Baker, S. N.; McCleskey, T. M.; Baker, G. A., The large scale synthesis of pure imidazolium and pyrrolidinium
ionic liquids. Green Chemistry 2007, 9, (5), 449-454.35. Aakeroy, C. B.; Evans, T. A.; Seddon, K. R.; Páinkó, I., The C-H...Cl hydrogen bond: does it exist? New Journal of Chemistry 1999, 23, 145-
152.36. Newman, P. J.; MacFarlane, D. R., Preparation of CdSe Quantum Dots in Ionic Liquids. Zeitschrift für Physikalische Chemie 2006, 220, 1473–
1481.37. Cytec CYPHOS IL 101 phosphonium ionic liquid; 2/9/2005, 2005.38. Cytec, CYPHOS IL 109 phosphonium ionic liquid. 2005.39. Calderon, J. U.; Lennox, B.; Kamal, M. R., Thermally stable phosphonium-montmorillonite organoclays. Applied Clay Science 2008, 40, (1-4),
90-98.40. Bain, C. D.; Troughton, E. B.; Tao, Y. T.; Evall, J.; Whitesides, G. M.; Nuzzo, R. G., Formation of monolayer films by the spontaneous
assembly of organic thiols from solution onto gold. Journal of the American Chemical Society 1989, 111, (1), 321-335.41. Gannon, G.; Larsson, J. A.; Greer, J. C.; Thompson, D., Quantification of Ink Diffusion in Microcontact Printing with Self-Assembled
Monolayers. Langmuir 2008, 25, (1), 242-247.42. Abdallah, D. J.; Bachman, R. E.; Perlstein, J.; Weiss, R. G., Crystal Structures of Symmetrical Tetra-n-Alkyl Ammonium and Phosphonium
Halides. Dissection of Competing Interactions Leading to “Biradial†and “Tetraradial†Shapes. � � The Journal of Physical Chemistry B 1999, 103, (43), 9269-9278.
43. Dupont, J., On the Solid, Liquid and Solution Organization of Imidazolium Ionic Liquids. Journal of the Brazilian Chemical Society 2004, 15, (3), 341-350.
44. Laidler, K. J.; Meiser, J. H., Physical Chemistry. 3rd edition ed.; Houghton Mifflin: Boston, 1999.
29


















