
RoleofmRNA translational efficiency Escherichia · MIA(6); the Ipp promoter is located in the...
5
Proc. Natl. Acad. Sci. USA Vol. 81, pp. 5403-5407, September 1984 Biochemistry Role of mRNA translational efficiency in bovine growth hormone expression in Escherichia coli (runaway replicon/mRNA secondary structure/cistron) BRIGITTE E. SCHONER, HANSEN M. HSIUNG, RAMA M. BELAGAJE, NANCY G. MAYNE, AND RONALD G. SCHONER Division of Molecular and Cellular Biology, Lilly Research Laboratories, Indianapolis, IN 46285 Communicated by Hamilton 0. Smith, May 29, 1984 ABSTRACT The conditions necessary for high-level expression of methionyl bovine growth hormone (Met-bGH) in Escierichia coli were investigated. Plasmids were constructed that contain a thermoinducible runaway replicon and either the E. coli tryptophan or lipoprotein promoter and ribosome binding sites, which served as transcriptional and translational initiation sites for the expression of the bGH gene. The expres- sion of Met-bGH was low with either system. However, expression levels of up to 30% of total cell protein were ob- tained after the introduction of additional codons 3' to the ini- tiating AUG codon, thus altering the NH2-terminal amino acid sequence of bGH. To obtain high-level expression of Met-bGH a two-cistron system was constructed in which the codons that enhanced the expression of bGH were incorporated into the first cistron, and the coding region for Met-bGH was incorpo- rated into the second cistron. This approach may be generally applicable to achieving high-level expression of a gene that contains NH2-terminal sequences that do not allow for its effi- cient expression. Analyses of the stabilities of the bGH deriva- tives and their transcripts in vivo suggested that the variations in the level of expression were due to variations in the efficien- cy of mRNA translation. High-level expression of a cloned gene in Escherichia coli generally involves the incorporation of the gene into a multi- copy replicon, transcription of the gene from a strong pro- moter, and efficient translation of the mRNA (1-3). In this paper, we present our studies on the conditions necessary for high-level expression of bovine growth hormone (bGH) in E. coli. A cDNA clone for the full-length bovine growth pre-hormone was isolated by Miller et al. (4), and the coding region for mature bGH (5) was then cloned into pBR322 (un- published data). Our initial approach to obtain high-level expression of bGH was to increase the gene dosage. This was achieved by using the thermoinducible runaway-replication plasmid pl- MIA (6). This plasmid was derived from pKN402 (7) and has a copy number of 10-15 per cell below 30'C and 1000-2000 per cell at 370C. pIMIA contains a kanamycin-resistance marker and the E. coli lipoprotein (lpp) promoter and ribo- some binding site. When the bGH gene was cloned into pI- MIA, behind the lpp promoter, we observed that only small amounts of Met-bGH were synthesized. The poor expres- sion of Met-bGH was due to the poor translational efficiency of the mRNA encoding Met-bGH, which was overcome by inserting additional codons 3' to the AUG initiation codon. To obtain high-level expression of Met-bGH, without the in- troduction of extra codons, we constructed a genetic system consisting of two contiguous cistrons. The first cistron con- tains the codons that enhanced the translational efficiency of Met-bGH, and the second cistron contains the Met-bGH coding sequence. We refer to this system as a two-cistron construction, and we propose that this approach will be gen- erally applicable to improving gene expression in E. coli. MATERIALS AND METHODS Bacterial Strains and Media. E. coli K-12 RV308 [su-, AlacX74, gal IS II::OP308, strA] (8) was the host strain for all of the recombinant plasmids. The source of the trp PO region was pHI7 (9). pIMIA (6) was obtained from M. Inou- ye. The bGH-cDNA clone was obtained from J. D. Baxter (4). In all experiments, RV308 cells containing recombinant plasmids were grown in TY broth (Difco) with 50 Mg of kana- mycin per ml (Sigma) at temperatures indicated in the text. Chemical Synthesis of DNA Linker Sequences. All deoxyri- booligonucleotides were synthesized either manually by the modified phosphotriester method (10) or by the DNA syn- thesizers [Biosearch (San Rafael, CA) Sam I or Applied Bio- systems (Foster City, CA) model 380A] according to the pro- cedures recommended by the manufacturers. The oligonu- cleotides were purified by HPLC using a Whatman Partisil- 10 SAX column (11). The linkers were prepared by joining these oligonucleotides enzymatically using T4 DNA ligase (12). Plasmid Constructions. Conditions for all restriction endo- nuclease and ligation reactions were those recommended by the manufacturers (New England Biolabs or Boehringer Mannheim). For plasmid constructions, gel-purified restric- tion fragments (13) were ligated and transformed into CaCI2- treated RV308 cells (14). Transformants were selected on TY agar plates containing 50 Mg of kanamycin per ml. Plas- mid DNA from the transformants was extracted (15) and ex- amined by restriction analysis. Appropriate restriction frag- ments containing synthetic linkers were sequenced by the method of Maxam and Gilbert (16). Polyacrylamide Gel Electrophoresis. Polyacrylamide gels (17) were used to analyze cells for bGH accumulation. Cell pellets were dissolved in modified sample buffer: 0.125 M Tris HCl, pH 6.8/2% NaDodSO4/30% (vol/vol) glycerol/i M 2-mercaptoethanol/6 M urea, and boiled for 3 min before loading. After staining with Coomassie blue (Sigma), gels were analyzed with a Shimadzu (Columbia, MD) 910 scanner on line with a Hewlett-Packard 2100 computer that integrat- ed the areas under the peaks. For pulse-chase experiments, cells grown at 250C to early logarithmic phase were shifted to 370C for 3 hr prior to the addition of 20 ,uCi of [355]methio- nine per ml (1240 Ci/mmol; 1 Ci = 37 GBq; Amersham). After 40 min of labeling, a 50,000-fold excess of unlabeled methionine over [35S]methionine was added to the culture, and cells were removed at the indicated times for analysis. RNA Isolation and Blotting. Cells grown at 250C to early logarithmic phase were shifted to 370C for 3 hr and total cel- lular RNA was isolated as described by Young and Furano (18). For RNA blots, the RNA was transferred from 1.2% Abbreviation: Met-bGH, methionyl bovine growth hormone. 5403 The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. §1734 solely to indicate this fact. Downloaded by guest on January 6, 2020
Transcript of RoleofmRNA translational efficiency Escherichia · MIA(6); the Ipp promoter is located in the...

Proc. Natl. Acad. Sci. USAVol. 81, pp. 5403-5407, September 1984Biochemistry
Role of mRNA translational efficiency in bovine growth hormoneexpression in Escherichia coli
(runaway replicon/mRNA secondary structure/cistron)
BRIGITTE E. SCHONER, HANSEN M. HSIUNG, RAMA M. BELAGAJE, NANCY G. MAYNE,AND RONALD G. SCHONERDivision of Molecular and Cellular Biology, Lilly Research Laboratories, Indianapolis, IN 46285
Communicated by Hamilton 0. Smith, May 29, 1984
ABSTRACT The conditions necessary for high-levelexpression of methionyl bovine growth hormone (Met-bGH) inEscierichia coli were investigated. Plasmids were constructedthat contain a thermoinducible runaway replicon and eitherthe E. coli tryptophan or lipoprotein promoter and ribosomebinding sites, which served as transcriptional and translationalinitiation sites for the expression of the bGH gene. The expres-sion of Met-bGH was low with either system. However,expression levels of up to 30% of total cell protein were ob-tained after the introduction of additional codons 3' to the ini-tiating AUG codon, thus altering the NH2-terminal amino acidsequence of bGH. To obtain high-level expression of Met-bGHa two-cistron system was constructed in which the codons thatenhanced the expression of bGH were incorporated into thefirst cistron, and the coding region for Met-bGH was incorpo-rated into the second cistron. This approach may be generallyapplicable to achieving high-level expression of a gene thatcontains NH2-terminal sequences that do not allow for its effi-cient expression. Analyses of the stabilities of the bGH deriva-tives and their transcripts in vivo suggested that the variationsin the level of expression were due to variations in the efficien-cy of mRNA translation.
High-level expression of a cloned gene in Escherichia coligenerally involves the incorporation of the gene into a multi-copy replicon, transcription of the gene from a strong pro-moter, and efficient translation of the mRNA (1-3). In thispaper, we present our studies on the conditions necessaryfor high-level expression of bovine growth hormone (bGH)in E. coli. A cDNA clone for the full-length bovine growthpre-hormone was isolated by Miller et al. (4), and the codingregion for mature bGH (5) was then cloned into pBR322 (un-published data).Our initial approach to obtain high-level expression of
bGH was to increase the gene dosage. This was achieved byusing the thermoinducible runaway-replication plasmid pl-MIA (6). This plasmid was derived from pKN402 (7) and hasa copy number of 10-15 per cell below 30'C and 1000-2000per cell at 370C. pIMIA contains a kanamycin-resistancemarker and the E. coli lipoprotein (lpp) promoter and ribo-some binding site. When the bGH gene was cloned into pI-MIA, behind the lpp promoter, we observed that only smallamounts of Met-bGH were synthesized. The poor expres-sion of Met-bGH was due to the poor translational efficiencyof the mRNA encoding Met-bGH, which was overcome byinserting additional codons 3' to the AUG initiation codon.To obtain high-level expression of Met-bGH, without the in-troduction of extra codons, we constructed a genetic systemconsisting of two contiguous cistrons. The first cistron con-tains the codons that enhanced the translational efficiency ofMet-bGH, and the second cistron contains the Met-bGH
coding sequence. We refer to this system as a two-cistronconstruction, and we propose that this approach will be gen-erally applicable to improving gene expression in E. coli.
MATERIALS AND METHODSBacterial Strains and Media. E. coli K-12 RV308 [su-,
AlacX74, gal IS II::OP308, strA] (8) was the host strain forall of the recombinant plasmids. The source of the trp POregion was pHI7 (9). pIMIA (6) was obtained from M. Inou-ye. The bGH-cDNA clone was obtained from J. D. Baxter(4). In all experiments, RV308 cells containing recombinantplasmids were grown in TY broth (Difco) with 50 Mg of kana-mycin per ml (Sigma) at temperatures indicated in the text.Chemical Synthesis ofDNA Linker Sequences. All deoxyri-
booligonucleotides were synthesized either manually by themodified phosphotriester method (10) or by the DNA syn-thesizers [Biosearch (San Rafael, CA) Sam I or Applied Bio-systems (Foster City, CA) model 380A] according to the pro-cedures recommended by the manufacturers. The oligonu-cleotides were purified by HPLC using a Whatman Partisil-10 SAX column (11). The linkers were prepared by joiningthese oligonucleotides enzymatically using T4 DNA ligase(12).Plasmid Constructions. Conditions for all restriction endo-
nuclease and ligation reactions were those recommended bythe manufacturers (New England Biolabs or BoehringerMannheim). For plasmid constructions, gel-purified restric-tion fragments (13) were ligated and transformed into CaCI2-treated RV308 cells (14). Transformants were selected onTY agar plates containing 50 Mg of kanamycin per ml. Plas-mid DNA from the transformants was extracted (15) and ex-amined by restriction analysis. Appropriate restriction frag-ments containing synthetic linkers were sequenced by themethod of Maxam and Gilbert (16).Polyacrylamide Gel Electrophoresis. Polyacrylamide gels
(17) were used to analyze cells for bGH accumulation. Cellpellets were dissolved in modified sample buffer: 0.125 MTris HCl, pH 6.8/2% NaDodSO4/30% (vol/vol) glycerol/iM 2-mercaptoethanol/6 M urea, and boiled for 3 min beforeloading. After staining with Coomassie blue (Sigma), gelswere analyzed with a Shimadzu (Columbia, MD) 910 scanneron line with a Hewlett-Packard 2100 computer that integrat-ed the areas under the peaks. For pulse-chase experiments,cells grown at 250C to early logarithmic phase were shifted to370C for 3 hr prior to the addition of 20 ,uCi of [355]methio-nine per ml (1240 Ci/mmol; 1 Ci = 37 GBq; Amersham).After 40 min of labeling, a 50,000-fold excess of unlabeledmethionine over [35S]methionine was added to the culture,and cells were removed at the indicated times for analysis.RNA Isolation and Blotting. Cells grown at 250C to early
logarithmic phase were shifted to 370C for 3 hr and total cel-lular RNA was isolated as described by Young and Furano(18). For RNA blots, the RNA was transferred from 1.2%
Abbreviation: Met-bGH, methionyl bovine growth hormone.
5403
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Dow
nloa
ded
by g
uest
on
Janu
ary
6, 2
020

5404 Biochemistry: Schoner et al.
agarose gels to nitrocellulose paper; for dot blots, the RNAwas applied directly to the paper (19). The conditions fornick-translation, hybridization, and washing have been de-scribed (20).
RESULTSStructure of bGlI Expression Vectors. pIMIA was used as a
starting plasmid to construct a series of expression vectorsfor bGH that differed primarily with respect to the promoter(E. coli lpp or trp) and the DNA sequence at the 5' end of thebGH coding region (Fig. 1). The sequence between the pro-moter and the beginning of the bGH coding region (Fig. 2)was essentially that of the native E. coli trp operon (21) orlpp gene (22) 5'-untranslated region, which included the ribo-some binding site and the ATG initiation codon. The bGHgene was flanked by the initiation codon and a TAG termina-tion codon that overlapped with the unique BamHI site. Thesequence 3' to the BamHI site was part of the pIMIA plas-mid and codes for the 3' end of the E. coli Ipp gene and thelpp transcriptional terminator, which is located 150 basepairs downstream from the BamHI site (22).
Plasmids with altered sequences at the 5' end of the bGHgene were constructed by ligating synthetic DNA oligonucle-otides between the Taq I and HgiAI sites of the trp promoterplasmids and between the Xba I and HgiAI sites of the lpppromoter plamids, as shown in Fig. 1. The sequences ofthese oligonucleotides are shown in Fig. 2. In each case, thesequence of the newly synthesized and cloned oligonucleo-tide was confirmed by DNA sequencing.
A
NdeI
NdeI
B
NdeIBgl IIHind IIINde I
Bst E LI
FIG. 1. Structure of bGH expression plasmids. (A) The 'ppexpression plasmids are 10.8 kilobases and were derived from pl-MIA (6); the Ipp promoter is located in the region between EcoRIand Xba I. (B) The trp expression plasmids are also 10.8-kilobasederivatives of pIMIA but contain the trp promoter from pHI7 (9),which is located in the region between EcoRI and Taq I. The loca-tion and direction of transcription of the bGH genes and the kana-
mycin resistance (Kan') gene are indicated by the arrows.
IIII
Relative Expression of bGH Derivatives. The bGH expres-sion plasmids were transformed into E. coli RV308, and theresulting cultures were grown at 250C to early logarithmicphase. To amplify the synthesis of the bGH derivatives en-coded by these plasmids, the growth temperature was shift-ed to 370C for 6 hr, which induces runaway replication ofthese plasmids. After amplification of the trp promoter plas-mids, bGH synthesis was no longer repressed by tryptophan(data not shown) because of titration of the trp repressor bythe trp operator. The cells were harvested, lysed in samplebuffer, and the lysates were subjected to NaDodSO4/poly-acrylamide gel electrophoresis. The gels were stained withCoomassie blue and scanned. The results are shown in Figs.2 and 3. We confirmed that these cells were synthesizingbGH by radioimmunoassay, HPLC analysis, and bioassay(data not shown).The amount of bGH synthesized from similar bGH genes
varied dramatically and did not depend on the promoterused. For example, low-level expression was obtained withcells harboring pCZ104 or pCZ110, plasmids in which theMet-bGH gene is transcribed from the ipp and the trp pro-moters, respectively (Fig. 3, lanes 3 and 6). In these plas-mids, five of the first seven codons in the bGH gene werereplaced by those shown in Fig. 2. They specify the sameamino acids as do the natural bGH codons but are those pre-ferred in E. coli (23). A slightly higher level of Met-bGHexpression was observed with pCZ108 (lane 5) in which thenatural bGH codons are retained. However, a substantial in-crease in bGH expression was obtained (from 1.7% to -30%of total cell protein) in cells harboring pCZ101 and pCZ105(lanes 2 and 4), which differ from pCZ108 in that they con-tain eight additional codons 3' to the initiation codon. Thisresult suggested that high-level expression obtained withthese plasmids depends on the eight extra codons and is in-dependent of the promoter and the 5'-untranslated region ofthe mRNA.The low bGH expression obtained with pCZ110 could be
increased to "30% of cell protein by adding three of the ex-tra eight codons present in pCZ105, generating pCZ112 (lane7), or by adding just one codon (GAT), generating pCZ115(lane 9). However, adding one codon (GCT) to pCZ104 toconstruct pCZ100 did not result in high-level expression ofbGH (lane 1). It should be noted that GCT was not amongthe eight 5' codons used in pCZ101 and pCZ105. Thus, thereappears to be some specificity in the codons that can serveto increase the level of bGH expression.To obtain high-level expression of Met-bGH without intro-
ducing extra codons, we converted the coding sequence ofpCZ101 to a two-cistron expression system for Met-bGH(designated pCZ114) as shown in Fig. 4. The minimum num-ber of base changes were introduced into the 5' end of thecoding sequence of pCZ101 to create a termination codon(TAA) and an initiation codon (ATG). One additional basechange (T to G) served to create a Shine-Dalgarno sequencefor the second cistron. Two products should be encoded bypCZ114, a seven-amino acid polypeptide, the product of thefirst cistron, and Met-bGH, the product of the second cis-tron. The DNA sequence of the Met-bGH cistron in pCZ114is identical to that of the Met-bGH gene in pCZ108. Wefound that the Met-bGH encoded by pCZ114 was expressedat a high level, - 19% of total cell protein (Fig. 3, lane 8).These results indicated that the low-level expression of Met-bGH from pCZ108 was not due to an inherent problem intranslating the codons in this gene. When we changed theATi initiator codon for the first cistron in pCZ114 (Fig. 4) toan ATT codon or to an ATC codon, the level of Met-bGHexpression was <0.5% of cell protein (data not shown).Thus, initiation of translation at the first cistron appears tobe obligatory for efficient translation of the second cistron.
These results also suggested that Met-bGH is stable in E.
Proc. NatL Acad Sci. USA 81 (1984)
Dow
nloa
ded
by g
uest
on
Janu
ary
6, 2
020

Proc. Natl. Acad. Sci USA 81 (1984) 5405
5' UNTRANSLATED REGION MET
- ATG-TTC-CCA-TTG-IGATI4-AAG
ATG
ATG
ATG GCT
ATG-TTC-CCA-TT6- OGAT14-AAG
TG
ATG GAT
ATG GAT-OAT-AAG
bGH CODING SEQUENCEPHE-PRO-ALA-MET-SER-LEU-SER-GLY- LEU-PHE- ALA-ASN-ALA-VAL-LEU
PLASMID PERCENTbGH
pCZIO1
pCZ108
pCZ104
pCZIOO
pCZ105
pCZ10
pCZI15
pCZ112
30
1.7
<0.5
<0.5
34
< 0.5
32
33
FIG. 2. DNA sequence variations around the 5' end of the bGH gene in the expression plasmids. The first four lines represent the DNAsequence corresponding to the 5' end of the Ipp mRNA extending to the HgiAI site within the bGH coding region of the lpp expression plasmids(see Fig. 1). The next four lines represent the sequence of the 5' end of the trp mRNA in the trp expression plasmids. The sequences beyond theHgiAI site are identical in all plasmids. The amounts of bGH accumulated by cells containing the plasmids listed are expressed as percentage oftotal cell protein as determined by scanning Coomassie blue-stained polyacrylamide gels. The NH2-terminal codons of the bGH gene in pCZ101and pCZ105 were chosen initially to create a derivative of Met-bGH with eight additional NH2-terminal amino acids, which provide a cleavagesite for enterokinase. The use of enterokinase allows for the enzymatic removal of the initiator methionine and the next eight amino acids toyield mature bGH. SD, Shine-Dalgarno sequence.
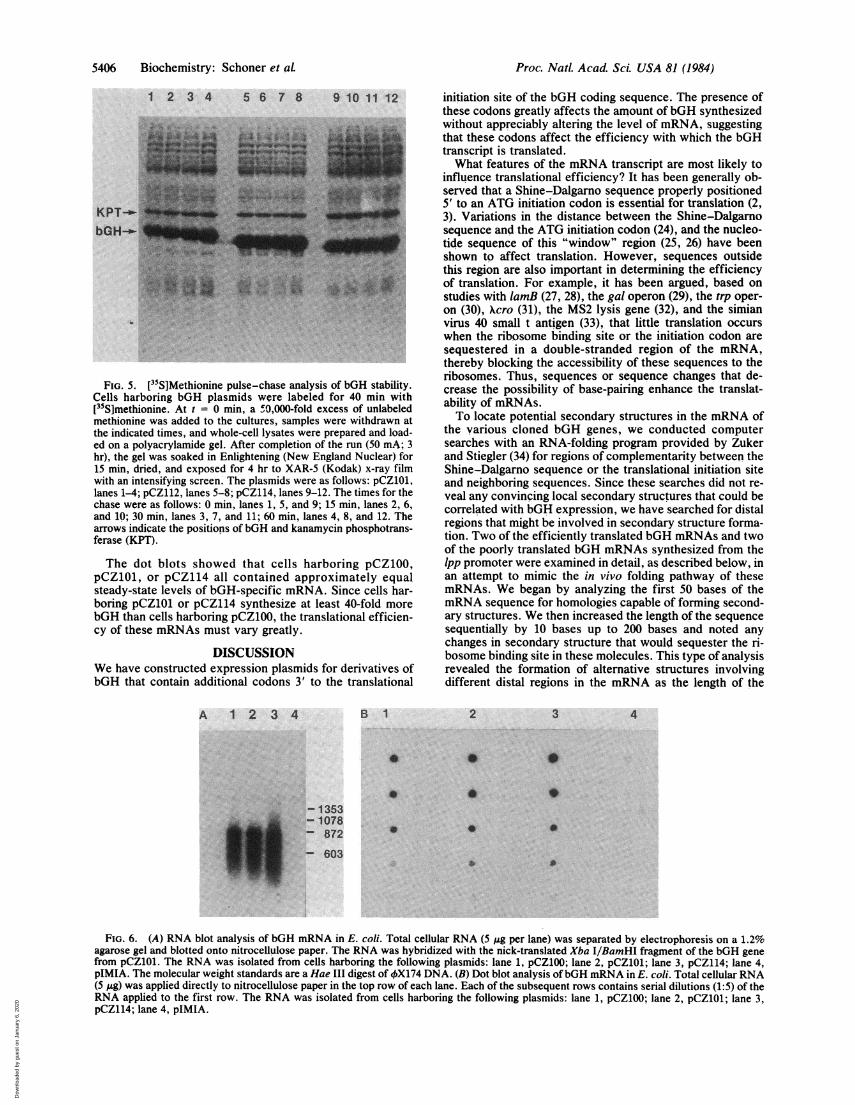
coli. The in vivo stabilities of Met-bGH and two other bGHderivatives were confirmed in pulse-chase experimentswith [35S]methionine, as shown in Fig. 5. Little degradationof the bGH derivatives encoded by pCZ101, pCZ112, and
1 2 3 4 5 6 7 8 9 10
.n..I .> lt! .......... t.
.- KPT
= bGH
pCZ114 occurred during a 60-min chase with unlabeled me-thionine.
Steady-State bGJI mRNA Pool Size in E. coli. Since differ-ences in the protein stabilities did not appear to cause theobserved variation in bGH accumulation, we asked whetherdifferences exist in the relative stabilities of the bGH tran-scripts of three bGH genes.RNA blots (Fig. 6A) and dot blots (Fig. 6B) compared the
steady-state levels of bGH-specific mRNA in cells accumu-lating large quantities ofbGH (pCZ101 and pCZ114) or smallquantities of bGH (pCZ100). In each plasmid, transcriptionof the bGH gene was initiated at the lpp promoter. The RNAblots demonstrated that cells harboring pCZ100, pCZ101, orpCZ114 all synthesized a mRNA that hybridized to the bGHgene. The size of the mRNA, -800 base pairs, was that pre-dicted for the bGH mRNA. As expected, cells harboring pl-MIA did not synthesize a bGH-specific mRNA. The lowermolecular weight mRNA species shown in Fig. 6A could bedue to degradation of the full-length mRNA, or it could rep-resent mRNA molecules that are not yet completely tran-scribed.
1 MET-PHE-PRO-LEU-ASP-ASP-ASP-ASP-LYS-PHE . . . bGH
2 GAGGGTATTAATA-ATG-TTC-CCA-TTB-IAT-OAT-GAT-GAT-AAG-TTC ...
SD
MET-PHE-PRO-LEU-GLU-ASP-ASP MET-PHE ... bGH
FIG. 3. Polyacrylamide gel analysis of bGH synthesis. Lysateswere prepared from 1 ml of cell culture harboring the bGH plasmidsdescribed in Fig. 2 after 6 hr of growth at 370C. The lysates wereloaded on a 12.5% polyacrylamide gel (1.5 mm x 18 cm x 18 cm)and run at 50 mA for 3 hr. Lanes: 1, pCZ100; 2, pCZ101; 3, pCZ104;4, pCZ105; 5, pCZ108; 6, pCZ110; 7, pCZ112; 8, pCZ114; 9,pCZ115; 10, molecular weight markers were phosphorylase B(92,500), bovine serum albumin (66,200), ovalbumin (45,000), car-bonic anhydrase (31,000), soybean trypsin inhibitor (21,500), and ly-sozyme (14,400). The arrows indicate the positions of bGH andkanamycin phosphotransferase (KPT).
4 GAGGGTATTAATA-ATG-TTC-CCA-TTG-GAG-GAT-GAT-TAA-ATG-TTC ...
SD SD
FIG. 4. Two-cistron construction for Met-bGH expression. Line1 shows the NH2-terminal amino acid sequence of bGH in pCZ101.Line 2 shows the corresponding DNA sequence and the region 5' tothe initiator codon. Lines 3 and 4 show the changes in the amino acidsequence and the DNA sequence (underlined bases) that were madeto convert the bGH gene into two cistrons (as in pCZ114). SD) de-notes Shine-Dalgarno sequences for the first and second cistrons.
Ipp
trp
SD5'- GCTACATGGAGATTAACTCAATCTAGAGGOTATTAAT
XbaII - SD5'- GCTACATGGAGATTAACTCAATCTAOAGOGTATTAATA
Xbal_> SD
5'- GCTACATGGAGATTAACTCAATCTAGAGdGTATTAATAXbalI i-:Al SD
5'- GCTACATGGAGATTAACTCAATCTAGAOGGTATTAATAXbal
SD5'- AAGTTCACGTAAAAAGGGTATCGAC
SD Taql5'- AAGTTCACGTAAAAAGGGTATCOACA
SD Taql5'- AAGTTCACGTAAAAAGOGTATCGACC
SD Taql
5'- AAGTTCACGTAAAAAGOOTATCGACCTaql
TTC-CCA-GCC-ATG-TCC-TTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-CTG-CTCHgiAI-
TTC-CCA-GCC-ATG-TCC-TTG-TCC-GOC-CTG-TTT-GCC-AAC-GCT-GTG-CTCH9iAI-
TTT-CCG-GCT-ATG-TCT-CTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-OTO-CTCHgIAI-
TTT-CCG-GCT-ATG-TCT-CTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-GTG-CTCHgiAI.._
TTC-CCA-GCC-ATG-TCC-TTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-GTG-CTCHgIAI _
TTT-CCG-GCT-ATG-TCT-CTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-GT-CTCTHgIAI --
TTT-CCG-GCT-ATG-TCT-CTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-GTO-CTCH91AI
TTT-CCG-GCT-ATG-TCT-CTG-TCC-GGC-CTG-TTT-GCC-AAC-GCT-OTG-CTCH91AI _
.w'..3
n -,.P-,vf:*.- JPOAW *:y:Kaw. low -. 000 0 I :.," "'a, 'O
SIM0400 now we'
Biochemistry: Schoner et aL
I
Dow
nloa
ded
by g
uest
on
Janu
ary
6, 2
020
5406 Biochemistry: Schoner et al.
1 2 3 4 5 6 7 8 9 10 11 12
KPT-"bGH-.
U:.
FIG. 5. [35S]Methionine pulse-chase analysis of bGH stability.Cells harboring bGH plasmids were labeled for 40 min with[35S]methionine. At t = 0 min, a '0,OOO-fold excess of unlabeledmethionine was added to the cultures, samples were withdrawn atthe indicated times, and whole-cell lysates were prepared and load-ed on a polyacrylamide gel. After completion of the run (50 mA; 3hr), the gel was soaked in Enlightening (New England Nuclear) for15 min, dried, and exposed for 4 hr to XAR-5 (Kodak) x-ray filmwith an intensifying screen. The plasmids were as follows: pCZ101,lanes 1-4; pCZ112, lanes 5-8; pCZ114, lanes 9-12. The times for thechase were as follows: 0 min, lanes 1, 5, and 9; 15 min, lanes 2, 6,and 10; 30 min, lanes 3, 7, and 11; 60 min, lanes 4, 8, and 12. Thearrows indicate the positions of bGH and kanamycin phosphotrans-ferase (KPT).
The dot blots showed that cells harboring pCZ100,pCZ101, or pCZ114 all contained approximately equalsteady-state levels of bGH-specific mRNA. Since cells har-boring pCZ101 or pCZ114 synthesize at least 40-fold morebGH than cells harboring pCZ100, the translational efficien-cy of these mRNAs must vary greatly.
DISCUSSIONWe have constructed expression plasmids for derivatives ofbGH that contain additional codons 3' to the translational
initiation site of the bGH coding sequence. The presence ofthese codlons greatly affects the amount ofbGH synthesizedwithout appreciably altering the level of mRNA, suggestingthat these codons affect the efficiency with which the bGHtranscript is translated.What features of the mRNA transcript are most likely to
influence translational efficiency? It has been generally ob-served that a Shine-Dalgarno sequence properly positioned5' to an ATG initiation codon is essential for translation (2,3). Variations in the distance between the Shine-Dalgarnosequence and the ATG initiation codon (24), and the nucleo-tide sequence of this "window" region (25, 26) have beenshown to affect translation. However, sequences outsidethis region are also important in determining the efficiencyof translation. For example, it has been argued, based onstudies with lamB (27, 28), the gal operon (29), the trp oper-on (30), Xcro (31), the MS2 lysis gene (32), and the simianvirus 40 small t antigen (33), that little translation occurswhen the ribosome binding site or the initiation codon aresequestered in a double-stranded region of the mRNA,thereby blocking the accessibility of these sequences to theribosomes. Thus, sequences or sequence changes that de-crease the possibility of base-pairing enhance the translat-ability of mRNAs.To locate potential secondary structures in the mRNA of
the various cloned bGH genes, we conducted computersearches with an RNA-folding program provided by Zukerand Stiegler (34) for regions of complementarity between theShine-Dalgarno sequence or the translational initiation siteand neighboring sequences. Since these searches did not re-veal any convincing local secondary structures that could becorrelated with bGH expression, we have searched for distalregions that might be involved in secondary structure forma-tion. Two of the efficiently translated bGH mRNAs and twoof the poorly translated bGH mRNAs synthesized from theIpp promoter were examined in detail, as described below, inan attempt to mimic the in vivo folding pathway of thesemRNAs. We began by analyzing the first 50 bases of themRNA sequence for homologies capable of forming second-ary structures. We then increased the length of the sequencesequentially by 10 bases up to 200 bases and noted anychanges in secondary structure that would sequester the ri-bosome binding site in these molecules. This type of analysisrevealed the formation of alternative structures involvingdifferent distal regions in the mRNA as the length of the
A 1 2 3 4 B 1 2 3 4
- 1353- 1078- 872- 603
FIG. 6. (A) RNA blot analysis of bGH mRNA in E. coli. Total cellular RNA (5 Ag per lane) was separated by electrophoresis on a 1.2%agarose gel and blotted onto nitrocellulose paper. The RNA was hybridized with the nick-translated Xba I/BamHI fragment of the bGH genefrom pCZ101. The RNA was isolated from cells harboring the following plasmids: lane 1, pCZ100; lane 2, pCZ101; lane 3, pCZ114; lane 4,pIMIA. The molecular weight standards are a Hae III digest of 4X174 DNA. (B) Dot blot analysis ofbGH mRNA in E. coli. Total cellular RNA(5 ,g) was applied directly to nitrocellulose paper in the top row of each lane. Each of the subsequent rows contains serial dilutions (1:5) of theRNA applied to the first row. The RNA was isolated from cells harboring the following plasmids: lane 1, pCZ100; lane 2, pCZ101; lane 3,pCZ114; lane 4, pIMIA.
Proc. NatL Acad Sci. USA 81 (1984)
:::f ..,
Dow
nloa
ded
by g
uest
on
Janu
ary
6, 2
020

Proc. NatL Acad. Sci. USA 81 (1984) 5407
mRNA sequence was increased. For the two poorly translat-ed mRNAs, the computer generated a greater number ofsuch base-paired molecules compared to the efficientlytranslated mRNAs. Thus, while such a computer analysis isrelatively consistent with our expression data, we have notdemonstrated that these structures indeed exist in vivo.Seeburg et al. (35) have achieved high-level Met-bGH
expression by changing the first 24 bGH codons to a se-quence that is similar to the 5' end of a synthetic humangrowth hormone gene sequence that expressed well in E.coli. Since our results show that the bGH gene with naturalcodons can be expressed at a high level (in RV308/pCZ114),we infer that the native bGH codons are not a barrier to effi-cient translation but that by changing the bGH codons asdescribed by Seeburg et al. (35), the formation of secondarystructure(s) inhibitory to translation may be blocked, thusallowing for high-level Met-bGH expression.Our data show that poor translation of the native bGH
transcripts is also improved by the addition of extra codons.However, the presence of these codons results in the synthe-sis of altered proteins. Therefore, we have used an alterna-tive approach, the two-cistron construction, which does notsuffer from these limitations. The sequences that allow forefficient translation initiation are placed into a separate cis-tron in front of the weakly expressed bGH genes. In thisrespect, the two-cistron construction may be functionallyanalogous to the translation of single-stranded bacteriophagemRNAs, for example MS2, where the expression of the lysiscistron depends on the translation of the preceding coat pro-tein cistron (32). The ribosomes initiate at the first cistronand disrupt the inhibitory secondary structures of the secondcistron mRNA as they translate the first cistron. Because thetwo-cistron construction gives maximal flexibility in the se-quence of the first cistron, we believe that such construc-tions will be of general utility for maximizing gene expres-sion in E. coli.
We thank J. Paul Burnett, Thomas Ingolia, and Richard Jaskunasfor helpful discussions; John Sharp for suggesting the sequentialanalysis of bGH mRNA secondary structure; Dennis Smith andCharles Brush for technical assistance; and Jennifer Shrote andCheryl Alexander for their outstanding effort in manuscript prepara-tion. We thank Michael Zuker for making his RNA-folding programavailable to us.
1. Rosenberg, M. & Court, D. (1979) Annu. Rev. Genet. 13, 319-353.
2. Gold, L., Pribnow, D., Schneider, T., Shinedling, S., Singer,B. S. & Stormo, G. (1981) Annu. Rev. Microbiol. 35, 365-403.
3. Shine, J. & Dalgarno, L. (1974) Proc. Natl. Acad. Sci. USA 71,1342-1346.
4. Miller, W. L., Martial, J. A. & Baxter, J. D. (1980) J. Biol.Chem. 255, 7521-7524.
5. Dayhoff, M. O., ed. (1972) Atlas of Protein Sequence andStructure (Natl. Biomed. Res. Found., Washington, DC), Vol.5.
6. Masui, Y., Coleman, J. & Inouye, M. (1983) in ExperimentalManipulation ofGene Expression, ed. Inouye, M. (Academic,New York), pp. 15-32.
7. Uhlin, B. E., Molin, S., Gustafsson, P. & Nordstrom, K.(1979) Gene 6, 91-106.
8. Maurer, R., Meyer, B. J. & Ptashne, M. (1980) J. Mol. Biol.139, 147-161.
9. Rosteck, P. R. & Hershberger, C. L. (1983) Gene 25, 29-38.10. Narang, S. A., Hsiung, H. M. & Brousseau, R. (1979) Meth-
ods Enzymol. 68, 90-109.11. Newton, C. R., Greene, A. R., Heathcliffe, G. R., Atkinson,
T. C., Holland, D., Markham, A. F. & Edge, M. D. (1983)Anal. Biochem. 129, 22-30.
12. Brown, E. L., Belagaje, R., Ryan, M. J. & Khorana, H. G.(1979) Methods Enzymol. 68, 109-151.
13. Danner, D. B. (1982) Anal. Biochem. 125, 139-142.14. Wensink, P. C., Finnegan, D. J., Donelson, J. E. & Hogness,
D. S. (1974) Cell 3, 315-325.15. Birnboim, H. C. & Doly, J. (1979) Nucleic Acids Res. 7, 1513-
1523.16. Maxam, A. M. & Gilbert, W. (1980) Methods Enzymol. 65,
499-560.17. Laemmli, U. K. (1970) Nature (London) 227, 680-685.18. Young, F. S. & Furano, A. V. (1981) Cell 24, 695-706.19. Thomas, P. S. (1983) Methods Enzymol. 110, 255-265.20. Schoner, B. & Schoner, R. G. (1981) Gene 16, 347-352.21. Yanofsky, C., Platt, T., Crawford, I. P., Nichols, B. P., Chris-
tie, G. E., Horowitz, H., VanCleemput, M. & Wu, A. M.(1981) Nucleic Acids Res. 9, 6647-6668.
22. Nakamura, K. & Inouye, M. (1979) Cell 18, 1109-1117.23. Konigsberg, W. & Godson, G. N. (1983) Proc. Natl. Acad.
Sci. USA 80, 687-691.24. Shepard, M. G., Yelverton, E. & Goeddel, D. V. (1982) DNA
1, 125-131.25. de Boer, H. A., Hui, A., Comstock, L. J., Wong, E. &
Vasser, M. (1983) DNA 2, 231-235.26. Hui, A., Hayflick, J., Dinkelspiel, K. & de Boer, H. A. (1984)
EMBO J. 3, 623-629.27. Hall, M. N., Gabay, J., Debarbouille, M. & Schwartz, M.
(1982) Nature (London) 295, 616-618.28. Schwartz, M., Roa, M. & Debarbouille, M. (1981) Proc. Natl.
Acad. Sci. USA 78, 2937-2941.29. Queen, C. & Rosenberg, M. (1981) Cell 25, 241-249.30. Das, A., Urbanowski, J., Weissbach, H., Nestor, J. & Yan-
ofsky, C. (1983) Proc. Natl. Acad. Sci. USA 80, 2879-2883.31. Iserentant, D. & Fiers, W. (1980) Gene 9, 1-12.32. Kastelein, R. A., Berkhout, B. & van Duin, J. (1983) Nature
(London) 305, 741-743.33. Gheysen, D., Iserentant, D., Derom, C. & Fiers, W. (1982)
Gene 17, 55-63.34. Zuker, M. & Stiegler, P. (1981) Nucleic Acids Res. 9, 133-148.35. Seeburg, P. H., Sias, S., Adelman, J., de Boer, H., Hayflick,
J., Jhurani, P., Goeddel, D. V. & Heynecker, H. L. (1983)DNA 2, 37-45.
Biochemistry: Schoner et aL
Dow
nloa
ded
by g
uest
on
Janu
ary
6, 2
020


















