
RNA structure prediction. RNA functions RNA functions as –mRNA –rRNA –tRNA –Nuclear export...
45
RNA structure prediction
-
date post
19-Dec-2015 -
Category
Documents
-
view
217 -
download
0
Transcript of RNA structure prediction. RNA functions RNA functions as –mRNA –rRNA –tRNA –Nuclear export...

RNA structure prediction

RNA functions
• RNA functions as– mRNA– rRNA– tRNA– Nuclear export– Spliceosome– Regulatory molecules (RNAi)– Enzymes– Virus– Retrotransposons– Medicine
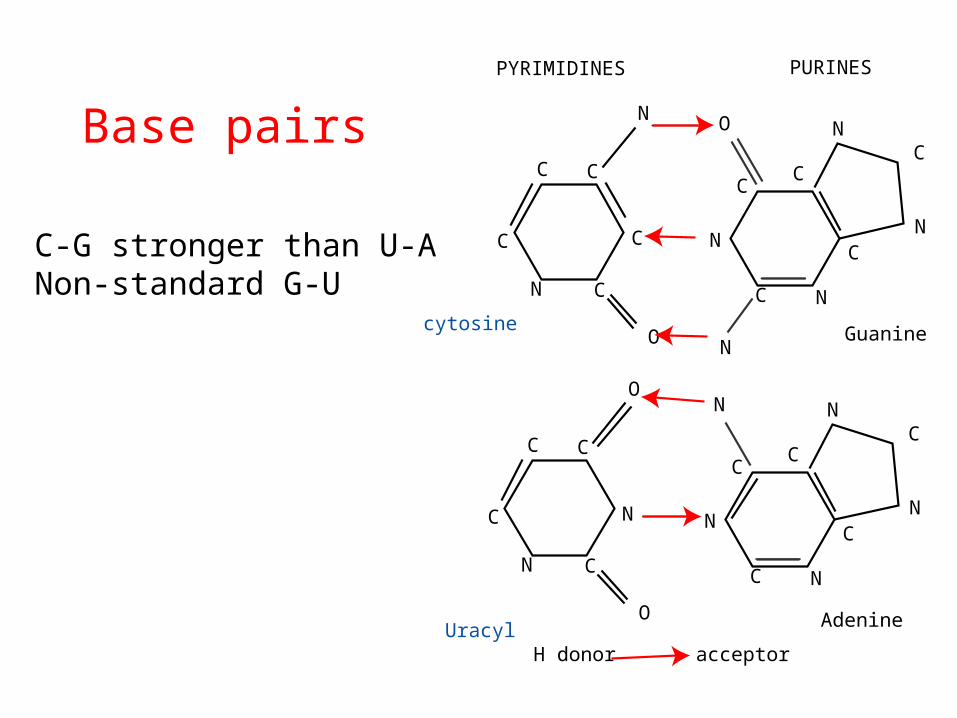
Base pairs
• C-G stronger than U-A• Non-standard G-U
CC
C
N
N C
O
C
CC
C
O
N C
O
N
cytosine
Uracyl
N
CC
C
NC
NC
N
N
O
N
CC
C
NC
NC
N
N
Adenine
Guanine
PYRIMIDINES PURINES
H donor acceptor

• Base-pairs are usually coplanar
• are almost always stacked
• stems – continuous stacks
• 3D structure of a stack is a helix
hairpin
Stacking

Predictable structures

Hard-to-predict structures
• Pseudoknots, kissing hairpins, hairpin-bulge

Secondary structure notations

Structure representation

Tertiary structure


RNAi


Known RNA Structureshttp://www.rnabase.org/metaanalysis/ httpp://www.sanger.ac.uk/Software/rfam http://www.scor.lbl,gov
Rfam – database of RNA alignments and secondary structure models
Scor - database of RNA experimentally solved structures


Main approaches to RNA secondary structure prediction
• Energy minimization – dynamic programming approach– does not require prior sequence alignment– require estimation of energy terms
contributing to secondary structure
• Comparative sequence analysis– Using sequence alignment to find conserved
residues and covariant base pairs.– most trusted
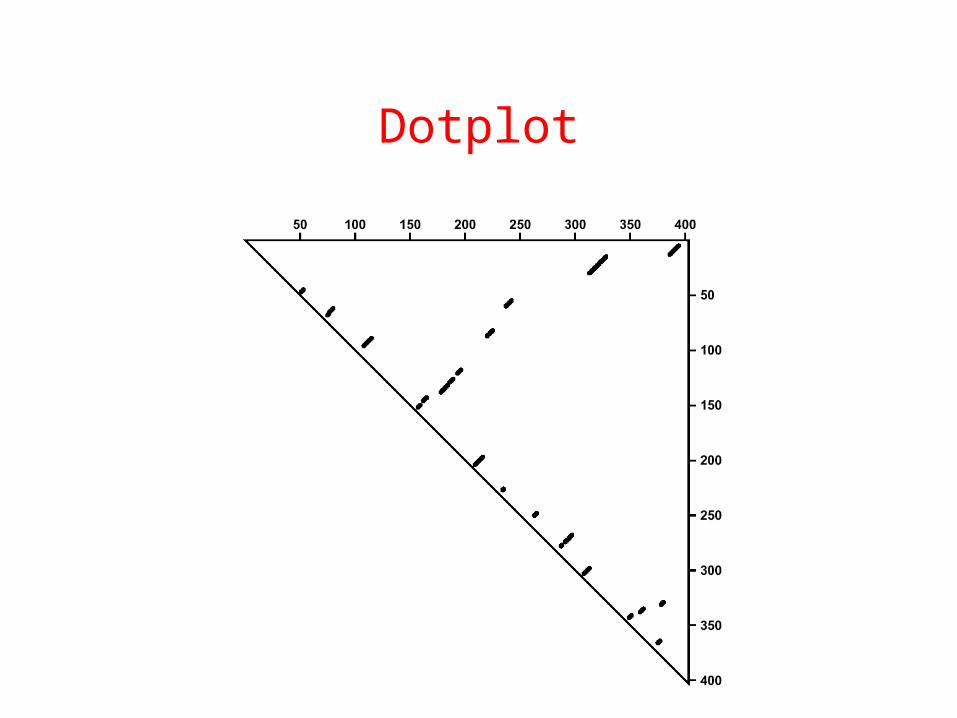
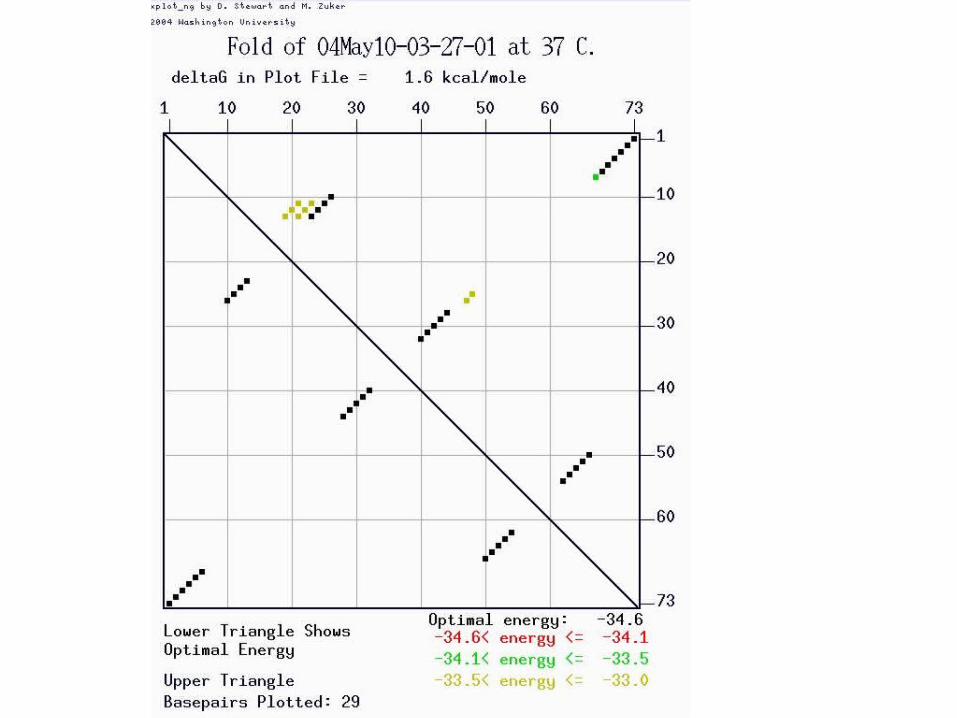
Dotplot

Think!
• Make a dotplot of an RNA molecule– Sequence : GGGAAAUCC
• What is the secondary structure?

Dynamic programming approach
• Nussinov algorithm
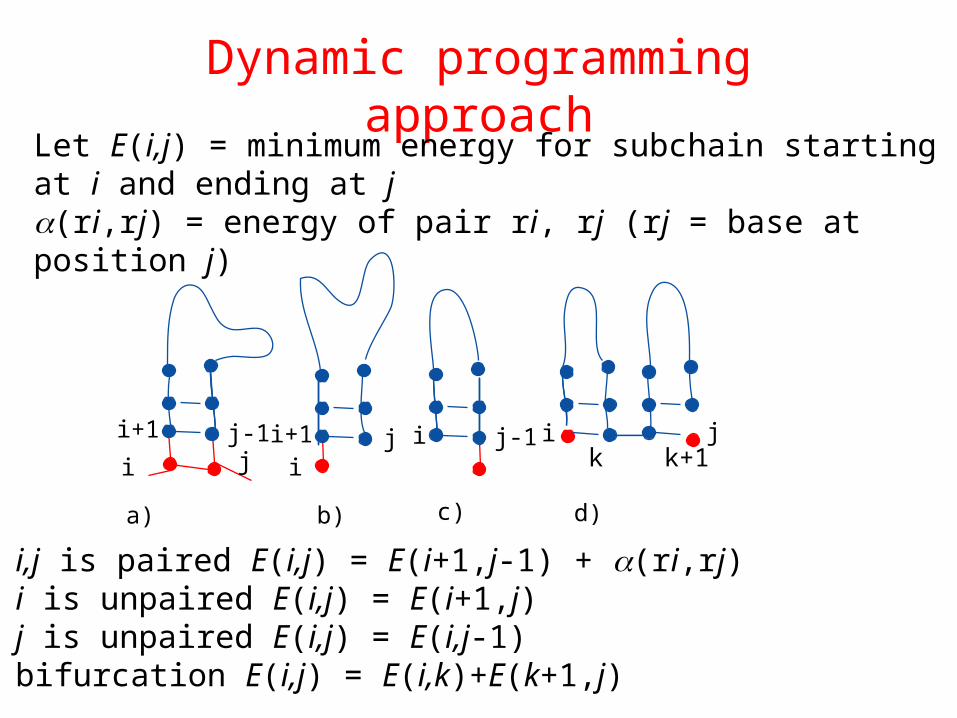
Dynamic programming approach
a) i,j is paired E(i,j) = E(i+1,j-1) + (ri,rj)b) i is unpaired E(i,j) = E(i+1,j) c) j is unpaired E(i,j) = E(i,j-1)d) bifurcation E(i,j) = E(i,k)+E(k+1,j)
i+1 j-1 i+1 j ji j-1i j i
ik k+1
a) b) c) d)
Let E(i,j) = minimum energy for subchain starting at i and ending at j(ri,rj) = energy of pair ri, rj (rj = base at position j)

RNA secondary structure algorithm
• Given: RNA sequence x1,x2,x3,x4,x5,x6,…,xL
• Initialization: E(i, i-1) = 0 for i = 2 to LE(i, i) = 0 for i = 1 to L
Recursion: for n = 2 to L # iteration over length
E(i,j) = min { E(i+1, j), E(i, j-1), E(i+1, j-1)+ (ri,rj) ,
min i<k<j {E(i,k)+E(k+1, j)} }
• Cost: O(n3)

ExampleLet (ri,rj) = -1 if ri,rj form a base pair and 0 otherwise Input : GGAAAUCC
G G A A A U C C
G 0
G 0 0
A 0 0
A 0 0
A 0 0
U 0 0
C 0 0
C 0 0
E(i,j) = lowestenergy conformation for subchain from i to j
i
j
Here we should have min energy for AAAUC

Example-continued
G G A A A U C C
G 0 0
G 0 0 0
A 0 0 0
A 0 0 0
A 0 0 -1
U 0 0 0
C 0 0 0
C 0 0
GGA (i=2, j=3)min { 0,
0,0+ (GA)
} = 0
AAU (i=5, j=6)min { 0,
0,0+ (AU)
} = -1
-1
0
i
j

Recovering the structure from the DP table
Main difference to sequence alignment – we are tracing back a tree-like structure not a single optimal path (bifurcation introduces branch points).
Method 1: Leave pointers as you compute the table: for each element of the table store (at most two) pointers to the subsequences used in the solution.
Method 2: Recover history based on numerical values in the table.Stacking – check value along diagonalBifurcation - find k such that E(i,k)+E(k+1,j) =
E(i,j)




• Base-pairs are usually coplanar
• are almost always stacked
• stems – continuous stacks
• 3D structure of a stack is a helix
hairpin
Stacking

More realistic energy function

Stacking energies


Covariance method
In a correct multiple alignment RNAs, conserved base pairs are often revealed by the presence of frequent correlated compensatory mutations.
Two boxed positions are covarying to maintain Watson-Crick complementary. This covariation implies a base pair which may then be extended in both directions.
GCCUUCGGGCGACUUCGGUCGGCUUCGGCC
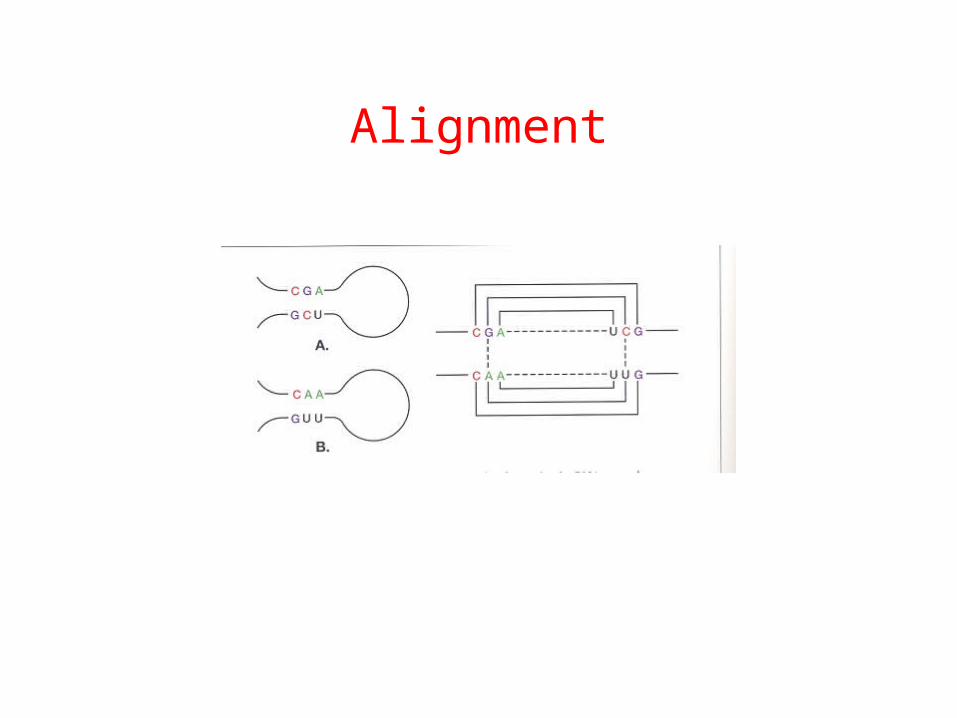
Alignment



Measure of pairwise sequence covariation
Mutual information Mij between two aligned columns i, j
Mij = i,j fxixj log2 (fxixj/fxi fxj)where fxixj frequency of the pair (observed)
fxi frequency of nucleotide xi at position i
Observations: 0 <= Mij <=2
i,j uncorrelated Mij = 0

MI: examples
A
A
C
G
U
U
G
C
fAi = .5fCi = .25fGi = .25fUj = .5fCj = .25fGj = .25
fAU = .5fCG = .25fGC = .25
Mij = xixj fxixj log2 (fxixj/fxi fxj) =
.5 log2 (.5/(.5*.5))+2*.25 log2 (.25/(.25*.25))=
.5 *1 +.5*2 = 1.5A
A
A
A
U
U
U
UMij = 1 log 1 = 0
U
A
C
G
A
U
G
C
Mij = 4*.25 log 4 = 2
i j


Other methods
• HMMs• Stochastic context free grammars
• Allow for modeling complex structures.
• Allow incorporation of additional info:– Phylogenetic distances– Biochemical properties

sno-RNA HMM

Stochastic Grammars
S -> aSa -> abSba -> abaaba
i. Start with S. Production rules:S --> (0.3)aT (0.7)bS T --> (0.2)aS (0.4)bT (0.2)
S -> aT -> aaS –> aabS -> aabaT -> aaba
ii. S--> (0.3)aSa (0.5)bSb (0.1)aa (0.1)bb
*0.3
*0.3 *0.2 *0.7 *0.3 *0.2
*0.5 *0.1
Derivation:


Conclusion
• RNA secondary structure prediction– Single sequence:
• Dot-plot• Nussinov dynamic programming• Energy function
– Covariance analysis• Mutual information• Hidden Markov Models • SCFGs


















